
Submitted:
08 January 2026
Posted:
09 January 2026
You are already at the latest version
Abstract
This paper does not present new experimental data, but it offers unique insights into the replication mechanism and DNA topology based on ambidextrous double helix model.
Analysis of positively and negatively supercoiled plasmids revealed that the differences between them could be reasonably explained by the ambidextrous double helix model, but not by the classical double helix model.
The superhelical structure of DNA has been understood and explained in an unprecedented way, which may help us better unravel the mysteries of nature.
Acquiring knowledge is important, but finding the right way of thinking that breaks with tradition and inspires new ideas is even more crucial.
Keywords:
positive supercoiling
; gyrase
; ambidextrous double-helix model
; DNA topology
; DNA replication
Introduction
After more than 70 years of in-depth research, most scientists believe that DNA exists primarily in three forms: A-DNA, B-DNA, and Z-DNA. Under physiological conditions, the widely accepted B-DNA form, the famous double helix model proposed in 1953, has become a fundamental pillar of molecular biology [1,2,3]
The DNA double helix model mainly consists of three parts: 1. Both DNA strands are composed of four types of nucleotides arranged linearly, and run in opposite directions; 2. The two strands are connected by hydrogen bonds between complementary base pairs; 3. The two strands are tightly coiled together, always forming a right-handed helix structure.
The discovery of the double helix structure revolutionized biology, revealing how genetic information is stored and transmitted, thus giving rise to molecular biology and genetics. It is considered one of the greatest discoveries in the history of biology, ranking alongside the achievements of Darwin and Mendel.
The double helix structure has achieved remarkable success in fields such as the Human Genome Project, genetic engineering, forensic science, agriculture, and medicine, leading people to believe it is entirely correct and flawless. Therefore, most people naturally overlook the limitations of the double helix structure.
Detailed analysis shows that all these achievements are attributable to the correctness of the first and second parts of the double helix structure, that is, the primary structure of DNA. To date, no one has questioned the validity of the first and second parts of the double helix structure. Only the third part has been questioned by some, including the authors of this paper.
More than forty years ago, we discovered that, under specially designed conditions, the two circular strands of a supercoiled or relaxed plasmid could be separated and clearly visualized under an electron microscope [4,5].
This discovery was further confirmed by the identification of topoisomer with zero linking number, which contradicts the third part of the classic double helix model—that the two circular DNA strands are inseparable. In addition to this direct visual evidence, a wealth of evidence and indirect evidence from numerous laboratories around the world also cast doubt on the validity of the classic double helix structure [6,7]
Everyone can notice unusual phenomena, but not everyone has the time, courage, or willingness to investigate their causes and potential consequences. We believe this rare and surprising discovery is highly significant, and its potential implications warrant further investigation! This is because it challenges a fundamental concept in molecular biology: the validity of the classic double helix model. More importantly, it breaks the taboo of questioning the validity of the double helix structure, encouraging a rethinking of other possibilities for DNA secondary structure.
In the field of DNA structure research, a typical example that every challenger or dissenter encounters are the immense pressure they face from experts or organizations that support the classic Watson-Crick Model.*
*Dr. Wu Dede, a tenured professor at Cornell Medical School, was forced to switch from DNA research to immunology and lost his research funding simply because his research questioned the validity of the double helix structure. He expressed his anger and despair by posting comments online: "Politics determines scientific truth." Yet, despite his expertise, he found himself marginalized, illustrating how institutional consensus can override empirical challenge. Furthermore, under the halo of the Nobel Prize, the Watson-Crick Model intimidated anyone who dared to question its validity.
When venturing into uncharted territory, different people may have different ideas, sometimes even contradictory ones. What matters is not the volume of the voice, power, age, qualifications, courage, or any other factor, but rather wisdom and verifiable evidence.
DNA has a double helix structure, and almost all the relevant details can be found in textbooks. However, are all the theories described in textbooks completely accurate? Our answer is no. We have reason to question the accuracy of the double helix structure.
In terms of epistemology, Donald Rumsfeld's words are quite insightful: “Reports that claim something didn’t happen always interest me, because, as we know, there are known knowns; there are things we know we know. We also know that there are known unknowns; that is, there are things we know we don’t know. But there are also unknown unknowns—things we don’t know we don’t know.”
Clearly, he overlooked another possibility: the known unknowns. This combination is far more dangerous and deserves more attention, because we don't know that what we think we understand might be partially wrong, or even completely unknown, and artificial intelligence is powerless in such situations. The problem is that this might lead people to stop thinking about these known unknowns altogether.
The Achilles Heel of the Double Helix
In 1869, Swiss biochemist Friedrich Miescher isolated a substance from the nucleus of white blood cells, calling it "nuclein." At that time, no one knew what it was. After generations of research, it was eventually classified as a unique macromolecule and named DNA. Its structure was finally discovered by Watson and Crick in 1953.
Interestingly, Watson and Crick were not initially professional scientists specializing in DNA research. They undertook this research primarily out of intense curiosity and personal ambition. Surprisingly, they didn't conduct any experiments of their own, but instead gathered a wealth of useful evidence from various sources and proposed a seemingly ingenious structural model that could logically explain the relationship been DNA structure and its replication. Even more astonishingly, the key components of the double helix model were subsequently confirmed by numerous experiments and have remained the accepted model of DNA structure in mainstream science for the past seventy years.
As many experts have pointed out, the double helix structure should be considered a hypothesis, and under the current peer-review system, their first manuscript should have been rejected by the publisher, requiring more evidence [8].
Various sources indicate that the winding pattern of the double helix structure (i.e., the way the helical chains intertwine) was initially questioned by many scientists, including Watson and Crick:
- On May 1953, Delbrück send a letter to Watson saying: “I am willing to bet that the plectonemic coiling of the chains in your structure is radically wrong, because (1) The difficulties of untangling the chains do seem, after all, insuperable to me. (2) The X-ray data suggest only coiling but not specially your kind of coiling [9].
- Rich once pointed out that a few sports of “fiber diffraction could not ‘prove’ a structure” [10].
- As Holmes pointed out in his book that Watson himself “suffered from periodic fears that the structure might be wrong and that he had made an ass of himself. ... Watson really did harbor serious doubts about the validity of their structure for DNA, before and after he and Crick published their first paper in Nature” [11].
- After obtaining X-ray crystallographic evidence of oligonucleotides, Crick admitted: “The double-helical structure of DNA was thus finally confirmed only in the early 1980s. It took over entry-five years for our model of DNA to go from being rather plausible, to being very plausible (as a result of the detailed work on DNA fibers), and from there to being virtually certainly correct.”[12].
This clearly shows that the Watson-Crick Model, like Achilles' heel, has a fatal flaw. In fact, this flaw is so deeply hidden that it is difficult to detect even today.
The Watson-Crick Model Cannot Solve the Problems in Replication
DNA replication is an extremely complex process. Through many years of in-depth research by numerous outstanding scientists, all the components and proteins involved in DNA replication have been discovered, and their functions are clearly understood. Detailed explanations of this important biological function can be found in various textbooks and videos. The success of understanding the DNA replication mechanism made us feel like everything worth doing had already been done. It seemed there was nothing new left to discover.
Bacterial physiology tells us that, under suitable conditions, each DNA replication cycle in E. coli can be completed within 40 minutes. The E. coli chromosomal DNA contains approximately 4.6 million base pairs and is located within the cell wall, measuring about 1-2 micrometers in size. Therefore, the DNA replication rate can be calculated as: 4.6 × 10⁶ bp / 40 minutes / 2 replication forks ≈ 60,000 base pairs per minute per replication fork, or 1 kb per second per replication fork.
Currently, there are two main models regarding the movement of parental DNA and ribosomes: the "factory model" and the "train model." In the "factory model," the replisome is stationary; while in the "train model," the replisome moves along the DNA like a train. Both models are supported by substantial evidence and appear to be quite plausible [1][15].
Nevertheless, both analogies are misleading. The replisome is neither a factory nor a train. It is not fixed in a specific location within the cell like a factory, nor can it easily move from one place to another like a train. It is a large and complex replication machine, containing helicase, DNA polymerase, primase, sliding clamp, clamp loader protein, single-stranded DNA binding protein, ligase, and many DNA building blocks, namely deoxyribonucleoside triphosphates (dNTPs) and adenosine triphosphate (ATP). These components work together to ensure that daughter DNA is quickly and accurately replicated from parental DNA. The large size of the replisome makes it difficult to move quickly in the viscous cytoplasm.
In reality, both models are partially correct. Since both replisomes begin replication at the same oriC site in the center of the cell and continue working until one replisome reaches a Ter site and stops, while the other replication fork continues replicating the remaining portion until it reaches the Ter site at the other end of the cell, the short DNA segment containing the Ter site may take about one minute to move from one end of the cell to the other. It is important to understand that due to chromosome movement, the same Ter region may be located at different poles of the cell at different times.
As shown in Figure 1, the parental DNA enters the replisome at a speed of up to 60 kb per minute or 20 μm per minute, while the replisome itself (moving from the center of the cell towards the periphery) moves at a speed of less than 1 μm / 40 minutes = 0.025 μm/minute. Therefore, compared to the rapidly entering DNA, the replisome is essentially stationary.
Having understood the relative movement between the parental DNA and the replisome, we can now focus on the detailed mechanism by which the tightly intertwined parental DNA strands are separated.
The replication of E. coli DNA requires the correct distribution of the long, circular parental DNA into two daughter cells that is so called“the semi conservative bidirectional replication”. This problem became particularly important after the discovery of circular DNA. In the late 1970s, many different DNA models were proposed. Albeit, all of these parallel, or "side-by-side," (SBS) models, while theoretically plausible, were never experimentally verified or confirmed [16,17,18].
In 1979, Crick et al. published a review article admitted that“The original model for the double helix was right-handed. The experimental evidence for this feature is suggestive but not yet completely compelling.” [19]
If the DNA is in the B-DNA form, then its linking number should decrease from 4 × 10⁻⁵ to 0 after replication. This unwinding process can only be accomplished by gyrase and topoisomerase IV, because they possess the unique ability to alter the topological properties of the circular chromosomal DNA of E. coli.
Multiple experiments have shown that DNA gyrase is the main enzyme responsible for the initiation and elongation of DNA synthesis, while topoisomerase IV is responsible for unwinding the entangled DNA molecules at the end of replication. Both enzymes belong to type II topoisomerases and are essential for the survival of E. coli cells. Each reaction reduces the linking number by 2. Studies have shown that they preferentially bind to and cleave sites that are unevenly distributed on the E. coli chromosomal DNA. These sites are dispersed mosaic elements (BIMEs) or topoisomerase binding sites (toposites).
Studies have shown that the reaction rate of DNA gyrase is very slow, approximately only six reactions per minute, and the reaction rate of topoisomerase IV is similarly slow. This result is reasonable considering the high complexity of the protein reaction mechanisms. However, this finding makes it difficult to separate the parental DNA strands when the E. coli DNA is in the B-DNA conformation. [7,20]
On the other hand, the function of helicase is to cut the hydrogen bonds between the complementary strands with the help of ATP, but it cannot change the linking number.
Based on these reliable facts and data, we can conclude that the Watson-Crick Model is unable to solve the problem of replication. The reasons are as follows:
- The rapid replication process requires the parental DNA to unwind at a rate of 60,000 base pairs per minute at each replication fork, equivalent to 6,000 helical turns per minute at each replication fork. This is a very high rotation speed, which could potentially cause the mechanically sensitive parental DNA to break.
- DNA molecules are highly hydrophilic, and their rapid rotation can cause hydrodynamic shear forces in the cytoplasm, generating a large amount of heat, which may damage the cell.
- Due to the rapid unwinding of parental DNA, the positive supercoils generated ahead of each replisome must be immediately removed; otherwise, replication cannot continue [21]. A key question arises: if each helical turn requires a separate gyrase-mediated strand passage, then up to 3 000 such events would be required per minute at each fork—a demand that appears incompatible with the measured catalytic turnover of gyrase in vivo.
- According to the Watson-Crick Model, the answer is straightforward: 6000 turns / 6 reactions / minute / 2 linkages / gyrase reaction = 500 DNA gyrase molecules per minute per replication fork. However, in vitro experiments by Stracy et al. [22] showed that approximately 5 topoisomerase IV and 10 DNA gyrase molecules are possibly working there. This number is still far below the theoretically required number to maintain high-speed replication.
- Even with so many gyrase and topoisomerases IV enzymes available in front of each replication fork to unwind the rapidly replicating DNA, how these enzymes cooperate to avoid over or under-twisting the parental DNA remains a serious problem. Each enzyme molecule has to independently perform its task and consistently follows the cell's instructions is another question that urgently needs to be addressed.
- A rapidly growing E. coli cell produces approximately 4,285 different proteins, around 50-60 different t-RNAs (with a total of about 375,000 functional molecules), and 3 distinct ribosomal RNAs (16S, 23S, and 5S). The chromosomal DNA of E.coli also has some parts are in the supercoiled form. All of these proteins and their associated substances prevent DNA from rotating rapidly around its own axis.
- A surprising finding is that DNA gyrase can react with positively supercoiled DNA at a rate up to 10 times faster than under normal conditions [23,24]. We don't understand how the complex "cutting-and-ligation" process in the DNA gyrase reaction leads to such dramatic changes in reaction rate just depending on the supercoiling state, increasing from 6 reactions per minute to 60 reactions per minute. Even so, successful unwinding of the parental DNA still requires the coordinated action of multiple new DNA gyrases, operating at maximum efficiency throughout the entire 40-minute process. Furthermore, It is unlikely the clusters of toposites should be distributed across the entire chromosome.
From the above description, it is clear that the possibility of rapid unwinding of parental DNA has been completely refuted. All reliable data and quantitative analyses contradict the fallacy that the problem of DNA replication can be solved by relying on the miraculous abilities of gyrase.
Whereas, if DNA adopted a side-by-side model or ambidextrous double-helix model, then all the problems encountered during the rapid replication of E. coli would immediately disappear.
In fact, E. coli or its ancestors have survived for millions of years and have never stopped reproducing. All of the above problems stem from the theory that the two DNA strands are tightly coiled together in the form of B-DNA. Clearly, when a theory fails to explain the facts, it is the theory that should be changed, not the facts themselves.
Now, it's time to completely abandon the dogma of the third part of the Watson-Crick Model and seek a new way to understand the structure and function of DNA.
An Alternative Way of Unwinding the Double-Stranded DNA
To understand the detailed process of parental DNA unwinding during replication, we must re-examine and update some old concepts that are deeply ingrained in most people's minds:
- The two strands of DNA are not actually tightly wound in the way of B-DNA with 10 base pairs per turn.
- The name "helicase" is misleading because its main function is to break the hydrogen bonds between base pairs of the two complementary strands, rather than to rotate the DNA along the axis of DNA.
- With the help of ATP, helicase can break hydrogen bonds and pull the DNA into the replication complex for subsequent processes, but there is no evidence that it can change the linking number of the DNA. Therefore, it cannot solve the topological problems in DNA replication on its own.
- On the other hand, helicase catalyzes the separation of parental DNA strands, leading to positive supercoiling of the DNA ahead of the replication fork. This excessive positive supercoiling might be misinterpreted as the helicase having the ability to rotate DNA. In reality, this is merely a consequence of helicase activity, not an inherent rotational capability.
- The positive supercoils generated during replication must be removed by DNA gyrase. The question is, how many DNA gyrase molecules are needed to successfully unwind the DNA during DNA replication? A study by Stracy et al. showed that there are approximately 5 DNA gyrase molecules and 10 DNA topoisomerase molecules around each replication fork (located about 10 kb ahead of the replication fork).
The evidence and facts accumulated in literature helps to illustrate a clear dynamic image as shown in Figure 2.
Instead, helicase just breaks the hydrogen bonds between the paired base pairs with the help of ATP. This rapid reaction leads the formation of some positive supercoils in the parental DNA ahead of the replication fork. These supercoils can be quickly removed by DNA gyrases.
The quantitative implication of this mechanism—namely that a single gyrase molecule may suffice per fork—has not previously been emphasized. but it can reasonably explain the topological problems in DNA replication. To better understand the meaning of this figure, a few points should be clarified:
- According to the B-DNA model, rapid unwinding of double-stranded DNA would generate 6000 positive supercoils per minute ahead of each replication fork. Whereas, according to the ambidextrous DNA model, the number of positive supercoils should be much less than this value. Because the supercoiling is caused from the differences been right-handed DNA and left-handed DNA in the corresponding sections of parental DNA. Data from plasmids shows that the number of supercoils in a plasmid containing N base pairs is two to three orders of magnitude smaller than N/10.
- Since the formation of positive supercoils depends on the degree of negative supercoiling in the region ahead of the replication fork, their number may vary, but it always remains at a very low level. This possibly means that only one gyrase molecule is sufficient to complete the unwinding work during the replication process.
- Once DNA gyrase detects positive supercoiling, it begins to act, thereby avoiding potential over-unwinding or the need for cooperative action between multiple gyrase molecules. The dynamic nature of replication prevents gyrase from remaining fixed ahead of the replication fork. Each gyrase is quickly turning around after finished its work.
We must recognize that this diagram is merely a hypothetical snapshot of the rapid replication process, and everything is constantly in motion.
There is no reason to believe that so many toposites are always present at every location on the E. coli DNA. These enzymes are likely simply moving freely in the cell, waiting for an opportunity to bind to the appropriate site on the DNA. Some of them may bind to DNA, but it may not exert its function. This situation is analogous to a fisherman reeling in his line, causing the distance between the floats to shorten, making them appear to be squeezed together
During the replication, perhaps only one DNA gyrase around each replication fork is sufficient to solve the unwinding problem. This is not because the reaction rate of DNA gyrase can be extremely high, exceeding 6 times per minute, but because such a high reaction rate is not necessary if the total number of linkages in E. coli is less than 6 × 40 × 2 x 2 = 1920. In other words, the positive supercoiling generated is less than 12 / m per replication fork.
Because DNA replication is extremely fast, we cannot expect the same one gyrase molecule to perform all the unwinding work. It is likely that many DNA gyrase molecules work together, but they unwind the DNA in a sequential, one-after-another manner. The claim that DNA gyrase and topoisomerase IV are located 10 kb ahead of the replication fork requires further verification, as a DNA length of 10 kb is approximately 3 micrometers, which exceeds the length of the cell itself. Furthermore, these data are not derived directly from experimental measurements but are calculated values. The authors acknowledge that the in vitro experimental results were obtained using synchronously cultured E. coli cells, which may not accurately reflect the actual processes in vivo.
The DNA unwinding process described above is purely speculative. In addition, based on existing evidence and data, it provides a reasonable explanation for solving the topological problems in E.coli DNA replication. It is different from all the proposals and deserves careful examination.
Mathematics Helps Resolve the Debate Over Entanglement in the Double Helix
Many people may be surprised to learn that mathematics is not a science, because it is based on axioms and uses strict logical rules to arrive at irrefutable conclusions. Mathematical conclusions are always correct and cannot be falsified. Perhaps it was for this reason that Alfred Nobel did not include mathematics as a science in his will.
We should admire Nobel's wisdom, as this design effectively avoids debates about which mathematical conjectures should be rewarded for their great achievements, or which doomsday prophecies should be considered genuine threats to humanity. It is often thought that biological research does not require mathematics. Yet, mathematics can help biology move from qualitative descriptions to quantitative analysis of complex phenomena. Gregor Mendel, the father of genetics, used probability theory and statistics. Topology and Gaussian distributions have also proven their value in the study of DNA structure [25].
Topological Evidence from Plasmids Is Undeniable
Topological rules were first proposed by Vinograd et al. after the discovery of circular DNA. The properties of plasmids are particularly well-suited for quantitative analysis of their three main characteristics: the linking number Lk (the number of links between the two circular strands), the total twist number Tw, and the writhe number Wr. A simple formula is Lk = Tw + Wr, or L = T + W.
Although this formula is simple, truly understanding it remains one of the most difficult topics for many doctoral students. The tertiary mathematical problems are rather difficult to visualize or solve, even a mathematician can make mistakes [26].
For example, when the hydrogen bonds between the two circular DNA strands of a plasmid in an aqueous solution break, countless different shapes are produced, and these shapes are also influenced by many surrounding environmental conditions. Only topological rules can be used to study them.
Among all set of topoisomers of a plasmid, there is a special one with zero linking number, in which its two circular strands can be completely separated when the hydrogen bonds are broken. The experimental result of such finding is not depending on any kind of DNA structure models but can clearly prove the two strands are not linked [6].
There are several different explanations for this finding. The most common reaction is to dismiss it as an artifact, believing it must be incorrect. However, electron microscopy is a powerful and indispensable tool in scientific research. Its observations are generally reliable and reproducible, greatly expanding our ability to observe microscopic objects invisible to the naked eye. Provided the correct methods and sample preparation techniques are used, its observations should be considered conclusive evidence.
Another reason for opposing this discovery is the typical Pavlovian conditioned response. This is a cognitive bias stemming from knowledge repeatedly instilled by textbooks and mentors, as well as concerns that the potential consequences of this report could shake the classic double-helix model that the mainstream scientific community has accepted for decades.
This evidence clearly demonstrates that the two DNA strands are not always tightly intertwined. This topological evidence comes directly from experimental results, not theoretical calculations. Numerous direct and indirect experimental reports from around the world support this evidence and have become important grounds for challenging the third part of the double helix structure [6,7,25].
The Formation of Circular DNA Follows the Rule of Gaussian Distribution
Plasmid supercoiling is affected by enzymatic activity, such as topoisomerases, and by physical processes like transcription and replication, which generate torsional stress. Cellular and environmental factors like temperature, DNA-binding proteins (e.g., HU protein), and the composition of the growth medium also play a role in regulating the degree of supercoiling.
It is worth noting that the peaks of the topoisomers observed in the experiment followed a Gaussian distribution. Many studies have indicated that the number of topoisomers is less than 10.
Gaussian (normal) distribution has been applied to the study of plasmids.
In 1975, Keller discovered that the degree of supercoiling of pure SV40 DNA (a plasmid containing 5224 base pairs) could be determined using agarose gel electrophoresis (AGE). He was a pioneer in the field of DNA topology [27].
Purified supercoiled SV40 DNA typically exists in the form of a set of topoisomers, exhibiting a Gaussian distribution centered around on a mean value. By contrast, there are significant differences in the interpretation of this mean value between the B-DNA model and the ambidextrous double- helix model.
As shown in Figure 3, we attempt to explain the implications of these experimental results from two perspectives, so that readers can weigh their validity and credibility.
If DNA contains 10 base pairs per turn, as in the Watson-Crick Model, then anyone can easily determine that the relaxed SV40 topoisomer in Figure 3a lane 1 has a linking number of 522 (L = 522) and a superhelical number of zero (W=0). Whereas, according to ambidextrous model, both the linking number and the superhelical number of this relaxed plasmid are zero.
As shown in Figure 3b. electrophoresis at 4 °C increases helical twist relative to the relaxation temperature (37 °C), producing an estimated +6 superhelical turns under the conditions used.”
The 4 ºC decrease caused this number is calculated according to ratio of temperature difference in two AGE tests to the differences of temperature at relaxation reaction and test. (Suppose the test of AGE is carried at 22 ºC. )
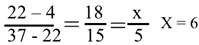
This rotation can also be calculated by the way of Depew, D. E. & Wang, J. C. [28]
Increasing the EthBr during relaxation reaction may cause the two strands twist left ward but when the sample was made and during examination, its two strands should turn back to more negatively supercoiled form as shown in figure c and figure d.
Increasing the EthBr in the agarose gel may cause the DNA winding right handedly and their total twisting number decreases.
Using Gaussian distribution, the topological characteristics of SV40 DNA can be clearly revealed. The presented data show significant differences between the two DNA models. This helps interested parties determine which model better reflects the actual situation.
The Old Definitions in DNA Topology Needs to Be Updated
Superhelical density (σ, sigma) is defined as a value used to estimate the degree of supercoiling in plasmid.[21,26] In traditional DNA topology, σ = (L – L0) / L0. L is the actual linking number (number of strand crossings), and L0 is the linking number in the relaxed state. According to the Watson-Crick Model, for a plasmid with N base pairs, assuming that each turn of the DNA contains 10 base pairs, the linking number of the plasmid in the relaxed state is always a large integer, i.e., L0 = T + W ≈ N/10 + W >> 0.
Interestingly, the statement "any number divided by it equals one" is not entirely correct, because mathematics does not allow zero to be used as a denominator. Since the discovery of zero-linking number topoisomer, this definition of superhelical density σ seems outdated and mathematically unsound.
In order to study the supercoiling of plasmid, supercoiled index (Si) is proposed to replace the superhelical density. It relates to two parameters: the number of twists number W and base pairs number N of the plasmid. i.e., Si = Wr / (N/10.4). The reported σ value can still be used as Si value that is a good indication for comparing the supercoiling of any topoisomers of different plasmids.
Therefore, this is the first rule in the old DNA topology that needs to be updated.
The second rule in the old DNA topology model, which relates to the orientation of DNA, is difficult to discern but also needs updating.
The conventional assignment of right-handed DNA as ‘positive’ rests on an analogy in which a ladder rotated clockwise around its axis generates a right-handed helix. While pedagogically convenient, this analogy conflates extrinsic rotation with intrinsic helical sense and should be abandoned in rigorous topological discussions [25].
Figure 4 provides a detailed analysis of the problems related to DNA orientation. In the Euclidean coordinate system, each intersection point can be defined as +1 for a right turn and -1 for a left turn.
Because the two strands are antiparallel, according to Euclidean coordinates, their total twist number should be –N/10 << 0, as shown in Figure 4B. This old practice is unacceptable because it violates both the facts and mathematical rules.
Numerous facts demonstrate that many long-serving frontline workers are more likely to encounter new problems they have never faced before, or to invent new methods prohibited by the rules to overcome difficulties.
Even the most brilliant minds make mistakes when exploring uncharted territory. Therefore, we should maintain a clear head, listen carefully to different opinions, and adopt reasonable suggestions, rather than becoming complacent and clinging to outdated ideas.
Making such changes is no easy task, as old ideas are deeply ingrained, and even the most educated people need time to adapt to the new rules. However, in today's rapidly developing technological world, everyone must constantly update their knowledge to avoid falling behind.
The benefits of changing the rules may not be immediately apparent, but it can standardize the terminology used in scientific fields and avoid contradictions. Once people become accustomed to the new rules, there will be no problems.
The Reasons for DNA Supercoiling
Once you understand the two new rules of DNA topology mentioned above, it becomes easy to understand the reasons for DNA supercoiling.
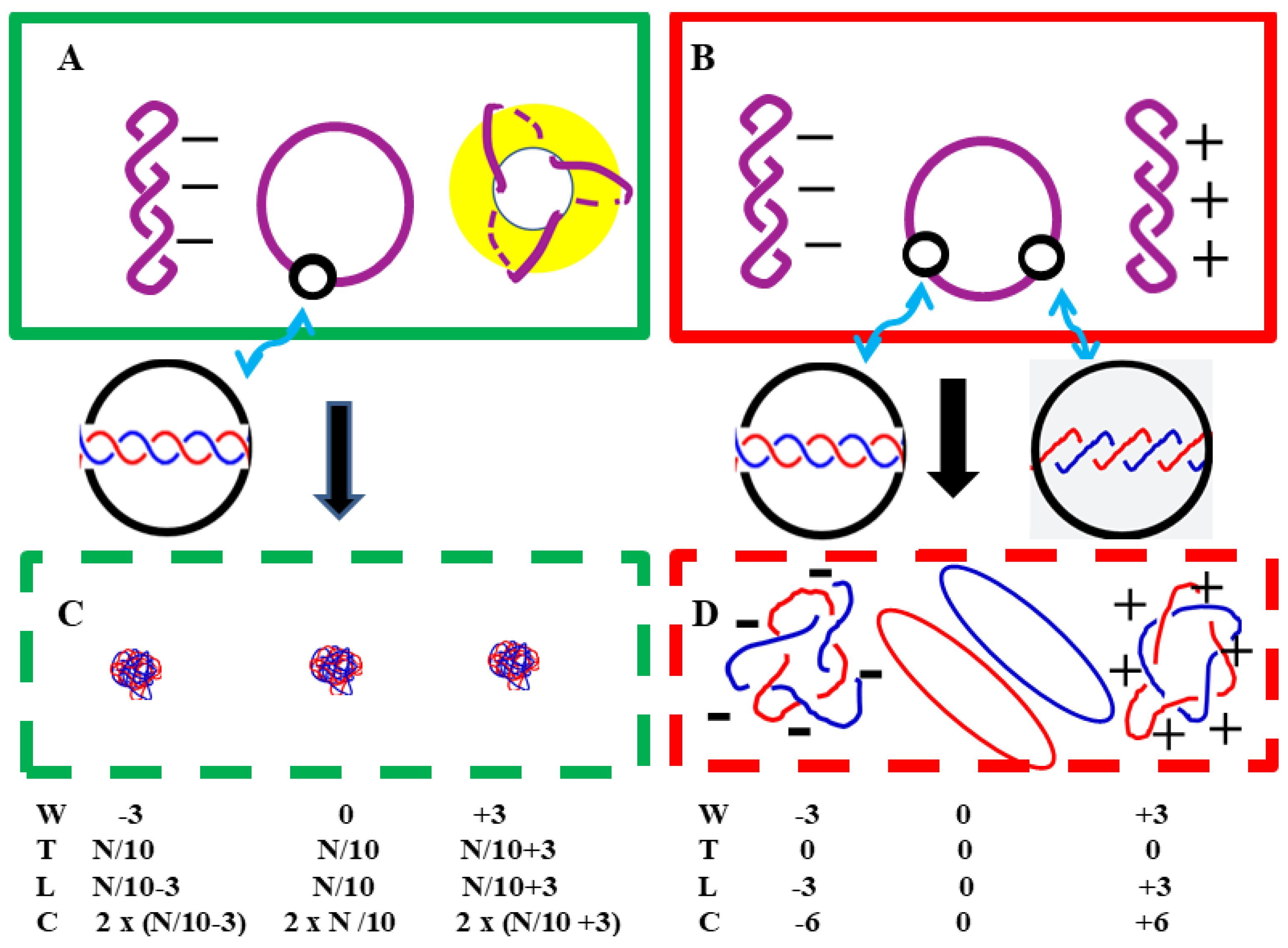
According to the Watson-Crick Model, in negatively supercoiled plasmids, the total twist number T is less than the normal value, i.e., T < N/10; while in positively supercoiled plasmids, the opposite is true, i.e., T > N/10. The question then arises: to what extent can the double helix structure be twisted in positively supercoiled plasmid? Since DNA is composed of carbon, hydrogen, oxygen, nitrogen, and phosphorus atoms, these atoms are incompressible. This question has puzzled researchers ever since the existence of highly positively supercoiled pBR322 was reported and confirmed by two-dimensional agarose gel electrophoresis [29]. After careful consideration, another explanation is reasonable enough to explain why positive supercoiling can exist. Figure 5 shows a schematic diagram of the relaxed plasmid in the two DNA models.
As shown in Figure 5, the differences in appearance between the relaxed plasmids in the two structural models are obvious. The reason for this difference is simple: their total twist number T and the linking number L are different.
In aqueous solution, the morphology of negatively supercoiled plasmids was similar in both DNA models. The DNA duplex is rotating to the right.
There are significant differences in the structure of the positively supercoiled plasmids in the two models (A and B). In the Watson-Crick Model, the DNA duplex states in a toroidal form. In the ambidextrous model, the DNA duplex coil in plectonemic way turning leftward.
Even the AI's answer to the question "What would happen if all the hydrogen bonds in the plasmid were broken?" was very formal, lacking depth, and superficial, and did not match the experimental results.
In B-DNA, when the two denatured single-stranded circular chains of a plasmid are projected onto a plane, the number of crossover points between the two strands is C = 2 × (N/10). Normally a plasmid contains hundreds or thousands of base pairs, so this number should be very large, ultimately leading to the two single-stranded circular DNA molecules becoming tightly intertwined, forming a random coil structure as shown in Figure 5C.
However, what we found is the two strands of denatured supercoiled pBR322 DNA appeared as a pair of loosely tangled rings.[6] Surprisingly under the EM, the denatured relaxed pBR322 DNA with 3 negative supercoiling looks just have 6 crossings per plasmid as schematically shown in Figure 5D.
These facts indicate that the two DNA strands in the plasmid are actually coiled in opposite directions, and this opposite coiling results in a very low total twist number. When a plasmid is completely relaxed (W = 0), its two circular strands can be completely separated, i.e., L = 0.
Additional experimental results show that left-handed DNA is slightly different from right-handed DNA, resembling Z-DNA with 12 bp per turn. [30]
Experiments from other laboratories indicated that topoisomers of a 336 bp mini-plasmid with the same supercoiling index but opposite supercoiling differ in shape, electrophoretic mobility, or chemical properties. [31] Surprisingly, the electrophoretic mobility (μ) of the 336 bp plasmid showed significant differences between topoisomers with the same but oppositely supercoiling. In other words:
μ ( L = -1) >> μ ( L = +1)
μ ( L = -2) ≈μ ( L = +2)
μ ( L = -3) << μ ( L = +3)
These phenomena cannot be explained by the Watson-Crick Model. It indicates that each pair of plasmids is not a pair of enantiomers.
The research published by Li et al. also shows that positively supercoiled plasmids and negatively supercoiled plasmids have opposite winding directions.[32] They react very differently to T7 endonuclease 1.
Under atomic force microscopy (AFM), the DNA duplex of negative pBR322 DNA can be clearly seen, and appeared as turning right-handedly. But DNA duplex of positive pBR322 DNA appeared as turning left handedly. This is consistent with the claims of the ambidextrous model as shown in Figure 5 B.
Furthermore, in pBR322 DNA, the length of positive supercoils is greater than the length of negative supercoils. These results were obtained through direct observation using atomic force microscopy (AFM) and are independent of any theoretical model. This is consistent with our previous findings that there may be more left-handed DNA containing 12 base pairs per turn in positively supercoiled pBR322 DNA.
Therefore, these results reliably demonstrate that the ambidextrous model accurately reflects the real-world situation.
Discussion
As the blueprint for all life, DNA is crucial because it is intimately involved in many biological functions. Scientists have figured out how genetic information is stored, how it is accurately replicated during cell division, and how this information is translated into proteins and various RNAs.
However, the structure and function of DNA are very complex and must be considered as a whole; theoretically, it should be unified and capable of explaining all the facts.
Since the discovery of the double helix structure, experts in various fields have published countless research papers. No one has the time or ability to read or understand all the techniques, methods, or interpretations of the results. Most scientists can only keep up with a portion of the advancements in their own field, and information bias is inevitable.
Thanks to years of research by numerous scientists, the structure and function of DNA have become increasingly clear, leaving a profound impression on almost every educated citizen. However, the challenge of fully unraveling the double helix structure remains unsolved.
There are several reasons why the problems in DNA structure went undiscovered:
- Due to the explosive growth of knowledge, scientists had many other interesting and important research topics besides studying DNA structure.
- Information bias led scientists to focus on their own research tasks without noticing that their research might be related to the secondary or tertiary structure of DNA. A famous example is Chargaff's mistake; his diligent work did not lead him to discover the base pairing rules or the double helix structure.
- Experts suffered from confirmation bias; they were overly confident in their previously acquired knowledge and unable to accept any views that contradicted their own.
- Expressing an opinion that contradicts widely accepted views requires immense courage, and this courage stems from determination, which in turn stems from strong self-confidence. Ultimately, this self-confidence comes from personal experience and a deep trust in the firsthand observations of frontline workers or researchers. Few people possess both strong mental and physical resilience and a willingness to take risks.
- Few people are willing to invest the time and effort into a challenging project that doesn't guarantee any results or rewards.
Not all challenges are worth pursuing. For anyone seriously seeking truth, choosing the right challenge at the right time, in the right place, and within the right social context is crucial. Typically, before deciding to accept a challenge, scientists must assess whether the challenge is meaningful and whether they have the capacity to meet it.
For many scientists, studying the secondary or tertiary structure of DNA seems extremely difficult because there are currently no instruments capable of clearly observing the details of plasmids or chromosomes. Even the most advanced atomic force microscopes can only obtain blurry images of the DNA double helix structure. We should strive to find some better methods to solve this problem.
Conducting scientific research is by no means easy, but it can be viewed as participating in an exciting jigsaw puzzle. Not all the puzzle pieces are readily available; most are scattered throughout the literature worldwide. Sometimes, you even need to find or create the missing pieces yourself. Furthermore, there are no standard answers, and no one can tell you exactly what to do. You need to collaborate with others, working together to ultimately piece all the fragments together perfectly, finally completing a flawless and comprehensive picture.
Currently, this DNA structure puzzle has been assembled, but it is not yet perfect and still contains some errors. We hope this project will be completed in the near future, providing a reasonable solution to the entanglement problem during DNA replication. Furthermore, all children will be able to learn a clear explanation of DNA structure and its functions from this project.
Perhaps the impact of publishing this paper will be negligible, like a tree falling in a forest—almost no one will notice. Questions about DNA structure and its replication still remain. We cannot expect deeply ingrained old ideas to change overnight.
Most people prefer to hear what they want to hear. Empirical evidence that contradicts long-standing models may initially be met with skepticism or disregard, a response that underscores the importance of reproducible data and open discourse. The views expressed in this article may offend some experts and professionals, as they challenge the very foundation of their confidence.
Scientific knowledge is objective, universal, and also provisional. All scientific claims, methods, and results should be free from personal bias, cultural influence, or subjective interpretation. Any evidence that contradicts existing theories should be carefully examined, and flawed theories should be revised or discarded.
In summary, after more than seventy years of research, the questions regarding the secondary structure of DNA and how the two strands unwind remain unanswered. We are attempting to address this problem using different approaches.
The role of scientists is not to persuade others to believe their own opinions, but to convince people of the validity with evidence. Topological evidence convincingly demonstrates that the two strands are not permanently intertwined in a canonical B-form helix; the precise geometric path of the duplex in vivo remains to be fully elucidated.
This article only provides some clues for those willing to seek the truth. As a modification to the third part of the Watson-Crick Model, it remains a scientific hypothesis that needs to be verified.
In 2011, we propose a double-helix conjecture to test our hypothesis. [33] If the hypothesis about secondary structure is correct, then it should be possible to find a topoisomer with a linking number of zero in any type of plasmid. However, achieving this may be very difficult, and so far, no one has successfully verified this prediction.
To alleviate the difficulties encountered in the experiments, two additional predictions were proposed [25]:
1. The melting point of the acidic form of plasmids in the relaxed or supercoiled state can be easily measured.
2. There are significant differences in the dissociation kinetics of the acidic form of plasmids between the supercoiled and relaxed states.
These predictions are reasonable inferences based on the ambidextrous double-helix model and these results could not be obtained from the Watson-Crick Model. Verifying these predictions will help us distinguish between correct and incorrect theories.
The mysteries of DNA structure and function pose a tremendous challenge to our understanding of the brain! Hopefully, some clever new ideas or breakthroughs in methodology will help us unravel this unsolved mystery, this "known but unknown" problem.
Acknowledgments
The author sincerely thanks all the scholars, family members, and friends who provided him with help, support, and encouragement during this independent research project. Thanks Zhang, K. J. and Xu, L. for their meticulous review and valuable suggestions.
References
References
- Watson, J. D. & Crick & F. H. C., (1953) Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature, 171, 737. [CrossRef]
- Hernandez, V. (2018) The debate over DNA replication before the Meselson-Stahl experiment (1953-1957). Embryo Project Encyclopedia. ISSN: 1940-5030.
- Ciurea, A. V. et al. (2024) Celebrating 70 years of DNA discovery: exploring the Blueprint of Life. J. Med. Life, Apr;17(4):387–391. [CrossRef]
- Xu Y.C. Qian, L. and Tao, Z. J.(1982) A hypothesis of DNA structure. Scientia Sinaca, 25B, 827—836.
- Xu Y. C. and Qian, L.(1983) Determination of the linking number of pBR322 DNA. Scientia Sinica, 26B, 602—613.
- Xu, Y. C.(2009) Finding of a zero linking number topoisomer. Biochimica et Biophysica Acta 1790, 126- 133. Y. C.
- Xu, Y. C. (2019) Evidence falsifying the double helix model. Symmetry, 11,1445. (December 30,2020 Reprinted in eBook 《Prime Archives in Symmetry》. [CrossRef]
- Vale, R. D. (2015) Accelerating scientific publication in biology. Proc. Nat. Acad. Sci. USA Vol. 112, 13439-13446. [CrossRef]
- Wang, J. C. (2009) Untangling the double helix. Cold spring harbor laboratory Press. [CrossRef]
- A. Rich, (2004) The excitement of discovery. Ann. Rev. Biochem. 73, 1-37. 2004.
- Holmes, F. L. (2001)Meselson, Stahl, and the Replication of DNA: A History of 'The Most Beautiful Experiment in Biology ' Yale University Press.
- F. R. C. Crick (1990) What mad pursuit Penguin Books.
- Lovett, S. T & Segall, A. M. (2004) New views of the bacterial chromosome . The EMBO Reports, Volume 5, pages 860–864. [CrossRef]
- Japaridze, A. et al., (2020) Direct observation of independently moving replisomes in Escherichia coli. Nature communication 11, 3109.
- Gras, k. et al., (2024) The Escherichia coli chromosome moves to the replisome. Nature communication 10, 1038.
- Cyriax, B. and Gath, R. (1978). The Conformation of Double-Stranded DNA. Naturwissenschaften 65, 106108. [CrossRef]
- Rodley, G. A. et al. (1976). A possible conformation for double stranded polynucleotides. Proc. Natl. Acad. Sci. USA, 73, 2959-2963. [CrossRef]
- Sasisekharan, V. Pattabiraman, N. and Goutam, G. (1978). Some implications of an alternative structure for DNA, Proc. Natl. Acad. Sci. USA 75, 4092-4096. [CrossRef]
- Crick, F. R. C. et al., (1979) Is DNA Really a Double Helix ? J. Mol. Biol. (1979), 129, 449—461. [CrossRef]
- Ullsperger, C & Cozzarelli, N.R. (1996). Contrasting enzymatic activities of topoisomerase IV and gyrase from Escherichia coli. J. Biol. Chem., 271, 31549-31555.
- Wang, J. C. (2009) Untangling the double helix. Cold spring harbor laboratory Press. [CrossRef]
- Stracy, M., et al., (2019) Single-molecule imaging of DNA gyrase activity in living Escherichia coli. Nucleic Acids Research, 47, 210-220. [CrossRef]
- Schvartzman, J. B., (2019) Closing the DNA replication cycle: from simple circular molecules to supercoiled and knotted DNA. Nucleic Acids Research, 2019, 47(14), 7182-7198. [CrossRef]
- Ashley, R. E. et al. (2017) Activities of gyrase and topoisomerase IV on positively supercoiled DNA. Nucleic Acids Research, 45(16), 9611-9824.
- Xu, YC, (2025). Some More Facts that the Watson-Crick Model Cannot Explain. Enliven: J Genet Mol Cell Biol. 14(1): 001.
- Sumners, D. W., (1987), The role of knot theory in DNA research. In Geometry and topology, Ed. McCrory,C & Shifrin, T. Mercel Dekker INC. [CrossRef]
- Keller, W. ( 1975) Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc. Nat. Acad. Sci. USA Vol. 72, No. 12, pp. 4876-4880. [CrossRef]
- Depew, D.E. & Wang, J.C. (1975). Conformation fluctuations of DNA helix. Proc. Natl. Acad. Sci. USA, 72, 4275-4279. [CrossRef]
- Lockshon, D. & Morris, D.R. (1983). Positively supercoiled DNA is produced by treatment of Escherichia coli with DNA gyrase inhibitor. Nucleic Acids Res. 11, 2999-3017. [CrossRef]
- Xu, YC, (2014) Helical repeats of left-handed DNA, Open journal of Molecular and integrative physiology, 4, 23-26. [CrossRef]
- Irobalieva, et al. (2015). Structural diversity of supercoiled DNA. Nature communication, volume 6, Article number: 8440. [CrossRef]
- Li, D. et al., (2017). Direct observation of positive supercoils introduced by reverse gyrase through atomic force microscopy. Bioorg Med Che Lett. 27 (17), 4086-4090.
- Xu, Y. C, (2011) Replication Demands an Amendment of the Double Helix. In book: (Seligmann, H., Ed., DNA Replication-Current Advances, InTech, Rijeka, 29-56.
Figure 1.
Schematic drawing of relative movement of parental DNA and replisome. Note: The length of DNA with 3000 base pairs is about 1μ.
Figure 1.
Schematic drawing of relative movement of parental DNA and replisome. Note: The length of DNA with 3000 base pairs is about 1μ.
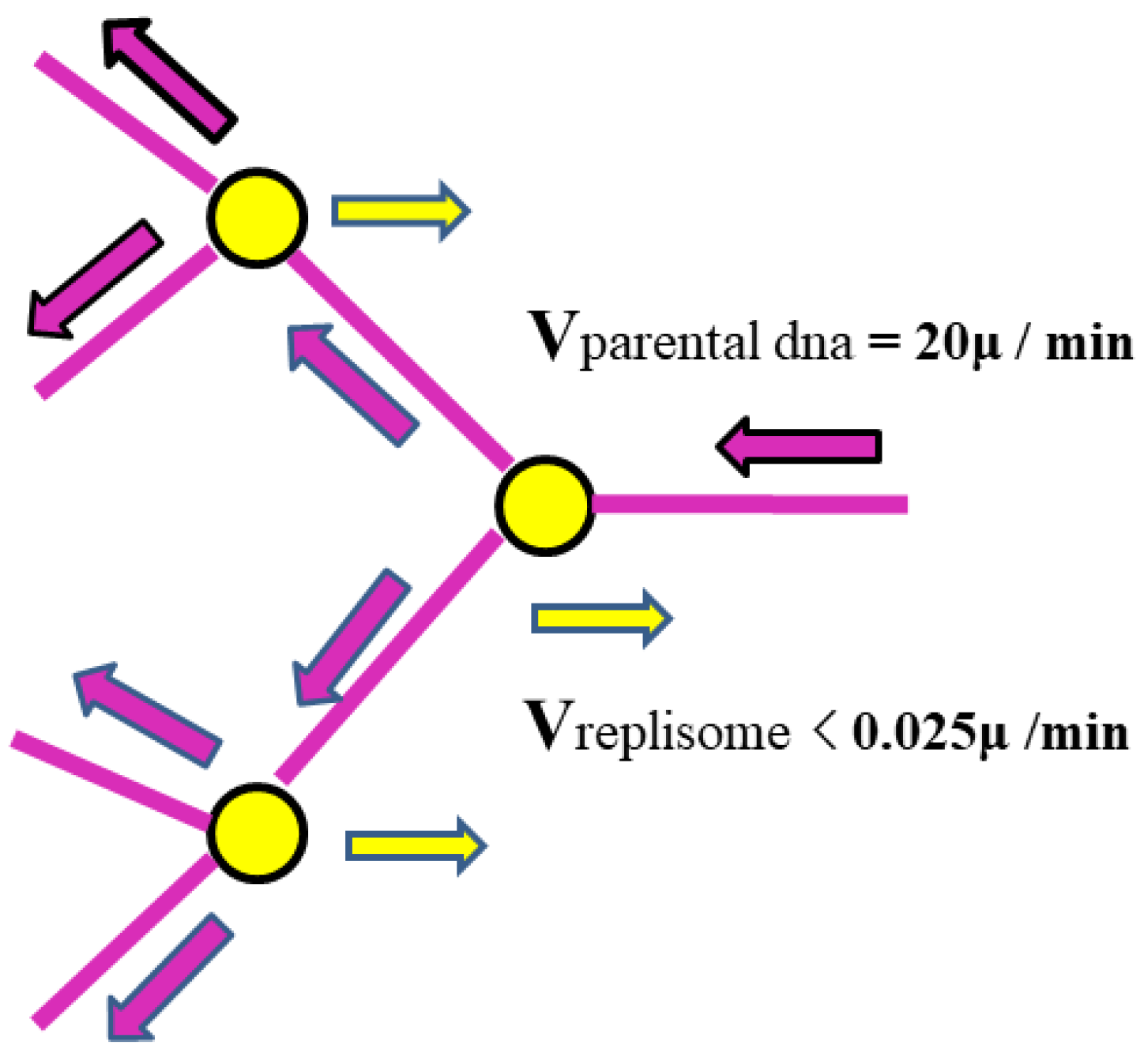
Figure 2.
shows the way helicase separates parental DNA. It does not rapidly unwind the two strands at a rate of 6000 turns per minute, as required by the Watson-Crick Model, because no such rule is stipulated in the ambidextrous double-helix DNA model.
Figure 2.
shows the way helicase separates parental DNA. It does not rapidly unwind the two strands at a rate of 6000 turns per minute, as required by the Watson-Crick Model, because no such rule is stipulated in the ambidextrous double-helix DNA model.
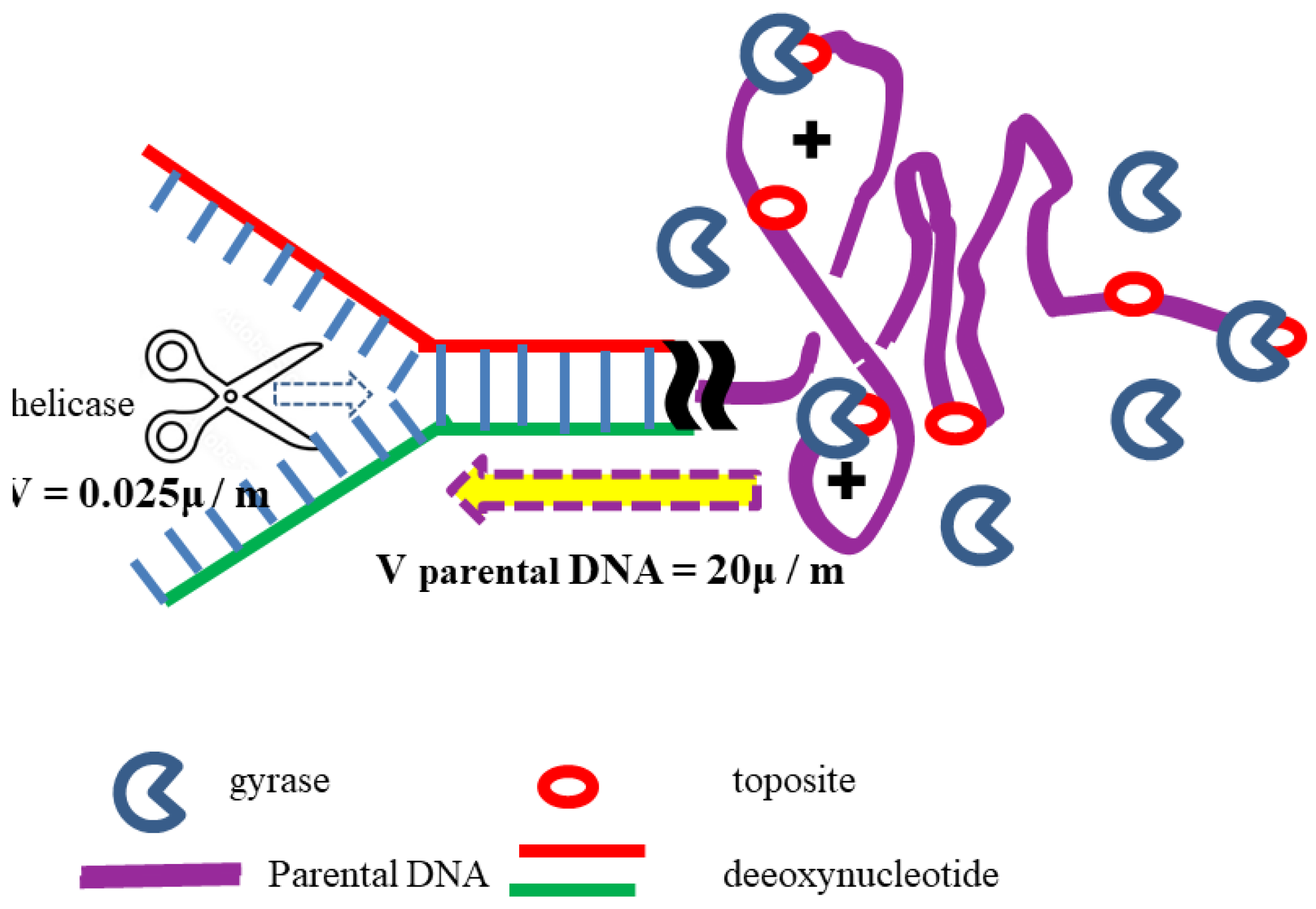
Figure 3.
Topological properties of SV40. Image excerpted from Keller's research [27]. In this figure, native SV40 DNA was relaxed with topoisomerase at 37°C and analyzed by agarose gel electrophoresis (AGE) on a 1.4% agarose gel at 4 V/cm. The gel contained various concentrations of EtdBr as indicated; electrophoresis was performed primarily at room temperature, with one run at 4°C. Lanes 1 to 10 were relaxed by topoisomerase, with buffer containing 0, 0.25, 0.5, 1.0, 1.5, 2.5, 3.5, 5, and 6.9 μM EtdBr respectively; lane 11 is native SV40 DNA. The data in the green box are estimated values based on the ambidextrous model. The data in the red dashed box are estimated values based on the Watson-Crick Model.
Figure 3.
Topological properties of SV40. Image excerpted from Keller's research [27]. In this figure, native SV40 DNA was relaxed with topoisomerase at 37°C and analyzed by agarose gel electrophoresis (AGE) on a 1.4% agarose gel at 4 V/cm. The gel contained various concentrations of EtdBr as indicated; electrophoresis was performed primarily at room temperature, with one run at 4°C. Lanes 1 to 10 were relaxed by topoisomerase, with buffer containing 0, 0.25, 0.5, 1.0, 1.5, 2.5, 3.5, 5, and 6.9 μM EtdBr respectively; lane 11 is native SV40 DNA. The data in the green box are estimated values based on the ambidextrous model. The data in the red dashed box are estimated values based on the Watson-Crick Model.
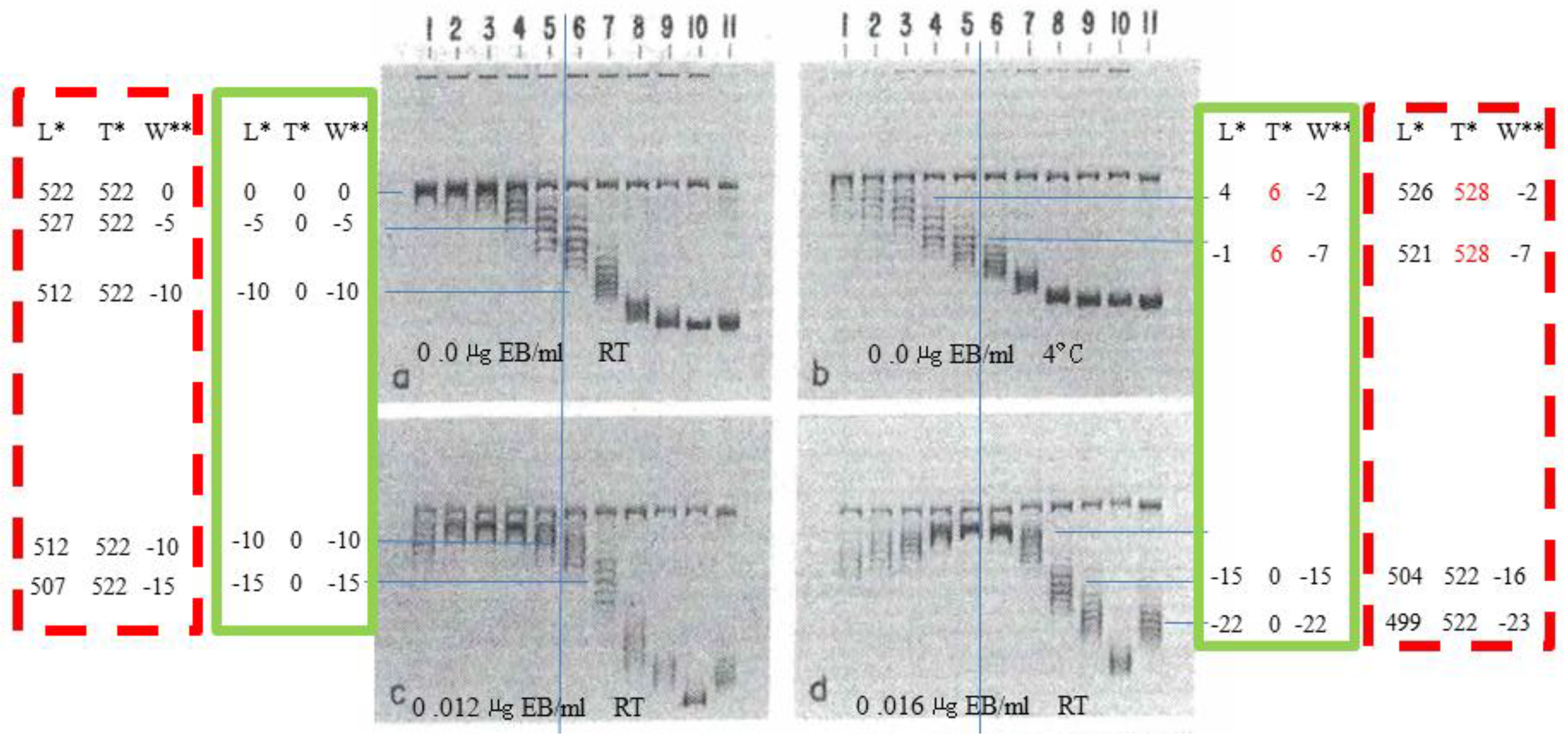
Figure 4.
The orientation of right-handed helical DNA is usually assumed to be positive, but it should actually be considered negative. A. Traditionally, right-handed helical DNA is considered positive; B. Scientific analysis of right-handed helical DNA suggests that it should be defined as negative; C. When the two strands have the same direction, the right-handed double helix structure can be considered positive.
Figure 4.
The orientation of right-handed helical DNA is usually assumed to be positive, but it should actually be considered negative. A. Traditionally, right-handed helical DNA is considered positive; B. Scientific analysis of right-handed helical DNA suggests that it should be defined as negative; C. When the two strands have the same direction, the right-handed double helix structure can be considered positive.
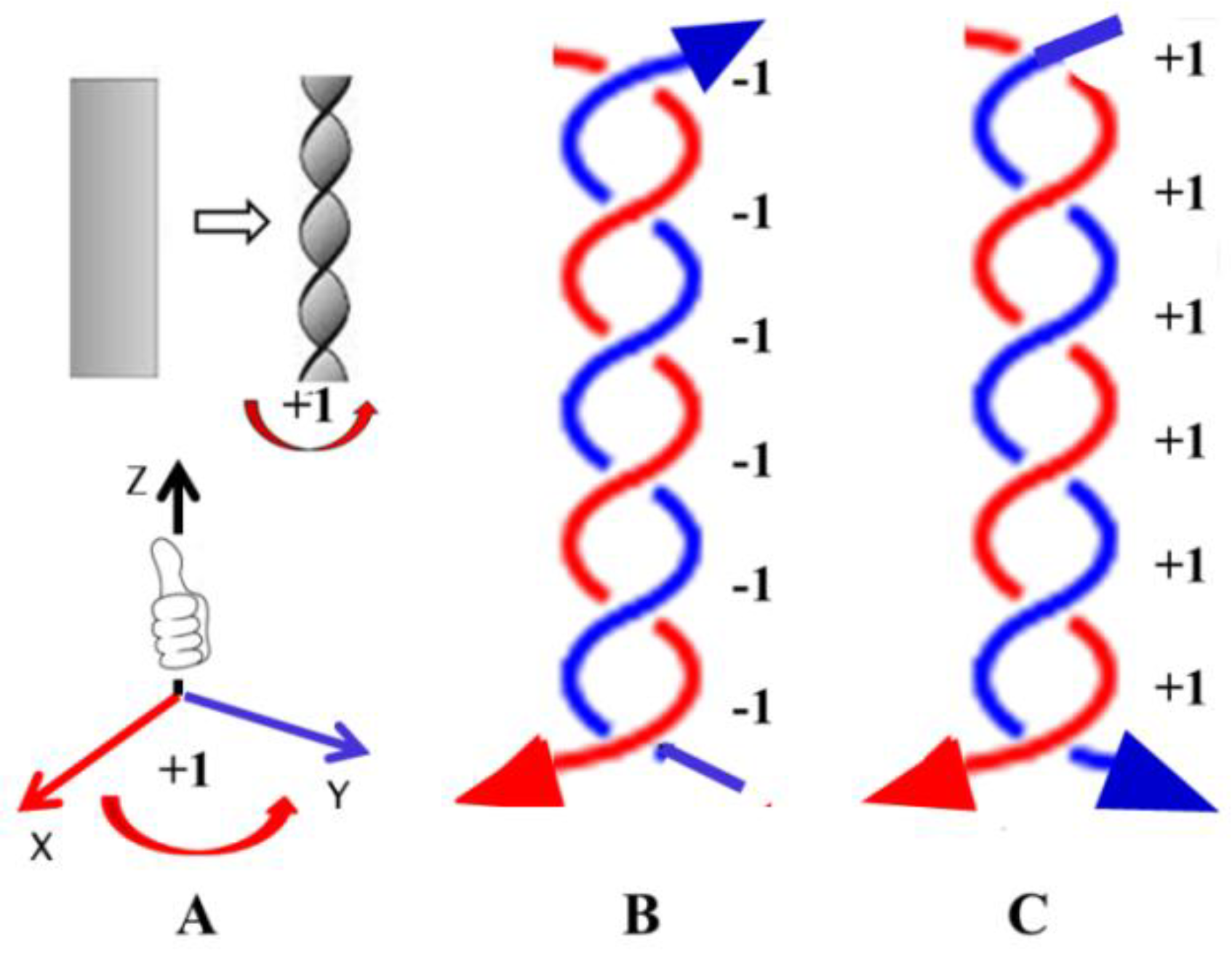
Figure 5.
Schematic diagrams of relaxed plasmids in two DNA models. A and B represent plasmids in an aqueous solution; C and D represent plasmids where all hydrogen bonds between complementary strands have been broken; green boxes A and C represent DNA conforming to the Watson-Crick Model, with 10 base pairs per turn; and boxes B and D represent DNA conforming to the ambidextrous double-helix model. The black circles represent magnified portions of the DNA double-stranded fragments.
Figure 5.
Schematic diagrams of relaxed plasmids in two DNA models. A and B represent plasmids in an aqueous solution; C and D represent plasmids where all hydrogen bonds between complementary strands have been broken; green boxes A and C represent DNA conforming to the Watson-Crick Model, with 10 base pairs per turn; and boxes B and D represent DNA conforming to the ambidextrous double-helix model. The black circles represent magnified portions of the DNA double-stranded fragments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



