
Submitted:
22 December 2025
Posted:
23 December 2025
You are already at the latest version
Abstract
The rapid diffusion of high-throughput sequencing technologies has generated a vast repertoire of protein-coding se- quences whose biological roles remain unknown. This discrepancy between sequence availability and functional under- standing has led to the definition of the dark proteome, comprising proteins or protein regions that lack experimentally resolved structures and reliable functional annotations. Classical sequence-based approaches often fail to characterize these targets due to extreme sequence divergence, intrinsic disorder, or membrane localization. Here, we present an inte- grated, structure-centric computational framework that leverages recent advances in artificial intelligence to enable func- tional inference in the human dark proteome. By combining deep learning–based protein structure prediction, large-scale structural alignment, and machine learning–driven surface pocket analysis, we uncover remote evolutionary relationships and conserved functional features that remain invisible to traditional bioinformatics pipelines. Our results demonstrate that artificial intelligence provides a powerful strategy to bridge the gap between genomic information and biological function, opening new avenues for systematic exploration of uncharacterized regions of the human proteome.
Keywords:
artificial intelligence
; protein function analysis
; dark proteome
1. Introduction
The sequencing of the human genome represented a defining milestone in modern biology, providing a comprehensive inventory of protein-coding genes. However, more than two decades after its completion, the functional characterization of the encoded proteome remains strikingly incomplete. While the cost and speed of DNA sequencing have improved exponentially, experimental techniques for determining protein structure and function have not scaled at the same rate. As a consequence, a substantial fraction of proteins lacks reliable functional annotation.
This imbalance has given rise to the concept of the dark proteome, defined as the subset of proteins or protein regions whose three-dimensional structure and biological role remain unknown or poorly characterized. Analyses of UniProt and Protein Data Bank (PDB) entries reveal that only a small fraction of known protein sequences is associated with experimentally determined structures, highlighting a persistent knowledge gap between sequence space and structure–function understanding [1].
Traditional protein annotation pipelines rely primarily on sequence homology transfer, under the assumption that evolutionary conservation at the sequence level implies conservation of function. Tools such as BLAST have been instrumental in annotating newly discovered proteins by identifying similarity to known sequences. However, protein evolution tends to conserve tertiary structure more strongly than primary sequence. As a result, proteins with low sequence similarity may still adopt highly similar folds and perform related biological functions. This phenomenon renders sequence-based approaches ineffective for detecting remote homologies, particularly in the context of the dark proteome [2].
Dark proteins are frequently enriched in intrinsically disordered regions, transmembrane segments, or lineage-specific domains, all of which pose challenges for both experimental and computational characterization. Moreover, many of these proteins are weakly expressed, embedded in complex interaction networks, or conditionally active, further complicating their study [3].
Recent advances in artificial intelligence (AI), and deep learning in particular, have fundamentally reshaped the landscape of structural bioinformatics. The introduction of AlphaFold2 demonstrated that protein structures can be predicted with near-experimental accuracy directly from amino acid sequences [4]. This breakthrough effectively removed one of the most significant bottlenecks in protein science and enabled a paradigm shift from sequence-centric to structure-centric functional inference.
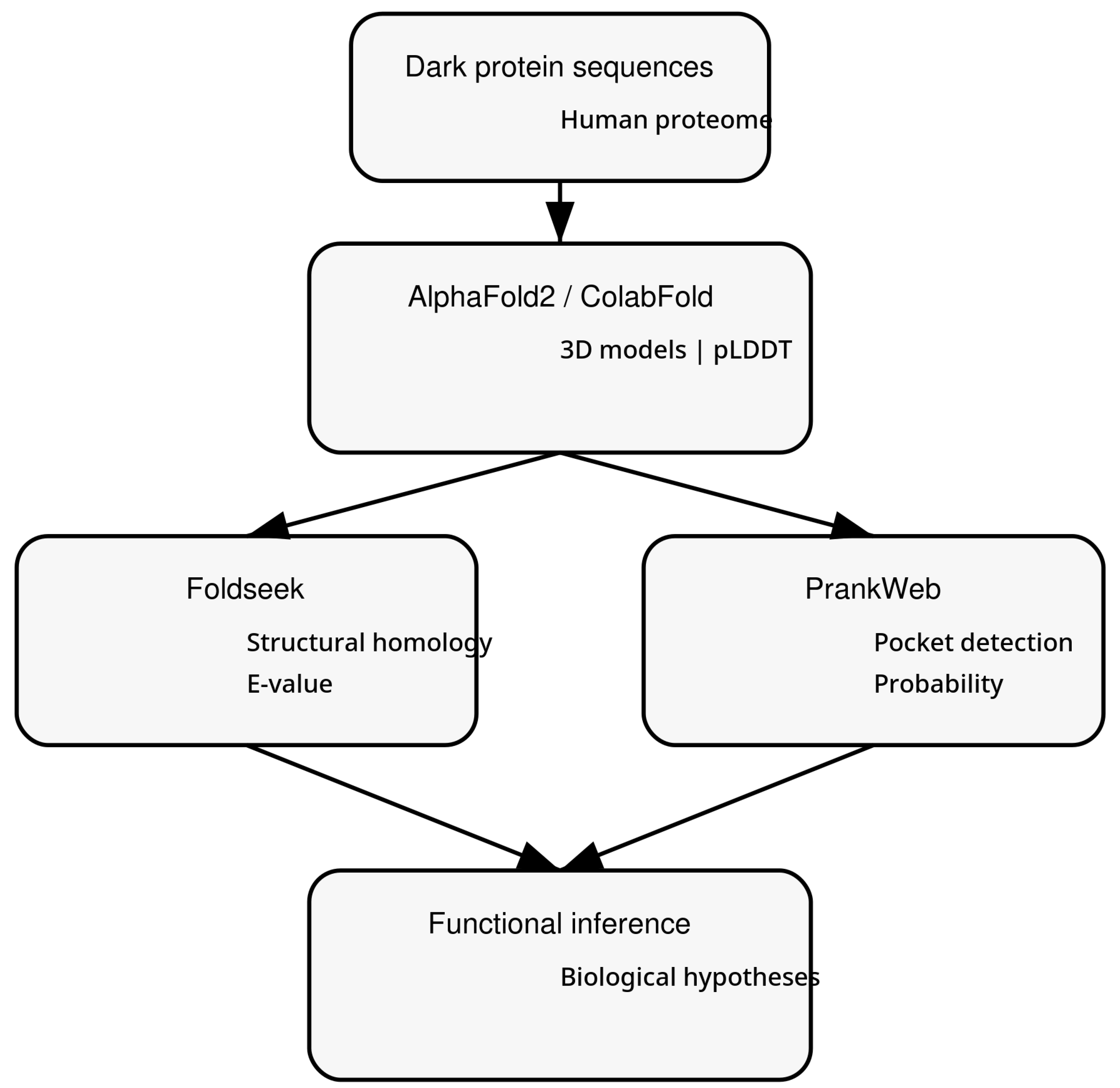
In this work, we exploit this shift to address the challenge of dark proteome characterization. We present a computational pipeline that integrates AI-driven structure prediction, large-scale structural mining, and surface-based functional analysis to infer plausible biological roles for uncharacterized human proteins. Figure 1 depicts the workflow of this article.
2. Artificial Intelligence in Structural Bioinformatics
Artificial intelligence has become an essential component of modern computational biology, enabling the analysis of complex, high-dimensional datasets that exceed the capabilities of traditional rule-based methods. Machine learning, a central subfield of AI, focuses on the development of algorithms that improve performance through exposure to data rather than explicit programming [5]. Among machine learning approaches, deep learning has proven particularly effective for biological applications due to its ability to learn hierarchical representations.
Early computational approaches to protein structure prediction relied on physics-based force fields and statistical potentials, often combined with homology modeling. While these methods achieved moderate success, they struggled with scalability and accuracy, particularly for proteins lacking close homologs. The introduction of deep neural networks, and more recently transformer-based architectures, has dramatically improved predictive performance.
Transformers, originally developed for natural language processing, rely on self-attention mechanisms to capture long-range dependencies within sequences [6]. These properties make them particularly well suited for modeling protein sequences, where interactions between distant residues play a crucial role in determining three-dimensional structure.
AlphaFold2 represents a landmark application of transformer-based deep learning to protein structure prediction [4]. By integrating evolutionary information from multiple sequence alignments with attention-based neural networks, AlphaFold2 predicts inter-residue distances and orientations with unprecedented accuracy. The release of AlphaFold-predicted structures for entire proteomes has fundamentally altered the scope of structural biology.
Beyond structure prediction, AI has enabled advances in structural comparison and functional site detection. Tools such as Foldseek exploit compact structural representations to perform fast and sensitive similarity searches across millions of protein structures [7]. Similarly, machine learning–based surface analysis methods can identify ligand-binding pockets and functional sites by integrating geometric, physicochemical, and evolutionary features [8].
Together, these developments suggest that AI-driven structural analysis provides a powerful framework for systematic exploration of the dark proteome.
3. Methods
3.1. Protein Structure Prediction
Protein structures were predicted using AlphaFold2 through the ColabFold framework [9]. ColabFold accelerates multiple sequence alignment generation by replacing traditional profile construction with MMseqs2-based searches, substantially reducing computational cost while preserving predictive accuracy. For each protein target, five structural models were generated and ranked according to confidence metrics.
The predicted Local Distance Difference Test (pLDDT) score was used to assess per-residue confidence. Regions with high pLDDT values were interpreted as structurally ordered, whereas regions with low confidence were considered potentially disordered or flexible.
3.2. Structural Homology Detection
To identify remote evolutionary relationships, predicted protein structures were compared against large structural databases using Foldseek [7]. Foldseek encodes three-dimensional protein conformations into a discrete structural alphabet that captures local residue–residue interaction patterns. This encoding enables the use of fast sequence alignment algorithms for structural comparison.
Statistical significance was evaluated using alignment scores and E-values. Structural matches with low E-values and consistent fold overlap were considered indicative of shared evolutionary origin and potential functional similarity, even in the absence of detectable sequence homology.
3.3. Surface and Pocket Analysis
For proteins lacking strong global structural homologs, surface-based functional analysis was performed using PrankWeb [8]. This tool is based on the P2Rank algorithm, which employs a random forest classifier trained on known protein–ligand complexes.
PrankWeb identifies and ranks potential ligand-binding pockets by integrating geometric descriptors, physicochemical properties, and evolutionary conservation. Predicted pockets were evaluated based on probability scores, residue composition, and conservation patterns, providing additional evidence for potential molecular function.
4. Results
Application of the proposed pipeline revealed that many proteins previously classified as dark exhibit well-defined structural cores with high prediction confidence. Structural alignment using Foldseek frequently identified remote homologs associated with known protein families, suggesting that structural conservation persists despite extensive sequence divergence.
In cases where global structural similarity was weak or ambiguous, surface analysis revealed conserved binding pockets suggestive of enzymatic or ligand-binding activity. These findings indicate that a significant fraction of the dark proteome may be functionally constrained rather than intrinsically non-functional or purely disordered.
The combined use of structure prediction, structural mining, and surface analysis enabled the formulation of coherent functional hypotheses for proteins that previously lacked any meaningful annotation.
5. Discussion
Our results highlight the transformative impact of artificial intelligence on proteome annotation. By shifting the focus from primary sequence to three-dimensional structure, it becomes possible to uncover functional relationships that are invisible to traditional bioinformatics approaches.
The proposed pipeline illustrates how generative and analytical AI tools can be integrated into a unified framework for hypothesis generation. Importantly, this strategy does not aim to replace experimental validation but rather to guide it by prioritizing targets and suggesting plausible functional roles.
As predicted structural databases continue to expand, structure-centric approaches are expected to become increasingly central to functional genomics and systems biology.
6. Conclusion
We present an artificial intelligence–driven framework for the structural and functional exploration of the human dark proteome. By integrating deep learning–based structure prediction, large-scale structural alignment, and machine learning–based surface analysis, our approach bridges the gap between genomic sequence data and biological function. This work demonstrates that AI provides a powerful and scalable strategy for illuminating previously inaccessible regions of the human proteome.
References
- Zitnik, Marinka; Li, Michelle M; Wells, Aydin; Glass, Kimberly; Morselli Gysi, Deisy; Krishnan, Arjun; Murali, T_M; Radivojac, Predrag; Roy, Sushmita; Baudot, Anaïs; et al. Current and future directions in network biology. 2024. [Google Scholar]
- Cannataro, Mario; Guzzi, Pietro Hiram; Mazza, Tommaso; Tradigo, Giuseppe; Veltri, Pierangelo. Using ontologies for preprocessing and mining spectra data on the grid. Future Generation Computer Systems 2007, 23(1), 55–60. [Google Scholar]
- Cannataro, Mario; Guzzi, Pietro H; Veltri, Pierangelo. Impreco: Distributed prediction of protein complexes. Future Generation Computer Systems 2010, 26(3), 434–440. [Google Scholar]
- Jumper, John; Evans, Richard; Pritzel, Alexander; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar]
- Mitchell, Tom M. Machine Learning; McGraw-Hill, 1997. [Google Scholar]
- Vaswani, Ashish; Shazeer, Noam; Parmar, Niki; et al. Attention is all you need. Advances in Neural Information Processing Systems 2017, 30. [Google Scholar]
- van Kempen, Michel; Kim, Stephanie; Tumescheit, Christian; et al. Fast and accurate protein structure search with foldseek. Nature Biotechnology 2023. [Google Scholar]
- Jendele, Lukas; Krivak, Radka; Skoda, Petr; et al. Prankweb: a web server for ligand binding site prediction. Nucleic Acids Research 2019, 47, W345–W349. [Google Scholar]
- Mirdita, Milot; Schütze, Konstantin; Moriwaki, Yosuke; et al. Colabfold: making protein folding accessible to all. Nature Methods 2022, 19, 679–682. [Google Scholar]
Figure 1.
Artificial intelligence–driven workflow for functional inference in the human dark proteome. Uncharacterized human protein sequences are converted into three-dimensional structures using deep learning–based prediction. Predicted models are analyzed through large-scale structural homology mining and machine learning–based surface pocket detection, integrating confidence metrics to generate functional hypotheses and prioritize experimental validation targets.
Figure 1.
Artificial intelligence–driven workflow for functional inference in the human dark proteome. Uncharacterized human protein sequences are converted into three-dimensional structures using deep learning–based prediction. Predicted models are analyzed through large-scale structural homology mining and machine learning–based surface pocket detection, integrating confidence metrics to generate functional hypotheses and prioritize experimental validation targets.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



