
Submitted:
15 January 2026
Posted:
16 January 2026
You are already at the latest version
Abstract
The antibiotic pipeline has stalled: most recent approvals reflect incremental modifications of existing scaffolds, while antimicrobial resistance continues to outpace discovery. Antimicrobial peptides (AMPs) offer a compelling alternative because of rapid, multi-modal activity, but clinical translation has been limited by toxicity, serum instability, and the prohibitive cost of synthesizing and testing large libraries. Recent progress in protein language models (pLMs) changes the computational landscape by providing embeddings that capture sequence context and biophysical regularities from massive unlabeled datasets. However, pLMs alone are not a design solution. We propose a technique coupling pLM-derived representations to diffusion or discrete flow-based generative models that can explore non-homologous regions of peptide space while being steered by multi-objective guidance. This framework supports direct optimization for potency, selectivity, and developability during generation, compressing hit discovery and early optimization into a single in silico loop. Conditioning generation on target and safety predictors could shift AMPs from membrane-lytic ‘blunt instruments’ toward more selective, target-aware therapeutics.
Keywords:
antibiotic discovery
; antimicrobial peptides
; protein language models
; AI drug discovery
The Innovation Gap in the Antibiotics Pipeline
Following a peak in antibiotic discovery between 1940 and 1960, the rate of new antibiotic classes reaching the market has significantly declined [1]. The pipeline has largely transitioned from the discovery of new classes to the iterative refinement of known scaffolds, leaving a deficit of first-in-class candidates. Simultaneously, a growing number of infections are unable to be treated with existing antibiotics worldwide, making antimicrobial resistance (AMR) a major public health crisis.
Antimicrobial peptides (AMPs) are a promising alternative to conventional antibiotics, exhibiting fast-acting, multi-target activity that can reduce the propensity for rapid resistance [2]. While many conventional small-molecule drugs are characterized by their affinity for specific protein targets, AMPs as a class employ diverse mechanisms of action; individual peptides may exert their effects through membrane permeabilization, biofilm degradation, or the inhibition of specific intracellular biosynthetic pathways [3]. Compared to small molecules, which can only bind to defined enzymatic pockets, AMPs are larger and can form extended contacts with protein surfaces and membranes. This allows them to bind more complex targets including membrane interfaces and outer membrane proteins. However, the clinical translation of AMPs is frequently stalled by serum instability and host toxicity, leading to high attrition rates. Screening large libraries to identify new leads requires peptide synthesis—often costing $200–500 per candidate [4]. Thus, high-throughput wet-lab validation can be prohibitively expensive.
To address this issue, other therapeutic areas have begun to prize in silico methods to improve efficiency by generating lead compounds, particularly for peptides and biologics. Across biologics, generative and representation-learning methods are now producing credible preclinical and clinical candidates, which raises an uncomfortable question for AMPs: why is AMP discovery still dominated by expensive brute-force screening? For one, applying these AI methods to AMP discovery faces unique challenges due to the scarcity of high-quality, non-homologous training data. For the latest AI tools, such as those emerging from advances in deep learning, large datasets are often a prerequisite. Alas, antimicrobial compound datasets emerge mostly from academic projects and are relatively fragmented, inconsistent, and limited in scale. The leading APD3 database contains fewer than 3,000 validated natural peptides, limiting the effectiveness of standard supervised learning [5]. Consequently, there is a need for the development of computational approaches that can overcome the limitations of data scarcity.
From Homology Mining to Generative Peptide Design
Antimicrobial peptide discovery has relied on screening natural variants of known AMPs or mining sequences with recognizable physicochemical motifs [6]. This paradigm of homology-based discovery has benefitted from advancements in gene sequencing and AI, allowing researchers to search AMP-like sequences in previously unstudied organisms. However, the number of experimentally validated AMPs remains small relative to the landscape of possible peptides, thus constraining discovery by similarity-based approaches.
Protein language models (pLMs) offer a transformative alternative to traditional methods: they treat protein design as a generative linguistic task, accessing “dark” regions of sequence space that evolution and homology modeling have yet to explore. Models like ESM-2 or ProtT5 are trained on hundreds of millions of unannotated sequences using a Masked Language Modeling (MLM) objective [7]. To understand this process, imagine a human reading a sentence where 15% of the words are missing. If that person can accurately fill in the blanks based on the surrounding context, you can conclude they have a deep understanding of that language. By training pLMs under a similar objective—forcing the model to predict hidden amino acids within a sequence—this article proposes a promising pathway for generating representations that encode vital biophysical properties, such as charge, hydrophobicity, and binding sites.
These models generate rich high-dimensional embeddings or latent vectors that capture deep biophysical rules, demonstrating state-of-the-art performance on tasks like protein folding, predicting protein-DNA binding, and de novo protein design [7]. However, simply fine-tuning these models often results in mode collapse or high similarity to training data. Recent work demonstrates that pLM embeddings can serve as effective inputs for diffusion-based generation of AMPs [8]. In Torres et al., latent diffusion over ESM-derived embeddings enabled the generation of thousands of non-homologous peptide sequences, several of which demonstrated promising in vivo activity against drug-resistant bacterial pathogens with favorable cytotoxicity profiles. In our opinion, coupling pLM representations with generative flow methodologies will help access truly novel regions of the representation space. This approach represents a promising strategy to decouple antibiotic discovery from the constraints of homology and transform the search for new peptides from a stochastic mining process into programmable biology.
Multi-Objective Optimization for Selectivity and Novel Targets
pLMs are best viewed as representation learners: they provide embeddings that can be used as priors for diffusion or discrete flow models that can perform peptide generation. We believe these representations can be used to design for undruggable microbial targets that have traditionally eluded both small-molecule and natural AMP interventions (Figure 1). Natural AMPs often act as blunt instruments, disrupting the entire bacterial membrane through relatively non-specific electrostatic and hydrophobic interactions [9]. This mechanism often drives host toxicity and poor therapeutic indices. Because pLMs are trained on massive, diverse sequence corpora, their embeddings encode contextual patterns correlated with structure and interaction propensity, including motifs enriched at interfaces and in disordered regions. We can therefore steer diffusion models toward peptides that bind to and inhibit complex, flexible, or disordered bacterial proteins. This strategy is already bearing fruit in other therapeutic classes through the development of de novo peptide-based binders and modulators. Recent frameworks like FusOn-pLM suggest that pLM-derived representations can support in silico ‘guide’ peptide design against previously inaccessible targets, including disordered fusion oncoproteins such as SS18::SSX1 and PAX3::FOXO1 [10].
Figure 1.
Multi-objective generative design of antimicrobial peptides via flow matching.
(a) A self-supervised protein language model extracts general-purpose biophysical embeddings from unannotated protein sequences. (b) Generative flow matching steers latent trajectories from noise to protein structure using simultaneous gradient guidance to maximize potency and minimize toxicity. (c) The pipeline outputs structurally novel de novo candidates with high predicted activity and low host toxicity for wet-lab validation.
Figure 1.
Multi-objective generative design of antimicrobial peptides via flow matching.
(a) A self-supervised protein language model extracts general-purpose biophysical embeddings from unannotated protein sequences. (b) Generative flow matching steers latent trajectories from noise to protein structure using simultaneous gradient guidance to maximize potency and minimize toxicity. (c) The pipeline outputs structurally novel de novo candidates with high predicted activity and low host toxicity for wet-lab validation.
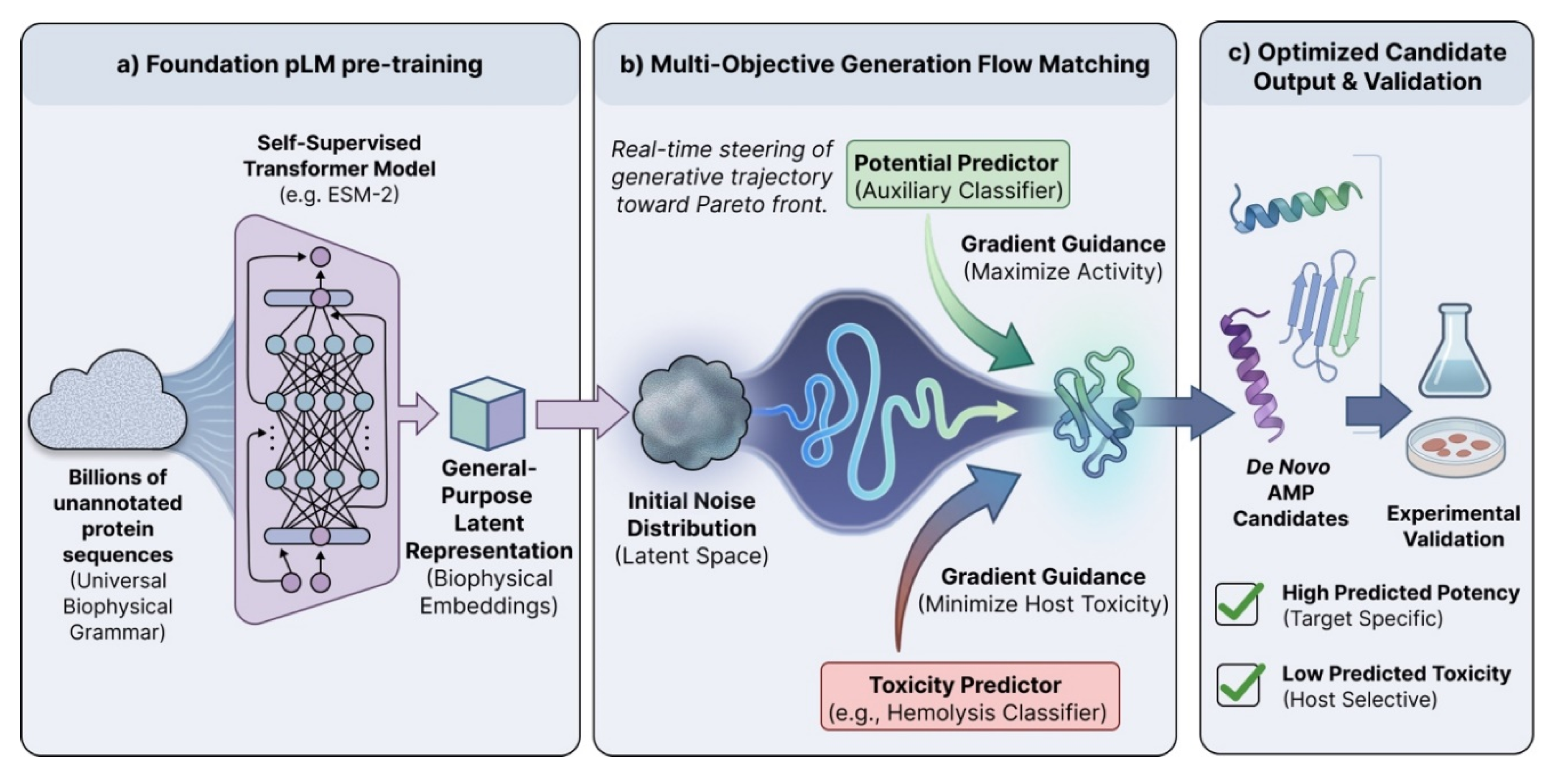
Direct sequence sampling from pLMs, whether autoregressive decoding or MLM, tend to optimize for plausibility, not for clinical constraints like hemolysis, stability, and selectivity. One usually discovers toxicity, aggregation, or instability after generation. These models also lack an intrinsic mechanism to enforce multiple objectives (such as “high antimicrobial potency” and “low hemolysis”) during generation. We believe that research targeting discrete flow matching offers a solution. Rather than committing to tokens one-by-one, diffusion and flow-based methods learn a trajectory from noise to a peptide representation, either in a continuous latent embedding space or via discrete flows over categorical amino acids [11]. That trajectory can be steered online with guidance signals, making constraints part of generation. Recent advances demonstrate that such models enable multi-objective optimization (MOO) through classifier guidance during generation of peptides [12]. We believe MOO can bear fruit in AMP generation by integrating an auxiliary toxicity predictor in the generation process. At each step of the diffusion or flow trajectory, one can calculate the gradient of predicted toxicity metrics with respect to the peptide. The generative model can then dynamically push the sequence away from toxic regions and toward regions predictive of high antimicrobial activity. Crucially, this guidance occurs while the sequence is being formed. Our proposed technique would yield candidates that are pre-optimized for high selectivity. By the time an in silico peptide reaches the final output, it has effectively navigated a Pareto front balancing potency and safety.
This presents an opportunity to (1) leverage pLMs to learn a prior; (2) use diffusion or discrete flow models to turn that prior into an AMP generator; (3) optimize during sampling for potency, selectivity, and developability.
Interpretability in AMP Design
The ‘black box’ problem of AI, particularly deep learning methods, has long been discussed. When it’s difficult for human researchers to understand design choices and features, there is a greater perceived risk and uncertainty about results. There are already many variables contributing to failed antibiotic programs: toxicity, instability, and unclear mechanisms. Adding an uninterpretable AI generator could further limit informed iteration during downstream development. To mitigate this, we propose mechanistic interpretability techniques using sparse autoencoders (SAEs) trained on internal activations of a pLM (Figure 2).
Figure 2.
Mechanistic interpretability of antimicrobial peptide design using Sparse Autoencoders.
(a) High-dimensional, dense activations from the AI model (e.g., pLM backbone) represent the input sequence (KLRSKTR..AFI...KW...RL) in an entangled, non-interpretable state. (b) A trained Sparse Autoencoder (SAE) processes these activations, enforcing sparsity to decompose the dense representation into distinct, independent features. (c) The SAE recovers interpretable biophysical features characterized by specific activation patterns, such as periodic firing corresponding to helix turns (Feature 1), distributed activation across hydrophobic regions (Feature 2), or specific residue motifs (Feature 3). (d) These features are mapped directly onto the generated sequence and structure such as the polar/cationic face (blue), amphipathic core (yellow), and Tryptophan membrane anchor (purple).
Figure 2.
Mechanistic interpretability of antimicrobial peptide design using Sparse Autoencoders.
(a) High-dimensional, dense activations from the AI model (e.g., pLM backbone) represent the input sequence (KLRSKTR..AFI...KW...RL) in an entangled, non-interpretable state. (b) A trained Sparse Autoencoder (SAE) processes these activations, enforcing sparsity to decompose the dense representation into distinct, independent features. (c) The SAE recovers interpretable biophysical features characterized by specific activation patterns, such as periodic firing corresponding to helix turns (Feature 1), distributed activation across hydrophobic regions (Feature 2), or specific residue motifs (Feature 3). (d) These features are mapped directly onto the generated sequence and structure such as the polar/cationic face (blue), amphipathic core (yellow), and Tryptophan membrane anchor (purple).
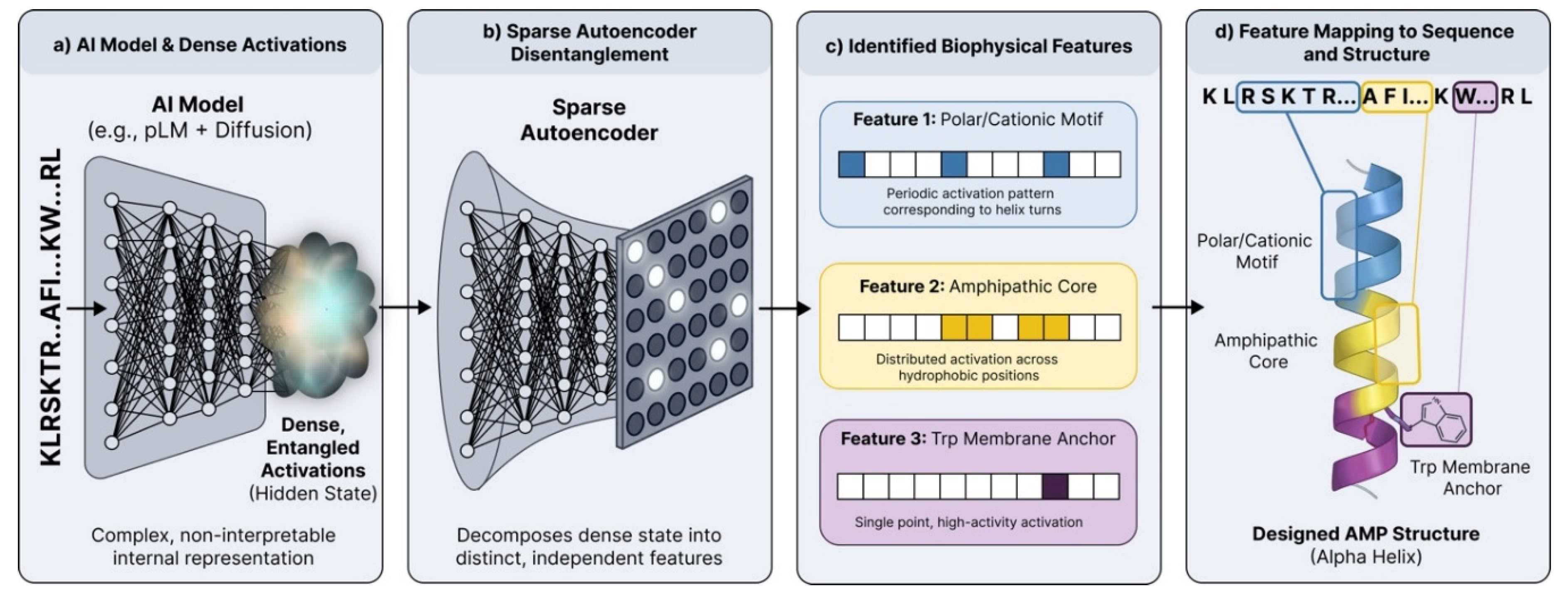
SAEs are a type of neural network designed to learn efficient representations of data by training the network to reconstruct its input while enforcing sparsity, meaning that only a few neurons are active at any given time. This sparse representation can highlight key features of the input data, making it easier to understand which aspects are most important for certain predictions. By applying SAEs to the internal activations of a protein language model, we can potentially identify which specific features or patterns are driving the model’s predictions. This could provide insights into why certain AMP candidates are being selected over others, and what structural or biophysical characteristics are associated with potency and selectivity. In recent work, SAE-identified features corresponded to secondary-structure elements, conserved motifs, full domains, and biochemical patterns tied to structural context [13]. These features could be systematically categorized by activation pattern (point, periodic, motif, whole-domain) and by whether they are broadly used versus family-specific. This moves beyond “attention heatmaps” toward a dictionary of reusable, inspectable features that can be visualized directly on sequences and structures that researchers and clinicians are familiar with.
In an AMP setting, we expect highly weighted features to align with design-relevant properties such as amphipathic helices, cationic residue clustering, transmembrane-like segments, disulfide-forming cysteine patterns, or N-terminal signal-like motifs. Even when a predictive feature lacks an obvious biological label, it still provides a concrete hypothesis to test, rather than an ungrounded score. By understanding which features are most influential in driving the model’s decisions, researchers can adjust inputs or modify the model’s architecture to emphasize or de-emphasize certain characteristics. The interpretability gained through these techniques might also facilitate regulatory approval, as it allows developers to better communicate the basis of their designs. Regulatory agencies often require a clear understanding of how a drug works, and AI-powered drug discovery is no exception. By providing insight into the model’s decision-making process, we can build confidence in AI-driven candidates, ensuring that they are not only innovative but also safe and effective.
Reducing Costs and De-Risking Pre-Clinical Development
A pLM-guided antibiotic discovery pipeline deviates from the typical cost structure of drug development. Foundation protein models learn directly from unlabeled sequence data, avoiding the need for expensive task-specific datasets or high-throughput in vitro screens to train each new model. Instead of screening millions of compounds in wet labs—a process that can cost on the order of $0.5–2 million to set up—the pLM approach generates a focused set of peptide candidates in silico. Compared to the current process costing up to $10 million and ~4.5 years for a new lead antibiotic, an AI-driven generative platform could cut that timeline to ~2.5 years while slashing cost by two-thirds [14]. The savings come from minimizing brute-force experimentation and late-stage attrition. Our framework for designing pLMs can prioritize candidates with desirable properties (high potency, low toxicity) before synthesis, leading to fewer peptides that need to be made and tested.
Beyond reducing the number of experiments, automation and smart workflows further amplify cost benefits. Many steps in antibiotic discovery, from peptide synthesis to microbial growth inhibition assays, are labor-intensive and difficult to scale. Now, these tasks are becoming increasingly automated. Robotic systems are being developed to execute and monitor biological assays continuously, with a throughput unattainable by human scientists [15]. Crucially, the economic gains from automation are not generic to drug discovery; they become magnified when paired with pLM-guided design. Evaluation of AMPs requires tightly coupled, multi-axis testing across MIC, selectivity, hemolysis, and serum stability. These assays are individually inexpensive but can become combinatorially prohibitive without strong pre-filtering. Because pLMs output discrete, synthesis-ready peptide sequences, candidates flow directly into standardized, parallel solid-phase synthesis (SPPS) and high-throughput bacterial testing with less bespoke chemistry. When combined with the automation loop, we suggest a fully autonomous discovery cycle in which experimental throughput scales without proportional increases in cost or personnel.
Lowering early discovery costs de-risks antibiotic development financially. This does not, by itself, solve the revenue-side market failure in antibiotics, where reimbursement is delinked from societal value and stewardship suppresses sales. Nonetheless, reducing discovery and preclinical costs meaningfully mitigates the economic “valley of death,” where promising programs often stall due to lack of capital before reaching proof-of-concept. By lowering the cost required to advance candidates toward an Investigative New Drug (IND) application, pLM-driven pipelines can make antibiotic programs more attractive to investors and pharmaceutical partners who have historically been wary of poor returns in this space. Because most program cost accrues during clinical development, economics improve disproportionately if AI-enabled discovery also increases the probability of success in clinical trials; preliminary evidence of AI-discovered molecules tested in clinical trials suggests this may indeed be the case [16]. In practical terms, even modest gains in clinical success rates could substantially reduce expected development costs, lowering the capital required for a small biotech to advance an AI-designed peptide into efficacy studies. Lower cost also implies more flexibility to pursue novel targets (which may have been deemed too risky or time-consuming before). Additionally, the oft-cited concern that peptide therapeutics are inherently more expensive than small molecules is becoming outdated. Advances in SPPS and “green chemistry” have significantly driven down manufacturing costs [17]. Modern automated synthesizers can produce dozens of peptide variants simultaneously, and convergent purification techniques further economize the process. Again, pLM-focused design helps reduce cost because the number of peptides that must be made is relatively small. Collectively, these efficiencies support a more sustainable antibiotic development pipeline, even as broader policy and reimbursement interventions remain necessary to fully address market incentives of antibiotic development.
Outlook
Antibiotic discovery is at an inflection point. Although AMPs offer mechanistic advantages over small molecules, their development has been constrained by toxicity, instability, and the high cost of empirical screening. We argue that these barriers largely reflect outdated discovery paradigms rather than intrinsic limitations of peptides. By coupling protein language model representations to diffusion or discrete flow–based generative models, AMP design can be reframed as a programmable, multi-objective process. During this process, features such as potency, selectivity, and developability can be optimized during generation. This approach is particularly urgent for pathogens designated by the World Health Organization as critical priorities, including carbapenem-resistant Acinetobacter baumannii and Pseudomonas aeruginosa, where conventional pipelines have failed to deliver new therapeutic classes. Realizing this opportunity will require coordinated action. Machine learning researchers must prioritize controllable and interpretable generative systems; microbiologists and clinicians must define biologically grounded objectives and validation standards; and funding agencies, regulators, and biotechnology companies must invest in integrated, automated discovery platforms that close the loop. Without such alignment, advances in generative modeling will remain largely academic. With it, guided generative design could help re-establish a sustainable, innovation-driven antibiotic pipeline where it is most urgently needed.
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Current Opinion in Microbiology 2019, 51, 72–80. [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Frontiers in Microbiology 2020, 11. [CrossRef] [PubMed]
- Kim, S.H.; Min, Y.-H.; Park, M.C. Microorganisms 2025, 13, 2574.
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Drug Discovery Today 2022, 28, 103464. [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. Nucleic Acids Research 2015, 44, 1087–1093. [CrossRef] [PubMed]
- Santos-Júnior, C.D.; Torres, M.D.T.; Duan, Y.; Rodríguez Del Río, Á.; Schmidt, T.S.B.; Chong, H.; Fullam, A.; Kuhn, M.; Zhu, C.; Houseman, A.; Somborski, J.; Vines, A.; Zhao, X.-M.; Bork, P.; Huerta-Cepas, J.; de la Fuente-Nunez, C.; Coelho, L.P. Cell 2024, 187, 3761–3778. [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; Rives, A. Science 2023, 379, 1123–1130. [CrossRef] [PubMed]
- DTTorres, M.; Chen, T.; Wan, F.; Chatterjee, P.; de la Fuente-Nunez, C. Cell Biomaterials 2025, 1, 100183.
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Frontiers in Chemistry 2018, 6, 204. [CrossRef] [PubMed]
- Vincoff, S.; Goel, S.; Kholina, K.; Pulugurta, R.; Vure, P.; Chatterjee, P. Nature Communications 2025, 16, 1436. [CrossRef] [PubMed]
- Stark, H.; Jing, B.; Wang, C.; Corso, G.; Berger, B.; Barzilay, R.; Jaakkola, T. Preprint at. 2024. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, Y.; Chatterjee, P. Preprint. 2024. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.; Bai, L.; Lee, M.; Yu, Y.; Al Quraishi, M. Preprint at. 2025. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, A. Stanford Social Innovation Review (2024). [CrossRef]
- Lan, Z.; Jiang, Y.; Wang, R.; Xie, X.; Zhang, R.; Zhu, Y.; Li, P.; Yang, T.; Chen, T.; Gao, H.; Yang, X.; Li, X.; Zhang, H.; Mu, Y.; Luo, P. Preprint at. 2025. [Google Scholar] [CrossRef] [PubMed]
- Jayatunga, M.K.P.; Ayers, M.; Bruens, L.; Meier, C. Drug Discovery Today 2024, 29, 104009. [CrossRef] [PubMed]
- Kiss, K.; Ránky, S.; Gyulai, Z.; Molnár, L. SLAS Technology 2023, 28, 89–97. [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



