
Submitted:
06 January 2026
Posted:
09 January 2026
You are already at the latest version
Abstract
The global rise of multidrug-resistant (MDR) bacterial infections in pediatric populations poses an alarming challenge to effective clinical management and antimicrobial stewardship. Two-component systems (TCSs), comprising sensor kinases and response regulators, play a pivotal role in bacterial adaptation, virulence, and resistance mechanisms, making them promising targets for diagnostic and therapeutic innovation. This study employs a machine learning-driven bioinformatics pipeline to identify and prioritize potential TCS biomarkers across MDR pediatric pathogens for integration into next-generation diagnostic biosensors. Genomic datasets from clinically relevant MDR bacteria were curated and analyzed to extract TCS-associated gene and protein signatures. Using Pfam domain features, multiple supervised learning models were trained, with XGBoost, Random Forest, and a Stacking Ensemble achieving high overall accuracies (0.9883-0.9885). While the dominant Non-TCS class was predicted with near-perfect accuracy, minority subclasses exhibited variable detection due to severe class imbalance, particularly for rare groups such as CpxA-like and EnvZ-like proteins (n=2 each). Moderate F1-scores were obtained for generic response regulators and OmpR-like proteins. Feature importance analysis identified a small set of highly discriminative domains, including PF01339, PF00702, PF07679, PF03997, and PF04886, associated with conserved regulatory and signaling motifs. These results demonstrate that Pfam domain signatures offer biologically meaningful features for TCS classification, while highlighting the need for expanded datasets or embedding-based features to improve minority-class prediction. Overall, this work provides a scalable, AI-driven foundation for TCS biomarker discovery, aiming to develop diagnostic biosensors for MDR pediatric pathogens.
Keywords:
two-component systems
; multidrug resistance
; pediatric pathogens
; machine learning
; biosensors
; biomarker discovery
Introduction
The rapid rise of multidrug-resistant (MDR) bacterial infections represents one of the most critical global health threats, with an especially severe impact on pediatric populations. Children particularly neonates, infants, and immunocompromised patients are disproportionately vulnerable to MDR pathogens due to their developing immune systems, frequent hospital exposures, and limited availability of age-appropriate antimicrobial therapies (1, 2). As resistance to frontline and last-line antibiotics steadily increases, the need for early diagnostics and targeted therapeutic interventions has become urgent. A key challenge in addressing MDR infections understands how bacteria sense and respond to clinical and environmental stresses, particularly those imposed by antimicrobial agents.
Two-component systems (TCSs) are fundamental regulatory circuits that enable bacteria to detect external signals and mount adaptive responses. Typically composed of a membrane-bound histidine kinase and a cytoplasmic response regulator, TCSs orchestrate essential processes such as virulence, biofilm formation, host colonization, metabolic adaptation, and antimicrobial resistance (3, 4). Several TCSs including PhoP/PhoQ, EnvZ/OmpR, and CpxA/CpxR play direct roles in resistance to polymyxins, β-lactams, and aminoglycosides in clinically significant MDR pathogens such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae (5). Understanding the architecture and diversity of TCS proteins is therefore vital for identifying conserved regulatory elements that may serve as biomarkers or therapeutic targets. With advances in genomic sequencing and protein annotation, databases such as Pfam have become invaluable for identifying conserved protein domains associated with TCSs across bacterial species (6). Pfam domain signatures provide insights into evolutionary relationships and functional classifications; however, interpreting domain combinations and mapping them to biological roles remains complex. Simple domain presence or absence does not fully capture the functional nuances of TCS subclasses, regulatory hierarchies, or pathogen-specific adaptations. Thus, translating large Pfam-annotated datasets into actionable biological insights requires sophisticated computational approaches capable of modeling nonlinear interactions.
Machine learning (ML) offers powerful tools for classification, pattern recognition, and feature prioritization in microbial genomics (7). ML models such as Random Forests and XGBoost can integrate domain features to predict TCS classes, regulatory functions, or pathway assignments with high accuracy across diverse bacterial lineages. Furthermore, feature importance allows transparent interpretation of model predictions, enabling researchers to identify the most influential domains contributing to classification outcomes. This combination of predictive accuracy and interpretability makes ML particularly suited for analyzing TCS domain architecture. The integration of ML with Pfam-based TCS annotation establishes a systematic workflow (Figure 1) for biomarker discovery: (i) extracting and encoding domain signatures; (ii) training ML classifiers on labeled TCS classes; (iii) evaluating performance using cross-validation; (iv) applying feature importance to identify key functional domains; and (v) prioritizing conserved, pathogen-specific TCS elements as potential diagnostic biomarkers. These biomarkers have promising applications in next-generation biosensor systems. Because TCSs naturally function as environmental detectors, their modular sensor-regulator architecture can be repurposed to engineer whole-cell or cell-free biosensors capable of detecting specific microbial signals, host metabolites, or antimicrobial stressors. Given the growing burden of MDR pediatric infections and the urgent need for rapid, reliable diagnostics, ML-driven analysis of TCS protein domain architecture offers a powerful approach for identifying clinically relevant biomarkers. Such biomarkers can support the development of advanced biosensor platforms, improving early detection, guiding treatment decisions, and ultimately enhancing clinical outcomes for vulnerable pediatric populations.
Materials and Methods
The proteomics sequences of selected bacterial pathogens Pseudomonas aeruginosa (ID: UP000002438; No. of proteins: 5563), Acinetobacter baumannii (ID: UP000498640; No. of proteins: 3713), and Klebsiella pneumoniae (ID: UP000019183; No. of proteins: 5959) were retrieved from Uniprot Proteome database. Using the Galaxy-PfamScan plug-in, the FASTA sequences of the retrieved proteomes were searched for protein domains against the Pfam HMM library. The output of PfamScan was used to build a ML framework for classifying TCS proteins into biologically meaningful functional subclasses. The ML input dataset consisted of three variables sequence ID, Pfam domains, and TCS class. The Pfam domain field, originally stored as a string representation of lists, was converted into Python lists, de-duplicated, and expanded by exploding multi-domain entries into separate rows.
These domain features were transformed into a multi-hot encoded representation using a Multi-Label Binarizer, thereby generating a binary matrix indicating the presence or absence of each Pfam domain per sequence. The target labels representing TCS subclasses (CpxA-like, EnvZ-like, OmpR-like, generic response regulator (GRR), and non-TCS) were encoded using a label encoder. The dataset was split into stratified training and test sets, and two baseline models Random Forest (RF) and XGBoost (XGB) were first trained to capture nonlinear associations between domain architectures and TCS class. To improve predictive performance, both models were combined into a meta-learning framework using a Stacking Classifier, where RF and XGB served as base learners and a logistic regression classifier acted as the final estimator. Model performance was assessed using accuracy, classification reports, confusion matrices, and cross-validation. To interpret the biological relevance of learned features, feature importance analysis was performed on both the XGB and RF models, generating global and local explanations that highlight the contribution of specific Pfam domains to TCS class discrimination. The final stacked model and all preprocessing encoders were saved to enable consistent prediction on new protein sequences. This integrative workflow provides a transparent, interpretable, and modular ML pipeline for TCS biomarker discovery using domain-level protein signatures.
Results and Discussion
ML models developed in this study demonstrated high overall accuracy in classifying Pfam-annotated proteins into five TCS categories, with RF achieving an accuracy of 0.9883 and both XGB and the Stacking Ensemble reaching 0.9885. This strong performance was primarily driven by the dominant non-TCS class, which constituted nearly 98% of the dataset and was predicted with near-perfect precision, recall, and F1-scores across all models. In contrast, classification performance varied among the TCS subclasses: GRR were identified with moderate F1-scores (0.52-0.58), and OmpR-like proteins achieved F1-scores around 0.65, reflecting the presence of moderately distinct domain signatures. However, none of the models were able to correctly classify the extremely rare CpxA-like and EnvZ-like proteins (n=2 each), resulting in zero precision and recall for these categories. This outcome is consistent with the severe class imbalance and the high degree of sequence similarity shared among histidine kinase subclasses, which limits the ability of classical ML algorithms to learn discriminative patterns from very small sample sizes. Although the Stacking model slightly improved detection of GRR, the rare subclasses remained challenging, emphasizing the need for techniques such as oversampling, expanded datasets, or integration of advanced protein embedding models to enhance minority-class detection.
The XGB feature importance analysis revealed that a small subset of Pfam domains contributed disproportionately to the model’s predictive power, highlighting clear domain-level signatures associated with TCS classification. The top-ranked domains PF01339, PF00702, PF07679, PF03997, and PF04886 showed markedly higher relative importance scores, indicating their strong association with distinguishing TCS-related proteins from non-TCS categories (Figure 2). Many of these domains correspond to conserved regulatory or signaling motifs commonly present in response regulators and histidine kinases, suggesting that the model effectively captured biologically meaningful features underlying TCS architecture. Lower-ranked domains, although contributing less individually, collectively provided complementary signals for refining class boundaries, especially for subclasses with partially overlapping motifs such as GRR and OmpR-like proteins.
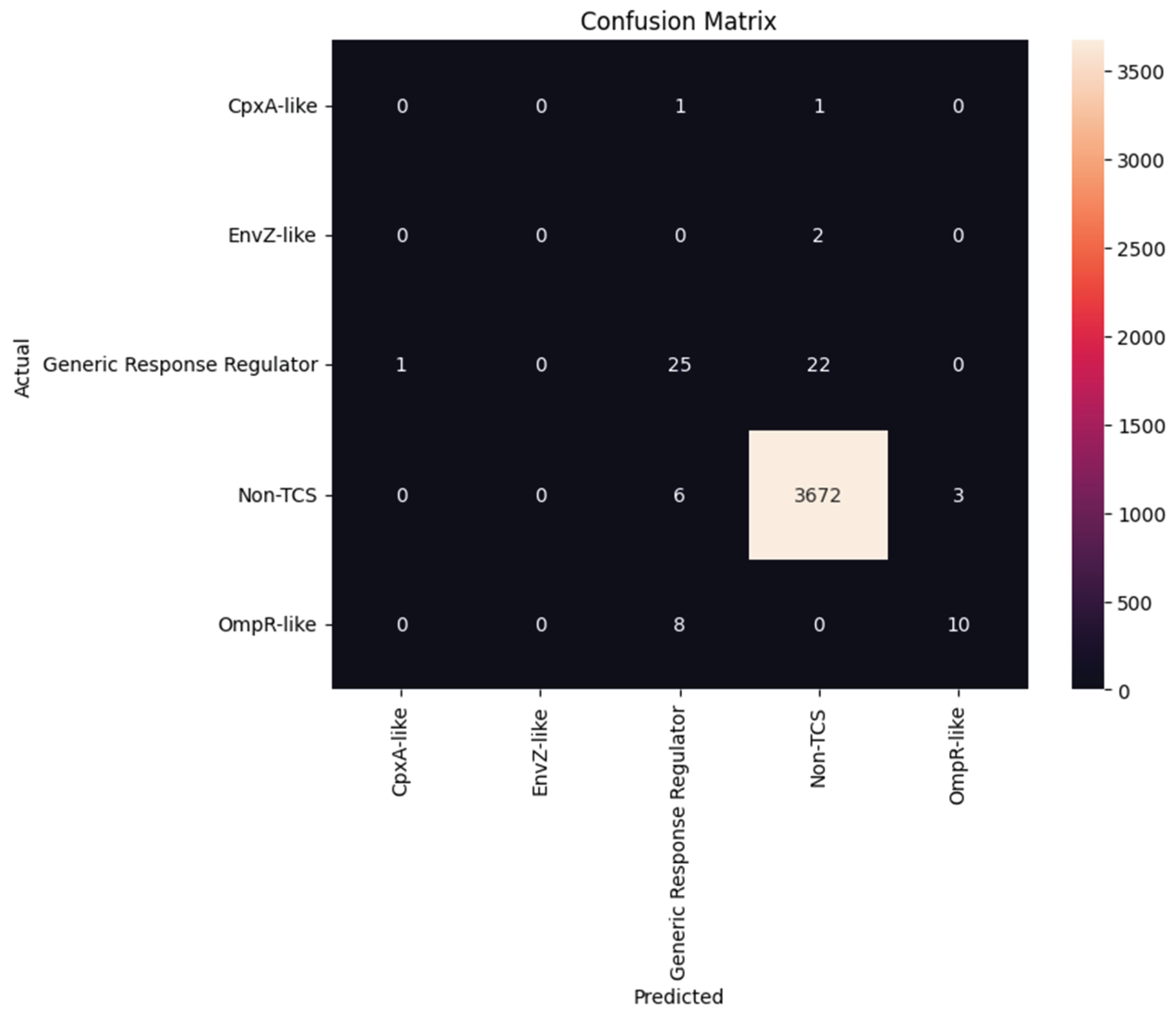
The confusion matrix further contextualizes these findings by illustrating how the model performs across the five TCS classes (Figure 3). As expected from the class imbalance in the dataset, the model achieved near-perfect accuracy for the overwhelmingly dominant non-TCS category, misclassifying only a handful of instances. However, detection of rare subclasses such as CpxA-like and EnvZ-like remained challenging, with both categories showing complete misclassification due to having only two samples each insufficient for the model to generalize their domain signatures. The GRR class displayed moderate performance, with notable confusion between this class and non-TCS proteins, reflecting partial domain overlap or incomplete domain signal. Similarly, OmpR-like proteins were often misclassified as GRR, consistent with the shared receiver domains that characterize these regulatory families. Despite these challenges, the model demonstrated clear ability to identify strong, discriminative Pfam domain drivers, and the feature importance distribution underscores the biological relevance of domain-based learning for TCS classification. These findings collectively suggest that while domain signatures are highly informative for abundant classes, improving representation of rare TCS subclasses or integrating sequence embeddings may further enhance discriminative power in future work. Despite these limitations, the models robustly captured discriminative domain features for the major TCS-related groups, enabling reliable identification of biologically meaningful signatures. These results highlight the utility of ML-driven Pfam domain analysis for prioritizing potential TCS biomarkers and underscore its value as a scalable framework for guiding the development of next-generation biosensors aimed at rapid detection of multidrug-resistant pathogens in pediatric clinical settings.
Limitations
Despite the strong overall performance of the ML models particularly the XGB and stacking ensemble several limitations must be acknowledged. First, the dataset is highly imbalanced, with the non-TCS class overwhelmingly dominating the sample size. This imbalance leads to biased learning, causing the model to achieve near-perfect accuracy while underperforming on minority classes such as CpxA-like and EnvZ-like, both of which contain only two instances each. As reflected in the classification reports, precision, recall, and F1-scores for these classes are zero, indicating that the model fails to learn meaningful patterns for rare categories. Second, the features used in this study are derived solely from Pfam domain annotations. While domain-level features are biologically meaningful, they may not fully capture the complexity of TCS proteins, whose classification could benefit from additional structural, evolutionary, or physicochemical descriptors. Third, although feature importance was used to interpret feature contributions, its insights are constrained by the limited diversity of the input representations. Finally, the current models assume that domain presence is sufficient for classification; however, closely related domains or domain combinations may require more granular sequence-level embeddings (e.g., transformer-based protein language models) to improve resolution for subtle class distinctions.
Conclusion
This study demonstrates that ML methods can effectively classify proteins into TCS categories using domain-level features extracted from Pfam annotations. The RF, XGB, and stacking ensemble models all achieved high overall accuracy (~0.988), with XGB and the stacked model showing slightly improved performance in detecting minority classes such as GRR and OmpR-like proteins. The use of domain frequency encoding combined with robust ML classifiers provided a scalable, interpretable, and computationally efficient workflow for TCS classification. However, the persistent misclassification of small classes underscores the need for larger and better-balanced datasets, alongside richer sequence representations. Future work could integrate deep-learning-based protein embeddings, structural modeling outputs, and evolutionary profiles to enhance predictive performance, particularly for low-abundance TCS subclasses. Overall, the study provides a foundational ML framework for identifying biosensor-related TCS proteins, offering a valuable step toward automated discovery pipelines in microbial genomics and synthetic biology.
Funding Information
The author(s) received no financial support for this article's research, authorship, and/or publication.
Acknowledgments
The authors are acknowledged to the Department of Medical Biotechnology, AVMC&H, Vinayaka Mission’s Research Foundation (Deemed to be University), Puducherry Campus for providing all the required facilities to complete this study.
Conflicts of Interest
The authors have none to declare.
Disclosures
None.
References
- Laxminarayan, R; Duse, A; Wattal, C; Zaidi, AK; Wertheim, HF; Sumpradit, N; Vlieghe, E; Hara, GL; Gould, IM; Goossens, H; Greko, C. Antibiotic resistance the need for global solutions. The Lancet infectious diseases 2013, 13(12), 1057–98. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C. L. The antibiotic resistance crisis: Part 1. Pharmacy and Therapeutics 2015, 40(4), 277–283. [Google Scholar] [PubMed]
- Stock, A. M.; Robinson, V. L.; Goudreau, P. N. Two-component signal transduction. Annual Review of Biochemistry 2000, 69, 183–215. [Google Scholar] [CrossRef] [PubMed]
- Groisman, EA. Feedback control of two-component regulatory systems. Annual review of microbiology 2016, 70(1), 103–24. [Google Scholar] [CrossRef] [PubMed]
- Gooderham, WJ; Hancock, RE. Regulation of virulence and antibiotic resistance by two-component regulatory systems in Pseudomonas aeruginosa. FEMS microbiology reviews 2009, 33(2), 279–94. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J; Chuguransky, S; Williams, L; Qureshi, M; Salazar, GA; Sonnhammer, EL; Tosatto, SC; Paladin, L; Raj, S; Richardson, LJ; Finn, RD. The protein families database in 2021. Nucleic Acids Res. 2021, 49(D1), D412–9. [Google Scholar] [CrossRef] [PubMed]
- Camacho, DM; Collins, KM; Powers, RK; Costello, JC; Collins, JJ. Next-generation machine learning for biological networks. Cell 2018, 173(7), 1581–92. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic work-flow diagram of the study.
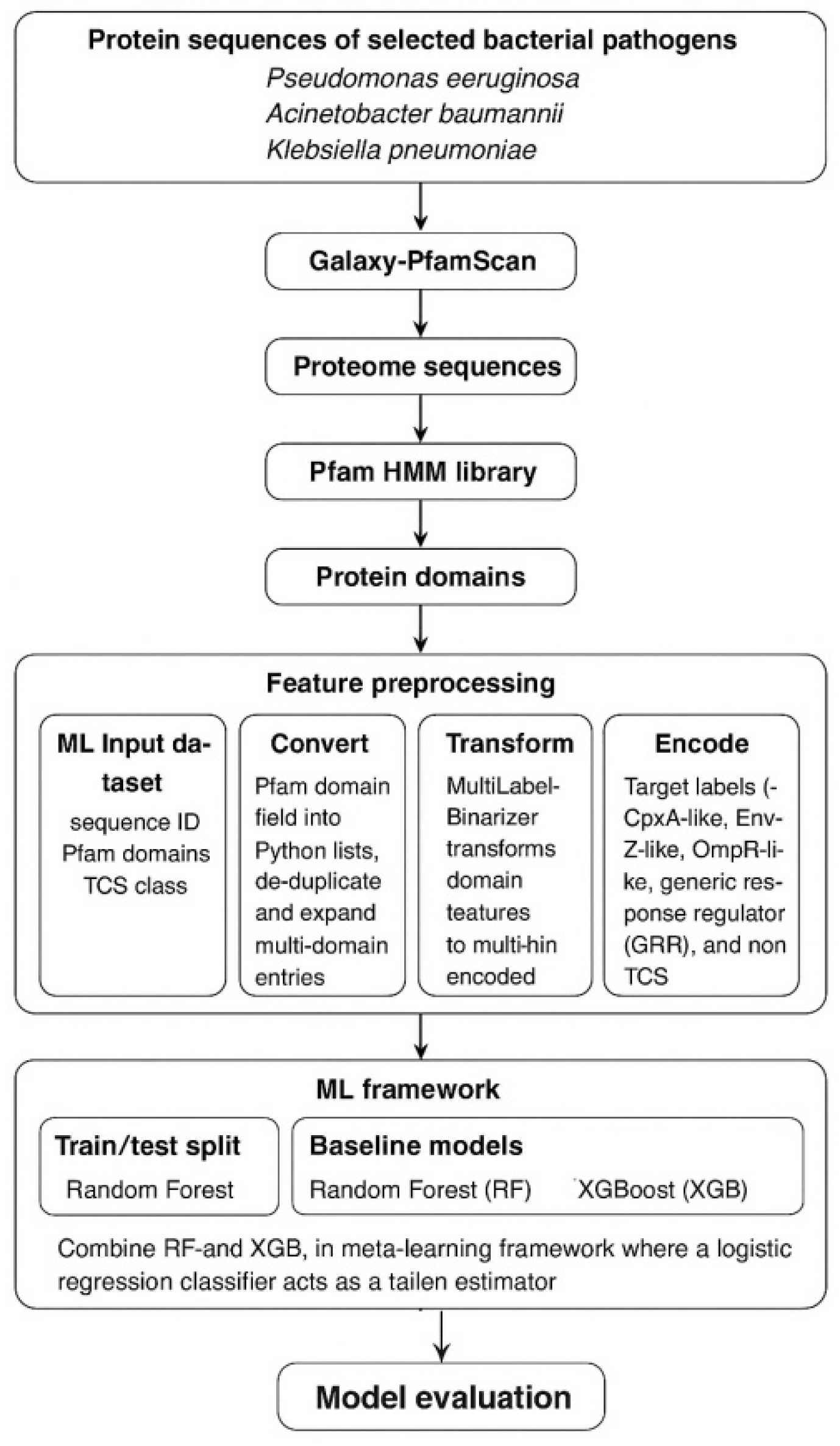
Figure 2.
Top 20 Pfam Domain Features Ranked by XGBoost Importance. This bar plot shows the 20 most important Pfam domains contributing to the XGBoost classification model. Higher importance values indicate stronger influence on predicting TCS classes. Dominant domains such as PF01339, PF00702, and PF07679 demonstrate their key role in distinguishing signaling and regulatory protein families.
Figure 2.
Top 20 Pfam Domain Features Ranked by XGBoost Importance. This bar plot shows the 20 most important Pfam domains contributing to the XGBoost classification model. Higher importance values indicate stronger influence on predicting TCS classes. Dominant domains such as PF01339, PF00702, and PF07679 demonstrate their key role in distinguishing signaling and regulatory protein families.
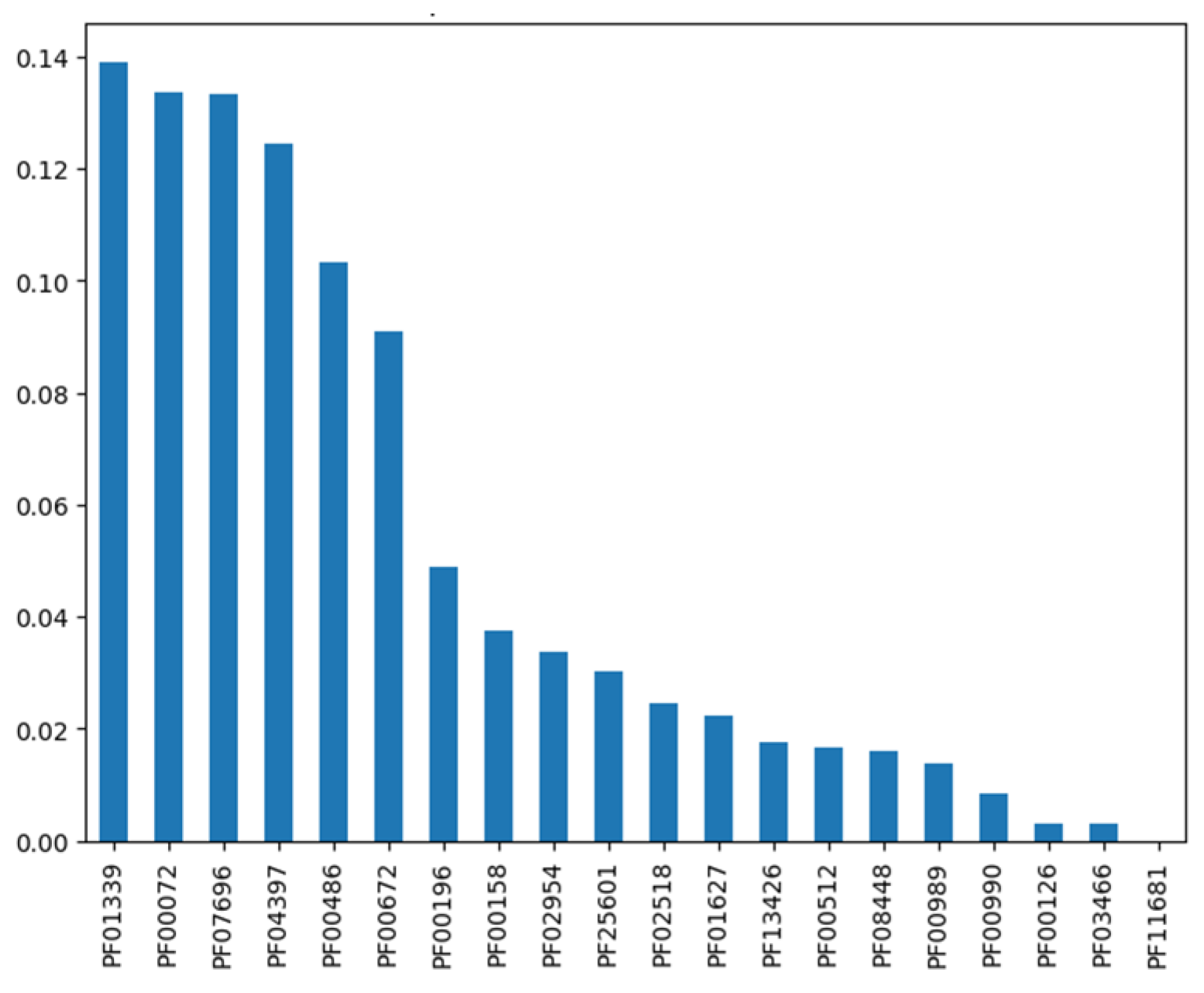
Figure 3.
Confusion matrix of the XGB classification model. The confusion matrix displays the classification performance across the five TCS classes: CpxA-like, EnvZ-like, GRR, non-TCS, and OmpR-like. Correct predictions appear along the diagonal. The model performs strongly for the Non-TCS class but shows misclassification in underrepresented classes such as CpxA-like and EnvZ-like due to limited sample size.
Figure 3.
Confusion matrix of the XGB classification model. The confusion matrix displays the classification performance across the five TCS classes: CpxA-like, EnvZ-like, GRR, non-TCS, and OmpR-like. Correct predictions appear along the diagonal. The model performs strongly for the Non-TCS class but shows misclassification in underrepresented classes such as CpxA-like and EnvZ-like due to limited sample size.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



