Submitted:
24 December 2025
Posted:
26 December 2025
You are already at the latest version
Abstract
Helicobacter pylori (H. pylori) infection is recognized as one of the most common bacterial infections worldwide. Its role in infection-associated cancers, such as in gastric cancer and MALT-lymphoma, is well known. Mitochondrial alterations in these malignancies are less documented. Mitochondria is a key organelle in maintaining cellular homeostasis under normal and pathological conditions. They regulate complex cellular processes, and they are key players in carcinogenesis and cancer progression in these H.pylori-associated malignancies. In this review, we summarize the role of mitochondrial stress in H. pylori infection, in gastric cancer, and MALT-lymphoma.
Keywords:
1. General Aspects of Helicobacter Pylori Associated Malignancies
1.2. Pathological Classification of Helicobacter Pylori Associated Malignancies
1.2.1. Gastric Cancer
1.2.2. MALT Lymphoma
1.2. Introduction to Classical Molecular Mechanisms of H. pylori-Driven Carcinogenesis
1.2.1. Gastric Cancer
1.2.2. MALT Lymphoma
2. H. pylori Infection and Mitochondrial Stress
2.1. H. pylori Infection on the Mitochondrial Functions of Gastric Epithelial Cells
2.2. Molecular Mechanisms of Virulence Factors
2.2.1. Vacuolating Cytotoxin A (VacA)
2.2.2. Cag Pathogenicity Island (cagPAI) and Cytotoxin-Associated Gene A (CagA)
2.2.3. Interactions Between VacA and CagA

| NF-κB Level | CagA+VacA+ | CagA-only | VacA-only | Pathogenic Outcome |
|---|---|---|---|---|
| p65/RelA (canonical) | Moderate ↑ | High ↑↑↑ | Low | Proliferation ✓ |
| RelB/p52 (non-canonical) | Moderate ↑ | Low | High ↑↑↑ | Recruitment ✓ |
| Total NF-κB activity | Optimal | Excessive(cell cycle arrest) | Ineffective (apoptosis) |
Persistence✓ |
2.3. VacA and CagA in ROS/RNS-Induced Oxidative Cascade
3. H. pylori Induces Mitochondrial Dynamics Imbalance
4. Autophagy—Mitophagy
5. Apoptosis
5. mtDNA Mutations
6. Mitochondrial Bioenergetics During Helicobacter Pylori Infection
7. Helicobacter Pylori Induced Dysregulation of Immune cells
7. The role of Mitochondrial Stress in H. pylori-Linked Gastric Cancer
7.1. Different Haplotypes Polymorphism and Strains of H. pylori and Their Pathogenic Features
7.2. Gastric Cancer-Specific Mitochondrial Alterations Beyond Acute Infection
7.3. mtROS-Mediated Apoptosis Evasion and Tumorigenesis
7.4. Mitochondria-Targeted Therapeutic Strategies for H. pylori-Associated GC
8. Mitochondrial Stress in Helicobacter pylori-Associated MALT Lymphoma
8.1. Helicobacter pylori Infection in MALT Lymphoma: Pathogenic Significance
8.2. Mitochondrial Stress in MALT Lymphoma
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Conflicts of Interest
Copyright confirmation
Abbreviations
References
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet Lond. Engl. 1983, 1, 1273–1275. [Google Scholar]
- Li, Y.; Hahn, A.I.; Laszkowska, M.; Jiang, F.; Zauber, A.G.; Leung, W.K. Global burden of young-onset gastric cancer: a systematic trend analysis of the global burden of disease study 2019. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2024, 27, 684–700. [Google Scholar] [CrossRef]
- Chen, Y.C.; Malfertheiner, P.; Yu, H.T.; Kuo, C.L.; Chang, Y.Y.; Meng, F.T.; Wu, Y.X.; Hsiao, J.L.; Chen, M.J.; Lin, K.P.; et al. Global Prevalence of Helicobacter pylori Infection and Incidence of Gastric Cancer Between 1980 and 2022. Gastroenterology 2024, 166, 605–619. [Google Scholar] [CrossRef]
- Lin, J.L.; Lin, J.X.; Lin, G.T.; Huang, C.M.; Zheng, C.H.; Xie, J.W.; Wang, J.B.; Lu, J.; Chen, Q.Y.; Li, P. Global incidence and mortality trends of gastric cancer and predicted mortality of gastric cancer by 2035. BMC Public Health 2024, 24, 1763. [Google Scholar] [CrossRef] [PubMed]
- Grgov, S.; Katic, V.; Krstic, M.; Nagorni, A.; Radovanovic-Dinic, B.; Tasic, T. Treatment of low-grade gastric MALT lymphoma using Helicobacter pylori eradication. Vojn. Pregl 2015, 72, 431–436. [Google Scholar] [CrossRef]
- Lim, N.R.; Chung, W.C. Helicobacter pylori-associated Chronic Atrophic Gastritis and Progression of Gastric Carcinogenesis. Korean J Gastroenterol 2023, 82, 171–179. [Google Scholar] [CrossRef]
- Du, M.Q. Molecular biology of gastric MALT lymphoma: application in clinical management. Hematology 2002, 7, 339–344. [Google Scholar] [CrossRef]
- Dorflinger, B.; Badr, M.T.; Haimovici, A.; Fischer, L.; Vier, J.; Metz, A.; Eisele, B.; Bronsert, P.; Aumann, K.; Hoppner, J.; et al. Mitochondria supply sub-lethal signals for cytokine secretion and DNA-damage in H. pylori infection. Cell Death Differ 2022, 29, 2218–2232. [Google Scholar] [CrossRef]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef]
- Diaz, P.; Valenzuela Valderrama, M.; Bravo, J.; Quest, A.F.G. Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front Microbiol 2018, 9, 5. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Yang, H.; Jielili, A.; Cao, Z.; Yuan, T. Clinical features & treatment of early-stage gastric mucosa-associated lymphoid tissue lymphoma. Indian J. Med. Res. 2021, 154, 504–508. [Google Scholar] [CrossRef]
- Merino, F.; Vázquez Martinez, C.; Zubillaga, I.; Sánchez Aniceto, G.; Ballestín, C. MALT Lymphoma occurring in the maxillofacial region: A review of the literature and case report. Oral Maxillofac. Surg. Cases 2017, 3, 70–75. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Calvet, X. Review article: common misconceptions in the management of Helicobacter pylori-associated gastric MALT-lymphoma: Review: H. pylori and gastric MALT lymphoma. Aliment. Pharmacol. Ther. 2011, 34, 1047–1062. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P. Helicobacter pylori Infection, Chronic Inflammation, and Genomic Transformations in Gastric MALT Lymphoma. Mediators Inflamm. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef]
- Nakamura, M.; Øverby, A.; Michimae, H.; Matsui, H.; Takahashi, S.; Mabe, K.; Shimoyama, T.; Sasaki, M.; Terao, S.; Kamada, T.; et al. PCR analysis and specific immunohistochemistry revealing a high prevalence of non- Helicobacter pylori Helicobacters in Helicobacter pylori -negative gastric disease patients in Japan: High susceptibility to an Hp eradication regimen. Helicobacter 2020, 25, e12700. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, H.; Yuge, R.; Masaki, S.; Otani, R.; Kadota, H.; Naito, T.; Hayashi, R.; Urabe, Y.; Oka, S.; Tanaka, S.; et al. Involvement of non-Helicobacter pylori helicobacter infections in Helicobacter pylori-negative gastric MALT lymphoma pathogenesis and efficacy of eradication therapy. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2021, 24, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hojo, M. Diagnosis and Treatment for Gastric Mucosa-Associated Lymphoid Tissue (MALT) Lymphoma. J. Clin. Med. 2022, 12, 120. [Google Scholar] [CrossRef]
- Xie, Y.-L.; He, C.-Y.; Wei, S.-Q.; Guan, W.-J.; Jiang, Z. Clinical efficacy of the modified Helicobacter pylori eradication therapy for Helicobacter pylori-negative gastric mucosa-associated lymphoid tissue lymphoma: a meta analysis. Chin. Med. J. (Engl.) 2020, 133, 1337–1346. [Google Scholar] [CrossRef]
- Kim, J.S.; Kang, S.H.; Moon, H.S.; Sung, J.K.; Jeong, H.Y. Clinical Outcome of Eradication Therapy for Gastric Mucosa-Associated Lymphoid Tissue Lymphoma according to H. pylori Infection Status. Gastroenterol. Res. Pract. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Nakamura, S. Helicobacter pylori and gastric mucosa-associated lymphoid tissue lymphoma: Recent progress in pathogenesis and management. World J. Gastroenterol. 2013, 19, 8181. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-T.; Lin, J.-T.; Tai, J.J.; Chen, G.-H.; Yeh, H.-Z.; Yang, S.-S.; Wang, H.-P.; Kuo, S.-H.; Sheu, B.-S.; Jan, C.-M.; et al. Long-Term Results of Anti– Helicobacter pylori Therapy in Early-Stage Gastric High-Grade Transformed MALT Lymphoma. JNCI J. Natl. Cancer Inst. 2005, 97, 1345–1353. [Google Scholar] [CrossRef]
- Sugizaki, K.; Tari, A.; Kitadai, Y.; Oda, I.; Nakamura, S.; Yoshino, T.; Sugiyama, T. Anti- Helicobacter pylori therapy in localized gastric mucosa-associated lymphoid tissue lymphoma: A prospective, nationwide, multicenter study in Japan. Helicobacter 2018, 23, e12474. [Google Scholar] [CrossRef]
- Cuciureanu, D.; Maria Vladareanu, A.; et al.; Violeta Filip; P; “Carol Davila” University of Medicine and Pharmacy; Bucharest; Department of Internal Medicine II and Gastroenterology; Emergency University Hospital; Bucharest; Sorina Diaconu; L; “Carol Davila” University of Medicine and Pharmacy; Bucharest; Department of Internal Medicine II and Gastroenterology; Emergency University Hospital; Bucharest MALT lymphoma: epidemiology, clinical diagnosis and treatment. J. Med. Life 2018, 11, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, Y.; Zhang, X.; Fu, K. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori infection: a review of current diagnosis and management. Biomark. Res. 2016, 4, 15. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, Y.; Zhang, H.; Yang, W.; Yang, J.; Sun, L.; Jiang, M.; Wang, Q.; Wang, Q.; Ye, X.; et al. Mechanism of regulation of the Helicobacter pylori Cagβ ATPase by CagZ. Nat. Commun. 2023, 14, 479. [Google Scholar] [CrossRef] [PubMed]
- Poppe, M.; Feller, S.M.; Romer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007, 26, 3462–3472. [Google Scholar] [CrossRef]
- Shiota, S.; Suzuki, R.; Yamaoka, Y. The significance of virulence factors in Helicobacter pylori. J. Dig. Dis. 2013, 14, 341–349. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors-Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef]
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef]
- Chen, B.; Liu, X.; Yu, P.; Xie, F.; Kwan, J.S.H.; Chan, W.N.; Fang, C.; Zhang, J.; Cheung, A.H.K.; Chow, C.; et al. H. pylori-induced NF-κB-PIEZO1-YAP1-CTGF axis drives gastric cancer progression and cancer-associated fibroblast-mediated tumour microenvironment remodelling. Clin. Transl. Med. 2023, 13, e1481. [Google Scholar] [CrossRef]
- Huang, S.; Bucana, C.D.; Van Arsdall, M.; Fidler, I.J. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene 2002, 21, 2504–2512. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Liu, B.; Bukhari, I.; Li, F.; Ren, F.; Xia, X.; Hu, B.; Liu, H.; Meyer, T.F.; Marshall, B.J.; Tay, A.; et al. Enhanced LRP8 expression induced by Helicobacter pylori drives gastric cancer progression by facilitating β-Catenin nuclear translocation. J. Adv. Res. 2025, 69, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Gao, Z.; Bao, Y.; Chen, L.; Huang, Y.; Liu, Y.; Dong, Q.; Wei, X. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol.J Hematol Oncol 2024, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Qin, Y.; Zhang, X.; Deng, S.; Yuan, Y.; Feng, X.; Chen, W.; Hu, F.; Gao, Y.; He, J.; et al. MiRNA-20b/SUFU/Wnt axis accelerates gastric cancer cell proliferation, migration and EMT. Heliyon 2021, 7, e06695. [Google Scholar] [CrossRef]
- Duan, Y.; Xu, Y.; Dou, Y.; Xu, D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol 2025, 18, 10. [Google Scholar] [CrossRef]
- Greiner, A.; Knörr, C.; Qin, Y.; Sebald, W.; Schimpl, A.; Banchereau, J.; Müller-Hermelink, H.K. Low-grade B cell lymphomas of mucosa-associated lymphoid tissue (MALT-type) require CD40-mediated signaling and Th2-type cytokines for in vitro growth and differentiation. Am. J. Pathol. 1997, 150, 1583–1593. [Google Scholar]
- Ye, H.; Liu, H.; Attygalle, A.; Wotherspoon, A.C.; Nicholson, A.G.; Charlotte, F.; Leblond, V.; Speight, P.; Goodlad, J.; Lavergne-Slove, A.; et al. Variable frequencies of t(11;18)(q21;q21) in MALT lymphomas of different sites: significant association with CagA strains of H pylori in gastric MALT lymphoma. Blood 2003, 102, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Simonitsch-Klupp, I.; Müllauer, L.; Lamprecht, A.; Huber, D.; Siebert, R.; Stolte, M.; Trautinger, F.; Lukas, J.; Püspök, A.; et al. Variable frequencies of MALT lymphoma-associated genetic aberrations in MALT lymphomas of different sites. Leukemia 2004, 18, 1722–1726. [Google Scholar] [CrossRef]
- Willis, T.G.; Jadayel, D.M.; Du, M.Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef]
- Zhang, Q.; Siebert, R.; Yan, M.; Hinzmann, B.; Cui, X.; Xue, L.; Rakestraw, K.M.; Naeve, C.W.; Beckmann, G.; Weisenburger, D.D.; et al. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat. Genet. 1999, 22, 63–68. [Google Scholar] [CrossRef]
- Ye, H.; Dogan, A.; Karran, L.; Willis, T.G.; Chen, L.; Wlodarska, I.; Dyer, M.J.; Isaacson, P.G.; Du, M.Q. BCL10 expression in normal and neoplastic lymphoid tissue. Nuclear localization in MALT lymphoma. Am. J. Pathol. 2000, 157, 1147–1154. [Google Scholar] [CrossRef]
- Arjunan, A.; Venkatraman, G.; Joseph, L.D.; Perumalsamy, L.R. Mitochondrial homeostasis and their impact on gastric carcinoma. Int J Biochem Cell Biol 2025, 186, 106827. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Kidane, D. Decoding mitochondrial DNA damage and repair associated with H. pylori infection. Front Cell Infect Microbiol 2024, 14, 1529441. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, Y.; Hu, B. Potential role of mitochondria in gastric cancer detection: Fission and glycolysis. Oncol Lett 2021, 21, 439. [Google Scholar] [CrossRef]
- Mazumder, S.; De, R.; Debsharma, S.; Bindu, S.; Maity, P.; Sarkar, S.; Saha, S.J.; Siddiqui, A.A.; Banerjee, C.; Nag, S.; et al. Indomethacin impairs mitochondrial dynamics by activating the PKCzeta-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem 2019, 294, 8238–8258. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shu, X.; Wang, J. Helicobacter pylori-Mediated Oxidative Stress and Gastric Diseases: A Review. Front. Microbiol. 2022, 13, 811258. [Google Scholar] [CrossRef]
- Fiorentino, M.; Ding, H.; Blanchard, T.G.; Czinn, S.J.; Sztein, M.B.; Fasano, A. Helicobacter pylori-induced disruption of monolayer permeability and proinflammatory cytokine secretion in polarized human gastric epithelial cells. Infect Immun 2013, 81, 876–883. [Google Scholar] [CrossRef]
- Wang, J.; Fan, X.; Lindholm, C.; Bennett, M.; O’Connoll, J.; Shanahan, F.; Brooks, E.G.; Reyes, V.E.; Ernst, P.B. Helicobacter pylori modulates lymphoepithelial cell interactions leading to epithelial cell damage through Fas/Fas ligand interactions. Infect Immun 2000, 68, 4303–4311. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.; Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Effect of Helicobacter pylori on gastric epithelial cells. World J Gastroenterol 2014, 20, 12767–12780. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S. Helicobacter pylori virulence factors except CagA. Nihon Rinsho 2009, 67, 2251–2256. [Google Scholar]
- Kumar, S.; Dhiman, M. Inflammasome activation and regulation during Helicobacter pylori pathogenesis. Microb Pathog 2018, 125, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qian, J.; Zhang, X.; Zou, Q. Outer membrane inflammatory protein A, a new virulence factor involved in the pathogenesis of Helicobacter pylori. Mol Biol Rep 2014, 41, 7807–7814. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc Jpn Acad Ser B Phys Biol Sci 2017, 93, 196–219. [Google Scholar] [CrossRef]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol 2005, 3, 320–332. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Harrer, A.; Rottner, K.; Backert, S. Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation. Cancers Basel 2021, 13. [Google Scholar] [CrossRef]
- Necchi, V.; Sommi, P.; Vanoli, A.; Fiocca, R.; Ricci, V.; Solcia, E. Natural history of Helicobacter pylori VacA toxin in human gastric epithelium in vivo: vacuoles and beyond. Sci Rep 2017, 7, 14526. [Google Scholar] [CrossRef]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol 2012, 2, 92. [Google Scholar] [CrossRef]
- Wang, L.; Yi, J.; Yin, X.Y.; Hou, J.X.; Chen, J.; Xie, B.; Chen, G.; Wang, Q.F.; Wang, L.N.; Wang, X.Y.; et al. Vacuolating Cytotoxin A Triggers Mitophagy in Helicobacter pylori-Infected Human Gastric Epithelium Cells. Front Oncol 2022, 12, 881829. [Google Scholar] [CrossRef]
- Chatre, L.; Fernandes, J.; Michel, V.; Fiette, L.; Ave, P.; Arena, G.; Jain, U.; Haas, R.; Wang, T.C.; Ricchetti, M.; et al. Helicobacter pylori targets mitochondrial import and components of mitochondrial DNA replication machinery through an alternative VacA-dependent and a VacA-independent mechanisms. Sci Rep 2017, 7, 15901. [Google Scholar] [CrossRef]
- Foo, J.H.; Culvenor, J.G.; Ferrero, R.L.; Kwok, T.; Lithgow, T.; Gabriel, K. Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J Mol Biol 2010, 401, 792–798. [Google Scholar] [CrossRef]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J 2000, 19, 6361–6370. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, H.; Li, S.; Pintilie, G.D.; Mou, T.C.; Gao, Y.; Zhang, Q.; van den Bedem, H.; Schmid, M.F.; Au, S.W.N.; et al. Cryo-EM structures of Helicobacter pylori vacuolating cytotoxin A oligomeric assemblies at near-atomic resolution. Proc Natl Acad Sci U A 2019, 116, 6800–6805. [Google Scholar] [CrossRef]
- Gonzalez-Rivera, C.; Gangwer, K.A.; McClain, M.S.; Eli, I.M.; Chambers, M.G.; Ohi, M.D.; Lacy, D.B.; Cover, T.L. Reconstitution of Helicobacter pylori VacA toxin from purified components. Biochemistry 2010, 49, 5743–5752. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.C.; Peek, R.M.; Tham, K.T.; Cover, T.L.; Blaser, M.J. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997, 112, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.C.; Cao, P.; Peek, R.M.; Tummuru, M.K.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, M.; Lehn, N.; Neumayer, N.; Boren, T.; Rad, R.; Schepp, W.; Miehlke, S.; Classen, M.; Prinz, C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U A 1999, 96, 12778–12783. [Google Scholar] [CrossRef]
- McClain, M.S.; Cao, P.; Iwamoto, H.; Vinion-Dubiel, A.D.; Szabo, G.; Shao, Z.; Cover, T.L. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J Bacteriol 2001, 183, 6499–6508. [Google Scholar] [CrossRef]
- Jones, K.R.; Jang, S.; Chang, J.Y.; Kim, J.; Chung, I.S.; Olsen, C.H.; Merrell, D.S.; Cha, J.H. Polymorphisms in the intermediate region of VacA impact Helicobacter pylori-induced disease development. J Clin Microbiol 2011, 49, 101–110. [Google Scholar] [CrossRef]
- Liu, X.; He, B.; Cho, W.C.; Pan, Y.; Chen, J.; Ying, H.; Wang, F.; Lin, K.; Peng, H.; Wang, S. A systematic review on the association between the Helicobacter pylori vacA i genotype and gastric disease. FEBS Open Bio 2016, 6, 409–417. [Google Scholar] [CrossRef]
- Kimura, M.; Goto, S.; Wada, A.; Yahiro, K.; Niidome, T.; Hatakeyama, T.; Aoyagi, H.; Hirayama, T.; Kondo, T. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb Pathog 1999, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.C.; Bryant, K.N.; Cover, T.L. The Helicobacter pylori cag pathogenicity island as a determinant of gastric cancer risk. Gut Microbes 2024, 16, 2314201. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Lacy, D.B.; Ohi, M.D. The Helicobacter pylori Cag Type IV Secretion System. Trends Microbiol 2020, 28, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Poppe, M.; Feller, S.M.; Romer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007, 26, 3462–3472. [Google Scholar] [CrossRef]
- Padda, J.; Khalid, K.; Cooper, A.C.; Jean-Charles, G. Association Between Helicobacter pylori and Gastric Carcinoma. Cureus 2021, 13, e15165. [Google Scholar] [CrossRef]
- Alipour, M. Molecular Mechanism of Helicobacter pylori-Induced Gastric Cancer. J Gastrointest Cancer 2021, 52, 23–30. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Gong, Y.; Yuan, Y. Association of CagA EPIYA-D or EPIYA-C phosphorylation sites with peptic ulcer and gastric cancer risks: A meta-analysis. Med. Baltim. 2017, 96, e6620. [Google Scholar] [CrossRef]
- Keikha, M.; Karbalaei, M. EPIYA motifs of Helicobacter pylori cagA genotypes and gastrointestinal diseases in the Iranian population: a systematic review and meta-analysis. New Microbes New Infect 2021, 41, 100865. [Google Scholar] [CrossRef]
- Fujii, Y.; Murata-Kamiya, N.; Hatakeyama, M. Helicobacter pylori CagA oncoprotein interacts with SHIP2 to increase its delivery into gastric epithelial cells. Cancer Sci 2020, 111, 1596–1606. [Google Scholar] [CrossRef]
- Ji, X.; Wu, Q.; Cao, X.; Liu, S.; Zhang, J.; Chen, S.; Shan, J.; Zhang, Y.; Li, B.; Zhao, H. Helicobacter pylori East Asian type CagA hijacks more SHIP2 by its EPIYA-D motif to potentiate the oncogenicity. Virulence 2024, 15, 2375549. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Murata-Kamiya, N.; Hatakeyama, M. Helicobacter pylori CagA oncoprotein interacts with SHIP2 to increase its delivery into gastric epithelial cells. Cancer Sci 2020, 111, 1596–1606. [Google Scholar] [CrossRef]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef]
- Nishikawa, H.; Hayashi, T.; Arisaka, F.; Senda, T.; Hatakeyama, M. Impact of structural polymorphism for the Helicobacter pylori CagA oncoprotein on binding to polarity-regulating kinase PAR1b. Sci Rep 2016, 6, 30031. [Google Scholar] [CrossRef] [PubMed]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog 2009, 5, e1000603. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Blanke, S.R. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol 2012, 2, 37. [Google Scholar] [CrossRef]
- Ghanbarian, M.; Bakhtiari, R.; Mirbagheri, S.Z.; Rezaei, F.; Rahimi Foroushani, A.; Alebouyeh, M. Transcriptional alteration of NF-kappaB-associated long noncoding RNAs in the stomach of Helicobacter pylori-infected and non-infected patients. J Infect Dev Ctries 2023, 17, 1556–1565. [Google Scholar] [CrossRef]
- Kim, I.J.; Lee, J.; Oh, S.J.; Yoon, M.S.; Jang, S.S.; Holland, R.L.; Reno, M.L.; Hamad, M.N.; Maeda, T.; Chung, H.J.; et al. Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593 e8. [Google Scholar] [CrossRef]
- Huang, X.W.; Luo, R.H.; Zhao, Q.; Shen, Z.Z.; Huang, L.L.; An, X.Y.; Zhao, L.J.; Wang, J.; Huang, Y.Z. Helicobacter pylori induces mitochondrial DNA mutation and reactive oxygen species level in AGS cells. Int J Med Sci 2011, 8, 56–67. [Google Scholar] [CrossRef]
- Sah, D.K.; Arjunan, A.; Lee, B.; Jung, Y.D. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxid. Basel 2023, 12. [Google Scholar] [CrossRef]
- Sah, D.K.; Arjunan, A.; Lee, B.; Jung, Y.D. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxid. Basel 2023, 12. [Google Scholar] [CrossRef]
- Sierra, J.C.; Piazuelo, M.B.; Luis, P.B.; Barry, D.P.; Allaman, M.M.; Asim, M.; Sebrell, T.A.; Finley, J.L.; Rose, K.L.; Hill, S.; et al. Spermine oxidase mediates Helicobacter pylori-induced gastric inflammation, DNA damage, and carcinogenic signaling. Oncogene 2020, 39, 4465–4474. [Google Scholar] [CrossRef]
- Dzikowiec, M.; Galant, S.; Lik, P.; Goralska, K.; Nejc, D.; Piekarski, J.; Majos, A.; Brzezianska-Lasota, E.; Pastuszak-Lewandoska, D. Analysis of Spermine Oxidase gene and proinflammatory cytokines expression in gastric cancer patients with and without Helicobacter pylori infection - A pilot study in Polish population. Adv Med Sci 2024, 69, 443–450. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.M.; Sierra, J.C.; Latour, Y.L.; Hawkins, C.V.; Asim, M.; Williams, K.J.; Barry, D.P.; Allaman, M.M.; Zagol-Ikapitte, I.; Luis, P.B.; et al. Spermine oxidase promotes Helicobacter pylori-mediated gastric carcinogenesis through acrolein production. Oncogene 2025, 44, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Cherdantseva, L.A.; Potapova, O.V.; Sharkova, T.V.; Belyaeva, Y.Y.; Shkurupiy, V.A. Association of Helicobacter pylori and iNOS production by macrophages and lymphocytes in the gastric mucosa in chronic gastritis. J Immunol Res 2014, 2014, 762514. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Barry, D.P.; Frye, J.W.; Casero, R.A.; Wilson, K.T. Spermine oxidase is a regulator of macrophage host response to Helicobacter pylori: enhancement of antimicrobial nitric oxide generation by depletion of spermine. Amino Acids 2014, 46, 531–542. [Google Scholar] [CrossRef]
- Ding, S.Z.; Minohara, Y.; Fan, X.J.; Wang, J.; Reyes, V.E.; Patel, J.; Dirden-Kramer, B.; Boldogh, I.; Ernst, P.B.; Crowe, S.E. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun 2007, 75, 4030–4039. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, L.; Liu, X.; Wang, Q.; Gui, S.; Bao, L.; Wang, Z.; He, X.; Zhao, Y.; Zhou, J.; et al. Helicobacter pylori CagA mediated mitophagy to attenuate the NLRP3 inflammasome activation and enhance the survival of infected cells. Sci Rep 2024, 14, 21648. [Google Scholar] [CrossRef]
- Jain, P.; Luo, Z.Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci U A 2011, 108, 16032–16037. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Hu, B. Potential role of mitochondria in gastric cancer detection: Fission and glycolysis. Oncol Lett 2021, 21, 439. [Google Scholar] [CrossRef]
- Chen, X.J.; Cui, Q.X.; Wang, G.L.; Li, X.L.; Zhou, X.L.; Zhao, H.J.; Zhang, M.Q.; Li, M.J.; He, X.J.; Zheng, Q.S.; et al. Sanggenon C Suppresses Tumorigenesis of Gastric Cancer by Blocking ERK-Drp1-Mediated Mitochondrial Fission. J Nat Prod 2022, 85, 2351–2362. [Google Scholar] [CrossRef]
- Tanprasert, P.; Limpakan Yamada, S.; Chattipakorn, S.C.; Chattipakorn, N.; Shinlapawittayatorn, K. Targeting mitochondria as a therapeutic anti-gastric cancer approach. Apoptosis 2022, 27, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ghatak, D.; Dutta, R.; Goswami, D.; De, R. PINK1 insufficiency can be exploited as a specific target for drug combinations inducing mitochondrial pathology-mediated cell death in gastric adenocarcinoma. Arch Biochem Biophys 2024, 759, 110110. [Google Scholar] [CrossRef]
- Luo, J.; Bai, L.; Tao, J.; Wen, Y.; Li, M.; Zhu, Y.; Luo, S.; Pu, G.; Ma, L. Autophagy induced by H. pylori VacA regulated the survival mechanism of the SGC7901 human gastric cancer cell line. Genes Genomics 2021, 43, 1223–1230. [Google Scholar] [CrossRef]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef]
- Wang, L.; Yi, J.; Yin, X.Y.; Hou, J.X.; Chen, J.; Xie, B.; Chen, G.; Wang, Q.F.; Wang, L.N.; Wang, X.Y.; et al. Vacuolating Cytotoxin A Triggers Mitophagy in Helicobacter pylori-Infected Human Gastric Epithelium Cells. Front Oncol 2022, 12, 881829. [Google Scholar] [CrossRef]
- Kim, I.J.; Lee, J.; Oh, S.J.; Yoon, M.S.; Jang, S.S.; Holland, R.L.; Reno, M.L.; Hamad, M.N.; Maeda, T.; Chung, H.J.; et al. Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593 e8. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hirai, M. Helicobacter pylori infection and oxidative stress. J Clin Biochem Nutr 2024, 75, 178–182. [Google Scholar] [CrossRef]
- Dang, Y.; Zhang, Y.; Xu, L.; Zhou, X.; Gu, Y.; Yu, J.; Jin, S.; Ji, H.; Shu, Y.; Zhang, G.; et al. PUMA-mediated epithelial cell apoptosis promotes Helicobacter pylori infection-mediated gastritis. Cell Death Dis 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, K.; Lu, F.; Zhai, C.; Cheng, F. Programmed cell death in Helicobacter pylori infection and related gastric cancer. Front Cell Infect Microbiol 2024, 14, 1416819. [Google Scholar] [CrossRef]
- Yahiro, K.; Akazawa, Y.; Nakano, M.; Suzuki, H.; Hisatune, J.; Isomoto, H.; Sap, J.; Noda, M.; Moss, J.; Hirayama, T. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Radin, J.N.; Gonzalez-Rivera, C.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect Immun 2011, 79, 2535–2543. [Google Scholar] [CrossRef]
- Zhu, P.; Xue, J.; Zhang, Z.J.; Jia, Y.P.; Tong, Y.N.; Han, D.; Li, Q.; Xiang, Y.; Mao, X.H.; Tang, B. Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis 2017, 8, 3207. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, J.S.; Lee, J.Y.; Sim, Y.S.; Kim, Y.J.; Oh, Y.K.; Yoon, H.J.; Kang, J.S.; Youn, J.; Kim, N.; et al. Dual effects of Helicobacter pylori vacuolating cytotoxin on human eosinophil apoptosis in early and late periods of stimulation. Eur J Immunol 2010, 40, 1651–1662. [Google Scholar] [CrossRef]
- Ding, S.Z.; Smith, M.F.; Goldberg, J.B. Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J Gastroenterol Hepatol 2008, 23, e67-78. [Google Scholar] [CrossRef]
- Choi, I.J.; Kim, J.S.; Kim, J.M.; Jung, H.C.; Song, I.S. Effect of inhibition of extracellular signal-regulated kinase 1 and 2 pathway on apoptosis and bcl-2 expression in Helicobacter pylori-infected AGS cells. Infect Immun 2003, 71, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Palrasu, M.; Zaika, E.; Paulrasu, K.; Caspa Gokulan, R.; Suarez, G.; Que, J.; El-Rifai, W.; Peek, R.M.; Garcia-Buitrago, M.; Zaika, A.I. Helicobacter pylori pathogen inhibits cellular responses to oncogenic stress and apoptosis. PLoS Pathog 2022, 18, e1010628. [Google Scholar] [CrossRef]
- Kim, K.M.; Lee, S.G.; Kim, J.M.; Kim, D.S.; Song, J.Y.; Kang, H.L.; Lee, W.K.; Cho, M.J.; Rhee, K.H.; Youn, H.S.; et al. Helicobacter pylori gamma-glutamyltranspeptidase induces cell cycle arrest at the G1-S phase transition. J Microbiol 2010, 48, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Garcia-Saez, A.J. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol 2023, 24, 732–748. [Google Scholar] [CrossRef]
- Lin, W.C.; Tsai, H.F.; Liao, H.J.; Tang, C.H.; Wu, Y.Y.; Hsu, P.I.; Cheng, A.L.; Hsu, P.N. Helicobacter pylori sensitizes TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human gastric epithelial cells through regulation of FLIP. Cell Death Dis 2014, 5, e1109. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; You, B.; Dong, S.; Zhou, C. FRA-1 suppresses apoptosis of Helicobacter pylori infected MGC-803 cells. Mol Biol Rep 2021, 48, 611–621. [Google Scholar] [CrossRef]
- Liu, J.F.; Guo, D.; Kang, E.M.; Wang, Y.S.; Gao, X.Z.; Cong, H.Y.; Liu, P.; Zhang, N.Q.; Wang, M.Y. Acute and chronic infection of H. pylori caused the difference in apoptosis of gastric epithelial cells. Microb Pathog 2021, 150, 104717. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Zhao, H.; Chen, X.; Li, J.; Li, B. Early apoptosis of monocytes induced by Helicobacter pylori infection through multiple pathways. Dev Comp Immunol 2017, 73, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.; Potthoff, A.; Ledig, S.; Cornberg, M.; Jandl, O.; Manns, M.P.; Kubicka, S.; Flemming, P.; Athmann, C.; Beil, W.; et al. Effect of H. pylori on the expression of TRAIL, FasL and their receptor subtypes in human gastric epithelial cells and their role in apoptosis. Helicobacter 2004, 9, 371–386. [Google Scholar] [CrossRef]
- Lee, S.; Shin, M.G.; Jo, W.H.; Kim, M.J.; Kim, H.R.; Lee, W.S.; Park, D.H.; Won, J.H.; Shin, J.H.; Suh, S.P.; et al. Association between Helicobacter pylori-related peptic ulcer tissue and somatic mitochondrial DNA mutations. Clin Chem 2007, 53, 1390–1392. [Google Scholar] [CrossRef]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207. [Google Scholar] [CrossRef]
- Shahi, A.; Kidane, D. Decoding mitochondrial DNA damage and repair associated with H. pylori infection. Front Cell Infect Microbiol 2024, 14, 1529441. [Google Scholar] [CrossRef] [PubMed]
- Simsek, D.; Jasin, M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 2011, 10, 3636–3644. [Google Scholar] [CrossRef]
- Machado, A.M.; Desler, C.; Boggild, S.; Strickertsson, J.A.; Friis-Hansen, L.; Figueiredo, C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection affects mitochondrial function and DNA repair, thus, mediating genetic instability in gastric cells. Mech Ageing Dev 2013, 134, 460–466. [Google Scholar] [CrossRef]
- Shen, Q.Y.; Zhou, Y.Y.; Wang, Y.L.; Qi, Y.; Lian, D.B.; Li, Z.H.; Chen, M.F.; Zhang, X.L.; Zhang, Y.; Song, J.; et al. Blocking dopamine receptor 2 decreases gastrin levels in H. pylori-infected mice through increasing gastric somatostatin content. Am J Physiol Cell Physiol 2025, 328, C1409–C1422. [Google Scholar] [CrossRef]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.-I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef]
- Okuyama, K.; Kitajima, Y.; Egawa, N.; Kitagawa, H.; Ito, K.; Aishima, S.; Yanagihara, K.; Tanaka, T.; Noshiro, H. Mieap-induced accumulation of lysosomes within mitochondria (MALM) regulates gastric cancer cell invasion under hypoxia by suppressing reactive oxygen species accumulation. Sci. Rep. 2019, 9, 2822. [Google Scholar] [CrossRef]
- Alikhani, M.; Saberi, S.; Esmaeili, M.; Michel, V.; Tashakoripour, M.; Abdirad, A.; Aghakhani, A.; Eybpoosh, S.; Vosough, M.; Mohagheghi, M.A.; et al. Mitochondrial DNA Copy Number Variations and Serum Pepsinogen Levels for Risk Assessment in Gastric Cancer. Iran Biomed J 2021, 25, 323–333. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, T.; Han, Y.; Ji, Z.; Ye, H. Dynamics of mitochondrial DNA copy number regulation in relation to gastric cancer survival. Discov Oncol 2025, 16, 1090. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shangguan, T.; Xu, Y.; Chen, C.; Wang, K. Unveiling the enigma: Investigating the controversy surrounding mitochondrial DNA copy number and gastric cancer using Mendelian randomization analysis. Med. Baltim. 2025, 104, e43916. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Wang, J.; Chen, C.; Tang, X.; Zhu, J.; Liu, J. Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. Onco Targets Ther 2019, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Seeger, A.Y.; Zaidi, F.; Alhayek, S.; Jones, R.M.; Zohair, H.; Holland, R.L.; Kim, I.J.; Blanke, S.R. Host cell sensing and restoration of mitochondrial function and metabolism within Helicobacter pylori VacA intoxicated cells. mBio 2023, 14, e0211723. [Google Scholar] [CrossRef]
- Luo, B.; Wang, M.; Hou, N.; Hu, X.; Jia, G.; Qin, X.; Zuo, X.; Liu, Y.; Luo, K.; Song, W.; et al. ATP-Dependent Lon Protease Contributes to Helicobacter pylori-Induced Gastric Carcinogenesis. Neoplasia 2016, 18, 242–252. [Google Scholar] [CrossRef]
- Duan, Y.; Xu, Y.; Dou, Y.; Xu, D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol 2025, 18, 10. [Google Scholar] [CrossRef]
- Li, Y.; Liu, G.; Zhou, L.; Wang, Y.; Sun, Y.; Chen, Y.; Chen, L.; Xiao, J. Helicobacter Pylori-Induced Apoptosis in Gastric Diseases: Mechanisms, Implications, and Diagnostic Applications. Int J Gen Med 2025, 18, 2995–3009. [Google Scholar] [CrossRef]
- Dooyema, S.D.R.; Noto, J.M.; Wroblewski, L.E.; Piazuelo, M.B.; Krishna, U.; Suarez, G.; Romero-Gallo, J.; Delgado, A.G.; Peek, R.M. Helicobacter pylori actively suppresses innate immune nucleic acid receptors. Gut Microbes 2022, 14, 2105102. [Google Scholar] [CrossRef]
- Kranzer, K.; Sollner, L.; Aigner, M.; Lehn, N.; Deml, L.; Rehli, M.; Schneider-Brachert, W. Impact of Helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect Immun 2005, 73, 4180–4189. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Cadamuro, A.C.; Rossi, A.F.; Maniezzo, N.M.; Silva, A.E. Helicobacter pylori infection: host immune response, implications on gene expression and microRNAs. World J Gastroenterol 2014, 20, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.J. The number of Foxp3-positive regulatory T cells is increased in Helicobacter pylori gastritis and gastric cancer. Pathol Res Pr. 2010, 206, 34–38. [Google Scholar] [CrossRef]
- Moyat, M.; Velin, D. Immune responses to Helicobacter pylori infection. World J Gastroenterol 2014, 20, 5583–5593. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Asim, M.; Barry, D.P.; Singh, K.; de Sablet, T.; Boucher, J.L.; Gobert, A.P.; Chaturvedi, R.; Wilson, K.T. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol 2010, 184, 2572–2582. [Google Scholar] [CrossRef]
- Gobert, A.P.; Cheng, Y.; Wang, J.Y.; Boucher, J.L.; Iyer, R.K.; Cederbaum, S.D.; Casero, R.A.; Newton, J.C.; Wilson, K.T. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol 2002, 168, 4692–4700. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.W.; Lai, Y.C. The Role of Helicobacter pylori Neutrophil-Activating Protein in the Pathogenesis of H. pylori and Beyond: From a Virulence Factor to Therapeutic Targets and Therapeutic Agents. Int J Mol Sci 2022, 24. [Google Scholar] [CrossRef]
- Hansen, P.S.; Madsen, P.H.; Petersen, S.B.; Nielsen, H. Inflammatory activation of neutrophils by Helicobacter pylori; a mechanism insensitive to pertussis toxin. Clin Exp Immunol 2001, 123, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, T.; Fukuda, S.; Liu, Q.; Nakaji, S.; Fukuda, Y.; Sugawara, K. Production of chemokines and reactive oxygen species by human neutrophils stimulated by Helicobacter pylori. Helicobacter 2002, 7, 170–174. [Google Scholar] [CrossRef]
- Pachathundikandi, S.K.; Blaser, N.; Bruns, H.; Backert, S. Helicobacter pylori Avoids the Critical Activation of NLRP3 Inflammasome-Mediated Production of Oncogenic Mature IL-1beta in Human Immune Cells. Cancers Basel 2020, 12. [Google Scholar] [CrossRef]
- Koch, K.N.; Muller, A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes 2015, 6, 382–387. [Google Scholar] [CrossRef]
- Pachathundikandi, S.K.; Blaser, N.; Bruns, H.; Backert, S. Helicobacter pylori Avoids the Critical Activation of NLRP3 Inflammasome-Mediated Production of Oncogenic Mature IL-1beta in Human Immune Cells. Cancers Basel 2020, 12. [Google Scholar] [CrossRef]
- Kim, J.K.; Sapkota, A.; Roh, T.; Jo, E.K. The intricate interactions between inflammasomes and bacterial pathogens: Roles, mechanisms, and therapeutic potentials. Pharmacol Ther 2025, 265, 108756. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.E.; Peniche, A.G. Helicobacter pylori Deregulates T and B Cell Signaling to Trigger Immune Evasion. Curr Top Microbiol Immunol 2019, 421, 229–265. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Fu, Z.; Zhang, R.; Zhu, W.; Zhao, Q.; Wang, P.; Hu, C.; Cheng, X. The role of reactive oxygen species in gastric cancer. Cancer Biol Med 2024, 21, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Usui, Y.; Taniyama, Y.; Endo, M.; Koyanagi, Y.N.; Kasugai, Y.; Oze, I.; Ito, H.; Imoto, I.; Tanaka, T.; Tajika, M.; et al. Helicobacter pylori, Homologous-Recombination Genes, and Gastric Cancer. N Engl J Med 2023, 388, 1181–1190. [Google Scholar] [CrossRef]
- Figura, N.; Marano, L.; Moretti, E.; Ponzetto, A. Helicobacter pylori infection and gastric carcinoma: Not all the strains and patients are alike. World J Gastrointest Oncol 2016, 8, 40–54. [Google Scholar] [CrossRef]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Liu, L.; Chen, J.; Zhu, W.; Zhao, Y.; Wu, H. TRAP1 induced cisplatin resistance in gastric cancer cells by regulating oxidative stress. Front. Mol. Biosci. 2025, 12, 1676811. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Siddon, A.J.; Huntington, S.F.; Jain, D. Helicobacter pylori –negative mucosa-associated lymphoid tissue (MALT) lymphoma of the stomach: A clinicopathologic analysis. Am. J. Clin. Pathol. 2023, 160, 612–619. [Google Scholar] [CrossRef]
- Floch, P.; Mégraud, F.; Lehours, P. Helicobacter pylori Strains and Gastric MALT Lymphoma. Toxins 2017, 9, 132. [Google Scholar] [CrossRef]
- Troppan, K.; Wenzl, K.; Neumeister, P.; Deutsch, A. Molecular Pathogenesis of MALT Lymphoma. Gastroenterol Res Pr. 2015, 2015, 102656. [Google Scholar] [CrossRef]
- Pereira, M.I.; Medeiros, J.A. Role of Helicobacter pylori in gastric mucosa-associated lymphoid tissue lymphomas. World J Gastroenterol 2014, 20, 684–698. [Google Scholar] [CrossRef]
- Cogswell, P.C.; Kashatus, D.F.; Keifer, J.A.; Guttridge, D.C.; Reuther, J.Y.; Bristow, C.; Roy, S.; Nicholson, D.W.; Baldwin, A.S. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem 2003, 278, 2963–2968. [Google Scholar] [CrossRef]
- Carra, G.; Avalle, L.; Secli, L.; Brancaccio, M.; Morotti, A. Shedding Light on NF-kappaB Functions in Cellular Organelles. Front Cell Dev Biol 2022, 10, 841646. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Ivanova, I.G.; Perkins, N.D. Hypoxia induces rapid, STAT3 and ROS dependent, mitochondrial translocation of RelA(p65) and IκBα. Biosci Rep 2019, 39. [Google Scholar] [CrossRef] [PubMed]
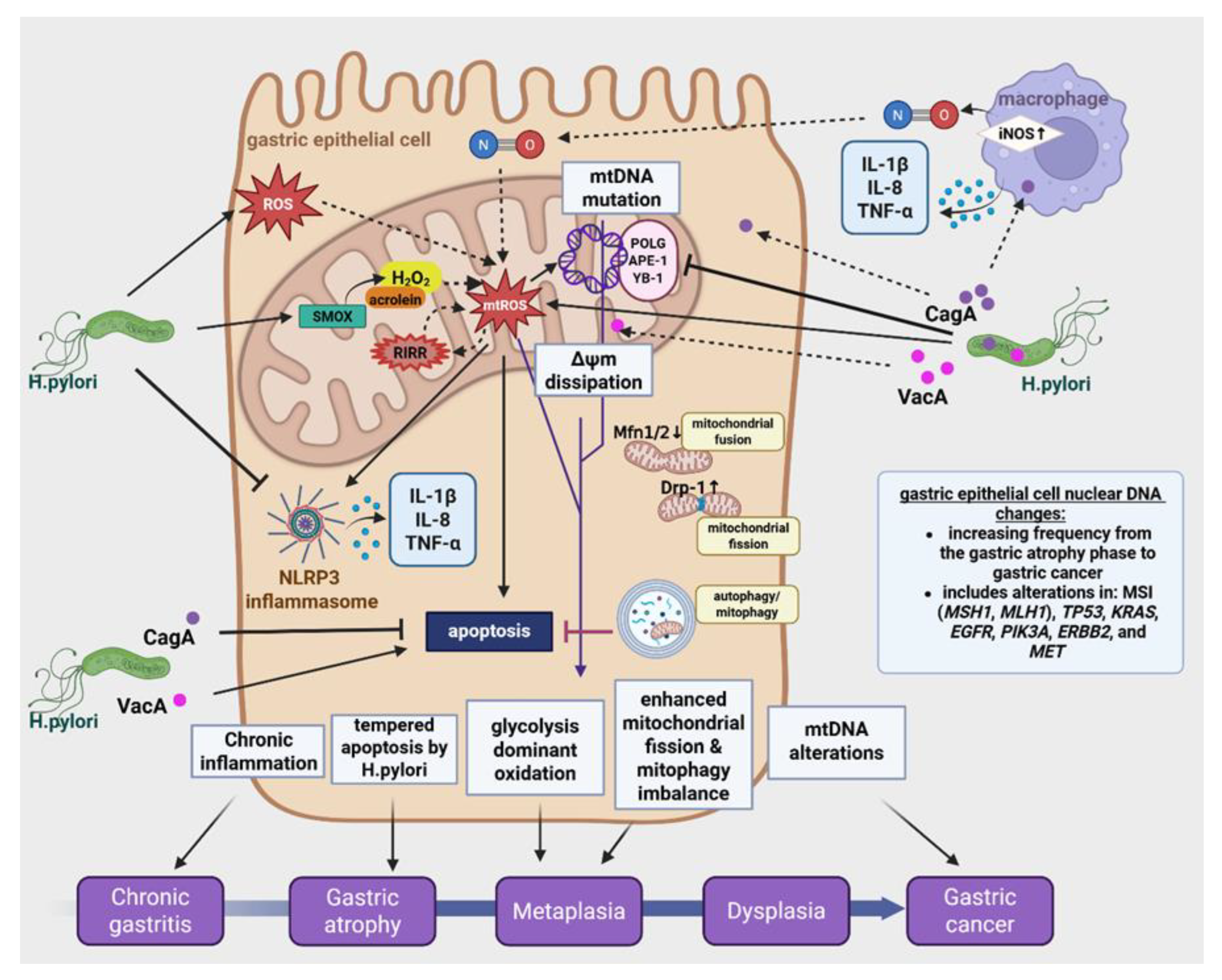
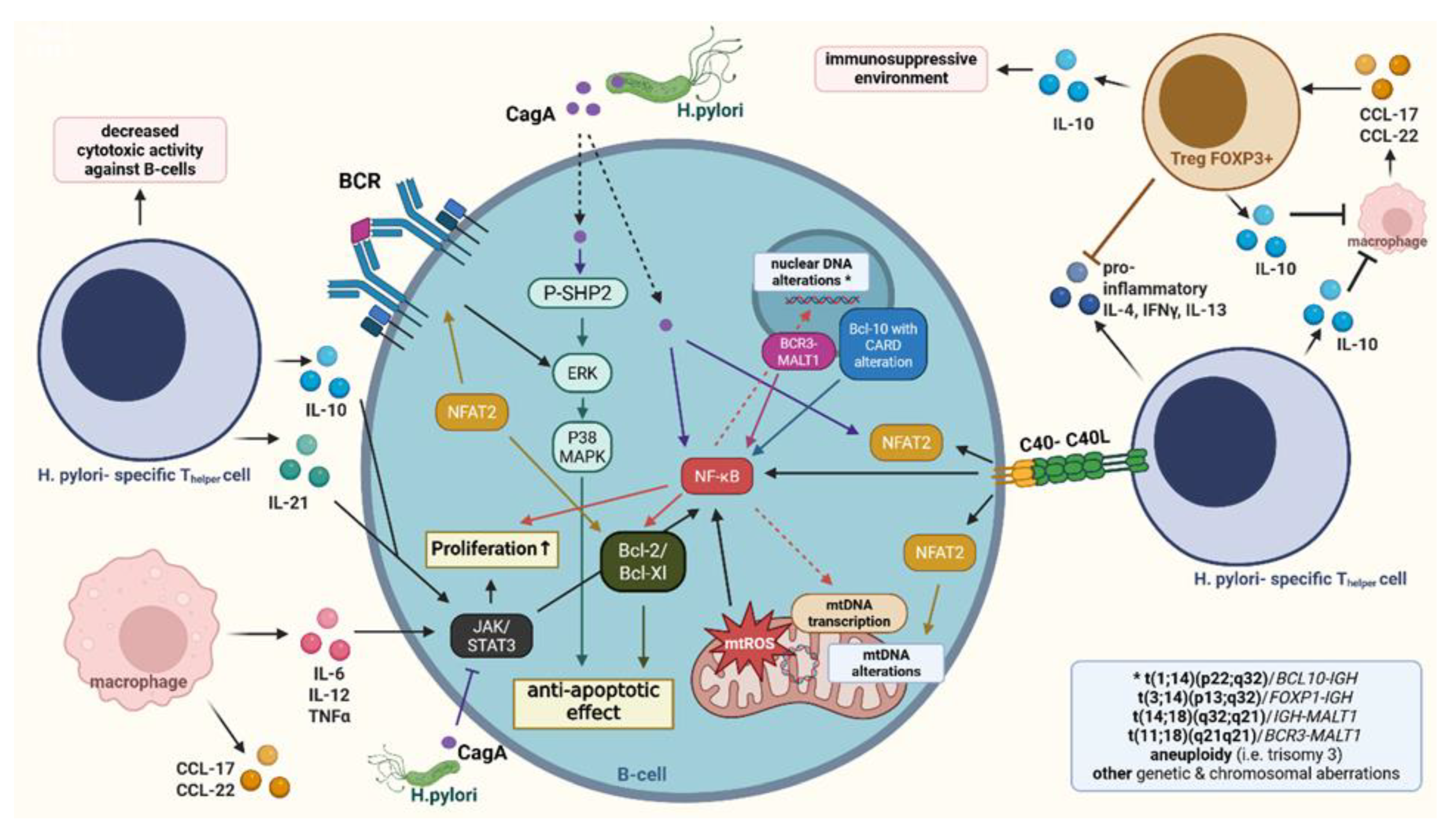
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



