
Submitted:
19 January 2026
Posted:
20 January 2026
You are already at the latest version
Abstract
Prostate cancer remains a leading cause of male cancer mortality, principally due to the inevitable transition from androgen-sensitive disease to lethal castration-resistant prostate cancer (CRPC). This review synthesizes the multifaceted molecular mechanisms driving this therapeutic failure, moving beyond a catalogue of signaling nodes to a structural and kinetic analysis of resistance networks. We dissect the reactivation of Androgen Receptor (AR) signaling through the lens of key kinase cascades, specifically the PI3K/AKT/mTOR and MAPK axes, and their regulation of the steroidogenic gatekeeper CYP17A1. Special emphasis is placed on the structural determinants of CYP17A1 activity, including the critical Asn202 gating mechanism and the "backdoor" pathway of dihydrotestosterone (DHT) biosynthesis that bypasses canonical testosterone intermediates. Furthermore, we integrate the "hydrophobic spine" hypothesis of kinase activation to explain the efficacy and failure of Type I and Type II kinase inhibitors. In parallel, we unravel the regulatory complexity of non-coding RNAs (ncRNAs), detailing how upregulated transcripts like LINC00675 and Lnc-ZNF30-3 function as dynamic scaffolds and competing endogenous RNAs (ceRNAs) to stabilize AR and promote epithelial-mesenchymal transition (EMT). By converging these insights with emerging therapeutic strategies, such as next-generation lipid nanoparticle (LNP)-mediated RNA interference utilizing hexagonal HII phase endosomal escape, we provide a comprehensive roadmap for dismantling the molecular fortresses of advanced prostate cancer.
Keywords:
prostate cancer
; steroids
; MAPK
; androgen receptor
; ncRNA
1. Introduction (Overview)
Despite the initial efficacy of Androgen Deprivation Therapy (ADT), the transition to lethal castration-resistant prostate cancer (CRPC) remains a pervasive clinical reality, driven by complex molecular adaptations [1,2,3]. The disease is primarily driven by elevated androgen levels, a condition known as hyperandrogenism [4]. Men over 65 are at higher risk of developing prostate cancer [5]. Androgen biosynthesis involves multiple enzymes, particularly cytochrome P450 enzymes, which regulate different stages of metabolism. A critical step in generating key androgen precursors, such as dihydrotestosterone (DHT) and dehydroepiandrosterone (DHEA), occurs in the adrenal gland and gonads and is catalyzed by CYP17A1 and HSD3β1 [6]. CYP17A1 is a membrane-associated hemoprotein located in the endoplasmic reticulum [7]. Crystallographic data reveals that substrate binding is highly specific; physiological substrates such as pregnenolone and progesterone bind within the active site at an angle of approximately 60 degrees relative to the plane of the heme group [8]. This orientation is not trivial; it is the structural determinant that dictates the site of oxidation. The enzyme's dual functionality, acting as both a 17α-hydroxylase and a 17,20-lyase, is governed by subtle variations in this substrate positioning [9]. CYP17A1 is a major regulator of androgen production, exhibiting two distinct activities: hydroxylase and lyase [10,11,12]. CYP17A1 is indispensable for catalyzing critical steps in both the canonical and the alternative 'backdoor' pathways of androgen biosynthesis, positioning it as a metabolic linchpin in the reprogramming of prostate tumors. Through its hydroxylase activity, CYP17A1 hydroxylates the 17-carbon (C17) position of pregnenolone and progesterone, producing 17α-hydroxypregnenolone and 17α-hydroxyprogesterone, respectively [13]. Its 17,20-lyase activity then facilitates the conversion of these intermediates into DHEA and androstenedione, key precursors in testosterone synthesis [1]. Furthermore, CYP17A1 plays a crucial role in intratumoral androgen biosynthesis in castration-resistant prostate cancer (CRPC), emphasizing its significance in the treatment of advanced prostate cancer [14]. In the canonical pathway, cholesterol proceeds through pregnenolone to progesterone to androstenedione to testosterone to DHT. The backdoor pathway represents a metabolic detour that allows for the synthesis of potent androgens without passing through testosterone intermediates. This pathway is initiated by the 5α-reduction of 17α-hydroxyprogesterone by SRD5A1, a step that commits the steroid to the backdoor route. Subsequently, the reductive 3α-hydroxysteroid dehydrogenases (AKR1C2 and AKR1C4) convert 5α-pregnan-17α-ol-3,20-dione into 17OH-allopregnanolone. It is this intermediate that serves as the substrate for the CYP17A1 lyase reaction, yielding androsterone, often dismissed as a weak androgen or waste product, becomes the primary precursor in this pathway [15,16]. In CRPC, the upregulation of AKR1C3 facilitates the conversion of androsterone directly to androstanediol and finally oxidized to Dihydrotestosterone (DHT) by oxidative 3α-HSDs (like AKR1C3), thereby bypassing the therapeutic blockade of canonical testosterone synthesis [9]. In CRPC, tumors often upregulate AKR1C3 and SRD5A1. This allows the tumor to synthesize the most potent androgen, DHT, without ever producing testosterone and without relying on the adrenal precursors targeted by standard therapies. This explains why patients with undetectable serum testosterone can still have high intratumoral DHT and progressing disease.
The distinction between hydroxylase and lyase activity is the holy grail of CYP17A1 inhibition [17,18]. The 17,20-lyase reaction, which is the gateway to androgen production (converting C21 steroids to C19 androgens), requires an additional interaction with cytochrome b5 (cyt b5) [7,12,19]. The selectivity of CYP17A1 for 17α-hydroxypregnenolone over 17α-hydroxyprogesterone in the lyase reaction is not merely a matter of binding affinity (Kd), but of catalytic efficiency (kcat). The presence of cytochrome b5 acts as an allosteric effector that accelerates the lyase reaction by orders of magnitude. This protein-protein interaction induces a conformational change in CYP17A1, likely widening the substrate access channel or altering the redox potential of the heme to favor the lyase reaction. The structural basis for this is likely the alteration of the redox potential of the heme iron, making it a more potent oxidant capable of attacking the C17-C20 bond. However, the Asn202 residue creates a hydrogen bond network that rigidly holds the substrate. For the lyase reaction to proceed, the substrate must possess a degree of flexibility to align with the activated oxygen species. The backdoor substrates, such as 17OH-allopregnanolone, possess a 3α-hydroxyl group and a planar A/B ring fusion (trans) that fits optimally into the active site even in the absence of cyt b5 facilitation, explaining why the backdoor pathway remains active even when canonical regulation is disrupted. Current inhibitors like abiraterone bind to the heme iron via a pyridine nitrogen, mimicking the substrate but blocking catalysis. However, because abiraterone inhibits both hydroxylase and lyase activities competitively, it leads to a buildup of upstream mineralocorticoids (like deoxycorticosterone), causing hypertension and hypokalemia. Future drug design must target the interface between CYP17A1 and cyt b5, or exploit the subtle metastable binding sites used during the multistep ligand entry process, to achieve lyase-specific inhibition. This would cut off androgen supply without disrupting cortisol biosynthesis, obviating the need for prednisone co-administration.
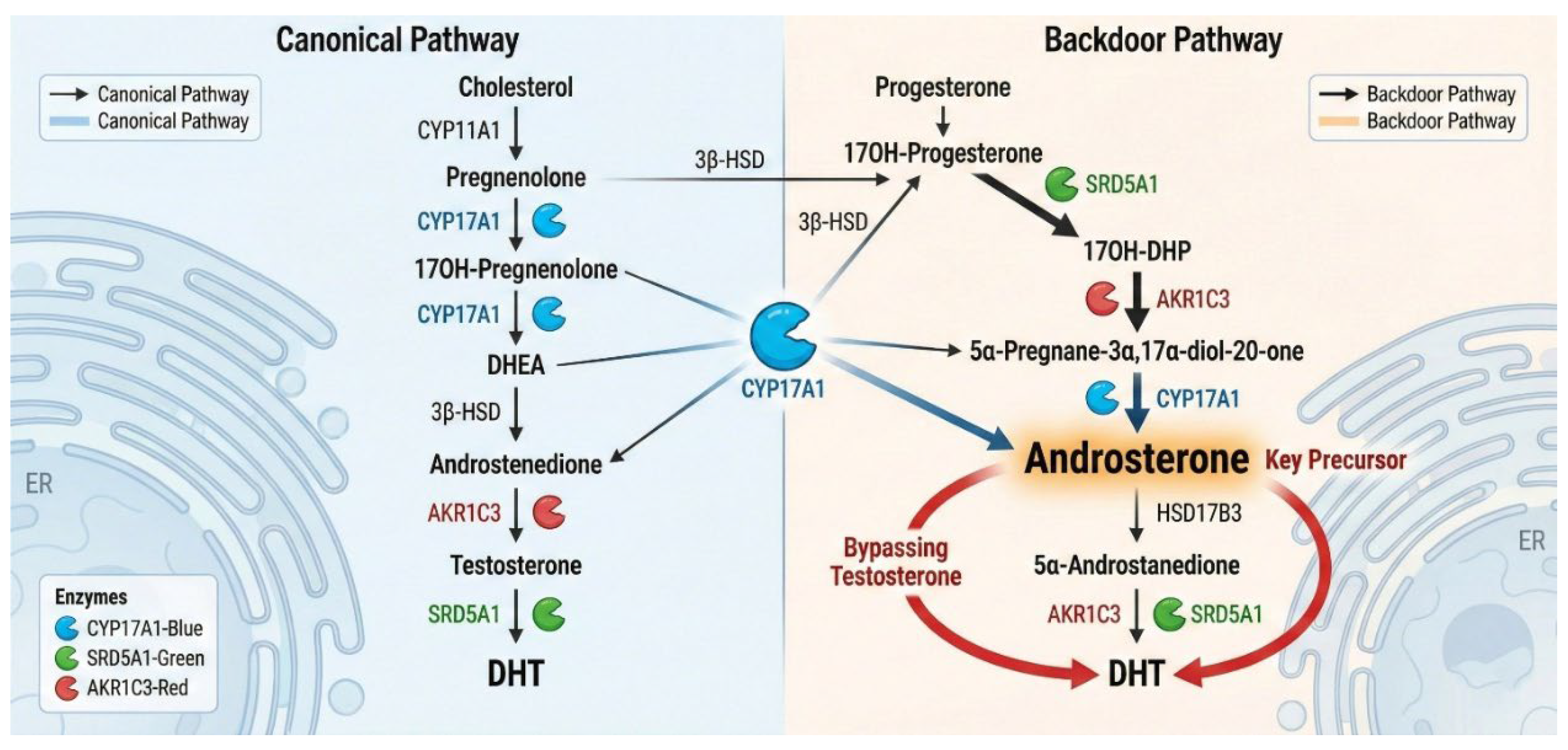
Figure 1.
The "Backdoor" Pathway of Dihydrotestosterone (DHT) Biosynthesis. Metabolic flux diagram illustrating the alternative "backdoor" route prevalent in CRPC. Unlike the canonical pathway (left), which requires testosterone as an intermediate, the backdoor pathway (right) converts 17OH-Progesterone to DHT via 5α-reduction (SRD5A1) and reductive 3α-hydroxysteroid dehydrogenases (AKR1C2/4). The molecule androsterone serves as the key precursor in this pathway, ultimately oxidized by AKR1C3 to produce potent DHT without elevating serum testosterone levels.
Figure 1.
The "Backdoor" Pathway of Dihydrotestosterone (DHT) Biosynthesis. Metabolic flux diagram illustrating the alternative "backdoor" route prevalent in CRPC. Unlike the canonical pathway (left), which requires testosterone as an intermediate, the backdoor pathway (right) converts 17OH-Progesterone to DHT via 5α-reduction (SRD5A1) and reductive 3α-hydroxysteroid dehydrogenases (AKR1C2/4). The molecule androsterone serves as the key precursor in this pathway, ultimately oxidized by AKR1C3 to produce potent DHT without elevating serum testosterone levels.
Early-stage prostate cancer can be effectively treated with surgery or radiation therapy. However, once the cancer metastasizes, hormonal therapy, such as androgen deprivation therapy (ADT), becomes the primary treatment [20,21]. Despite the initial efficacy of ADT, the development of therapeutic resistance is a pervasive clinical reality. This resistance precipitates the transition to castration-resistant prostate cancer (CRPC), a lethal phenotype characterized by the reactivation of AR signaling despite castrate serum testosterone levels [22]. This resistance arises due to mechanisms such as androgen receptor (AR) mutations, abnormal activation, amplification, and alternative splicing of AR variants, which restore AR signaling and drive androgen-independent prostate cancer [2].
To address these challenges, research efforts have focused on developing alternative and combination treatments, leading to the targeting of androgen receptor pathways with therapeutic drugs like enzalutamide and apalutamide. Additionally, promising treatments such as PROTACs, PARP inhibitors, HDAC inhibitors, and immunotherapy are being explored [23,24]. Abiraterone, approved by the FDA in 2011, is used to treat prostate cancer by inhibiting CYP17A1 activity [25]. It is a selective inhibitor of both the lyase and hydroxylase activities of CYP17A1. The use of abiraterone has been shown to reduce prostate-specific antigen (PSA) levels, alleviate pain, and improve both survival rates and overall health in prostate cancer patients [26]. However, blocking CYP17A1 hydroxylase activity leads to excessive mineralocorticoid production, resulting in adverse effects such as hypertension, hypokalemia, and cardiovascular disorders [1,27,28].
Posttranslational phosphorylation of CYP17A1 at the serine/threonine domain by MAPK14 has been proposed to play a significant role in androgen biosynthesis by enhancing CYP17A1 17,20-lyase activity and promoting its interaction with cytochrome P450 oxidoreductase (POR) [29]. This molecular event is one of many downstream outcomes of the deregulated intracellular signaling cascades that underpin prostate cancer progression. Key oncogenic pathways include the PI3K/AKT/mTOR signaling axis, which is frequently activated due to loss of PTEN, a negative regulator of PI3K activity. PTEN loss, observed in a significant proportion of prostate cancer patients, leads to constitutive activation of AKT, promoting cell proliferation, metabolic adaptation, and resistance to apoptosis and ADT [30,31]. Simultaneously, AR signaling remains central to prostate tumor biology. Upon activation by androgens, AR translocates to the nucleus and regulates transcription of genes involved in growth, differentiation, and survival. Even under androgen-deprived conditions such as after ADT prostate cancer cells often reactivate AR signaling through gene amplification, ligand-independent activation, or expression of constitutively active splice variants, sustaining tumor progression in the CRPC [32,33]. The EGFR/FGFR–MEK–ERK pathway also contributes significantly to disease progression, promoting cell cycle progression, proliferation, and survival via RAS-dependent and -independent mechanisms. The activation of this cascade supports mitogenic signaling and has been linked to therapeutic resistance[34].
Moreover, the TGF-β/SMAD signaling pathway exerts complex, stage-specific roles in prostate cancer. While it acts as a tumor suppressor in early stages by inducing cell cycle arrest and apoptosis, it paradoxically promotes epithelial–mesenchymal transition (EMT), immune escape, and metastasis during advanced disease. These dynamic signaling networks do not function in isolation; rather, they exhibit substantial crosstalk and feedback regulation, collectively driving tumor evolution, metastasis, and resistance to therapy [35,36].
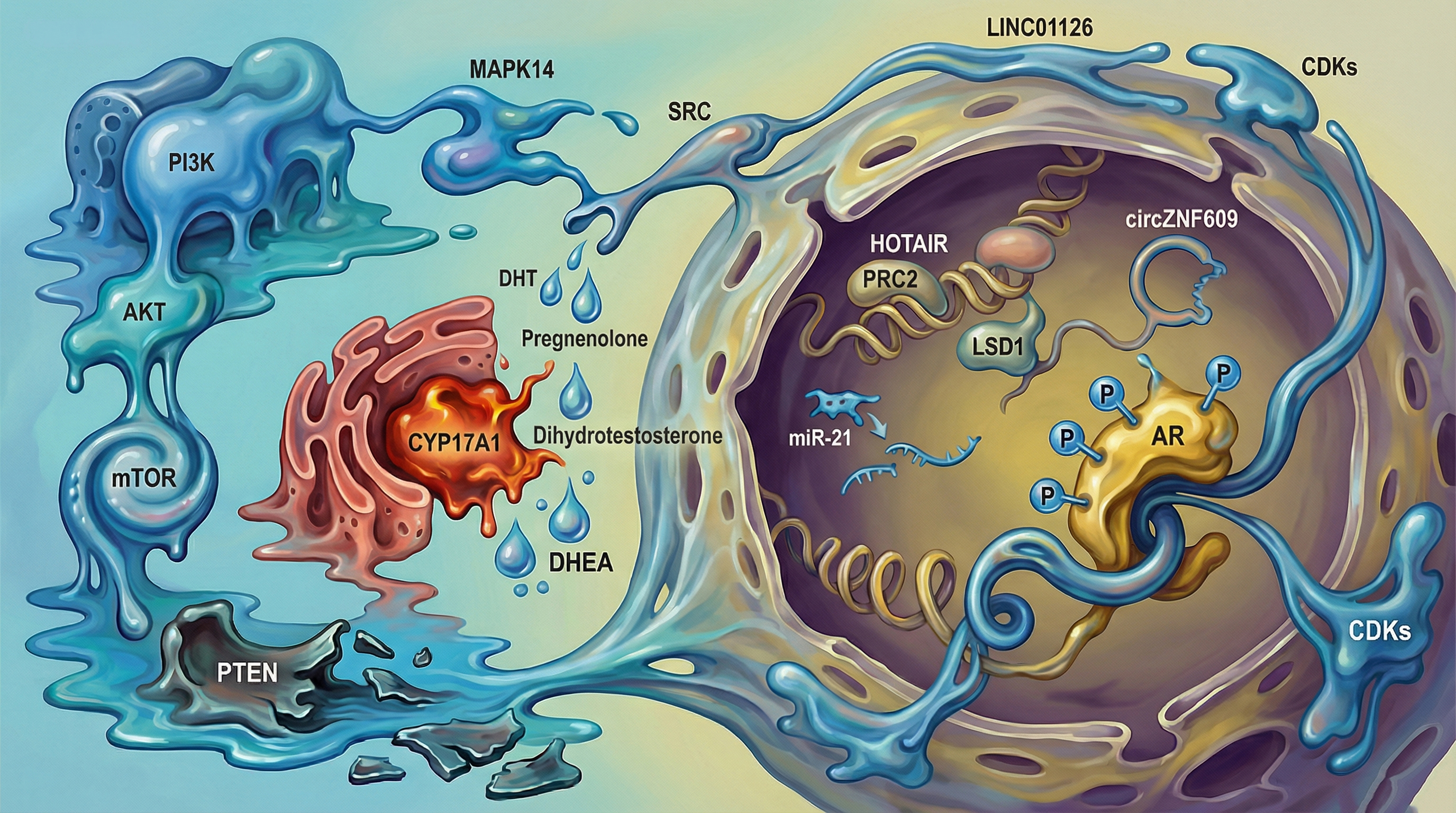
Figure 2.
The Molecular Landscape of Castration-Resistant Prostate Cancer. A comprehensive visualization of the intracellular signaling networks driving CRPC. The diagram highlights the hyperactivation of the PI3K/AKT/mTOR cascade (blue) and its crosstalk with the Androgen Receptor (AR). The cytochrome P450 enzyme CYP17A1 enzymes (orange) on the Endoplasmic Reticulum, facilitates intratumoral steroidogenesis. In the nucleus, long non-coding RNAs (lncRNAs) act as scaffolds for chromatin-modifying complexes (PRC2), altering the epigenetic landscape to sustain survival gene expression.
Figure 2.
The Molecular Landscape of Castration-Resistant Prostate Cancer. A comprehensive visualization of the intracellular signaling networks driving CRPC. The diagram highlights the hyperactivation of the PI3K/AKT/mTOR cascade (blue) and its crosstalk with the Androgen Receptor (AR). The cytochrome P450 enzyme CYP17A1 enzymes (orange) on the Endoplasmic Reticulum, facilitates intratumoral steroidogenesis. In the nucleus, long non-coding RNAs (lncRNAs) act as scaffolds for chromatin-modifying complexes (PRC2), altering the epigenetic landscape to sustain survival gene expression.
Over the past decade, the field of RNA biology has been transformed by the discovery and functional characterization of non-coding RNAs (ncRNAs), including small interfering RNA (siRNA) and micro-RNA (miRNA), which are now recognized as regulators of gene expression. These gene expression control mechanisms occur at multiple levels, including post-transcriptional modification, mRNA processing, degradation, and translation . Based on protein-coding potential, RNA molecules are broadly categorized into coding and non-coding RNAs. Non-coding RNAs can be further categorized into regulatory RNAs which include short and long ncRNAs and housekeeping RNAs, including ribosomal RNA (rRNA) and transfer RNA (tRNA) [37]. In the context of prostate cancer, ncRNAs have gained increasing attention for their roles in tumorigenesis, progression, and therapeutic resistance. They modulate oncogenic and tumor suppressive pathways, including AR signaling, PI3K/AKT/MTOR, MAPK, P53, TGF-ß1 and mechanisms controlling mRNA stability. Furthermore, ncRNAs influence post-translational modifications, proteasome-mediated ubiquitination, and the degradation of AR and other regulatory proteins [38,39]. These multifaceted functions show their potential as both therapeutic targets and diagnostic biomarkers in advanced prostate cancer. Therefore, this review aims to explore the interplay between kinase signaling pathways and ncRNA-mediated regulation in prostate cancer, with a focus on therapeutic implications and future opportunities for precision medicine.
2. Signaling Through Protein Phosphorylation
Signal transduction within the cell is driven by various stress stimulus and is tightly regulated to control essential cellular functions. These processes are important for cell survival under both normal and pathological conditions. Mediators of these signals can activate or deactivate signaling cascades, thereby regulating cellular mechanisms such as growth, replication, survival, and apoptosis to maintain cellular homeostasis [40]. Aberrant protein phosphorylation is a hallmark of CRPC, driving tumor plasticity and drug resistance. This signaling deregulation is not merely a downstream effect but a driver of the transition to androgen independence. Protein phosphorylation is a reversible post-translational modification that involves the transfer of a phosphate group to a specific amino acid residue on a substrate [41]. In contrast, dephosphorylation removes a phosphate group from the substrate, often transferring it to water molecules to deactivate its function. The dynamic interplay between phosphorylation and dephosphorylation acts as a molecular switch, modulating the structural and functional protein conformation, interactions with other molecules, and subcellular localization in response to intracellular and extracellular stimuli [40]. This regulatory circuit is essential for controlling numerous cellular events, including enzyme activity, cell division, aging, protein synthesis, metabolism, and signal transduction [42].
2.1. Protein Kinases
Protein kinases (PKs) constitute approximately 2% of the eukaryotic genome yet regulate the post-translational modification of over 30% of the cellular proteome [43,44]. The catalytic efficiency of protein kinases in prostate cancer oncogenesis is governed by the dynamic assembly of the 'Regulatory (R) Spine,' a non-contiguous vertical stack of four hydrophobic residues (RS1-RS4) that connects the N-terminal and C-terminal lobes. In the inactive state, this spine is disassembled; upon activation, often driven by upstream signaling or oncogenic mutation, the spine residues align to stabilize the active conformation, facilitating ATP binding and substrate phosphorylation. Understanding this 'spine switch' is critical for the design of next-generation kinase inhibitors in PCa. Type I inhibitors target the assembled spine (active conformation), often leading to adaptive resistance, whereas Type II inhibitors stabilize the disassembled spine (inactive conformation), potentially offering more durable suppression of oncogenic drivers like BRAF and AKT [45]. Protein kinases are broadly grouped into three major classes based on the amino acid domain they phosphorylate: serine, threonine and tyrosine. Additionally, dual specificity kinases have also been identified as kinases that can act as serine, threonine and tyrosine [45,46]. PKs contain a catalytic domain which comprises C- and N- subdomains that act together as an active site of the protein [47]. This catalytic domain is normally inactive due to inward rotation of C-and N- lobes but upon activation via phosphorylation induces rearrangements on the protein, allowing non-catalytic domain of the protein to interact and recruit the substrates and activate other signaling proteins or cascades [41].
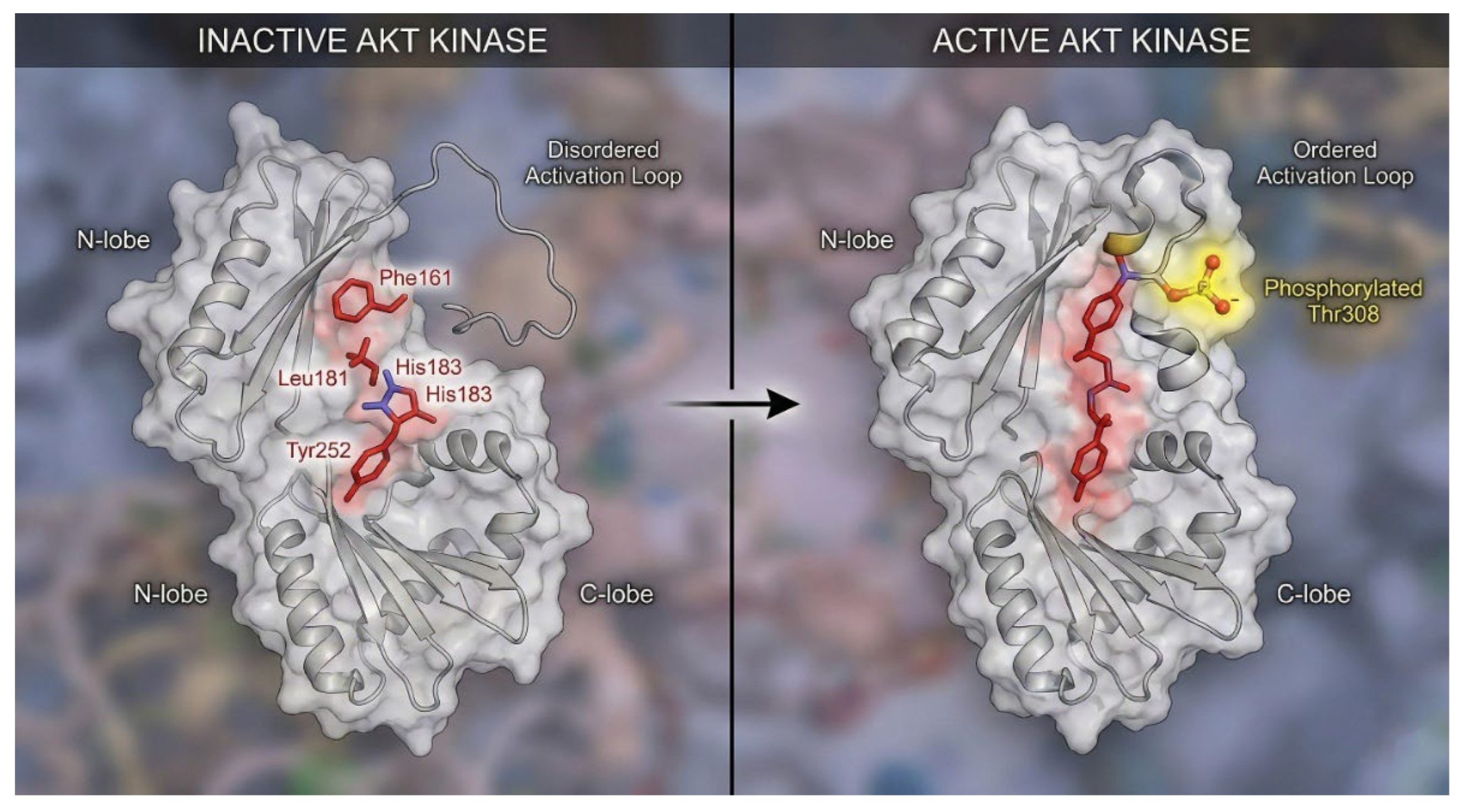
Figure 3.
Structural Dynamics of Kinase Activation: The Hydrophobic Spine. Comparison of the inactive (left) and active (right) conformations of the AKT kinase domain. In the inactive state, the Regulatory (R) spine residues are misaligned, and the activation loop is disordered. Upon phosphorylation (yellow glow), the N-lobe and C-lobe close, forcing the R-spine residues into a continuous vertical stack. This "spine assembly" rigidifies the catalytic cleft, enabling ATP binding and substrate phosphorylation.
Figure 3.
Structural Dynamics of Kinase Activation: The Hydrophobic Spine. Comparison of the inactive (left) and active (right) conformations of the AKT kinase domain. In the inactive state, the Regulatory (R) spine residues are misaligned, and the activation loop is disordered. Upon phosphorylation (yellow glow), the N-lobe and C-lobe close, forcing the R-spine residues into a continuous vertical stack. This "spine assembly" rigidifies the catalytic cleft, enabling ATP binding and substrate phosphorylation.
2.1.1. Serine/Threonine Kinases
Serine/threonine kinases which constitute the larger group of protein kinases, are known to phosphorylate through the hydrolysis of OH group and transferring of phosphate to the serine/threonine residues of the substrates. They can be activated in response to signals such as DNA damage or Ca[2]+ mediated chemical signaling [41]. The major classes of serine/threonine kinases include second messenger dependent kinases (cAMP, protein C, cGMP, and Ca[2]+ or calmodulin kinases), MAP kinases (ERKS, JNKs), MAP Kinase-regulating kinases (MEKs, SEKs, Raf, MEK kinases), CDKs (Cdc-2) CDK-regulating kinases (CAK, CAK kinase) . Their activation is via N-and C- termini lobes, where N- terminal is responsible for binding and hydrolyzing ATP, while C-terminal binds to the substrate to transfer the phosphate group; thereby, activating the downstream cascades.
2.1.2. Tyrosine Kinases
Tyrosine kinases (TKs) catalyze the transfer of the γ-phosphate from ATP to the phenolic hydroxyl group of tyrosine residues. This modification frequently generates high-affinity docking sites for SH2 and PTB domain-containing proteins, thereby nucleating multiprotein signaling complexes at the plasma membrane [48,49]. TKs, which are regulated by posttranslational modification are classified into two major subfamilies: receptor tyrosine kinases (e.g. EGF, VEGF, PDGF) and non-receptor tyrosine kinases [50]. TKs contains ligand binding and transmembrane domains, which enables them to interact with ligands and other proteins through outer membrane to activate tyrosine domain that is critical in mediating their intracellular signaling pathways [51,52,53]. Furthermore, TKs are involved in the activation of many signaling cascades, hence mutations or any abnormalities in the regulation of these signals has been linked with several diseases including atherosclerosis, diabetes, inflammations, cancer and angiogenesis [54]. In the context of prostate cancer, non-receptor tyrosine kinases (NRTKs) such as Src and Etk play pivotal roles in androgen-independent growth. Src kinase, for instance, physically interacts with and phosphorylates the Androgen Receptor (AR) at Tyr534, a modification that sustains AR transcriptional activity even in androgen-depleted environments. This Src-AR axis represents a major escape mechanism from ADT, rendering the receptor sensitive to low levels of adrenal androgens or even non-androgen ligands.
2.2. Protein Phosphatases
Protein phosphatases (PPs) are hydrolases that antagonize protein kinases via hydrolysis of removing the phosphate from substrates to activate or regulate their specific activity. Just like PKs, PPs contain tyrosine and serine/threonine residues with approximately 107 of phosphates having tyrosine and 30 phosphatases with serine/threonine residues, respectively. Protein serine/threonine phosphatases can further be classified into three subfamilies: phosphoprotein phosphatases (PPPs) [such as protein phosphatases (PP1, PP2, PP4, PP5)], metal-dependent protein phosphatases (PPMs) (such as PP2C, which depend on magnesium Mg[2]+/ manganese Mn[2]+ ions) and aspartate-based phosphatases [55]. Due to their roles in helping to maintain the molecular switching between phosphorylated and unphosphorylated state, PPs have been postulated to regulate many important signals including the process of tumorigenesis. For instance, PTEN, which is negatively regulated by PI3K, can act as tumor suppressor as its downregulation has associated with tumorigenesis progression in many cancer types [56]. Similarly, the downregulation of PP2A and SrC homolog region 2 domain-containing phosphatase-2 (SHP2) in tumorigenesis has been well documented. SHP2 are involved in the regulation of downstream signal of growth receptor like RAS [57], while PP2A acts a tumor suppressor via dephosphorylation of kinase-induced signaling leading to progression of cancer. Therefore, dysregulation of these phosphatases can promote tumorigenesis, making them a good druggable candidate against cancer [58,59].
2.3. In Vitro and In Vivo Assays Targeting Kinases in Human Diseases
The ability to measure kinases in vitro activity is important to assay for their capability to transfer y-phosphate and phosphorylate substrates from the donor substrates to acceptor protein [60,61]. Several commercially available phosphor-specific antibodies are used to stimulate phosphorylation sites within a given kinase [62]. This is important to determine if the kinase used is responsible for catalyzing the phosphotransfer by using a standardized kinase assay [63].
The role of protein kinases in tumorigenesis has been well documented and their identification has helped to explain the evolution of many cancer types, which makes them an ideal target for cancer treatment. For instance, many classes of phosphoinositide kinases, most especially, PI3K have been targeted in vivo and in vitro for the treatment of human diseases including neurodegenerative, viral infections, and prostate and breast cancer [64]. This is due to their crucial roles in trafficking and signaling transduction across membranes. Similarly, tyrosine kinases including JAK, SYK, TEC families and Rho kinase, ABL kinase and NAK kinase has been targeted for treatment due to their modulating roles in signaling pathways involve in immunodeficiency diseases including autoimmune disorders, hematological malignancy, allergic disorders and covid-19 [65,66].
3. Non-Coding RNAs (ncRNAs)
RNA interference (RNAi) technology is a powerful molecular tool used to regulate post-transcriptional processes by silencing mRNA in a sequence-specific manner. Its therapeutic potential lies in its ability to precisely target and inhibit specific mRNA, leading to gene silencing. This approach has shown promise in treating various human diseases, including cancers, viral infections, and genetic disorders, particularly those involving peptide products that are traditionally considered undruggable [67,68].
3.1. Small Non-Coding RNA (sncRNAs)
sncRNAs are the heterogenous group of untranslated transcripts, which include siRNA, miRNA and piwi-interacting (piRNA). This classification is based on their structures, origins, biological roles, and associated proteins [69]. Recently, many studies have been oriented towards sncRNAs because of their diverse roles in regulating gene expression. While the housekeeping sncRNA such as tRNA and rRNA are involved in the maintenance of ribosomal formations, the critical roles of regulatory sncRNA such as siRNA, miRNA and piRNA in modulating gene expression has been well documented [69,70].
3.1.1. Micro-RNA (miRNA)
miRNA encompass a single-strand and functional RNA that does not code for proteins but is produced to play critical roles in the regulation and repression of many biological processes at a genomic level. miRNA is the most studied class of sncRNA, and it has been demonstrated that a single miRNA can bind and regulate many mRNAs translation through its multiple target sites, which increases repression efficiency [71,72]. In prostate cancer, miRNAs act as tumor suppressors or oncogenes:
Tumor-suppressive miRNAs: miR-34a, let-7b, miR-141, miR-15a/miR-16-1 cluster, and miR-133a-3p inhibit PCa cell proliferation, invasion, and metastasis, often by inducing apoptosis, cell cycle arrest, or targeting pathways such as EGFR/AKT [73,74,75]. miR-195 and miR-203 similarly suppress invasion and EMT in PCa.
3.1.2. siRNA
siRNA is a non-coding, double stranded RNA oligonucleotide with roughly 20-24 base pairs (bp) in length and its production is catalyzed by a dicer enzyme, which catalyzes the cleavage of poly A tail. Upon activation, siRNA can specifically target and prevent the translation of many mRNA transcripts [81]. Since the first discovery in plants and later in roundworm (c. elegans) and subsequently in mammalian cells, siRNA has attracted serious interest because of its ability to specifically target and inhibit a particular gene in several human diseases including cancer. This makes it an ideal therapeutic target in the treatment of these diseases. The phenomenon of using dsRNA to silence or inhibit post-transcription of genes or to mediate mRNA degradation is called RNA interfering (RNAi) technology.
3.1.3. piRNAs
piRNAs are small RNAs that associate with Piwi proteins to regulate gene expression epigenetically. In PCa, PIWIL2 is upregulated and enhances EMT, invasion, and metastasis, contributing to tumor progression [82].
3.2. Long Non-Coding RNAs (lncRNAs)
lncRNAs includes enhancer RNAs (eRNAs), circular (ciRNAs), intergenic RNAs and natural antisense transcripts (NATs) have attracted a wide range of research interest due their roles in chromatin remodeling, RNA splicing, transcription, and translation [83]. In addition, lncRNAs has been implicated in VDJ recombination and class switching of immune cells, p53 mediated DNA damage responses, regulation of cholesterol metabolism as well as developmental stage in animals, making them an ideal target in human diseases [84].
In PCa, lncRNAs can be oncogenic or tumor-suppressive. Oncogenic lncRNAs: HOTAIR, Lnc-ZNF30-3, CRNDE, MALAT1, TERC, PCAT1, PCAT6, DANCR, and LINC00675 are upregulated, promoting proliferation, invasion, metastasis, and therapy resistance via chromatin modification, Wnt/β-catenin, PI3K/AKT, and NF-κB pathways [82,85,92].
Tumor-suppressive lncRNAs: PCAT29, DRAIC, GAS5, and MEG3 are downregulated and inhibit migration, metastasis, and NF-κB signaling, acting as negative regulators of PCa progression [86]. LINC00675 has a dual role, where its downregulation contributes to castration-resistant PCa and therapy resistance [89].
Due to their various functions in many important physiological events, alteration in their gene expression pattern has implications in the development of many diseases making them an ideal biological marker, which holds therapeutic strategies [93]. The roles of lncRNA in prostate cancer and progression to CRPC have been documented, and it is mediated through several mechanisms including chromatin remodeling, recruitment of chromatin modifiers and post-transcriptional regulation via interaction with proteins or RNAs.
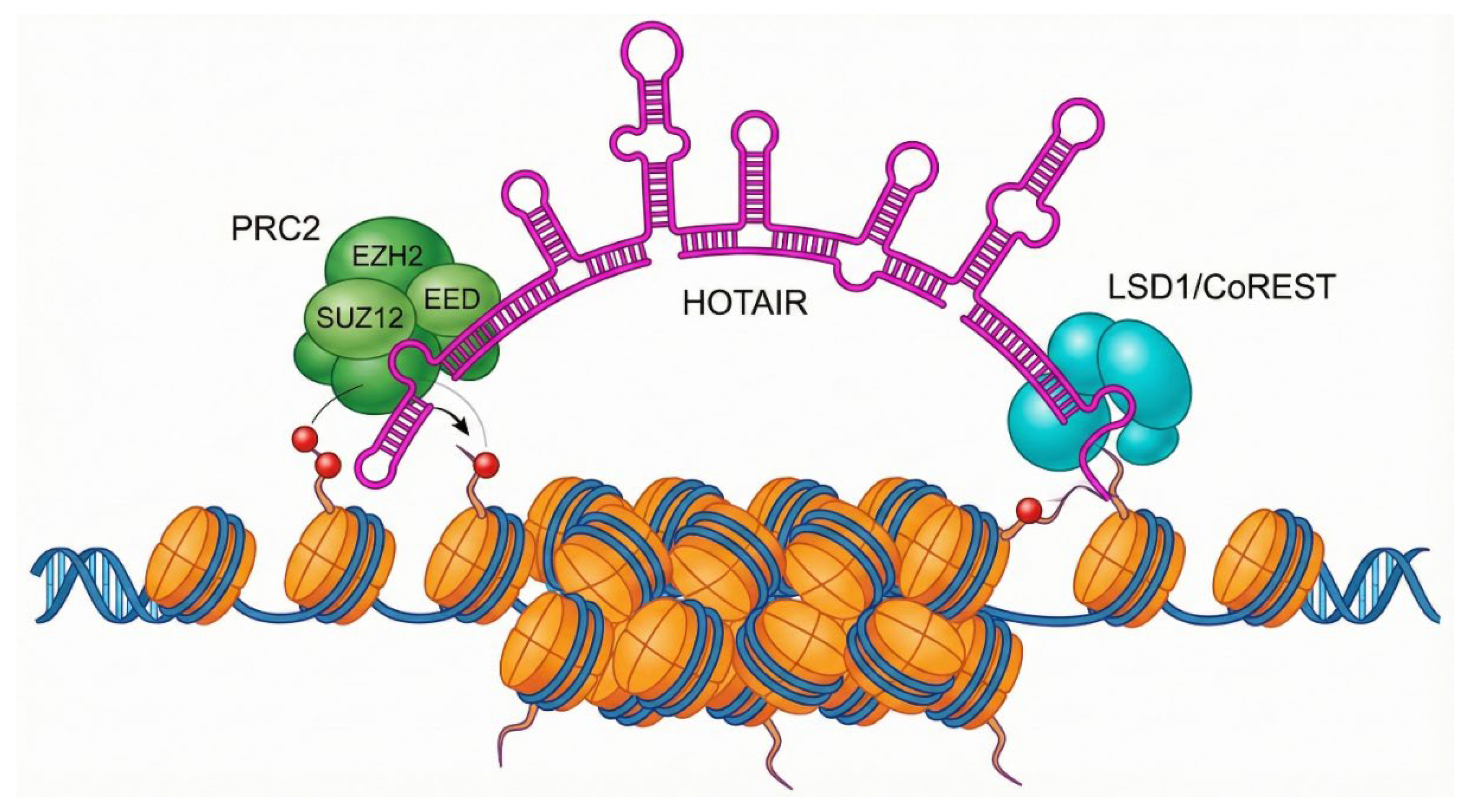
Long non-coding RNAs (lncRNAs) like HOTAIR do not merely float in the nucleus; they act as flexible scaffolds. HOTAIR possesses specific secondary structures (hairpins) that serve as docking sites for protein complexes. The 5' domain of HOTAIR binds to EZH2, the catalytic methyltransferase subunit of the Polycomb Repressive Complex 2 (PRC2). Simultaneously, the 3' domain can bind to the LSD1/CoREST/REST complex (a demethylase). By bridging these two complexes, HOTAIR coordinates a double hit on chromatin: it guides PRC2 to deposit repressive H3K27me3 marks (trimethylation of lysine 27 on histone H3) while LSD1 removes active H3K4me2 marks. This creates a massive block of heterochromatin, silencing tumor suppressor genes. Recent data suggests this is not just random recruitment; RNA matchmaking occurs where HOTAIR interacts with the target gene's nascent RNA transcripts via base-pairing, stabilizing the PRC2 complex at that specific locus. This RNA-RNA interaction relieves the autoinhibition of PRC2, activating its methyltransferase activity specifically at the target site. This insight suggests that blocking the RNA-RNA interaction surface could be a more specific therapy than global EZH2 inhibitors. A variety of non-coding RNAs, including microRNAs, long non-coding RNAs, piRNAs, and circular RNAs, have been implicated in prostate cancer, with 9 upregulated and 6 downregulated molecules identified (Table 1).
3.3. Mechanism of Action of RNAi Silencing
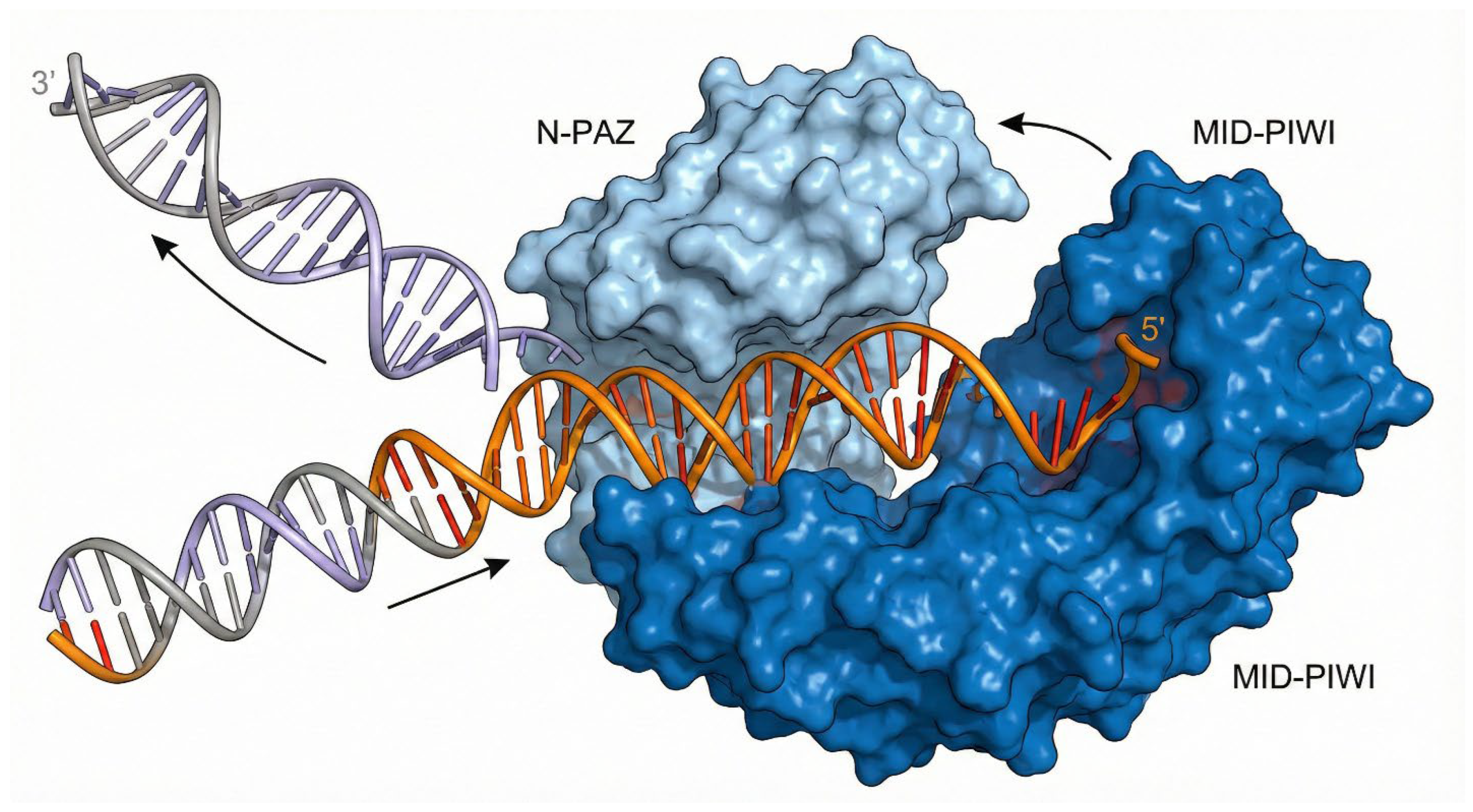
RNAi is a cellular natural phenomenon that mediates gene expression silencing using dsRNA to promote sequence specific mRNA degradation. Using the knowledge obtained from c. elegans, it was demonstrated that plants possess a specialized RNA-dependent RNA polymerase (RDRP) when triggered by high cellular level of messenger RNA. In response, RDRP creates dsRNA to shut down gene expression at posttranscriptional level in petunia hybrida plant [95,96]. Subsequently, a similar process was observed in mammalian cells [97]. In recent years, the knowledge and understanding of underlying mechanisms of RNAi technology has substantially grown and is well understood. It is now understood that ncdsRNA either biologically transcribed or artificially incorporated into the cells can be cleaved by the Dicer enzyme (called ribonuclease III) into short base pairs of about 21-25 nucleotide sequence called siRNA [98]. Upon activation, siRNA can interact and activate a multiprotein complex called RNA-induced silencing complex (RISC) or RISC loading complex [99]. RISC comprises endonuclease argonaute 2 (AGO2) and other RISC associated proteins such as TAR RNA binding protein (TRBP). AGO2 facilitates the thermodynamic unwinding and subsequent endonucleolytic cleavage of the passenger strand. AGO2 is the catalytic heart of RNAi. It possesses a bilobal architecture comprising four domains: N, PAZ, MID, and PIWI. The molecule is shaped roughly like a crescent or a duck. The MID domain contains a specific binding pocket that anchors the 5' phosphate of the miRNA or siRNA guide strand. This anchoring is critical; it sets the "register" for determining the seed sequence. The PAZ domain anchors the 3' end of the small RNA. Loading the RISC is a dynamic physical process. Structural studies reveal that when AGO2 binds a target mRNA that is complementary to the guide strand, the protein undergoes a dramatic conformational change. The PAZ domain releases the 3' end of the guide RNA, and the N-PAZ lobe rotates away from the MID-PIWI lobe. This "widening" of the central channel allows the guide RNA and target mRNA to propagate their double-helix pairing beyond the seed region without steric hindrance [100,101]. Thereafter, the antisense strand directs the RISC complex to its complementary target mRNA and effectively causes specific gene silencing at posttranscriptional level [99,102]. This structural flexibility is what makes RNAi potent but also vulnerable. In PCa cells, oncogenic environments (hypoxia, stress) can lead to post-translational modifications of AGO2 (e.g., phosphorylation by MAPK) that might impair this conformational flexibility. Furthermore, the slicer activity resides in the PIWI domain, which folds like RNase H. If the pairing is perfect (as with siRNA therapeutics), the PIWI domain positions the mRNA backbone exactly near catalytic aspartate residues for cleavage. If pairing is imperfect (as with most endogenous miRNAs), slicing does not occur, and translational repression follows. Therapeutic siRNAs must be designed to maximize this slicer conformation.
Figure 4.
Conformational Dynamics of the RISC Loading Complex. Mechanism of Argonaute 2 (AGO2) loading. The AGO2 protein (blue) is shown in its bilobal open state. As the double-stranded small RNA enters the central cleft, the PAZ domain anchors the 3' end while the MID domain anchors the 5' phosphate. The protein undergoes a structural widening to separate the passenger strand (peeling away) from the guide strand (orange), which remains threaded in the channel to direct gene silencing.
Figure 4.
Conformational Dynamics of the RISC Loading Complex. Mechanism of Argonaute 2 (AGO2) loading. The AGO2 protein (blue) is shown in its bilobal open state. As the double-stranded small RNA enters the central cleft, the PAZ domain anchors the 3' end while the MID domain anchors the 5' phosphate. The protein undergoes a structural widening to separate the passenger strand (peeling away) from the guide strand (orange), which remains threaded in the channel to direct gene silencing.
3.4. Advantages and Disadvantages of RNAi Technology
RNAi technology remains a cornerstone and widely used technology in biochemical and biological research to effectively knockdown a target mRNA sequence. It offers several advantages over conventional methods of gene silencing vectors, which include highly specific suppression of gene expression and less off-target effects [103]. Despite the progress that have been made with application of RNAi technology to effectively inhibit a specific gene and combat human diseases, it has been faced with several challenges including off-target silencing, degradation of closely related that are not identical mRNA genes, inability to pass through cellular membrane due to net negative charge, less success rate of 1 in 3 ratios and delivery issues [104,105]. To overcome these barriers, Lipid Nanoparticles (LNPs) have been engineered to facilitate endosomal escape. When Lipid Nanoparticles (LNPs) carrying siRNA are endocytosed, they are trapped in endosomes. As the endosome matures and acidifies, the ionizable lipids in the LNP become protonated (positively charged). This allows them to interact electrostatically with the anionic lipids of the endosomal membrane. This interaction induces a phase transition in the lipids from a laminar (bilayer) phase to a hexagonal HII phase. This non-bilayer structure is fusogenic, it disrupts the integrity of the endosomal membrane, creating transient "fusion pores" through which the RNA cargo can escape into the cytosol. [106]. However, research shows this escape efficiency is remarkably low (often <2%). Most LNPs are degraded in lysosomes or recycled/exocytosed. The next-generation LNPs are being engineered with specific lipids (like phytosterols or specific ionizable tails) that lower the energy barrier for this hexagonal phase transition, thereby enhancing cytosolic bioavailability.
3.5. Roles of Non-Coding RNA in Prostate Cancer
Over the years, the paradigm shifts towards targeting RNAi gene silencing has been propelled by the identification of non-coding RNA cancer-related genes for their oncogenic or tumor suppressor potential. The roles of ncRNA in different types of cancer have been highlighted. For instance, overexpression of long non-coding RNA (HOTAIR) and dysregulation of piwi-interacting (piRNA) have been linked with breast cancer, while deregulation of microRNA (miR-15 and miR-16) has been associated with chronic lymphocytic leukemia [107]. Similarly, the identification of lncRNAs (PRCR1 and PCGEM1) binding to androgen to induce the transcription of over 200 AR-responsive genes correlates with the aggression and progression of the PCa [108]. In mouse model, deletion of miR-15a-miR-16-1 cluster resulted in PCa cells survival, proliferation, and invasion, whereas overexpression led to regression characterized by increased apoptosis and cell cycle arrest [107].
In bone metastasis PCa, upregulation of miRNAs (such as miR-133a-3p, miR-466, miR-210 and miR-375) were found to control the inhibition or promotion of the important signaling pathways including wnt signal, transforming growth factor β (TGF-β), AKT, nuclear transcription factor-kB (NF-kB), which are crucial for PCa progression, recurrence and response to therapy [109,110]. Importantly, the oncogenic miR-21 promotes tumorigenesis by targeting PTEN and driving persistent PI3K/AKT signaling, while the tumor suppressor miR-34a regulates MET and NOTCH1, thereby modulating apoptosis and epithelial-to-mesenchymal transition (EMT), further augmenting the context-specific role of ncRNAs in prostate cancer biology [111,112]. The increased expression of Lnc-ZNF30-3 in PCa was associated with poor prognosis, possibly via its interaction with AGO2 complex and miR145-5p and mediating gene silencing [91]. Through the regulation of miR-146a-5p and suppression of miR-101 mediated inhibition of Ras-related oncogenic protein 1A, lncRNA CRNDE can inhibit apoptosis while up-regulating the metalloproteases like MPP-2 and MPP-9, thereby promoting cell invasion and proliferation in PCa cells [113]. The knock-down of SNHG4 which regulate RRM2 expression led to significant increase in cell senescence and cell cycle arrest, whereas overexpression resulted PCa cells resistance to enzalutamide in in vitro and mouse model [114]. The roles of ncRNAs in PCa in bone metastasis cannot be overemphasized as overexpression of these RNAs has been reported, which correspond with either invasion or inhibition of PCa progression. PCa progression coupled with epithelial-to-mesenchymal transition and resistance to therapy has been attributed to circular non-coding RNA (hsa-circ-0005276/FUS axis) via the upregulation of XIAP [115]. In addition, circZNF609 promotes cell proliferation by sponging miR-186-5p and activating AKT signaling, adding another layer of circRNA-mediated oncogenic regulation [116].
Figure 5.
lncRNA HOTAIR as a Chromatin Scaffold. Schematic of the long non-coding RNA HOTAIR functioning as a molecular bridge. The 5' domain of HOTAIR recruits the Polycomb Repressive Complex 2 (PRC2), responsible for H3K27 trimethylation (repressive mark), while the 3' domain recruits the LSD1 demethylase complex. This simultaneous recruitment coordinates the formation of heterochromatin, silencing tumor suppressor genes in metastatic prostate cancer cells.
Figure 5.
lncRNA HOTAIR as a Chromatin Scaffold. Schematic of the long non-coding RNA HOTAIR functioning as a molecular bridge. The 5' domain of HOTAIR recruits the Polycomb Repressive Complex 2 (PRC2), responsible for H3K27 trimethylation (repressive mark), while the 3' domain recruits the LSD1 demethylase complex. This simultaneous recruitment coordinates the formation of heterochromatin, silencing tumor suppressor genes in metastatic prostate cancer cells.
Overexpression of MALAT1 PCa was associated with metastasis and progression of castration-resistance prostate cancer, whereas its downregulation using siRNA has been reported to inhibit PCa invasion and progression marked with increased cell cycle arrest in G0/G1 phase in both in vivo and in vitro [117,118]. Nevertheless, the downregulation and low-level expression of lncRNAs (PCAT29 and DRAIC) has been associated with progression to CRPC in androgen-dependent manner in PCa patients at high risk of biochemical recurrence [86,117]. Similarly, overexpression of GAS5 in LnCaP, 22-RV-1 and PC-3 cells was associated with reduced cell viability, which sensitized cells to chemotherapy via induction of apoptosis [119]. The lncRNA PCA3, one of the most clinically validated PCa biomarkers, is highly prostate-specific and detectable in urine, where it has been shown to improve diagnostic specificity compared to PSA [120]. Beyond its diagnostic utility, PCA3 enhances androgen receptor signaling, thereby supporting tumor progression [121]. Similarly, HOTAIR promotes oncogenesis through recruitment of the PRC2 complex and epigenetic silencing of tumor suppressor genes, further reinforcing its link to PCa progression and metastases [122,123]. Their biological roles in PCa tumor microenvironment have highlighted their unique importance in diagnosis and therapeutic options in the treatment of PCa. ncRNAs contribute to the regulation of tumor immunosurveillance microenvironment through the regulation of the expression, development and differentiation of several immune cells including macrophages activation, natural killer cells, dendritic cells, regulatory T cells and cytotoxic T cells, which participate in the PCa cell invasion and metastasis [124].
LINC00675 has emerged as a critical modulator of AR stability in CRPC. Unlike in gastric cancers where it acts as a tumor suppressor, in prostate cancer, LINC00675 expression is elevated and correlates with poor prognosis. Mechanistically, LINC00675 binds to the N-terminal domain (NTD) of the Androgen Receptor, sterically hindering the binding of the E3 ubiquitin ligase MDM2. This blockade prevents ubiquitination and subsequent proteasomal degradation of AR, thereby maintaining high levels of receptor protein even under therapeutic stress. This LINC00675/MDM2/AR axis represents a direct link between the non-coding transcriptome and protein stability networks driving resistance [89]. Downregulation of miR-143, miR-21 and miR-200 have been reported to induce resistant to docetaxel therapy in CRPC via the upregulation of VEGF (vascular endothelial growth factor), which activate KRAS oncogene downstream signaling pathways that promote cell survival while suppressing the apoptotic mediated cell death [125,126]. Using Insilico analysis, Mbeje and colleagues predicted the differential expression of lncRNAs in androgen sensitive (LnCaP) and androgen insensitive prostate cancer cells, where the aberrant expression of lnRNA (TERC) correlates with AR signaling mediated tumorigenesis [127]. This is mediated via the interaction with tumor suppressor miRNAs, which activates TGF signaling that favors tumor progression [127]. Altogether, mis-regulation of ncRNAs is linked to tumor formation through epigenic modifications including hypermethylation of CpG island promoter leading to the transcription of oncogenic genes that promotes the modification of prostate cancer and progression to CRPC [128].
Figure 6.
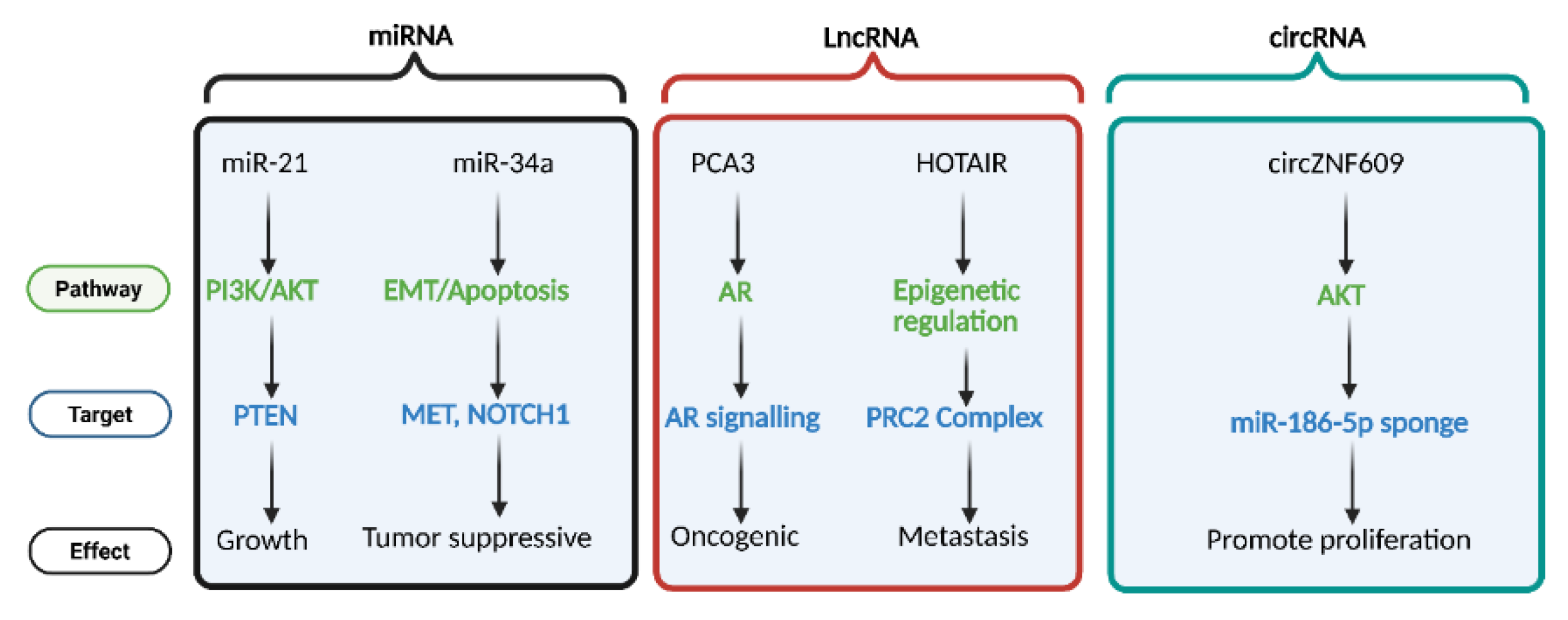
The different classes of non-coding RNAs microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) modulate prostate cancer progression via multiple targets. miR-21 and miR-34a regulate key pathways such as PI3K/AKT and EMT/Apoptosis by targeting PTEN, MET, and NOTCH1, influencing tumor growth and suppression. lncRNAs PCA3 and HOTAIR modulate androgen receptor (AR) signaling and epigenetic regulation through interactions with the PRC2 complex, contributing to oncogenesis and metastasis. circRNA circZNF609 promotes cell proliferation by acting as a sponge for miR-186-5p, influencing AKT signaling. Pathways (green), molecular targets (blue), and cellular effects (black) are color-coded accordingly.
Figure 6.
The different classes of non-coding RNAs microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) modulate prostate cancer progression via multiple targets. miR-21 and miR-34a regulate key pathways such as PI3K/AKT and EMT/Apoptosis by targeting PTEN, MET, and NOTCH1, influencing tumor growth and suppression. lncRNAs PCA3 and HOTAIR modulate androgen receptor (AR) signaling and epigenetic regulation through interactions with the PRC2 complex, contributing to oncogenesis and metastasis. circRNA circZNF609 promotes cell proliferation by acting as a sponge for miR-186-5p, influencing AKT signaling. Pathways (green), molecular targets (blue), and cellular effects (black) are color-coded accordingly.
4. Interplay Between Kinase Signaling and Non-Coding RNAs in Prostate Cancer Progression
Mechanisms that restore AR signaling after ADT therapy are the critical drivers of metastasis and drug resistance in CRPC. Persistent AR signaling due to overexpression, somatic mutations, intra-tumoral androgen biosynthesis, de novo androgen synthesis, genomic amplification and altered post-translational regulation is strongly associated with metastatic progression and AR-addicted CRPC [129,130]. AR activity is tightly controlled through post-transcriptional and posttranslational modifications including acetylation, ubiquitination and phosphorylation [131]. These modifications are regulated by the kinase-mediated signaling cascades such as mechanistic target of rapamycin complex-1 (mTORC1), PI3K, protein kinase B (AKT), and mitogen-activated protein kinases (MAPK) which are dysregulated in over 70% of metastatic PCa, compared to about 40% in localized disease . In advanced PCa, treatment with salinomycin inhibits the mTORC/AKT pathway, resulting in loss of AR phosphorylation, downregulation of steroidogenic enzymes CYP17A1 and HSD3β, and less proliferation of PCa in vivo [132]. While the direct phospho-regulation of HSD3β remains under investigation, compelling evidence demonstrates that MAPK14-mediated phosphorylation of CYP17A1 significantly enhances its 17,20-lyase activity, thereby prioritizing androgen synthesis over mineralocorticoid production.
Figure 7.
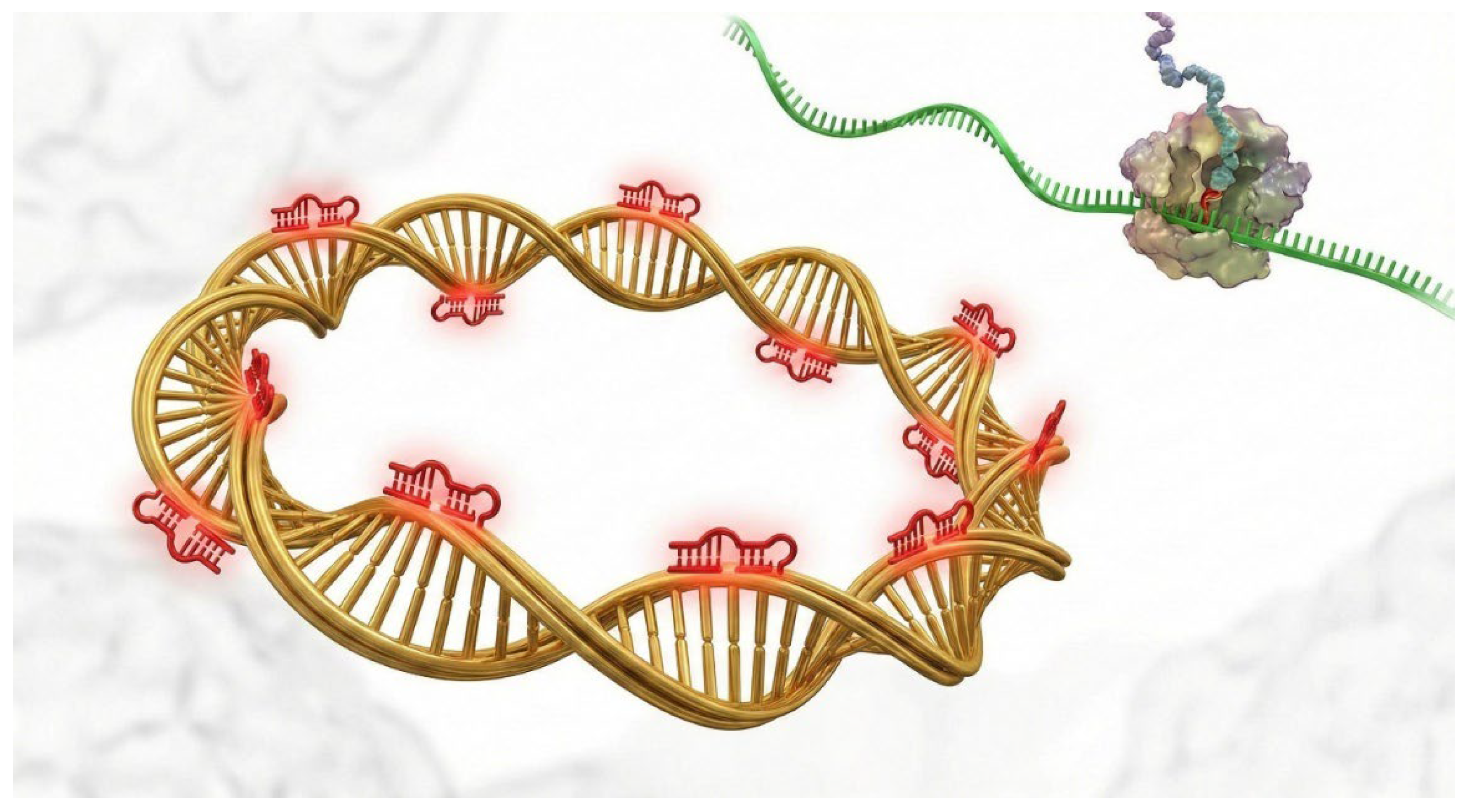
The Sponge Mechanism of Circular RNAs. Illustration of circular RNA (circRNA) function in the cytoplasm. The closed-loop circRNA structure (gold) confers stability against exonucleases. It functions as a competing endogenous RNA (ceRNA) by containing multiple binding sites for specific microRNAs (red clips). By sequestering these miRNAs, the circRNA prevents them from repressing their target mRNAs (green), thereby effectively upregulating oncogenic proteins.
Figure 7.
The Sponge Mechanism of Circular RNAs. Illustration of circular RNA (circRNA) function in the cytoplasm. The closed-loop circRNA structure (gold) confers stability against exonucleases. It functions as a competing endogenous RNA (ceRNA) by containing multiple binding sites for specific microRNAs (red clips). By sequestering these miRNAs, the circRNA prevents them from repressing their target mRNAs (green), thereby effectively upregulating oncogenic proteins.
The concept of lncRNAs acting as sponges for miRNAs is widely cited but rarely scrutinized quantitatively. In the context of Lnc-ZNF30-3 and miR-145-5p in prostate cancer, the stoichiometry is critical. For a lncRNA to effectively derepress a target like TWIST1, its abundance must be comparable to the pool of the miRNA it is sequestering. In CRPC, the amplification of the Lnc-ZNF30-3 locus often leads to copy number variations that support this high expression level. However, the sponge effect is likely non-linear; it operates as a threshold mechanism. Below a certain concentration, the lncRNA has no effect on the miRNA-target interaction. Once the lncRNA concentration exceeds the free miRNA pool, a sudden de-repression of TWIST1 occurs, triggering a rapid phenotypic switch to EMT. This digital response mechanism explains the explosive metastatic potential often seen in late-stage disease.

Table 2.
The Structural Logic of Kinase Inhibitors in Prostate Cancer.
| Inhibitor Class | Binding Conformation | R-Spine State | Mechanism of Action | Resistance Mechanism in PCa |
| Type I (e.g., Dasatinib) | Active (DFG-in) | Assembled | Binds to the ATP pocket when the kinase is in its catalytically active state. | "Gatekeeper" mutations (e.g., T315I) that sterically block the pocket; upregulation of upstream activators. |
| Type II (e.g., Cabozantinib) | Inactive (DFG-out) | Disassembled | Stabilizes the inactive conformation by occupying the hydrophobic pocket adjacent to the ATP site. | Mutations that favor the active conformation (R-spine assembly), preventing the inhibitor from binding. |
| Allosteric (e.g., Trametinib) | Inactive | Disassembled | Binds outside the ATP pocket, preventing the conformational change required for R-spine assembly. | Feedback loop activation of alternative pathways (e.g., PI3K activation following MAPK inhibition). |
Previous studies have shown that MAPK14-mediated phosphorylation of CYP17A1 at threonine/serine residues is vital for enhancing its lyase activity and promoting interaction with POR, an important step in androgen biosynthesis [29,133,134]. Given CYP17A1’s pivotal role in both classical and “backdoor” androgen biosynthesis pathways, and especially it involvement in intratumoral androgen production in CRPC [1], its dysregulation may contribute to sustained AR signaling, metastatic progression, and resistance to anti-androgen therapy. Upregulation of CYP17A1 expression, combined with the emergence of AR splice variants, has been implicated in resistance to abiraterone, a CYP17A1 inhibitor, by compensating for systemic androgen deprivation [135]. Despite the therapeutic value of targeting CYP17A1, existing inhibitors including abiraterone lack specificity and often affect other steroidogenic enzymes, leading to undesirable side effects such as cortisol suppression [133,136,138]. Hence, more precise approaches are needed, including targeting CYP17A1 post-translational regulation, such as its phosphorylation, or designing non-steroidal small molecule inhibitors to improve efficacy and reduce adverse effects[22]. Phosphorylation of AR itself plays a key role in AR reactivation during CRPC progression. For instance, phosphorylation of AR at Ser81 has been shown to restore AR signaling following ARD1 knockdown, contributing to castration resistance [139]. Inhibition of CYP17A1 using galeterone not only disrupts AR signaling but also promotes degradation of full-length and variant AR isoforms (f-ARVs). Additionally, PI3K-mediated phosphorylation of AR at Ser81 facilitates its recognition and proteasomal degradation via MDM2, a tumor suppressor E3 ubiquitin ligase [140]. A series of mechanistic studies by Tee and Miller confirmed that MAPK14, a member of the MAP kinase family, phosphorylates CYP17A1 and enhances its lyase activity in non-steroidogenic COS-1 cells, further confirming the enzyme’s regulation through phosphorylation [29].
Beyond CYP17A1 and AR, protein kinases broadly contribute to prostate cancer proliferation, metastasis, and resistance to ADT, likely through complex interactions with epigenetic regulators and chromatin remodeling enzymes that influence AR transcriptional programs [141,142,143]. Upon androgen binding, AR translocates from the cytoplasm to the nucleus, where it activates the transcription of androgen-responsive genes (ARGs) that support cancer cell proliferation and survival. Phosphoproteomic profiling has revealed over 10 phosphotyrosine residues on AR in metastatic prostate cancer cells, especially when co-expressed with SRC, a non-receptor tyrosine kinase, supporting the role of hyperphosphorylated AR in driving CRPC [144,145]. Additionally, AKT-mediated phosphorylation of AR at Ser213 and Ser791 enhances prostate cancer cell survival, further implicating kinase pathways in AR regulation [146].
Recent discoveries highlight non-coding RNAs (ncRNAs) including miRNAs and lncRNAs as important regulators of the kinase-AR axis. Their expression is often upregulated in late-stage prostate cancer, where they contribute to therapy resistance and sustained AR activity. Elevated levels of miR-197 and miR-361-3p have been reported in enzalutamide-resistant prostate cancer xenografts [147]. Similarly, LINC01126, a long non-coding RNA induced by ADT, promotes cancer cell survival, recurrence, and metastasis in CRPC. Mechanistically, LINC01126 enhances AR Ser81 phosphorylation, facilitating its nuclear translocation, stabilization, and interaction with cyclin-dependent kinases (CDKs), ultimately worsening disease prognosis [148]. The miR-302/367 cluster, when overexpressed in vitro and in vivo, promotes prostate cancer growth and ADT resistance. However, these effects can be reversed via antisense knockdown or by co-expressing the tumor suppressor LATS2, indicating a phosphorylation-dependent mechanism [149]. Phosphorylation of AR by the MAPK/mTOR/AKT pathway stabilizes its structure and facilitates the recruitment of co-activators, leading to AR hypersensitization in CRPC. Pharmacological blockade of these pathways in preclinical models significantly improves therapeutic outcomes [150]. miR-205 plays an opposing role by disrupting AR co-activator assembly and downregulating AR phosphorylation, making it a potential anti-metastatic agent that limits cell invasion, migration, and growth in prostate cancer [129]. In another example, systemic delivery of miR-124, in combination with enzalutamide, induces cell cycle arrest and promotes apoptosis in prostate tumor cells [151]. lncRNASAT1, a lncRNA associated with supraphysiological androgen levels (SAL), acts as a tumor suppressor in LNCaP cell lines by reducing AKT phosphorylation. In ex vivo patient-derived prostate samples, SAL treatment induces senescence characterized by elevated p16INK[4]a and p15INK[4]b expression. This senescent phenotype is abolished upon lncRNASAT1 knockdown, indicating its pivotal role in modulating AKT signaling and maintaining SAL-induced tumor suppression [152].
5. Therapeutic Potential and Future Perspectives
Although ncRNAs lack protein-coding ability, they hold significant therapeutic potential in CRPC due to their ability to regulate AR gene expression. Through this regulation, ncRNAs can influence critical cellular processes including proliferation, invasion, metastasis, and the transition from PCa to CRPC often mediated via Wnt/MAPK/mTOR/AKT signaling pathways. For instance, inhibition of miR-182 and miR-744 using siRNA has been shown to drastically suppress the growth of PCa xenografts [110], highlighting their value as potential therapeutic targets. Additionally, Leite and colleagues reported that loss of miR-100-5p facilitates the transition from localized PCa to metastatic disease [153]. miR-100-5p exerts anti-tumor effects by downregulating the mTOR pathway [154], emphasizing its role in key signaling cascades. Interestingly, while miR-100-5p may act as a tumor suppressor, its upregulation has also been observed in androgen deprivation therapy (ADT)-refractory patients at higher risk of relapse post-prostatectomy, suggesting a dual role in the context of ADT resistance [153]. lncRNAs are also central to CRPC pathogenesis. They influence transcriptional and post-transcriptional regulation of AR signaling and are involved in epigenetic modifications [93,155,156]. Takayama et al. demonstrated that CRPC-associated lncRNAs (CRPC-Lncs) are upregulated in disease and promote AR signaling. siRNA-mediated knockdown of these lncRNAs led to reduced expression of AR variants, splicing machinery, and downstream AR-regulated genes [24]. Other ncRNAs, such as miR-21, have been associated with CRPC aggressiveness. miR-21 represses the expression of tumor suppressors like B-cell translocation gene 2 (BTG2) and PTEN when recruited to AR promoters in androgen-dependent cells [157]. Similarly, miR-12B targets pro-apoptotic genes such as p53, reinforcing its oncogenic role in prostate tumor progression and CRPC development [158]. lncRNAs such as SNHG3, LINC00152, and AK022798 have been implicated in resistance to anti-androgen therapies like enzalutamide by upregulating Notch signaling, thus presenting themselves as crucial therapeutic targets [159].
5.1. Combining Kinase Inhibitors with RNA-Based Therapies
Given the central role of kinase-mediated phosphorylation and non-coding RNAs (ncRNAs) in sustaining androgen receptor (AR) signaling and driving resistance to anti-androgen therapies in CRPC, a novel therapeutic paradigm could involve co-targeting both axes. Specifically, combining kinase inhibitors (e.g., AKT, mTOR, or MAPK14 inhibitors) with RNA-based interventions such as miRNA mimics, antisense oligonucleotides (ASOs), or siRNA constructs may provide synergistic effects by disrupting feedback loops that promote tumor plasticity, androgen biosynthesis, and metastasis[149,160]. For example, miR-205 or miR-124 mimics, when co-administered with kinase inhibitors, could suppress AR phosphorylation, limit co-activator recruitment, and sensitize resistant cells to treatment [160,161]. This dual-targeted approach offers the promise of overcoming acquired resistance and enhancing therapeutic efficacy in patients with advanced disease.
5.2. Advancing Precision Medicine Through Multi-Omics and Functional Genomics
The integration of multi-omics profiling (including transcriptomics, phosphoproteomics, and epigenomics) and CRISPR/Cas9-based functional screens offers powerful tools to identify patient-specific vulnerabilities in prostate cancer. These platforms can reveal oncogenic ncRNAs, essential kinases, or resistance pathways that can be exploited for precision-targeted therapies. For instance, genome-wide CRISPR knockout studies have uncovered synthetic lethal interactions involving AR splice variants, mTOR pathway components, and epigenetic regulators, enabling the design of individualized therapeutic strategies [162,163,164]. Furthermore, integrated omics analysis can stratify patients based on their ncRNA expression signatures, enabling the rational selection of RNA-based or kinase-targeted drugs for tailored intervention.
Figure 8.
Integrated Multi-Omics Precision Medicine Workflow. A conceptual framework for patient stratification. Data streams from genomics (DNA mutations), transcriptomics (RNA expression), proteomics (protein abundance), and metabolomics are integrated via machine learning algorithms. This multi-dimensional analysis identifies patient-specific vulnerabilities, such as a specific kinase hyperactivity or lncRNA upregulation, guiding the selection of targeted therapies (e.g., PARP inhibitors, kinase inhibitors) for personalized treatment.
Figure 8.
Integrated Multi-Omics Precision Medicine Workflow. A conceptual framework for patient stratification. Data streams from genomics (DNA mutations), transcriptomics (RNA expression), proteomics (protein abundance), and metabolomics are integrated via machine learning algorithms. This multi-dimensional analysis identifies patient-specific vulnerabilities, such as a specific kinase hyperactivity or lncRNA upregulation, guiding the selection of targeted therapies (e.g., PARP inhibitors, kinase inhibitors) for personalized treatment.
5.3. Nanoparticle-Based Delivery Systems for RNA and Kinase Inhibitors
One of the major limitations of current RNA-based therapies and kinase inhibitors is their poor pharmacokinetics, off-target effects, and limited tumor penetrance. Nanoparticle delivery systems, including lipid nanoparticles (LNPs) and polymeric nanocarriers, offer innovative solutions by protecting nucleic acids from degradation, enhancing cellular uptake, and enabling tumor-specific targeting. Encapsulation of siRNAs targeting CYP17A1 or AR variants, or delivery of kinase inhibitors like AKT/mTOR inhibitors, via ligand-decorated nanoparticles could greatly enhance the selectivity and effectiveness of CRPC treatment [165,166,167]. Moreover, nanoparticle platforms could be engineered to deliver combination payloads, such as miRNA mimics and kinase inhibitors, in a coordinated and controlled release, further enhancing therapeutic synergy. The efficacy of siRNA therapeutics is contingent upon cytosolic bioavailability, a process heavily dependent on endosomal escape. Lipid Nanoparticles (LNPs) utilize ionizable cationic lipids that become protonated in the acidifying environment of the late endosome (pH ~5.5). This protonation triggers an electrostatic interaction with anionic endosomal phospholipids (e.g., phosphatidylserine), inducing a phase transition from the lamellar bilayer (Lα) to the inverted hexagonal phase (HII). The HII phase is highly fusogenic, disrupting the endosomal membrane integrity and creating transient pores through which the RNA cargo is released into the cytosol to engage the RISC machinery. Optimizing the pKa of these ionizable lipids is the current frontier in enhancing the endosomal escape efficiency, which currently remains below 2-5% for standard formulations.
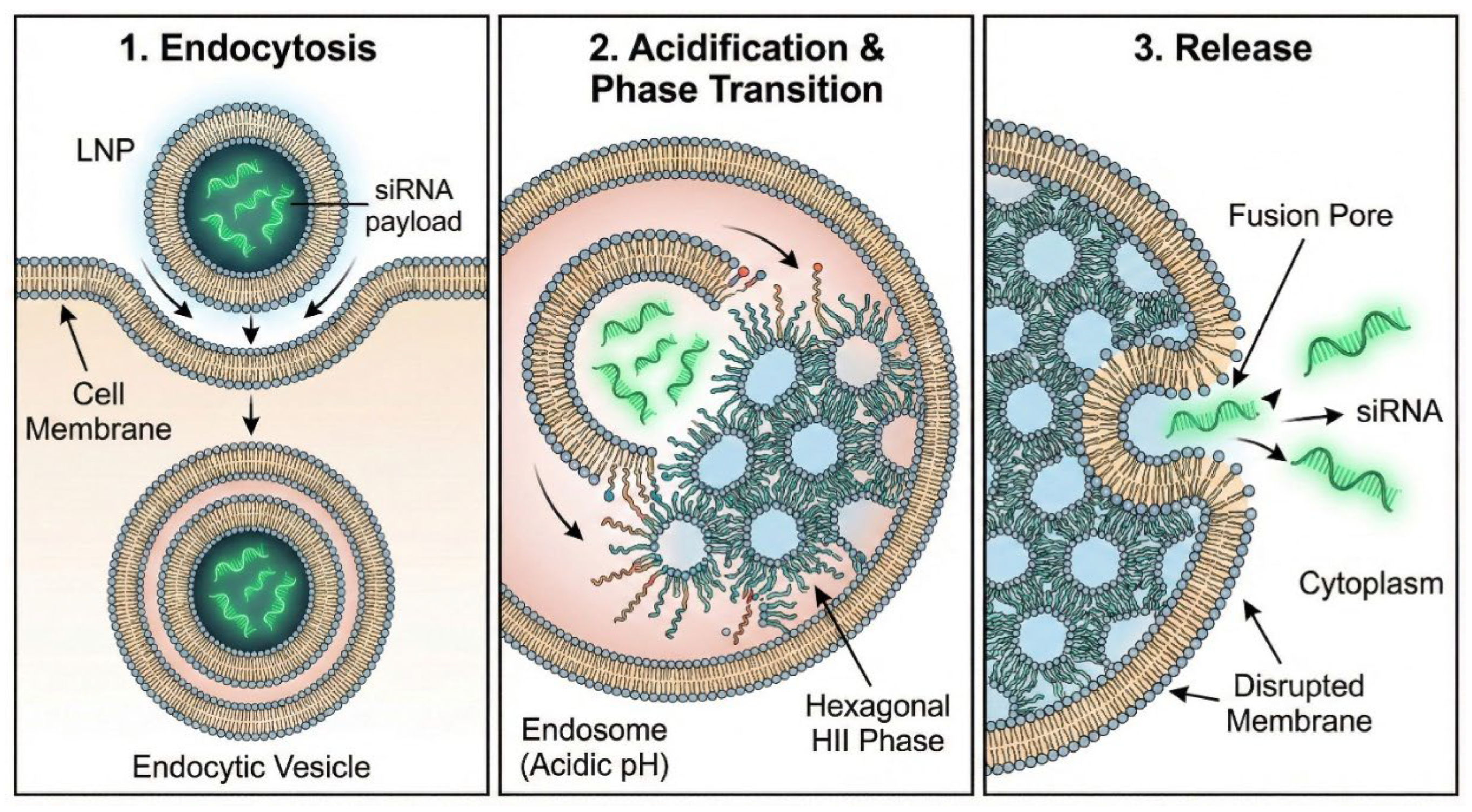
Figure 9.
Lipid Nanoparticle (LNP) Endosomal Escape. The physicochemical mechanism of RNA delivery via LNPs. (Left) Endocytosis of the LNP. (Middle) As the endosome acidifies, ionizable lipids within the LNP become protonated, interacting with anionic endosomal lipids to induce a transition from a laminar bilayer to a hexagonal HII phase. (Right) This membrane disruption creates transient fusion pores, allowing the therapeutic siRNA cargo to escape into the cytosol to engage the RNAi machinery.
Figure 9.
Lipid Nanoparticle (LNP) Endosomal Escape. The physicochemical mechanism of RNA delivery via LNPs. (Left) Endocytosis of the LNP. (Middle) As the endosome acidifies, ionizable lipids within the LNP become protonated, interacting with anionic endosomal lipids to induce a transition from a laminar bilayer to a hexagonal HII phase. (Right) This membrane disruption creates transient fusion pores, allowing the therapeutic siRNA cargo to escape into the cytosol to engage the RNAi machinery.
5.4. ncRNAs as Biomarkers for Prognosis and Treatment Monitoring
Several ncRNAs, including miR-197, miR-361-3p, miR-205, and LINC01126, have emerged as promising biomarkers for CRPC progression, therapeutic response, and disease recurrence[148,149,160,168]. For instance, elevated circulating levels of miR-197 and LINC01126 have been associated with enzalutamide resistance and poor prognosis. Monitoring the expression of these molecules in liquid biopsies (e.g., blood or urine samples) could enable real-time tracking of therapeutic efficacy and early identification of resistance phenotypes, thus facilitating timely treatment adjustments. Their incorporation into clinical diagnostic panels may also improve risk stratification and help tailor treatment strategies in a non-invasive, dynamic manner.
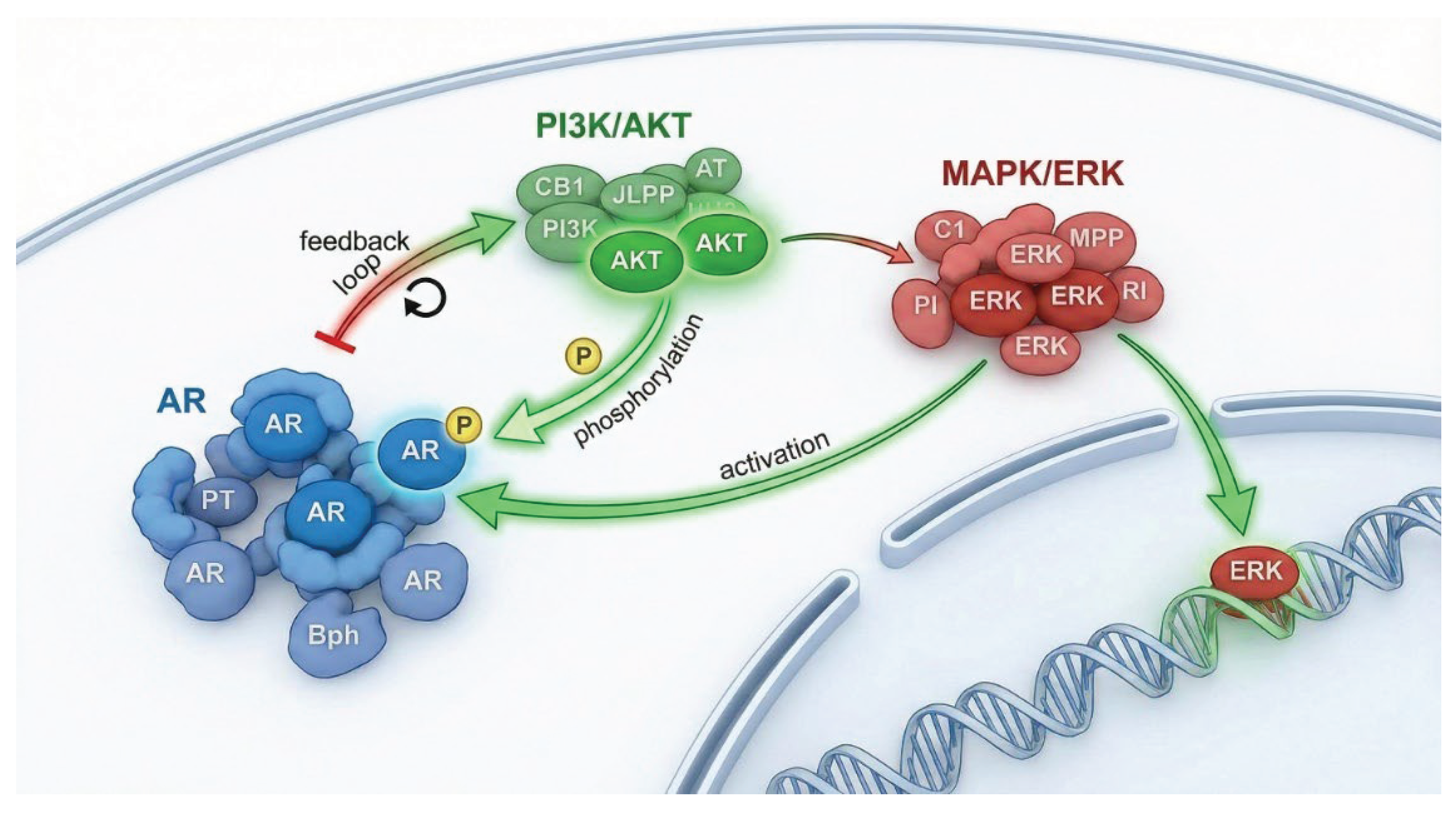
Figure 10.
Crosstalk Networks: AR, PI3K, and MAPK. A network topology diagram visualizing the reciprocal feedback loops between major oncogenic pathways. Inhibition of the Androgen Receptor (AR) relieves negative feedback on the PI3K/AKT pathway, leading to compensatory survival signaling. Conversely, activated MAPK/ERK can phosphorylate AR directly, facilitating ligand-independent nuclear translocation. These interconnected nodes illustrate the necessity of combination therapies to prevent adaptive resistance.
Figure 10.
Crosstalk Networks: AR, PI3K, and MAPK. A network topology diagram visualizing the reciprocal feedback loops between major oncogenic pathways. Inhibition of the Androgen Receptor (AR) relieves negative feedback on the PI3K/AKT pathway, leading to compensatory survival signaling. Conversely, activated MAPK/ERK can phosphorylate AR directly, facilitating ligand-independent nuclear translocation. These interconnected nodes illustrate the necessity of combination therapies to prevent adaptive resistance.
6. Conclusion
This review highlights the intricate and multifactorial roles of signaling molecules particularly kinase pathways and non-coding RNAs (ncRNAs) in the development, progression, and therapeutic resistance of prostate cancer, especially in castration-resistant prostate cancer (CRPC). These molecular regulators orchestrate critical events in androgen receptor (AR) signaling, steroidogenesis, tumor cell proliferation, and survival. Beyond being transcriptional "junk," ncRNAs have emerged as functional regulators with roles in chromatin remodeling, mRNA stability, alternative splicing, and post-transcriptional gene silencing [169]. Increasing evidence suggests that miRNAs and lncRNAs directly modulate AR expression and its phosphorylation status, influence the availability of co-activators, and even regulate upstream kinases that control post-translational modifications. This positions them as pivotal nodes within the regulatory circuitry driving therapy resistance and disease aggressiveness. Kinase-mediated phosphorylation of AR and CYP17A1 also contributes to AR reactivation under androgen-deprived conditions, thereby sustaining tumor growth despite treatment. These pathways are frequently upregulated in advanced disease and often interact with ncRNAs, forming feedback loops that complicate therapeutic targeting. Despite these advances, major challenges persist. These include the context-dependent roles of ncRNAs, limited knowledge of kinase-ncRNA crosstalk, off-target effects of current inhibitors, and inter-patient heterogeneity that limits the applicability of standard treatments. In this regard, future research should focus on multi-omics integration, CRISPR-based functional screens, and preclinical validation of candidate ncRNA and kinase targets. Moreover, the development of nanoparticle-based delivery systems for RNA therapeutics and kinase inhibitors, alongside real-time biomarker monitoring through liquid biopsies, holds promise for advancing precision oncology in prostate cancer. A shift toward patient-specific molecular profiling and the co-targeting of both signaling and regulatory RNA networks may ultimately redefine treatment paradigms and improve outcomes for patients with advanced prostate cancer.
Funding statement
This research was supported by the CANCER RESEARCH SWITZERLAND grant number KFS-5557-02-2022 to AVP. PCI is funded in part by a Swiss Government Excellence Scholarship award number 2023.0441.
Disclosure statement
The authors report there are no competing interests to declare.
References
- He, Y. et al. Targeting signaling pathways in prostate cancer: mechanisms and clinical trials. Signal Transduct Target Ther 7, 198 (2022). [CrossRef]
- Maekawa, S., Takata, R. & Obara, W. Molecular Mechanisms of Prostate Cancer Development in the Precision Medicine Era: A Comprehensive Review. Cancers (Basel) 16 (2024). [CrossRef]
- Perdana, N. R., Mochtar, C. A., Umbas, R. & Hamid, A. R. The Risk Factors of Prostate Cancer and Its Prevention: A Literature Review. Acta Med Indones 48, 228–238 (2016).
- Zhang, H. et al. Androgen Metabolism and Response in Prostate Cancer Anti-Androgen Therapy Resistance. Int J Mol Sci 23 (2022). [CrossRef]
- Stangelberger, A., Waldert, M. & Djavan, B. Prostate cancer in elderly men. Rev Urol 10, 111–119 (2008).
- Snaterse, G. et al. Prostate cancer androgen biosynthesis relies solely on CYP17A1 downstream metabolites. J Steroid Biochem Mol Biol 236, 106446 (2024). [CrossRef]
- Miller, W. L. & Auchus, R. J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 32, 81–151 (2011). [CrossRef]
- Petrunak, E. M., DeVore, N. M., Porubsky, P. R. & Scott, E. E. Structures of human steroidogenic cytochrome P450 17A1 with substrates. J Biol Chem 289, 32952–32964 (2014). [CrossRef]
- Kmetova Sivonova, M. et al. The role of CYP17A1 in prostate cancer development: structure, function, mechanism of action, genetic variations and its inhibition. Gen Physiol Biophys 36, 487–499 (2017). [CrossRef]
- Ferraldeschi, R., Sharifi, N., Auchus, R. J. & Attard, G. Molecular pathways: Inhibiting steroid biosynthesis in prostate cancer. Clin Cancer Res 19, 3353–3359 (2013). [CrossRef]
- Pandey, A. V., Mellon, S. H. & Miller, W. L. Protein phosphatase 2A and phosphoprotein SET regulate androgen production by P450c17. J Biol Chem 278, 2837–2844 (2003). [CrossRef]
- Pandey, A. V. & Miller, W. L. Regulation of 17,20 lyase activity by cytochrome b5 and by serine phosphorylation of P450c17. J Biol Chem 280, 13265–13271 (2005). [CrossRef]
- Yoshimoto, F. K. & Auchus, R. J. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J Steroid Biochem Mol Biol 151, 52–65 (2015). [CrossRef]
- Alex, A. B., Pal, S. K. & Agarwal, N. CYP17 inhibitors in prostate cancer: latest evidence and clinical potential. Ther Adv Med Oncol 8, 267–275 (2016). [CrossRef]
- Flück, C. E. et al. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am J Hum Genet 89, 201–218 (2011). [CrossRef]
- Biason-Lauber, A., Miller, W. L., Pandey, A. V. & Flück, C. E. Of marsupials and men: "Backdoor" dihydrotestosterone synthesis in male sexual differentiation. Mol Cell Endocrinol 371, 124–132 (2013). [CrossRef]
- Wrobel, T. M. et al. Non-steroidal CYP17A1 Inhibitors: Discovery and Assessment. J Med Chem 66, 6542–6566 (2023). [CrossRef]
- Wrobel, T. M. et al. Pyridine indole hybrids as novel potent CYP17A1 inhibitors. J Enzyme Inhib Med Chem 40, 2463014 (2025). [CrossRef]
- Auchus, R. J., Lee, T. C. & Miller, W. L. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J Biol Chem 273, 3158–3165 (1998).
- Klein, E. A., Ciezki, J., Kupelian, P. A. & Mahadevan, A. Outcomes for intermediate risk prostate cancer: are there advantages for surgery, external radiation, or brachytherapy? Urol Oncol 27, 67–71 (2009). [CrossRef]
- Knudsen, K. E. & Penning, T. M. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 21, 315–324 (2010). [CrossRef]
- Wrobel, T. M. et al. Discovery of Novel Non-Steroidal Cytochrome P450 17A1 Inhibitors as Potential Prostate Cancer Agents. Int J Mol Sci 21, 4868 (2020). [CrossRef]
- Wang, B. R. et al. Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease. Biomedicines 10 (2022). [CrossRef]
- Obinata, D. et al. Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front Oncol 10, 581515 (2020). [CrossRef]
- Li, Z. et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature 533, 547–551 (2016). [CrossRef]
- Fizazi, K. et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol 20, 686–700 (2019). [CrossRef]
- Attard, G., Reid, A. H., Olmos, D. & de Bono, J. S. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res 69, 4937–4940 (2009). [CrossRef]
- Lee, Y. H. A. et al. Major adverse cardiovascular events of enzalutamide versus abiraterone in prostate cancer: a retrospective cohort study. Prostate Cancer Prostatic Dis 27, 776–782 (2024). [CrossRef]
- Tee, M. K. & Miller, W. L. Phosphorylation of human cytochrome P450c17 by p38alpha selectively increases 17,20 lyase activity and androgen biosynthesis. J Biol Chem 288, 23903–23913 (2013). [CrossRef]
- Mulholland, D. J. et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19, 792–804 (2011). [CrossRef]
- Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [CrossRef]
- Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371, 1028–1038 (2014). [CrossRef]
- Watson, P. A., Arora, V. K. & Sawyers, C. L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711 (2015). [CrossRef]
- Davies, M. A. & Samuels, Y. Analysis of the genome to personalize therapy for melanoma. Oncogene 29, 5545–5555 (2010). [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 134, 215–230 (2008). [CrossRef]
- Ikushima, H. & Miyazono, K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 10, 415–424 (2010). [CrossRef]
- Li, C. & Wand, M. Approximate Translational Building Blocks for Image Decomposition and Synthesis. Acm Transactions on Graphics 34, 1–16 (2015). [CrossRef]
- Elimam, H. et al. Natural products and long non-coding RNAs in prostate cancer: insights into etiology and treatment resistance. Naunyn Schmiedebergs Arch Pharmacol 398, 6349–6368 (2025). [CrossRef]
- Kohvakka, A. et al. Long noncoding RNA EPCART regulates translation through PI3K/AKT/mTOR pathway and PDCD4 in prostate cancer. Cancer Gene Ther 31, 1536–1546 (2024). [CrossRef]
- Narla, G., Sangodkar, J. & Ryder, C. B. The impact of phosphatases on proliferative and survival signaling in cancer. Cell Mol Life Sci 75, 2695–2718 (2018). [CrossRef]
- Ardito, F., Giuliani, M., Perrone, D., Troiano, G. & Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med 40, 271–280 (2017). [CrossRef]
- Cheng, H. C., Qi, R. Z., Paudel, H. & Zhu, H. J. Regulation and function of protein kinases and phosphatases. Enzyme Res 2011, 794089 (2011). [CrossRef]
- Taylor, S. S., Herberg, F. W., Veglia, G. & Wu, J. Edmond Fischer's kinase legacy: History of the protein kinase inhibitor and protein kinase A. IUBMB Life 75, 311–323 (2023). [CrossRef]
- Hanks, S. K. & Hunter, T. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification 1. The FASEB journal 9, 576–596 (1995).
- Schwartz, P. A. & Murray, B. W. Protein kinase biochemistry and drug discovery. Bioorg Chem 39, 192–210 (2011). [CrossRef]
- Dhanasekaran, N. & Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 17, 1447–1455 (1998). [CrossRef]
- Adeyelu, T. et al. KinFams: De-Novo Classification of Protein Kinases Using CATH Functional Units. Biomolecules 13 (2023). [CrossRef]
- Robinson, M. J., Xu, B.-e., Stippec, S. & Cobb, M. H. Different Domains of the Mitogen-activated Protein Kinases ERK3 and ERK2 Direct Subcellular Localization and Upstream Specificityin Vivo. Journal of Biological Chemistry 277, 5094–5100 (2002).
- Robinson, D. R., Wu, Y. M. & Lin, S. F. The protein tyrosine kinase family of the human genome. Oncogene 19, 5548–5557 (2000). [CrossRef]
- Torrecilla, I. et al. Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J Cell Biol 177, 127–137 (2007). [CrossRef]
- Collins, S. J. Exploiting Water-Mediated Interactions to Inhibit Mutationally Activated HER2, State University of New York at Stony Brook, (2022).
- Doublet, P., Grangeasse, C., Obadia, B., Vaganay, E. & Cozzone, A. J. Structural organization of the protein-tyrosine autokinase Wzc within Escherichia coli cells. J Biol Chem 277, 37339–37348 (2002). [CrossRef]
- Lu, Q. et al. TREM (Triggering Receptor Expressed on Myeloid Cells)-1 Inhibition Attenuates Neuroinflammation via PKC (Protein Kinase C) delta/CARD9 (Caspase Recruitment Domain Family Member 9) Signaling Pathway After Intracerebral Hemorrhage in Mice. Stroke 52, 2162–2173 (2021). [CrossRef]
- Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010). [CrossRef]
- Shi, Y. Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 (2009). [CrossRef]
- Cully, M., You, H., Levine, A. J. & Mak, T. W. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6, 184–192 (2006). [CrossRef]
- Zhou, X. D. & Agazie, Y. M. Inhibition of SHP2 leads to mesenchymal to epithelial transition in breast cancer cells. Cell Death Differ 15, 988–996 (2008). [CrossRef]
- Ruvolo, P. P. Role of protein phosphatases in the cancer microenvironment. Biochim Biophys Acta Mol Cell Res 1866, 144–152 (2019). [CrossRef]
- Vainonen, J. P., Momeny, M. & Westermarck, J. Druggable cancer phosphatases. Sci Transl Med 13, eabe2967 (2021). [CrossRef]
- Crespo, P. & Gutkind, J. S. Activation of MAPKs by G protein-coupled receptors. Methods Mol Biol 250, 203–210 (2004). [CrossRef]
- Marinissen, M. J. et al. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell 14, 29–41 (2004). [CrossRef]
- Hilder, T. L., Malone, M. H. & Johnson, G. L. Hyperosmotic induction of mitogen-activated protein kinase scaffolding. Methods Enzymol 428, 297–312 (2007). [CrossRef]
- Black, M. H., Gradowski, M., Pawłowski, K. & Tagliabracci, V. S. in Methods in enzymology Vol. 667 575–610 (Elsevier, 2022).
- Burke, J. E., Triscott, J., Emerling, B. M. & Hammond, G. R. V. Beyond PI3Ks: targeting phosphoinositide kinases in disease. Nat Rev Drug Discov 22, 357–386 (2023). [CrossRef]
- Castelo-Soccio, L. et al. Protein kinases: drug targets for immunological disorders. Nat Rev Immunol 23, 787–806 (2023). [CrossRef]
- Narayanan, J. et al. Role and mechanistic actions of protein kinase inhibitors as an effective drug target for cancer and COVID. Arch Microbiol 205, 238 (2023). [CrossRef]
- Kim, D. H. & Rossi, J. J. Strategies for silencing human disease using RNA interference. Nat Rev Genet 8, 173–184 (2007). [CrossRef]
- Mahmoodi Chalbatani, G. et al. Small interfering RNAs (siRNAs) in cancer therapy: a nano-based approach. Int J Nanomedicine 14, 3111–3128 (2019). [CrossRef]
- Carthew, R. W. & Sontheimer, E. J. Origins and Mechanisms of miRNAs and siRNAs. Cell 136, 642–655 (2009). [CrossRef]
- Ozata, D. M., Gainetdinov, I., Zoch, A., O'Carroll, D. & Zamore, P. D. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20, 89–108 (2019). [CrossRef]
- Rottiers, V. & Naar, A. M. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol 13, 239–250 (2012). [CrossRef]
- Farberov, L. et al. Multiple Copies of microRNA Binding Sites in Long 3'UTR Variants Regulate Axonal Translation. Cells 12 (2023). [CrossRef]
- Bonci, D. et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 14, 1271–1277 (2008). [CrossRef]
- Ostadrahimi, S. et al. miR-1266-5p and miR-185-5p Promote Cell Apoptosis in Human Prostate Cancer Cell Lines. Asian Pac J Cancer Prev 19, 2305–2311 (2018). [CrossRef]
- Strand, S. H. et al. Validation of the four-miRNA biomarker panel MiCaP for prediction of long-term prostate cancer outcome. Sci Rep 10, 10704 (2020). [CrossRef]
- Liu, C. et al. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res 72, 3393–3404 (2012). [CrossRef]
- Gan, J. et al. MicroRNA-375 is a therapeutic target for castration-resistant prostate cancer through the PTPN4/STAT3 axis. Exp Mol Med 54, 1290–1305 (2022). [CrossRef]
- Konoshenko, M. & Laktionov, P. The miRNAs involved in prostate cancer chemotherapy response as chemoresistance and chemosensitivity predictors. Andrology 10, 51–71 (2022). [CrossRef]
- Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004). [CrossRef]
- Place, R. F., Li, L. C., Pookot, D., Noonan, E. J. & Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 105, 1608–1613 (2008). [CrossRef]
- Roberts, A. M., Ward, C. C. & Nomura, D. K. Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr Opin Biotechnol 43, 25–33 (2017). [CrossRef]
- Weng, W., Li, H. & Goel, A. Piwi-interacting RNAs (piRNAs) and cancer: Emerging biological concepts and potential clinical implications. Biochim Biophys Acta Rev Cancer 1871, 160–169 (2019). [CrossRef]
- Wu, H., Yang, L. & Chen, L. L. The Diversity of Long Noncoding RNAs and Their Generation. Trends Genet 33, 540–552 (2017). [CrossRef]
- Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol 24, 430–447 (2023). [CrossRef]
- Cayuela, M. L., Flores, J. M. & Blasco, M. A. The telomerase RNA component Terc is required for the tumour-promoting effects of Tert overexpression. EMBO Rep 6, 268–274 (2005). [CrossRef]
- Sakurai, K., Reon, B. J., Anaya, J. & Dutta, A. The lncRNA DRAIC/PCAT29 Locus Constitutes a Tumor-Suppressive Nexus. Mol Cancer Res 13, 828–838 (2015). [CrossRef]
- Feng, Y. et al. Circular RNA circ0005276 promotes the proliferation and migration of prostate cancer cells by interacting with FUS to transcriptionally activate XIAP. Cell Death Dis 10, 792 (2019). [CrossRef]
- Hao, T. et al. MALAT1 knockdown inhibits prostate cancer progression by regulating miR-140/BIRC6 axis. Biomed Pharmacother 123, 109666 (2020). [CrossRef]
- Yao, M. et al. LINC00675 activates androgen receptor axis signaling pathway to promote castration-resistant prostate cancer progression. Cell Death Dis 11, 638 (2020). [CrossRef]
- Fu, D. et al. Long non-coding RNA CRNDE regulates the growth and migration of prostate cancer cells by targeting microRNA-146a-5p. Bioengineered 12, 2469–2479 (2021). [CrossRef]
- Le Hars, M. et al. Pro-tumorigenic role of lnc-ZNF30-3 as a sponge counteracting miR-145-5p in prostate cancer. Biol Direct 18, 38 (2023). [CrossRef]
- Cantile, M. et al. The role of HOTAIR in the modulation of resistance to anticancer therapy. Front Mol Biosci 11, 1414651 (2024). [CrossRef]
- Statello, L., Guo, C. J., Chen, L. L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22, 96–118 (2021). [CrossRef]
- Ren, D. et al. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-kappaB signaling pathway. Mol Cancer 16, 117 (2017). [CrossRef]
- Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998). [CrossRef]
- Pushparaj, P. N., Aarthi, J. J., Manikandan, J. & Kumar, S. D. siRNA, miRNA, and shRNA: in vivo applications. J Dent Res 87, 992–1003 (2008). [CrossRef]
- Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001). [CrossRef]
- Kim, D. H. et al. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23, 222–226 (2005). [CrossRef]
- Lam, J. K., Chow, M. Y., Zhang, Y. & Leung, S. W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids 4, e252 (2015). [CrossRef]
- Penner, P. E., Cohen, L. H. & Loeb, L. A. RNA-dependent DNA polymerase: presence in normal human cells. Biochem Biophys Res Commun 42, 1228–1234 (1971). [CrossRef]
- Agrawal, N. et al. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67, 657–685 (2003). [CrossRef]
- Yokota, T. et al. Unique structure of Ascaris suum b5-type cytochrome: an additional alpha-helix and positively charged residues on the surface domain interact with redox partners. Biochem J 394, 437–447 (2006). [CrossRef]
- Mocellin, S. & Provenzano, M. RNA interference: learning gene knock-down from cell physiology. J Transl Med 2, 39 (2004). [CrossRef]
- Moreira, D., Pereira, A. M., Lopes, A. L. & Coimbra, S. The best CRISPR/Cas9 versus RNA interference approaches for Arabinogalactan proteins' study. Mol Biol Rep 47, 2315–2325 (2020). [CrossRef]
- Moreira, V. J. V. et al. In planta RNAi targeting Meloidogyne incognita Minc16803 gene perturbs nematode parasitism and reduces plant susceptibility. Journal of Pest Science 97, 411–427 (2024).
- Pecot, C. V., Calin, G. A., Coleman, R. L., Lopez-Berestein, G. & Sood, A. K. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer 11, 59–67 (2011). [CrossRef]
- Ramalho-Carvalho, J., Fromm, B., Henrique, R. & Jeronimo, C. Deciphering the function of non-coding RNAs in prostate cancer. Cancer Metastasis Rev 35, 235–262 (2016). [CrossRef]
- Ling, H. et al. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 34, 5003–5011 (2015). [CrossRef]
- Sang, H. et al. The regulatory process and practical significance of non-coding RNA in the dissemination of prostate cancer to the skeletal system. Front Oncol 14, 1358422 (2024). [CrossRef]
- Bu, T., Li, L. & Tian, J. Unlocking the role of non-coding RNAs in prostate cancer progression: exploring the interplay with the Wnt signaling pathway. Front Pharmacol 14, 1269233 (2023). [CrossRef]
- Imani, S. et al. MicroRNA-34a targets epithelial to mesenchymal transition-inducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget 8, 21362–21379 (2017). [CrossRef]
- Chawra, H. S. et al. MicroRNA-21's role in PTEN suppression and PI3K/AKT activation: Implications for cancer biology. Pathol Res Pract 254, 155091 (2024). [CrossRef]
- Zhang, H., Hamdane, D., Im, S. C. & Waskell, L. Cytochrome b5 inhibits electron transfer from NADPH-cytochrome P450 reductase to ferric cytochrome P450 2B4. J Biol Chem 283, 5217–5225 (2008). [CrossRef]
- Dong, Q., Qiu, H., Piao, C., Li, Z. & Cui, X. LncRNA SNHG4 promotes prostate cancer cell survival and resistance to enzalutamide through a let-7a/RREB1 positive feedback loop and a ceRNA network. J Exp Clin Cancer Res 42, 209 (2023). [CrossRef]
- Guarnerio, J. et al. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 165, 289–302 (2016). [CrossRef]
- Wang, L. et al. Downregulation of circ-ZNF609 Promotes Heart Repair by Modulating RNA N(6)-Methyladenosine-Modified Yap Expression. Research (Wash D C) 2022, 9825916 (2022). [CrossRef]
- Xu, Y. H., Deng, J. L., Wang, G. & Zhu, Y. S. Long non-coding RNAs in prostate cancer: Functional roles and clinical implications. Cancer Lett 464, 37–55 (2019). [CrossRef]
- Ren, S. et al. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J Urol 190, 2278–2287 (2013). [CrossRef]
- Pickard, M. R., Mourtada-Maarabouni, M. & Williams, G. T. Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim Biophys Acta 1832, 1613–1623 (2013). [CrossRef]
- Marks, L. S. & Bostwick, D. G. Prostate Cancer Specificity of PCA3 Gene Testing: Examples from Clinical Practice. Rev Urol 10, 175–181 (2008).
- Merola, R. et al. PCA3 in prostate cancer and tumor aggressiveness detection on 407 high-risk patients: a National Cancer Institute experience. J Exp Clin Cancer Res 34, 15 (2015). [CrossRef]
- Li, T. et al. Long noncoding RNA HOTAIR regulates the invasion and metastasis of prostate cancer by targeting hepaCAM. Br J Cancer 124, 247–258 (2021). [CrossRef]
- Zhang, A. et al. LncRNA HOTAIR Enhances the Androgen-Receptor-Mediated Transcriptional Program and Drives Castration-Resistant Prostate Cancer. Cell Rep 13, 209–221 (2015). [CrossRef]
- Yang, F. et al. Non-coding RNAs: Emerging roles in the characterization of immune microenvironment and immunotherapy of prostate cancer. Biochem Pharmacol 214, 115669 (2023). [CrossRef]
- Pudova, E. et al. Non-Coding RNAs and the Development of Chemoresistance to Docetaxel in Prostate Cancer: Regulatory Interactions and Approaches Based on Machine Learning Methods. Life (Basel) 13 (2023). [CrossRef]
- Liu, Y. et al. Function of microRNA-124 in the pathogenesis of cancer (Review). Int J Oncol 64 (2024). [CrossRef]
- Mbeje, M. et al. In Silico Bioinformatics Analysis on the Role of Long Non-Coding RNAs as Drivers and Gatekeepers of Androgen-Independent Prostate Cancer Using LNCaP and PC-3 Cells. Curr Issues Mol Biol 45, 7257–7274 (2023). [CrossRef]
- Fu, A., Jacobs, D. I., Hoffman, A. E., Zheng, T. & Zhu, Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis 36, 1094–1102 (2015). [CrossRef]
- Shih, J. W., Wang, L. Y., Hung, C. L., Kung, H. J. & Hsieh, C. L. Non-Coding RNAs in Castration-Resistant Prostate Cancer: Regulation of Androgen Receptor Signaling and Cancer Metabolism. Int J Mol Sci 16, 28943–28978 (2015). [CrossRef]
- Missaghian, E. et al. Role of DNA methylation in the tissue-specific expression of the CYP17A1 gene for steroidogenesis in rodents. J Endocrinol 202, 99–109 (2009). [CrossRef]
- Fu, M. et al. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity. J Biol Chem 279, 29436–29449 (2004). [CrossRef]
- Bridges, A. et al. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem 273, 17036–17049 (1998). [CrossRef]
- Udhane, S. S. & Fluck, C. E. Regulation of human (adrenal) androgen biosynthesis-New insights from novel throughput technology studies. Biochem Pharmacol 102, 20–33 (2016). [CrossRef]
- Zhu, W. et al. Oxidative stress increases the 17,20-lyase-catalyzing activity of adrenal P450c17 through p38alpha in the development of hyperandrogenism. Mol Cell Endocrinol 484, 25–33 (2019). [CrossRef]
- Argos, P. & Mathews, F. S. The structure of ferrocytochrome b5 at 2.8 A resolution. J Biol Chem 250, 747–751 (1975). [CrossRef]
- Yin, L. & Hu, Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol 11, 32–42 (2014). [CrossRef]
- Mostaghel, E. A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag Res 6, 39–51 (2014). [CrossRef]
- Ang, J. E., Olmos, D. & de Bono, J. S. CYP17 blockade by abiraterone: further evidence for frequent continued hormone-dependence in castration-resistant prostate cancer. Br J Cancer 100, 671–675 (2009). [CrossRef]
- Pozas, J. et al. Androgen Receptor Signaling Inhibition in Advanced Castration Resistance Prostate Cancer: What Is Expected for the Near Future? Cancers (Basel) 14 (2022). [CrossRef]
- Bonfils, C., Balny, C. & Maurel, P. Direct evidence for electron transfer from ferrous cytochrome b5 to the oxyferrous intermediate of liver microsomal cytochrome P-450 LM2. J Biol Chem 256, 9457–9465 (1981). [CrossRef]
- Xu, R. & Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed Pharmacother 121, 109679 (2020). [CrossRef]
- Chau, V., Madan, R. A. & Aragon-Ching, J. B. Protein kinase inhibitors for the treatment of prostate cancer. Expert Opin Pharmacother 22, 1889–1899 (2021). [CrossRef]
- Bagheri, S. et al. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 14 (2022). [CrossRef]
- Guo, Z. et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10, 309–319 (2006). [CrossRef]
- Nunes-Xavier, C. E., Mingo, J., Lopez, J. I. & Pulido, R. The role of protein tyrosine phosphatases in prostate cancer biology. Biochim Biophys Acta Mol Cell Res 1866, 102–113 (2019). [CrossRef]
- Wang, Y., Kreisberg, J. I. & Ghosh, P. M. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets 7, 591–604 (2007). [CrossRef]
- Andl, T., Ganapathy, K., Bossan, A. & Chakrabarti, R. MicroRNAs as Guardians of the Prostate: Those Who Stand before Cancer. What Do We Really Know about the Role of microRNAs in Prostate Biology? Int J Mol Sci 21 (2020). [CrossRef]
- Cai, Y. et al. Androgen-repressed lncRNA LINC01126 drives castration-resistant prostate cancer by regulating the switch between O-GlcNAcylation and phosphorylation of androgen receptor. Clin Transl Med 14, e1531 (2024). [CrossRef]
- Guo, Y. et al. miR-302/367/LATS2/YAP pathway is essential for prostate tumor-propagating cells and promotes the development of castration resistance. Oncogene 36, 6336–6347 (2017). [CrossRef]
- Kinkade, C. W. et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest 118, 3051–3064 (2008). [CrossRef]
- Yang, Y., Liu, K. Y., Liu, Q. & Cao, Q. Androgen Receptor-Related Non-coding RNAs in Prostate Cancer. Front Cell Dev Biol 9, 660853 (2021). [CrossRef]
- Mirzakhani, K. et al. The androgen receptor-lncRNASAT1-AKT-p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene 41, 943–959 (2022). [CrossRef]
- Leite, K. R. et al. MicroRNA-100 expression is independently related to biochemical recurrence of prostate cancer. J Urol 185, 1118–1122 (2011). [CrossRef]
- Nabavi, N. et al. miR-100-5p inhibition induces apoptosis in dormant prostate cancer cells and prevents the emergence of castration-resistant prostate cancer. Sci Rep 7, 4079 (2017). [CrossRef]
- He, R. Z., Luo, D. X. & Mo, Y. Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis 6, 6–15 (2019). [CrossRef]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol Proced Online 16, 11 (2014). [CrossRef]
- Coppola, V. et al. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial-mesenchymal transition. Oncogene 32, 1843–1853 (2013). [CrossRef]
- Takayama, K. et al. RUNX1, an androgen- and EZH2-regulated gene, has differential roles in AR-dependent and -independent prostate cancer. Oncotarget 6, 2263–2276 (2015). [CrossRef]
- Shi, L. et al. Review on Long Non-Coding RNAs as Biomarkers and Potentially Therapeutic Targets for Bacterial Infections. Curr Issues Mol Biol 46, 7558–7576 (2024). [CrossRef]
- Hagman, Z. et al. miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br J Cancer 108, 1668–1676 (2013). [CrossRef]
- Shi, X. B. et al. miR-124 and Androgen Receptor Signaling Inhibitors Repress Prostate Cancer Growth by Downregulating Androgen Receptor Splice Variants, EZH2, and Src. Cancer Res 75, 5309–5317 (2015). [CrossRef]
- Lei, Y. & Lai, M. Epigenetic Regulation and Therapeutic Targeting of Alternative Splicing Dysregulation in Cancer. LID - 10.3390/ph18050713 [doi] LID - 713.
- Wadosky, K. M. & Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 8, 18550–18576 (2017). [CrossRef]
- Shalem, O., Sanjana, N. E. & Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311 (2015). [CrossRef]
- Ashrafizadeh, M. et al. Progress in Delivery of siRNA-Based Therapeutics Employing Nano-Vehicles for Treatment of Prostate Cancer. Bioengineering (Basel) 7 (2020). [CrossRef]
- Mainini, F. & Eccles, M. R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 25 (2020). [CrossRef]
- Zhang, S., Zhi, D. & Huang, L. Lipid-based vectors for siRNA delivery. J Drug Target 20, 724–735 (2012). [CrossRef]
- He, M. X. et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat Med 27, 426–433 (2021). [CrossRef]
- Anil, P., Dastidar, S. G. & Banerjee, S. Unravelling the role of long non-coding RNAs in prostate carcinoma. Advances in Cancer Biology-Metastasis 6, 100067 (2022). [CrossRef]
Table 1.
Differential Expression and Functional Roles of Non-Coding RNAs in Prostate Cancer Progression.
Table 1.
Differential Expression and Functional Roles of Non-Coding RNAs in Prostate Cancer Progression.
| RNA Type | Name | Classification | Expression in PCa | Functional Roles | Reference(s) |
|---|---|---|---|---|---|
| miRNA | miR-34a, let-7b, miR-141, miR-106 | Tumor-suppressive microRNAs | Downregulated | Inhibit prostate cancer (PCa) cell invasion by inducing apoptosis and arresting the cell cycle | [74] |
| miRNA | miR-452, miR-301b | Oncogenic microRNAs | Upregulated | Enhance invasion and stemness of cancer stem cells (CSCs) | [76,78] |
| miRNA | miR-15a/miR-16-1 cluster | Tumor-suppressive microRNAs | Downregulated | Induce apoptosis and cell cycle arrest in PCa cells via targeting BCL2 | [73] |
| miRNA | miR-133a-3p | Tumor-suppressive microRNA | Downregulated | Inhibits viability and invasion of PCa cells by promoting apoptosis; may regulate EGFR/AKT pathway | [75] |
| miRNA | miR-210-3p | Hypoxia-induced oncogenic microRNA | Upregulated | Drives epithelial-mesenchymal transition (EMT) and bone metastasis via NF-κB pathway activation |
[94] |
| miRNA | miR-375 | Oncogenic microRNA | Upregulated | Facilitates PCa progression, possibly via targeting SEC23A | [77] |
| lncRNA | HOTAIR | Oncogenic lncRNA | Upregulated | Enhances PCa cell proliferation and invasion; modulates chromatin state via PRC2 complex | [92] |
| piRNA | PIWIL2 | Oncogenic piRNA-associated protein | Upregulated | Promotes EMT, invasion, and metastasis; potential role in epigenetic silencing | [82] |
| lncRNA | Lnc-ZNF30-3 | Oncogenic lncRNA | Upregulated | Enhances PCa cell survival and correlates with poor prognosis in patients. acting as a competitive endogenous RNA (ceRNA) sponge for miR-145-5p, thereby derepressing the EMT driver TWIST1 | [91] |
| lncRNA | CRNDE | Oncogenic lncRNA | Upregulated | Stimulates PCa cell proliferation, migration, and invasion through Wnt/β-catenin signaling | [90] |
| circRNA | circ-0005276 | Oncogenic circular RNA | Upregulated | Promotes proliferation and invasion of PCa cells, likely via miRNA sponging | [87] |
| lncRNA | MALAT1 | Oncogenic lncRNA | Upregulated | Inhibits apoptosis and promotes PCa invasion by regulating EMT-related genes | [88] |
| lncRNA | PCAT29, DRAIC | Tumor-suppressive lncRNAs | Downregulated | Inhibit migration and metastasis of PCa cells; DRAIC interacts with IKK complex to suppress NF-κB | [86] |
| lncRNA | LINC00675 | Dual function lncRNA in CRPC | Upregulated | Promotes castration-resistant prostate cancer (CRPC) progression and therapy resistance. Direct binding to the AR N-terminal domain, which blocks MDM2 interaction, preventing ubiquitination and degradation | [89] |
| lncRNA | TERC (Telomerase RNA Component) | Oncogenic lncRNA | Upregulated | Supports tumor progression by maintaining telomere integrity and enhancing proliferation | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



