
Submitted:
30 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Synaptic plasticity relies on precise spatial and temporal compartmentalization of signaling within dendritic spines, presynaptic terminals, and axonal domains. This compartmentalization is progressively reinforced by experience through activity-dependent remodeling of spine geometry, cytoskeletal scaffolds, calcium handling, and local protein synthesis, allowing plasticity signals to remain localized and terminate appropriately. Here, a unifying framework is proposed in which neurodegenerative diseases emerge when the capacity to maintain and renew these compartments declines. In this state, plasticity induction remains largely preserved, but signaling becomes spatially diffuse and temporally prolonged, imposing chronic structural and energetic stress on synapses and axons. Proteins such as tau and α-synuclein, which normally support cytoskeletal organization and dynamic phase-separated assemblies, become destabilized under these conditions leading to pathological aggregation. This framework provides an explanation for early synaptic dysfunction, selective neuronal vulnerability, long presymptomatic phases, network-level disease propagation, the protective effects of education and cognitive engagement, and the limited efficacy of proteinopathy centric therapeutic strategies. Neurodegeneration is thus best conceptualized as a failure of experience-built synaptic compartmentalization, with protein aggregation arising downstream of this primary vulnerability.
Keywords:
synaptic plasticity
; neurodegeneration
; Alzheimer’s disease
; Parkinson’s disease
Introduction
Neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD) have traditionally been framed as disorders of protein misfolding and aggregation, mitochondrial failure, or impaired clearance.[1] Evidence from human imaging, electrophysiology, genetics, and systems neuroscience suggests that synaptic dysfunction precedes proteinopathy, neuronal loss, and clinical decline.[2,3] In fact synaptic failure is considered a hallmark of Alzheimer’s disease.[4] A unifying framework for neurodegeneration is proposed here which originates from failure of mechanisms that normally ensure spatial confinement and temporal termination of synaptic plasticity signals. The human brain is a high-throughput associative learning network and puts demands on dendritic and presynaptic compartmentalization. When these compartmentalization systems erode, plasticity signals become spatially diffuse and temporally persistent within a neuron, generating false-positive or non-terminating long term potentiation (LTP) or long term depression (LTD) states. Cytoskeletal remodeling proteins such as tau and α-synuclein emerge downstream as structural stress responders rather than primary toxins. This framework reconciles synapse-first vulnerability, selective regional degeneration, calcium dysregulation, network-based disease spread, and the limited efficacy of proteinopathy centric therapeutics.
Why Are Synaptic Microdomains Relevant in Neurodegeneration?
Synaptic plasticity is evolutionarily conserved and robust across species, yet progressive, neurodegeneration is overwhelmingly human.[5] Processes which are typically enhanced in humans include lifelong associative learning, long chains of abstract cognition, persistent replay of learned representations and prolonged survival. This combination creates an increased lifetime plasticity load. Although, plasticity itself is not pathological certain mechanisms make plasticity local, transient, and resolvable. LTP and LTD are microdomain-restricted processes, typically confined to a single dendritic spine head or presynaptic bouton, milliseconds-to-minutes Ca2+ transients, locally translated proteins and highly localized cytoskeletal remodeling.[6,7] This precision is achieved by numerous microdomain mechanisms which are discussed below.
Restricted Diffusion of Calcium
Dendritic spines possess minimal intrinsic calcium-buffering capacity, enabling calcium concentrations to rise to high levels and change with exceptional speed following synaptic activation.[8] Under normal physiological conditions, calcium movement through the spine neck is highly restricted. As a result, the spine head behaves as a functionally independent microdomain over prolonged time scales, permitting the accumulation of calcium locally within the spine head during repeated or sustained synaptic stimulation often without significant diffusion into the parent dendrite.
Liquid–Liquid Phase Separation
Spatial compartmentalization of the downstream signalling molecules adds to another layer to the microdomain. Liquid–liquid phase separation (LLPS) has emerged as a fundamental principle by which cells dynamically organise signalling molecules and achieve spatial compartmentalization of molecular signals.[9] Unlike classical modes of intracellular organisation such as sequestration of key components within membrane-bound organelles like the nucleus or mitochondria, or regulation of molecular diffusion through cytoskeletal barriers, LLPS enables the spontaneous segregation of selected biomolecules, most commonly proteins and nucleic acids, into coexisting condensed and dilute phases. This process gives rise to membrane-less droplets that locally concentrate signalling components within specific cellular regions. LLPS is primarily driven by proteins containing intrinsically disordered regions and multiple interaction motifs, whose multivalent protein–protein and protein–nucleic acid interactions promote the formation of dynamic molecular condensates. Crucially, phase separation is reversible, allowing these assemblies to rapidly form and dissolve in response to changing cellular conditions, thereby providing a highly flexible and responsive mechanism for organising intracellular signalling.
Alternative splicing of G protein coupled receptors adds an additional layer of regulation to LLPS. Specific splice variants of latrophilin-3 that contain a PDZ-binding motif can drive the phase separation and clustering of key postsynaptic scaffold proteins, including SHANK, PSD-95, GKAP, and HOMER. This organisation is further strengthened in the presence of extracellular ligands such as teneurin-2 and FLRT3, indicating that LLPS contributes directly to synaptic structural and signalling architecture. Importantly, neuronal activity can alter the splicing pattern of latrophilin-3, suggesting that activity-dependent splicing may dynamically regulate LLPS and, in turn, fine-tune synaptic signalling and plasticity.[10]
The Distinct Cytoskeleton of the Dendritic Spine
Unlike the dendritic shaft, which is rich in microtubules, dendritic spines are primarily supported by an actin-based cytoskeleton organized as a dense, branched network within the spine head and neck. Actin here is dynamic, cycling between monomeric G-actin and polymeric F-actin through ATP-dependent assembly and disassembly.[11] Because actin filaments are polar, with a fast-growing barbed end and a slower pointed end, rapid remodeling can occur, allowing spines to respond quickly to synaptic activity. Both G-actin and F-actin coexist within spines, and their relative balance strongly influences spine shape and synaptic function. The spine cytoskeleton lies just beneath the postsynaptic density (PSD), a specialized protein complex that anchors glutamate receptors and signaling molecules to support synaptic transmission and plasticity. Key PSD scaffold proteins, including SHANK and PSD-95, are physically linked to the actin network via actin-binding proteins such as cortactin and α-actinin. Dendritic spines contain two functionally distinct F-actin pools: a highly dynamic pool near the membrane that interacts with receptors and signaling complexes, and a more stable internal pool that maintains spine architecture.
Synaptic Compartmentalization Is Energy Dependent
Synaptic compartmentalization during plasticity is inherently energy-dependent, relying on continuous ATP expenditure to localize and terminate signaling within dendritic spines, presynaptic terminals, and axonal domains. Spatial confinement of calcium signals, a prerequisite for input-specific LTP and LTD, is maintained by ATP-driven calcium extrusion through plasma membrane Ca²⁺-ATPases (PMCAs).[12] Np65 (Neuroplastin-65) regulates postsynaptic calcium dynamics by controlling PMCA expression and GluA1-containing AMPA receptor levels, thereby coordinating PMCA and iGluR (ionotropic glutamate receptor) interactions and GluN2A-dependent synaptic plasticity, a process disrupted in Np65 deficiency and associated with retrograde memory impairment.[13] Sequestration by SERCA (sarco-endoplasmic reticulum Ca2+-ATPase) pumps, also actively limit the amplitude and duration of postsynaptic Ca²⁺ transients.[14,15]
The diffusion barrier discussed previously is imposed by spine neck geometry and sustained through ongoing actin and septin cytoskeletal remodeling, an ATP-consuming process essential for spine maturation and biochemical isolation.[16] Local ionic homeostasis is required to preserve synapse-specific excitability and prevent signal spillover, depends heavily on Na⁺/K⁺-ATPase activity, one of the dominant energy sinks in the brain.
Consistent with these demands, mitochondria are actively recruited and stabilized near dendritic spines and presynaptic boutons, where they provide localized ATP necessary for sustained plasticity, cytoskeletal dynamics, and signal termination.[17] Vesicle-associated membrane protein–associated protein (VAP), which has been implicated in amyotrophic lateral sclerosis, stabilizes mitochondria near dendritic spines through interactions with the actin cytoskeleton. In fact, VAP was found to be a spatial organizer of mitochondrial compartments, maintaining their stability for approximately 60 minutes and defining a dendritic domain of roughly 30 μm within which synaptic plasticity can be energetically supported.[18]
Together, these observations identify synaptic compartmentalization as an energetically maintained state (Figure 1). Modest energy insufficiency would be expected to broaden plasticity signals spatially and temporally, providing a mechanism by which metabolic stress and vascular risk factors amplifies synaptic instability and predisposes to neurodegeneration.[19]
Leaky Synaptic Microdomains as the Earliest Marker of Neurodegeneration
In humans, plasticity microdomains are:activated more frequently, distributed across large association networks and maintained across decades. This pushes microdomain compartment maintenance systems close to their functional limits.
Neurodegeneration likely begins when microdomain compartmentalisation degrades, leading to: 1. Spatial leakage: Plasticity signals (Ca²⁺, cAMP, Ras/ERK, CaMKII activity) escape the spine head into the dendritic shaft or adjacent spines. 2. Temporal failure: Signals fail to terminate, remaining active beyond their adaptive window. 3. False tagging: Synapses behave as if learning has occurred even in the absence of meaningful information continually requesting consolidation resources. 4.Chronic repair state:Cytoskeletal remodeling, transport demand, and local translation become constitutive.
Tau and α-Synuclein Appear Central but Might Not Be the Drivers
Tau and α-synuclein are intrinsically disordered proteins that undergo reversible LLPS under physiological conditions, a property increasingly recognized as central to synaptic and cytoskeletal compartmentalization during plasticity. In vitro and cellular studies demonstrate that tau forms dynamic condensates capable of concentrating tubulin and promoting microtubule assembly, while remaining highly sensitive to phosphorylation state, ionic strength, and molecular crowding, indicating that tau LLPS functions as a regulatable, activity-responsive scaffold rather than a static structural element.[20,21] Similarly, α-synuclein undergoes LLPS driven by electrostatic and hydrophobic interactions, forming liquid condensates that localize presynaptically and modulate synaptic vesicle clustering and calcium-dependent release dynamics.[22,23] In both cases, LLPS precedes and accelerates fibrillization in a concentration and time-dependent manner. Since, synaptic plasticity is characterized by intense, spatially restricted biochemical flux, particularly calcium transients, kinase activation, and cytoskeletal remodeling, these conditions can promote phase separation of intrinsically disordered proteins. These observations support a model in which tau- and α-synuclein–mediated condensates contribute to synaptic microdomain governance by transiently organizing cytoskeletal elements, trafficking machinery, and signaling complexes within dendritic spines and presynaptic boutons. Chronic activity, impaired signal termination, or age and mutation-related reductions in condensate reversibility would be expected to prolong condensate lifetime, increase local protein concentration, and bias phase-separated states toward gelation or aggregation. Thus, protein aggregation in neurodegenerative diseases can be modelled as a failure of physiological LLPS-based compartmentalisation mechanisms rather than being causal.[24]
How This Model Explains Selective Vulnerability
Different neurons employ distinct plasticity architectures such as spine-rich vs spine-poor neurons, different Ca²⁺ channel compositions, varying buffering capacities and distinct cytoskeletal designs. Interneurons enriched in buffers such as parvalbumin or calbindin exhibit faster, spatially constrained Ca²⁺ transients, whereas spines of pyramidal neurons maintain low buffering to permit large, rapid, and highly localized Ca²⁺ elevations.[25]
These signaling differences have another layer of distinct cytoskeletal frameworks: actin-dominated spine architectures enable rapid structural remodeling, while shaft-based synapses are supported by more stable microtubule–actin arrangements. At the systems level, highly connected cortical hubs, particularly within association cortex and default-mode–like networks operate under sustained baseline activity and high metabolic load making them disproportionately vulnerable when microdomain failure occurs.
Why Indulgence in Cognitive Activities Can Be Protective
Synaptic compartmentalization is not an innate, static property but an experience-dependent feature that strengthens with repeated, structured neural activity. Immature or weakly used synapses are characterized by thin spines with wide necks, high diffusional coupling to the parent dendrite, poor calcium confinement, and transient, easily reversible plasticity.[26] Repeated, meaningful activation drives spine head enlargement, progressive narrowing and elongation of the spine neck, actin and septin cytoskeletal reinforcement, improved localization of calcium and second-messenger signaling, and more efficient local protein synthesis, resulting in cleaner termination of plasticity signals. Experimental and imaging studies demonstrate that spine neck geometry dynamically regulates biochemical isolation during LTP, and activity-dependent spine maturation enhances microdomain fidelity.[27] This architecture initial leakiness (probably for early life exploration) followed by use-dependent stabilization, explains why structured learning and education improve plasticity resolution, reduce background synaptic noise, and increase network efficiency, thereby building a durable “microdomain reserve.” Epidemiological and longitudinal data linking education and cognitive engagement to delayed dementia onset are thus explained by improved capacity to localize and consolidate plasticity signals. With aging, the ability to maintain and rebuild these compartmental barriers progressively declines, rendering the same cognitive load increasingly unsafe and predisposing to false plasticity signals, cytoskeletal stress, and downstream proteinopathy.
Microglia, Complement, and Genetics
Building on this model, microglia and complement systems act as quality-control enforcers. When synapses repeatedly signal abnormal plasticity states, they are tagged for elimination. Genetic risk factors modulate how aggressively this pruning response is executed, transforming local microdomain failure into network-wide synapse loss.
Thus, persistent microdomain stress increases activity-dependent release of misfolded proteins and vesicular cargo.
Predictions That Distinguish This Framework
This framework predicts that before plaques, tangles, or cell death, vulnerable neurons show certain features which include prolonged and spatially broadened plasticity signaling kinetics, impaired termination of Ca²⁺ and kinase activity after synaptic events, disrupted sleep-dependent synaptic downscaling, cytoskeletal stress signatures preceding overt aggregation and activity-weighted, network-specific propagation patterns. Table 1 shows how the model predicts the current observations in neurodegeneration.
Future Strategies
A test of this microdomain failure hypothesis requires experiments that (i) quantify spatial leakage and temporal non-termination of plasticity signals at single synapses, (ii) show these changes precede proteinopathy/synapse loss, and (iii) demonstrate causal rescue by restoring compartmentalisation without directly clearing aggregates. Using targeted therapies to restore this compartmentalization and spine geometry (e.g., enhancing spine-neck diffusion barriers via cytoskeletal/septin stabilization, improving Ca²⁺ extrusion/buffering) and testing whether this normalizes leakage/termination metrics and preserves synapse integrity even when amyloid/tau/α-syn burden increases, needs further studies.
This model predicts the existence of two coupled, measurable compartment thresholds: a synaptic threshold and a neuronal threshold. At the synaptic level, each excitatory synapse can be assigned a quantitative microdomain integrity index derived from experimentally observable features, including the spatial spread of calcium from spine head to dendritic shaft, the decay kinetics of calcium- and kinase-dependent signaling, and diffusional coupling across the spine neck. When this index persistently exceeds a defined synaptic threshold, the synapse enters a microdomain-failing state characterized by impaired signal localization and termination, rendering it unstable and prone to elimination.
At the neuronal level, degeneration might not be triggered by failure of any single synapse but by the cumulative burden of such microdomain-failing synapses within a neuron. The neuronal threshold is therefore a function of both the number and distribution of synapses that exceed the synaptic threshold, such that clustered or strategically located failures disproportionately increase vulnerability. This model is experimentally testable by longitudinal single-synapse imaging to define the synaptic threshold that predicts future synapse loss or immune tagging, followed by per-neuron quantification of failing synapses to determine the burden at which dendritic, axonal, or cellular degeneration ensues.
This model explains why synaptic dysfunction precedes neuronal loss, why vulnerability is heterogeneous across neurons, and why early intervention must target synaptic microdomain stability rather than neuronal survival. A summary of potential targets that can be studied are listed in Table 2.
Conclusion
Neurodegeneration may thus be a disease of synaptic compartmentalization failure with secondary protein aggregation as a consequence.
References
- Toader, C; Tataru, CP; Munteanu, O; Serban, M; Covache-Busuioc, R-A; Ciurea, AV; Enyedi, M. Decoding Neurodegeneration: A Review of Molecular Mechanisms and Therapeutic Advances in Alzheimer’s, Parkinson’s, and ALS. International Journal of Molecular Sciences 2024, 25(23), 12613. [Google Scholar] [CrossRef] [PubMed]
- Mecca, AP; Chen, MK; O'Dell, RS; Naganawa, M; Toyonaga, T; Godek, TA; Harris, JE; Bartlett, HH; Zhao, W; Nabulsi, NB; Wyk, BCV; Varma, P; Arnsten, AFT; Huang, Y; Carson, RE; van Dyck, CH. In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimers Dement 2020, 16(7), 974–982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meftah, S.; Gan, J. Alzheimer’s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Frontiers in Synaptic Neuroscience 2023, 15, 1129036. [Google Scholar] [CrossRef]
- Selkoe, DJ. Alzheimer's disease is a synaptic failure. Science 2002, 298(5594), 789–91. [Google Scholar] [CrossRef] [PubMed]
- Glanzman, DL. Common mechanisms of synaptic plasticity in vertebrates and invertebrates. Curr Biol 2010, 20(1), R31–6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lur, G.; Higley, M. J. Glutamate receptor modulation is restricted to synaptic microdomains. Cell Reports 2015, 12(2), 326–334. [Google Scholar] [CrossRef]
- Grunditz, A; Holbro, N; Tian, L; Zuo, Y; Oertner, TG. Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J Neurosci 2008, 28(50), 13457–66. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sabatini, B. L.; Oertner, T. G.; Svoboda, K. The life cycle of CA2+ ions in dendritic spines. Neuron 2002, 33(3), 439–452. [Google Scholar] [CrossRef]
- Folkmanaite, M.; Zaccolo, M. Compartmentalisation in cAMP signalling: A phase separation perspective. British Journal of Pharmacology 2025. [Google Scholar] [CrossRef]
- Langenhan, T. Modularization of adhesion G protein-coupled receptor (aGPCR) structure: How alternative splicing regulates synaptogenesis. Signal Transduction and Targeted Therapy 2024, 9, 106. [Google Scholar] [CrossRef]
- Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic spines in Alzheimer’s disease: how the actin cytoskeleton contributes to synaptic failure. Int J Mol Sci. 2020, 21(3), 908. [Google Scholar] [CrossRef] [PubMed]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E. J. The plasma membrane calcium ATPasesand their role as major new players in human disease. Physiological Reviews 2017, 97(3), 1089–1125. [Google Scholar] [CrossRef] [PubMed]
- Malci, A.; Lin, X.; Sandoval, R.; Gundelfinger, E. D.; Naumann, M.; Seidenbecher, C. I.; Herrera-Molina, R. Ca2+ signaling in postsynaptic neurons: Neuroplastin-65 regulates the interplay between plasma membrane Ca2+ ATPases and ionotropic glutamate receptors. Cell Calcium 2022, 106, 102623. [Google Scholar] [CrossRef] [PubMed]
- Davi, V.; Parutto, P.; Crapart, C.; Zhang, Y.; Konno, T.; Chambers, J.; Franklin, J. P.; Maddison, D.; Awadelkareem, M. A.; Devine, M. J.; Koslover, E.; Avezov, E. Endoplasmic reticulum geometry dictates neuronal bursting via calcium store refill rates. In bioRxiv; Cold Spring Harbor Laboratory, 2025. [Google Scholar] [CrossRef]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. In Calcium Signaling. Advances in Experimental Medicine and Biology; Islam, M., Ed.; Springer: Cham, 2020; vol 1131. [Google Scholar] [CrossRef]
- Hotulainen, P; Hoogenraad, CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol Epub. 2010, 189(4), 619–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rangaraju, V; Calloway, N; Ryan, TA. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156(4), 825–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bapat, O.; Purimetla, T.; Kruessel, S.; et al. VAP spatially stabilizes dendritic mitochondria to locally support synaptic plasticity. Nat Commun 2024, 15, 205. [Google Scholar] [CrossRef]
- Patel, V; Edison, P. Cardiometabolic risk factors and neurodegeneration: a review of the mechanisms underlying diabetes, obesity and hypertension in Alzheimer's disease. J Neurol Neurosurg Psychiatry 2024, 95(6), 581–589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hernández-Vega, A.; Braun, M.; Scharrel, L.; Jahnel, M.; Wegmann, S.; Hyman, B. T.; Alberti, S.; Diez, S.; Hyman, A. A. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Reports 2017, 20(10), 2304–2312. [Google Scholar] [CrossRef]
- Wegmann, S; Eftekharzadeh, B; Tepper, K; Zoltowska, KM; Bennett, RE; Dujardin, S; Laskowski, PR; MacKenzie, D; Kamath, T; Commins, C; Vanderburg, C; Roe, AD; Fan, Z; Molliex, AM; Hernandez-Vega, A; Muller, D; Hyman, AA; Mandelkow, E; Taylor, JP; Hyman, BT. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 2018, 37(7), e98049. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mukherjee, S.; Sakunthala, A.; Gadhe, L.; Poudyal, M.; Sawner, A. S.; Kadu, P.; Maji, S. K. Liquid-liquid Phase Separation of α-Synuclein: A New Mechanistic Insight for α-Synuclein Aggregation Associated with Parkinson’s Disease Pathogenesis. Journal of Molecular Biology 2022, 435(1), 167713. [Google Scholar] [CrossRef]
- Hoffmann, C.; Sansevrino, R.; Morabito, G.; Logan, C.; Vabulas, R. M.; Ulusoy, A.; Ganzella, M.; Milovanovic, D. Synapsin condensates recruit alpha-Synuclein. Journal of Molecular Biology 2021, 433(12), 166961. [Google Scholar] [CrossRef] [PubMed]
- Espay, A. J.; Okun, M. S. Abandoning the proteinopathy paradigm in Parkinson disease. JAMA Neurology 2022, 80(2), 123. [Google Scholar] [CrossRef]
- Higley, MJ; Sabatini, BL. Calcium signaling in dendritic spines. Cold Spring Harb Perspect Biol 2012, 4(4), a005686. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grunditz, A; Holbro, N; Tian, L; Zuo, Y; Oertner, TG. Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J Neurosci 2008, 28(50), 13457–66. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tønnesen, J.; Katona, G.; Rózsa, B.; et al. Spine neck plasticity regulates compartmentalization of synapses. Nat Neurosci 2014, 17, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A.; Núñez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Paños, D.; Colom-Cadena, M.; Gomez-Giro, G.; Muñoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; Rami, L.; Lladó, A.; Molinuevo, J. L.; Tainta, M.; Clarimón, J.; Spires-Jones, T.; Blesa, R.; Fortea, J.; Martínez-Lage, P.; Belbin, O. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid. Molecular & Cellular Proteomics 2019, 18(3), 546–560. [Google Scholar] [CrossRef]
- Ahnaou, A.; Moechars, D.; Raeymaekers, L.; et al. Emergence of early alterations in network oscillations and functional connectivity in a tau seeding mouse model of Alzheimer’s disease pathology. Sci Rep 2017, 7, 14189. [Google Scholar] [CrossRef] [PubMed]
- Landau, SM; Lee, J; Murphy, A; Ward, TJ; Harrison, TM; Baker, SL; DeCarli, C; Harvey, D; Tosun, D; Weiner, MW; Koeppe, RA; Jagust, WJ. Alzheimer's Disease Neuroimaging Initiative. Individuals with Alzheimer's disease and low tau burden: Characteristics and implications. Alzheimers Dement 2024, 20(3), 2113–2127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sedghi, F.; Foroughi, E.; Sheikhzadeh, F.; Youshanlui, M. A.; Kohnehshahri, A. A.; Karimzadeh, O.; Sadat-Madani, S.; Ghadri, H.; Parhiz, P.; Amirbeyk, A.; Afshari, S.; Ebrahimnia, Y.; Soleimanzadeh, M.; Anar, M. A.; Borzabad, P. A.; Deravi, N. Association between educational attainment and amyloid deposition across the spectrum from normal cognition to dementia: A meta-analysis. IBRO Neuroscience Reports 2025, 19, 133–142. [Google Scholar] [CrossRef]
- Owen, JE; Veasey, SC. Impact of sleep disturbances on neurodegeneration: Insight from studies in animal models. Neurobiol Dis. 2020, 139, 104820. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, X.; Ma, Z.; Lian, P.; Xu, Y.; Cao, X. Common mechanisms underlying axonal transport deficits in neurodegenerative diseases: a mini review. Frontiers in Molecular Neuroscience 2023, 16, 1172197. [Google Scholar] [CrossRef]
- Buccellato, FR; D'Anca, M; Serpente, M; Arighi, A; Galimberti, D. The Role of Glymphatic System in Alzheimer's and Parkinson's Disease Pathogenesis. Biomedicines 2022, 10(9), 2261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Imbimbo, BP; Balducci, C; Ippati, S; Watling, M. Initial failures of anti-tau antibodies in Alzheimer's disease are reminiscent of the amyloid-β story. Neural Regen Res. 2023, 18(1), 117–118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Espay, A. J.; Herrup, K.; Kepp, K. P.; Daly, T. The proteinopenia hypothesis: Loss of Aβ42 and the onset of Alzheimer’s Disease. Ageing Research Reviews 2023, 92, 102112. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, D.; Allen, N. E.; Kuźma, E.; Littlejohns, T. J. Metabolic syndrome and risk of incident all-cause dementia, Alzheimer’s disease and vascular dementia: a systematic review and meta-analysis of longitudinal studies. Alzheimer S Research & Therapy 2025, 17(1), 198. [Google Scholar] [CrossRef]
- Qiu, S.; Zhang, D.; Ma, L.; Li, Q.; Wang, L.; Wang, Y.; Wang, Y.; Xiong, S.; Tan, L. Associations of metabolic syndrome with risks of dementia and cognitive impairment: A systematic review and meta-analysis. Journal of Alzheimer S Disease 2025, 105(1), 15–27. [Google Scholar] [CrossRef] [PubMed]
- Kinney, JW; Bemiller, SM; Murtishaw, AS; Leisgang, AM; Salazar, AM; Lamb, BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y) 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Figure 1.
Mechanisms of synaptic compartmentalization. Legend: a)Glutamate release from the presynaptic bouton activates postsynaptic receptors, leading to calcium influx into the dendritic spine head and activation of calcium–calmodulin–dependent signaling pathways, including CaMKII, that drive long-term potentiation (LTP). b) Dendritic spines act as discrete biochemical microdomains in which actin-based cytoskeletal architecture and spine neck geometry restrict calcium diffusion and confine plasticity-related signaling. Transient microtubule interactions support targeted cargo delivery without disrupting compartmental integrity. Vesicle-associated membrane protein–associated protein (VAP) stabilizes mitochondria near spines through actin interactions, providing local energetic support during synaptic plasticity. Within the spine, cytoskeletal and signaling proteins can assemble into dynamic liquid–liquid phase–separated (LLPS) compartments that further enhance spatial organization. Together, these mechanisms maintain localized, self-terminating plasticity signals and preserve synaptic stability.Adapted from Servier Medical Art (https://smart.servier.com), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Figure 1.
Mechanisms of synaptic compartmentalization. Legend: a)Glutamate release from the presynaptic bouton activates postsynaptic receptors, leading to calcium influx into the dendritic spine head and activation of calcium–calmodulin–dependent signaling pathways, including CaMKII, that drive long-term potentiation (LTP). b) Dendritic spines act as discrete biochemical microdomains in which actin-based cytoskeletal architecture and spine neck geometry restrict calcium diffusion and confine plasticity-related signaling. Transient microtubule interactions support targeted cargo delivery without disrupting compartmental integrity. Vesicle-associated membrane protein–associated protein (VAP) stabilizes mitochondria near spines through actin interactions, providing local energetic support during synaptic plasticity. Within the spine, cytoskeletal and signaling proteins can assemble into dynamic liquid–liquid phase–separated (LLPS) compartments that further enhance spatial organization. Together, these mechanisms maintain localized, self-terminating plasticity signals and preserve synaptic stability.Adapted from Servier Medical Art (https://smart.servier.com), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
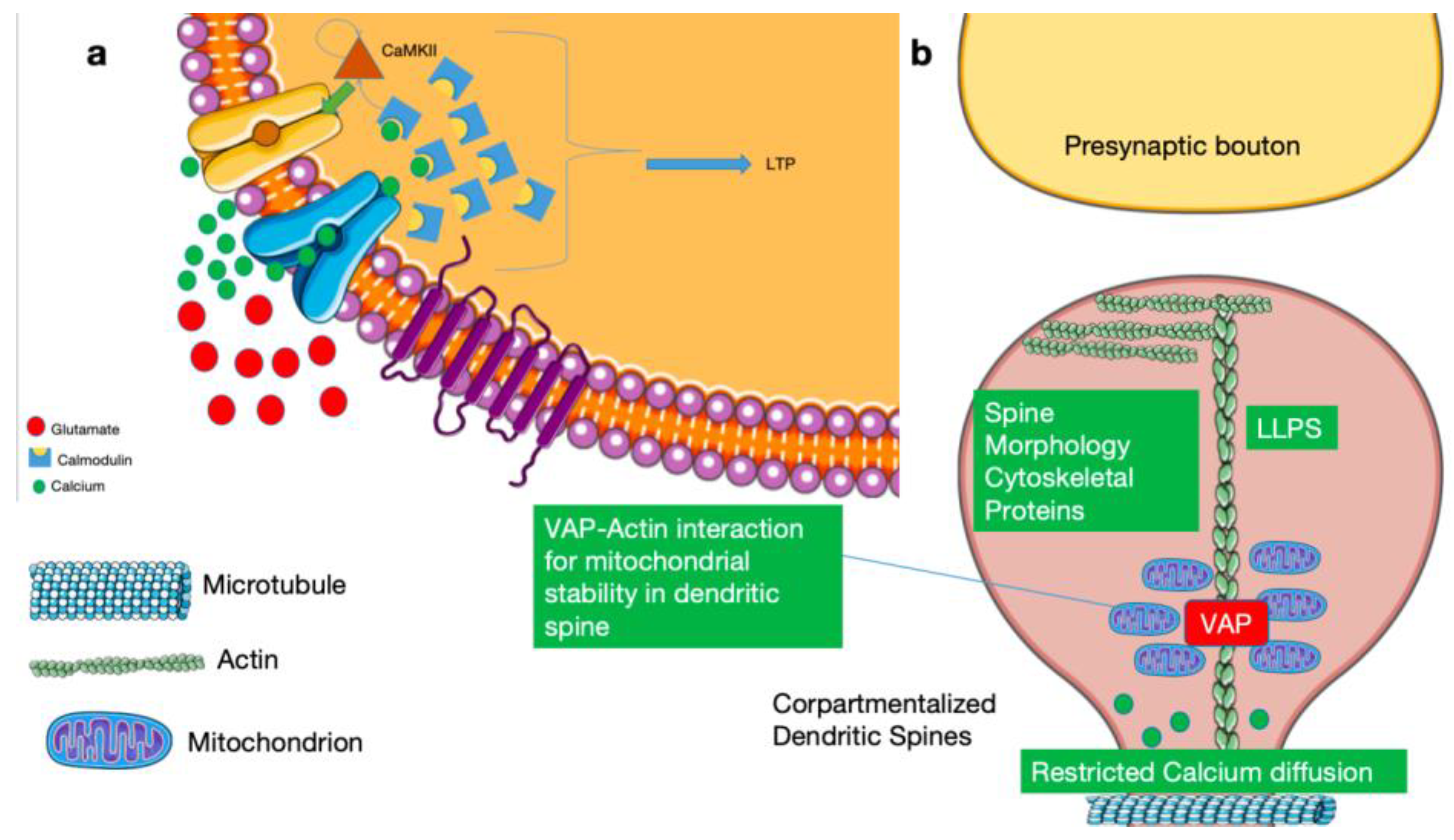

Table 1.
Integration of Established Neurodegenerative Observations within a Synaptic Compartmentalization Model.
Table 1.
Integration of Established Neurodegenerative Observations within a Synaptic Compartmentalization Model.
| What is observed | How this model explains it |
|---|---|
| Synapse loss precedes neuronal death (AD and PD) [28] | Microdomain failure is defined at synapses; neurons tolerate some synapse loss until a burden threshold is crossed |
| Early functional changes occur before overt pathology (hyperexcitability, network instability) [29] | Loss of spatial/temporal confinement causes Ca²⁺ spillover and prolonged signaling before structural loss |
| Selective neuronal vulnerability | Different neuron types have distinct microdomain architectures, energy demands, and repair capacity (different thresholds). |
| Poor correlation between protein burden and cognition [30] | Cognitive function tracks synaptic microdomain integrity and synapse count, not aggregate load |
| Education and cognitive activity delay dementia onset [31] | Structured learning strengthens compartmentalisation and raises synaptic/neuronal thresholds |
| Sleep disruption accelerates neurodegeneration [32] | Sleep is required for plasticity termination and metabolic reset; loss increases microdomain stress |
| Axonal transport deficits appear early [33] | Actin/microtubule systems required for transport are destabilized by chronic microdomain Ca²⁺ and cytoskeletal stress |
| ‘Dying-back’ pattern of degeneration | Synapses are highest-stress microdomains; failure propagates to axon and soma |
| Glial activation follows neuronal stress | Glia act as compensatory sinks and regulators |
| Complement-mediated synapse pruning | Dysfunctional synapses are tagged for destruction. |
| Glymphatic clearance is beneficial [34] | Clearance of potential toxins improves microdomain stability |
| Failure of protein-removal therapies to halt disease [35,36] | Removing aggregates does not fix upstream compartmentalisation failure |
| Neuronal collapse after prolonged stability | Nonlinear neuronal threshold reached after cumulative synaptic failure (Likely a power law dynamics) |
| Energy metabolism strongly influences disease risk [37,38] | Compartmentalisation is ATP-dependent (Ca²⁺ pumps, cytoskeleton, transport) |
| Inflammation accelerates progression [39] | Inflammation increases Ca²⁺ stress, lowering microdomain reserve |
Table 2.
Potential therapeutic targets for prodromal neurodegeneration that can be considered based on this framework.
Table 2.
Potential therapeutic targets for prodromal neurodegeneration that can be considered based on this framework.
| Functional domain | Representative targets | Spine location | Role | Conceptual therapeutic implications |
|---|---|---|---|---|
| Calcium termination | PMCA, SERCA | Spine head, spine base, presynapse | Rapid clearance and sequestration of Ca²⁺ after synaptic activity | Strengthen signal termination without suppressing induction |
| Spine neck diffusion barrier | Actin regulators (cofilin/profilin balance), septins (e.g., Sept7) | Spine neck | Control spine geometry and molecular diffusion between spine and shaft | Restore compartmental isolation at vulnerable synapses |
| Mitochondrial positioning & energy microdomains | VAP | Spine base, dendritic shaft | Anchor and regulate mitochondrial mobility near synapses | Raise neuronal threshold by stabilizing local ATP supply |
| LLPS regulation / condensate dynamics | Tau, α-synuclein (physiological state), kinases, phosphatases, chaperones (Hsp70/90) | Spine head, presynaptic terminal | Organize signaling proteins into dynamic, reversible condensates | Preserve fluid LLPS states, prevent solidification |
| Presynaptic release containment | Active zone scaffolds, Ca²⁺ buffers | Presynaptic bouton | Shape vesicle release probability and presynaptic Ca²⁺ microdomains | Reduce induction overload |
| Glial buffering (modifiers) | Astrocytic EAATs, K⁺ buffering | Perisynaptic | Limit extracellular glutamate and ionic spillover | Delay threshold crossing |
Abbreviations: PMCA:Plasma Membrane Calcium Adenosine Triphosphatase; SERCA: Sarco/Endoplasmic Reticulum Calcium Adenosine Triphosphatase; VAP:Vesicle-Associated Membrane Protein–Associated Protein; LLPS: Liquid–Liquid Phase Separation; EAATs: Excitatory Amino Acid Transporters; Hsp:Heat Shock Protein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



