
Submitted:
12 January 2026
Posted:
14 January 2026
You are already at the latest version
Abstract
Autism spectrum disorder (ASD) is a prevalent and largely idiopathic developmental disorder with relatively widespread etiology. Currently there are no validated diagnostic or screening biomarkers for ASD, besides addressing the associated comorbidities. ASD is primarily diagnosed based on behavioral motor and cognitive characteristics. Until recently, the cerebellum had been particularly implicated in motor control, and under-researched for its potential role in the development of ASD. However, cerebellar circuitry is altered in ASD, impacting its brain interconnections, affecting brain development, and social and behavioral outcomes associated with ASD. We review the potential role of the cerebellum in ASD, how its dysfunction during development or its early postnatal injury may impact the maturation of other connected circuits, and play a role in the development of core ASD symptoms. We address cerebellar changes that may alter synaptic pruning, immune cells’ function, neurotransmitters, blood brain barrier permeability, and potential signaling pathways involved in ASD and how all these changes interplay may contribute to ASD pathophysiology. Understanding of these interactions, may provide novel therapeutic options specifically targeted to the cerebellum.
Keywords:
autism
; cerebellum
; astroglia
; autism linked genes
; autism signaling pathways
; neurodevelopmental disease
Introduction
Autism Spectrum Disorders (ASD) are neurodevelopmental diseases of varying severity, resulting from complex genetic, environmental and immunological interactions. ASD is diagnosed by atypical social behavior and interactions, limited intellectual and interest skills, repetitive/stereotypic behaviors, and variable levels of cognitive and intellectual impairments [1]. Individuals with ASD may also present with other symptoms such as seizures, anxiety, apraxia of speech, sleep disorders, and gastrointestinal complications. According to CDC, about 1 in 44 children have been identified with ASD corresponding to estimates from CDC’s Autism and Developmental Disabilities Monitoring (ADDM) network [2]. While most cases are idiopathic, multiple genetic mutations were identified in patients and are widely studied in animal models, also implicating immune dysregulation and environmental factors, as all these could interact to bring about ASD [1,3,4]. There are currently no validated biomarkers for diagnostic or screening for ASD, besides addressing the related comorbidities. Therefore, understanding the fundamental mechanisms of the disease is of utmost importance to develop ASD-specific targeted treatment strategies.
Most studies have focused on various neural networks associated with social behavior, language, and cognition. Post-mortem studies of ASD patients with genetic predisposition and showing clinical evidence, and of animal models have implicated cerebellar development and dysfunction in ASD etiology [5,6,7]. The cerebellum, previously mostly associated with motor functions, is now recognized for its involvement in cognitive functions, due to its complex connections with other brain areas [8,9,10,11]. Cerebellar structural, functional and neurochemical abnormalities have been identified in ASD, and lesions in particular cerebellar sites yield effects all throughout the brain via network connections [1,9,12]. Cerebellar dysfunction during development may affect the maturation of other connected cerebral circuits that have been shown to regulate several ASD-relevant behaviors [7,10,13]. Multiple animal models of early life cerebellar damage display ASD behavioral symptoms. Indeed, cerebellar defects if they arise by birth or during early postnatal period are often sufficient to induce ASD behavioral symptoms [5].
This review article offers a comprehensive overview of different aspects of cerebellar involvement with respect to brain development and function, enabling the onset of ASD (Figure 1), highlighting earlier studies and new potential areas of research. Thorough understanding of the cerebellar function in ASD may lead to novel strategies regulating the underlying mechanisms to elaborate treatments that are more precisely aimed at cerebellar dysfunction in ASD individuals.
2. Cerebellum structure and function
The highly conserved brain structure and neuronal developmental trajectory of the vertebrate cerebellum develop following a conserved chronological sequence of neurogenesis, preserving its function and internal circuitry [14]. During the evolution, the size of the mammalian cerebellum has expanded when compared to the other areas of the brain, especially the cerebellar posterior lobe, which is important for the processing of cognitive as well as language skills [1,15,16,17]. While the cerebellum occupies just 10% of the total brain volume, it contains more neurons than the rest of the brain because of its substantial number of granule cells [18,19].
The orderly spaced parallel grooves of the cerebellar cortex are organized in three layers, the outer molecular layer (ML), Purkinje cell layer (PCL) and the inner granular layer (GL). The neuronal organization of Purkinje cells (PC) and granule cells (GC) brings about an immense and complex signal processing potential. However, the output signal from the cerebellar cortex goes out through a set of cells called cerebellar deep nuclei sitting in the white matter of the cerebellum [20]. Mossy fibers, climbing fibers and parallel fibers are the three types of axons playing a major role in the cerebellar circuit. The molecular layer, the outermost layer of the cerebellar cortex, is comprised of PC dendritic branches and parallel fibers, which are the axons of the granule cells. The molecular layer contains two types of cells, the stellate cells and the basket cells, which are inhibitory interneurons forming GABAergic synapses with PC dendrites [19]. PC are unique neurons in the brain with large cell bodies arranged into a single cell layer of the cerebellar cortex. The PC dendrites receive inputs from parallel fibers, estimated around 200,000 dendritic spines on a single human PCs, and are inhibitory in nature [19]. Loss of PC, consistent with impaired cerebellar function is typically identified in ASD and may contribute to the excitatory/inhibitory imbalance that characterizes the disorder [13,21]. Thus, PC play a critical role in providing downstream cerebellar nuclei with efferent inhibitory output [22,23]. Granular cells are small and seen in large number in the human brain, estimated around 50 billion in total number and are mainly excitatory in nature as glutamate is their neurotransmitter [19]. Granular cells receive input from mossy fibers while PCs receive input from climbing fibers.
Cerebellar deep nuclei consist of three nuclei embedded within the white matter, the fastigial (medial) nucleus, the interposed nucleus, and the dentate (lateral) nucleus. These nuclei together form the exclusive output of the cerebellum, and it has been estimated that the total number of cerebellar deep nuclei neurons in cerebellum are about 50-100,000 [24] and include both excitatory projection neurons [25,26,27] and inhibitory projection neurons [28,29]. Therefore, disruption of GC/PC and cerebellar deep nuclei output may contribute to excitatory/inhibitory imbalance underlying ASD neuropathophysiology.
The cerebellar deep nuclei are closely associated with the cerebellar functions related to the sensorimotor region, limbic system etc., Besides their role in motor function, it has been reported that the cerebellar deep nuclei are also involved in cognitive and linguistic functions [30], as evidenced by neuropsychological, neurophysiological and imaging studies.
Evidence shows that the cerebellum may coordinate communication and contribute to sensory motor deficits in ASD [7,13,23,31]. For instance, while the primary cause of apraxia is damage to the cortex, cerebellar damage can also contribute to apraxia of speech [30,32]. A recent study from Milton S. Hershey Medical Center, recognizes apraxia as a common incidence in ASD, involving both speech and communication. Apraxia of speech affects children’s brain pathways accountable for performing movement associated with speech production and their ability to align motor movements to deliver speech, despite having fully functional muscles [1]. In a previously reported study, 64% of the children with ASD diagnosis also exhibited apraxia of speech, and 37% of the children with an apraxia diagnosis also had ASD [33]. Because of the failure in coordinating tongue, lips, mouth and jaw, the same word would be pronounced differently every time it was spoken [33].
Localized lesions in the cerebellum yield effects throughout the brain via its network connections [1]. Patients with cerebellar damage failed to control their thought process, as the cerebellum plays a crucial role in regulating cognitive processes [8,34]. Although ASD is behaviorally defined, brain structural differences may be implicated. In neuroimaging and neuromodulation studies, specific sub-cerebellar regions differences have been associated with ASD, both in humans and animal studies [7,10,21,23]. Van Overwalle et al reported decreased cerebellar volume in the posterior vermis, bilateral Crus II and right VI and Crus I/II in children with autism, that correlated with social interaction and communication scores. Reduced gray matter volume has been found in the posterior vermis, Crus I/II, inferior cerebellar vermis (lobule IX), left lobule VIIIB and right Crus I in autism. ASD children also displayed differences in cerebellar activation in lobule VII, including Crus I/II. Among other differences, cerebellar volume and reduced grey matter volume, white matter and activation in specific cerebellar regions have been observed in ASD patients compared to controls [7,10,35,36,37].
Studies also documented a significant reduction in Purkinje cells in the autistic brains [38,39,40,41]. Understanding how these cerebellar dysfunctions are associated with neuroinflammation and neuroimmune alterations seen in ASD, would contribute to targeted intervention strategies.
Cerebellar astroglial involvement in ASD
Cerebellar astrocytes involvement
Astrocytes, the most numerous glial cell type and accounting for one third of brain mass, are involved in the maintenance of the blood–brain barrier (BBB), regulation of water, ion homeostasis and amino acid neurotransmitter metabolism, as well as energy and nutrient support of neurons. Neuron-glia bidirectional communication is associated with the proliferation, migration and differentiation of neural precursor cells and is essential for normal functioning of the brain during early neurodevelopment and throughout life. Altered expression of astroglial markers, such as GFAP, aquaporin-4 (AQP4), and connexin 43 (CX43) have been reported in postmortem studies of ASD patients [46,47,48]. During neuroinflammation, GFAP expression is upregulated when astrocytes are hypertrophic and proliferate. Other studies in animal models of ASD have also reported an upregulation of GFAP gene expression, and an increase in GFAP protein level in the cerebellum [44,46,47,49,50]. Mounting evidence has illustrated that glial cells have a key role in synaptic pruning through phagocytosis in health and disease [51,52,53,54,55]. To uncover the involvement of glial phagocytosis in synaptic pruning, Morizawa et al., created a genetic strategy for visualizing phagocytic events [56]. In naive healthy mouse cerebellar cortex, they found that Bergmann glia (BG) has a high phagocytic capacity and the BG engulfment of neuronal structures, spines and the dendrites were characterized by three-dimensional electron microscopy (3D-EM) analysis. Mice undergoing cerebellum-dependent motor learning, tissue examination revealed enhancement of nibbling of both presynaptic and postsynaptic structures by BG, including postsynaptic spine volume reduction [56].
CX43 is a major protein component in the astrocytic gap junctions and is primarily expressed in BG in the cerebellum. CX43 is responsible for regulating cellular growth and cell-cell adhesion, which also allow for gap junction intercellular communication between cells to regulate cell death, proliferation, and differentiation. Studies reported a higher expression level of CX43 in ASD patients [57], which could suggest enhancement of glial- neuronal communication in the brain. Cx43 on astrocytes can be involved in influencing neurotransmitter transport, thus inhibiting hyperactivation of the nervous system [58]. Cx43-mediated gap junction overactivation can trigger excitotoxicity, neuroinflammation and other pathological conditions [59,60]. Excitation/Inhibition (E/I) Imbalance has been observed in patients with ASD, generally there is less inhibition to balance the overexcitation [61,62]. Abnormal Cx43 expression causes (E/I) Imbalance and excitotoxicity which in turn can substantially hamper synaptic pruning through triggering synapse loss, disturbing neural connections, causing network dysfunction, and contributing to neurological disorders [63,64]. However, conflicting findings report that CX43 decreased as a result of AQP4 depletion [65]. Earlier findings enable us to assume that correcting the AQP4 changes in the brain could also lead to the normalized function and expression of CX43 in the gap junctions.
We hypothesize that reactive astrocytes and alterations in GFAP, AQP4 and CX43 expression in the cerebellum not only compromise the neuron-glia communication but also facilitate and aggravate the effects of neuro-inflammation, edematous condition and hinder synaptic sprouting during early development. Cerebellar abnormalities may also be responsible for dysfunctions within the motor system associated with autism. Understanding the effects of astrocytes and especially how the alterations in any of the GFAP, AQP4 or CX43 markers in the cerebellum may contribute to the development of therapeutic advances in ASD patients.
AQP4 is widely expressed in astrocyte terminal feet enwrapping blood vessels that constitute the blood brain barrier (BBB) and is the most abundant water channel in the CNS. AQP4 has many functions, especially in a developing brain, such as accelerating cell migration by facilitating the transmembrane water fluxes that mediate migrating cells rapid changes in shape and volume as they move through a developing brain [66]. Decreased cerebellar AQP4 expression may mean that the cell structure, cell volume and ionic homeostasis are compromised.
In addition, AQP4 regulates the water transport in various organs such as the formation of cerebrospinal fluid in the brain, formation of urine and aqueous humor of the eye [67,68,69]. As reported, water accumulation was greater in AQP4 knockout mice than in wild type mice in brain tumor edema and vasogenic edema [70,71]. Bloch et al., 2006 [72] demonstrated that the brain swelling was accelerated in obstructive hydrocephalus condition in AQP4 null mice. Studies also report AQP4’s involvement in seizures, as seizures duration and their intensity increased in AQP4 deficient mice [73]. This phenomenon is supported by studies showing that delayed K+ reuptake by AQP4 deficient astrocytes leads to neuroexcitation [73,74]. In addition to synaptic alterations, dysregulation of AQP4 leads to compromised brain homeostasis and BBB development and BBB breach that have been associated with autism and Fragile X through overlapping pathways of neuroinflammation and synaptic dysfunction [75].
Furthermore, several studies support the hypothesis that AQP4 has a key role in regulating synaptic plasticity, including compelling data suggesting a promising link between defective long-term potentiation (LTP) and the downregulation of glutamate transporter-1 (GLT-1), with AQP4 and GLT-1 co-localized expression [76,77,78,79,80,81,82,83]. Zhang et al and others have evidenced that decreased GLT-1 expression GLT-1 in AQP4-null mice which lead to the reduced glutamate uptake by astrocytes [83,84].
Another potential mechanism that could be linked to synaptic plasticity and learning and memory impairment is that AQP4 deficiency promoted a decrease in BDNF signaling [85,86]. Studies found that the release of mature BDNF leads to LTP [87], and the inhibition of LTD [88], hence, the delayed LTP in AQP4−/−mice was a remarkable finding [76]. These data suggest a tight control of BDNF release for either LTP or LTD and that BDNF release in adaptable synaptic plasticity may be modified by AQP4.
Broad opportunities remain for the development of AQP4-based diagnostics and therapeutics. several anti-epileptic drugs, including zonisamide, lamotrigine, phenytoin, and topiramate, were also found to inhibit AQP4 [89]. In addition, 2-(nicotinamide)-1,3,4-thiadiazole (TGN-020) appears to be a more selective inhibitor and has demonstrated promising results in preclinical studies [90] Inhibitors of AQP4 and small molecule aquaporin modulators are predicted to reduce brain swelling in cytotoxic edema, potentially offering neuroprotection following brain injury, ischemic stroke, epilepsy, infection, neuroinflammation and thus could also alleviate ASD symptoms.
Cerebellar microglia, macrophage, monocyte and neutrophil involvement
Previous reports have documented increased microglia and Bergmann glia reactivity within the PC layer and accompanying white matter in ASD patients’ brain. Activated microglia are detected in the vicinity of degenerating PCs and granule cells [44]. The microglia and astrocyte activation extends beyond the cerebellum to mid-frontal and cingulate gyrus as measured by the increased expression of cell surface major histocompatibility complex molecule HLA-DR and glial fibrillary acidic protein. Activated microglia and astrocytes are also accompanied by monocyte and macrophage accumulation. Such accumulation may underlie the phagocytic capacity of synaptosomes by human macrophages derived from peripheral blood monocytes, macrophage polarity [91]. Higher microglia activation in white matter is detected in ASD individuals with a history of epileptic seizures compared to non-epileptic ASD population. The microglia activation results in increased production of several cytokine and chemokines such as interleukin (IL-6), transforming growth factor beta 1 (TGFβ1), C–C motif ligand 2 (CCL2), and CCL17 [44,92,93,94]. Gene expression analysis from ASD patients further confirms a significantly increased and/or activated MET, NF-κB, IL-1 receptor, TOLL, and TNF receptor 2 immune system-related genetic pathways. Whether the altered immune activity is causal or in response to the neuronal damage induced during early onset of ASD is unknown [95]. Beyond cerebellum, the immune activation is also a hallmark factor that is compounded with elevated plasma cytokine levels (IL-1β, IL-6, IL-12, and TNFα), immunoglobulin levels, complement proteins and chemokines (CCL2, CCL5, CCL11) within the periphery. The natural killer cells, monocytes and T cells that show higher activation in response to immunological challenges disrupt cellular functions in ASD patients in parallel to decreased production of regulatory cytokines such as TGFβ1 and IL-10 implying a shift in inflammatory profile [96,97,98,99]. The high production of cytokines also correlates with higher activation of monocyte, macrophage, mast cells and microglia [100]. Applying CIBERSORT algorithm, Li et al., [101] showed that ASD patient group exhibit a significantly higher estimated proportion of resting/activated dendritic cells, M0/M2 macrophages, and monocytes in children and M0 macrophages, resting mast cells, and resting/activated NK cell group in adults with a higher percentage or increasing trend of apoptosis in monocytes [102]. Preclinical animal models support clinical studies especially with respect to monocyte infiltration [103] and neutrophil density [104]. We have previously shown increased microglial activation in the Sema3F-KO mouse model of autism [45]. The idiopathic BTBR autism mouse model that exhibits ASD phenotypes display increased IL-33, IL-18 and IL-6 cytokines [105]. More recent results show that the brain region specific knockout of PTEN that results in macrocephaly, motor coordination defects, cellular hypertrophy and autistic phenotype in mice show enhanced phagocytic capacity of microglia and aberrant microglia activation [106]. Beyond glial activation, children with ASD exhibit a higher neutrophil to lymphocyte ratio (NLR) primarily mediated by acute immune infections, autoimmune diseases [107] and superoxide dismutase mediated dysregulated enzymatic antioxidant network [108]. In summary, both human and mice with ASD phenotype show increased generalized and cerebellar microglia activation that leads to altered cytokine and chemokine levels, and enhanced monocyte and macrophage accumulation and higher neutrophil density.
Several immune-regulating compounds have demonstrated a potential effect decreasing some of the inflammatory ASD symptoms. Several anti-inflammatory compound such as a COX2 inhibitor (Celecoxib) and inflammatory cytokines inhibitor (Pentoxifyllin) have been shown to improve social deficits but have not yet been proven as significant therapies [109,110,111,112].
Neurotransmitters and cerebellar connectivity association with ASD
The neurobiological characteristic of ASD comprise abnormal synaptic connectivity, imbalances in excitatory inhibitory signaling, and alterations in neurotransmitters such as glutamate, gamma aminobutyric acid (GABA), dopamine and serotonin [113,114]. Thus far, accruing evidence points out to the hypothesis that fundamental features of ASD emerge from irregularities in the excitatory inhibitory balance within neural circuits [115,116,117]. Among the neurotransmitters involved, the abnormal signaling of Glutamate and GABA have been steadily reported to be the most affected in ASD [118,119,120]. A study by Purcell et al. reported a decrease in glutamatergic neurotransmission in ASD patients’ cerebellum when compared to the neurotypical ones [49]. Both motor and cognitive impairment in ASD directs attention to an excitatory/inhibitory imbalance within the cerebellum [121].
Cerebellar coordination of cerebral activity involvement in ASD.
Among the main three layers of the cerebellar cortex, the outer molecular layer is composed of inhibitory neurons such as stellate cells and basket cells, and the Purkinje layer consists of inhibitory Purkinje cells. The inner granular layer is composed of both excitatory granule cells and inhibitory Golgi cells. The mossy fibers and the climbing fibers are the primary input pathways entering the cerebellum, where in the granular layer mossy fibers synapse on the dendrites of granular cells, whose axons lead to the molecular layer where they form parallel fibers [122,123]. Mossy fiber axons derive from multiple sources in the brain stem and spinal cord neurons, including pontine nuclei, vestibular nuclei, and reticular formation, providing main excitatory input to the cerebellum by synapsing onto granule cells. The granule cells then project parallel fibers, which go up to the molecular layer forming excitatory synapses onto Purkinje cells [124,125,126,127,128]. Parallel fibers and climbing fibers send excitatory signals to Purkinje cells projecting their neurons to deep cerebellar nuclei neurons [129], which in turn give the cerebellar final output by incorporating both inhibitory and excitatory inputs from Purkinje cell axons, mossy fiber and climbing fibers [130].
Several anatomical studies have reported that cerebellum is interconnected with the cortex, hippocampus and amygdala, shaping cognitive, affective, and social behavior [12,37,131,132,133,134]. Coactivation of amygdala and cerebellum was reported during the demonstration of facial expressions in human subjects [135], and its connection with the cingulate cortex indicates the participation in motivational and emotional processing [136]. Studies have reported electrical stimulation of deep cerebellar nuclei in rodents to induce the release of dopamine in the medial prefrontal cortex [137,138,139]. Previous studies have shown that the cerebellum regulates the prefrontal cortical function and the disturbances in this cerebellar-prefrontal circuitry directs to deficits in executive functioning and social cognition in ASD individuals [37,140]. Overall, these reports show that the dysfunctions in the cerebellar cortical network, typically associated with ASD symptoms, could be linked to a compromised connectivity between the cerebellum and cortical social areas in the brain.
Earlier studies in ASD patients’ cerebellum reported a decrease in PC of glutamic acid decarboxylase 67 (GAD76) mRNA, a basic enzyme converting glutamate to GABA [141,142]. On the other hand, in another study, Yip et al. found increased GAD67 mRNA expression in the cerebellar molecular layer interneurons, indicating the presence of an upregulation process to counterbalance the altered inhibition of Purkinje cells [143]. Studies have reported that deep cerebellar nuclei GABAergic neurons, which project precisely to the Inferior olive showed a decrease in GAD65 mRNA expression [144,145,146]. Hence, changes in GABAergic neurotransmission in the deep cerebellar nuclei could intensely disturb the Purkinje cell activity. Other studies have also reported that serotonin concentrations were altered in the cerebellum of ASD individuals [147,148]. Serotonin, an inhibitory neurotransmitter, has a significant role in neurodevelopment, neuronal survival, controlling cellular migration, proliferation, neurite outgrowth, and synaptogenesis [149,150,151]. Chugani et al. showed lowered serotonin levels in the thalamus and the frontal cortex accompanying with elevated serotonin concentration in the deep cerebellar nuclei using PET scanning with tracer for serotonin synthesis in ASD individuals [147,148]. Fatemi and colleagues reported that the reelin expression was decreased in the cerebellum of ASD individuals [141]. Reelin is a glycoprotein encoded by the RELN gene, that can regulate the development of inhibitory synapses, proper cortex lamination, neuronal migration during development in the cerebellum and adult life maintaining cell signaling and synaptic function [152,153,154]. RELN genetic alteration and single nucleotide polymorphisms (SNPs) have been reported to affect brain development and contribute to ASD [154]. Mutations in RELN gene have been found associated with individuals having ASD symptoms (SFARI Gene), as autistic postmortem brains show an impaired reelin signaling in the cortex and cerebellum [153]. RELN mouse mutants exhibit cerebellar hypoplasia, ataxia, reduced GC numbers, PC migration deficits, with motor coordination, and balance deficits, co-relating with human RELN mutations with intense cerebellar hypoplasia [155,156].
Multiple drugs modulating neurotransmitter’s function are currently under study or at different stages of clinical trials. Baclofen and Arbaclofen, selective GABA-B agonists, appear to improve ASD-relevant behavior [157,158]. Serotonin receptors antagonists such as Risperidone and Aripiprazole or SSRI drugs such as Clomipramine and Fluoxetine are in clinical use and show improvement in aggression, anxiety and obsessive behavior [111]. Memantine, a noncompetitive NMDA antagonist, shows improvement in language function, social behavior, self-stimulatory behavior and hyperactivity [111,158]. Several metabotropic mGluR antagonists (Fenobam, JNJ16259685, MPEP) improve social behavior and reduce repetitive behavior [111,159]. In conclusion these findings spotlight the vital role of GABAergic neurotransmission, GAD enzymes, reelin and serotonin concentration in the cerebellum of ASD patients, however more studies are required to better assess the mechanisms underlying excitatory and inhibitory imbalance in ASD.
Taken together, these observations suggest that these cerebellar processes are the center of active chronic neural dysfunction in ASD. Elucidating potential cerebellar signaling pathways disrupted by ASD and affecting cerebellar circuitry, could provide promising therapeutic interventions.
Cerebellar signaling involved in ASD pathogenesis.
Studies highlight the cerebellum as a pathological brain area in ASD patients, as hundreds of identified and validated autism genes have been shown to have important functions in cerebellar development and could provide precise molecular targets for the treatment of ASD symptoms [13,160,161,162,163]. Multiple genes mutation can lead to dysregulation of signaling pathways that may affect cerebellar development, synapse function, elimination or plasticity, and could interact to lead to variable intensity of ASD symptoms. Manipulating these pathways may connect them more specifically to certain phenotypes and allow mitigation of ASD symptoms.
Mammalian target of rapamycin (mTOR)
The mammalian target of rapamycin (mTOR) signaling pathway is an important regulator with a critical role mediating various cellular processes involving protein synthesis and synaptic plasticity [164,165]. The PI3K/Akt/ mTOR pathway is expressed as two types of complexes, mTORC1, controlling cellular metabolism and autophagy, and mTORC2, controlled by TSC1/2 (tuberous sclerosis complex 1 and 2) and up-stream PI3K signaling. mTORC2 Akt phosphorylation results in mTORC1 activation. Pathogenic variants of genes of the PI-3K/Akt/mTOR signaling pathway, including FMR1, PTEN, TSC1, and TSC2, have been associated with ASD [112,166]. Reports on various neurodegenerative diseases show that overactivation of mTOR is associated with BBB disruption, enhanced ROS, superoxide and eNOS uncoupling, and exacerbate patients’ vascular cognitive impairment and worsen the disease [167,168]. In a normal brain, mTOR regulates BBB integrity by inhibiting the downstream effector of mTOR, rpS6, reducing superoxide production and enhancing NO production [169]. We and others have previously reported increased BBB permeability, neuroinflammation, oxidative stress, and iNOS expression in the Sema3F KO and the BTBR pre-clinical models of ASD [45,170]. Multiple studies report that enhanced mTOR signaling may play a significant role in the pathophysiology of ASD and that pharmacological intervention with mTOR signaling could rescue behavioral ASD symptoms as well as some ASD-related brain morphological changes [171,172,173]. Mitigating mTOR with rapamycin in a mouse model of Alzheimer’s disease (AD) improves cognitive function, cerebral blood flow and microvascular endothelial function [174,175,176,177,178]. Altogether, the neuroinflammatory response associated with BBB breach, mTOR activation and abnormal trafficking of BBB tight junction proteins could promote BBB dysfunction in ASD.
The level of cerebellar contribution to the pathogenesis of ASD still remains unclear. Tuberous Sclerosis Complex (TSC) is a genetic disorder with increased proportions of co-morbid ASD consequences of mutation of either TSC1 or TSC2, whose protein products dimerize and negatively regulate mTOR signaling [179]. TSC mutation is an interesting model to study the cerebellar involvement in the underlying pathogenesis of ASD, as recent reports in TSC patients reveal cerebellar pathology and associate cerebellar pathology with increased ASD symptomatology [180,181,182]. Although, Tsc1's roles and its dysfunction in the cerebellum have not been well explored, Tsai et al., show that both heterozygous and homozygous loss of Tsc1 in mouse cerebellar PCs results in autistic-like behaviors, including abnormal social interaction, and repetitive behavior [183]. They also found that treatment of mutant mice with the mTOR inhibitor, rapamycin, reversed the pathological and behavioral deficits. Thus, PC Tsc1 mutants can deliver a study model to explore the effects of PC dysfunction on neuronal networks and other mechanisms causative of ASD pathogenesis, to assess potential therapeutic strategies attenuating the effect of mTOR overactivation and BBB dysfunction and that could further be used to alleviate the cognitive dysfunction and behavioral impairment in children with ASD. However, mTOR inhibitors also depress autophagy and have some immunosuppressive effects, impairing Treg cells differentiation, therefore may not be applicable to ASD populations unless some concomitant treatment alleviates this side effect [112]. It is important to study different ASD models using pharmacological mTOR inhibitor, rapamycin and /or a specific amino acid diet aimed at attenuating mTOR signaling pathway in the cerebellum.
Additional ASD-linked genes associated with cerebellar development.
Mutations of the gene encoding the chromatin remodeler chromodomain helicase DNA-binding protein 8 (CHD8) are substantial risk factors for ASD [184,185]. Patients with CHD8 mutations regularly display cognitive deficits, gastrointestinal illnesses, anxiety, macrocephaly, craniofacial abnormalities, in addition CHD8 also regulates the expression of ASD risk genes associated with synaptic function and neurodevelopment [186,187,188]. A study reported by Kawamura et al reveals CHD8 plays a critical role in cerebellar development, with its substantial associations of this brain area to manifest ASD symptoms [189]. The study shows that the removal of the CHD8 in neural precursor/stem cells or granular neuron progenitor (GNP) specific deletion of CHD8 in mice, results in cerebellar malformation and motor function defects. Additionally, they uncovered CHD8 regulation of local chromatin accessibility, thus activating the expression of several neuronal genes in GNPs. Engrailed homeobox 2 (EN2) has a crucial role in the early and late embryonic development of cerebellar neurons as mice deficient of EN2 exhibit abnormal cerebellar development [162,190]. A few studies have found a genetic correlation between the gene encoding the transcription factor EN2 and autism, as the EN2 knockout mice exhibit behavioral impairments that are related to autism, including social deficits and increased grooming [163,191,192,193].
Several studies have reported that transcription factors such as forkhead box 2 (Foxp2) and RAR-related orphan receptor alpha (ROR-alpha) are crucial for the cerebellar PC development [194,195]. Foxp2 gene deficient mice and mice model with language disorders show impairment in cerebellar cell development, migration, cell morphology and cerebellar synaptic deficits, impaired motor learning [196,197,198]. Foxp2 gene mutations in humans cause developmental speech and language deficits are associated with autism (SFARI Gene, Autism KB).
A study by Li et al., suggests that Jun proto-oncogene (JUN) and platelet derived growth factor receptor alpha (PDGFRA) are critical genes in the cerebellum of individuals with ASD [199]. They have indicated that the activation of cerebellar JUN and PDGFRA in ASD children may be significantly associated with the inflammatory response, as they have highlighted the role of the IL17 signaling pathway in the activation of the immune response in ASD. In addition, JUN plays an important role in the BBB integrity and function and with the release of a variety of inflammatory mediators including IL-6, IL-1β, TNF-α etc., as it regulates the transcription of proinflammatory genes [200,201].
Further investigations are needed to explore this area and provide more insights into the molecular mechanisms underlying these altered pathways in cerebellum, and how they relate to ASD, to enable novel therapeutic strategies.
Conclusion
Several studies have reported that the cerebellum is an important brain area that affects ASD pathology [13,161,202]. Additionally, the concurrence of early behavioral defects in ASD, including motor, cognitive, and behavioral impairments, suggests that the cerebellar abnormalities are an important factor affecting individuals with ASD [202]. More studies need to be conducted to explore the cerebellum pathology and its impairment to the ability to coordinate the communication between neuronal groups in social, cognitive, and corticostriatal networks, thus the appearance of autistic behaviors in children with ASD. Molecular studies to evaluate the therapeutic efficacy of different compounds targeting interventions including both pharmacological and dietary aspects, may offer a new approach in the management of ASD.
Further investigations are needed to better understand the complex interactions between social brain areas, connectivity, frequency bands, and physiological aspects (i.e., roles of specific cell types, maturational processes, receptors) and how they relate to different cognitive processes.
Author Contributions
Investigation and original draft of the manuscript R.J.; part of the manuscript writing I.T. and N.N.; Writing-review and editing G.B. and E.G.
Acknowledgments
Pilot grant from the National Institute of Environmental Health Sciences (P30ES030283) and Department of Defense grant (AR230178P1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hampson, D. R.; Blatt, G. J. Autism spectrum disorders and neuropathology of the cerebellum. Front Neurosci 2015, vol. 9, pp. 420. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M. J.; Shaw, K. A.; Bakian, A. V.; Bilder, D. A.; Durkin, M. S.; Esler, A.; Furnier, S. M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; Hughes, M. M.; Patrick, M.; Pierce, K.; Poynter, J. N.; Salinas, A.; Shenouda, J.; Vehorn, A.; Warren, Z.; Constantino, J. N.; DiRienzo, M.; Fitzgerald, R. T.; Grzybowski, A.; Spivey, M. H.; Pettygrove, S.; Zahorodny, W.; Ali, A.; Andrews, J. G.; Baroud, T.; Gutierrez, J.; Hewitt, A.; Lee, L. C.; Lopez, M.; Mancilla, K. C.; McArthur, D.; Schwenk, Y. D.; Washington, A.; Williams, S.; Cogswell, M. E. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill Summ 2021, vol. 70(no. 11), 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, D. I.; Ververi, A.; Dafoulis, V.; Kalyva, E.; Vargiami, E. Autism spectrum disorders: the quest for genetic syndromes. Am J Med Genet B Neuropsychiatr Genet 2013, vol. 162b(no. 4), 327–66. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.; Ashwood, P. Evidence supporting an altered immune response in ASD. Immunol Lett 2015, vol. 163(no. 1), 49–55. [Google Scholar] [CrossRef]
- Wang, S. S.; Kloth, A. D.; Badura, A. The cerebellum, sensitive periods, and autism. Neuron 2014, vol. 83(no. 3), 518–32. [Google Scholar] [CrossRef]
- Mosconi, M. W.; Wang, Z.; Schmitt, L. M.; Tsai, P.; Sweeney, J. A. The role of cerebellar circuitry alterations in the pathophysiology of autism spectrum disorders. Front Neurosci 2015, vol. 9, pp. 296. [Google Scholar] [CrossRef]
- Stoodley, C. J.; D'Mello, A. M.; Ellegood, J.; Jakkamsetti, V.; Liu, P.; Nebel, M. B.; Gibson, J. M.; Kelly, E.; Meng, F.; Cano, C. A.; Pascual, J. M.; Mostofsky, S. H.; Lerch, J. P.; Tsai, P. T. Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat Neurosci 2017, vol. 20(no. 12), 1744–1751. [Google Scholar] [CrossRef]
- Schmahmann, J. D. The cerebellum and cognition. Neurosci Lett 2019, vol. 688, 62–75. [Google Scholar] [CrossRef]
- Marien R., P. a. B. Chapter 11 - Language and the cerebellum. In Handbook of Clinical Neurology; 2018; pp. ^pp. 182–202. [Google Scholar]
- Van Overwalle, F.; Manto, M.; Cattaneo, Z.; Clausi, S.; Ferrari, C.; Gabrieli, J. D. E.; Guell, X.; Heleven, E.; Lupo, M.; Ma, Q.; Michelutti, M.; Olivito, G.; Pu, M.; Rice, L. C.; Schmahmann, J. D.; Siciliano, L.; Sokolov, A. A.; Stoodley, C. J.; van Dun, K.; Vandervert, L.; Leggio, M. Consensus Paper: Cerebellum and Social Cognition. Cerebellum 2020, vol. 19(no. 6), 833–868. [Google Scholar] [CrossRef]
- Fatemi, S. H.; Aldinger, K. A.; Ashwood, P.; Bauman, M. L.; Blaha, C. D.; Blatt, G. J.; Chauhan, A.; Chauhan, V.; Dager, S. R.; Dickson, P. E.; Estes, A. M.; Goldowitz, D.; Heck, D. H.; Kemper, T. L.; King, B. H.; Martin, L. A.; Millen, K. J.; Mittleman, G.; Mosconi, M. W.; Persico, A. M.; Sweeney, J. A.; Webb, S. J.; Welsh, J. P. Consensus paper: pathological role of the cerebellum in autism. Cerebellum 2012, vol. 11(no. 3), 777–807. [Google Scholar] [CrossRef]
- Mapelli, L.; Soda, T.; D'Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int J Mol Sci 2022, vol. 23(no. 7). [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Meng, F.; Fujita, H.; Morgado, F.; Kazemi, Y.; Rice, L. C.; Ren, C.; Escamilla, C. O.; Gibson, J. M.; Sajadi, S.; Pendry, R. J.; Tan, T.; Ellegood, J.; Basson, M. A.; Blakely, R. D.; Dindot, S. V.; Golzio, C.; Hahn, M. K.; Katsanis, N.; Robins, D. M.; Silverman, J. L.; Singh, K. K.; Wevrick, R.; Taylor, M. J.; Hammill, C.; Anagnostou, E.; Pfeiffer, B. E.; Stoodley, C. J.; Lerch, J. P.; du Lac, S.; Tsai, P. T. Regulation of autism-relevant behaviors by cerebellar-prefrontal cortical circuits. Nat Neurosci 2020, vol. 23(no. 9), 1102–1110. [Google Scholar]
- Rueda-Alana, E.; Garcia-Moreno, F. Time in Neurogenesis: Conservation of the Developmental Formation of the Cerebellar Circuitry. Brain Behav Evol 2022, vol. 97(no. 1-2), 33–47. [Google Scholar] [CrossRef] [PubMed]
- D. M. Broussard, The Cerebellum: Learning Movement, Language, and Social Skills.; John Wiley & Sons, Inc.: Chichester, 2014.
- Sereno, M. I.; Diedrichsen, J.; Tachrount, M.; Testa-Silva, G.; d'Arceuil, H.; De Zeeuw, C. The human cerebellum has almost 80% of the surface area of the neocortex. Proc Natl Acad Sci U S A 2020, vol. 117(no. 32), 19538–19543. [Google Scholar]
- Weaver, A. H. Reciprocal evolution of the cerebellum and neocortex in fossil humans. Proc Natl Acad Sci U S A 2005, vol. 102(no. 10), 3576–80. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Front Neuroanat 2010, vol. 4, pp. 12. [Google Scholar]
- Llinas, R. R.; Walton, K.D.; Lang, E.J. Ch. 7 Cerebellum; Shepherd, GM, Ed.; Oxford University Press: The Synaptic Organization of the Brain, 2004. [Google Scholar]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Hall, W.C.; LaMantia, A.S.; White, L.E. Neuroscience, 5th ed.; Sinauer: Sunderland, Mass., 2011. [Google Scholar]
- Baizer, J. S. Neuroanatomy of autism: what is the role of the cerebellum? Cereb Cortex 2024, vol. 34(no. 13), 94–103. [Google Scholar]
- Ito, M.; Kano, M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett 1982, vol. 33(no. 3), 253–8. [Google Scholar] [CrossRef]
- Kelly, E.; Escamilla, C. O.; Tsai, P. T. Cerebellar Dysfunction in Autism Spectrum Disorders: Deriving Mechanistic Insights from an Internal Model Framework. Neuroscience 2021, vol. 462, 274–287. [Google Scholar] [CrossRef]
- Green, J. T.; Arenos, J. D.; Dillon, C. J. The effects of moderate neonatal ethanol exposure on eyeblink conditioning and deep cerebellar nuclei neuron numbers in the rat. Alcohol 2006, vol. 39(no. 3), 135–50. [Google Scholar] [CrossRef]
- Dum, R. P.; Strick, P. L. An unfolded map of the cerebellar dentate nucleus and its projections to the cerebral cortex. J Neurophysiol 2003, vol. 89(no. 1), 634–9. [Google Scholar]
- Middleton, F. A.; Strick, P. L. Cerebellar projections to the prefrontal cortex of the primate. J Neurosci 2001, vol. 21(no. 2), 700–12. [Google Scholar] [CrossRef] [PubMed]
- Teune, T. M.; van der Burg, J.; van der Moer, J.; Voogd, J.; Ruigrok, T. J. Topography of cerebellar nuclear projections to the brain stem in the rat. Prog Brain Res 2000, vol. 124, 141–72. [Google Scholar]
- de Zeeuw, C. I.; Holstege, J. C.; Ruigrok, T. J.; Voogd, J. Ultrastructural study of the GABAergic, cerebellar, and mesodiencephalic innervation of the cat medial accessory olive: anterograde tracing combined with immunocytochemistry. J Comp Neurol 1989, vol. 284(no. 1), 12–35. [Google Scholar]
- Fredette, B. J.; Mugnaini, E. The GABAergic cerebello-olivary projection in the rat. Anat Embryol (Berl) 1991, vol. 184(no. 3), 225–43. [Google Scholar]
- Habas, C.; Dun, K.T.; Manto, M.; Marien, P. The Linguistic Cerebellum, Chapter 13 - Deep Cerebellar Nuclei (DCN) and Language; 2016. [Google Scholar]
- McAfee, S. S.; Liu, Y.; Sillitoe, R. V.; Heck, D. H. Cerebellar Coordination of Neuronal Communication in Cerebral Cortex. Front Syst Neurosci 2021, vol. 15, pp. 781527. [Google Scholar]
- Marien, P.; van Dun, K.; Verhoeven, J. Cerebellum and apraxia. Cerebellum 2015, vol. 14(no. 1), 39–42. [Google Scholar] [CrossRef]
- Tierney, C.; Mayes, S.; Lohs, S. R.; Black, A.; Gisin, E.; Veglia, M. How Valid Is the Checklist for Autism Spectrum Disorder When a Child Has Apraxia of Speech? J Dev Behav Pediatr 2015, vol. 36(no. 8), 569–74. [Google Scholar]
- Schmahmann, J. D. Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J Neuropsychiatry Clin Neurosci 2004, vol. 16(no. 3), 367–78. [Google Scholar]
- Ecker, C.; Marquand, A.; Mourao-Miranda, J.; Johnston, P.; Daly, E. M.; Brammer, M. J.; Maltezos, S.; Murphy, C. M.; Robertson, D.; Williams, S. C.; Murphy, D. G. Describing the brain in autism in five dimensions--magnetic resonance imaging-assisted diagnosis of autism spectrum disorder using a multiparameter classification approach. J Neurosci 2010, vol. 30(no. 32), 10612–23. [Google Scholar] [CrossRef]
- Riva, D.; Annunziata, S.; Contarino, V.; Erbetta, A.; Aquino, D.; Bulgheroni, S. Gray matter reduction in the vermis and CRUS-II is associated with social and interaction deficits in low-functioning children with autistic spectrum disorders: a VBM-DARTEL Study. Cerebellum 2013, vol. 12(no. 5), 676–85. [Google Scholar] [CrossRef] [PubMed]
- D'Mello, A. M.; Crocetti, D.; Mostofsky, S. H.; Stoodley, C. J. Cerebellar gray matter and lobular volumes correlate with core autism symptoms. Neuroimage Clin 2015, vol. 7, 631–9. [Google Scholar]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. Brain 1998, vol. 121 Pt 5, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.; Kemper, T. L. Histoanatomic observations of the brain in early infantile autism. Neurology 1985, vol. 35(no. 6), 866–74. [Google Scholar] [CrossRef]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional alterations in purkinje cell density in patients with autism. PLoS One 2014, vol. 9(no. 2), pp. e81255. [Google Scholar] [CrossRef]
- Whitney, E. R.; Kemper, T. L.; Bauman, M. L.; Rosene, D. L.; Blatt, G. J. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum 2008, vol. 7(no. 3), 406–16. [Google Scholar] [CrossRef]
- Pardo, A.; Vargas, D. L.; Zimmerman, A. W. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry 2005, vol. 17(no. 6), 485–95. [Google Scholar]
- Takano, T. Role of Microglia in Autism: Recent Advances. Dev Neurosci 2015, vol. 37(no. 3), 195–202. [Google Scholar]
- Vargas, L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A. W.; Pardo, C. A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 2005, vol. 57(no. 1), 67–81. [Google Scholar]
- Jagadapillai, R.; Qiu, X.; Ojha, K.; Li, Z.; El-Baz, A.; Zou, S.; Gozal, E.; Barnes, G. N. Potential Cross Talk between Autism Risk Genes and Neurovascular Molecules: A Pilot Study on Impact of Blood Brain Barrier Integrity. Cells 2022, vol. 11(no. 14). [Google Scholar]
- Laurence, J. A.; Fatemi, S. H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, vol. 4(no. 3), 206–10. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol Autism 2014, vol. 5(no. 1), pp. 3. [Google Scholar]
- Deckmann, I.; Santos-Terra, J.; Fontes-Dutra, M.; Korbes-Rockenbach, M.; Bauer-Negrini, G.; Schwingel, G. B.; Riesgo, R.; Bambini-Junior, V.; Gottfried, C. Resveratrol prevents brain edema, blood-brain barrier permeability, and altered aquaporin profile in autism animal model. Int J Dev Neurosci 2021, vol. 81(no. 7), 579–604. [Google Scholar]
- Purcell, A. E.; Jeon, O. H.; Zimmerman, A. W.; Blue, M. E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, vol. 57(no. 9), 1618–28. [Google Scholar] [CrossRef]
- Crawford, D.; Chandley, M. J.; Szebeni, K.; Szebeni, A.; Waters, B.; Ordway, G. A. Elevated GFAP Protein in Anterior Cingulate Cortical White Matter in Males With Autism Spectrum Disorder. Autism Res 2015, vol. 8(no. 6), 649–57. [Google Scholar]
- Chung, W. S.; Clarke, L. E.; Wang, G. X.; Stafford, B. K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L. C.; Thompson, A.; Chen, C.; Smith, S. J.; Barres, B. A. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, vol. 504(no. 7480), 394–400. [Google Scholar] [CrossRef]
- Schafer, P.; Lehrman, E. K.; Kautzman, A. G.; Koyama, R.; Mardinly, A. R.; Yamasaki, R.; Ransohoff, R. M.; Greenberg, M. E.; Barres, B. A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, vol. 74(no. 4), 691–705. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V. F.; Nfonoyim, B. M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K. M.; Shi, Q.; Rosenthal, A.; Barres, B. A.; Lemere, C. A.; Selkoe, D. J.; Stevens, B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, vol. 352(no. 6286), 712–716. [Google Scholar] [CrossRef]
- Morizawa, Y. M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; Okano, H.; Koizumi, S. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun 2017, vol. 8(no. 1), pp. 28. [Google Scholar]
- Lee, H.; Kim, J. Y.; Noh, S.; Lee, H.; Lee, S. Y.; Mun, J. Y.; Park, H.; Chung, W. S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2021, vol. 590(no. 7847), 612–617. [Google Scholar]
- Morizawa, Y. M.; Matsumoto, M.; Nakashima, Y.; Endo, N.; Aida, T.; Ishikane, H.; Beppu, K.; Moritoh, S.; Inada, H.; Osumi, N.; Shigetomi, E.; Koizumi, S.; Yang, G.; Hirai, H.; Tanaka, K.; Tanaka, K. F.; Ohno, N.; Fukazawa, Y.; Matsui, K. Synaptic pruning through glial synapse engulfment upon motor learning. Nat Neurosci 2022, vol. 25(no. 11), 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S. H.; Folsom, T. D.; Reutiman, T. J.; Lee, S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse 2008, vol. 62(no. 7), 501–7. [Google Scholar] [CrossRef] [PubMed]
- Figiel, M.; Allritz, C.; Lehmann, C.; Engele, J. Gap junctional control of glial glutamate transporter expression. Mol Cell Neurosci 2007, vol. 35(no. 1), 130–7. [Google Scholar]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverria, C.; Orellana, J. A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; Saez, J. C.; Retamal, M. A. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J 2012, vol. 26(no. 9), 3649–57. [Google Scholar]
- Peng, B.; Xu, C.; Wang, S.; Zhang, Y.; Li, W. The Role of Connexin Hemichannels in Inflammatory Diseases. Biology (Basel) 2022, vol. 11(no. 2). [Google Scholar] [CrossRef]
- El-Ansary, A.; Al-Ayadhi, L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J Neuroinflammation 2014, vol. 11, pp. 189. [Google Scholar]
- Horder, J.; Petrinovic, M. M.; Mendez, M. A.; Bruns, A.; Takumi, T.; Spooren, W.; Barker, G. J.; Kunnecke, B.; Murphy, D. G. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry 2018, vol. 8(no. 1), pp. 106. [Google Scholar]
- Henstridge, C. M.; Tzioras, M.; Paolicelli, R. C. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front Cell Neurosci 2019, vol. 13, pp. 63. [Google Scholar]
- Hernandez, D. E.; Salvadores, N. A.; Moya-Alvarado, G.; Catalan, R. J.; Bronfman, F. C.; Court, F. A. Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J Cell Sci 2018, vol. 131(no. 22). [Google Scholar]
- Li, X.; Kong, H.; Wu, W.; Xiao, M.; Sun, X.; Hu, G. Aquaporin-4 maintains ependymal integrity in adult mice. Neuroscience 2009, vol. 162(no. 1), 67–77. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Saadoun, S.; Verkman, A. S. Aquaporins and cell migration. Pflugers Arch 2008, vol. 456(no. 4), 693–700. [Google Scholar]
- Umenishi, F.; Schrier, R.W. Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by the activation of MAPK pathways and hypertonicity-responsive element in the AQP1 gene. J Biol Chem 2003, vol. 278(no. 18), 15765–70. [Google Scholar]
- Zhang, D.; Vetrivel, L.; Verkman, A. S. Aquaporin deletion in mice reduces intraocular pressure and aqueous fluid production. J Gen Physiol 2002, vol. 119(no. 6), 561–9. [Google Scholar] [CrossRef] [PubMed]
- Speake, T.; Freeman, L. J.; Brown, P. D. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim Biophys Acta 2003, vol. 1609(no. 1), 80–6. [Google Scholar] [CrossRef]
- Bloch, O.; Papadopoulos, M. C.; Manley, G. T.; Verkman, A. S. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J Neurochem 2005, vol. 95(no. 1), 254–62. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Manley, G. T.; Krishna, S.; Verkman, A. S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 2004, vol. 18(no. 11), 1291–3. [Google Scholar] [CrossRef]
- Bloch, O.; Auguste, K. I.; Manley, G. T.; Verkman, A. S. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J Cereb Blood Flow Metab 2006, vol. 26(no. 12), 1527–37. [Google Scholar]
- Binder, D. K.; Yao, X.; Verkman, A. S.; Manley, G. T. Increased seizure duration in mice lacking aquaporin-4 water channels. Acta Neurochir Suppl 2006, vol. 96, 389–92. [Google Scholar]
- Padmawar, P.; Yao, X.; Bloch, O.; Manley, G. T.; Verkman, A. S. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Methods 2005, vol. 2(no. 11), 825–7. [Google Scholar] [CrossRef]
- Azizi, F.; Chan, W.K.; Ardalan, M. Aquaporins: Bridging Normal Brain Development and Neurodevelopmental Disorder Mechanisms. Dev Neurosci 2025, 1–15. [Google Scholar] [CrossRef]
- Skucas, V. A.; Mathews, I. B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A. M.; Verkman, A. S.; Hempstead, B. L.; Wood, M. A.; Binder, D. K.; Scharfman, H. E. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci 2011, vol. 31(no. 17), 6392–7. [Google Scholar]
- Fan, Y.; Liu, M.; Wu, X.; Wang, F.; Ding, J.; Chen, J.; Hu, G. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct Funct 2013, vol. 218(no. 1), 39–50. [Google Scholar]
- Yang, J.; Li, M. X.; Luo, Y.; Chen, T.; Liu, J.; Fang, P.; Jiang, B.; Hu, Z. L.; Jin, Y.; Chen, J. G.; Wang, F. Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology 2013, vol. 75, 213–22. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. K.; Wang, F.; Wang, W.; Luo, Y.; Wu, P. F.; Xiao, J. L.; Hu, Z. L.; Jin, Y.; Hu, G.; Chen, J. G. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology 2012, vol. 37(no. 8), 1867–78. [Google Scholar] [CrossRef]
- Bear, M. F.; Malenka, R. C. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 1994, vol. 4(no. 3), 389–99. [Google Scholar]
- Perea, M. Navarrete; Araque, A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 2009, vol. 32(no. 8), 421–31. [Google Scholar] [CrossRef]
- Ben Achour, S.; Pascual, O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int 2010, vol. 57(no. 4), 440–5. [Google Scholar]
- Zeng, X. N.; Sun, X. L.; Gao, L.; Fan, Y.; Ding, J. H.; Hu, G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci 2007, vol. 34(no. 1), 34–9. [Google Scholar]
- Papadopoulos, M. C.; Verkman, A. S. Aquaporin water channels in the nervous system. Nat Rev Neurosci 2013, vol. 14(no. 4), 265–77. [Google Scholar]
- Hubbard, A.; Szu, J. I.; Binder, D. K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res Bull 2018, vol. 136, 118–129. [Google Scholar]
- Scharfman, H. E.; Binder, D. K. Aquaporin-4 water channels and synaptic plasticity in the hippocampus. Neurochem Int 2013, vol. 63(no. 7), 702–11. [Google Scholar]
- Kang, H.; Schuman, E. M. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 1995, vol. 267(no. 5204), 1658–62. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Akaneya, Y.; Hata, Y.; Tsumoto, T. Long-term depression is not induced by low-frequency stimulation in rat visual cortex in vivo: a possible preventing role of endogenous brain-derived neurotrophic factor. J Neurosci 2003, vol. 23(no. 9), 3761–70. [Google Scholar] [CrossRef] [PubMed]
- Huber, V. J.; Tsujita, M.; Kwee, I. L.; Nakada, T. Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg Med Chem 2009, vol. 17(no. 1), 418–24. [Google Scholar]
- Igarashi, H.; Huber, V. J.; Tsujita, M.; Nakada, T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci 2011, vol. 32(no. 1), 113–6. [Google Scholar]
- Nishi, Y.; Toritsuka, M.; Takada, R.; Ishikawa, M.; Ishida, R.; Kayashima, Y.; Yamauchi, T.; Okumura, K.; Takeda, T.; Yamamuro, K.; Ikehara, M.; Noriyama, Y.; Kamikawa, K.; Murayama, S.; Ichikawa, O.; Nagata, H.; Okano, H.; Iwata, N.; Makinodan, M. Impaired synaptosome phagocytosis in macrophages of individuals with autism spectrum disorder. Mol Psychiatry 2025, vol. 30(no. 8), 3837–3845. [Google Scholar] [CrossRef]
- Chez, M. G.; Dowling, T.; Patel, P. B.; Khanna, P.; Kominsky, M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol 2007, vol. 36(no. 6), 361–5. [Google Scholar]
- Li, X.; Chauhan, A.; Sheikh, A. M.; Patil, S.; Chauhan, V.; Li, X. M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J Neuroimmunol 2009, vol. 207(no. 1-2), 111–6. [Google Scholar] [CrossRef]
- Wei, H.; Zou, H.; Sheikh, A. M.; Malik, M.; Dobkin, C.; Brown, W. T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation 2011, vol. 8, pp. 52. [Google Scholar]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis 2008, vol. 30(no. 3), 303–311. [Google Scholar]
- Enstrom, A.; Krakowiak, P.; Onore, C.; Pessah, I. N.; Hertz-Picciotto, I.; Hansen, R. L.; Van de Water, J. A.; Ashwood, P. Increased IgG4 levels in children with autism disorder. Brain Behav Immun 2009, vol. 23(no. 3), 389–95. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, M.; Onore, C. E.; Van de Water, J. A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun 2010, vol. 24(no. 1), 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Schauer, J.; Pessah, I. N.; Van de Water, J. Preliminary evidence of the in vitro effects of BDE-47 on innate immune responses in children with autism spectrum disorders. J Neuroimmunol 2009, vol. 208(no. 1-2), 130–5. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 2011, vol. 25(no. 1), 40–5. [Google Scholar] [CrossRef]
- Lampiasi, N.; Bonaventura, R.; Deidda, I.; Zito, F.; Russo, R. Inflammation and the Potential Implication of Macrophage-Microglia Polarization in Human ASD: An Overview. Int J Mol Sci 2023, vol. 24(no. 3). [Google Scholar]
- Li, H.; Xu, Y.; Li, W.; Zhang, L.; Zhang, X.; Li, B.; Chen, Y.; Wang, X.; Zhu, C. Novel insights into the immune cell landscape and gene signatures in autism spectrum disorder by bioinformatics and clinical analysis. Front Immunol 2022, vol. 13, pp. 1082950. [Google Scholar]
- Lopez-Cacho, M.; Gallardo, S.; Posada, M.; Aguerri, M.; Calzada, D.; Mayayo, T.; Lahoz, C.; Cardaba, B. Characterization of immune cell phenotypes in adults with autism spectrum disorders. J Investig Med 2016, vol. 64(no. 7), 1179–85. [Google Scholar]
- Chen, H. R.; Chen, C. W.; Mandhani, N.; Short-Miller, J. C.; Smucker, M. R.; Sun, Y. Y.; Kuan, C. Y. Monocytic Infiltrates Contribute to Autistic-like Behaviors in a Two-Hit Model of Neurodevelopmental Defects. J Neurosci 2020, vol. 40(no. 49), 9386–9400. [Google Scholar]
- Nadeem, A.; Ahmad, S. F.; Al-Harbi, N. O.; Attia, S. M.; Bakheet, S. A.; Ibrahim, K. E.; Alqahtani, F.; Alqinyah, M. Nrf2 activator, sulforaphane ameliorates autism-like symptoms through suppression of Th17 related signaling and rectification of oxidant-antioxidant imbalance in periphery and brain of BTBR T+tf/J mice. Behav Brain Res 2019, vol. 364, 213–224. [Google Scholar]
- Xiao, R.; Zhong, H.; Li, X.; Ma, Y.; Zhang, R.; Wang, L.; Zang, Z.; Fan, X. Abnormal Cerebellar Development Is Involved in Dystonia-Like Behaviors and Motor Dysfunction of Autistic BTBR Mice. Front Cell Dev Biol 2020, vol. 8, pp. 231. [Google Scholar] [CrossRef]
- Kwon, A. H.; Zhu, X.; Zhang, J.; Knoop, L. L.; Tharp, R.; Smeyne, R. J.; Eberhart, C. G.; Burger, P. C.; Baker, S. J. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 2001, vol. 29(no. 4), 404–11. [Google Scholar] [CrossRef] [PubMed]
- Ellul, P.; Maruani, A.; Peyre, H.; Vantalon, V.; Hoareau, D.; Tiercelin, H.; Rosenzwajg, M.; Klatzmann, D.; Delorme, R. Abnormal neutrophil-to-lymphocyte ratio in children with autism spectrum disorder and history of maternal immune activation. Sci Rep 2023, vol. 13(no. 1), pp. 22424. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S. F.; Attia, S. M.; Al-Ayadhi, L. Y.; Al-Harbi, N. O.; Bakheet, S. A. Dysregulated enzymatic antioxidant network in peripheral neutrophils and monocytes in children with autism. Prog Neuropsychopharmacol Biol Psychiatry 2019, vol. 88, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Boris, M.; Kaiser, C.C.; Goldblatt, A.; Elice, M.W.; Edelson, S.M.; Adams, J.B.; Feinstein, D.L. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J Neuroinflammation 2007, vol. 4, pp. 3. [Google Scholar] [CrossRef]
- Asadabadi, M.; Mohammadi, M. R.; Ghanizadeh, A.; Modabbernia, A.; Ashrafi, M.; Hassanzadeh, E.; Forghani, S.; Akhondzadeh, S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: a randomized, double-blind, placebo-controlled trial. Psychopharmacology (Berl) 2013, vol. 225(no. 1), 51–9. [Google Scholar] [CrossRef]
- Lim, S.; Lee, S. Chemical Modulators for Targeting Autism Spectrum Disorders: From Bench to Clinic. Molecules 2022, vol. 27(no. 16). [Google Scholar] [CrossRef]
- Jyonouchi, H. Autism spectrum disorder and a possible role of anti-inflammatory treatments: experience in the pediatric allergy/immunology clinic. Front Psychiatry 2024, vol. 15, pp. 1333717. [Google Scholar] [CrossRef]
- Gogolla, N.; Leblanc, J.J.; Quast, K.B.; Südhof, T.C.; Fagiolini, M.; Hensch, T. K. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 2009, vol. 1(no. 2), 172–81. [Google Scholar] [CrossRef]
- Tripathi, M. K.; Ojha, S. K.; Kartawy, M.; Hamoudi, W.; Choudhary, A.; Stern, S.; Aran, A.; Amal, H. The NO Answer for Autism Spectrum Disorder. Adv Sci (Weinh) 2023, vol. 10(no. 22), pp. e2205783. [Google Scholar] [CrossRef]
- Hegarty, J. P., 2nd; Weber, D. J.; Cirstea, C. M.; Beversdorf, D. Q. Cerebro-Cerebellar Functional Connectivity is Associated with Cerebellar Excitation-Inhibition Balance in Autism Spectrum Disorder. J Autism Dev Disord 2018, vol. 48(no. 10), 3460–3473. [Google Scholar] [CrossRef]
- Rubenstein, J. L.; Merzenich, M. M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2003, vol. 2(no. 5), 255–67. [Google Scholar] [CrossRef]
- Hussman, J. P. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord 2001, vol. 31(no. 2), 247–8. [Google Scholar]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol Psychiatry 2017, vol. 81(no. 10), 838–847. [Google Scholar] [CrossRef] [PubMed]
- Culotta, L.; Penzes, P. Exploring the mechanisms underlying excitation/inhibition imbalance in human iPSC-derived models of ASD. Mol Autism 2020, vol. 11(no. 1), pp. 32. [Google Scholar]
- Canitano, R.; Palumbi, R. Excitation/Inhibition Modulators in Autism Spectrum Disorder: Current Clinical Research. Front Neurosci 2021, vol. 15, pp. 753274. [Google Scholar]
- Leiner, H. C.; Leiner, A. L.; Dow, R. S. Does the cerebellum contribute to mental skills? Behav Neurosci 1986, vol. 100(no. 4), 443–54. [Google Scholar]
- Oberdick, J.; Sillitoe, R. V. Cerebellar zones: history, development, and function. Cerebellum 2011, vol. 10(no. 3), 301–6. [Google Scholar] [CrossRef]
- Prestori, F.; Mapelli, L.; D'Angelo, E. Diverse Neuron Properties and Complex Network Dynamics in the Cerebellar Cortical Inhibitory Circuit. Front Mol Neurosci 2019, vol. 12, pp. 267. [Google Scholar]
- Kandel, E. R. Principles of Neural Science, 5th ed.; McGraw-Hill, 2013. [Google Scholar]
- Voogd, J.; Glickstein, M. The anatomy of the cerebellum. Trends Neurosci 1998, vol. 21(no. 9), 370–5. [Google Scholar]
- Watson, C. The Spinal Cord: A Christopher and Dana Reeve Foundation Text and Atlas; 2009. [Google Scholar]
- Voogd, J.; Barmack, N. H. Oculomotor cerebellum. Prog Brain Res 2006, vol. 151, 231–68. [Google Scholar]
- Ruigrok, J. Ins and outs of cerebellar modules. Cerebellum 2011, vol. 10(no. 3), 464–74. [Google Scholar]
- Ito, M. Error detection and representation in the olivo-cerebellar system. Front Neural Circuits 2013, vol. 7, pp. 1. [Google Scholar]
- Bengtsson, F.; Jorntell, H. Specific relationship between excitatory inputs and climbing fiber receptive fields in deep cerebellar nuclear neurons. PLoS One 2014, vol. 9(no. 1), pp. e84616. [Google Scholar] [CrossRef] [PubMed]
- Snider, R. S.; Maiti, A. Cerebellar contributions to the Papez circuit. J Neurosci Res 1976, vol. 2(no. 2), 133–46. [Google Scholar]
- Anand, K.; Malhotra, C. L.; Singh, B.; Dua, S. Cerebellar projections to limbic system. J Neurophysiol 1959, vol. 22(no. 4), 451–7. [Google Scholar]
- Terburg, T.; van Honk, J.; Schutter, D. Doubling down on dual systems: A cerebellum-amygdala route towards action- and outcome-based social and affective behavior. Cortex 2024, vol. 173, 175–186. [Google Scholar] [CrossRef]
- Froula, J. M.; Hastings, S. D.; Krook-Magnuson, E. The little brain and the seahorse: Cerebellar-hippocampal interactions. Front Syst Neurosci 2023, vol. 17, pp. 1158492. [Google Scholar]
- Morris, J. S.; Frith, C. D.; Perrett, D. I.; Rowland, D.; Young, A. W.; Calder, A. J.; Dolan, R. J. A differential neural response in the human amygdala to fearful and happy facial expressions. Nature 1996, vol. 383(no. 6603), 812–5. [Google Scholar] [CrossRef]
- Ernst, M.; Brol, A. E.; Gratz, M.; Ritter, C.; Bingel, U.; Schlamann, M.; Maderwald, S.; Quick, H. H.; Merz, C. J.; Timmann, D. The cerebellum is involved in processing of predictions and prediction errors in a fear conditioning paradigm. Elife 2019, vol. 8. [Google Scholar] [CrossRef]
- Rogers, T. D.; Dickson, P. E.; Heck, D. H.; Goldowitz, D.; Mittleman, G.; Blaha, C. D. Connecting the dots of the cerebro-cerebellar role in cognitive function: neuronal pathways for cerebellar modulation of dopamine release in the prefrontal cortex. Synapse 2011, vol. 65(no. 11), 1204–12. [Google Scholar] [CrossRef]
- Mittleman, G.; Goldowitz, D.; Heck, D. H.; Blaha, C. D. Cerebellar modulation of frontal cortex dopamine efflux in mice: relevance to autism and schizophrenia. Synapse 2008, vol. 62(no. 7), 544–50. [Google Scholar] [CrossRef]
- Watson, T. C.; Becker, N.; Apps, R.; Jones, M. W. Back to front: cerebellar connections and interactions with the prefrontal cortex. Front Syst Neurosci 2014, vol. 8, pp. 4. [Google Scholar]
- Kelly, R. M.; Strick, P. L. Cerebellar loops with motor cortex and prefrontal cortex of a nonhuman primate. J Neurosci 2003, vol. 23(no. 23), 8432–44. [Google Scholar]
- Fatemi, S. H.; Stary, J. M.; Halt, A. R.; Realmuto, G. R. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J Autism Dev Disord 2001, vol. 31(no. 6), 529–35. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.; Soghomonian, J. J.; Blatt, G. J. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol 2007, vol. 113(no. 5), 559–68. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J. J.; Blatt, G. J. Increased GAD67 mRNA expression in cerebellar interneurons in autism: implications for Purkinje cell dysfunction. J Neurosci Res 2008, vol. 86(no. 3), 525–30. [Google Scholar]
- Chan-Palay, V.; Palay, S.L.; Brown, J.T.; Van Itallie, C. Sagittal organization of olivocerebellar and reticulocerebellar projections: autoradiographic studies with 35S-methionine. Exp Brain Res 1977, vol. 30(no. 4), 561–76. [Google Scholar] [CrossRef]
- R. L. Llinas, E.; Makarenko, V. The Olivo-Cerebellar Circuit as a Universal Motor Control System. IEEE Journal of Oceanic Engineering 2004, vol. 29(no. 3), 631–639. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J. J.; Blatt, G. J. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: an in situ hybridization study. Autism Res 2009, vol. 2(no. 1), 50–9. [Google Scholar]
- Chugani, C.; Muzik, O.; Rothermel, R.; Behen, M.; Chakraborty, P.; Mangner, T.; da Silva, E. A.; Chugani, H. T. Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann Neurol 1997, vol. 42(no. 4), 666–9. [Google Scholar] [CrossRef]
- Chugani, D. C. Role of altered brain serotonin mechanisms in autism. Mol Psychiatry 2002, vol. 7 Suppl 2, S16–7. [Google Scholar] [CrossRef]
- Vichier-Guerre, C.; Parker, M.; Pomerantz, Y.; Finnell, R. H.; Cabrera, R. M. Impact of selective serotonin reuptake inhibitors on neural crest stem cell formation. Toxicol Lett 2017, vol. 281, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Fricker, A. D.; Rios, C.; Devi, L. A.; Gomes, I. Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Brain Res Mol Brain Res 2005, vol. 138(no. 2), 228–35. [Google Scholar]
- Khozhai, L. I.; Otellin, V. A. Synaptogenesis in the dorsal raphe nucleus of rat medulla oblongata in serotonin deficiency. Morfologiia 2012, vol. 142(no. 6), 20–4. [Google Scholar]
- Rice, D. S.; Curran, T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci 2001, vol. 24, 1005–39. [Google Scholar]
- Fatemi, S. H.; Snow, A. V.; Stary, J. M.; Araghi-Niknam, M.; Reutiman, T. J.; Lee, S.; Brooks, A. I.; Pearce, D. A. Reelin signaling is impaired in autism. Biol Psychiatry 2005, vol. 57(no. 7), 777–87. [Google Scholar] [CrossRef]
- Scala, M.; Grasso, E. A.; Di Cara, G.; Riva, A.; Striano, P.; Verrotti, A. The Pathophysiological Link Between Reelin and Autism: Overview and New Insights. Front Genet 2022, vol. 13, pp. 869002. [Google Scholar]
- Mariani, J.; Crepel, F.; Mikoshiba, K.; Changeux, J. P.; Sotelo, C. Anatomical, physiological and biochemical studies of the cerebellum from Reeler mutant mouse. Philos Trans R Soc Lond B Biol Sci 1977, vol. 281(no. 978), 1–28. [Google Scholar]
- Hong, S. E.; Shugart, Y. Y.; Huang, D. T.; Shahwan, S. A.; Grant, P. E.; Hourihane, J. O.; Martin, N. D.; Walsh, C. A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 2000, vol. 26(no. 1), 93–6. [Google Scholar]
- Parellada, M.; San Jose Caceres, A.; Palmer, M.; Delorme, R.; Jones, E. J. H.; Parr, J. R.; Anagnostou, E.; Murphy, D. G. M.; Loth, E.; Wang, P. P.; Charman, T.; Strydom, A.; Arango, C. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy, Safety, and Tolerability of Arbaclofen Administered for the Treatment of Social Function in Children and Adolescents With Autism Spectrum Disorders: Study Protocol for AIMS-2-TRIALS-CT1. Front Psychiatry 2021, vol. 12, pp. 701729. [Google Scholar]
- Jiang, C. C.; Lin, L. S.; Long, S.; Ke, X. Y.; Fukunaga, K.; Lu, Y. M.; Han, F. Signalling pathways in autism spectrum disorder: mechanisms and therapeutic implications. Signal Transduct Target Ther 2022, vol. 7(no. 1), pp. 229. [Google Scholar] [CrossRef]
- Aguilar-Valles, A.; Matta-Camacho, E.; Khoutorsky, A.; Gkogkas, C.; Nader, K.; Lacaille, J. C.; Sonenberg, N. Inhibition of Group I Metabotropic Glutamate Receptors Reverses Autistic-Like Phenotypes Caused by Deficiency of the Translation Repressor eIF4E Binding Protein 2. J Neurosci 2015, vol. 35(no. 31), 11125–32. [Google Scholar] [CrossRef] [PubMed]
- Carta, I.; Chen, C.H.; Schott, A.L.; Dorizan, S.; Khodakhah, K. Cerebellar modulation of the reward circuitry and social behavior. Science 2019, vol. 363(no. 6424). [Google Scholar] [CrossRef] [PubMed]
- Vacher, C. M.; Lacaille, H.; O'Reilly, J. J.; Salzbank, J.; Bakalar, D.; Sebaoui, S.; Liere, P.; Clarkson-Paredes, C.; Sasaki, T.; Sathyanesan, A.; Kratimenos, P.; Ellegood, J.; Lerch, J. P.; Imamura, Y.; Popratiloff, A.; Hashimoto-Torii, K.; Gallo, V.; Schumacher, M.; Penn, A. A. Placental endocrine function shapes cerebellar development and social behavior. Nat Neurosci 2021, vol. 24(no. 10), 1392–1401. [Google Scholar] [CrossRef]
- Kuemerle, B.; Zanjani, H.; Joyner, A.; Herrup, K. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci 1997, vol. 17(no. 20), 7881–9. [Google Scholar] [CrossRef] [PubMed]
- Brielmaier, J.; Matteson, P. G.; Silverman, J. L.; Senerth, J. M.; Kelly, S.; Genestine, M.; Millonig, J. H.; DiCicco-Bloom, E.; Crawley, J. N. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One 2012, vol. 7(no. 7), pp. e40914. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol 2015, vol. 28(no. 2), 91–102. [Google Scholar] [CrossRef]
- Wang, H.; Doering, L. C. Reversing autism by targeting downstream mTOR signaling. Front Cell Neurosci 2013, vol. 7, pp. 28. [Google Scholar] [CrossRef]
- Thomas, S. D.; Jha, N. K.; Ojha, S.; Sadek, B. mTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, vol. 28(no. 4). [Google Scholar]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P. H. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab 2010, vol. 30(no. 12), 1939–50. [Google Scholar]
- Zhao, Z.; Hu, J.; Gao, X.; Liang, H.; Liu, Z. Activation of AMPK attenuates lipopolysaccharide-impaired integrity and function of blood-brain barrier in human brain microvascular endothelial cells. Exp Mol Pathol 2014, vol. 97(no. 3), 386–92. [Google Scholar]
- Rajapakse, A. G.; Yepuri, G.; Carvas, J. M.; Stein, S.; Matter, C. M.; Scerri, I.; Ruffieux, J.; Montani, J. P.; Ming, X. F.; Yang, Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS One 2011, vol. 6(no. 4), pp. e19237. [Google Scholar]
- Li, Z.; Jagadapillai, R.; Gozal, E.; Barnes, G. Deletion of Semaphorin 3F in Interneurons Is Associated with Decreased GABAergic Neurons, Autism-like Behavior, and Increased Oxidative Stress Cascades. Mol Neurobiol 2019, vol. 56(no. 8), 5520–5538. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D. J.; Ramesh, V.; Silva, A. J. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med 2008, vol. 14(no. 8), 843–8. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C. G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D. B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; Major, F.; Lasko, P.; Ruggero, D.; Nader, K.; Lacaille, J. C.; Sonenberg, N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, vol. 493(no. 7432), 371–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; de Theije, C.G.M.; da Silva, S.L.; Abbring, S.; van der Horst, H.; Broersen, L.M.; Willemsen, L.; Kas, M.; Garssen, J.; Kraneveld, A.D. Dietary interventions that reduce mTOR activity rescue autistic-like behavioral deficits in mice. Brain Behav Immun 2017, vol. 59, 273–287. [Google Scholar] [CrossRef]
- Lin, A. L.; Zheng, W.; Halloran, J. J.; Burbank, R. R.; Hussong, S. A.; Hart, M. J.; Javors, M.; Shih, Y. Y.; Muir, E.; Solano Fonseca, R.; Strong, R.; Richardson, A. G.; Lechleiter, J. D.; Fox, P. T.; Galvan, V. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer's disease. J Cereb Blood Flow Metab 2013, vol. 33(no. 9), 1412–21. [Google Scholar]
- Lin, L.; Jahrling, J. B.; Zhang, W.; DeRosa, N.; Bakshi, V.; Romero, P.; Galvan, V.; Richardson, A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer's disease. J Cereb Blood Flow Metab 2017, vol. 37(no. 1), 217–226. [Google Scholar] [CrossRef]
- Majumder, S.; Caccamo, A.; Medina, D. X.; Benavides, A. D.; Javors, M. A.; Kraig, E.; Strong, R.; Richardson, A.; Oddo, S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell 2012, vol. 11(no. 2), 326–35. [Google Scholar]
- Spilman, P.; Podlutskaya, N.; Hart, M. J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One 2010, vol. 5(no. 4), pp. e9979. [Google Scholar]
- Van Skike, E.; Jahrling, J. B.; Olson, A. B.; Sayre, N. L.; Hussong, S. A.; Ungvari, Z.; Lechleiter, J. D.; Galvan, V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol 2018, vol. 314(no. 4), H693–H703. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S. S.; Sahin, M.; Bolton, P.; Ploubidis, G. B.; Humphrey, A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol 2008, vol. 23(no. 5), 520–5. [Google Scholar] [CrossRef] [PubMed]
- Ertan, G.; Arulrajah, S.; Tekes, A.; Jordan, L.; Huisman, T. A. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J Neuroradiol 2010, vol. 37(no. 4), 231–8. [Google Scholar]
- Weber, A. M.; Egelhoff, J. C.; McKellop, J. M.; Franz, D. N. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord 2000, vol. 30(no. 6), 511–7. [Google Scholar] [CrossRef]
- Eluvathingal, T. J.; Behen, M. E.; Chugani, H. T.; Janisse, J.; Bernardi, B.; Chakraborty, P.; Juhasz, C.; Muzik, O.; Chugani, D. C. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol 2006, vol. 21(no. 10), 846–51. [Google Scholar]
- Tsai, T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A. R.; Leech, J. M.; Steinberg, J.; Crawley, J. N.; Regehr, W. G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, vol. 488(no. 7413), 647–51. [Google Scholar] [CrossRef]
- Talkowski, E.; Rosenfeld, J. A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A. M.; Pereira, S.; Ruderfer, D.; Kirby, A.; Ripke, S.; Harris, D. J.; Lee, J. H.; Ha, K.; Kim, H. G.; Solomon, B. D.; Gropman, A. L.; Lucente, D.; Sims, K.; Ohsumi, T. K.; Borowsky, M. L.; Loranger, S.; Quade, B.; Lage, K.; Miles, J.; Wu, B. L.; Shen, Y.; Neale, B.; Shaffer, L. G.; Daly, M. J.; Morton, C. C.; Gusella, J. F. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, vol. 149(no. 3), 525–37. [Google Scholar] [CrossRef]
- Neale, M.; Kou, Y.; Liu, L.; Ma'ayan, A.; Samocha, K. E.; Sabo, A.; Lin, C. F.; Stevens, C.; Wang, L. S.; Makarov, V.; Polak, P.; Yoon, S.; Maguire, J.; Crawford, E. L.; Campbell, N. G.; Geller, E. T.; Valladares, O.; Schafer, C.; Liu, H.; Zhao, T.; Cai, G.; Lihm, J.; Dannenfelser, R.; Jabado, O.; Peralta, Z.; Nagaswamy, U.; Muzny, D.; Reid, J. G.; Newsham, I.; Wu, Y.; Lewis, L.; Han, Y.; Voight, B. F.; Lim, E.; Rossin, E.; Kirby, A.; Flannick, J.; Fromer, M.; Shakir, K.; Fennell, T.; Garimella, K.; Banks, E.; Poplin, R.; Gabriel, S.; DePristo, M.; Wimbish, J. R.; Boone, B. E.; Levy, S. E.; Betancur, C.; Sunyaev, S.; Boerwinkle, E.; Buxbaum, J. D.; Cook, E. H., Jr.; Devlin, B.; Gibbs, R. A.; Roeder, K.; Schellenberg, G. D.; Sutcliffe, J. S.; Daly, M. J. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, vol. 485(no. 7397), 242–5. [Google Scholar] [CrossRef]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; Sheridan, S. D.; Stortchevoi, A.; Kellis, M.; Haggarty, S. J.; Katsanis, N.; Gusella, J. F.; Talkowski, M. E. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A 2014, vol. 111(no. 42), E4468–77. [Google Scholar]
- Cotney, J.; Muhle, R. A.; Sanders, S. J.; Liu, L.; Willsey, A. J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; Reilly, S. K.; Tebbenkamp, A. T.; Bichsel, C.; Pletikos, M.; Sestan, N.; Roeder, K.; State, M. W.; Devlin, B.; Noonan, J. P. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 2015, vol. 6, pp. 6404. [Google Scholar]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H. A.; Coe, B. P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A. T.; Schuurs-Hoeijmakers, J. H.; Fichera, M.; Bosco, P.; Buono, S.; Alberti, A.; Failla, P.; Peeters, H.; Steyaert, J.; Vissers, L.; Francescatto, L.; Mefford, H. C.; Rosenfeld, J. A.; Bakken, T.; O'Roak, B. J.; Pawlus, M.; Moon, R.; Shendure, J.; Amaral, D. G.; Lein, E.; Rankin, J.; Romano, C.; de Vries, B. B. A.; Katsanis, N.; Eichler, E. E. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, vol. 158(no. 2), 263–276. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Katayama, Y.; Kakegawa, W.; Ino, D.; Nishiyama, M.; Yuzaki, M.; Nakayama, K. I. The autism-associated protein CHD8 is required for cerebellar development and motor function. Cell Rep 2021, vol. 35(no. 1), pp. 108932. [Google Scholar] [CrossRef] [PubMed]
- Millen, K. J.; Wurst, W.; Herrup, K.; Joyner, A. L. Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development 1994, vol. 120(no. 3), 695–706. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.; Kollu, R.; Banerjee-Basu, S. AutDB: a gene reference resource for autism research. Nucleic Acids Res 2009, vol. 37, no. Database issue, D832–6. [Google Scholar] [CrossRef]
- Xu, M.; Li, J. R.; Huang, Y.; Zhao, M.; Tang, X.; Wei, L. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res 2012, vol. 40, no. Database issue, D1016–22. [Google Scholar] [CrossRef]
- Cheh, A.; Millonig, J. H.; Roselli, L. M.; Ming, X.; Jacobsen, E.; Kamdar, S.; Wagner, G. C. En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res 2006, vol. 1116(no. 1), 166–76. [Google Scholar] [CrossRef]
- Boukhtouche, F.; Janmaat, S.; Vodjdani, G.; Gautheron, V.; Mallet, J.; Dusart, I.; Mariani, J. Retinoid-related orphan receptor alpha controls the early steps of Purkinje cell dendritic differentiation. J Neurosci 2006, vol. 26(no. 5), 1531–8. [Google Scholar]
- Gold, A.; Gent, P. M.; Hamilton, B. A. ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain Res 2007, vol. 1140, 19–25. [Google Scholar]
- Shu, W.; Cho, J. Y.; Jiang, Y.; Zhang, M.; Weisz, D.; Elder, G. A.; Schmeidler, J.; De Gasperi, R.; Sosa, M. A.; Rabidou, D.; Santucci, A. C.; Perl, D.; Morrisey, E.; Buxbaum, J. D. Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci U S A 2005, vol. 102(no. 27), 9643–8. [Google Scholar]
- Fujita, E.; Tanabe, Y.; Shiota, A.; Ueda, M.; Suwa, K.; Momoi, M. Y.; Momoi, T. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci U S A 2008, vol. 105(no. 8), 3117–22. [Google Scholar]
- Groszer, M.; Keays, D. A.; Deacon, R. M.; de Bono, J. P.; Prasad-Mulcare, S.; Gaub, S.; Baum, M. G.; French, C. A.; Nicod, J.; Coventry, J. A.; Enard, W.; Fray, M.; Brown, S. D.; Nolan, P. M.; Paabo, S.; Channon, K. M.; Costa, R. M.; Eilers, J.; Ehret, G.; Rawlins, J. N.; Fisher, S. E. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol 2008, vol. 18(no. 5), 354–62. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Hu, C.; Li, H.; Xu, Z.; Lei, P.; Luo, X.; Hao, Y. JUN and PDGFRA as Crucial Candidate Genes for Childhood Autism Spectrum Disorder. Front Neuroinform 2022, vol. 16, pp. 800079. [Google Scholar] [CrossRef]
- Byrns, C. N.; Perlegos, A. E.; Miller, K. N.; Jin, Z.; Carranza, F. R.; Manchandra, P.; Beveridge, C. H.; Randolph, C. E.; Chaluvadi, V. S.; Zhang, S. L.; Srinivasan, A. R.; Bennett, F. C.; Sehgal, A.; Adams, P. D.; Chopra, G.; Bonini, N. M. Senescent glia link mitochondrial dysfunction and lipid accumulation. Nature 2024, vol. 630(no. 8016), 475–483. [Google Scholar] [CrossRef]
- Liao, B.; Geng, L.; Zhang, F.; Shu, L.; Wei, L.; Yeung, P. K. K.; Lam, K. S. L.; Chung, S. K.; Chang, J.; Vanhoutte, P. M.; Xu, A.; Wang, K.; Hoo, R. L. C. Adipocyte fatty acid-binding protein exacerbates cerebral ischaemia injury by disrupting the blood-brain barrier. Eur Heart J 2020, vol. 41(no. 33), 3169–3180. [Google Scholar] [CrossRef]
- Sathyanesan, A.; Zhou, J.; Scafidi, J.; Heck, D. H.; Sillitoe, R. V.; Gallo, V. Emerging connections between cerebellar development, behaviour and complex brain disorders. Nat Rev Neurosci 2019, vol. 20(no. 5), 298–313. [Google Scholar] [CrossRef]
Figure 1.
ASD-associated cerebellar alterations that may lead to ASD symptoms development. (Created with BioRender.com).
Figure 1.
ASD-associated cerebellar alterations that may lead to ASD symptoms development. (Created with BioRender.com).
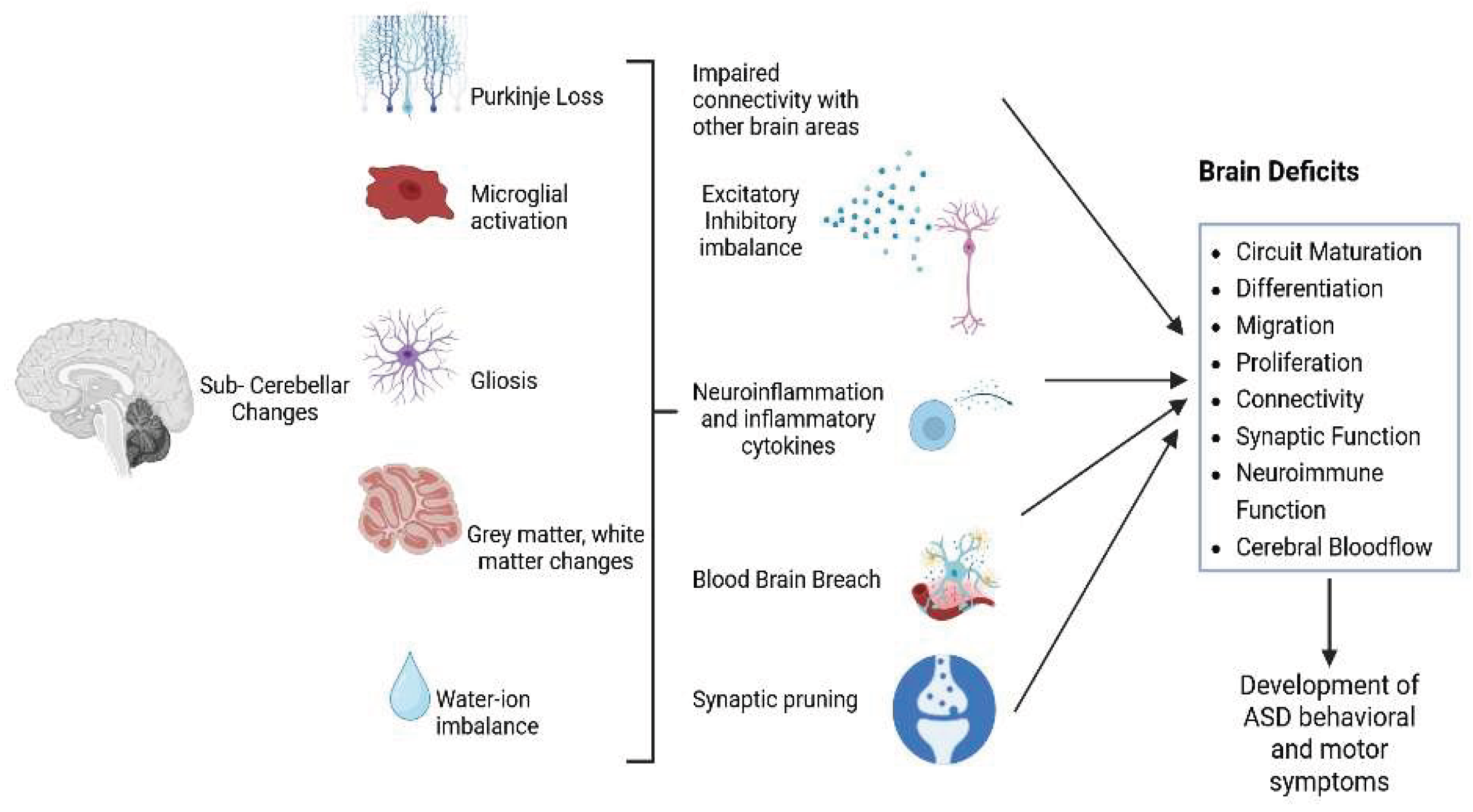
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



