Submitted:
20 January 2026
Posted:
21 January 2026
You are already at the latest version
Abstract
Background: Pharmaceutical innovation plays a vital role in advancing global health. This study evaluates the landscape of pharmaceutical innovation in the European Union (EU) over the 2011-2020 decade. Methods: A retrospective analysis was performed on new medicinal products containing new active substances (NAS) authorized between 2011 and 2020 through the centralized procedure. Products were categorized in first-in-class, advance-in-class, and addition-to-class. Trends in therapeutic areas (ATC codes), orphan designation, technology platforms etc), administration routes, and dosage forms were analyzed. Results: Across the decade, 357 new medicinal products received authorization. Of these, 56% were designated as first in class and 28% as advance in class, indicating that 84% of new products contributed substantive therapeutic innovation. Small molecules remained the predominant technology (63.5%), yet the decade also wit-nessed a pronounced expansion in monoclonal antibodies and the introduction of cell and gene therapies (Advanced Therapy Medicinal Products, ATMPs). The increased use of subcutaneous delivery systems, particularly for monoclonal antibodies, suggests a broad-er move toward patient centered administration routes. Conclusion: Between 2011 and 2020, pharmaceutical innovation in the EU was distinguished by a high rate of break-through innovation and a transition to more sophisticated biologic therapies. EU could boost innovation further by refining expedited approval pathways (e.g., PRIME).
Keywords:
pharmaceutical innovation
; innovation metrics
; EMA
; first-in-class
; advance-in-class
; addition-to-class
; marketing authorizations
1. Introduction
Pharmaceutical innovation plays a key role in enhancing population health outcomes by extending life expectancy, improving quality of life, expanding the range of available treatments, reducing costs, and elevating the overall efficiency of healthcare systems [1]. Pharmaceutical innovation plays a partial yet meaningful role in shaping population health over the long term. In the health of pharmaceutical companies is more directly tied to their ability to bring new drugs to market [2].
Multiple definitions of innovation have been advanced across time, with different focal points, extents, and perspectives. For instance, in a somewhat classical definition, “innovation consists of the generation of a new idea and its implementation into a new product, process or service, leading to the dynamic growth of the national economy and the increase of employment as well as to a creation of pure profit for the innovative business enterprise.”[3] Such a definition is useful from an economic perspective, but not optimal from a scientific or medical standpoint. For patients and healthcare professionals it is likely that development of a new product fulfilling a therapeutic need is much more relevant than causing a “dynamic growth of the national economy”, because “pharmaceutical products have no intrinsic value to patients or to society; rather, their value lies in the health outcomes they generate” [4]. It has therefore been argued that neither novelty alone, nor effectiveness on its own are sufficient to define pharmaceutical innovation, and combining the two does not necessarily result in defining it [4]. We move beyond this limited perspective, as it is broadly recognized that impactful innovation often stems from numerous small advancements. For example, Orville Wright’s initial 37-meter (120 feet) flight in a heavier-than-air machine [5] paved the way, through many incremental improvements, for routine transatlantic flights today. The journey to modern high-fidelity sound began with the Edison phonograph, a basic device whose initial recordings were dismal (with much background noise) and very short-lived; it was only through a series of gradual, cumulative enhancements that today’s sophisticated instruments were achieved. The first human kidney transplant was performed by Dr. Yurii Voronoy in Ukraine on April 3, 1933, with the patient surviving for just 2 days; similar pioneering attempts in Boston, Chicago, and Paris also ended in failure [6]. By incremental advances, though, this failed innovation made solid organ transplantation as we know it possible. Successful innovation is preferable, but innovation, even when failing, is still preferable to the status quo; and this should be equally applicable for pharmaceutical innovation. An ineffective drug won’t clear regulatory hurdles, but its innovative features could be enhanced to develop new products that achieve regulatory approval.
Back in 2005, there was a common view that the pharmaceutical sector had little motivation to pursue real innovation, as the majority of newly introduced drugs were “me-too” products rather than truly novel therapies [7]. In 2011, two U.S. researchers expressed what they considered justified worries that too few products were making it to market to fund future innovation amid steeply climbing research and development (R&D) expenses [8]. A 2009 analysis covering a 60-year span noted that while pharmaceutical R&D investment has surged dramatically, reaching $50 billion annually, the number of new medicines approved each year is no higher today than it was decades ago. In fact, only 21 new drugs received marketing approval in the United States in 2008, a figure considered by the author far below what was needed to ensure the long-term viability of the pharmaceutical industry. At that time, over 4,300 companies were involved in pharmaceutical innovation, but only 261 of them (around 6%) had successfully registered at least one new molecular entity (NME) since 1950 [9]. In this context, analyzing approved medicines from the next decade (2011 to 2020) is relevant, because it reveals whether earlier concerns about limited innovation, rising R&D costs, and stagnant approval rates have improved. This period also reflects positive advances, such as the successful completion of the Human Genome Project, which has enabled more targeted and personalized drug development and there were already expectations that this second decade will result in an incremental progress as compared with the previous decade (although it was accepted that improvements in healthcare effectiveness would be still limited and only bloom in the 2021-2030 decade) [10].
While quantifying innovation is relatively simple, measuring its quality with appropriate metrics proves much harder. To address this, industry critics have invented systems that group innovations into labeled tiers (e.g., “highly innovative,” “moderately innovative”), sometimes basing these labels on the FDA’s assessment of a drug’s value at the time of its NDA (New Drug Application). Yet this approach remains undependable and is likewise subjective and selective as informal, story-based descriptions of innovation [7]. How then can pharmaceutical innovation be measured? A variety of metrics have been proposed across time, from market size (as a proxy for future R&D spending) to the extent of clinical trials, and from the numbers of relevant journal articles or disease regimens to new drugs launched [11]. One metric often used has been the number of new molecular entities (NME, i.e., new active substances) authorized by a regulatory authority in a defined interval [12]. However, this is obviously not a sufficient metric in itself, as even when medicinal products are commercially very successfully, their degree of innovation may be very modest, well-known block-busters being, in fact, mere me-too products with little innovative features [13]. Therefore, finer-grained criteria are needed to more accurately assess innovation.
Along this line, pharmaceutical innovation has been conventionally classified in “radical” or “incremental”, where “radical innovations are generally taken to be a new molecular entity (NME)”, whereas “incremental innovations” refers to new developments that provide either a modest improvement or smaller, additional health benefits when compared to currently available treatments [1]; it would include me-too drugs and reformulations [14]. However, such a definition is rather confusing, because me-too drugs are most often NMEs, though non-NMEs may also be classified as me-too (e.g., enantiomers versus an authorized racemic or new dosage forms for an older active ingredient).
A four-tier classification has also been advanced, classifying general innovation (not specifically pharmaceutical innovation) as follows: (i) incremental innovation (minor technology adjustments and limited benefit to consumers); (ii) market breakthroughs (slight technology changes but significant consumer benefits); (iii) technology breakthroughs (pioneering technology but low consumer gain); and (iv) radical innovation (integrating a novel technology with high consumer value) [15]. While this provides a helpful overarching framework, it has not generally been adopted in the pharmaceutical field, possibly due to the distinct nature of drug development, regulatory processes, treatment effects, and patient health outcomes. For pharmaceuticals, a tripartite model has been proposed in 2013 by Lanthier et al. [12]. Although all NMEs are chemically unique, their level of innovation differs. To reflect this, the researchers categorized them as first-in-class, advance-in-class, or addition-to-class, using drug class, approval date, and priority review status as criteria. First-in-class medications constitute genuine pharmacological innovation, as each establishes a previously unused biological pathway for disease treatment. Although follow-on agents within the same drug class may eventually surpass the pioneer in performance, the original first-in-class compound is distinguished by its fundamentally novel therapeutic strategy. Advance-in-class drugs are (in the view of the cited authors) those that, while not pioneering a new mechanism, are granted priority review due to their potential to deliver major clinical improvements; thus, the main criterion of classification becomes the existence of a priority review decision granted by FDA. Addition-to-class drugs, by comparison, exhibit functional similarity to existing therapies and lack substantial enhancements in efficacy or safety [12]. A similar classification, but more focused on the molecule shape and scaffold of drug active ingredients distinguishes between pioneers, settlers and colonists [16].
The main goal of this paper is to investigate how medical progress can be measured by developing a rigorous methodological framework, applying it to European regulatory data, and describing the resulting patterns of pharmaceutical innovation between 2011 and 2020. More specifically, the paper addresses three key research questions: (1) What was the overall scale and magnitude of therapeutic innovation introduced to the EU market during 2011–2020? (2) What were the principal characteristics and distinctive features of the new medicines approved by the European Commission based on the recommendation of the European Medicines Agency (EMA) over this period? And (3) In what ways can the assessment of pharmaceutical innovation be refined to capture more nuanced dimensions?
2. Materials and Methods
A dataset on marketing authorizations (MAs) granted by EMA for medicinal products containing new active substances (new molecular entities, NMEs) between 2011 and 2020 was compiled through manual extraction and synthesis of publicly available regulatory information from the European Medicines Agency (EMA). The primary source was the Annex 9 or 10 of EMA’s Annual Reports (2011-2020) [17], which provided structured summaries of newly authorized medicinal products, including therapeutic class, innovation status, and regulatory milestones. Supplementary data, particularly for defining the innovation class for each product were retrieved from the European Public Assessment Reports (EPARs) [18], the Union Register of medicinal products [19], and relevant scientific literature searched through PubMed and Google Scholar. The year of authorization reflects the date of initial EU marketing approval. This approach of focusing on NMEs has its strengths but also its own limitations, as orphan medicinal products with old active substances were not considered (for instance cholic acid in the treatment of hereditary disorders that impair the synthesis of primary bile acids, authorized as an orphan medicinal product in 2013, was not included in the data set). Constructing such a data set involves a degree of subjectivity, as there are value judgements to be made about the qualification of certain products as having or not a new active substances, particularly in the case of vaccines and a few other biological products. In the case of vaccines, where generally the concept of ‘generics” is not applied, we only retained vaccines considered as first-in-class or at least advance-in-class (e.g., Fluenz Tetra, containing a live attenuated tetravalent influenza vaccine was included, as a trivalent Fluenz authorized by the European Comission/EMA in 2011). Fixed-dose combinations (FDCs) were included only if they contained at least one new active substance. FDCs made entirely of previously authorized active substances were excluded. In many cases, the approval of a new active substance was accompanied within the same year by the authorization of one or more fixed-dose combinations. For consistency, we considered these clusters around the same active substance as a single product, instead of classifying them as separate products. For instance, alogliptin was authorized in 2013, but in the same year two of its FDCs, one with metformin and another with pioglitazone were authorized, increasing the number of alogliptin products to three, although two are rather FDCs of the same new active ingredient); for this reason, with decided to keep only alogliptin in this case, leaving aside its two combinations. The analysis dataset was restricted to products granted a Marketing Authorization (MA). For example, although Folcepri (etarfolatide) received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP, the main committee involved in the assessment of MA dossiers), it was excluded from the study because the applicant withdrew the application before the European Commission issued a final authorization. The situation was similar for Vynfinity (vintafolide).
Cobicistat was initially authorized in May 2013 as one of the components of the four-drug fixed-dose combination Stribild. A subsequent authorization for cobicistat as a single-ingredient product (Tybost) was granted in September 2013. As cobicistat no longer qualified as a new active substance at that time, only Stribild was retained in the analysis. By contrast, tenofovir alafenamide was first approved in 2015 within the Genvoya combination (elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide) and did not receive a stand-alone authorization (Vemlidy) until 2017. Because it remained a new active substance in 2017—having not been previously authorized independently—both marketing authorizations were included, together with Descovy, another fixed-dose combination containing emtricitabine and tenofovir alafenamide that had been approved before 2017. Eylea obtained EU authorization on 21 November 2012. Consequently, when Zaltrap was authorized on 1 February 2013, its active substance was no longer new. We therefore included Zaltrap in the dataset and excluded Eylea.
Dinutuximab and dinutuximab beta raised an interesting classification challenge: the former (under the brand name Unituxin) was issued a marketing authorization in August 2015, but in 2017 its MA was withdrawn by the European Commission at the request of the marketing authorization holder (MAH), due to the company’s inability to adequately supply Unituxin globally in the near future. In 2017, under a different MAH, a new MA was granted for what was in essence an improved version of the same antibody (dinutuximab beta). Within the process, the applicant argued that dinutuximab beta is a different substance from dinutuximab, as the new antibody “has a different manufacturing process and differs significantly in properties with regard to safety and/or efficacy from the already authorised active substance” [20]. Indeed, while the two antibodies share the same core mechanism (targeting the GD2 ganglioside) and therapeutic indication (neuroblastoma), one was produced in murine SP2/o cells and the other in Chinese hamster ovary cells (CHO); also, the former was administered as short infusions (in association with GM-CSF, IL-2, and isotretinoin), whereas the latter is used in a safer manner with a continuous 10-day infusion or once-daily infusions for a duration of 5 days [21]. The two were treated therefore in this paper as two different active substances, although they have much in common and in essence one is the improved version of the other; since one replaced the other, though, one could argue that the two should be only counted once as a single new active substance.
To assess the degree of pharmaceutical innovation, each product was classified using the tripartite framework proposed by Lanthier et al. (2013) [12], which distinguishes between first-in-class (introducing a novel mechanism of action), advance-in-class (offering improved efficacy or safety within an existing class), and addition-to-class (providing similar therapeutic benefit as prior agents). While regulatory designations such as FDA Priority Review [22] and EMA’s PRIority MEdicines (PRIME) scheme [23] offer valuable signals of clinical promise and unmet medical need, we deliberately avoided using such decisions as metrics for innovation level. These designations are influenced not only by the drug’s inherent novelty or merit, but also by contextual factors such as submission timing, sponsor engagement, and internal resource allocation within regulatory agencies. As such, they may reflect strategic or administrative considerations beyond the intrinsic novelty or therapeutic advancement of the product. For instance, the EMA guidance document for PRIME applicants explicitly acknowledges that novel medicines could successfully add alternatives or incremental benefits to current treatment options, while not meeting the PRIME eligibility criteria. The key reason for this is often a timing issue: if the drug development plans are already in an advanced stage or the pivotal trials have already started, the benefit of the program’s support becomes irrelevant. Therefore, in such cases being denied PRIME status simply reflects the timing of the application and should not be necessarily taken as a critique of the products’s value; the product may still qualify for accelerated assessment [24]. Furthermore, because innovation assessments are often context-dependent and geographically specific, the significant global disparity in the utilization of expedited regulatory pathways must be considered. Currently, the EU employs these pathways less frequently than other major regions. [25]. According to a 2020 retrospective cohort analysis, 57% of drugs approved by the FDA between 2007 and 2017 received at least one expedited designation, whereas only 15% of EMA-approved drugs met similar criteria [26]. Relying on PRIME or similar designations as a primary benchmark for innovation would, therefore, lead to a disproportionate misclassification of truly innovative therapies due to this low utilization rate. As noted in the literature, Priority Review represents a preliminary regulatory judgment of a drug’s value based on limited early data, rather than a definitive label of breakthrough status [7].
Therefore, the classification of “advance-in-class” was based on the authors’ informed judgment, supported by peer-reviewed literature and comparative assessments of pharmacodynamic mechanisms, clinical efficacy and safety data, and therapeutic positioning. This approach allowed for a more consistent and mechanistically grounded evaluation of innovation across products, independent of regulatory procedural designations. Classification discrepancies among authors were resolved via discussion until consensus was reached. The entire dataset is available in Table SI, which also includes a short rationale explaining the classification of each individual product.
Because the dataset includes only products containing a new active substance (NAS), Vargatef was retained while Ofev was excluded. Although both products contain nintedanib, Vargatef received its EU marketing authorization in November 2014, making it the first NAS-containing product. When Ofev was authorized in 2015, nintedanib was no longer considered a new active substance, and therefore Ofev did not meet the inclusion criteria.
The classification of innovation degrees was based on the European Union regulatory perspective. For example, Opdivo (nivolumab) received its EU marketing authorization on 19 June 2015, preceding Keytruda (pembrolizumab), which was authorized on 17 July 2015. In contrast, Keytruda obtained its first FDA approval on 4 September 2014, whereas Opdivo was first approved by the FDA on 22 December 2014. Thus, Opdivo is considered first-in-class in the EU, while Keytruda holds that position in the United States. Instead, Yescarta (axicabtagene ciloleucel) and Kymriah (tisagenlecleucel), both CAR-T therapies authorized in related indications were both labeled as first-in-class, as both were authorized on the same day (August 23, 2018).
3. Results
3.1. Distribution by Innovation Degree
A total of 357 medicinal products containing new active substances were authorized by the European Commission via the centralized procedure between 2011 and 2020 (Figure 1). Of these, 204 were classified as first-in-class (Table 1). The remaining 160, while based on new active ingredients, did not qualify as first-in-class and were therefore considered to represent a lower level of innovation. Within this group, 57 were categorized as additions-to-class and 103 as advances-in-class (Table 2). Overall, these data indicate that roughly 56% of new drug approvals introduced groundbreaking therapeutic approaches, while the remaining 446% provided incremental advances or alternatives within existing classes. On the other hand, the same data can be interpreted as involving at least some form of advancement in about 84% of the cases, whereas they represented mere additions to existing classes in about 16% of cases.
3.2. Time Evolution
The year-by-year pattern (Figure 2) of newly authorized products indicates a general upward tendency over the decade, but with intermittent variability marked by two pronounced drops in 2016 and 2019, each followed by substantial recovery in the subsequent year. The lowest number (30) of new MAs granted was 2011, while the highest number (41) was granted in 2018 and 2020. With the exception of 2013, first-in-class products outnumbered advance-in-class products in every year of the dataset.
The 357 medicines vary widely by therapeutic class, as shown in Table 1. The most widely represented class is that of antineoplastic and immunomodulating agents, which alone cover 36.1% of total; in other words, out of every three newly authorized medicinal products, on average one was an anticancer or immunomodulating agent. At the opposite pole, only 3 new medicines were part of the systemic hormonal preparations.
The 129 antineoplastic and immunomodulating agents authorized over the last decade are distributed unevenly across the four major ATC classes. Antineoplastic agents (L01) dominate the landscape with 92 products, followed by 29 immunosuppressants (L04). In contrast, endocrine therapies (L02) and immunostimulants (L03) represent a much smaller share of the market, with only four approvals each. Among the 92 antineoplastic agents, 53 were first-in-class, 21 advance-in-class, and 18 addition-to-class.
Within the category of antiinfectives for systemic use (ATC code J), antivirals (J05) were the most prevalent with 28 products, followed by vaccines (J07, n=18) and antibiotics (J01, n=13). The J06 class (immune sera and immunoglobulins) was limited to two (first-in-class) monoclonal antibodies: Zinplava (bezlotoxumab) and Nyxthracis (obiltoxaximab). Innovation in other subclasses was also limited; only two antimycobacterials (Sirturo and Dovprela, bedaquiline and pretomanid, respectively) and a single systemic antimycotic (Cresemba - isavuconazole) were authorized during this period.
Among the products for the alimentary tract and metabolism (ATC code A), the most widely represented were those of the subclass A10 (drugs used in diabetes, 12 products), and A16 (other alimentary tract and metabolism products, n=15).
3.3. Variation by Technology Class
A synthetic presentation of the technology classes to which the 357 medicinal products belong is shown in Table 2.
We were also interested to know whether these technology classes followed certain time trends across the analyzed decade. Between 2011 and 2020, the number of small-molecule medicine authorizations in the EU showed no sustained long-term trend, fluctuating considerably year-to-year. This moderate variability included peaks in 2013 and 2020 and a sharp dip in 2016 (Figure S1).
In contrast, monoclonal antibodies (mAbs) displayed a clear upward dynamic over the decade. Approvals were modest early on (2–3 per year from 2011 to 2013) but began a rapid expansion starting in 2014–2015, driven by a sharp increase in 2015 (9 approvals). This sustained high output in the mid-2010s led to a plateau of consistently higher approval numbers in the latter part of the decade (Figure S2). The proportion of mAbs among the first-in-class medicinal products (22.06%) was almost equal with the proportion of mAbs among addition-to-class products (22.81%), but it was lower among those classified as advance-in-class (Table 2).
The number of proteins and polypeptides authorized in the EU between 2011 and 2020 exhibited substantial year-to-year variability without a sustained directional trend. The general pattern for proteins and polypeptides was a fluctuating but generally stable landscape, marked by random increases and decreases rather than a consistent upward or downward trend. The number of proteins and peptides authorized each ear across the decade was also relatively stable, fluctuating around an annual mean of 5.1 (Figure S3). Proteins and polypeptides represented a larger fraction of the first-in-class medicinal products compared to those classified as either advance-in-class or addition-to-class (Table 2).
The mean annual number of FDCs involving at least one new active substance was 3.0 (s.d. 1.7), with variations between 1 and 6 (Figure S4). These data exclude FDCs based on previously authorized active ingredients (i.e., this was limited to authorizations based on art. 8(3) of the Directive 2001/83/EC, excluding those authorized on the basis of Article 10b of Directive No 2001/83/EC). The annual number of authorized vaccines varied between 1 and 4, with a mean of 2.0 (s.d. 1.1) (Figure S5). The fraction of Fixed-Dose Combinations (FDCs) was lowest in the first-in-class products, but this proportion was highest in the advance-in-class products (Table 2).
Only in five of the 10 years considered here were radiopharmaceuticals authorized (2012-2014 and 2016-2017), and in each of those years the number of approved radiopharmaceuticals was two, except for 2016, when only one was authorized. Out of the 9 radiopharmaceuticals approved during this period, seven were first-in-class, while two were considered addition-to-class.
More innovative technologies were authorized in the analyzed decade compared with the previous time. For instance, a number of 5 cell-based Advanced Therapy Medicinal Products (ATMPs) were authorized between 2014 and 2019, whereas before this decade a single somatic-cell therapy medicines (sCTMP) had been authorized within EU, i.e., ChondroCelect (in 2009). A number of four other products are also based on somatic cells, for instance Zalmoxis (allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (-LNGFR) and the herpes simplex I virus thymidine kinase HSV-TK Mut2)) or the three CAR-T cell therapies, but they are legally classified (Under EU Regulation (EC) No 1394/2007) as gene therapy medicinal products (GTMPs), because the T cells contained by those products are genetically modified. To those four gene therapies, a product based on an oncolytic virus (Imlygic, with the active ingredient Talimogene laherparepvec, a genetically modified, oncolitic herpes simplex virus), two gene therapy using adeno-associated viruses (Luxturna, voretigene neparvovec, and Zolgensma, onasemnogene abeparvovec), as well as an ex vivo gene therapy (Libmeldy, atidarsagene autotemcel) should be added, increasing the total number of gene therapies authorized in this period to eight. From only one before the beginning this decade, the progress is rather spectacular. Although not classified as gene therapies, but only as biologicals, three antisense oligonucleotide medicinal products have been authorized in this period (whereas before only one had reached regulatory approval and had in 2002 been withdrawn). Also, four siRNA (small interference RNA) medicinal products and have been approved as medicines based on a new technology platform, for the first time in this decade, opening new ways of treating a variety of diseases. All such sophisticated products were categorized as first-in-class, zero were classified as advance-in-class or addition-to-class.
A small number of the new medicinal products had complex active ingredients, four of non-herbal origin and three of an herbal origin. The active ingredient of Velphoro is sucroferric oxyhydroxide, a complex made of polynuclear iron(III)-oxyhydroxide combined with sucrose and starches, which functions as a phosphate binder within the gastrointestinal tract, whereas ferumoxytol consists of superparamagnetic iron oxide nanoparticles enveloped by a carbohydrate coating made of polyglucose sorbitol carboxymethyl ether. Colestilan (the active ingredient of BindRen) and patiromer (Veltassa) are polymers with binding properties for phosphates and potassium ions, respectively. In the analyzed decade only three new products were based on complex herbal extracts: a defatted powder of peanuts (Arachis hypogaea L.) used in the treatment of peanut allergies (), a birch bark extract (Episalvan) authorized in the treatment of partial-thickness wounds (and withdrawn after several years for commercial reasons), and a bromelain enriched proteolytic enzyme preparation from Ananas comosus (Nexobrid) authorized for the enzymatic debridement of deep burns. Defibrotide (the active substance of Defitelio) is a mixture of oligonucleotides derived from porcine intestinal mucosa.
A number of three lipoglycopeptide antibiotics (a subgroup of the larger glycopeptide class) have been authorized in this period: Vibativ (telavancin), Xydalba (dalbavancin), and Orbactiv (Oritavancin). The rise of vancomycin-non-susceptible strains spurred development of lipoglycopeptides, which are semisynthetic glycopeptides with an added lipid tail, which prolongs their half-lives and increases their efficacy against Gram-positive cocci. All three are bactericidal agents given intravenously, despite having distinct dosing schedules and share a common bactericidal mechanism (interference with cell-wall synthesis via binding to terminal D-Ala-D-Ala residues of precursors). Their efficacy is targeted primarily against susceptible Gram-positive bacteria, such as S. aureus (including MRSA) and streptococci, making them ineffective for most Gram-negative infections. Telavancin was the first in class, whereas dalbavancin and oritavancin advanced the class, mostly by improving the PK profile and the administration convenience [27].
3.4. Administration Routes
By far the most widely used administration route was oral (158 products), followed by intravenous (106 products); combined, these cover 73.95% of the dataset (however, as shown below, parenteral routs collectively outweigh the oral route). The pursuit of oral formulations remains a major driver of pharmaceutical innovation, with first-in-class drugs exemplifying this trend and reinforcing the historical preference for oral administration. The substantial number of first-in-class oral products indicates ongoing innovation in overcoming the key challenges of drug development represented by bioavailability and first-pass metabolism. IV administration has traditionally been reserved for drugs requiring rapid onset, high bioavailability, or those unstable in the gastrointestinal tract, particularly various proteins, including mAbs. Since peptides, proteins and mABs represent about one third of all data set, the high number of IV products (29.69%) should not be surprising. These two routes were followed by subcutaneous (44 products + one subcutaneous implant) and intramuscular (13 products) routes. The high number of medicines administered subcutaneously, particularly the first in class (28 out of 41 such medicines), indicates a shift towards SC delivery of biologics, as before 2010 the SC route tended to be used especially for vaccines and insulin; now both monoclonal antibodies and a variety of other biological products use SC as the main delivery route. Several products offered more flexible administration options, such as oral + IV (4 products), oral + IM, IV for the initial dose followed by SC for subsequent doses, and even ID + SC + intratumoral + peritumoral injection following radiolabelling (Lymphoseek/tilmanocept). Other parenteral routes (such as intra-articular, intracerebroventricular, and intralesional) were largely reserved for rare first-in-class products. Other administration routes included three nasal vaccines, one vaginal pessary (Intrarosa), and five topical (cutaneous) treatments. Ophthalmic innovations featured unique delivery methods, including the Holoclar ocular surface implant and Luxturna’s subretinal injection (Table 3).
To facilitate a structured analysis of drug delivery trends, we have also categorized administration routes into four distinct groups: those involving systemic injections, infusions or localized internal delivery (e.g., intravenous, subcutaneous, intrathecal) were grouped as Parenteral; treatments applied to external or mucosal surfaces, such as topical, intranasal, and ophthalmic routes, were consolidated under Topical/Local; and products involving multiple delivery methods were designated as Mixed. When the products are thus grouped, the distribution is as follows: 172 parenteral, 158 oral, 15 topical/local, 9 with mixed delivery routes, and 3 others (two implants and one sui generis, in vitro radiolabelling, following by approved route). This more synthetic distribution of medicinal products by administration route and innovation category is shown in Table 4.
As shown in Table 4, oral administration dominates the lower-innovation categories (addition-to-class and advance-in-class), while the higher-innovation first-in-class products favor the parenteral route.
3.5. Pharmaceutical Forms
Although pharmaceutical forms are contingent upon the route of administration, multiple forms are frequently available for a single route. It is consequently of interest to analyze the patterns in their use for newly authorized drugs during the period under review. In a number of cases, MAHs later submitted one or more alternative dosage forms. Since our analysis was restricted to the date of first authorization, subsequent dosage forms were not included. A synthesis of the main dosage forms used in the products analyzed and their frequencies is shown in Table 5.
Pharmaceutical forms generally aligned with their respective administration routes. Within the oral category, film-coated tablets were the predominant dosage form (102 products). In contrast, non-coated tablets and prolonged-release tablets were rare, each appearing in only six products for non-coated tablets (Dovprela - pretomanid, Edarbi/Ipreziv - azilsartan medoxomil, Jakavi - ruxolitinib, Sirturo - bedaquiline, Spedra - avanafil, and Zytiga - abiraterone acetate), and four products for prolonged-release tablets (Betmiga - mirabegron, Fampyra - fampridine, Rinvoq - upadacitinib, and Rukobia – fostemsavir). Hard capsules were the second most common oral formulation (39 products), significantly outperforming soft capsules (4 products). Liquid-reconstitution forms were minimal: two products were powders for oral suspension (Lokelma - sodium zirconium cyclosilicate and Veltassa - patiromer), and one offered both effervescent and suspension formulations (Vaxchora - oral live cholera vaccine). No new active substances were authorized as powders for oral solution during this decade.
Parenteral pharmaceutical forms exhibited greater diversity than those for oral routes. Solutions for injection were most prevalent (36 products), followed by concentrates for solution for infusion (35) and powder for concentrate for solution for infusion (23). Other common formulations included powder and solvent kits for solution for injection (21), dispersions for infusion (7), powder for solution for injection and and solution for injection (pre-filled pen) (6 each).
Topical (cutaneous) formulations were limited to four products: three standard gels and one powder and gel for gel (where the powder is first mixed with the gel to obtain the final gel that will be applied on the wound area). Only two products were formulated as eye drops: Oxervate (cenegermin) and Rhokiinsa (netarsudil).
A small number of products occurred twice in the list, while a number of 31 products belonged to a diverse range of formulations, each one occuring only once. They can be grouped as follows:
Oral solid formulations: chewable tablets; effervescent powder and powder for oral suspension; granules for oral suspension; oral powder in capsules for opening or sachet; gastro-resistant hard capsules. Such formulations reflect attempts to improve patient acceptability (chewable, effervescent) or bioavailability (gastro-resistant). Granules and powders in sachets are often pediatric-friendly, but only one of the two was indicated in children (Palforzia - defatted powder peanuts (Arachis hypogaea L.), semen, intended for children with peanut allergy - age 1-17); the other was formulated as oral powder in capsules for opening or sachet because it is a single-dose diagnostic agent (Macimorelin Aeterna Zentaris - macimorelin).
Oral liquid formulations: one oral liquid (RAVICTI - glycerol phenylbutyrate, a product which contains only the active substance as a pure liquid, with no excipients) and one oral solution. Despite their traditional importance in pediatrics and geriatrics, oral liquids are rare among products with new active substances, likely due to stability and logistical challenges associated with liquid formulations.
Various parenteral preparations: Concentrate and solvent for solution for infusion; concentrate and solvent for solution for injection; powder and solvent for suspension for injection; powder and suspension for injection; powder for concentrate and solution for solution for infusion; powder for concentrate for solution for injection/infusion; powder for solution for injection (subcutaneous use); solution for injection/infusion. Such formulations reflect stability challenges, as many proteins (including monoclonal antibodies) or chemically sensitive molecules require reconstitution or dilution before administration.
Pre-filled and device-linked injections: solution for injection (vial, pre-filled pen, syringe, or cartridge). Such device-linked formulations reflect a patient-centric orientation in drug development, as they are apt for self-administration and come with increased convenience.
Combinations of dosage forms or hybrid forms: film-coated tablet + concentrate/solution combinations, hard capsule + powder for concentrate for solution for infusion, film-coated tablet + oral solution + solution for intravenous injection etc (eight such combinations in total among the whole data set). They correspond to a relatively small number of products with multi-route strategies: oral maintenance plus parenteral initiation or rescue.
Implantable and advanced delivery systems: implant, implantation suspension, and living tissue equivalent, transparent circular sheet. They represent advanced formulations, closer to the frontier of regenerative medicine (Spherox - spheroids of human autologous matrix-associated chondrocytes for implantation suspended in isotonic sodium chloride solution, used for the treatment of cartilage defects, and Holoclar - expanded autologous human corneal epithelial cells, indicated in the treatment of limbal stem cell deficiency), or intended to perform sustained release (Scenesse – afamelanotide, ensuring a sustained effect for about two months).
Topical and localized forms: nasal spray suspension, ointment, pessary (vaginal), powder and gel for gel (for topical use). Such delivery systems are well-established in the field, but their single occurrence indicate that major innovation is focused on systemic therapies for major indications (oncology, immunology, infectious disease), whereas occasionally niche innovations make use of topical therapy.
Radiopharmaceuticals: a kit for radiopharmaceutical preparation, and a radiopharmaceutical precursor, solution. Radiopharmaceutical kits are inherently specialized, requiring on-site preparation and strict handling, whereas a precursor has to be subject to a later radiolabelling step. Several other radiopharmaceuticals have been formulated as solutions for injections, whereas these two are distinct formulations, tailored to their specific preparation and use requirements.
3.6. Orphan Versus Non-Orphan Products
The distribution of orphan innovation status among the study participants is shown in Figure 3. Of the total sample (N = 357), about one-third (32.49%, n = 116) had an orphan status, while the remaining two-thirds (67.51%, n = 241) were non-orphan. Looking at the first-in-class products, the number of non-orphans was slightly superior to that of orphans (115 versus 89, 32.21% out of total versus 24.93% out of total). Among the orphan products the first-in-class had the highest number, whereas among the non-orphan medicines the distribution was more evenly spaced (although the first-in-class was still the largest subcategory). We would like to clarify that this analysis reflects the orphan status assigned at the time each marketing authorization was granted. A comparison with the current EU Register of Medicinal Products reveals a markedly different picture, as the orphan status of many products has since changed, either at the request of the MAH or following withdrawal of the marketing authorization. We are preparing a separate paper that examines these post-authorization changes for the products in this dataset.
4. Discussion
4.1. Innovation Levels and Competition Landscape
Our findings reveal a notable level of innovation among medicinal products containing new active substances (NAS) authorized through the European Commission’s centralized procedure from 2011 to 2020. Of the 357 NAS approvals, 204 (approximately 57%) were classified as first-in-class, representing groundbreaking therapeutic approaches with novel mechanisms of action, while the remaining 153 included 55 additions to class (mere alternatives within existing classes) and 98 advances in class (incremental improvements offering clinical or at least patient convenience advantages).
First-in-class drugs represent 56.0% of the total, indicating a substantial achievement of innovation during this decade. These agents introduce novel mechanisms of action and frequently address unmet medical needs or new therapeutic pathways. [28]. These findings align with the EMA’s centralized procedure requirements. Because this pathway is compulsory for biotechnological and advanced therapy medicinal products (ATMPs), it serves as a primary pipeline for the most innovative therapeutic developments in Europe [29]. Although directly comparable statistics on first-in-class drug approvals at the EMA during this specific decade are not available, evidence from analyses covering an overlapping period of the FDA–EMA approvals suggests a resilient European pipeline of novel mechanisms. However, EMA seems to utilize expedited pathways for such innovations considerably less frequently than the FDA (30% versus 81% of first-in-class approvals receiving expedited status between 2013 and 2023) [30]. The high first-in-class share within the analyzed decade supports the idea that Europe’s regulatory systems prioritize innovative therapies, especially in oncology and rare diseases, where novel targets have increasingly proliferated [31]. The proportion of first-in-class is broadly in line with the tendency reported in the last two decades of domination by “pioneers” (at 60–80% in the 2010s) and a reduction in the share of “colonists” (to a 10–25% share) [16].
While groundbreaking therapies justifiably are drawing most of the attention, the distribution of approvals (15.5% as additions to class and 27.5% as advances in class) suggest a robust landscape of incremental innovation. The vast majority of NAS approvals (about 84%) offer at least some degree of therapeutic advancement over existing options. Among the advance-in-class products have been grouped those able to deliver meaningful improvements in efficacy, safety, tolerability, pharmacokinetics, or patient convenience. New products within a specific therapeutic class can bring significant benefits even when they are only additions-to-class. Discrete clinical nuances, such as the different safety profiles of SSRIs or the selectivity properties of beta-blockers can result in high adoption rates for what are called ‘me-too’ drugs. Limited evidence indicates that sometimes even when priced above the first-in-class pioneer, these follow-on therapies command a large market share and are chosen by health professionals because they offer tailored options for patient-specific needs [32]. Moreover, among the Essential Medicines List developed by the World Health Organization (WHO), about 60% belong to the “me-too” drugs (i.e., additions-to-class or at most advances-in-class). However, their availability can contribute to the protection against shortages, as they allow professionals in the context of a shortage to substitute one product substituting with another from the same class [33]. Our data show that most non-first-in-class drugs in Europe represent tangible progress rather than unnecessary alternatives, with only 16% being classified as additions without any advancement in care. This limited share of mere additions-to-class reflects that fact that the market appetite for drugs with minimal differences from reference products is dwindling and the demand is increasing for products bringing superior safety or efficacy benefits over existing therapeutic options [34].
The balanced distribution of products with NAS among first-in-class and follow-on (advance-in-class or addition-to-class) is likely to reflect intense development competition for the same target, rather than a servile, low-risk imitation of a successful new product. Historically, the first drug to market in a class was often not the first to begin clinical testing [35]. Similarly, in about half of the cases, follow-on drugs were patented (in the U.S. or worldwide) before the pioneering first-in-class drug had secured any patent protection [36]. Therefore, regarding follow-on products as mere, low-quality imitators, overly simplistic and unfounded.
4.2. Time Evolution
As shown above, the temporal distribution of MAs for medicinal products containing NAS via the centralized procedure between 2011 and 2020 reveals a general upward trend in approvals, increasing from a low of 30 in 2011 to a peak of 41 in 2018 and 2020, albeit with notable fluctuations including marked declines in 2016 and 2019 (followed by recoveries). This general ascent reflects the growing productivity in the European pipeline for cutting-edge medicines over this period, driven by greater R&D investments and breakthroughs in multiple therapeutic fields, particularly oncology and rare diseases, as well as in that of certain technology platforms, such as the now mature monoclonal antibody technology [37], but also newer ones, from siRNAs[38] to CAR-T cells [39] or to progresses in cell research and advanced therapies [40]. For instance, under the 7th Framework Programme (2007-2013) and Horizon 2020 (2014-2020), the EU allocated €3.2 billion to over 550 cross-border research collaborations dedicated to rare diseases [41]. Multiple EU initiatives and national projects within EU have provided funding for cancer research, both at the basic and the translational levels [42].
The significant declines observed in 2016 and 2019, followed by immediate recoveries, are in line with the known natural volatility of annual regulatory approvals, typically seen in both EU and other regulatory regions [43,44]. Similarly low rates of approval in 2016 have also been reported in the US [43,45]. Time series analyses of US data have found cyclical patterns in FDA drug approvals, with regulatory, economic, and policy factors likely serving as the key influences [46]. The 2018 EU peak in approvals had an FDA counterpart, as the US agency approved 59 novel drugs in that year, marking a US record. This was fueled by widespread adoption of expedited procedures (namely Fast Track, Breakthrough Therapy, Priority Review, and Accelerated Approval) [47]. The 2020 peak is also consistent with one of the FDA’s most productive years: 49 novel therapeutics were approved (nearly 23% above the 2011–2020 annual average of 40) [48].
Annual authorization rates in the EU this decade are somewhat higher than those observed in the United States during the 1990s, yet remain below the peak levels seen in the U.S. in 2002 and 2003 [7]. Nevertheless, they exceed the total U.S. new molecular entity approvals from 2004–2009[8]. For context, the number of new active substances (NAS) authorized annually in the EU between 2002 and 2010 ranged from 14 to 36, surpassing 35 only in 2007 and exceeding 25 in just two years (2007 and 2009) [49].
To conclude, the 2011–2020 data illustrates a robust and increasingly dynamic European regulatory environment for novel therapies, defined by long-term growth despite natural annual fluctuations. These findings align with international pharmaceutical trends, indicating that European approval rates have kept pace with the accelerating rate of global scientific discovery in the field of drug development.
4.3. Therapeutic Classes
The dominance of antitumor therapies among newly approved products highlights the ongoing focus on oncology in global pharmaceutical innovation. Antineoplastic agents (ATC L01) accounted for the largest share of approvals over the last decade, reflecting both the high burden of cancer and the progress in developing a variety of new(er) antitumor approaches, from diverse targeted therapies such as small kinase inhibitors to a variety of immunotherapies, and monoclonal antibodies, including antibody-drug conjugates (ADCs). Antine drugs remain central to therapeutic development efforts of the pharmaceutical companies and public research, consistent with broader analyses showing their dominant role among oncologic approvals in recent years and in line with the perceived “strong unmet need” for both common and rare tumors [50]. This might also be to a certain extent driven by the perceived willingness of health systems to pay more for oncological products than for medicines intended for non-oncological diseases, even when the latter have relatively high DALY burdens [51]. Oncology has been a major focus of accelerated development pathways and regulatory incentives, such as access to expedite development procedures (Fast Track, Priority Review etc), and thus it is more appealing and may seem more rewarding and less risky than other therapeutic areas.
A paper based on USA data reported that even with 57 anticancer drug approvals occurring in a single year (2020), only 46 approvals with a new biological target were authorized over a 12-year period examined, implying that on average FDA is authorizing under four novel targets each year [52]. A direct comparison is difficult to make because our focus was not on novel biological targets, but rather on first-in-class (the two do not necessarily coincide); besides, defining what constitutes a “novel biological target” is more complex than it would seem at first sight, as discussed in the introductory section.
It has been stated that “antibody development in oncology driven by collaborative work from academia and industry has surpassed the growth of other cancer-targeting agents such as kinase inhibitors, hormonal drugs, cancer vaccines, and chemotherapies by a wide margin” [53]. Within the ten-year period analyzed, our data do not corroborate this assertion. Among the 92 antineoplastic agents approved, 55 were small molecules compared with just 28 monoclonal antibodies, meaning that the latter represented only about half the number of the former. It remains possible that the subsequent decade (now partly completed) will align more closely with this statement.
The relatively lower approval volume in other L-class ATC level-2 subgroups, including endocrine therapies (L02), immunostimulants (L03), and immunosuppressants (L04), likely reflect both a combination of market demand and different R&D priorities. The progress in the field of immunosuppressants, while vital for transplant and autoimmune care, often centers on well-characterized targets and established therapeutic classes, and is less focused on novel breakthroughs, although “no immunosuppression regimen has proven to be without a potential pitfall” [54]. Compared with antineoplastic agents, endocrine therapies focus on hormone-driven malignancies or defined metabolic axes, which naturally imply a narrower field of application and this probably explains the substantially lower number of products from this subclass. As for the immunostimulants, the development of immunosuppressive agents for oncology continues to be difficult, largely due to the immunosuppressive nature of the tumor microenvironment and the complexity of immune-evasion mechanisms such as hypoxia, increased glutathione expression, and other pathways that dampen immune activity [55]. There have been multiple obstacles in the pathway of developing new immunostimulants, but with recent progresses this might change in future, as indicated by the progresses with the CAR-T therapies and checkpoint inhibitors [56].
Within ATC code J (antiinfectives for systemic use), antivirals (J05) were the most prevalent, followed by vaccines and antibacterials. The 28 antiviral agents cover a broad spectrum of viral targets, including HIV, HCV, influenza, and SARS-CoV-2, highlighting both persistent unmet needs and pandemic-driven momentum in antiviral development. Many approvals involve fixed-dose HIV-1 combination therapies such as Genvoya, Biktarvy, Delstrigo, and Stribild, which represent incremental innovation aimed at streamlined, once-daily regimens that enhance adherence and strengthen resistance barriers in long-term HIV management. In contrast to monotherapy, drug combinations deliver enhanced efficacy, ensuring continued viral suppression, safeguarding T-cell levels, and minimizing side effects. These benefits have made combination regimens a major focus of HIV research and pharmaceutical development. Their flexibility allows patients to switch therapies more readily in response to toxicity or the need for simpler dosing, and they significantly curb the development of resistance that commonly occurs with single-drug approaches [57]. The large proportion of HIV therapies, including second generation integrase inhibitors (Tivicay), next generation of non-nucleoside reverse transcriptase inhibitors (Pifeltro), and the first first CD4-directed post-attachment inhibitor (Trogarzo), useful in multi-drug-resistant HIV-1, reflects a long-standing evolution of antiviral research and development, where about half of all antiviral approvals in recent decades have targeted HIV [58]. As a post-attachment entry inhibitor, the monoclonal antibody Trogarzo (ibalizumab) exemplifies the use of biologics in managing multi-drug resistant HIV[59]. Although small-molecule therapies still dominate the market, this class highlights a strategic move toward non-traditional antiviral platforms, at least for challenging, multidrug resistance forms of the disease.
Another key group of antiviral products authorized within this decade included several hepatitis C virus (HCV) direct-acting antiviral combinations such as Harvoni (sofosbuvir/ledipasvir), Epclusa (sofosbuvir/velpatasvir), Viekirax (ombitasvir/paritaprevir/ritonavir), and Maviret (glecaprevir/pibrentasvir), alongside earlier-generation protease inhibitors like Incivo (telaprevir) and Victrelis (boceprevir), authorized at the beginning of the decade. They exemplify the significant advances in HCV therapy over the past decade [58], with potent pangenotypic efficacy and impressive cure rates (quasi-total) compared with interferon-based regimens, even in hard to treat populations, such as cirrhosis patients infected with genotype 3 [60].
Two influenza antiviral products were authorized in this interval, Alpivab (peramivir) and Xofluza (baloxavir marboxil). Peramivir offers the benefit of single-dose administration, though via a more invasive route, and demonstrates better safety and efficacy than oseltamivir in pediatric patients, but not in adults. [61,62]. As a first-in-class inhibitor of cap-dependent endonuclease activity, Baloxavir marboxil represents a rare mechanistic breakthrough in the treatment of acute respiratory viral infections. By disrupting the influenza virus’s cap-snatching machinery, it employs a mode of action fundamentally distinct from neuraminidase inhibition (furthermore, Baloxavir has demonstrated superior safety and efficacy profiles compared to oseltamivir [63]).
The COVID-19 pandemic expanded the range of systemic antiviral therapies. Veklury (remdesivir), initially created for other viral diseases and later redirected toward SARS-CoV-2, is as a broad-spectrum RNA polymerase inhibitor [64] authorized to reduce disease progression in patients with COVID-19.
In the ATC A category, new drug development is heavily concentrated in two areas: (type 2) diabetes (A10) and rare metabolic diseases (A16). This pattern highlights the two biggest drivers of recent innovation in this field, the (r)evolution of diabetes care [65] and a progress in targeted medicines for ultra-rare conditions [66]. The inclusion of 14 agents in subclass A10 reflects a period of rapid transformation in diabetes pharmacotherapy in the analyzed period, particularly for type 2 of the disease. This group of medicines is dominated by two therapeutic classes: SGLT2 inhibitors (empagliflozin, dapagliflozin, canagliflozin, ertugliflozin, sotagliflozin)[67] and GLP-1 receptor agonists (e.g., semaglutide, dulaglutide, albiglutide, lixisenatide)[68]. Two DPP-4 inhibitors (alogliptin, linagliptin), were authorized in the first part of the decade (2011 and 2013), suggesting a higher interest for new approaches in diabetes treatment. The list of subclass A16 is primarily composed of enzyme replacement therapies (ERTs) (e.g., sebelipase alfa for lysosomal acid lipase (LAL) deficiency, asfotase alfa for hypophosphatasia, elosulfase alfa for mucopolysaccharidosis type IVA (MPS IVA)), small molecule substrate reduction or chaperone therapies (e.g., eliglustat for Gaucher disease, migalastat for Fabry disease), and siRNA molecules targeting specific enzymes in the heme and oxalate biosynthesis pathways for acute hepatic porphyria and primary hyperoxaluria type 1 (givosiran and lumasiran).
4.4. Administration Routes
The examination of administration routes makes evident the tight linkage between delivery routes and the level of pharmaceutical innovation. Across the dataset, oral and intravenous formulations predominate, representing nearly three-quarters of all products and reaffirming their longstanding prominence in drug development. The oral route persists as the predominant mode of administration industry-wide [69], preferred for its established profile in patient compliance, convenience, and appropriateness for extended treatment regimens [70]. Its dominance among both addition-to-class and advance-in-class agents suggests that much incremental innovation is built on existing pharmacological and pharmaceutical foundations, wherein the obstacles to effective oral delivery have been previously resolved or are reasonably manageable. The substantial number of first-in-class products addressed to the oral route indicates that this delivery approach is not limited to incremental developments. Instead, it continues to be a major driver of innovation, with new molecular entities successfully overcoming classical barriers such as poor solubility, limited permeability, and extensive first-pass metabolism [71]. This finding reinforces the notion that advances in medicinal chemistry, formulation technologies, and enabling approaches (e.g., prodrugs and absorption enhancers) have expanded the scope of what can be achieved via the oral route, even for novel mechanisms of action.
Although oral dosing is the predominant route in general, parenteral administration, especially IV delivery, is widely used for first-in-class drugs. This is consistent with the fact that numerous novel and complex biologic agents, such as therapeutic proteins and monoclonal antibodies, and even some small molecules, are administered parenterally because they are poorly absorbed and unstable in the gastrointestinal environment when taken orally [72,73,74]. By bypassing first-pass metabolism and entering systemic circulation directly, parenteral routes offer predictable pharmacokinetic profiles and consistent systemic drug levels (high bioavailability levels), both essential for medicines with novel pharmacological mechanisms [75,76].
While intravenous infusion remains the standard route for the majority of therapeutic antibodies, there is a trend toward diversifying parenteral delivery. Advancements in formulation are increasingly allowing for subcutaneous administration, which facilitates self-care and reduces the burden on healthcare facilities. Small molecules like opioids have been administered subcutaneously since the 1800s, but this route only became common for monoclonal antibodies with adalimumab’s introduction, gaining widespread adoption after 2009. Within our dataset, among the 42 products for which the administration route was subcutaneous, half (21) were monoclonal antibodies. Today, subcutaneous delivery is preferred for managing chronic conditions like rheumatoid arthritis because patients can administer the medication themselves, improving adherence to treatment [77]. Compared to the IV delivery, the SC route offers a less invasive alternative with significantly shorter administration times. Transitioning monoclonal antibodies to SC formats is a strategic priority aimed at alleviating the pressure on healthcare systems, lowering costs, and enhancing the quality of life for both patients and their families [78]. The substantial share of SC products within the first-in-class subgroup (28 out of 42) in our dataset is consistent with this shift, in line with the move toward more patient-centric delivery formats for biologics.
Beyond standard oral and parenteral routes, the innovative products include a number of 15 that employ mixed administration strategies (e.g., oral plus IV, IV plus SC, IM plus SC,), reflecting adaptable delivery designs that aim to pair rapid therapeutic onset with convenient long-term dosing (e.g., Sivextro, administered by the IV route for a rapid effect, and by the oral route otherwise) or simply alternative ways of administration (for instance Lymphoseek, in the meantime withdrawn from the market for commercial reasons, could be administered by intradermal, subcutaneous, intratumoural, or peritumoural injection) [79]. These hybrid approaches are commonly selected when clinical needs call for an immediate effect followed by sustained management suitable for outpatient care [80,81]. Such flexible paradigms are gaining prominence in clinical development and formulation science as part of broader efforts to enhance both therapeutic performance and patient adherence [81].
Specialized administration routes (such as intralesional or subretinal) are predominantly utilized for high-innovation therapies or orphan drugs [82,83]; the intranasal delivery has been adopted for the live attenuated influenza vaccine, offering distinct advantages compared with traditional inactivated, split-virion formulations [84]. This aligns with established research indicating that local delivery is the preferred strategy for novel targets or approaches requiring site-specific access [85]. Such precision is particularly critical for gene therapies and localized biologics, where therapeutic success depends on direct entry into specific tissue compartments.
Although cutaneous and other local routes represent a smaller share of products, they encompass some of the most distinctive delivery innovations. Examples like ocular surface implants (which has clear benefits over traditional techniques for occular drug administration) and retinal injections for ophthalmic therapies indicate that at least in some cases the administration route can itself be a critical component of pharmaceutical innovation for certain therapeutic fields [86,87,88].
4.5. Pharmaceutical Forms
The range of pharmaceutical forms for the analyzed new products spans various administration routes, indicating both traditional drug delivery methods and newer solutions designed to enhance bioavailability, safety, and patient convenience. Oral solid and parenteral formulations dominate the list, serving as the foundation of pharmaceutical development. Besides, a smaller segment corresponds to incremental technological improvements rather than groundbreaking innovations.
Film-coated tablets are the most popular oral dosage form, representing 98 products, a finding that highlights their advantages in both production and patient preference. The coating offers multiple benefits: facilitate swallowing and compliance, improves taste by creating a physical barrier between the taste buds and the active ingredient, shields the active substance from biological degradation or environmental damage, and allows for distinctive branding [89]. With only four non-coated tablets identified in the dataset, it is obvious that the vast majority of newly approved drugs when in tablet form, use tablet coatings, for the mentioned advantages.
The low number of prolonged-release tablets (only four products) is worth noting. It probably reflects the intricate relationship between the pharmacokinetic profiles of novel molecular entities and the difficulties inherent in their formulation. On one hand, certain modern drug candidates, especially in oncology and immunology but not only, feature narrow therapeutic indices, wide inter-patient pharmacokinetic variability or particular absorption patterns [90,91,92,93], that are less suited to controlled-release approaches, as inadequate control of drug release from such formulations may lead to diminished therapeutic efficacy or an increased risk of toxicity [94], and such risks are considerably higher for drugs with a narrow therapeutic index. On the other hand, creating modified-release dosage forms demands significant extra resources in formulation development and clinical testing, resources that developers may choose not to allocate during initial approval when standard immediate-release versions are sufficient to prove efficacy (and coming later with a prolonged-delivery solution provides further market advantage). For instance, a recent study estimated that in Thailand the R&D costs associated with developing an incrementally modified sustained-release drug range between USD 1.5 and 3.0 million when development is limited to Phase I studies, and between USD 18.6 and 20.2 million for a full clinical development program [95]. Besides, the development of extended-release formulations from initially authorized immediate-release products is a widely used strategy in the pharmaceutical industry to prolong the commercial life cycle of medicines [96].
The predominance of hard capsules (36 products) over soft capsules (4 products) among the new products is consistent with the typical physicochemical profiles of the active pharmaceutical ingredients. Hard capsules come with strong manufacturing advantages (simplicity and flexibility) [97] and are compatible with a broad spectrum of enabling technologies for poorly soluble active ingredients, allowing incorporation of multiparticulate systems, liquid and semi-solid fills, interactive inhalation blends, and mini-tablet units in a single dosage form [98]. Soft capsules are well suited to deliver lipid-based formulations, due to their ability to hold liquids or semi-solids, flexible excipient choices, protective sealing, and quick dissolution in the body [99].
Parenteral administration presented significantly more varied formulations than the oral route, reflecting the key therapeutic areas that have driven recent regulatory approvals. Solutions for injection (35 products) remained widely used when chemical stability is achievable, benefiting from ease of manufacturing and administration. Yet, the high count of formulations requiring reconstitution, including concentrates for solution for infusion (33), powder for concentrate for solution for infusion (23), and powder and solvent kits (19) indicate stability challenges associated with many new biopharmaceuticals. Many protein therapies, including monoclonal antibodies, face challenges in maintaining stability in aqueous media over time [100,101]. The instability of proteins in solution, coupled with their general sensitivity, necessitates lyophilization to create stable therapeutic products. Freeze-drying (lyophilization) remains the dominant strategy to ensure their long-term storage stability, despite additional complexities in manufacturing, distribution, and final use [102,103], although similar technologies with anticipated better results are being explored [104]. The continued dominance of lyophilized products suggests that emerging alternatives such as liquid formulations with novel stabilizers (different families of excipients), those obtained by liquid-liquid phase separation or other buffer-free and high concentration formulations [105,106,107], have not yet overcome these stability hurdles for the majority of therapeutic proteins.
The emergence of pre-filled pens and syringes (generally limited to two or three variants per device type in our data set) reflects a shift toward more patient-focused drug delivery. These integrated formulations improve adherence, minimize dosing errors, and empower patients to self-administer treatments for chronic diseases. Their limited adoption, however, indicates they are chiefly used for therapies designed for long-term outpatient care, where the higher development costs are offset by better health outcomes and competitive advantage.
Pre-filled pens and syringes (integrated formulations including a drug plus an injecting device) are more user-centric and can easily overcome the key challenge associated with the drug delivery using parenteral routes: convenience issues, costs and logistics, measurement accuracy, sterility, risks associated with the break of the skin barrier etc. They provide a number of key advantages, such as ease of use, precise dosing, and reduced medication waste; they also enhance patient safety by minimizing needle-stick risks and hazardous drug exposure. By eliminating the overfill required with vials, they improve efficiency and are particularly valuable for high-cost or low-volume biologic therapies [108,109].
The paucity of topical (cutaneous) formulations (4 products) and eye drops (2 products) reflects partly the pharmaceutical industry’s strategic prioritization of high-value therapeutic areas such as oncology, immunology, and rare diseases, where systemic therapies command premium pricing and address unmet medical needs. It has been recently stated that the widespread belief that dermatological drug products offer limited commercial viability has hampered innovation and fostered a cautious development strategy. For instance, efforts have largely centered on repurposing or reformulating established active pharmaceutical ingredients (APIs) to optimize them specifically for topical administration [110]. However, topical and ophthalmic drug delivery routes encounter unique physiological obstacles, which may also account for their limited representation. For example, eye drops typically achieve very low ocular bioavailability (less than 5%) because of rapid tear clearance and drainage through the nasolacrimal duct resulting in a short residence time for the active ingredient, as well as corneal resistance, restricting their effectiveness, especially in reaching deeper eye tissues [111]. While innovative delivery systems are being explored to address these barriers, the inherent pharmacokinetic challenges reduce the likelihood of new active substances being successfully developed for topical or ophthalmic use, regardless of therapeutic priorities. However, there remains a continuous interest for topical formulations and in the last few years new such therapies have been developed in major markets for atopic dermatitis [112], psoriasis [113], and other diseases. It should also be kept in mind that although topical/local are of interest for EMA when sufficiently innovative, there is a high likelihood that such products are authorized mostly through national regulatory routes [114].
Many of the products occurring only twice and the 33 products featuring unique formulations that appear only a single time in the dataset (“Others” in Table 5), exemplify the forefront of pharmaceutical innovation and address highly specialized therapeutic needs. Several patterns could be identified among this heterogeneous group:
- Pediatric-oriented formulations, such as chewable tablets and granules for oral suspension in sachets, remain underrepresented despite regulatory incentives for pediatric drug development [115]. The limited presence of gastro-resistant dosage forms suggests that few new active substances need shielding from gastric acid, or that acceptable bioavailability can generally be achieved without enteric protection. This could be a consequence of current discovery efforts emphasizing compounds with robust absorption profiles or the deployment of alternative tools like acid-resistant prodrug designs. On the other hand, while enteric coating can optimize a drug’s stability and release profile, its actual impact on oral bioavailability remains debatable, as its effectiveness often fluctuates based on a complex interplay between the drug’s chemical properties and the physiology factors such as gastric transit speed and pH fluctuations [116].
- Innovative delivery systems, including implants (e.g., Scenesse/alfamelanotide [117]) and tissue-engineered therapies (e.g., Spherox, Holoclar) [117,118], are the result of the fusion of pharmaceutical innovation and regenerative medicine. While such products are still relatively rare, they reflect the rise of personalized [119] and long-acting therapies[120]. Scenesse’s implant, which achieves two months of continuous drug release, illustrates how such advanced technologies can redefine treatment approaches by minimizing the frequency of administration.
- While most radiopharmaceuticals approved during this period were formulated as conventional forms (such as solutions for injection, included in the products discussed earlier), three products were formulated as radiopharmaceutical precursor solutions (two) and a kit for radiopharmaceutical preparation. They reflect the unique requirements of nuclear medicine, where the radiopharmaceutical precursor must undergo radiolabeling at the clinical site, and kits must enable rapid, reliable preparation while maintaining radiochemical purity, labeling efficiency, and overall quality of the final radiopharmaceutical [121,122]. The predominance of ready-to-use solutions among radiopharmaceuticals indicates progress in developing stable formulations that can be centrally manufactured and distributed, as well as improved cold-chain logistics and generator-based radionuclides (e.g., 68Ga, 99ᵐTc) [123,124,125], though the persistence of precursors and kits highlights situations where on-site preparation remains necessary, particularly due to the short physical half-lives of certain radionuclides or the complexity of the associated radiolabeling chemistry.
4.6. Orphan Status
The observation that roughly one third of the products (32.49%) carried an orphan designation is consistent with wider trends highlighting the increasing role of orphan medicines in contemporary pharmaceutical development. Orphan status is granted to treatments intended for rare diseases, which have traditionally benefited of limited treatment options because of limited patient populations and reduced profitability. To address this, regulatory initiatives such as the U.S. Orphan Drug Act and the European Union’s Orphan Drug Regulation have introduced regulatory incentives, such as periods of market exclusivity, fiscal benefits, and regulatory stimuli [126,127,128]. The sizeable share of products with an orphan status within our dataset underscores the effectiveness of these measures in encouraging therapeutic innovation for areas of high unmet medical need.
The relatively small difference among the proportions of first-in-class products in orphan (24.5%) and non-orphan (31.6%) designations suggests that major therapeutic innovation occurs with similar frequency in both rare and common therapeutic areas, a finding similar to what was reported in the US in the same period [129]. This also reflects a commitment of the pharmaceutical industry to targeting previously unaddressed therapeutic needs, regardless of disease prevalence. This pattern likely stems from progress in fields such as multi-omics, precision medicine and biomarker identification, which facilitate the development of more specific and novel treatment approaches [130,131].
While orphan drugs benefit from regulatory and financial incentives, their high costs and limited patient populations can pose challenges for healthcare systems and payers. Orphan drugs frequently deliver greater health benefits compared to non-orphan drugs, but their significantly higher prices often result in lower cost-effectiveness. This creates a dilemma for healthcare payers, who must balance ensuring patient access to these vital therapies with the need to maximize the value derived from pharmaceutical expenditures [132]. However, it should be taken into account that traditional cost-effectiveness analysis (CEA), primarily based on the cost per quality-adjusted life year (QALY) metric, frequently is devoid of the necessary sensitivity for evaluating orphan medicinal products. These standard frameworks are often ill-equipped to address the specific complexities of the rare disease landscape, particularly the interplay between high acquisition costs, the inherent limitations of small clinical datasets, as well as social equity goals, such as prioritizing interventions for patients with the greatest medical need [133]. A significant gap between medical innovation and global access has been reported, as fewer than 5% of approximately 7,000 rare conditions have authorized treatments, and prohibitive costs further restrict use in low- and middle-income countries (LMICs). These disparities are rooted in systemic challenges, including financial barriers, inadequate diagnostic infrastructure, and a lack of cohesive regulatory oversight [134]. The concentration of first-in-class innovation in the orphan category underscores the need for continued policy support to ensure that breakthrough therapies reach patients who need them, without compromising affordability or equitable access. Treatment costs rise sharply as disease prevalence falls, especially for ultra-orphan conditions. When generics existed, branded orphan drugs may cost up to 82,000 times more than their generic equivalents, underscoring generics’ role in improving affordability [135]. However, this cost-reduction offered by generics depends entirely on upstream innovation: reference products must first be developed and approved before generics can emerge. Although various frameworks, including global solidarity funds, regional pooled procurement, and equity-driven subsidies, have been proposed to harmonize orphan drug access across diverse economic landscapes, their operationalization is frequently stalled by localized legal, fiscal, and administrative limitations [134].
4.7. Limitations
Although our analysis maps EU pharmaceutical innovation from 2011-2020 in detail, a number of limitations should be acknowledged. First, the scope of the analysis was limited to medicinal products with NAS approved through the centralized procedure, handled by EMA. Because of this limitation, orphan medicinal products based on “old” active substances, despite their potential for meaningful therapeutic innovation (e.g., mercaptamine, which was first authorized within EU in 1997) were excluded. Besides, innovative therapies authorized via national procedures within EU Member States were not included in our dataset.
A second limitation refers to a certain extent of subjectivity in classification. Classifying innovation as “first-in-class,” “advance-in-class,” or “addition-to-class” carries inherent subjectivity, especially in assessing clinical advancements or mechanism novelty. Whereas FDA’s Priority Review is often used as a proxy for innovation assessment, and the EU PRIME scheme could be used similarly for this purpose, none is sufficient in itself for the classification of innovation, as shown above. Therefore, the categories here were based on the authors’ expert assessment, informed by scientific literature and EPARs.
To prevent artificial overcounting, we clustered products based on a common active substance (authorized simultaneously), including fixed-dose combinations co-authorized with the parent NAS in the same month and year as a single product. This mitigates overcounting but might not fully capture the pharmaceutical innovation and development resources dedicated to fixed-dose combinations.
A further limitation stems from our geopolitical perspective: innovation status (e.g., “first-in-class”) was assessed solely from a European regulatory viewpoint. Since global approval dates differ, a drug deemed “first-in-class” in the EU may have been a follow-on product elsewhere, as illustrated by the nivolumab and pembrolizumab, whose innovative status differ between EU and USA.
4.8. Future Perspectives and Conclusions
The trends observed over the evaluated decade, together with early evidence from the current one, point to several directions for future research and industry development. Notably, technology platforms are expected to continue evolving and improving. The remarkable advances seen in gene therapies, siRNA, and ATMPs toward the end of the previous decade are likely to persist. Future research is expected to examine whether and to what extent these advanced platforms can extend beyond rare diseases into wider therapeutic areas. Artificial intelligence (AI) is expected to reshape substantially pharmaceutical innovation across the next decade, revolutionizing every phase of drug discovery, development, and delivery. Areas like target identification and validation, de novo drug design, clinical trial optimization, drug repurposing, manufacturing and supply chain management, personalized medicine, real-world evidence and pharmacovigilance, collaboration and knowledge sharing, plus regulatory compliance, could all see major improvements or accelerations through AI integration.
As the boundary between “pioneers” and “followers” becomes increasingly nuanced, there is a growing need for robust, objective measures of innovation at both the authorization stage and during post-marketing use. We are currently developing a structured metric for initial innovation assessment. Post-marketing evaluation is expected to integrate long-term health outcomes, cost-effectiveness, and other relevant dimensions, moving beyond initial regulatory status or mechanism of action.
Subcutaneous (SC) administration of biologics and the growing popularity of pre-filled devices indicate a transition toward increased patient-centered care approaches. It is likely that in the future, gradually, pharmaceutical innovation will increasingly focus on developing treatments that lessen the load on healthcare systems while improving convenience in administration and adherence for patients managing chronic disease.
With approved treatments available for less than 5% of the over 7000 rare conditions, future policy and R&D initiatives must overcome systemic hurdles, such as exorbitant expenses and access to appropriate diagnostic equipment, to ensure pioneering therapies are accessible to all patients, including those in low- and middle-income nations.
Our analysis of 364 new medicinal products authorized in the EU from 2011 to 2020 reveals a robust and growing innovation pipeline. The EU regulatory landscape stands to benefit from adopting efficiencies observed in other jurisdictions; modernizing the regulatory framework to facilitate easier access to accelerated approval tracks could create a more robust innovation ecosystem, providing the necessary incentives for companies to prioritize the European market for new drug launches.
Over half (56%) were first-in-class, with breakthroughs concentrated in oncology and immunomodulation, antiinfectives for systemic use, alimentary tract and metabolism, and rare diseases across different therapeutic areas. Although small molecules are still the most common technology, the surge in monoclonal antibodies and the emergence of gene and cell therapies reflects a transformative change in the pharmaceutical landscape.
Moreover, the analysis confirms that innovation extends beyond radical novelty and has different degrees; most (84%) new active substances provided at least some degree of therapeutic advancement or clinical convenience over the current options. While the industry meets therapeutic needs via novel chemical structures or biological approaches, mechanisms of actions and delivery systems, a critical future challenge lies in reconciling the costs of such innovation with healthcare affordability and global distributive justice in access to the new products.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, RA; methodology, RA; software, RA; validation, RA and MD; formal analysis, RA, DD, BEL, MD; investigation, RA, DD, BEL, MD; resources, DD and MD; data curation, RA and BEL; writing—original draft preparation, RA and MD; writing—BEL and DD.; visualization, RA; supervision, DD and MD; project administration, DD; funding acquisition, MD. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable..
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
R.A. received consultancy or speakers’ fees in the past from Merck, UCB, Sandoz, Abbvie, Zentiva, Teva, Laropharm, CEGEDIM, Angelini, Biessen Pharma, Hofigal, AstraZeneca, and Stada.
Abbreviations
The following abbreviations are used in this manuscript:
| ADC | Antibody-drug conjugates |
| AI | Artificial intelligence |
| API | Active pharmaceutical ingredients |
| ATMP | Advanced Therapy Medicinal Product |
| CEA | Cost-effectiveness analysis |
| DALY | Disability-Adjusted Life Year |
| EMA | European Medicines Agency |
| EPAR | European Public Assessment Report |
| ERT | Enzyme replacement therapies |
| EU | European Union |
| FDA | Food and Drug Administration |
| FDC | Fixed-dose combination |
| GTMP | Gene therapy medicinal product |
| HCV | Hepatitis C virus |
| HIV | Human immunodeficiency virus |
| ID | Intradermal |
| IM | Intramuscular |
| IV | Intravenous |
| LAL | Lysosomal acid lipase |
| LMIC | Low- and middle-income countries |
| mAb | Monoclonal antibody |
| MAH | Marketing Authorization Holder |
| MPS IVA | Mucopolysaccharidosis type IVA |
| NAS | New active substance |
| NDA | New Drug Application |
| NME | New Molecular Entity |
| QALY | Quality-adjusted life year |
| R&D | Research and Development |
| SC | Subcutaneous |
| sCTMP | Somatic-cell therapy medicines |
| SGLT2 | Sodium/glucose cotransporter 2 |
| siRNA | Small interference RNA |
| WHO | World Health Organization |
References
- Wakutsu, N.; Hirose, E.; Yonemoto, N.; Demiya, S. Assessing Definitions and Incentives Adopted for Innovation for Pharmaceutical Products in Five High-Income Countries: A Systematic Literature Review. Pharmaceut Med 2023, 37, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Horrobin, D.F. Innovation in the Pharmaceutical Industry. Journal of the Royal Society of Medicine 2000, 93, 341–345. [Google Scholar] [CrossRef]
- Urabe, K.; Child, J.; Kagono, T. Innovation and Management: International Comparisons. In De Gruyter Studies in Organization Ser; De Gruyter, Inc: Berlin/Boston, 2018; ISBN 978-3-11-086451-9. [Google Scholar]
- Morgan, S.; Lopert, R.; Greyson, D. Toward a Definition of Pharmaceutical Innovation. Open Med 2008, 2, e4-7. [Google Scholar]
- National Museum of the United States Air Force Conquering the Sky: Dec. 17; 1903. Available online: https://www.nationalmuseum.af.mil/Visit/Museum-Exhibits/Fact-Sheets/Display/Article/196027/conquering-the-sky-dec-17-1903/ (accessed on 15.01.2026).
- Nordham, K.D.; Ninokawa, S. The History of Organ Transplantation. Proc (Bayl Univ Med Cent) 2022, 35, 124–128. [Google Scholar] [CrossRef]
- Cohen, F.J. Macro Trends in Pharmaceutical Innovation. Nature Reviews Drug Discovery 2005, 4, 78–84. [Google Scholar] [CrossRef]
- Kaitin, K.I.; DiMasi, J.A. Pharmaceutical Innovation in the 21st Century: New Drug Approvals in the First Decade, 2000–2009. Clin Pharmacol Ther 2011, 89, 183–188. [Google Scholar] [CrossRef]
- Munos, B. Lessons from 60 Years of Pharmaceutical Innovation. Nat Rev Drug Discov 2009, 8, 959–968. [Google Scholar] [CrossRef]
- Green, E.D.; Guyer, M.S.; National Human Genome Research Institute. Charting a Course for Genomic Medicine from Base Pairs to Bedside. Nature 2011, 470, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.; De Mouzon, O.; Scott-Morton, F.; Seabright, P. Market Size and Pharmaceutical Innovation. The RAND Journal of Economics 2015, 46, 844–871. [Google Scholar] [CrossRef]
- Lanthier, M.; Miller, K.L.; Nardinelli, C.; Woodcock, J. An Improved Approach To Measuring Drug Innovation Finds Steady Rates Of First-In-Class Pharmaceuticals, 1987–2011. Health Affairs 2013, 32, 1433–1439. [Google Scholar] [CrossRef]
- Light, D.W. Global Drug Discovery: Europe Is Ahead: A Reanalysis of Data from 1982–2003 Contradicts the Claim That U.S. Drug Firms Overtook European Firms in Pharmaceutical Innovation. Health Affairs 2009, 28, w969–w977. [Google Scholar] [CrossRef]
- Battaglia, L.E. Drug Reformulation Regulatory Gaming: Enforcement and Innovation Implications. European Competition Journal 2011, 7, 379–405. [Google Scholar] [CrossRef]
- Sternitzke, C. Knowledge Sources, Patent Protection, and Commercialization of Pharmaceutical Innovations. Research Policy 2010, 39, 810–821. [Google Scholar] [CrossRef]
- Wills, T.J.; Lipkus, A.H. Structural Approach to Assessing the Innovativeness of New Drugs Finds Accelerating Rate of Innovation. ACS Med. Chem. Lett. 2020, 11, 2114–2119. [Google Scholar] [CrossRef]
- European Medicine Agency Annual Reports and Work Programmes. Available online: https://www.ema.europa.eu/en/about-us/annual-reports-work-programmes (accessed on 15.01.2026).
- European Medicine Agency European Public Assessment Reports (EPARs). Available online: https://www.ema.europa.eu/en/medicines (accessed on 15.01.2026).
- European Commission Public Health - Union Register of Medicinal Products. Available online: https://ec.europa.eu/health/documents/community-register/html/index_en.htm (accessed on 15.01.2026).
- European Medicine Agency Assessment Report. Dinutuximab Beta Apeiron International Non-Proprietary Name: Dinutuximab Beta 2017.
- McKeage, K.; Lyseng-Williamson, K.A. Dinutuximab Beta in High-Risk Neuroblastoma: A Profile of Its Use. Drugs Ther Perspect 2018, 34, 281–287. [Google Scholar] [CrossRef]
- Michaeli, D.T.; Michaeli, T.; Albers, S.; Boch, T.; Michaeli, J.C. Special FDA Designations for Drug Development: Orphan, Fast Track, Accelerated Approval, Priority Review, and Breakthrough Therapy. The European Journal of Health Economics 2024, 25, 979–997. [Google Scholar] [CrossRef]
- Del Grosso, V.; Perini, M.; Casilli, G.; Costa, E.; Boscaro, V.; Miglio, G.; Genazzani, A.A. Performance of the Accelerated Assessment of the European Medicines Agency. British Journal of Clinical Pharmacology 2025, 91, 1439–1448. [Google Scholar] [CrossRef]
- European Medicine Agency European Medicines Agency. EMA/7872/2021 2024; Guidance for Applicants Seeking Access to PRIME Scheme.
- Franco, P.; Jain, R.; Rosenkrands-Lange, E.; Hey, C.; Koban, M.U. Regulatory Pathways Supporting Expedited Drug Development and Approval in ICH Member Countries. Ther Innov Regul Sci 2023, 57, 484–514. [Google Scholar] [CrossRef]
- Hwang, T.J.; Ross, J.S.; Vokinger, K.N.; Kesselheim, A.S. Association between FDA and EMA Expedited Approval Programs and Therapeutic Value of New Medicines: Retrospective Cohort Study. BMJ 2020, 371, m3434. [Google Scholar] [CrossRef]
- Liu, F.; Rajabi, S.; Shi, C.; Afifirad, G.; Omidi, N.; Kouhsari, E.; Khoshnood, S.; Azizian, K. Antibacterial Activity of Recently Approved Antibiotics against Methicillin-Resistant Staphylococcus Aureus (MRSA) Strains: A Systematic Review and Meta-Analysis. Ann Clin Microbiol Antimicrob 2022, 21, 37. [Google Scholar] [CrossRef]
- Eder, J.; Sedrani, R.; Wiesmann, C. The Discovery of First-in-Class Drugs: Origins and Evolution. Nat Rev Drug Discov 2014, 13, 577–587. [Google Scholar] [CrossRef]
- Kohler, M. Regulatory Pathways in the European Union. MAbs 2011, 3, 241–242. [Google Scholar] [CrossRef]
- Han, J.; Kesselheim, A.S. First-In-Class Drugs Experienced Different Regulatory Treatment In The US And Europe. Health Aff (Millwood) 2025, 44, 265–273. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Sullivan, R.; Blagden, S. UK Oncology Approvals in 2022: Global Regulatory Collaboration and New Regulatory Pathways Deliver New Anti-Cancer Therapies. The Lancet Oncology 2023, 24, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Jena, A.B.; Calfee, J.E.; Mansley, E.C.; Philipson, T.J. “Me-Too” Innovation in Pharmaceutical Markets. Forum Health Econ Policy 2009, 12(5), 1558–9544.1138. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K.; Green, A.R. Me-too Pharmaceutical Products: History, Definitions, Examples, and Relevance to Drug Shortages and Essential Medicines Lists. Brit J Clinical Pharma 2020, 86, 2114–2122. [Google Scholar] [CrossRef]
- Okuyama, R. Chronological Analysis of First-in-Class Drugs Approved from 2011 to 2022: Their Technological Trend and Origin. Pharmaceutics 2023, 15, 1794. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Paquette, C. The Economics of Follow-on Drug Research and Development: Trends in Entry Rates and the Timing of Development. PharmacoEconomics 2004, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Faden, L.B. Competitiveness in Follow-on Drug R&D: A Race or Imitation? Nature Reviews Drug Discovery 2011, 10, 23–27. [Google Scholar]
- Lyu, X.; Zhao, Q.; Hui, J.; Wang, T.; Lin, M.; Wang, K.; Zhang, J.; Shentu, J.; Dalby, P.A.; Zhang, H.; et al. The Global Landscape of Approved Antibody Therapies. Antib Ther 2022, 5, 233–257. [Google Scholar] [CrossRef]
- Guerriaud, M.; Kohli, E. RNA-Based Drugs and Regulation: Toward a Necessary Evolution of the Definitions Issued from the European Union Legislation. Front. Med. 2022, 9, 1012497. [Google Scholar] [CrossRef]
- Matsuda, K.; Nonami, A.; Shinohara, K.; Nagai, S. Regulatory Approval of CAR-T Cell and BsAb Products for Lymphoid Neoplasms in the US, EU, and Japan. Clin Pharma and Therapeutics 2025, 118, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Bellino, S.; La Salvia, A.; Cometa, M.F.; Botta, R. Cell-Based Medicinal Products Approved in the European Union: Current Evidence and Perspectives. Front. Pharmacol. 2023, 14, 1200808. [Google Scholar] [CrossRef]
- European Commission Rare Diseases. EU Research on Rare Diseases. Available online: https://research-and-innovation.ec.europa.eu/research-area/health/rare-diseases_en?utm_source=copilot.com (accessed on 15.01.2026).
- Ringborg, U.; Berns, A.; Celis, J.E.; Heitor, M.; Tabernero, J.; Schüz, J.; Baumann, M.; Henrique, R.; Aapro, M.; Basu, P.; et al. The Porto European Cancer Research Summit 2021. Molecular Oncology 2021, 15, 2507–2543. [Google Scholar] [CrossRef] [PubMed]
- Batta, A.; Kalra, B.S.; Khirasaria, R. Trends in FDA Drug Approvals over Last 2 Decades: An Observational Study. J Family Med Prim Care 2020, 9, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rodier, C.; Bujar, M.; McAuslane, N.; Liberti, L.; Munro, J. New Drug Approvals in Six Major Authorities 2010–2019: Focus on Facilitated Regulatory Pathways and Internationalisation. R&D Briefing 2020, 77, 1–24. [Google Scholar]
- US Food & Drug Administration 2016 Novel Drugs Summary. Available online: https://fda.report/media/102618/2016-Novel-Drugs-Summary.pdf?utm_source=copilot.com (accessed on 15.01.2026).
- Daizadeh, I. US FDA Drug Approvals Are Persistent and Polycyclic: Insights into Economic Cycles, Innovation Dynamics, and National Policy. Ther Innov Regul Sci 2021, 55, 743–754. [Google Scholar] [CrossRef]
- Ebied, A.M.; Na, J.; Cooper-DeHoff, R.M. New Drug Approvals in 2018 - Another Record Year! Am J Med 2019, 132, 1038–1043. [Google Scholar] [CrossRef]
- Mitra-Majumdar, M.; Gunter, S.J.; Kesselheim, A.S.; Brown, B.L.; Joyce, K.W.; Ross, M.; Pham, C.; Avorn, J.; Darrow, J.J. Analysis of Supportive Evidence for US Food and Drug Administration Approvals of Novel Drugs in 2020. JAMA Netw Open 2022, 5, e2212454. [Google Scholar] [CrossRef]
- Wang, T.; McAuslane, N. New Drug Approvals in ICH Countries: 2002–2011. Center for Innovation in Regulatory Science, White Paper. Abgerufen. 2012, 11.
- Falcone, R.; Lombardi, P.; Filetti, M.; Duranti, S.; Pietragalla, A.; Fabi, A.; Lorusso, D.; Altamura, V.; Paroni Sterbini, F.; Scambia, G.; et al. Oncologic Drugs Approval in Europe for Solid Tumors: Overview of the Last 6 Years. Cancers 2022, 14, 889. [Google Scholar] [CrossRef]
- Yu, S.-R.; Choi, S. New Drug Expenditure by Therapeutic Area in South Korea: International Comparison and Policy Implications. Healthcare 2025, 13, 468. [Google Scholar] [CrossRef]
- Olivier, T.; Haslam, A.; Prasad, V. Anticancer Drugs Approved by the US Food and Drug Administration From 2009 to 2020 According to Their Mechanism of Action. JAMA Netw Open 2021, 4, e2138793. [Google Scholar] [CrossRef]
- Paul, S.; Zhou, S. Six Events That Shaped Antibody Approvals in Oncology. Front Immunol 2025, 16, 1533796. [Google Scholar] [CrossRef]
- Wojciechowski, D.; Wiseman, A. Long-Term Immunosuppression Management: Opportunities and Uncertainties. Clinical Journal of the American Society of Nephrology 2021, 16, 1264–1271. [Google Scholar] [CrossRef]
- Guo, G.; Zhong, W.; Zhao, H.; An, Y.; Dong, X.; Li, Z.; Qin, S.; Xu, G.; Yue, X.; Wang, X.; et al. Multifunctional Nano Immunostimulant: Overcoming Immunosuppressive Microenvironment for Antitumor Immunotherapy. Advanced Science 2025, e17480. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.; Denlinger, N.; Yang, Y. Recent Advances and Challenges in Cancer Immunotherapy. Cancers 2022, 14, 3972. [Google Scholar] [CrossRef]
- Frontiers in Clinical Drug Research: HIV; Atta-ur-Rahman, Ed.; Frontiers in Clinical Drug Research: HIV; BENTHAM SCIENCE PUBLISHERS, 2021; Vol. 5, ISBN 978-981-14-6445-4. [Google Scholar]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and Trends in the Development and Approval of Antiviral Medicines: 1987-2017 and Beyond. Antiviral Res 2018, 155, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Chahine, E.B.; Durham, S.H. Ibalizumab: The First Monoclonal Antibody for the Treatment of HIV-1 Infection. Ann Pharmacother 2021, 55, 230–239. [Google Scholar] [CrossRef]
- Zarębska-Michaluk, D.; Jaroszewicz, J.; Parfieniuk-Kowerda, A.; Janczewska, E.; Dybowska, D.; Pawłowska, M.; Halota, W.; Mazur, W.; Lorenc, B.; Janocha-Litwin, J.; et al. Effectiveness and Safety of Pangenotypic Regimens in the Most Difficult to Treat Population of Genotype 3 HCV Infected Cirrhotics. JCM 2021, 10, 3280. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; An, S.; Zhang, Z. Efficacy and Safety of Intravenous Peramivir versus Oral Oseltamivir in the Treatment of Influenza in Children: A Meta-Analysis. Journal of Clinical Virology Plus 2024, 4, 100179. [Google Scholar] [CrossRef]
- Fang, Y.-H.; Hsu, T.-H.; Lin, T.-Y.; Liu, C.-H.; Chou, S.-C.; Wu, J.-Y.; Perng, P.-C. Comparing Intravenous Peramivir with Oral Oseltamivir for Patients with Influenza: A Meta-Analysis of Randomized Controlled Trials. Expert Review of Anti-infective Therapy 2021, 19, 1039–1046. [Google Scholar] [CrossRef]
- Shiraishi, C.; Kato, H.; Hagihara, M.; Asai, N.; Iwamoto, T.; Mikamo, H. Comparison of Clinical Efficacy and Safety of Baloxavir Marboxil versus Oseltamivir as the Treatment for Influenza Virus Infections: A Systematic Review and Meta-Analysis. Journal of Infection and Chemotherapy 2024, 30, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Lee, H.W.; Tchesnokov, E.P.; Perry, J.K.; Feng, J.Y.; Bilello, J.P.; Porter, D.P.; Götte, M. Efficient Incorporation and Template-Dependent Polymerase Inhibition Are Major Determinants for the Broad-Spectrum Antiviral Activity of Remdesivir. Journal of Biological Chemistry 2022, 298, 101529. [Google Scholar] [CrossRef]
- Gastaldi, G.; Lucchini, B.; Thalmann, S.; Alder, S.; Laimer, M.; Brändle, M.; Wiesli, P.; Lehmann, R. Working Group Of The Sged/Ssed Swiss Recommendations of the Society for Endocrinology and Diabetes (SGED/SSED) for the Treatment of Type 2 Diabetes Mellitus (2023). Swiss Med Wkly 2023, 153, 40060. [Google Scholar] [CrossRef]
- Hahl, E.; Kurko, T.; Koskinen, H.; Airaksinen, M.; Sarnola, K. EMA Approved Orphan Medicines since the Implementation of the Orphan Legislation. Orphanet J Rare Dis 2025, 20, 266. [Google Scholar] [CrossRef]
- Wright, E.M. SGLT2 Inhibitors: Physiology and Pharmacology. Kidney360 2021, 2, 2027–2037. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.; Kristensen, S.L.; Branch, K.R.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.; Lopes, R.D.; McMurray, J.J.; Pratley, R.E.; et al. Cardiovascular, Mortality, and Kidney Outcomes with GLP-1 Receptor Agonists in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomised Trials. The lancet Diabetes & endocrinology 2021, 9, 653–662. [Google Scholar]
- Lou, J.; Duan, H.; Qin, Q.; Teng, Z.; Gan, F.; Zhou, X.; Zhou, X. Advances in Oral Drug Delivery Systems: Challenges and Opportunities. Pharmaceutics 2023, 15, 484. [Google Scholar] [CrossRef]
- Shahiwala, A. Formulation Approaches in Enhancement of Patient Compliance to Oral Drug Therapy. Expert Opinion on Drug Delivery 2011, 8, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Basit, A.W. Gastrointestinal Stability of Therapeutic Anti-TNF α IgG1 Monoclonal Antibodies. International Journal of Pharmaceutics 2016, 502, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Skalko-Basnet, N. Biologics: The Role of Delivery Systems in Improved Therapy. Biologics 2014, 8, 107–114. [Google Scholar] [CrossRef]
- Panchal, K.; Katke, S.; Dash, S.K.; Gaur, A.; Shinde, A.; Saha, N.; Mehra, N.K.; Chaurasiya, A. An Expanding Horizon of Complex Injectable Products: Development and Regulatory Considerations. Drug Deliv Transl Res 2023, 13, 433–472. [Google Scholar] [CrossRef]
- Hedaya, M.A. Routes of Drug Administration. In Pharmaceutics; Elsevier, 2024; pp. 537–554. ISBN 978-0-323-99796-6. [Google Scholar]
- Stielow, M.; Witczyńska, A.; Kubryń, N.; Fijałkowski, Ł.; Nowaczyk, J.; Nowaczyk, A. The Bioavailability of Drugs-The Current State of Knowledge. Molecules 2023, 28, 8038. [Google Scholar] [CrossRef]
- Pitiot, A.; Heuzé-Vourc’h, N.; Sécher, T. Alternative Routes of Administration for Therapeutic Antibodies-State of the Art. Antibodies (Basel) 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.D.; Bravo Padros, M.; Conrado, D.J.; Ganguly, S.; Guan, X.; Hassan, H.E.; Hazra, A.; Irvin, S.C.; Jayachandran, P.; Kosloski, M.P.; et al. Subcutaneous Administration of Monoclonal Antibodies: Pharmacology, Delivery, Immunogenicity, and Learnings From Applications to Clinical Development. Clin Pharma and Therapeutics 2024, 115, 422–439. [Google Scholar] [CrossRef]
- Surasi, D.S.; O’Malley, J.; Bhambhvani, P. 99mTc-Tilmanocept: A Novel Molecular Agent for Lymphatic Mapping and Sentinel Lymph Node Localization. J Nucl Med Technol 2015, 43, 87–91. [Google Scholar] [CrossRef]
- Rodriguez-Pardo, D.; Pigrau, C.; Campany, D.; Diaz-Brito, V.; Morata, L.; De Diego, I.C.; Sorlí, L.; Iftimie, S.; Pérez-Vidal, R.; García-Pardo, G.; et al. Effectiveness of Sequential Intravenous-to-Oral Antibiotic Switch Therapy in Hospitalized Patients with Gram-Positive Infection: The SEQUENCE Cohort Study. Eur J Clin Microbiol Infect Dis 2016, 35, 1269–1276. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Lapteva, L.; Purohit-Sheth, T.; Serabian, M.; Puri, R.K. Clinical Development of Gene Therapies: The First Three Decades and Counting. Mol Ther Methods Clin Dev 2020, 19, 387–397. [Google Scholar] [CrossRef]
- Palomo, G.M.; Pose-Boirazian, T.; Naumann-Winter, F.; Costa, E.; Duarte, D.M.; Kalland, M.E.; Malikova, E.; Matusevicius, D.; Vitezic, D.; Larsson, K.; et al. The European Landscape for Gene Therapies in Orphan Diseases: 6-Year Experience with the EMA Committee for Orphan Medicinal Products. Mol Ther 2023, 31, 3414–3423. [Google Scholar] [CrossRef]
- Avanthay, R.; Garcia-Nicolas, O.; Ruggli, N.; Grau-Roma, L.; Párraga-Ros, E.; Summerfield, A.; Zimmer, G. Evaluation of a Novel Intramuscular Prime/Intranasal Boost Vaccination Strategy against Influenza in the Pig Model. PLoS Pathog 2024, 20, e1012393. [Google Scholar] [CrossRef]
- Carvalho, M.; Sepodes, B.; Martins, A.P. Regulatory and Scientific Advancements in Gene Therapy: State-of-the-Art of Clinical Applications and of the Supporting European Regulatory Framework. Front. Med. 2017, 4, 182. [Google Scholar] [CrossRef]
- Giri, B.R.; Jakka, D.; Sandoval, M.A.; Kulkarni, V.R.; Bao, Q. Advancements in Ocular Therapy: A Review of Emerging Drug Delivery Approaches and Pharmaceutical Technologies. Pharmaceutics 2024, 16, 1325. [Google Scholar] [CrossRef]
- Gade, S.; So, Y.; Mishra, D.; Baviskar, S.M.; Assiri, A.A.; Glover, K.; Sheshala, R.; Vora, L.K.; Thakur, R.R.S. Ocular Drug Delivery: Emerging Approaches and Advances. Pharmaceutics 2025, 17, 599. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.W.; Akotoye, C.; Harris, A.; Ciulla, T.A. Beyond the Injection: Delivery Systems Reshaping Retinal Disease Management. Expert Opin Pharmacother 2025, 26, 939–952. [Google Scholar] [CrossRef]
- Zaid, A.N. A Comprehensive Review on Pharmaceutical Film Coating: Past, Present, and Future. Drug Des Devel Ther 2020, 14, 4613–4623. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Martin, J.H.; Mclachlan, A.J.; Boddy, A.V. Precision Dosing of Targeted Anticancer Drugs—Challenges in the Real World. Transl. Cancer Res 2017, 6, S1500–S1511. [Google Scholar] [CrossRef]
- Reyner, E.; Lum, B.; Jing, J.; Kagedal, M.; Ware, J.A.; Dickmann, L.J. Intrinsic and Extrinsic Pharmacokinetic Variability of Small Molecule Targeted Cancer Therapy. Clinical Translational Sci 2020, 13, 410–418. [Google Scholar] [CrossRef]
- Habet, S. Narrow Therapeutic Index Drugs: Clinical Pharmacology Perspective. Journal of Pharmacy and Pharmacology 2021, 73, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Lee, Y.; Zhou, T. Pharmacokinetic/Pharmacodynamic Modeling of Therapeutic Antibodies in Oncology: Current Advances and Future Perspectives. J. Pharm. Investig. 2025. [Google Scholar] [CrossRef]
- Chen, M.-L.; Shah, V.P.; Ganes, D.; Midha, K.K.; Caro, J.; Nambiar, P.; Rocci, M.L.; Thombre, A.G.; Abrahamsson, B.; Conner, D.; et al. Challenges and Opportunities in Establishing Scientific and Regulatory Standards for Assuring Therapeutic Equivalence of Modified-Release Products: Workshop Summary Report. European Journal of Pharmaceutical Sciences 2010, 40, 148–153. [Google Scholar] [CrossRef]
- Laichapis, M.; Sakulbumrungsil, R.; Udomaksorn, K.; Kessomboon, N.; Nerapusee, O.; Hongthong, C.; Poonpolsub, S. Financial Feasibility of Developing Sustained-Release Incrementally Modified Drugs in Thailand’s Pharmaceutical Industry: Mixed Methods Study. JMIRx Med 2025, 6, e65978. [Google Scholar] [CrossRef]
- Berger, J.; Dunn, J.D.; Johnson, M.M.; Karst, K.R.; Shear, W.C. How Drug Life-Cycle Management Patent Strategies May Impact Formulary Management. Am J Manag Care 2016, 22, S487–S495. [Google Scholar]
- Stegemann, S.; Tian, W.; Morgen, M.; Brown, S. CHAPTER 2. Hard Capsules in Modern Drug Delivery. In Drug Discovery; Tovey, G.D., Ed.; Royal Society of Chemistry: Cambridge, 2018; pp. 21–51. ISBN 978-1-84973-941-2. [Google Scholar]
- Tovey, G. Pharmaceutical Formulation: The Science and Technology of Dosage Forms; RSC Drug discovery series; Royal Society of Chemistry: Cambridge, 2018; ISBN 978-1-84973-941-2. [Google Scholar]
- Savla, R.; Browne, J. Softgel Formulations - Lipid-Based Drug Delivery System to Bring Poorly Soluble Drugs to Market. Drug development & Delivery. 2016. Available online: https://drug-dev.com/softgel-formulations-lipid-based-drug-delivery-system-to-bring-poorly-soluble-drugs-to-market/ (accessed on 15.01.2026).
- Sarvepalli, S.; Pasika, S.R.; Verma, V.; Thumma, A.; Bolla, S.; Nukala, P.K.; Butreddy, A.; Bolla, P.K. A Review on the Stability Challenges of Advanced Biologic Therapeutics. Pharmaceutics 2025, 17, 550. [Google Scholar] [CrossRef] [PubMed]
- Le Basle, Y.; Chennell, P.; Tokhadze, N.; Astier, A.; Sautou, V. Physicochemical Stability of Monoclonal Antibodies: A Review. Journal of Pharmaceutical Sciences 2020, 109, 169–190. [Google Scholar] [CrossRef]
- Cheng, Y.; Duong, H.T.T.; Hu, Q.; Shameem, M.; Tang, X. (Charlie) Practical Advice in the Development of a Lyophilized Protein Drug Product. Antibody Therapeutics 2025, 8, 13–25. [Google Scholar] [CrossRef]
- Butreddy, A.; Janga, K.Y.; Ajjarapu, S.; Sarabu, S.; Dudhipala, N. Instability of Therapeutic Proteins — An Overview of Stresses, Stabilization Mechanisms and Analytical Techniques Involved in Lyophilized Proteins. International Journal of Biological Macromolecules 2021, 167, 309–325. [Google Scholar] [CrossRef]
- Chandrababu, K.B.; Kannan, A.; Savage, J.R.; Stadmiller, S.; Ryle, A.E.; Cheung, C.; Kelley, R.F.; Maa, Y.; Saggu, M.; Bitterfield, D.L. Stability Comparison Between Microglassification and Lyophilization Using a Monoclonal Antibody. Journal of Pharmaceutical Sciences 2024, 113, 1054–1060. [Google Scholar] [CrossRef]
- Castañeda Ruiz, A.J.; Shetab Boushehri, M.A.; Phan, T.; Carle, S.; Garidel, P.; Buske, J.; Lamprecht, A. Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates. Pharmaceutics 2022, 14, 2575. [Google Scholar] [CrossRef] [PubMed]
- Bramham, J.E.; Davies, S.A.; Podmore, A.; Golovanov, A.P. Stability of a High-Concentration Monoclonal Antibody Solution Produced by Liquid-Liquid Phase Separation. MAbs 2021, 13, 1940666. [Google Scholar] [CrossRef]
- Bas, T.G. Innovative Formulation Strategies for Biosimilars: Trends Focused on Buffer-Free Systems, Safety, Regulatory Alignment, and Intellectual Property Challenges. Pharmaceuticals 2025, 18, 908. [Google Scholar] [CrossRef]
- Makwana, S.; Basu, B.; Makasana, Y.; Dharamsi, A. Prefilled Syringes: An Innovation in Parenteral Packaging. Int J Pharm Investig 2011, 1, 200–206. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs 2018, 32, 425–440. [Google Scholar] [CrossRef]
- Hsiao, W.-K.; Herbig, M.E.; Newsam, J.M.; Gottwald, U.; May, E.; Winckle, G.; Birngruber, T. Opportunities of Topical Drug Products in a Changing Dermatological Landscape. European Journal of Pharmaceutical Sciences 2024, 203, 106913. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lockwood, A. Topical Ocular Drug Delivery Systems: Innovations for an Unmet Need. Experimental Eye Research 2022, 218, 109006. [Google Scholar] [CrossRef] [PubMed]
- Freitas, E.; Gooderham, M.; Torres, T. New Topical Therapies in Development for Atopic Dermatitis. Drugs 2022, 82, 843–853. [Google Scholar] [CrossRef]
- Lie, E.; Choi, M.; Wang, S.-P.; Eichenfield, L.F. Topical Management of Pediatric Psoriasis: A Review of New Developments and Existing Therapies. Pediatr Drugs 2024, 26, 9–18. [Google Scholar] [CrossRef]
- Morgan-Warren, P.J.; Morarji, J.B. Trends in Licence Approvals for Ophthalmic Medicines in the United Kingdom. Eye 2020, 34, 1856–1865. [Google Scholar] [CrossRef]
- Vieira, I.; Sousa, J.J.; Vitorino, C. Paediatric Medicines – Regulatory Drivers, Restraints, Opportunities and Challenges. Journal of Pharmaceutical Sciences 2021, 110, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Maderuelo, C.; Lanao, J.M.; Zarzuelo, A. Enteric Coating of Oral Solid Dosage Forms as a Tool to Improve Drug Bioavailability. Eur J Pharm Sci 2019, 138, 105019. [Google Scholar] [CrossRef]
- Gerth, A.; Opitz, C.H.; Lee, K. Manufacturing, Approval and Market Launch of Tissue Engineered Products in Europe. Cytotherapy 2024, 26, S107–S108. [Google Scholar] [CrossRef]
- Pellegrini, G.; Ardigò, D.; Milazzo, G.; Iotti, G.; Guatelli, P.; Pelosi, D.; De Luca, M. Navigating Market Authorization: The Path Holoclar Took to Become the First Stem Cell Product Approved in the European Union. Stem Cells Translational Medicine 2018, 7, 146–154. [Google Scholar] [CrossRef]
- Taghi, H.S.; Al-hakeem, M.A.; Almajidi, Y.Q. The Evolution of Drug Delivery Systems: Historical Advances and Future Directions. Journal of Pharmacology and Drug Development 2025, 3, 13–32. [Google Scholar]
- Bassand, C.; Villois, A.; Gianola, L.; Laue, G.; Ramazani, F.; Riebesehl, B.; Sanchez-Felix, M.; Sedo, K.; Ullrich, T.; Duvnjak Romic, M. Smart Design of Patient-Centric Long-Acting Products: From Preclinical to Marketed Pipeline Trends and Opportunities. Expert Opinion on Drug Delivery 2022, 19, 1265–1283. [Google Scholar] [CrossRef]
- Pijarowska-Kruszyna, J.; Garnuszek, P.; Decristoforo, C.; Mikołajczak, R. Radiopharmaceutical Precursors. Theranostics: An Old Concept in New Clothing 2021, 3. [Google Scholar]
- Gillings, N.; Hjelstuen, O.; Ballinger, J.; Behe, M.; Decristoforo, C.; Elsinga, P.; Ferrari, V.; Peitl, P.K.; Koziorowski, J.; Laverman, P.; et al. Guideline on Current Good Radiopharmacy Practice (cGRPP) for the Small-Scale Preparation of Radiopharmaceuticals. EJNMMI radiopharmacy and chemistry 2021, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Ioppolo, J.A.; Alvarez de Eulate, E.; Cullen, D.R.; Mohamed, S.; Morandeau, L. Development of a High-Performance Liquid Chromatography Method for Rapid Radiochemical Purity Measurement of [18F] PSMA-1007, a PET Radiopharmaceutical for Detection of Prostate Cancer. Journal of Labelled Compounds and Radiopharmaceuticals 2023, 66, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P. Theranostics in the Indian Subcontinent: Opportunities, Challenges, and the Path Forward. Nuclear Medicine and Molecular Imaging 2025, 1–11. [Google Scholar] [CrossRef]
- Feng, Y.; Shao, Y.; Li, Z.; Luo, M.; Xu, D.; Ma, L. Research Progress on Major Medical Radionuclide Generators. Processes 2025, 13, 521. [Google Scholar] [CrossRef]
- Hahl, E.; Kurko, T.; Koskinen, H.; Airaksinen, M.; Sarnola, K. EMA Approved Orphan Medicines since the Implementation of the Orphan Legislation. Orphanet J Rare Dis 2025, 20, 266. [Google Scholar] [CrossRef]
- Rodriguez-Monguio, R.; Spargo, T.; Seoane-Vazquez, E. Ethical Imperatives of Timely Access to Orphan Drugs: Is Possible to Reconcile Economic Incentives and Patients’ Health Needs? Orphanet J Rare Dis 2017, 12, 1. [Google Scholar] [CrossRef]
- Sarpatwari, A.; Beall, R.F.; Abdurrob, A.; He, M.; Kesselheim, A.S. Evaluating The Impact Of The Orphan Drug Act’s Seven-Year Market Exclusivity Period. Health Aff (Millwood) 2018, 37, 732–737. [Google Scholar] [CrossRef]
- Gu, J.; Wu, Q.; Zhang, Q.; You, Q.; Wang, L. A Decade of Approved First-in-Class Small Molecule Orphan Drugs: Achievements, Challenges and Perspectives. Eur J Med Chem 2022, 243, 114742. [Google Scholar] [CrossRef]
- Kantor, A.; Haga, S.B. The Potential Benefit of Expedited Development and Approval Programs in Precision Medicine. JPM 2021, 11, 45. [Google Scholar] [CrossRef]
- Cacoub, E.; Lefebvre, N.B.; Milunov, D.; Sarkar, M.; Saha, S. Quantifying Hope: An EU Perspective of Rare Disease Therapeutic Space and Market Dynamics. Front. Public Health 2025, 13, 1520467. [Google Scholar] [CrossRef]
- Chambers, J.D.; Silver, M.C.; Berklein, F.C.; Cohen, J.T.; Neumann, P.J. Orphan Drugs Offer Larger Health Gains but Less Favorable Cost-Effectiveness than Non-Orphan Drugs. J Gen Intern Med 2020, 35, 2629–2636. [Google Scholar] [CrossRef]
- Postma, M.J.; Noone, D.; Rozenbaum, M.H.; Carter, J.A.; Botteman, M.F.; Fenwick, E.; Garrison, L.P. Assessing the Value of Orphan Drugs Using Conventional Cost-Effectiveness Analysis: Is It Fit for Purpose? Orphanet J Rare Dis 2022, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Alum, E.U.; Ekpang Ii, J.E.; Ekpang, P.O.; Ainebyoona, C.; Nwagu, K.E.; Nwuruku, O.A.; Muhammad, K. Toward an Ethical Future for Orphan Drugs: Balancing Access, Affordability, and Innovation. J Med Econ 2025, 28, 1869–1886. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Spencer, E.A.; Thompson, M.J.; Heneghan, C.J. Effectiveness, Safety and Costs of Orphan Drugs: An Evidence-Based Review. BMJ Open 2015, 5, e007199. [Google Scholar] [CrossRef]
Figure 1.
Distribution of the 357 medicinal products by their innovation status: first-in-class, advance-in-class or addition to class.
Figure 1.
Distribution of the 357 medicinal products by their innovation status: first-in-class, advance-in-class or addition to class.
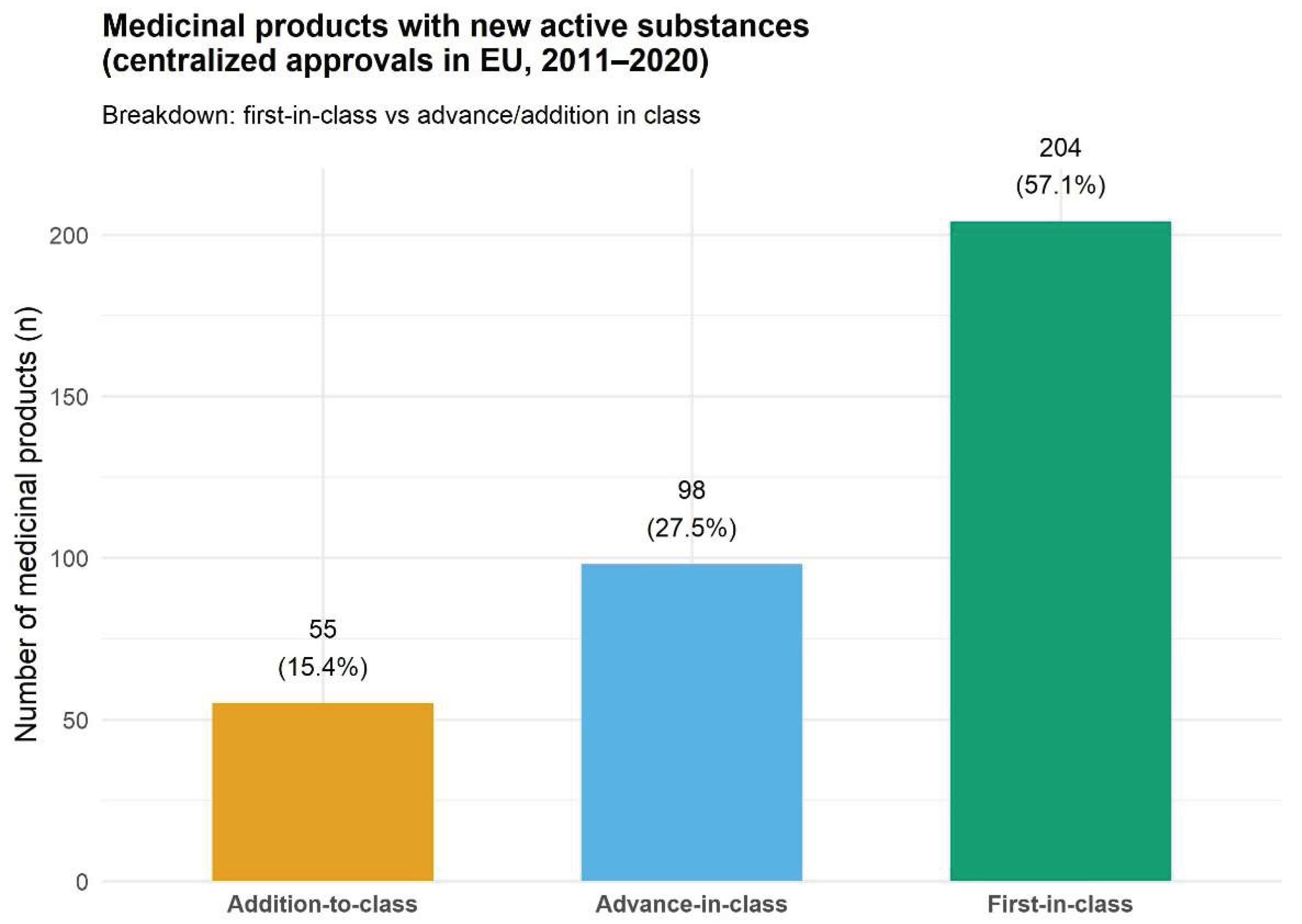
Figure 2.
Number of new marketing authorizations (MAs) granted yearly by the European Commission between 2011 and 2020 within the European Union, showing year-to-year variation and an overall upward trend despite intermittent declines.
Figure 2.
Number of new marketing authorizations (MAs) granted yearly by the European Commission between 2011 and 2020 within the European Union, showing year-to-year variation and an overall upward trend despite intermittent declines.
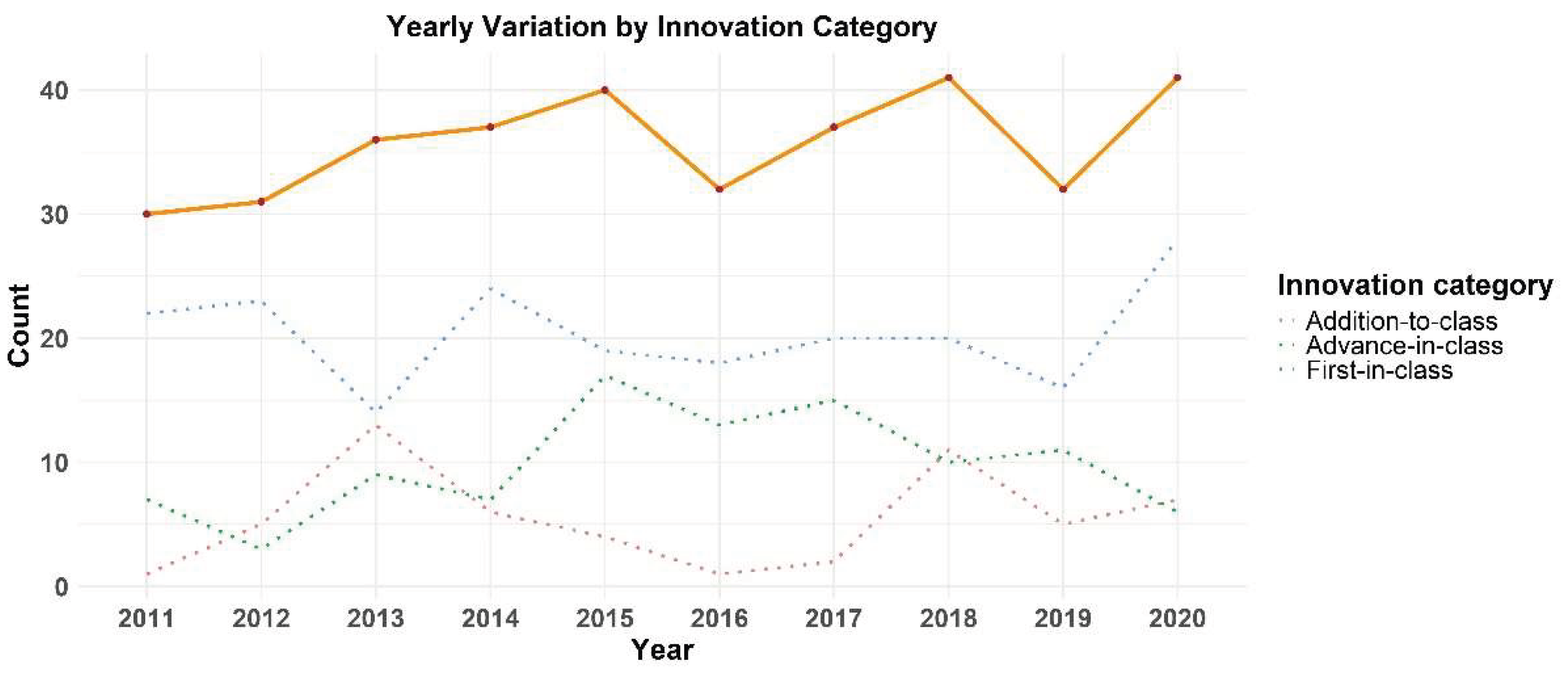
Figure 3.
Distribution of innovation types by orphan drug status (counts and corresponding percentages).
Figure 3.
Distribution of innovation types by orphan drug status (counts and corresponding percentages).
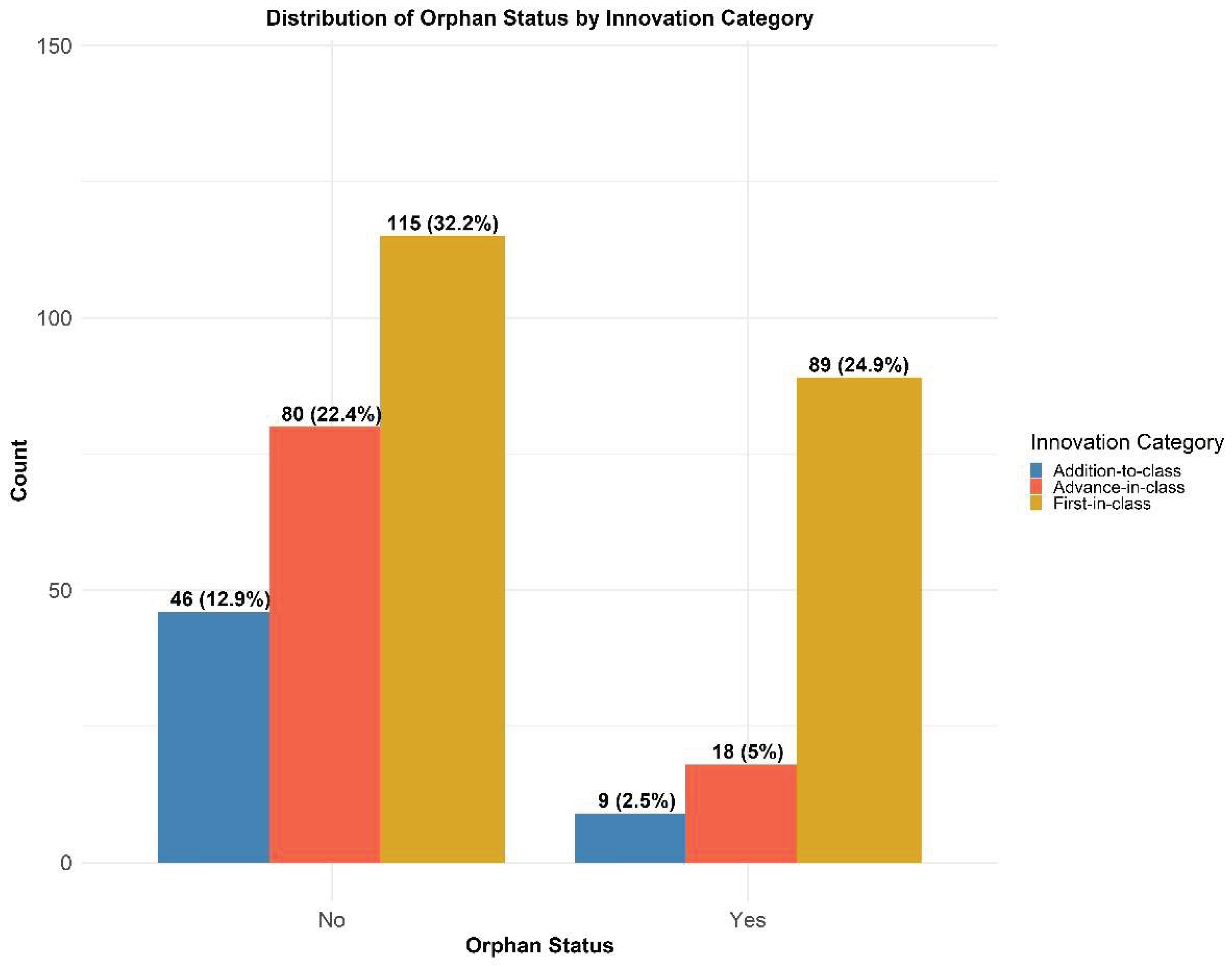

Table 1.
Distribution of the 357 new medicinal products by the ATC code.
| ATC Code | Category | Drug Count |
|---|---|---|
| L | Antineoplastic and immunomodulating agents | 129 |
| J | Antiinfectives for systemic use | 67 |
| A | Alimentary tract and metabolism | 38 |
| B | Blood and blood forming organs | 34 |
| N | Nervous system | 24 |
| V | Various | 18 |
| C | Cardiovascular system | 11 |
| M | Musculo-skeletal system | 10 |
| R | Respiratory system | 10 |
| D | Dermatologicals | 7 |
| S | Sensory organs | 7 |
| G | Genito-urinary system and sex hormones | 5 |
| H | Systemic hormonal preparations | 3 |
Table 2.
Distribution of the 357 medicinal products by the technology class used.
| Technology class | Number of medicinal products | First in class (% within column) |
Advance in class (% within column) | Addition to class (% within column) |
|---|---|---|---|---|
| Small molecules | 157 | 80 (39.22%) | 50 (51.02%) | 27 (49.09%) |
| Monoclonal antibodies | 64 | 45 (22.06%) | 6 (6.12%) | 13 (23.64%) |
| Proteins/polypeptides | 51 | 33 (16.18%) | 11 (11.22%) | 7 (12.73%) |
| FDCs | 27 | 4 (1.96%) | 18 (18.37%) | 5 (9.09%) |
| Vaccines | 18 | 9 (4.41%) | 9 (9.18%) | 0 (0.00%) |
| Radiopharmaceuticals | 9 | 7 (3.43%) | 0 (0.00%) | 2 (3.64%) |
| ATMP cell therapies | 5 | 5 (2.45%) | 0 (0.00%) | 0 (0.00%) |
| Gene therapies (excluding CAR-T cells) | 5 | 5 (2.45%) | 0 (0.00%) | 0 (0.00%) |
| siRNA | 4 | 4 (1.96%) | 0 (0.00%) | 0 (0.00%) |
| Complex, non-herbal | 5 | 2 (0.98%) | 2 (2.04%) | 1 (1.82%) |
| CAR-T cells | 3 | 3 (1.47%) | 0 (0.00%) | 0 (0.00%) |
| Antisense oligonucleotides | 3 | 3 (1.47%) | 0 (0.00%) | 0 (0.00%) |
| Complex, herbal | 3 | 3 (1.47%) | 0 (0.00%) | 0 (0.00%) |
| Glycopeptides | 3 | 1 (0.49%) | 2 (2.04%) | 0 (0.00%) |
| Total | 357 | 204 (57.14%) | 98 (27.45%) | 55 (15.41%) |
Table 3.
Distribution of the 357 products by administration route and the innovation level.
| Administration_route | Addition-to-class | Advance-in-class | First-in-class |
|---|---|---|---|
| Oral | 28 | 57 | 73 |
| IV | 14 | 17 | 75 |
| SC | 7 | 9 | 28 |
| IM | 1 | 7 | 5 |
| Topical | 0 | 0 | 5 |
| Intralesional | 0 | 0 | 3 |
| Ophthalmic | 0 | 0 | 2 |
| Oral + IV | 0 | 3 | 1 |
| Nasal | 0 | 2 | 1 |
| Intravitreal | 1 | 0 | 1 |
| ID + SC + intratumoral + peritumoral injection (following radiolabelling) | 0 | 0 | 1 |
| In vitro radiolabelling of medicinal products, which are subsequently administered by the approved route | 0 | 0 | 1 |
| Intra-articular | 0 | 0 | 1 |
| Intracerebroventricular | 0 | 0 | 1 |
| Intrathecal | 0 | 0 | 1 |
| IV (first dose) + SC (subsequent doses) | 0 | 0 | 1 |
| Ocular (surgical implantation onto the corneal surface / under the conjunctiva) | 0 | 0 | 1 |
| SC (implant) | 0 | 0 | 1 |
| Subretinal | 0 | 0 | 1 |
| Vaginal | 0 | 0 | 1 |
| Inhalation | 3 | 1 | 0 |
| Oral + gastroenteral | 0 | 1 | 0 |
| Oral + IM | 0 | 1 | 0 |
| Oral + enteral | 1 | 0 | 0 |
Table 4.
Distribution of medicinal products by grouped administration routes across innovation classes. Individual delivery methods were consolidated into four primary categories: Parenteral (injections, infusions, and implants), Oral, Mixed (two or more alternative or concomitant routes), and Topical/Local (ophthalmic, intranasal, inhalation, and vaginal routes).
Table 4.
Distribution of medicinal products by grouped administration routes across innovation classes. Individual delivery methods were consolidated into four primary categories: Parenteral (injections, infusions, and implants), Oral, Mixed (two or more alternative or concomitant routes), and Topical/Local (ophthalmic, intranasal, inhalation, and vaginal routes).
| Administration_route | Addition-to-class (% within column) |
Advance-in-class (% within column) |
First-in-class (% within column) |
|---|---|---|---|
| Mixed | 1 | 5 | 3 |
| Oral | 28 | 57 | 73 |
| Parenteral | 23 | 33 | 116 |
| Topical/Local | 3 | 3 | 9 |
| Other | 0 | 0 | 3 |
Table 5.
Frequency distribution of pharmaceutical forms for the 357 medicinal products, sorted in descending order of occurrence. Pharmaceutical forms appearing a single time are consolidated under the “Others” category.
Table 5.
Frequency distribution of pharmaceutical forms for the 357 medicinal products, sorted in descending order of occurrence. Pharmaceutical forms appearing a single time are consolidated under the “Others” category.
| Dosage_form | Count |
|---|---|
| Film-coated tablet | 98 |
| Hard capsule | 38 |
| Concentrate for solution for infusion | 35 |
| Solution for injection | 35 |
| Others | 33 |
| Powder for concentrate for solution for infusion | 23 |
| Powder and solvent for solution for injection | 21 |
| Dispersion for infusion | 7 |
| Powder for solution for injection | 6 |
| Tablet | 6 |
| Solution for infusion | 5 |
| Suspension for injection | 5 |
| Prolonged-release tablet | 4 |
| Soft capsule | 4 |
| Gel | 3 |
| Powder for solution for infusion | 3 |
| Solution for injection (pre-filled pen) | 3 |
| Solution for injection in pre-filled pen | 3 |
| Solution for injection in pre-filled syringe | 3 |
| Dispersion for injection | 2 |
| Eye drops, solution | 2 |
| Film-coated tablet + powder for concentrate for solution for infusion | 2 |
| Inhalation powder, pre-dispensed | 2 |
| Nasal spray, suspension | 2 |
| Powder for concentrate and solution for solution for infusion | 2 |
| Powder for concentrate for solution for injection/infusion | 2 |
| Powder for oral suspension | 2 |
| Radiopharmaceutical precursor, solution | 2 |
| Solution for injection (pre-filled pen/syringe) | 2 |
| Suspension for injection (pre-filled syringe) | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



