Submitted:
09 January 2026
Posted:
12 January 2026
You are already at the latest version
Abstract
Background: MicroRNAs (miRNAs) are an important class of non-coding RNAs that regulate gene expression by binding to target mRNAs and influencing cellular processes such as differentiation, proliferation, and apoptosis. Dysregulation in miRNA expression has been reported to be implicated in many human diseases, including cancer, cardiovascular, and neurodegenerative disorders. Identifying disease-related miRNAs is therefore essential for understanding disease mechanisms and supporting biomarker discovery, but time and costs of experimental validation are the main limitations. Methods: We present a graph-based learning framework that models the complex relationships between miRNAs, diseases, and related biological entities within a heterogeneous network. The model employs a message-passing neural architecture to learn structured embeddings from multiple node and edge types, integrating biological priors from curated resources. This network representation enables the inference of novel miRNA–disease associations, even in sparsely annotated regions of the network. The approach was trained and validated on a dataset benchmark using ten replicated experiments to ensure robustness. Results: The method achieved an average AUC–ROC of ~98%, outperforming previously reported computational approaches on the same dataset. Moreover, predictions were consistent across validation folds and robustness analyses were conducted to evaluate stability and highlight the most important information. Conclusions: Integrating heterogeneous biological information and representing them through graph neural representation learning offers a powerful and generalizable way to predict relevant associations, including miRNA–disease, and provide a robust computational framework to support biomedical discovery and translational research.
Keywords:
microRNA
; miRNA–disease association
; graph neural network
1. Introduction
MicroRNAs (miRNAs) are short, endogenous RNA molecules, usually 19–25 nucleotides long that play a key role in regulating gene expression [1,2,3]. They act by incorporating into the RISC complex, which then binds to target messenger RNAs (mRNAs) at the 3′ untranslated regions (UTRs) through sequence complementarity, leading to gene silencing [4]. The resulting interaction normally leads to the inhibition of the target mRNA [5], although cases of translational activation have also been found in literature [6]. In this way a single miRNA is able to regulate hundreds of gene transcripts, and, on a genome-wide scale, it is estimated that miRNAs are able to control expression of up to 60% of genes in the human genome [7], affecting virtually every physiological process. Starting from the initial discoveries of lin-4 and let-7 [8], the number of known miRNAs has increased rapidly in recent years, with the latest miRbase (Release 22.1) counting about 38,000 entries [9], underlining the evolutionary conservation and functional significance of these regulators. Dysregulation of miRNAs is associated to the pathogenesis of numerous complex human diseases [10], in particularly in cancer [11], cardiovascular [12], neurodegenerative [13], and metabolic diseases [14]. For these reasons, identifying specific miRNA–disease associations (MDAs) is a useful step for understand disease mechanisms and for developing novel therapeutic strategies [15]. Limitations of experimental approaches (PCR and high-throughput sequencing), which are typically resource-intensive, expensive, and time-consuming on a large scale [16], have motivated the development of computational methods to predict potential miRNA–disease associations (MDAs) [17,18].
Many approaches exploit the large amount of of public data available today (like HMDD V2.0/V3.0 [19,20], dbDEMC [21], and miR2Disease [22] rely on the widely accepted principle that functionally similar miRNAs are likely to be associated with diseases exhibiting similar phenotypes [23,24].
Early computational approaches, which inferred MDAs by leveraging known interactions between miRNAs and their target genes or between target genes and diseases, often suffered from the incompleteness and noise of current miRNA–target interaction datasets. This category includes models based on Random Walk over protein–protein interaction (PPI) networks [25] and methods like miRPD [26] that uses intermediate networks to identify functional links between miRNAs and diseases.
To overcome these limitations, similarity-based network models were developed [27,28,29,30,31,32,33], integrating miRNA functional and disease semantic or phenotypic similarities with known MDAs. Approaches like HDMP [34], used local similarity metrics, which proved ineffective for diseases lacking any known associated miRNAs ("new diseases"). This led to the development of sophisticated global network methods, for example those employing the Random Walk with Restart (RWR) algorithm (RWRMDA [35], MIDP/MIDPE [36]). By traversing the entire network RWR, provides a global view of connectivity, significantly improving performance. More advanced approaches involved integrating Gaussian Interaction Profile (GIP) Kernel similarity with functional/semantic similarity. Examples in this categories include WBSMDA [37] and HGIMDA [38], allowed for the calculation of similarity for new entities (miRNA or diseases) without prior associations, marking a significant leap toward predicting associations for both new miRNAs and new diseases.
Modern Machine Learning (ML) techniques provided more powerful tools to approach MDA prediction [39,40,41]. They range from supervised classifiers like Support Vector Machines (SVMs) [42] and Restricted Boltzmann Machines (RBMs) [43] to semi-supervised methods. A critical challenge for supervised learning is the difficulty in accurately obtaining reliable negative MDA samples. Addressing this, semi-supervised models like RLSMDA (Regularized Least Squares [44]) and Matrix Completion (MC) methods, such as MCMDA [45] were proposed. MCMDA, for instance, is highly efficient, operating only on the known positive MDA matrix by leveraging the assumption that the underlying adjacence matrix is low-rank, thereby inherently avoiding the need for negative samples. The high predictive power of MC methods was demonstrated by MCMDA, which achieved high AUC (87.49%) and a strong confirmation rate (up to 90% of top 50 predictions for diseases like prostate neoplasms). More recently, ensemble learning approaches such as ELMDA [46] have been proposed, which do not rely on known associations to calculate miRNA and disease similarities and use multi-classifier voting for prediction, achieving an average AUC of 92.29% on HMDD v2.0, confirming the potential of ensemble strategies in accurately predicting disease-associated miRNAs.
The continuous development of these models now involves various forms of Deep Learning and Network Embedding and Graph Attention Networks (GAT) [47,48,49] to capture complex, non-linear relationships within the integrated biological data.
Despite advances in computational prediction of miRNA–disease associations, key challenges remain. In particular, integrating heterogeneous biological data and capturing complex, non-linear relationships across miRNAs, diseases, and associated patterns is still difficult. Furthermore, limitations in data completeness and the dynamic nature of biological networks constrain model generalizability. Graph-based approaches, especially those leveraging message passing on heterogeneous networks, offer a natural framework to address these issues by propagating information across nodes and edges of multiple types, effectively learning embeddings that encode functional and phenotypic similarities.
In this work, we propose a Heterogeneous Graph Neural Network that models miRNA-disease associations by leveraging a multi-node, multi-edge approach to integrate diverse sources of information. Our model employs message passing and node-specific linear transformations to generate embeddings, enabling accurate prediction of miRNA–disease associations while efficiently handling new entities and heterogeneous network structures. Comparison with existing methods from the literature demonstrates improved performance in terms of AUC ROC in the prediction of true miRNA-disease associations.
2. Materials and Methods
2.1. Dataset
The dataset used in this study integrates multiple layers of biological information derived from curated repositories, ontology mapping, and sequence-level analyses. Experimentally validated miRNA–disease associations were obtained from the HMDD database [19,20] (version 2, 3.2 and 4). All miRNA identifiers were manually curated to ensure consistency across resources, including resolving deprecated or ambiguous names, normalizing letter case, and harmonizing naming conventions. The nucleotide sequences of all miRNAs were retrieved from miRBase [9].
Disease names reported in HMDD were manually normalized to match DisGeNET [50] terminology. This included removing formatting inconsistencies, resolving synonyms, and applying uniform rules before mapping each disease to its corresponding UMLS Concept Unique Identifier (CUI) [51]. Based on these CUIs, disease–gene associations were obtained from DisGeNET and represented as binary vectors indicating the presence or absence of gene relationships for each disease.
Similarity graph between miRNAs were computed using pairwise sequence alignments obtained with the well known Needleman-Wunsch method [52]. Two miRNAs were considered similar if their alignment identity score is more than 60%, generating a binary miRNA–miRNA adjacency matrix. Furthermore, k-mer frequency vectors (k = 2 and 3) were extracted to obtain sequence-based embeddings. Finally short sequence motifs of length 5 were computed to derive miRNA–pattern matrix, providing an additional sequence-derived relational layer.
For HMDD version 4 the final dataset consists of 1183 miRNAs and 2114 distinct diseases, collectively forming 24074 miRNA–disease positive associations (0.96% matrix density). After harmonizing disease names with DisGeNET, we obtained a set of 6356 genes, which resulted in 18653 disease–gene associations mapped through UMLS CUIs. Sequence alignment produced miRNA–miRNA adjacency with 209186 entries, while the k-mer analysis yielded a structured representation of each miRNA through 80 embedding features. The extraction of motifs of length 4 generated 256 distinct patterns, leading to moderately dense adjacency matrices: 73515 miRNA–pattern associations (~24%). Table 1 describes the details of graph size for the three version of HMDD datasets. Figure 1 and Figure 2 provide a graphical representation of selected portions of the data graph for illustrative purposes, highlighting the structure and relationships among miRNAs, diseases, genes, and sequence-derived patterns.
Analysis of the Venn diagrams (Figure 3) reveals that successive HMDD versions contain an increasing number of miRNAs and diseases. Notably, a substantial portion of the entries in earlier versions is retained in later releases, reflecting continuity and expansion of the curated data.
2.2. Graph Neural Network Architecture
We model the prediction of miRNA–disease associations using a heterogeneous graph neural network based on message passing [53]. The heterogeneous graph includes multiple node types, each associated with its own feature space, and multiple edge types capturing the biological relations among them (Figure 4). For each edge type , the network learns a distinct linear transformation , that governs how messages are propagated across that relation. Let denote the resulting heterogeneous graph, where the each node has its own type (miRNA, disease, gene and sequence pattern) and each edge has a type representing the nodes it connects (see figure 4)
For a node its embedding at layer is computed using a message-passing rule:
where is the set of neighbors of under relation e, a non-linear activation function, usually the ReLU function.
This mechanism allows the model to integrate heterogeneous biological signals-sequence-derived miRNA features, similarity relations, and gene-level mechanistic information—into a unified latent representation. This architecture enables end-to-end learning of latent biological relationships across the heterogeneous network.
After message-passing layer, the model computes node embeddings for miRNAs and diseases and the association score for each miRNA–disease pair is then predicted by computing the dot product of the corresponding embeddings
During training, only edges of the miRNA–disease type contribute to the supervised loss. Nonetheless, all other edge types influence the node embeddings through relation-specific message passing, enabling the model to combine sequence-derived miRNA features, similarity networks, and gene-level signals into a unified latent representation.
2.3. Training and Validation
The evaluation follows a 10-fold cross-validation scheme, repeated 10 times with different random partitions. In each fold, the set of miRNAs is divided into a train and validation subset. All nodes and all edges not involving miRNA–disease associations remain visible to both splits, in order to preserve the overall graph structure.
For the train/validation separation of miRNA–disease edges: all miRNA–disease links involving validation miRNAs are removed from the training graph, and all links involving training miRNAs are removed from the validation graph. Thus, the model must infer associations for miRNAs never seen in training, based solely on graph topology and message passing. A similar masking strategy is applied to pattern–disease associations: pattern–disease edges are included in the training graph only if the pattern connects to a disease through at least one miRNA-disease association belonging to the training set. If a pattern is linked to a disease exclusively through a validation miRNA–disease pair, the edge is removed from the training graph (and symmetrically for the validation graph). This ensures that no label leakage occurs through sequence patterns. Each fold is trained independently using the Adam optimizer with early stopping based on validation loss.
3. Results
3.1. Evaluation Metrics
Performance was primarily assessed using AUC-ROC, which is the standard evaluation metrics in MDA prediction due to the highly imbalanced nature of MDA datasets (< 3% of positive samples). For comparison with other approach, we additionally computed the area under the precision-recall curve (AUPR), Precision, Recall, and F1-score, defined as:
We first evaluated the proposed heterogeneous graph neural network on two benchmark datasets: HMDD v4.0, the most recent release, and HMDD v3.2 and v.2, widely used in prior computational studies, to facilitate direct comparison with the state of the art. All experiments were performed using the 10-fold cross-validation strategy described in Section 2.3, with the entire evaluation repeated 10 times using different random partitions.
Across all repetitions on HMDD v4.0, the presented model achieved an average AUC-ROC of ~98% and an AUPR of ~95%, demonstrating strong discriminative capability also in the presence of class imbalance. Similar results were obtained on HMDD v3.2, where the AUC-ROC reached ~97–98% and the AUPR remained consistently above 94%.
Figure 5 reports the mean ROC and PR curves aggregated over all replications. The narrow confidence bands observed in both curves indicate high stability across validation folds and independent experiments.
3.2. Comparison with Existing Methods
To position the presented approach to current computational models, we compared it against several representative methods evaluated on HMDD v2. We first considered four widely used and powerful machine learning methods that are known to be able to handle complex features, but still constrained to vectorized feature representations: Support Vector Machine (SVM), a margin-based classifier effective in high-dimensional settings; the Gradient Boosting Decision Trees (GBDT), a sequential ensemble of decision trees using boosting to reduce errors; Random Forest (RF), an ensemble of decision trees; and eXtreme Gradient Boosting (XGBoost), a regularized boosting method offering strong predictive performance.
Traditional machine learning methods remain highly effective for structured biological prediction tasks, especially when relying on engineered similarity features or association profiles. Nevertheless, their inherent tabular representation of featues, inder their ability to capture heterogeneous, multi-relational graph structures. This limits their capacity to exploit the full topology of miRNA–disease–gene–pattern networks—an aspect naturally handled by graph neural architectures.
Next, we included in the comparison six more specialized tools: MDA-CF [54], which leverages weighted hypergraph-based generalized matrix factorization to integrate multi-omics features of microbes and drugs, effectively predicting novel microbe-drug associations; TCRWMDA [55], employing hypergraph-based logistic matrix factorization to capture higher-order relationships between metabolites and diseases, enabling accurate identification of disease-related metabolites; WBSMDA [47], an attention-aware multi-view graph convolutional network combined with hypergraph learning to model miRNA–disease associations by integrating multiple similarity networks and fusing node information from diverse perspectives; ABMDA [56], which explores miRNA-mediated mechanisms underlying disease progression and drug resistance, providing experimentally informed predictions of functional miRNA-disease links; ICFMDA [57], a computational framework exploiting functional similarity and network inference to uncover potential miRNA–disease interactions; and ELMDA [46], an ensemble learning approach that does not rely on known associations to calculate miRNA and disease similarities, combining multiple classifiers via voting to robustly predict disease-related miRNAs across diverse validation settings.
Compared to the most competitive methods—MDA-CF (AUC 92.58%) and ELMDA (AUC 92.29)—our heterogeneous graph-based approach improves performance by a substantial margin, highlighting the benefits of: Integrating heterogeneous biological relationships (miRNA–miRNA, disease–gene, miRNA–pattern, pattern–disease), Using message-passing to propagate functional signals across the network, and learning embedding representations directly from multiple node and edge types, rather than depending on pre-defined similarity kernels. These results indicate that the proposed model captures non-linear relationships more effectively than similarity-based or feature-engineering-based models.
Table 2 reports the performance metrics of the methods compared in this study, including AUC and AUPR and when available Precision, Recall and F1-score.
3.3. Analysis of Newly Predicted Associations
In order to evaluate the ability of our models to predict novel miRNA–disease associations, we performed a comparative analysis using two graph neural models trained on two different versions of the HMDD dataset: Model 3 was trained on HMDD v3.2, containing only the associations known at that time, whereas Model 4 was trained on the more comprehensive HMDD v4.0, which includes additional associations reported after v3.2. Both models were then applied to predict association scores for all new miRNA–disease pairs, those not previously observed by Model 3. Since Model 3 is built on a smaller knowledge base, we expect it to perform less accurately. The Pearson correlation between the prediction scores of the two models, shows a moderate correlation across all pairs (r ≈ 0.45), indicating that Model 3 is partially able to anticipate novel associations present in version 4 (Figure 6). Moreover, the AUCROC for Model 3 considering only the previously unseen positive associations was 89%, indicating that the model successfully discriminates the majority of novel miRNA–disease links, further supporting its ability to anticipate associations absent from the training dataset.
To further analyze these differences, we examined a subset of representative positive associations and visualized the corresponding prediction scores from both models (Figure 7). Beyond simply contrasting the two score distributions, several patterns emerge: in many cases Model 4 assigns consistently higher confidence, reflecting the additional knowledge introduced in HMDD v4.0, while a number of pairs show near-identical scores, indicating that Model 3 successfully anticipates future annotations. Conversely, a few outliers (~15%) exhibit substantial divergence (aboslute score difference more than 0.5) between the two models, suggesting either overgeneralization by Model 3 or revised evidence incorporated in the updated dataset.
3.4. Ablation Analysis
Our heterogeneous graph neural network integrates multiple node types and relation-specific message passing to learn latent embeddings for miRNAs, diseases, genes, and sequence patterns. While the network is trained using the full set of edges, we observed that the model’s performance remains largely stable even when certain edge types are removed or perturbed. This indicates that the learned node embeddings capture significant information from node features themselves, and that the graph structure primarily provides additional contextual information rather than being strictly necessary for high predictive performance. Consequently, the predictive accuracy of miRNA–disease associations is robust with respect to partial or noisy graph information.
On the other hand we observe a different picture when the graph is perturbed before training. Specifically, ablating certain edge types prior to model training leads to significant drops in predictive performance, highlighting the importance of relational information during embedding learning. The quantitative effects of these pre-training ablations are reported in Table 3, showing that edge information is crucial for guiding the model to capture biologically meaningful associations.
4. Discussion and Conclusions
In this study, we presented a heterogeneous graph neural network framework for predicting miRNA–disease associations, leveraging the rich relational structure inherent in biological networks. Biological entities such as miRNAs, genes, diseases, and sequence motifs are naturally represented as nodes in a graph, with interactions forming edges of multiple types. Traditional machine learning approaches, which rely on tabular representations of features, often struggle to fully capture these complex, high-order relationships. In contrast, graph-based models excel at encoding both local and global structural patterns, allowing the integration of multiple types of biological information—ranging from sequence-derived features to gene–disease associations—within a unified latent space. This capability enables the model to infer indirect associations, identify hidden patterns, and generalize to previously unseen nodes with high accuracy.
Message-passing mechanisms within the network allow for effective propagation and aggregation of information, ensuring that the contribution of neighboring nodes is weighted according to their relevance, while attention-based layers enhance interpretability and robustness. Our experiments demonstrated that the model consistently achieves high predictive performance across multiple HMDD datasets, with narrow confidence intervals, confirming stability and reproducibility. Ablation and perturbation studies further highlighted the model’s sensitivity to network structure and its robustness to small levels of noise, underscoring the importance of accurately modeling heterogeneous interactions.
Despite these promising results, several limitations remain. While the model integrates diverse sources of biological information, the construction of the graph relies on pre-defined similarity measures and curated associations, which may overlook emerging or context-specific relationships. Future work could explore alternative strategies for graph construction, incorporating additional biological knowledge such as miRNA–target gene interactions, expression profiles across tissues or conditions, and temporal dynamics of disease progression. Moreover, integrating multi-omics data or environmental factors could enrich node features and edge relationships, improving prediction accuracy and providing deeper mechanistic insights. Advances in graph neural network architectures, including more sophisticated message-passing schemes or hierarchical graph representations, also offer avenues for performance enhancement and better interpretability.
Overall, this study confirms the strength of graph-based learning for miRNA–disease association prediction, demonstrating that modeling biological entities and their relationships as a heterogeneous network allows for accurate, robust, and generalizable inference. The proposed framework not only achieves state-of-the-art performance compared with existing methods, but also provides a flexible and extensible approach for future investigations, supporting the discovery of novel associations and facilitating hypothesis generation in translational research.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created in this study.
Acknowledgments
CNRBIOMICS PON R&I PIR01_00017.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, DP. Metazoan MicroRNAs. Cell. 2018, 173(1), 20–51. [Google Scholar] [CrossRef]
- Eulalio, A; Huntzinger, E; Izaurralde, E. Getting to the Root of miRNA-Mediated Gene Silencing. Cell. 2008, 132, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- de Rooij, LA; Mastebroek, DJ; Ten Voorde, N; van der Wall, E; van Diest, PJ; Moelans, CB. The microRNA lifecycle in health and cancer. Cancers 2022, 14(23), 5748. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S; Saini, HK; van Dongen, S; Enright, AJ. miRBase: tools for microRNA genomics. Nucleic acids research 2008, 36, D154–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alaimo, S; Giugno, R; Pulvirenti, A. ncPred: ncRNA-Disease Association Prediction through Tripartite Network-Based Inference. Front Bioeng Biotechnol. 2014, 2, 71. [Google Scholar] [CrossRef]
- Calin, GA; Croce, CM. MicroRNA signatures in human cancers. Nature reviews Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, SS; Jin, JP; Wang, JQ; Zhang, ZG; Freedman, JH; Zheng, Y; Cai, L. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacologica Sinica 2018, 39(7), 1073–84. [Google Scholar] [CrossRef] [PubMed]
- Li, S; Lei, Z; Sun, T. The role of microRNAs in neurodegenerative diseases: a review. Cell biology and toxicology 2023, 39(1), 53–83. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V; Näär, AM. MicroRNAs in metabolism and metabolic disorders. Nature reviews Molecular cell biology 2012, 13(4), 239–50. [Google Scholar] [CrossRef]
- Liu, Z; Sall, A; Yang, D. MicroRNA: An emerging therapeutic target and intervention tool. International journal of molecular sciences 2008, 9, 978–999. [Google Scholar] [CrossRef] [PubMed Central]
- Ye, JW; Xu, MC; Tian, XK; Cai, S; Zeng, S. Research advances in the detection of miRNA. J Pharm Anal. 2019, 9(4), 217–26. [Google Scholar] [CrossRef]
- Chen, X; Xie, D; Zhao, Q; You, ZH. MicroRNAs and complex diseases: from experimental results to computational models. Briefings in bioinformatics 2019, 20, 515–539. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X; Ding, N; Rodríguezpatón, A; Lin, Z; Ju, Y. Prediction of MicroRNA–disease Associations by Matrix Completion. Current Proteomics 2016, 13, 151–157. [Google Scholar] [CrossRef]
- Li, Y; Qiu, C; Tu, J; Geng, B; Yang, J; et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic acids Res. 2014, 42, D1070–1074. [Google Scholar] [CrossRef]
- Huang, Z; Shi, JC; Gao, YX; Cui, CM; Zhang, S; Li, JW; Zhou, Y; Cui, QH. HMDD v3.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47(D1), D1013–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z; Ren, F; Liu, CN; He, SM; Sun, G; Gao, QA; Yao, L; Zhang, YD; Miao, RY; Cao, Y; et al. dbDEMC: a database of differentially expressed miRNAs in human cancers. Bmc Genom 2010, 11. [Google Scholar] [CrossRef]
- Jiang, Q; Wang, Y; Hao, Y; Juan, L; Teng, M; et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic acids research 2009, 37, D98–104. [Google Scholar] [CrossRef]
- Pasquier, C; Gardes, J. Prediction of miRNA-disease associations with a vector space model. Scientific reports 2016, 6, 27036. [Google Scholar] [CrossRef]
- Bandyopadhyay, S; Mitra, R; Maulik, U; Zhang, MQ. Development of the human cancer microRNA network. Silence 2010, 1, 6. [Google Scholar] [CrossRef]
- Shi, H; Xu, J; Zhang, G; Xu, L; Li, C; et al. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. Bmc Systems Biology 2013, 7, 101. [Google Scholar] [CrossRef] [PubMed Central]
- Mørk, S; Pletscherfrankild, S; Palleja, CA; Gorodkin, J; Jensen, LJ. Protein-driven inference of miRNA-disease associations; Bioinformatics: Oxford, England, 2014; Volume 30, pp. 392–397. [Google Scholar]
- Chen, X; Jiang, ZC; Xie, D; Huang, DS; Zhao, Q; et al. A novel computational model based on super-disease and miRNA for potential miRNA-disease association prediction. Molecular bioSystems 2017, 13, 1202–1212. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, X; Wu, QF; Yan, GY. RKNNMDA: ranking-based KNN for MiRNA-disease association prediction. RNA Biol. 2017, 14(7), 952–62. [Google Scholar] [CrossRef] [PubMed]
- Ha, J. SMAP: Similarity-based matrix factorization framework for inferring miRNA-disease association. Knowl. Based Syst. 2023, 263, 110295. [Google Scholar] [CrossRef]
- Jiang, Q; Hao, Y; Wang, G; Juan, L; Zhang, T; et al. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC systems biology 2010, 4 Suppl 1, pmid:20522252. [Google Scholar] [CrossRef]
- You, ZH; Huang, ZA; Zhu, Z; Yan, GY; Li, ZW; et al. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS computational biology 2017, 13, pmid:28339468. [Google Scholar] [CrossRef]
- Yu, H; Chen, X; Lu, L. Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm. Scientific reports 2017, 7, 43792. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, Q. Generalized matrix factorization based on weighted hypergraph learning for microbe-drug association prediction. Computers in Biology and Medicine 2022, 145, 105503. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P; Han, K; Guo, MZ; Guo, YH; Li, JB; Ding, J; Liu, Y; Dai, QG; Li, J; Teng, ZX; et al. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8(8), pmid:23950912. [Google Scholar] [CrossRef]
- Chen, X; Liu, MX; Yan, GY. RWRMDA: predicting novel human microRNA-disease associations. Molecular Biosystems 2012, 8, 2792–2798. [Google Scholar] [CrossRef] [PubMed Central]
- Xuan, P; Han, K; Guo, Y; Li, J; Li, X; et al. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics (Oxford, England) 2015, 31, 1805–1815. [Google Scholar] [CrossRef] [PubMed Central]
- Chen, X; Yan, CC; Zhang, X; You, ZH; Deng, LX; Liu, Y; Zhang, YD; Dai, QH. WBSMDA: within and between score for MiRNA-disease association prediction. Sci Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Chen, X; Clarence, YC; Zhang, X; You, ZH; Huang, YA; et al. HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget 2016, 7, 65257–65269. [Google Scholar] [CrossRef]
- Chen, X; Zhou, Z; Zhao, Y. ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. RNA biology 2018, 15, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Ha, J. Graph Convolutional Network with Neural Collaborative Filtering for Predicting miRNA-Disease Association. Biomedicines 2025, 9, 1152. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, M.; Tang, C.; Zheng, X.; Zhang, W.; Sha, X.; An, S. Predicting miRNA-disease association via graph attention learning and multiplex adaptive modality fusion. Comput. Biol. Med. 2024, 169, 107904. [Google Scholar] [CrossRef]
- Xu, J; Li, CX; Lv, JY; Li, YS; Xiao, Y; et al. Prioritizing Candidate Disease miRNAs by Topological Features in the miRNA Target-Dysregulated Network: Case Study of Prostate Cancer. Molecular Cancer Therapeutics 2011, 10, 1857–1866. [Google Scholar] [PubMed Central]
- Chen, X; Yan, CC; Zhang, X; Li, Z; Deng, L; et al. RBMMMDA: predicting multiple types of disease-microRNA associations. Scientific reports 2015, 5, 13877. [Google Scholar]
- Chen, X; Yan, GY. Semi-supervised learning for potential human microRNA-disease associations inference. Sci Rep. 2014, 4, pmid:24975600. [Google Scholar] [CrossRef]
- Li, JQ; Rong, ZH; Chen, X; Yan, GY; You, ZH. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187–21199. [Google Scholar] [CrossRef] [PubMed Central]
- Gu, C; Li, X. Prediction of disease-related miRNAs by voting with multiple classifiers. BMC bioinformatics 2023, 24(1), 177. [Google Scholar] [CrossRef]
- Ning, Q; Zhao, YM; Gao, J; Chen, C; Li, X; Li, TT; Yin, MH. AMHMDA: attention aware multi-view similarity networks and hypergraph learning for miRNA-disease associations identification. Brief Bioinform. 2023, 24(2). [Google Scholar]
- Peng, JJ; Hui, WW; Li, QQ; Chen, BL; Hao, JY; Jiang, QH; Shang, XQ; Wei, ZY. A learning-based framework for miRNA-disease association identification using neural networks. Bioinformatics 2019, 35(21), 4364–71. [Google Scholar] [PubMed]
- Yu, L.; Yu, Z.G.; Han, G.S.; Li, J.; Anh, V. Heterogeneous types of miRNA-disease associations stratified by multi-layer network embedding and prediction. Biomedicines 2021, 9, 1152. [Google Scholar] [CrossRef]
- Available online: https://www.disgenet.org/.
- Bodenreider, O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic acids research 2004, 32 suppl_1, D267–70. [Google Scholar] [CrossRef]
- Needleman, SB; Wunsch, CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology 1970, 48(3), 443–53. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, J; Schoenholz, SS; Riley, PF; Vinyals, O; Dahl, GE. Neural message passing for quantum chemistry. InInternational conference on machine learning 2017 Jul 17 (pp. 1263-1272). Pmlr.
- Ma, YJ. DeepMNE: deep multi-network embedding for lncRNA-disease association prediction. IEEE J Biomed Health 2022, 26(7), 3539–49. [Google Scholar] [CrossRef]
- Ma, Y; Ma, Y. Hypergraph-based logistic matrix factorization for metabolite–disease interaction prediction. Bioinformatics 2022, 38(2), 435–43. [Google Scholar] [CrossRef] [PubMed]
- Barbato, A; Iuliano, A; Volpe, M; D’Alterio, R; Brillante, S; Massa, F; De Cegli, R; Carrella, S; Salati, M; Russo, A; Russo, G. Integrated genomics identifies miR-181/TFAM pathway as a critical driver of drug resistance in melanoma. International Journal of Molecular Sciences 2021, 22(4), 1801. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y; Xu, W; Yang, Y; Zhang, Z. miRNA-93-5p promotes gemcitabine resistance in pancreatic cancer cells by targeting the PTEN-mediated PI3K/Akt signaling pathway. Annals of Clinical & Laboratory Science 2021, 51(3), 310–20. [Google Scholar]
Figure 1.
Graphical representation of a portion of the heterogeneous biological network. Nodes correspond to miRNAs, diseases, genes, and sequence-derived patterns, while edges represent miRNA–disease associations, disease–gene links, sequence similarity, and motif-derived connections. Node colors indicate entity type, and edge colors indicate relationship type.
Figure 1.
Graphical representation of a portion of the heterogeneous biological network. Nodes correspond to miRNAs, diseases, genes, and sequence-derived patterns, while edges represent miRNA–disease associations, disease–gene links, sequence similarity, and motif-derived connections. Node colors indicate entity type, and edge colors indicate relationship type.
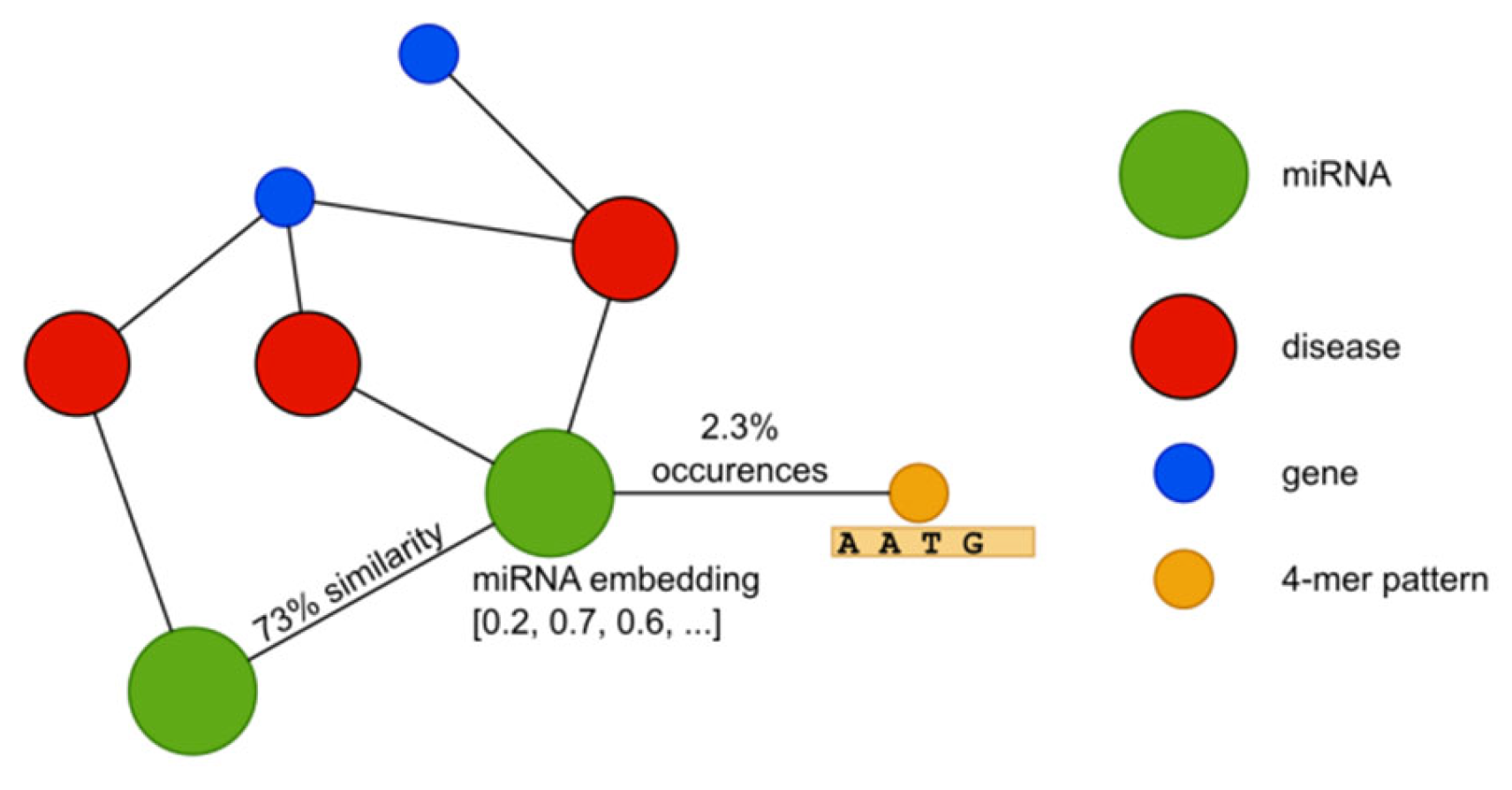
Figure 2.
Network representation of a selected miRNA and its associations. The central green node represents the target miRNA. Red nodes indicate diseases associated with the miRNA, while blue nodes correspond to genes linked to those diseases. Orange nodes represent sequence patterns connected to the miRN.- Edges indicate relationships between nodes: miRNA–disease, disease–gene, miRNA–pattern and miRNA–miRNA,.
Figure 2.
Network representation of a selected miRNA and its associations. The central green node represents the target miRNA. Red nodes indicate diseases associated with the miRNA, while blue nodes correspond to genes linked to those diseases. Orange nodes represent sequence patterns connected to the miRN.- Edges indicate relationships between nodes: miRNA–disease, disease–gene, miRNA–pattern and miRNA–miRNA,.
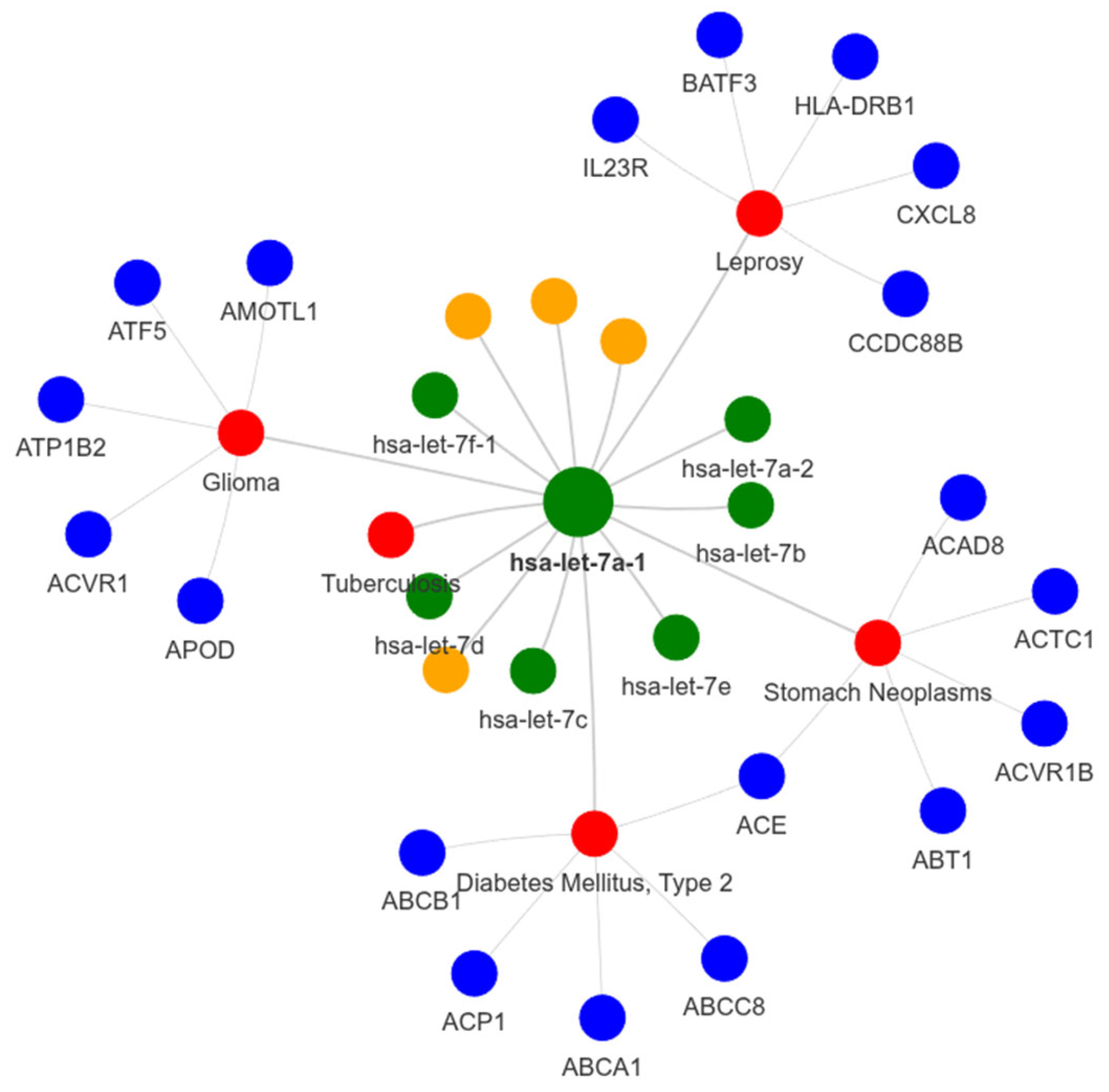
Figure 3.
Venn diagrams showing the overlap of miRNAs (left) and diseases (right) across three versions of the HMDD database (v2, v3, and v4). The diagrams illustrate the number of shared and unique elements in each dataset version.
Figure 3.
Venn diagrams showing the overlap of miRNAs (left) and diseases (right) across three versions of the HMDD database (v2, v3, and v4). The diagrams illustrate the number of shared and unique elements in each dataset version.
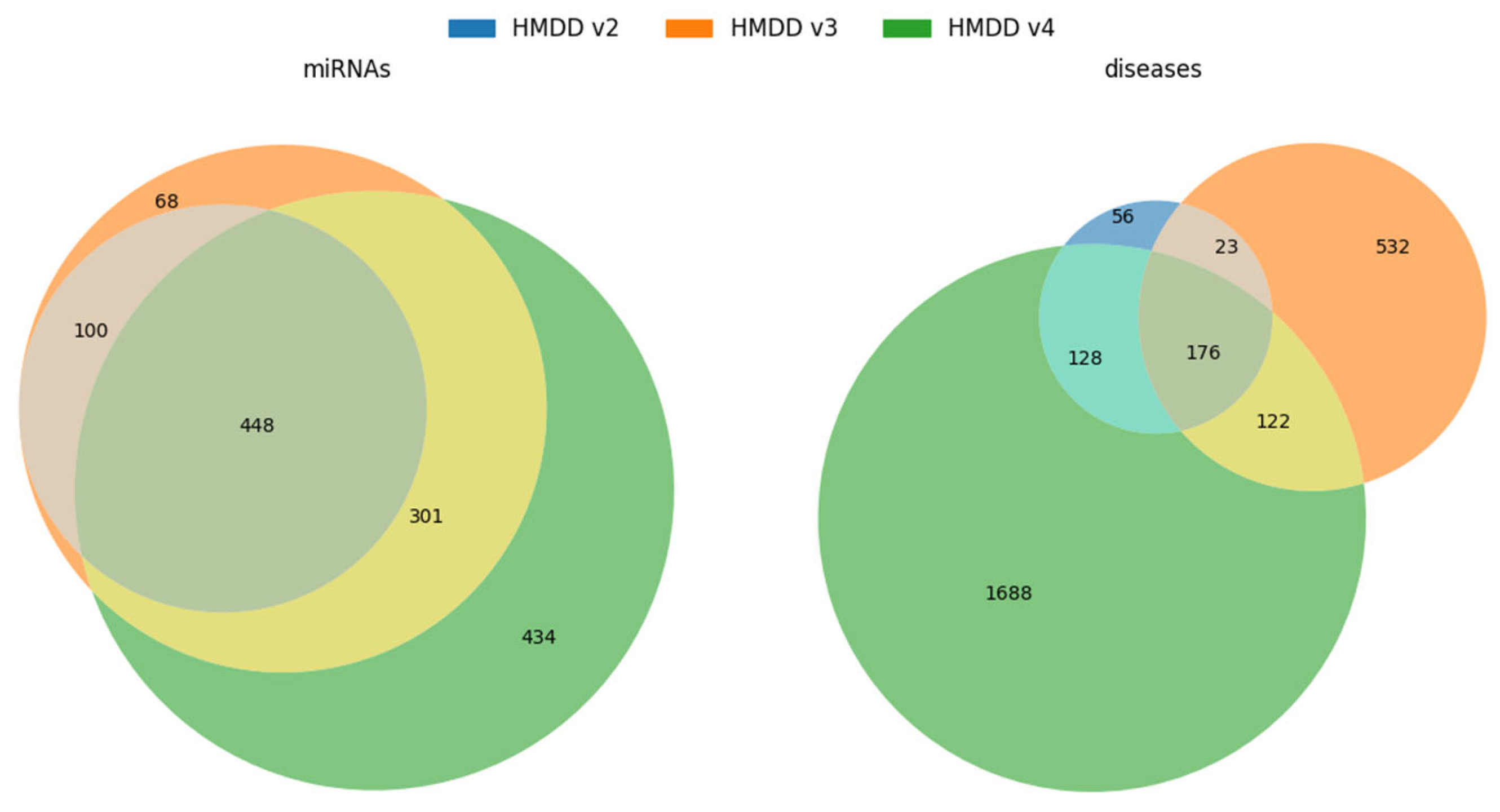
Figure 4.
Overview of the heterogeneous graph neural network architecture used for miRNA–disease association prediction. The model integrates four node types (miRNAs, diseases, genes, and sequence patterns) connected through multiple biologically meaningful edge types. For each relation, a dedicated transformation matrix enables relation-specific message passing across the graph. Node embeddings are updated layer by layer through aggregation of messages from typed neighbors, followed by non-linear activation. After L message-passing layers, the learned embeddings of miRNAs and diseases are combined through a dot-product decoder to generate association scores.
Figure 4.
Overview of the heterogeneous graph neural network architecture used for miRNA–disease association prediction. The model integrates four node types (miRNAs, diseases, genes, and sequence patterns) connected through multiple biologically meaningful edge types. For each relation, a dedicated transformation matrix enables relation-specific message passing across the graph. Node embeddings are updated layer by layer through aggregation of messages from typed neighbors, followed by non-linear activation. After L message-passing layers, the learned embeddings of miRNAs and diseases are combined through a dot-product decoder to generate association scores.
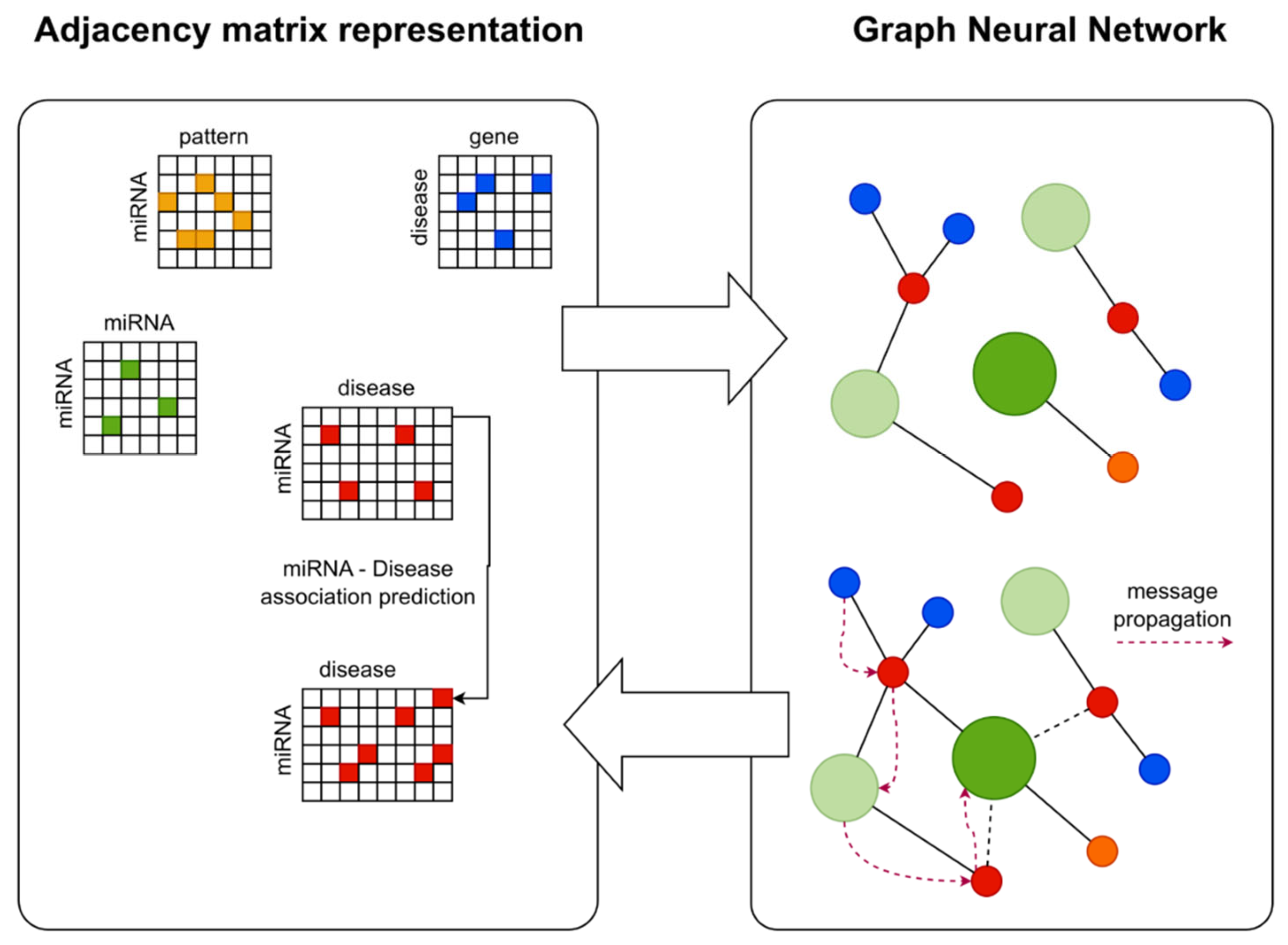
Figure 5.
Receiver Operating Characteristic (ROC) and Precision–Recall (PR) curves obtained for the three evaluation datasets (HMDD v3.2, HMDD v4.0 and the combined heterogeneous dataset). Each curve represents the mean performance aggregated over all 10×10 replicated experiments, while the shaded regions denote the corresponding confidence bands. The tight variability observed across replications indicates the high stability of the model. The legend reports, for each dataset, the average AUC and AUPR (± their variance), confirming consistently strong predictive accuracy across all evaluation settings.
Figure 5.
Receiver Operating Characteristic (ROC) and Precision–Recall (PR) curves obtained for the three evaluation datasets (HMDD v3.2, HMDD v4.0 and the combined heterogeneous dataset). Each curve represents the mean performance aggregated over all 10×10 replicated experiments, while the shaded regions denote the corresponding confidence bands. The tight variability observed across replications indicates the high stability of the model. The legend reports, for each dataset, the average AUC and AUPR (± their variance), confirming consistently strong predictive accuracy across all evaluation settings.
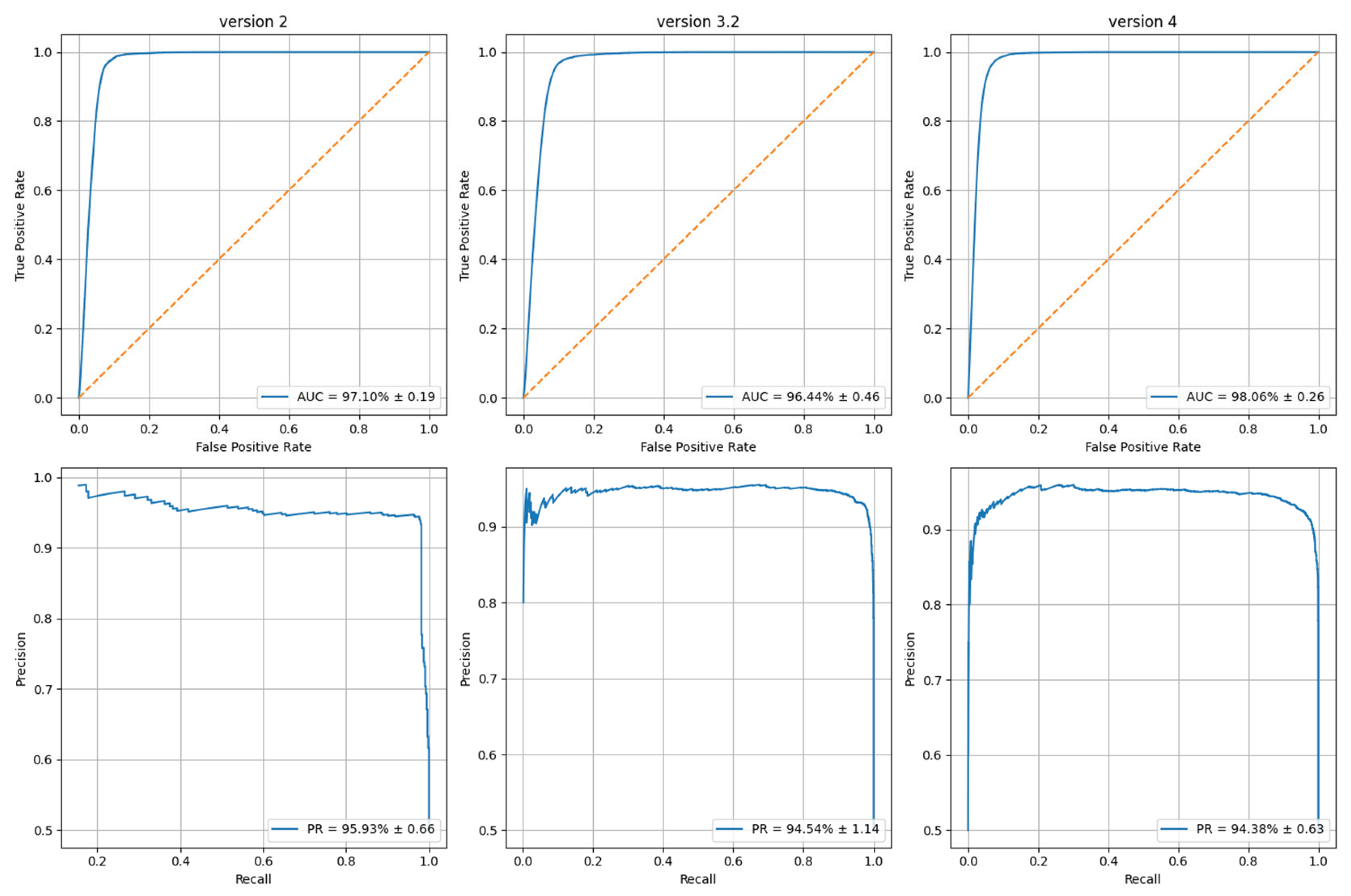
Figure 6.
Comparison of prediction scores from Model 3 and Model 4 for selected miRNA–disease associations. Each point represents a miRNA–disease pair included in the analysis. The x-axis shows the prediction scores from Model 3, trained on HMDD v3.2, and the y-axis shows scores from Model 4, trained on the more complete HMDD v4.0 dataset. The Pearson correlation coefficient (r = 0.45) is reported in the legend, indicating moderate agreement between the two models while also revealing differences in predictions for novel associations.
Figure 6.
Comparison of prediction scores from Model 3 and Model 4 for selected miRNA–disease associations. Each point represents a miRNA–disease pair included in the analysis. The x-axis shows the prediction scores from Model 3, trained on HMDD v3.2, and the y-axis shows scores from Model 4, trained on the more complete HMDD v4.0 dataset. The Pearson correlation coefficient (r = 0.45) is reported in the legend, indicating moderate agreement between the two models while also revealing differences in predictions for novel associations.
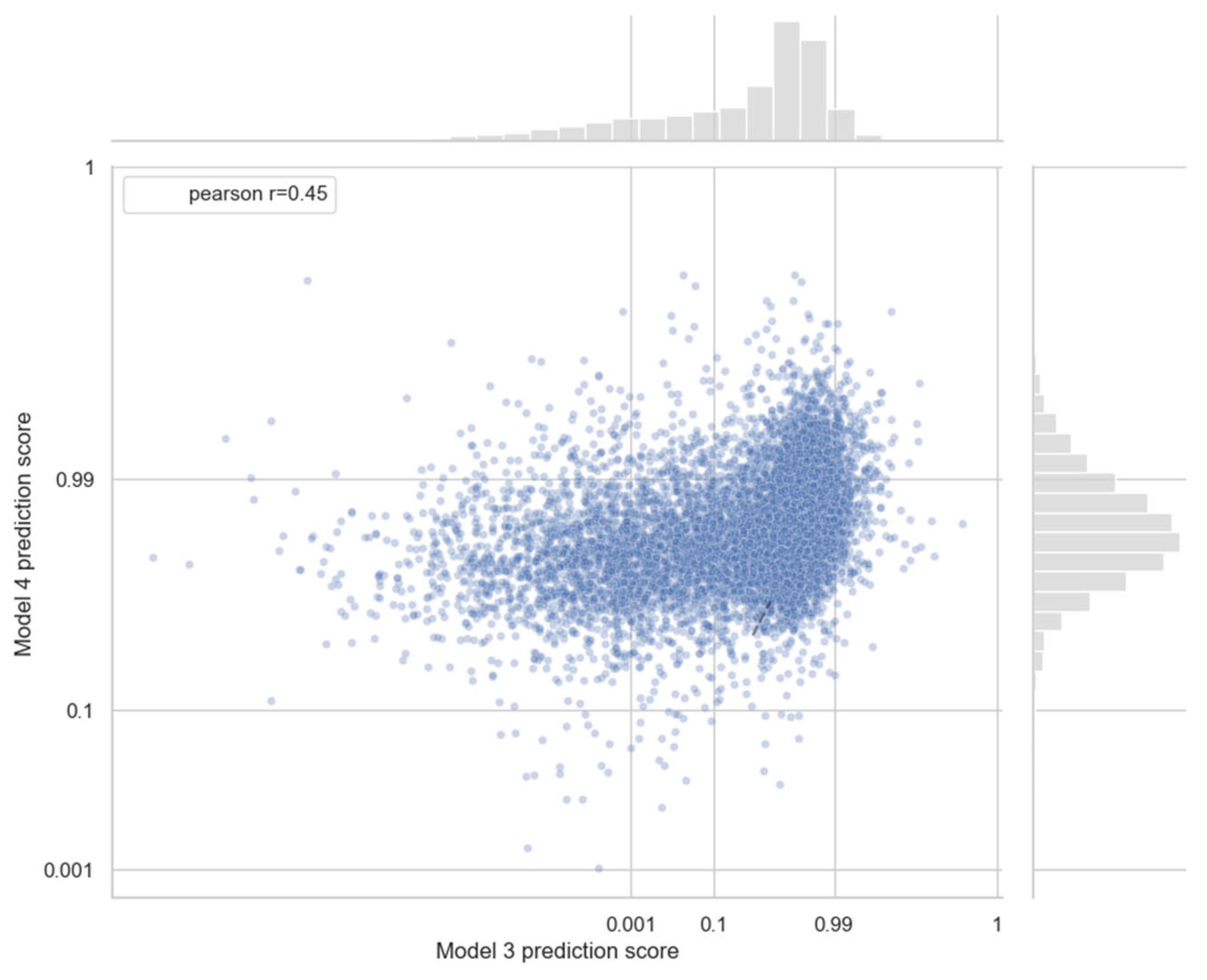
Figure 7.
Bar plot comparing the prediction scores of Model 3 (trained on HMDD v3.2) and Model 4 (trained on HMDD v4.0) for a selected subset of positive miRNA–disease associations. Each row represents a single miRNA–disease pair, with the left bar indicating the score from Model 3 (blue) and the right bar the score from Model 4 (red). This visualization highlights both cases where Model 3 anticipates associations later reported in HMDD v4.0 and cases where the two models diverge, illustrating the models’ predictive behavior across heterogeneous scenarios.
Figure 7.
Bar plot comparing the prediction scores of Model 3 (trained on HMDD v3.2) and Model 4 (trained on HMDD v4.0) for a selected subset of positive miRNA–disease associations. Each row represents a single miRNA–disease pair, with the left bar indicating the score from Model 3 (blue) and the right bar the score from Model 4 (red). This visualization highlights both cases where Model 3 anticipates associations later reported in HMDD v4.0 and cases where the two models diverge, illustrating the models’ predictive behavior across heterogeneous scenarios.
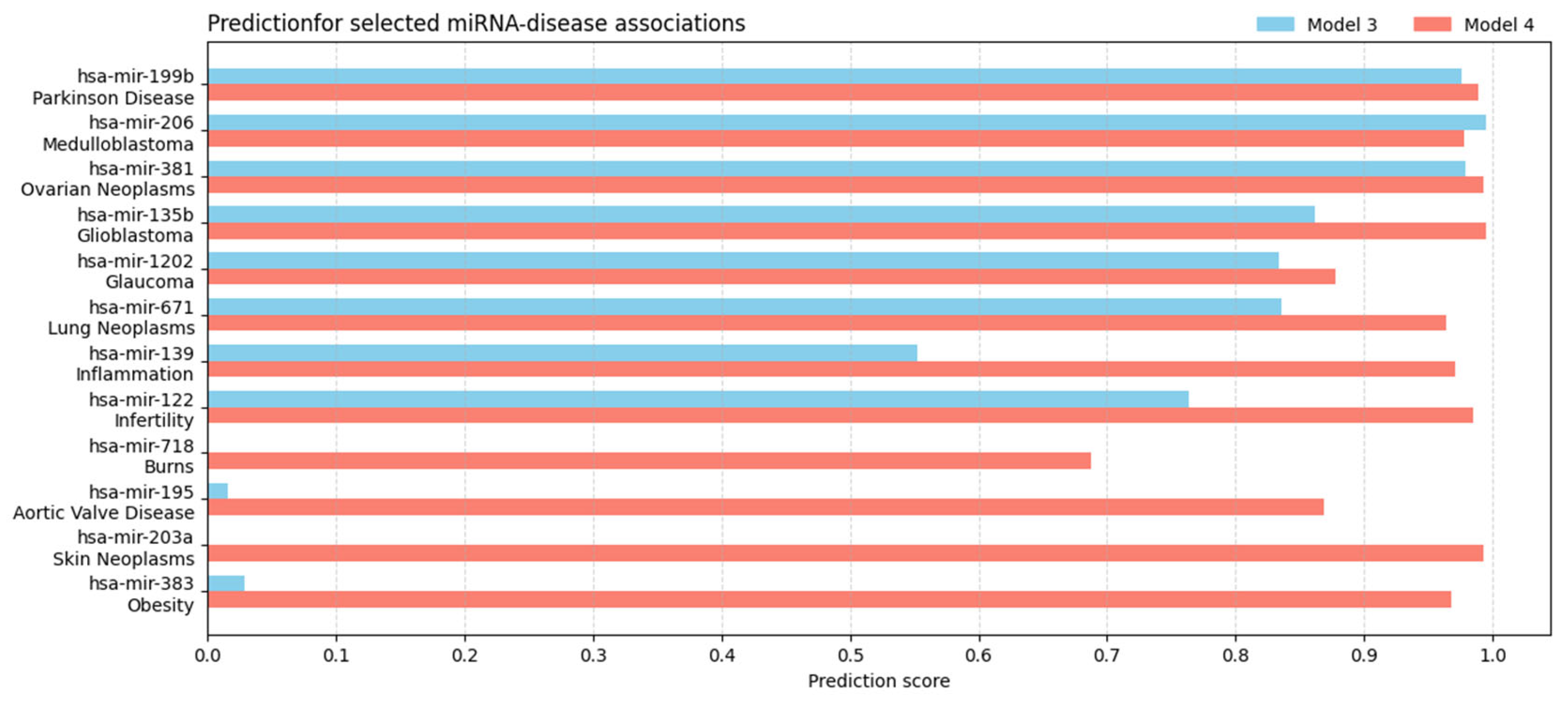

Table 1.
Summary of the graph size for the three HMDD dataset versions. For each version, the number of miRNAs, diseases, genes, patterns, and the total number of edges in each relationship type are reported. Edge density is expressed as the percentage of observed associations over all possible associations.
Table 1.
Summary of the graph size for the three HMDD dataset versions. For each version, the number of miRNAs, diseases, genes, patterns, and the total number of edges in each relationship type are reported. Edge density is expressed as the percentage of observed associations over all possible associations.
| version 2 | version 3.2 | version 4 | |
| nodes | |||
| miRNAs | 548 | 917 | 1183 |
| diseases | 383 | 853 | 2114 |
| genes | 6356 | 6356 | 6356 |
| patterns (4-mers) | 256 | 256 | 256 |
| edges | |||
| miRNA–disease | 6331 (3.02%) |
15161 (1.94%) |
24074 (0.96%) |
| miRNA–miRNA similarity |
58814 (19.58%) |
133958 (15.93%) |
209186 (14.95%) |
| disease–gene |
11977 0.49%) |
13683 (0.25%) |
18617 (0.14%) |
| miRNA–pattern |
36602 (24.27%) |
58695 (25.0%) |
73515 (26.09%) |
Table 2.
Comparison of different Machine Learning approaches proposed in literature reported from [46]. The proposed approach (P.A. in the table) has been reported for all the three versions of the dataset. All other methods are evaluated on version 2.
Table 2.
Comparison of different Machine Learning approaches proposed in literature reported from [46]. The proposed approach (P.A. in the table) has been reported for all the three versions of the dataset. All other methods are evaluated on version 2.
| Method | Precision | Recall | F1-Score | AUCROC | AUPR |
| SVM | 83.69 ± 0.85 | 83.71 ± 1.43 | 83.70 ± 0.75 | 90.91 ± 0.31 | 90.57 ± 0.36 |
| GBDT | 83.69 ± 1.07 | 84.90 ± 0.57 | 84.29 ± 0.54 | 91.72 ± 0.34 | 91.38 ± 0.39 |
| RF | 84.24 ± 1.08 | 83.54 ± 1.31 | 83.88 ± 0.91 | 91.41 ± 0.49 | 91.23 ± 0.47 |
| XGBoost | 84.71 ± 0.90 | 84.86 ± 0.99 | 84.78 ± 0.76 | 91.91 ± 0.39 | 91.65 ± 0.45 |
| ELMDA | 84.85 ± 1.39 | 85.36 ± 1.01 | 85.10 ± 0.94 | 92.29 ± 0.35 | 92.17 ± 0.31 |
| MDA-CF | - | - | - | 92.58 | - |
| TCRWMDA | - | - | - | 92.09 | - |
| WBSMDA | - | - | - | 81.85 | - |
| ABMDA | - | - | - | 90.45 | - |
| ICFMDA | N.A. | N.A. | N.A. | 90.23 | N.A. |
| P.A. v2 | 92.02 ± 0.90 | 96.19 ± 0.90 | 94.06 ± 0.63 | 97.10 ± 0.19 | 95.93 ± 0.66 |
| P.A. v3 | 91.49 ± 1.09 | 94.79 ± 1.02 | 93.11 ± 0.92 | 96.44 ± 0.46 | 94.54 ± 1.14 |
| P.A. v4 | 94.94 ± 0.57 | 90.56 ± 1.79 | 92.70 ± 1.01 | 98.06 ± 0.26 | 94.38 ± 0.63 |
Table 3.
Comparison of different Machine Learning approaches proposed in literature reported from [46]. The proposed approach (P.A. in the table) has been reported for all the three versions of the dataset. All other methods are evaluated on version 2.
Table 3.
Comparison of different Machine Learning approaches proposed in literature reported from [46]. The proposed approach (P.A. in the table) has been reported for all the three versions of the dataset. All other methods are evaluated on version 2.
| Dropped edge | Decrease AUC |
| miRNA–miRNA similarity | 5.4 % |
| disease–gene | 11.2 % |
| miRNA–pattern | 3.4 % |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



