Submitted:
05 May 2026
Posted:
07 May 2026
You are already at the latest version
Abstract
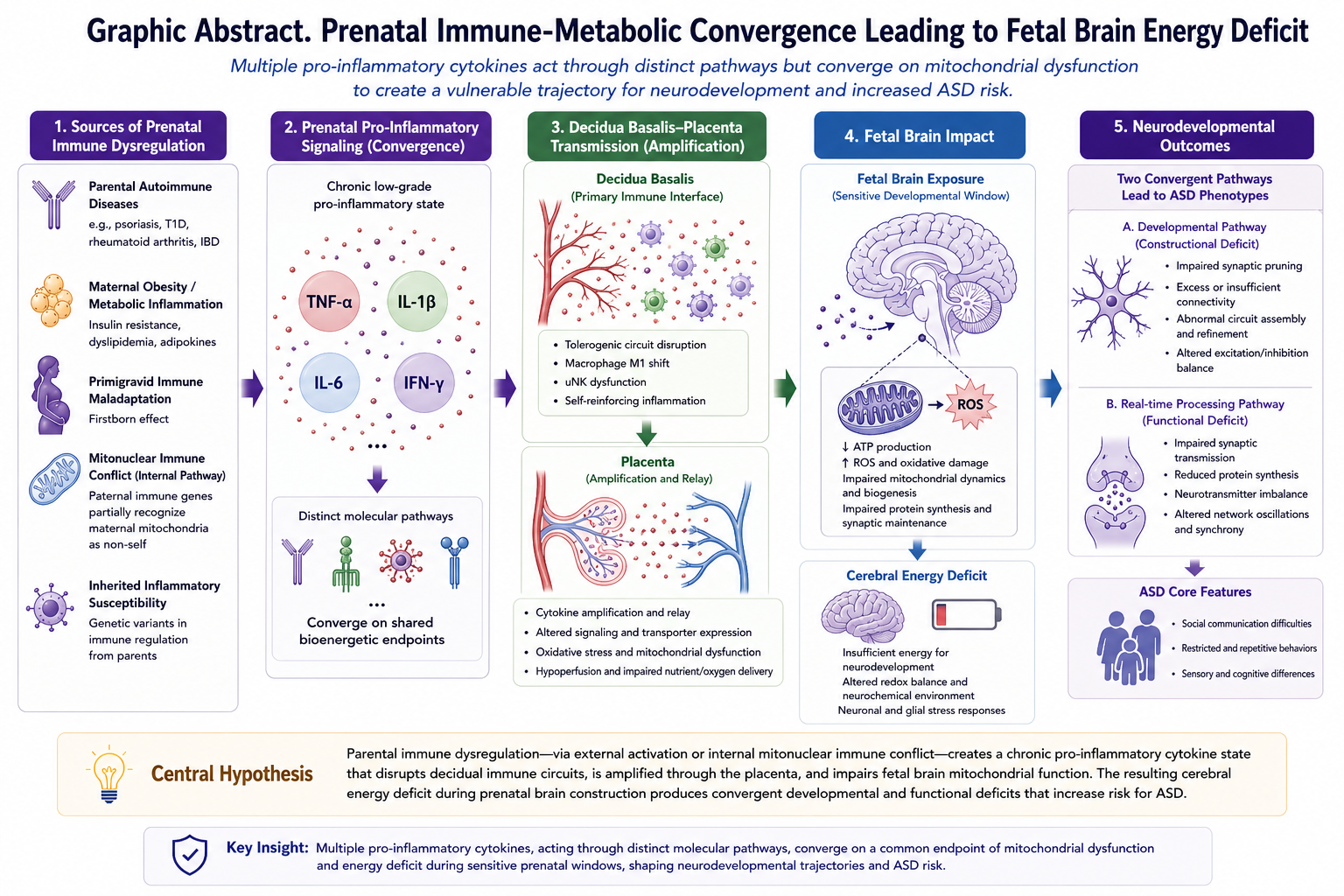
Background: Autism spectrum disorder (ASD) affects approximately 1-2% of children worldwide, yet its etiology remains incompletely understood. Emerging evidence suggests that offspring of parents with autoimmune diseases show elevated autism prevalence. Notably, children of parents with psoriasis (OR 1.59), type 1 diabetes (OR 1.49-2.36), and rheumatoid arthritis (OR 1.51) demonstrate particularly strong associations.Hypothesis: I propose that autism may be conceptualized as an immune-metabolic disorder in which multiple pro-inflammatory cytokines—including TNF-α, IL-6, IL-1β, and IFN-γ—act through distinct molecular pathways yet converge on a common endpoint of mitochondrial dysfunction and cerebral energy deficiency. This convergence implies that it is the cumulative prenatal inflammatory burden, rather than any single cytokine, that drives the energy deficit. The resulting energy shortage may impair three critical processes: (1) synaptic pruning during neurodevelopment, (2) real-time social cognition including gaze processing and emotion recognition, and (3) protein synthesis of critical synaptic scaffolding molecules.The proposed mechanism is a chronic low-grade pro-inflammatory cytokine state—clinically silent, yet biologically consequential—arising from inherited inflammatory susceptibility and/or direct fetal exposure to elevated maternal inflammatory signaling during pregnancy. Unlike high-grade inflammatory states in which maternal and fetal survival are acutely threatened, low-grade cytokine elevations may proceed without conspicuous symptoms or detectable clinical signs, particularly when chronic. Although seemingly quiet, such a state may be insufficient to endanger maternal or fetal survival, yet sufficient to disrupt fetal brain bioenergetics during sensitive gestational windows—producing neonates who appear outwardly healthy at term while their neurodevelopmental trajectories have already been altered.I further propose that the well-documented "firstborn effect" in autism reflects maternal immune maladaptation during primigravid pregnancies. Additionally, for cases without parental autoimmune history, a speculative secondary mechanism is proposed: mitonuclear immune conflict, where paternal immune genes may partially recognize maternal mitochondria as non-self, generating endogenous pro-inflammatory signaling.Implications: This framework may provide an integrative account of disparate observations about autism pathophysiology and suggests that pro-inflammatory immune pathways and mitochondrial protection strategies merit further investigation for potential risk modification, particularly in pregnancies identified as high-risk through parental autoimmune or inflammatory disease. If supported by sufficient subsequent evidence, prenatal cytokine monitoring and corresponding clinical management—currently not part of routine obstetric care—may merit consideration by the medical community as a candidate strategy for autism risk reduction.
Keywords:
autism spectrum disorder
; multi-cytokine convergence
; mitochondrial dysfunction
; chronic low-grade inflammation
; prenatal origin
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



