
Submitted:
31 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Genetic variation underlies the capacity of populations to adapt, yet what drives how this variation is generated and maintained in natural populations remains poorly understood. Fundamental processes such as mutation, ploidy, and recombination are known to shape genetic variation and adaptive potential but are typically studied in isolation and under controlled laboratory conditions. How these processes act together under varying environmental conditions to structure genetic variation across complex natural populations remains unresolved. In yeasts, these processes are dependent on reproductive mode, ploidy shifts, and environmental stressors which jointly shape genomic stability and adaptive potential. Here we review our current knowledge on the roles of mutation, ploidy, and recombination in adaptation in the model yeasts Saccharomyces cerevisiae and the human pathogenic Cryptococcus. We highlight heterogeneity in mutation rates, recombination, and ploidy states across strains, environments, and populations, challenging the assumption that these parameters are uniform. We argue that fluctuating environments, increasingly driven by climate change, are likely to intensify interactions among these processes in ways that remain difficult to predict. Integrating population genomics with ecologically realistic frameworks will be essential for understanding natural evolutionary dynamics and anticipating fungal adaptation and disease emergence.
Keywords:
Baker’s yeast
; cryptococcosis
; mutation accumulation
; whole-genome sequencing
; mitotic recombination
; sexual reproduction
; haploidy
; diploidy
1. Introduction
Evolution is the change in genetic material over time and is a fundamental component of biology. Even before heredity was understood at the molecular level, Darwin introduced life as a dynamic system shaped by descent with modification, fundamentally altering how biological diversity was viewed[1]. Yet it still took until the mid-20th century for Avery, MacLeod, and McCarty to demonstrate that DNA is the hereditary material underlying biological variation which allows evolution to occur[2]. As genetics and molecular biology continue to advance, our appreciation for the complexity of this system as only increased. In 1968, Kimura, followed by King and Jukes, independently proposed the Neutral Theory of Evolution, which argues that most genetic changes are selectively neutral[3,4]. This theory highlights spontaneous mutation and genetic drift as playing central roles in evolution, shifting the framework away from selection as the dominant force. However, though stochastic processes are increasing recognized as key to understanding natural genetic variations, adaptation driven by selection continues to be a fundamental aspect of evolutionary change[5]. Adaptation is the process where a population increases its fitness relative to others in the immediate environment by acting on genetic variations among individuals[5]. Thus, a population’s capacity for adaptation is directly related to the genetic variation present. This variation is shaped by many biological processes including mutation, recombination and ploidy level.
Yeasts are unicellular fungi. They are phylogenetically diverse and have been found to inhabit a wide range of ecological niches, including those associated with human activities[6,7,8]. For example, domesticated nearly 9,000 years ago for alcoholic fermentation, the budding or baker’s yeast Saccharomyces cerevisiae has since become central to baking, brewing, cheese making, and many other applications[9,10,11,12]. However, the same adaptive potential also contributes to the evolution of pathogenicity which establishes many yeast species as human pathogens[13,14,15]. Although uncommon, isolates of S. cerevisiae are also observed in clinics [16,17]. Invasive fungal infections caused by yeasts are estimated to contribute to more than one million deaths worldwide annually[18]. Among these, the human pathogenic Cryptococcus (HPC), which comprises seven species, can cause cryptococcal meningitis, a leading cause of mortality among HIV/AIDS+ individuals [19,20]. More recently, the emergence of the multidrug-resistant yeast Candidozyma auris (syn. Candida auris), a species first reported in 2009 and its global spread illustrates how yeasts can evolve into critical public health threats[21]. Climate change is expected to amplify these evolutionary pressures by shifting environmental conditions and altering selective pressures, such as temperature and humidity[22]. These shifts are predicted to influence both pathogenic and domesticated species, underscoring the need to understand how yeast populations respond to stress to predict future disease risk and antifungal resistance.
Increased genetic variation in natural pathogen populations can increase adaptive potential, enabling rapid evolutionary responses to host immune defenses and antifungal use. Elevated genetic variation increases the likelihood that drug-resistant or ecologically-adapted genotypes are present prior to infection-treatment intervention, allowing pathogens to quickly respond to treatments or the host immune response[23,24]. This genetic heterogeneity complicates treatment strategies and pathogen surveillance while cryptic and unsampled variation in natural reservoirs reduces the predictability of pathogen evolution. As a result, public health responses which must defend against ongoing and pre-existing adaptive mutations can lag pathogen adaptation. Thus, increased natural genetic variation creates a challenge for management of pathogenic yeast populations. Yet, how genetic variation is increased and maintained across natural reservoirs remain largely unresolved. Additionally, understanding how genetic variation is generated and contributes to adaptation in response to stress is fundamental for our understanding of evolution. Mutation, ploidy and recombination are not independent processes but rather interact with each other to influence genetic variation. The divergent yeasts S. cerevisiae and the HPC represent model yeasts from which to investigate the combined impact of these processes on evolution.
Both S. cerevisiae and HPC have well-annotated genomes, robust laboratory strains and are recognized as model systems used to study molecular genetics. Each yeast can reproduce both asexually and sexually, are observed to undergo ploidy variation and contain pathogenic and non-pathogenic strains. In addition, S. cerevisiae and HPC represent the two largest divergent fungal phyla Ascomycota and Basidiomycota, respectively. They differ in their natural ploidy state and are associated with distinct ecological and anthropological conditions, subjecting them to different selective pressures. These shared and divergent traits make S. cerevisiae and HPC strong candidates to compare conserved evolutionary processes while contrasting between key parameters such as natural ploidy state and environmental pressures. Laboratory experiments of S. cerevisiae are central for understanding fundamental principles of yeast genetics while HPC provides a system for evaluating these principles in the context of human pathogens.
This review aims to outline the life cycle of S. cerevisiae and HPC, discuss the process of mutation, the influence of ploidy levels and the role of recombination in shaping their adaptive potentials. Understanding these mechanisms is critical as we anticipate increased rate of evolution under changing environmental conditions.
2. Life Cycle
To help understand the three processes to be reviewed in this study, we first outline the life cycles of S. cerevisiae and HPC, as these cycles provide the framework for describing when mutations arise and how recombination and ploidy shifts occur. Figure 1 provides a visualize summary of this section. Both yeasts possess two mating types, MATa and MATα, specified at the mating-type locus (MAT)[25,26].
The life cycle of S. cerevisiae involves regular alternation between haploid and diploid states, with the diploid state considered the more stable one of the two[26]. In nutrient-rich conditions, both haploid and diploid cells can proliferate mitotically by budding. Under nutrient limitation, diploid mitotic growth will arrest and transition to meiosis producing haploid ascospores. When conducive conditions arise, mating between MATα and MATa haploid cells will occur, fusing to restore diploidy[26]. Both heterothallic and homothallic strains of S. cerevisiae occur in nature. Heterothallic strains produce daughter cells of the same mating type, whereas homothallic strains switch mating type, enabling mating and self-fertilization between recently divided sister cells containing opposite mating types[26].
In contrast, HPC cells are predominantly haploid, with a heterothallic mating system, meaning there is no mating-type switching[25]. The canonical sexual cycle involves fusion between MATa and MATα cells followed by meiosis, though accumulating evidence indicates that (α–α) unisexual reproduction also occurs in nature[25]. Initiation of the sexual cycle is generally associated with stressful conditions. Diploid cells of Cryptococcus spp. are rare and often associated with hybridization between divergent lineages where sequence divergence results in disrupted meiosis[25].
For both yeasts, most reproduction is thought to be clonal, through asexual budding or selfing, with outcrossing being rare[26,27]. However, the relative rates of clonal versus sexual reproduction in natural populations remain largely unknown. This mixture of reproductive modes and shift in ploidy creates a complex evolutionary system where sexual reproduction can generate genetically diverse progeny for selection to act, while clonal proliferation then amplifies genotypes with advantageous mutations or allelic combinations. The occasional return to sexual reproduction then allows for recombining genetic materials which can facilitate adaptation and reduce the accumulation of deleterious mutations[28,29]. Taken together, the reproductive strategies of S. cerevisiae and HPC shape the timing and frequency of genetic change. Because the life cycle is thought to be dependent on environmental conditions and changing conditions are predicted to increase the ecological range of lineage boundaries, increasing the likelihood of divergent strains interacting, climate change may alter natural rates of sexual reproduction, further contributing to increased genetic variation. To understand how these differences translate into evolutionary potential, the following sections explore the three key processes that generate genetic variation: mutation, ploidy, and recombination.
3. Mutation
Mutations are the ultimate source of genetic variation that enable evolutionary change. Although most mutations are neutral or deleterious, rare beneficial mutations are produced and necessary for adaptation. Mutations include both structural changes in the genome such as deletions, insertions, inversions, translocations, and duplications as well as single nucleotide changes[30]. Mutations may arise spontaneously through imperfect DNA replication or repair, or by exposure to environmental mutagens[30].
Spontaneous mutations arise due to limitations in the efficiency of a cell’s DNA replication and repair machinery. Replication fidelity depends on DNA polymerases correctly selecting base pairs and removing incorrect nucleotides during proofreading. In addition, the mismatch repair system detects and corrects helical distortions caused by replication errors, playing a critical role in maintaining genomic integrity[31,32,33]. Mutations can arise when these repair mechanisms are disrupted which can increase the overall mutation rate[34,35]. Mutated genes that increase mutation rates are referred to as mutator alleles and are often considered detrimental as the increased accumulation of deleterious mutations can have profound effects on the fitness of an organism. However, in large populations, mutator alleles are also thought to accelerate adaptation in a dynamic environment by increasing the rate of novel genetic diversity for selection to act on[36,37]. Mutations may also arise through stress-induced mutagenesis, in which exogenous stressors disrupt repair systems. Such stressors include proteotoxic and oxidative stress, chemical agents, temperature shock, and exposure to antifungal drugs[38,39,40,41]. However, it is difficult to distinguish whether mutations arise from intrinsic DNA replication and repair fidelity or from environmental stress. Thus, identifying the source can be challenging without comparative analyses across controlled environments that examine changes in mutation rate and mutational spectrum which link these patterns to specific DNA damage or repair pathways.
Mutation rate refers to the number of mutations per site per generation and reflects how frequently genetic changes arise. It is a key parameter in evolutionary biology because it directly influences the rate at which populations accumulate genetic variation [42]. Mutation rates are commonly estimated using laboratory-based experiments, such as Luria–Delbrück fluctuation assays, mutation accumulation experiments, as well as be inferred from patterns of genetic polymorphism in natural populations[33,35,43,44,45]. Luria–Delbrück fluctuation assays estimate mutation rates based on selectable phenotypes arising in clonally expanding populations subjected to artificial selective pressure at the end. This method captures only mutations that confer a detectable phenotype under selection and reflect a non-neutral subset of mutations[44]. In contrast, mutation accumulation experiments aim to estimate the baseline neutral mutation rate by imposing replicate lines through repeated single-cell bottlenecks, thereby minimizing the efficacy of selection. After many generations, accumulated mutations are identified by comparing evolved genomes to their ancestral state[46]. Although mutation accumulation experiments provide a more direct estimate of spontaneous mutation rates, they are time-intensive and may still underestimate mildly deleterious mutations that are purged over the course of the experiment.
Although informative, the above two laboratory-based estimates do not capture the ecological and demographic complexity of natural populations. As an alternative, mutation rates can be inferred from observed genetic variation in natural populations[45]. These estimates are sensitive to assumptions about selection, demographic history, and effective population size. Events such as population bottlenecks and expansions can strongly bias inferred rates because genetic diversity scales with effective population size. Thus, errors in estimating effective population size or variation in effective population size can further influence mutation rate inference[36]. Consequently, inferred mutation rates typically represent long-term averages and may not reflect short-term environmentally or context-dependent change. As growing empirical evidence challenges the assumption that mutation rate should be treated as a constant parameter, accurately inferring natural mutation rates and understanding the factors shaping their temporal and population-level variability has become important in population genetic studies. It is increasingly recognized that mutation rates vary substantially among species, populations, and even among strains within a species[35,37,47]. Gou et al. (2019) demonstrated variation across seven strains of S. cerevisiae ranging from 1.1 × 10−7 to 5.8 × 10−7 [48]. Similarly, Jiang et al. (2021) reported a 10-fold range across 16 haploid, unstressed S. cerevisiae strains[35]. In Cryptococcus spp. Xu et al. (2001) observed mutation rates ranging across 21 strains from 0.41 × 10−9 to 3,135.36 × 10−9, with replicates of the same strain varying from 23.52 × 10−9 to 272.55 × 10−9 [49].
Two theoretical frameworks have been proposed to explain this variation: the drift barrier hypothesis and the model of stabilizing selection. The drift barrier hypothesis proposes that, because most mutations are neutral or deleterious, selection acts to minimize mutation rates until further increases in fidelity are outweighed by the energetic cost of maintaining highly accurate replication and repair[37]. Under this model, the lower bound of mutation rate is determined by the strength of genetic drift. Thus, mutation rates are primarily shaped by selection against the accumulation of deleterious mutations, whereas selection for beneficial mutations or the cost for fidelity is assumed to be inconsequential[36,37,50].
Alternatively, the model of stabilizing selection proposes that an optimal mutation rate is determined by balancing multiple selective pressures[51]. Here, mutation rate is jointly influenced by both genotype and environment, and reflects selection acting on deleterious mutations, beneficial mutations, and the energetic costs of replication fidelity[51]. Two forms of positive selection can modify mutation rate. First-order selection acts directly on alleles that modify the cost of fidelity through DNA replication and repair mechanisms. And Second-order selection increases mutation rate when mutator alleles hitchhike with linked beneficial mutations[3,51]. If selection favouring an increased mutation rate is strong, the optimal mutation rate may be maintained above the drift barrier [35,51]. Likewise, if the pressure to maintain a high mutation rate is removed and the accumulation of deleterious mutations outweighs the benefit the mutation rate will decrease[51]. The outcome of these opposing forces is highly context-dependent, with environmental variation altering both the costs and benefits associated with mutation rate. Under this framework, it is expected that a higher mutation rate will be maintained under fluctuating environmental conditions and selected for in the presence of stress[51].
Empirical evidence supports the stabilizing selection model in S. cerevisiae. Liu et al. (2021) accumulated mutations across replicate lines, observing 19 lines with a significantly higher and 13 lines with a significantly lower mutation rate than the progenitor with some lines reducing mutation rate by 40-50%[51]. Under a strict drift barrier hypothesis this level of reduction should not occur as it suggests the initial mutation rate was maintained above the drift barrier[37]. Researchers calculated S. cerevisiae’s mutation rate to be more than 3000 times higher than expected under the drift barrier[51]. Liu et al. (2021) further identified PSP2 as a mutator gene, its knockout nearly halved mutation rate, suggesting the function of PSP2 contributes to an increased mutation rate[51].
Second-order selection is particularly relevant in organisms that reproduce asexually as mutator alleles can persist without reshuffling from meiosis[52]. Hypermutator strains, defined as strains with significantly increased mutation rates, commonly arise in laboratory evolution experiments[53,54,55]. These strains are often observed to emerge under stress where they happen to provide a selective advantage by increasing the supply of adaptive mutations. For example, exposure to mildly stressful conditions such as ferulic acid and lithium chloride significantly increase the mutation rate of S. cerevisiae[40]. However, it is unclear if this is due to a regulated increase in mutation rate or a result of an advantageous coincidence. Regardless, this pattern is repeatedly observed across laboratory experiments and recently in natural populations[35].
Hypermutators are highly relevant in human pathogenic yeasts. It is hypothesized that hypermutator phenotypes may facilitate colonization of a host by accelerating adaptation to the harsh host environment[32,56]. Laboratory-derived mutants of HPC lacking functional MSH2, MLH1, or PMS1 genes exhibit mutation frequencies approximately 200-fold higher than those observed in wild-type strains[32]. Clinical isolates of HPC have been identified with mutations in MSH2 that result in significantly elevated antifungal resistance rates, exceeding 120-fold increases in certain context[56]. Because stress may drive selection for hypermutator strains, environmental change and antifungal exposure may elevate rates among pathogenic populations, further increasing adaptative potential. Currently, our knowledge of what drives mutator alleles in nature or how long they persist is incomplete. Jiang et al. (2021) recently identified the first natural hypermutator strain of S. cerevisiae associated with mosaic beer fermentation revealing a mutation in the OGG1 that drove the increased mutation rate[35]. Additionally, by analyzing 93 clinical isolates of S. cerevisiae, Strope et al. (2015) identified four strains to contain a MLH1 and PMS1 allele combination that is engineered in laboratory strains to artificially increase the mutation rate 40 fold above the baseline rate[57,58]. However, direct mutation-rate estimates obtained by Skelly et al. (2017) using fluctuation assays revealed only a 5.6-fold increase in mutation rate in these strains relative to non-mutator backgrounds, substantially lower than expected based on engineered laboratory mutators[59]. Although these clinical isolates did not exhibit mutator phenotypes in the diploid state, Raghavan et al. (2018) generated haploid spore clones from three strains and observed an approximately 340-fold range in mutation rates [60]. These results highlight the relationship between mutation and ploidy state. Table 1 summarizes various estimates of mutation rates for S. cerevisiae and HPC.
Further understanding how hypermutators emerge and persist in natural populations could strengthen our ability to predict the emergence of drug resistance and disease outbreaks, as climate change alters temperature, nutrient availability, and host–pathogen interactions in ways that may favour rapid evolution. The evolutionary consequences of mutation greatly depend on ploidy state and rates of recombination reshuffling new variants. Thus, mutation must be considered in the context of these two processes to determine how genetic variation is generated and maintained.
4. Ploidy
Populations of haploid and diploid yeast frequently exhibit distinct evolutionary dynamics in experimental systems. However, factors underlying these differences remain unclear. It’s likely that differences in repair pathways, replication fidelity, and selection pressures between ploidy states contribute to this pattern, but their relative contributions remain to be elucidated[63,72,73]. Comparative studies of spontaneous mutation rate across ploidy revealed that diploids do not simply accumulate twice as many mutations as haploids, indicating that ploidy influences the mechanisms of DNA replication and repair as well as the adaptive potential of cells[63]. For example, in one study spanning 51 strains across 33 environmental conditions, the relative fitness advantage of haploids versus diploids depended strongly on the type of stressor[74]. It is hypothesized that recessive deleterious mutations would be masked in diploids. However, accumulated single nucleotide mutations demonstrated a stronger negative effect on diploids than on haploids in mutation accumulation experiments, leading to a reduced average growth rate in diploid lines. An explanation for this pattern could be that selective pressure against mutations could be more uniformly applied or stronger in haploid than in diploids[63]. Haploid S. cerevisiae cells accumulate approximately 40% more spontaneous single nucleotide mutations per nucleotide site than genetically identical diploid cells, consistent with enhanced replication fidelity in diploids[63]. Sharp et al. (2018) further demonstrated that the genomic distribution of single nucleotide mutations differs significantly between haploid and diploid S. cerevisiae strains, suggesting that ploidy influences both the rate and the spatial pattern of mutations[63].
In contrast to point mutations, structural mutations are substantially more common in diploids than in haploids, with some experiments reporting nearly twice as many whole-chromosome changes per cell division in diploids[63]. Diploid S. cerevisiae cells are also expected to undergo approximately one loss-of-heterozygosity event per mitotic division, and in heterozygous diploids these events can rapidly generate homozygosity for advantageous alleles[75]. Similar patterns are observed in HPC, where disrupted meiosis during hybridization frequently results in diploid or aneuploid hybrids[76]. Zhu et al. (2016) identified a third of clinical S. cerevisiae isolates analyzed to be aneuploids (>2n) [77]. Aneuploidy in both S. cerevisiae and HPC contributes substantial genome plasticity and has been repeatedly associated with adaptive responses. In HPC, aneuploidy has been shown to enhance drug resistance through gene dosage effects mediated by increased chromosome copy number[78]. By shifting ecological ranges which bring previously isolated populations of HPC together, climate change has the potential to increase the likelihood of hybridization. Increased hybridization events can further drive genomic alterations and potentially accelerate evolutionary responses to host immune and antifungal treatments[79,80]. Understanding how ploidy and hybridization operate alongside mutation and recombination will be critical for predicting disease emergence from yeast populations.
5. Recombination
Recombination can occur during both mitotic and meiotic replications. During mitotic replication, recombination contributes to loss of heterozygosity events while during meiosis, sexual recombination can accelerate adaptation by reshuffling genetic material. Meiotic recombination creates novel allele combinations, facilitates the purge of deleterious mutations and increases the pool of genetic variation available for selection to act. Experimental evolution of S. cerevisiae has demonstrated that sexual recombination can increase the efficacy of natural selection in adapting populations. However, this advantage is strongly dependent on the environment[81]. If a clonal genotype has optimized fitness in relation to the environment, recombination is not only an energetically costly event, but it can risk decreasing the fitness level[81]. Meiosis alone can elevate the mutation rate by approximately 4–21 times relative to the mitotic rate, additionally expanding the mutational supply[67]. The initiation of sexual reproduction is frequently associated with stressful environmental conditions, such as nutrient depletion, high temperatures, and oxidative stress. These conditions likely favour the production of spores for long-term survival while simultaneously generating a pool of diverse genotypes with different fitness spectra across environments through recombination and assortment of genetic material [82].
Genome-wide analyses demonstrate that meiotic recombination hot- and cold-spots shift with temperature which highlight that recombination patterns can be sensitive to the environment[83]. The formation of interspecies hybrids within HPC enables divergent genomes to mix, producing offspring that can display hybrid vigour by producing novel phenotypes which enhance stress tolerance[76,84,85]. These traits can result in a fitness advantage in new or challenging environments, such as within a host or in the presence of antifungal treatments. The increasing prevalence of hybrids in clinical settings suggests that hybridization is an ongoing evolutionary force driven by the enhanced adaptive potential of hybrid genotypes [80,86,87]. For example, a whole-genome analysis of 144 S. cerevisiae strains identified three clinical isolates contained genomic regions of two closely related species Saccharomyces paradoxus and Saccharomyces kudriavzevii[77]. However, accurately estimating recombination rates in natural populations remains challenging. Recombination rates are often heterogeneous across genomes, varying among chromosomes, genomic regions, strains, life stages, and environmental conditions, which complicates efforts to define a single, representative population-level rate[88,89]. In natural populations, recombination is typically inferred indirectly from patterns of linkage disequilibrium or phylogenetic incompatibility, approaches that rely on assumptions such as the infinite sites model[90]. Violations to these assumptions and past demographic events can bias recombination signals[91]. As well, a comprehensive understanding of environmental triggers that initiate recombination is largely unresolved in most species, making it difficult to determine how frequently recombination occurs in natural populations and how it varies across environments.
Although meiotic recombination appears infrequent in natural populations of S. cerevisiae and HPC, genomic data provide evidence that it does occur[27,92,93,94]. Recent analyses have revealed signatures of recombination in a natural population of HPC composed almost exclusively of a single mating type (MATα), indicating that sexual reproduction may occur more frequently than previously expected[95]. The most recent outbreak of cryptococcosis, which primarily impacted immunocompetent individuals, is thought to have resulted from clonal expansion of a genotype derived from sexual reproduction. It is hypothesized that recombination between two lineages produced a genotype able to expand into Vancouver Island as a new ecological niche [96,97]. These strains also demonstrated increased fertility and virulence, illustrating how meiotic recombination can drive the emergence of novel pathogenic strains[98,99,100]. Additionally, clinical isolates of S. cerevisiae demonstrate higher levels of heterozygosity compared to non-clinical isolates, consistent with the importance of outcrossing of the human pathogenic population of this model yeast[75].
In addition to meiotic processes, mitotic recombination which occurs during vegetative growth of diploid or aneuploid cells can contribute to loss of heterozygosity, gene conversion, or chromosomal rearrangements[101,102]. Under stress, these chromosomal changes can fix advantageous alleles and drop deleterious alleles in diploid cells without requiring meiosis, allowing for quick adaptation. This mechanism is often observed in the presence of antifungal and oxidative pressure [76,80,103,104]. Thus, recombination is a powerful evolutionary force generating diversity under stress and enabling rapid adaptation, yet the frequency and drivers of recombination in natural populations remain unresolved for these and other fungi, making it difficult to understand how recombination, mutation and ploidy interact to shape adaptive potential.
6. Future Directions
Even though ample work has been completed to characterize the role of mutation, ploidy and recombination in maintaining genetic variation, many unresolved questions remain. These processes are typically studied independently under controlled laboratory conditions which fail to reflect the true complexity of natural populations. As this review highlights, these processes are interdependent and additionally influenced by life cycle, environment and population history, and the interaction of these factors determines a population’s ability to adapt. Thus, it is important that future research aims to address how these factors act beyond controlled, single parameter estimates to improve our capacity for predicting how genetic variation is maintained across changing environments in complex natural systems.
Although fundamental to our understanding, dependence on laboratory-derived parameter estimates alone restricts the extent to which current knowledge can be generalized to natural systems. Due to the nature of experimental design, often mutation, ploidy and recombination are tested under a constant environment with fixed environmental parameters. While such designs can provide a sense for how these processes work, they fail to capture the true complexity of the system. In addition, current designs often rely on single-colony transfers that impose relaxed selection through population bottlenecks and are conducted under relatively short timeframes. Even with artificially induced selection, most experiments often investigate a single adaptative trait and are restricted to clonal lineages, simplifying how adaptation is governed under natural conditions.
Considering that both S. cerevisiae and HPC only undergo sexual reproduction under specific conditions, which are not fully elucidated in nature, designing experiments which reflect natural life cycle variation remains difficult. If sexual reproduction does not occur within an experimental design, it is unclear if this reflects a biological constraint or if it is due to conditions which do not reflect what occurs in nature. In these species, outcrossing is expected to occur infrequently over a long period and depends on mating type compatibility which may not be fully captured in a typical laboratory experiment. In addition, research often restricts experiments to haploid, clonal lineages with manipulated genetic backgrounds that aim to supress the likelihood of meiosis. For example, most experiments of S. cerevisiae are done using haploid strains, although the diploid state is considered more frequent in nature. Ploidy changes can influence both reproductive mode and genetic diversity, yet most experiments aim to limit variation in ploidy state by starting with a defined ploidy background. Ploidy shifts which occur during an experiment are often excluded and treated as experimental noise, generating a biased representation of how variation in ploidy across a population may influence genetic variation.
Additionally, most research is completed using a sub-set of laboratory strains which reflect a similar genetic background. These strains are often optimized for controlled laboratory conditions which decreases pressure for adaptative processes. Long-term culturing of these strains can lead to an accumulation of laboratory-specific traits which do not reflect local adaptation and demographic history of natural strains. Therefore, strains used for experimental design do not represent the true diversity and population structure of natural populations and overlook divergent sub-populations. For example, a natural S. cerevisiae population was found to demonstrate a highly elevated rate of C -> A mutations that distinguishes them from traditional lab strains[35]. To address this limitation, environmental sampling of yeast populations across heterogenous ecological niches is required. By doing so, researchers can identify rare genotypes and begin to capture a more realistic representation of genetic variation across natural populations. Additionally, whole-genome sequencing of natural isolates is required to elucidate cryptic variation and detect rare signals of recombination and ploidy changes. Combining whole-genome polymorphic data analysis with comprehensive sampling data and in vitro examination of natural strains will greatly improve our overall understanding of how these processes shape genetic variation.
The role of lateral gene transfer in fungal evolution should also be critically examined further. Although traditionally considered rare in eukaryotes, increasing genomic evidence suggests that horizontal gene transfer may contribute to rapid adaptation in fungal pathogens[105]. Lateral gene transfer complicates inference of mutation, ploidy and recombination because it can lead to signals which bias the interpretation of these processes. Thus, understanding how lateral gene transfer interacts with mutation, recombination, and ploidy will be critical for fully elucidating how genetic variation is maintained across natural populations.
As climate change continues to increase the instability of environmental conditions, it is expected that altering temperature and humidity averages, shifting ecological boundaries and increasing overlap among previously isolated populations will continue to drive the adaptation of natural yeast populations, including human pathogens[22,106,107,108,109]. This makes it increasingly more important to recognize how mutation, ploidy and recombination work interdependently to maintain genetic variation and drive adaptation to heterogenous environmental conditions.
7. Conclusion
Mutation, ploidy, and recombination are not intrinsically beneficial but have the capacity to increase adaptive potential of organisms in specific environment. In a well-adapted population across a stable environment, increases in mutation rate, recombination, or ploidy shifts can decrease fitness by disrupting genome stability and locally - adapted genotypes. Here, it is expected that selection will favour mechanisms that reduce genetic change. Yet, under stressful conditions these same processes are essential for success by favouring increased genetic variation conducive for adaptation. Pathogen populations often experience dynamic and stressful environments such as host immunity and antimicrobial treatments which likely contribute to maintained genetic variation within hosts. Although controlled laboratory experiments contribute to our understanding of how mutation, ploidy changes and recombination occur, they are insufficient for understanding the complexity of these processes across natural populations. Increasing evidence supports that rates of mutation, recombination and ploidy shifts vary across environments, populations and strains. The variability of these processes increases the sophistication of pathogen adaptation, including traits that influence pathogenicity and antifungal resistance. A comprehensive understanding of how genetic variation is generated and maintained across complex and changing environments is central to combating disease emergence.
Author Contributions
writing—original draft preparation, M.H.; writing—review and editing, J.X.; visualization, M.H., J.X..; supervision, J.X.; project administration, J.X.; funding acquisition, J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science and Engineering Research Council (NSERC) of Canada grant RGPIN-2020-05732 (J.X.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
M.H. acknowledges financial support from the Ontario Graduate Scholarship.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Darwin, C. The origin of species. William Collins. (2011).
- Avery, O.T.; MacLeod, C.M.; McCarty, M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types. in Die Entdeckung der Doppelhelix: Die grundlegenden Arbeiten von Watson, Crick und anderen (ed. Nickelsen, K.) 97–120 (Springer, Berlin, Heidelberg, 2017). Nickelsen, K. (Ed.). [CrossRef]
- Kimura, M. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res. 1967, 9, 23–34. [Google Scholar] [CrossRef]
- King, J.L.; Jukes, T.H. Non-Darwinian evolution. Science 1969, 164, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. Understanding Natural Selection: Essential Concepts and Common Misconceptions. Evo Edu Outreach 2009, 2, 156–175. [Google Scholar] [CrossRef]
- Marsit, S.; Dequin, S. Diversity and adaptive evolution of Saccharomyces wine yeast: A review. FEMS Yeast Res 2015, 15, fov067. [Google Scholar] [CrossRef]
- Giannakou, K.; Cotterrell, M.; Delneri, D. Genomic Adaptation of Saccharomyces Species to Industrial Environments. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Bai, F.-Y.; Han, D.-Y.; Duan, S.-F.; Wang, Q.-M. The Ecology and Evolution of the Baker’s Yeast Saccharomyces cerevisiae. Genes 2022, 13, 230. [Google Scholar] [CrossRef]
- McGovern, P.E.; et al. Fermented beverages of pre- and proto-historic China. Proceedings of the National Academy of Sciences (USA) 2004, 101, 17593–17598. [Google Scholar] [CrossRef]
- Parapouli, M.; Vasileiadis, A.; Afendra, A.-S.; Hatziloukas, E. Saccharomyces cerevisiae and its industrial applications. AIMS Microbiol 2020, 6, 1–31. [Google Scholar] [CrossRef]
- Lahue, C.; Madden, A.; Dunn, R.R.; Smukowski Heil, C. History and Domestication of Saccharomyces cerevisiae in Bread Baking. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Fröhlich-Wyder, M.-T.; Arias-Roth, E.; Jakob, E. Cheese yeasts. Yeast 2019, 36, 129–141. [Google Scholar] [CrossRef]
- Jeffery-Smith, A.; et al. Candida auris: A Review of the Literature. Clin Microbiol Rev 2018, 31, e00029-17. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action, World Health Organization. (Available, Geneva, Switzerland, 2022).
- Hazen, K.C. New and emerging yeast pathogens. Clin Microbiol Rev 1995, 8, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torrado, R.; Querol, A. Opportunistic Strains of Saccharomyces cerevisiae: A Potential Risk Sold in Food Products. Front Microbiol 2016, 6, 1522. [Google Scholar] [CrossRef] [PubMed]
- Clemons, K.V.; McCusker, J.H.; Davis, R.W.; Stevens, D.A. Comparative Pathogenesis of Clinical and Nonclinical Isolates of Saccharomyces cerevisiae. J Infect Dis 1994, 169, 859–867. [Google Scholar] [CrossRef]
- Denning, D.W. Global incidence and mortality of severe fungal disease. Lancet Infect Dis 2024, 24, e428–e438. [Google Scholar] [CrossRef]
- Hagen, F.; et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol 2015, 78, 16–48. [Google Scholar] [CrossRef]
- Dao, A.; Kim, H.Y.; Garnham, K.; et al. Cryptococcosis—A systematic review to inform the World Health Organization Fungal Priority Pathogens List. Med Mycol. 2024, 62, myae043. [Google Scholar] [CrossRef]
- Kim, J.-S.; Cha, H.; Bahn, Y.-S. Comprehensive Overview of Candida auris: An Emerging Multidrug-Resistant Fungal Pathogen. J Microbiol Biotechnol 2024, 34, 1365–1375. [Google Scholar] [CrossRef]
- Seidel, D.; et al. Impact of climate change and natural disasters on fungal infections. The Lancet. Microbe 2024, 5, e594–e605. [Google Scholar] [CrossRef]
- Souque, C.; González Ojeda, I.; Baym, M. From Petri Dishes to Patients to Populations: Scales and Evolutionary Mechanisms Driving Antibiotic Resistance. Annual Review of Microbiology 2024, 78, 361–382. [Google Scholar] [CrossRef]
- Wilson, B.A.; Garud, N.R.; Feder, A.F.; Assaf, Z.J.; Pennings, P.S. The population genetics of drug resistance evolution in natural populations of viral, bacterial and eukaryotic pathogens. Mol Ecol 2016, 25, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lin, J.; Fan, Y.; Lin, X. Life Cycle of Cryptococcus neoformans. Annu. Rev. Microbiol. 2019, 73, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, I. Life cycle of the budding yeast Saccharomyces cerevisiae. Microbiol Rev 1988, 52, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Feretzaki, M.; Sun, S.; Wang, X.; Heitman, J. Sex in fungi. Annu Rev Genet 2011, 45, 405–430. [Google Scholar] [CrossRef]
- Sun, S.; Coelho, M.A.; David-Palma, M.; Priest, S.J.; Heitman, J. The Evolution of Sexual Reproduction and the Mating-Type Locus: Links to Pathogenesis of Cryptococcus Human Pathogenic Fungi. Annu. Rev. Genet. 2019, 53, 417–444. [Google Scholar] [CrossRef]
- Gray, J.C.; Goddard, M.R. Sex enhances adaptation by unlinking beneficial from detrimental mutations in experimental yeast populations. BMC Evol Biol 2012, 12, 43. [Google Scholar] [CrossRef]
- Loewe, L.; Hill, W.G. The population genetics of mutations: Good, bad and indifferent. Philos Trans R Soc Lond B Biol Sci 2010, 365, 1153–1167. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annual Review of Biochemistry 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Boyce, K.J.; et al. Mismatch Repair of DNA Replication Errors Contributes to Microevolution in the Pathogenic Fungus Cryptococcus neoformans. mBio 2017, 8, e00595-17. [Google Scholar] [CrossRef]
- Foster, P. Methods for Determining Spontaneous Mutation Rates. in Methods in Enzymology (Academic Press ) 409, 195–213 (2006).
- Melde, R.H.; Bao, K.; Sharp, N.P. Recent insights into the evolution of mutation rates in yeast. Current Opinion in Genetics & Development 2022, 76, 101953. [Google Scholar] [CrossRef]
- Jiang, P.; et al. A modified fluctuation assay reveals a natural mutator phenotype that drives mutation spectrum variation within Saccharomyces cerevisiae. eLife 2021, 10, e68285. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Evolution of the mutation rate. Trends Genet 2010, 26, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; et al. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Shor, E.; Fox, C.A.; Broach, J.R. The Yeast Environmental Stress Response Regulates Mutagenesis Induced by Proteotoxic Stress. PLoS Genetics 2013, 9, e1003680. [Google Scholar] [CrossRef]
- Akasaka, S.; Yamamoto, K. Hydrogen peroxide induces G:C to TA and G:C to C:G transversions in the supF gene of Escherichia coli. Molec. Gen. Genet. 1994, 243, 500–505. [Google Scholar] [CrossRef]
- Tervo, A.A. Average mutation rates of Saccharomyces cerevisiae in industrially relevant stressful environments. MSc thesis (https://urn.fi/URN:NBN:fi:aalto-202412117741.
- Li, K.-J.; et al. Spontaneous and environment induced genomic alterations in yeast model. Cell Insight 2025, 4, 100209. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Lang, G.I.; Murray, A.W. Estimating the Per-Base-Pair Mutation Rate in the Yeast Saccharomyces cerevisiae. Genetics 2008, 178, 67–82. [Google Scholar] [CrossRef]
- Luria, S.E.; Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943, 28, 491–511. [Google Scholar] [CrossRef]
- Zhu, Y.O.; Siegal, M.L.; Hall, D.W.; Petrov, D.A. Precise estimates of mutation rate and spectrum in yeast. Proceedings of the National Academy of Sciences 2014, 111, E2310–E2318. [Google Scholar] [CrossRef]
- Halligan, D.L.; Keightley, P.D. Spontaneous Mutation Accumulation Studies in Evolutionary Genetics. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 151–172. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J. Yeast Spontaneous Mutation Rate and Spectrum Vary with Environment. Current Biology 2019, 29, 1584–1591.e3. [Google Scholar] [CrossRef] [PubMed]
- Gou, L.; Bloom, J.S.; Kruglyak, L. The Genetic Basis of Mutation Rate Variation in Yeast. Genetics 2019, 211, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; et al. Dynamic and Heterogeneous Mutations to Fluconazole Resistance in Cryptococcus neoformans. Antimicrob Agents Chemother 2001, 45, 420–427. [Google Scholar] [CrossRef]
- Sung, W.; Ackerman, M.S.; Miller, S.F.; Doak, T.G.; Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci USA 2012, 109, 18488–18492. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J. The rate and molecular spectrum of mutation are selectively maintained in yeast. Nat Commun 2021, 12, 4044. [Google Scholar] [CrossRef]
- Taddei, F.; et al. Role of mutator alleles in adaptive evolution. Nature 1997, 387, 700–702. [Google Scholar] [CrossRef]
- Lang, G.I.; Desai, M.M. The spectrum of adaptive mutations in experimental evolution. Genomics 2014, 104, 412–416. [Google Scholar] [CrossRef]
- Raynes, Y.; Sniegowski, P.D. Experimental evolution and the dynamics of genomic mutation rate modifiers. Heredity 2014, 113, 375–380. [Google Scholar] [CrossRef]
- Healey, K.R.; et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat Commun 2016, 7, 11128. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Clancey, S.A.; Heitman, J. Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii. eLife 2017, 6, e28802. [Google Scholar] [CrossRef] [PubMed]
- Strope, P.K.; et al. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 2015, 25, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Heck, J.A.; Argueso, J.L.; Gemici, Z.; et al. Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair. Proc Natl Acad Sci USA 2006, 103, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Magwene, P.M.; Meeks, B.; Murphy, H.A. Known mutator alleles do not markedly increase mutation rate in clinical Saccharomyces cerevisiae strains. Proc Biol Sci 2017, 284, 20162672. [Google Scholar] [CrossRef]
- Raghavan, V.; et al. Incompatibilities in Mismatch Repair Genes MLH1-PMS1 Contribute to a Wide Range of Mutation Rates in Human Isolates of Baker’s Yeast. Genetics 2018, 210, 1253–1266. [Google Scholar] [CrossRef]
- Lynch, M.; Sung, W.; Morris, K.; et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci USA 2008, 105, 9272–9277. [Google Scholar] [CrossRef]
- Nishant, K.T.; et al. The Baker’s Yeast Diploid Genome Is Remarkably Stable in Vegetative Growth and Meiosis. PLoS Genetics 2010, 6, e1001109. [Google Scholar] [CrossRef]
- Sharp, N.P.; Sandell, L.; James, C.G.; Otto, S.P. The genome-wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proceedings of the National Academy of Sciences 2018, 115, E5046–E5055. [Google Scholar] [CrossRef]
- Dutta, A.; et al. Genome Dynamics of Hybrid Saccharomyces cerevisiae During Vegetative and Meiotic Divisions. G3 Genes|Genomes|Genetics 2017, 7, 3669–3679. [Google Scholar] [CrossRef]
- Pankajam, A.; et al. Loss of Heterozygosity and Base Mutation Rates Vary Among Saccharomyces cerevisiae Hybrid Strains. G3 Genes|Genomes|Genetics 2020, 10, 3309–3319. [Google Scholar] [CrossRef]
- Ohnishi, G.; et al. Spontaneous mutagenesis in haploid and diploid Saccharomyces cerevisiae. Biochemical and Biophysical Research Communications 2004, 325, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Rattray, A.; Santoyo, G.; Shafer, B.; Strathern, J.N. Elevated Mutation Rate during Meiosis in Saccharomyces cerevisiae. PLoS Genetics 2015, 11, e1004910. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Estimating the spontaneous mutation rate of loss of sex in the human pathogenic fungus Cryptococcus neoformans. Genetics 2002, 162, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Genotype-Environment Interactions of Spontaneous Mutations for Vegetative Fitness in the Human Pathogenic Fungus Cryptococcus neoformans. Genetics 2004, 168, 31177–31188. [Google Scholar] [CrossRef]
- Priest, S.J.; et al. Factors enforcing the species boundary between the human pathogens Cryptococcus neoformans and Cryptococcus deneoformans. PLoS Genet 2021, 17, e1008871. [Google Scholar] [CrossRef]
- Roe, C.; et al. Dating the Cryptococcus gattii Dispersal to the North American Pacific Northwest. MSphere 2018, 3, 10.1128. [Google Scholar] [CrossRef]
- Chen, V.; et al. Evolution of haploid and diploid populations reveals common, strong, and variable pleiotropic effects in non-home environments. eLife 2023, 12, e92899. [Google Scholar] [CrossRef]
- Zeyl, C. Experimental studies of ploidy evolution in yeast. FEMS Microbiol Lett 2004, 233, 187–192. [Google Scholar] [CrossRef]
- Sharp, N.P.; Otto, S.P. Evolution: Zeroing in on the Rate of Genome Doubling. Curr Biol 2018, 28, R320–R322. [Google Scholar] [CrossRef]
- Magwene, P.M.; et al. Outcrossing, mitotic recombination, and life-history trade-offs shape genome evolution in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences USA 2011, 108, 1987–1992. [Google Scholar] [CrossRef]
- Samarasinghe, H.; You, M.; Jenkinson, T.S.; Xu, J.; James, T.Y. Hybridization Facilitates Adaptive Evolution in Two Major Fungal Pathogens. Genes 2020, 11, 101. [Google Scholar] [CrossRef]
- Zhu, Y.O.; Sherlock, G.; Petrov, D.A. Whole Genome Analysis of 132 Clinical Saccharomyces cerevisiae Strains Reveals Extensive Ploidy Variation. G3 Genes|Genomes|Genetics 2016, 6, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Sionov, E.; Lee, H.; Chang, Y.C.; Kwon-Chung, K.J. Cryptococcus neoformans Overcomes Stress of Azole Drugs by Formation of Disomy in Specific Multiple Chromosomes. PLoS Pathogens 2010, 6, e1000848. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Xu, J. What Are the Best Parents for Hybrid Progeny? An Investigation into the Human Pathogenic Fungus Cryptococcus. JoF 2021, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; You, M.; Xu, J. Genetic Changes in Experimental Populations of a Hybrid in the Cryptococcus neoformans Species Complex. Pathogens 2020, 9, 3. [Google Scholar] [CrossRef]
- Goddard, M.R.; Godfray, H.C.J.; Burt, A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature 2005, 434, 636–640. [Google Scholar] [CrossRef]
- Lee, S.C.; Ni, M.; Li, W.; Shertz, C.; Heitman, J. The Evolution of Sex: A Perspective from the Fungal Kingdom. Microbiol Mol Biol Rev 2010, 74, 298–340. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, X.-C.; Zheng, D.-Q.; Petes, T.D. Effects of Temperature on the Meiotic Recombination Landscape of the Yeast Saccharomyces cerevisiae. mBio 2017, 8, e02099-17. [Google Scholar] [CrossRef]
- Samarasinghe, H.; Xu, J. Hybrids and hybridization in the Cryptococcus neoformans and Cryptococcus gattii species complexes. Infection, Genetics and Evolution 2018, 66, 245–255. [Google Scholar] [CrossRef]
- Li, W.; et al. Genetic Diversity and Genomic Plasticity of Cryptococcus neoformans AD Hybrid Strains. G3 Genes|Genomes|Genetics 2012, 2, 83–97. [Google Scholar] [CrossRef]
- Cogliati, M.; et al. Multi-locus sequence typing and phylogenetics of Cryptococcus neoformans AD hybrids. Fungal Genetics and Biology 2024, 170, 103861. [Google Scholar] [CrossRef]
- Viviani, M.A.; et al. Molecular analysis of 311 Cryptococcus neoformans isolates from a 30-month ECMM survey of cryptococcosis in Europe. FEMS Yeast Res 2006, 6, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Variation in recombination frequency and distribution across eukaryotes: Patterns and processes. Philos Trans R Soc Lond B Biol Sci 2017, 372, 20160455. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Sun, S.; James, T.Y. Evolution of fungal sexual reproduction. Mycologia 2013, 105, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef]
- Wall, J.D. Recombination and the power of statistical tests of neutrality. Genet. Res. 1999, 74, 65–79. [Google Scholar] [CrossRef]
- Hitchcock, M.; Xu, J. Global Analyses of Multi-Locus Sequence Typing Data Reveal Geographic Differentiation, Hybridization, and Recombination in the Cryptococcus gattii Species Complex. J Fungi 2023, 9, 276. [Google Scholar] [CrossRef]
- Hitchcock, M.; Xu, J. Analyses of the Global Multilocus Genotypes of the Human Pathogenic Yeast Cryptococcus neoformans Species Complex. Genes 2022, 13, 2045. [Google Scholar] [CrossRef]
- Fischer, G.; Liti, G.; Llorente, B. The budding yeast life cycle: More complex than anticipated? Yeast 2021, 38, 5–11. [Google Scholar] [CrossRef]
- Hitchcock, M.; et al. Genomic evidence for a-α heterothallic and α-α unisexual mating and recombination in an environmental Cryptococcus deneoformans population. PLoS Genetics 2025, 21, e1011844. [Google Scholar] [CrossRef]
- Fraser, J.A.; et al. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 2005, 437, 1360–1364. [Google Scholar] [CrossRef]
- Kidd, S.E.; et al. Cryptococcus gattii Dispersal Mechanisms, British Columbia, Canada. Emerg. Infect. Dis. 2007, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Engelthaler, D.M.; et al. Cryptococcus gattii in North American Pacific Northwest: Whole-Population Genome Analysis Provides Insights into Species Evolution and Dispersal. mBio 2014, 5, e01464-14. [Google Scholar] [CrossRef]
- Billmyre, R.B.; et al. Highly Recombinant VGII Cryptococcus gattii Population Develops Clonal Outbreak Clusters through both Sexual Macroevolution and Asexual Microevolution. mBio 2014, 5, e01494-14. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, E.J.; et al. Emergence and Pathogenicity of Highly Virulent Cryptococcus gattii Genotypes in the Northwest United States. PLoS Pathog 2010, 6, e1000850. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; et al. Genome-wide mapping of spontaneous genetic alterations in diploid yeast cells. Proceedings of the National Academy of Sciences 2020, 117, 28191–28200. [Google Scholar] [CrossRef]
- Vogan, A.A.; Khankhet, J.; Xu, J. Evidence for mitotic recombination within the basidia of a hybrid cross of Cryptococcus neoformans. PLoS ONE 2013, 8, e62790. [Google Scholar] [CrossRef]
- Forche, A.; et al. Stress alters rates and types of loss of heterozygosity in Candida albicans. mBio 2011, 2, e00129-11. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, D.-Q.; Sui, Y.; Qi, L.; Petes, T.D. Genome-wide analysis of genomic alterations induced by oxidative DNA damage in yeast. Nucleic Acids Res 2019, 47, 3521–3535. [Google Scholar] [CrossRef]
- Fitzpatrick, D.A. Horizontal gene transfer in fungi. FEMS Microbiol Lett 2012, 329, 1–8. [Google Scholar] [CrossRef]
- Fisher, M.C.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat Rev Microbiol 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Garcia-Solache, M.A.; Casadevall, A. Global warming will bring new fungal diseases for mammals. mBio 2010, 1, e00061-10. [Google Scholar] [CrossRef]
- Casadevall, A.; Kontoyiannis, D.P.; Robert, V. On the Emergence of Candida auris: Climate Change, Azoles, Swamps, and Birds. mBio 2019, 10, e01397-19. [Google Scholar] [CrossRef]
- Xu, J. Assessing Global Fungal Threats to Humans. mLife 2022, 1, 223–240. [Google Scholar] [CrossRef]
Figure 1.
Schematic life cycles of the two model yeasts reviewed in this study. (A) The life cycle of Saccharomyces cerevisiae including both heterothallic and homothallic cycles. (B) The life cycle of the human pathogenic Cryptococcus representing both a-α and α-α mating.
Figure 1.
Schematic life cycles of the two model yeasts reviewed in this study. (A) The life cycle of Saccharomyces cerevisiae including both heterothallic and homothallic cycles. (B) The life cycle of the human pathogenic Cryptococcus representing both a-α and α-α mating.
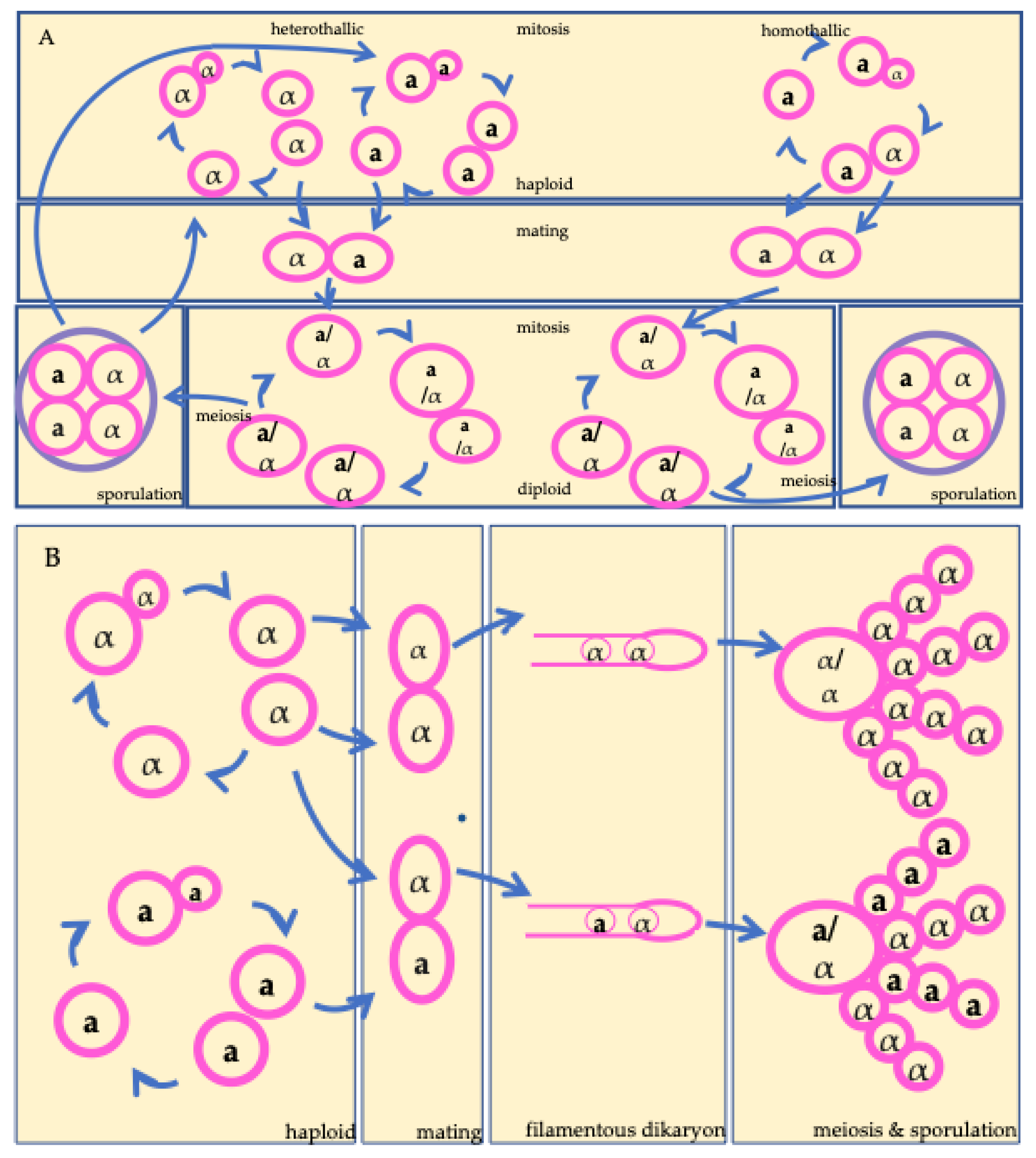

Table 1.
Summary of reported mutations rate for Saccharomyces cerevisiae and the human pathogenic Cryptococcus.
Table 1.
Summary of reported mutations rate for Saccharomyces cerevisiae and the human pathogenic Cryptococcus.
| Species | Strain | Experimental design | Ploidy | Reported rate | Rate calculated based on | Meiosis | Growth conditions |
Source |
|---|---|---|---|---|---|---|---|---|
| S. cerevisiae | FY10 | Mutation accumulation | Haploid | 3.3 × 10−10 | nucleotide site per generation | No | YPD, 30 °C | Lynch et al. 2008[61] |
| S. cerevisiae | EAY2531 | Mutation accumulation | Diploid | 2× 10−10 - 3.8×10−10 | nucleotide site per generation | No | YPD, 30 °C | Nishant et al. 2010[62] |
| S. cerevisiae | Lab strain | Mutation accumulation | Diploid | 1.67 ×10−10 | nucleotide site per generation | No | YPD | Zhu et al. 2014[45] |
| S. cerevisiae | SEY6211 derivatives | Mutation accumulation | Haploid | 4.04 ×10−10 | nucleotide site per generation | No | YPD + 40 mg/L adenine sulfate,30 °C | Sharp et al. 2018[63] |
| S. cerevisiae | SEY6211 derivatives | Mutation accumulation | Diploid | 2.89 × 10−10 | nucleotide site per generation | No | YPD + 40 mg/L adenine sulfate,30 °C | Sharp et al. 2018[63] |
| S. cerevisiae | S288C × YJM789 | Mutation accumulation | Diploid | 7.3 × 10−9 – 2.92 x10-10 | nucleotide site per generation | No | YPD, 30 °C | Dutta et al. 2017[64] |
| S. cerevisiae | S288C × YJM789 | Mutation accumulation | Diploid | 9.8 × 10−9 | nucleotide site per generation | No | YPD, 30 °C | Pankajam et al. 2020[65] |
| S. cerevisiae | S288C x RM11-1a | Mutation accumulation | Diploid | 1.7 × 10−9 | nucleotide site per generation | No | YPD, 30 °C | Pankajam et al. 2020[65] |
| S. cerevisiae | S288C | Mutation accumulation | Diploid | 1.35 ×10−10 | nucleotide site per generation | No | YPD, 30 °C | Pankajam et al. 2020[65] |
| S. cerevisiae | RM11-1a | Mutation accumulation | Diploid | 5.4 × 10−9 | nucleotide site per generation | No | YPD, 30 °C | Pankajam et al. 2020[65] |
| S. cerevisiae | YJM789 | Mutation accumulation | Diploid | 1.16 ×10−10 | nucleotide site per generation | No | YPD, 30 °C | Pankajam et al. 2020[65] |
| S. cerevisiae | GIL104 | Fluctuation assay (URA3 & CAN1) | Haploid | 3.07 x 10-6 | a-Factor phenotypic resistance | No | Synthetic complete medium, 30 °C | Lang & Murray 2008[43] |
| S. cerevisiae | GIL104 | Fluctuation assay (URA3 & CAN1) | Haploid | 1.52 x 10-7 | 10x canavanine resistance | No | Synthetic complete medium, 30 °C | Lang & Murray 2008[43] |
| S. cerevisiae | GIL104 | Fluctuation assay (URA3 & CAN1) | Haploid | 5.43 x 10-8 | 5-FOA phenotypic resistance | No | Synthetic complete medium, 30 °C | Lang & Murray 2008[43] |
| S. cerevisiae | Natural isolates | Fluctuation assay (CAN1) | NA | 1.1 × 10−7 - 5.8 × 10−7 | Canavanine phenotypic resistance | No | Synthetic complete medium, 30 °C | Gou et al. 2019[48] |
| S. cerevisiae | YAS101, YAS106 | Fluctuation assay (CAN1) | Haploid | 9.08 × 10−7 | Canavanine phenotypic resistance | No | YPD, 30 °C | Ohnishi et al. 2004[66] |
| S. cerevisiae | YAS3001 (YAS101 x YAS106) | Fluctuation assay (CAN1) | Diploid | 1.03 × 10−4 | Canavanine phenotypic resistance | No | YPD, 30 °C | Ohnishi et al. 2004[66] |
| S. cerevisiae | GRY2691 | Fluctuation assay (CAN1) | Haploid | 2.8×10−8 | Canavanine phenotypic resistance | No | YPD, 30 °C | Rattray et al. 2015[67] |
| S. cerevisiae | GRY3262 | Fluctuation assay (CAN1) | Diploid | 37×10−8 | Canavanine phenotypic resistance | Yes | YPD, 30 °C | Rattray et al. 2015[67] |
| S. cerevisiae | Natural isolates | Fluctuation assay (CAN1) | Haploid | 2.1⨉10–7 - 2.1⨉10–6 | Canavanine phenotypic resistance | No | YPD, 30 °C | Jiang et al. 2021[35] |
| Cryptococcus spp. (C. neoformans) | Clinical isolates | Mutation accumulation | Haploid | 0.41 × 10−9 to 3,135.36 × 10−9 | Fluconazole phenotypic resistance | No | YEPD+ fluconazole, 37 °C | Xu et al. 2001[49] |
| Cryptococcus spp. (C. neoformans) | JEC50, MCC3 | Mutation accumulation | Haploid | 3.6 × 10−3 -2.32 × 10−2 | Filamentation phenotype | No | YEPD, 25 °C | Xu 2002[68] |
| Cryptococcus spp. (C. neoformans) | JEC50, MCC3 | Mutation accumulation | Diploid | 1.72 × 10−2 - 7.72 × 10−2 | Filamentation phenotype | Yes | YEPD, 25 °C | Xu 2002[68] |
| Cryptococcus spp. (C. deneoformans) | JEC21 | Mutation accumulation | Haploid | 5.662x10-3 | Vegetative growth | Yes | YEPD, 25 °C | Xu 2004[69] |
| Cryptococcus spp. (C. deneoformans) | JEC21 | Mutation accumulation | Haploid | 5.332x10-3 | Vegetative growth | Yes | YEPD, 37 °C | Xu 2004[69] |
| Cryptococcus spp. (C. deneoformans) | JEC20a | Fluctuation assay (FRR1) | Haploid | 8.59 × 10−8 | Rapamycin+FK506 phenotypic resistance | No | YPD + rapamycin+FK506, 37 °C | Priest et al. 2021[70] |
| Cryptococcus spp. (C. gattii) | 134 natural isolates | Polymorphic data | Haploid | 1.59×10−8 – 2.70×10−8 | nucleotide site per generation | NA | Bayesian evolutionary analysis by sampling trees (BEAST) | Roe et al. 2018[71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



