
Submitted:
16 January 2026
Posted:
19 January 2026
You are already at the latest version
Abstract
Here we discuss three veterinary alphaherpesviruses—pseudorabies virus, equid alphaherpesvirus 1, and bovine alphaherpesvirus 1—that were instrumental in uncovering the true extent of transcriptome complexity through long-read RNA sequencing, which earlier short-read approaches could not resolve. We focus on three major transcriptomic features whose discovery and characterization relied heavily on these viral models: (i) widespread transcriptional overlaps that complicate read assignment and may drive transcriptional interference; (ii) diverse transcript isoforms arising from alternative 5′ and 3′ transcript termini, as well as splicing; and (iii) non-coding RNAs clustered near replication origins that illuminate replication–transcription interactions on a shared nuclear template. Long-read viromics in these veterinary systems has additionally served as a stringent benchmark for transcript callers and annotation pipelines, because the extreme density of overlaps and co-terminal transcript families exposes errors that often go unnoticed in typical mammalian transcriptomes. This has made veterinary herpesvirus datasets disproportionately influential in shaping best practices for full-length isoform calling, transcript end mapping, and artifact-robust cDNA library handling. We also discuss animal gammaherpesviruses as proxies for human gammaherpesviruses, allowing experimental investigation of viral programs difficult to study in human infection. Finally, we describe pseudorabies virus applications as a retrograde transneuronal tracer.
Keywords:
veterinary herpesviruses
; alphaherpesvirus
; gammaherpesvirus
; transcriptomics
; longread sequencing
; nanopore
; RNA isoforms
; non-coding RNAs
; replication origin
; transcriptional interference
; transneuronal tract-tracing
1. Introduction
Herpesviruses (order Herpesvirales) are large double-stranded DNA viruses with a shared virion architecture and a nuclear replication cycle. Among veterinary alphaherpesviruses, pseudorabies virus (PRV) causes Aujeszky’s disease in swine, characterized by respiratory illness, neurological signs, and high mortality in young piglets [1]. Equid alphaherpesvirus 1 (EHV-1) is a major pathogen of horses, causing respiratory disease, abortion, and neurological disorders that result in significant economic losses worldwide [2]. Bovine alphaherpesvirus 1 (BoHV-1) is responsible for infectious bovine rhinotracheitis (IBR), leading to respiratory and reproductive disorders in cattle [3]. In contrast, caviid gammaherpesvirus 1 (CaGHV-1) represents the gammaherpesvirus subfamily and serves as a tractable small-animal model for studying latency, immune evasion, and cross-species gammaherpesvirus biology [4]. Together, these four veterinary herpesviruses provide accessible experimental models for investigating fundamental principles of herpesvirus transcriptome organization, regulation, and replication across different subfamilies.
Following entry, their genomes are delivered to the nucleus, where viral gene expression is driven largely by host RNA polymerase II and associated processing pathways, and is coordinated with viral DNA replication to produce progeny virions [5,6]. Their genomes are large by viral standards yet highly compact and transcriptionally crowded. Herpesvirus genes are frequently arranged in tandem clusters and are transcribed from both DNA strands, generating pervasive transcriptional overlaps. This genome architecture produces large 3′-co-terminal transcripts that share common polyadenylation sites, widespread antisense RNAs, and multigenic transcripts, together yielding a transcriptomic landscape of remarkable structural complexity. Herpesvirus gene expression is temporally regulated. The classical immediate-early, early and late cascade provides a useful conceptual framework, yet pervasive read-through transcription, alternative termination and time-dependent promoter usage frequently blur these categories. Transcript architectures therefore differ substantially across infection stages, making time-resolved sampling essential. Latency and reactivation-hallmark features of herpesvirus biology-involve highly restricted transcriptional programs dominated by non-coding RNAs (ncRNAs) that are often low-abundance, cell-type-specific or spatially confined. These ncRNAs regulate viral persistence, immune evasion and reactivation rather than serving as mere transcriptional by-products [7]. Given the pervasive transcriptional overlaps in herpesvirus genomes, transcription complexes are expected to collide frequently as they traverse shared genomic regions. Viral DNA replication and transcription occur simultaneously on the same nuclear template, creating opportunities for direct mechanistic interactions between the replication and transcription machineries. Notably, replication origins (Oris) have a special regulatory role, as they are key control points for initiating and modulating viral DNA synthesis. Herpesviruses thus offer experimentally accessible systems for studying fundamental principles of genome regulation that extend beyond virology [8].
2. Sequencing Technologies and Analytical Framework
Short-read RNA sequencing (srRNA-seq) has long been the backbone of transcriptomic studies, computationally assembling transcript structures from fragmented reads while delivering robust quantification and high throughput [9]. This computational assembly, however, faces inherent ambiguity in herpesvirus genomes characterized by extensive overlap and polygenic transcription. Alternative transcription start sites (TSSs) and transcription end sites (TESs), antisense RNAs and short ncRNAs are often poorly resolved. Long-read RNA sequencing (lrRNA-seq) has addressed these limitations by enabling capture of full-length RNA molecules [10]. Implemented mainly on Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) platforms, lrRNA-seq allows direct experimental identification of transcript boundaries, isoform structures and multigenic RNAs [8].
ONT direct RNA sequencing (dRNA-seq) uniquely reads native RNA molecules without reverse transcription or amplification [11]. In addition to avoiding cDNA-associated artifacts, dRNA-seq enables detection of epitranscriptomic modifications such as N6-methyladenosine (m6A) [12], 5-methylcytosine (m5C) [13] and pseudouridine [14] via characteristic nanopore current disruptions, and allows inference of adenosine-to-inosine (A-to-I) RNA editing from systematic basecalling discrepancies [15,16]. Its limitations include a requirement for polyadenylated transcripts and coverage biases toward intact RNA molecules [17]. PacBio-based long-read approaches, which typically sequence cDNA, provide highly accurate consensus transcript sequences, but RNA base modifications are generally not preserved through cDNA synthesis; however, SMRT assays that monitor reverse-transcription kinetics can indirectly report certain RNA modifications and structure-dependent RT behavior [18].
A recurring lesson from veterinary herpesvirus datasets is that “platform choice” cannot be separated from “question choice”. If the goal is isoform discovery, transcript boundary mapping, and detection of polygenic RNAs, full-length reads are indispensable. If the goal is fine-grained differential expression with high statistical power across many conditions, short reads remain useful—provided the annotation is already trustworthy. Notably, recent advances in lrRNA-seq technology now enable high-coverage quantification, increasingly blurring this traditional division. In practice, the highest-confidence herpesvirus transcriptomes have been built by using lrRNA-seq to define isoforms and transcript ends. Another practical consideration is library bias. Poly(A)-selection enriches for canonical mRNAs and many herpesvirus RNAs, but can under-represent non-polyadenylated species and some immature or decay intermediates. Conversely, total RNA approaches increase breadth but introduce rRNA-dominated backgrounds and may complicate accurate end calling. For herpesviruses, where closely spaced TESs and frequent read-through transcription are defining features, end-precision—often improved by cap-based or end-enrichment protocols—can matter as much as total depth. Finally, artifact awareness is essential. Template switching, internal priming, and reverse-transcription drop-off can create “ghost isoforms” that appear as alternative polyadenylation or splicing events. Veterinary herpesviruses have been instrumental in identifying these pitfalls because dense co-terminal transcript families provide many opportunities for false-positive isoforms unless filtering rules are explicit and validated across platforms.
Because cDNA-based methods may introduce artifacts such as template switching, current best practice often relies on hybrid strategies combining long-read sequencing (LRS) for structural resolution with short-read sequencing (SRS) for deep quantitative support, alongside orthogonal validation methods such as cap analysis of gene expression (CAGE) (Pardo-Palacios et al., 2024). Bioinformatically, Minimap2 is widely used for long-read alignment [19], FLAIR [20], Bambu [21] support isoform discovery, NAGATA provides nanopore direct RNA–guided transcriptome annotation (particularly useful for compact, gene-dense viral genomes) [22] and the LoRTIA pipeline provides integrated transcriptome annotation [23]. For viral genome assembly from long reads, tools such as Flye [24] enable de novo reconstruction, and in virome/metagenome settings viralFlye extends this workflow by assembling viral contigs from ONT/PacBio long reads and optionally using short reads for polishing when available. Finally, ONT’s Dorado basecaller (RNA004) includes pre-trained models that enable de novo calling of selected RNA modifications (e.g., pseudouridine/Ψ, m6A, inosine and m5C) during direct RNA basecalling [25,26].
3. Massive Transcriptional Overlaps
One of the most striking features of herpesvirus transcriptomes is the extent of transcriptional overlap, where transcripts frequently overlap in convergent, divergent or tandem orientations and antisense transcription is widespread (Figure 1).
Such pervasive overlap complicates transcript annotation and quantification, particularly for srRNA-seq, where individual reads may map equally well to multiple transcripts. Importantly, transcriptional overlaps are not merely technical nuisances but may have biological significance. Convergent and antisense transcription can give rise to transcriptional interference [27], altered termination efficiency and polymerase collisions [8]. lrRNA-seq studies in veterinary alphaherpesviruses—most notably PRV [28]—revealed that extensive overlap is a defining organizational principle rather than a rare exception. These analyses uncovered dense networks of overlapping coding and non-coding transcripts that were largely invisible to short-read approaches [29]. Similar conclusions emerged from comprehensive EHV-1 [30] and BoHV-1 [31] transcriptome maps, underscoring that overlap must be interpreted as a functional feature of herpesvirus genome organization [32]. In fact, similar patterns of overlapping transcription were observed in other virus families, such as poxviruses [33], baculoviruses [34], and flaviviruses [35].
In PRV, lrRNA-seq has revealed that overlap patterns extend well beyond the long-known parallel overlaps within tandem gene clusters: divergent gene pairs can generate extensive head-to-head overlaps through very long alternative 5′-untranslated regions (UTRs), while convergent loci frequently produce readthrough RNAs that embed antisense segments relative to the partner gene [36]. Two major types of convergent overlap can be distinguished: “hard” overlaps occur when the canonical transcripts of both genes intrinsically overlap—that is, the TES of one canonical transcript lies within the neighboring convergent gene—whereas “soft” overlaps arise from occasional readthrough events extending into the partner gene. Although convergent genes are often separated by relatively long, commonly repetitive intergenic regions, soft overlaps via readthrough occur in essentially every convergent gene pair, while hard overlaps (e.g., PRV ul7/8, ul30/31, ul50/51 gene pairs) remain rare exceptions [36].
EHV-1 provides an even more overlap-dense example: nearly every divergent gene pair produces transcripts with extensive head-to-head overlap, and the transcriptome contains very long overlaps spanning multiple genes. Canonical convergent transcripts usually show only “soft” overlaps via occasional readthrough, but the ORF29/30 (ortholog of PRV ul31/30) pair represents a rare “hard” overlap exception [30]. In BoHV-1, time-course long-read analyses further revealed the complexity of this overlapping meshwork organization [32].
4. Transcript Isoforms: Splice Variants and 5′/3′ Termini Diversity
A second major layer of herpesvirus transcriptome complexity arises from transcript isoform diversity. Individual genomic loci often give rise to multiple transcripts initiated from distinct TSSs, terminated at alternative TESs, and in some cases processed by splicing. Although the prevalence of splicing varies among herpesvirus subfamilies, alternative transcript boundaries and nested transcript organization are nearly universal. Alternative TSS usage can reflect multiple promoters within a locus, temporal shifts in promoter activity during infection, or processing of read-through transcripts. Herpesviruses exhibit substantially greater TSS variability than TES variability—unlike poxviruses [33], for example, where the opposite pattern is observed. TES variability and alternative polyadenylation generate co-terminal transcript families with different 3′ untranslated region lengths, potentially influencing RNA stability, localization and post-transcriptional regulation. While srRNA-seq can detect splice junctions, it faces two fundamental limitations in isoform reconstruction: (1) it cannot unambiguously assign spliced reads to specific TSS–TES combinations, and (2) when transcripts contain two or more introns, it cannot reliably determine which introns co-occur within the same mature transcript molecule. lrRNA-seq overcomes this limitation by capturing complete transcript structures in single reads. In PRV, long-read transcriptome mapping revealed extensive previously unrecognized isoform diversity and demonstrated that variation in TSSs and TESs contributes more to overall complexity than splicing itself [29].
PRV illustrates the scale of transcript-termini diversity particularly clearly: the updated atlas reported numerous TSS isoforms with variable 5′-UTR lengths and fewer TES variants altering 3′-UTR boundaries under stringent filtering [36]. A particularly informative observation is that some long TES isoforms traverse the intergenic region (e.g., UL27-AT, UL35-AT, UL44-AT, CTO-S-AT, US2-AT), potentially impacting downstream gene regulation or creating novel transcript overlaps. Crucially, PRV also revealed a large class of 5′-truncated RNAs that are 3′-co-terminal with canonical mRNAs: many of these initiate within ORFs, carry in-frame ATGs, and share the canonical stop codon—consistent with the potential for N-terminally truncated polypeptides—whereas the remainder are likely non-coding [36]. This nested/co-terminal architecture blurs the boundary between “TSS isoforms” and distinct coding units and directly motivates LRS-based full-length validation when interpreting intragenic initiation [36]. Replication-origin loci further highlight how TES/TSS heterogeneity interacts with regulatory hubs: CTO-S is extremely abundant in PRV, and the Ori-associated region contains additional low-copy isoforms and antisense transcripts, including CTO-as and UL21-as, alongside novel CTO-S variants (e.g., a novel TES isoform CTO-S-AT2 and CTO-S-cx RNA) [36].
BoHV-1 time-course long-read profiling adds two concrete isoform principles that are hard to recover with SRS alone. First, a single promoter can generate multiple TSSs with gene-specific distribution patterns that remain reproducible across timepoints, providing a quantitative handle on promoter “micro-heterogeneity” [32]. Second, the bicp4 locus shows unusually rich transcript-end diversity. TSS isoforms initiate from both the 5′-UTR and within the ORF, while TES isoforms—rare for herpesvirus genes—terminate in both the 3′-UTR and within the ORF. The locus also generates 3′-truncated ncRNAs and ncRNAs overlapping long UTR isoforms [32]. In parallel, bicp22 exhibits one of the most complex architectures, combining TSS polymorphism, multiple splice patterns, very long readthroughs spanning much of the US region, and detectable antisense transcription; its short versus long TSS isoforms also differ in upstream ORF (uORF) content, providing a plausible route for translational modulation layered onto transcript-structure diversity [32]. EHV-1 extends isoform complexity into both splicing and termination logic. The hybrid atlas reported abundant fusion/chimeric transcripts, some compatible with chimeric proteins. Notably, many upstream genes within tandem clusters possess their own TESs in addition to shared co-terminal ends—an organization far less prominent in related alphaherpesviruses [30]. EHV-1 encodes a CTO-S homolog near OriL and detects CTO-L as a TES isoform of the ul21 homolog (ORF40) co-terminal with canonical CTO-S.
5. Replication Origin-Associated RNAs: CTO and NOIR Families
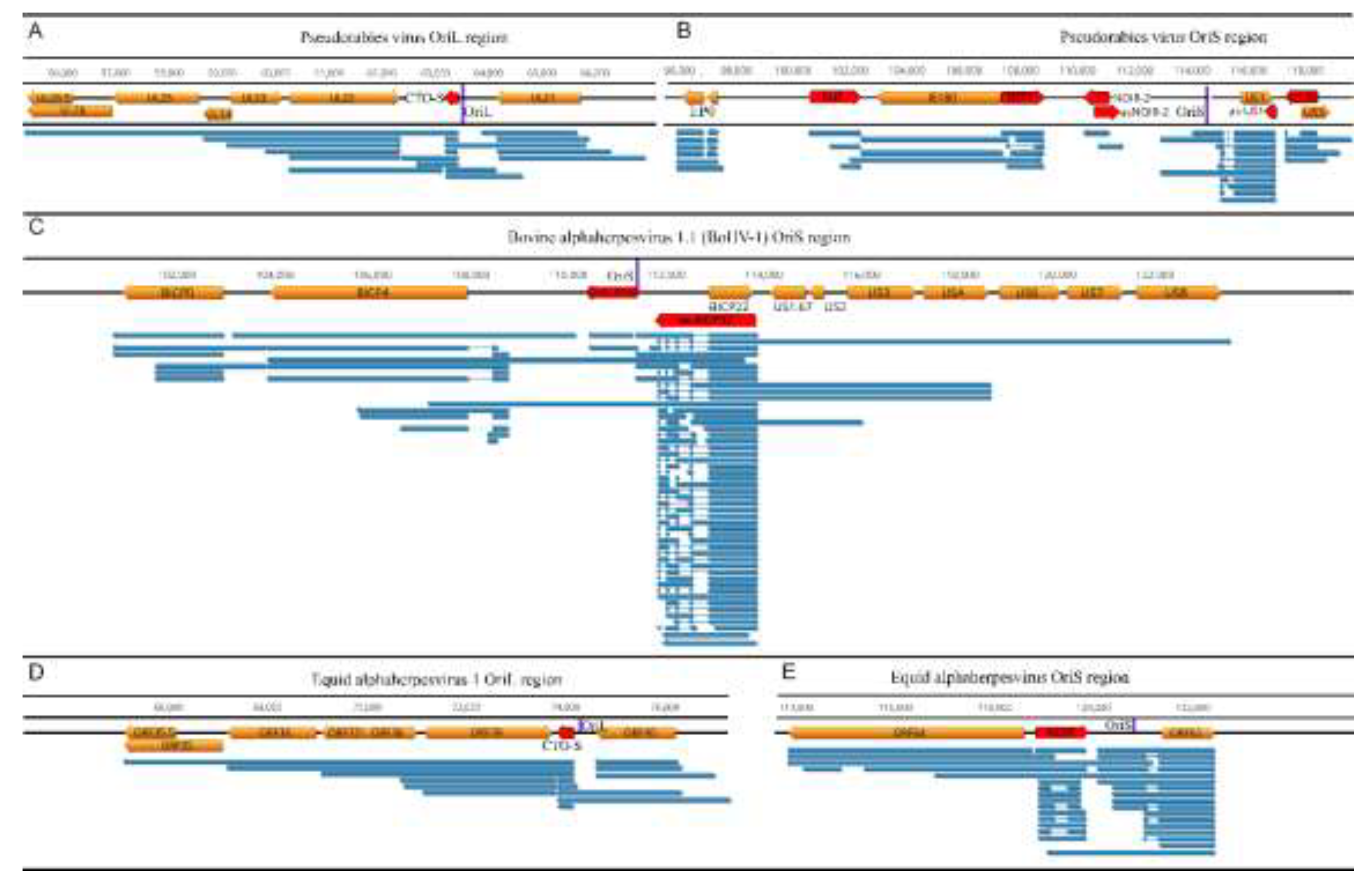
A particularly distinctive contribution of veterinary herpesvirus models concerns transcription around the Oris (Figure 2A-E).
lrRNA-seq studies revealed that Ori regions are frequently embedded within dense transcriptional landscapes enriched for ncRNAs. In alphaherpesviruses, several classes of replication origin-associated RNAs (raRNAs) have been described, including transcript families referred to as CTO and NOIR, characterized by their proximity to OriL and OriS, respectively [37,38]. CTO transcripts have been identified in PRV and EHV-1 but are absent in BoHV-1, which lacks OriL. In EHV-1, the longer CTO-S isoform’s TATA box is co-localized with OriL [30]. In contrast, NOIR-like transcriptional activity at OriS appears to be a conserved feature of varicelloviruses: NOIR homologues or functionally analogous transcripts have been detected in all varicellovirus genomes subjected to comprehensive LRS. For example, in varicella-zoster virus and simian varicella virus, NOIR-like RNAs are present, while in BoHV-1, which lacks a canonical NOIR gene, a very long 5′-UTR isoform of the us1 homologue may fulfill this function [38]. In PRV, the OriS region harbors not only NOIR-1 but also NOIR-2, which is transcribed convergently to NOIR-1. In BoHV-1, OriS-RNA may mediate similar NOIR-2-like activity [31]. These ncRNAs form structured transcriptional environments around the Ori, which may influence replication initiation, local chromatin organization, or replication–transcription interference.
In alphaherpesviruses, OriS is flanked by the two master transcriptional regulators—icp4 and us1—whose extended TSS isoforms span the Ori itself. In BoHV-1, the OriS region harbors an exceptionally dense regulatory architecture, including long, oppositely oriented IE transcripts overlapping OriS: long TSS isoforms of bICP4 and bICP22. Notably, the bICP22 promoter/TSS region itself overlaps OriS, reinforcing the idea that replication-initiation and local transcription initiation can physically and functionally intersect [32]. These long isoforms overlap both OriS and each other, creating a multi-layered transcriptional architecture. This genomic configuration suggests that the OriS region functions as a super-regulatory hub where DNA replication and global transcriptional control are spatially integrated and likely subject to reciprocal interference. These observations have broader implications for understanding how replication and transcription are coordinated on compact eukaryotic DNA genomes.
A key open question is whether Ori-associated transcription actively triggers replication initiation or merely reflects the high regulatory activity concentrated at these genomic hubs. Several non-mutually exclusive mechanisms have been proposed based on existing transcript maps [38]. First, raRNAs may directly regulate Ori function by recruiting or displacing Ori-binding proteins through RNA–protein interactions. Second, the act of transcription through Ori may mechanically alter local chromatin structure—changing supercoiling and nucleosome positioning—thereby modulating Ori accessibility. Third, transcriptional polymerase traffic may create regulated collisions that control when and where replication forks can enter. Fourth, raRNAs may act as molecular decoys, sequestering host RNA-binding proteins that would otherwise inhibit viral replication. Supporting a direct regulatory role, functional studies in betaherpesviruses [39] and gammaherpesviruses [40] have demonstrated that replication-associated RNAs control DNA synthesis initiation through formation of RNA:DNA hybrids. Veterinary herpesviruses provide ideal systems for testing these models experimentally, as Ori regions and their flanking promoters can be genetically manipulated and the consequences measured through replication kinetics and transcriptome profiling.
Another underappreciated implication is that Ori-regions are hotspots for annotation artifacts unless end calling is precise. Because multiple long isoforms can traverse Ori regions from both directions, incomplete cDNAs may appear as distinct short ncRNAs. Direct RNA sequencing and cross-platform confirmation in PRV/EHV-1/BoHV-1 have therefore been critical not only for the precise characterization of CTO/NOIR isoforms but also for establishing stringent criteria for calling authentic raRNAs.
6. Caviid Gammaherpesvirus-1 as a Gammaherpesvirus Model
Human gammaherpesviruses, such as Epstein–Barr virus (EBV) [41,42,43] and Kaposi’s sarcoma-associated herpesvirus (KSHV) [44,45], are major pathogens associated with malignancies and lifelong latency. Despite their clinical importance, experimental study is constrained by ethical and technical limitations: limited access to human tissues, restricted invasive sampling, and infeasibility of longitudinal studies. In vitro systems often fail to recapitulate the natural infection cycle, particularly latency, reactivation and host immunity. Animal gammaherpesviruses, such as murine gammaherpesvirus 68 (MHV68) [46,47], overcome these barriers through controlled infection and genetic manipulation. However, MHV68 has important limitations: limited sequence homology to human gammaherpesviruses, lack of key regulatory elements found in KSHV and EBV, and significant physiological differences in latency establishment.
Caviid gammaherpesvirus-1 (CaGHV-1), originally identified in 1969 [48] and recently reclassified as a rhadinovirus [4], exhibits remarkable genomic and functional similarity to KSHV. The virus encodes 75 predicted ORFs, including orthologues of key KSHV oncogenes: ORF73 (LANA), ORF50 (RTA), and the PAN ncRNA essential for lytic replication in KSHV but absent in MHV68. Torma and colleagues (2025) [49] conducted the first comprehensive lrRNA-seq analysis of CaGHV-1, revealing extensive transcript complexity mirroring KSHV. The study identified monogenic mRNAs, polygenic transcripts, complex transcripts, and antisense RNAs. Using LoRTIA [23], TSSs were mapped at single-nucleotide resolution, identifying TATA boxes and the TATTWAA motif essential for late gene transcription in KSHV, EBV, and HCMV [50]. Splicing patterns in key regulatory genes showed remarkable conservation with KSHV: CaGHV-1 ORF50 (RTA) contains four exons matching KSHV ORF50, and ORF57 splicing mirrors its KSHV orthologue. Additionally, extensive transcriptional complexity was identified in the G4-G5 region and ORF63-64 locus, with multiple splice variants paralleling KSHV [45]. Furthermore, genome-wide transcriptional overlaps were revealed between convergent, divergent, and co-oriented genes. “Hard” overlaps were identified in convergent clusters (ORF18-ORF19, ORF64-ORF65, ORF74-ORF75), and “soft” overlaps in others (ORF10-K3, G4-ORF52)—patterns strikingly similar to EBV [43] and KSHV [45]. Numerous raRNAs were detected near both lytic Oris, including transcripts overlapping origins and long complex RNAs encompassing Oris, phenomena documented in KSHV, EBV, and HCMV [38].
The guinea pig model offers practical benefits: lower cost, established husbandry, and fewer ethical constraints than non-human primate models, while maintaining closer biological similarity to human gammaherpesviruses than MHV68. CaGHV-1 is naturally oncogenic in guinea pigs, inducing lymphoproliferative disease analogous to KSHV-associated malignancies [4], providing a physiologically relevant system for studying gammaherpesvirus-driven oncogenesis and enabling investigations infeasible in human studies.
7. Pseudorabies Virus as a Transneuronal Tracer
Beyond its role as a veterinary pathogen and transcriptomic model, PRV has become a cornerstone tool in modern neuroscience. Attenuated and genetically engineered PRV strains are widely used as retrograde transneuronal tracers for mapping neural circuits, owing to their ability to infect neurons, replicate efficiently, and spread across synaptically connected networks in a directionally controlled manner [51,52]. Several biological properties make PRV particularly well suited for this application. First, PRV exhibits robust neurotropism and efficient axonal transport, allowing it to traverse multisynaptic pathways with high fidelity. Second, the temporal progression of infection can be experimentally controlled using replication-competent, attenuated or replication-deficient strains, enabling time-resolved dissection of neuronal connectivity. Third, PRV tolerates substantial genome engineering, permitting insertion of reporter genes or fluorescent activity markers without compromising viral spread.
PRV-based circuit tracing has been successfully applied to diverse neural systems. The virus has been used to map autonomic pathways controlling peripheral organs such as brown adipose tissue, bone marrow, and the cardiovascular system [53,54]. In the visual system, PRV injection into the eye has revealed retinopetal connections between the pineal body and retina in hamsters [55]. Motor system studies have demonstrated PRV-based visualization of cortical reorganization following facial nerve injury [56]. Additionally, PRV has been instrumental in delineating descending pathways from cortex to spinal motor circuits and in identifying sympathetic premotor neurons in medullary raphe regions mediating thermoregulatory functions [54].
Beyond traditional anatomical tracing, recombinant PRV strains have been engineered to carry genetically encoded activity sensors such as ratiometric calcium indicators, enabling optical monitoring of neural activity in virally labeled circuits [57]. These activity sensor PRVs permit researchers to both identify synaptically connected neurons and simultaneously record their functional responses to physiological or pharmacological stimuli. Timer PRVs expressing two fluorescent proteins with different kinetics have been developed to define temporal windows early after infection, allowing functional interrogation during periods when neuronal physiology remains relatively intact. Furthermore, multicolor rainbow PRVs expressing spectrally distinct fluorescent proteins facilitate the simultaneous tracing and differentiation of multiple parallel circuits within complex brain regions. PRV has also been explored as a gene delivery vector for experimental neuroscience and gene function studies [57]. Compared with non-viral delivery systems, PRV-based vectors provide high transgene expression levels, efficient neuronal infection and the ability to target defined neural circuits. Unlike human herpesviruses, PRV can be used in animal models with fewer biosafety and ethical constraints, while still preserving key biological properties relevant to alphaherpesvirus biology.
The dual role of PRV—as both a model alphaherpesvirus for transcriptomic discovery and a practical neurobiological tool—highlights the broader impact of veterinary herpesvirus research. Insights gained from transcriptome analysis not only refine our understanding of viral gene regulation but also directly inform applied uses of these viruses as experimental reagents.
8. Concluding Remarks
Veterinary herpesviruses have played a pivotal role in transforming not only herpesvirus transcriptomics but also broader eukaryotic transcriptomics from an inference-driven discipline into an experimentally grounded field. Alphaherpesvirus models have been central to identifying three defining features of herpesvirus transcriptomes: massive transcriptional overlaps, extensive transcript isoform diversity, and complex arrays of Ori-associated ncRNAs, including the CTO and NOIR families. In parallel, animal gammaherpesviruses provide indispensable systems for studying gammaherpesvirus transcription under conditions that are ethically and technically inaccessible in humans. As lrRNA-seq technologies continue to mature, veterinary herpesviruses are likely to remain central in understanding viral gene regulation. Beyond virology, PRV has been repurposed as a retrograde transsynaptic neural circuit tracer. Looking forward, inducible systems enabling temporal control over viral spread represent a promising perspective for enhancing the precision of PRV-based circuit mapping.
Funding
National Research, Development and Innovation Office grants: K 142674 and ADVANCED 152705 to Zsolt Boldogkői, and FK 142676 to Dóra Tombácz.
Data Availability Statement
No new datasets were generated in this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular Biology of Pseudorabies Virus: Impact on Neurovirology and Veterinary Medicine. Microbiol Mol Biol Rev 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Heldens, J. Equine Herpesviruses 1 (EHV-1) and 4 (EHV-4)--Epidemiology, Disease and Immunoprophylaxis: A Brief Review. Vet J 2005, 170, 14–23. [Google Scholar] [CrossRef]
- Nandi, S.; Kumar, M.; Manohar, M.; Chauhan, R.S. Bovine Herpes Virus Infections in Cattle. Anim Health Res Rev 2009, 10, 85–98. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Ruiz, E.; Chouljenko, V.N.; Kousoulas, K.G. Guinea Pig Herpes like Virus Is a Gamma Herpesvirus. Virus Genes 2024, 60, 148–158. [Google Scholar] [CrossRef]
- Davison, A.J. Herpesvirus Systematics. Veterinary Microbiology 2010, 143, 52–69. [Google Scholar] [CrossRef]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. Journal of Virology 2017, 92. [Google Scholar] [CrossRef]
- Hancock, M.H.; Skalsky, R.L. Roles of Non-Coding RNAs During Herpesvirus Infection. In Roles of Host Gene and Non-coding RNA Expression in Virus Infection; Tripp, R.A., Tompkins, S.M., Eds.; Springer International Publishing: Cham, 2018; pp. 243–280. ISBN 978-3-030-05369-7. [Google Scholar]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-Read Sequencing – A Powerful Tool in Viral Transcriptome Research. Trends in Microbiology 2019, 27, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Long-Read Sequencing. Nat Methods 2023, 20, 6–11. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Rihtman, B.; Michniewski, S.; Redgwell, T.; Clokie, M.; Stekel, D.J.; Chen, Y.; Scanlan, D.J.; Hobman, J.L.; et al. The Long and Short of It: Benchmarking Viromics Using Illumina, Nanopore and PacBio Sequencing Technologies. Microbial Genomics 2024, 10, 001198. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA Sequencing on Nanopore Arrays Redefines the Transcriptional Complexity of a Viral Pathogen. Nat Commun 2019, 10, 754. [Google Scholar] [CrossRef]
- Zhong, Z.-D.; Xie, Y.-Y.; Chen, H.-X.; Lan, Y.-L.; Liu, X.-H.; Ji, J.-Y.; Wu, F.; Jin, L.; Chen, J.; Mak, D.W.; et al. Systematic Comparison of Tools Used for m6A Mapping from Nanopore Direct RNA Sequencing. Nat Commun 2023, 14, 1906. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA Modifications Detection by Comparative Nanopore Direct RNA Sequencing. Nat Commun 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Begik, O.; Lucas, M.C.; Pryszcz, L.P.; Ramirez, J.M.; Medina, R.; Milenkovic, I.; Cruciani, S.; Liu, H.; Vieira, H.G.S.; Sas-Chen, A.; et al. Quantitative Profiling of Pseudouridylation Dynamics in Native RNAs with Nanopore Sequencing. Nat Biotechnol 2021, 39, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore Native RNA Sequencing of a Human Poly(A) Transcriptome. Nat Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct RNA Sequencing Enables m6A Detection in Endogenous Transcript Isoforms at Base-Specific Resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly Parallel Direct RNA Sequencing on an Array of Nanopores. Nat Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Vilfan, I.D.; Tsai, Y.-C.; Clark, T.A.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA Base Modification and Structural Rearrangement by Single-Molecule Real-Time Detection of Reverse Transcription. J Nanobiotechnology 2013, 11, 8. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-Length Transcript Characterization of SF3B1 Mutation in Chronic Lymphocytic Leukemia Reveals Downregulation of Retained Introns. Nat Commun 2020, 11, 1438. [Google Scholar] [CrossRef]
- Chen, Y.; Sim, A.; Wan, Y.K.; Yeo, K.; Lee, J.J.X.; Ling, M.H.; Love, M.I.; Göke, J. Context-Aware Transcript Quantification from Long-Read RNA-Seq Data with Bambu. Nat Methods 2023, 20, 1187–1195. [Google Scholar] [CrossRef]
- Abebe, J.S.; Alwie, Y.; Fuhrmann, E.; Leins, J.; Mai, J.; Verstraten, R.; Schreiner, S.; Wilson, A.C.; Depledge, D.P. Nanopore Guided Annotation of Transcriptome Architectures. mSystems 2024, 9, e00505-24. [Google Scholar] [CrossRef]
- Balázs, Z.; Tombácz, D.; Csabai, Z.; Moldován, N.; Snyder, M.; Boldogkői, Z. Template-Switching Artifacts Resemble Alternative Polyadenylation. BMC Genomics 2019, 20, 824. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat Biotechnol 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.I.; Rübsam, F.N.M.; Sun, Y.; Ehrenhofer-Murray, A.E. Evaluation of Dorado v5.2.0 de Novo Basecalling Models for the Detection of tRNA Modifications Using RNA004 Chemistry 2025, 2025.12.09.693013.
- Rübsam, F.N.M.; Liu-Wei, W.; Sun, Y.; Patel, B.I.; van der Toorn, W.; Piechotta, M.; Dieterich, C.; von Kleist, M.; Ehrenhofer-Murray, A.E. MoDorado: Enhanced Detection of tRNA Modifications in Nanopore Sequencing by off-Label Use of Modification Callers. Nucleic Acids Res 2025, 53, gkaf795. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, N.; Wang, L.; Fang, O.; Leach, L.J.; Hu, X.; Luo, Z. 3’ Untranslated Regions Mediate Transcriptional Interference between Convergent Genes Both Locally and Ectopically in Saccharomyces Cerevisiae. PLoS Genet 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Balázs, Z.; Csabai, Z.; Moldován, N.; Szűcs, A.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of the Dynamic Transcriptome of a Herpesvirus with Long-Read Single Molecule Real-Time Sequencing. Sci Rep 2017, 7, 43751. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Balázs, Z.; Likó, I.; Zsigmond, L.; Sharon, D.; Snyder, M.; Boldogkői, Z. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLOS ONE 2016, 11, e0162868. [Google Scholar] [CrossRef]
- Tombácz, D.; Torma, G.; Gulyás, G.; Fülöp, Á.; Dörmő, Á.; Prazsák, I.; Csabai, Z.; Mizik, M.; Hornyák, Á.; Zádori, Z.; et al. Hybrid Sequencing Discloses Unique Aspects of the Transcriptomic Architecture in Equid Alphaherpesvirus 1. Heliyon 2023, 9, e17716. [Google Scholar] [CrossRef]
- Moldován, N.; Torma, G.; Gulyás, G.; Hornyák, Á.; Zádori, Z.; Jefferson, V.A.; Csabai, Z.; Boldogkői, M.; Tombácz, D.; Meyer, F.; et al. Time-Course Profiling of Bovine Alphaherpesvirus 1.1 Transcriptome Using Multiplatform Sequencing. Sci Rep 2020, 10, 20496. [Google Scholar] [CrossRef]
- Tombácz, D.; Kakuk, B.; Torma, G.; Csabai, Z.; Gulyás, G.; Tamás, V.; Zádori, Z.; Jefferson, V.A.; Meyer, F.; Boldogkői, Z. In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing. Viruses 2022, 14, 1289. [Google Scholar] [CrossRef]
- Nagy, G.Á.; Tombácz, D.; Prazsák, I.; Csabai, Z.; Dörmő, Á.; Gulyás, G.; Kemenesi, G.; Tóth, G.E.; Holoubek, J.; Růžek, D.; et al. Exploring the Transcriptomic Profile of Human Monkeypox Virus via CAGE and Native RNA Sequencing Approaches. mSphere 2024, 9, e00356-24. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Moldován, N.; Szűcs, A.; Tombácz, D. Transcriptome-Wide Analysis of a Baculovirus Using Nanopore Sequencing. Sci Data 2018, 5, 180276. [Google Scholar] [CrossRef] [PubMed]
- Olasz, F.; Tombácz, D.; Torma, G.; Csabai, Z.; Moldován, N.; Dörmő, Á.; Prazsák, I.; Mészáros, I.; Magyar, T.; Tamás, V.; et al. Short and Long-Read Sequencing Survey of the Dynamic Transcriptomes of African Swine Fever Virus and the Host Cells. Frontiers in Genetics 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Torma, G.; Tombácz, D.; Csabai, Z.; Göbhardter, D.; Deim, Z.; Snyder, M.; Boldogkői, Z. An Integrated Sequencing Approach for Updating the Pseudorabies Virus Transcriptome. Pathogens 2021, 10, 242. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-Read Sequencing - A Powerful Tool in Viral Transcriptome Research. Trends Microbiol 2019, 27, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Torma, G.; Tombácz, D.; Csabai, Z.; Almsarrhad, I.A.A.; Nagy, G.Á.; Kakuk, B.; Gulyás, G.; Spires, L.M.; Gupta, I.; Fülöp, Á.; et al. Identification of Herpesvirus Transcripts from Genomic Regions around the Replication Origins. Sci Rep 2023, 13, 16395. [Google Scholar] [CrossRef]
- Tai-Schmiedel, J.; Karniely, S.; Lau, B.; Ezra, A.; Eliyahu, E.; Nachshon, A.; Kerr, K.; Suárez, N.; Schwartz, M.; Davison, A.J.; et al. Human Cytomegalovirus Long Noncoding RNA4.9 Regulates Viral DNA Replication. PLoS Pathog 2020, 16, e1008390. [Google Scholar] [CrossRef]
- Rennekamp, A.J.; Lieberman, P.M. Initiation of Epstein-Barr Virus Lytic Replication Requires Transcription and the Formation of a Stable RNA-DNA Hybrid Molecule at OriLyt. J Virol 2011, 85, 2837–2850. [Google Scholar] [CrossRef]
- O’Grady, T.; Wang, X.; Höner zu Bentrup, K.; Baddoo, M.; Concha, M.; Flemington, E.K. Global Transcript Structure Resolution of High Gene Density Genomes through Multi-Platform Data Integration. Nucleic Acids Res 2016, 44, e145. [Google Scholar] [CrossRef]
- Shekhar, R.; O’Grady, T.; Keil, N.; Feswick, A.; Amador, D.A.M.; Tibbetts, S.A.; Flemington, E.K.; Renne, R. High-Density Resolution of the Kaposi’s Sarcoma Associated Herpesvirus Transcriptome Identifies Novel Transcript Isoforms Generated by Long-Range Transcription and Alternative Splicing. Nucleic Acids Research 2024, 52, 7720–7739. [Google Scholar] [CrossRef]
- Fülöp, Á.; Torma, G.; Moldován, N.; Szenthe, K.; Bánáti, F.; Almsarrhad, I.A.A.; Csabai, Z.; Tombácz, D.; Minárovits, J.; Boldogkői, Z. Integrative Profiling of Epstein–Barr Virus Transcriptome Using a Multiplatform Approach. Virol J 2022, 19, 7. [Google Scholar] [CrossRef]
- Chandriani, S.; Xu, Y.; Ganem, D. The Lytic Transcriptome of Kaposi’s Sarcoma-Associated Herpesvirus Reveals Extensive Transcription of Noncoding Regions, Including Regions Antisense to Important Genes. Journal of Virology 2010, 84, 7934–7942. [Google Scholar] [CrossRef]
- Prazsák, I.; Tombácz, D.; Fülöp, Á.; Torma, G.; Gulyás, G.; Dörmő, Á.; Kakuk, B.; McKenzie Spires, L.; Toth, Z.; Boldogkői, Z. KSHV 3.0: A State-of-the-Art Annotation of the Kaposi’s Sarcoma-Associated Herpesvirus Transcriptome Using Cross-Platform Sequencing. mSystems 2024, 9, e01007-23. [Google Scholar] [CrossRef]
- Virgin, H.W.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; Dal Canto, A.J.; Speck, S.H. Complete Sequence and Genomic Analysis of Murine Gammaherpesvirus 68. J Virol 1997, 71, 5894–5904. [Google Scholar] [CrossRef]
- Rajcáni, J.; Blaskovic, D.; Svobodová, J.; Ciampor, F.; Hucková, D.; Staneková, D. Pathogenesis of Acute and Persistent Murine Herpesvirus Infection in Mice. Acta Virol 1985, 29, 51–60. [Google Scholar]
- Hsiung, G.D.; Kaplow, L.S. Herpeslike Virus Isolated from Spontaneously Degenerated Tissue Culture Derived from Leukemia-Susceptible Guinea Pigs. J Virol 1969, 3, 355–357. [Google Scholar] [CrossRef]
- Torma, G.; Dörmő, Á.; Fülöp, Á.; Tombácz, D.; Mizik, M.; Pretory, A.M.; Lee, S.-C.; Toth, Z.; Boldogkői, Z. Long-Read Transcriptomics of Caviid Gammaherpesvirus 1: Compiling a Comprehensive RNA Atlas. mSystems 2025, 10, e01678-24. [Google Scholar] [CrossRef] [PubMed]
- Dremel, S.E.; Didychuk, A.L. Better Late than Never: A Unique Strategy for Late Gene Transcription in the Beta- and Gammaherpesviruses. Seminars in Cell & Developmental Biology 2023, 146, 57–69. [Google Scholar] [CrossRef]
- Boldogköi, Z.; Sı́k, A.; Dénes, Á.; Reichart, A.; Toldi, J.; Gerendai, I.; Kovács, K.J.; Palkovits, M. Novel Tracing Paradigms—Genetically Engineered Herpesviruses as Tools for Mapping Functional Circuits within the CNS: Present Status and Future Prospects. Progress in Neurobiology 2004, 72, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Card, J.P.; Enquist, L.W. Transneuronal Circuit Analysis with Pseudorabies Viruses. Curr Protoc Neurosci 2014, 68, 1.5.1–1.5.39. [Google Scholar] [CrossRef] [PubMed]
- Dénes, Á.; Boldogkoi, Z.; Uhereczky, G.; Hornyák, Á.; Rusvai, M.; Palkovits, M.; Kovács, K.J. Central Autonomic Control of the Bone Marrow: Multisynaptic Tract Tracing by Recombinant Pseudorabies Virus. Neuroscience 2005, 134, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Matsumura, K.; Hübschle, T.; Nakamura, Y.; Hioki, H.; Fujiyama, F.; Boldogköi, Z.; König, M.; Thiel, H.-J.; Gerstberger, R.; et al. Identification of Sympathetic Premotor Neurons in Medullary Raphe Regions Mediating Fever and Other Thermoregulatory Functions. J Neurosci 2004, 24, 5370–5380. [Google Scholar] [CrossRef]
- Csáki, Á.; Vígh, B.; Boldogkői, Z.; Vereczki, V.; Szél, Á.; Köves, K. Is a Neuronal Chain between the Pineal Body and the Retina in Rats and Hamsters? Transneural Tracing Studies. Neuroscience Letters 2015, 588, 1–6. [Google Scholar] [CrossRef]
- Horváth, S.; Kis, Z.; Boldogköi, Z.; Nógrádi, A.; Toldi, J. Oestrogen-Dependent Tracing in the Rat CNS after Pseudorabies Virus Infection. Eur J Neurosci 2002, 15, 937–943. [Google Scholar] [CrossRef]
- Boldogkoi, Z.; Balint, K.; Awatramani, G.B.; Balya, D.; Busskamp, V.; Viney, T.J.; Lagali, P.S.; Duebel, J.; Pásti, E.; Tombácz, D.; et al. Genetically Timed, Activity-Sensor and Rainbow Transsynaptic Viral Tools. Nat Methods 2009, 6, 127–130. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genome-wide transcriptional overlaps across the caviid gammaherpesvirus 1 (CaGHV-1) genome. Strand-specific long-read RNA-seq read alignments displayed in IGV illustrate transcription from the forward (red) and reverse (blue) strands along the complete viral genome. Overlay of signals from the two strands (color blending/shading) highlights pervasive convergent, divergent, and parallel transcriptional overlaps that extend across coding loci and intergenic intervals.
Figure 1.
Genome-wide transcriptional overlaps across the caviid gammaherpesvirus 1 (CaGHV-1) genome. Strand-specific long-read RNA-seq read alignments displayed in IGV illustrate transcription from the forward (red) and reverse (blue) strands along the complete viral genome. Overlay of signals from the two strands (color blending/shading) highlights pervasive convergent, divergent, and parallel transcriptional overlaps that extend across coding loci and intergenic intervals.
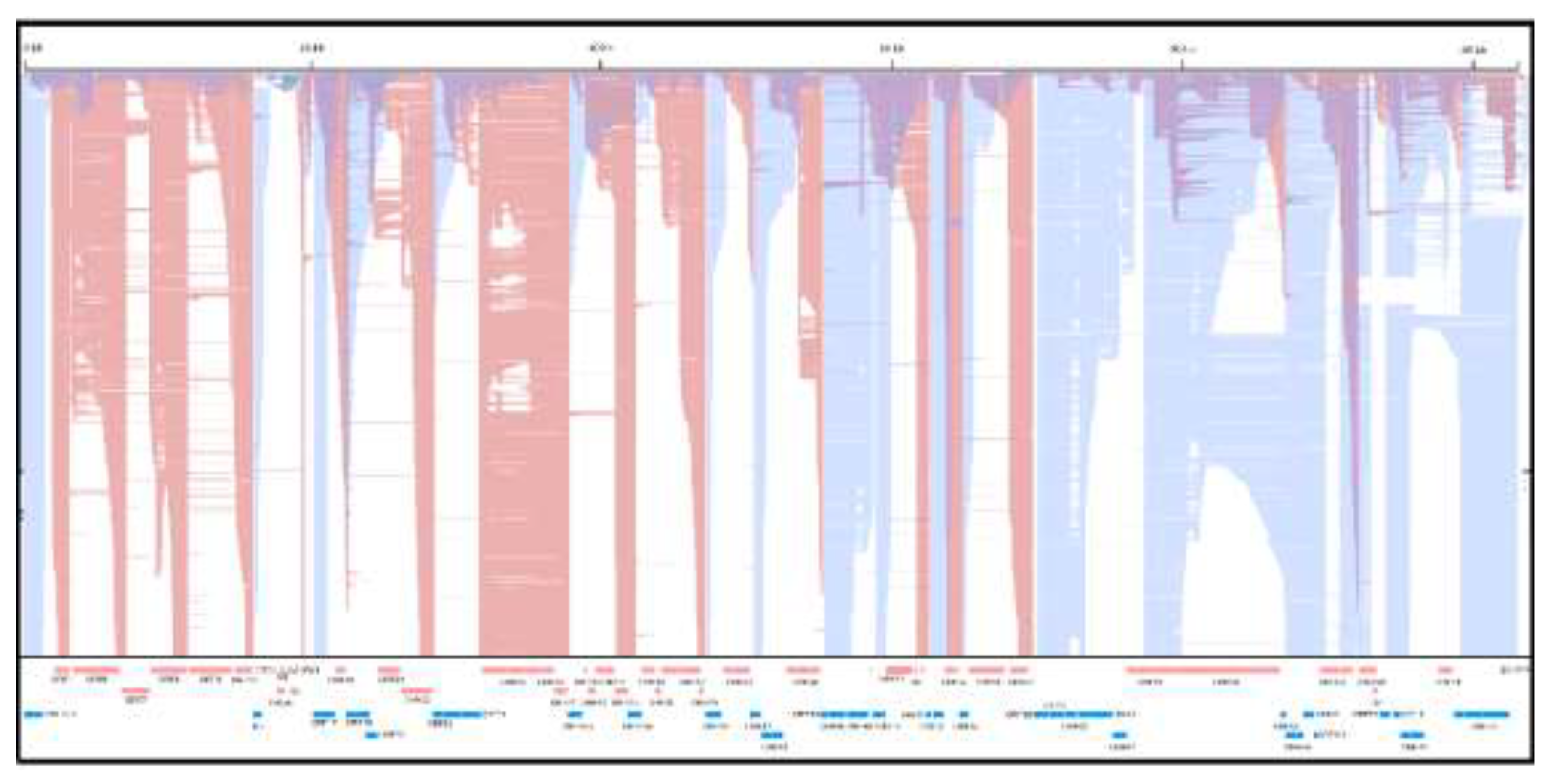
Figure 2.
Transcriptional architectures surrounding lytic replication origins (OriL/OriS) in varicelloviruses. Panels show (A) pseudorabies virus (PRV) OriL, (B) PRV OriS, (C) bovine alphaherpesvirus 1.1 (BoHV-1) OriS, (D) equid alphaherpesvirus 1 (EHV-1) OriL, and (E) EHV-1 OriS. Protein-coding genes (orange), replication-origin-associated RNAs (red), and origin positions (purple) are shown together with long-read-defined transcript isoforms (blue). The comparison highlights that Ori loci are embedded in dense, frequently overlapping transcription, with multiple long isoforms traversing or flanking the origins.
Figure 2.
Transcriptional architectures surrounding lytic replication origins (OriL/OriS) in varicelloviruses. Panels show (A) pseudorabies virus (PRV) OriL, (B) PRV OriS, (C) bovine alphaherpesvirus 1.1 (BoHV-1) OriS, (D) equid alphaherpesvirus 1 (EHV-1) OriL, and (E) EHV-1 OriS. Protein-coding genes (orange), replication-origin-associated RNAs (red), and origin positions (purple) are shown together with long-read-defined transcript isoforms (blue). The comparison highlights that Ori loci are embedded in dense, frequently overlapping transcription, with multiple long isoforms traversing or flanking the origins.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



