
Submitted:
16 January 2026
Posted:
19 January 2026
You are already at the latest version
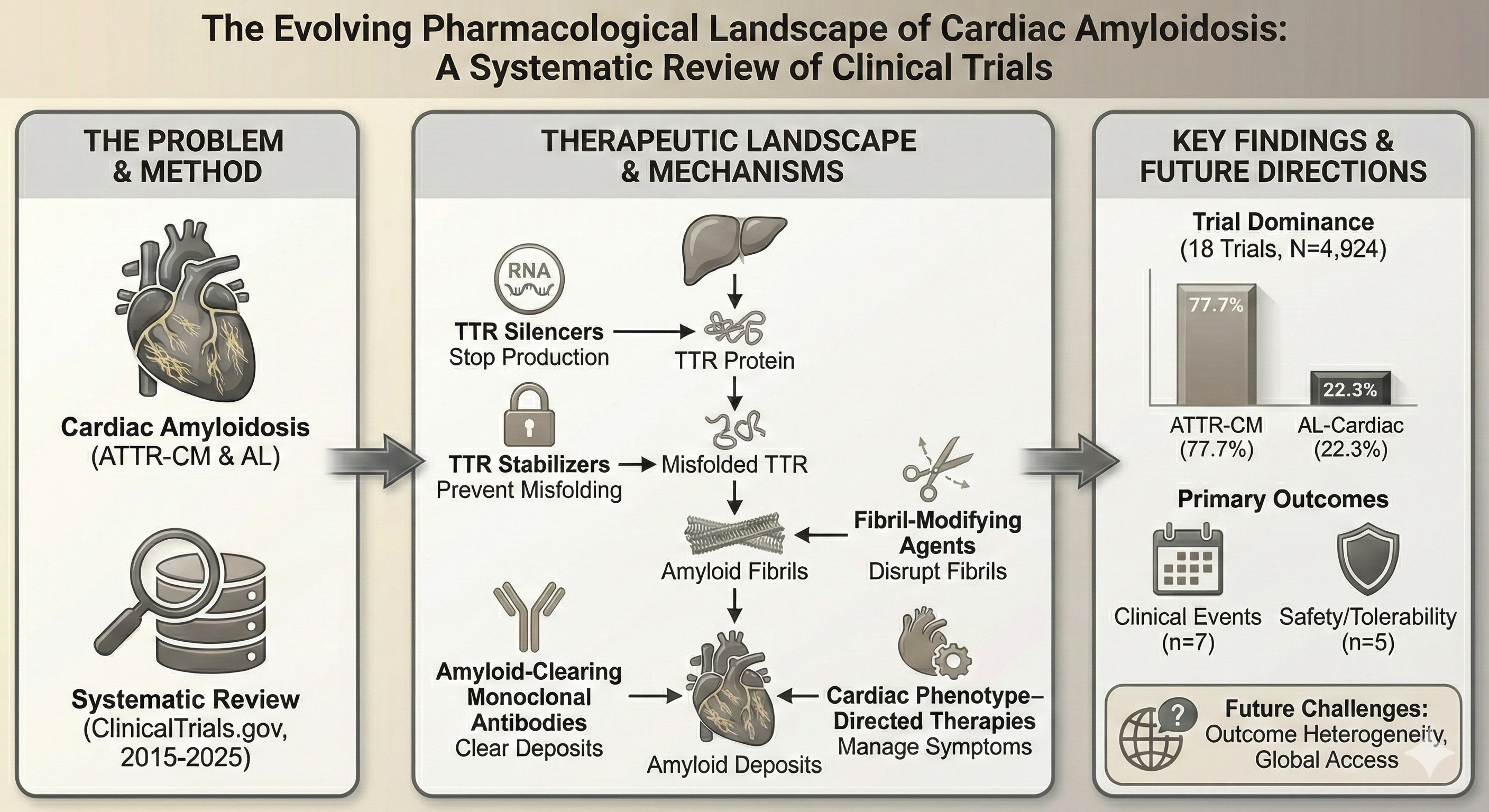
Abstract
Introduction: Cardiac amyloidosis, primarily comprising transthyretin amyloid cardiomyopathy (ATTR-CM) and light-chain (AL) subtypes, is an increasingly recognized contributor to the global heart failure burden. Management has shifted from supportive care to disease-modifying agents targeting specific stages of the amyloid cascade. This systematic review qualitatively characterizes the current pharmacological clinical trial landscape through a registry-based analysis. Methods: A qualitative systematic analysis of ClinicalTrials.gov was conducted for interventional trials registered between January 2015 and November 2025. Following PRISMA principles, studies were screened to include pharmacological interventions with explicit cardiac targeting while excluding neuropathy-dominant amyloidosis. Trial-level data regarding therapeutic classes, study phases, enrollment, and primary outcome domains were extracted and synthesized. Results: Eighteen trials met the inclusion criteria (14 ATTR-CM; 4 AL-cardiac), representing a total enrollment of 4,924 participants across 11 unique agents. Five therapeutic classes were identified: amyloid-clearing monoclonal antibodies (44.4% of trials), TTR silencers, TTR stabilizers, fibril-modifying agents, and cardiac phenotype–directed therapies. Monoclonal antibodies represented the largest class by both trial count and enrollment (3,075 participants). Clinical events (n=7) and safety/tolerability (n=5) were the most frequent primary outcome domains. ATTR-CM trials dominated the landscape, accounting for 77.7% of the total study population, while parallel-group placebo-controlled designs were the most common study architecture (n=10). Conclusion: The therapeutic landscape for cardiac amyloidosis is transitioning toward stage-specific, mechanism-based interventions. While ATTR-CM currently dominates research efforts, the expansion of silencers and monoclonal antibodies reflects an increasing capacity to intercept the amyloid cascade at distinct molecular checkpoints. However, significant heterogeneity in outcome measures and the shift toward diagnosing milder disease pose challenges for demonstrating clinical efficacy. Future priorities include standardized progression markers and addressing barriers to global access for these high-cost therapies.
Keywords:
cardiac amyloidosis
; transthyretin amyloid cardiomyopathy
; light-chain amyloidosis
; protein misfolding
1. Introduction
In the past, cardiac amyloidosis was regarded as a rare and atypical diagnosis. It is now recognized as an important contributor to the global burden of restrictive cardiomyopathy and heart failure.[1] Reported prevalence and mortality have increased substantially over the past two decades; as a result of improved detection and the widespread adoption of noninvasive diagnostic modalities, including bone scintigraphy and cardiac magnetic resonance imaging, rather than a true rise in disease incidence.[2,3] Earlier and more accurate diagnosis has consequently altered the clinical spectrum of cardiac amyloidosis, enabling identification of patients at less advanced stages of disease compared with historical cohorts.[3,4]
Transthyretin amyloid cardiomyopathy (ATTR-CM) and light-chain amyloidosis with cardiac involvement (AL-cardiac) are the two main types of cardiac amyloidosis. They have different pathophysiological causes and clinical courses. ATTR-CM is caused by misfolding of transthyretin, a transport protein produced by the liver, into insoluble fibrils that accumulate within the myocardium, producing a slowly progressive infiltrative cardiomyopathy that predominantly affects older adults.[5] In contrast, AL-cardiac amyloidosis arises from a plasma cell dyscrasia characterized by overproduction of monoclonal immunoglobulin light chains, which exert direct cardiotoxic effects in addition to forming myocardial deposits.[6]
In the past, the only management of cardiac amyloidosis available was supportive heart failure therapy and symptom control.[5] There were limited disease-modifying pharmacological options were at that time. Progress in understanding the molecular and cellular mechanisms underlying amyloid formation lead to the development of pharmacological strategies that target multiple stages of the amyloid cascade.[7] Contemporary approaches include transthyretin stabilizers that inhibit tetramer dissociation, gene-silencing therapies that reduce hepatic production of amyloidogenic precursor proteins, and amyloid-clearing monoclonal antibodies designed to promote removal of established myocardial deposits.[8] The emergence of these mechanism-based therapies has resulted in a rapidly expanding and increasingly heterogeneous clinical trial landscape, underscoring the need for structured synthesis of ongoing pharmacological development efforts.
In rare and evolving disease areas such as cardiac amyloidosis, registry-based analyses play a critical role in contextualizing therapeutic innovation and identifying research trends that may not yet be reflected in the published literature.[9,10,11] The ClinicalTrials.gov registry provides a comprehensive and standardized platform for capturing global interventional activity, facilitating evaluation of trial design, therapeutic mechanisms, and outcome priorities as new pharmacological strategies emerge.
Accordingly, the aim of this review is to perform a qualitative, registry-based analysis of interventional clinical trials registered on ClinicalTrials.gov to characterize current pharmacological strategies for cardiac amyloidosis, with particular emphasis on therapeutic drug classes, primary outcome domains, and the frequency of investigational agents.
2. Materials and Methods
This review was conducted as a qualitative, registry-based analysis of interventional clinical trials evaluating pharmacological treatment strategies for cardiac amyloidosis. The methodology followed the principles of the PRISMA statement to ensure transparent reporting of study identification, screening, eligibility assessment, and inclusion. The objective was to synthesize trial-level evidence describing emerging and established pharmacological approaches relevant to cardiac amyloidosis without performing quantitative meta-analysis.
Clinical trial data were obtained exclusively from the ClinicalTrials.gov registry, a publicly accessible database maintained by the U.S. National Library of Medicine. This registry was selected as the sole data source due to its comprehensive coverage of global interventional trials, standardized reporting structure, and suitability for systematic identification and qualitative synthesis of ongoing and completed pharmacological studies.
A structured search of ClinicalTrials.gov was performed using predefined condition-related terms relevant to cardiac amyloidosis. The initial search combined the terms “cardiac amyloidosis,” “ATTR cardiomyopathy,” “transthyretin cardiomyopathy,” and “AL amyloidosis.” The search was restricted to interventional studies involving adult participants, including both adults aged 18–64 years and older adults aged 65 years or above. Eligible study phases included Early Phase 1, Phase 1, Phase 2, Phase 3, Phase 4, and studies categorized as not applicable. Trials with recruitment statuses of not yet recruiting, recruiting, active but not recruiting, completed, enrolling by invitation, or unknown status were included. The search was limited to trials first posted between January 1, 2015 and November 30, 2025. This initial search yielded 187 records.
To improve specificity for cardiac amyloidosis and to exclude neuropathy-dominant forms of transthyretin amyloidosis, a refined search was subsequently conducted using Boolean exclusion terms. Records containing the terms neuropathy, polyneuropathy, FAP, or familial amyloid polyneuropathy were excluded. Application of these exclusion criteria reduced the dataset to 102 records, which were subjected to manual screening.
The remaining records were screened by reviewing study titles, condition descriptions, intervention types, and brief summaries to assess relevance to pharmacological treatment of cardiac amyloidosis. Studies were excluded at this stage if they were observational, diagnostic, imaging-only, device-based, procedural, or focused solely on supportive or symptomatic management. Additional exclusions included pediatric studies, trials targeting localized or non-systemic amyloidosis, systemic light-chain amyloidosis trials without explicit cardiac relevance, and studies lacking sufficient methodological information for qualitative assessment. Full registry entries of potentially eligible trials were then reviewed in detail to confirm cardiac disease relevance, pharmacological intent, and eligibility. Following this screening and eligibility assessment process, 18 interventional clinical trials were included in the final qualitative synthesis (Figure 1).
For each included study, relevant trial-level data were extracted from ClinicalTrials.gov and recorded in a structured spreadsheet. Extracted information included the trial registration number, study title, amyloidosis subtype, investigational drug or biological agent, therapeutic mechanism of action, study phase, recruitment status, planned or actual enrolment, primary outcome domain, and study design with comparator characteristics. Therapeutic class assignment was based on the dominant mechanism of action of the investigational intervention, allowing classification into transthyretin stabilizers, transthyretin silencers, amyloid-clearing monoclonal antibodies, amyloid disruptors or fibril-modifying agents, and cardiac phenotype–directed therapies relevant to transthyretin amyloid cardiomyopathy.
A descriptive qualitative synthesis was performed to summarize the characteristics of the included trials according to therapeutic class, outcome domains, and study design features. Given the heterogeneity of pharmacological interventions, trial designs, and outcome measures, no quantitative synthesis or meta-analysis was undertaken. As this study involved analysis of publicly available registry data without individual patient-level information, ethical approval and informed consent were not required. Gemini AI was used to generate graphical figure of amyloid cascade and relevant therapeutic targets (Figure 2).
3. Results
Study Selection
After initial registry filtering, systematic screening of the ClinicalTrials.gov dataset yielded 102 records for review. Following application of predefined inclusion and exclusion criteria focused on interventional pharmacological trials with explicit cardiac targeting or disease-modifying intent, 18 trials were selected for inclusion in this review.
General Characteristics of Included Trials
The 18 included trials primarily targeted two disease subtypes: transthyretin amyloid cardiomyopathy (ATTR-CM) and light-chain amyloidosis with cardiac involvement (AL-cardiac). As detailed in Table 1, fourteen trials focused on ATTR-CM, while four trials enrolled patients with AL-cardiac disease. The studies spanned clinical phases 1 through 4, with phase 3 trials representing the most common design. Planned or actual enrolment varied widely, ranging from small exploratory studies enrolling approximately 15 participants to large phase 3 trials enrolling up to 1,400 participants (Table 1). Enrolment was not reported for one included trial.
Pharmacological Strategies Evaluated
Five distinct therapeutic classes were identified among the pharmacological strategies evaluated. These included transthyretin (TTR) stabilizers, TTR silencers (small interfering RNA or antisense oligonucleotides), amyloid-clearing monoclonal antibodies, amyloid disruptors or fibril-modifying agents, and cardiac phenotype–directed therapies in ATTR-CM that did not directly target amyloid deposition. Classification was based on the dominant mechanism of action of each investigational intervention, as summarized in Table 1.
Frequency of Investigational Agents
The distribution of individual investigational agents across the included trials is presented in Table 2. CAEL-101 and eplontersen were the most frequently evaluated agents, each studied in three trials. ALXN2220, AG10/ALXN2060, and doxycycline were each evaluated in two trials. Other agents including NI006, coramitug, AT-02, tafamidis, trimetazidine, and empagliflozin were each assessed in a single interventional study.
Distribution of Therapeutic Drug Classes
As shown in Table 3, amyloid-clearing monoclonal antibodies constituted the largest therapeutic class, accounting for 8 of the 18 trials (44.4%) and the highest total reported enrolment (3,075 participants). This class included four ATTR-CM trials and four AL-cardiac trials. TTR silencers (n=3) and TTR stabilizers (n=3) followed, with total reported enrolments of 1,481 and 127 participants, respectively. Amyloid disruptors or fibril-modifying agents were evaluated in two trials enrolling 202 participants, while cardiac phenotype–directed therapies were assessed in two trials with a combined enrolment of 39 participants.
Primary Outcome Domains
Primary endpoints across the included trials were categorized into six outcome domains, as summarized in Table 4. Clinical events including all-cause mortality and hospitalization were the most frequently reported primary outcome domain, used in seven trials. Safety and tolerability were the primary endpoints in five trials, including the safety-focused CAEL-101 study. Biomarker or pharmacodynamic endpoints, such as transthyretin levels or stabilization, were primary outcomes in three trials. Cardiac structural or functional assessment using imaging modalities (e.g., echocardiography, cardiac magnetic resonance) served as the primary outcome in two trials, while functional or health-status measures (six-minute walk distance) were the primary endpoint in one trial. None of the included trials used hematologic or organ response as the sole primary outcome domain.
Trial Design and Comparator Characteristics
Trial design and comparator characteristics are detailed in Table 5. Parallel-group placebo-controlled designs were the most common, utilized in ten trials. Six trials employed a single-arm, open-label design without a reported comparator. Parallel-group active-controlled designs were used in two trials, both comparing investigational therapy with standard of care. One trial utilized a crossover design with a placebo comparator.
Summary of Results
In summary, 18 interventional clinical trials met the eligibility criteria for this review, representing a total reported enrolment of 4,924 participants across 11 unique pharmacological agents. Amyloid-clearing monoclonal antibodies were the most prevalent therapeutic class by both trial count and enrolment volume. Clinical events and safety were the most common primary outcome domains, and parallel-group placebo-controlled designs predominated. The distribution of included trials was weighted toward ATTR-CM, which accounted for 77.7% of the study population.
4. Discussion
The systematic search across the clinical trials registry identified therapeutic strategies targeting different stages of the amyloid cascade. Transthyretin (TTR) silencers act by degrading messenger RNA (mRNA) and thereby reducing hepatic synthesis of the amyloid precursor protein.[1] TTR stabilizers act by slowing the formation of new amyloid deposits through binding to the natural tetramer preventing its breakdown to monomers.[12] In contrast, amyloid-clearing monoclonal antibodies are designed to recognize epitopes on deposited fibrils, promoting immune-mediated clearance of established myocardial amyloid.[13] Fibril-modifying agents act by interfering with fibril assembly or altering fibril structure to enhance their clearance.[14] Cardiac phenotype-directed therapies represent a distinct category, since they do not directly target amyloid pathology but focus on myocardial function and the alleviation of heart failure symptoms within the framework of infiltrative cardiomyopathy.[15]
Clinical trial representation between the two types of cardiac amyloidosis, ATTR-CM and AL-cardiac shows an imbalance, reflecting significant differences pathophysiology and reflects fundamental differences in disease biology and clinical progression.[16] ATTR-CM typically follows a slowly progressive course over many years, creating a relatively stable platform for long-duration cardiovascular trials centered on clinical outcomes.[12] AL-cardiac amyloidosis, on the other hand, is often an aggressive disease with a high early death mortality.In contrast, AL-cardiac amyloidosis often presents as an aggressive disease with high early mortality. Consequently, AL-cardiac frequently necessitates urgent hematologic intervention and limits the feasibility of placebo-controlled designs or delayed treatment initiation.[17] Furthermore, the therapeutic development for each type has taken different courses. For AL, research has been driven by hematology-led plasma cell dyscrasia trials, whereas ATTR-CM studies have followed a cardiology-focused model targeting a single, organ-specific amyloidogenic protein.[18]
The diversity of primary outcomes in cardiac amyloidosis clinical trials adds some difficulty in interpreting current trials. In cases of clinical trials of rare diseases, it is challenging to follow clinical event endpoints as they require large sample sizes and prolonged follow-up.[19] Functional assessments, including six-minute walk distance and health-status questionnaires, offer sensitivity to change but may be influenced by non-cardiac factors such as age, frailty, or coexisting neuropathy.[20] Moreover, earlier diagnosis and improved disease awareness have shifted trial populations toward milder baseline disease, resulting in slower progression within placebo groups compared with historical cohorts. Consequently, detectable differences between active treatment and control arms may be less relative to earlier landmark studies.[16,21]
Clinical positioning of available and emerging therapies appears closely linked to disease stage along both biological and clinical progression.[1,22] In early or pre-symptomatic stages, limiting amyloid formation by upstream precursor suppression (TTR silencers) is most relevant to prevent irreversible myocardial injury.[23] Meanwhile, TTR stabilizers and amyloid-clearing antibodies serve as foundational mid-cascade intervention in symptomatic patients or positioned as adjunctive therapies in individuals with substantial residual amyloid burden, respectively.[24] In AL-cardiac amyloidosis, rapid suppression of the plasma cell clone remains the primary therapeutic priority, with fibril-targeting agents considered potential adjuncts only after adequate hematologic control has been achieved.[25,26] Although conceptual sequencing strategies including early combination approaches have been proposed, registry data indicate that robust comparative evidence to guide such strategies remains limited.[8,27]
The key unmet needs in pharmacological treatment of cardiac amyloidosis direct us to the future research focus. Although contemporary therapies attenuate disease progression, they do not provide consistent reversal of myocardial damage.[28,29] To improve care, researchers are focusing on several key areas; there is a need for highly sensitive biomarkers to define therapeutic response [30]. Multiple blood-based candidates are under evaluation in ATTR, including circulating TTR concentration, retinol-binding protein 4, and TTR aggregate–specific probes such as TAD1.[31,32] It is critical to set standards for disease progression and for functional and quality of life measures, as consensus-driven, prospectively validated standards are needed.[33,34,35] Many of these new life-changing drugs are extremely expensive, which prevents many patients around the world from getting fair and equal access to treatment. Because this disease affects many parts of the body, heart specialists (cardiologists) and blood specialists (hematologists) must work together more closely. This teamwork will help design clinical trials that focus on what matters most to the patient: feeling better, staying independent, and enjoying a higher quality of life.
Reviews that are based on registry-based data have a few inherent limitations, which must be considered when interpreting these findings.[11] This review relied exclusively on the ClinicalTrials.gov registry, there is a chance we missed some early-phase or locally sponsored international studies. Public registries also suffer from variable data quality, inconsistent coding and missing entries, as we found in one of the included studies did not report enrollment numbers. There is also a real risk that these public records make the research landscape look more advanced than it actually is, as registries aren’t always updated to show when a trial is terminated or ends in a negative result. Significant trial heterogeneity further complicates the synthesis, particularly regarding the evolving baseline characteristics of the study populations. Newer trials like HELIOS-B and ATTRibute-CM are enrolling “healthier” participants who were diagnosed at much earlier stages than the people in older landmark studies like ATTR-ACT. This makes finding measurable differences between drug and placebo arms harder and smaller to detect. Finally, the diversity of primary outcome domains that exists between studies (Table 4), limits the ability to perform direct comparisions across different pharmacological classes.
5. Conclusions
The shift in the pharmacological management of cardiac amyloidosis transitioning from purely palliative symptomatic care toward stage-specific, disease-modifying strategies represents a fundamental change in clinical practice. Recent advances in transthyretin silencers and the development of amyloid-clearing monoclonal antibodies suggest a broader capacity to intercept the amyloid cascade at distinct molecular checkpoints. While refined diagnostic protocols now facilitate detection in earlier, more milder phases of the disease, this improved prognostic outlook complicates the selection of clinical trial endpoints and the interpretation of long-term therapeutic efficacy. This situation highlights why we need solid, data-driven guidelines on which drugs to use first, how to combine them safely, and how to set clear standards for tracking a patient’s health over time.
Author Contributions
Conceptualization, M.H.H. and Y.A.A.; methodology, M.H.H; validation, Y.A.A; formal analysis, Y.A.A.; investigation, M.H.H.; writing—original draft preparation, M.H.H.; writing—review and editing, Y.A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable to studies analyzing publicly available registry data.
Informed Consent Statement
Not applicable.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments). Where GenAI has been used for purposes such as generating text, data, or graphics, or for study design, data collection, analysis, or interpretation of data, please add “During the preparation of this manuscript/study, the author(s) used [tool name, version information] for the purposes of [description of use]. The authors have reviewed and edited the output and take full responsibility for the content of this publication.”.
Conflicts of Interest
Declare conflicts of interest or state “The authors declare no conflicts of interest.” Authors must identify and declare any personal circumstances or interest that may be perceived as inappropriately influencing the representation or interpretation of reported research results. Any role of the funders in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results must be declared in this section. If there is no role, please state “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
Abbreviations
The following abbreviations are used in this manuscript:
| Abbreviation | Definition |
| 6MWD | Six-minute walk distance |
| AL | Light-chain amyloidosis |
| AL-cardiac | Light-chain amyloidosis with cardiac involvement |
| ASO | Antisense oligonucleotide |
| ATTR-CM | Transthyretin amyloid cardiomyopathy |
| CMR | Cardiac magnetic resonance |
| ECV | Extracellular volume |
| GLS | Global longitudinal strain |
| HF | Heart failure |
| KCCQ | Kansas City Cardiomyopathy Questionnaire |
| LV | Left ventricular |
| mAb | Monoclonal antibody |
| mRNA | Messenger RNA |
| NCT | National Clinical Trial identifier |
| NT-proBNP | N-terminal pro–B-type natriuretic peptide |
| PCWP | Pulmonary capillary wedge pressure |
| SAE | Serious adverse event |
| siRNA | Small interfering RNA |
| TEAE | Treatment-emergent adverse event |
| TTR | Transthyretin |
References
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L.; et al.; American Heart Association Heart; F Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e7–e22. [Google Scholar] [CrossRef]
- Oladiran, O.D.; Oladunjoye, A.O.; Dhital, R.; Oladunjoye, O.O.; Nwosu, I.; Licata, A. Hospitalization Rates, Prevalence of Cardiovascular Manifestations and Outcomes Associated With Amyloidosis in the United States. Cureus 2021, 13, e14177. [Google Scholar] [CrossRef]
- Ioannou, A.; Patel, R.K.; Razvi, Y.; Porcari, A.; Sinagra, G.; Venneri, L.; Bandera, F.; Masi, A.; Williams, G.E.; O’Beara, S.; et al. Impact of Earlier Diagnosis in Cardiac ATTR Amyloidosis Over the Course of 20 Years. Circulation 2022, 146, 1657–1670. [Google Scholar] [CrossRef]
- Argon, A.; Nart, D.; Yilmazbarbet, F. Cardiac Amyloidosis: Clinical Features, Pathogenesis, Diagnosis, and Treatment. Turk Patoloji Derg 2024, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Fontana, M.; Berk, J.L.; Drachman, B.; Garcia-Pavia, P.; Hanna, M.; Lairez, O.; Witteles, R. Changing Treatment Landscape in Transthyretin Cardiac Amyloidosis. Circ Heart Fail 2025, 18, e012112. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Aimo, A.; Emdin, M.; Porcari, A.; Solomon, S.D.; Hawkins, P.N.; Gillmore, J.D. Transthyretin amyloid cardiomyopathy: from cause to novel treatments. Eur Heart J 2026, 47, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dimza, M.; Vasilakis, G.; Grodin, J.L. Transthyretin Amyloid Cardiomyopathy: The Plot Thickens as Novel Therapies Emerge. US Cardiol 2025, 19, e21. [Google Scholar] [CrossRef]
- Gitt, A.K.; Bueno, H.; Danchin, N.; Fox, K.; Hochadel, M.; Kearney, P.; Maggioni, A.P.; Opolski, G.; Seabra-Gomes, R.; Weidinger, F. The role of cardiac registries in evidence-based medicine. Eur Heart J 2010, 31, 525–529. [Google Scholar] [CrossRef]
- Dawson, L.P.; Biswas, S.; Lefkovits, J.; Stub, D.; Burchill, L.; Evans, S.M.; Reid, C.; Eccleston, D. Characteristics and Quality of National Cardiac Registries: A Systematic Review. Circ Cardiovasc Qual Outcomes 2021, 14, e007963. [Google Scholar] [CrossRef]
- Meltzer, S.N.; Weintraub, W.S. The Role of National Registries in Improving Quality of Care and Outcomes for Cardiovascular Disease. Methodist Debakey Cardiovasc J 2020, 16, 205–211. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Higaki, J.N.; Chakrabartty, A.; Galant, N.J.; Hadley, K.C.; Hammerson, B.; Nijjar, T.; Torres, R.; Tapia, J.R.; Salmans, J.; Barbour, R.; et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid 2016, 23, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Martins, D.; Ribeiro, T.; Merlini, G.; Saraiva, M.J. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Senigarapu, S.; Driscoll, J.J. A review of recent clinical trials to evaluate disease-modifying therapies in the treatment of cardiac amyloidosis. Front Med (Lausanne) 2024, 11, 1477988. [Google Scholar] [CrossRef] [PubMed]
- Oubari, S.; Naser, E.; Papathanasiou, M.; Luedike, P.; Hagenacker, T.; Thimm, A.; Rischpler, C.; Kessler, L.; Kimmich, C.; Hegenbart, U.; et al. Impact of time to diagnosis on Mayo stages, treatment outcome, and survival in patients with AL amyloidosis and cardiac involvement. Eur J Haematol 2021, 107, 449–457. [Google Scholar] [CrossRef]
- Poledniczek, M.; Schmid, L.M.; Kronberger, C.; Ermolaev, N.; Rettl, R.; Binder, C.; Camuz Ligios, L.; Eslami, M.; Nitsche, C.; Hengstenberg, C.; et al. Applicability of phase 3 trial selection criteria to real-world transthyretin amyloid cardiomyopathy patients. Sci Rep 2025, 15, 37893. [Google Scholar] [CrossRef]
- Maurer, M.S.; Dunnmon, P.; Fontana, M.; Quarta, C.C.; Prasad, K.; Witteles, R.M.; Rapezzi, C.; Signorovitch, J.; Lousada, I.; Merlini, G. Proposed Cardiac End Points for Clinical Trials in Immunoglobulin Light Chain Amyloidosis: Report From the Amyloidosis Forum Cardiac Working Group. Circ Heart Fail 2022, 15, e009038. [Google Scholar] [CrossRef]
- Dorbala, S.; Adigun, R.; Alexander, K.M.; Brambatti, M.; Cuddy, S.A.M.; Dispenzieri, A.; Dunnmon, P.; Emdin, M.; Abou Ezzeddine, O.F.; Falk, R.H.; et al. Development of Imaging Endpoints for Clinical Trials in AL and ATTR Amyloidosis: Proceedings of the Amyloidosis Forum. JACC Cardiovasc Imaging 2025, 18, 602–617. [Google Scholar] [CrossRef]
- Vogel, J.; Carpinteiro, A.; Luedike, P.; Buehning, F.; Wernhart, S.; Rassaf, T.; Michel, L. Current Therapies and Future Horizons in Cardiac Amyloidosis Treatment. Curr Heart Fail Rep 2024, 21, 305–321. [Google Scholar] [CrossRef]
- Adam, R.D.; Coriu, D.; Jercan, A.; Badelita, S.; Popescu, B.A.; Damy, T.; Jurcut, R. Progress and challenges in the treatment of cardiac amyloidosis: a review of the literature. ESC Heart Fail 2021, 8, 2380–2396. [Google Scholar] [CrossRef]
- Stein-Merlob, A.F.; Swier, R.; Vucicevic, D. Evolving Strategies in Cardiac Amyloidosis: From Mechanistic Discoveries to Diagnostic and Therapeutic Advances. Cardiol Clin 2025, 43, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez, E.; Lopez-Sainz, A.; Garcia-Pavia, P. Diagnosis and Treatment of Transthyretin Cardiac Amyloidosis. Progress and Hope. Rev Esp Cardiol (Engl Ed) 2017, 70, 991–1004. [Google Scholar] [CrossRef]
- Quarta, C.C.; Solomon, S.D.; Uraizee, I.; Kruger, J.; Longhi, S.; Ferlito, M.; Gagliardi, C.; Milandri, A.; Rapezzi, C.; Falk, R.H. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation 2014, 129, 1840–1849. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N Engl J Med 2003, 349, 583–596. [Google Scholar] [CrossRef]
- Nguyen, D.M.; Ramazani, N.; Sodhi, G.; Tak, T. Cardiac Amyloidosis: Tribulations and New Frontiers. J Pers Med 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Brownell, D.; Pillai, A.J.; Nair, N. Cardiac Amyloidosis: A Contemporary Review of Medical and Surgical Therapy. Curr Cardiol Rev 2024, 20, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Lacy, M.Q.; Gastineau, D.A.; Inwards, D.J.; Chen, M.G.; Tefferi, A.; Kyle, R.A.; Litzow, M.R. Blood stem cell transplantation as therapy for primary systemic amyloidosis (AL). Bone Marrow Transplant 2000, 26, 963–969. [Google Scholar] [CrossRef]
- Arvanitis, M.; Koch, C.M.; Chan, G.G.; Torres-Arancivia, C.; LaValley, M.P.; Jacobson, D.R.; Berk, J.L.; Connors, L.H.; Ruberg, F.L. Identification of Transthyretin Cardiac Amyloidosis Using Serum Retinol-Binding Protein 4 and a Clinical Prediction Model. JAMA Cardiol 2017, 2, 305–313. [Google Scholar] [CrossRef]
- Martinez Valle, F.; Perez Bocanegra, C. Biomarkers in transthyretin amyloidosis. Present and future. Med Clin (Barc) 2025, 164, 106939. [Google Scholar] [CrossRef] [PubMed]
- Gulati, J.S.; Pedretti, R.; Hendren, N.; Kozlitina, J.; Saelices, L.; Roth, L.R.; Grodin, J.L. Biomarkers in Subclinical Transthyretin Cardiac Amyloidosis. Curr Heart Fail Rep 2025, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Chrysohoou, C.; Panagiotakos, D.; Tsiachris, D.; Dimitriadis, K.; Lazaros, G.; Tsioufis, K.; Stefanadis, C. A simplified cardiac amyloidosis score predicts all-cause and cardiovascular disease mortality and morbidity in the general population: the Ikaria Study. Arch Med Sci 2025, 21, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Willixhofer, R.; Contini, M.; Emdin, M.; Magri, D.; Bonomi, A.; Salvioni, E.; Celeste, F.; Del Torto, A.; Passino, C.; Capelle, C.D.J.; et al. Exercise limitations in amyloid cardiomyopathy assessed by cardiopulmonary exercise testing-A multicentre study. ESC Heart Fail 2025, 12, 1326–1335. [Google Scholar] [CrossRef]
- Chacko, L.; Karia, N.; Venneri, L.; Bandera, F.; Passo, B.D.; Buonamici, L.; Lazari, J.; Ioannou, A.; Porcari, A.; Patel, R.; et al. Progression of echocardiographic parameters and prognosis in transthyretin cardiac amyloidosis. Eur J Heart Fail 2022, 24, 1700–1712. [Google Scholar] [CrossRef]
Figure 1.
PRISMA flow diagram summarizing the identification and selection of interventional clinical trials of pharmacological therapies for cardiac amyloidosis from ClinicalTrials.gov.
Figure 1.
PRISMA flow diagram summarizing the identification and selection of interventional clinical trials of pharmacological therapies for cardiac amyloidosis from ClinicalTrials.gov.
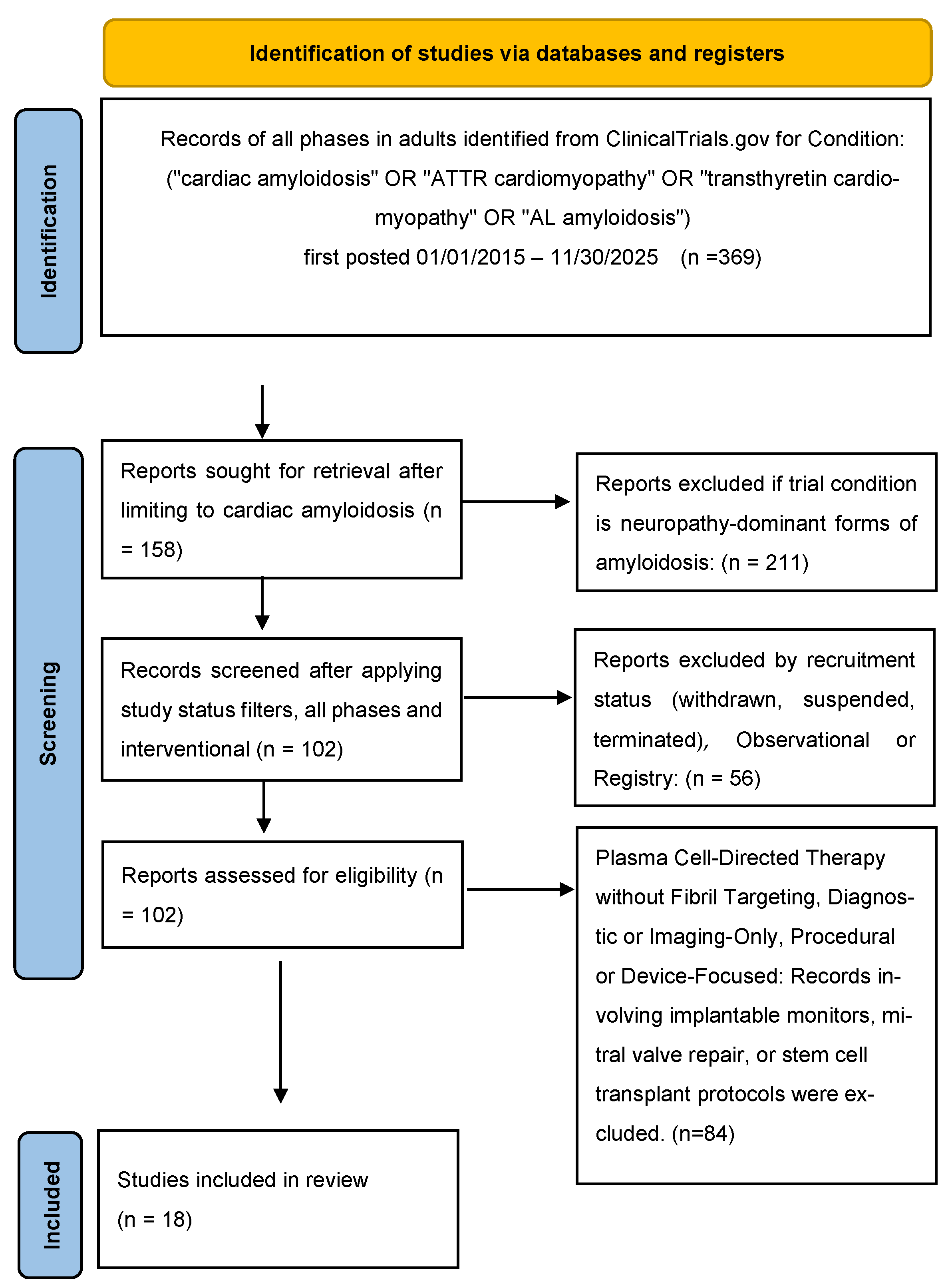
Figure 2.
Pharmacological intervention points in ATTR-CM versus AL amyloidosis with cardiac involvement. Comparison of the amyloid cascades and therapeutic targets in transthyretin amyloid cardiomyopathy (ATTR-CM) and light-chain amyloidosis with cardiac involvement (AL-cardiac). In ATTR-CM, liver-derived transthyretin misfolds following tetramer dissociation, aggregates into amyloid fibrils, and deposits within the myocardium, leading to restrictive cardiomyopathy. Therapeutic strategies target distinct stages of this cascade, including TTR silencers, TTR stabilizers, fibril-modifying agents, amyloid-clearing monoclonal antibodies, and downstream phenotype-directed cardiac therapies. In AL-cardiac disease, a bone marrow plasma cell clone produces toxic monoclonal light chains that drive myocardial injury and amyloid deposition; upstream plasma cell–directed therapy is the cornerstone of treatment, with fibril-targeting strategies and supportive cardiac care serving adjunctive roles.
Figure 2.
Pharmacological intervention points in ATTR-CM versus AL amyloidosis with cardiac involvement. Comparison of the amyloid cascades and therapeutic targets in transthyretin amyloid cardiomyopathy (ATTR-CM) and light-chain amyloidosis with cardiac involvement (AL-cardiac). In ATTR-CM, liver-derived transthyretin misfolds following tetramer dissociation, aggregates into amyloid fibrils, and deposits within the myocardium, leading to restrictive cardiomyopathy. Therapeutic strategies target distinct stages of this cascade, including TTR silencers, TTR stabilizers, fibril-modifying agents, amyloid-clearing monoclonal antibodies, and downstream phenotype-directed cardiac therapies. In AL-cardiac disease, a bone marrow plasma cell clone produces toxic monoclonal light chains that drive myocardial injury and amyloid deposition; upstream plasma cell–directed therapy is the cornerstone of treatment, with fibril-targeting strategies and supportive cardiac care serving adjunctive roles.
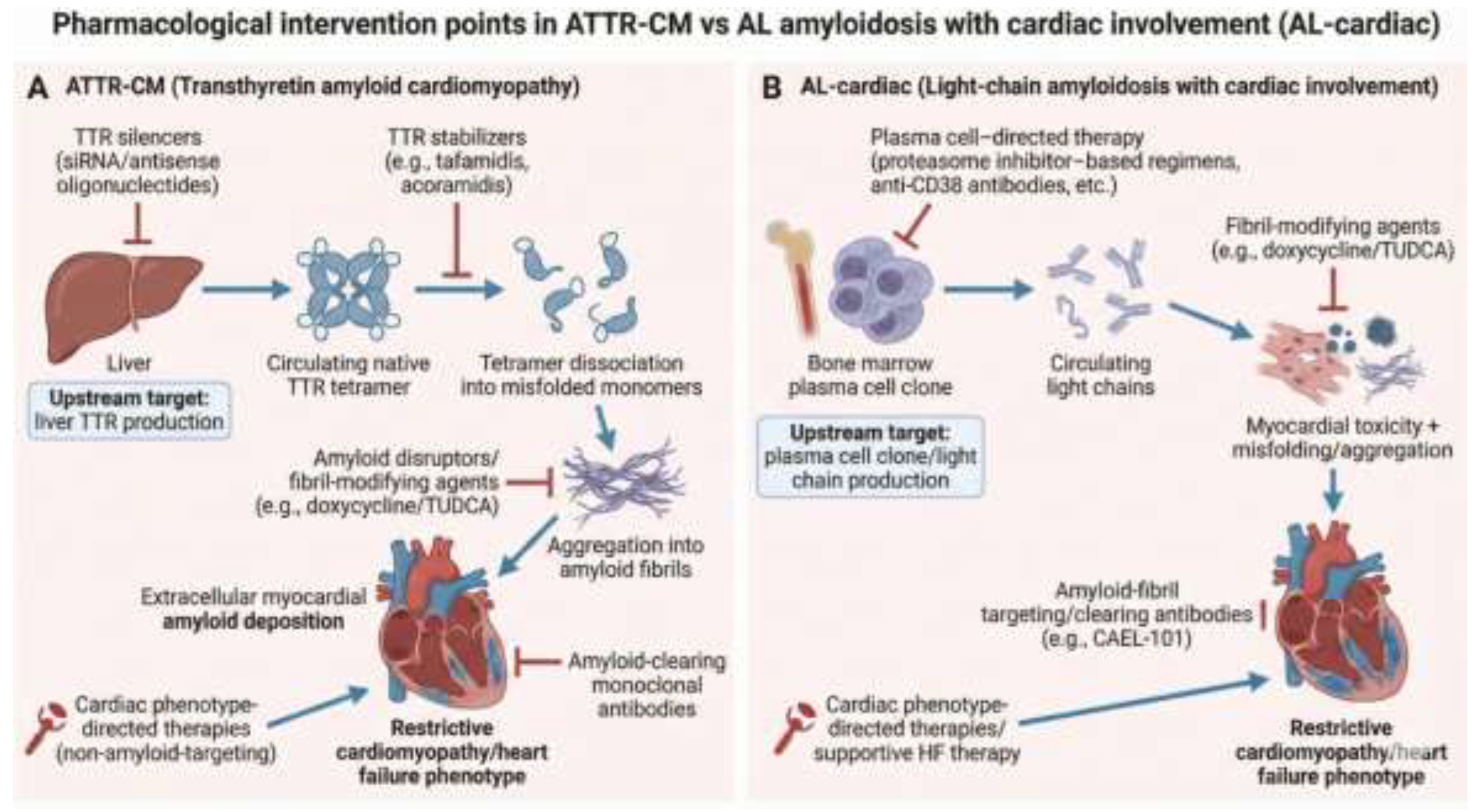

Table 1.
Summary of Included Interventional Clinical Trials for Cardiac Amyloidosis.
| NCT number | Study title | Amyloidosis subtype (ATTR-CM or AL-cardiac) | Intervention | Therapeutic class | Study phase | Enrolment (planned or actual) |
| NCT07213583 | Study of Re-Treatment With ALXN2220 in Patients With ATTR-CM | ATTR-CM | ALXN2220 | Amyloid-clearing monoclonal antibodies | Phase 2 | 35 |
| NCT04512235 | A Study to Evaluate the Efficacy and Safety of CAEL-101 in Patients With Mayo Stage IIIa AL Amyloidosis (CARES) | AL-cardiac | CAEL-101 | Amyloid-clearing monoclonal antibodies | Phase 3 | 125 |
| NCT04504825 | A Study to Evaluate the Efficacy and Safety of CAEL-101 in Patients With Mayo Stage IIIb AL Amyloidosis (CARES) | AL-cardiac | CAEL-101 | Amyloid-clearing monoclonal antibodies | Phase 3 | 284 |
| NCT04304144 | A Study to Evaluate the Safety and Tolerability of CAEL-101 in Patients With AL Amyloidosis | AL-cardiac | CAEL-101 | Amyloid-clearing monoclonal antibodies | Phase 2 | 25 |
| NCT05633563 | The Effect of Trimetazidine on Mitochondrial Function and Myocardial Performance in ATTR-CM | ATTR-CM | Trimetazidine | Cardiac phenotype–directed therapies in ATTR-CM (non–amyloid-targeting) | Phase 4 | 24 |
| NCT04360434 | First-in-Human Study of NI006 in Patients With ATTR-CM | ATTR-CM | NI006 | Amyloid-clearing monoclonal antibodies | Phase 1 | 46 |
| NCT03481972 | Doxycycline/TUDCA Plus Standard Supportive Therapy Versus Standard Supportive Therapy Alone in Cardiac ATTR Amyloidosis | ATTR-CM | Doxycycline/TUDCA | Amyloid disruptors / fibril-modifying agents | Phase 3 | 102 |
| NCT03458130 | Study of AG10 in Amyloid Cardiomyopathy | ATTR-CM | AG10 | TTR stabilizers | Phase 2 | 49 |
| NCT04843020 | ION-682884 in Patients With TTR Amyloid Cardiomyopathy | ATTR-CM | ION 682884 | TTR silencers (siRNA / antisense oligonucleotides) | Phase 2 | 17 |
| NCT07207811 | CLEOPATTRA Coramitug Study in ATTR-CM | ATTR-CM | Coramitug | Amyloid-clearing monoclonal antibodies | Phase 3 | 1280 |
| NCT06194825 | EPIC-ATTR: Eplontersen in Chinese Subjects With ATTR-CM | ATTR-CM | Eplontersen | TTR silencers (siRNA / antisense oligonucleotides) | Phase 3 | 64 |
| NCT06183931 | Study of ALXN2220 Versus Placebo in Adults With ATTR-CM | ATTR-CM | ALXN2220 | Amyloid-clearing monoclonal antibodies | Phase 3 | 1160 |
| NCT04814186 | Tafamidis in Chinese Participants With ATTR-CM | ATTR-CM | Tafamidis | TTR stabilizers | Phase 3 | 53 |
| NCT04622046 | A Phase 3 Study of ALXN2060 in Japanese Participants With Symptomatic ATTR-CM | ATTR-CM | ALXN2060 | TTR stabilizers | Phase 3 | 25 |
| NCT04136171 | CARDIO-TTRansform: Eplontersen in Participants With ATTR-CM | ATTR-CM | Eplontersen | TTR silencers (siRNA / antisense oligonucleotides) | Phase 3 | 1400 |
| NCT03401372 | BCD With or Without Doxycycline in Mayo Stage II-III AL | AL-cardiac | Doxycycline | Amyloid disruptors / fibril-modifying agents | not reported | not reported |
| NCT05233163 | SGLT2 Inhibitors in Transthyretin Amyloid Cardiomyopathy | ATTR-CM | Empagliflozin | Cardiac phenotype–directed therapies in ATTR-CM (non–amyloid-targeting) | Phase 4 | 15 |
| NCT05951049 | A Study of AT-02 in Subjects With Systemic Amyloidosis | AL-cardiac | AT-02 | Amyloid-clearing monoclonal antibodies | Phase 2 | 120 |
Abbreviations: AL, light-chain amyloidosis; AL-cardiac, light-chain amyloidosis with cardiac involvement; ATTR-CM, transthyretin amyloid cardiomyopathy; ASO, antisense oligonucleotide; HF, heart failure; mAb, monoclonal antibody; NCT, National Clinical Trial identifier; TTR, transthyretin. Therapeutic class assignment was based on the dominant mechanism of action of the investigational agent as specified in the ClinicalTrials.gov registry entry. Trials enrolling mixed amyloidosis populations were classified according to the primary cardiac-targeted intervention. Enrollment numbers reflect planned or reported enrollment at the time of registry extraction.
Table 2.
Frequency of Pharmacological Agents Evaluated in Cardiac Amyloidosis Trials.
| Drug/agent | Therapeutic class | Number of trials |
| CAEL-101 | Amyloid-clearing monoclonal antibodies | 3 |
| Eplontersen | TTR silencers (siRNA / antisense oligonucleotides) | 3 |
| ALXN2220 | Amyloid-clearing monoclonal antibodies | 2 |
| AG10 (ALXN2060) | TTR stabilizers | 2 |
| Doxycycline | Amyloid disruptors / fibril-modifying agents | 2 |
| NI006 | Amyloid-clearing monoclonal antibodies | 1 |
| Coramitug | Amyloid-clearing monoclonal antibodies | 1 |
| AT-02 | Amyloid-clearing monoclonal antibodies | 1 |
| Tafamidis | TTR stabilizers | 1 |
| Trimetazidine | Cardiac phenotype–directed therapies in ATTR-CM (non–amyloid-targeting) | 1 |
| Empagliflozin | Cardiac phenotype–directed therapies in ATTR-CM (non–amyloid-targeting) | 1 |
Table 3.
Distribution of Therapeutic Drug Classes in Interventional Cardiac Amyloidosis Trials.
| Drug Class | Number of trials (n/N) | Total enrolment | ATTR-CM vs AL-cardiac (counts) | Primary outcome focus (brief) | Typical design/comparator (brief) | Notes (1 line max) |
| TTR stabilizers | 3/18 | 127 | 3 ATTR / 0 AL | Biomarkers/pharmacodynamics; functional/health status | Randomized parallel; placebo | Targets protein tetramer stability to prevent amyloidogenesis. |
| TTR silencers (ASO/siRNA) | 3/18 | 1481 | 3 ATTR / 0 AL | Clinical events; biomarkers/pharmacodynamics | Randomized parallel; placebo | Evaluates reduction in hepatic TTR production via genetic silencing. |
| Amyloid-clearing monoclonal antibodies | 8/18 | 3075 | 4 ATTR / 4 AL | Clinical events; imaging; safety/tolerability | Randomized parallel; placebo | Includes safety-focused study for CAEL-101 and trials for cardiac-staged AL. |
| Amyloid disruptors/fibril-modifying agents | 2/18 | 202 | 1 ATTR / 1 AL | Clinical events | Randomized parallel; standard-of-care | Classified by doxycycline (fibril-modifying) component; background plasma cell therapy SoC. |
| Cardiac phenotype–directed therapies in ATTR-CM (non–amyloid-targeting) | 2/18 | 39 | 2 ATTR / 0 AL | Functional/health status; imaging; safety/tolerability | Randomized crossover; placebo or single group | Focuses on myocardial performance and heart failure symptom management. |
Abbreviations: AL, light-chain amyloidosis; AL-cardiac, light-chain amyloidosis with cardiac involvement; ATTR-CM, transthyretin amyloid cardiomyopathy; ASO, antisense oligonucleotide; mAb, monoclonal antibody; siRNA, small interfering RNA; TTR, transthyretin. Therapeutic drug classes were defined according to the principal biological target within the amyloid cascade. Cardiac phenotype–directed therapies were defined as pharmacological agents that improve myocardial function or heart failure symptoms without directly targeting amyloid production, aggregation, or clearance. Percentages were calculated using the total number of included trials as the denominator.
Table 4.
Primary Outcome Domains in Interventional Cardiac Amyloidosis Trials.
| Outcome domain | Frequency (number of trials) | Example outcomes/measures (brief) |
| Clinical events (mortality/hospitalization) | 7 | All-cause mortality, cardiovascular-related hospitalizations, and progression-free survival. |
| Functional/health status (6MWD/KCCQ) | 2 | Distance walked during the 6-minute walk test (6MWT). |
| Biomarkers/pharmacodynamics (NT-proBNP, TTR levels) | 3 | Percent stabilization of TTR tetramers and reduction in serum TTR concentration. |
| Cardiac imaging/structure (echo/GLS, CMR/ECV/LV mass, scintigraphy) | 2 | Change in myocardial amyloid burden assessed by echocardiography, cardiac magnetic resonance (ECV/LV mass), or nuclear scintigraphy. |
| Safety/tolerability (TEAEs/SAEs) | 5 | Incidence and severity of treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs). |
| Hematologic/organ response (AL-cardiac) | 0 | N/A (Included AL-cardiac trials prioritized clinical events or safety as primary endpoints). |
Abbreviations: 6MWD, six-minute walk distance; CMR, cardiac magnetic resonance imaging; ECV, extracellular volume; GLS, global longitudinal strain; HF, heart failure; KCCQ, Kansas City Cardiomyopathy Questionnaire; LV, left ventricular; NT-proBNP, N-terminal pro–B-type natriuretic peptide; PCWP, pulmonary capillary wedge pressure; SAE, serious adverse event; TEAE, treatment-emergent adverse event. Primary outcome domain classification was based on the prespecified primary endpoint listed in the ClinicalTrials.gov registry. Trials reporting multiple primary endpoints were categorized according to the dominant clinical or biological outcome. Hematologic response endpoints were considered distinct from cardiac-specific outcome domains.
Table 5.
Trial Design and Comparator Characteristics.
| Study title | NCT number | Design type | Comparator type |
| Study of Re-Treatment With ALXN2220 in Patients With ATTR-CM | NCT07213583 | single-arm/open-label | none/not reported |
| A Study to Evaluate the Efficacy and Safety of CAEL-101 in Patients With Mayo Stage IIIa AL Amyloidosis (CARES) | NCT04512235 | parallel-group placebo-controlled | placebo |
| A Study to Evaluate the Efficacy and Safety of CAEL-101 in Patients With Mayo Stage IIIb AL Amyloidosis (CARES) | NCT04504825 | parallel-group placebo-controlled | placebo |
| A Study to Evaluate the Safety and Tolerability of CAEL-101 in Patients With AL Amyloidosis | NCT04304144 | single-arm/open-label | none/not reported |
| The Effect of Trimetazidine on Mitochondrial Function and Myocardial Performance in ATTR-CM | NCT05633563 | crossover/other | placebo |
| First-in-Human Study of NI006 in Patients With ATTR-CM | NCT04360434 | parallel-group placebo-controlled | placebo |
| Doxycycline and Tauroursodeoxycholic Acid (Doxy/TUDCA) in Cardiac ATTR Amyloidosis | NCT03481972 | parallel-group active-controlled | standard of care (± placebo) |
| Study of AG10 in Amyloid Cardiomyopathy | NCT03458130 | parallel-group placebo-controlled | placebo |
| ION-682884 in Patients With TTR Amyloid Cardiomyopathy | NCT04843020 | single-arm/open-label | none/not reported |
| CLEOPATTRA Coramitug Study in ATTR-CM | NCT07207811 | parallel-group placebo-controlled | placebo |
| EPIC-ATTR: Eplontersen in Chinese Subjects With ATTR-CM | NCT06194825 | parallel-group placebo-controlled | placebo |
| Study of ALXN2220 Versus Placebo in Adults With ATTR-CM | NCT06183931 | parallel-group placebo-controlled | placebo |
| A Study to Assess the Safety and Efficacy Of Tafamidis In Chinese Participants With ATTR-CM | NCT04814186 | single-arm/open-label | none/not reported |
| A Phase 3 Study of ALXN2060 in Japanese Participants With Symptomatic ATTR-CM | NCT04622046 | single-arm/open-label | none/not reported |
| CARDIO-TTRansform: Eplontersen in Participants With ATTR-CM | NCT04136171 | parallel-group placebo-controlled | placebo |
| BCD With or Without Doxycycline in Mayo Stage II-III AL | NCT03401372 | parallel-group active-controlled | standard of care |
| SGLT2 Inhibitors in Transthyretin Amyloid Cardiomyopathy | NCT05233163 | single-arm/open-label | none/not reported |
| A Study of AT-02 in Subjects With Systemic Amyloidosis | NCT05951049 | single-arm/open-label | none/not reported |
Abbreviations: AL, light-chain amyloidosis; ATTR-CM, transthyretin amyloid cardiomyopathy; HF, heart failure; NCT, National Clinical Trial identifier. Design type and comparator classification were extracted directly from registry-reported study design fields. “Standard of care” comparators refer to background therapy appropriate for the disease subtype at the time of trial registration. Single-arm studies were defined as interventional trials without an active or placebo comparator.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



