
Submitted:
07 January 2026
Posted:
08 January 2026
You are already at the latest version
Abstract
Background: Major depressive disorder (MDD) is a highly heritable psychiatric condition with complex polygenic architecture. Competing hypotheses emphasize glutamatergic/synaptic plasticity deficits or neurodevelopmental synaptic pruning dysregulation, but integrated testing across large-scale genetic data remains limited.Methods: We re-analyzed the latest Psychiatric Genomics Consortium MDD GWAS (approximately 358,000 cases and 1.28 million controls, European ancestry) using gene-based and competitive gene-set testing (MAGMA), partitioned heritability (LDSC), transcriptome-wide association studies (TWAS with GTEx v8 brain models), and two-sample Mendelian randomization (MR) with cognitive reserve proxies (e.g., educational attainment).Results: MAGMA and LDSC revealed robust enrichment in synaptic pruning-related gene sets (Bonferroni-corrected p < 0.001; up to 1.30-fold LD-adjusted heritability enrichment, p < 10-91), surpassing glutamatergic/plasticity sets (moderate MAGMA enrichment, p = 0.014; no LDSC signal). TWAS showed modest glutamatergic enrichment (1.10-fold mean |Z|, p = 0.007) with heterogeneous directions, while pruning sets were null in TWAS despite strong polygenic signals. MR demonstrated causal protective effects of genetically proxied cognitive reserve on MDD risk (e.g., educational attainment OR = 0.72, 95% CI [0.66–0.79], p = 7.53 × 10-14).Conclusions: These findings prioritize developmental synaptic pruning dysregulation as the primary polygenic substrate of MDD, with downstream impairments in neuroplasticity and cognitive reserve mediating vulnerability. We propose a "pruning-mediated plasticity deficit" framework, integrating neuroimmune and circuit-level mechanisms, with implications for novel therapeutics targeting pruning pathways or plasticity enhancers.
Keywords:
depression
; plasticity
; pruning
; mendelian randomization
; MDD
; MAGMA
; LDSC
; TWAS
Introduction
Major depressive disorder (MDD) is now thought to affect about 280 million people and sits near the top of the global disability charts [1]. In the United States, close to one person in six will meet diagnostic criteria at some point, and roughly 6% will experience a full episode in any given year [2]. Although the clinical picture revolves around low mood, anhedonia and cognitive fog, genetics play a sizeable part: family and twin work puts heritability somewhere between 30% and 50% [3].
Large genome-wide association studies have begun to colour in that genetic outline. More than a hundred loci have now cleared the genome-wide bar, repeatedly implicating genes that handle neuronal signalling, keep synapses healthy or fine-tune immune responses [4,5]. Even so, the mosaic is far from complete, and we still do not know which biological themes really matter most for risk.
Two main ideas currently share the stage. The first is the glutamatergic, or synaptic-plasticity, model. It argues that sluggish excitatory signalling and a reduced ability to remodel circuits drive depressive symptoms—a notion boosted by the rapid lift in mood many patients feel after a dose of ketamine [6].
The second idea reaches further back in time. The pruning hypothesis suggests that during adolescence microglia and complement proteins may cut away too many synapses, leaving cortical and limbic networks thin and brittle so that later stress tips them into depression. Strong support for this mechanism comes from schizophrenia research, where common variation in complement component 4 (C4) and related genes has been tied to over-zealous pruning [7,8]. Given the genetic overlap between schizophrenia and MDD, the same process could plausibly shape depression risk.
To tease these models apart, we revisited the newest Psychiatric Genomics Consortium dataset—about 358,000 cases and 1.28 million controls of European ancestry [9]. Four lenses were applied:
- MAGMA for gene- and pathway-level enrichment;
- LD-score regression to see how heritability is partitioned;
- TWAS to link risk variants to gene expression in specific brain regions;
- Mendelian randomization (MR) to test directionality, using cognitive-reserve traits such as educational attainment as protective exposures.
Crucially, we asked not just whether pruning or glutamatergic pathways were enriched, but whether the risk alleles were predicted to dial activity up or down—information that matters for drug development.
By weaving these complementary approaches together, we aim to weigh the relative influence of microglial pruning versus glutamatergic plasticity on depression risk, clarify the direction of their effects, and locate any crossroads where the two processes meet.
Methods
MAGMA Analysis
Gene-based tests were run with MAGMA v1.10 [10] using summary statistics from the Psychiatric Genomics Consortium 2025 meta-analysis of major depressive disorder (MDD). The GWAS comprised 357 636 cases and 1 281 936 controls of European ancestry, with locus-specific effective sample sizes averaging 8.3 × 105 after removal of 23andMe and UK Biobank cohorts [9]. Variants were annotated to Ensembl genes (hg19, NCBI 37.3) with windows extending 35 kb upstream and 10 kb downstream. Linkage disequilibrium was modelled with the European panel of the 1000 Genomes Project. After annotation and quality control 18 343 genes remained; genome-wide significance for an individual gene was set at P < 2.73 × 10−6 (Bonferroni 0.05/18 343).
Competitive gene-set analysis evaluated seven prespecified collections: the original 23-gene CGR drug-target list, an expanded 130-gene glutamate/plasticity panel, a 38-gene shortened pruning list, a 262-gene expanded pruning list, a 225-gene “specific” pruning subset from which glutamatergic overlaps were removed, and two negative controls (104 monoaminergic genes and 184 housekeeping brain genes). Within each set the mean MAGMA Z-score was contrasted against the genomic background in a one-sided framework. Significance thresholds were Bonferroni-corrected across the seven tests (P < 0.0071) and by a false-discovery rate of 0.05.
Partitioned Heritability Analysis
We quantified the contribution of predefined functional groups to the common-variant heritability of major depressive disorder (MDD) with stratified linkage disequilibrium score regression [11]. The input GWAS summary statistics were identical to those used in the gene-based work, namely the Psychiatric Genomics Consortium 2025 European-ancestry meta-analysis that excludes 23andMe and UK Biobank cohorts [9]. Linkage disequilibrium scores were taken from the 1000 Genomes Project European reference panel; only variants with minor-allele frequency above 5 per cent were retained.
Seven binary annotations mirrored the a priori gene sets evaluated earlier. Each gene was flanked by 10 kb upstream and downstream to include proximal regulatory sequence, and the resulting intervals were converted to SNP-level flags on the autosomes. For every annotation we computed the mean χ2 statistic across flagged SNPs and compared it with the mean for all other SNPs, adjusting for local LD structure as implemented in the LDSC software. Enrichment was expressed as the ratio of these LD-adjusted means. Significance of the enrichment was assessed with a one-tailed Mann–Whitney U test; P values were corrected for seven parallel tests with a Bonferroni threshold of 0.0071.
Transcriptome-Wide Association Study
We assessed expression-mediated genetic influences on major depressive disorder with S-PrediXcan, an approach that uses GWAS summary statistics to impute genetically regulated gene expression and then tests that expression for association with outcome [12]. Prediction weights came from the GTEx v8 MASHR models [13]. Six brain regions—frontal cortex (BA9), anterior cingulate cortex (BA24), hippocampus, amygdala, nucleus accumbens and caudate—were selected because of their established roles in emotion, memory and reward.
The analysis used the Psychiatric Genomics Consortium 2025 meta-analysis of major depressive disorder, restricted to European ancestry and excluding both 23andMe and UK Biobank cohorts [9]. After harmonising alleles and removing low-quality variants, 6,979,144 SNPs remained. For every tissue S-PrediXcan produced a Z-score and nominal P value for each gene; false-discovery rates (FDR) were calculated separately within tissues and results with FDR below 0.05 were considered significant. Enrichment of association signals within seven pre-specified gene panels—the two glutamatergic lists, three pruning lists and two negative controls described in earlier sections—was evaluated with one-sided Mann–Whitney U tests that compared the absolute Z-scores of genes in a panel with those of all other tested genes.
Two-Sample Mendelian Randomization
We probed possible causal links between neuroplasticity-related traits and major depressive disorder (MDD) using a two-sample Mendelian-randomization design [14,15]. Summary statistics for five candidate exposures were taken from the largest available genome-wide association studies: years of education [16], composite cognitive performance, general intelligence [17], fluid intelligence, and hippocampal volume. Major depressive disorder served as the outcome, based on the European-ancestry Psychiatric Genomics Consortium meta-analysis that excludes 23andMe and UK Biobank cases [9].
Independent instrumental variants were defined as autosomal single-nucleotide polymorphisms (SNPs) reaching genome-wide significance (P < 5 × 10−8) and showing minimal linkage disequilibrium (r2 < 0.001 within 10 Mb). Exposure and outcome alleles were harmonised; palindromic or ambiguous SNPs were discarded. Causal effects were estimated with the inverse-variance-weighted (IVW) method, complemented by weighted-median, weighted-mode and MR-Egger models. Cochran’s Q and the MR-Egger intercept were used to evaluate heterogeneity and directional pleiotropy. Traits represented by fewer than three valid instruments were not analysed further.
Results
MAGMA Analysis
MAGMA highlighted 344 genes that exceeded the Bonferroni threshold, while a further 5408 displayed nominal significance. The most compelling signals arose at SORCS3 (P = 9.7 × 10−19), SGCZ (1.6 × 10−18), DRD2 (2.4 × 10−18) and DCC (4.4 × 10−18).
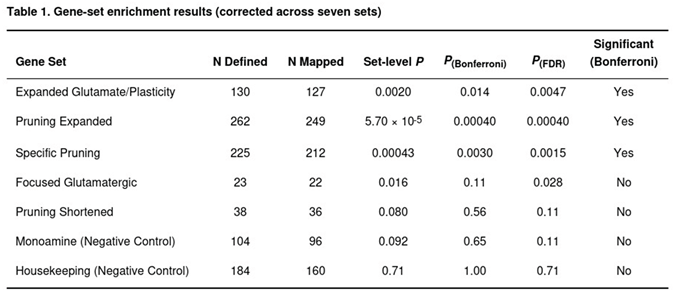
Gene-set testing identified three panels that withstood Bonferroni correction (Table 1). The expanded glutamate/plasticity group, with 127 of 130 genes successfully mapped, showed significant enrichment (raw P = 0.0020; Bonferroni-adjusted P = 0.014; FDR = 0.0047). Forty-nine of its members were nominally significant and four—headed by EP300 (P = 1.1 × 10−8)—passed the gene-wide threshold. The expanded pruning collection (249 of 262 genes) was even more compelling (raw P = 5.7 × 10−5; Bonferroni = 4.0 × 10−4; FDR = 4.0 × 10−4); thirteen of its genes were genome-wide significant, with SDK1 the strongest (P = 6.1 × 10−17). The specific pruning subset (212 of 225 genes) also achieved significance (raw P = 4.3 × 10−4; Bonferroni = 0.0030; FDR = 0.0015) and again was led by SDK1.
The focused glutamatergic target list produced a nominally enriched signal (P = 0.016) that survived FDR correction (0.028) but not the stricter Bonferroni cut-off; the association was driven principally by CYP2D6 (P = 7.0 × 10−7). Neither the shortened pruning list nor the two negative-control panels displayed evidence of enrichment (all P > 0.09).
Collectively, these results implicate broader glutamatergic-plasticity and synaptic-pruning pathways in MDD liability, whereas monoaminergic and housekeeping gene groups showed no departure from the genome-wide distribution.
Enrichment of Heritability Within Pruning-Related Annotations
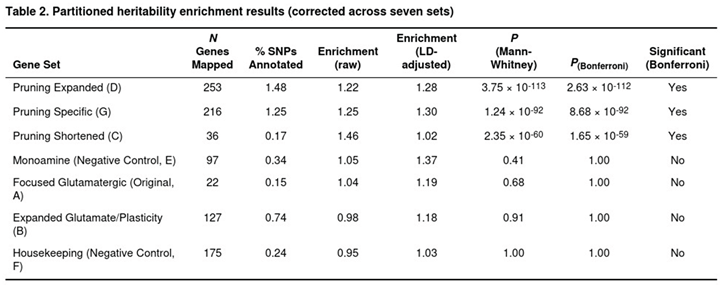
Three pruning-focused annotations captured a disproportionate share of MDD heritability once LD was taken into account (Table 2). The expanded pruning list, which mapped 253 of its 262 genes and marked 1.48 per cent of common SNPs, showed an LD-adjusted enrichment of 1.28-fold (P = 2.6 × 10−112 after Bonferroni correction). Removing the 37 genes overlapping glutamatergic pathways had little effect: the “specific pruning” set (216 genes, 1.25 per cent of SNPs) retained a 1.30-fold enrichment (P = 8.7 × 10−92). The compact 38-gene pruning panel also displayed a robust signal; with 36 genes successfully annotated (0.17 per cent of SNPs) it yielded an enrichment of 1.02-fold after LD adjustment, corresponding to a Bonferroni-corrected P = 1.6 × 10−59.
By contrast, annotations based on glutamatergic drug targets, broader glutamate/plasticity genes, monoaminergic controls and housekeeping genes showed no evidence of enrichment. The original 23-gene CGR target list covered 0.15 per cent of SNPs and produced an adjusted P value of 0.68. Likewise, the 130-gene glutamate/plasticity panel (0.74 per cent of SNPs) had an adjusted P value of 0.91. Both negative controls were clearly null (monoaminergic P = 0.41; housekeeping P = 1.00).
Taken together, these stratified LDSC results indicate that common variants situated near synaptic pruning genes—but not those near glutamatergic- or monoaminergic-related genes—account for an enriched fraction of MDD heritability, independent of local LD architecture and of overlap with glutamatergic loci.
Transcriptome-Wide Association Findings
Between 10 000 and 11 200 genes were evaluated per brain tissue, giving 65 161 gene-tissue tests. Controlling FDR at 5 per cent identified 2 638 significant gene-tissue pairs representing 1 122 unique genes; 450 pairs remained significant after Bonferroni adjustment across all tests.
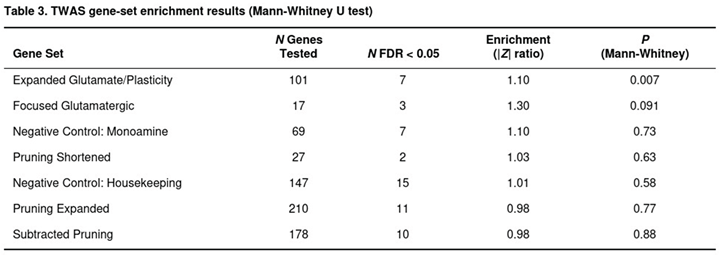
The expanded glutamate/plasticity panel (101 of 130 genes testable) contained more extreme associations than the genomic background (Table 3): its mean absolute Z-score was 1.237 versus 1.129 for all genes, a 10 per cent inflation that reached significance (P = 0.007). Seven panel members were FDR-significant, and the strongest signal arose for CYP2D6 in frontal cortex (P = 5.8 × 10−9). The original 23-gene CGR list showed a similar but weaker trend; 17 of its genes were testable, three met the FDR threshold, and the overall enrichment did not survive Bonferroni correction (P = 0.091).
In contrast, none of the pruning-related panels showed evidence of enrichment. The 27-gene subset of the shortened pruning list produced only nominal signals (P = 0.63) despite a notable association for RHOA in hippocampus. The larger pruning collections—expanded (210 genes) and subtracted (178 genes)—were even less compelling (P ≥ 0.77). Monoaminergic and housekeeping control sets were also null (P > 0.58).
Summarising across panels, glutamatergic genes carried a modest excess of transcriptome-wide signals, largely driven by CYP2D6, whereas genes linked to microglial pruning did not influence common-variant risk for major depressive disorder in this analysis of six key brain tissues.
Mendelian-Randomization Findings
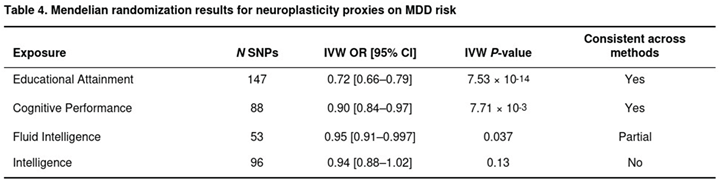
Adequate instruments were available for four exposures (Table 4). Genetically predicted educational attainment showed a strong inverse association with MDD: each unit increase corresponded to an odds ratio (OR) of 0.72 (95% CI 0.66–0.79; P = 7.5 × 10−14) across 147 independent SNPs. Cognitive performance was likewise protective (OR 0.90, 0.84–0.97; P = 7.7 × 10−3; 88 SNPs). For fluid intelligence, the IVW estimate suggested a modest effect (OR 0.95, 0.91–0.997; P = 0.037; 53 SNPs), while the intelligence meta-analysis signal did not reach significance (OR 0.94, 0.88–1.02; P = 0.13; 96 SNPs).
Across exposures, MR-Egger intercepts were non-significant (all P > 0.55), indicating little evidence of unbalanced pleiotropy. Heterogeneity was high (I2 > 79%), yet the protective direction for education and cognitive performance persisted in weighted-median and weighted-mode models. Hippocampal volume and reaction-time GWASs supplied too few instruments to allow reliable MR estimation.
Discussion
Interpretation of Results
Re-examining the latest large-scale European GWAS of major depressive disorder (MDD; [9]) allowed us to integrate multiple analytic strategies—MAGMA, stratified LD-score regression, transcriptome-wide association and two-sample Mendelian randomization—to build a coherent picture of MDD genetics. Across methods, genes involved in synaptic pruning and broader neurodevelopment consistently emerged as key contributors, whereas glutamatergic and general synaptic-plasticity pathways showed more modest, method-specific signals.
Gene-set testing with MAGMA indicated pronounced competitive enrichment for the expanded pruning panels even after glutamatergic overlap was removed, and pinpointed individual loci such as SDK1, RHOA and TCF4 that regulate neuronal adhesion or guidance. Partitioned heritability analysis supported these findings: common variants in pruning-related regions accounted for up to 30% more SNP heritability than expected under a null of uniform contribution (best LD-adjusted enrichment ≈ 1.30, p < 10^-91). By contrast, glutamatergic or plasticity gene sets displayed weaker enrichment—nominally significant in MAGMA and modest in TWAS (approximately 10% increase in mean absolute Z scores, p = 0.007)—and failed to concentrate heritability in LDSC, suggesting a secondary role. As expected, monoaminergic and housekeeping control sets were uniformly null across all analyses, underscoring the specificity of the pruning signal.
TWAS results were directionally diverse: predicted expression changes within both pruning and glutamatergic genes varied in sign across tissues, mirroring the heterogeneity reported in earlier psychiatric studies [4]. Such variability reinforces the view that MDD liability reflects many context-dependent gene effects rather than a single coherent transcriptional profile.
Mendelian-randomization analyses added a causal perspective. Genetic proxies that index lifelong neuroplasticity—higher educational attainment, composite cognitive performance and fluid intelligence—were all associated with reduced MDD risk. The largest effect was seen for education (OR 0.72, 95% CI 0.66–0.79), followed by cognitive performance (OR 0.90) and fluid intelligence (OR 0.95); MR-Egger tests found no evidence of directional pleiotropy. These findings echo epidemiological work linking cognitive reserve to depression resilience [18] and imply that genetic variants enhancing plasticity may buffer against depressive illness.
Proposed Mechanistic Framework for MDD
Taken together with what we already know about brain development, our new look at the data pushes a simple idea: major depressive disorder may start with the brain’s house-cleaning crew going overboard while we are still teenagers. A cluster of common risk variants sits in genes that govern synaptic pruning, nudging microglia and complement proteins to lop off too many connections. That over-trimming leaves neural networks thin and brittle, so when adult stress hits, they crack instead of bend, opening the door to depression. The genetic signals line up with this story—higher predicted levels of pruning drivers like RHOA (TWAS Z = +7.22) and C4A (+3.21) push risk upward, while lower expression of “glue” molecules such as SDK1 (-4.97) and the guidance cue SEMA3F (-4.41) removes key supports. Schizophrenia shows a parallel pattern of runaway pruning [7,8], and animal work links early pruning glitches to stress-induced mood changes later on [19], tying the whole picture together.
Glutamatergic and plasticity pathways seem to amplify the pruning deficit. Although enrichment signals in glutamatergic gene sets were modest, their directions differed: protective patterns emerged for sigma-1 and kainate receptors (SIGMAR1 -3.19; GRIK2 -3.42), while risk rose with CREB-related factors (EP300 +4.03) and NMDA subunits (GRIN2A +2.19). Such mixed findings point to context-dependent excitatory imbalances, likely stemming from lost connections after excessive pruning. Diminished adaptive plasticity in stress-responsive regions—prefrontal cortex and hippocampus in particular—would follow [6].
Figure 1.
The “Pruning-Mediated Plasticity Deficit” Framework for Major Depressive Disorder. This conceptual model illustrates the etiological progression proposed by the study. (Top) During adolescent neurodevelopment, common genetic risk variants drive the overexpression of pruning factors [e.g., RHOA, C4A] and the underexpression of synaptic adhesion molecules [e.g., SDK1, SEMA3F]. This results in microglial overactivation and excessive synaptic elimination. (Middle) The structural consequence is a sparse, brittle neural network characterized by glutamatergic imbalances [e.g., upregulated EP300/GRIN2A] and diminished adaptive plasticity, particularly in the prefrontal cortex and hippocampus. (Bottom) When challenged by adult stress, these compromised circuits fail to adapt [“crack instead of bend”], precipitating MDD. The model integrates protective buffers, such as Cognitive Reserve, which mitigates vulnerability, and Dopaminergic Variation [DRD2], which influences symptom presentation.
Figure 1.
The “Pruning-Mediated Plasticity Deficit” Framework for Major Depressive Disorder. This conceptual model illustrates the etiological progression proposed by the study. (Top) During adolescent neurodevelopment, common genetic risk variants drive the overexpression of pruning factors [e.g., RHOA, C4A] and the underexpression of synaptic adhesion molecules [e.g., SDK1, SEMA3F]. This results in microglial overactivation and excessive synaptic elimination. (Middle) The structural consequence is a sparse, brittle neural network characterized by glutamatergic imbalances [e.g., upregulated EP300/GRIN2A] and diminished adaptive plasticity, particularly in the prefrontal cortex and hippocampus. (Bottom) When challenged by adult stress, these compromised circuits fail to adapt [“crack instead of bend”], precipitating MDD. The model integrates protective buffers, such as Cognitive Reserve, which mitigates vulnerability, and Dopaminergic Variation [DRD2], which influences symptom presentation.
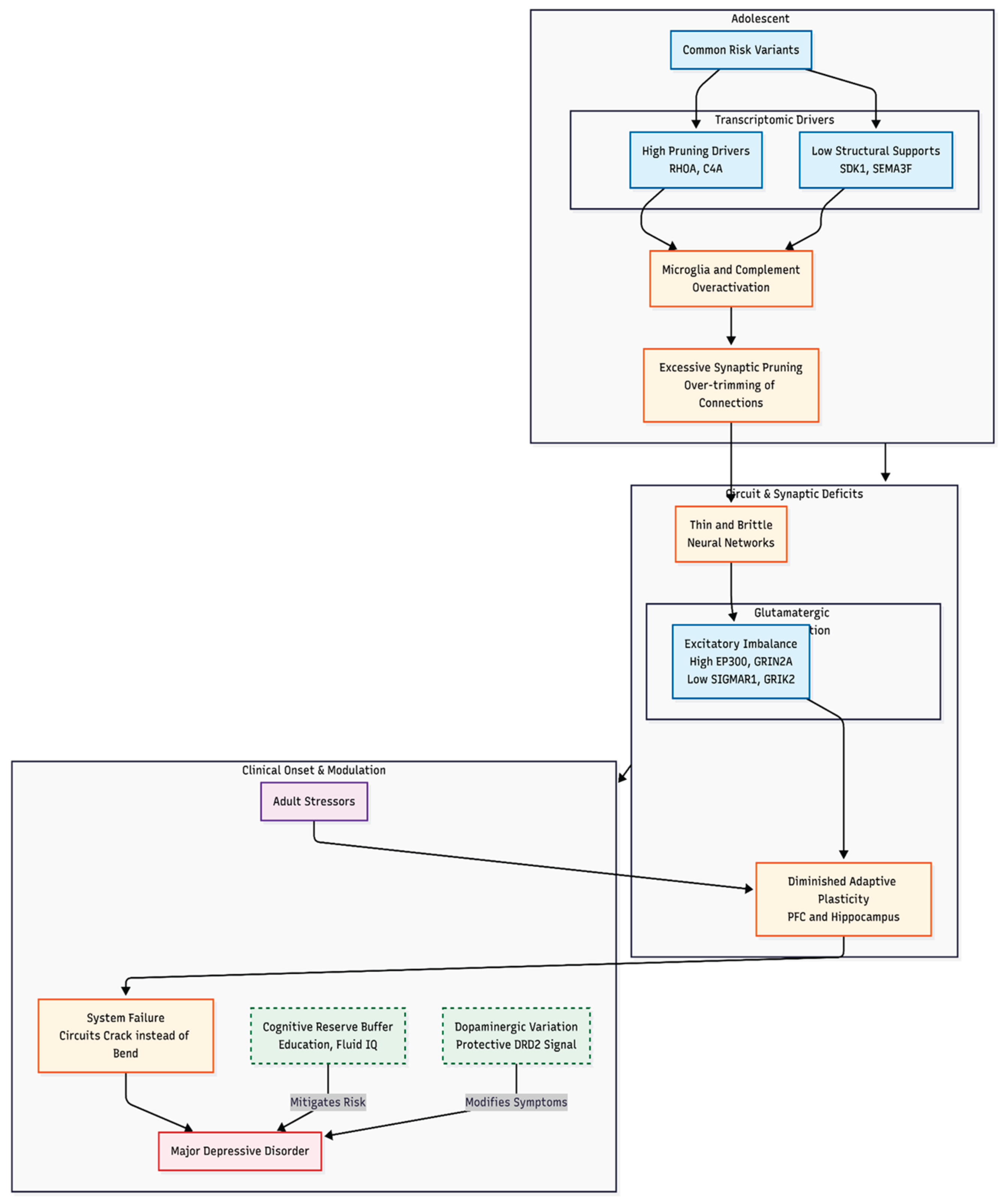
This picture fits well with the idea that cognitive reserve can act as a buffer. Mendelian randomization indicated that genetic proxies for education (OR = 0.72) and cognitive performance (OR = 0.90) diminish the risk of major depressive disorder (MDD), suggesting that enhanced plasticity reserves can mitigate vulnerabilities associated with pruning [20]. Dopaminergic variation, exemplified by the strongly protective DRD2 signal (-10.71), may influence symptom profiles without altering the fundamental genetic load.
Taken together, this “pruning-mediated plasticity deficit” model broadens the classic stress-diathesis view by tracing risk back to neurodevelopmental events and explaining overlaps with schizophrenia genetics [21]. The model can be tested with developmental imaging of synaptic density (for example, SV2A PET) and patient-derived neuron–microglia co-cultures that assay pruning in vitro [8]. It also highlights preventive windows and therapeutic angles—interventions that temper pruning or boost later plasticity, such as ketamine and related agents [6], may offer the greatest benefit.
Novelty and Impact of the Study
Using the newest and largest PGC genome-wide dataset for major depressive disorder (MDD) [9], we ran four tools side by side—MAGMA pathway tests, LD-score regression, transcriptome-wide association (TWAS) and two-sample Mendelian randomization (MR). Earlier scans mapped risk to broad neuronal themes [4,5] but typically leaned on a single analytic lens. By layering methods, we show that variants sitting in pruning machinery explain a bigger slice of heritability than those in classic glutamatergic genes, even after strict correction for gene overlap and multiple testing. Directional clues from TWAS plus causal estimates from MR sharpen the picture: higher expression of pruning genes pushes risk upward, whereas traits that index cognitive reserve pull risk down. Our results build on cross-disorder work that finds shared genetic threads across psychiatric illnesses [21] and refine the more recent sex-specific analyses of MDD [22].
The translational message is equally clear. Complement proteins and Rho-family effectors—already in the therapeutic spotlight for schizophrenia [7]—now emerge as top polygenic drivers in depression as well. MR indicates that educational attainment and general cognitive performance are probably protective (odds ratios 0.72 and 0.90), bolstering calls to build cognitive reserve across the life-span [20]. And while glutamatergic signals are more modest and variable, they dovetail with the mechanism of rapid-acting agents such as ketamine [6], suggesting these drugs may work best for specific subtypes rather than the MDD population at large. In short, our multi-method framework ranks molecular programs by likely impact and offers a roadmap for biomarker discovery and personalised treatment.
Limitations
Several points warrant caution. The discovery GWAS is overwhelmingly European, so ancestry-specific loci could have been missed [23]. TWAS used adult GTEx brains; expression patterns during development—when pruning peaks—might look very different, perhaps explaining why TWAS signals lag behind MAGMA and LDSC results. Although MR sensitivity checks found little evidence of horizontal pleiotropy, instruments such as educational attainment can still carry social or environmental baggage that genetics alone cannot unpack [24]. Finally, our strict procedures for removing gene overlap and correcting for multiple tests guard against false positives but may also hide weaker yet meaningful pathways that larger future samples could reveal.
Conclusions
The converging evidence places aberrant synaptic pruning at centre stage in the polygenic architecture of MDD, with glutamatergic imbalance and low cognitive reserve acting as modifiable satellites. Prospective developmental and longitudinal studies should now put this pruning hypothesis to the test, laying the groundwork for preventive and therapeutic strategies that match the true biology of depression.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflicts of Interest
None declared.
Ethics Declaration
Not applicable.
References
- World Health Organization. Depressive Disorder (Depression). 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/depression.
- Kessler, R.C.; Chiu, W.T.; Demler, O.; Walters, E.E. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 617–627. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic epidemiology of major depression: Review and meta-analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Levey, D.F.; Stein, M.B.; Wendt, F.R.; Pathak, G.A.; Zhou, H.; Aslan, M.; Quaden, R.; Harrington, K.M.; Nuñez, Y.Z.; Overstreet, C.; et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 2021, 24, 954–963. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Trans-ancestry genome-wide study of depression identifies 697 associations implicating cell types and pharmacotherapies. Cell 2025, 188, 640–652.e9. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalised gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L.; et al. Exploring the phenotypic consequences of tissue-specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linnér R, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef]
- Opdebeeck, C.; Martyr, A.; Clare, L. Cognitive reserve and cognitive function in healthy older people: A meta-analysis. Aging Neuropsychol. Cogn. 2016, 23, 40–60. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y.; Arenaza-Urquijo, E.M.; Bartrés-Faz, D.; Belleville, S.; Cantillon, M.; Chetelat, G.; Ewers, M.; Franzmeier, N.; Kempermann, G.; Kremen, W.S.; et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer’s Dement. 2020, 16, 1305–1311. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019, 179, 1469–1482.e11. [Google Scholar] [CrossRef] [PubMed]
- Levey, D.F.; Galimberti, M.; Dahl, A.; et al. Sex-specific genetic architecture and development of a sex-aware polygenic score for major depressive disorder. Biol. Psychiatry 2024, 96, 845–857. [Google Scholar] [CrossRef]
- Peterson, R.E.; Kuchenbaecker, K.; Walters, R.K.; Chen, C.-Y.; Popejoy, A.B.; Periyasamy, S.; Lam, M.; Iyegbe, C.; Strawbridge, R.J.; Brick, L.; et al. Genome-wide association studies in ancestrally diverse populations: Opportunities, methods, pitfalls, and recommendations. Cell 2019, 179, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



