
Submitted:
20 January 2026
Posted:
21 January 2026
You are already at the latest version
Abstract
Background: Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune demyelinating disease with important disability accumulation. Early-onset NMOSD, defined as disease onset before age 50, exhibits distinct clinical characteristics compared to late-onset disease. We present a case series of patients with first symptom onset before age 30.
Methods: Retrospective review of 10 patients diagnosed with NMOSD at our center in San Luis Potosí, Mexico, with disease onset before age 30. Clinical presentation, imaging findings, AQP4 antibody status, treatment response, and disability outcomes were analyzed.
Results: Mean age at onset was 18.6 years (range 6-30). Area postrema syndrome was the most common presentation (40%), followed by acute myelitis and optic neuritis (30% each). All tested patients were AQP4-positive. Mean EDSS at follow-up was 6.6, indicating severe disability. Most patients received rituximab with variable response rates.
Conclusions: Our cohort showed higher disability than reported in other early-onset series, emphasizing the need for prompt diagnosis and aggressive treatment in this population.
Keywords:
neuromyelitis optica
; demyelinating disease
; area postrema syndrome
; optic neuritis
1. Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune inflammatory disorder of the central nervous system characterized by severe attacks of optic neuritis, longitudinally extensive transverse myelitis, and area postrema syndrome.[1,2,3] The disease affects predominantly female gender, with a female-to-male ratio reaching 23:1 during fertile years, and has an estimated global incidence of 0.029-0.880 per 100,000 population.[4,5]
While NMOSD typically presents between ages 30 and 40, early-onset disease (before age 50) accounts for a significant proportion of cases and demonstrates distinct clinical features.[6,7] Early-onset NMOSD is associated with a higher risk of severe visual impairment, more frequent non-opticospinal presentations, and distinct laboratory and imaging features compared to late-onset disease.[8,9,10]
The pathophysiology involves anti-aquaporin-4 (AQP4) antibodies targeting water channels predominantly expressed on astrocyte foot processes, leading to complement-mediated astrocyte destruction, with a secondary oligodendrocyte injury, and neuronal damage.[11,12] This cascade results in severe demyelination and necrosis, particularly affecting the optic nerves and spinal cord.
Despite growing recognition of early-onset NMOSD, data from Latin American populations remain limited. We present a case series of 10 patients from San Luis Potosí, Mexico, with NMOSD onset before age 30, highlighting their clinical characteristics and outcomes.
2. Methods
We conducted a retrospective review of patient files from the Neurology Service in a Hospital in San Luis Potosí, Mexico, between 2015 and 2024. The inclusion criteria were: (1) diagnosis of NMOSD according to the 2015 International Panel criteria, (2) first symptom onset before age 30, and (3) at least 6 months of follow-up.
Data collected included demographic information, age at onset, principal presenting symptoms, clinical course, MRI findings, AQP4 antibody status (assessed by cell-based assay), prescribed treatment, and disability outcomes measured using the Expanded Disability Status Scale (EDSS). Longitudinally extensive myelitis was defined as spinal cord lesions with a long extension over three or more vertebral segments (periependymal lesions surrounding the ventricles or involving corticospinal tracts).
Statistical analysis was performed using descriptive statistics. Mean values and ranges were calculated for continuous variables. The institutional review board approved the study (Registration Number 82-24).
3. Results
3.1. Patient Demographics and Clinical Characteristics
Ten patients (9 females, one male) met the inclusion criteria. The mean age at disease onset was 18.6 years (range 6-30 years), with a mean current age of 32.1 years. The female-to-male ratio was 9:1. Mean disease duration was 13.5 years. (Table 1).
3.2. Clinical Presentations
The most common initial presentation was area postrema syndrome (4 patients, 40%), characterized by intractable vomiting and hiccups lasting 3-10 days. Three patients (30%) presented with acute myelitis manifesting as paraparesis or quadriparesis with sensory levels ranging from T7 to T12. Three patients (30%) had optic neuritis as the initial symptom, with bilateral involvement in 2 cases.
Multiple clinical syndromes were observed in 3 patients (30%) at disease onset. The mean interval between initial presentation and diagnosis was 3.2 years (range 0-10 years). (Table 1)
3.3. Laboratory and Imaging Findings
All patients tested for AQP4 antibodies (10/10) were positive by cell-based assay. Titers ranged from 1:100 to 1:10,000. Oligoclonal bands were tested in 2 patients and were negative in both.
MRI findings included longitudinally extensive myelitis in 7 patients (70%), with lesions ranging from 3 to 9 vertebral segments. Cervical cord involvement was present in 5 patients (50%). Brain lesions were identified in 4 patients (40%), including periependymal lesions around the fourth ventricle in 3 cases and area postrema hyperintensity in 4 cases. Optic nerve atrophy was documented in 4 patients with a history of optic neuritis.
3.4. Treatment and Outcomes
Acute treatment consisted of intravenous methylprednisolone (1g daily for 5 days) in all patients, with partial response in 7 cases (70%). Three patients required plasma exchange due to insufficient steroid response, with 2 showing improvement.
For maintenance therapy, nine patients received rituximab (1g every 6 months), with six patients (67%) remaining relapse-free. One patient was switched to tocilizumab due to ongoing relapses on rituximab. The mean EDSS at last follow-up was 6.6 (range 3.5-9.0), indicating severe disability requiring assistance for ambulation in most cases.
3.5. Case Description
3.5.1. Case 1
A 26-year-old female presented with paresthesia in the four limbs. She referred two episodes of sudden vision loss in both eyes. The first began 10 years ago, characterized by a visual acuity of 20/200 in the left eye, discromatopsia, 5 days later, involvement of the right eye, and she was treated with intravenous methylprednisolone with partial recovery. Five years later, a new episode was presented with sudden blindness. Magnetic resonance imaging was performed, during which multiple cortical lesions were found, bilateral optic nerve atrophy (Figure 1). In the spinal cord, longitudinally extensive myelitis was found at C3-C6. The Aqp 4 was positive.
3.5.2. Case 2
A 19-year-old female presented with multiple episodes of vomiting of alimentary content, occurring more than 10 times per day, unrelated to intestinal infection and unresponsive to treatment. Two weeks later, she developed weakness in the right lower limb that prevented ambulation, along with paresthesia. Five years earlier, she had experienced a decrease in monocular visual acuity to less than 20/200. MRI revealed longitudinally extensive myelitis with no brain lesions. AQP4 antibodies were positive. Treatment with rituximab was initiated and was well tolerated; no new relapses occurred (EDSS 4).
3.5.3. Case 3
A 24-year-old pregnant female at 19.6 weeks of gestation presented with weakness in the left lower limb, leading to a fall from standing height. Five days later, she developed weakness in the right lower limb, progressing to an inability to walk and becoming bedridden. On examination, she showed 0/5 strength in both lower limbs and a sensory level at T12. MRI revealed longitudinally extensive myelitis from C6 to T5 with a central pattern. AQP4 antibodies were positive. The patient was started on intravenous methylprednisolone pulses without clinical improvement, so plasma exchange was initiated for five cycles, resulting in recovery of strength to 3/5. Subsequently, she was started on rituximab 1 g IV.
3.5.4. Case 4
A 30-year-old female began her illness at age six with atypical optic neuritis characterized by vision loss in the left eye, followed by decreased visual acuity in the right eye (20/100), for which she did not receive treatment. At age 22, she developed lower limb weakness that rendered her bedridden, accompanied by paresthesias; during this episode, she received steroid pulses for five days. AQP4 antibodies were positive, and MRI showed optic nerve atrophy and longitudinally extensive myelitis (Figure 2). She is currently receiving rituximab 1 g IV every six months, with no new relapses (EDSS 6).
3.5.5. Case 5
A 36-year-old female, living in a group home for the past 15 years, with a history of depressive disorder, right lower limb venous thrombosis, and recurrent urinary tract infections, presented to the neurology clinic with paresthesia in the upper limbs and weakness. On detailed questioning, she reported bilateral visual acuity loss (<20/200) at age 10, with partial recovery. At age 12, she experienced paraparesis that led to wheelchair dependence, and multiple sclerosis was diagnosed during that episode. At age 20, she had another episode of optic neuritis, resulting in blindness. MRI revealed optic nerve and spinal cord atrophy. AQP4 antibody was positive (1:10,000). Treatment with rituximab 1 g IV every six months was initiated.
3.5.6. Case 6
A 13-year-old female presented with recurrent episodes of hiccups and vomiting lasting approximately three days. Four months later, she developed decreased visual acuity in the left eye (<20/200) and milder involvement of the right eye (20/70), accompanied by ocular pain. She was initially diagnosed with multiple sclerosis; oligoclonal bands were negative. She experienced a new episode characterized by bilateral visual acuity loss, for which she did not receive treatment. Rituximab was started thereafter, with no subsequent relapses. AQP4 antibody testing later returned positive.
3.5.7. Case 7
A 32-year-old female began her illness with decreased visual acuity in the right eye (counting fingers), and fundoscopy revealed blurred nasal disc margins. Lower limb strength was 0/5 with hyperreflexia, a sensory level at T9, and acute urinary retention. She reported a similar episode at age 25, during the second trimester of her first pregnancy, characterized by paraparesis and loss of sphincter control, treated with steroid pulses. In 2017, she experienced another episode of lower limb weakness that prevented ambulation, for which interferon therapy was initiated. During the current evaluation, she received methylprednisolone 1 g daily for 5 days, achieving partial recovery. MRI showed longitudinally extensive myelitis from T7 to T12. Treatment with rituximab 1 g every 24 hours was initiated.
3.5.8. Case 8
A 35-year-old female began her illness at age 8 with decreased visual acuity in the right eye (20/200), followed days later by left eye involvement. She was treated with methylprednisolone, achieving full recovery. One year later, she developed quadriparesis with partial recovery, resulting in wheelchair dependence. Methotrexate therapy was started; however, she continued to experience recurrent episodes of bilateral optic neuritis and myelitis. Her last relapse occurred in 2022, leading to right eye blindness and only light perception in the left eye. MRI revealed optic nerve and thoracolumbar spinal cord atrophy. Rituximab was initiated in 2020, but due to new relapses, therapy was switched to tocilizumab, after which no further relapses occurred.
3.5.9. Case 9
A 26-year-old female with no previous medical history began her illness four months earlier with persistent nausea and vomiting occurring more than six times per day. One week later, she developed vertical diplopia on binocular gaze and was evaluated by general medicine and gastroenterology, with partial symptom improvement lasting one week. She subsequently presented with persistent hiccups, paresthesias in the arms and legs, and cerebellar ataxia. MRI revealed hyperintensity in the area postrema (Figure 3). AQP4 antibodies were positive. One month later, she developed painful facial and leg spasms, followed by quadriparesis two weeks later. Treatment with methylprednisolone 1 g daily for 5 days resulted in partial recovery. Rituximab 1 g IV was then initiated.
3.5.10. Case 10
A 39-year-old female presented to the emergency department with lower limb weakness, low back pain, and dyspnea. Her family reported an episode four months earlier of intractable vomiting and hiccups, as well as a similar episode ten years prior. On examination, oxygen saturation was 61% with the use of accessory respiratory muscles. Neurological evaluation revealed right-sided cranial nerve III and VI palsies. Multiplex PCR in CSF was negative. During hospitalization, she developed upper limb weakness (strength 2/5). MRI showed longitudinally extensive myelitis from the ventral medulla to T1. AQP4 antibodies were positive. Plasma exchange was initiated, and during her hospital course, she was diagnosed with severe ARDS secondary to SARS-CoV-2 infection.
4. Discussion
This case series highlights the severe disability burden of early-onset NMOSD in a Mexican cohort. Our mean EDSS of 6.5 is substantially higher than reported in other early-onset series, where mean EDSS typically ranges from 2.0-4.0.[13,14,15] This discrepancy may reflect diagnostic delays, limited access to high-efficacy treatments, or population-specific factors.
The predominance of area postrema syndrome (40%) as the initial presentation differs from that in other cohorts, where optic neuritis or myelitis typically predominate.[16,17] Area postrema syndrome is often misdiagnosed as gastrointestinal illness, potentially contributing to diagnostic delays. The mean 3.2-year delay from symptom onset to diagnosis in our cohort emphasizes the need for increased awareness of NMOSD presentations.
Consistent with the literature, early-onset NMOSD in our cohort showed marked female predominance (9:1) and universal AQP4 seropositivity.[18] The high frequency of cervical cord involvement (50%) and periependymal brain lesions (30%) aligns with reported imaging patterns in early-onset disease.[19,20]
Treatment outcomes were suboptimal, with only 67% of rituximab-treated patients achieving relapse freedom. This may reflect treatment initiation after significant disability accumulation, emphasizing the importance of early aggressive immunosuppression. The high disability despite treatment underscores the unmet need for more effective therapies in resource-limited settings.
Study limitations in our report include a small sample size, a retrospective design, and a single-center design. Selection bias toward more severe cases presenting to our tertiary center may explain the high disability levels. Prospective multicenter studies are needed to better characterize early-onset NMOSD in Latin American populations.
In conclusion, early-onset NMOSD in this Mexican cohort is associated with severe disability, highlighting the critical need for early recognition and treatment. Area postrema syndrome should be considered in young patients with unexplained vomiting and hiccups. Further research is needed to identify factors contributing to poor outcomes in this population.
Author Contributions
DMR coordinated patient enrollement, cases analysis and writing of manuscript draft; MFCZ secured ethical approval and assisted with data validation; AMM, clinical supervision and imaging review; IRL, conceived the study and supervised data collection; MEJC, analysis, discussion and writing.
Funding
This work was completed without external funding, using institutional resources only for routine clinical care and data collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wingerchuk DM, Lucchinetti CF. Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2022;387(7):631-639. [CrossRef] [PubMed]
- Jarius S, Paul F, Weinshenker BG, Levy M, Kim HJ, Wildemann B. Neuromyelitis optica. Nat Rev Dis Primers. 2020;6(1):85. [CrossRef] [PubMed]
- Huda S, Whittam D, Bhojak M, et al. Neuromyelitis Optica Spectrum Disorders. Clin Med (Lond). 2019;19(2):169-176. [CrossRef] [PubMed]
- Hor JY, Asgari N, Nakashima I, et al. Epidemiology of Neuromyelitis Optica Spectrum Disorder and Its Prevalence and Incidence Worldwide. Front Neurol. 2020;11:501. [CrossRef] [PubMed]
- Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler. 2015;21(7):845-853. [CrossRef] [PubMed]
- Collongues N, Marignier R, Zéphir H, et al. Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology. 2010;75(12):1084-1088. [CrossRef] [PubMed]
- Fragoso YD, Sousa NAC, Saad T, et al. Clinical characteristics of patients with neuromyelitis optica spectrum disorders with early onset. J Child Neurol. 2019;34(9):487–490. [CrossRef] [PubMed]
- Macaron G, Khoury J, Bena J, et al. Early age of onset predicts severity of visual impairment in patients with neuromyelitis optica spectrum disorder. Mult Scler. 2021;27(11):1749-1759. [CrossRef] [PubMed]
- Zhang LJ, Yang LN, Li T, et al. Distinctive characteristics of early-onset and late-onset neuromyelitis optica spectrum disorders. Int J Neurosci. 2017;127(4):334-338. [CrossRef] [PubMed]
- Hu Y, Sun Q, Yi F, et al. Age of onset correlates with clinical characteristics and prognostic outcomes in neuromyelitis optica spectrum disorder. Front Immunol. 2022;13:1056944. [CrossRef] [PubMed]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–477. [CrossRef] [PubMed]
- Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11(6):535–544. [CrossRef] [PubMed]
- Chitnis T, Ness J, Krupp L, et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology. 2016;86(3):245-252. [CrossRef] [PubMed]
- Fragoso YD, Ferreira ML, Oliveira EM, et al. Neuromyelitis optica with onset in childhood and adolescence. Pediatr Neurol. 2014;50(1):66-68. [CrossRef] [PubMed]
- Tarhan B, Rempe T, Rahman S, et al. A comparison of pediatric- and adult-onset aquaporin-4 immunoglobulin G-positive neuromyelitis optica spectrum disorder. J Child Neurol. 2022;37(8-9):727-737. [CrossRef] [PubMed]
- Li R, Lu D, Li H, et al. Neuromyelitis optica spectrum disorders with non-opticospinal manifestations as initial symptoms: a long-term observational study. BMC Neurol. 2021;21(1):35. [CrossRef] [PubMed]
- Li Z, Yang M, Pan Y, Fang Q. Neuromyelitis optica spectrum disorder with acute brainstem manifestations as initial symptoms. Heliyon. 2024;10(12):e32539. [CrossRef] [PubMed]
- Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation. 2012;9:14. [CrossRef] [PubMed]
- Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84(11):1165-1173. [CrossRef] [PubMed]
- Bennett JL, Costello F, Chen JJ, et al. Optic neuritis and autoimmune optic neuropathies: advances in diagnosis and treatment. Lancet Neurol. 2023;22(1):89-100. [CrossRef] [PubMed]
Figure 1.
Axial Brain MRI FLAIR/T2 sequence shows optic chiasm hyperintensity due to acute inflammation, accompanied by bilateral optic nerve atrophy representing chronic damage from previous optic neuritis in NMOSD.
Figure 1.
Axial Brain MRI FLAIR/T2 sequence shows optic chiasm hyperintensity due to acute inflammation, accompanied by bilateral optic nerve atrophy representing chronic damage from previous optic neuritis in NMOSD.
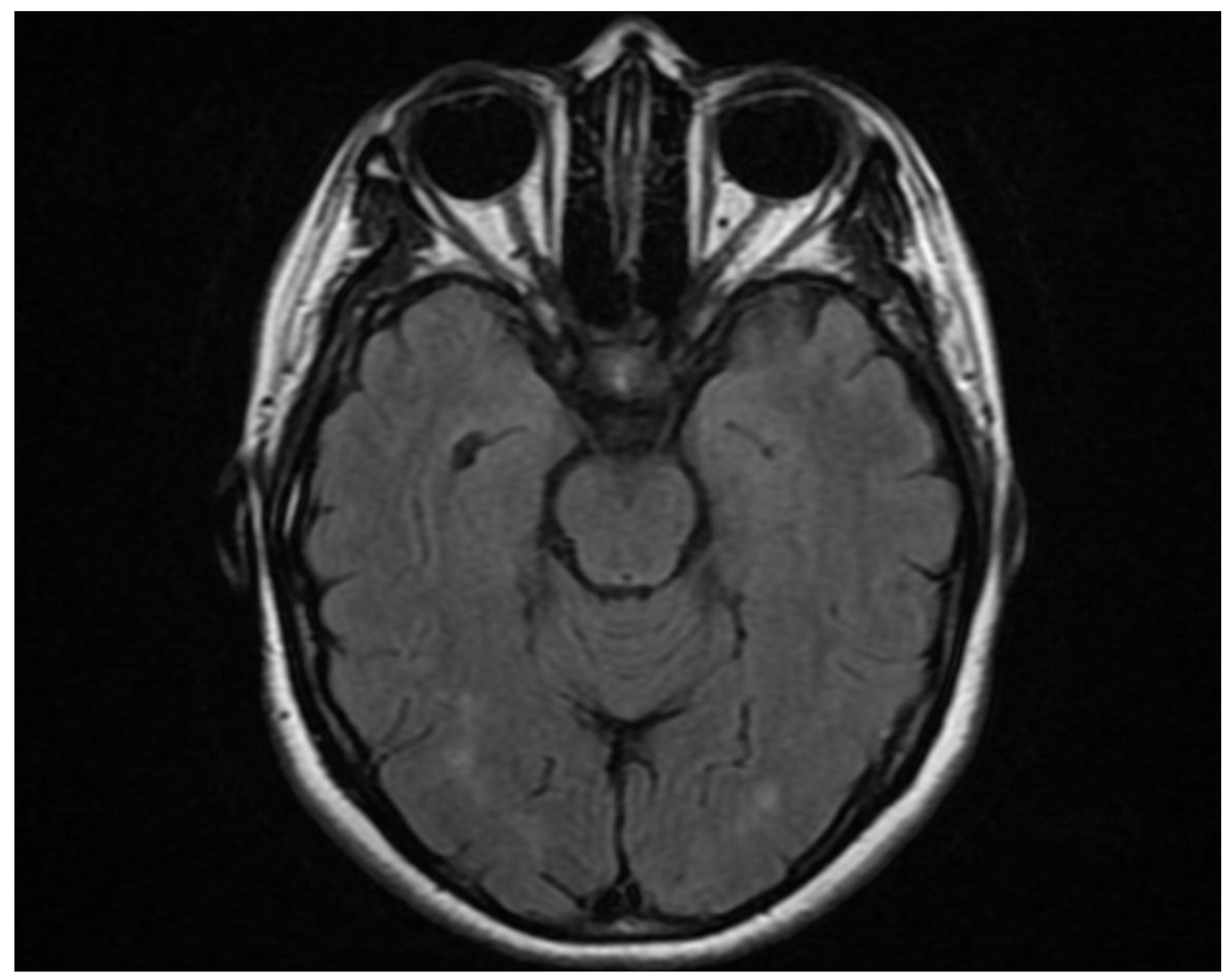
Figure 2.
Sagital cervical and proximal thoracic MRI T2. The image demontrastes a longitudinal lesion involving C2-C5 segments.
Figure 2.
Sagital cervical and proximal thoracic MRI T2. The image demontrastes a longitudinal lesion involving C2-C5 segments.
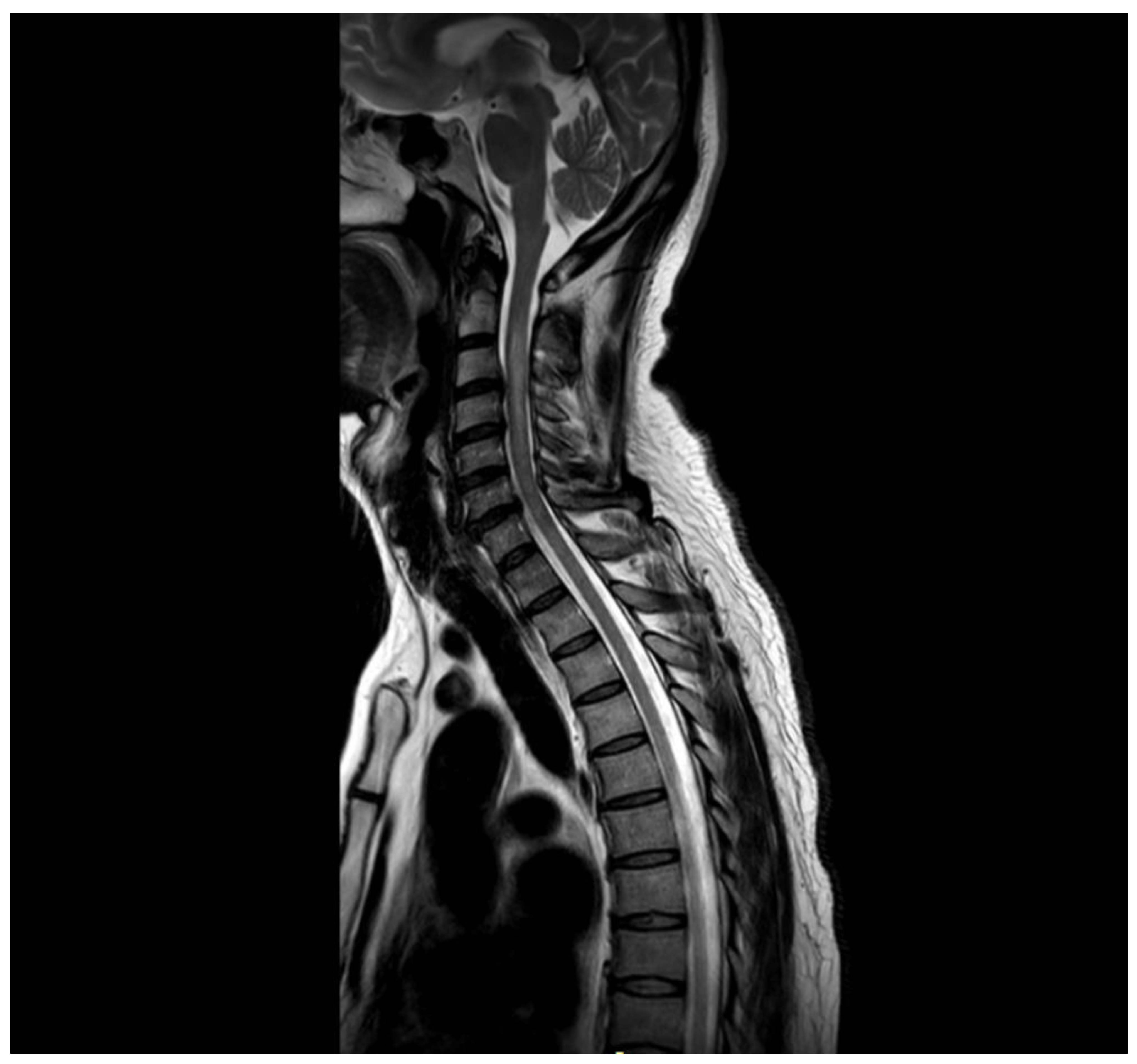
Figure 3.
Sagittal Brain MRI, FLAIR/T2 sequence. It revealed a focal hyperintense lesion involving the area postrema.
Figure 3.
Sagittal Brain MRI, FLAIR/T2 sequence. It revealed a focal hyperintense lesion involving the area postrema.
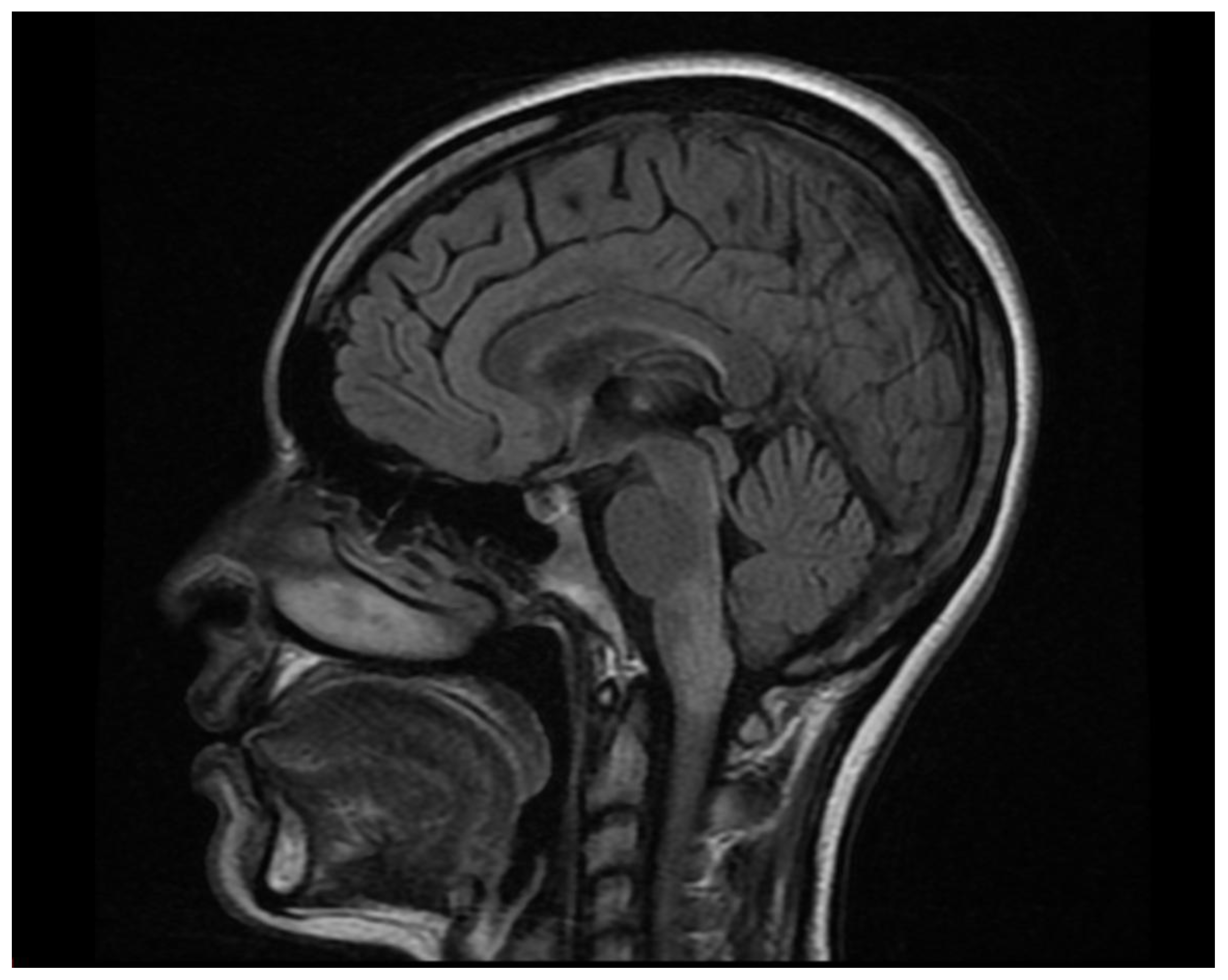

Table 1.
Demographic and Clinical Characteristics.
| Case | Age at Onset | Time since diagnosis | Initial Presentation | Treatment | Current EDSS |
| 1 | 16 | 10 | Bilateral ON | Rituximab | 7.5 |
| 2 | 14 | 5 | APS + ON | Rituximab | 4.0 |
| 3 | 24 | 1 | LETM | Rituximab | 6.5 |
| 4 | 6 | 24 | ON | Rituximab | 6.0 |
| 5 | 10 | 26 | Bilateral ON | Rituximab | 8.5 |
| 6 | 13 | 6 | APS | Rituximab | 3.5 |
| 7 | 25 | 7 | LETM | Rituximab | 7.0 |
| 8 | 8 | 27 | ON | Tocilizumab | 9.0 |
| 9 | 26 | 1 | APS | Rituximab | 6.5 |
| 10 | 29 | 10 | APS + LETM | Rituximab | 8.0 |
ON: optic neuritis; LETM: longitudinally extensive transverse myelitis; APS: area postrema syndrome; EDSS: Expanded Disability Status Scale.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



