
Submitted:
31 December 2025
Posted:
01 January 2026
You are already at the latest version
Abstract
Staphylococcus cohnii, a coagulase-negative staphylococcus (CoNS), is a human skin microbiome constituent with significant antibacterial potential due to its production of bacteriocins. This study aims to characterize the genome of S. cohnii 148-XN2B 18.2 isolated from healthy human skin, focusing on its bacteriocin genes, phylogenetic relationships, and potential antimicrobial applications. Whole-genome sequencing (WGS) was conducted by integration of Illumina and Oxford Nanopore platforms. Bacteriocin genes were identified using BLASTp against reference databases, while phylogenetic analysis was conducted using MAFFT and IQ-TREE. Comparative genomic analysis of bacteriocin clusters was performed using Clinker. The genome of S. cohnii 18.2 consists of a circular chromosome of 2,768,657 bp with a GC content of 32.7%. Functional annotation revealed 5 putative bacteriocin genes. Phylogenetic analysis confirmed a close evolutionary relationship with S. arlettae. Comparative genomics showed high conservation of bacteriocin clusters including Colicin V-like, Enterocin B-like, Pyocin AP41-like and Bacteriocin 28b-like between S. cohnii and S. arlettae.
Keywords:
Staphylococcus cohnii
; whole-genome sequencing
; bacteriocins
; antibacterial potential
; human skin microbiome
1. Introduction
Finding effective antibiotics to treat bacterial infections is a major concern, as antibiotic resistance (AMR) is becoming increasingly common, threatening treatment capabilities. The misuse of antibiotics has led to the rise of multidrug-resistant (MDR) bacteria, reducing the effectiveness of conventional antimicrobial drugs. The World Health Organization (WHO) has warned that the situation of antibiotic resistance is worsening, while the Centers for Disease Control and Prevention (CDC) has declared that humanity is now living in a "post-antibiotic era" [1].
The human skin microbiome contains hundreds of different types of bacteria that serve as the body's first defense against the outside environment [2]. Coagulase-Negative Staphylococci (CoNS) are bacteria generally considered non-pathogenic and are commonly found as part of the human skin microbiome [3]. These bacteria create heat-stable, ribosomal synthesized antimicrobial peptides called bacteriocins, which can have both broad and narrow range inhibitory spectra. These bacteriocin have no antagonistic effects on the cells that make them [4,5].
Staphylococcins produced by Staphylococci display remarkable functional diversity, being effective not only against other staphylococci but also against a wide range of microorganisms. Likewise, "bacteriocin-like inhibitory substances" (BLIS) refer to biomolecules which inhibit widely range of bacteria including both Gram-positive and Gram-negative bacteria [6]. Gram-positive bacteriocins are categorized into three main classes based on their molecular structure and post-translational modifications. Class I, also called lantibiotics, includes molecules under 5 kDa that are thermostable and feature significant post-translational modifications, with distinct amino acid compositions like lanthionine and methyl-lanthionine forming intramolecular rings; this class is further divided into Class Ia, which are polar with a positive net charge, and Class Ib, which are neutral or negatively charged. Class II comprises small molecules (<10 kDa) with minimal or no post-translational modifications and is subdivided into four subclasses based on their structure and mode of action: IIa, pediocin-like peptides with conserved N-terminal YGNG(V/L)XC sequence; IIb, unmodified two-component bacteriocins; IIc, circular peptides; and IId, other single-peptide unmodified bacteriocins [7]. Class III encompasses large proteinaceous bacteriocins (>30 kDa) with complex, thermolabile structures, some of which can also be produced by Gram-negative bacteria [8]. In our previous study, phenotypic assays revealed significant antibacterial activity of the Staphylococci strain habitated from human epidermis against several bacterial species, including Staphylococcus aureus ATCC 29213, Staphylococcus aureus ATCC 43300, Enterococcus faecalis ATCC 29212, Enterococcus faecalis ATCC 51299, Enterococcus casseliflavus ATCC 700327, and Staphylococcus saprophyticus ATCC BAA-750 (data not shown). Thus, the present study aims to: (1) characterize the complete genome of a Staphylococci strain isolated from healthy human skin; (2) identify and analyze the propebacteriocin genes within its genome, with an emphasis on their potential antimicrobial properties; (3) investigate the phylogenetic relationships between the Staphylococci strain and other related Staphylococcus species to elucidate its evolutionary dynamics.
2. Materials and Methods
2.1. Bacterial Strain and Culture Condition
Tested strains were isolated from healthy human skin and cultured under aerobic conditions at 37oC in BHA medium (Brain heart agar) for 24h in an incubator. Isolated colonies were subjected to Gram staining to differentiate between cocci, bacilli, and fungi, and underwent catalase, tube coagulase tests, and oxidase testing to distinguish between coagulase-positive Staphylococcus (CoPS), coagulase-negative Staphylococcus (CoNS), and Micrococcus spp [9]. Participation was voluntary, and every participant signed a written informed consent form. The study was approved by the Ethics Committee in Biomedical Research, Hue University of Medicine and Pharmacy, Hue University (Approval Code H2023/062; May 20, 2023).
2.1. Genome Sequencing and Assembly
DNA extraction:
Genomic DNA extraction was carried out using the Monarch HMW DNA extraction kit for tissue (NEB, USA). DNA concentration was determined by fluorescence method by Qubit DNA assay (Thermo Fisher Scientific) while the OD260 and OD280 values were determined by Nanodrop spectrophotometer. DNA size was assessed with 1% agarose gel electrophoresis. Extracted genomes must had a concentration of at least 50.00 ng/μL, OD260/OD280 ≥ 1.50. DNA size < 1000 bp will be considered carefully.
Library preparation and sequencing
To overcome the drawbacks of both short- and long-read sequencing, we employed a hybrid assembly strategy that integrates the high accuracy of Illumina reads with the long-range continuity provided by Oxford Nanopore. Paired-end short-read libraries with 150 bp reads were prepared using the NEBNext dsDNA Fragmentase, NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, USA) while 8000 bp long-read libraries were carried out by Ligation Sequencing Kit (Oxford Nanopore Technologies, USA) according to the manufacturer's instructions. Short-read libraries were sequenced using next-generation sequencing on Novaseq (Illumina) and long-read libraries were sequenced using next-generation sequencing on Flongle (Oxford Nanopore Technologies). The size of short-read and long-read libraries were obtained within 0.4 Gb and 0.15 Gb, respectively.
Genome assembly
To ensure high-quality data, raw sequencing reads were first cleaned and filtered. Short reads were processed with fastp v0.23.1 [10], while long reads were handled using filtlong v0.2.1. Low quality nucleotides and ambiguous bases ("N"s) were removed based on Phred quality scores. Long reads were further filtered by length. The resulting high-quality short and long reads were then assembled into a genome using two independent de novo approaches: Unicycler v0.4.8 [11] and a pipeline consisting of Flye v2.9 – medaka v1.4.3 -polca v4.0.9 [12,13]. The best assembly, chosen by comparing the outputs of these methods, was then selected for further analysis. Assembly quality was assessed using several methods, including Quast v5.2.0 [14] for quantitative metrics and checkM v1.2.1 for genome completeness [15]. To refine the assembly, Dnaapler was used to correct the chromosome start position.
2.4. Gene Content Analysis
Genome annotation
The assembled genome was annotated using Prokka v1.14.6 [16] in genus-specific mode to identify genes and other genomic features. To access the completeness of the genome assembly and gene annotation, BUSCO v5.8.2 [17] was used with “genome” and “proteins” modes and the lineage dataset of “bacteria_odb12”.
Amino acid sequences of the annotated genes were used as input for the COGclassifier (https://github.com/moshi4/COGclassifier) and GhostKOALA [18] software packages to classify them into different functional groups. GhostKOALA classification results were visualized using “ggplot2” and “ggpubr” R packages [19,20].
Bacteriocin encoding gene identification and analysis
BLASTp [21] was used to identify the suspected bacteriocin with custom database of reference bacteriocin sequences from previous studies [22]. The E-value cutoff for BLASTp was set as 0.01. The amino acid sequences having the best BLAST hits based on bit scores, E-value, and nucleotide were considered as hypothetical bacteriocins and used for further analysis.
2.5. Phylogenetic Analysis
16S rDNA phylogenetic tree construction
The 16S rDNA region of the collected isolate was amplified by using universal primer pairs of 27F (5´-AGAGTTTGATCCTGGCTCAG-3´) and 1492R (5´-GGTTACCTTGTTACGACTT-3´) on genomic DNA template as described by Ha et al [23]. Then, the 16S rDNA sequences of the isolate and other Staphylococcus species, including S. haemolyticus, S. epidermidis, S. aureus, S. saprophyticus, S. capitis and S. hominis were aligned using MAFFT v7.526 [24] with the alignment strategy set as “auto”. The alignment file was then used as input for IQ-TREE v2.3.6 (-m MFP -B 5000 -bnni -alrt 1000 -seed 31081997 -st DNA) [25].
Bacteriocin gene phylogenetic construction
To investigate the origins of hypothetical bacteriocin genes, Orthofinder v.3.0.1b1 [26] was used to find orthologs of these genes in 14 other Staphylococcus species, with default parameters. The Staphylococcus species from which 14 genomes were downloaded from EsemblBacteria (https://bacteria.ensembl.org/index.html) include S. agnetis (GCA_003040835), S. argenteus (GCA_003967115), S. arlettae (GCA_900457375), S. aureus (GCA_000638495), S. auricularis (GCA_002902455), S. capitis (GCA_001179785), S. caprae (GCA_000160215), S. carnosus (GCA_003970565), S. chromogenes (GCA_003040195), S. condimenti (GCA_001618885), S. croceilyticus (GCA_002902575), S. debuckii (GCA_003718735), S. delphini (GCA_002369735), and S. devriesei (GCA_002902625). The ortho-groups containing a single gene for each species were concatenated and aligned using MAFFT v7.526 [27] with the alignment strategy set as “auto”. The alignment file was then used as input for IQ-TREE v2.3.6 (-m MFP -B 5000 -bnni -alrt 1000 -seed 31081997 -st AA) [25].
The GFF3 and genome FASTA files of the Staphylococcus species having bacteriocin orthologs that are the most identical to the ones in our isolate were downloaded from EnsemblBacteria. Annotation features of the bacteriocin orthologs were then extracted from the GFF3 file. The extracted annotation features for bacteriocin orthologs and the genome sequences were used to generate gbk files using the “seqret” tools of EMBOSS v.6.6 [28]. The identity of bacteriocin orthologs were visualized using Clinker v0.0.31 [29] and the script “Genbank_slicer.py” (https://github.com/jrjhealey/bioinfo-tools/blob/master/Genbank_slicer.py).
Phylogenetic tree visualization
All generated Maximum-likelihood trees were visualized using the “ggtree” package from the R programming language [30].
3. Results
3.1. 16S Phylogenetic Analysis
Presumptive coagulase-negative Staphylococci isolates were identified from the skin swabs based on exhibiting coagulase-negative and oxidase-negative phenotypes. To identify the Staphylococci isolate (denoted by 148-XN2B 18.2), the 16S rDNA of the strain were amplified and compared with the 16S rDNA sequence of S. haemolyticus, S. epidermidis, S. aureus, S. saprophyticus, S. capitis and S. hominis. The resulting phylogeny tree (Figure 1) showed that strain 148-XN2B 18.2 has a high similarity to Staphylococcus cohnii ATCC 29974 (OR801636) and S. cohnii subsp. cohnii (D83361.1), with a sequence identity of 100%. Additionally, strain 148-XN2B 18.2 also exhibited 100% identity with strains isolated in other studies, including S. cohnii GH137 and S. cohnii GS29, and 99.93% similarity with strain S. cohnii CTB46.
3.2. Genome Analysis of S. Cohnii 148-XN2B 18.2
Regarding short-read sequencing metrics, the total raw reads reached 3,376,387, with an average GC content of 32.7% and the rate of reads with Q30 was 96.5%. The GC content of the genome was 32.7%. For long-read, a total of 47,648 raw reads was achieved, with the rate of reads reaching Q7 was 100%. Genome assembly was performed using both short and long reads, and revealed a total of 4 contigs. The complete genome of S. cohnii 148-XN2B 18.2 was identified to have a single circular chromosome with a total length of 2,768,657 bp and no plasmids (Figure 2). Within the theoretical coding regions, 2,756 genes were predicted, including 2,676 CDS (protein-coding regions), which accounted for the majority of the genome's total length (Table 1). The complete genome sequence has been deposited in NCBI with the project accession SAMN54131608.
3.3. Gene Annotation and Functional Annotation
The functional classification of annotated genes was performed using GhostKOALA, and the results were visualized in Figure 3. A total of 19 functional groups were identified, in which the largest group was Genetic Information Processing (A), comprising over 400 genes, followed by Signaling and Cellular Processes (B) with approximately 200 genes and Carbohydrate Metabolism (C) with over 150 genes. In addition, genes associated with Metabolism (D), Environmental Information Processing (E) and Amino Acid Metabolism (F) were also significantly represented, each containing around 100 genes. Meanwhile, other categories had fewer genes. Notably, the Unclassified group (I) accounted for a notable number of genes, approximately 100.
3.4. Identification of Bacteriocin Genes in S. cohnii 148-XN2B 18.2
A previous study on genome analysis of bacteriocin-producing Staphylococci showed that the their genomes contain various gene encoding for bacteriocin class I and class II [31]. Thus, the genome of S. cohnii were searched for the potential genes encode for bacteriocin class I and class II. The results present in Table 2 indicated 5 potential bacteriocin-related genes identified in the genome of S. cohnii, along with their reference bacteriocin IDs and associated information. Among these, S. cohnii carries one gene for class I and four genes for class II.
3.5. Phylogenetic Relationship and Comparative Genomic Analysis of S. cohnii and S. arlettae
To find the genetic context of S. cohnii isolate, an ortholog phylogenetic tree was constructed between different Staphylococci. The phylogenetic analysis based on the orthogroups of different Staphylococci demonstrates a close evolutionary relationship between tested S. cohnii and S. arlettae, supported by a bootstrap value of 95.2%. This analysis highlights the strong genetic similarities between these two species (Figure 4), which would be investigated for the diversity of their bacteriocin contents.
The comparative genomic analysis of S. cohnii and S. arlettae’ revealed three bacteriocin-related gene clusters with conserved and homologous regions between the two species. The identified clusters ranged in size from 0.3 kb to 2.5 kb, with sequence identity values represented in grayscale for comparative visualization. Colicin V-like gene cluster was identified in S. cohnii (coordinates: 1,734,743–1,735,624) and exhibited high sequence identity with the corresponding region in S. arlettae (coordinates: 1,821,949–1,822,940). Besides, Enterocin B-like gene cluster was observed in S. cohnii (coordinates: 2,216,019–2,215,730) and its homologous region in S. arlettae (coordinates: 2,216,590–2,190,969), which also encoded a Pyocin AP41-like gene. A Bacteriocin 28b-like gene cluster was also identified in S. cohnii (coordinates: 2,386,002–2,386,520) and S. arlettae (coordinates: 2,448,002–2,448,651), with strong sequence similarity (Figure 5).
To provide a broader context for the genomic data of an isolated Staphylococci from healthy human skin, a 16S rRNA phylogenetic tree was constructed between S. cohnii and related Staphylococcus spp, which showed the 148-XN2B 18.2 isolate to be closely related to commensal coagulase-negative S. cohnii strains, such as S. cohnii ATCC29974 and GS29, while differed from other common coagulase-positive Staphylococcus species like S. aureus and S. epidermidis . The functional annotation of S. cohnii 148-XN2B 18.2 genes reveals a genome structured to prioritize essential processes, particularly genetic information processing pathways, signaling and cellular processes, and different metabolism pathways. This focus aligns with the needs of a commensal organism, where maintaining transcriptional, translational, and replicative fidelity is crucial for survival under diverse environmental conditions [32,33,34,35,36,37]. Class II bacteriocins were also identified, comprising Acidocin 8912, Enterocin B, Bacteriocin 28b and Colicin V, which exert antimicrobial effects through mechanisms such as pore formation and membrane destabilization in target bacteria. Identified class II bacteriocins had demonstrated a wide range of target, from Gram-positive bacteria (Acidocin 8912, Enterocin B) [4,38], Gram-negative bacteria (Colicin V) [39], to both targets (Bacteriocin 28b) [40]. The coexistence of these three potent bacteriocin-encoding genes in S. cohnii underscores its ecological adaptability and biotechnological potential in applications such as food preservation and therapeutic development [41]. In addition, a Class I Nif11-like peptide family was also identified, which are notable for their structural stability and potential applications in synthetic biology due to their post-translational modifications [42]. The discovery of such genes highlights S. cohnii’s potential to produce highly stable and functionally diverse antimicrobial peptides.
Based on the orthogroup phylogenetic tree between S. cohnii 148-XN2B and different Staphylococcus spp., a close phylogenetic relationship between S. cohnii and S. arlettae was unveiled. This clustering aligns with previous studies that have identified S. cohnii and S. arlettae as part of a group of coagulase-negative staphylococci (CoNS) frequently isolated from similar host environments, such as human and animal skin microbiomes [43,44]. The comparative genomic analysis emphasized the conservation of the Colicin V-like gene cluster, Enterocin B-like cluster, Pyocin AP41-like gene and Bacteriocin 28b-like cluster between S. cohnii and S. arlettae, which may reflect their shared ecological niches and evolutionary pressures, suggesting that both S. cohnii and S. arlettae may rely on similar mechanisms to inhibit competing bacteria in their environments [45]. Additionally, the functional conservation of these bacteriocin gene clusters may have implications for the development of novel antimicrobial agents. The shared genetic architecture between S. cohnii and S. arlettae highlights their potential as reservoirs for diverse bacteriocins with therapeutic applications.
However, there were some limitations in this study. While the classification tools COGclassifier and GhostKOALA provided valuable insights, they rely on existing reference datasets, which might limit the detection of novel functional genes. Additionally, targeted analysis of underrepresented categories, such as secondary metabolite biosynthesis, could reveal further insights into the organism's biosynthetic potential. Besides, the functional roles of several bacteriocin genes identified in this study have not been experimentally validated, which makes it challenging to confirm their precise contribution to the observed antimicrobial activity. Moreover, translating these findings into clinical applications may face hurdles such as scaling up bacteriocin production, navigating regulatory requirements, and addressing variability in efficacy due to genetic differences among strains or environmental factors. Despite these challenges, future studies focusing on functional assays, optimization of production processes, and exploration of synergistic applications with other antimicrobial agents could significantly advance the utility of S. cohnii-derived bacteriocins.
4. Conclusions
This study provides a comprehensive genomic and functional analysis of S. cohnii 148-XN2B 18.2, emphasizing its potential as a reservoir of bacteriocins. The evolutionary conservation and demonstrated antimicrobial activity of its bacteriocins make it a promising candidate for future applications in addressing antimicrobial resistance.
Author Contributions
Conceptualization, T.D.B..; methodology, N.DH and N.T.C.A.; software, N.D.H. and N.T.C.A; validation, L.V.A and T.D.B; formal analysis, N.D.H, N.T.C.A, T.H.M.N, and N.T.M.N.; investigation, N.T.C.A, T.H.M.N, N.T.T, and N.TN.M.; resources, L.V.A.; data curation, T.D.B, N.D.H, N.T.T, D.T.N.M, D.T.H, N.T.K.L and L.V.A; writing—original draft preparation, N.D.H, N.T.C.A and N.T.M.N; writing—review and editing, T.D.B, N.D.H, and N.T.C.A; visualization, N.D.H., N.T.C.A, and T.H.M.N.; supervision, T.D.B; project administration, T.D.B., and N.T.C.A; funding acquisition, T.D.B. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
This study is a result of the Ministerial-level Science and Technology Project, project code B2023-DHH-09. We would like to express our sincere gratitude to all the members of the research team for their valuable contributions and to the Department of Science and Technology of Hue University for their kind support.
References
- Michael, C. A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: Causes, consequences, and management. Front Public Health 2014, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Natsuga, K. Epidermal barriers. Cold Spring Harb Perspect Med 2014, 4, a018218. [Google Scholar] [CrossRef] [PubMed]
- Nagase, N.; Sasaki, A.; Yamashita, K. Isolation and species distribution of staphylococci from animal and human skin. Journal of Veterinary Medical Science 2002, 64, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Diep, D.; Nes, I. Ribosomally Synthesized Antibacterial Peptides in Gram Positive Bacteria. Curr Drug Targets 2005, 3, 107–122. [Google Scholar] [CrossRef]
- Jack, R. W.; Tagg, J. R.; Ray, B. Bacteriocins of gram-positive bacteria. Microbiol Rev 1995, 59, 171–200. [Google Scholar] [CrossRef]
- Daw, M. A.; Falkiner, F. R.; Background, A. Nature, Function and Structure; Bacteriocins, 1997; p. 4328. [Google Scholar]
- Antoshina, D. V; Balandin, S. V; Ovchinnikova, T. V. Structural Features, Mechanisms of Action, and Prospects for Practical Application of Class II Bacteriocins. Biochemistry (Moscow) 2022, 87, 1387–1403. [Google Scholar] [CrossRef]
- Solis-Balandra, M. A.; Sanchez-Salas, J. L. Classification and Multi-Functional Use of Bacteriocins in Health, Biotechnology, and Food Industry. Antibiotics 2024, 13. [Google Scholar] [CrossRef]
- Baker, J. S. Comparison of Various Methods for Differentiation of Staphylococci and Micrococci. J Clin Microbiol 1984. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wick, R. R.; Judd, L. M.; Gorrie, C. L.; Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 2017, 13, 1–22. [Google Scholar] [CrossRef]
- Krasnov, G. S.; Pushkova, E. N.; Novakovskiy, R. O. High-Quality Genome Assembly of Fusarium oxysporum f. sp. lini. Front Genet 2020, 11, 959. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A. V; Salzberg, S. L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput Biol 2020, 16, e1007981. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Parks, D. H.; Imelfort, M.; Skennerton, C. T.; Hugenholtz, P.; Tyson, G. W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Simão, F. A.; Waterhouse, R. M.; Ioannidis, P.; Kriventseva, E. V; Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kassambara, A. ggplot2 Based Publication Ready Plots. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 31 Oct 2024 2023).
- Wickham, H; Chang, W; Henry, L; Pedersen, TL; Takahashi, K; Wilke, C; Woo, K; Yutani, H; Dunnington, D; Posit, P. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. Available online: https://cran.r-project.org/web/packages/ggplot2/index.html (accessed on 16 Jun 2024 2023).
- Camacho, C.; Coulouris, G.; Avagyan, V. BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Akhter, S.; Miller, J. H. BPAGS: a web application for bacteriocin prediction via feature evaluation using alternating decision tree, genetic algorithm, and linear support vector classifier. Frontiers in Bioinformatics 2024, 3, 1284705. [Google Scholar] [CrossRef]
- Thanh Ha, D. T.; Kim Thoa, L. T.; Phuong Thao, T. T. Production of extracellular agarase from Priestia megaterium AT7 and evaluation on marine algae hydrolysis. Enzyme Microb Technol 2024, 172, 110339. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H. A.; Von Haeseler, A.; Minh, B. Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Emms, D. M.; Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Hancock, J. M.; Bishop, M. J. EMBOSS (The E uropean M olecular B iology O pen S oftware S uite). In Dictionary of bioinformatics and computational biology; 2004. [Google Scholar]
- Gilchrist, C. L. M.; Chooi, Y.-H. clinker & clustermap.js: automatic generation of gene cluster comparison figures. In Bioinformatics; Robinson, P., Ed.; 2021; Volume 37, pp. 2473–2475. [Google Scholar]
- Yu, G.; Smith, D. K.; Zhu, H.; Guan, Y.; Lam, T. T. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. In Methods Ecol Evol; McInerny, G., Ed.; 2017; Volume 8, pp. 28–36. [Google Scholar]
- Onyango, L. A.; Alreshidi, M. M. Adaptive Metabolism in Staphylococci: Survival and Persistence in Environmental and Clinical Settings. J Pathog 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Solis-Balandra, M. A.; Sanchez-Salas, J. L. Classification and Multi-Functional Use of Bacteriocins in Health, Biotechnology, and Food Industry. Antibiotics 2024, 13. [Google Scholar] [CrossRef]
- Pérez-Ramos, A.; Madi-Moussa, D.; Coucheney, F.; Drider, D. Current knowledge of the mode of action and immunity mechanisms of lab-bacteriocins. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- Gargis, S. R.; Gargis, A. S.; Heath, H. E. Zif, the zoocin A immunity factor, is a FemABX-like immunity protein with a novel mode of action. Appl Environ Microbiol 2009, 75, 6205–6210. [Google Scholar] [CrossRef]
- Sano, Y.; Kageyama, M. Purification and properties of an S-type pyocin, pyocin AP41. J Bacteriol 1981, 146, 733–739. [Google Scholar] [CrossRef]
- Le, M. N.-T.; Nguyen, T. H.-H.; Trinh, V. M. Comprehensive Analysis of Bacteriocins Produced by the Hypermucoviscous Klebsiella pneumoniae Species Complex. Microbiol Spectr 2023, 11, 1–20. [Google Scholar] [CrossRef]
- Tahara, T.; Kanatani, K.; Yoshida, K.; Miura, H.; Sakamoto, M.; Oshimura, M. Purification and some properties of acidocin 8912, a novel bacteriocin produced by Lactobacillus acidophilus TK8912. Biosci Biotechnol Biochem 1992, 56, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Cascales, E.; Buchanan, S. K.; Duché, D. Colicin Biology. Microbiology and Molecular Biology Reviews 2007, 71, 158–229. [Google Scholar] [CrossRef]
- Guasch, J. F.; Enfedaque, J.; Ferrer, S.; Gargallo, D.; Regué, M. Bacteriocin 28b, a chromosomally encoded bacteriocin produced by most Serratia marcescens biotypes. Res Microbiol 1995, 146, 477–483. [Google Scholar] [CrossRef]
- Alvarez-Cisneros, Y.; Espuñes, T. Enterocins: Bacteriocins with applications in the food industry. Formatex.Info 2011, 1330–1341. [Google Scholar]
- Arnison, P. G.; Bibb, M. J.; Bierbaum, G. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin Microbiol Rev 2014, 27, 870–926. [Google Scholar] [CrossRef]
- Schleifer, K. H.; Kilpper-Bälz, R.; Devriese, L. A. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. k1oosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst Appl Microbiol 1984, 5, 501–509. [Google Scholar] [CrossRef]
- Newstead, L. L.; Varjonen, K.; Nuttall, T.; Paterson, G. K. Staphylococcal-produced bacteriocins and antimicrobial peptides: Their potential as alternative treatments for staphylococcus aureus infections. Antibiotics 2020, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
16S rRNA phylogenetic tree of Staphylococci spp. *The percentage at the nodes indicate the levels of bootstrap support based on 1000 replications. Taxa in the box indicates closely related species to strain 148-XN2B 18.2.
Figure 1.
16S rRNA phylogenetic tree of Staphylococci spp. *The percentage at the nodes indicate the levels of bootstrap support based on 1000 replications. Taxa in the box indicates closely related species to strain 148-XN2B 18.2.
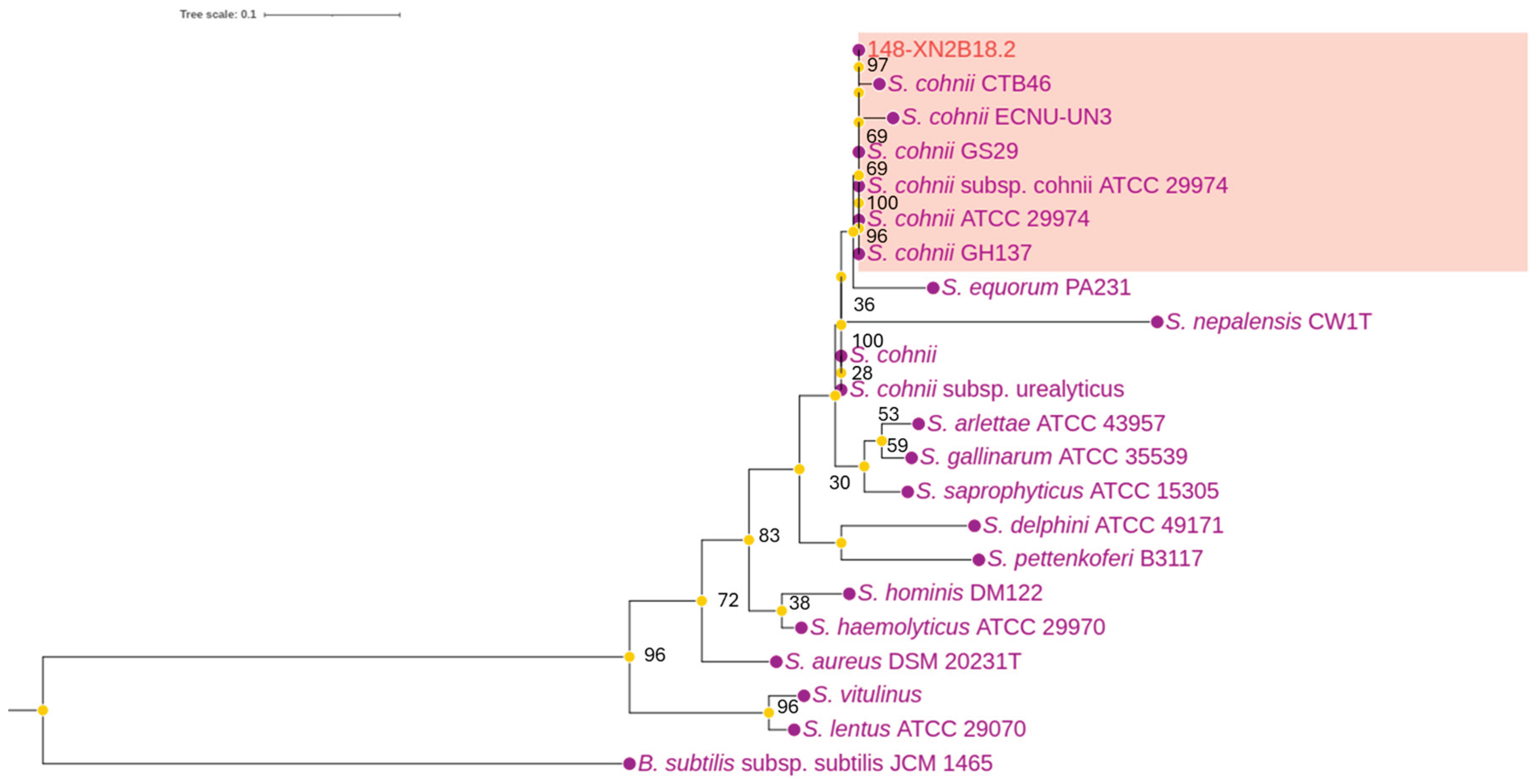
Figure 2.
The circular genome map of S. cohnii 18.2. *Displays key information organized from the outermost to the innermost circles, as follows: the first circle represents the scale of the genome; the second and the third circles represent the positions of CDS (Forward and Reverse), the fourth and the fifth circles showing the positions of tRNA; and rRNA on the genome; the sixth circle illustrates the GC content (positive and negative values); and the seventh circles shows the GC skew (positive and negative values).
Figure 2.
The circular genome map of S. cohnii 18.2. *Displays key information organized from the outermost to the innermost circles, as follows: the first circle represents the scale of the genome; the second and the third circles represent the positions of CDS (Forward and Reverse), the fourth and the fifth circles showing the positions of tRNA; and rRNA on the genome; the sixth circle illustrates the GC content (positive and negative values); and the seventh circles shows the GC skew (positive and negative values).
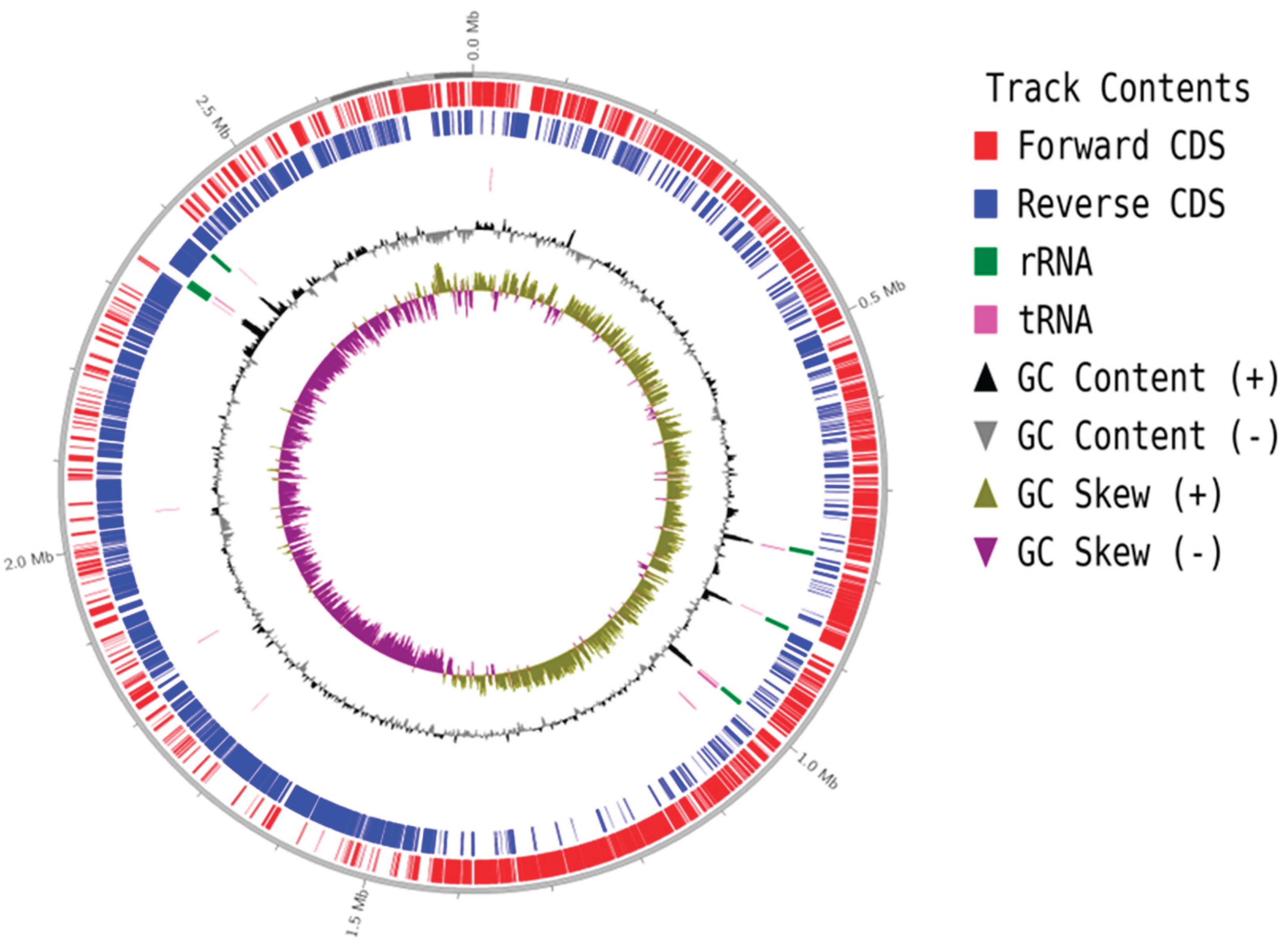
Figure 3.
Function classification in COG (Cluster of Orthologous Groups of proteins) of S. cohnii 18.2.
Figure 3.
Function classification in COG (Cluster of Orthologous Groups of proteins) of S. cohnii 18.2.
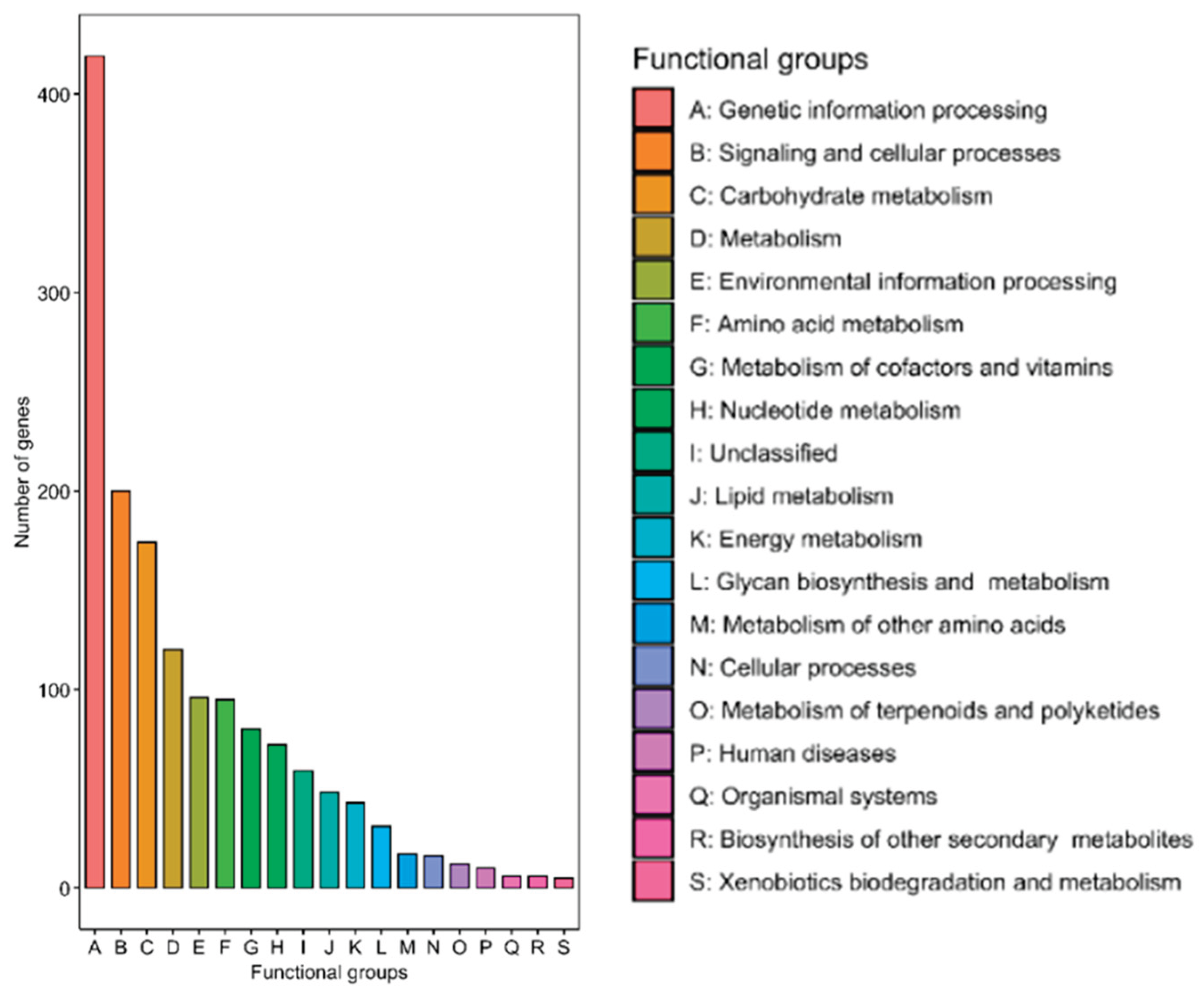
Figure 4.
Phylogenetic Tree of Staphylococcus Species Based on Maximum Likelihood Analysis.
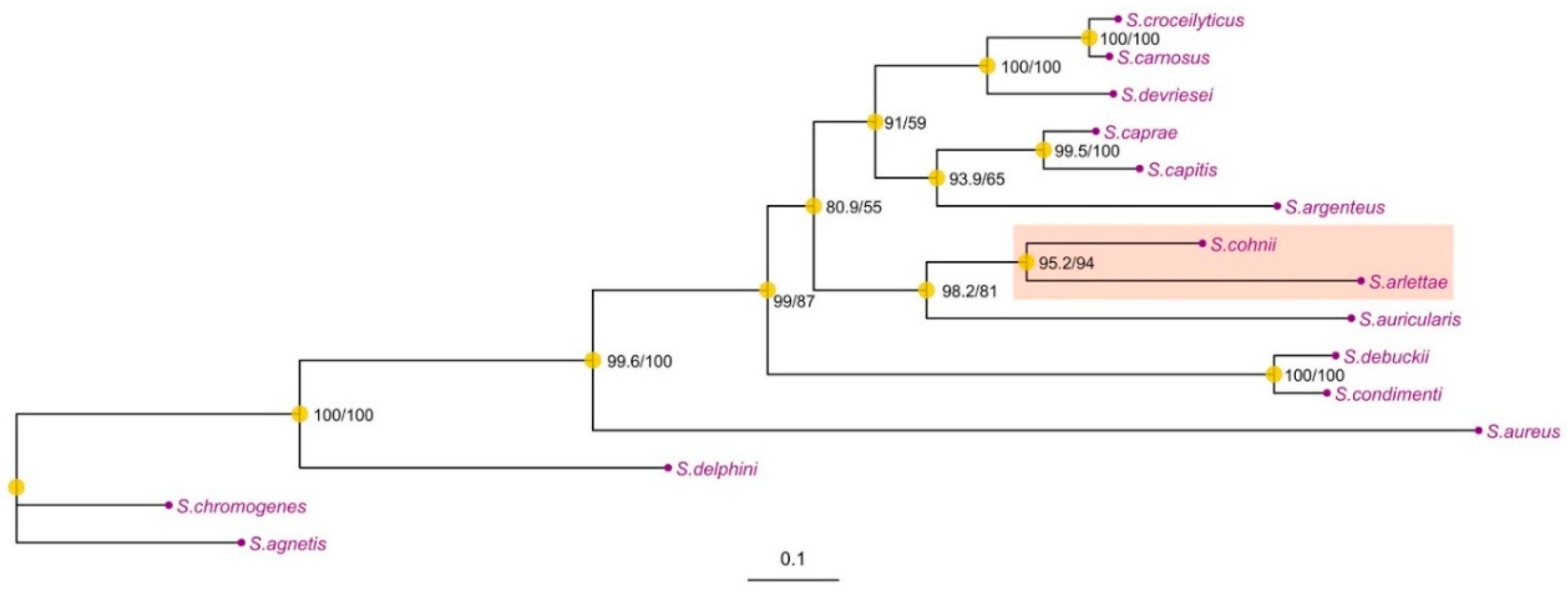
Figure 5.
Bacteriocin gene clusters in Staphylococcus cohnii and Staphylococcus arlettaeDiscussion.
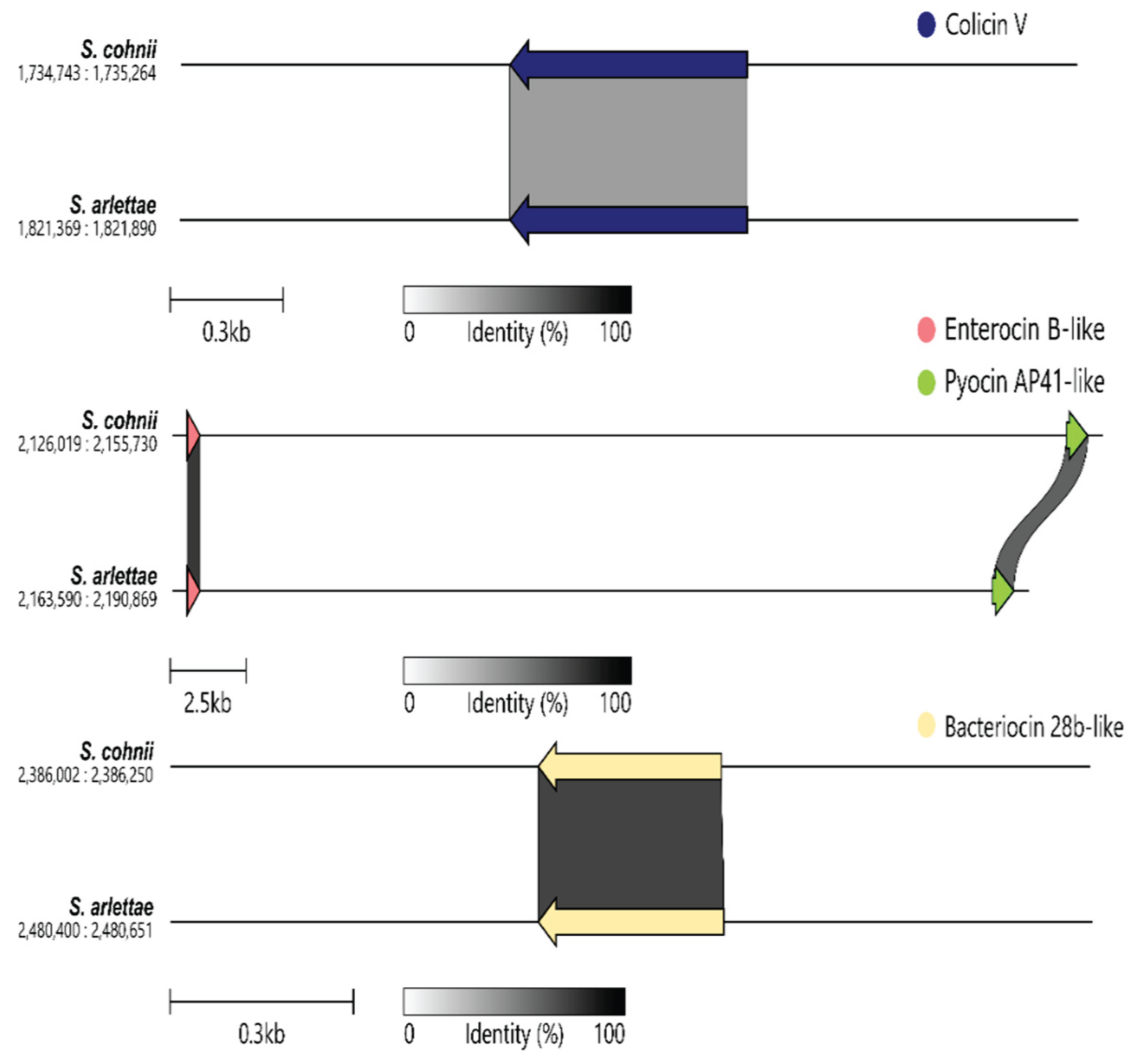

Table 1.
Basic genome information of test strains.
| Strain | S. cohnii 18.2 |
|---|---|
| Genomic size | 2,768,657 |
| G + C content | 32.7%. |
| Number of ORFs | 2676 |
| Gene | 2756 |
| Number of tRNA | 60 |
| Number of rRNA | 19 |
| Number of tm RNA | 1 |
| Number of repeat regions | 4 |
Table 2.
Functional Annotation of Bacteriocin-Related Genes in Staphylococcus cohnii.
| No | Gene id | Reference bacteriocin ID | Class & Name | Per. Ident (%) | Query coverage (%) |
| 1 | WH1322402A02_00901_gene | NP_604414.1 | Class II Acidocin 8912 |
33.3 | 95.2 |
| 2 | WH1322402A02_02122_gene | AAD28234.1 | Class II: Enterocin B | 27.7 | 91.5 |
| 3 | WH1322402A02_02267_gene | WP_120446043.1 | Class I: RiPP (Ribosomally synthesized and post-translationally modified peptides) | 31.6 | 93.9 |
| 4 | WH1322402A02_02390_gene | CAA44310.1 | Class II: Bacteriocin 28b | 31.3 | 98.5 |
| 5 | WH1322402A02_01720_gene | Class II: Colicin V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



