
Submitted:
13 January 2026
Posted:
14 January 2026
You are already at the latest version
Abstract
Cancer remains one of the most significant health challenges facing humanity today. Extensive oncological research has demonstrated that cancer progression is not solely driven by malignant cells but also by the tumor microenvironment (TME), which plays a crucial role in tumor development, immune evasion and metastasis. As a result, the TME has emerged as a promising therapeutic target. Nanotechnology has revolutionized cancer diagnosis and treatment, with metallic nanoparticles (mNPs) being extensively studied. However, their effects on the TME remain poorly understood. While some molecular pathways through which mNPs influence the TME have been identified, these findings likely represent only a small fraction of the underlying mechanisms, as analyzed in this review. Furthermore, a major challenge in studying these interactions is the lack of physiologically relevant models, as currently available cell culture and in vivo systems often fail to accurately replicate the complex and dynamic interactions of the TME. These limitations underscore the urgent need for more comprehensive research to establish the TME as a viable therapeutic target for treatment strategies involving NPs. Specifically, a deeper understanding of how mNPs interact with the TME at multiple levels, including immune modulations, stromal remodeling and metabolic reprogramming, is essential toward optimizing the therapeutic potential of mNPs in cancer care.
Keywords:
metallic nanoparticles
; cancer
; tumor microenvironment (TME)
; cancer-associated fibroblasts (CAFs)
; tumor-associated macrophages (TAMs)
; nanoparticle-cell interactions
Introduction
The focus of this review is to provide a comprehensive and up-to-date overview of the complex interactions between metallic nanoparticles (mNPs) and various cell types within the TME. Understanding these interactions is critical, as the TME influences tumor behavior and response to therapy. By examining mNP interactions with stromal and immune components of the TME, as well as the subsequent effects these altered cells exert on cancer cells, this review scrutinize the potential of mNPs to modulate tumor progression, reshape intercellular communication and improve therapeutic outcomes.
1.1. Cancer and The Tumor Microenvironment
Cancer has recently and appropriately been described as a disease of uncontrolled proliferation by transformed cells subjected to Darwinian evolution by natural selection [1], resulting in complex ecosystems that emerge and evolve under robust selective pressure. This pressure promotes the diversification of both malignant and non-malignant cells, culminating in a significant degree of intratumoral heterogeneity, even across distinct regions of the same tumor or along its temporal evolution [2,3]. Taking into account intratumoral heterogeneity and comparing it with intertumoral heterogeneity, which can be anticipated as higher, highlights the profound variability and complexity in tumor biology [4]. Although there are numerous classified cancer types, each exhibiting distinct molecular mechanisms driving carcinogenesis and varying metastatic dissemination patterns [5], they all share a common set of biological capabilities known as the “Hallmarks of Cancer” [6]. As our understanding of cancer mechanisms continues to advance, additional facets of the disease are emerging, refining this conceptualization. Currently, the fourteen hallmarks provide a comprehensive overview: sustaining proliferative signaling; evading growth suppressors; non-mutational epigenetic reprogramming; avoiding immune destruction; enabling replicative immortality; tumor-promoting inflammation; polymorphic microbiomes; activating invasion and metastasis; inducing/accessing vasculature; senescent cells; genome instability and mutation; resisting cell death; deregulating cellular metabolism; and unlocking cellular plasticity. It is worth mentioning that some “Hallmarks of Cancer” are not met by hematological malignancies such as leukemia or lymphoma, as they do not form a solid mass (primary tumor) [7]. Furthermore, despite significant progress in cancer research, the foundational origins of these diseases remain largely and unfortunately enigmatic [8]. Based on this statement and in alignment with the strategic principles articulated by Sun Tzu (The Art of War), “an inadequate comprehension of the adversary leads to a scenario where every victory obtained is counterbalanced by an equivalent defeat”. Consequently, in addition to the development of new therapeutic approaches, one of the main objectives of cancer research must be to thoroughly and precisely understand this devastating disease, encompassing all research fields.
Despite significant advances in our understanding of the TME, some researchers still mistakenly perceive tumors as mere collections of cancer cells, failing to grasp their full complexity [9]. This narrow perspective overlooks the fact that tumor tissue is not just a cluster of malignant cells but a highly dynamic and heterogeneous ecosystem composed of infiltrating and resident host cells (e.g., stromal fibroblasts, endothelial cells and immune cells such as: macrophages, lymphocytes, and microglia), secreted factors (including cytokines, chemokines, growth factors, inflammatory mediators, matrix-remodeling enzymes, etc.) and extracellular matrix (ECM) components (such as collagen, fibronectin, hyaluronan and laminin), all of which vary across tumor types [10] (Figure 1). Notably, tumor cells actively reshape their microenvironment, inducing profound molecular, cellular and structural changes that promote tumor growth and progression [11], Through intricate signaling networks, cancer cells manipulate both cellular and non-cellular components of the TME, recruiting and reprogramming non-malignant host cells while remodeling the vasculature and ECM to sustain proliferation, evade immune surveillance and enhance metastasis [12]. Emerging evidence continues to reveal additional layers of complexity in these interactions, emphasizing the need for a comprehensive analysis to fully elucidate tumor progression mechanisms and identify novel therapeutic targets. In this context, the above-mentioned intercellular communication within the TME is orchestrated through multiple mechanisms, including direct cell-cell contact and paracrine signaling [13].
TME cells and their secreted molecules play critical roles in cancer pathogenesis, making them attractive therapeutic targets. While most cancer therapies have traditionally focused on directly targeting tumor cells, the genetic stability of stromal cells within the TME renders them less prone to developing therapeutic resistance [14]. Additionally, given the extensive heterogeneity observed in cancer cells at multiple levels, targeting the TME presents a promising alternative strategy to overcome resistance and improve treatment efficacy [15].
Finaly, to get a more comprehensive information on the TME and their elements, readers are encouraged to search for other review articles [16].
1.2. Cancer Associated Fibroblast
CAFs in solid tumors can be defined as a heterogeneous group of mesenchymal cells associated to tumor tissues [17] (Figure 2). CAFs are a highly heterogeneous population, originating primarily from the activation of normal fibroblasts near cancer cells [18] and undergoing distinct phenotypic and epigenetic alterations. The intrinsic heterogenicity of CAFs is further shaped by their numerous cellular origins and diverse modes of activation [19]. Interactions between CAFs and cancer cells are bidirectional, reinforcing tumor progression through reciprocal signaling [20]. Unlike normal fibroblasts, CAFs remain perpetually activated [21], secreting a wide array of cytokines, chemokines, growth factors, enzymes and ECM components [22]. These factors remodel the TME, promoting cancer cell survival, angiogenesis, therapy resistance, invasion and metastasis [23,24]. Within the tumor tissue, CAFs modulate immune cell recruitment and function, fostering an immunosuppressive milieu that facilitates tumor immune evasion [25]. Moreover, CAFs are known drivers of the epithelial-to-mesenchymal transition (EMT) process in cancer cells. In EMT, tumor cells undergo E-cadherin repression, resulting in loss of cell-cell junctions and apical-basal polarity, resulting in a motile mesenchymal-like cell phenotype [26]. This transformation enhances the metastatic potential of malignant cells [27]. In contrast, normal fibroblasts favor the maintenance of epithelial-like phenotype of tumor cells that suppresses metastasis [28]. One of the most intriguing properties of CAFs is their ability to co-travel with tumor cells in the bloodstream, effectively bringing their own “soil” to distant metastatic sites and enhancing extravasation and colonization [29]. The presence of CAFs in the TME is associated with poor prognosis in multiple cancers [30]. With the advent of single-cell sequencing, the complexity of CAFs biology during tumor evolution and across different tumor entities has become increasingly apparent [31]. Although initially perceived as a homogenous cell population, CAFs are now recognized as a diverse mixture of fibroblast phenotypes with distinct behaviors, including both tumor-promoting and tumor-restraining subtypes [32,33]. Despite the proven existence of CAF diversity in both preclinical and clinical settings, the context-dependent roles of the various CAF populations and their interchangeable plasticity remain largely unknown. On one hand, CAF heterogeneity and plasticity pose challenges for therapeutic targeting. On the other hand, these same characteristics also provide promising opportunities for novel therapeutic interventions [34]. Hence, a deeper understanding of CAF-mediated interactions within the TME is essential for developing strategies aimed at disrupting tumor-stroma crosstalk and overcoming treatment resistance.
1.3. Tumor Associated Macrophages
Macrophages infiltrating tumor tissues or residing within the microenvironment of solid tumors are classified as TAMs. Extensive evidence supports their causal role in cancer initiation, given their central function as key mediators of inflammation [35]. Moreover, TAMs contribute to tumor progression by promoting growth, facilitating angiogenesis, modulating immune responses, inducing chemoresistance, and enhancing metastasis [36]. Initially, macrophages involved in early cancer-associated inflammation exhibit immune activation; nonetheless, as tumors become established, these macrophages undergo reprogramming, adopting a protumoral phenotype [37]. Macrophages, including TAMs, display remarkable plasticity, adopting distinct and often opposing phenotypes in response to their microenvironment. They are traditionally classified into M1 (classically activated) and M2 (alternatively activated) macrophages. M1-macrophages promote inflammation and anti-tumor immunity, whereas M2-macrophages exert immunosuppressive functions, facilitating tissue repair and tumor progression [38]. These subsets differ in surface markers, metabolic profiles, and gene expression patterns [39]. Transition between M1 (anti-tumor) and M2 (pro-tumor) states, a process known as “macrophage polarization”, is orchestrated by tumor (and stroma) derived signals, including cytokines and growth factors [40]. However, TAMs rarely conform strictly to either phenotype, highlighting their functional complexity beyond this binary classification [41]. It was long believed that TAMs were exclusively recruited from outside the tumor through chemotactic signals and originated from local monocytic precursors. Nevertheless, new evidence suggests that, at least certain tumors, resident macrophages derived from embryonic tissues can also infiltrate the TME, highlighting a previously underrecognized source of TAMs [42]. TAMs mainly accumulate at the invasive front and within hypoxic, avascular regions of the tumor [43]. Furthermore, a subset of TAMs localizes along the abluminal side of blood vessels [44]. Notably, clinical observations consistently show that the accumulation of macrophages in the TME is strongly correlated with poorer disease outcomes [45]. Among all TAMs roles, two will be discussed in greater detail, as they are critical determinants of patient life expectancy and interconnected with angiogenesis and metastasis. On one hand, tumor vasculature supplies primary tumors with nutrients and oxygen while also serving as a primary route for metastasis. TAMs are critical regulators of the angiogenic switch, forming clusters in intratumoral regions and invasive fronts, which are hotspots for both angiogenesis and metastasis [46]. TAMs influence the formation of new tumor vessels and stimulate the remodeling of established vasculature into a more tortuous and leaky form that favors tumor dissemination [47]. In contrast, the absence of TAMs significantly reduces vessel density by 40% [47]. On the other hand, TAMs play a crucial role in the formation of pre-metastatic niches (PMNs), which are organ-specific microenvironments primed by primary tumors to facilitate metastatic colonization before tumor cells even arrive [48]. TAMs are considered major drivers of PMNs establishment, preparing secondary sites for tumor cell invasion through immune modulation and ECM remodeling [49]. Beyond PMNs formation, TAMs regulate EMT [50], by secreting a wide array of proteases and enzymes involved in ECM degradation and remodeling, thereby facilitating tumor cell detachment and migration [51]. Through interactions with ECM components such as fibronectin and vitronectin, tumor cells migrate along ECM fibers toward tumor vasculature [52]. Additionally, TAM-derived secreted protein acidic and rich in cysteine (SPARC) generates traction forces along ECM fibers, accelerating tumor cell movement within the stroma and toward blood vessels [53,54], thus, enhancing tumor cell intravasation and dissemination [52].. Tumor-immune hybrid cells influenced by TAMs are crucial to cancer progression [55]. These hybrid cells have been associated with disease stage, patient survival, and treatment response, indicating their potential clinical significance [56,57]. Finally, once lodged in capillaries of metastatic sites, tumor cells rely on TAM assistance to attach to endothelial walls and migrate into the surrounding tissue, further promoting metastatic outgrowth [58]. Remarkably, TAMs serve as key regulators of numerous processes within TME (among other important functions), many of which critically influence patient prognosis and, consequently, TAMs must be considered as primary targets for the development of advanced therapeutic strategies.
1.4. Immune cells of the TME
In the complex landscape of the TME, various immune cells interact to influence cancer progression and response to therapy. In this regard, the most evident examples are, Dendritic Cells (DC), Natural Killer (NK) cells and T cells, which represent members of the innate and adaptive immune systems.
Dendritic cells (DCs) are a diverse group of specialized antigen-presenting cells (APCs) derived from hematopoietic stem cells (HSCs) in the bone marrow, forming a widely distributed cellular system throughout the body and are the most efficient and potent APCs of the immune system [59]. DCs play a crucial role in initiating and regulating both innate and adaptive immune responses by activating naïve T cells and maintaining immune tolerance under homeostatic conditions [60]. DCs are central components of the TME and play a crucial role in promoting antitumor T-cell responses. Although DCs do not exert direct antitumor activity, they facilitate tumor control through a mutualistic relationship with T cells. However, an immunosuppressive TME can impair DC function, altering their phenotype and promoting dysfunction and tolerogenicity. These effects are mediated through various mechanisms, including soluble mediators and direct cell-to-cell interactions [61]. Worth of mention, DCs play a central role in immune modulation critical, with a especial role in the therapeutic response to immune checkpoint inhibitors (ICIs), and thus represent promising targets for cancer immunotherapy [62].
Natural killer (NK) cells are effector lymphocytes within the innate lymphoid cell (ILC) family [63], recognized for their ability to eliminate tumor cells without prior sensitization [64]. However, NK cell dysfunction (driven by tumor-derived molecules, tumor-educated stromal cells and direct suppression by cancer cells) has emerged as a key factor in tumor progression, not only promoting tumor cell proliferation but also facilitating distant metastasis [65]. Beyond immune suppression, NK cell impairment is also attributed to hostile metabolic conditions within the TME, including hypoxia, nutrient deprivation and the accumulation of tumor-derived products such as lactate, which contributes to an unfavorable acidic milieu [66]. Importantly, the specific role of NK cells in cancer remains controversial, as their impact varies significantly across different tumor types [67]. Even within the same cancer type, NK cells exhibit considerable heterogeneity, influenced by the diversity of surface receptors and the complexity of tumor-intrinsic signaling pathways [68].
Regulatory T cells (Tregs) are a critical subset of the T cell family, essential for maintaining immune homeostasis [69]. However, within the TME, they play a pivotal role in immune suppression, thereby facilitating tumor immune evasion. Tregs exert their immunosuppressive effects through multiple mechanisms, including the secretion of inhibitory cytokines, suppression of cytolytic activity and induction of metabolic disruption [70]. Their ability to inhibit effector T cells, NK cells and dendritic cells [71] is a key factor in enabling tumors to escape immune surveillance, posing a significant challenge to the efficacy of cancer immunotherapies [72] and allowing tumor progression. Additionally, the presence of an immunosuppressive TME and prolonged antigen exposure drive T cell exhaustion, characterized by diminished cytotoxic function and impaired cytokine production, further contributing to immune dysfunction and tumor progression [73].
2. Nanotechnology in Cancer Treatment
The long-standing standard treatment modalities for cancer include surgery, chemotherapy, targeted therapies addressing druggable mutations, immunotherapy and radiotherapy [74,75]. The sequence and combination of these treatments are tailored to each specific condition of the patient. Typically, a multimodal approach involving two or more treatments is employed to effectively manage the primary tumor and to prevent or delay recurrence and/or metastasis [76].
Over the past three decades, nanotechnology in cancer treatment (or more precisely, nanomedicine applied to oncology) has evolved from a conceptual framework into a powerful tool for advancing both diagnostic [77] and/or therapeutic approaches [78]. This rapidly growing field stands at the forefront of biomedical research, integrating both diagnostics and therapeutics into a single nanoscale platform, a concept known as theranostics. Among the most widely utilized tools in this domain are metallic nanoparticles (mNPs), which have demonstrated significant potential in both cancer imaging and/or therapy [79,80]. Certainly, successful and highly promising therapeutic strategies have been achieved using mNPs following intravenous administration in preclinical models, either through sophisticated core design [81] or by leveraging metabolically driven active targeting to achieve unprecedented efficiency [82]. For a more comprehensive exploration of tumor cell targeting in cell culture and preclinical models, associated therapies, and cancer nanomedicine candidates currently in clinical trials (all of them falling outside the scope of this review) readers are encouraged to refer to [83,84]. Remarkably, a cornerstone in nanomedicine applied to oncology is the concepts of “tumor targeting” and “tumor cell targeting”, which are often intertwined [85]. This intertwining, especially regarding active tumor targeting with cell targeting, may lead to design flaws and, consequently, resulting at the end in failed antitumoral processes. Nevertheless, the potential of targeting/treating the TME rather than the cancer cells themselves in nanomedicine has been relatively underexplored.
To conclude the section on nanomedicine applied to oncology, several remarks are pertinent. Notably, with great foresight, Professor Lammers highlighted a fundamental problem in this research field some years ago [86]. Upon reviewing the basic principles of drug targeting to pathological sites and critically assessing the feasibility and translational value of proposed concepts, it becomes evident that most novel nanomaterials are more “art” than “smart”, as their complex, multi-component designs (often larger than 100 nm) pose significant challenges for reproducible synthesis, scalability and clinical translation. Recently, Lammers and collaborators nicely proposed “smart” cancer nanomedicine as an umbrella term for rational and realistic “Strategies and Materials to Advance and Refine Treatments”, with four main directions to enhance nanomedicine performance and exploitation: 1) smart patient stratification, 2) smart drug selection, 3) smart combination therapies and 4) smart immunomodulation [87]. Also worth mentioning is a recent work published by a team of leading researchers in nanotechnology for oncology applications, where the future of healthcare research hinges on the evolution of collaborative approaches across multiple levels [88].
2.1. Nanotechnology and the TME
With the information presented so far, the critical role of the TME in cancer progression is evident. Regarding impact on mNPs following intravenous administration in animals/humans, a groundbreaking study by Professor Chan and collaborators demonstrated that TAMs sequester the vast majority of mNPs, effectively competing with cancer cells for mNPs uptake within the tumor [89]. This dominant sequestration is primarily driven by spatial distribution, as TAMs are highly concentrated near blood vessels. Consequently, upon extravasation from the tumor vasculature, mNPs are more likely to interact with TAMs before reaching cancer cells. Therefore, and without being redundant, these findings underscore the importance of considering TME dynamics rather than viewing the tumor merely as a cluster of cancer cells when designing mNP-based therapeutic approaches to enhance tumor uptake values and increase NPs efficacy.
Considering the influence of non-malignant cells of the TME on therapeutic approaches against cancer using mNPs, the next section will provide a comprehensive review of the use of mNPs targeting main cells of the TME.
2.1.1. mNPs and CAFs
Gadolinium (Gd) mNPs and CAFs
Gd@C82(OH)22 NPs were investigated in both cell culture and in vivo settings [90]. The in vivo findings, derived from a previous study, demonstrated that specific NPs may significantly enhance the synthesis of collagen types I and III in human pancreatic tumor xenografts, ultimately delaying tumor progression. Subsequently, in a separate set of cell culture experiments using fibrosarcoma and primary lung CAFs, Gd@C82(OH)22 NPs were shown to upregulate transcriptional expression of collagen types I and III in a dose-dependent manner. The underlying mechanism was identified as the activation of the factor receptor 2 (TNFR2)/p38 mitogen-activated protein kinase (MAPK) signaling pathway, mediated by enhanced binding of tumor necrosis factor-alpha (TNFα) to TNFR2, leading to increased collagen expression. An in vivo validation for fibrosarcoma and lung cancer models to confirm the proposed mechanism in a physiological context would be necessary.
Gold (Au) mNPs and CAFs
In contrast to Gd mNPs, intravenous administration of gold nanoparticles (AuNPs) in a colorectal cancer model led to a reduction in collagen type I, transforming growth factor beta-1 (TGF-β1), connective tissue growth factor (CTGF), vascular endothelial growth factor (VEGF) and CAFs [91]. This tumor-modulating effect also facilitated enhanced accumulation of systemically administered cisplatin, resulting in a significant delay in tumor progression. Cell culture studies (monocultures) further identified that downregulation of TGF-β1, CTGF and VEGF was mediated through an Akt-dependent pathway; however, this mechanism was not validated in vivo. Notably, authors used vernier calipers to monitor the tumor growth, which has several drawbacks compared to imaging techniques such as MRI or CT [92].
As another example, the presence of AuNPs was associated with increased lipid accumulation in CAFs, leading to reversion of activated cells to a quiescent state. This effect was mediated by upregulation of key lipogenesis-related genes, including fatty acid synthase (FASN), sterol regulatory element-binding protein 2 (SREBP2) and fatty acid-binding protein 3 (FABP3), in both immortalized and primary patient-derived CAFs [93]. This transformation could potentially contribute to tumor growth delay, although this effect has not been conclusively confirmed.
To provide another perspective, in a remarkable preclinical study, AuNPs of varying hydrodynamic diameters exhibited a size-dependent effect on CAFs, with smaller (~3 nm) AuNPs demonstrating the highest uptake efficiency [94]. This uptake resulted in significant downregulation of key CAF markers, including Vimentin, α-SMA, fibroblast-specific protein-1 (FSP-1) and N-cadherin. Additionally, CAF-mediated secretion of multiple pro-tumorigenic factors, such as hepatocyte growth factor (HGF), VEGF, interleukin-6 (IL-6), interleukin-8 (IL-8), TGF-β1, and platelet-derived growth factor-AA (PDGF-αα), was markedly reduced following AuNP treatment (24 hours). Interestingly, conditioned media derived from AuNP-treated CAFs enhanced proliferation and migration rates for oral squamous cell carcinoma (OSCC) cells, suggesting a complex interaction between CAFs and cancer cells. In vivo tumor models were established by co-injecting CAFs and cancer cells, revealing that presence of CAFs significantly promoted tumor engraftment and growth (also demonstrated by some of us in [95], but it is worth noting that, based on our experience, injected CAFs tend to disappear from tumors within just a few days post-transplantation), but AuNP treatment reversed this effect by downregulating N-cadherin, Vimentin, α-SMA and FSP-1. Notably, in tumor models generated solely with cancer cells, AuNPs failed to inhibit cancer cell proliferation, reinforcing the notion that their primary impact occurs through CAF modulation rather than direct cytotoxicity.
In contrast to all previously described studies on AuNPs and CAFs, the functionalization of these NPs with polyethylene glycol (PEG) and subsequent functionalization with the peptide RGD did not cause any damage to CAFs [96]. This necessitated the use of radiotherapy (2 Gy), with no significant difference observed between irradiated cells (in monoculture) exposed to AuNPs and those not exposed, except for some DNA damage. Therefore, these results strongly emphasize that surface coating significantly determines the intracellular behavior, as previously pointed out [97]. The same authors, later, described that these AuNPs were internalized by CAFs at a rate >10% higher than that of tumor cells in cell culture experiments [98]. The corresponding in vivo study revealed that ~10% of the injected AuNPs accumulated within the tumor 24 hours post-injection, whereas an impressive (and striking) >70% was measured 48 hours post-injection. Controversially, these accumulations at 48 hours post-injection was two orders of magnitude higher compared to the average (0.7%) accumulation rate described in a meta-analysis encompassing more than 100.000 works [99]. The increased accumulation suggests that the NPs may remain in circulation for an extended period and/or be sequestered by certain organs and subsequently released (either in their intact form or as degradation products).. Of note, PEGylated iron oxide nanoparticles (IONPs) have been shown to remain in the circulation for a maximum of 24 hours [100]. After exposing a 3D cell culture model (spheroids) to the same AuNPs-PEG-RGD nanostructure along with a single dose of radiotherapy (2 Gy), a significant decrease in diameter was observed in both monoculture (cancer cells or CAFs) and co-culture (cancer cells + CAFs) spheroids compared to using radiation alone. Notably, the results were similar when comparing 3D models composed of cancer cells alone or in combination with CAFs and, consequently, the presence of CAFs in this specific experimental setup did not contribute significantly [101].
Silver (Ag) and Core-Shell Au@Ag mNPs and CAFs
Beyond AuNPs, silver nanoparticles (AgNPs) and core-shell Au@AgNPs were also investigated for their effects on tumor-stroma interactions in a co-culture system of fibroblasts and tumor cells exposed to such NPs for 24 hours [102]. In this model, the presence of CAFs significantly enhanced tumor cell migration, whereas treatment with AgNPs or Au@AgNPs effectively suppressed this pro-migratory effect. Additionally, conditioned media from untreated CAFs promoted cancer cell migration, but media derived from nanoparticle-treated CAFs mitigated this effect, indicating a potential impact on CAF-secreted factors. Histological analysis of tumor sections further revealed a higher proliferation of cancer cells in fibroblast-enriched regions, a phenomenon that was significantly reduced following Au@AgNP treatment. These findings suggest that Au@AgNPs can effectively inhibit fibroblast-driven tumor cell proliferation and migration, both in cell culture and in vivo. Nonetheless, there is a lack of analysis regarding pathways involved in the suppression of the migration, as well as secreted factors present in the conditioned medium that drive the different cancer cell responses. Furthermore, a comprehensive analysis of the tumor sections is required to elucidate the observed behavior.
Multicomponent mNPs and CAFs
In addition to AuNPs, multicomponent nanoparticles (MCNPs) have gained significant interest because they can integrate the properties of various inorganic materials into a single nanoscale entity [103]. Multifunctional iron oxide nanoflowers decorated with gold nanoparticles (GIONFs) have demonstrated ability to significantly reduce tumor stiffness and achieve complete tumor regression following three sessions of mild hyperthermia in animal tumor models. This effect was induced by irradiation with a near-infrared (NIR) laser (808 nm, 2 W/cm2) for 10 minutes [104]. In a co-culture model, GIONFs were preferentially internalized by hTERT-HSC cells and RAW264.7 macrophages, compared to cancer cells. During in vivo settings, their administration led to a reduction in α-Smooth Muscle Actin (α-SMA) expression, a key marker of CAFs. Worth to mention that hTERT-HSC cells, while used as a CAF model, are immortalized hepatic stellate cells rather than bona fide CAFs.
Summary and Reflections About mNPs and CAFs
In summary, the impact of mNPs on CAFs has been well-documented, with AuNPs demonstrating particularly significant effects. However, these effects primarily target CAFs and, consequently, the ECM, thereby restricting cancer cell migration rather than directly influencing the tumor cells themselves. Notably, conditioned media from NPs treated CAFs lead to uncontrolled cancer cell growth, suggesting that NPs treatment of CAFs could be a double-edged sword. As a result, the potential mechanism underlying interactions between NPs, CAFs and cancer cells remains incompletely understood and requires further investigation. It is worth mentioning that several works have not been mentioned in this review, as the unique CAF-like cells selected were normal fibroblast, which are fundamentally different from tumor resident CAFs. Moreover, most studies have not confirmed the direct accumulation mNPs in tumor tissue (by ICP-MS for instance), suggesting that their effects may result from either direct nanoparticle action or indirect systemic responses. As a final point, a methodological concern arises regarding the measurement of tumor growth. The vast majority of studies (nearly all those discussed) rely on caliper-based measurements, which, as previously noted, can introduce substantial inaccuracies and may compromise the reliability of tumor response assessments. Additionally, most studies employed intratumoral drug administration, which, while effective in experimental settings, may limit the translational relevance of the findings to clinical practices.

Table 1.
Summary of selected studies describing interactions between NPs and CAFs discussed in this review. ‘N.A.’ indicates information not available.
Table 1.
Summary of selected studies describing interactions between NPs and CAFs discussed in this review. ‘N.A.’ indicates information not available.
| NPs | Cancer type | Cell culture |
Animal model |
Pathway / Mechanism |
Effects | Ref |
| Gd@C82(OH)22 NPs | Human pancreatic, fibrosarcoma and primary lung CAFs | 2D of primary cells (human) in all cases | Subcutaneous | TNFR2/p38 MAPK | Increase the synthesis of collagen types I and III | [90] |
| AuNPs (15 nm) | Colorectal cancer | 2D of SW620 cancer cells (human) | Subcutaneous | Akt | Decrease of collagen I, CAF density and stromal factors in vivo. Increase the drug-uptake (cisplatin) | [91] |
| AuNPs (20 nm) | Pancreatic cancer | 2D of primary CAFs (human) and CAF-19 cells (human) | N.A. | Lipogenesis-related genes | Lipid accumulation in CAFs, transforming them to a quiescent state | [93] |
| AuNPs (3 nm to 80 nm) | Oral squamous cell carcinoma | 2D of primary CAFs (human) | Subcutaneous | Reduction in expression of critical proteins and interleukins | Delay in tumor growth by co-inoculation of CAFs and cancer cells | [94] |
| AuNPs-PEG-RGD | Cervical cancer | 2D of Hs.895.T CAFs (human), Hs.895.Sk fibroblast (human) and HeLa cancer cells (human) | N.A. | N.A. | No effects over CAFs | [96] |
| AuNPs-PEG-RGD | Pancreatic cancer | 2D of CAF-98 cells (human), primary NPF-98 cells (human) and MIA-PaCa-2 and PANC-1cancer cells (both human) | Subcutaneous | N.A. | Higher retention in CAFs in cell culture and within tumor in vivo | [98] |
| AuNPs-PEG-RGD | Pancreatic cancer | 3D of CAF-98 cells (human) and MIA-PaCa-2 cancer cells (human) | N.A. | N.A. | No effects due to presence of CAFs | [101] |
| AuNPs (8 nm), AgNPs and Au@Ag NPs (11 nm) | Breast cancer | Co-culture of primary CAFs (human), NIH/3T3 fibroblasts (murine) and 4T1 cancer cells (murine) and human MCF-7 cancer cells (human) | Orthotopic | N.A. | CAFs exposed to NPs or the resulting conditioned media mitigated cancer cell migration | [102] |
| GIONFs | Desmoplastic cholangiocarcinoma | 2D and Co-culture of hTERT-HSC cells (human), RAW264.7 TAM cells (murine) and EGI-1 cancer cells (human) |
Subcutaneous | N.A. | Reduction of tumor stiffness and complete tumor regression | [104] |
2.1.2. mNPs and TAMs
Au mNPs and TAMs
One of the most significant preclinical studies in recent years regarding interaction between mNPs and TAMs explored particles based on Human Serum Albumin (HSA)-Au(III) thiosemicarbazone NPs [105]. From a physicochemical perspective, the design and development of these NPs were outstanding. In cell culture, the NPs preferentially accumulated in both cancer cells and TAMs, showing a three-fold increase compared to healthy cells, inducing reactive oxygen species (ROS) production in macrophages. This oxidative stress led to significant upregulation of nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) and inducible nitric oxide synthase (iNOS), both of which drive macrophage polarization toward the M1 phenotype. Conversely, expression of myosuppressin receptor 2 (MsR2) and signal transducer and activator of transcription 3 (STAT3), key regulators of M2 polarization, was suppressed. Western blot analysis further confirmed that these NPs significantly downregulated phosphorylated STAT3 (p-STAT3), total STAT3 and Programmed Cell Death Protein 1 (PD-1) (a marker associated with M2 macrophages) while markedly increasing TNF-α levels. In vivo, these NPs exhibited remarkable tumor accumulation and potent antitumor effects, alongside suppression of MsR2 and STAT3. Immunohistochemical analysis revealed a significant increase in M1 macrophage infiltration within the TME. It would be valuable to provide details on the methodology used to isolate TAMs and the characterization of their gene expression profile.
Beyond their role as a component of protein-based NPs, AuNPs have been also investigated for their interactions with TAMs. A study explored the effects of 5-fluorouracil-conjugated AuNPs on macrophages and tumor models [106]. Interestingly, cell culture experiments demonstrated that TAMs efficiently internalized these NPs, without causing cellular damage or altering molecular pathways. In contrast, in an in vivo metastatic model, the administration of these NPs via intraperitoneal injection resulted in significant increase in TAMs and a pronounced recruitment of CD3+ T lymphocytes. Moreover, TAMs (with a substantially greater proportion of M1-like macrophages observed compared to controls) exhibited high NPs uptake. Despite these promising immunomodulatory effects, the study lacked molecular pathway analyses both in cell culture and in vivo settings.
Similarly, furin-responsive aggregated AuNPs loaded with doxorubicin and hydroxychloroquine, well-known chemotherapy drugs, effectively promoted macrophage repolarization toward the M1 phenotype in cell culture in both IL-4-pretreated bone marrow-derived macrophages (BMDMs) and TAMs [107]. This was evidenced by an increase in CD86 expression, reduction in CD206 expression, decrease in IL-10 secretion and elevated secretion of TNF-α and IL-6. These effects over protein expression were also confirmed in vivo following intravenous administration, as well as a significant delay in tumor growth.
Regarding other AuNPs, polyaniline-based glyco-coated AuNPs of varying hydrodynamic diameters (~50 and ~250 nm) were evaluated, with the smallest variant proving most effective at reprogramming M2-polarized macrophages back to the M1 phenotype. This was evidenced by increased CD86 expression (a well-established M1 marker) in IL-4-pretreated RAW264.7 macrophages in cell culture, as well as the highest secretion of interferon-gamma-induced protein 10 (IP-10) and the lowest secretion of IL-10 [108]. Mechanistically, macrophages exposed to these NPs upregulated NF-κB and iNOS, while downregulating signal transducer and activator of transcription 6 (STAT6) and arginase 1 (ARG1), an M2-associated marker. These molecular changes aligned with the observed shift toward a M1 phenotype and the corresponding cytokine secretion profile. In a subcutaneous lung cancer model, these NPs significantly delayed tumor growth. Given their lack of direct cytotoxicity against cancer cells, this effect was attributed to the remodeling of the TME. This was further corroborated by increased CD86 expression and reduced CD206 expression in TAMs, alongside a cytokine shift, characterized by elevated levels of interferon-gamma (IFN-γ), TNF-α and interleukin-12 (IL-12), coupled with decreased secretion of immunosuppressive cytokines IL-4, IL-10 and interleukin-13 (IL-13). Of note, this effect was even more pronounced when these NPs were co-administered with an anti-PD-1 checkpoint inhibitor. Further, in an orthotopic lung cancer model, a similar TME remodeling effect was observed, with TAMs shifting to a M1-like phenotype, increased recruitment of CD8+ T cells and DCs; and a reduction in Tregs.
Finally, another group used macrophages derived from THP-1 (human monocyte-derived macrophages) were co-cultured with two different prostate cancer cell lines. Upon exposure to AuNPs, these macrophages exhibited a decrease in M2-associated markers, including IL-10, TGF-β and ARG1 mRNA, while M1-associated markers such as IL-6, TNF-α and iNOS mRNA were upregulated [109]. Flow cytometry analysis further confirmed reduction in the proportion of CD163+ macrophages (an M2 marker) in both co-culture systems. Western blot analysis revealed changes in autophagy-related protein expression, including alterations in the LC3-II/GAPDH and SQSTM1/GAPDH ratios. In addition, several autophagy-related genes, including ATG5, ATG7, ATG12 and BECN1, were downregulated in TAMs co-cultured with cancer cells. In vivo, intravenous administration of AuNPs resulted in a decrease in CD68+ CD163+ macrophages (M2 markers) within the TME, alongside a significant tumor growth delay. While the findings are promising, NPs accumulation in tumors peaked at 4 hours and declined to baseline within 24 hours, which contrasts with the prolonged retention generally expected from the enhanced permeability and retention (EPR) effect [110].
Multicomponent mNPs and TAMs
Au-manganese oxide (Au-MnO) NPs have demonstrated the ability to reprogram TAMs, specifically reverting the M2 phenotype back to the M1 phenotype. This reprogramming is mediated by a reduction in levels of superoxide (O2−), nitric oxide (NO) and ROS in primary TAMs, while having no discernible effect on either healthy or tumor-bearing splenic macrophages [111]. This TAM modulation also resulted in a significant downregulation of hypoxia-inducible factor-1 alpha (HIF-1α), accompanied by a shift in cytokine secretion patterns (specifically, a decrease in interleukin-10 (IL-10) and TNF-α levels, with a concomitant increase in IL-12).
As other illustration, a multifunctional nanoplatform was developed by coating IONPs with poly(lactic-co-glycolic acid) (PLGA), followed by the formation of AuNPs on the PLGA and functionalization with a programmed death-ligand 1 (PD-L1) antibody (antiPD-L1-SPIOs@PLGA@Au). These NPs demonstrated tumor accumulation by MRI and, upon radiotherapy (15 Gy in 1 fraction), effectively modulated the TME, leading to a significant delay in tumor growth in vivo [112]. Under irradiation, these NPs induced an increase in ROS, promoted a shift in TAM polarization towards the M1 phenotype (as evidenced by increased CD86+ and decreased CD206+ cell populations). Furthermore, elevated levels of IFN-γ, TNF-α and IL-12 were detected in the blood serum, collectively contributing to tumor growth inhibition. A critical aspect that warrants further investigation is whether these effects on TAM polarization and TME modulation could be achieved without irradiation, allowing for a more precise evaluation of the intrinsic immunomodulatory properties of these NPs. In addition, the observed ROS dynamics contrast with findings from previous studies, raising an important question regarding the specific ROS thresholds required for effective TAM polarization. Combined treatment modalities is the way to go, as monotherapies generally are faring worse in patient/clinical outcome.
Metal-Organic Framework (MOF) NPs and TAMs
An outstanding study reported the use of iron-containing metal-organic framework (MOF) NPs loaded with erastin to modulate TAMs, which, in turn, regulate CAFs [113]. These iron-based NPs exhibited a dose-dependent cytotoxic effect on cultured cancer cells, driven by two key mechanisms (Figure 3): 1) Erastin-induced ferroptosis via inhibition of System Xc− (the cystine/glutamate transporter), leading to reduced intracellular glutathione (GSH) levels and disruption of redox homeostasis, and 2) NP-mediated Fe3+ release, which catalyzed the Fenton reaction, generating ROS and promoting lipid peroxidation, consequently amplifying ferroptosis. These mechanisms were rigorously validated in the study. Additionally, the NPs induced polarization of TAMs from a M2 to M1 phenotype, resulting in decreased secretion of TGF-β. The conditioned medium from these reprogrammed macrophages, when applied to fibroblasts, led to a significant reduction in α-SMA expression, indicative of a shift towards a quiescent CAF/NF state. This transition reduced tumor stiffness, enhanced NPs penetration, as confirmed both in cell culture and in vivo through fluorescence imaging. Moreover, polarization of TAMs to the M1 phenotype and a significant tumor growth delay were also observed in vivo. Despite these promising findings, a critical aspect remains unaddressed, as an excessive reduction in ECM density may facilitate metastatic dissemination, which was not explored in the study.
IONPs and TAMs
Besides previous NPs, IONPs are widely used in biomedical applications, especially those related to cancer, due to their exceptional properties as contrast agents (CA) and/or therapeutic platforms [114] and by far, these NPs are the most widely used to explore their interactions with TAMs. In this context, fluorescent silica-coated F-IONPs have been utilized for labeling TAMs in vitro and for delineating the margins of glioblastoma tissue in vivo. Notably, immunofluorescence analysis of excised tissues confirmed that the majority of these NPs were internalized by TAMs, highlighting their potential as imaging agents for precise tumor margin identification [115].
As another example, Hyaluronic acid (HA)-modified doxorubicin-conjugated IONPs exhibited significantly higher uptake efficiency and cytotoxic effects in both macrophages and cancer cells compared to free DOX and non-modified doxorubicin IONPs, while these modified NPs demonstrated enhanced antitumor and anti-metastatic effects in a breast cancer model, potentially due to in vivo M1 macrophage polarization, although a direct effect on tumor cells cannot be excluded [116].
Building on the application of functionalized IONPs, a study investigated the use of arginine-loaded hollow IONPs to modulate TAMs [117]. These NPs were efficiently internalized by TAMs and successfully promoted their polarization from the M2 to the M1 phenotype. This shift was accompanied by an increase in TNF-α levels and iNOS expression, leading to a subsequent rise in NO production in cell culture. Of note, in a co-culture model, treatment of TAMs with these NPs resulted in elevated TNF-α and NO levels, which in turn reduced cancer cell viability in a dose-dependent manner. In vivo, arginine-loaded hollow IONPs demonstrated remarkable tumor accumulation, even with a core size of >200 nm, but sufficient to induce TAM polarization toward the M1 phenotype while reducing the presence of regulatory Tregs within the TME. These immune modulations ultimately led to effective tumor growth control.
As an additional case, IONPs were co-encapsulated with colony-stimulating factor 1 (CSF-1) inhibitor within liposomes functionalized with the TAT peptide, a well-known cell-penetrating peptide. These NPs effectively promoted polarization of BMDMs in cell culture, as evidenced by increased CD86 expression and reduced CD206 expression. In vivo, under an alternating magnetic field (AMF), these NPs significantly delayed tumor growth and induced an increase in iNOS and TNF-α, further confirming TAM polarization toward the M1 phenotype. Additionally, a decrease in ARG1 was observed, reinforcing the shift in macrophage phenotype [118]. Worth to mention that the TAT peptide functions as a general cell-penetrating molecule rather than a tumor-specific targeting ligand, meaning that all cells in the organism were susceptible to increased NPs uptake, potentially affecting biodistribution.
To provide another instance, polyaniline-coated IONPs effectively promoted the polarization of macrophages toward the M1 phenotype, as indicated by increased CD86 expression, in both 2D and 3D cell culture models. The 3D model, consisted of co-cultures of cancer cells, fibroblasts and macrophages, but the underlying molecular pathways were not analyzed [119].
As another example, IONPs coated with a catechol-derivative ligand and further functionalized with HA induced macrophage polarization toward the M1 phenotype when exposed to non-pretreated macrophages. This polarization was evidenced by the upregulation of key M1-associated genes, including C-X-C motif chemokine ligand 11 (CXCL11), CD68, CD80, iNOS, interleukin-1β (IL-1β) and TNF-α [120]. In vivo, these NPs promoted a significant increase in protein expression levels of CD80 and CD86, markers indicative of TAM polarization, as well as CD4, suggesting enhanced CD4+ T-helper cell infiltration. Interestingly, the most effective formulation in terms of tumor growth was the larger and non-HA-functionalized NPs (despite the fact that these NPs did not exhibit the most pronounced gene expression changes in cell culture).
Taking it one step further, enzyme-responsive mannose-grafted IONPs exhibited significant cytotoxicity in cell culture, particularly against J774A macrophage cells (a murine TAMs model derived from sarcoma), compared to both cancer cells and healthy fibroblasts [121]. Notably, in M1 macrophages stimulated with lipopolysaccharide (LPS) and IFN-γ, these NPs upregulated IL-6 expression at low doses, whereas higher doses led to a decrease in IL-6 and an increase in a ARG1. Interestingly, the opposite trend was observed in M2 macrophages stimulated with IL-4 [122]. No data were provided regarding their impact on untreated macrophages.
By way of illustration, nanodisc-shaped IONPs were investigated. These NPs induced cancer cell death upon NIR irradiation in cell culture. The conditioned medium from these treated cancer cells, when used to culture non-pretreated macrophages, led to increased CD86 and decreased CD206 protein expression (M1 phenotype). This was accompanied by upregulated mRNA levels of CD86, TNF-α and IL-1β, along with a reduction in CD206 and ARG1 mRNA expression. Moreover, the secretion profile revealed increased TNF-α for group receiving IONPs + NIR medium and decreased IL-4 levels for both IONPs alone and IONPs combined with NIR exposure [123]. Of note, conditioned medium from IONPs without NIR exposure resulted in elevated IL-4 and IL-10 mRNA levels, which returned to baseline when using the medium from IONPs + NIR-treated cells. Additionally, IL-6 levels decreased in the presence of IONPs alone but were restored to baseline when NIR was applied. Interestingly, TNF-α secretion remained unaffected by IONPs without NIR irradiation. In vivo, these NPs significantly delayed tumor growth, with the most pronounced effect observed in the group receiving IONPs + NIR in combination with the intraperitoneal administration of BMS-1, a small-molecule inhibitor of PD-1/PD-L1 interaction.
As another example, preconditioning osteosarcoma-bearing mice with an anti-CD47 antibody resulted in a higher tumor accumulation of PEGylated IONPs, as the treatment significantly increased the number of CD80+ macrophages (indicative of the M1 phenotype) compared to CD206+ macrophages, regardless of the administration of IONPs or an iron compound [124]. The authors suggested that anti-CD47 enhances M1 macrophage-mediated phagocytosis of cancer cells. Nonetheless, a previous study discussed in this review indicated that M1 macrophages can also influence CAFs, leading to decreased TME stiffness, which could be an alternative explanation for the enhanced accumulation of IONPs.
Finally, non-pretreated macrophages were exposed to IONPs, aluminum oxide NPs AONPs and zinc oxide NPs ZnONPs. Among these, IONPs induced the highest combined secretion of ATP and HMGB1 from macrophages without compromising their viability. Regarding macrophage polarization, all three NPs slightly increased the expression of CD86 and iNOS. However, CD206 expression was only affected by AONPs and ZnONPs. None of the NPs significantly altered the secretion of TNF-α or IL-10. Conditioned media from macrophages incubated with these NPs were then used to treat bone marrow-derived dendritic cells (BMDCs). Notably, only the medium from macrophages exposed to IONPs induced BMDC maturation, as evidenced by increased CD86 and CD80 expression. Furthermore, in a co-culture system of B16F10-OVA cells and BMDCs, only the medium derived from IONP-treated macrophages promoted the expression of H-2Kb/SIINFEKL on the surface of DCs. In vivo, intratumoral injection of BMDMs pre-exposed to IONPs, combined with radiotherapy (5 Gy), led to significant tumor growth delay and TME remodeling. This included an increase in intratumoral M1 macrophages and a reduction in M2 macrophages, enhanced infiltration of cytotoxic T lymphocytes (CTLs) and an increase in mature DCs and activated CTLs [125]. Since data on a non-irradiated control group were not provided, the specific contribution of BMDMs encapsulating IONPs remains unclear.
Other mNPs and TAMs
Regarding the application of other types of NPs, bone-targeting immunostimulatory metal-organic framework (BT-isMOF) NPs, functionalized with zoledronic acid (ZOL) and CpG oligonucleotides, effectively induced macrophage polarization toward the M1 phenotype both in cell culture and in vivo, as well as, modulated the interleukin secretion profile in cell culture [126]. Because of their strong bone-targeting capability and therapeutic effect, protected mice from the detrimental effects of bone metastatic osteolysis and destruction while simultaneously reducing tumor growth and progression.
Another study about other type of mNPs presents a well-designed and comprehensive approach using chromium (CrNPs) and small interfering RNA (siYTHDF1) adsorbed onto chitosan (CTS), encased in carboxymethyl mannose (Man-COOH), and decorated with RGD-modified DSPE. The work effectively demonstrates macrophage polarization and TME remodeling in both cell culture and in vivo models. The knockdown of YTHDF1 was expected to reduce M2 phenotype formation, which was later confirmed by Western blot analysis in two different TAMs cell lines. In IL-4/IL-13-stimulated THP-1 cells, M1-associated genes (NOS2, TNF-α, IL-1β and IL-12) were significantly upregulated, whereas M2-associated genes (ARG1, IL-10 and TGF-β) showed a downward trend. Similarly, in BMDMs, the absence of YTHDF1 led to an upregulation of M1 inflammatory markers and downregulation of M2 markers specifically in M2 phenotype BMDMs. Gene expression analysis revealed 677 altered genes, with particular relevance to decreased STAT3 expression and phosphorylation, alongside increased STAT1 expression and phosphorylation (both indicative of M1 polarization) [127]. In vivo, the combination of intravenous administration of NPs and NIR irradiation produced the greatest tumor growth delay, accompanied by a proinflammatory shift in the TME, marked by increased M1 TAMs, reduced M2 TAMs and Tregs, elevated proinflammatory cytokines, and decreased IL-10 levels.
Summary and Reflections About mNPs and TAMs
In summary, and from a critical perspective, much of our current understanding of the interaction between mNPs and TAMs is based on experimental models that do not accurately reflect true human TAMs, as they often involve non-human cells (mainly RAW264.7 cells) or macrophages that are not bona fide TAMs. Additionally, macrophages in these studies are frequently conditioned to adopt an M2 phenotype, which does not fully capture the functional heterogeneity of TAMs in the TME. On one hand, TAMs rarely conform strictly to the M1 or M2 phenotypic classification, reflecting their complex and dynamic nature. However, the in vivo reprogramming of macrophages toward an M1-like phenotype is often observed, partially validating these cell culture models, however, the in vivo molecular pathways involved in these processes have been scarcely elucidated. On the other hand, many studies aim to convert M2 macrophages into M1, assuming that M1 macrophages exert exclusively anti-tumoral effects. This perspective simplifies tumor tissues as a mere combination of cancer cells and TAMs, overlooking critical interactions with other stromal components. Notably, some reports indicate that M1 TAMs can influence CAFs, leading to modifications in the ECM that may promote metastasis [128]. Another crucial observation is that, in many cases, the effects observed in tumors are not directly linked to the confirmed presence of mNPs (for instance, while the fluorescent signal from a specific component of the nanostructure was detected in the tumor, the presence of the metallic core was not confirmed). This raises the question of whether the observed therapeutic effects stem from the entire nanostructure reaching the tumor or from active therapeutic components that are released elsewhere (partial NPs degradation?) and later act on the tumor site. Furthermore, the vast majority of studies (nearly all of those discussed in this section) rely on caliper-based measurements to assess tumor growth. Finally, several studies have reported that in vivo polarization toward the M1 phenotype enhances the recruitment of CD4+ and CD8+ T cells.
Table 2.
Summary of selected studies describing interactions between NPs and TAMs discussed in this review. ‘N.A.’ indicates information not available.
Table 2.
Summary of selected studies describing interactions between NPs and TAMs discussed in this review. ‘N.A.’ indicates information not available.
| NPs | Cancer type | Cell culture |
Animal model |
Pathway / Mechanism |
Effects | Ref |
| Human Serum Albumin (HSA)−Au(III) thiosemicarbazone NPs | Gastric cancer | 2D of RAW264.7 TAM cells (murine) and MGC-803 cancer cells (human) | Subcutaneous | NF-κB, iNOS, MsR2, STAT3, p-STAT3 and PD-1 | Remarkable tumor accumulation and potent antitumor effects | [105] |
| AuNPs conjugated with 5-fluorouracil (16 nm) |
Colorectal cancer and peritoneal metastasis | 2D of RAW264.7 TAM cells (murine) and CT26 cancer cells (murine) | Subcutaneous and a model of metastasis | N.A. | Following intraperitoneal administration of NPs, noticeable increase in TAMs (polarized to M1 phenotype) and CD3+T lymphocyte and high uptake of NPs by TAMs, in the metastatic model | [106] |
| Furin-responsive aggregated AuNPs loaded with doxorubicin and hydroxychloroquine (in the range 40-50 nm) |
Breast cancer | 2D of RAW264.7 TAM cells (murine), primary BMDM cells (murine) and MCF-7 cancer cells (human) | Subcutaneous | TNF-α, IL-6 and IL-10 | Polarization of TAMs and tumor growth delay | [107] |
| Polyaniline-based glyco-coated AuNPs (18-32 nm) |
Lung cancer | 2D of RAW264.7 TAM cells (murine), 3T3-L1 cells (murine) and MRC-5 cells (human) | Subcutaneous and orthotopic | Cell culture: NF-κB, iNOS, STAT6 and ARG1. Different pattern of interleukins secretion | Polarization of TAMs in cell culture and in vivo; tumor growth delay. Increase in CD8+ T cells and DC within the tumor and a reduction in Tregs | [108] |
| AuNPs (62 nm) |
Prostate cancer | 2D and Co-culture of THP-1 cancer cells (human), LNCaP cancer cells (human) and PC3 cancer cells (human) |
Orthotopic | IL-10, TGF-β, ARG1 IL-6, TNF-α, iNOS, CD163, LC3-II, GAPDH, SQSTM1, GAPDH. ATG5, ATG7, ATG12 and BECN1 | Polarization of TAMs | [109] |
| Au-manganese oxide NPs | Fibrosarcoma | 2D of primary murine TAMs | Subcutaneous (to obtain TAMs) | O2−, NO, ROS and HIF-1α Different pattern of interleukins secretion | Polarization of TAMs | [111] |
| antiPD-L1-SPIOs@PLGA@Au (>300 nm) |
Melanoma | 2D and Co-culture of BMDM (murine), Human Umbilical Vein Endothelial Cells and B16F10 cancer cells (murine) | Orthotopic | Increased in ROS levels | Application of radiotherapy, lead to polarization of TAMs. Increase in CD4+ and CD8+ T cells within the tumor. Tumor growth delay | [112] |
| Iron-containing metal-organic framework (MOF) NPs loaded with erastin | Pancreatic cancer | 3D of RAW264.7 TAM cells (murine), NIH3T3 cells (murine) and KPC1199 cancer cells (murine) | Subcutaneous | Antitumoral effect by composition and polarization of TAMs by different ways | The polarization of TAMs transforms CAFs to a quiescent state, both of which lead to delayed tumor growth in vivo | [113] |
| F-IONPs | Glioblastoma | 2D of RAW264.7 TAM cells (murine), CCD-986sk cells (human) and u87 cancer cells (human) | Subcutaneous | N.A. | Delineation of tumor margins | [115] |
| Hyaluronic acid-modified doxorubicin IONPs (>200nm) |
Breast cancer | 2D of RAW264.7 TAM cells (murine) and 4T1 cancer cells (murine) | Orthotopic | N.A. | Higher uptake efficiency and cytotoxic in cell culture. Both antitumor and anti-metastatic effects in vivo | [116] |
| Arginine-loaded hollow IONPs (>200nm) | Breast cancer | 2D and Co-culture of RAW264.7 TAM cells (murine) and 4T1 cancer cells (murine) | Subcutaneous | Cell culture: TNF-α, iNOS and NO. In vivo: TNF-α and NO |
Treated TAMs impact cancer cell viability both in cell culture and in vivo. In vivo, there is an increase in CD4+ and CD8+ T cells within the tumor and a reduction in Tregs | [117] |
| IONPs encapsulated with an inhibitor of CSF-1 in liposomes functionalized with TAT | Colorectal cancer | 2D of BMDM (murine) and CT26 cancer cells (murine) | Subcutaneous | CD86, CD206, iNOS, TNF-α and ARG1 | Polarization of TAMs. Tumor growth delay |
[118] |
| Polyaniline-coated IONPs (38 nm) |
Breast cancer | 2D and 3D of fibroblast (hMF) (human), primary monocytes (human) and MCF-7 cancer cells (human) |
N.A. | CD86 | Polarization of TAMs | [119] |
| IONPs coated with a catechol ligand and functionalized with HA | Breast cancer | 2D of RAW264.7 TAM cells (murine) and 4T1 cancer cells (murine) | Orthotopic | Cell culture: CXCL11, CD68, CD80, iNOS, IL-1β and TNF-α |
Polarization of TAMs. Tumor growth delay |
[120] |
| Enzyme-responsive mannose-grafted IONPs | Breast cancer and hepatic cancer | 2D of J774A TAM cells (murine), NIH/3T3 fibroblasts (murine), MCF-7 cancer cells (human) and HepG2 cancer cells (human) | N.A. | IL-6 and ARG1 | Keep the M1 phenotype at low dose. At high dose, keep the M2 phenotype | [121] |
| nanodisc-shaped IONPs | Head and neck squamous carcinoma | 2D of RAW264.7 TAM cells (murine) and SCC7 cancer cells (murine) | Subcutaneous | CD86, CD206, TNF-α, IL-1β, ARG1 and IL-4. | Polarization of TAMs in cell culture. Tumor growth delay in vivo. | [123] |
| PEGylated IONPs | Murine and human osteosarcoma | N.A. | Orthotopic | N.A. | Polarization of TAMs (induced by anti-CD47 rather than by IONPs) | [124] |
| Macrophages exposed to IONPs, AONPs, ZnONPs (≈30 nm) |
Melanoma | 2D and Co-culture of RAW264.7 TAM cells (murine), BMDCs (murine), 4T1 cancer cells (murine), CT26 cancer cells (murine) and B16F10-OVA cancer cells (murine) |
Orthotopic | CD86 and iNOS. | Polarization of TAMs. Tumor growth delay | [125] |
| Bone-targeting immunostimulatory metal-organic framework (BT-isMOF) NPs functionalized with zoledronic acid (ZOL) and CpG oligonucleotides | Breast cancer (Bone metastasis in vivo) | 2D of RAW264.7 TAM cells (murine), BMDM cells (murine) and MDA-MB-231 cancer cells (human) | Orthotopic (Metastatic model) | N.A. | Polarization of TAMs. Decrease of bone metastatic osteolysis and reduction of tumor growth and progression |
[126] |
| Chromium nanoparticles (Cr NPs) and siYTHDF1 were loaded onto chitosan, coated with carboxymethyl mannose, and functionalized with DSPE-modified RGD. | Hepatic cancer | 2D of RAW264.7 TAM cells (murine), BMDM cells (murine), THP-1 cancer cells (human) and Hepa1-6 cancer cells (murine) | Subcutaneous | NOS2, TNF-α, IL-1β, IL-12 ARG1, IL10, TGF-β, STAT3 and STAT1. | Polarization of TAMs. Tumor growth delay. Increase in CD4+ and CD8+ T cells within tumors |
[127] |
2.1.3. mNPs and Other Non-Malignant Cells of the TME
This section will focus primarily on endothelial cells, which play two critical roles in cancer: 1) promoting angiogenesis to supply nutrients and oxygen to the tumor mass, and 2) acting as a physical barrier that impedes the delivery of chemotherapeutic agents and NPs (both organic and inorganic). The process of angiogenesis is tightly regulated by a complex signaling network, in which vascular endothelial growth factor (VEGF) is a well-established modulator and a key biological driver [129]. Consequently, the vast majority of therapeutic strategies (whether based on conventional drugs, mNPs, or their combination) have been designed to target the VEGF pathway. These include the use of monoclonal antibodies against VEGF or its receptors (VEGFRs), as well as inhibitors of intracellular tyrosine kinase signaling. This topic has been comprehensively and recently reviewed; therefore, readers are encouraged to consult those works for further details [130]. Nonetheless, a noteworthy study reported that AuNPs of varying sizes could induce endothelial leakiness, thereby enhancing the delivery efficiency of anticancer therapeutics [131]. In cell culture, using a monolayer of endothelial cells as model, smaller-sized AuNPs were shown to increase intercellular gap formation (ranging from 5–20 μm) by binding to transmembrane VE-cadherin and disrupting the homophilic interactions of these adherent junction proteins. In vivo, increased vascular permeability and deeper tumor penetration were observed in both orthotopic breast cancer (4T1 cancer cells, murine) and pancreatic cancer (Panc2 cancer cells, human) models, as well as in a subcutaneous breast cancer model.
2.1.4. Direct and Indirect Effects of mNP on the TME
In addition to the intended tumoricidal effects that mNPs may exert on cancer cells, some groups have explored the potential of this treatment modality to trigger activation of anti-tumor immune responses which ultimately could render into enhanced therapeutic effects or could serve to potentiate systemic responses in combination with immunotherapies. Harnessing of the immune system and overcoming peripheral tolerance in the context of cancer treatment normally happens through activation of immunogenic cell death (ICD). Immunogenic cell death (ICD) is a form of regulated cell death that elicits an immune response against dying cells. Unlike apoptosis, which is typically non-immunogenic, ICD is characterized by the release of tumor specific antigens (antigenicity) and damage-associated molecular patterns (DAMPs) together with the exposure of calreticulin on the cell surface (adjuvanticity). These signals facilitate the recruitment and activation of dendritic cells and other antigen-presenting cells, leading to the presentation of tumor antigens to T cells. Consequently, ICD plays a crucial role in anti-tumor immunity, In the following chapter, we delve into studies that have explored mNP-mediated anti-tumor immune regulation.
AuNPs and the Tumor Immune Microenvironment
PLGA microspheres co-encapsulating hollow gold nanoshells and metformin were employed in a model of metastatic progression to delay tumor growth in primary and distant tumors [132]. This effect was achieved through intratumoral administration of the NPs into the primary tumor, followed by phototherapy. The treatment triggered release of tumor antigens (TAs) and damage-associated molecular patterns, which induced ICD and subsequently activated DCs and effector T cells. Thus, these NPs did not directly target DCs or T cells. Caution should be taken when the intended use of mNPs is to stimulate the immune system as excessive engagement of the innate immune system in the form of inflammatory reactions could exert detrimental effects on healthy tissue/organ surrounding the tumor lesions. In the presented study, the so-called metastatic model used consisted of implanting two tumors rather than employing a true metastatic model, such as the 4T1 model, which more accurately recapitulates metastatic progression.
As further evidence, Glycoadjuvant@AuNPs promoted BMDC maturation by upregulating major histocompatibility complex class II (MHC II) and CD86 expression in cell culture. In vivo, intratumoral administration of these NPs led to tumor growth delay, along with increased infiltration of CD8+ T cells in the primary tumor and a reduction in metastasis [133]. The treatment also resulted in a decreased population of T reg cells, a polarization of TAMs toward the M1 phenotype, a reduction in myeloid-derived suppressor cells (MDSCs) and elevated concentrations of IFN-γ and TNF-α within the primary tumor. Nevertheless, key mechanistic aspects remain unexplored. The specific role of AuNPs in these effects was not clearly defined, and the temporal sequence of changes within the TME was not analyzed. For instance, it is unclear whether DCs were directly affected by NPs, leading to subsequent TAM polarization, or if TAMs were influenced first, indirectly promoting DC differentiation. Additionally, the source of alterations in the interleukin secretion pattern was not addressed, leaving open questions regarding the exact pathways mediating the observed immune modulation.
As another example, Zwitterion-functionalized dendrimer-entrapped AuNPs loaded with CpG oligodeoxynucleotides enhanced the expression of CD80, CD86 and MHC-II in BMDCs, thereby promoting their maturation. These matured BMDCs, in turn, increased CD4+ and CD8+ expression on the surface of T cells in a co-culture system [134]. Besides, the conditioned medium from these activated T cells exhibited an anti-cancer effect in cell culture.
AgNPs and the Tumor Immune Microenvironment
As other type of mNPs, β-D-Glucose-reduced AgNPs enhanced CD8+ T cells, memory T cells and innate effector T cells while reducing CD4+ T cells and Tregs in in vivo experiments. Also, these NPs increased TNF-α, IFN-γ and IL-6 levels while decreasing IL-2, IL-4 and IL-10. This immune modulation resulted in tumor growth delay following peritumoral administration [135]. Worth of mention, these NPs differed from most studies, as they led to a decrease in CD4+ T cells. Furthermore, the exact mechanism underlying their antitumoral effects remains unclear, whether attributable to a direct action of the NPs themselves or to the resultant immune activation.
As an additional instance, AgNPs of varying sizes (5 nm and 50 nm) and different surface coatings, including polyvinylpyrrolidone (PVP) and citrate, altered the interleukin secretion profile of cancer cells in cell culture. In vivo, these AgNPs increased CD8+ T cell infiltration within the tumor and induced a tumor growth delay, as assessed using 2D imaging techniques [136].
IONPs and the Tumor Immune Microenvironment
The only study applying IONPs to non-malignant cells within the TME reported that these NPs, functionalized with polydopamine (PDA), RGD and anisamide (AA); and physically adsorbing glucose oxidase (GOx), were capable of maturing BMDCs in a co-culture setup after affecting cancer cells [137]. In vivo, intravenous administration of these NPs, combined with phototherapy and antiPD-L1 treatment, resulted in tumor growth delay. Nonetheless, BMDCs were not exposed to mNPs; the control group receiving phototherapy and antiPD-L1 alone was absent; and the immune response analysis focused on lymph nodes and the spleen rather than the tumor itself.
Manganese (Mn) mNPs the Tumor Immune Microenvironment
Cancer cells were exposed to a nanostructure composed of MnO2, iron atoms (Fe3+) and doxorubicin encapsulated within PEG-polyphenols. After washing the NPs, these treated cancer cells were co-cultured with BMDCs, leading to a significant increase in DC maturation, as indicated by higher CD11c+CD80+CD86+ expression, along with an elevated secretion of IL-6 and TNF-α [138]. In vivo, these NPs peaked at 8 hours post-intravenous administration. Regardless of prior tumor sensitization with anti-PD1 treatment, the NPs promoted DC maturation, increased M1 macrophage phenotype, higher infiltration of CD8+ T cells, CD8+IFN-γ+ T cells and memory T cells, while reducing the number of Tregs.
As an additional instance, Mn molybdate nanodots promoted BMDC maturation (CD80+ and CD86+) in cell culture and, following intravenous administration, increased the number of mature DCs within the tumor in vivo. This was accompanied by TAM polarization toward the M1 phenotype, an increase in CD8+ T cells and decrease in Tregs and MDSCs, ultimately leading to tumor growth delay [139]. Nevertheless, molecular changes were analyzed in serum rather than in tumors or cell cultures. Remarkably, some successful DC maturation experiments were conducted using Mn ions alone, raising questions about whether the observed intratumoral effects were truly mediated by intact mNPs.
As an additional instance, MnO2 NPs and attenuated Salmonella were intravenously administered to tumor-bearing mice. The bacteria selectively colonized the TME, leading to recruitment of neutrophils, as evidenced by the increased expression of neutrophil markers CD11b and Ly6G. Concurrently, the Mn ions released from the NPs polarized neutrophils toward an N1 phenotype, as indicated by the upregulation of CD54 and CD95. This polarization was accompanied by recruitment and activation of CD8+ T cells, along with elevated levels of CCL3 and TNF-α within the tumor. Ultimately, this strategy resulted in significant tumor growth delay [140]. However, how will other organs respond to the introduction of even attenuated bacteria or to the exposure to Mn ions? Additionally, will it be possible to achieve sufficient bacterial concentration in the tumor without adversely affecting the surrounding organs?.
Other mNPs and the Tumor Immune Microenvironment
From a novel perspective, tumor lysates processed by DCs were presented to CD8+ T cells, forming adoptive T cell vectors (ATVs). These ATVs were then exposed to mineralized metal-organic frameworks (MOFs) encapsulating perforin and granzyme B, coupled with a lysosome-targeting aptamer (CD63-aptamer). The ATVs subsequently internalized and processed the NPs, releasing the therapeutic agents and enhancing cancer cell death compared to CD8+ T cells alone [141]. In vivo, higher fluorescence signals were observed in the tumor at 24 hours for both ATVs encapsulating NPs and ATVs alone, indicating that NPs internalization did not alter ATV tumor accumulation. Nonetheless, a significant portion of the NPs was cleared by the liver and spleen, surpassing their accumulation in the tumor. Ultimately, the ATV-NP formulation delayed tumor growth and increased CD8+ T cell recruitment, which can be partially due to polarization of TAMs, as mentioned previously in this review.
Regarding other MOFs, these nanostructured loaded with GOx and an indoleamine 2,3-dioxygenase inhibitor (1-methyltryptophan) enhanced the immune response in vivo by increasing the number of CD8+ T cells, matured DCs, B cells and NK cells, while reducing Tregs in orthotopic melanoma and breast cancer models, which lead at the end in a tumor growth control and a decrease in the metastatic lesions, after intravenous administration [142].
Similarly, MOFs functionalized with bovine serum albumin (BSA) and folic acid (FA) and loaded with triptolide (TPL), Fe3+ and tannic acid (TA), promoted an increase in matured DCs as well as CD8+ and CD4+ T cells in vivo after intravenous administration. This immune activation ultimately led to tumor growth delay and a reduction in metastasis [143].
As a further illustration, conditioned medium from cancer cells exposed to TiO2 NPs functionalized with a Ruthenium complex, conjugated with small interfering RNA (siRNA), and subjected to irradiation (visible light at 525 nm), stimulated IFN-γ expression in both CD4+ and CD8+ T cells derived from peripheral blood mononuclear cells (PBMCs) of patients [144]. Specifically, PBMCs exposed to IL-24 exhibited a similar response. In vivo, the intratumoral administration of these NPs effectively controlled tumor growth in both patient-derived and induced tumor models.
Multicomponent mNPs and the Tumor Immune Microenvironment
A remarkable study demonstrated the ability of an anti-PD-L1-immobilized magnetic gold nanohut nanostructure to remodel the TME [145]. After 24 hours (optimal timeframe for the EPR effect [146]) and the point at which NPs accumulation peaked, as confirmed via gamma counter analysis, tumors were irradiated with an NIR laser (808 nm, 0.2 W/cm2) for 10 minutes. This weak irradiation triggered two key effects: 1) a localized increase in temperature within the tumor microenvironment (though not directly assessed in tumor cells) and 2) the controlled release of anti-PD-L1. Of note, administering the NPs and anti-PD-L1 separately failed to produce comparable results, underscoring the role of the NPs as a targeted delivery vehicle. The combined effects led to an increase in tumor-infiltrating immune cells (including dendritic cells, antigen-presenting cells, cytotoxic T cells, helper T cells and memory T cells) (Figure 4) and a concurrent decrease in immunosuppressive populations (such as TAMs and Tregs) (Figure 4), ultimately leading to a delayed tumor growth and prolonged survival in treated animals.
Finally, an IONPs core was functionalized with an optimal number of AuNPs, onto which clusters of HER2 B/CD4 T cell epitopes were conjugated, forming the construct known as ACNVax. This nanovaccine promoted B cell antigen presentation and enhanced the activation of CD4+ T cells in cell culture [147]. In vivo, the immune cell composition in draining lymph nodes was analyzed in healthy mice; however, no data were reported for tumor-bearing models. When combined with anti-PD-1 therapy, these NPs significantly delayed tumor growth and induced a marked increase in the frequency of B cells, CD4+ T cells (in contradiction with previous report), CD8+ T cells and memory T cells (both CD4+ and CD8+). Additionally, they reduced Tregs and upregulated expression of genes: B-cell lymphoma 6 (BCL-6), IFN-γ, TNF-α, chemokine receptor type 4 (CXCR4), CXCR5, CC-chemokine receptor 7 (CCR7), L-selectin, CD11a, VLA-4 and IL-21; as well as the levels of chemotactic factors CC motif chemokine ligand 19 (CCL19), CCL21a, CXC motif chemokine ligand 13 (CXCL13) and CCL2. Despite these promising results, the study did not clarify the route of administration, which is a critical factor for evaluating the translational potential of this approach.
Summary and Reflections About Direct and Indirect Effects of mNP on Tumor Immune Cells
Among the topics covered in this review, the molecular pathways underlying the interaction between mNPs and non-macrophage immune cells within the TME remain among the least explored, representing a significant knowledge gap that must be addressed in future research. Collectively, many studies demonstrate adequate engagement of the immune system when mNPs of different natures are used. Observed effects in many instances are related to the induction of ICD or the release of pro-inflammatory factors from tumor cells exposed to mNPs. A number of studies have also shown synergistic effects when mNPs are combined with ICBs, highlighting the potential of this combinatory treatment strategy to boost anti-cancer immune responses.
Table 3.
Summary of selected studies describing interactions between NPs and Other non-malignant cells of the TME discussed in this review. ‘N.A.’ indicates information not available.
Table 3.
Summary of selected studies describing interactions between NPs and Other non-malignant cells of the TME discussed in this review. ‘N.A.’ indicates information not available.
| NPs | Cancer type | Cell culture |
Animal model |
Pathway / Mechanism |
Effects | Ref |
| PLGA microspheres co-encapsulated with hollow gold nanoshells | Melanoma and lymphoma | 2D of splenic lymphocytes (murine), B16F10 cancer cells (murine) and EG7-OVA cancer cells (murine) | Orthotopic and subcutaneous | N.A. | Tumor growth delay in the primary and metastatic mass by NPs and phototherapy leading to activation of DC and T cells | [132] |
| Glycoadjuvant AuNPs | Melanoma | 2D of BMDC (murine) and B16-OVA cancer cells (murine) | Orthotopic | Cell culture: MHC II and CD86. In vivo: IFN-γ and TNF-α |
Tumor growth delay and inhibition of metastasis. Polarization of TAMs toward a M1 phenotype. Increased CD8+ T cells. Decreased T reg cells. Decreased MDSCs. |
[133] |
| Zwitterion-functionalized dendrimer-entrapped AuNPs loaded with CpG | Breast cancer | 2D and Co-culture of BMDCs (murine) and 4T1 cancer cells (murine) | N.A. | N.A. | Maturation of BMDCs and activation of DC. Anti tumoral effect | [134] |
| β-D-Glucose-reduced AgNPs | Breast cancer | N.A. | Subcutaneous | TNF-α, IFN-γ, IL-6, IL-2, IL-4 and IL-10 | Tumor growth delay. Increased levels of CD8+ cells, memory T cells and innate effector T cells. Decreased levels of CD4+ cells and Treg | [135] |
| AgNPs (5 nm and 50 nm) coated with PVP or citrate | Renal carcinoma | N.A. | Subcutaneous | N.A. | Tumor growth delay. Increased levels of CD8+ cells |
[136] |
| IONPs functionalized with PDA, subsequently with RGD and AA; with GOx physically absorbed | Colorectal Cancer | Co-culture of BMDCs (murine) and CT26 cancer cells (murine) | Subcutaneous | Ferroptosis of cancer cells induced BMDCs maturation in cell culture | BMDCs maturation in cell culture. Tumor growth delay |
[137] |
| MnO2 + Irom atoms (Fe3+) + Doxorubicin; encapsulated within PEG-polyphenols | Melanoma | 2D and Co-culture of BMDCs (murine) and B16–F10 cancer cells (murine) |
Orthotopic | CD11c, CD80, CD86, IL-6 and TNF-α | Tumor growth delay and metastatic control | [138] |
| Mn molybdate nanodots | Colorectal, melanoma and breast cancer | Co-culture of BMDCs (murine), CT26 cancer cells (murine) B16F10 cancer cells (murine), and 4T1 cancer cells (murine) | Subcutaneous and Orthotopic | N.A. | DCs maturation, TAM polarization toward the M1 phenotype, increased CD8+ T cells. Decreased Tregs and MDSCs, Tumor growth delay |
[139] |
| MnO2 NPs | Breast cancer | N.A. | Subcutaneous | CCL3 and TNF-α | Recruitment of neutrophils and their subsequent polarization. Increased CD8+ T cells. Tumor growth delay |
[140] |
| Mineralized MOF, encapsulating Perforin and Granzyme B, coupled with a lysosome-targeting aptamer (CD63-aptamer) | Breast cancer | 2D of T cells (murine) and 4T1 cancer cells (murine) | Orthotopic | Novel ATVs to improve NPs drug targeting and increase cancer cell specificity | Increasing cancer cell death in cell culture. Tumor growth delay and higher recruitment of CD8+ T cells | [141] |
| MOFs loaded with GOx and an indoleamine 2,3-dioxygenase inhibitor (1-methyltryptophan) | Melanoma and breast cancer | N.A. | Orthotopic | N.A. | Increased the number of CD8+ T cell, matured DC, B cells and NK cells and decreased Treg | [142] |
| MOFs functionalized with bovine serum albumin (BSA) and folic acid (FA), and loaded with triptolide (TPL), Fe3+ and tannic acid (TA) | Melanoma | N.A. | Orthotopic | N.A. | Increased matured DC; CD8+ and CD4+ cells | [143] |
| TiO2 NPs functionalized with a Ruthenium complex, followed by conjugation with siRNA | Head and neck squamous cell carcinoma | 2D of PBMCs (human) and HN6 cancer cells (human) | Subcutaneous (patient derived cells) and Orthotopic (induced model) |
IFN-γ | Tumor growth delay | [144] |
| anti-PD-L1- magnetic gold nanohut | Hepatocellular carcinoma | 2D of Hep55.1c cancer cells (murine) | Orthotopic | N.A. | Direct treatment with NPs and remodeling of the TME in vivo | [145] |
| ACNVax | Breast cancer | N.A. | Subcutaneous | BCL-6, IFN-γ, TNF-α, CXCR4, CXCR5, CCR7, L-selectin, CD11a, VLA-4, IL-21, CCL19, CCL21a, CXCL13 and CCL2 | Tumor growth delay. Increased of B cells, CD4+ T cells, CD8+ T cells and memory T cells |
[147] |
Conclusion
One of the main limitations in the fight against cancer is our incomplete understanding of the disease across multiple levels, including cellular populations, metabolism and signaling pathways. Given this complexity, fully elucidating how mNPs influence the TME is an immense challenge that requires significantly greater efforts from the scientific community. As an example, while nearly 5.5 million studies have been published on cancer throughout history (PUBMED), approximately 75.000 focus on NPs in cancer and only 7.300 specifically examine the effects of NPs on the TME. When considering mNPs, this number is even lower. Another major challenge lies in the models used for studying mNPs in the TME. For instance, many studies employ normal macrophages or immortalized macrophage cell lines (often from a different species) as models for TAMs, without accounting for the interactions among the diverse cellular populations within the TME. In many cases, researchers attempt to polarize these “TAMs” toward an M1 phenotype, despite evidence that M1 macrophages can influence CAFs, leading to a less dense ECM and potentially promoting metastasis. This highlights the necessity of studying cancer as a complex and dynamic ecosystem rather than as a single-cell lineage. The TME consists of multiple interacting cellular populations in addition to the ECM, all of which must be considered in experimental models. Methodological limitations further hinder progress in this field. Additionally, the frequent use of intratumoral injection may lead to the accumulation of mNPs in necrotic regions (areas of dead tissue that do not require treatment) thus limiting their therapeutic impact and resulting in suboptimal treatment strategies.
Regarding the effects of mNPs on non-tumoral cells within the TME, multiple studies report the upregulation of NF-κB and iNOS gene expression, increased levels of IL-6, TNF-α and IFN-γ; and downregulation of IL-4, IL-10 and TGF-β. However, these findings likely represent only a subset of the mechanisms involved, with many others yet to be identified. Furthermore, it remains largely unknown whether these mNP-induced changes in non-tumoral TME cells directly affect cancer cells. The majority of studies describe ECM modifications (low collagen secretion, changes in the secretion patterns, etc) or tumor size reduction without clarifying the underlying mechanisms or how cancer cells themselves respond. Indeed, there is a lack of preclinical transplantable tumor models that fully recapitulate the complexity and composition of the tumor stroma, which limits our ability to realistically assess how mNPs interact with this critical component of the TME. Finally, and not least important are the administration routes of mNPs, which are often not delivered intravenously (the primary route of administration in clinical settings) and, consequently, delay the potential clinical translation of the findings.
In conclusion, the overall impact of mNPs on the TME and their subsequent effects on cancer cells remain inadequately understood, with numerous unresolved aspects that need to be addressed in the coming years (Figure 5). Given the critical role of the TME in primary tumor progression, future research should prioritize elucidating these complex interactions to advance the development of more effective therapeutic strategies.
Ultimately, tumor size reduction or elimination remains the primary goal of oncology research. Fortunately, many cancer patients are successfully cured or are able to live long lives with cancer, despite the fact that researchers and clinicians do not yet fully understand all the molecular mechanisms driving the therapeutic response.
Acknowledgments
This work was supported by Tromsø Forskningsstiftelse (19_PET-NUKL)/180°N/Norwegian-Nuclear-Medicine-Consortium.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol Cancer Res 2023, 21, 1142–1147. [Google Scholar] [CrossRef]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front Oncol 2023, 13, 1164535. [Google Scholar] [CrossRef]
- Onken, J.S.; Fekonja, L.S.; Wehowsky, R.; Hubertus, V.; Vajkoczy, P. Metastatic dissemination patterns of different primary tumors to the spine and other bones. Clin Exp Metastasis 2019, 36, 493–498. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Trosko, J.E. On the potential origin and characteristics of cancer stem cells. Carcinogenesis 2021, 42, 905–912. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal 2020, 18, 59. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Medicine 2023, 12, 11149–11165. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, A.; Chelstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther 2021, 221, 107753. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr Biol 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Pradip, D.; Jennifer, A.; Nandini, D. Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, Y.; Wu, B.; Li, Y.; Liu, Z.; Liu, Z.; Gao, Y.; Gao, L.; Song, Q.; Zheng, Z.; et al. Irradiation enhances the malignancy-promoting behaviors of cancer-associated fibroblasts. Front Oncol 2022, 12, 965660. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin Transl Radiat Oncol 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Berzaghi, R.; Gundersen, K.; Dille Pedersen, B.; Utne, A.; Yang, N.; Hellevik, T.; Martinez-Zubiaurre, I. Immunological signatures from irradiated cancer-associated fibroblasts. Front Immunol 2024, 15, 1433237. [Google Scholar] [CrossRef]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.T.; Yang, J. Epithelial–mesenchymal transition in tumor metastasis. Molecular Oncology 2017, 11, 28–39. [Google Scholar] [CrossRef]
- Dumont, N.; Liu, B.; Defilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A 2010, 107, 21677–21682. [Google Scholar] [CrossRef]
- Hellevik, T.; Berzaghi, R.; Lode, K.; Islam, A.; Martinez-Zubiaurre, I. Immunobiology of cancer-associated fibroblasts in the context of radiotherapy. J Transl Med 2021, 19, 437. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J Exp Med 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: a versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev 2024, 43, 1095–1116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiao, X.; Yi, Y.; Wang, X.; Zhu, L.; Shen, Y.; Lin, D.; Wu, C. Tumor initiation and early tumorigenesis: molecular mechanisms and interventional targets. Signal Transduct Target Ther 2024, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 2019, 12, 76. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Karimova, A.F.; Khalitova, A.R.; Suezov, R.; Markov, N.; Mukhamedshina, Y.; Rizvanov, A.A.; Huber, M.; Simon, H.U.; Brichkina, A. Immunometabolism of tumor-associated macrophages: A therapeutic perspective. Eur J Cancer 2025, 220, 115332. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593 e1578. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol 2020, 11, 583084. [Google Scholar] [CrossRef]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol 2008, 84, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol 2022, 19, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, S.; Pandha, H.S.; Morgan, R. Antiangiogenic effects of zoledronate on cancer neovasculature. Future Oncol 2011, 7, 1325–1333. [Google Scholar] [CrossRef]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res 2007, 67, 5064–5066. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res 2008, 68, 9050–9059. [Google Scholar] [CrossRef]
- Barker, T.H.; Baneyx, G.; Cardo-Vila, M.; Workman, G.A.; Weaver, M.; Menon, P.M.; Dedhar, S.; Rempel, S.A.; Arap, W.; Pasqualini, R.; et al. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J Biol Chem 2005, 280, 36483–36493. [Google Scholar] [CrossRef]
- Bradshaw, A.D. The role of SPARC in extracellular matrix assembly. J Cell Commun Signal 2009, 3, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: biology and clinical significance. Signal Transduct Target Ther 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.L.; Patel, R.K.; Anderson, A.N.; Bowden, S.G.; Whalen, R.; Giske, N.R.; Wong, M.H. Circulating Cells with Macrophage-like Characteristics in Cancer: The Importance of Circulating Neoplastic-Immune Hybrid Cells in Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Bates, M.; Mohamed, B.M.; Ward, M.P.; Kelly, T.E.; O’Connor, R.; Malone, V.; Brooks, R.; Brooks, D.; Selemidis, S.; Martin, C.; et al. Circulating tumour cells: The Good, the Bad and the Ugly. Biochim Biophys Acta Rev Cancer 2023, 1878, 188863. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 2009, 4, e6562. [Google Scholar] [CrossRef]
- Zanna, M.Y.; Yasmin, A.R.; Omar, A.R.; Arshad, S.S.; Mariatulqabtiah, A.R.; Nur-Fazila, S.H.; Mahiza, M.I.N. Review of Dendritic Cells, Their Role in Clinical Immunology, and Distribution in Various Animal Species. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cellular & Molecular Immunology 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol 2021, 52, 101481. [Google Scholar] [CrossRef] [PubMed]
- Stokic-Trtica, V.; Diefenbach, A.; Klose, C.S.N. NK Cell Development in Times of Innate Lymphoid Cell Diversity. Front Immunol 2020, 11, 813. [Google Scholar] [CrossRef] [PubMed]
- Coenon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell Death Dis 2024, 15, 614. [Google Scholar] [CrossRef]
- Terren, I.; Borrego, F. Role of NK Cells in Tumor Progression. Exp Suppl 2022, 113, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Dorner, B.G.; Smith, H.R.; French, A.R.; Kim, S.; Poursine-Laurent, J.; Beckman, D.L.; Pingel, J.T.; Kroczek, R.A.; Yokoyama, W.M. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol 2004, 172, 3119–3131. [Google Scholar] [CrossRef]
- Ahn, J.; Kim, B.; Bello, A.B.; Moon, J.J.; Arai, Y.; Lee, S.H. Regenerative Functions of Regulatory T Cells and Current Strategies Utilizing Mesenchymal Stem Cells in Immunomodulatory Tissue Regeneration. Tissue Eng Regen Med 2025, 22, 167–180. [Google Scholar] [CrossRef]
- Goldmann, O.; Nwofor, O.V.; Chen, Q.; Medina, E. Mechanisms underlying immunosuppression by regulatory cells. Front Immunol 2024, 15, 1328193. [Google Scholar] [CrossRef]
- Terme, M.; Chaput, N.; Combadiere, B.; Ma, A.; Ohteki, T.; Zitvogel, L. Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+ self-reactive T cells. J Immunol 2008, 180, 4679–4686. [Google Scholar] [CrossRef]
- Zhang, A.; Fan, T.; Liu, Y.; Yu, G.; Li, C.; Jiang, Z. Regulatory T cells in immune checkpoint blockade antitumor therapy. Mol Cancer 2024, 23, 251. [Google Scholar] [CrossRef]
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin Cancer Res 2021, 27, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Davern, M.; Lysaght, J. Cooperation between chemotherapy and immunotherapy in gastroesophageal cancers. Cancer Lett 2020, 495, 89–99. [Google Scholar] [CrossRef]
- Pallares, R.M.; Abergel, R.J. Nanoparticles for targeted cancer radiotherapy. Nano Research 2020, 13, 2887–2897. [Google Scholar] [CrossRef]
- Marotta, C.B.; Haber, T.; Berlin, J.M.; Grubbs, R.H. Surgery-Guided Removal of Ovarian Cancer Using Up-Converting Nanoparticles. ACS Appl Mater Interfaces 2020, 12, 48371–48379. [Google Scholar] [CrossRef]
- Kitsios, K.; Sharifi, S.; Mahmoudi, M. Nanomedicine Technologies for Diagnosis and Treatment of Breast Cancer. ACS Pharmacology & Translational Science 2023, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Paez-Munoz, J.M.; Gamez, F.; Fernandez-Afonso, Y.; Gallardo, R.; Pernia Leal, M.; Gutierrez, L.; de la Fuente, J.M.; Caro, C.; Garcia-Martin, M.L. Optimization of iron oxide nanoparticles for MRI-guided magnetic hyperthermia tumor therapy: reassessing the role of shape in their magnetocaloric effect. J Mater Chem B 2023, 11, 11110–11120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Mishra, P.K.; Talegaonkar, S.; Vaidya, B. Metal nanoparticles: a theranostic nanotool against cancer. Drug Discov Today 2015, 20, 1143–1151. [Google Scholar] [CrossRef]
- Xulu, J.H.; Ndongwe, T.; Ezealisiji, K.M.; Tembu, V.J.; Mncwangi, N.P.; Witika, B.A.; Siwe-Noundou, X. The Use of Medicinal Plant-Derived Metallic Nanoparticles in Theranostics. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Caro, C.; Guzzi, C.; Moral-Sanchez, I.; Urbano-Gamez, J.D.; Beltran, A.M.; Garcia-Martin, M.L. Smart Design of ZnFe and ZnFe@Fe Nanoparticles for MRI-Tracked Magnetic Hyperthermia Therapy: Challenging Classical Theories of Nanoparticles Growth and Nanomagnetism. Adv Healthc Mater 2024, 13, e2304044. [Google Scholar] [CrossRef]
- Caro, C.; Paez-Munoz, J.M.; Pernia Leal, M.; Carayol, M.; Feijoo-Cuaresma, M.; Garcia-Martin, M.L. Metabolically-Driven Active Targeting of Magnetic Nanoparticles Functionalized with Glucuronic Acid to Glioblastoma: Application to MRI-Tracked Magnetic Hyperthermia Therapy. Adv Healthc Mater 2025, 14, e2404391. [Google Scholar] [CrossRef]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in cancer therapy. Signal Transduct Target Ther 2023, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hu, S.; Teng, Y.; Chen, J.; Wang, H.; Xu, Y.; Wang, K.; Xu, J.; Cheng, Y.; Gao, X. Current advance of nanotechnology in diagnosis and treatment for malignant tumors. Signal Transduct Target Ther 2024, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Urbano-Gamez, J.D.; Guzzi, C.; Bernal, M.; Solivera, J.; Martinez-Zubiaurre, I.; Caro, C.; Garcia-Martin, M.L. Tumor versus Tumor Cell Targeting in Metal-Based Nanoparticles for Cancer Theranostics. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T. Smart drug delivery systems: back to the future vs. clinical reality. Int J Pharm 2013, 454, 527–529. [Google Scholar] [CrossRef]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat Nanotechnol 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Prasad, R.; Ghosh, A.; Patel, V.; Peng, B.; Mendes, B.B.; Win, E.H.A.; Delogu, L.G.; Wong, J.Y.; Pischel, K.J.; Bellare, J.R.; et al. Voices of Nanomedicine: Blueprint Guidelines for Collaboration in Addressing Global Unmet Medical Needs. ACS Nano 2025, 19, 2979–2991. [Google Scholar] [CrossRef]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C.W. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef]
- Liu, J.; Kang, S.G.; Wang, P.; Wang, Y.; Lv, X.; Liu, Y.; Wang, F.; Gu, Z.; Yang, Z.; Weber, J.K.; et al. Molecular mechanism of Gd@C(82)(OH)(22) increasing collagen expression: Implication for encaging tumor. Biomaterials 2018, 152, 24–36. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, J.; Li, W.; Yang, W.; Qin, L.; Pan, Y. Gold nanoparticles enhance cisplatin delivery and potentiate chemotherapy by decompressing colorectal cancer vessels. Int J Nanomedicine 2018, 13, 6207–6221. [Google Scholar] [CrossRef]
- Caro, C.; Pourmadadi, M.; Eshaghi, M.M.; Rahmani, E.; Shojaei, S.; Paiva-Santos, A.C.; Rahdar, A.; Behzadmehr, R.; García-Martín, M.L.; Díez-Pascual, A.M. Nanomaterials loaded with Quercetin as an advanced tool for cancer treatment. Journal of Drug Delivery Science and Technology 2022, 78, 103938. [Google Scholar] [CrossRef]
- Hossen, M.N.; Rao, G.; Dey, A.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticle Transforms Activated Cancer-Associated Fibroblasts to Quiescence. ACS Appl Mater Interfaces 2019, 11, 26060–26068. [Google Scholar] [CrossRef]
- Xia, C.; Pan, J.; Wang, J.; Pu, Y.; Zhang, Q.; Hu, S.; Hu, Q.; Wang, Y. Functional blockade of cancer-associated fibroblasts with ultrafine gold nanomaterials causes an unprecedented bystander antitumoral effect. Nanoscale 2020, 12, 19833–19843. [Google Scholar] [CrossRef] [PubMed]
- Grinde, M.T.; Vik, J.; Camilio, K.A.; Martinez-Zubiaurre, I.; Hellevik, T. Ionizing radiation abrogates the pro-tumorigenic capacity of cancer-associated fibroblasts co-implanted in xenografts. Sci Rep 2017, 7, 46714. [Google Scholar] [CrossRef]
- Bromma, K.; Cicon, L.; Beckham, W.; Chithrani, D.B. Gold nanoparticle mediated radiation response among key cell components of the tumour microenvironment for the advancement of cancer nanotechnology. Sci Rep 2020, 10, 12096. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, A.; Di Silvio, D.; Llarena, I.; Murray, R.A.; Marelli, M.; Lay, L.; Polito, L.; Moya, S.E. Influence of surface coating on the intracellular behaviour of gold nanoparticles: a fluorescence correlation spectroscopy study. Nanoscale 2017, 9, 14730–14739. [Google Scholar] [CrossRef]
- Alhussan, A.; Bromma, K.; Bozdogan, E.P.D.; Metcalfe, A.; Karasinska, J.; Beckham, W.; Alexander, A.S.; Renouf, D.J.; Schaeffer, D.F.; Chithrani, D.B. Investigation of Nano-Bio Interactions within a Pancreatic Tumor Microenvironment for the Advancement of Nanomedicine in Cancer Treatment. Curr Oncol 2021, 28, 1962–1979. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials 2016, 1, 16014. [Google Scholar] [CrossRef]
- Pernia Leal, M.; Caro, C.; Garcia-Martin, M.L. Shedding light on zwitterionic magnetic nanoparticles: limitations for in vivo applications. Nanoscale 2017, 9, 8176–8184. [Google Scholar] [CrossRef] [PubMed]
- Alhussan, A.; Jackson, N.; Calisin, R.; Morgan, J.; Beckham, W.; Chithrani, D.B. Utilizing Gold Nanoparticles as Prospective Radiosensitizers in 3D Radioresistant Pancreatic Co-Culture Model. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Kovacs, D.; Igaz, N.; Marton, A.; Ronavari, A.; Belteky, P.; Bodai, L.; Spengler, G.; Tiszlavicz, L.; Razga, Z.; Hegyi, P.; et al. Core-shell nanoparticles suppress metastasis and modify the tumour-supportive activity of cancer-associated fibroblasts. J Nanobiotechnology 2020, 18, 18. [Google Scholar] [CrossRef]
- Christou, E.; Pearson, J.R.; Beltran, A.M.; Fernandez-Afonso, Y.; Gutierrez, L.; de la Fuente, J.M.; Gamez, F.; Garcia-Martin, M.L.; Caro, C. Iron-Gold Nanoflowers: A Promising Tool for Multimodal Imaging and Hyperthermia Therapy. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Boluda, A.; Vaquero, J.; Laurent, G.; Renault, G.; Bazzi, R.; Donnadieu, E.; Roux, S.; Fouassier, L.; Gazeau, F. Photothermal Depletion of Cancer-Associated Fibroblasts Normalizes Tumor Stiffness in Desmoplastic Cholangiocarcinoma. ACS Nano 2020, 14, 5738–5753. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, M.; Li, S.; Zhang, Z.; Sun, H.; Yang, F.; Liang, H. Developing a Novel Anticancer Gold(III) Agent to Integrate Chemotherapy and Immunotherapy. J Med Chem 2021, 64, 6777–6791. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Nicolas-Boluda, A.; Pinto, A.; Balfourier, A.; Carn, F.; Silva, A.K.A.; Pocard, M.; Gazeau, F. Tumor-Selective Immune-Active Mild Hyperthermia Associated with Chemotherapy in Colon Peritoneal Metastasis by Photoactivation of Fluorouracil-Gold Nanoparticle Complexes. ACS Nano 2021, 15, 3330–3348. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Ruan, S.; Liu, J.; Qin, L.; Yang, C.; Tong, F.; Lei, T.; Shevtsov, M.; Gao, H.; Qin, Y. Furin-instructed aggregated gold nanoparticles for re-educating tumor associated macrophages and overcoming breast cancer chemoresistance. Biomaterials 2021, 275, 120891. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.; Chang, L.C.; Song, W.H.; Yang, L.X.; Wang, L.C.; Chia, Z.C.; Chin, Y.C.; Shan, Y.S.; Huang, C.C.; Yeh, C.S. Polyaniline-Based Glyco-Condensation on Au Nanoparticles Enhances Immunotherapy in Lung Cancer. ACS Appl Mater Interfaces 2022, 14, 24144–24159. [Google Scholar] [CrossRef]
- Hao, Y.; Duan, F.; Dong, X.; Bi, R.; Wang, Y.; Zhu, S.; Hu, J. Gold Nanoparticle Inhibits the Tumor-Associated Macrophage M2 Polarization by Inhibiting m(6)A Methylation-Dependent ATG5/Autophagy in Prostate Cancer. Anal Cell Pathol (Amst) 2025, 2025, 6648632. [Google Scholar] [CrossRef]
- Sykes, E.A.; Chen, J.; Zheng, G.; Chan, W.C. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano 2014, 8, 5696–5706. [Google Scholar] [CrossRef]
- Nath, A.; Pal, R.; Singh, L.M.; Saikia, H.; Rahaman, H.; Ghosh, S.K.; Mazumder, R.; Sengupta, M. Gold-manganese oxide nanocomposite suppresses hypoxia and augments pro-inflammatory cytokines in tumor associated macrophages. Int Immunopharmacol 2018, 57, 157–164. [Google Scholar] [CrossRef]
- Du, C.; Jiang, J.; Wan, C.; Pan, G.; Kong, F.; Zhai, R.; Hu, C.; Ying, H. AntiPD-L1 antibody conjugated Au-SPIOs nanoplatform for enhancing radiosensitivity and triggering anti-tumor immune response. Sci Rep 2022, 12, 19542. [Google Scholar] [CrossRef]
- Huang, A.; Li, Q.; Shi, X.; Gao, J.; Ma, Y.; Ding, J.; Hua, S.; Zhou, W. An iron-containing nanomedicine for inducing deep tumor penetration and synergistic ferroptosis in enhanced pancreatic cancer therapy. Mater Today Bio 2024, 27, 101132. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Guo, Z.; Gao, F.; Gao, Q.; Wang, D.; Liaw, B.S.; Cai, Q.; Sun, X.; Wang, X.; Zhao, L. Shape-, size- and structure-controlled synthesis and biocompatibility of iron oxide nanoparticles for magnetic theranostics. Theranostics 2018, 8, 3284–3307. [Google Scholar] [CrossRef]
- Lee, C.; Kim, G.R.; Yoon, J.; Kim, S.E.; Yoo, J.S.; Piao, Y. In vivo delineation of glioblastoma by targeting tumor-associated macrophages with near-infrared fluorescent silica coated iron oxide nanoparticles in orthotopic xenografts for surgical guidance. Sci Rep 2018, 8, 11122. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Dong, Z.; Fu, Y.; Gong, T.; Deng, L.; Zhang, Z. Hyaluronic acid modified doxorubicin loaded Fe(3)O(4) nanoparticles effectively inhibit breast cancer metastasis. J Mater Chem B 2019, 7, 5861–5872. [Google Scholar] [CrossRef]
- Wu, X.; Cheng, Y.; Zheng, R.; Xu, K.; Yan, J.; Song, P.; Wang, Y.; Rauf, A.; Pan, Y.; Zhang, H. Immunomodulation of Tumor Microenvironment by Arginine-Loaded Iron Oxide Nanoparticles for Gaseous Immunotherapy. ACS Appl Mater Interfaces 2021, 13, 19825–19835. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; He, Y.; Wu, C.; Zhang, M.; Gu, Z.; Zhang, J.; Liu, E.; Xu, Q.; Asrorov, A.M.; Huang, Y. Magnetism-mediated targeting hyperthermia-immunotherapy in “cold” tumor with CSF1R inhibitor. Theranostics 2021, 11, 6860–6872. [Google Scholar] [CrossRef]
- Nascimento, C.; Castro, F.; Domingues, M.; Lage, A.; Alves, E.; de Oliveira, R.; de Melo, C.; Eduardo Calzavara-Silva, C.; Sarmento, B. Reprogramming of tumor-associated macrophages by polyaniline-coated iron oxide nanoparticles applied to treatment of breast cancer. Int J Pharm 2023, 636, 122866. [Google Scholar] [CrossRef]
- Hu, A.; Pu, Y.; Xu, N.; Cai, Z.; Sun, R.; Fu, S.; Jin, R.; Guo, Y.; Ai, H.; Nie, Y.; et al. Controlled intracellular aggregation of magnetic particles improves permeation and retention for magnetic hyperthermia promotion and immune activation. Theranostics 2023, 13, 1454–1469. [Google Scholar] [CrossRef]
- Darya, G.H.; Zare, O.; Karbalaei-Heidari, H.R.; Zeinali, S.; Sheardown, H.; Rastegari, B. Enzyme-responsive mannose-grafted magnetic nanoparticles for breast and liver cancer therapy and tumor-associated macrophage immunomodulation. Expert Opin Drug Deliv 2024, 21, 663–677. [Google Scholar] [CrossRef]
- Arnosa-Prieto, A.; Diaz-Rodriguez, P.; Gonzalez-Gomez, M.A.; Garcia-Acevedo, P.; de Castro-Alves, L.; Pineiro, Y.; Rivas, J. Magnetic-driven Interleukin-4 internalization promotes magnetic nanoparticle morphology and size-dependent macrophage polarization. J Colloid Interface Sci 2024, 655, 286–295. [Google Scholar] [CrossRef]
- Sun, H.; Wang, X.; Guo, Z.; Hu, Z.; Yin, Y.; Duan, S.; Jia, W.; Lu, W.; Hu, J. Fe(3)O(4) Nanoparticles That Modulate the Polarisation of Tumor-Associated Macrophages Synergize with Photothermal Therapy and Immunotherapy (PD-1/PD-L1 Inhibitors) to Enhance Anti-Tumor Therapy. Int J Nanomedicine 2024, 19, 7185–7200. [Google Scholar] [CrossRef] [PubMed]
- Roudi, R.; Pisani, L.; Pisani, F.; Kiru, L.; Daldrup-Link, H.E. Novel Clinically Translatable Iron Oxide Nanoparticle for Monitoring Anti-CD47 Cancer Immunotherapy. Invest Radiol 2024, 59, 391–403. [Google Scholar] [CrossRef]
- Zhan, S.; Cao, Z.; Li, J.; Chen, F.; Lai, X.; Yang, W.; Teng, Y.; Li, Z.; Zhang, W.; Xie, J. Iron Oxide Nanoparticles Induce Macrophage Secretion of ATP and HMGB1 to Enhance Irradiation-Led Immunogenic Cell Death. Bioconjug Chem 2025, 36, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Fu, Y.; Li, C.; Wu, Z.; Cao, W.; Hu, X.; Sun, X.; He, W.; Cao, X.; Ling, D.; et al. Metal-Organic Framework Nanoparticles for Ameliorating Breast Cancer-Associated Osteolysis. Nano Lett 2020, 20, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; He, Y.; Huang, X.; Shen, Y.; Zou, Q.; Yang, G.; Fu, L.; Liu, Q.; Luo, D. Photosensitive and dual-targeted chromium nanoparticle delivering small interfering RNA YTHDF1 for molecular-targeted immunotherapy in liver cancer. J Nanobiotechnology 2024, 22, 348. [Google Scholar] [CrossRef]
- Yu, S.; Wang, S.; Wang, X.; Xu, X. The axis of tumor-associated macrophages, extracellular matrix proteins, and cancer-associated fibroblasts in oncogenesis. Cancer Cell Int 2024, 24, 335. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Khafaga, A.F.; Gaballa, M.M.S.; Karam, R.; Shoulah, S.A.; Shamma, R.N.; Khalifa, N.E.; Farrag, N.E.; Noreldin, A.E. Synergistic therapeutic strategies and engineered nanoparticles for anti-vascular endothelial growth factor therapy in cancer. Life Sci 2024, 341, 122499. [Google Scholar] [CrossRef]
- Setyawati, M.I.; Wang, Q.; Ni, N.; Tee, J.K.; Ariga, K.; Ke, P.C.; Ho, H.K.; Wang, Y.; Leong, D.T. Engineering tumoral vascular leakiness with gold nanoparticles. Nat Commun 2023, 14, 4269. [Google Scholar] [CrossRef]
- Luo, L.; Qin, B.; Jiang, M.; Xie, L.; Luo, Z.; Guo, X.; Zhang, J.; Li, X.; Zhu, C.; Du, Y.; et al. Regulating immune memory and reversing tumor thermotolerance through a step-by-step starving-photothermal therapy. J Nanobiotechnology 2021, 19, 297. [Google Scholar] [CrossRef]
- Xu, X.; Gan, M.; Ge, Y.; Yi, C.; Feng, T.; Liu, M.; Wu, C.; Chen, X.; Zhang, W.; Zhao, L.; et al. Multifaceted glycoadjuvant@AuNPs inhibits tumor metastasis through promoting T cell activation and remodeling tumor microenvironment. J Nanobiotechnology 2021, 19, 376. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Li, L.; Guo, R.; Shi, X.; Cao, X. Effective CpG Delivery Using Zwitterion-Functionalized Dendrimer-Entrapped Gold Nanoparticles to Promote T Cell-Mediated Immunotherapy of Cancer Cells. Biosensors (Basel) 2022, 12. [Google Scholar] [CrossRef]
- Felix-Pina, P.; Franco Molina, M.A.; Garcia Coronado, P.L.; Prado-Garcia, H.; Zarate-Trivino, D.G.; Castro-Valenzuela, B.E.; Moreno-Amador, K.A.; Uscanga Palomeque, A.C.; Rodriguez Padilla, C. beta-D-Glucose-Reduced Silver Nanoparticles Remodel the Tumor Microenvironment in a Murine Model of Triple-Negative Breast Cancer. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Sargsian, A.; Koutsoumpou, X.; Girmatsion, H.; Egil, C.; Buttiens, K.; Luci, C.R.; Soenen, S.J.; Manshian, B.B. Silver nanoparticle induced immunogenic cell death can improve immunotherapy. J Nanobiotechnology 2024, 22, 691. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Xia, Q.; Shang, J.; He, Y.; Li, Z.; Chen, Y.; Gao, F.; Yu, X.; Yuan, Z.; et al. Photothermal Fe(3)O(4) nanoparticles induced immunogenic ferroptosis for synergistic colorectal cancer therapy. J Nanobiotechnology 2024, 22, 630. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wang, G.; Sang, W.; Li, J.; Zhang, Z.; Li, W.; Yan, J.; Zhao, Q.; Dai, Y. Phenolic immunogenic cell death nanoinducer for sensitizing tumor to PD-1 checkpoint blockade immunotherapy. Biomaterials 2021, 269, 120638. [Google Scholar] [CrossRef]
- Lei, H.; Li, Q.; Li, G.; Wang, T.; Lv, X.; Pei, Z.; Gao, X.; Yang, N.; Gong, F.; Yang, Y.; et al. Manganese molybdate nanodots with dual amplification of STING activation for “cycle” treatment of metalloimmunotherapy. Bioact Mater 2024, 31, 53–62. [Google Scholar] [CrossRef]
- Lu, S.; Mi, Z.; Liu, P.; Ding, J.; Ma, Y.; Yang, J.; Rong, P.; Zhou, W. Repolarizing neutrophils via MnO(2) nanoparticle-activated STING pathway enhances Salmonella-mediated tumor immunotherapy. J Nanobiotechnology 2024, 22, 443. [Google Scholar] [CrossRef]
- Zhao, Q.; Gong, Z.; Li, Z.; Wang, J.; Zhang, J.; Zhao, Z.; Zhang, P.; Zheng, S.; Miron, R.J.; Yuan, Q.; et al. Target Reprogramming Lysosomes of CD8+ T Cells by a Mineralized Metal-Organic Framework for Cancer Immunotherapy. Adv Mater 2021, 33, e2100616. [Google Scholar] [CrossRef]
- Dai, L.; Yao, M.; Fu, Z.; Li, X.; Zheng, X.; Meng, S.; Yuan, Z.; Cai, K.; Yang, H.; Zhao, Y. Multifunctional metal-organic framework-based nanoreactor for starvation/oxidation improved indoleamine 2,3-dioxygenase-blockade tumor immunotherapy. Nat Commun 2022, 13, 2688. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Q.; Xu, R.; Lin, P.; Deng, G.; Xia, X. Combination of ferroptosis and pyroptosis dual induction by triptolide nano-MOFs for immunotherapy of Melanoma. J Nanobiotechnology 2023, 21, 383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Wang, W.J.; Zhang, C.Y.; Ling, Y.Y.; Hong, X.J.; Su, Q.; Li, W.G.; Mao, Z.W.; Cheng, B.; Tan, C.P.; et al. Ru(II)-modified TiO(2) nanoparticles for hypoxia-adaptive photo-immunotherapy of oral squamous cell carcinoma. Biomaterials 2022, 289, 121757. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; Lee, W.; Hsu, F.T.; Lai, Y.H.; Huang, S.R.; Lim, C.S.H.; Lin, Z.K.; Hsu, S.C.; Chiang, C.S.; Jeng, L.B.; et al. Manipulating the Crosstalk between Cancer and Immunosuppressive Cells with Phototherapeutic Gold-Nanohut for Reprogramming Tumor Microenvironment. Adv Sci (Weinh) 2024, 11, e2404347. [Google Scholar] [CrossRef] [PubMed]
- Caro, C.; Pozo, D. Polysaccharide Colloids as Smart Vehicles in Cancer Therapy. Curr Pharm Des 2015, 21, 4822–4836. [Google Scholar] [CrossRef]
- Li, C.; Clauson, R.; Bugada, L.F.; Ke, F.; He, B.; Yu, Z.; Chen, H.; Jacobovitz, B.; Hu, H.; Chuikov, P.; et al. Antigen-Clustered Nanovaccine Achieves Long-Term Tumor Remission by Promoting B/CD 4 T Cell Crosstalk. ACS Nano 2024, 18, 9584–9604. [Google Scholar] [CrossRef]
Figure 1.
The TME plays a critical role in cancer progression and metastasis. It consists of various components, including immune cells, CAFs, endothelial cells, and the ECM, which interact with the tumor and evolve alongside it. Initially, the normal tissue environment can suppress tumor growth, but as cancer progresses, it manipulates the TME to support tumor development by promoting cell proliferation, invasion, and spread. The TME also contributes to preparing distant sites for metastasis, aiding cancer cell survival in the bloodstream, and supporting metastatic colonization and outgrowth. Copyright (2023) Elsevier as referenced in [10].
Figure 1.
The TME plays a critical role in cancer progression and metastasis. It consists of various components, including immune cells, CAFs, endothelial cells, and the ECM, which interact with the tumor and evolve alongside it. Initially, the normal tissue environment can suppress tumor growth, but as cancer progresses, it manipulates the TME to support tumor development by promoting cell proliferation, invasion, and spread. The TME also contributes to preparing distant sites for metastasis, aiding cancer cell survival in the bloodstream, and supporting metastatic colonization and outgrowth. Copyright (2023) Elsevier as referenced in [10].
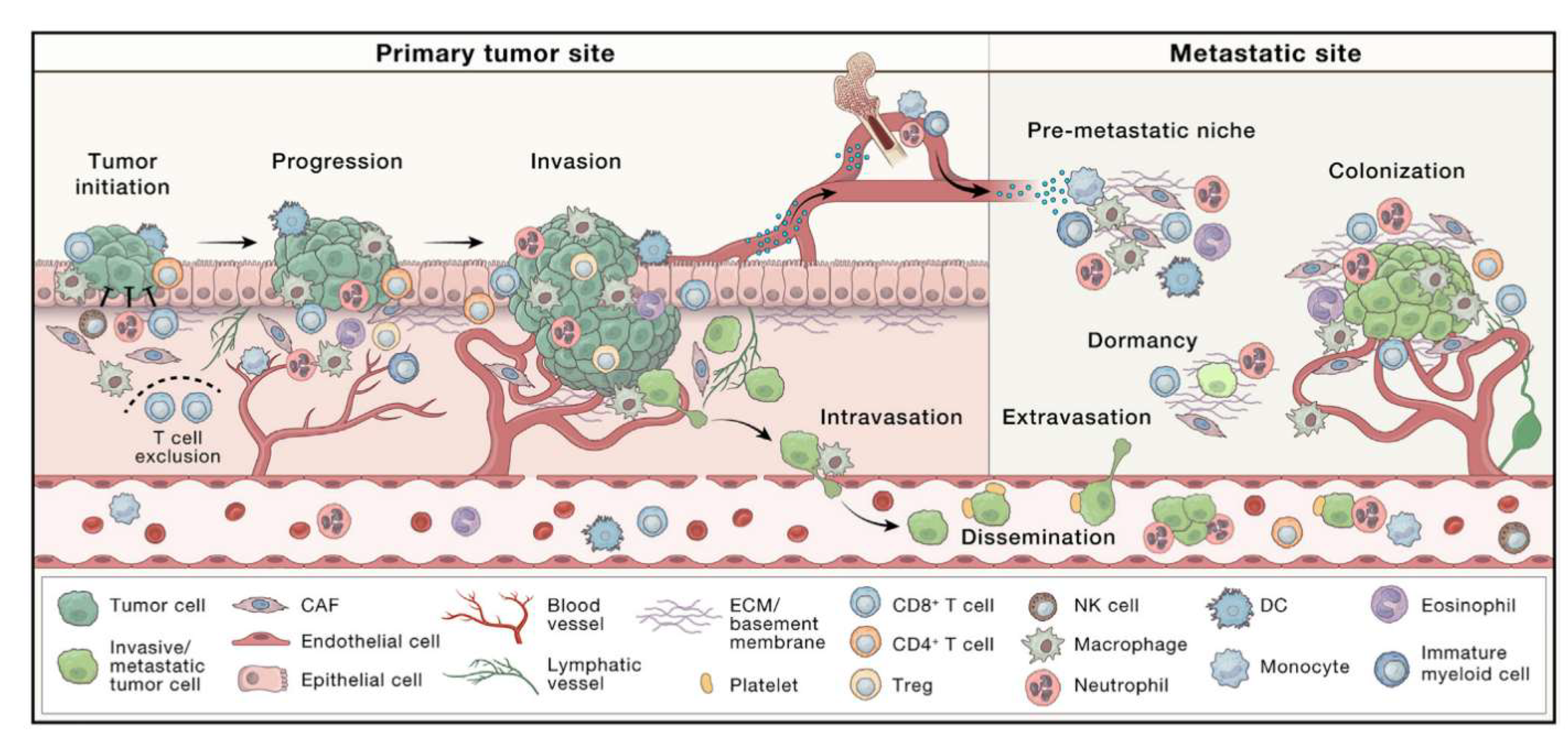
Figure 2.
CAFs have dynamic, bidirectional interactions with tumor cells and other TME components (namely, the immune and vascular systems). These interactions, while occurring in close physical proximity, are functionally compartmentalized at the cellular level. Communication occurs through secreted factors (secretomes), autocrine/paracrine/angiocrine signaling, and direct cell-to-cell contact. All three TME components influence each other and the tumor, and these interactions can change in real time (for example), in response to drug treatment (potentially contributing to therapy resistance). The diagram also highlights CAF heterogeneity, showing their complex and distinct roles within the TME. Copyright (2021) MDPI as referenced in [17].
Figure 2.
CAFs have dynamic, bidirectional interactions with tumor cells and other TME components (namely, the immune and vascular systems). These interactions, while occurring in close physical proximity, are functionally compartmentalized at the cellular level. Communication occurs through secreted factors (secretomes), autocrine/paracrine/angiocrine signaling, and direct cell-to-cell contact. All three TME components influence each other and the tumor, and these interactions can change in real time (for example), in response to drug treatment (potentially contributing to therapy resistance). The diagram also highlights CAF heterogeneity, showing their complex and distinct roles within the TME. Copyright (2021) MDPI as referenced in [17].
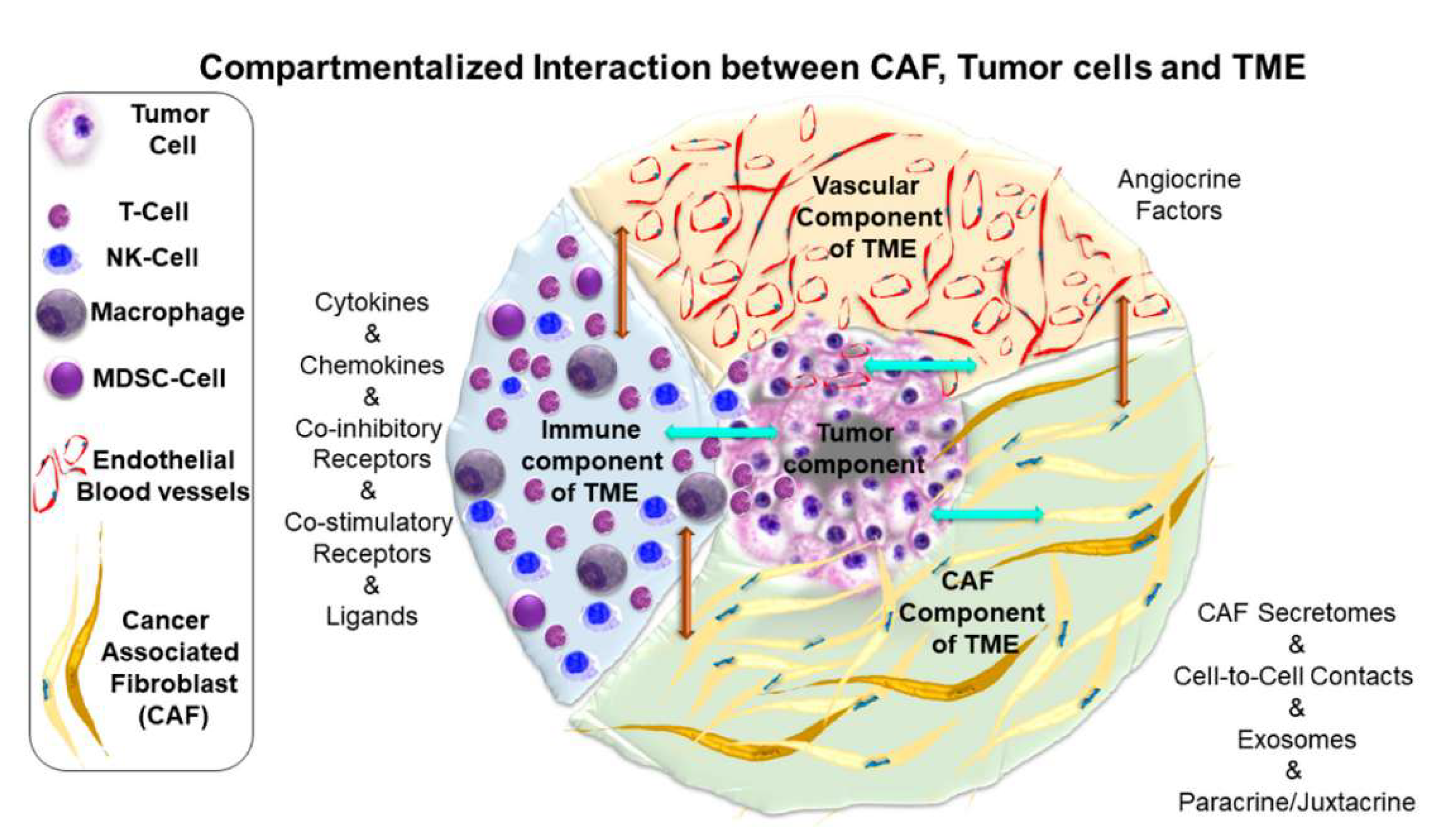
Figure 3.
Schematic illustration of the preparation and mechanism of action for the PTFE. Ferroptosis inducer Erastin is encapsulated by a MOF shell formed by Fe3+ coordination with TA to eventually form PTFE. PTFE regulates TAFs and tumor dense stroma by inducing macrophage polarization, thereby improving nanoparticle permeability. Meanwhile, PTFE disrupts the redox balance at the tumor site and induces pronounced ferroptotic damage. Copyright (2024) Elsevier as referenced in [113].
Figure 3.
Schematic illustration of the preparation and mechanism of action for the PTFE. Ferroptosis inducer Erastin is encapsulated by a MOF shell formed by Fe3+ coordination with TA to eventually form PTFE. PTFE regulates TAFs and tumor dense stroma by inducing macrophage polarization, thereby improving nanoparticle permeability. Meanwhile, PTFE disrupts the redox balance at the tumor site and induces pronounced ferroptotic damage. Copyright (2024) Elsevier as referenced in [113].
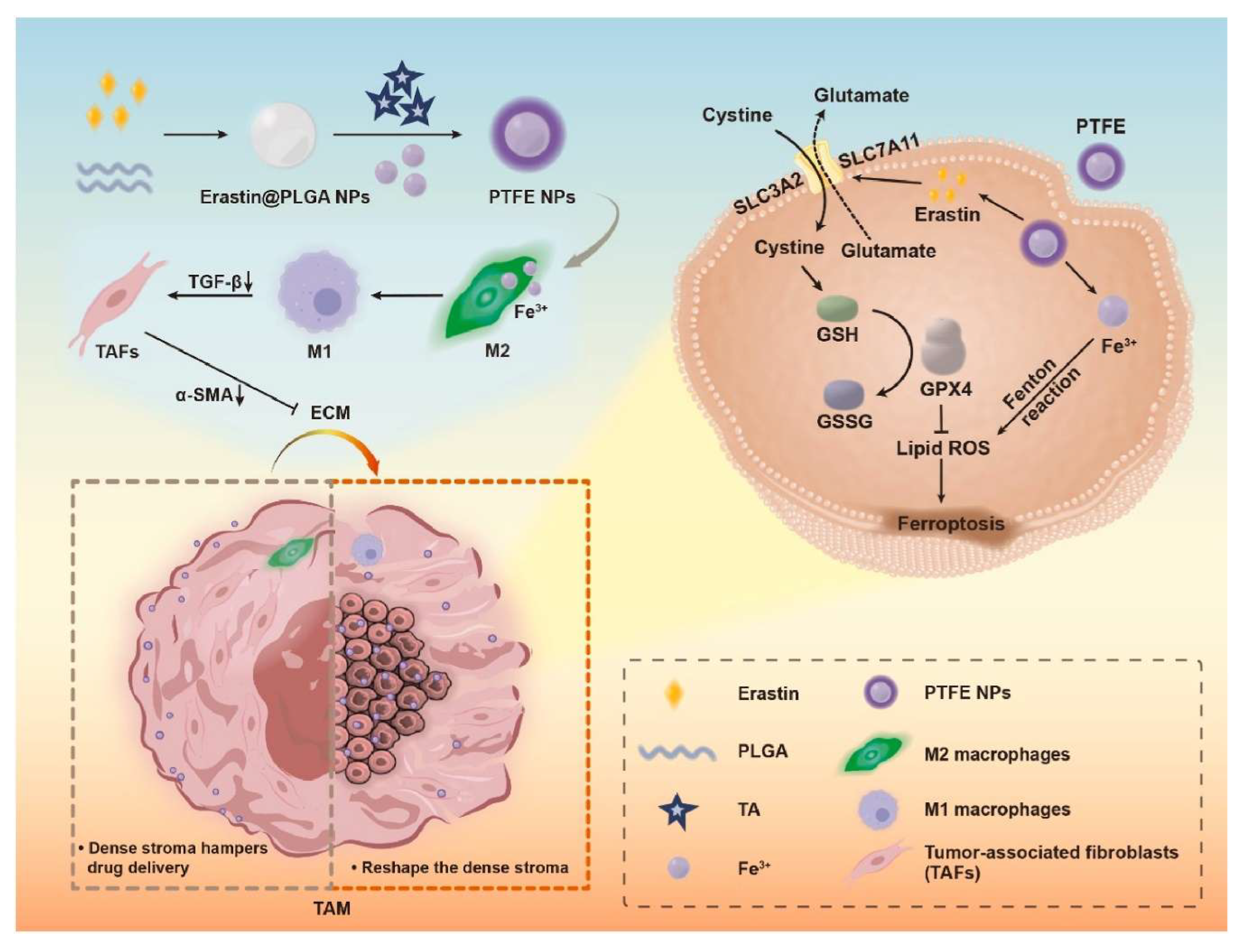
Figure 4.
Photoimmunotherapy with AuNH-Ab treatment. a) Schematic illustration of the immune response during AuNH-2-Ab treatment. Quantitative assessment of immune cells including b) DC (CD11c+CD40+ CD11c+CD80+ and CD11c+CD86+), c) APC (CD11c+MHC-II+), d) TAMs, e) Tregs, f) T cytotoxic cells (CD3+CD8+, CD45+CD8+ IFN-γ), g) T helper cells (CD3+CD4+, CD45+CD4+IFN-γ), h) Granzyme B+ positive cell, and i) memory T cells (CD8+CD44+IFN-γ) in the TIME after various treatments at four weeks after tumor inoculation. All data were presented in mean ± SD. One-way ANOVA with the Tukey’s post-hoc test; n = 6. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control group and #p < 0.05, ##p < 0.01 and ###p < 0.001 between groups. Copyright (2024) Wiley as referenced in [145].
Figure 4.
Photoimmunotherapy with AuNH-Ab treatment. a) Schematic illustration of the immune response during AuNH-2-Ab treatment. Quantitative assessment of immune cells including b) DC (CD11c+CD40+ CD11c+CD80+ and CD11c+CD86+), c) APC (CD11c+MHC-II+), d) TAMs, e) Tregs, f) T cytotoxic cells (CD3+CD8+, CD45+CD8+ IFN-γ), g) T helper cells (CD3+CD4+, CD45+CD4+IFN-γ), h) Granzyme B+ positive cell, and i) memory T cells (CD8+CD44+IFN-γ) in the TIME after various treatments at four weeks after tumor inoculation. All data were presented in mean ± SD. One-way ANOVA with the Tukey’s post-hoc test; n = 6. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control group and #p < 0.05, ##p < 0.01 and ###p < 0.001 between groups. Copyright (2024) Wiley as referenced in [145].
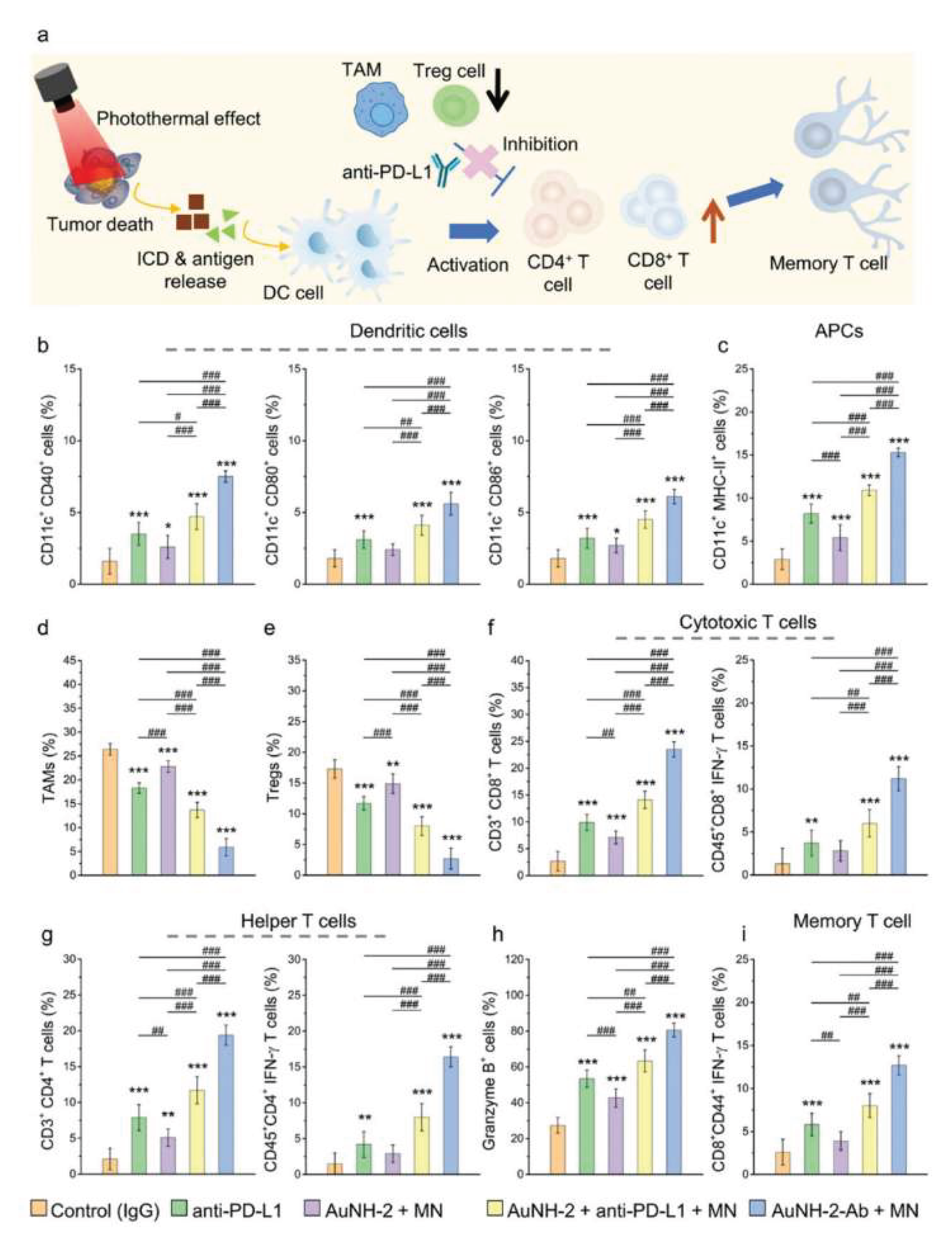
Figure 5.
A schematic representation highlighting the current, albeit partial, understanding (illustrated by yellow shading) and existing knowledge gaps (depicted by grey shading) regarding the interaction between mNPs and non-malignant cells within the TME, both in cell culture and in vivo, as well as the subsequent effects on cancer cells.
Figure 5.
A schematic representation highlighting the current, albeit partial, understanding (illustrated by yellow shading) and existing knowledge gaps (depicted by grey shading) regarding the interaction between mNPs and non-malignant cells within the TME, both in cell culture and in vivo, as well as the subsequent effects on cancer cells.
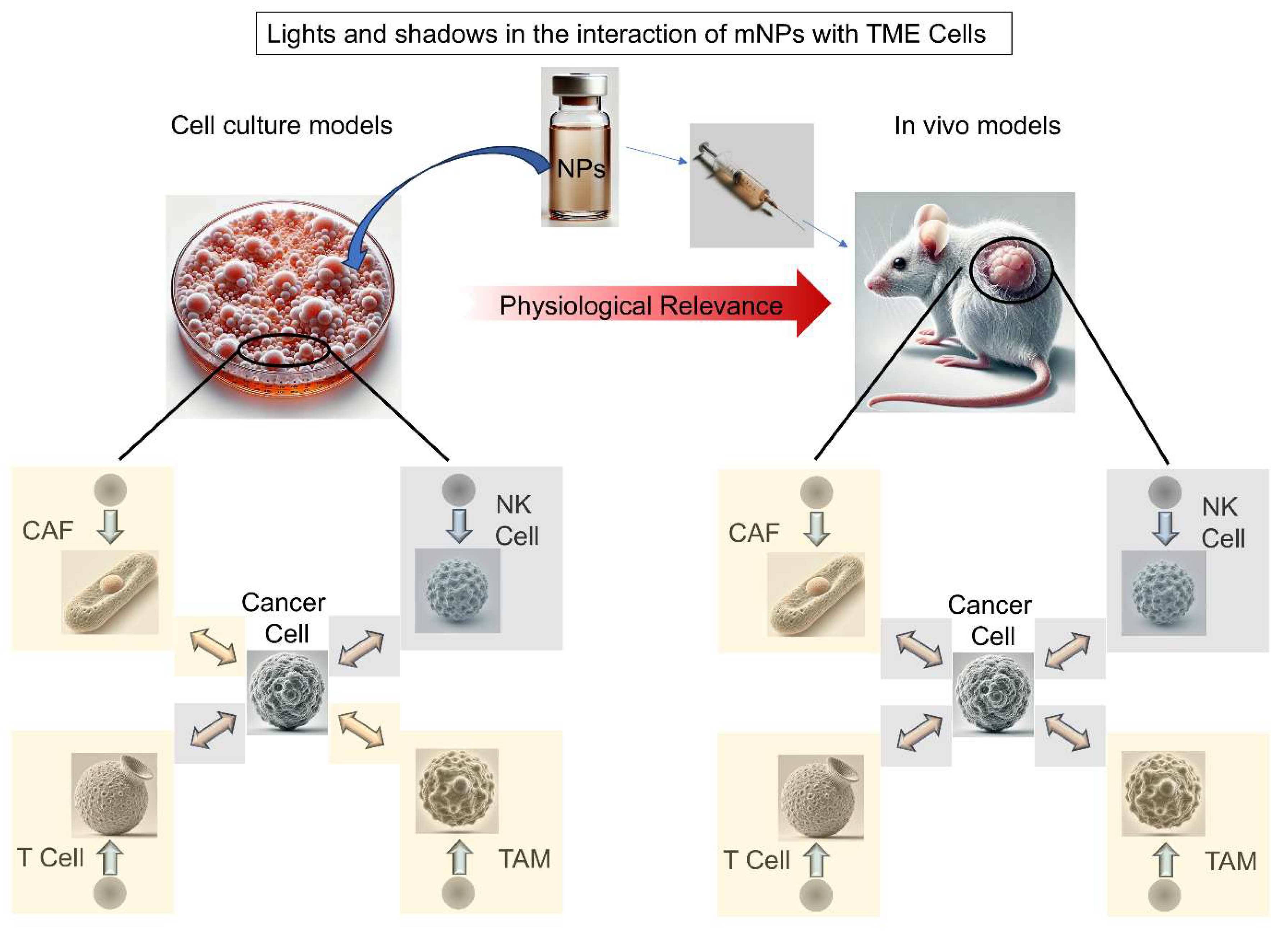
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



