
Submitted:
02 February 2026
Posted:
03 February 2026
You are already at the latest version
Abstract
Mitochondria govern energy transfer, redox balance, and cell fate. Tryptophan catabolism generates kynurenines (KYNs) that can tune mitochondrial function, with growing evidence that G protein coupled receptor 35 (GPR35), aryl hydrocarbon receptor (AhR), and N-methyl-D-aspartate receptors (NMDA receptors) link extracellular cues to adenosine 5 prime triphosphate (ATP) maintenance, calcium (Ca2+) handling, mitophagy, and inflammasome control. In parallel, quinolinic acid (QA)-driven de novo nicotinamide adenine dinucleotide (NAD+) synthesis connects KYN flux to tricarboxylic acid (TCA) cycle activity and sirtuin programs across tissues. Key gaps remain: receptor pharmacology is rarely integrated with NAD+ economics and respiration, and clinical workflows still lack single run assays that quantify both kynurenine and TCA nodes. We therefore integrate receptor proximal signaling, QA-driven NAD+ supply, and unified liquid chromatography mass spectrometry (LC-MS) measurement into one translational framework spanning kynurenic acid (KYNA), KYN, 3-hydroxykynurenine (3-HK), and QA, using mitochondrial endpoints as the common readout. We synthesize evidence for mitochondrial GPR35 signaling that preserves ATP, AhR programs that tune oxidative defenses and mitophagy, and NMDA receptor antagonism that limits excitotoxic stress. These mechanisms are linked to QA-dependent NAD+ biogenesis and alpha-ketoglutarate control points, then aligned with chromatography and ionization choices suited to routine LC-MS workflows. This receptor-to-organelle framework couples KYN flux to respiratory control and provides a practical roadmap for standardized single-run LC-MS panels. It can strengthen target validation in ischemia, neurodegeneration, psychiatry, and oncology while improving biomarker qualification through harmonized analytics and decision-grade readouts.
Keywords:
kynurenic acid (KYNA)
; mitochondria
; tricarboxylic acid (TCA) cycle
; nicotinamide adenine dinucleotide (NAD⁺)
; metabolomics
; liquid chromatography–mass spectrometry (LC-MS)
; receptors
; G-protein-coupled receptors
; aryl hydrocarbon receptor (AhR)
; N-methyl-D-aspartate (NMDA)
; mitophagy
1. Introduction
Mitochondria choreograph energy flux, redox poise, and fate decisions through tightly coupled metabolism, signaling, and quality control (QC) [1,2]. Nuclear factor erythroid 2–related factor 2 (Nrf2) and nuclear respiratory factor 1 (Nrf1) align bioenergetics with antioxidant defenses, tuning respiration, detoxification, and ROS setpoints to prevent maladaptive stress responses [3,4]. Proton leak via uncoupling proteins subtly tempers superoxide, reshaping signaling without collapsing adenosine 5′-triphosphate (ATP) supply [5,6]. Dynamic cycles of fission, fusion, biogenesis, and mitophagy purge damage and license apoptosis or survival, thereby preserving tissue function across development and aging [1,7,8]. In stem and neuronal lineages, mitochondrial metabolites and ROS act as instructive cues that program transcription and differentiation while guarding viability [2,9,10]. These convergent circuits constitute a master key for health and disease [11,12].
Tryptophan (Trp) catabolism feeds the kynurenine (KYN) metabolic pathway, yielding kynurenic acid (KYNA) that operates as a pleiotropic signal aligning mitochondrial respiration, redox poise, and cellular metabolism [13,14,15]. KYNA engages G protein–coupled receptor 35 (GPR35) and AMP-activated protein kinase (AMPK) to modulate bioenergetics, thermogenesis, and lipid handling across adipose and muscle, with exercise-driven KYN aminotransferases (KATs) activity boosting KYNA output and efficiency [16,17,18]. Genetic or pharmacologic perturbation of KAT enzymes reveals KYNA’s necessity for ATP synthesis and mitochondrial stability in brain and peripheral tissues [13,19,20,21]. At stress frontiers, KYNA preserves ATP, curbs mtROS, and licenses mitophagy, thereby limiting inflammasome activation and ischemic injury [21,22,23,24]. Pathway flux also supports NAD⁺ economy and neuroprotection, linking immune tone to mitochondrial longevity and disease modification [14,25,26,27,28,29,30].
KYNA coordinates receptor signaling that feeds directly into mitochondrial control. As outlined in Section 2, KYNA GPR35 signaling can interface with ATPIF1 to limit ATP synthase reverse activity and protect inner membrane function, so downstream disease effects are interpreted here as readouts of that conserved energy preservation module [22,31]. GPR35 signaling restrains calcium (Ca2+) mobilization, limits NLRP3 activation, and enables autophagic disposal of inflammasomes, linking immunity to mitochondrial QC [32,33]. As an AhR ligand, it reprograms redox and apoptotic set points across neural and cardiovascular contexts [34,35]. Antagonism at N-methyl-D-aspartate (NMDA) receptors and mitochondrial nicotinic acetylcholine receptors containing α7 subunits (α7nAChR) rewires excitatory drive and metabolic coupling, tuning respiration and protecting tissue function [36,37]. Additional endogenous ligands at GPR35 add complexity to this regulatory axis [38,39].
Trp degradation through the KYN metabolism supplies de novo nicotinamide adenine dinucleotide (NAD⁺), the redox currency that feeds the tricarboxylic acid cycle (TCA) and the electron transport chain, thereby tuning respiration at its core [14,40]. Flux through this pathway sets mitochondrial NAD⁺/NADH ratios and ROS thresholds via redox-active intermediates, stabilizing oxidative phosphorylation and ATP output [15,41]. Immune and tissue contexts reveal causality: macrophages require pathway-derived NAD⁺ for oxidative metabolism, while ischemia-reperfusion diverts flux and collapses antioxidant capacity until NAD⁺ is restored [42,43]. Enzyme control points are actionable. ACMSD inhibition elevates NAD⁺; kynurenine 3-monooxygenase (KMO) modulation redirects carbon to sustain TCA activity; pathway blockade diminishes SIRT1 signaling and viability [44,45]. These circuitries extend to microbiota, cardio-metabolic risk, T-cell bioenergetics, cancer, and aging [45,46,47,48,49].
The KYN metabolism links inflammation to mitochondrial bioenergetics, and its clinical footprint spans neurology, psychiatry, ischemic injury, metabolism, and cancer [26,50]. In neurodegeneration, skewed production of neurotoxic versus protective metabolites accelerates oxidative stress, excitotoxicity, and decline, while enzyme targeting can tilt the balance toward resilience [51,52,53]. Psychiatric syndromes display immune driven pathway activation with measurable biomarker shifts and actionable enzymatic nodes [50,54,55,56,57,58,59]. Cardiovascular and systemic contexts reveal redox and immune dysregulation that worsens tissue injury and aligns with ischemic vulnerability [60,61]. Metabolic disease reflects chronic low-grade inflammation that routes Trp away from homeostasis [26,62]. Tumors exploit pathway derived NAD⁺ and immunosuppression, creating therapeutic entry points under active clinical evaluation [63,64].
Capturing KYN metabolites and tricarboxylic acid intermediates in the same clinical sample remains a moving target [65,66,67]. Targeted LC-MS workflows for KYN species vary widely in extraction, chromatography, and calibration, and no protocol robustly spans all key metabolites or matrices [65,66,68]. Matrix effects, polarity extremes, and poor chromatographic behavior of compounds such as quinolinic acid (QA) confound accuracy and comparability [65,69]. Parallel LC-MS assays for TCA intermediates add further hurdles due to instability and matrix-dependent losses [65,70]. Alternative readouts help but fragment the picture: voltammetry and immunostrips deliver speed at the cost of scope, while capillary electrochromatography trades coverage for protracted runs [71,72]. Clinically useful panels will demand matrix-specific preparation, isotope-labeled standards, and harmonized cross-platform validation [65,73].
Current evidence offers vivid mechanistic snapshots, yet the mosaic remains disjointed across species, cell types, and measurement scales [21,55]. Elegant studies in lupus-prone T cells link Rab4A trafficking to mitophagy, cluster of differentiation 98 (CD98), and KYN-sensitive mTOR, but insights are constrained by model specificity and temporal windows [47,74]. Clinical syntheses in depression expose state-dependent metabolite signatures alongside striking heterogeneity in cohorts and methods [50,75]. Cross-phyla work in Lymnaea underscores evolutionary conservation while complicating translation to humans [76,77]. Analytical platforms further splinter datasets, with electrochemical and chromatographic approaches optimized for different matrices and targets [78,79]. Reviews connecting the pathway to NAD+ and aging highlight gaps between molecular flux, organelle dynamics, and outcomes that matter clinically [50,77,80,81,82].
Existing reviews have typically advanced one axis at a time. Neurology and psychiatry focused syntheses emphasize pathway shifts and biomarker associations, often framing KYNA and QA as neuroprotective versus neurotoxic signals without fully resolving how extracellular receptor engagement is translated into organelle level control of ATP, Ca2+, mitophagy, and inflammasome tone [83,84,85]. Aging and NAD+ centered reviews clarify the logic of quinolinate driven de novo NAD+ supply, sirtuin programs, and redox buffering, yet they rarely connect these metabolic steps to receptor pharmacology or to clinical grade analytics that can quantify both KYN and TCA nodes in a single workflow [86,87]. Immunometabolism and oncology oriented discussions outline IDO and TDO driven tolerance circuits and AhR dependent immune rewiring, but mitochondrial mechanism is often treated as background physiology rather than a defined causal layer [88,89]
This review contributes three integration moves that are not usually delivered together. First, it uses a receptor to mitochondria framework that treats GPR35 trafficking, AhR programs, and NMDA-linked Ca2+ gating as proximal controllers of mitochondrial decision points, rather than downstream correlates [22,90]. Second, it formalizes a QA to NAD⁺ to TCA scaffold that links de novo NAD⁺ supply to respiratory control and redox set points, and then identifies actionable checkpoints where flux and signaling intersect [91]. Third, it connects analytics to clinics by evaluating LC-MS designs that can co-quantify KYN and TCA intermediates in one run, and by outlining quality control and harmonization steps that make multi cohort translation realistic.

Table 1.
How this review differs from prior review themes in the KYN pathway literature.
| Prior review theme (typical emphasis) | What is usually well covered | What is commonly underdeveloped | What this review adds | Reference |
|---|---|---|---|---|
| Neurology and psychiatry | Metabolite patterns, QA and KYNA balance, clinical associations | Receptor proximal steps that explain how signals reach mitochondria | Receptor to mitochondria causal chain with ATP, Ca2+, mitophagy, inflammasome endpoints | [50,92] |
| NAD⁺ and aging biology | Quinolinate to NAD⁺ logic, sirtuins, redox and longevity framing | How receptor pharmacology reshapes NAD+ economics in specific tissues | QA to NAD⁺ to TCA integration tied to mitochondrial control nodes | [93,94] |
| Immunometabolism and oncology | IDO and TDO tolerance circuits, AhR mediated immune programming | Mitochondrial mechanism often implicit rather than specified | Organellar mechanisms positioned as selectable therapeutic levers | [47,95] |
| Analytical and metabolomics reviews | KYN panels, targeted LC-MS methods, matrix effects | Co-measurement of KYN plus TCA plus NAD relevant nodes in one clinical run | Analytics to clinics workflow with single run design choices and harmonization checkpoints | [96,97] |
AhR, aryl hydrocarbon receptor; ATP, adenosine triphosphate; Ca2+, calcium ion; IDO, indoleamine 2,3-dioxygenase; KYN, kynurenine; KYNA, kynurenic acid; LC-MS, liquid chromatography mass spectrometry; NAD+, nicotinamide adenine dinucleotide; QA, quinolinic acid; TCA, tricarboxylic acid; TDO, tryptophan 2,3-dioxygenase.
Despite striking mechanistic vignettes, four gaps impede synthesis. First, temporal dynamics remain under-sampled: circadian, acute, and chronic windows yield nonoverlapping readouts that are rarely integrated [98]. Second, signaling is compartment-specific across tissues, cell types, and subcellular locales, complicating translation from regionally restricted or model-bound observations [99]. Third, causal links from receptors such as AhR or GPR35 to mitochondrial remodeling are inferred more often than demonstrated longitudinally in vivo [22]. Fourth, multi-analyte assays lack harmonization across matrices, throttling cross-study comparability and biomarker qualification [66]. Addressing these deficits will require time-resolved, compartment-aware designs that couple receptor activation to mitochondrial endpoints while deploying standardized, multiplexed metabolomic and signaling panels across preclinical and clinical cohorts [81,98].
Objectives are fourfold. First, chart receptor-specific mitochondrial actions of KYNA by resolving GPR35-dependent Ca2+ control, ATP preservation, and inflammasome restraint, and by contrasting AhR-driven stress programs and synaptic α7nAChR modulation. Second, delineate pathway–cycle crosstalk by linking enzyme localization and NAD+ biogenesis to respiratory control and organelle dynamics across tissues and time. Third, appraise integrated analytics that fuse targeted LC-MS panels with multi-omics, isotope tracing, and trafficking readouts to capture mechanism and flux in matched samples. Finally, synthesize translational implications in ischemic protection, neurodegeneration, cancer immunity, and network-level metabolic resilience to guide trial design and biomarker qualification.
Bridging correlation to cure requires two pillars. First, standardized, validated quantification across matrices and cohorts so biomarker signals mean the same thing in every lab [100,101]. Second, causal mechanistic experiments that link receptor and enzyme perturbations to mitochondrial dynamics and clinical outcomes [102]. Harmonized LC-MS/MS panels and fit-for-purpose QC will enable longitudinal, multi-analyte readouts in neurology, psychiatry, cardiometabolic disease, and oncology, converting meta-analytic heterogeneity into actionable thresholds [103]. Interventional studies that pair enzyme inhibition or pathway rerouting with mitochondrial endpoints can validate target engagement and refine patient selection [104]. The sections that follow track these objectives in order, moving from receptor to mitochondria mechanisms, to QA linked NAD+ and TCA control, to assay design choices, and finally to translational decision points.
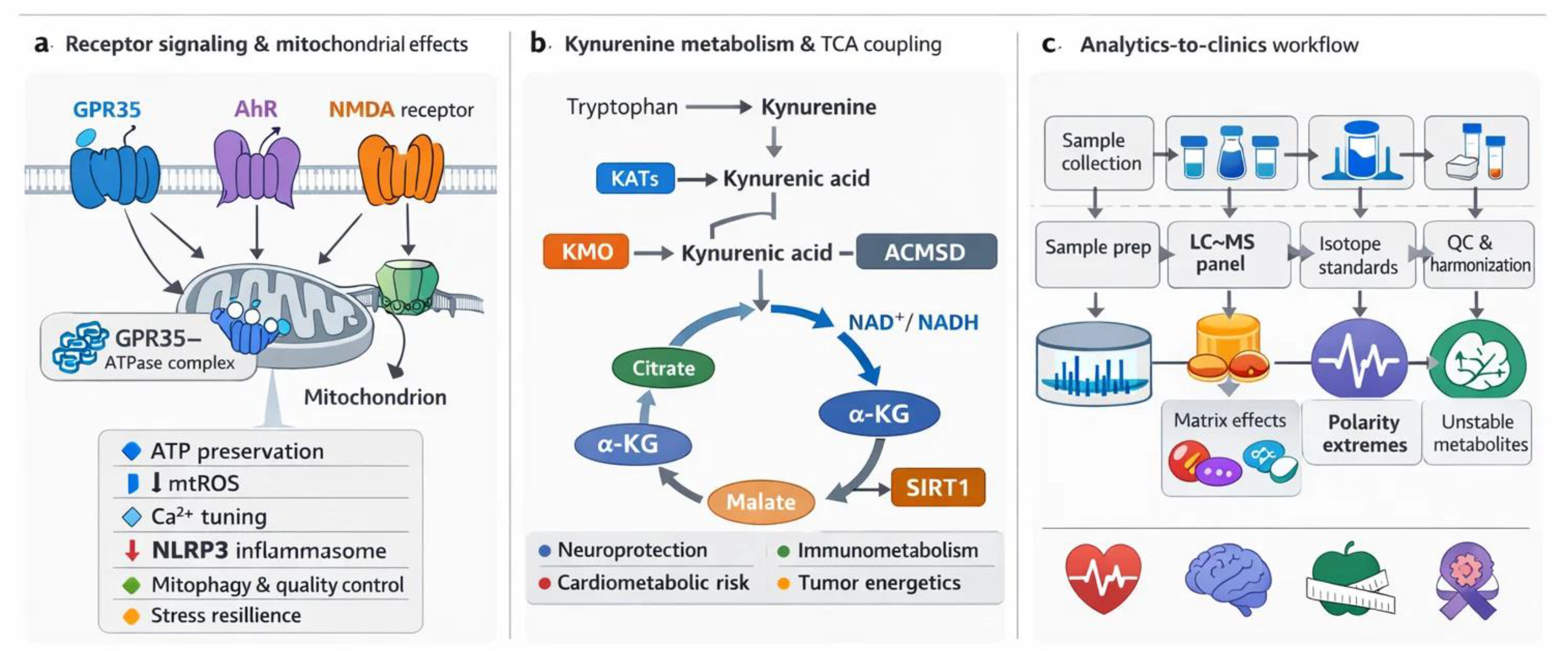
Figure 1.
KYN pathway metabolites as mitochondrial gatekeepers: receptor signaling, QA-driven NAD⁺ coupling, and an analytics-to-clinics workflow. (A) Receptor landscape and mitochondrial endpoints linked to KYNA, highlighting GPR35, AhR, NMDA-R, and α7nAChR signaling. (B) KYN pathway to TCA coupling, showing how branch points such as KATs, KMO, and ACMSD route flux toward KYNA signaling versus QA-to-NAD⁺ supply and shape mitochondrial redox state and respiratory throughput. (C) Analytics-to-clinics workflow summarizing LC-MS panel design, matrix and stability constraints, and QC steps needed for reproducible clinical measurement. Mechanistic and metabolic links are developed in Section 2 and Section 3, while measurement and translation are expanded in Section 4 and Section 5. ACMSD, alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase; AhR, aryl hydrocarbon receptor; α7nAChR, alpha-7 nicotinic acetylcholine receptor; GPR35, G protein–coupled receptor 35; KATs, kynurenine aminotransferases; KMO, kynurenine 3-monooxygenase; KYN, kynurenine; KYNA, kynurenic acid; LC-MS, liquid chromatography–mass spectrometry; NAD⁺, nicotinamide adenine dinucleotide; NMDA-R, N-methyl-D-aspartate receptor; QA, quinolinic acid; QC, quality control; TCA, tricarboxylic acid.
Figure 1.
KYN pathway metabolites as mitochondrial gatekeepers: receptor signaling, QA-driven NAD⁺ coupling, and an analytics-to-clinics workflow. (A) Receptor landscape and mitochondrial endpoints linked to KYNA, highlighting GPR35, AhR, NMDA-R, and α7nAChR signaling. (B) KYN pathway to TCA coupling, showing how branch points such as KATs, KMO, and ACMSD route flux toward KYNA signaling versus QA-to-NAD⁺ supply and shape mitochondrial redox state and respiratory throughput. (C) Analytics-to-clinics workflow summarizing LC-MS panel design, matrix and stability constraints, and QC steps needed for reproducible clinical measurement. Mechanistic and metabolic links are developed in Section 2 and Section 3, while measurement and translation are expanded in Section 4 and Section 5. ACMSD, alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase; AhR, aryl hydrocarbon receptor; α7nAChR, alpha-7 nicotinic acetylcholine receptor; GPR35, G protein–coupled receptor 35; KATs, kynurenine aminotransferases; KMO, kynurenine 3-monooxygenase; KYN, kynurenine; KYNA, kynurenic acid; LC-MS, liquid chromatography–mass spectrometry; NAD⁺, nicotinamide adenine dinucleotide; NMDA-R, N-methyl-D-aspartate receptor; QA, quinolinic acid; QC, quality control; TCA, tricarboxylic acid.
2. Receptor- and Metabolite-Driven Mitochondrial Regulation Within the Kynurenine (KYN) Pathway
KYNA sits at a neat signaling crossroads, but it is not the only KYN that can steer mitochondrial behavior in a mechanistically meaningful way. KYN itself can act through the aryl hydrocarbon receptor (AhR)-biased programs that reshape oxidative defenses and tune mitophagy, so the pathway branch point already carries receptor-linked mitochondrial consequences. Step downstream and the chemistry sharpens. 3-Hydroxykynurenine (3-HK) can tip redox balance toward oxidative stress, while QA can couple de novo NAD⁺ supply to bioenergetic control yet also intensify excitotoxic pressure when it accumulates. Taken together, these metabolites create a layered control system in which receptor signaling and metabolite driven stress converge on shared endpoints such as respiration, reactive oxidative species handling, Ca2+ homeostasis, and mitochondrial quality control, with tissue specific fingerprints across brain, immune cells, and tumors.
2.1. Kynurenic Acid (KYNA) and G protein–Coupled Receptor 35 (GPR35): Energy Homeostasis and Ischemic Protection
GPR35 has emerged as a regulator of metabolic stress responses [105,106,107,108,109]. It is ex-pressed in the gastrointestinal tract, immune cells, central nervous system, and heart, with expression upregulated in pathological conditions including ischemia–reperfusion injury, hypoxia, stroke, and heart failure [110,111,112]. Although GPR35 is associated with cytoprotection, evidence remains inconsistent regarding whether receptor activation or inhibition is beneficial [110,111,113,114]. Furthermore, a GPR35-dependent gut–microbe–brain metabolic axis has been identified, linking receptor activity to neuroimmune regulation and depressive-like behavior [115,116].
GPR35 interacts with several G protein families, enabling the regulation of diverse intracellular pathways with both pro- and anti-inflammatory effects [114,117,118,119]. Coupling to Gαi/o suppresses adenylate cyclase, decreases cyclic adenosine monophosphate (cAMP) levels [120], and reduces extracellular signal-regulated kinase (ERK) activity [121], which can limit extracellular signal-regulated kinases (ERKs) driven pro-inflammatory transcription [117]. Conversely, Gβγ subunits released from Gαi/o activate PLCβ, driving phosphoinositide hydrolysis, phosphoinositide 3-kinase (PI3K)/ protein kinase B (AKT) signaling, and nuclear factor kappa-light-chain-enhancer of activated b cells (NF-κB) activation, thereby promoting inflammatory gene expression [117]. Interaction with Gα12/13 stimulates ras homolog family protein (Rho)-dependent cytoskeletal remodeling, enhancing immune cell chemotaxis and reinforcing pro-inflammatory signaling. In addition, Gαq coupling exerts dual actions: it restricts PI3K-mediated AKT activation while facilitating ERK signaling through a PLCβ/Ca²⁺/Src pathway [122]. These diverse and occasionally opposing mechanisms underscore the context-dependent role of GPR35 in coordinating G protein signaling and regulating mitochondrial responses [32,123,124,125]. Activation of these receptors has also been described to contribute to organellar damage via calpain-mediated proteolysis under various pathophysiological conditions. For example, Ca2+ overload activates calpains, which translocate to intracellular organelles, degrade target proteins, destabilize nuclear, lysosomal, and mitochondrial membranes, and release cathepsins and pro-apoptotic factors, ultimately leading to cell death [126]. Evidence implicates calpain-1 and calpain-2 in mitochondrial damage following cardiac ischemia/reperfusion [113,127,128]. Notably, GPR35 is strongly upregulated after myocardial ischemia, and its inhibition attenuates ROS production, reduces mitochondrial apoptosis, and preserves contractile function [129,130]. Consistently, blockade of GPR35 downregulates calpain-1 and calpain-2 expression and activity, attenuating calpain-mediated mitochondrial injury. The detrimental effects of calpain-driven proteolysis affect multiple mitochondrial sites, including increasing mitochondrial membrane permeability, inducing cytochrome c release, and initiating apoptosis [131]. In addition, calpain-1 can cleave the ATP synthase α-subunit, reducing ATP production and exacerbating oxidative stress [131].
Wyant and colleagues (2022) demonstrated that KYNA exerts cardioprotective effects during ischemia/reperfusion by acting on GPR35 [22]. Their study identified GPR35 as both necessary and sufficient for mediating KYNA-induced ischemic protection, a process tightly coupled to mitochondrial remodeling. Upon ligand binding, GPR35 activates Gi- and G12/13-dependent signaling cascades and translocates to the outer mitochondrial membrane, where it associates, likely indirectly, with ATP synthase inhibitory factor subunit 1 (ATPIF1) [118,132]. Through this interaction, activated GPR35 promotes ATP synthase dimerization and modulates oxidative phosphorylation to preserve cellular ATP content and maintain energy homeostasis under ischemic stress [133,134,135]. ATP synthase normally generates ATP from the proton gradient; however, during ischemia, it can reverse and hydrolyze ATP, resulting in energy loss and mitochondrial dysfunction [136,137,138]. ATPIF1 prevents this reverse mode without affecting ATP synthesis [110]. By stabilizing ATP synthase dimers, ATPIF1 also supports cristae integrity and prevents mitochondrial permeability transition pore (mPTP) opening [22,139]. In more detail, phosphorylation of ATPIF1 deactivates the protein, thereby permitting ATP hydrolysis, whereas dephosphorylation at Ser39 activates ATPIF1 and suppresses ATPase activity [22,140]. Mitochondrial GPR35 signaling dampens cAMP production by inhibiting adenylyl cyclase, thereby reducing PKA-mediated phosphorylation of ATPIF1 and maintaining it in its active, dephosphorylated state [22]. Collectively, these findings delineate a mitochondrial GPR35 in coordinating energy conservation and structural stability.
The mitochondrial membrane potential (ΔΨm) serves as the primary driving force for ATP synthesis, generated by the proton gradient across the inner mitochondrial membrane [141,142,143]. A decrease in ΔΨm weakens the proton motive force, leading to diminished or even halted ATP production. Sustained depolarization of the mitochondrial membrane results in cellular energy depletion, metabolic disturbances, and ultimately, cell death [144,145]. Preservation of ATP levels under ischemic conditions is therefore closely dependent on maintaining ΔΨm, which is essential for cell viability and the physiological function of organs [145]. A direct link between GPR35 modulation and alterations in ΔΨm has also been demonstrated under pathological conditions. Specifically, inhibition of GPR35 was shown to mitigate mitochondrial dysfunction not only by enhancing oxidative phosphorylation but also by preserving ΔΨm [113]. In neonatal murine ventricular myocytes (NMVMs), JC-1 assays revealed a higher ΔΨm under ischemic or anoxic stress when GPR35 was inhibited compared to control conditions, indicating a protective mitochondrial effect [113].
Beyond its well-established role in ischemic protection, accumulating evidence indicates that KYNA–GPR35 signaling constitutes a critical regulatory axis in systemic energy homeostasis [16,146,147], particularly in the regulation of lipid catabolism [17]. Following three days of KYNA administration (a single daily intraperitoneal dose of 5 mg/kg body weight) in C57BL/6J mice, increased oxygen consumption, carbon dioxide production, and heat generation were observed, indicating enhanced metabolic activity and energy expenditure [16]. Moreover, KYNA administered on consecutive days significantly reduced white adipose tissue mass, including both inguinal and visceral (epididymal) depots, without exerting any measurable effect on brown adipose tissue mass. In adipose tissue, activation of GPR35 induces thermogenic and anti-inflammatory transcriptional programs, and thereby mitigates high-fat diet–induced adiposity while simultaneously improving glucose tolerance [16]. At the molecular level, this pathway upregulates PGC-1α expression and enhances mitochondrial oxidative capacity, thereby promoting mitochondrial biogenesis [16]. The anti-inflammatory mechanism involves KYNA signaling, which increases the expression of type 2 cytokines such as IL-4, IL-10, IL-13, and IL-33, while reducing pro-inflammatory markers such as TNFα [16,148]. This cytokine shift promotes a type 2 immune environment, supporting the resolution of inflammation and improving insulin sensitivity [17]. The KYNA–Gpr35 pathway has been shown to enhance the presence and activity of regulatory T cells (Tregs) and type 2 innate lymphoid cells (ILC2s), thereby contributing to anti-inflammatory signaling and adipose tissue being [16].
The KYNA–Gpr35 axis has emerged as a critical regulator of adipose tissue metabolism and inflammation, with considerable therapeutic relevance [16,17,34]. KYNA exerts a dual modulatory effect on metabolic efficiency and immune homeostasis by promoting adipose tissue being and enhancing mitochondrial oxidative capacity to support thermogenesis [16]. In parallel, KYNA modulates the inflammatory milieu, directing the immune balance toward an anti-inflammatory phenotype [17,147,149]. These coordinated actions integrate metabolic and immunoregulatory mechanisms, suggesting that pharmacological activation of Gpr35 may constitute a promising therapeutic approach to augment systemic energy expenditure and mitigate metabolic disorders associated with chronic low-grade inflammation and disrupted energy balance, including obesity, type 2 diabetes, and metabolic syndrome [16].
2.2. Kynurenic Acid (KYNA) and Aryl Hydrocarbon Receptor (AhR): Mitophagy and Organelle Quality Control (QC)
The AhR functions as a ligand-activated transcription factor that remains localized in the cytoplasm under basal conditions [150,151,152]. Upon ligand binding, AhR undergoes conformational changes that promote its nuclear translocation and the transcriptional regulation of a broad array of genes involved in cellular homeostasis [95,153,154,155]. AhR is well recognized for its central role in detecting xenobiotics and regulating their metabolism through cytochrome P450 enzymes (e.g., CYP1A1, CYP1A2, and CYP1B1) [156,157]. Moreover, increasing evidence indicates that AhR participates in a wide range of physiological processes, including immune regulation and embryogenesis [158,159,160]. However, its contribution to mitochondrial regulation in association with the Trp-KYN pathway has only recently come to light.
The first study identifying KYNA as an endogenous agonist of AhR was reported in the early 2010s [161,162]. Like KYNA, xanthurenic acid (XA)—another metabolite of the Trp–KYN pathway—has been shown to function as a ligand that can activate the AhR at physiologically relevant concentrations [161,163]. Recent evidence has demonstrated that AhR directly contributes to hepatic energy preservation by modulating mitophagy [155]. Mitophagy is a selective form of autophagy occurring within lysosomes and is responsible for the removal of damaged mitochondria [164,165,166,167,168]. This process reduces mitochondrial ROS overproduction, limits inflammasome activation, decreases apoptosis, maintains proper ATP synthesis, and promotes mitochondrial turnover [169,170,171]. In both AhR knockout mice and hepatocyte models, the loss of AhR expression resulted in impaired mitochondrial respiration, decreased substrate utilization, and dysregulation of mitochondria-associated gene networks [155,171]. Under pathophysiological conditions, a study conducted in intestinal porcine enterocytes (IPEC-J2 cells) further illustrated that activation of the AhR by Trp ameliorates lipopolysaccharide (LPS)-induced inflammatory responses [172,173]. This work also provided mechanistic insight into the interplay between AhR activation and mitochondrial QC by demonstrating that AhR functions as a direct transcriptional activator of PINK1 [155,172,174].
Mitophagy, in addition to being initiated through the ubiquitin-dependent PINK1–Parkin pathway, can also occur via receptor-mediated mechanisms involving BCL2-interacting protein 3 (BNIP3) and NIX [175,176,177,178,179]. In this pathway, LC3 binds to these receptors through its LC3-interacting region (LIR), ensuring the targeted recruitment of autophagosomes to mitochondria and enabling selective mitochondrial degradation [180]. Fasting strongly induced Bnip3 expression in livers obtained from wild-type samples, whereas this response was completely absent in AhR-deficient mice [155]. Activation of AhR by its endogenous ligand KYN significantly increased Bnip3 mRNA and protein levels in primary hepatocytes and AML12 cells [155]. Moreover, inhibition of AhR elevated mitochondrial ROS production—an effect entirely reversed by BNIP3 overexpression—and decreased LC3A expression [155]. Together, these results indicate that AhR plays a crucial role in regulating receptor-mediated mitophagy in the liver.
Excessive mitochondrial ROS promote mitochondrial dysfunction [181,182,183], a mechanism previously linked to AhR activation and AhR-dependent ROS generation induced by dioxins [184]. One potential source of these ROS is the accumulation of damaged or dysfunctional mitochondria, highlighting the importance of their removal for proper cellular function, including ATP production [185,186]. Physiological AhR activity appears to play a significant role in controlling mitochondrial stress responses, as its inhibition or loss has been associated with ROS imbalance and disturbance in oxidative metabolism [184,187]. In the liver, results suggests that AhR contributes to hepatic metabolic adaptation by maintaining mitochondrial efficiency and redox balance under nutrient, environmental, or hypoxic stress. Activation of AhR supports oxidative metabolism and preserves energy homeostasis, whereas its inhibition disrupts metabolic balance and promotes ROS accumulation, which can reduce cell viability [155].
The AhR plays a cellular-context–dependent role in mitochondrial biology, mediating both protective and deleterious outcomes. Under physiological conditions, AhR supports mitochondrial homeostasis: in hepatocytes, AhR loss impairs mitophagy, increases mitochondrial ROS, and reduces electron transport system function [155]. Similarly, in human melanocytes following H₂O₂-induced oxidative injury, AhR activation promotes mitochondrial biogenesis, mtDNA synthesis, and ATP production via NRF1 upregulation [188]. Conversely, toxic or xenobiotic ligands, such as PM2.5 or elevated KYN, trigger AhR-mediated mtROS production, mPTP opening, and ΔΨm collapse, leading to apoptosis in mouse neuronal cells and zebrafish heart [189,190]. These findings raise key questions: how do cell type, tissue context, and metabolic state dictate whether AhR signaling is protective or deleterious? How do ligand characteristics, dose, and exposure time affect mitochondrial outcomes? Resolving these issues is critical for understanding AhR’s divergent role in mitochondrial physiology.
2.3. Kynurenic Acid (KYNA) and N-Methyl-D-Aspartate (NMDA) Receptors: Ca2+ Regulation and Excitotoxicity
NMDA-R are glutamate receptors and ligand-gated channels, widely recognized for their roles in synaptic signaling, Ca2+ regulation, and excitotoxicity. Although they are predominantly expressed in the central nervous system, receptor subunits have also been detected in peripheral organs, including the heart, stomach, and intestine. Emerging evidence indicates that NMDA-R localization is not restricted to the plasma membrane, as these receptors have also been identified in the inner mitochondrial membrane, suggesting a potential role in regulating mitochondrial function.
KYNA acts as an NMDA-R antagonist by exerting its inhibitory effect at the glycine co-agonist site of the receptor [41,191,192], resulting in pleiotropic protective effects that include the modulation of mitochondrial function through this mechanism [13,193,194].
NMDA R overdrive elevates cytosolic Ca2+ and can push mitochondria past their buffering range. When Ca2+ loading coincides with oxidative pressure, pore sensitivity rises, ΔΨm drops, and apoptosis-linked signaling accelerates. In this context, KYNA antagonism at NMDA-R functions less like a single target trick and more like a gatekeeper that lowers Ca2+ entry, which reduces downstream mitochondrial destabilization and inflammatory spillover.
By limiting NMDA-R–mediated Ca2+ influx, KYNA may indirectly reduce the probability of mPTP opening, thereby preventing the release of both cytochrome c and mtDNA into the cytosol [13,195]. This dual action not only attenuates pro-apoptotic signaling but also mitigates inflammatory processes associated with mitochondrial damage, emphasizing the potential cyto- and mitoprotective roles of KYNA. Recent studies further support the anti-apoptotic effects linked to its mitoprotective actions, although these mechanisms are not solely mediated by NMDA-Rs. In H9c2 cells and primary rat cardiomyocytes exposed to simulated ischemia/reperfusion [31,125], KYNA exerted a dose-dependent effect on cell viability, with the most effective concentration being 64 µM [125]. Specifically, KYNA attenuated intramitochondrial Ca²⁺ accumulation, reduced ROS generation, and alleviated alterations in mitochondrial network architecture following simulated ischemia/reperfusion. Moreover, apoptosis markers such as caspase-3/7 and BAX (a pro-apoptotic modulator) were reduced, while the expression of the anti-apoptotic protein Bcl-XL was increased following KYNA treatment [125]. Consistent with these mitoprotective and anti-apoptotic effects, a reduction of neuronal apoptosis in microglial cultures via attenuation of CXCL10 expression by KYNA and its analogue (SZR-104) cannot be ruled out [196].
NMDA-Rs are not limited to the plasma membrane but have also been detected in mitochondria [197]. It was reported that the presence of NR1 and NR2B subunits, together with GABAA (alpha-6) and GABAB (R2) receptors, in rat heart mitochondria. Besides, extensive NR2a subunit immunoreactivity was observed on hippocampal mitochondria using immunogold electron microscopy [198]. Although the precise role of these receptors within mitochondria remains unclear, they are hypothesized to regulate Ca2+ fluxes, modulate ROS production, and contribute to metabolic adaptation under hypoxic or ischemic conditions [198,199]. An additional layer of regulation may involve direct interactions between NMDA-R subunits and mitochondrial complex I components, such as ND2, mediated via Src adaptor proteins [200,201]. This receptor–complex I crosstalk establishes a direct molecular link between receptor activity and mitochondrial energy metabolism, providing a mechanistic framework for how glutamatergic signaling influences mitochondrial bioenergetics. Collectively, these findings suggest that mitochondrial NMDA-Rs may serve as critical modulators of organelle function, integrating cellular signaling with energy generation and oxidative stress responses, with potential implications for pathophysiological conditions such as ischemia, hypoxia, and neurodegeneration.
Taken together, these findings suggest that KYNA exerts multifaceted, receptor-mediated effects on mitochondria. Through interactions with GPR35, AhR, and NMDA-Rs, KYNA helps preserve energy homeostasis, supports mitochondrial quality control, and protects against Ca2+-induced mitochondrial dysfunction—highlighting its therapeutic potential in conditions associated with mitochondrial stress or impairment [30,202,203,204,205]. Mitochondrial dysfunction has been implicated in various pathologies, including neurodegenerative diseases such as Alzheimer’s, Huntington’s, and Parkinson’s diseases, as well as psychiatric disorders linked to mood disturbances, such as bipolar depression and migraine [13,41,206,207,208,209,210]. Therefore, maintaining mitochondrial homeostasis appears to be a promising therapeutic strategy for these conditions [174].
Future studies should investigate the role of mitochondrial NMDA-Rs and their modulation by KYNA and its analogues, as it remains unclear whether these channels are sensitive to conventional NMDA-R inhibitors. Likewise, the potential for targeting mitochondrial GPR35 and AhR receptors under stress conditions warrants further exploration, particularly regarding how their trafficking influences mitochondrial membrane dynamics and transport processes. Elucidating these mechanisms may provide crucial insights into how KYNA and its signaling pathways regulate mitochondrial function and could reveal novel therapeutic targets for diseases characterized by impaired bioenergetics or oxidative stress.
2.4. Nicotinic Acetylcholine Receptors (α7nAChR)-like
α7nAChRs, a type of ligand-gated ion channel previously thought to be localized only to the plasma membrane, are also present in the outer mitochondrial membrane [211]. Electron microscopy and binding assays (α-bungarotoxin, α-cobratoxin) have confirmed their presence in this organelle; however, their precise role remains to be characterized.
These α7nAChRs are thought to interact with voltage-dependent anion channels (VDAC1). Kalashnyk et al. confirmed this interaction, identifying α7 nAChR–Bax and α7 nAChR–VDAC1 complexes in mitochondria isolated from human glioblastoma astrocytoma cells [212]. Pharmacological studies demonstrated that inhibition of α7nAChRs using antagonists such as methyllycaconitine or α7-specific antibodies suppressed mitochondrial cytochrome c release, whereas stimulation with the receptor agonist PNU 282987 enhanced it in isolated mouse liver mitochondria [213]. Furthermore, mitochondrial ROS production was reduced following both receptor inhibition (methyllycaconitine) and stimulation with acetylcholine [213]. The α7nAChRs exhibit relatively high permeability to Ca2+ ions, which can influence mitochondrial function by stimulating Ca2+ influx and efflux through the mitochondrial Ca2+ uniporter and mPTPs [214]. In addition, modulation of various kinase pathways—including PI3K/Akt, Ca2+–calmodulin, and Src kinase–dependent signaling—appears to influence mPTP activity via these receptors, thereby supporting the OXPHOS machinery and maintaining cellular energy production [213].
Surprisingly, KYNA has also been reported to inhibit α7nAChRs; however, these findings remain controversial [37], as several research groups have failed to reproduce the original results. One possible explanation for these discrepancies may arise from the distinct pharmacological profiles of the kynurenate analogues most frequently used, such as 7-chloro-kynurenic acid (7-CKA) and 5,7-dichloro-kynurenic acid, which act at different sites on the NMDA-R. A similar mechanism might account for the observed inconsistencies in the context of α7nAChRs as well.
It remains an important question, given conflicting experimental data [215], whether modulation of the α7nAChR—by inhibition or activation—can provide sustained neuroprotection and anti-inflammatory effects, and how these mechanisms interact with each other. At the same time, the potential role of mitochondrial α7nAChRs in mediating the cellular and mitoprotective effects of KYNA or its synthetic analogues has yet to be comprehensively characterized and requires further investigation to bridge the gap between cellular and subcellular mechanisms.
2.5. Other Potential Kynurenic Acid (KYNA)-Sensitive Sites
Beyond the canonical receptor set, KYNA likely engages a wider mitochondrial “sensing” network. Additional receptors and redox-tunable targets—especially those shaping ion homeostasis and respiratory control—could help explain KYNA-linked phenotypes in ischemia and inflammation, and may open testable directions for neurodegenerative and psychiatric disease mechanisms.
Mitochondrial ATP-sensitive potassium (mitoKATP) channels are crucial mediators of cardioprotection induced by ischemic preconditioning and neuroprotection following cerebral ischemia-reperfusion. Pharmacological blockade of these channels with 5-hydroxydecanoate abolishes cardioprotective effects, whereas activation with the mitoKATP opener diazoxide confers neuroprotection [216,217]. Mechanistically, mitoKATP channel opening attenuates mitochondrial Ca²⁺ overload and delays or inhibits the opening of mPTP. Given that ROS and elevated Ca²⁺ are major triggers of mPTP opening, and that GPR35 modulates Ca²⁺ flux while ROS can induce mitoKATP opening and upregulate GPR35 [218], a potential crosstalk between these two mitochondrial receptors—both of which influence mPTP function—may underlie coordinated cytoprotective signaling in these disease conditions.
Mitochondrial ROS originate from 16 distinct redox sites within the organelle, with Complexes I and III of the electron transport system representing the principal ROS generators [219,220]. Redox-sensitive cysteine residues in Complex I subunits, such as NDUFS1, NDUFS2, and ND3, fine-tune ROS generation (superoxide and H₂O₂) by acting as redox switches. In contrast to excessive ROS production, transient Complex I–derived ROS induction enhances stress tolerance and lifespan in C. elegans [221]. KYNA, which scavenges hydroxyl radicals and superoxide [222], may reduce mitochondrial ROS at Complexes I and III by preserving electron transport integrity.
2.6. Kynurenine (KYN) as an Aryl Hydrocarbon Receptor (AhR)-linked Mitochondrial Program Driver
KYN is not a passive precursor [50]. It can function as an AhR ligand in its own right, shifting transcriptional programs that regulate BNIP3 and PINK1 linked mitophagy, oxidative metabolism, and ROS buffering [155,172]. This means receptor to mitochondria coupling is not exclusive to KYNA [155,190]. It can also arise from the pathway branch point metabolite that rises during inflammation, cancer, and chronic stress states [50,223,224].
2.7. 3-Hydroxykynurenine (3-HK), Redox Cycling, and Mitochondrial Injury Endpoints
3-HK behaves like a spark in dry grass [15]. In some contexts, it amplifies oxidative chemistry through autoxidation and glutathione depletion, then the mitochondrion pays the bill via impaired TCA flux, ROS escalation, and apoptosis [15,51]. That redox pressure can also reprogram immune signaling, so 3 HK links mitochondrial damage to immunometabolic state rather than merely reporting it [47,225].
2.8. Quinolinic acid (QA), N-methyl-D-aspartate (NMDA) Signaling, and QA to Nicotinamide Adenine Dinucleotide (NAD⁺) Plus Constraints
QA sits at a double junction [14,94]. It is a precursor for de novo NAD⁺ synthesis, yet it can also drive excitotoxic and oxidative stress through NMDA-linked Ca2+ loading and downstream mitochondrial dysfunction when it accumulates or when conversion capacity is limited [14,198,206]. The mechanistic question in Section 2 is whether QA is being cleared into NAD plus or accumulating as a mitochondrial stressor, while Section 3 details how this branch choice rewires NAD plus pools, TCA throughput, and redox ratios at scale [14,94].
Figure 2.
KYNA-linked receptor signaling converges on mitochondrial bioenergetics, redox balance, calcium handling, and quality control during metabolic or ischemic stress. (A) GPR35 coupled energy preservation module that supports ATP maintenance and inner membrane stability. (B) AhR linked redox tuning and mitophagy programs that promote selective clearance of damaged mitochondria. (C) NMDA-R linked calcium entry control that lowers excitotoxic pressure and reduces pore sensitivity. (D) Mitochondrial alpha7nAChR linked modulation of VDAC associated calcium flux and respiratory ROS output. (E) Additional KYNA sensitive mitochondrial nodes, including mitoKATP channels and redox responsive ETC sites, that can fine tune stress thresholds and tissue resilience. Detailed mechanisms are described in Section 2, while QA to NAD+ and TCA coupling is developed in Section 3. AhR, aryl hydrocarbon receptor; alpha7nAChR, alpha7 nicotinic acetylcholine receptor; ATP, adenosine triphosphate; ETC, electron transport chain; GPR35, G protein coupled receptor 35; KYNA, kynurenic acid; mitoKATP, mitochondrial ATP sensitive potassium channel; NAD+, nicotinamide adenine dinucleotide; NMDA-R, N methyl D aspartate receptor; QA, quinolinic acid; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; VDAC, voltage dependent anion channel.
Figure 2.
KYNA-linked receptor signaling converges on mitochondrial bioenergetics, redox balance, calcium handling, and quality control during metabolic or ischemic stress. (A) GPR35 coupled energy preservation module that supports ATP maintenance and inner membrane stability. (B) AhR linked redox tuning and mitophagy programs that promote selective clearance of damaged mitochondria. (C) NMDA-R linked calcium entry control that lowers excitotoxic pressure and reduces pore sensitivity. (D) Mitochondrial alpha7nAChR linked modulation of VDAC associated calcium flux and respiratory ROS output. (E) Additional KYNA sensitive mitochondrial nodes, including mitoKATP channels and redox responsive ETC sites, that can fine tune stress thresholds and tissue resilience. Detailed mechanisms are described in Section 2, while QA to NAD+ and TCA coupling is developed in Section 3. AhR, aryl hydrocarbon receptor; alpha7nAChR, alpha7 nicotinic acetylcholine receptor; ATP, adenosine triphosphate; ETC, electron transport chain; GPR35, G protein coupled receptor 35; KYNA, kynurenic acid; mitoKATP, mitochondrial ATP sensitive potassium channel; NAD+, nicotinamide adenine dinucleotide; NMDA-R, N methyl D aspartate receptor; QA, quinolinic acid; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; VDAC, voltage dependent anion channel.
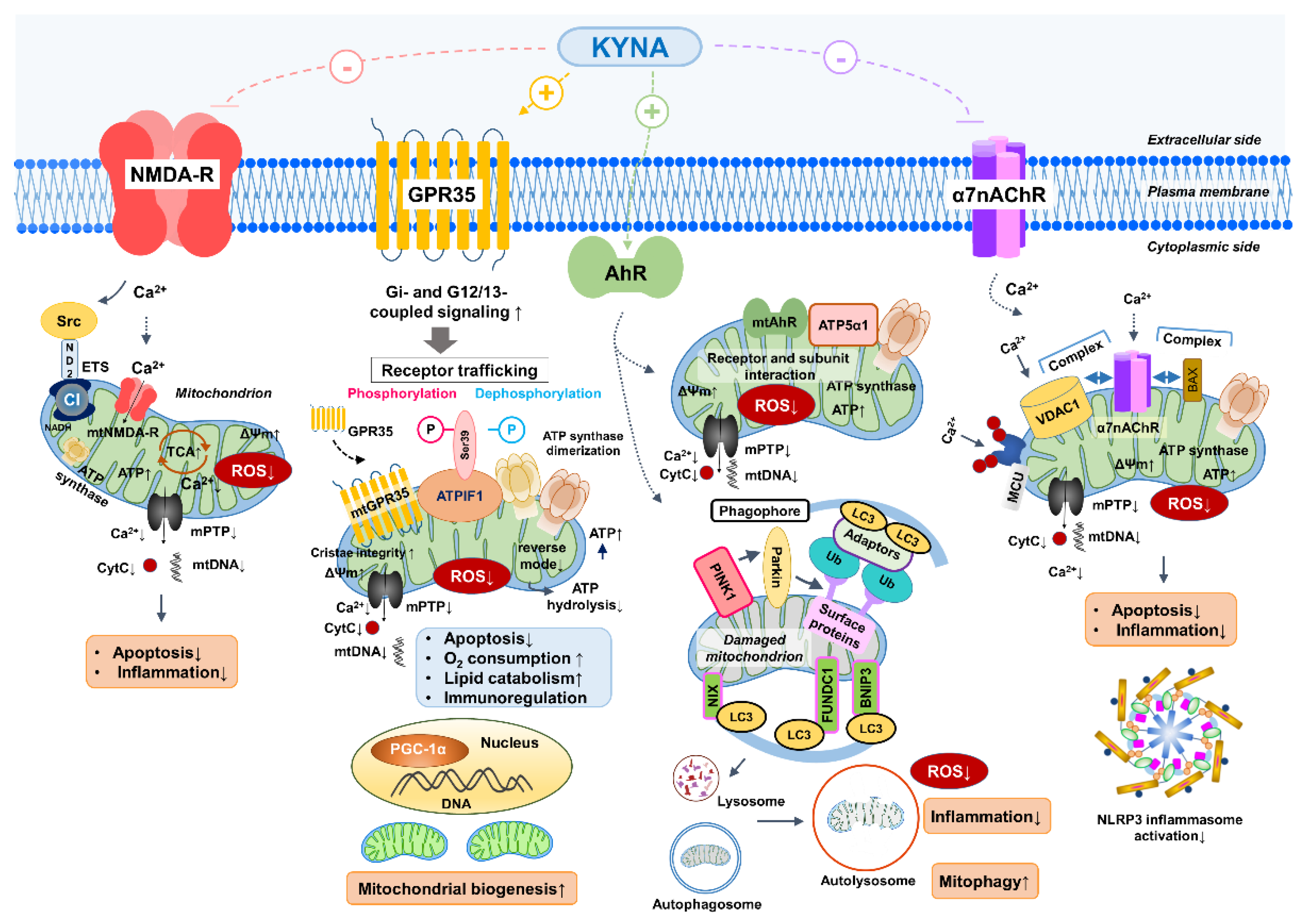
Table 2.
Receptor targets of KYNA and metabolite driven-mitochondrial endpoints of KYNs
| Target | Mitochondrial effects | Animal model/Cell type | Key findings | References |
|---|---|---|---|---|
| GPR35 | -ATP preservation -ATP turnover ↑ -Mitochondrial oxidative capacity ↑ -ROS production ↓ -mPTP opening inhibition -Calpain-1/2 activity ↓ |
Myocardial ischemia/reperfusion (rat, NMVMs) Myocardial ischemia/reperfusion (mouse, heart tissue) C57BL/6J mice, adipose tissue |
-Receptor activation stabilizes ATP synthase, maintains ΔΨm, prevents ATP hydrolysis, limits ROS and apoptosis -GPR35 blockade reduces calpain-mediated proteolysis, preserves mitochondrial integrity, and mitigates oxidative stress -KYNA enhances PGC-1α expression, promotes mitochondrial biogenesis, increases O₂ consumption, and initiate anti-inflammatory cytokine production |
[16,22,110,126,139,218] |
| mt GPR35 |
-ATP synthase dimerization -ATP preservation via the inhibition of ATP hydrolysis |
GPR35 knoclout mice and neonaatal cardiomyocytes | -Binds to ATPIF1 and associates with the mitochondrial outer membrane -Inhibits mitochondrial adenylate cyclase and thereby PKA -Allows ATPIF1 to promote ATP synthase dimerization and prevent ATP hydrolysis |
[22,139] |
| AhR |
-Mitophagy (BNIP/PINK1–Parkin) ↑ -ROS production ↓ -ATP preservation -Oxidative metabolism ↑ |
Hepatocytes, AML12 cells, IPEC-J2 cells, AhR knockout mice, primary hepaocytes |
-KYNA and KYN activate AhR to induce PINK1 and BNIP3 expression, promoting mitophagy and preserving mitochondrial respiration under stress -Loss of AhR impairs mitochondrial quality control and increases ROS accumulation, disrupting energy metabolism |
[155,172,184,226,227] |
| mtAhR |
-ATP synthase regulation -Fine-tuning ROS production |
Mitochondrial fraction, liver cells | -Mitochondrial AhR interacts with ATP5α1; its localization and activity depend on ligand status, possibly influencing ATP synthesis and redox balance | [226] |
| NMDA-R |
-Ca²⁺ influx ↓ -mPTP opening ↓ -Cytochrome c release ↓ -Bcl-XL expression ↑ -Apoptosis ↓ -Complex I coupling |
Neurons, Microglia, neuronal cultures |
-KYNA blocks NMDA-R at the glycine site, limits Ca²⁺ overload, prevents mPTP opening, and protects against apoptosis -Potential crosstalk with complex I regulates bioenergetics |
[196] |
| mt NMDA-R | -Ca²⁺ flux modulation -Fine-tuning ROS production |
Rat heart mitochondria |
-NR1/NR2B subunits detected in mitochondria; -Regulation of ROS production and Ca²⁺ level under hypoxia/ischemia |
[197] |
| α7nAChR |
-Regulates Ca²⁺ flux, ROS production, and cytochrome c release via interaction with VDAC1 -Influences mPTP opening and OXPHOS activity through kinase signaling (PI3K/Akt, CaM, Src). -Limits apoptosis through the regulation of Bcl-2/Bcl-xL and caspases. |
Isolated mouse liver mitochondria; U373 human glioblastoma astrocytoma cells KAT II knockout (KAT II⁻/⁻) mice |
-α7nAChR–VDAC1 and α7nAChR–Bax complexes identified; receptor inhibition (methyllycaconitine) suppresses cytochrome c release; stimulation (PNU 282987) enhances it; acetylcholine reduces ROS. -Decreased KYNA levels increase α7nAChR activity; α7nAChR activation linked to neuroprotection and anti-apoptotic signaling; KYNA may physiologically regulate the receptor |
[37,213,215,228,229] |
| mitoKATP channels (?) |
-Channel opening reduces mitochondrial Ca²⁺ overload, delays mPTP opening, and supports cellular survival during ischemic or oxidative stress; -Its function is modulated by GPR35 and ROS signaling |
In vivo ischemia–reperfusion models: cardiac and neuronal mitochondria |
-Diazoxide (mitoKATP opener) confers neuro- and cardioprotection; inhibition (5-hydroxydecanoate) abolishes protective effects; -ROS and GPR35 signaling may crosstalk to regulate mitoKATP and mPTP. |
[216,217,218] |
| Complex I / Complex III redox sites (?) |
-Redox-sensitive cysteine residues regulate ROS generation (superoxide, H₂O₂); transient ROS acts as signaling for stress adaptation; -KYNA may scavenge radicals and preserves electron transport. |
Isolated mitochondria; C. elegans |
-KYNA may reduce ROS at Complex I and III independent of receptor mechanisms; mild Complex I ROS prolongs lifespan in C. elegans; antioxidant effects support mitochondrial stability. |
[219,220,221,222] |
Abbreviations: GPR35, G protein–coupled receptor 35; KYNA, kynurenic acid; ATPIF1, ATP synthase inhibitory factor 1; KYN, kynurenine; KYNA, kynurenic acid; PKA, protein kinase A; ΔΨm, mitochondrial membrane potential; ROS, reactive oxygen species; mPTP, mitochondrial permeability transition pore; PGC-1α, per-oxisome proliferator–activated receptor gamma coactivator 1-alpha; AhR, aryl hydrocarbon re-ceptor; mtAhR, mitochondrial aryl hydrocarbon receptor; BNIP3, Bcl-2/adenovirus E1B 19 kDa–interacting protein 3; PINK1, PTEN-induced kinase 1; NMDA-R, N-methyl-D-aspartate receptor; mtNMDA-R, mitochondrial N-methyl-D-aspartate receptor; Bcl-XL, B-cell lymphoma extra-large (anti-apoptotic protein); VDAC1, Voltage-dependent anion channel 1; α7nAChR, α7 nicotinic acetylcholine receptor; mitoKATP (mKATP), mitochondrial ATP-sensitive potassium channel; OXPHOS, oxidative phosphorylation; CI / CIII, Complex I / Complex III (electron transport chain).
3. Crosstalk Between the KYN Metabolism and the TCA Cycle
Section 3 addresses Objective 2 by focusing on shared metabolic control points. We connect QA fueled NAD+ biogenesis to TCA wiring, redox ratios, and respiratory throughput, then use these nodes to explain how shifts in KYN pathway flux become mitochondrial phenotypes in immune and neural contexts.
3.1. Shared Metabolic Intermediates and Redox Balance
Shared intermediates choreograph redox balance across compartments. TCA cycle flux supplies reducing power and precursors, while anaplerosis preserves pool sizes that sustain NADPH and glutathione buffering [3,8]. When flux falters, ATF4 programs rewire amino acid and antioxidant metabolism [1]. Malic enzyme-1 senses malate to pyruvate and tunes NADPH output [11]. Meanwhile, ROS encode signals that reshape metabolic set points and defenses [18,19,20].
Mitochondrial respiration reads the NAD+/NADH ratio as a control signal that tunes flux through the TCA cycle, the electron transport chain, and fate decisions in diverse lineages [1,5]. Pool size and localization matter. Mitochondrial carrier transporting NAD⁺ (MCART1) imports NAD⁺ to sustain matrix reactions, while deficits collapse dehydrogenase activity and oxygen consumption [4,6]. Oxaloacetate generated by malate dehydrogenase 2 (MDH2) selectively restrains complex II and reroutes electron flow, thereby reshaping the redox couple in real time [10]. Nicotinamide nucleotide transhydrogenase coordinates nicotinamide adenine dinucleotide (NADH) with nicotinamide adenine dinucleotide phosphate (NADPH) demand, stabilizing redox poise during shifts in glutamine and glucose use [15]. When respiration stalls, serine-driven NADH accumulates and throttles biosynthesis [11].
Immune and disease contexts expose the same logic. LKB1 dependent mitochondrial programs and thioredoxin circuits sculpt NADH turnover, with consequences for chromatin state and T cell effector function [2,13]. Cells with succinate dehydrogenase (SDH) lesions adopt alternative aspartate synthesis routes that hinge on matrix NAD+/NADH, salvaging growth despite impaired cycling [7]. De novo NAD+ from the KYN metabolism supports macrophage respiration and systemic redox communication, linking Trp catabolism to respiratory control across tissues [3,12,18]. ROS then operate as graded messengers downstream of the ratio, reinforcing or reprogramming signaling pathways [19,20]. Therapeutically, targeted manipulation of NAD+/NADH can complement defective electron transport and may slow age related decline [8,16].
QA sits at the fulcrum of de novo NAD⁺ synthesis, converting Trp catabolism into respiratory capacity. In macrophages, intact quinolinate to NAD⁺ flux sustains complex I driven oxidation and immune effector programs; aging and inflammation magnify the dependence, and pathway blockade collapses oxygen consumption [1]. In tissues under ischemia reperfusion, diversion away from quinolinate depletes NAD⁺, weakens antioxidant defenses, and heightens oxidative injury, all reversible with NAD⁺ augmentation [3]. Genetic and clinical data converge on QPRT as a bottleneck whose repression or loss yields quinolinate accumulation, reduced NAD⁺, and vulnerability to damage [9,13]. Astrocytes and neurons similarly require quinolinate conversion to maintain SIRT activity, viability, and mitochondrial function during neuroinflammation [8].
Evolution supplies redundancy and reach. Yeast can secrete and reimport quinolinate to stabilize NAD⁺ pools, and, when canonical steps fail, UMPS can substitute to complete synthesis [4,11]. Engineered circuits that route quinolinate toward NAD⁺ raise cellular NAD(H) and bolster electron transport, illustrating design principles for metabolic support [5]. Across physiology and disease, rising mitochondrial work rates associate with higher circulating quinolinate and nicotinamide, consistent with coupled biogenesis and electron transport chain (ETC) demand [10]. Yet excess or chronic pathway activation is harmful, impairing bioenergetics in neurons and reshaping tumor growth constraints, where ubiquinol oxidation remains a nonnegotiable requirement beyond NAD+ regeneration alone [12,17]. Together, these findings position QA as both a sensor and supplier that links Trp flux to NAD+ homeostasis, redox poise, and sustained respiratory throughput [2,6,7,14,15,16,18,19].
α-Ketoglutarate sits at the crossroads of carbon flow and immune control, shaping how cells route Trp into the KYN metabolic pathway. Through IDH2 dependent reductive carboxylation, α-ketoglutarate fuels isocitrate and citrate production, generates NADPH, and thereby tunes redox poise that can favor or restrain IDOs and TDO activity through cofactor availability and metabolic context [1,2]. Inflammatory signaling rewires this hub. Type I interferon inhibits isocitrate dehydrogenase, distorting the citrate to α-ketoglutarate ratio, shifting mitochondrial electron supply, and altering the local redox environment that licenses Trp catabolic flux [3].
Macrophage polarization provides a vivid example. Network integration reveals a metabolic break at IDH in M1 cells, fragmenting the TCA cycle and lowering α-ketoglutarate regeneration; the result is constrained anaplerosis, altered NADPH production, and a redox profile conducive to heightened immune effector programs that intersect with IDOs induction [5]. In tumors, nutrient competition and hypoxia reshape the same nodes. The microenvironment modulates α-ketoglutarate levels and the balance between oxidative and reductive TCA routing, which in turn conditions IDO1 and TDO2 expression and the effectiveness of their pharmacologic blockade [4]. Across aging and chronic inflammation, these α-ketoglutarate centered adjustments integrate energy metabolism with Trp fate, coupling respiratory control to immunoregulatory enzyme flux [2].
Table 3.
Integration points linking the KYN metabolism to the TCA Cycle and immune signaling. concise map of biochemical “nodes” where KYN-pathway flux or signaling intersects mitochondrial control, NAD⁺/NADH balance, anaplerosis, and immune/ROS outcomes.
Table 3.
Integration points linking the KYN metabolism to the TCA Cycle and immune signaling. concise map of biochemical “nodes” where KYN-pathway flux or signaling intersects mitochondrial control, NAD⁺/NADH balance, anaplerosis, and immune/ROS outcomes.
| Node (e.g., QA → NAD⁺) | Enzymes/regulators | Directionality | Impact on NAD⁺/NADH or anaplerosis | Immune/ROS consequence | Evidence class | References |
|---|---|---|---|---|---|---|
| QA → NAD⁺ | QPRT → NMNAT → NADS; modulation by aging/inflammation | KYN → NAD⁺ biogenesis → ETC | Increases cellular/matrix NAD(H); sustains complex I oxidation and O₂ consumption | Supports macrophage respiration; QPRT loss ↓NAD⁺, ↑injury/ROS; neuroinflammation requires conversion for SIRT activity | Mechanistic (cells/animals), clinical/genetic | [14,42,230] |
| KYNA → GPR35 → mitochondrial nodes (incl. MAS) | KATs (KYNA production), GPR35; MAS components | KYNs ligands → receptor → mitochondria/TCA | Tunes ETC throughput and shuttle-coupled redox set-points | Sets immune tone; receptor-proximal control of inflammatory signaling | Mechanistic receptor signaling | [16,22,132] |
| IDO1/TDO2 rate-setting → α-KG availability | IDO1, TDO2; IFN, hypoxia, nutrient status | Immune cues → KYNs flux → TCA carbon | Pulls carbon from Trp; constrains or supports α-KG–linked anaplerosis | Biases tolerance vs activation; conditions efficacy of IDOs/TDO blockade | Multi-omic/tumor-inflammation frameworks | [26,223,224] |
| MCART1-mediated NAD⁺ import (cytosol → matrix) | MCART1 (SLC25 family) | Cytosol → mitochondria | Maintains matrix NAD⁺ for dehydrogenases; prevents collapse of OXPHOS | Preserves respiratory control; supports T-cell effector programs | Mechanistic (transport/respiration) | [231,232,233] |
| MDH2 → oxaloacetate restraint of Complex II | MDH2; OAA | TCA intermediate → ETC modulation | Re-routes electron flow; dynamically resets NAD⁺/NADH coupling | Shapes graded ROS signaling downstream of ratio | Mechanistic ETC control | [219,220,234] |
| NNT couples NADH ↔ NADPH demand | Nicotinamide nucleotide transhydrogenase (NNT) | Matrix redox coupling | Balances NADH oxidation with NADPH generation; stabilizes redox poise | Supports antioxidant defenses; buffers ROS during substrate shifts | Mechanistic redox coupling; | [235,236,237] |
| ME1 (malic enzyme-1): malate → pyruvate (NADPH) | ME1 | Cytosol anaplerosis ↔ redox | Raises NADPH; protects glutathione/NADPH buffering | Reinforces antioxidant capacity; feeds redox-encoded signaling | Mechanistic redox metabolism | [238,239] |
| Serine catabolism → NADH accumulation when respiration stalls | One-carbon/serine axis | Amino acid metabolism → redox | Builds cytosolic/mitochondrial NADH when ETC is limited; throttles biosynthesis | Constrains proliferative programs under low respiration | Mechanistic metabolic control | [2,9,240] |
| SDH lesion → alternative aspartate synthesis (matrix NAD⁺/NADH-dependent) | SDH/Complex II context; aspartate pathways | ETC defect → rerouted biosynthesis | Forces aspartate generation routes that depend on matrix NAD⁺/NADH | Salvages growth despite impaired cycling | Mechanistic pathology | [241,242,243] |
| De novo NAD⁺ from KYNs supports macrophage respiration | QPRT→NMNAT→NADS; macrophage programs | KYNs → NAD⁺ → OXPHOS | Expands NAD(H) pool; sustains respiratory control across tissues | Coordinates systemic redox communication; tunes inflammatory effectors | Mechanistic + systems | [14,42,244] |
| Type I IFN → IDH inhibition → citrate/α-KG ratio shift | IDH1/IDH2; Type I IFN | Immune signal → TCA wiring → KYNs context | Alters NADPH generation/redox milieu that licenses IDOs/TDO activity | Reprograms Trp catabolism vs defense state | Mechanistic immunometabolism | [26,221,234] |
| M1 macrophage “IDH break” → α-KG drop | IDH node; network integration | Polarization cue → TCA fragmentation | ↓Anaplerosis; altered NADPH; redox favoring effector programs and IDOs induction | Heightened inflammatory activation | Network/multi-omic | [234,245] |
| Sirtuin activity sustained by quinolinate-derived NAD⁺ | SIRTs; QPRT/NMNAT/NADS | KYNs → NAD⁺ → sirtuin deacylases | Preserves mitochondrial protein deacylation and efficiency | Supports neuronal viability; mitigates inflammatory stress | Mechanistic neuroinflammation | [14,25,94] |
| Ischemia–reperfusion diversion away from quinolinate | Pathway branch choice; NAD⁺ augmentation | Stress → KYN branch → NAD⁺ | NAD⁺ depletion when diverted; restoration rescues antioxidant capacity | Less oxidative injury with NAD⁺ repletion | Mechanistic/therapeutic modulation | [22,31,43] |
| UMPS bypass completing NAD⁺ synthesis when canonical steps fail | UMPS (bypass), salvage enzymes | Engineered/alternative route → NAD⁺ | Raises total NAD(H) when QPRT or steps are compromised | Enhances ETC throughput in designed systems | Engineering proof-of-principle | [246,247,248] |
| Quinolinate/NAM rise tracks mitochondrial work/biogenesis | Systemic KYNs NAD⁺ salvage | Workload/biogenesis → KYNs output | Correlated elevation of circulating quinolinate & nicotinamide with ETC demand | Links tissue respiratory programs to systemic KYNs tone | Integrative physiology; | [94,249,250] |
| Ubiquinol (CoQH₂) oxidation requirement beyond NAD⁺ regeneration | ETC Complex III/CoQ cycle | ETC constraint → metabolic outcome | NAD⁺ repletion alone insufficient if CoQ oxidation is limited | Governs tumor growth constraints; bioenergetic bottleneck | Mechanistic tumor bioenergetics | [104,219,220] |
| LKB1 programs & thioredoxin circuits sculpt NADH turnover | LKB1, TRX/thioredoxin | Kinase/antioxidant systems → NADH flux | Adjusts NADH oxidation and chromatin-linked NAD⁺ usage | Sets T-cell effector capacity | Mechanistic immune control | [251,252,253] |
α-KG, alpha-ketoglutarate; CoQH₂, ubiquinol; ETC, electron transport chain; GPR35, G protein–coupled receptor 35; IDO, indoleamine 2,3-dioxygenase; IDO1, indoleamine 2,3-dioxygenase 1; IDH, isocitrate dehydrogenase; IFN, interferon; KAT, kynurenine aminotransferase; KYN, kynurenine; KYNA, kynurenic acid; LKB1, liver kinase B1; MAS, malate–aspartate shuttle; MCART1, mitochondrial carrier of NAD⁺ transporter 1; MDH2, malate dehydrogenase 2; ME1, malic enzyme 1; NAM, nicotinamide; NADS, NAD⁺ synthetase; NADH, nicotinamide adenine dinucleotide (reduced form); NADPH, nicotinamide adenine dinucleotide phosphate (reduced form); NMNAT, nicotinamide mononucleotide adenylyltransferase; NNT, nicotinamide nucleotide transhydrogenase; OAA, oxaloacetate; OXPHOS, oxidative phosphorylation; QA, quinolinic acid; QPRT, quinolinate phosphoribosyltransferase; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SIRT, sirtuin; SLC25, solute carrier family 25; TCA, tricarboxylic acid cycle; TDO, tryptophan 2,3-dioxygenase; Trp, tryptophan; TRX, thioredoxin; UMPS, uridine monophosphate synthase.
3.2. Immunometabolic Integration
Immune activation tilts Trp fate toward the KYN axis, lowering substrate and generating metabolites that reprogram effector circuits [10,15,19]. KYN and allied ligands engage AhR, dampen T cell proliferation, and favor regulatory programs, shaping tolerance in infection, autoimmunity, and cancer [1,3,4,11,18]. Context matters. Inflammaging sustains this reflex, while serotonin–KYN balance and metabolic feedback refine outcomes [12,14,20]. KYN to Trp ratios index pathway load, and Rab4A dependent mTOR signaling links mitochondrial metabolism to KYN sensitivity [5,13].
Succinate links carbon flux to inflammatory licensing. In lipopolysaccharide challenged macrophages, accumulation of succinate stabilizes hypoxia-inducible factor 1 alpha (HIF-1α), boosts glycolysis, and elevates IL-1β transcription and release [2,10]. Mitochondrial control is pivotal. Inhibition or retuning of SDH rewires electron flow, sustains HIF-1α, and aggravates tissue injury, while SUCNR1 signaling extends succinate’s reach to endothelium and epithelium, amplifying cytokines and vascular pathology [1,6,9,13,15]. Parallel cues converge. STING activation raises succinate and locks HIF-1α–dependent effector programs during infection; L-2-hydroxyglutarate similarly enforces the HIF-IL-1β axis [3,11,14].
Context shapes outcome. Proinflammatory cytokines further potentiate HIF-1α, embedding a feedforward loop, yet SDH also supports STAT3 driven IL-10, revealing countervailing anti-inflammatory circuitry within the same module [12,13,17]. Upstream metabolic governors, including SIRT6, adjust succinate levels and HIF linked glycolysis in pathogen challenged macrophages [18]. In vascular and synovial beds, pharmacologic or nutraceutical inhibitors that blunt succinate accumulation or HIF-1α activation reduce IL-1β output, neovascularization, and plaque inflammation, illustrating therapeutic tractability across diseases characterized by immunometabolic stress [4,16,19,20,28,203,254]. Together these data position the succinate–HIF-1α axis as a tunable rheostat that integrates mitochondrial respiration with inflammatory effector commitment.
#-HK sits at a volatile intersection of metabolism and immunity, where its redox cycling can both ignite and quench oxidative chemistry. Autoxidation and dimerization generate ROS that deplete glutathione, derail the TCA cycle, and trigger apoptosis, yet context permits radical scavenging and neuroprotective outcomes [3,4,5,6,7]. In vascular, renal, and neuroinflammatory states, elevated 3-HK tracks with oxidative stress and endothelial dysfunction, positioning this metabolite as a sentinel of immunometabolic strain [1,5,6].
These redox swings feed signaling loops that tune inflammatory tone. ROS linked to 3-HK amplify IL-6 and IL-1–driven programs, prime monocytes through mTOR dependent circuits, and shape T cell activation thresholds and lineage decisions [2,8,9,10,11,12]. Feedback control is nuanced. Mitochondrial injury can proceed with or without early ROS surges, implying parallel mitochondria centered sensors and effector arms downstream of 3-HK [13]. Collectively, 3-HK orchestrates bidirectional crosstalk between metabolism and immunity, coupling Trp flux to ROS governed transcriptional and epigenetic checkpoints that determine tolerance versus tissue injury [1,4,7,10,11,12].
At the interface of Trp catabolism and the TCA cycle, a set of metabolic checkpoints governs whether immunity accelerates or brakes. Enzymes such as IDO1, TDO, and KMO gate substrate flow to KYN and downstream ligands that converge on AhR, adjusting effector programs, tolerance, and chronic inflammation [1,2,3]. This control integrates with organelle level routing of carbon and reducing equivalents, where cytosol–mitochondria handoffs shape glycolysis–TCA coupling and enforce redox thresholds for immune activation [8,9,11]. De novo NAD⁺ synthesis from KYN intermediates adds another lever, feeding respiratory capacity and feedback to transcriptional fate decisions in aging and cancer [6,12].
Checkpoint behavior is contextual. In T cells, Rab4A controlled endosomal traffic intersects with KYN sensitive mTOR signaling to couple mitophagy, nutrient transport, and lineage specification [4]. Tumors exploit depletion and signaling in parallel, creating an immunosuppressive niche that often resists single agent IDOs or TDO blockade, arguing for combinations that co-target metabolic nodes and canonical immune checkpoints [2,5,13,15,18]. Microbial Trp products reinforce AhR dependent suppression in tumor associated macrophages, linking diet and microbiota to checkpoint tone [16]. Stress programs that arise when TCA flux is curtailed activate ATF4 and remodel amino acid and redox metabolism, thereby re-indexing sensitivity to Trp pathway control [14]. Multi omic maps now resolve these modules in human macrophages and guide rational intervention across inflammatory disease and oncology [10,17,19].
3.3. Pathological Implications
From circuits to clinics, dysregulated Trp–KYN and TCA nodes shape disease trajectories across brain, vasculature, metabolism, and cancer. Imbalanced metabolites drive neurotoxicity, immune exhaustion, and cardiometabolic risk, while altered NAD+ supply and ATF4 programs expose vulnerabilities [3,4,5,6,8,9,10,12,13,14,15,16,17,18]. Translational paths now pair pathway modulation with tumor bioenergetic targets and immune recalibration [6,12,13,14,16].
Excess QA and diminished KYNA form a pathogenic redox and excitotoxic dyad in neurodegeneration. QA engages NMDA-Rs, drives mitochondrial ROS, and depresses respiratory capacity, creating a feedforward loop of bioenergetic failure and inflammation [2,5,16]. By contrast, KYNA buffers glutamatergic stress and restores antioxidant defenses, including Nrf2 signaling, yet is frequently reduced in Alzheimer’s and Parkinson’s disease [3,5,6,19]. Clinical and biomarker studies converge on elevated QA to KYNA ratios across aging and disease, linking this imbalance to tau and amyloid burden, neuronal dysfunction, and faster progression [49,53,255]. Therapeutically, redirecting flux away from QA and enhancing KYNA, for example with KMO inhibition or pathway modulation, offers a rational strategy to stabilize mitochondria and slow neurodegeneration [4,18,20].
Psychiatric disorders display a characteristic coupling of immune tone and bioenergetics, centered on a rerouting of Trp toward KYN metabolism [49,256,257]. Meta analyses and cohort studies reveal reduced Trp and KYN with imbalanced neurotoxic and neuroprotective metabolites, tracking mood, psychosis, and cognitive deficits [2,3,4,6]. Immune activation accelerates this shift, depresses serotonin, and favors QA, with state dependent oscillations across acute and remitted phases [10,11,12,18,19]. These molecular changes map onto mitochondrial and synaptic function, and show moderate blood–brain concordance for select metabolites that can index symptom burden and progression [13,16]. Mechanistically, KYN reprogramming may be compensatory or pathogenic, often both; therapeutic strategies now target enzymes and flux control while integrating microbiome sensitive modulators and trial readouts [1,5,7,14,15,258].
Tumors co-opt Trp catabolism to choreograph immune escape while fueling growth. IDO1 and TDO2 divert substrate toward KYN, which activates AhR, expands Tregs, and exhausts cytotoxic T cells; clinical experience shows that single-enzyme blockade often falters, underscoring redundant wiring and the need for multi-target strategies [1,2,3,4,6,12,15]. Beyond initiation, downstream nodes such as KMO and kynureninase (KYNU) shape metastatic behavior, stromal crosstalk, and chemoresistance, particularly in aggressive breast and renal cancers [5,7,8,14,16].
Therapeutic concepts increasingly pair immune checkpoint inhibitors with metabolic rewiring. KYNU depots deplete intratumoral KYN and synergize with programmed cell death protein 1 (PD-1) blockade, while small molecules like icariside I attenuate AhR signaling and restore effector function [9,10]. Remodeling the glutathathione peroxidase 4 (GPX4)–KYNU axis or broader tumor microenvironment (TME) metabolism reprograms macrophage polarization and dismantles suppressive niches, offering tractable paths to overcome resistance [11,13,18,19,20].
Table 4.
Clinical and preclinical patterns connecting KYN metabolism–TCA crosstalk to disease phenotypes. Summary of disease-domain patterns linking KYN-pathway metabolites to mitochondrial phenotypes and clinical/functional readouts. Study type/size reflects the level of evidence stated in the text (meta-analyses/cohorts, biomarker studies, preclinical models, early clinical combination strategies) without inventing sample counts.
Table 4.
Clinical and preclinical patterns connecting KYN metabolism–TCA crosstalk to disease phenotypes. Summary of disease-domain patterns linking KYN-pathway metabolites to mitochondrial phenotypes and clinical/functional readouts. Study type/size reflects the level of evidence stated in the text (meta-analyses/cohorts, biomarker studies, preclinical models, early clinical combination strategies) without inventing sample counts.
| Indication (neurodegeneration/psychiatry/oncology) | Metabolite signature KYNA, QA, KYN, 3-HK | Mitochondrial phenotype | Clinical/functional readouts | Study type/size | References |
|---|---|---|---|---|---|
| Neurodegeneration | KYNA ↓, QA ↑; KYN context-dependent; 3-HK not primary in text | NMDA-driven ROS; depressed respiratory capacity; feed-forward bioenergetic failure and inflammation | Higher QA:KYNA ratios track tau/amyloid burden, neuronal dysfunction, faster progression | Clinical & biomarker studies; aging/disease cohorts; preclinical mechanistic work | [51,52,80] |
| Neurodegeneration (therapeutic angle) | Shift flux away from QA; raise KYNA (e.g., KMO modulation) | Mitochondrial stabilization; restored antioxidant defenses (incl. Nrf2 signaling) | Slower neurodegeneration trajectory; reduced excitotoxic stress | Preclinical + translational strategy proposals; early-phase targeting concepts | [3,8,207] |
| Psychiatry | Trp ↓, KYN ↓ (cohorts/meta-analyses); QA favored under immune activation; KYNA/KYN/QA show state-dependent oscillations; 3-HK not emphasized | Immune-bioenergetic coupling; mitochondrial/synaptic function shifts; serotonin depression when KYN metabolism is upshifted | Mood, psychosis, cognitive deficits; moderate blood–brain metabolite concordance indexing symptom burden/progression | Meta-analyses and multi-cohort studies; biomarker cohorts; mechanistic frameworks | [54,75,92] |
| Psychiatry (therapeutic angle) | Enzyme/flux control targeting; microbiome-sensitive modulators | Rebalancing KYNs–TCA redox and neurotransmission coupling | Symptom modulation and progression tracking via peripheral KYNs panels | Translational strategies; trial-readout integration (sizes vary) | [49,50,63] |
| Oncology (tumor immune escape) | KYN ↑ via IDO1/TDO2 → AhR activation; downstream KMO/KYNU shape invasive traits; KYNA/QA context-specific | Bioenergetic rewiring supporting growth; TME-conditioned redox | Treg expansion, CD8⁺ exhaustion; metastatic behavior, stromal crosstalk, chemoresistance | Clinical experience with IDOs/TDO blockade; preclinical tumor models; biomarker studies | [223,224,259] |
| Oncology (combination therapy) | KYN depletion (KYNU depots); AhR attenuation (e.g., small molecules) | Reprogrammed mitochondrial/immune metabolism; macrophage repolarization (e.g., GPX4–KYNU axis) | Synergy with PD-1 blockade; dismantling suppressive niches; overcoming resistance | Preclinical synergy studies; early translational combinations; emerging clinical strategies | [74,223,244] |
3-HK, 3-hydroxykynurenine; AhR, aryl hydrocarbon receptor; CD8⁺, cluster of differentiation 8 positive; GPX4, glutathione peroxidase 4; IDO1, indoleamine 2,3-dioxygenase 1; KMO, kynurenine 3-monooxygenase; KYN, kynurenine; KYNA, kynurenic acid; KYNU, kynureninase; Nrf2, nuclear factor erythroid 2–related factor 2; PD-1, programmed cell death protein 1; QA, quinolinic acid; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; TDO2, tryptophan 2,3-dioxygenase 2; TME, tumor microenvironment; Trp, tryptophan; Treg, regulatory T cell.,
Figure 3.
Bridging KYN metabolism and the TCA cycle via NAD+ biogenesis, alpha KG feedback, and inflammatory checkpoints. (A) QA feeds the QPRT NMNAT NADS axis to expand NAD(H) pools and tune NAD+/NADH, dehydrogenase flux, CI driven oxidation, and respiratory throughput; impaired QA to NAD+ conversion promotes QA accumulation, NAD+ depletion, and oxidative vulnerability during inflammation or ischemia. (B) α-KG acts as a flux governor linking carbon routing and redox context to IDO TDO rate setting and KYN output, with immune cues, IDH inhibition, and TME constraints reshaping alpha KG regeneration and NADPH buffering. (C) Succinate HIF 1 alpha and 3 HK redox cycling amplify glycolysis, IL 1 beta, SDH and SUCNR1 driven inflammatory tone, and ROS glutathione stress that shifts mTOR linked activation thresholds. (D) Disease wheel maps these nodes onto neuropsychiatry and oncology via IDO TDO AhR rewiring and QA KYNA imbalance. 3-HK, 3-hydroxykynurenine; AhR, aryl hydrocarbon receptor; CI, Complex I; HIF-1α, hypoxia-inducible factor 1 alpha; IDH, isocitrate dehydrogenase; IDO, indoleamine 2,3-dioxygenase; IL-1β, interleukin 1 beta; KYN, kynurenine; KYNA, kynurenic acid; mTOR, mechanistic target of rapamycin; NAD(H), nicotinamide adenine dinucleotide (oxidized and reduced forms); NAD+, nicotinamide adenine dinucleotide; NADS, NAD synthetase; NMNAT, nicotinamide mononucleotide adenylyltransferase; QA, quinolinic acid; QPRT, quinolinate phosphoribosyltransferase; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SUCNR1, succinate receptor 1; TCA, tricarboxylic acid cycle; TDO, tryptophan 2,3-dioxygenase; TME, tumor microenvironment; alpha KG, alpha-ketoglutarate.
Figure 3.
Bridging KYN metabolism and the TCA cycle via NAD+ biogenesis, alpha KG feedback, and inflammatory checkpoints. (A) QA feeds the QPRT NMNAT NADS axis to expand NAD(H) pools and tune NAD+/NADH, dehydrogenase flux, CI driven oxidation, and respiratory throughput; impaired QA to NAD+ conversion promotes QA accumulation, NAD+ depletion, and oxidative vulnerability during inflammation or ischemia. (B) α-KG acts as a flux governor linking carbon routing and redox context to IDO TDO rate setting and KYN output, with immune cues, IDH inhibition, and TME constraints reshaping alpha KG regeneration and NADPH buffering. (C) Succinate HIF 1 alpha and 3 HK redox cycling amplify glycolysis, IL 1 beta, SDH and SUCNR1 driven inflammatory tone, and ROS glutathione stress that shifts mTOR linked activation thresholds. (D) Disease wheel maps these nodes onto neuropsychiatry and oncology via IDO TDO AhR rewiring and QA KYNA imbalance. 3-HK, 3-hydroxykynurenine; AhR, aryl hydrocarbon receptor; CI, Complex I; HIF-1α, hypoxia-inducible factor 1 alpha; IDH, isocitrate dehydrogenase; IDO, indoleamine 2,3-dioxygenase; IL-1β, interleukin 1 beta; KYN, kynurenine; KYNA, kynurenic acid; mTOR, mechanistic target of rapamycin; NAD(H), nicotinamide adenine dinucleotide (oxidized and reduced forms); NAD+, nicotinamide adenine dinucleotide; NADS, NAD synthetase; NMNAT, nicotinamide mononucleotide adenylyltransferase; QA, quinolinic acid; QPRT, quinolinate phosphoribosyltransferase; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SUCNR1, succinate receptor 1; TCA, tricarboxylic acid cycle; TDO, tryptophan 2,3-dioxygenase; TME, tumor microenvironment; alpha KG, alpha-ketoglutarate.
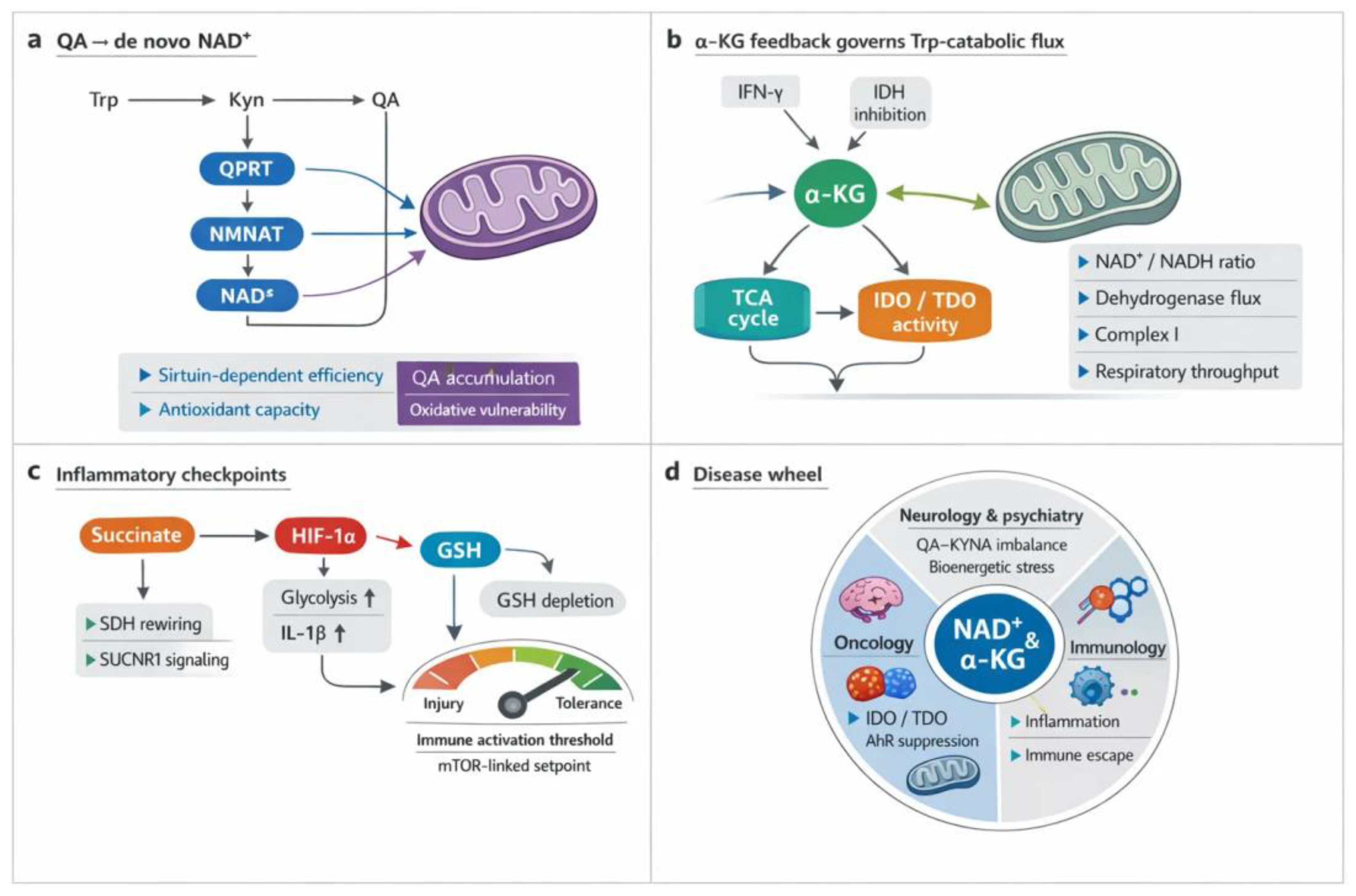
4. Analytical Strategies for Simultaneous Quantification
The combined measurement of the TCA and KYN metabolic pathway in a single method has attracted significant interest in the fields of neurology, oncology, and mitochondrial biology [13,14,260]. While the two metabolic pathways are typically studied separately, combined studies are increasingly sought after [65,261,262,263]. This allows for the investigation of interactions between pathways underlying disease phenotypes in a single method, without separate preparations, and by eliminating inter-assay variance. However, designing a single analytical method requires consideration of chromatographic incompatibilities, ionization, and polarity differences [264,265,266]. In addition, several orders of magnitude variations in biological samples for some metabolites must be taken into account, which weakens the usefulness of an already well-developed analytical method [267,268,269]. This section explains the fundamental challenges of the simultaneous measurement and presents practical engineering solutions. Finally, we would like to highlight the validation standards required in clinical research, similar to diagnostic applications, where the methods used must meet serious criteria, for which appropriate guidelines are available [270,271,272]
4.1. Rationale for Unified Measurement
The scientific and clinical promise of simultaneous quantification lies in minimizing fragmentation in experimental workflows [273,274,275]. Instead of separate assays for TCA intermediates and KYNs, a unified measurement allows for a simultaneous overview of cellular energetic processes and neuroactive metabolism [276,277]. Common measurement improves efficiency, reduces sample burden, and makes meaningful comparisons between groups more effective [278,279,280].
Metabolomic studies often struggle with limited availability. In cerebrospinal fluid (CSF), where lumbar punctures yield only a few hundred microliters, or in animal studies, where only a few microliters are available, distributing aliquots to multiple specialized assays reduces the measurement options, thereby compromising the results or the statistical power of the work [281,282,283]. Similarly, plasma samples collected in efficacy studies may be limited by ethical and logistical considerations, while tissue biopsies are often only available in milligram quantities [284,285,286]. When TCA intermediates and KYN derivatives are measured in separate analytical runs, each uses a portion of the already small amount of biological sample, which reduces measurement efficiency and may lead to the possibility of missing biologically important correlations [287,288,289,290]. Simultaneous measurements in single-run assays eliminate the need to split the sample volume into multiple aliquots, and metabolite relationships within it can be examined more reliably [268,288,291,292]. These measurements also allow the application of multivariate statistical models that integrate data collected from mitochondrial and KYN metabolites and increase the potential for cross-metabolic pathway analysis, allowing for more reliable quantification of ratios such as succinate/quinolinate ratios or citrate-KYNA correlations in psychiatric and neurological diseases. [13,14,234,245,293,294,295,296,297,298].
A major obstacle to reproducibility in biomarker research is interassay variability. Separate LC-MS methods, independently optimized for TCA intermediates and KYNs, differ in chromatographic stationary phases, gradient conditions, derivatization techniques, and ionization polarity [96,263,299,300]. Harmonized measurement reduces technical heterogeneity by incorporating both metabolic pathways into a single analytical measurement, ensuring that co-eluting matrix components and ion suppression phenomena are consistent across analytes [268,300,301,302]. This is particularly important for meta-analyses and longitudinal cohort integration, where cumulative error from multiple platforms can mask biologically significant differences [97,303,304]. Unified workflows also simplify QC procedures and validations, as pooled reference samples, system proficiency tests, and internal standards can be used globally [270,305,306,307]. This ultimately results in improved reproducibility, increased statistical reliability, and comparability between stronger studies – key requirements for moving beyond exploratory research to regulatory-level biomarker validation [308,309].
In addition to consistency within a single assay, standardized analytical workflows also facilitate inter-center comparability, allowing for standardization and the establishment of international cutoffs instead of results with diverse and high variance [73,310]. They are also characterized by long-term reproducibility and reliability [311,312,313]. When laboratories use identical chromatographic conditions, calibration strategies, and internal standard placements, the correction of batch-to-batch drift and instrument-specific biases becomes significantly more reliable, although in practice this is almost impossible due to the large variety of chromatographic systems, columns, and mass spectrometers available [314,315,316,317].
4.2. Chromatographic and Mass Spectrometric Challenges
Simultaneous detection of TCA intermediates and KYNs can be analytically challenging, if not impossible [263,289,318,319]. TCA acids are highly polar, have low molecular weight, break easily at low voltages, are poorly retained in reversed-phase systems, and are often measured in negative electrospray mode [263,320]. In contrast, KYNs exhibit different polarity, aromaticity, and proton affinity, and are typically measured in positive electrospray ionization (ESI) mode [68,289,321,322,323]. Reconciliation of these different physicochemical properties defines the fundamental challenge of chromatography and ionization [322,324,325,326].
TCA intermediates, such as citrate, α-ketoglutarate, and malate, are highly polar, low molecular weight organic acids that elute early on conventional C18 columns and exhibit poor retention [290,327,328]. Ion-pairing agents or hydrophilic interaction chromatography (HILIC) are often required for proper separation [329,330,331,332]. However, metabolites of KYN metabolism range from more neutral, aromatic amino acid derivatives (KYN, KYNA) to highly acidic, low molecular weight, simple molecular structures with few transitions (QA, picolinic acid), requiring a broader range of chromatographic selectivity [299,333,334,335]. Choosing a mobile phase that is capable of retaining both classes of molecules requires a balance between retention and ionization efficiency [265,336]. Formic acid is a common additive used for positive ion detection, while ammonium acetate or ammonium formate buffers at neutral-basic pH help negative mode detection by enhancing ionization [265,337,338]. Buffers with high ionic strength can suppress the electrospray response, especially for aromatic KYNs [322,339,340]. Consequently, hybrid strategies slightly buffered aqueous phases with carefully screened organic modifiers—are used to retain acids without unduly compromising positive mode sensitivity [265,341,342,343]. Such trade-offs reflect the trade-off between chromatographic retention and MS compatibility [265,341,344].
The polarity of electrospray ionization fundamentally affects the quality of the signal, and some molecules only give signals in one polarity mode [322,345,346,347]. TCA cycle intermediates ionize best in the negative mode, forming stable deprotonated species [263,290,348]. KYNs, especially KYN and KYNA, favor the positive mode through protonation or ammonium adduct formation [68,249,349]. Trying to quantify both in a single run either forces a polarity switch within a gradient or the acceptance of one polarity, which can cause a decrease in sensitivity of up to several orders of magnitude for some analytes [266,350,351]. The polarity switch causes residence time losses when the analytes elute closely [266,352,353]. Furthermore, the ion source parameters – desolvation temperature, gas, and capillary voltage – should be set differently for acids and aromatics [354,355,356,357]. Adduct formation further complicates the process: sodium and potassium adducts are common for citrate and succinate, while ammonium adducts affect KYNs [358,359,360]. Without careful control, the diversity of ionic species reduces quantitative reproducibility [361,362,363,364]. Thus, the challenge is not only chromatographic but also electrochemical, requiring source-level design or the use of stable isotope-labeled internal standards to normalize for adduct heterogeneity [365,366,367,368].
Another critical difficulty arises from concentration differences. TCA intermediates such as lactate, pyruvate, citrate, malate and succinate often circulate in the plasma in the high micromolar and millimolar range, while downstream KYNs such as 3-hydroxykynurenine, xanthurenic acid, anthranilic acid, QA or picolinic acid are present in the nanomolar range [249,276,369,370,371]. This difference spans four to six orders of magnitude, exceeding the linear dynamic range of almost all detectors used in mass spectrometers [341,372,373]. Injecting samples to achieve adequate sensitivity for low amounts of KYNs carries the risk of saturating the signals of high amounts of TCA metabolites, leading to peak distortion and poor quantitation, while diluting samples to TCA concentrations below the detection limit of kynurenes can reduce detection limits [341,374,375]. Signal compression and ion suppression further exacerbate the problem when high concentrations of acids dominate the spray cloud [376,377,378,379]. These concentration dynamics require either double injections (concentrated and many-fold diluted) or special detector linearization approaches that carefully span the entire range [380,381,382]. Without such adaptations, single-run assays run the risk of erroneously measuring either low-abundance KYN metabolites or concentrated TCA intermediates [240,383].
4.3. Practical Solutions
Co-measurements with physicochemical and quantitative differences between TCA and KYN metabolites require considerable foresight and planning [68,384]. To solve this problem, advanced chromatographic approaches such as HILIC, mixed mode or multidimensional separations are available, but advanced use of the mass spectrometer is also essential, such as polarity switching and properly optimized ion source conditions [266,385,386,387]. The guiding principle is comprehensive metabolite coverage without sacrificing sensitivity or reproducibility.
Strong retention of polar acids provides better separation of TCA intermediates, while less polar KYNs can be adequately retained by selecting the appropriate eluent composition, gradients, column temperature, eluent composition, and pH. Mixed-mode columns, which combine ion-exchange and reversed-phase characteristics, offer another option, improving resolution across different chemical classes (e.g., ZIC-cHILIC), BEH Amide, BEH HILIC HILIC Silica, or Accucore HILIC) [68,279,388,389]. Even more precise, but more complex, are two-dimensional LC (2D-LC) systems, where an initial HILIC or ion-exchanger binds the highly polar acids, followed by reversed-phase separation of the aromatic kynurenes [390,391]. Although technically demanding, 2D-LC allows the analysis of both metabolite classes in a single workflow [392,393,394]. They also have the disadvantage of being complex, extremely error-prone and difficult to implement, especially in biological matrices [391,395]. A high degree of chromatographic and engineering expertise is required to design and operate such a complex system efficiently [396,397,398]. The piping, switching valves, and dead volume must be optimized to preserve peak shape and sensitivity [399]. Nevertheless, these approaches illustrate how chromatographic ingenuity can reduce polarity bias and expand assay coverage [329,400,401].
A practical compromise is to design two complementary LC-MS runs instead of forcing all analytes into a single chromatographic measurement [269,402,403]. For example, an optimized and validated run in negative mode for TCA intermediates and a second run in positive mode for KYNs allows for adequate sensitivity in both cases [73,263,404,405]. Although this doubles the run time, it can solve the problem of multiple sample preparation and sample volumes, and it also maintains quantitative reliability, but in return avoids retention or ionization compromises [287,395,404,406]. Hybrid approaches also exist: a single-run method that uses precisely timed polarity reversals at appropriate times in the gradient can measure both acidic and aromatic metabolites with acceptable reproducibility [264,407,408,409,410]. Advances in high-speed electronics optimization have reduced the time-dependent degradation of polarity reversal measurements, making single-run hybrids increasingly feasible [266,350,402]. The choice between two-run and single-run strategies depends on the scale of the study, throughput requirements, and instrumentation, with hybrid methods being preferred for exploratory studies and two-run strategies for large clinical cohorts where reproducibility overrides throughput [268,269,411,412,413].
Internal standards (IS) labeled with stable isotopes (D, C13, N15) are essential for reliable and reproducible mass spectrometry measurements [366,414,415]. They correct for matrix effects, ion suppression and adduct variability in both metabolite classes [414,416,417]. The choice of IS should be carefully considered due to their extremely high cost and availability [305,418]. Optimally, each metabolite to be measured would have its own stable isotope-labeled IS, but this would significantly increase the cost [415,419,420]. It is worth considering what the goal is, which metabolites can be combined under a common IS, incorporating them at the earliest possible step, ideally during sample extraction, compensates for losses during preparation and ensures normalization between runs [421,422,423,424]. For studies spanning a wide concentration range, multiple concentration-matched calibration curves recorded by IS prevent saturation in the high range while maintaining sensitivity for trace analytes [425,426,427]. Without this strategic use of isotopes, uniform workflows risk systematic bias in both chemical classes [422,428,429].
Figure 4.
Decision framework and validation pathways for LC–MS/MS quantification of KYN pathway metabolites and TCA intermediates. Practical decision framework and validation roadmap for targeted LC–MS/MS quantification of KYN pathway metabolites and TCA intermediates. Method choice is guided by sample volume, polarity range, quantitative rigor, and chromatographic feasibility. Single-run hybrid LC–MS/MS suits limited-volume clinical matrices but may compromise retention of highly polar acids. Dual-run strategies (ESI⁺ for KYNs; ESI⁻ for TCA acids) maximize sensitivity and chromatographic performance at the cost of runtime and QC burden. For extreme polarity or isomeric overlap, 2D-LC provides orthogonal separation with added complexity. Validation includes matrix-matched calibration, defined LLOQ/ULOQ, accuracy and precision across ≥4 QC levels, and assessment of interferences, stability, recovery, carryover, and matrix effects. Ionization mode selection is critical, with challenging analytes requiring tailored retention and suppression control. 2D-LC, two-dimensional liquid chromatography; ESI⁺, positive electrospray ionization; ESI⁻, negative electrospray ionization; KYN, kynurenine; LC–MS/MS, liquid chromatography–tandem mass spectrometry; LLOQ, lower limit of quantification; QC, quality control; TCA, tricarboxylic acid (cycle); ULOQ, upper limit of quantification.
Figure 4.
Decision framework and validation pathways for LC–MS/MS quantification of KYN pathway metabolites and TCA intermediates. Practical decision framework and validation roadmap for targeted LC–MS/MS quantification of KYN pathway metabolites and TCA intermediates. Method choice is guided by sample volume, polarity range, quantitative rigor, and chromatographic feasibility. Single-run hybrid LC–MS/MS suits limited-volume clinical matrices but may compromise retention of highly polar acids. Dual-run strategies (ESI⁺ for KYNs; ESI⁻ for TCA acids) maximize sensitivity and chromatographic performance at the cost of runtime and QC burden. For extreme polarity or isomeric overlap, 2D-LC provides orthogonal separation with added complexity. Validation includes matrix-matched calibration, defined LLOQ/ULOQ, accuracy and precision across ≥4 QC levels, and assessment of interferences, stability, recovery, carryover, and matrix effects. Ionization mode selection is critical, with challenging analytes requiring tailored retention and suppression control. 2D-LC, two-dimensional liquid chromatography; ESI⁺, positive electrospray ionization; ESI⁻, negative electrospray ionization; KYN, kynurenine; LC–MS/MS, liquid chromatography–tandem mass spectrometry; LLOQ, lower limit of quantification; QC, quality control; TCA, tricarboxylic acid (cycle); ULOQ, upper limit of quantification.
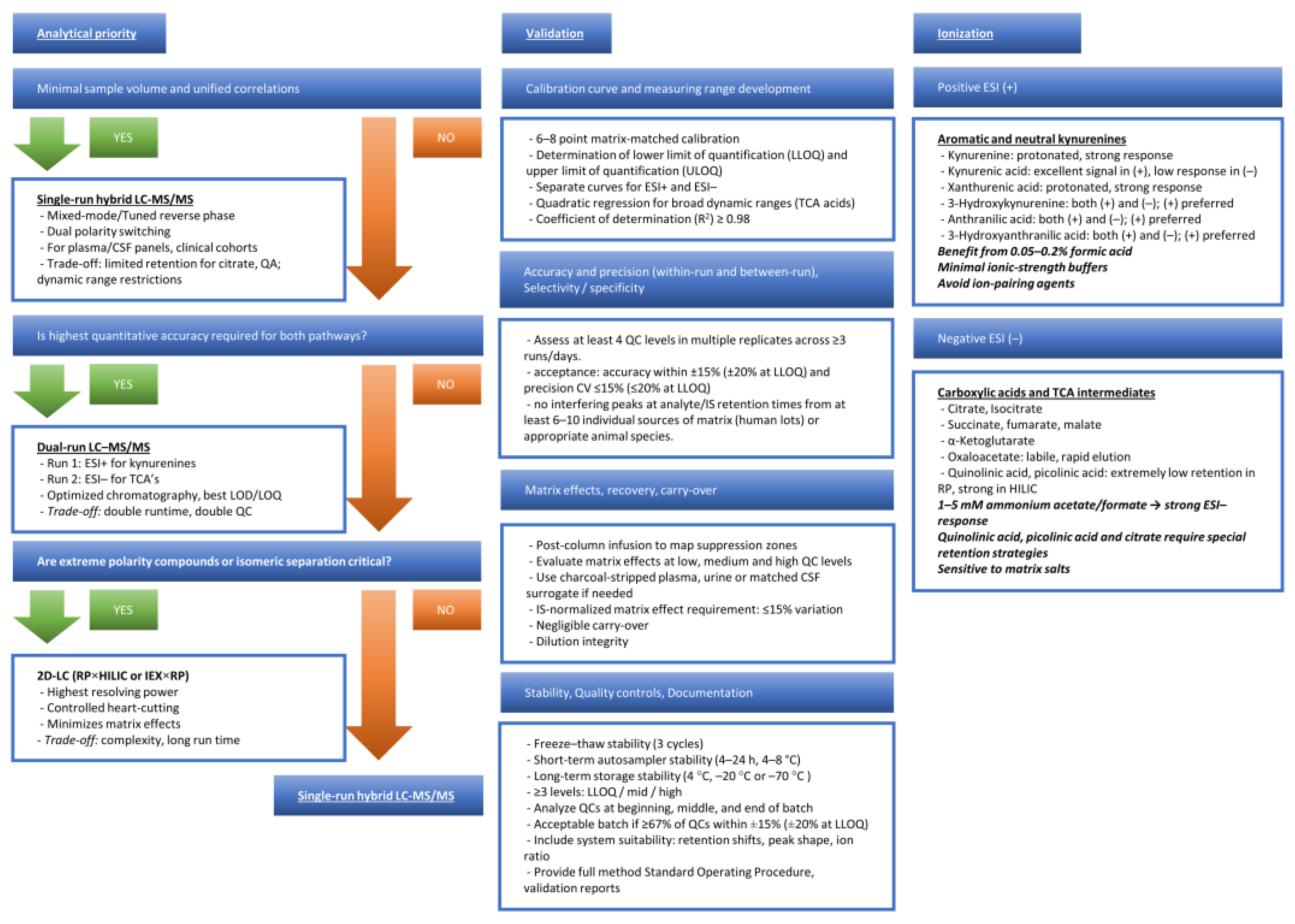
4.4. Validation and Clinical Feasibility
Rigorous validation is required for a simultaneous TCA-KYN metabolite assay to become a clinical or research method [263,288,402,430]. Guidelines governing mass spectrometry methods require reproducibility testing, both inter- and intraday, linearity, limit of detection, lower limit of quantification (LLQ) determination, carry-over, precision and accuracy, matrix effect, recovery, selectivity and stability testing [431,432,433].
An important step in validation is the linearity of the calibration curve, the determination of dynamic ranges, the measurement of matrix effects, and the stability of the samples [434,435,436]. The linearity range should be excellent (R2 > 0.98) from the nanomolar range to the millimolar range depending on the metabolite [402,437,438,439]. To calculate the matrix effect, a standard solution or different matrices need to be spiked for each matrix to be measured [440,441,442,443]. A mandatory element of stability studies is the performance of multiple freeze-thaw tests, and it is also important to test the stability of samples under different conditions (e.g. room temperature, 4 °C, -20 °C, -80 °C) [444,445,446,447,448]. Reproducibility is used to determine the differences between tests performed on the same day and on different days, which is an important basis for long-term QC [449,450,451,452]. Validations are accompanied by rigorous documentation, where changing any of the steps can cause critical differences in the results, so a validated system should not be modified without compelling reason, or only with caution and revalidation [453,454].
Several studies have shown that HILIC methods can be suitable for the analysis of both TCA and KYN metabolites with appropriate sensitivity and selectivity [400,409,455,456]. Methods using anion exchange stationary phase exploit the almost unlimited potential of buffers, which can be properly applied by a well-trained analyst [457,458,459,460,461]. Hybrid HILIC-AEX systems are promising for both clinical and research purposes, which can be used with high-resolution MS systems [279,462]. Although suitable assay KITs are not yet available on the market for a wide range of metabolites, especially for TCA and PK, their appearance in the future should be expected due to the high demand [305,463,464,465]. With the continuous optimization of diverse stationary phases, polarity switching protocols and isotope calibration, combined assays may soon provide robust, validated tools for integrative mitochondrial and immuno-metabolic diagnostics [264,466,467,468,469].
Table 5.
LC-MS/MS bioanalytical validation criteria for the simultaneous measurement of KYNs and TCA cycle metabolites.
Table 5.
LC-MS/MS bioanalytical validation criteria for the simultaneous measurement of KYNs and TCA cycle metabolites.
| Parameter | Acceptance Threshold | Notes / Mitigations | References |
|---|---|---|---|
| Calibration model | R² ≥ 0.98; Back-calculated concentrations within ±15% (±20% at LLOQ) | Use matrix-matched standards; weighted (1/x or 1/x²) regression for wide dynamic ranges; verify linearity and curvature. | [470] |
| Linearity / Dynamic range | At least 6–8 calibration levels; deviation ≤15% (≤20% at LLOQ) | For TCA acids with steep response, consider quadratic fit; re-optimize injection volume to avoid saturation. | [471] |
| LLOQ / ULOQ | Signal ≥5× noise; precision ≤20%; accuracy 80–120% | Confirm LLOQ in actual matrix (plasma/CSF). Check for ion suppression at LLOQ. | [472] |
| Carryover | <20% of LLOQ signal and <5% of IS signal in blank after highest standard | Insert strong wash; consider needle wash with ACN/MeOH/water + 0.1% FA; extend gradient reequilibration. | [473] |
| Matrix effect (ME) | IS-normalized ME CV ≤15% across ≥6 donors | Perform post-column infusion; evaluate ME at low/mid/high QC; use isotopologues matched by polarity and retention. | [440,473,474] |
| Extraction recovery | 80–120% with CV ≤15% | Compare pre-spike vs post-spike; optimize precipitation solvent and pH; avoid ion-pairing agents in precipitant. | [440,475] |
| Precision – intra-day (repeatability) | CV ≤15% (≤20% at LLOQ) | Analyze ≥5 replicates each QC level; monitor retention shifts and ion ratio stability. | [472] |
| Precision – inter-day (reproducibility) | CV ≤15% | Include replicate QCs on ≥3 days; reinject archived QCs to detect long-term drift. | [471] |
| Accuracy | 85–115% (80–120% at LLOQ) | Compare to spiked reference material; evaluate both absolute and IS-normalized values. | [470] |
| Short-term autosampler stability | ≤15% change over 4–24 h at 4–8 °C | KYNs TCAs, may degrade – use immediate analysis or stabilizing additives. | [476] |
| Freeze–thaw stability | ≤15% change after 3 cycles | Avoid repeated thawing; aliquot samples; confirm TCA acids stability separately. | [473] |
| Long-term stability (–70 °C) | ≤15% deviation | Validate ≥1–3 months | [477] |
| Processed sample stability | ≤15% after 6–12 h in autosampler | For unstable analytes use rapid acquisition. | [478] |
| System suitability | Retention time shift <0.2–0.3 min; IS area CV ≤10%; ion ratio within ±20% | Verify before each batch; monitor column pressure, peak shape, and polarity switching efficiency. | [479] |
| QC frequency per batch | ≥3 levels (LQC/MQC/HQC); QCs at start, every 10–15 samples, and end | Large cohorts: insert pooled QC every 10–12 injections. | [472] |
| Batch acceptance criteria | ≥67% of all QCs and ≥50% at each level must be within ±15% (±20% at LLOQ) | If failure: investigate ME shifts, IS suppression, column fouling, or calibration instability. | [471] |
| Inter-batch comparability | QC CV ≤15% across batches | Use pooled QC and IS-normalized drift correction. | [480] |
R2: Coefficient of determination; LLOQ: lower limit of quantification; KYNs: kynurenines; TCA: tricarboxylic acid cycle; ULOQ: upper limit of quantification; CSF: cerebrospinal fluid; ACN: acetonitrile; MeOH: methanol; FA: formic acid; ME: matrix effect; IS: internal standard; QC: quality control; CV: coefficient of variation; LQC/MQC/HQC low/medium/high quality control.
4.5. Potential Clinical Deployment of a Harmonized Tricyclic acid (TCA)–Kynurenine (KYN) Assay
A coordinated LC-MS-based assay targeting the most important metabolites of the TCA cycle and KYN pathway could be used in a number of clinical studies, such as mitochondrial dysfunction and immunometabolic dysregulation [73,89,288,481]. This section moves from analytical diversity to convergence [73,482]. Rather than exhaustively reviewing all reported LC–MS approaches, we define a minimal, clinically deployable LC–MS/MS strategy for simultaneous quantification of KYNs and selected TCA intermediates, highlighting decision points relevant for routine laboratories [483]. Table 6 is intended as a minimal, trial-ready panel rather than an exhaustive analytical overview [73,482]. Given the differential stability of QA, 3-HK and selected TCA intermediates, harmonized pre-analytical handling (rapid quenching, antioxidant stabilization, limited freeze–thaw cycles) and the use of isotope-labeled internal standards are essential prerequisites for interlaboratory comparability. All targeted metabolites can be accommodated using HILIC-based or mixed-mode chromatographic separation, enabling simultaneous coverage of highly polar KYNs and TCA cycle intermediates within a harmonized analytical workflow. KYN pathway metabolites are preferentially detected in positive electrospray ionization (ESI+), whereas TCA cycle intermediates exhibit superior sensitivity and robustness in negative ion mode (ESI−), supporting polarity-switching or dual-mode acquisition strategies [288,484].
In neuropsychiatric disorders such as major depression, plasma sampling at baseline and post-treatment could be used to monitor Trp, KYNe, KYNA, QA, and selected TCA intermediates, with pharmacodynamic data including KYN/Trp ratios, pathway branching shifts, and markers of systemic metabolism [50,92,206]. It could also be used in acute inflammatory or infectious conditions, including sepsis [488,489,490]. Sampling at multiple time points over 24–48 hours can reveal dynamic changes in KYN pathway activation and TCA cycle disruption, reflecting immune activation and metabolic stress [491,492]. In cases of acute or chronic kidney injury, combined plasma and urine testing is essential for accurate assessment [493,494,495,496]. Together, these scenarios illustrate how standardized sampling times and pathway-based readouts allow for the use of a harmonized assay for longitudinal monitoring, assessment of response to treatment, and comparability across studies in different clinical settings [497,498,499,500].
5. Discussion: Strengths, Limitations, and Future Perspectives
This section integrates mechanism with measurement. Strong signals emerge where high quality LC–MS workflows align with biology: KYN metabolite panels show diagnostic and prognostic promise, including KYNA in depression and cardiometabolic risk stratification [92,288,501]. Yet uncertainty persists. Static metabolite levels obscure flux and compartmentation, cell lines incompletely mirror tumors, and heterogeneous sampling blurs psychiatric associations [502,503]. Next steps should prioritize standardized protocols and reference materials, longitudinal and tissue matched cohorts, and fluxomics with stable isotopes to resolve directionality [504,505]. Improved annotation and pathway contextualization, combined with targeted validation and mechanistic interventions, will convert metabolite signatures into clinically actionable tools [506,507].
A final point of positioning matters. Prior syntheses have been strong within their lanes, whether they center on neuropsychiatric signatures, immunoregulatory enzyme control, or NAD+ centered metabolic theory. Here the emphasis is different. We treat mitochondrial readouts as the common currency and then ask which receptor signals and which metabolic branch points most plausibly move those readouts in patients. That framing is why the analytics discussion sits beside receptor biology and de novo NAD⁺ biochemistry rather than being relegated to a technical appendix.
KYNA appears to choreograph mitochondrial resilience through a convergent receptor network [22]. Engagement of GPR35 preserves ATP under stress, tempers Ca2+ influx, and restrains NLRP3 signaling, linking membrane cues to organelle protection in ischemia, infection, and metabolic inflammation [22,32]. Parallel signaling through the AhR modulates oxidative programs and inflammatory tone, with context specific outcomes across brain and cardiovascular systems [190,508]. At excitatory synapses, NMDA-R antagonism by KYNA lowers excitotoxic drive and secondarily shields mitochondrial function [193,509]. α7 nicotinic receptor interactions remain debated, yet may contribute to immune–neuronal crosstalk in select settings [37,214]. Receptor crosstalk is likely, as GPR35 and AhR signals intersect Ca2+ handling, bioenergetics, and transcriptional defenses, while gut derived KYNA can prime GPR35 positive myeloid cells or coordinate epithelial protection via combined AhR–GPR35 sensing [16,510]. Therapeutically, receptor selective modulation offers leverage to enhance mitochondrial robustness without broad immunosuppression [16,22].
Modern LC–MS has moved the KYN and Try–NAD+ networks from piecemeal panels to near-complete coverage, enabling coordinated profiling across plasma, CSF, urine, peritoneal fluid, tissues, and even subcellular fractions [68,299]. Single-run or low-cost targeted assays quantify core KYN metabolites alongside central carbon nodes, while hybrid targeted–untargeted and solvent-switching workflows expand breadth without sacrificing precision [65,511]. Organelle-resolved strategies using chemical labeling now map compartmental metabolomes, a prerequisite for linking receptor cues to mitochondrial readouts in vivo [512,513]. Comparative cell-line and clinical applications illustrate scalability, drug screening utility, and disease stratification potential, including acute kidney injury and diabetes [481,514]. Limitations persist. Protocol heterogeneity, matrix effects, and incomplete annotation still blunt cross-study synthesis, motivating harmonized methods, reference materials, and improved pathway enrichment analytics [250,515]. The path forward integrates stable-isotope flux tracing with multi-organ, multi-compartment LC–MS to resolve directionality and translate signatures into actionable endpoints [516,517].
Coupling of the KYN metabolism to the TCA cycle provides a unifying scaffold for NAD+ governance and redox control across tissues [14,50]. KYN flux supplies quinolinate for de novo NAD+ and tunes mitochondrial function, intersecting with sirtuins, DNA repair, and stress programs [15,94]. Tissue context dictates outcome [14,225]. In liver ischemia–reperfusion, diversion from quinolinate depletes NAD+, weakens antioxidant defenses, and heightens injury, whereas augmentation restores resilience [43,518]. Macrophages require KYN derived NAD+ to maintain respiratory capacity and phagocytic competence, especially with aging [14,42]. Neurons and astrocytes similarly depend on KYN metabolites inputs to preserve viability and redox poise [45,230]. Kidney and brain exhibit disease specific deficits or sex dependent shifts in KYNs and NAD+ pools, linking metabolic imbalance to dysfunction [519,520]. These nodes communicate with NADPH–glutathione circuits that relay mitochondrial workload to the endoplasmic reticulum, completing a cross organ redox axis [15,521]. Cancer exploits the same wiring to fuel growth and immune escape, emphasizing context precise intervention [224,244].
Generalization falters when biology and measurement drift by model and tissue. LC–MS workflows vary in extraction, ionization, and matrices, so results hinge on protocol and normalization choices; no single assay spans all key metabolites across compartments [522]. Biological context adds another filter [523]. Enzyme expression, receptor density, and circadian control reshape flux in organ specific ways, while inflammation rewires the same nodes differently across tissues [524]. Plasma signals often diverge from tumor or CNS readouts, and correlations between serum, CSF, urine, and viscera are imperfect, urging multi-compartment designs with matrix specific validation [525]. Model choice matters. Disease stage, timing, and targeted enzyme alter outcomes in autoimmune and cancer settings, and cell line metabolism incompletely recapitulates in vivo states [526,527]. Standards are therefore essential. Anchored designs, reference materials, harmonized extraction, and data driven normalization should precede interpretation, enabling cross study synthesis and credible biomarker development [314,528].
Progress toward single-run LC–MS coverage of the KYN and Trp–NAD networks is real yet fragmented [66,250]. Panels quantifying 9–11 metabolites in CSF, plasma, urine, or peritoneal fluid prove feasibility, but validation remains matrix specific and inter-laboratory comparability is limited [68,288]. Methods for broader coverage or alternative separations extend analyte space while introducing longer run times, partial validation, or unresolved matrix effects [250,289]. Hybrid targeted–untargeted assays in large cohorts show precision, yet harmonized adoption and cross-site calibration are uncommon [65]. Cell-based workflows accelerate mechanism and screening but are not configured for clinical interchangeability [72,511]. The path forward is methodological convergence: clinical and laboratory standards institute (CLSI) C62-A–aligned development, isotopologue-rich internal standards, commutable multi-matrix calibrators, external quality assessment, and anchored normalization across batches and sites [289,299]. Reporting standards should include matrix-specific recovery, LLOQ, and carryover, plus retention-time locking and shared reference materials, so signatures translate across studies and into routine diagnostics [73,529].
Temporal and spatial resolution remain the weakest links in vivo. Most datasets provide static plasma or CSF snapshots that miss rapid fluxes and compartment shifts documented by disease meta-analyses and multi-compartment comparisons [530,531]. Maternal–fetal gradients and region-specific brain profiles illustrate strong spatial heterogeneity, yet longitudinal and organelle-resolved measurements are rare [532,533]. Cell-type time courses show inducible, phase-specific enzyme and metabolite waves, underscoring dynamics that bulk tissues blur [534,535]. Chronic KMO loss reshapes circulating metabolites without altering whole-body energetics, implying that acute transients, not steady states, drive phenotype [536]. Cross-kingdom exemplars point the way: subcellular fractionation and flux analysis resolve fast, compartment-locked control, often dictated by metabolite levels rather than enzyme abundance [537,538]. Physicochemical constraints and receptor transport further complicate KYNA partitioning, while KMO also governs mitochondrial morphology independent of catalysis [539,540]. Closing these blind spots will require synchronized sampling, stable-isotope fluxomics, spatial metabolomics, and organelle-targeted assays integrated across compartments and time.
A credible standardization roadmap rests on shared materials, shared methods, and shared language. First, anchor quantification with pooled multi-matrix reference materials, isotopologue-rich internal standards, and external proficiency testing to harmonize calibration and batch correction across sites [506,541]. Second, codify cross-lab standard operating procedures (SOPs) that cover preanalytics, extraction, chromatography, ionization, and QC frequency, drawing on best-practice tutorials and ultra-high-performance liquid chromatography (UHPLC)-high-resolution mass spectrometry (HRMS) guidance to minimize matrix effects and maximize metabolome coverage [542,543]. Third, mandate harmonized reporting: adopt community annotation levels, findable, accessible, interoperable, and reusable (FAIR) data deposition, and transparent identification criteria with library spectra, retention-time locking, and predictive models for MS1 features [317,544]. Open, referenceable workflows should be executable end-to-end and versioned in platforms such as MetaboAnalystR and Workflow4Metabolomics, with simulated and real benchmark datasets for software validation and interlaboratory comparability [545,546]. Finally, extend standardization to organelle-resolved assays using immunopurified mitochondria and absolute quantification, ensuring that pathway claims translate across compartments and cohorts [547,548].
Longitudinal, multi-layer cohorts can convert metabolomic snapshots into mechanistically anchored, prognostic readouts of mitochondrial health [549,550]. Population platforms such as translational metabolic cohort study (TMCS) and consortium of metabolomics studies (COMETS) already pair deep phenotyping with serial biospecimens, enabling stable-isotope substudies and genetic instruments to infer directionality between metabolite shifts and organelle function [549,551]. Coupling these frameworks to targeted mitochondrial phenotyping—multiplexed respiratory flux assays, immune-cell subtype profiling, and organelle-resolved metabolomics—links pathway panels to actionable defects in energy transfer, biogenesis, and redox control [468]. Prognostic models in sepsis, diabetic kidney disease, glioma, and mitochondrial disorders illustrate translational yield when pathway signatures are integrated with outcomes and imaging [552].
Genomic and microbiome layers further de-risk translation by prioritizing causal metabolites, resolving ancestry-specific effects, and exposing host–microbe axes that shape mitochondrial phenotypes [93,553]. Systems proteomics in liver and cross-species maps of mitochondrial gene function provide mechanistic scaffolds to interpret cohort signals and nominate interventions [554,555]. The immediate agenda is practical: harmonize preanalytics, embed reference materials, schedule repeated sampling, and co-capture mitochondrial function with metabolomics at scale [556]. Such integrated phenotyping will upgrade risk prediction, clarify therapeutic targets, and shorten the path from discovery to clinic [468,557].
Medicinal chemistry around KYNA is poised to pivot from blunt modulation to receptor precise control [558]. Decades of structure–activity work already yielded high affinity glycine site antagonists at NMDA-Rs and clarified competitive versus noncompetitive mechanisms, setting a template for selective central nervous system probes [228,558]. Parallel advances nominate GPR35 as a mitochondrial-proximal target for ischemic protection and inflammasome control, motivating small molecules with tuned efficacy and residence at this GPCR [16,22]. AhR active KYN scaffolds expand the chemical space for immunometabolic intervention, while a critical appraisal of purported α7 nicotinic activity focuses prioritization on validated targets [223,559]. Next steps should embrace biased agonism: leverage GPCR structural insights, cryptic allosteric pockets, and pathway selective screening to favor mitochondrial Ca2+ restraint and anti inflammatory outputs over broad immunosuppression [560,561]. Integration with enzyme directed KYN-pathway modulators will enable orthogonal control of flux and signaling, improving therapeutic index in neurodegeneration and oncology [110,562].
With the mechanistic map and measurement blueprint established, the translational question becomes operational: which lever is druggable, what mitochondrial endpoint should move, and what assay can verify it in patients. This section distills ligand and study design principles that follow directly from the receptor and QA to NAD+ architecture developed earlier.
6. Conclusion
This review converges on a simple logic with wide reach: KYN metabolic flux, receptor signaling, and TCA cycle control co-determine mitochondrial resilience, redox poise, and immune tone. The authors integrate receptor biology with NAD⁺ economics and analytics, showing how KYNA–GPR35, AhR, and NMDA interfaces can be read alongside unified LC–MS panels [14,563]. The practical output is a mechanistic map that predicts mitochondrial endpoints from receptor and flux states, paired with an assay blueprint that can test those predictions in clinical cohorts. The take-home message is pragmatic. Mechanism and measurement must travel together to turn associative metabolomics into causal, intervention-ready biology. Three priorities emerge. First, clinically validated, harmonized assays that quantify KYN and TCA intermediates in single runs across matrices, with isotope-labeled standards and cross-lab QC. Second, causal trials that pair receptor-selective ligands or enzyme modulators with mitochondrial endpoints such as ATP preservation, ΔΨm stability, and mitophagy readouts. Third, longitudinal, compartment-aware cohorts linking pathway state to outcomes in neurology, psychiatry, cardiometabolic disease, and oncology [49]. These steps will de-risk translation, refine patient selection, and enable adaptive designs that use pathway ratios as eligibility and pharmacodynamic markers [63,66]. The authors’ comprehensive synthesis underscores a tractable path from circuits to clinics: receptor-selective KYNA analogs, de novo NAD⁺ support, and standardized analytics. Uncertainties remain around temporal dynamics, tissue specificity, and receptor–mitochondria causality in vivo [224]. Future work should integrate isotope tracing, organelle profiling, and receptor bias pharmacology within harmonized workflows. With these elements aligned, mechanistic precision can mature into durable therapies and decision-grade biomarkers.
Author Contributions
Conceptualization, M.T.; methodology, L.J., Z.G., and M.T.; software, L.J., Z.G., and M.T.; validation, L.J., Z.G., and M.T..; formal analysis, L.J., Z.G., and M.T.; investigation, L.J., Z.G., and M.T.; resources, L.J., Z.G., and M.T.; data curation, L.J., Z.G., and M.T.; writing—original draft preparation, L.J., Z.G., and M.T.; writing—review and editing, L.J., Z.G., M.T., and L.V.; visualization, L.J., Z.G., and M.T.; supervision, M.T. and L.V.; project administration, M.T. and L.V.; funding acquisition, M.T. and L.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research, Development, and Innovation Office—NKFIH K138125, SZTE SZAOK-KKA No: 2022/5S729, SZTE SZAOK-KKA SZGYA No: 2023/5S775, the HUN-REN Hungarian Research Network, and University of Szeged Open Access Fund 8317.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The authors acknowledge the limited use of artificial intelligence tools during manuscript preparation. English language style was partly refined based on suggestions generated by AI, and all revisions were reviewed and approved by the authors, who retain full responsibility for the final content. AI tools assisted with reference discovery and preliminary searches, while all cited sources were independently selected and evaluated by the authors. _Some figure schematics were crafted by the authors from an initial draft and design created with AI assistance, with substantial modifications made by the authors to reflect original scientific concepts. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| α7nAChR | Nicotinic acetylcholine receptors containing α7 subunits |
| ACMSD | alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase |
| AhR | aryl hydrocarbon receptor |
| AKT | protein kinase B |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| ATPIF1 | ATP synthase inhibitory factor 1 |
| BNIP3 | BCL2 interacting protein 3 |
| cAMP | cyclic adenosine monophosphate |
| CD98 | cluster of differentiation 98 |
| CLSI | clinical and laboratory standards institute |
| COMETS | consortium of metabolomics studies |
| CSF | cerebrospinal fluid |
| ERK | extracellular signal-regulated kinase |
| ETC | electron transport chain |
| FAIR | findable, accessible, interoperable, and reusable |
| GPR35 | G protein–coupled receptor 35 |
| GPX4 | glutathione peroxidase 4 |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HILIC | hydrophilic interaction liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| IDO | indoleamine 2,3-dioxygenase |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IDO2 | indoleamine 2,3-dioxygenase 2 |
| KATs | kynurenine aminotransferases |
| KMO | kynurenine 3-monooxygenase |
| KYN | kynurenine |
| KYNA | kynurenic acid |
| KYNU | kynureninase |
| LC–MS | liquid chromatography–mass spectrometry |
| LLOQ | lower limit of quantitation |
| MCART1 | mitochondrial carrier transporting NAD⁺ |
| MDH2 | malate dehydrogenase 2 |
| mPTP | mitochondrial permeability transition pore |
| NAD+ | nicotinamide adenine dinucleotide (oxidized form) |
| NADH | nicotinamide adenine dinucleotide (reduced form) |
| NADPH | nicotinamide adenine dinucleotide phosphate (reduced form) |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated b cells |
| NMDA | N-methyl-D-aspartate |
| NMDA-R | N-methyl-D-aspartate receptor |
| NRF1 | nuclear respiratory factor 1 |
| NRF2 | nuclear factor erythroid 2–related factor 2 |
| PD-1 | programmed cell death protein 1 |
| PI3K | phosphoinositide 3-kinase |
| QC | quality control |
| QA | quinolinic acid |
| Rho | ras homolog family protein |
| ROS | reactive oxygen species |
| SAR | structure–activity relationship |
| SDH | succinate dehydrogenase |
| SOP | standard operating procedure |
| TCA | tricarboxylic acid |
| TDO | tryptophan 2,3-dioxygenase |
| TMCS | translational metabolic cohort study |
| TME | tumor microenvironment |
| Trp | tryptophan |
| UHPLC | ultra-high-performance liquid chromatography |
References
- Liu, B.H.; Xu, C.Z.; Liu, Y.; Lu, Z.L.; Fu, T.L.; Li, G.R.; Deng, Y.; Luo, G.Q.; Ding, S.; Li, N.; et al. Mitochondrial quality control in human health and disease. Mil Med Res 2024, 11, 32. [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [CrossRef]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic Biol Med 2022, 189, 136–153. [CrossRef]
- Hu, S.; Feng, J.; Wang, M.; Wufuer, R.; Liu, K.; Zhang, Z.; Zhang, Y. Nrf1 is an indispensable redox-determining factor for mitochondrial homeostasis by integrating multi-hierarchical regulatory networks. Redox Biol 2022, 57, 102470. [CrossRef]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid Redox Signal 2018, 29, 667–714. [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front Cell Dev Biol 2020, 8, 467. [CrossRef]
- Tanaka, M.; Szatmári, I.; Vécsei, L. Quinoline Quest: Kynurenic Acid Strategies for Next-Generation Therapeutics via Rational Drug Design. Pharmaceuticals (Basel) 2025, 18. [CrossRef]
- Tan, D.Q.; Suda, T. Reactive Oxygen Species and Mitochondrial Homeostasis as Regulators of Stem Cell Fate and Function. Antioxid Redox Signal 2018, 29, 149–168. [CrossRef]
- Tanaka, M. Neurogenesis and Neuroinflammation in Dialogue: Mapping Gaps, Modulating Microglia, Rewiring Aging. Cells 2026, 15, 78. [CrossRef]
- Murakami, S.; Kusano, Y.; Okazaki, K.; Akaike, T.; Motohashi, H. NRF2 signalling in cytoprotection and metabolism. Br J Pharmacol 2023. [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol Appl Pharmacol 2018, 359, 24–33. [CrossRef]
- Juhász, L.; Spisák, K.; Szolnoki, B.Z.; Nászai, A.; Szabó, Á.; Rutai, A.; Tallósy, S.P.; Szabó, A.; Toldi, J.; Tanaka, M.; et al. The Power Struggle: Kynurenine Pathway Enzyme Knockouts and Brain Mitochondrial Respiration. J Neurochem 2025, 169, e70075. [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp Gerontol 2020, 132, 110841. [CrossRef]
- González Esquivel, D.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; Pérez de la Cruz, V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112, 331–345. [CrossRef]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab 2018, 27, 378–392.e375. [CrossRef]
- Zhen, D.; Liu, J.; Zhang, X.D.; Song, Z. Kynurenic Acid Acts as a Signaling Molecule Regulating Energy Expenditure and Is Closely Associated With Metabolic Diseases. Front Endocrinol (Lausanne) 2022, 13, 847611. [CrossRef]
- Sánchez Chapul, L.; Pérez de la Cruz, G.; Ramos Chávez, L.A.; Valencia León, J.F.; Torres Beltrán, J.; Estrada Camarena, E.; Carillo Mora, P.; Ramírez Ortega, D.; Baños Vázquez, J.U.; Martínez Nava, G.; et al. Characterization of Redox Environment and Tryptophan Catabolism through Kynurenine Pathway in Military Divers’ and Swimmers’ Serum Samples. Antioxidants (Basel) 2022, 11. [CrossRef]
- Guidetti, P.; Amori, L.; Sapko, M.T.; Okuno, E.; Schwarcz, R. Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem 2007, 102, 103–111. [CrossRef]
- Wyckelsma, V.L.; Lindkvist, W.; Venckunas, T.; Brazaitis, M.; Kamandulis, S.; Pääsuke, M.; Ereline, J.; Westerblad, H.; Andersson, D.C. Kynurenine aminotransferase isoforms display fiber-type specific expression in young and old human skeletal muscle. Exp Gerontol 2020, 134, 110880. [CrossRef]
- Szabó, Á.G., Z.; Spekker, E.; Martos, D.; Szűcs, M.; Fejes-Szabó, A.; Fehér, Á.; Takeda, K.; Ozaki, K.; Inoue, H.; et al. Behavioral Balance in Tryptophan Turmoil: Regional Metabolic Rewiring in Kynurenine Aminotransferase II Knockout Mice. Cells 2025, 2025, 1711.
- Wyant, G.A.; Yu, W.; Doulamis, I.P.; Nomoto, R.S.; Saeed, M.Y.; Duignan, T.; McCully, J.D.; Kaelin, W.G., Jr. Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 2022, 377, 621–629. [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol 2019, 26, 101254. [CrossRef]
- Braidy, N.; Liu, Y. NAD+ therapy in age-related degenerative disorders: A benefit/risk analysis. Exp Gerontol 2020, 132, 110831. [CrossRef]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9. [CrossRef]
- Tanaka, M.; Battaglia, S. Dualistic Dynamics in Neuropsychiatry: From Monoaminergic Modulators to Multiscale Biomarker Maps. Biomedicines 2025, 13. [CrossRef]
- Figueiredo Godoy, A.C.; Frota, F.F.; Araújo, L.P.; Valenti, V.E.; Pereira, E.; Detregiachi, C.R.P.; Galhardi, C.M.; Caracio, F.C.; Haber, R.S.A.; Fornari Laurindo, L.; et al. Neuroinflammation and Natural Antidepressants: Balancing Fire with Flora. Biomedicines 2025, 13. [CrossRef]
- Barbalho, S.M.; Laurindo, L.F.; de Oliveira Zanuso, B.; da Silva, R.M.S.; Gallerani Caglioni, L.; Nunes Junqueira de Moraes, V.B.F.; Fornari Laurindo, L.; Dogani Rodrigues, V.; da Silva Camarinha Oliveira, J.; Beluce, M.E.; et al. AdipoRon’s Impact on Alzheimer’s Disease-A Systematic Review and Meta-Analysis. Int J Mol Sci 2025, 26. [CrossRef]
- Tanaka, M.; Szabó, Á.; Vécsei, L. Redefining Roles: A Paradigm Shift in Tryptophan-Kynurenine Metabolism for Innovative Clinical Applications. Int J Mol Sci 2024, 25. [CrossRef]
- Gáspár, R.; Nógrádi-Halmi, D.; Demján, V.; Diószegi, P.; Igaz, N.; Vincze, A.; Pipicz, M.; Kiricsi, M.; Vécsei, L.; Csont, T. Kynurenic acid protects against ischemia/reperfusion injury by modulating apoptosis in cardiomyocytes. Apoptosis 2024, 29, 1483–1498. [CrossRef]
- Sun, T.; Xie, R.; He, H.; Xie, Q.; Zhao, X.; Kang, G.; Cheng, C.; Yin, W.; Cong, J.; Li, J.; et al. Kynurenic acid ameliorates NLRP3 inflammasome activation by blocking calcium mobilization via GPR35. Front Immunol 2022, 13, 1019365. [CrossRef]
- Zheng, X.; Hu, M.; Zang, X.; Fan, Q.; Liu, Y.; Che, Y.; Guan, X.; Hou, Y.; Wang, G.; Hao, H. Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress. Brain Behav Immun 2019, 79, 244–255. [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front Immunol 2017, 8, 1957. [CrossRef]
- Grishanova, A.Y.; Perepechaeva, M.L. Kynurenic Acid/AhR Signaling at the Junction of Inflammation and Cardiovascular Diseases. Int J Mol Sci 2024, 25. [CrossRef]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci 2013, 34, 136–143. [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J Neurochem 2020, 152, 627–649. [CrossRef]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem Biophys Res Commun 2010, 395, 232–237. [CrossRef]
- Deng, H.; Hu, H.; Fang, Y. Multiple tyrosine metabolites are GPR35 agonists. Sci Rep 2012, 2, 373. [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int J Tryptophan Res 2017, 10, 1178646917691938. [CrossRef]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23. [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol 2019, 20, 50–63. [CrossRef]
- Xu, B.; Zhang, P.; Tang, X.; Wang, S.; Shen, J.; Zheng, Y.; Gao, C.; Mi, P.; Zhang, C.; Qu, H.; et al. Metabolic Rewiring of Kynurenine Pathway during Hepatic Ischemia-Reperfusion Injury Exacerbates Liver Damage by Impairing NAD Homeostasis. Adv Sci (Weinh) 2022, 9, e2204697. [CrossRef]
- Yang, Y.; Borel, T.; de Azambuja, F.; Johnson, D.; Sorrentino, J.P.; Udokwu, C.; Davis, I.; Liu, A.; Altman, R.A. Diflunisal Derivatives as Modulators of ACMS Decarboxylase Targeting the Tryptophan-Kynurenine Pathway. J Med Chem 2021, 64, 797–811. [CrossRef]
- Braidy, N.; Guillemin, G.J.; Grant, R. Effects of Kynurenine Pathway Inhibition on NAD Metabolism and Cell Viability in Human Primary Astrocytes and Neurons. Int J Tryptophan Res 2011, 4, 29–37. [CrossRef]
- Xue, C.; Li, G.; Zheng, Q.; Gu, X.; Shi, Q.; Su, Y.; Chu, Q.; Yuan, X.; Bao, Z.; Lu, J.; et al. Tryptophan metabolism in health and disease. Cell Metab 2023, 35, 1304–1326. [CrossRef]
- Stone, T.W.; Williams, R.O. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol Sci 2023, 44, 442–456. [CrossRef]
- Lu, Z.; Zhang, C.; Zhang, J.; Su, W.; Wang, G.; Wang, Z. The Kynurenine Pathway and Indole Pathway in Tryptophan Metabolism Influence Tumor Progression. Cancer Med 2025, 14, e70703. [CrossRef]
- Tanaka, M.; Vécsei, L. From Microbial Switches to Metabolic Sensors: Rewiring the Gut-Brain Kynurenine Circuit. Biomedicines 2025, 13. [CrossRef]
- Savitz, J. The kynurenine pathway: a finger in every pie. Mol Psychiatry 2020, 25, 131–147. [CrossRef]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10. [CrossRef]
- Pathak, S.; Nadar, R.; Kim, S.; Liu, K.; Govindarajulu, M.; Cook, P.; Watts Alexander, C.S.; Dhanasekaran, M.; Moore, T. The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies. Int J Mol Sci 2024, 25. [CrossRef]
- Tanaka, M.; Battaglia, S. From Biomarkers to Behavior: Mapping the Neuroimmune Web of Pain, Mood, and Memory. Biomedicines 2025, 13. [CrossRef]
- Strasser, B.; Becker, K.; Fuchs, D.; Gostner, J.M. Kynurenine pathway metabolism and immune activation: Peripheral measurements in psychiatric and co-morbid conditions. Neuropharmacology 2017, 112, 286–296. [CrossRef]
- Tanaka, M. Special Issue "Translating Molecular Psychiatry: From Biomarkers to Personalized Therapies". Int J Mol Sci 2025, 26. [CrossRef]
- Tanaka, M. Beyond the boundaries: Transitioning from categorical to dimensional paradigms in mental health diagnostics. Adv Clin Exp Med 2024, 33, 1295–1301. [CrossRef]
- Liloia, D.; Zamfira, D.A.; Tanaka, M.; Manuello, J.; Crocetta, A.; Keller, R.; Cozzolino, M.; Duca, S.; Cauda, F.; Costa, T. Disentangling the role of gray matter volume and concentration in autism spectrum disorder: A meta-analytic investigation of 25 years of voxel-based morphometry research. Neurosci Biobehav Rev 2024, 164, 105791. [CrossRef]
- Chehadi, A.C.; Pereira de Lima, E.; Detregiachi, C.R.P.; Santos de Argollo Haber, R.; Catharin, V.; Fornari Laurindo, L.; Engracia Valenti, V.; Machado Galhardi, C.; Tanaka, M.; Maria Barbalho, S. Harnessing Dietary Tryptophan: Bridging the Gap Between Neurobiology and Psychiatry in Depression Management. Int J Mol Sci 2026, 27. [CrossRef]
- Tanaka, M. From Monoamines to Systems Psychiatry: Rewiring Depression Science and Care (1960s-2025). Biomedicines 2025, 14. [CrossRef]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front Biosci (Landmark Ed) 2015, 20, 1116–1143. [CrossRef]
- Baumgartner, R.; Forteza, M.J.; Ketelhuth, D.F.J. The interplay between cytokines and the Kynurenine pathway in inflammation and atherosclerosis. Cytokine 2019, 122, 154148. [CrossRef]
- Kozieł, K.; Urbanska, E.M. Kynurenine Pathway in Diabetes Mellitus-Novel Pharmacological Target? Cells 2023, 12. [CrossRef]
- Pires, A.S.; Sundaram, G.; Heng, B.; Krishnamurthy, S.; Brew, B.J.; Guillemin, G.J. Recent advances in clinical trials targeting the kynurenine pathway. Pharmacol Ther 2022, 236, 108055. [CrossRef]
- Sforzini, L.; Nettis, M.A.; Mondelli, V.; Pariante, C.M. Inflammation in cancer and depression: a starring role for the kynurenine pathway. Psychopharmacology (Berl) 2019, 236, 2997–3011. [CrossRef]
- Eggertsen, P.P.; Hansen, J.; Andersen, M.L.; Nielsen, J.F.; Olsen, R.K.J.; Palmfeldt, J. Simultaneous measurement of kynurenine metabolites and explorative metabolomics using liquid chromatography-mass spectrometry: A novel accurate method applied to serum and plasma samples from a large healthy cohort. J Pharm Biomed Anal 2023, 227, 115304. [CrossRef]
- Hossen, M.M.; Fleiss, B.; Zakaria, R. The current state in liquid chromatography-mass spectrometry methods for quantifying kynurenine pathway metabolites in biological samples: a systematic review. Crit Rev Clin Lab Sci 2025, 62, 437–453. [CrossRef]
- Gomez-Gomez, A.; Olesti, E.; Montero-San-Martin, B.; Soldevila, A.; Deschamps, T.; Pizarro, N.; de la Torre, R.; Pozo, O.J. Determination of up to twenty carboxylic acid containing compounds in clinically relevant matrices by o-benzylhydroxylamine derivatization and liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 2022, 208, 114450. [CrossRef]
- Sadok, I.; Jędruchniewicz, K.; Rawicz-Pruszyński, K.; Staniszewska, M. UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients-Method Development and Validation. Int J Mol Sci 2021, 22. [CrossRef]
- Panuwet, P.; Hunter, R.E., Jr.; D’Souza, P.E.; Chen, X.; Radford, S.A.; Cohen, J.R.; Marder, M.E.; Kartavenka, K.; Ryan, P.B.; Barr, D.B. Biological Matrix Effects in Quantitative Tandem Mass Spectrometry-Based Analytical Methods: Advancing Biomonitoring. Crit Rev Anal Chem 2016, 46, 93–105. [CrossRef]
- Whiley, L.; Nye, L.C.; Grant, I.; Andreas, N.; Chappell, K.E.; Sarafian, M.H.; Misra, R.; Plumb, R.S.; Lewis, M.R.; Nicholson, J.K.; et al. Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry with Electrospray Ionization Quantification of Tryptophan Metabolites and Markers of Gut Health in Serum and Plasma-Application to Clinical and Epidemiology Cohorts. Anal Chem 2019, 91, 5207–5216. [CrossRef]
- Chawdhury, A.; Shamsi, S.A.; Miller, A.; Liu, A. Capillary electrochromatography-mass spectrometry of kynurenine pathway metabolites. J Chromatogr A 2021, 1651, 462294. [CrossRef]
- Sadok, I.; Rachwał, K.; Staniszewska, M. Simultaneous Quantification of Selected Kynurenines Analyzed by Liquid Chromatography-Mass Spectrometry in Medium Collected from Cancer Cell Cultures. J Vis Exp 2020. [CrossRef]
- Khetarpal, V.; Herbst, T.; Akhtar, S.; LaFayette, A.; Miller, D.; Farnham, J.; Steege, T.; Miao, Z.; Marks, B.; Ledvina, A.; et al. Validation of LC-MS/MS methods for quantitative analysis of kynurenine pathway metabolites in human plasma and cerebrospinal fluid. Bioanalysis 2023, 15, 637–651. [CrossRef]
- Huang, N.; Winans, T.; Wyman, B.; Oaks, Z.; Faludi, T.; Choudhary, G.; Lai, Z.W.; Lewis, J.; Beckford, M.; Duarte, M.; et al. Rab4A-directed endosome traffic shapes pro-inflammatory mitochondrial metabolism in T cells via mitophagy, CD98 expression, and kynurenine-sensitive mTOR activation. Nat Commun 2024, 15, 2598. [CrossRef]
- Ou, W.; Chen, Y.; Ju, Y.; Ma, M.; Qin, Y.; Bi, Y.; Liao, M.; Liu, B.; Liu, J.; Zhang, Y.; et al. The kynurenine pathway in major depressive disorder under different disease states: A systematic review and meta-analysis. J Affect Disord 2023, 339, 624–632. [CrossRef]
- Cristina, B.; Veronica, R.; Silvia, A.; Andrea, G.; Sara, C.; Luca, P.; Nicoletta, B.; M, C.B.; Silvio, B.; Fabio, T. Identification and characterization of the kynurenine pathway in the pond snail Lymnaea stagnalis. Sci Rep 2022, 12, 15617. [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [CrossRef]
- Sadok, I.; Staniszewska, M. Electrochemical Determination of Kynurenine Pathway Metabolites-Challenges and Perspectives. Sensors (Basel) 2021, 21. [CrossRef]
- Mrštná, K.; Krčmová, L.K.; Švec, F. Advances in kynurenine analysis. Clin Chim Acta 2023, 547, 117441. [CrossRef]
- Tanaka, M.; Battaglia, S.; Liloia, D. Navigating Neurodegeneration: Integrating Biomarkers, Neuroinflammation, and Imaging in Parkinson’s, Alzheimer’s, and Motor Neuron Disorders. Biomedicines 2025, 13. [CrossRef]
- Tanaka, M. From Serendipity to Precision: Integrating AI, Multi-Omics, and Human-Specific Models for Personalized Neuropsychiatric Care. Biomedicines 2025, 13. [CrossRef]
- Tanaka, M. Neurogenesis and Neuroinflammation in Dialogue: Mapping Gaps, Modulating Microglia, Rewiring Aging. Cells 2026, 15. [CrossRef]
- Ginsberg, M.D. The cerebral collateral circulation: Relevance to pathophysiology and treatment of stroke. Neuropharmacology 2018, 134, 280–292. [CrossRef]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 2002, 1, 609–620. [CrossRef]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol 2017, 35, 48–56. [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD(+) metabolism, from bench to bedside. Embo j 2017, 36, 2670–2683. [CrossRef]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004, 4, 762–774. [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019, 18, 379–401. [CrossRef]
- de Ridder, I.; Kerkhofs, M.; Lemos, F.O.; Loncke, J.; Bultynck, G.; Parys, J.B. The ER-mitochondria interface, where Ca(2+) and cell death meet. Cell Calcium 2023, 112, 102743. [CrossRef]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010, 31, 194–223. [CrossRef]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci Biobehav Rev 2018, 90, 16–25. [CrossRef]
- Xu, Y.; Ritchie, S.C.; Liang, Y.; Timmers, P.; Pietzner, M.; Lannelongue, L.; Lambert, S.A.; Tahir, U.A.; May-Wilson, S.; Foguet, C.; et al. An atlas of genetic scores to predict multi-omic traits. Nature 2023, 616, 123–131. [CrossRef]
- Moffett, J.R.; Arun, P.; Puthillathu, N.; Vengilote, R.; Ives, J.A.; Badawy, A.A.; Namboodiri, A.M. Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD(+) Synthesis During Inflammation and Infection. Front Immunol 2020, 11, 31. [CrossRef]
- Polonio, C.M.; McHale, K.A.; Sherr, D.H.; Rubenstein, D.; Quintana, F.J. The aryl hydrocarbon receptor: a rehabilitated target for therapeutic immune modulation. Nat Rev Drug Discov 2025, 24, 610–630. [CrossRef]
- Harrieder, E.M.; Kretschmer, F.; Böcker, S.; Witting, M. Current state-of-the-art of separation methods used in LC-MS based metabolomics and lipidomics. J Chromatogr B Analyt Technol Biomed Life Sci 2022, 1188, 123069. [CrossRef]
- Roth, H.E.; Powers, R. Meta-Analysis Reveals Both the Promises and the Challenges of Clinical Metabolomics. Cancers (Basel) 2022, 14. [CrossRef]
- da Rocha, A.L.; Pinto, A.P.; de Sousa Neto, I.V.; Muñoz, V.R.; Marafon, B.B.; da Silva, L.; Pauli, J.R.; Cintra, D.E.; Ropelle, E.R.; Simabuco, F.M.; et al. Exhaustive exercise abolishes REV-ERB-α circadian rhythm and shifts the kynurenine pathway to a neurotoxic profile in mice. J Physiol 2025, 603, 3923–3944. [CrossRef]
- O’Leary, K.M.; Slezak, T.; Kossiakoff, A.A. Conformation-specific synthetic intrabodies modulate mTOR signaling with subcellular spatial resolution. Proc Natl Acad Sci U S A 2025, 122, e2424679122. [CrossRef]
- Bader, J.M.; Albrecht, V.; Mann, M. MS-Based Proteomics of Body Fluids: The End of the Beginning. Mol Cell Proteomics 2023, 22, 100577. [CrossRef]
- Battaglia, S.; Tanaka, M. Screen, Sample, Stratify: Biomarkers and Machine Learning Compress Dementia Pathways. 2026, 14, 159.
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther 2023, 8, 333. [CrossRef]
- Rischke, S.; Hahnefeld, L.; Burla, B.; Behrens, F.; Gurke, R.; Garrett, T.J. Small molecule biomarker discovery: Proposed workflow for LC-MS-based clinical research projects. J Mass Spectrom Adv Clin Lab 2023, 28, 47–55. [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [CrossRef]
- Schneditz, G.; Elias, J.E.; Pagano, E.; Zaeem Cader, M.; Saveljeva, S.; Long, K.; Mukhopadhyay, S.; Arasteh, M.; Lawley, T.D.; Dougan, G.; et al. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci Signal 2019, 12. [CrossRef]
- Elias, J.E.; Debela, M.; Sewell, G.W.; Stopforth, R.J.; Partl, H.; Heissbauer, S.; Holland, L.M.; Karlsen, T.H.; Kaser, A.; Kaneider, N.C. GPR35 prevents osmotic stress induced cell damage. Commun Biol 2025, 8, 478. [CrossRef]
- Lin, L.C.; Quon, T.; Engberg, S.; Mackenzie, A.E.; Tobin, A.B.; Milligan, G. G Protein-Coupled Receptor GPR35 Suppresses Lipid Accumulation in Hepatocytes. ACS Pharmacol Transl Sci 2021, 4, 1835–1848. [CrossRef]
- Wei, X.; Yin, F.; Wu, M.; Xie, Q.; Zhao, X.; Zhu, C.; Xie, R.; Chen, C.; Liu, M.; Wang, X. G protein-coupled receptor 35 attenuates nonalcoholic steatohepatitis by reprogramming cholesterol homeostasis in hepatocytes. Acta pharmaceutica Sinica B 2023, 13, 1128–1144.
- Nam, S.-Y.; Park, S.-J.; Im, D.-S. Protective effect of lodoxamide on hepatic steatosis through GPR35. Cellular Signalling 2019, 53, 190–200.
- Nesci, S. GPR35, ally of the anti-ischemic ATPIF1-ATP synthase interaction. Trends Pharmacol Sci 2022, 43, 891–893. [CrossRef]
- Sharmin, O.; Abir, A.H.; Potol, A.; Alam, M.; Banik, J.; Rahman, A.; Tarannum, N.; Wadud, R.; Habib, Z.F.; Rahman, M. Activation of GPR35 protects against cerebral ischemia by recruiting monocyte-derived macrophages. Sci Rep 2020, 10, 9400. [CrossRef]
- Boleij, A.; Fathi, P.; Dalton, W.; Park, B.; Wu, X.; Huso, D.; Allen, J.; Besharati, S.; Anders, R.A.; Housseau, F.; et al. G-protein coupled receptor 35 (GPR35) regulates the colonic epithelial cell response to enterotoxigenic Bacteroides fragilis. Commun Biol 2021, 4, 585. [CrossRef]
- Chen, K.; He, L.; Li, Y.; Li, X.; Qiu, C.; Pei, H.; Yang, D. Inhibition of GPR35 Preserves Mitochondrial Function After Myocardial Infarction by Targeting Calpain 1/2. J Cardiovasc Pharmacol 2020, 75, 556–563. [CrossRef]
- Milligan, G. GPR35: from enigma to therapeutic target. Trends Pharmacol Sci 2023, 44, 263–273. [CrossRef]
- Cheng, L.; Wu, H.; Cai, X.; Zhang, Y.; Yu, S.; Hou, Y.; Yin, Z.; Yan, Q.; Wang, Q.; Sun, T.; et al. A Gpr35-tuned gut microbe-brain metabolic axis regulates depressive-like behavior. Cell Host Microbe 2024, 32, 227–243.e226. [CrossRef]
- Tanaka, M.; He, Z.; Han, S.; Battaglia, S. Editorial: Noninvasive brain stimulation: a promising approach to study and improve emotion regulation. Front Behav Neurosci 2025, 19, 1633936. [CrossRef]
- Wu, Y.; Zhang, P.; Fan, H.; Zhang, C.; Yu, P.; Liang, X.; Chen, Y. GPR35 acts a dual role and therapeutic target in inflammation. Front Immunol 2023, 14, 1254446. [CrossRef]
- Mackenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a Novel Therapeutic Target. Front Endocrinol (Lausanne) 2011, 2, 68. [CrossRef]
- Stone, T.W.; Darlington, L.G.; Badawy, A.A.-B.; Williams, R.O. The complex world of kynurenic acid: reflections on biological issues and therapeutic strategy. International Journal of Molecular Sciences 2024, 25, 9040.
- Brust, T.F.; Conley, J.M.; Watts, V.J. Gα(i/o)-coupled receptor-mediated sensitization of adenylyl cyclase: 40 years later. Eur J Pharmacol 2015, 763, 223–232. [CrossRef]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol 2002, 22, 3237–3246. [CrossRef]
- Zhang, L.; Shi, G. Gq-Coupled Receptors in Autoimmunity. J Immunol Res 2016, 2016, 3969023. [CrossRef]
- Takkar, S.; Sharma, G.; Kaushal, J.B.; Abdullah, K.M.; Batra, S.K.; Siddiqui, J.A. From orphan to oncogene: The role of GPR35 in cancer and immune modulation. Cytokine Growth Factor Rev 2024, 77, 56–66. [CrossRef]
- Poźniak, O.; Sitarz, R.; Sitarz, M.Z.; Kowalczuk, D.; Słoń, E.; Dudzińska, E. Utilization of AhR and GPR35 Receptor Ligands as Superfoods in Cancer Prevention for Individuals with IBD. Int J Mol Sci 2025, 26. [CrossRef]
- Nógrádi-Halmi, D.; Erdélyi-Furka, B.; Csóré, D.; Plechl, É.; Igaz, N.; Juhász, L.; Poles, M.Z.; Nógrádi, B.; Patai, R.; Polgár, T.F.; et al. Kynurenic Acid Protects Against Myocardial Ischemia/Reperfusion Injury by Activating GPR35 Receptors and Preserving Mitochondrial Structure and Function. Biomolecules 2025, 15. [CrossRef]
- García-Trevijano, E.R.; Ortiz-Zapater, E.; Gimeno, A.; Viña, J.R.; Zaragozá, R. Calpains, the proteases of two faces controlling the epithelial homeostasis in mammary gland. Front Cell Dev Biol 2023, 11, 1249317. [CrossRef]
- Hernando, V.; Inserte, J.; Sartório, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol 2010, 49, 271–279. [CrossRef]
- Chen, Q.; Lesnefsky, E.J. Heart mitochondria and calpain 1: Location, function, and targets. Biochim Biophys Acta 2015, 1852, 2372–2378. [CrossRef]
- Chen, M.; Won, D.J.; Krajewski, S.; Gottlieb, R.A. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem 2002, 277, 29181–29186. [CrossRef]
- Zheng, D.; Cao, T.; Zhang, L.L.; Fan, G.C.; Qiu, J.; Peng, T.Q. Targeted inhibition of calpain in mitochondria alleviates oxidative stress-induced myocardial injury. Acta Pharmacol Sin 2021, 42, 909–920. [CrossRef]
- Ni, R.; Zheng, D.; Xiong, S.; Hill, D.J.; Sun, T.; Gardiner, R.B.; Fan, G.C.; Lu, Y.; Abel, E.D.; Greer, P.A.; et al. Mitochondrial Calpain-1 Disrupts ATP Synthase and Induces Superoxide Generation in Type 1 Diabetic Hearts: A Novel Mechanism Contributing to Diabetic Cardiomyopathy. Diabetes 2016, 65, 255–268. [CrossRef]
- Jenkins, L.; Alvarez-Curto, E.; Campbell, K.; de Munnik, S.; Canals, M.; Schlyer, S.; Milligan, G. Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα₁₃ and β-arrestin-2. Br J Pharmacol 2011, 162, 733–748. [CrossRef]
- Domínguez-Zorita, S.; Romero-Carramiñana, I.; Santacatterina, F.; Esparza-Moltó, P.B.; Simó, C.; Del-Arco, A.; Núñez de Arenas, C.; Saiz, J.; Barbas, C.; Cuezva, J.M. IF1 ablation prevents ATP synthase oligomerization, enhances mitochondrial ATP turnover and promotes an adenosine-mediated pro-inflammatory phenotype. Cell Death Dis 2023, 14, 413. [CrossRef]
- Warnsmann, V.; Marschall, L.M.; Osiewacz, H.D. Impaired F(1)F(o)-ATP-Synthase Dimerization Leads to the Induction of Cyclophilin D-Mediated Autophagy-Dependent Cell Death and Accelerated Aging. Cells 2021, 10. [CrossRef]
- Domínguez-Zorita, S.; Romero-Carramiñana, I.; Cuezva, J.M.; Esparza-Moltó, P.B. The ATPase Inhibitory Factor 1 is a Tissue-Specific Physiological Regulator of the Structure and Function of Mitochondrial ATP Synthase: A Closer Look Into Neuronal Function. Front Physiol 2022, 13, 868820. [CrossRef]
- Nikolaou, P.E.; Lambrinidis, G.; Georgiou, M.; Karagiannis, D.; Efentakis, P.; Bessis-Lazarou, P.; Founta, K.; Kampoukos, S.; Konstantin, V.; Palmeira, C.M.; et al. Hydrolytic Activity of Mitochondrial F(1)F(O)-ATP Synthase as a Target for Myocardial Ischemia-Reperfusion Injury: Discovery and In Vitro and In Vivo Evaluation of Novel Inhibitors. J Med Chem 2023, 66, 15115–15140. [CrossRef]
- Mnatsakanyan, N.; Jonas, E.A. The new role of F(1)F(o) ATP synthase in mitochondria-mediated neurodegeneration and neuroprotection. Exp Neurol 2020, 332, 113400. [CrossRef]
- Acin-Perez, R.; Benincá, C.; Fernandez Del Rio, L.; Shu, C.; Baghdasarian, S.; Zanette, V.; Gerle, C.; Jiko, C.; Khairallah, R.; Khan, S.; et al. Inhibition of ATP synthase reverse activity restores energy homeostasis in mitochondrial pathologies. Embo j 2023, 42, e111699. [CrossRef]
- Pan, Y.; Ji, N.; Jiang, L.; Zhou, Y.; Feng, X.; Li, J.; Zeng, X.; Wang, J.; Shen, Y.Q.; Chen, Q. GPCRs identified on mitochondrial membranes: New therapeutic targets for diseases. J Pharm Anal 2025, 15, 101178. [CrossRef]
- García-Bermúdez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim Biophys Acta 2016, 1857, 1167–1182. [CrossRef]
- Kaim, G.; Dimroth, P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. Embo j 1999, 18, 4118–4127. [CrossRef]
- Dimroth, P.; Kaim, G.; Matthey, U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol 2000, 203, 51–59. [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal Biochem 2018, 552, 50–59. [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 2003, 10, 709–717. [CrossRef]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Zanin, C.; Vayssière, J.L.; Petit, P.X.; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med 1995, 181, 1661–1672. [CrossRef]
- Dadvar, S.; Ferreira, D.M.S.; Cervenka, I.; Ruas, J.L. The weight of nutrients: kynurenine metabolites in obesity and exercise. J Intern Med 2018, 284, 519–533. [CrossRef]
- Jung, T.W.; Park, J.; Sun, J.L.; Ahn, S.H.; Abd El-Aty, A.M.; Hacimuftuoglu, A.; Kim, H.C.; Shim, J.H.; Shin, S.; Jeong, J.H. Administration of kynurenic acid reduces hyperlipidemia-induced inflammation and insulin resistance in skeletal muscle and adipocytes. Mol Cell Endocrinol 2020, 518, 110928. [CrossRef]
- Mándi, Y.; Endrész, V.; Mosolygó, T.; Burián, K.; Lantos, I.; Fülöp, F.; Szatmári, I.; Lőrinczi, B.; Balog, A.; Vécsei, L. The Opposite Effects of Kynurenic Acid and Different Kynurenic Acid Analogs on Tumor Necrosis Factor-α (TNF-α) Production and Tumor Necrosis Factor-Stimulated Gene-6 (TSG-6) Expression. Front Immunol 2019, 10, 1406. [CrossRef]
- Fallarini, S.; Magliulo, L.; Paoletti, T.; de Lalla, C.; Lombardi, G. Expression of functional GPR35 in human iNKT cells. Biochem Biophys Res Commun 2010, 398, 420–425. [CrossRef]
- Richter, C.A.; Tillitt, D.E.; Hannink, M. Regulation of subcellular localization of the aryl hydrocarbon receptor (AhR). Arch Biochem Biophys 2001, 389, 207–217. [CrossRef]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol 2004, 36, 189–204. [CrossRef]
- Sorg, O. AhR signalling and dioxin toxicity. Toxicol Lett 2014, 230, 225–233. [CrossRef]
- Corre, S.; Tardif, N.; Mouchet, N.; Leclair, H.M.; Boussemart, L.; Gautron, A.; Bachelot, L.; Perrot, A.; Soshilov, A.; Rogiers, A.; et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat Commun 2018, 9, 4775. [CrossRef]
- Dai, S.; Qu, L.; Li, J.; Zhang, Y.; Jiang, L.; Wei, H.; Guo, M.; Chen, X.; Chen, Y. Structural insight into the ligand binding mechanism of aryl hydrocarbon receptor. Nat Commun 2022, 13, 6234. [CrossRef]
- Heo, M.J.; Suh, J.H.; Lee, S.H.; Poulsen, K.L.; An, Y.A.; Moorthy, B.; Hartig, S.M.; Moore, D.D.; Kim, K.H. Aryl hydrocarbon receptor maintains hepatic mitochondrial homeostasis in mice. Mol Metab 2023, 72, 101717. [CrossRef]
- Bahman, F.; Choudhry, K.; Al-Rashed, F.; Al-Mulla, F.; Sindhu, S.; Ahmad, R. Aryl hydrocarbon receptor: current perspectives on key signaling partners and immunoregulatory role in inflammatory diseases. Front Immunol 2024, 15, 1421346. [CrossRef]
- Nebert, D.W.; Karp, C.L. Endogenous functions of the aryl hydrocarbon receptor (AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem 2008, 283, 36061–36065. [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019, 19, 184–197. [CrossRef]
- Nacarino-Palma, A.; González-Rico, F.J.; Rejano-Gordillo, C.M.; Ordiales-Talavero, A.; Merino, J.M.; Fernández-Salguero, P.M. The aryl hydrocarbon receptor promotes differentiation during mouse preimplantational embryo development. Stem Cell Reports 2021, 16, 2351–2363. [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci 2010, 115, 89–97. [CrossRef]
- Moroni, F.; Cozzi, A.; Sili, M.; Mannaioni, G. Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J Neural Transm (Vienna) 2012, 119, 133–139. [CrossRef]
- Wu, X.; Wei, J.; Ran, W.; Liu, D.; Yi, Y.; Gong, M.; Liu, X.; Gong, Q.; Li, H.; Gao, J. The Gut Microbiota-Xanthurenic Acid-Aromatic Hydrocarbon Receptor Axis Mediates the Anticolitic Effects of Trilobatin. Adv Sci (Weinh) 2025, 12, e2412234. [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013, 20, 31–42. [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011, 12, 9–14. [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther 2023, 8, 304. [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front Genet 2012, 3, 297. [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 2016, 73, 775–795. [CrossRef]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat Metab 2023, 5, 2047–2061. [CrossRef]
- Peng, X.; Wang, K.; Zhang, C.; Bao, J.P.; Vlf, C.; Gao, J.W.; Zhou, Z.M.; Wu, X.T. The mitochondrial antioxidant SS-31 attenuated lipopolysaccharide-induced apoptosis and pyroptosis of nucleus pulposus cells via scavenging mitochondrial ROS and maintaining the stability of mitochondrial dynamics. Free Radic Res 2021, 55, 1080–1093. [CrossRef]
- Cao, J.; Bao, Q.; Hao, H. Indole-3-Carboxaldehyde Alleviates LPS-Induced Intestinal Inflammation by Inhibiting ROS Production and NLRP3 Inflammasome Activation. Antioxidants (Basel) 2024, 13. [CrossRef]
- Ma, M.; Guo, J.; Su, X.; Ma, B.; Wang, X.; Wangshao, M.; Zhong, K.; Wang, Y.; Yang, G.; Han, Y. Aryl hydrocarbon receptor (AhR) alleviates the LPS-induced inflammatory responses in IPEC-J2 cells by activating PINK1/Parkin-mediated mitophagy. Inflamm Res 2025, 74, 98. [CrossRef]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol 2017, 10, 1133–1144. [CrossRef]
- Tanaka, M. Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells 2025, 14. [CrossRef]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: molecular mechanisms and implications for human disease. Cell Death Dis 2021, 13, 14. [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012, 393, 547–564. [CrossRef]
- Antico, O.; Thompson, P.W.; Hertz, N.T.; Muqit, M.M.K.; Parton, L.E. Targeting mitophagy in neurodegenerative diseases. Nat Rev Drug Discov 2025, 24, 276–299. [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018, 20, 1013–1022. [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. Embo j 2021, 40, e104705. [CrossRef]
- Birgisdottir Å, B.; Lamark, T.; Johansen, T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013, 126, 3237–3247. [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther 2024, 9, 124. [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 2018, 17, 865–886. [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2014, 2, 702–714. [CrossRef]
- Senft, A.P.; Dalton, T.P.; Nebert, D.W.; Genter, M.B.; Puga, A.; Hutchinson, R.J.; Kerzee, J.K.; Uno, S.; Shertzer, H.G. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radic Biol Med 2002, 33, 1268–1278. [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial clearance: mechanisms and roles in cellular fitness. FEBS Lett 2021, 595, 1239–1263. [CrossRef]
- Garza-Lombó, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion 2020, 51, 105–117. [CrossRef]
- Huang, Y.; Zhang, J.; Tao, Y.; Ji, C.; Aniagu, S.; Jiang, Y.; Chen, T. AHR/ROS-mediated mitochondria apoptosis contributes to benzo[a]pyrene-induced heart defects and the protective effects of resveratrol. Toxicology 2021, 462, 152965. [CrossRef]
- Wang, X.; Li, S.; Liu, L.; Jian, Z.; Cui, T.; Yang, Y.; Guo, S.; Yi, X.; Wang, G.; Li, C.; et al. Role of the aryl hydrocarbon receptor signaling pathway in promoting mitochondrial biogenesis against oxidative damage in human melanocytes. J Dermatol Sci 2019, 96, 33–41. [CrossRef]
- Chen, J.; Zhang, M.; Zou, H.; Aniagu, S.; Jiang, Y.; Chen, T. PM2.5 induces mitochondrial dysfunction via AHR-mediated cyp1a1 overexpression during zebrafish heart development. Toxicology 2023, 487, 153466. [CrossRef]
- Ren, R.; Fang, Y.; Sherchan, P.; Lu, Q.; Lenahan, C.; Zhang, J.H.; Zhang, J.; Tang, J. Kynurenine/Aryl Hydrocarbon Receptor Modulates Mitochondria-Mediated Oxidative Stress and Neuronal Apoptosis in Experimental Intracerebral Hemorrhage. Antioxid Redox Signal 2022, 37, 1111–1129. [CrossRef]
- Majláth, Z.; Török, N.; Toldi, J.; Vécsei, L. Memantine and Kynurenic Acid: Current Neuropharmacological Aspects. Curr Neuropharmacol 2016, 14, 200–209. [CrossRef]
- Oliver, M.W.; Kessler, M.; Larson, J.; Schottler, F.; Lynch, G. Glycine site associated with the NMDA receptor modulates long-term potentiation. Synapse 1990, 5, 265–270. [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur J Pharmacol 1988, 154, 85–87. [CrossRef]
- Poles, M.Z.; Nászai, A.; Gulácsi, L.; Czakó, B.L.; Gál, K.G.; Glenz, R.J.; Dookhun, D.; Rutai, A.; Tallósy, S.P.; Szabó, A.; et al. Kynurenic Acid and Its Synthetic Derivatives Protect Against Sepsis-Associated Neutrophil Activation and Brain Mitochondrial Dysfunction in Rats. Front Immunol 2021, 12, 717157. [CrossRef]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Noh, Y.H.; Kim, D.H.; Lee, J.Y.; Cho, S.H.; Yoon, O.J.; Lee, W.B.; Kim, K.Y.; et al. Kynurenic acid attenuates MPP(+)-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur J Cell Biol 2008, 87, 389–397. [CrossRef]
- Szabo, M.; Lajkó, N.; Dulka, K.; Szatmári, I.; Fülöp, F.; Mihály, A.; Vécsei, L.; Gulya, K. Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains. Int J Mol Sci 2022, 23. [CrossRef]
- Nesterov, S.V.; Skorobogatova, Y.A.; Panteleeva, A.A.; Pavlik, L.L.; Mikheeva, I.B.; Yaguzhinsky, L.S.; Nartsissov, Y.R. NMDA and GABA receptor presence in rat heart mitochondria. Chem Biol Interact 2018, 291, 40–46. [CrossRef]
- Korde, A.S.; Maragos, W.F. Identification of an N-methyl-D-aspartate receptor in isolated nervous system mitochondria. J Biol Chem 2012, 287, 35192–35200. [CrossRef]
- Korde, A.S.; Maragos, W.F. Mitochondrial N-methyl-d-aspartate receptor activation enhances bioenergetics by calcium-dependent and -Independent mechanisms. Mitochondrion 2021, 59, 76–82. [CrossRef]
- Scanlon, D.P.; Bah, A.; Krzeminski, M.; Zhang, W.; Leduc-Pessah, H.L.; Dong, Y.N.; Forman-Kay, J.D.; Salter, M.W. An evolutionary switch in ND2 enables Src kinase regulation of NMDA receptors. Nat Commun 2017, 8, 15220. [CrossRef]
- Salter, M.W.; Kalia, L.V. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004, 5, 317–328. [CrossRef]
- Szabó, Á.; Galla, Z.; Spekker, E.; Szűcs, M.; Martos, D.; Takeda, K.; Ozaki, K.; Inoue, H.; Yamamoto, S.; Toldi, J.; et al. Oxidative and Excitatory Neurotoxic Stresses in CRISPR/Cas9-Induced Kynurenine Aminotransferase Knockout Mice: A Novel Model for Despair-Based Depression and Post-Traumatic Stress Disorder. Front Biosci (Landmark Ed) 2025, 30, 25706. [CrossRef]
- Barbalho, S.M.; Leme Boaro, B.; da Silva Camarinha Oliveira, J.; Patočka, J.; Barbalho Lamas, C.; Tanaka, M.; Laurindo, L.F. Molecular Mechanisms Underlying Neuroinflammation Intervention with Medicinal Plants: A Critical and Narrative Review of the Current Literature. Pharmaceuticals (Basel) 2025, 18. [CrossRef]
- de Lima, E.P.; Tanaka, M.; Lamas, C.B.; Quesada, K.; Detregiachi, C.R.P.; Araújo, A.C.; Guiguer, E.L.; Catharin, V.; de Castro, M.V.M.; Junior, E.B.; et al. Vascular Impairment, Muscle Atrophy, and Cognitive Decline: Critical Age-Related Conditions. Biomedicines 2024, 12. [CrossRef]
- Tanaka, M.; Vécsei, L. Revolutionizing our understanding of Parkinson’s disease: Dr. Heinz Reichmann’s pioneering research and future research direction. J Neural Transm (Vienna) 2024, 131, 1367–1387. [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov 2013, 12, 64–82. [CrossRef]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [CrossRef]
- Tan, L.; Yu, J.T.; Tan, L. The kynurenine pathway in neurodegenerative diseases: mechanistic and therapeutic considerations. J Neurol Sci 2012, 323, 1–8. [CrossRef]
- Németh, H.; Toldi, J.; Vécsei, L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl 2006, 285-304. [CrossRef]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? CNS Neurosci Ther 2022, 28, 19–35. [CrossRef]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS One 2012, 7, e31361. [CrossRef]
- Kalashnyk, O.; Lykhmus, O.; Uspenska, K.; Izmailov, M.; Komisarenko, S.; Skok, M. Mitochondrial α7 nicotinic acetylcholine receptors are displaced from complexes with VDAC1 to form complexes with Bax upon apoptosis induction. Int J Biochem Cell Biol 2020, 129, 105879. [CrossRef]
- Gergalova, G.; Lykhmus, O.; Komisarenko, S.; Skok, M. α7 nicotinic acetylcholine receptors control cytochrome c release from isolated mitochondria through kinase-mediated pathways. Int J Biochem Cell Biol 2014, 49, 26–31. [CrossRef]
- Abraham, S.M.; Suresh, S.; Komal, P. Targeting Neuronal Alpha7 Nicotinic Acetylcholine Receptor Upregulation in Age-Related Neurological Disorders. Cell Mol Neurobiol 2025, 45, 70. [CrossRef]
- Papke, R.L.; Horenstein, N.A. Therapeutic Targeting of α7 Nicotinic Acetylcholine Receptors. Pharmacol Rev 2021, 73, 1118–1149. [CrossRef]
- Wu, L.; Shen, F.; Lin, L.; Zhang, X.; Bruce, I.C.; Xia, Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett 2006, 402, 184–189. [CrossRef]
- Schultz, J.E.; Qian, Y.Z.; Gross, G.J.; Kukreja, R.C. The ischemia-selective KATP channel antagonist, 5-hydroxydecanoate, blocks ischemic preconditioning in the rat heart. J Mol Cell Cardiol 1997, 29, 1055–1060. [CrossRef]
- Chen, Z.; Jiao, Y.; Zhang, Y.; Wang, Q.; Wu, W.; Zheng, J.; Li, J. G-Protein Coupled Receptor 35 Induces Intervertebral Disc Degeneration by Mediating the Influx of Calcium Ions and Upregulating Reactive Oxygen Species. Oxid Med Cell Longev 2022, 2022, 5469220. [CrossRef]
- Hirst, J. Mitochondrial complex I. Annu Rev Biochem 2013, 82, 551–575. [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 2013, 1827, 1320–1331. [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol 2023, 67, 102926. [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol 2011, 33, 538–547. [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol Sci 2018, 39, 307–325. [CrossRef]
- Gouasmi, R.; Ferraro-Peyret, C.; Nancey, S.; Coste, I.; Renno, T.; Chaveroux, C.; Aznar, N.; Ansieau, S. The Kynurenine Pathway and Cancer: Why Keep It Simple When You Can Make It Complicated. Cancers (Basel) 2022, 14. [CrossRef]
- Joisten, N.; Ruas, J.L.; Braidy, N.; Guillemin, G.J.; Zimmer, P. The kynurenine pathway in chronic diseases: a compensatory mechanism or a driving force? Trends Mol Med 2021, 27, 946–954. [CrossRef]
- Tappenden, D.M.; Lynn, S.G.; Crawford, R.B.; Lee, K.; Vengellur, A.; Kaminski, N.E.; Thomas, R.S.; LaPres, J.J. The aryl hydrocarbon receptor interacts with ATP5α1, a subunit of the ATP synthase complex, and modulates mitochondrial function. Toxicol Appl Pharmacol 2011, 254, 299–310. [CrossRef]
- Brinkmann, V.; Ale-Agha, N.; Haendeler, J.; Ventura, N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Front Physiol 2019, 10, 1561. [CrossRef]
- Alkondon, M.; Pereira, E.F.; Yu, P.; Arruda, E.Z.; Almeida, L.E.; Guidetti, P.; Fawcett, W.P.; Sapko, M.T.; Randall, W.R.; Schwarcz, R.; et al. Targeted deletion of the kynurenine aminotransferase ii gene reveals a critical role of endogenous kynurenic acid in the regulation of synaptic transmission via alpha7 nicotinic receptors in the hippocampus. J Neurosci 2004, 24, 4635–4648. [CrossRef]
- Mugayar, A.A.; da Silva Guimarães, G.; de Oliveira, P.H.T.; Miranda, R.L.; Dos Santos, A.A. Apoptosis in the neuroprotective effect of α7 nicotinic receptor in neurodegenerative models. J Neurosci Res 2023, 101, 1795–1802. [CrossRef]
- Braidy, N.; Grant, R.; Brew, B.J.; Adams, S.; Jayasena, T.; Guillemin, G.J. Effects of Kynurenine Pathway Metabolites on Intracellular NAD Synthesis and Cell Death in Human Primary Astrocytes and Neurons. Int J Tryptophan Res 2009, 2, 61–69. [CrossRef]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [CrossRef]
- Kory, N.; Uit de Bos, J.; van der Rijt, S.; Jankovic, N.; Güra, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci Adv 2020, 6. [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gürtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat Commun 2020, 11, 6145. [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 2020, 11, 102. [CrossRef]
- Ronchi, J.A.; Francisco, A.; Passos, L.A.; Figueira, T.R.; Castilho, R.F. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. J Biol Chem 2016, 291, 20173–20187. [CrossRef]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic Biol Med 2013, 63, 446–456. [CrossRef]
- Fisher-Wellman, K.H.; Lin, C.T.; Ryan, T.E.; Reese, L.R.; Gilliam, L.A.; Cathey, B.L.; Lark, D.S.; Smith, C.D.; Muoio, D.M.; Neufer, P.D. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J 2015, 467, 271–280. [CrossRef]
- Simmen, F.A.; Alhallak, I.; Simmen, R.C.M. Malic enzyme 1 (ME1) in the biology of cancer: it is not just intermediary metabolism. J Mol Endocrinol 2020, 65, R77–R90. [CrossRef]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol 2021, 46, 102065. [CrossRef]
- Hernandez-Baixauli, J.; Abasolo, N.; Palacios-Jordan, H.; Foguet-Romero, E.; Suñol, D.; Galofré, M.; Caimari, A.; Baselga-Escudero, L.; Del Bas, J.M.; Mulero, M. Imbalances in TCA, Short Fatty Acids and One-Carbon Metabolisms as Important Features of Homeostatic Disruption Evidenced by a Multi-Omics Integrative Approach of LPS-Induced Chronic Inflammation in Male Wistar Rats. Int J Mol Sci 2022, 23. [CrossRef]
- Hart, M.L.; Quon, E.; Vigil, A.B.G.; Engstrom, I.A.; Newsom, O.J.; Davidsen, K.; Hoellerbauer, P.; Carlisle, S.M.; Sullivan, L.B. Mitochondrial redox adaptations enable alternative aspartate synthesis in SDH-deficient cells. Elife 2023, 12. [CrossRef]
- Lussey-Lepoutre, C.; Hollinshead, K.E.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun 2015, 6, 8784. [CrossRef]
- Esteban-Amo, M.J.; Jiménez-Cuadrado, P.; Serrano-Lorenzo, P.; de la Fuente, M.; Simarro, M. Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme. Biomedicines 2024, 12. [CrossRef]
- Wan, J.; Cheng, C.; Hu, J.; Huang, H.; Han, Q.; Jie, Z.; Zou, Q.; Shi, J.H.; Yu, X. De novo NAD(+) synthesis contributes to CD8(+) T cell metabolic fitness and antitumor function. Cell Rep 2023, 42, 113518. [CrossRef]
- Sadeghdoust, M.; Das, A.; Kaushik, D.K. Fueling neurodegeneration: metabolic insights into microglia functions. J Neuroinflammation 2024, 21, 300. [CrossRef]
- McReynolds, M.R.; Wang, W.; Holleran, L.M.; Hanna-Rose, W. Uridine monophosphate synthetase enables eukaryotic de novo NAD(+) biosynthesis from quinolinic acid. J Biol Chem 2017, 292, 11147–11153. [CrossRef]
- Ding, Y.; Li, X.; Horsman, G.P.; Li, P.; Wang, M.; Li, J.; Zhang, Z.; Liu, W.; Wu, B.; Tao, Y.; et al. Construction of an Alternative NAD(+) De Novo Biosynthesis Pathway. Adv Sci (Weinh) 2021, 8, 2004632. [CrossRef]
- Zhu, S.; Zhang, R.; Yao, L.; Lin, Z.; Li, Y.; Li, S.; Wu, L. De novo NAD(+) synthesis is ineffective for NAD(+) supply in axenically cultured Caenorhabditis elegans. Commun Biol 2025, 8, 545. [CrossRef]
- Trepci, A.; Imbeault, S.; Wyckelsma, V.L.; Westerblad, H.; Hermansson, S.; Andersson, D.C.; Piehl, F.; Venckunas, T.; Brazaitis, M.; Kamandulis, S.; et al. Quantification of Plasma Kynurenine Metabolites Following One Bout of Sprint Interval Exercise. Int J Tryptophan Res 2020, 13, 1178646920978241. [CrossRef]
- Wu, P.; Wang, W.; Huang, C.; Sun, L.; Wu, X.; Xu, L.; Xiao, P. A rapid and reliable targeted LC-MS/MS method for quantitative analysis of the Tryptophan-NAD metabolic network disturbances in tissues and blood of sleep deprivation mice. Anal Chim Acta 2024, 1328, 343125. [CrossRef]
- Baixauli, F.; Piletic, K.; Puleston, D.J.; Villa, M.; Field, C.S.; Flachsmann, L.J.; Quintana, A.; Rana, N.; Edwards-Hicks, J.; Matsushita, M.; et al. An LKB1-mitochondria axis controls T(H)17 effector function. Nature 2022, 610, 555–561. [CrossRef]
- Muri, J.; Heer, S.; Matsushita, M.; Pohlmeier, L.; Tortola, L.; Fuhrer, T.; Conrad, M.; Zamboni, N.; Kisielow, J.; Kopf, M. The thioredoxin-1 system is essential for fueling DNA synthesis during T-cell metabolic reprogramming and proliferation. Nat Commun 2018, 9, 1851. [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal 2013, 18, 1165–1207. [CrossRef]
- Nunes, Y.C.; Mendes, N.M.; Pereira de Lima, E.; Chehadi, A.C.; Lamas, C.B.; Haber, J.F.S.; Dos Santos Bueno, M.; Araújo, A.C.; Catharin, V.C.S.; Detregiachi, C.R.P.; et al. Curcumin: A Golden Approach to Healthy Aging: A Systematic Review of the Evidence. Nutrients 2024, 16. [CrossRef]
- de Lima, E.P.; Laurindo, L.F.; Catharin, V.C.S.; Direito, R.; Tanaka, M.; Jasmin Santos German, I.; Lamas, C.B.; Guiguer, E.L.; Araújo, A.C.; Fiorini, A.M.R.; et al. Polyphenols, Alkaloids, and Terpenoids Against Neurodegeneration: Evaluating the Neuroprotective Effects of Phytocompounds Through a Comprehensive Review of the Current Evidence. Metabolites 2025, 15. [CrossRef]
- Martos, D.; Lőrinczi, B.; Szatmári, I.; Vécsei, L.; Tanaka, M. Decoupling Behavioral Domains via Kynurenic Acid Analog Optimization: Implications for Schizophrenia and Parkinson’s Disease Therapeutics. Cells 2025, 14. [CrossRef]
- Tanaka, M. From Monoamines to Systems Psychiatry: Rewiring Depression Science and Care (1960s–2025). Biomedicines 2026, 14, 35. [CrossRef]
- Chehadi, A.C.P.d.L., E.; Detregiachi, C.R.P.; Santos de Argollo Haber, R.; Catharin, V.M.C.S.; Fornari Laurindo, L.; Engracia Valenti, V.; Machado Galhardi, C.; Tanaka, M.; Maria Barbalho, S. Harnessing Dietary Tryptophan: Bridging the Gap Between Neurobiology and Psychiatry in Depression Management. International Journal of Molecular Sciences 2026, 27, 465. [CrossRef]
- Seok, S.H.; Ma, Z.X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.A.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J Biol Chem 2018, 293, 1994–2005. [CrossRef]
- Palzkill, V.R.; Thome, T.; Murillo, A.L.; Khattri, R.B.; Ryan, T.E. Increasing plasma L-kynurenine impairs mitochondrial oxidative phosphorylation prior to the development of atrophy in murine skeletal muscle: A pilot study. Front Physiol 2022, 13, 992413. [CrossRef]
- Galla, Z.; Rajda, C.; Rácz, G.; Grecsó, N.; Baráth, Á.; Vécsei, L.; Bereczki, C.; Monostori, P. Simultaneous determination of 30 neurologically and metabolically important molecules: A sensitive and selective way to measure tyrosine and tryptophan pathway metabolites and other biomarkers in human serum and cerebrospinal fluid. Journal of Chromatography A 2021, 1635, 461775.
- Galla, Z.; Rácz, G.; Grecsó, N.; Baráth, Á.; Kósa, M.; Bereczki, C.; Monostori, P. Improved LC-MS/MS method for the determination of 42 neurologically and metabolically important molecules in urine. J Chromatogr B Analyt Technol Biomed Life Sci 2021, 1179, 122846. [CrossRef]
- Al Kadhi, O.; Melchini, A.; Mithen, R.; Saha, S. Development of a LC-MS/MS Method for the Simultaneous Detection of Tricarboxylic Acid Cycle Intermediates in a Range of Biological Matrices. J Anal Methods Chem 2017, 2017, 5391832. [CrossRef]
- Le, A.; Mak, J.; Cowan, T.M. Metabolic profiling by reversed-phase/ion-exchange mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2020, 1143, 122072. [CrossRef]
- Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O. Optimization of Mobile Phase Modifiers for Fast LC-MS-Based Untargeted Metabolomics and Lipidomics. Int J Mol Sci 2023, 24. [CrossRef]
- Carlsson, H.; Vaivade, A.; Emami Khoonsari, P.; Burman, J.; Kultima, K. Evaluation of polarity switching for untargeted lipidomics using liquid chromatography coupled to high resolution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2022, 1195, 123200. [CrossRef]
- Ren, J.L.; Zhang, A.H.; Kong, L.; Wang, X.J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv 2018, 8, 22335–22350. [CrossRef]
- Zhang, L.; Zheng, J.; Johnson, M.; Mandal, R.; Cruz, M.; Martínez-Huélamo, M.; Andres-Lacueva, C.; Wishart, D.S. A Comprehensive LC-MS Metabolomics Assay for Quantitative Analysis of Serum and Plasma. Metabolites 2024, 14. [CrossRef]
- Lee, H.J.; Kremer, D.M.; Sajjakulnukit, P.; Zhang, L.; Lyssiotis, C.A. A large-scale analysis of targeted metabolomics data from heterogeneous biological samples provides insights into metabolite dynamics. Metabolomics 2019, 15, 103. [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [CrossRef]
- Jenkins, R.; Duggan, J.X.; Aubry, A.F.; Zeng, J.; Lee, J.W.; Cojocaru, L.; Dufield, D.; Garofolo, F.; Kaur, S.; Schultz, G.A.; et al. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. Aaps j 2015, 17, 1–16. [CrossRef]
- .
- Thomas, S.N.; French, D.; Jannetto, P.J.; Rappold, B.A.; Clarke, W.A. Liquid chromatography-tandem mass spectrometry for clinical diagnostics. Nat Rev Methods Primers 2022, 2, 96. [CrossRef]
- Holbrook, J.H.; Kemper, G.E.; Hummon, A.B. Quantitative mass spectrometry imaging: therapeutics & biomolecules. Chem Commun (Camb) 2024, 60, 2137–2151. [CrossRef]
- Thorsteinsdóttir, U.A.; Thorsteinsdóttir, M. Design of experiments for development and optimization of a liquid chromatography coupled to tandem mass spectrometry bioanalytical assay. J Mass Spectrom 2021, 56, e4727. [CrossRef]
- Pocivavsek, A.; Schwarcz, R.; Erhardt, S. Neuroactive Kynurenines as Pharmacological Targets: New Experimental Tools and Exciting Therapeutic Opportunities. Pharmacol Rev 2024, 76, 978–1008. [CrossRef]
- Kurhaluk, N. Tricarboxylic Acid Cycle Intermediates and Individual Ageing. Biomolecules 2024, 14. [CrossRef]
- Tang, Y.; Wang, S.; Zhang, W.; Yang, R.; Yu, X.; Wang, X.; Mu, H.; Li, H.; Ji, F.; Chen, W. A single-run, rapid polarity switching method for simultaneous quantification of cardiovascular disease-related metabolites using liquid chromatography–tandem mass spectrometry. International Journal of Mass Spectrometry 2021, 461, 116500.
- Nakatani, K.; Izumi, Y.; Takahashi, M.; Bamba, T. Unified-Hydrophilic-Interaction/Anion-Exchange Liquid Chromatography Mass Spectrometry (Unified-HILIC/AEX/MS): A Single-Run Method for Comprehensive and Simultaneous Analysis of Polar Metabolome. Anal Chem 2022, 94, 16877–16886. [CrossRef]
- Quan, S.; Zhao, S.; Li, L. Study of Metabolite Detectability in Simultaneous Profiling of Amine/Phenol and Hydroxyl Submetabolomes by Analyzing a Mixture of Two Separately Dansyl-Labeled Samples. Metabolites 2025, 15. [CrossRef]
- Huan, T.; Xian, J.W.; Leung, W.N.; Li, L.; Chan, C.W. Cerebrospinal Fluid Metabolomics After Natural Product Treatment in an Experimental Model of Cerebral Ischemia. Omics 2016, 20, 670–680. [CrossRef]
- Yan, J.; Kuzhiumparambil, U.; Bandodkar, S.; Dale, R.C.; Fu, S. Cerebrospinal fluid metabolomics: detection of neuroinflammation in human central nervous system disease. Clin Transl Immunology 2021, 10, e1318. [CrossRef]
- Carlsson, H.; Abujrais, S.; Herman, S.; Khoonsari, P.E.; Åkerfeldt, T.; Svenningsson, A.; Burman, J.; Kultima, K. Targeted metabolomics of CSF in healthy individuals and patients with secondary progressive multiple sclerosis using high-resolution mass spectrometry. Metabolomics 2020, 16, 26. [CrossRef]
- Saoi, M.; Britz-McKibbin, P. New Advances in Tissue Metabolomics: A Review. Metabolites 2021, 11. [CrossRef]
- Unsihuay, D.; Phipps, W.S.; Paulovich, A.G.; Chapman, J.R.; Ducret, A.; Eberlin, L.S.; Spraggins, J.M.; Goodwin, R.J.A. Tissue Mass Spectrometry: How Solid Is Our Future? Clin Chem 2023, 69, 676–683. [CrossRef]
- Harstad, E.; Andaya, R.; Couch, J.; Ding, X.; Liang, X.; Liederer, B.M.; Messick, K.; Nguyen, T.; Schweiger, M.; Tarrant, J.; et al. Balancing Blood Sample Volume with 3Rs: Implementation and Best Practices for Small Molecule Toxicokinetic Assessments in Rats. Ilar j 2016, 57, 157–165. [CrossRef]
- Jeppesen, M.J.; Powers, R. Multiplatform untargeted metabolomics. Magn Reson Chem 2023, 61, 628–653. [CrossRef]
- Patel, V.D.; Shamsi, S.A.; Miller, A.; Liu, A.; Powell, M. Simultaneous separation and detection of nine kynurenine pathway metabolites by reversed-phase liquid chromatography-mass spectrometry: Quantitation of inflammation in human cerebrospinal fluid and plasma. Anal Chim Acta 2023, 1278, 341659. [CrossRef]
- Abusoglu, S.; Eryavuz Onmaz, D.; Abusoglu, G.; Humeyra Yerlikaya, F.; Unlu, A. Measurement of kynurenine pathway metabolites by tandem mass spectrometry. J Mass Spectrom Adv Clin Lab 2023, 28, 114–121. [CrossRef]
- Rathod, R.; Gajera, B.; Nazir, K.; Wallenius, J.; Velagapudi, V. Simultaneous Measurement of Tricarboxylic Acid Cycle Intermediates in Different Biological Matrices Using Liquid Chromatography-Tandem Mass Spectrometry; Quantitation and Comparison of TCA Cycle Intermediates in Human Serum, Plasma, Kasumi-1 Cell and Murine Liver Tissue. Metabolites 2020, 10. [CrossRef]
- Ivanisevic, J.; Want, E.J. From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites 2019, 9. [CrossRef]
- Wei, R.; Li, G.; Seymour, A.B. Multiplexed, quantitative, and targeted metabolite profiling by LC-MS/MRM. Methods Mol Biol 2014, 1198, 171–199. [CrossRef]
- Pukoli, D.; Vécsei, L. Kynurenines and Mitochondrial Disturbances in Multiple Sclerosis. Int J Mol Sci 2025, 26. [CrossRef]
- Gomez-Gomez, A.; Aguilera, P.; Langohr, K.; Casals, G.; Pavon, C.; Marcos, J.; To-Figueras, J.; Pozo, O.J. Evaluation of Metabolic Changes in Acute Intermittent Porphyria Patients by Targeted Metabolomics. Int J Mol Sci 2022, 23. [CrossRef]
- Hestad, K.; Alexander, J.; Rootwelt, H.; Aaseth, J.O. The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. Biomolecules 2022, 12. [CrossRef]
- Myint, A.M.; Halaris, A. Imbalances in Kynurenines as Potential Biomarkers in the Diagnosis and Treatment of Psychiatric Disorders. Front Psychiatry 2022, 13, 913303. [CrossRef]
- Boyd, A.; Boccara, F.; Meynard, J.L.; Ichou, F.; Bastard, J.P.; Fellahi, S.; Samri, A.; Sauce, D.; Haddour, N.; Autran, B.; et al. Serum Tryptophan-Derived Quinolinate and Indole-3-Acetate Are Associated With Carotid Intima-Media Thickness and its Evolution in HIV-Infected Treated Adults. Open Forum Infect Dis 2019, 6, ofz516. [CrossRef]
- Zhang, Y.M.; Qi, Y.B.; Gao, Y.N.; Chen, W.G.; Zhou, T.; Zang, Y.; Li, J. Astrocyte metabolism and signaling pathways in the CNS. Front Neurosci 2023, 17, 1217451. [CrossRef]
- Fuertig, R.; Ceci, A.; Camus, S.M.; Bezard, E.; Luippold, A.H.; Hengerer, B. LC-MS/MS-based quantification of kynurenine metabolites, tryptophan, monoamines and neopterin in plasma, cerebrospinal fluid and brain. Bioanalysis 2016, 8, 1903–1917. [CrossRef]
- Walvekar, A.; Rashida, Z.; Maddali, H.; Laxman, S. A versatile LC-MS/MS approach for comprehensive, quantitative analysis of central metabolic pathways. Wellcome Open Res 2018, 3, 122. [CrossRef]
- Iqbal, K.; Intemann, T.; Börnhorst, C.; Zhang, J.; Aleksandrova, K. Approaches for harmonization of biomarker data from multiple studies: a narrative methodological review. AJE Advances: Research in Epidemiology 2025, 1, uuaf005.
- Ivanovová, E.; Piskláková, B.; Dobešová, D.; Janečková, H.; Foltenová, H.; Kvasnička, A.; Prídavok, M.; Bouchalová, K.; de Sousa, J.; Friedecký, D. Wide metabolite coverage LC-MS/MS assay for the diagnosis of inherited metabolic disorders in urine. Talanta 2024, 271, 125699. [CrossRef]
- Sloan, A.; Cheng, C.; Rosner, B.; Ziegler, R.G.; Smith-Warner, S.A.; Wang, M. A repeated measures approach to pooled and calibrated biomarker data. Biometrics 2023, 79, 1485–1495. [CrossRef]
- Rhee, E.P.; Waikar, S.S.; Rebholz, C.M.; Zheng, Z.; Perichon, R.; Clish, C.B.; Evans, A.M.; Avila, J.; Denburg, M.R.; Anderson, A.H.; et al. Variability of Two Metabolomic Platforms in CKD. Clin J Am Soc Nephrol 2019, 14, 40–48. [CrossRef]
- Lippa, K.A.; Aristizabal-Henao, J.J.; Beger, R.D.; Bowden, J.A.; Broeckling, C.; Beecher, C.; Clay Davis, W.; Dunn, W.B.; Flores, R.; Goodacre, R.; et al. Reference materials for MS-based untargeted metabolomics and lipidomics: a review by the metabolomics quality assurance and quality control consortium (mQACC). Metabolomics 2022, 18, 24. [CrossRef]
- Rehder, C.; Bean, L.J.H.; Bick, D.; Chao, E.; Chung, W.; Das, S.; O’Daniel, J.; Rehm, H.; Shashi, V.; Vincent, L.M. Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021, 23, 1399–1415. [CrossRef]
- Broeckling, C.D.; Beger, R.D.; Cheng, L.L.; Cumeras, R.; Cuthbertson, D.J.; Dasari, S.; Davis, W.C.; Dunn, W.B.; Evans, A.M.; Fernández-Ochoa, A.; et al. Current Practices in LC-MS Untargeted Metabolomics: A Scoping Review on the Use of Pooled Quality Control Samples. Anal Chem 2023, 95, 18645–18654. [CrossRef]
- Mattsson-Carlgren, N.; Palmqvist, S.; Blennow, K.; Hansson, O. Publisher Correction: Increasing the reproducibility of fluid biomarker studies in neurodegenerative studies. Nat Commun 2021, 12, 196. [CrossRef]
- Chau, C.H.; Rixe, O.; McLeod, H.; Figg, W.D. Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res 2008, 14, 5967–5976. [CrossRef]
- Saito, K.; Goda, R.; Arai, K.; Asahina, K.; Kawabata, M.; Uchiyama, H.; Andou, T.; Shimizu, H.; Takahara, K.; Kakehi, M.; et al. Interlaboratory evaluation of LC-MS-based biomarker assays. Bioanalysis 2024, 16, 389–402. [CrossRef]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol 2009, 27, 633–641. [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal Chem 2019, 91, 14407–14416. [CrossRef]
- Fernández-Metzler, C.; Ackermann, B.; Garofolo, F.; Arnold, M.E.; DeSilva, B.; Gu, H.; Laterza, O.; Mao, Y.; Rose, M.; Vazvaei-Smith, F.; et al. Biomarker Assay Validation by Mass Spectrometry. Aaps j 2022, 24, 66. [CrossRef]
- Brunius, C.; Shi, L.; Landberg, R. Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 2016, 12, 173. [CrossRef]
- Wehrens, R.; Hageman, J.A.; van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.; Lommen, A.; van Eekelen, H.D.; Hall, R.D.; et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 2016, 12, 88. [CrossRef]
- Malinka, F.; Zareie, A.; Prochazka, J.; Sedlacek, R.; Novosadova, V. Batch alignment via retention orders for preprocessing large-scale multi-batch LC-MS experiments. Bioinformatics 2022, 38, 3759–3767. [CrossRef]
- Yao, Y.; Zhang, H.; Tu, L.; Yu, T.; Chen, B.; Huang, P.; Hu, Y.; Luan, T. Normalization Approach by a Reference Material to Improve LC-MS-Based Metabolomic Data Comparability of Multibatch Samples. Anal Chem 2023, 95, 1309–1317. [CrossRef]
- Siegel, D.; Permentier, H.; Reijngoud, D.J.; Bischoff, R. Chemical and technical challenges in the analysis of central carbon metabolites by liquid-chromatography mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2014, 966, 21–33. [CrossRef]
- Jankech, T.; Gerhardtova, I.; Majerova, P.; Piestansky, J.; Jampilek, J.; Kovac, A. Derivatization of carboxylic groups prior to their LC analysis - A review. Anal Chim Acta 2024, 1300, 342435. [CrossRef]
- Lu, W.; Clasquin, M.F.; Melamud, E.; Amador-Noguez, D.; Caudy, A.A.; Rabinowitz, J.D. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem 2010, 82, 3212–3221. [CrossRef]
- Vazquez, S.; Truscott, R.J.; O’Hair, R.A.; Weimann, A.; Sheil, M.M. A study of kynurenine fragmentation using electrospray tandem mass spectrometry. J Am Soc Mass Spectrom 2001, 12, 786–794. [CrossRef]
- Kiontke, A.; Oliveira-Birkmeier, A.; Opitz, A.; Birkemeyer, C. Electrospray Ionization Efficiency Is Dependent on Different Molecular Descriptors with Respect to Solvent pH and Instrumental Configuration. PLoS One 2016, 11, e0167502. [CrossRef]
- Steckel, A.; Schlosser, G. An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation. Molecules 2019, 24. [CrossRef]
- Liigand, J.; Laaniste, A.; Kruve, A. pH Effects on Electrospray Ionization Efficiency. J Am Soc Mass Spectrom 2017, 28, 461–469. [CrossRef]
- Hermans, J.; Ongay, S.; Markov, V.; Bischoff, R. Physicochemical Parameters Affecting the Electrospray Ionization Efficiency of Amino Acids after Acylation. Anal Chem 2017, 89, 9159–9166. [CrossRef]
- Ngere, J.B.; Ebrahimi, K.H.; Williams, R.; Pires, E.; Walsby-Tickle, J.; McCullagh, J.S.O. Ion-Exchange Chromatography Coupled to Mass Spectrometry in Life Science, Environmental, and Medical Research. Anal Chem 2023, 95, 152–166. [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [CrossRef]
- Wang, G.; Kakumanu, R.; Amer, B. Reversed phase LC-MS analysis of organic acids involved in the tricarboxylic acid cycle. 2025.
- Ovbude, S.T.; Sharmeen, S.; Kyei, I.; Olupathage, H.; Jones, J.; Bell, R.J.; Powers, R.; Hage, D.S. Applications of chromatographic methods in metabolomics: A review. J Chromatogr B Analyt Technol Biomed Life Sci 2024, 1239, 124124. [CrossRef]
- Keefer, J.F.; Schuster, S.M. Separation of citric acid cycle intermediates by high-performance liquid chromatography with ion pairing. J Chromatogr 1986, 383, 297–305. [CrossRef]
- Tang, D.Q.; Zou, L.; Yin, X.X.; Ong, C.N. HILIC-MS for metabolomics: An attractive and complementary approach to RPLC-MS. Mass spectrometry reviews 2016, 35, 574–600.
- Schwaiger, M.; Schoeny, H.; El Abiead, Y.; Hermann, G.; Rampler, E.; Koellensperger, G. Merging metabolomics and lipidomics into one analytical run. Analyst 2018, 144, 220–229. [CrossRef]
- Hiefner, J.; Rische, J.; Bunders, M.J.; Worthmann, A. A liquid chromatography-tandem mass spectrometry based method for the quantification of adenosine nucleotides and NAD precursors and products in various biological samples. Front Immunol 2023, 14, 1250762. [CrossRef]
- Meinitzer, A.; Tomaschitz, A.; Pilz, S.; Truber, M.; Zechner, G.; Gaksch, M.; Prietl, B.; Treiber, G.; Schwarz, M.; Baranyi, A. Development of a liquid chromatography-mass spectrometry method for the determination of the neurotoxic quinolinic acid in human serum. Clin Chim Acta 2014, 436, 268–272. [CrossRef]
- Tömösi, F.; Kecskeméti, G.; Cseh, E.K.; Szabó, E.; Rajda, C.; Kormány, R.; Szabó, Z.; Vécsei, L.; Janáky, T. A validated UHPLC-MS method for tryptophan metabolites: Application in the diagnosis of multiple sclerosis. J Pharm Biomed Anal 2020, 185, 113246. [CrossRef]
- Brookhart, A.; Arora, M.; McCullagh, M.; Wilson, I.D.; Plumb, R.S.; Vissers, J.P.; Tanna, N. Understanding mobile phase buffer composition and chemical structure effects on electrospray ionization mass spectrometry response. J Chromatogr A 2023, 1696, 463966. [CrossRef]
- Nshanian, M.; Lakshmanan, R.; Chen, H.; Ogorzalek Loo, R.R.; Loo, J.A. Enhancing Sensitivity of Liquid Chromatography-Mass Spectrometry of Peptides and Proteins Using Supercharging Agents. Int J Mass Spectrom 2018, 427, 157–164. [CrossRef]
- Walter, T.H.; Blaze, M.T.M.; Boissel, C. Electrospray ionization mass spectrometry ion suppression/enhancement caused by column bleed for three mixed-mode reversed-phase/anion-exchange high-performance liquid chromatography columns. Rapid Commun Mass Spectrom 2021, 35, e9098. [CrossRef]
- Sterling, H.J.; Batchelor, J.D.; Wemmer, D.E.; Williams, E.R. Effects of buffer loading for electrospray ionization mass spectrometry of a noncovalent protein complex that requires high concentrations of essential salts. J Am Soc Mass Spectrom 2010, 21, 1045–1049. [CrossRef]
- Zheng, J.J.; Lynch, E.D.; Unger, S.E. Comparison of SPE and fast LC to eliminate mass spectrometric matrix effects from microsomal incubation products. J Pharm Biomed Anal 2002, 28, 279–285. [CrossRef]
- Rakusanova, S.; Cajka, T. Tips and tricks for LC–MS-based metabolomics and lipidomics analysis. TrAC Trends in Analytical Chemistry 2024, 180, 117940.
- Silvester, S. Mobile phase pH and organic modifier in reversed-phase LC-ESI-MS bioanalytical methods: assessment of sensitivity, chromatography and correlation of retention time with in silico logD predictions. Bioanalysis 2013, 5, 2753–2770. [CrossRef]
- Liigand, J.; de Vries, R.; Cuyckens, F. Optimization of flow splitting and make-up flow conditions in liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom 2019, 33, 314–322. [CrossRef]
- Heinisch, S.; Rocca, J. Effect of mobile phase composition, pH and buffer type on the retention of ionizable compounds in reversed-phase liquid chromatography: application to method development. Journal of Chromatography A 2004, 1048, 183–193.
- Liigand, P.; Kaupmees, K.; Haav, K.; Liigand, J.; Leito, I.; Girod, M.; Antoine, R.; Kruve, A. Think Negative: Finding the Best Electrospray Ionization/MS Mode for Your Analyte. Anal Chem 2017, 89, 5665–5668. [CrossRef]
- Henriksen, T.; Juhler, R.K.; Svensmark, B.; Cech, N.B. The relative influences of acidity and polarity on responsiveness of small organic molecules to analysis with negative ion electrospray ionization mass spectrometry (ESI-MS). J Am Soc Mass Spectrom 2005, 16, 446–455. [CrossRef]
- Venter, P. The Effects of Solvation Enthalpy, Surface Tension, and Conductivity of Common Additives on Positive Electrospray Ionization in Selected Pharmaceuticals. Molecules 2025, 30. [CrossRef]
- Han, J.; Gagnon, S.; Eckle, T.; Borchers, C.H. Metabolomic analysis of key central carbon metabolism carboxylic acids as their 3-nitrophenylhydrazones by UPLC/ESI-MS. Electrophoresis 2013, 34, 2891–2900. [CrossRef]
- Huang, Y.; Louie, A.; Yang, Q.; Massenkoff, N.; Xu, C.; Hunt, P.W.; Gee, W. A simple LC-MS/MS method for determination of kynurenine and tryptophan concentrations in human plasma from HIV-infected patients. Bioanalysis 2013, 5, 1397–1407. [CrossRef]
- Breitkopf, S.B.; Ricoult, S.J.H.; Yuan, M.; Xu, Y.; Peake, D.A.; Manning, B.D.; Asara, J.M. A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 2017, 13. [CrossRef]
- Cai, R.; Mikkilä, J.; Bengs, A.; Koirala, M.; Mikkilä, J.; Holm, S.; Juuti, P.; Meder, M.; Partovi, F.; Shcherbinin, A.; et al. Extending the Range of Detectable Trace Species with the Fast Polarity Switching of Chemical Ionization Orbitrap Mass Spectrometry. Anal Chem 2024, 96, 8604–8612. [CrossRef]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 2012, 7, 872–881. [CrossRef]
- Kluger, B.; Bueschl, C.; Neumann, N.; Stückler, R.; Doppler, M.; Chassy, A.W.; Waterhouse, A.L.; Rechthaler, J.; Kampleitner, N.; Thallinger, G.G.; et al. Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching LC-ESI-HRMS. Anal Chem 2014, 86, 11533–11537. [CrossRef]
- Zherebker, A.; Kostyukevich, Y.; Kononikhin, A.; Roznyatovsky, V.A.; Popov, I.; Grishin, Y.K.; Perminova, I.V.; Nikolaev, E. High desolvation temperature facilitates the ESI-source H/D exchange at non-labile sites of hydroxybenzoic acids and aromatic amino acids. Analyst 2016, 141, 2426–2434. [CrossRef]
- Paíga, P.; Silva, L.M.; Delerue-Matos, C. Optimization of the Ion Source-Mass Spectrometry Parameters in Non-Steroidal Anti-Inflammatory and Analgesic Pharmaceuticals Analysis by a Design of Experiments Approach. J Am Soc Mass Spectrom 2016, 27, 1703–1714. [CrossRef]
- Pedro, L.; Van Voorhis, W.C.; Quinn, R.J. Optimization of Electrospray Ionization by Statistical Design of Experiments and Response Surface Methodology: Protein-Ligand Equilibrium Dissociation Constant Determinations. J Am Soc Mass Spectrom 2016, 27, 1520–1530. [CrossRef]
- Prothmann, J.; Molins-Delgado, D.; Braune, A.; Sandahl, M.; Turner, C.; Spégel, P. Examining functional group-dependent effects on the ionization of lignin monomers using supercritical fluid chromatography/electrospray ionization mass spectrometry. Anal Bioanal Chem 2024, 416, 4007–4014. [CrossRef]
- Kruve, A.; Kaupmees, K. Adduct Formation in ESI/MS by Mobile Phase Additives. J Am Soc Mass Spectrom 2017, 28, 887–894. [CrossRef]
- Flick, T.G.; Merenbloom, S.I.; Williams, E.R. Anion effects on sodium ion and acid molecule adduction to protein ions in electrospray ionization mass spectrometry. J Am Soc Mass Spectrom 2011, 22, 1968–1977. [CrossRef]
- Carnevale Neto, F.; Pascua, V.; Raftery, D. Formation of sodium cluster ions complicates liquid chromatography-mass spectrometry metabolomics analyses. Rapid Commun Mass Spectrom 2021, 35, e9175. [CrossRef]
- Stricker, T.; Bonner, R.; Lisacek, F.; Hopfgartner, G. Adduct annotation in liquid chromatography/high-resolution mass spectrometry to enhance compound identification. Anal Bioanal Chem 2021, 413, 503–517. [CrossRef]
- Birdsall, R.E.; Gilar, M.; Shion, H.; Yu, Y.Q.; Chen, W. Reduction of metal adducts in oligonucleotide mass spectra in ion-pair reversed-phase chromatography/mass spectrometry analysis. Rapid Commun Mass Spectrom 2016, 30, 1667–1679. [CrossRef]
- Erngren, I.; Nestor, M.; Pettersson, C.; Hedeland, M. Improved Sensitivity in Hydrophilic Interaction Liquid Chromatography-Electrospray-Mass Spectrometry after Removal of Sodium and Potassium Ions from Biological Samples. Metabolites 2021, 11. [CrossRef]
- Nash, W.J.; Ngere, J.B.; Najdekr, L.; Dunn, W.B. Characterization of Electrospray Ionization Complexity in Untargeted Metabolomic Studies. Anal Chem 2024, 96, 10935–10942. [CrossRef]
- Erngren, I.; Haglöf, J.; Engskog, M.K.R.; Nestor, M.; Hedeland, M.; Arvidsson, T.; Pettersson, C. Adduct formation in electrospray ionisation-mass spectrometry with hydrophilic interaction liquid chromatography is strongly affected by the inorganic ion concentration of the samples. J Chromatogr A 2019, 1600, 174–182. [CrossRef]
- Stokvis, E.; Rosing, H.; Beijnen, J.H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun Mass Spectrom 2005, 19, 401–407. [CrossRef]
- Kapoore, R.V.; Vaidyanathan, S. Towards quantitative mass spectrometry-based metabolomics in microbial and mammalian systems. Philos Trans A Math Phys Eng Sci 2016, 374. [CrossRef]
- Liigand, P.; Liigand, J.; Kaupmees, K.; Kruve, A. 30 Years of research on ESI/MS response: Trends, contradictions and applications. Anal Chim Acta 2021, 1152, 238117. [CrossRef]
- Chen, F.; Willenbockel, H.F.; Cordes, T. Mapping the Metabolic Niche of Citrate Metabolism and SLC13A5. Metabolites 2023, 13. [CrossRef]
- Dankel, S.N.; Kalleklev, T.L.; Tungland, S.L.; Stafsnes, M.H.; Bruheim, P.; Aloysius, T.A.; Lindquist, C.; Skorve, J.; Nygård, O.K.; Madsen, L.; et al. Changes in Plasma Pyruvate and TCA Cycle Metabolites upon Increased Hepatic Fatty Acid Oxidation and Ketogenesis in Male Wistar Rats. Int J Mol Sci 2023, 24. [CrossRef]
- Stone, T.W.; Darlington, L.G.; Badawy, A.A.; Williams, R.O. The Complex World of Kynurenic Acid: Reflections on Biological Issues and Therapeutic Strategy. Int J Mol Sci 2024, 25. [CrossRef]
- Tang, K.; Page, J.S.; Smith, R.D. Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry. J Am Soc Mass Spectrom 2004, 15, 1416–1423. [CrossRef]
- Bruin, M.A.C.; Rosing, H.; Lucas, L.; Wang, J.; Huitema, A.D.R.; Schinkel, A.H.; Beijnen, J.H. Development and validation of an LC-MS/MS method with a broad linear dynamic range for the quantification of tivozanib in human and mouse plasma, mouse tissue homogenates, and culture medium. J Chromatogr B Analyt Technol Biomed Life Sci 2019, 1125, 121723. [CrossRef]
- Liu, X.; Ser, Z.; Locasale, J.W. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem 2014, 86, 2175–2184. [CrossRef]
- Sands, C.J.; Gómez-Romero, M.; Correia, G.; Chekmeneva, E.; Camuzeaux, S.; Izzi-Engbeaya, C.; Dhillo, W.S.; Takats, Z.; Lewis, M.R. Representing the Metabolome with High Fidelity: Range and Response as Quality Control Factors in LC-MS-Based Global Profiling. Anal Chem 2021, 93, 1924–1933. [CrossRef]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin Chem 2003, 49, 1041–1044. [CrossRef]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom Rev 2001, 20, 362–387. [CrossRef]
- King, R.; Bonfiglio, R.; Fernandez-Metzler, C.; Miller-Stein, C.; Olah, T. Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom 2000, 11, 942–950. [CrossRef]
- Wu, Z.; Gao, W.; Phelps, M.A.; Wu, D.; Miller, D.D.; Dalton, J.T. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. Anal Chem 2004, 76, 839–847. [CrossRef]
- Cheng, W.L.; Markus, C.; Lim, C.Y.; Tan, R.Z.; Sethi, S.K.; Loh, T.P. Calibration Practices in Clinical Mass Spectrometry: Review and Recommendations. Ann Lab Med 2023, 43, 5–18. [CrossRef]
- Jeanne Dit Fouque, D.; Maroto, A.; Memboeuf, A. Internal Standard Quantification Using Tandem Mass Spectrometry of a Tryptic Peptide in the Presence of an Isobaric Interference. Anal Chem 2018, 90, 14126–14130. [CrossRef]
- Tkalec, Ž.; Koudelka, Š.; Klánová, J.; Price, E.J. Dual LC column characterization for mass spectrometry-based small molecule profiling of human plasma and serum. Anal Chim Acta 2025, 1356, 343942. [CrossRef]
- Wuli, W.; Tsai, S.T.; Chiou, T.W.; Harn, H.J. Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease. Int J Mol Sci 2020, 21. [CrossRef]
- Zhang, T.; Noll, S.E.; Peng, J.T.; Klair, A.; Tripka, A.; Stutzman, N.; Cheng, C.; Zare, R.N.; Dickinson, A.J. Chemical imaging reveals diverse functions of tricarboxylic acid metabolites in root growth and development. Nat Commun 2023, 14, 2567. [CrossRef]
- Vass, A.; Robles-Molina, J.; Pérez-Ortega, P.; Gilbert-López, B.; Dernovics, M.; Molina-Díaz, A.; García-Reyes, J.F. Study of different HILIC, mixed-mode, and other aqueous normal-phase approaches for the liquid chromatography/mass spectrometry-based determination of challenging polar pesticides. Anal Bioanal Chem 2016, 408, 4857–4869. [CrossRef]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)--a powerful separation technique. Anal Bioanal Chem 2012, 402, 231–247. [CrossRef]
- Kohler, I.; Verhoeven, M.; Haselberg, R.; Gargano, A.F. Hydrophilic interaction chromatography–mass spectrometry for metabolomics and proteomics: state-of-the-art and current trends. Microchemical Journal 2022, 175, 106986.
- Zhang, K.; Liu, X. Mixed-mode chromatography in pharmaceutical and biopharmaceutical applications. J Pharm Biomed Anal 2016, 128, 73–88. [CrossRef]
- Iwasaki, Y.; Sawada, T.; Hatayama, K.; Ohyagi, A.; Tsukuda, Y.; Namekawa, K.; Ito, R.; Saito, K.; Nakazawa, H. Separation technique for the determination of highly polar metabolites in biological samples. Metabolites 2012, 2, 496–515. [CrossRef]
- Chapel, S.; Rouvière, F.; Guillarme, D.; Heinisch, S. Reversed HILIC Gradient: A Powerful Strategy for On-Line Comprehensive 2D-LC. Molecules 2023, 28. [CrossRef]
- Grübner, M.; Dunkel, A.; Steiner, F.; Hofmann, T. Comparative evaluation of comprehensive offline 2D-LC strategies coupled to MS for untargeted metabolomic studies of human urine. Anal Bioanal Chem 2025. [CrossRef]
- Papatheocharidou, C.; Samanidou, V. Two-Dimensional High-Performance Liquid Chromatography as a Powerful Tool for Bioanalysis: The Paradigm of Antibiotics. Molecules 2023, 28. [CrossRef]
- Aly, A.A.; Górecki, T. Two-dimensional liquid chromatography with reversed phase in both dimensions: A review. J Chromatogr A 2024, 1721, 464824. [CrossRef]
- Lu, Y.; Qin, Q.; Pan, J.; Deng, S.; Wang, S.; Li, Q.; Cao, J. Advanced applications of two-dimensional liquid chromatography in quantitative analysis of natural products. J Chromatogr A 2025, 1743, 465662. [CrossRef]
- Ortmayr, K.; Hann, S.; Koellensperger, G. Complementing reversed-phase selectivity with porous graphitized carbon to increase the metabolome coverage in an on-line two-dimensional LC-MS setup for metabolomics. Analyst 2015, 140, 3465–3473. [CrossRef]
- Willemse, C.M.; Stander, M.A.; Tredoux, A.G.; de Villiers, A. Comprehensive two-dimensional liquid chromatographic analysis of anthocyanins. J Chromatogr A 2014, 1359, 189–201. [CrossRef]
- Nell, E.H.J.; Muller, M.; de Villiers, A. Advances in Online Comprehensive Two-Dimensional Liquid Chromatography Method Development. Annu Rev Anal Chem (Palo Alto Calif) 2025, 18, 359–381. [CrossRef]
- Pardon, M.; Reis, R.; de Witte, P.; Chapel, S.; Cabooter, D. Detailed comparison of in-house developed and commercially available heart-cutting and selective comprehensive two-dimensional liquid chromatography systems. J Chromatogr A 2024, 1713, 464565. [CrossRef]
- Malerod, H.; Lundanes, E.; Greibrokk, T. Recent advances in on-line multidimensional liquid chromatography. Analytical Methods 2010, 2, 110–122.
- Wernisch, S.; Pennathur, S. Evaluation of coverage, retention patterns, and selectivity of seven liquid chromatographic methods for metabolomics. Anal Bioanal Chem 2016, 408, 6079–6091. [CrossRef]
- Aarika, K.; Rajyalakshmi, R.; Nalla, L.V.; Gajula, S.N.R. From complexity to clarity: expanding metabolome coverage with innovative analytical strategies. Journal of Separation Science 2025, 48, e70099.
- Xu, K.; Berthiller, F.; Metzler-Zebeli, B.U.; Schwartz-Zimmermann, H.E. Development and Validation of Targeted Metabolomics Methods Using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for the Quantification of 235 Plasma Metabolites. Molecules 2025, 30. [CrossRef]
- Basov, N.V.; Rogachev, A.D.; Aleshkova, M.A.; Gaisler, E.V.; Sotnikova, Y.S.; Patrushev, Y.V.; Tolstikova, T.G.; Yarovaya, O.I.; Pokrovsky, A.G.; Salakhutdinov, N.F. Global LC-MS/MS targeted metabolomics using a combination of HILIC and RP LC separation modes on an organic monolithic column based on 1-vinyl-1,2,4-triazole. Talanta 2024, 267, 125168. [CrossRef]
- Lu, W.; Bennett, B.D.; Rabinowitz, J.D. Analytical strategies for LC-MS-based targeted metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci 2008, 871, 236–242. [CrossRef]
- Gamoh, K.; Saitoh, H.; Wada, H. Improved liquid chromatography/mass spectrometric analysis of low molecular weight carboxylic acids by ion exclusion separation with electrospray ionization. Rapid communications in mass spectrometry 2003, 17, 685–689.
- Zeki Ö, C.; Eylem, C.C.; Nemutlu, E. Optimization of GC-MS run time for untargeted metabolomics: Trade-offs between speed, coverage, and repeatability. J Pharm Biomed Anal 2025, 266, 117068. [CrossRef]
- Pinto, F.G.; Giddey, A.D.; Mohamed, N.; Almarri, R.S.B.; Murtaza, M.; Nassir, N.; Alkhnbashi, O.S.; Uddin, M.J.; Soares, N.C. Repurposing proteomic nanoLC-MS platforms for untargeted metabolomics: evaluating DIA and polarity switching performance in human plasma. Expert Rev Proteomics 2025, 22, 307–314. [CrossRef]
- Skogvold, H.B.; Sandås, E.M.; Østeby, A.; Løkken, C.; Rootwelt, H.; Rønning, P.O.; Wilson, S.R.; Elgstøen, K.B.P. Bridging the Polar and Hydrophobic Metabolome in Single-Run Untargeted Liquid Chromatography-Mass Spectrometry Dried Blood Spot Metabolomics for Clinical Purposes. J Proteome Res 2021, 20, 4010–4021. [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Vrhovsek, U.; Mattivi, F. Quantitative profiling of polar primary metabolites using hydrophilic interaction ultrahigh performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 2012, 1259, 121–127. [CrossRef]
- Violi, J.P.; Phillips, C.R.; Gertner, D.S.; Westerhausen, M.T.; Padula, M.P.; Bishop, D.P.; Rodgers, K.J. Comprehensive untargeted polar metabolite analysis using solvent switching liquid chromatography tandem mass spectrometry. Talanta 2025, 287, 127610. [CrossRef]
- Chen, L.; Zhong, F.; Zhu, J. Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches. Metabolites 2020, 10. [CrossRef]
- Cochran, D.; NourEldein, M.; Bezdekova, D.; Schram, A.; Howard, R.; Powers, R. A Reproducibility Crisis for Clinical Metabolomics Studies. Trends Analyt Chem 2024, 180. [CrossRef]
- Ivanisevic, J.; Zhu, Z.J.; Plate, L.; Tautenhahn, R.; Chen, S.; O’Brien, P.J.; Johnson, C.H.; Marletta, M.A.; Patti, G.J.; Siuzdak, G. Toward ’omic scale metabolite profiling: a dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism. Anal Chem 2013, 85, 6876–6884. [CrossRef]
- Arrivault, S.; Guenther, M.; Fry, S.C.; Fuenfgeld, M.M.; Veyel, D.; Mettler-Altmann, T.; Stitt, M.; Lunn, J.E. Synthesis and Use of Stable-Isotope-Labeled Internal Standards for Quantification of Phosphorylated Metabolites by LC-MS/MS. Anal Chem 2015, 87, 6896–6904. [CrossRef]
- Ulvik, A.; McCann, A.; Midttun, Ø.; Meyer, K.; Godfrey, K.M.; Ueland, P.M. Quantifying Precision Loss in Targeted Metabolomics Based on Mass Spectrometry and Nonmatching Internal Standards. Anal Chem 2021, 93, 7616–7624. [CrossRef]
- Mahmud, I.; Wei, B.; Veillon, L.; Tan, L.; Martinez, S.; Tran, B.; Raskind, A.; de Jong, F.; Liu, Y.; Ding, J.; et al. Ion suppression correction and normalization for non-targeted metabolomics. Nat Commun 2025, 16, 1347. [CrossRef]
- Ćeranić, A.; Bueschl, C.; Doppler, M.; Parich, A.; Xu, K.; Lemmens, M.; Buerstmayr, H.; Schuhmacher, R. Enhanced Metabolome Coverage and Evaluation of Matrix Effects by the Use of Experimental-Condition-Matched (13)C-Labeled Biological Samples in Isotope-Assisted LC-HRMS Metabolomics. Metabolites 2020, 10. [CrossRef]
- Bueschl, C.; Krska, R.; Kluger, B.; Schuhmacher, R. Isotopic labeling-assisted metabolomics using LC-MS. Anal Bioanal Chem 2013, 405, 27–33. [CrossRef]
- Bruheim, P.; Kvitvang, H.F.; Villas-Boas, S.G. Stable isotope coded derivatizing reagents as internal standards in metabolite profiling. J Chromatogr A 2013, 1296, 196–203. [CrossRef]
- Zhao, S. Development and Application of Chemical Isotope Labeling Methods and Metabolite Identification Solution for Liquid Chromatography-Mass Spectrometry-Based Metabolomics. 2018.
- Mathon, C.; Bovard, D.; Dutertre, Q.; Sendyk, S.; Bentley, M.; Hoeng, J.; Knorr, A. Impact of sample preparation upon intracellular metabolite measurements in 3D cell culture systems. Metabolomics 2019, 15, 92. [CrossRef]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Oresic, M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinformatics 2007, 8, 93. [CrossRef]
- Boysen, A.K.; Heal, K.R.; Carlson, L.T.; Ingalls, A.E. Best-Matched Internal Standard Normalization in Liquid Chromatography-Mass Spectrometry Metabolomics Applied to Environmental Samples. Anal Chem 2018, 90, 1363–1369. [CrossRef]
- Wu, Y.; Li, L. Sample normalization methods in quantitative metabolomics. J Chromatogr A 2016, 1430, 80–95. [CrossRef]
- Gu, H.; Zhao, Y.; DeMichele, M.; Zheng, N.; Zhang, Y.J.; Pillutla, R.; Zeng, J. In-Sample Calibration Curve Using Multiple Isotopologue Reaction Monitoring of a Stable Isotopically Labeled Analyte for Instant LC-MS/MS Bioanalysis and Quantitative Proteomics. Anal Chem 2019, 91, 2536–2543. [CrossRef]
- Mani, D.R.; Abbatiello, S.E.; Carr, S.A. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics 2012, 13 Suppl 16, S9. [CrossRef]
- Beger, R.D.; Goodacre, R.; Jones, C.M.; Lippa, K.A.; Mayboroda, O.A.; O’Neill, D.; Najdekr, L.; Ntai, I.; Wilson, I.D.; Dunn, W.B. Analysis types and quantification methods applied in UHPLC-MS metabolomics research: a tutorial. Metabolomics 2024, 20, 95. [CrossRef]
- Verhaeghe, T. Systematic internal standard variability and issue resolution: two case studies. Bioanalysis 2019, 11, 1685–1692. [CrossRef]
- Liao, H.W.; Cheng, Y.W.; Tang, S.C.; Kuo, C.H. Bias caused by incomplete metabolite extraction and matrix effect: Evaluation of critical factors for plasma sample preparation prior to metabolomics. J Pharm Biomed Anal 2022, 219, 114930. [CrossRef]
- Gao, S.; Wang, X.; Huang, J.; Zhu, Y.; Zhang, R.; He, J.; Abliz, Z. Development and validation of a sensitive and reliable targeted metabolomics method for the quantification of cardiovascular disease-related biomarkers in plasma using ultrahigh-performance liquid chromatography-tandem mass spectrometry. Rapid Commun Mass Spectrom 2022, 36, e9292. [CrossRef]
- Begou, O.; Gika, H.G.; Theodoridis, G.A.; Wilson, I.D. Quality Control and Validation Issues in LC-MS Metabolomics. Methods Mol Biol 2018, 1738, 15–26. [CrossRef]
- González-Domínguez, Á.; Estanyol-Torres, N.; Brunius, C.; Landberg, R.; González-Domínguez, R. QComics: Recommendations and Guidelines for Robust, Easily Implementable and Reportable Quality Control of Metabolomics Data. Anal Chem 2024, 96, 1064–1072. [CrossRef]
- Li, S.; Looby, N.; Chandran, V.; Kulasingam, V. Challenges in the Metabolomics-Based Biomarker Validation Pipeline. Metabolites 2024, 14. [CrossRef]
- Furlanello, T.; Bertolini, F.M.; Zoia, A.; Sanchez Del Pulgar, J.; Masti, R. Establishment and Validation of Sensitive Liquid Chromatography-Tandem Mass Spectrometry Method for Aldosterone Quantification in Feline Serum with Reference Interval Determination. Animals (Basel) 2025, 15. [CrossRef]
- Brêtas, J.M.; César, I.C.; Brêtas, C.M.; Teixeira Lde, S.; Bellorio, K.B.; Mundim, I.M.; Pianetti, G.A. Development and validation of an LC-ESI-MS/MS method for the simultaneous quantification of naproxen and sumatriptan in human plasma: application to a pharmacokinetic study. Anal Bioanal Chem 2016, 408, 3981–3992. [CrossRef]
- Prakash, K.; Adiki, S.K.; Kalakuntla, R.R. Development and validation of a liquid chromatography-mass spectrometry method for the determination of zileuton in human plasma. Sci Pharm 2014, 82, 571–583. [CrossRef]
- Nandania, J.; Peddinti, G.; Pessia, A.; Kokkonen, M.; Velagapudi, V. Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry. Metabolites 2018, 8. [CrossRef]
- Godoy, A.T.; Eberlin, M.N.; Simionato, A.V.C. Targeted metabolomics: Liquid chromatography coupled to mass spectrometry method development and validation for the identification and quantitation of modified nucleosides as putative cancer biomarkers. Talanta 2020, 210, 120640. [CrossRef]
- Luo, B.; Groenke, K.; Takors, R.; Wandrey, C.; Oldiges, M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J Chromatogr A 2007, 1147, 153–164. [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 2003, 75, 3019–3030. [CrossRef]
- Steiner, D.; Krska, R.; Malachová, A.; Taschl, I.; Sulyok, M. Evaluation of Matrix Effects and Extraction Efficiencies of LC-MS/MS Methods as the Essential Part for Proper Validation of Multiclass Contaminants in Complex Feed. J Agric Food Chem 2020, 68, 3868–3880. [CrossRef]
- Cortese, M.; Gigliobianco, M.R.; Magnoni, F.; Censi, R.; Di Martino, P.D. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review. Molecules 2020, 25. [CrossRef]
- Taylor, P.J. Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem 2005, 38, 328–334. [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Wilson, I.D. Liquid chromatography and ultra-performance liquid chromatography-mass spectrometry fingerprinting of human urine: sample stability under different handling and storage conditions for metabonomics studies. J Chromatogr A 2008, 1189, 314–322. [CrossRef]
- van de Merbel, N.; Savoie, N.; Yadav, M.; Ohtsu, Y.; White, J.; Riccio, M.F.; Dong, K.; de Vries, R.; Diancin, J. Stability: recommendation for best practices and harmonization from the Global Bioanalysis Consortium Harmonization Team. Aaps j 2014, 16, 392–399. [CrossRef]
- Briscoe, C. Assessing stability in bioanalysis: reflections on the last 10 years. Bioanalysis 2019, 11, 579–582. [CrossRef]
- Eilertsen, H.C.; Huseby, S.; Degerlund, M.; Eriksen, G.K.; Ingebrigtsen, R.A.; Hansen, E. The effect of freeze/thaw cycles on reproducibility of metabolic profiling of marine microalgal extracts using direct infusion high-resolution mass spectrometry (HR-MS). Molecules 2014, 19, 16373–16380. [CrossRef]
- Lee, J.E.; Kim, S.Y.; Shin, S.Y. Effect of Repeated Freezing and Thawing on Biomarker Stability in Plasma and Serum Samples. Osong Public Health Res Perspect 2015, 6, 357–362. [CrossRef]
- Tiwari, G.; Tiwari, R. Bioanalytical method validation: An updated review. Pharm Methods 2010, 1, 25–38. [CrossRef]
- Theodorsson, E.; Magnusson, B. Full method validation in clinical chemistry. Accreditation and Quality Assurance 2017, 22, 235–246.
- Gika, H.G.; Theodoridis, G.A.; Earll, M.; Wilson, I.D. A QC approach to the determination of day-to-day reproducibility and robustness of LC-MS methods for global metabolite profiling in metabonomics/metabolomics. Bioanalysis 2012, 4, 2239–2247. [CrossRef]
- Hoffman, D. Statistical considerations for assessment of bioanalytical incurred sample reproducibility. Aaps j 2009, 11, 570–580. [CrossRef]
- Ermer, J. Validation in pharmaceutical analysis. Part I: an integrated approach. J Pharm Biomed Anal 2001, 24, 755–767. [CrossRef]
- Verch, T.; Campa, C.; Chéry, C.C.; Frenkel, R.; Graul, T.; Jaya, N.; Nakhle, B.; Springall, J.; Starkey, J.; Wypych, J.; et al. Analytical Quality by Design, Life Cycle Management, and Method Control. Aaps j 2022, 24, 34. [CrossRef]
- Paglia, G.; Astarita, G. A High-Throughput HILIC-MS-Based Metabolomic Assay for the Analysis of Polar Metabolites. Methods Mol Biol 2022, 2396, 137–159. [CrossRef]
- Spagou, K.; Tsoukali, H.; Raikos, N.; Gika, H.; Wilson, I.D.; Theodoridis, G. Hydrophilic interaction chromatography coupled to MS for metabonomic/metabolomic studies. J Sep Sci 2010, 33, 716–727. [CrossRef]
- Kopaciewicz, W.; Regnier, F.E. Mobile phase selection for the high-performance ion-exchange chromatography of proteins. Anal Biochem 1983, 133, 251–259. [CrossRef]
- Choy, D.Y.; Creagh, A.L.; Haynes, C. Improved isoelectric focusing chromatography on strong anion exchange media via a new model that custom designs mobile phases using simple buffers. Biotechnol Bioeng 2014, 111, 552–564. [CrossRef]
- Jaćkowska, M.; Bocian, S.; Kosobucki, P.; Buszewski, B. Polymeric Functionalized Stationary Phase for Separation of Ionic Compounds by IC. Chromatographia 2010, 72, 611–616. [CrossRef]
- Studzińska, S.; Bocian, S.; Kilanowska, A.; Buszewski, B. Dendrimer Anion-Exchange Stationary Phase for Separation of Oligonucleotides. Molecules 2022, 27. [CrossRef]
- Law, B.; Hussain, M.A. Re-evaluation of anion-exchange HPLC for the analysis of acidic compounds. J Pharm Biomed Anal 2000, 22, 149–154. [CrossRef]
- Ikeda, K.; Takahashi, M.; Bamba, T.; Izumi, Y. Comparison of Amine-Modified Polymeric Stationary Phases for Polar Metabolomic Analysis Based on Unified-Hydrophilic Interaction/Anion Exchange Liquid Chromatography/High-Resolution Mass Spectrometry (Unified-HILIC/AEX/HRMS). Mass Spectrom (Tokyo) 2024, 13, A0143. [CrossRef]
- Yang, Q.; Zhang, A.H.; Miao, J.H.; Sun, H.; Han, Y.; Yan, G.L.; Wu, F.F.; Wang, X.J. Metabolomics biotechnology, applications, and future trends: a systematic review. RSC Adv 2019, 9, 37245–37257. [CrossRef]
- Castelli, F.A.; Rosati, G.; Moguet, C.; Fuentes, C.; Marrugo-Ramírez, J.; Lefebvre, T.; Volland, H.; Merkoçi, A.; Simon, S.; Fenaille, F.; et al. Metabolomics for personalized medicine: the input of analytical chemistry from biomarker discovery to point-of-care tests. Anal Bioanal Chem 2022, 414, 759–789. [CrossRef]
- Peng, B.; Li, H.; Peng, X.X. Functional metabolomics: from biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637. [CrossRef]
- Li, K.; Naviaux, J.C.; Bright, A.T.; Wang, L.; Naviaux, R.K. A robust, single-injection method for targeted, broad-spectrum plasma metabolomics. Metabolomics 2017, 13, 122. [CrossRef]
- Hosseinkhani, F.; Huang, L.; Dubbelman, A.C.; Guled, F.; Harms, A.C.; Hankemeier, T. Systematic Evaluation of HILIC Stationary Phases for Global Metabolomics of Human Plasma. Metabolites 2022, 12. [CrossRef]
- Fisher-Wellman, K.H.; Davidson, M.T.; Narowski, T.M.; Lin, C.T.; Koves, T.R.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep 2018, 24, 3593–3606.e3510. [CrossRef]
- Mordaunt, D.; Cox, D.; Fuller, M. Metabolomics to Improve the Diagnostic Efficiency of Inborn Errors of Metabolism. Int J Mol Sci 2020, 21. [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci Int 2007, 165, 216–224. [CrossRef]
- Viswanathan, C.T.; Bansal, S.; Booth, B.; DeStefano, A.J.; Rose, M.J.; Sailstad, J.; Shah, V.P.; Skelly, J.P.; Swann, P.G.; Weiner, R. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm Res 2007, 24, 1962–1973. [CrossRef]
- Shah, V.P.; Midha, K.K.; Findlay, J.W.; Hill, H.M.; Hulse, J.D.; McGilveray, I.J.; McKay, G.; Miller, K.J.; Patnaik, R.N.; Powell, M.L.; et al. Bioanalytical method validation--a revisit with a decade of progress. Pharm Res 2000, 17, 1551–1557. [CrossRef]
- Van Eeckhaut, A.; Lanckmans, K.; Sarre, S.; Smolders, I.; Michotte, Y. Validation of bioanalytical LC-MS/MS assays: evaluation of matrix effects. J Chromatogr B Analyt Technol Biomed Life Sci 2009, 877, 2198–2207. [CrossRef]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom Rev 2011, 30, 491–509. [CrossRef]
- Peters, F.T.; Remane, D. Aspects of matrix effects in applications of liquid chromatography-mass spectrometry to forensic and clinical toxicology--a review. Anal Bioanal Chem 2012, 403, 2155–2172. [CrossRef]
- Kostiainen, R.; Kauppila, T.J. Effect of eluent on the ionization process in liquid chromatography-mass spectrometry. J Chromatogr A 2009, 1216, 685–699. [CrossRef]
- Li, W.; Zhang, J.; Francis, L. Handbook of LC-MS bioanalysis: best practices, experimental protocols, and regulations. 2013.
- Jemal, M.; Schuster, A.; Whigan, D.B. Liquid chromatography/tandem mass spectrometry methods for quantitation of mevalonic acid in human plasma and urine: method validation, demonstration of using a surrogate analyte, and demonstration of unacceptable matrix effect in spite of use of a stable isotope analog internal standard. Rapid Commun Mass Spectrom 2003, 17, 1723–1734. [CrossRef]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat Protoc 2010, 5, 1005–1018. [CrossRef]
- Dunn, W.B.; Erban, A.; Weber, R.J.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J. Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66.
- Nadour, Z.; Simian, C.; Laprévote, O.; Loriot, M.A.; Larabi, I.A.; Pallet, N. Validation of a liquid chromatography coupled to tandem mass spectrometry method for simultaneous quantification of tryptophan and 10 key metabolites of the kynurenine pathway in plasma and urine: Application to a cohort of acute kidney injury patients. Clin Chim Acta 2022, 534, 115–127. [CrossRef]
- Sens, A.; Rischke, S.; Hahnefeld, L.; Dorochow, E.; Schäfer, S.M.G.; Thomas, D.; Köhm, M.; Geisslinger, G.; Behrens, F.; Gurke, R. Pre-analytical sample handling standardization for reliable measurement of metabolites and lipids in LC-MS-based clinical research. J Mass Spectrom Adv Clin Lab 2023, 28, 35–46. [CrossRef]
- Hodek, O.; Argemi-Muntadas, L.; Khan, A.; Moritz, T. Mixed-mode chromatography-mass spectrometry enables targeted and untargeted screening of carboxylic acids in biological samples. Anal Methods 2022, 14, 1015–1022. [CrossRef]
- Yamamoto, T.; Sato, K.; Yamaguchi, M.; Mitamura, K.; Taga, A. Development of simultaneous quantitative analysis of tricarboxylic acid cycle metabolites to identify specific metabolites in cancer cells by targeted metabolomic approach. Biochem Biophys Res Commun 2021, 584, 53–59. [CrossRef]
- Pawlak, K.; Kowalewska, A.; Mysliwiec, M.; Pawlak, D. Kynurenine and its metabolites--kynurenic acid and anthranilic acid are associated with soluble endothelial adhesion molecules and oxidative status in patients with chronic kidney disease. Am J Med Sci 2009, 338, 293–300. [CrossRef]
- Summers, B.S.; Thomas Broome, S.; Pang, T.W.R.; Mundell, H.D.; Koh Belic, N.; Tom, N.C.; Ng, M.L.; Yap, M.; Sen, M.K.; Sedaghat, S.; et al. A Review of the Evidence for Tryptophan and the Kynurenine Pathway as a Regulator of Stem Cell Niches in Health and Disease. Int J Tryptophan Res 2024, 17, 11786469241248287. [CrossRef]
- Pereira, J.B.; Westman, E.; Hansson, O. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol Aging 2017, 58, 14–29. [CrossRef]
- Zhang, J.; Liu, Y.; Zhi, X.; Xu, L.; Tao, J.; Cui, D.; Liu, T.F. Tryptophan catabolism via the kynurenine pathway regulates infection and inflammation: from mechanisms to biomarkers and therapies. Inflamm Res 2024, 73, 979–996. [CrossRef]
- Chiu, L.C.; Tang, H.Y.; Fan, C.M.; Lo, C.J.; Hu, H.C.; Kao, K.C.; Cheng, M.L. Kynurenine Pathway of Tryptophan Metabolism Is Associated with Hospital Mortality in Patients with Acute Respiratory Distress Syndrome: A Prospective Cohort Study. Antioxidants (Basel) 2022, 11. [CrossRef]
- Coutinho, L.G.; Christen, S.; Bellac, C.L.; Fontes, F.L.; Souza, F.R.; Grandgirard, D.; Leib, S.L.; Agnez-Lima, L.F. The kynurenine pathway is involved in bacterial meningitis. J Neuroinflammation 2014, 11, 169. [CrossRef]
- Hussain, H.; Vutipongsatorn, K.; Jiménez, B.; Antcliffe, D.B. Patient Stratification in Sepsis: Using Metabolomics to Detect Clinical Phenotypes, Sub-Phenotypes and Therapeutic Response. Metabolites 2022, 12. [CrossRef]
- Shrestha, D.; Pant, B.D.; Roychowdhury, S.; Gandhirajan, A.; Cross, E.; Chhabria, M.; Bauer, S.R.; Jeng, M.; Mitchell, M.; Mehkri, O.; et al. Immunometabolic chaos in septic shock. J Leukoc Biol 2025, 117. [CrossRef]
- Schley, G.; Köberle, C.; Manuilova, E.; Rutz, S.; Forster, C.; Weyand, M.; Formentini, I.; Kientsch-Engel, R.; Eckardt, K.U.; Willam, C. Comparison of Plasma and Urine Biomarker Performance in Acute Kidney Injury. PLoS One 2015, 10, e0145042. [CrossRef]
- Bhatraju, P.K.; Prince, D.K.; Mansour, S.; Ikizler, T.A.; Siew, E.D.; Chinchilli, V.M.; Garg, A.X.; Go, A.S.; Kaufman, J.S.; Kimmel, P.L.; et al. Integrated Analysis of Blood and Urine Biomarkers to Identify Acute Kidney Injury Subphenotypes and Associations With Long-term Outcomes. Am J Kidney Dis 2023, 82, 311-321.e311. [CrossRef]
- Sandokji, I.; Greenberg, J.H. Plasma and Urine Biomarkers of CKD: A Review of Findings in the CKiD Study. Semin Nephrol 2021, 41, 416–426. [CrossRef]
- Beier, U.H.; Hartung, E.A.; Concors, S.; Hernandez, P.T.; Wang, Z.; Perry, C.; Baur, J.A.; Denburg, M.R.; Hancock, W.W.; Gade, T.P.; et al. Tissue metabolic profiling shows that saccharopine accumulates during renal ischemic-reperfusion injury, while kynurenine and itaconate accumulate in renal allograft rejection. Metabolomics 2020, 16, 65. [CrossRef]
- Wen, Y.; Xu, L.; Melchinger, I.; Thiessen-Philbrook, H.; Moledina, D.G.; Coca, S.G.; Hsu, C.Y.; Go, A.S.; Liu, K.D.; Siew, E.D.; et al. Longitudinal biomarkers and kidney disease progression after acute kidney injury. JCI Insight 2023, 8. [CrossRef]
- Pickering, J.W.; Endre, Z.H. Acute kidney injury urinary biomarker time-courses. PLoS One 2014, 9, e101288. [CrossRef]
- Liu, C.; Debnath, N.; Mosoyan, G.; Chauhan, K.; Vasquez-Rios, G.; Soudant, C.; Menez, S.; Parikh, C.R.; Coca, S.G. Systematic Review and Meta-Analysis of Plasma and Urine Biomarkers for CKD Outcomes. J Am Soc Nephrol 2022, 33, 1657–1672. [CrossRef]
- Xu, B.; Zhu, Z.; Shu, H.; Wang, H.; Lu, X.; Zhu, X. Succinate links TCA cycle dysfunction to inflammation by activating HIF-1α signaling in acute kidney injury. Int Immunopharmacol 2025, 161, 115105. [CrossRef]
- Liu, H.; Ding, L.; Zhang, H.; Mellor, D.; Wu, H.; Zhao, D.; Wu, C.; Lin, Z.; Yuan, J.; Peng, D. The Metabolic Factor Kynurenic Acid of Kynurenine Pathway Predicts Major Depressive Disorder. Front Psychiatry 2018, 9, 552. [CrossRef]
- Pan, J.X.; Xia, J.J.; Deng, F.L.; Liang, W.W.; Wu, J.; Yin, B.M.; Dong, M.X.; Chen, J.J.; Ye, F.; Wang, H.Y.; et al. Diagnosis of major depressive disorder based on changes in multiple plasma neurotransmitters: a targeted metabolomics study. Transl Psychiatry 2018, 8, 130. [CrossRef]
- Bi, Q.; Zhao, J.; Nie, J.; Huang, F. Metabolic pathway analysis of tumors using stable isotopes. Semin Cancer Biol 2025, 113, 9–24. [CrossRef]
- Emwas, A.H.; Szczepski, K.; Al-Younis, I.; Lachowicz, J.I.; Jaremko, M. Fluxomics - New Metabolomics Approaches to Monitor Metabolic Pathways. Front Pharmacol 2022, 13, 805782. [CrossRef]
- Fan, T.W.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol Ther 2012, 133, 366–391. [CrossRef]
- Liu, K.H.; Nellis, M.; Uppal, K.; Ma, C.; Tran, V.; Liang, Y.; Walker, D.I.; Jones, D.P. Reference Standardization for Quantification and Harmonization of Large-Scale Metabolomics. Anal Chem 2020, 92, 8836–8844. [CrossRef]
- Gáspár, R.; Halmi, D.; Demján, V.; Berkecz, R.; Pipicz, M.; Csont, T. Kynurenine Pathway Metabolites as Potential Clinical Biomarkers in Coronary Artery Disease. Front Immunol 2021, 12, 768560. [CrossRef]
- Tan, Y.Q.; Wang, Y.N.; Feng, H.Y.; Guo, Z.Y.; Li, X.; Nie, X.L.; Zhao, Y.Y. Host/microbiota interactions-derived tryptophan metabolites modulate oxidative stress and inflammation via aryl hydrocarbon receptor signaling. Free Radic Biol Med 2022, 184, 30–41. [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur J Pharmacol 1988, 151, 313–315. [CrossRef]
- Sawa-Wejksza, K.; Parada-Turska, J.; Turski, W. The Pharmacological Evidences for the Involvement of AhR and GPR35 Receptors in Kynurenic Acid-Mediated Cytokine and Chemokine Secretion by THP-1-Derived Macrophages. Molecules 2025, 30. [CrossRef]
- Villani, S.; Fallarini, S.; Rezzi, S.J.; Di Martino, R.M.C.; Aprile, S.; Del Grosso, E. Selective inhibition of indoleamine and tryptophan 2,3-dioxygenases: Comparative study on kynurenine pathway in cell lines via LC-MS/MS-based targeted metabolomics. J Pharm Biomed Anal 2024, 237, 115750. [CrossRef]
- Zhao, S.; Luo, X.; Li, L. Chemical Isotope Labeling LC-MS for High Coverage and Quantitative Profiling of the Hydroxyl Submetabolome in Metabolomics. Anal Chem 2016, 88, 10617–10623. [CrossRef]
- Schönberger, K.; Mitterer, M.; Glaser, K.; Stecher, M.; Hobitz, S.; Schain-Zota, D.; Schuldes, K.; Lämmermann, T.; Rambold, A.S.; Cabezas-Wallscheid, N.; et al. LC-MS-Based Targeted Metabolomics for FACS-Purified Rare Cells. Anal Chem 2023, 95, 4325–4334. [CrossRef]
- Liu, Z.; Ma, Z.; Jin, L.; Nizhamuding, X.; Zeng, J.; Zhang, T.; Zhang, J.; Wang, J.; Zhao, H.; Zhou, W.; et al. Altered neopterin and IDO in kynurenine metabolism based on LC-MS/MS metabolomics study: Novel therapeutic checkpoints for type 2 diabetes mellitus. Clin Chim Acta 2024, 557, 117859. [CrossRef]
- Pang, Z.; Xu, L.; Viau, C.; Lu, Y.; Salavati, R.; Basu, N.; Xia, J. MetaboAnalystR 4.0: a unified LC-MS workflow for global metabolomics. Nat Commun 2024, 15, 3675. [CrossRef]
- Chen, J.; Zhang, P.; Qin, S.; Tan, B.; Li, S.; Tang, S.; Liao, C.; Zhang, Y.; Zhang, Z.; Xu, F. Stepwise solid phase extraction integrated with chemical derivatization for all-in-one injection LC-MS/MS analysis of metabolome and lipidome. Anal Chim Acta 2023, 1241, 340807. [CrossRef]
- Saigusa, D.; Okamura, Y.; Motoike, I.N.; Katoh, Y.; Kurosawa, Y.; Saijyo, R.; Koshiba, S.; Yasuda, J.; Motohashi, H.; Sugawara, J.; et al. Establishment of Protocols for Global Metabolomics by LC-MS for Biomarker Discovery. PLoS One 2016, 11, e0160555. [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid Redox Signal 2019, 30, 251–294. [CrossRef]
- Wee, H.N.; Liu, J.J.; Ching, J.; Kovalik, J.P.; Lim, S.C. The Kynurenine Pathway in Acute Kidney Injury and Chronic Kidney Disease. Am J Nephrol 2021, 52, 771–787. [CrossRef]
- Zhai, Y.; Chavez, J.A.; D’Aquino, K.E.; Meng, R.; Nawrocki, A.R.; Pocai, A.; Wang, L.; Ma, L.J. Kynurenine 3-monooxygenase limits de novo NAD(+) synthesis through dietary tryptophan in renal proximal tubule epithelial cell models. Am J Physiol Cell Physiol 2024, 326, C1423–C1436. [CrossRef]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int J Tryptophan Res 2019, 12, 1178646919852996. [CrossRef]
- Zhang, X.; Dong, J.; Raftery, D. Five Easy Metrics of Data Quality for LC-MS-Based Global Metabolomics. Anal Chem 2020, 92, 12925–12933. [CrossRef]
- Konakchieva, R.; Mladenov, M.; Konaktchieva, M.; Sazdova, I.; Gagov, H.; Nikolaev, G. Circadian Clock Deregulation and Metabolic Reprogramming: A System Biology Approach to Tissue-Specific Redox Signaling and Disease Development. Int J Mol Sci 2025, 26. [CrossRef]
- Froy, O.; Weintraub, Y. The circadian clock, metabolism, and inflammation-the holy trinity of inflammatory bowel diseases. Clin Sci (Lond) 2025, 139, 777–790. [CrossRef]
- Moaddel, R.; Candia, J.; Ubaida-Mohien, C.; Tanaka, T.; Moore, A.Z.; Zhu, M.; Fantoni, G.; Church, S.; D’Agostino, J.; Fan, J.; et al. Healthy Aging Metabolomic and Proteomic Signatures Across Multiple Physiological Compartments. Aging Cell 2025, 24, e70014. [CrossRef]
- Overgaard, N.H.; Fan, T.M.; Schachtschneider, K.M.; Principe, D.R.; Schook, L.B.; Jungersen, G. Of Mice, Dogs, Pigs, and Men: Choosing the Appropriate Model for Immuno-Oncology Research. Ilar j 2018, 59, 247–262. [CrossRef]
- Guerrero-López, P.; Martín-Pardillos, A.; Bonet-Aleta, J.; Mosseri, A.; Hueso, J.L.; Santamaria, J.; Garcia-Aznar, J.M. 2D versus 3D tumor-on-chip models to study the impact of tumor organization on metabolic patterns in vitro. Sci Rep 2025, 15, 19506. [CrossRef]
- Luan, H.; Ji, F.; Chen, Y.; Cai, Z. statTarget: A streamlined tool for signal drift correction and interpretations of quantitative mass spectrometry-based omics data. Anal Chim Acta 2018, 1036, 66–72. [CrossRef]
- Pedraz-Petrozzi, B.; Marszalek-Grabska, M.; Kozub, A.; Szalaj, K.; Trzpil, A.; Stachniuk, A.; Lamadé, E.K.; Gilles, M.; Deuschle, M.; Turski, W.A.; et al. LC-MS/MS-based quantification of tryptophan, kynurenine, and kynurenic acid in human placental, fetal membranes, and umbilical cord samples. Sci Rep 2023, 13, 12554. [CrossRef]
- Wang, G.; Heijs, B.; Kostidis, S.; Mahfouz, A.; Rietjens, R.G.J.; Bijkerk, R.; Koudijs, A.; van der Pluijm, L.A.K.; van den Berg, C.W.; Dumas, S.J.; et al. Analyzing cell-type-specific dynamics of metabolism in kidney repair. Nat Metab 2022, 4, 1109–1118. [CrossRef]
- Buergel, T.; Steinfeldt, J.; Ruyoga, G.; Pietzner, M.; Bizzarri, D.; Vojinovic, D.; Upmeier Zu Belzen, J.; Loock, L.; Kittner, P.; Christmann, L.; et al. Metabolomic profiles predict individual multidisease outcomes. Nat Med 2022, 28, 2309–2320. [CrossRef]
- Golubova, A.; Lanekoff, I. Quantitative approaches for spatial metabolomics with isomer differentiation using surface sampling capillary electrophoresis mass spectrometry. Talanta 2026, 296, 128482. [CrossRef]
- van der Walt, G.; Louw, R. Novel mitochondrial and cytosolic purification pipeline for compartment-specific metabolomics in mammalian disease model tissues. Metabolomics 2020, 16, 78. [CrossRef]
- Wang, Z.; Fu, W.; Huo, M.; He, B.; Liu, Y.; Tian, L.; Li, W.; Zhou, Z.; Wang, B.; Xia, J.; et al. Spatial-resolved metabolomics reveals tissue-specific metabolic reprogramming in diabetic nephropathy by using mass spectrometry imaging. Acta Pharm Sin B 2021, 11, 3665–3677. [CrossRef]
- Knitsch, R.; AlWahsh, M.; Raschke, H.; Lambert, J.; Hergenröder, R. In Vitro Spatio-Temporal NMR Metabolomics of Living 3D Cell Models. Anal Chem 2021, 93, 13485–13494. [CrossRef]
- van der Walt, G.; Lindeque, J.Z.; Mason, S.; Louw, R. Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome. Metabolites 2021, 11. [CrossRef]
- Qin, S.; Gao, M.; Zhang, Q.; Xiao, Q.; Fu, J.; Tian, Y.; Jiao, Y.; Zhang, Z.; Zhang, P.; Xu, F. High-Coverage Strategy for Multi-Subcellular Metabolome Analysis Using Dansyl-Labeling-Based LC-MS/MS. Anal Chem 2023, 95, 10034–10043. [CrossRef]
- More, T.H.; Hiller, K. Complexity of subcellular metabolism: strategies for compartment-specific profiling. Curr Opin Biotechnol 2022, 75, 102711. [CrossRef]
- Talmor-Barkan, Y.; Bar, N.; Shaul, A.A.; Shahaf, N.; Godneva, A.; Bussi, Y.; Lotan-Pompan, M.; Weinberger, A.; Shechter, A.; Chezar-Azerrad, C.; et al. Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat Med 2022, 28, 295–302. [CrossRef]
- Geier, B.; Sogin, E.M.; Michellod, D.; Janda, M.; Kompauer, M.; Spengler, B.; Dubilier, N.; Liebeke, M. Spatial metabolomics of in situ host-microbe interactions at the micrometre scale. Nat Microbiol 2020, 5, 498–510. [CrossRef]
- Yu, Y.; Zhang, N.; Mai, Y.; Ren, L.; Chen, Q.; Cao, Z.; Chen, Q.; Liu, Y.; Hou, W.; Yang, J.; et al. Correcting batch effects in large-scale multiomics studies using a reference-material-based ratio method. Genome Biol 2023, 24, 201. [CrossRef]
- Viant, M.R.; Ebbels, T.M.D.; Beger, R.D.; Ekman, D.R.; Epps, D.J.T.; Kamp, H.; Leonards, P.E.G.; Loizou, G.D.; MacRae, J.I.; van Ravenzwaay, B.; et al. Use cases, best practice and reporting standards for metabolomics in regulatory toxicology. Nat Commun 2019, 10, 3041. [CrossRef]
- Nian, M.; Chen, X.; Fang, M. Addressing Pitfalls of Metabolomics for Toxicology: A Call for Standardization, Reproducibility and Data Sharing. Chem Res Toxicol 2025, 38, 1150–1156. [CrossRef]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent Topics in Mass Spectrometry-Based Metabolomics and Lipidomics-Standardization, Coverage, and Throughput. Anal Chem 2021, 93, 519–545. [CrossRef]
- Garrett, R.; Ptolemy, A.S.; Pickett, S.; Kellogg, M.D.; Peake, R.W.A. Untargeted Metabolomics for Inborn Errors of Metabolism: Development and Evaluation of a Sustainable Reference Material for Correcting Inter-Batch Variability. Clin Chem 2024, 70, 1452–1462. [CrossRef]
- Long, N.P.; Nghi, T.D.; Kang, Y.P.; Anh, N.H.; Kim, H.M.; Park, S.K.; Kwon, S.W. Toward a Standardized Strategy of Clinical Metabolomics for the Advancement of Precision Medicine. Metabolites 2020, 10. [CrossRef]
- Malinowska, J.M.; Whelan, M. The important role of standards for the uptake of transcriptomics and metabolomics based in vitro methods in regulatory toxicology. Arch Toxicol 2025, 99, 3865–3875. [CrossRef]
- Liu, J. Computational aspects of psychometric methods with R by Patricia Martinková and Adéla Hladká, Chapman & Hall/CRC, 2023, ISBN: 9781003054313, https://doi.org/10.1201/9781003054313. Biometrics 2025. [CrossRef]
- Harada, S.; Iida, M.; Miyagawa, N.; Hirata, A.; Kuwabara, K.; Matsumoto, M.; Okamura, T.; Edagawa, S.; Kawada, Y.; Miyake, A.; et al. Study Profile of the Tsuruoka Metabolomics Cohort Study (TMCS). J Epidemiol 2024, 34, 393–401. [CrossRef]
- Sturm, G.; Monzel, A.S.; Karan, K.R.; Michelson, J.; Ware, S.A.; Cardenas, A.; Lin, J.; Bris, C.; Santhanam, B.; Murphy, M.P.; et al. A multi-omics longitudinal aging dataset in primary human fibroblasts with mitochondrial perturbations. Sci Data 2022, 9, 751. [CrossRef]
- Dukovski, I.; Bajić, D.; Chacón, J.M.; Quintin, M.; Vila, J.C.C.; Sulheim, S.; Pacheco, A.R.; Bernstein, D.B.; Riehl, W.J.; Korolev, K.S.; et al. A metabolic modeling platform for the computation of microbial ecosystems in time and space (COMETS). Nat Protoc 2021, 16, 5030–5082. [CrossRef]
- Weiss, E.; de la Peña-Ramirez, C.; Aguilar, F.; Lozano, J.J.; Sánchez-Garrido, C.; Sierra, P.; Martin, P.I.; Diaz, J.M.; Fenaille, F.; Castelli, F.A.; et al. Sympathetic nervous activation, mitochondrial dysfunction and outcome in acutely decompensated cirrhosis: the metabolomic prognostic models (CLIF-C MET). Gut 2023, 72, 1581–1591. [CrossRef]
- Alston, C.L.; Stenton, S.L.; Hudson, G.; Prokisch, H.; Taylor, R.W. The genetics of mitochondrial disease: dissecting mitochondrial pathology using multi-omic pipelines. J Pathol 2021, 254, 430–442. [CrossRef]
- Lane, A.N.; Tan, J.; Wang, Y.; Yan, J.; Higashi, R.M.; Fan, T.W. Probing the metabolic phenotype of breast cancer cells by multiple tracer stable isotope resolved metabolomics. Metab Eng 2017, 43, 125–136. [CrossRef]
- Roci, I.; Gallart-Ayala, H.; Schmidt, A.; Watrous, J.; Jain, M.; Wheelock, C.E.; Nilsson, R. Metabolite Profiling and Stable Isotope Tracing in Sorted Subpopulations of Mammalian Cells. Anal Chem 2016, 88, 2707–2713. [CrossRef]
- Timón-Gómez, A.; Doerrier, C.; Sumbalová, Z.; Garcia-Souza, L.F.; Baglivo, E.; Cardoso, L.H.D.; Gnaiger, E. Bioenergetic profiles and respiratory control in mitochondrial physiology: Precision analysis of oxidative phosphorylation. Exp Physiol 2025. [CrossRef]
- Seifert, J.; Taubert, M.; Jehmlich, N.; Schmidt, F.; Völker, U.; Vogt, C.; Richnow, H.H.; von Bergen, M. Protein-based stable isotope probing (protein-SIP) in functional metaproteomics. Mass Spectrom Rev 2012, 31, 683–697. [CrossRef]
- Rodriguez, P.; Laskowski, L.J.; Pallais, J.P.; Bock, H.A.; Cavalco, N.G.; Anderson, E.I.; Calkins, M.M.; Razzoli, M.; Sham, Y.Y.; McCorvy, J.D.; et al. Functional profiling of the G protein-coupled receptor C3aR1 reveals ligand-mediated biased agonism. J Biol Chem 2024, 300, 105549. [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 2001, 21, 7463–7473. [CrossRef]
- Sapko, M.T.; Guidetti, P.; Yu, P.; Tagle, D.A.; Pellicciari, R.; Schwarcz, R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp Neurol 2006, 197, 31–40. [CrossRef]
- Bock, A.; Bermudez, M. Allosteric coupling and biased agonism in G protein-coupled receptors. Febs j 2021, 288, 2513–2528. [CrossRef]
- Cadenas, S. Mitochondria rescue cells from ischemic injury. Science 2022, 377, 579–580. [CrossRef]
- Stone, T.W.; Williams, R.O. Tryptophan metabolism as a ’reflex’ feature of neuroimmune communication: Sensor and effector functions for the indoleamine-2, 3-dioxygenase kynurenine pathway. J Neurochem 2024, 168, 3333–3357. [CrossRef]
Table 6.
Proposed comprehensive LC–MS/MS panels covering the KYN pathway and TCA cycle for clinical and translational studies. Targeted LC–MS/MS panel covering key metabolites of the KYN pathway and TCA cycle in clinically relevant biological matrices. Analytes were selected based on biological relevance and analytical feasibility in targeted workflows. Recommended SIL-IS, chromatographic separation modes, and optimal ESI polarity are indicated. HILIC, including mixed-mode variants where appropriate, is suggested to enable retention of highly polar metabolites. Key stability considerations highlight compounds requiring particular attention during sample handling and processing.
Table 6.
Proposed comprehensive LC–MS/MS panels covering the KYN pathway and TCA cycle for clinical and translational studies. Targeted LC–MS/MS panel covering key metabolites of the KYN pathway and TCA cycle in clinically relevant biological matrices. Analytes were selected based on biological relevance and analytical feasibility in targeted workflows. Recommended SIL-IS, chromatographic separation modes, and optimal ESI polarity are indicated. HILIC, including mixed-mode variants where appropriate, is suggested to enable retention of highly polar metabolites. Key stability considerations highlight compounds requiring particular attention during sample handling and processing.
| Analyte | Matrix | Internal standard | Key stability issue | Clinical relevance |
Chromatography | Polarity | Reference |
|---|---|---|---|---|---|---|---|
| Trp | Plasma, serum | d5-Tryptophan | Stable; affected by hemolysis | Substrate availability; KYN/TRP ratio for IDO/TDO activity | HILIC compatible |
ESI(+) | [89] |
| KYN | Plasma, serum | d4-Kynurenine | Generally stable; protein binding may affect recovery | Central branch-point of KYN metabolism; immune activation marker | HILIC compatible |
ESI(+) |
[50] |
| KYNA | Plasma, serum, CSF | d5-Kynurenic acid | Light sensitivity; adsorption at low concentrations | NMDA, GPR35 signaling; neuroprotective and immunomodulatory roles | HILIC compatible |
ESI(+) | [206] |
| 3-HK | Plasma | d3-3-Hydroxy-kynurenine | Oxidation-prone; requires antioxidant stabilization | Pro-oxidant stress marker; neurotoxicity and redox imbalance | HILIC compatible |
ESI(+) |
[89] |
| AA | Plasma, urine | d4-Anthranilic acid | Generally stable | Alternative branch activity; inflammatory pathway balance | HILIC compatible |
ESI(+) |
[485] |
| 3-HAA | Plasma | d3-3-Hydroxy-anthranilic acid | Oxidation-sensitive; light exposure | Immunomodulation; redox-active intermediate | HILIC compatible |
ESI(+) | [486] |
| XA | Plasma, urine | d4-Xanthurenic acid | Moderate stability; pH dependent | Metabolic dysregulation; diabetes and mitochondrial stress associations | HILIC compatible |
ESI(+) | [487] |
| QA | Plasma, urine, CSF | d3-Quinolinic acid | Freeze–thaw sensitive; oxidative degradation | Excitotoxicity; NAD⁺ precursor flux; neuroinflammation | HILIC compatible |
ESI(+) | [89] |
| PA | Plasma, urine | d4-Picolinic acid | Generally stable | Metal chelation; immune modulation | HILIC compatible |
ESI(+) | [486] |
| Citrate | Plasma | ¹³C2-citrate | Moderate instability; delayed processing sensitive | Mitochondrial entry point; energy metabolism proxy | HILIC compatible |
ESI(−) | [234] |
| Isocitrate | Plasma | ¹³C2-citrate | Isomerization risk; low abundance | TCA flux regulation; redox coupling | HILIC compatible |
ESI(−) | [234] |
| α-Ketoglutarate | Plasma | ¹³C5-α-ketoglutarate | Rapid degradation; requires fast quenching | Central metabolic node; immunometabolic signaling | HILIC compatible |
ESI(−) | [234] |
| Succinate | Plasma | d4-Succinate | Rapid post-sampling increase | Inflammatory signaling; mitochondrial dysfunction | HILIC compatible |
ESI(−) | [234] |
| Fumarate | Plasma | ¹³C4-Fumarate | Generally stable | Oncometabolic and hypoxic signaling | HILIC compatible |
ESI(−) | [234] |
| Malate | Plasma | ¹³C4-malate | Moderate instability | NADH/NAD⁺ coupling; mitochondrial redox state | HILIC compatible |
ESI(−) | [234] |
3-HAA, 3-hydroxyanthranilic acid; 3-HK, 3-hydroxykynurenine; AA, anthranilic acid; CSF, cerebrospinal fluid; ESI, electrospray ionization; ESI(+), positive electrospray ionization; ESI(−), negative electrospray ionization; GPR35, G protein–coupled receptor 35; HILIC, hydrophilic interaction liquid chromatography; IDO, indoleamine 2,3-dioxygenase; KYN, kynurenine; KYNA, kynurenic acid; LC–MS/MS, liquid chromatography–tandem mass spectrometry; NAD⁺, nicotinamide adenine dinucleotide; NMDA, N-methyl-D-aspartate; PA, picolinic acid; QA, quinolinic acid; SIL-IS, stable isotope–labeled internal standards; TCA, tricarboxylic acid cycle; TDO, tryptophan 2,3-dioxygenase; TRP, tryptophan; XA, xanthurenic acid; α-KG, alpha-ketoglutarate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



