
Submitted:
17 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
Amino acid derivatives, such as β-keto esters and pyrrolones, were used as nucleophiles in organocatalyzed Michael additions to nitroalkene acceptors, while fatty acid derivatives acted as both nucleophiles (β-keto esters) and electrophile (nitroalkene acceptor). Bifunctional noncovalent organocatalysts were employed as asymmetric organocatalysts. Twenty compounds – including fatty acid and amino acid derivatives, as well as fatty acid–amino acid conjugates – were prepared with enantioselectivities of up to 98% ee. All novel products were fully characterized. This research demonstrates the ease of assembling readily available fatty acid and amino acid building blocks under ambient conditions.
Keywords:
fatty acids
; amino acids
; β-keto esters
; pyrrolones
; nitroalkene acceptors
; Michael addition
; asymmetric organocatalysis
; enantioselectivity
; diastereoselectivity
; tetronic acid
1. Introduction
The enantioselective construction of carbon–carbon and carbon–heteroatom bonds in modern organic synthesis is crucial, as enantiomerically pure compounds play a key role in natural products and pharmaceuticals. Asymmetric organocatalysis, the third pillar of enantioselective catalysis, is a powerful tool that enables the formation of complex products from simple building blocks in an asymmetric manner, without the use of potentially toxic metals and under mild, often non-inert reaction conditions [1,2]. Among numerous organocatalyzed transformations, the asymmetric 1,4-addition to electron-deficient alkenes stands out as a key reaction, serving as a platform for organocatalyst development, the formation of advanced, functionalized, enantiomerically pure building blocks, and target-oriented asymmetric synthesis [3,4,5,6,7,8]. Even after nearly 20 years of intensive research on asymmetric organocatalyzed 1,4-additions, the reaction remains relevant and is applied to various new nucleophile–electrophile starting building blocks [9,10,11,12,13,14,15,16,17,18,19,20].
While various amino acid derivatives, such as β-keto esters [21,22], pyrrolones [22,23,24], azlactones [25,26,27], glycine Schiff bases [28,29,30,31], α,β-unsaturated amino acids [32], α-aryl-α-isocyano acetates [33], α-amino acid-derived thiazolones [34], 1H-imidazol-4(5H)-ones [35], α-imino esters [36], and others, have been used as building blocks in asymmetric organocatalyzed transformations, only a few fatty acid building blocks have been reported in asymmetric organocatalysis. Among the few reports, the enantioselective organocatalytic synthesis of 3-hydroxy fatty acids and fatty γ-lactones is notable [37,38]. Bifunctional phosphorus-based organocatalysts have been used for the atom-economical reaction of CO2 with epoxidized oleochemicals [39], while amines and alkanolamines as organocatalysts have been used in the amidation of fatty acid methyl esters with 3-(dimethylamino)-1-propylamine [40]. Because fatty acid derivatives exhibit various biological activities, including antitumor [41] and antiproliferative activity [42], inhibition of glioma cell growth [43], activity as inhibitors against Gram-positive Staphylococcus aureus [44], and serving as biomarkers of oxidative stress [45], the demand for simple and efficient enantioselective syntheses – including organocatalytic methods – of fatty acid derivatives continues to grow. On the other hand, nitro fatty acids are an emerging class of bioactive fatty acids [46,47,48,49,50,51,52,53].
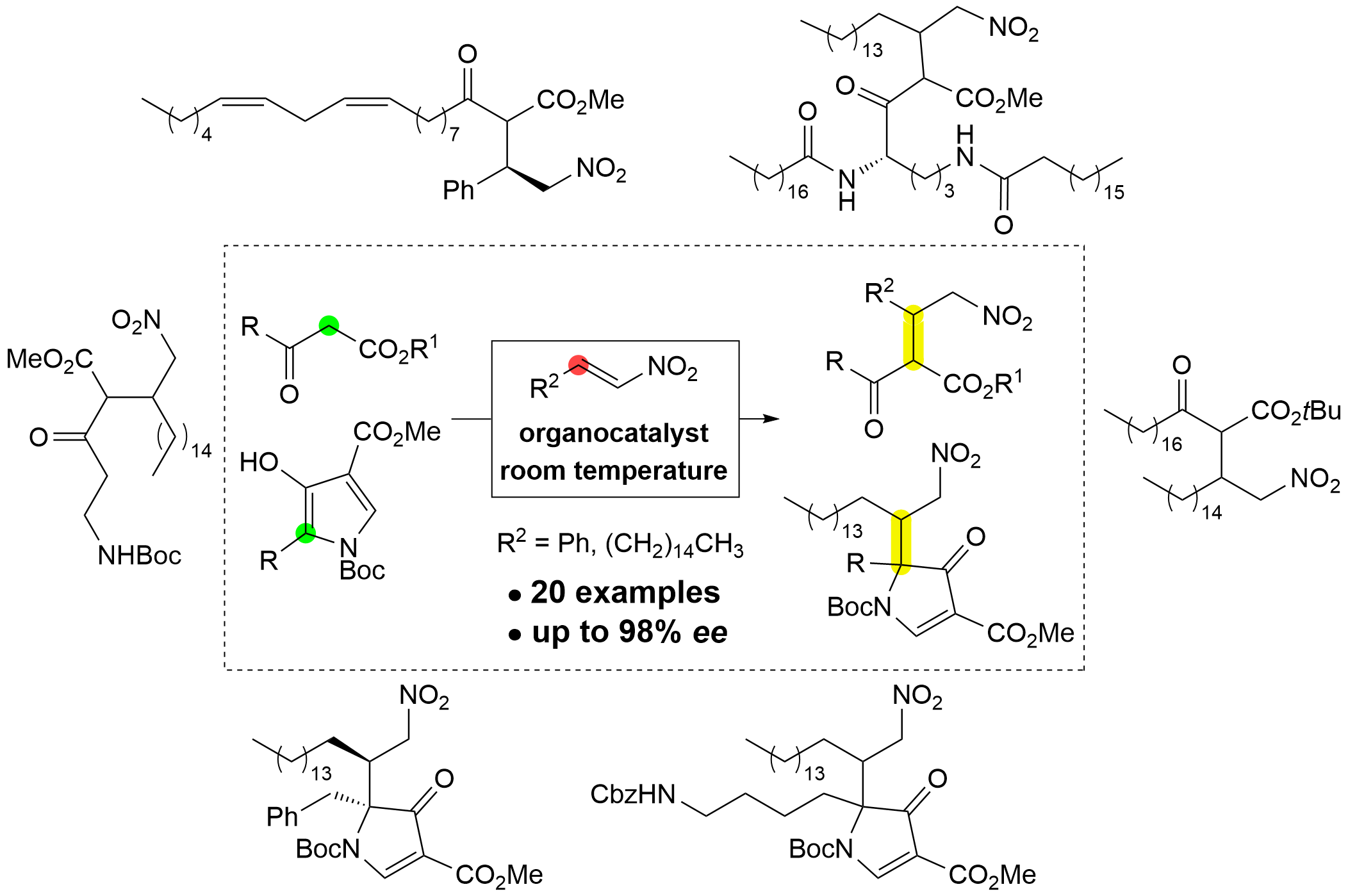
We report the synthesis of readily available β-keto ester derivatives of fatty acids and amino acids [22], as well as amino acid-derived pyrrolones used as nucleophiles in the (asymmetric) organocatalyzed Michael addition to fatty acid-derived nitroalkene and trans-β-nitrostyrene [22]. These reactions yield a small library of fatty acid and amino acid derivatives, doubly fatty acid derivatives, and amino acid–fatty acid conjugates. The results highlight the ease of forming these potentially interesting product classes, as well as the drawbacks and challenges encountered.
2. Results and Discussion
Synthesis. β-Keto ester nucleophiles were prepared by Masamune-Claisen homologation of carboxylic acids (Scheme 1) [22]. Activation of Boc-protected glycine 1a, Boc-protected β-alanine 1b, glycolic acid derivative 1c, and palmitic acid (1d), stearic acid (1e), and linoleic acid (1f) with 1,1’-carbonyldiimidazole (CDI) in anhydrous THF, followed by addition of alkyl (methyl, tert-butyl) potassium malonate in the presence of magnesium chloride, afforded the corresponding β-keto esters 2a–g in 45–91% yield. Except for compound 2f, the other β-keto esters are known in the literature and are individually referenced in the Experimental section. β-Keto esters 2h and 2i were prepared in three and five steps, respectively, from H-Orn(Boc)-OMe·HCl (3). Amidation of 3 with stearic acid (1e) gave amido ester 4, which was hydrolyzed under basic conditions to acid 5 and then homologated to β-keto ester 2h in 44% yield over three steps. For the preparation of 2i, compound 4 was Boc-deprotected using TFA to give ammonium salt 6, followed by amidation with stearic acid (1e) to yield diamido ester 7. Hydrolysis of 7 furnished acid 8, which was transformed into β-keto ester 2i in 20% yield over five steps (Scheme 1). Compounds 6–8 and 2i have low solubility in most solvents, including chloroform and dimethyl sulfoxide. Solubility in chloroform increases significantly with the addition of small amounts of trifluoroacetic acid (700 μL CHCl3 and 20 μL TFA to dissolve 10–20 mg of above mentioned compounds).
Similarly, the pyrrolone nucleophiles 11a–c were prepared from appropriately protected phenylalanine 9a, ornithine 9b, and glutamic acid 9c in a two-step procedure (Scheme 2) [22,23]. Initial homologation to the corresponding β-keto esters 10a–c was followed by treatment with N,N-dimethylformamide dimethyl acetal at elevated temperature, yielding the corresponding pyrrolones 11a–c in 57–62% yield after two steps. All attempts to prepare the corresponding pyrrolone 11d from amido β-keto ester 2h resulted in complex product mixtures (Scheme 2). In addition to commercially available trans-β-nitrostyrene (12), fatty acid-derived nitroalkene electrophile 16 was prepared in four steps from palmitic acid (1d) following procedures described in the literature [54,55,56]. Reduction of palmitic acid (1d) with LiAlH4 gave alcohol 13, which was oxidized with pyridinium chlorochromate (PCC) to aldehyde 14. The Henry reaction with nitromethane in the presence of tBuOK afforded β-nitro alcohol 15, which was dehydrated after treatment with trifluoroacetic anhydride (TFAA) and Et3N at –10°C to nitroalkene 16 in 38% yield over four steps (Scheme 2).
Next, the organocatalyzed 1,4-addition of Boc-β-alanine-derived β-keto ester 2b to trans-β-nitrostyrene (12), yielding adduct 17b, was used as a model reaction for catalyst optimization (Scheme 3) [21]. Noncovalent bifunctional organocatalysts I–IX (see ESI, Scheme S1) were tested, and achiral organocatalyst X was used to prepare the racemic product rac-17b. The best results for enantioselectivity, conversion, and reaction profile were obtained with the (+)-cinchonine-derived squaramide catalyst VIII (100% conversion, 85% yield, 95% ee for both diastereomers, (1R)-enantioselectivity), followed by the camphor-derived squaramide catalyst I, which showed the best complementary enantioselectivity (100% conversion, 92% and 93% ee for both diastereomers, (1S)-enantioselectivity). Screening of solvents (toluene, THF, EtOAc, MeCN) revealed broad compatibility of the model reaction catalyzed by VIII (ee between 94% and 96%), except for MeOH (81% and 83% ee) (Scheme 3; for details, see ESI, Scheme S2).
With optimal reaction conditions established (organocatalyst VIII, dichloromethane, 25 °C), the scope of the organocatalyzed 1,4-additions of β-keto esters 2 to trans-β-nitrostyrene (12) and fatty acid-derived nitroalkene 16 was explored. The achiral bifunctional noncovalent organocatalyst X was used efficiently to prepare all racemic adducts rac-17a–l in 37–88% yield. All doubly fatty acid-derived products rac-17g,i,k as well as products rac-17c and rac-17e, could not be separated by chiral HPLC. Only the racemic products rac-17a,b,d,f,h,j,l for which all four stereoisomers (two pairs of racemic diastereomers) were separated on a chiral HPLC column, were resynthesized using chiral noncovalent bifunctional organocatalyst VIII (Scheme 4); the corresponding chiral nonracemic products 17a,b,d,f,h,j,l were obtained in 47–89% yield, with high enantioselectivity (91–98% ee) and diastereoselectivity ranging from 50:50 to 64:36. Lastly, chiral nonracemic β-keto esters 2h and 2i were used for addition to trans-β-nitrostyrene (12) and fatty acid-derived nitroalkene 16. With the achiral organocatalyst X, addition of 2h to trans-β-nitrostyrene (12) and 16 gave the desired products 17m (71% yield) and 17n (77% yield), respectively, each as an inseparable mixture of four diastereomers in 27:21:24:28 and 26:28:25:21 ratios, respectively. Similarly, addition of 2i to 12 and 16 gave the desired products 17o (29% yield) and 17p (25% yield), respectively, each as an inseparable mixture of four diastereomers in 24:22:30:24 and 25:25:27:23 ratios, respectively. Application of chiral quinuclidine catalyst VIII for the synthesis of products 17n and 17p improved the diastereomer ratio, yielding compound 17n in 88% yield with a 45:11:8:36 diastereomer ratio and compound 17p in 23% yield with a 39:11:12:38 diastereomer ratio (Scheme 4). No attempts have been made to determine the diastereomeric or enantiomeric ratios of compounds 17m–n by HPLC. The stereochemical integrity (enantiomer ratio) of the starting β-keto esters 2h and 2i was not verified; nevertheless, this transformation highlights the challenges encountered – diastereoselectivity and separation of stereomers – in this simple, inherently diastereoselective reaction.
Next, phenylalanine- 11a, ornithine- 11b, and glutamic acid-derived pyrrolones 11c were tested as nucleophiles in the organocatalyzed addition to fatty acid nitroalkene 16 (Scheme 5). Reactions with racemic catalyst X proceeded smoothly, yielding the corresponding products rac-18a–c in 65–77% yields with high diastereoselectivity, ranging from 90:10 to 97:3. Application of the camphor-derived organocatalyst I in anhydrous toluene at room temperature, which were the optimal catalyst and conditions in our previous organocatalyzed additions of pyrrolone nucleophiles to nitroalkene acceptors [23], gave the corresponding chiral products 18a–c in 18–69% yield. For the phenylalanine-derived product 18a (69% yield), the diastereoselectivity remained unchanged at 97:3, with an enantioselectivity of 82% ee for the major diastereomer. For the ornithine-derived adduct 18b and the glutamic acid-derived adduct 18c, the diastereoselectivity with organocatalyst I not only decreased significantly but also reversed, changing from 90:10 to 26:74 for 18b and from 97:3 to 31:69 for 18c. For both diastereomers of product 11b, enantioselectivity could only be tentatively determined due to partial overlap (57% ee for the major diastereomer and 11% ee for the minor diastereomer). Similarly, for product 18c, enantioselectivity was 68% ee for the major diastereomer and 38% ee for the minor diastereomer. Finally, tetronic acid (19) was used as a nucleophile in the addition to nitroalkene 16, yielding the expected product 20 in 44% yield. All attempts to separate the stereoisomers of product 20 were unsuccessful (Scheme 5).
Structure determination. The structures of novel compounds 2f, 2h, 2i, 4–8, 16–18, and 20 were confirmed by spectroscopic methods (1H and 13C NMR, IR, and high-resolution mass spectrometry), while compound 15 was used as a crude product for further transformations; its structure was confirmed by 1H NMR. The diastereomers of compounds 17/rac-17, 18/rac-18, and 20 could not be separated by column chromatography and were characterized as mixtures of diastereomers. The diastereomeric ratios of compounds 17/rac-17 and 18/rac-18 were determined by proton spectra, in which the non-overlapping signals were integrated. Similarly, the keto-enol ratios of compounds 2, 10, 11, 17/rac-17, and 20 were determined from proton spectra. β-Keto esters 2, 10, and 17/rac-17 contain up to 17% (17% for compound 2i, see ESI) of the enol form, as indicated by the enol signal in the proton spectra (a singlet) at approximately 11.5–13.5 ppm. For details, see the ESI. In contrast, the enol form is the predominant tautomer for the pyrrolones 11a–c in DMSO-d6 (ranging from 55% for 11a′ to 63% for 11b′; see Scheme 2) [22,23]. The tetronic acid derivative 20 exists in CDCl3 solution exclusively in the enol form, as indicated by the enol-ester resonances in the 13C NMR spectra at 99.1 ppm, 177.4 ppm, and 177.9 ppm, and by the absence of a ketone signal above 200 ppm (see Scheme 5 and the ESI). The (E) configuration around the C=C bond of compound 16 was assigned based on the vicinal RHC=CHNO2 coupling constant (3J = 13.4 Hz). The structure and (1S,2S)-absolute configuration of the stereoisomer of adduct 17a (prepared with organocatalyst I) were determined by single-crystal X-ray diffraction analysis (Figure 1). The (1S)-absolute configuration is consistent with our previous findings [21]. Based on this, we assigned the (1R)-absolute configuration to the major enantiomer of both diastereomers of products 17a, 17b, 17d, 17f, 17h, 17j, and 17l prepared with organocatalyst VIII. The absolute configuration at the C-2 chiral center is labile due to rapid keto-enol tautomerization. The (1'S,5S)-absolute configuration of the major diastereomer of product 18a (see Scheme 5) was assigned based on our previous results in the series of 1,4-adducts of pyrrolones to nitroalkene acceptors [22].
4. Materials and Methods
Solvents for extractions and chromatography were of technical grade and were distilled prior to use. Extracts were dried over technical grade anhydrous Na2SO4. Melting points were determined on a Kofler micro hot stage and on SRS OptiMelt MPA100 – Automated Melting Point System (Stanford Research Systems, Sunnyvale, California, United States). The NMR spectra were obtained on a Bruker UltraShield 500 plus spektrometer and on a BRUKER AVANCE NEO 600 MHz NMR spektrometer (Bruker, Billerica, Massachusetts, United States) at 500 and 600 MHz for 1H and 126 and 150 MHz for 13C nucleus, respectively, using DMSO-d6 and CDCl3 with TMS as the internal standard, as solvents. Mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS and Agilent 6530 Q-TOF LC/MS coupled with Agilent 1260 Infinity2 HPLC (Agilent Technologies, Santa Clara, California, United States), IR spectra on a Perkin-Elmer Spectrum BX FTIR spectrophotometer (PerkinElmer, Waltham, Massachusetts, United States). Column chromatography (CC) was performed on silica gel (Silica gel 60, particle size: 0.035-0.070 mm (Sigma-Aldrich, St. Louis, Missouri, United States)). HPLC analyses were performed on an Agilent 1260 Infinity LC (Agilent Technologies, Santa Clara, California, United States) using CHIRALPAK IA-3 (0.46 cm ø × 25 cm), CHIRALPAK AD-H (0.46 cm ø × 25 cm), CHIRALCEL OD-H (0.46 cm ø × 25 cm), and CHIRALPAK AS-H (0.46 cm ø × 25 cm) as chiral column (CHIRAL TECHNOLOGIES, INC., West Chester, Pennsylvania, United States). All the commercially available chemicals used were purchased from Sigma-Aldrich (St. Louis, Missouri, United States).
Organocatalysts I [21], II [57], III [58], IV [58], VI [59], VII [60], VIII [61], and IX [62] were prepared following the literature procedures; organocatalyst V was purchased from Sigma-Aldrich.
Synthesis of β-keto esters 2 from carboxylic acids 1 – General procedure 1 (GP1)
To a solution or suspension of carboxylic acid 1 (10 mmol) in anhydrous THF (50 mL), 1,1'-carbonyldiimidazole (CDI; 12 mmol, ω = 0.97, 1.672 g) was added under argon, and the resulting reaction mixture was stirred for 2 h at room temperature. A solid mixture of MgCl2 (9.8 mmol, ω = 0.98, 952 mg) and methyl potassium malonate (15 mmol, 2.343 g) or tert-butyl potassium malonate (15 mmol, ω = 0.95, 3.130 g) was then added. The reaction mixture was stirred for a further 24 hours under argon at room temperature. The volatiles were evaporated in vacuo, the residue was dissolved in EtOAc (150 mL) and washed with NaHSO4 (1 M in H2O, 3×50 mL), NaHCO3 (aq. sat., 2×20 mL), and NaCl (aq. sat., 2×50 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles evaporated in vacuo. If necessary, the residue was purified by column chromatography (CC, Silica gel 60). The fractions containing the product were combined and the volatiles were evaporated in vacuo.
Synthesis of methyl 4-((tert-butoxycarbonyl)amino)-3-oxobutanoate (2a) [22]
Following GP1. Prepared from (tert-butoxycarbonyl)glycine (1a) (10 mmol, 1.752 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 2.10 g (9.1 mmol, 91 %) of yellowish oil. 1H-NMR (500 MHz, DMSO-d6): δ 1.39 (s, 9H), 3.60 (s, 2H), 3.63 (s, 3H), 3.86 (d, J=5.9 Hz, 2H), 7.13 (t, J=5.9 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 28.18, 45.91, 49.81, 51.93, 78.30, 155.81, 167.50, 200.67.
Synthesis of methyl 5-((tert-butoxycarbonyl)amino)-3-oxopentanoate (2b) [22]
Following GP1, prepared from Boc-β-alanine (1b) (10 mmol, 1.892 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 2.18 g (8.9 mmol, 89 %) of yellowish oil. 1H-NMR (500 MHz, DMSO-d6): δ 1.36 (s, 9H), 2.66 (t, J=6.9 Hz, 2H), 3.11 (q, J=6.9 Hz, 2H), 3.61 (s, 2H), 3.62 (s, 3H), 6.77 (t, J=5.6 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 28.24, 34.88, 42.40, 48.68, 51.84, 77.72, 155.53, 167.71, 202.42.
Synthesis of methyl 4-((3-methylbut-2-en-1-yl)oxy)-3-oxobutanoate (2c) [63]
Following GP1. Prepared from 2-((3-methylbut-2-en-1-yl)oxy)acetic acid (1c) [64] (10 mmol, 1.442 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 1.602 g (8.0 mmol, 80 %) of colorless oil. 1H-NMR (500 MHz, CDCl3): δ 1.69 (s, 3H), 1.77 (s, 3H), 3.55 (s, 2H), 3.74 (s, 3H), 4.04 (d, J=7.1 Hz, 2H), 4.09 (s, 2H), 5.29 – 5.36 (m, 1H). 13C-NMR (126 MHz, CDCl3): δ 18.11, 25.88, 45.77, 52.41, 67.89, 74.63, 119.97, 138.70, 167.62, 202.30.
Synthesis of methyl 3-oxooctadecanoate (2d) [65]
Following GP1. Prepared from palmitic acid (1d) (10 mmol, 2.564 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction and column chromatography (EtOAc/petroleum ether = 1:10). Yield: 2,717 g (8.70 mmol, 87 %) of white solid; m.p. = 49.2–50.5 °C. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.19 – 1.35 (m, 24H), 1.53 – 1.65 (m, 2H), 2.53 (t, J=7.4 Hz, 2H), 3.45 (s, 2H), 3.74 (s, 3H). 13C-NMR (126 MHz, CDCl3): δ 14.26, 22.83, 23.60, 29.14, 29.49, 29.50, 29.58, 29.73, 29.77, 29.79, 29.80, 29.82, 29.83, 32.06, 43.23, 49.15, 52.46, 167.85, 203.01.
Synthesis of methyl 3-oxoicosanoate (2e) [65]
Following GP1. Prepared from stearic acid (1e) (10 mmol, 2.845 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction and column chromatography (EtOAc/petroleum ether = 1:10). Yield: 2.282 g (6.70 mmol, 67 %) of white solid; m.p. = 49.5 –51.2 °C. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.18 – 1.34 (m, 28H), 1.55 – 1.63 (m, 2H), 2.53 (t, J=7.4 Hz, 2H), 3.45 (s, 2H), 3.74 (s, 3H). 13C-NMR (151 MHz, CDCl3): δ 14.25, 22.83, 23.60, 29.14, 29.49, 29.50, 29.58, 29.73, 29.78, 29.79, 29.80, 29.83, 32.06, 43.22, 49.14, 52.45, 167.84, 203.00 (3 signals missing due to overlapping).
Synthesis of tert-butyl 3-oxoicosanoate (2f)
Following GP1. Prepared from stearic acid (1e) (10 mmol, 2.845 g), tert-butyl potassium malonate (15 mmol, ω = 0.95, 3.130 g); isolation by extraction and column chromatography (EtOAc/petroleum ether = 1:15). Yield: 1.722 g (4.50 mmol, 45 %) of white solid; m.p. = 36.9–38.1 °C. EI-HRMS: m/z = 327.2885 (MH+-tBuOH); C20H39O3 requires: m/z = 327.2894 (MH+-tBuOH); νmax 2960, 2916, 2849, 1729, 1715, 1466, 1406, 1367, 1329, 1276, 1260, 1155, 1131, 1109, 1080, 947, 920, 842, 790, 723, 647 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.21 – 1.34 (m, 28H), 1.47 (s, 9H), 1.55 – 1.61 (m, 2H), 2.51 (t, J=7.4 Hz, 2H), 3.34 (s, 2H). 13C-NMR (151 MHz, CDCl3): δ 14.26, 22.83, 23.62, 28.10, 29.21, 29.50, 29.52, 29.59, 29.74, 29.78, 29.80, 29.81, 29.84, 32.07, 43.09, 50.81, 81.99, 166.69, 203.68 (3 signals missing due to overlapping).
Synthesis of methyl (11Z,14Z)-3-oxoicosa-11,14-dienoate (2g) [65]
Following GP1. Prepared from linoleic acid (1f) (10 mmol, 2.804 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction and column chromatography (EtOAc/petroleum ether = 1:5). Yield: 1.851 g (5.50 mmol, 55 %) of colorless oil. 1H-NMR (500 MHz, CDCl3): δ 0.89 (t, J=6.9 Hz, 3H), 1.23 – 1.40 (m, 14H), 1.54 – 1.64 (m, 2H), 1.97 – 2.09 (m, 4H), 2.53 (t, J=7.4 Hz, 2H), 2.77 (t, J=6.6 Hz, 2H), 3.45 (s, 2H), 3.74 (s, 3H), 5.28 – 5.43 (m, 4H). 13C-NMR (126 MHz, CDCl3): δ 14.21, 22.71, 23.57, 25.76, 27.31, 27.33, 29.10, 29.21, 29.39, 29.48, 29.72, 31.66, 43.20, 49.15, 52.46, 128.02, 128.19, 130.15, 130.35, 167.83, 202.95.
Synthesis of methyl (S)-4-((tert-butoxycarbonyl)amino)-3-oxo-5-phenylpentanoate (10a) [22]
Following GP1. Prepared from Boc-L-phenylalanine (9a) (10 mmol, 2.653 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 2.346 g (7.30 mmol, 73 %) of colorless oil. 1H-NMR (500 MHz, CDCl3): δ 1.40 (s, 9H), 2.98 (dd, J=7.5, 14.1 Hz, 1H), 3.14 (dd, J=6.2, 14.1 Hz, 1H), 3.46 (d, J=16.0 Hz, 1H), 3.52 (d, J=16.0 Hz, 1H), 3.71 (s, 3H), 4.56 (q, J=7.2 Hz, 1H), 4.96 – 5.07 (m, 1H), 7.14 – 7.20 (m, 2H), 7.21 – 7.35 (m, 3H).
Synthesis of methyl (S)-8-(((benzyloxy)carbonyl)amino)-4-((tert-butoxycarbonyl)amino)-3-oxooctanoate (10b) [22]
Following GP1. Prepared from Boc-Lys(Z)-OH (9b) (10 mmol, 3.804 g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 3.317 g (7.60 mmol, 76 %) of colorless oil. 1H-NMR (500 MHz, CDCl3): δ 1.30 – 1.65 (m, 5H), 1.43 (s, 9H), 1.81 – 1.93 (m, 1H), 3.12 – 3.27 (m, 2H), 3.54 (d, J=15.7 Hz, 1H), 3.59 (d, J=16.0 Hz, 1H), 3.73 (s, 3H), 4.27 – 4.36 (m, 1H), 4.93 (t, J=6.0 Hz, 1H), 5.05 – 5.17 (m, 2H), 5.27 (br d, J=7.7 Hz, 1H), 7.28 – 7.39 (m, 5H).
Synthesis of 7-benzyl 1-methyl (S)-4-((tert-butoxycarbonyl)amino)-3-oxoheptanedioate (10c) [23]
Following GP1. Prepared from Boc-Glu(OBzl)-OH (9c) (10 mmol, 3.374g), methyl potassium malonate (15 mmol, 2.343 g); isolation by extraction. Yield: 2.675 g (6.80 mmol, 68 %) of colorless oil. 1H-NMR (500 MHz, CDCl3): δ 1.43 (s, 9H), 1.80 – 1.91 (m, 1H), 2.22 – 2.32 (m, 1H), 2.38 – 2.56 (m, 2H), 3.55 – 3.65 (m, 2H), 3.73 (s, 3H), 4.38 – 4.45 (m, 1H), 5.12 (s, 2H), 5.23 (br d, J=8.1 Hz, 1H), 7.29 – 7.41 (m, 5H).
Synthesis of methyl (S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoate (4)
To a solution of stearic acid (1e) (20 mmol, 5.690 g) in anhydrous THF (50 mL) was added 1,1’-carbonyldiimidazole (CDI; 22 mmol, ω = 0.97, 3.678 g) under argon and the resulting reaction mixture was stirred for 2 h at room temperature. Then H-Orn(Boc)-OMe×HCl (3) (22 mmol, ω = 0.96, 6.480 g) and Et3N (22 mmol, 3.07 mL) were added. The reaction mixture was stirred for a further 24 hours under argon at room temperature. The volatiles were evaporated in vacuo, the residue was dissolved in CH2Cl2 (150 mL) and washed with NaHSO4 (1 M in H2O, 4×50 mL), NaHCO3 (aq. sat., 3×20 mL) and NaCl (aq. sat., 2×50 mL). The organic phase was dried over anhydrous Na2SO4, filtered and the volatiles evaporated in vacuo. Yield: 7.076 g (13.8 mmol, 69 %) of white solid; m.p. = 86.0–88.0 °C. EI-HRMS: m/z = 513.4272 (MH+); C29H57N2O5 requires: m/z = 513.4262 (MH+); νmax 3345, 2915, 2847, 1762, 1684, 1648, 1526, 1473, 1462, 1369, 1285, 1252, 1212, 1171, 1143, 1047, 994, 950, 871, 754, 729, 719, 654 cm-1. 1H-NMR (500 MHz, DMSO-d6): δ 0.85 (t, J=6.8 Hz, 3H), 1.14 – 1.31 (m, 30H), 1.37 (s, 9H), 1.42 – 1.57 (m, 3H), 1.61 – 1.69 (m, 1H), 2.09 (t, J=7.1 Hz, 2H), 2.89 (q, J=6.8 Hz, 2H), 3.60 (s, 3H), 4.14 – 4.23 (m, 1H), 6.79 (t, J=5.7 Hz, 1H), 8.14 (d, J=7.5 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 13.98, 22.11, 25.23, 26.02, 28.20, 28.26, 28.55, 28.72, 28.78, 28.97, 29.02, 29.05, 31.31, 34.94, 51.65, 51.70, 77.40, 155.58, 172.43, 172.78 (7 signals missing due to overlapping).
Synthesis of (S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoic acid (5)
To a solution/suspension of methyl (S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoate (4) (1.70 mmol, 872 mg) in a mixture of H2O (3.0 mL) and THF (3.0 mL) was added NaOH (15.0 mmol, 600 mg). The reaction mixture was stirred for 3 hours at room temperature. The mixture was acidified with HCl (aq. 1 M) to pH < 3 and extracted with EtOAc (3×30 mL). The combined organic layers were washed with brine (1×10 mL), dried over Na2SO4, filtered and the volatiles evaporated in vacuo. The residue was azeotropically evaporated with CHCl3 (3×30 mL) to give the anhydrous product 5. Yield: 763 mg (1.53 mmol, 90 %) of white solid; m.p. = 82.0–84.3 °C. EI-HRMS: m/z = 499.4113 (MH+); C28H55N2O5 requires: m/z = 499.4105 (MH+); νmax 3359, 2955, 2916, 2849, 1738, 1682, 1605, 1525, 1465, 1454, 1388, 1365, 1290, 1274, 1244, 1210, 1170, 1112, 1043, 1019, 957, 890, 860, 783, 727, 637 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.17 – 1.36 (m, 28H), 1.44 (s, 9H), 1.52 – 1.67 (m, 4H), 1.68 – 1.79 (m, 1H), 1.87 – 1.98 (m, 1H), 2.25 (t, J=7.8 Hz, 2H), 3.04 – 3.27 (m, 2H), 4.60 (td, J=4.9, 7.5 Hz, 1H), 4.86 (t, J=6.4 Hz, 1H), 6.73 (br d, J=7.4 Hz, 1H), 9.64 (br s, 1H). 13C-NMR (126 MHz, CDCl3): δ 14.27, 22.84, 25.79, 26.56, 28.52, 29.06, 29.42, 29.49, 29.51, 29.67, 29.80, 29.83, 29.86, 32.07, 36.57, 39.88, 52.22, 79.99, 156.91, 174.55, 174.80 (5 signals missing due to overlapping). 1H-NMR (500 MHz, DMSO-d6): δ 0.85 (t, J=6.9 Hz, 3H), 1.06 – 1.31 (m, 29H), 1.37 (s, 9H), 1.32 – 1.56 (m, 4H), 1.61 – 1.71 (m, 1H), 2.04 – 2.15 (m, 2H), 2.84 – 2.94 (m, 2H), 4.13 (td, J=5.0, 8.5 Hz, 1H), 6.77 (t, J=5.8 Hz, 1H), 7.98 (d, J=7.8 Hz, 1H), 12.40 (br s, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 13.91, 22.06, 25.24, 26.15, 28.23, 28.43, 28.57, 28.66, 28.77, 28.93, 28.97, 29.00, 31.26, 35.02, 51.54, 77.33, 155.54, 172.23, 173.71 (7 signals missing due to overlapping).
Synthesis of methyl (S)-7-((tert-butoxycarbonyl)amino)-3-oxo-4-stearamidoheptanoate (2h)
Following GP1, prepared from (S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoic acid (5) (1.5 mmol, 748 mg), CDI (1.8 mmol, ω = 0.97, 301 mg), MgCl2 (1.47 mmol, ω = 0.98, 143 mg), methyl potassium malonate (2.25 mmol, 351 mg); isolation by extraction and column chromatography (EtOAc/petroleum ether = 1:1). Yield: 591 mg (1.065 mmol, 71 %) of white solid; m.p. = 62.3–64.9 °C. EI-HRMS: m/z = 555.4384 (MH+); C31H59N2O6 requires: m/z = 555.4368 (MH+); νmax 3341, 2915, 2848, 1747, 1711, 1681, 1638, 1524, 1438, 1390, 1365, 1316, 1251, 1168, 1016, 886, 769, 719, 643 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.18 – 1.35 (m, 28H), 1.44 (s, 9H), 1.47 – 1.68 (m, 5H), 1.90 – 2.00 (m, 1H), 2.21 – 2.26 (m, 2H), 3.15 (q, J=6.7 Hz, 2H), 3.58 (s, 2H), 3.74 (s, 3H), 4.66 (br s, 1H), 4.68 – 4.74 (m, 1H), 6.43 (br d, J=7.4 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 14.27, 22.83, 25.73, 26.44, 27.58, 28.52, 29.44, 29.47, 29.50, 29.63, 29.77, 29.80, 29.84, 32.06, 36.62, 39.87, 46.26, 52.66, 58.11, 79.56, 156.40, 167.45, 173.54, 201.86 (6 signals missing due to overlapping). 1H-NMR (500 MHz, DMSO-d6): δ 0.85 (t, J=6.9 Hz, 3H), 1.08 – 1.30 (m, 29H), 1.37 (s, 9H), 1.31 – 1.54 (m, 4H), 1.62 – 1.75 (m, 1H), 2.12 (t, J=7.4 Hz, 2H), 2.89 (q, J=6.2 Hz, 2H), 3.58 (s, 2H), 3.61 (s, 3H), 4.23 (ddd, J=4.5, 7.3, 9.5 Hz, 1H), 6.78 (t, J=5.9 Hz, 1H), 8.16 (d, J=7.4 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 13.90, 22.05, 25.10, 25.88, 26.25, 28.22, 28.57, 28.65, 28.71, 28.89, 28.96, 28.99, 31.25, 34.91, 45.49, 51.76, 57.75, 77.36, 155.57, 167.46, 172.67, 202.73 (7 signals missing due to overlapping).
Synthesis of (S)-5-methoxy-5-oxo-4-stearamidopentan-1-aminium 2,2,2-trifluoroacetate (6)
Methyl (S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoate (4) (10 mmol, 5.128 g) was dissolved in a 1:1 mixture of CF3COOH and anhydrous CH2Cl2 (60 mL) under argon, and the reaction mixture was stirred for 3 hours at room temperature. Volatile components were evaporated in vacuo, and the residue was azeotropically evaporated with anhydrous toluene (3×100 mL) to give ammonium salt 6. Yield: 5.00 g (9.50 mmol, 95%) of white solid; m.p. = 93.0–95.7 °C. EI-HRMS: m/z = 413.3726 (MH+); C24H49N2O3+ requires: m/z = 413.3738 (MH+); νmax 3318, 2915, 2848, 1752, 1671, 1645, 1528, 1474, 1462, 1430, 1400, 1381, 1358, 1276, 1237, 1207, 1173, 1127, 1067, 1003, 970, 955, 893, 839, 800, 768, 747, 723, 668, 613 cm-1. 1H-NMR (500 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 0.88 (t, J=6.9 Hz, 3H), 1.09 – 1.35 (m, 26H), 1.53 – 1.63 (m, 2H), 1.69 – 1.85 (m, 3H), 1.91 – 2.01 (m, 1H), 2.23 – 2.33 (m, 2H), 3.01 – 3.20 (m, 2H), 3.76 (s, 3H), 4.48 – 4.57 (m, 1H), 6.86 (d, J=7.5 Hz, 1H), 7.61 (br s, 3H). 13C-NMR (126 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 14.25, 22.84, 23.37, 25.78, 29.22, 29.28, 29.31, 29.51, 29.59, 29.74, 29.80, 29.81, 29.86, 32.07, 36.22, 39.62, 51.65, 53.08, 115.46 (q, J=287.8 Hz), 161.08 (q, J=39.3 Hz), 172.10, 176.30 (3 signals missing due to overlapping).
Synthesis of methyl (S)-2,5-distearamidopentanoate (7)
To a solution of stearic acid (1e) (8.5 mmol, 2.416 g) in anhydrous THF (30 mL), 1,1’-carbonyldiimidazole (CDI; 9.35 mmol, ω = 0.97, 1.563 g) was added under argon, and the resulting reaction mixture was stirred for 2 h at room temperature. The resulting activated acid was transferred to a suspension of (S)-5-methoxy-5-oxo-4-stearamidopentan-1-aminium 2,2,2-trifluoroacetate (6) (9.35 mmol, 4.924 g) in anhydrous THF (30 mL) under argon. Et3N (9.35 mmol, 1.303 mL) was then added to the reaction mixture at room temperature. The reaction mixture was stirred at 40 °C for 12 hours. Volatile components were evaporated in vacuo. The residue was extracted with CH2Cl2 (100 mL), EtOAc (100 mL), Et2O (100 mL), and n-hexane (100 mL) using a laboratory ultrasonic bath (5 minutes each), followed by decanting, respectively, to remove unreacted starting material and small portions of the product. H2O (150 mL) was added to the residue, followed by ultrasonic bath treatment (15 minutes). The resulting precipitate was collected by filtration and thoroughly washed with H2O (3×70 mL). The residue was dried under high vacuum at 40 °C for 12 hours to give product 7. Yield: 3.476 g (5.270 mmol, 62 %) of white solid; m.p. = 106.7–108.4 °C. EI-HRMS: m/z = 679.6336 (MH+); C42H83N2O4 requires: m/z = 679.6347 (MH+); νmax 3305, 2914, 2848, 1742, 1639, 1542, 1470, 1420, 1385, 1277, 1259, 1239, 1205, 1173, 980, 717 cm-1. 1H-NMR (500 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 0.88 (t, J=6.9 Hz, 6H), 1.18 – 1.36 (m, 56H), 1.54 – 1.77 (m, 7H), 1.87 – 1.97 (m, 1H), 2.26 – 2.38 (m, 4H), 3.23 – 3.34 (m, 1H), 3.34 – 3.47 (m, 1H), 3.79 (s, 3H), 4.57 – 4.64 (m, 1H), 6.70 (t, J=6.1 Hz, 1H), 6.79 (d, J=7.7 Hz, 1H). 13C-NMR (126 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 14.25, 22.84, 24.97, 25.87, 25.99, 29.20, 29.23, 29.28, 29.51, 29.56, 29.72, 29.78, 29.81, 29.83, 29.85, 30.00, 32.07, 36.28, 36.35, 39.58, 52.17, 53.15, 172.43, 176.50, 177.11 (17 signals missing due to overlapping).
Synthesis of (S)-2,5-distearamidopentanoic acid (8)
To a suspension of methyl (S)-2,5-distearamidopentanoate (7) (5 mmol, 3.393 g) in a mixture of H2O (20 mL) and THF (5 mL), KOH (powder for synthesis, 50 mmol, 2.810 g) was added, and the reaction mixture was stirred at 90 °C for 12 hours. The mixture was cooled to room temperature, and HCl (aq., 2 M) was added under stirring until the pH reached 1–2. The precipitate was collected by filtration and thoroughly washed with H2O (3×100 mL). The residue was dried under high vacuum at 40 °C for 12 hours to give acid 8. Yield: 2.792 g (4.20 mmol, 84 %) of white solid; m.p. = 108.8–110.1 °C. EI-HRMS: m/z = 665.6191 (MH+); C41H81N2O4 requires: m/z = 665.6191 (MH+); νmax 3310, 2955, 2916, 2849, 1736, 1639, 1586, 1545, 1466, 1446, 1418, 1372, 1275, 1245, 1211, 1181, 1128, 970, 829, 720, 685, 633 cm-1. 1H-NMR (500 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 0.88 (t, J=6.8 Hz, 6H), 1.09 – 1.40 (m, 56H), 1.52 – 1.73 (m, 6H), 1.75 – 1.89 (m, 1H), 1.92 – 2.11 (m, 1H), 2.27 – 2.45 (m, 4H), 3.13 – 3.49 (m, 2H), 4.61 (q, J=6.7, 1H), 6.87 (s, 1H), 6.98 (d, J=7.3 Hz, 1H), 11.12 (br s, 1H). 13C-NMR (126 MHz, CDCl3 (700 μL) + TFA (20 μL)): δ 14.26, 22.84, 24.97, 25.88, 25.96, 29.14, 29.19, 29.21, 29.25, 29.27, 29.52, 29.55, 29.57, 29.73, 29.79, 29.82, 29.84, 29.86, 32.08, 36.09, 36.13, 39.82, 52.36, 175.92, 177.30, 177.72 (15 signals missing due to overlapping).
Synthesis of methyl (S)-3-oxo-4,7-distearamidoheptanoate (2i)
To a suspension of (S)-2,5-distearamidopentanoic acid (8) (2.5 mmol, 1.662 g) in anhydrous THF (25 mL), 1,1'-carbonyldiimidazole (CDI; 5 mmol, ω = 0.97, 836 mg) was added under argon, and the reaction mixture was stirred for 15 minutes at room temperature, 30 minutes at 90 °C, and 60 minutes at 50 °C. Then, a solid mixture of MgCl2 (2.5 mmol, ω = 0.98, 243 mg) and methyl potassium malonate (7.5 mmol, 1.171 g) was carefully added at 50 °C. The reaction mixture was stirred for 15 minutes at 50 °C, 30 minutes at 90 °C, and 24 hours at room temperature. The volatiles were evaporated in vacuo, and NaHSO4 (1 M in H2O, 100 mL) was added to the residue. The mixture was stirred at room temperature for 30 minutes. The precipitate was collected by filtration and thoroughly washed with H2O (3×100 mL). The residue was dried under high vacuum at 40 °C for 12 hours to give β-keto ester 2i. Yield: 1.099 g (1.525 mmol, 61 %) of white solid; m.p. = 92.1–94.7 °C. EI-HRMS: m/z = 721.6456 (MH+); C44H85N2O5 requires: m/z = 721.6453 (MH+); νmax 3306, 2916, 2849, 1748, 1717, 1638, 1539, 1463, 1378, 1328, 1258, 1239, 1223, 1204, 1147, 1013, 719 cm-1. 1H-NMR (600 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 6H), 1.18 – 1.39 (m, 56H), 1.51 – 1.67 (m, 7H), 1.88 – 1.97 (m, 1H), 2.17 (t, J=7.7 Hz, 2H), 2.25 (t, J=7.7 Hz, 2H), 3.22 – 3.38 (m, 2H), 3.58 (d, J=1.7 Hz, 2H), 3.74 (s, 3H), 4.65 – 4.72 (m, 1H), 5.87 (t, J=6.0 Hz, 1H), 6.67 (d, J=7.5 Hz, 1H). 13C-NMR (151 MHz, CDCl3): δ 14.26, 22.83, 25.72, 25.94, 25.98, 27.70, 29.45, 29.50, 29.52, 29.66, 29.78, 29.80, 29.81, 29.85, 32.07, 36.56, 36.96, 38.86, 46.22, 52.66, 58.21, 167.58, 173.81, 173.95, 201.90 (19 signals missing due to overlapping).
Synthesis of pyrrolones 11 from β-keto esters 10 – General procedure 2 (GP2)
To a solution of β-keto ester 10 (1.0 mmol) in anhydrous toluene (5 mL), DMFDMA (3 mmol, ω = 0.94, 424 μL) was added under argon and the resulting reaction mixture was stirred at 70 °C under argon until completion of the reaction, as judged by TLC analysis (1–3 hours). The volatiles were evaporated in vacuo and the residue was purified as quickly as possible by column chromatography (CC, Silica gel 60). The fractions containing the product 11 were combined and the volatiles were evaporated in vacuo. The product was immediately used for the following transformation or/and stored under argon at –20 °C.
Synthesis of 1-(tert-butyl) 3-methyl 5-benzyl-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11a) and 1-(tert-butyl) 3-methyl 5-benzyl-4-hydroxy-1H-pyrrole-1,3-dicarboxylate (11a’) [22]
Following GP2. Prepared from methyl (S)-4-((tert-butoxycarbonyl)amino)-3-oxo-5-phenylpentanoate (10a) (1 mmol, 321.4 mg), 45 minutes; CC (EtOAc/petroleum ether = 1:1). 11a/11a’ = 45:55 (in DMSO-d6). Yield: 268 mg (0.81 mmol, 81 %) of colorless oil. 1H-NMR (500 MHz, DMSO-d6) for 11a: δ 1.55 (s, 9H), 3.22 (dd, J=2.7, 13.8 Hz, 1H), 3.38 (dd, J=6.4, 13.8 Hz, 1H), 3.61 (s, 3H), 4.58 (dd, J=2.6, 6.3 Hz, 1H), 6.91 – 6.97 (m, 2H), 8.71 (s, 1H). 1H-NMR (500 MHz, DMSO-d6) for 11a': δ 1.34 (s, 9H), 3.76 (s, 3H), 4.14 (s, 2H), 6.98 – 7.04 (m, 2H), 7.11 – 7.28 (m, 3H), 7.57 (s, 1H), 8.26 (s, 1H).
Synthesis of 1-(tert-butyl) 3-methyl 5-(4-(((benzyloxy)carbonyl)amino)butyl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11b) and 1-(tert-butyl) 3-methyl 5-(4-(((benzyloxy)carbonyl)amino)butyl)-4-hydroxy-1H-pyrrole-1,3-dicarboxylate (11b’) [22]
Following GP2. Prepared from methyl (S)-8-(((benzyloxy)carbonyl)amino)-4-((tert-butoxycarbonyl)amino)-3-oxooctanoate (10b) (1 mmol, 436.5 mg), 1 hour; CC (EtOAc/petroleum ether = 1:1). 11b/11b’ = 37:63 (in DMSO-d6). Yield: 336 mg (0.82 mmol, 82 %) of colorless oil. 1H-NMR (500 MHz, DMSO-d6) for 11b: δ 0.98 – 1.08 (m, 1H), 1.08 – 1.19 (m, 1H), 1.50 (s, 9H), 1.83 – 1.93 (m, 1H), 1.97 – 2.08 (m, 1H), 2.93 (q, J=6.7 Hz, 2H), 3.68 (s, 3H), 4.30 (dd, J=3.1, 6.5 Hz, 1H), 8.98 (s, 1H). 1H-NMR (500 MHz, DMSO-d6) for 11b’: δ 1.28 – 1.46 (m, 4H), 1.54 (s, 9H), 2.70 (t, J=6.9 Hz, 2H), 2.98 (q, J=6.3 Hz, 2H), 3.73 (s, 3H), 4.99 (s, 2H), 7.23 (t, J=5.8 Hz, 1H), 7.26 – 7.41 (m, 5H), 7.49 (s, 1H), 7.95 (s, 1H).
Synthesis of 1-(tert-butyl) 3-methyl 5-(3-(benzyloxy)-3-oxopropyl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11c) and 1-(tert-butyl) 3-methyl 5-(3-(benzyloxy)-3-oxopropyl)-4-hydroxy-1H-pyrrole-1,3-dicarboxylate (11c’) [23]
Following GP2. Prepared from 7-benzyl 1-methyl (S)-4-((tert-butoxycarbonyl)amino)-3-oxoheptanedioate (10c) (1 mmol, 393.4 mg), 45 minutes; CC (EtOAc/petroleum ether = 1:1). 11c/11c’ = 42:58 (in DMSO-d6). Yield: 343 mg (0.85 mmol, 85 %) of colorless oil. 1H-NMR (500 MHz, DMSO-d6) for 11c: δ 1.50 (s, 9H), 3.68 (s, 3H), 4.35 – 4.40 (m, 1H), 5.06 (d, J=5.4 Hz, 2H), 8.92 (s, 1H). 1H-NMR (500 MHz, DMSO-d6) for 11c’: δ 1.53 (s, 9H), 2.51 – 2.57 (m, 2H), 2.99 – 3.05 (m, 2H), 3.73 (s, 3H), 5.08 (s, 2H), 7.30 – 7.40 (m, 5H), 7.49 (s, 1H), 8.11 (s, 1H).
Synthesis of hexadecan-1-ol (13) [54]
Palmitic acid (11d) (20 mmol, 5.128 g) was dissolved in anhydrous THF (80 mL) under argon and the solution was cooled in an ice bath (0 ºC). While stirring in the ice bath, LiAlH4 (2.4 M in THF, 80 mmol, 33.3 mL) was added and the reaction mixture was allowed to warm to room temperature over 1 hour. The reaction mixture was stirred for a further 24 hours under argon at room temperature, and then quenched by careful addition of NaOH (1 M in H2O, 60 mL). The reaction mixture was extracted with diethyl ether (2×70 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatile components evaporated in vacuo. Yield: 3.957 g (16.3 mmol, 81 %) of a white solid. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.19 – 1.39 (m, 27H), 1.52 – 1.61 (m, 2H), 3.64 (t, J=6.6 Hz, 2H).
Synthesis of palmitaldehyde (14) [55]
To a solution of hexadecan-1-ol (13) (16.3 mmol, 3.951 g) in anhydrous CH2Cl2 (100 mL), pyridinium chlorochromate (PCC, 24.5 mmol, ω = 0.98, 5.389 g) was added at room temperature and the reaction mixture was stirred for 16 h at room temperature. The solution was filtered through a plaque of Celite®, washed with CH2Cl2 and the volatiles evaporated in vacuo. The residue was purified by column chromatography (Silica gel 60, petroleum ether/ethyl acetate = 10:1). The fractions containing the pure product 14 were combined and the volatile components were evaporated in vacuo. Yield: 2.940 g (12.23 mmol, 75 %) of a colorless oil. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.26 (s, 24H), 1.58 – 1.67 (m, 2H), 2.42 (td, J=1.9, 7.4 Hz, 2H), 9.76 (t, J=1.9 Hz, 1H).
Synthesis of 1-nitroheptadecan-2-ol (15)
Prepared according to the literature procedure [56]. Palmitaldehyde (14) (11.44 mmol, 2.751 g) was dissolved in a mixture of anhydrous THF and anhydrous t-butanol in a 1:1 ratio (50 mL) under argon. Nitromethane (17.16 mmol, 930 μL) was then added at room temperature. The mixture was cooled to 0 °C, tBuOK (1.144 mmol, 128 mg) was added, and the reaction mixture was allowed to warm to room temperature over 1 hour. After 16 h at room temperature under argon, the reaction mixture was diluted with H2O (300 mL) and the product was extracted with diethyl ether (2×100 mL). The organic phase was washed with NaCl (aq. sat., 2×50 mL), dried over anhydrous Na2SO4, filtered, and the volatiles evaporated in vacuo. The crude product 15 was used for the following transformation without further purification. Yield: 2.794 g (9.267 mmol, 81 %) of a yellowish oil. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.18 – 1.42 (m, 26H), 1.44 – 1.57 (m, 2H), 2.61 (br s, 1H), 4.28 – 4.34 (m, 1H), 4.38 (dd, J=8.5, 13.0 Hz, 1H), 4.43 (dd, J=2.7, 13.0 Hz, 1H).
Synthesis of (E)-1-nitroheptadec-1-ene (16) [66]
Prepared according to the literature procedure [56]. 1-Nitroheptadecan-2-ol (15) (9.12 mmol, 2.75 g) was dissolved in anhydrous CH2Cl2 (25 mL) and cooled to –10 °C. With stirring, trifluoroacetic anhydride (TFAA, 9.12 mmol, 1.269 mL) was added dropwise and the cooled mixture (–10 °C) was stirred for another 2 minutes. Over the next 10 minutes, triethylamine (2.532 mL, 18.24 mmol) was added dropwise and the reaction mixture was stirred at –10 °C for another 30 minutes. The reaction mixture was then diluted with CH2Cl2 (100 mL) and washed with NaHSO4 (aq., 1 M, 200 mL). The aqueous phase was extracted with CH2Cl2 (2×40 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles evaporated in vacuo. The residue was purified by column chromatography (Silica gel 60; petroleum ether/EtOAc = 40:1). The fractions containing the pure product 16 were combined and the volatile components were evaporated in vacuo. Product 16 was stored under argon at 5 °C. Yield: 2.016 g (7.114 mmol, 78 %) of a white solid; m.p. = 25.0–25.7 °C. EI-HRMS: m/z = 306.2395 (MNa+); C17H34NNaO2 requires: m/z = 306.2404 (MNa+); νmax 2922, 2853, 1650, 1526, 1465, 1350, 960, 835, 723 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.19 – 1.37 (m, 24H), 1.51 (p, J=7.3 Hz, 2H), 2.27 (qd, J=1.5, 7.4 Hz, 2H), 6.98 (dt, J=1.6, 13.4 Hz, 1H), 7.23 – 7.33 (m, 1H). 13C-NMR (126 MHz, CDCl3): δ 14.27, 22.84, 27.86, 28.61, 29.24, 29.40, 29.51, 29.59, 29.72, 29.77, 29.80, 29.82, 29.84, 32.07, 139.69, 143.01 (1 signal missing due to overlapping).
Organocatalyzed Michael addition of β-keto esters 2 to nitroalkenes – General procedure for the preparation of racemic products rac-17 – General procedure 3 (GP3)
To a solution/suspension of nitroalkene 12 or 16 (0.2 mmol, 1.0 equivalent or 0.3 mmol, 1.5 equivalents) and the achiral organocatalyst X (0.04 mmol, 0.2 equivalents, 16.4 mg) in anhydrous CH2Cl2 (1 mL) under argon at room temperature, β-keto ester 2 (0.3 mmol, 1.5 equivalents or 0.2 mmol, 1.0 equivalent) was added and the resulting reaction mixture was stirred at room temperature for 24–72 hours. The volatiles were evaporated in vacuo and the residue was purified by column chromatography (Silica gel 60, mobile phase). The fractions containing the pure racemic product rac-17 were combined and the volatiles were evaporated in vacuo. The product rac-17 was fully characterized and analyzed by HPLC.
Organocatalyzed Michael addition of β-keto esters 2 to nitroalkenes – General procedure for the organocatalyzed asymmetric addition – General procedure 4 (GP4)
To a solution/suspension of nitroalkene 12 or 16 (0.2 mmol, 1.0 equivalent or 0.3 mmol, 1.5 equivalents) and the chiral organocatalyst VIII (0.02 mmol, 0.1 equivalents, 12.3 mg) or I (0.02 mmol, 0.1 equivalents, 10.9 mg) in anhydrous CH2Cl2 (1 mL) under argon at room temperature, β-keto ester 2 (0.3 mmol, 1.5 equivalents or 0.2 mmol, 1.0 equivalent) was added and the resulting reaction mixture was stirred at room temperature for 24–72 hours. The volatiles were evaporated in vacuo and the residue was purified by column chromatography (Silica gel 60, mobile phase). The fractions containing the pure chiral nonracemic product 17 were combined the volatiles were evaporated in vacuo. The product 17 was fully characterized and analyzed by HPLC.
Synthesis of methyl 4-((tert-butoxycarbonyl)amino)-2-((R)-2-nitro-1-phenylethyl)-3-oxobutanoate (17a)
Following GP3 and GP4. Prepared from methyl 4-((tert-butoxycarbonyl)amino)-3-oxobutanoate (2a) (0.2 mmol, 46.3 mg) and trans-β-nitrostyrene (12) (0.3 mmol, 44.7 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:4). rac-17a Yield: 41.8 mg (0.110 mmol, 55%, two diastereomers in a ratio of 59:41 in DMSO-d6) of white solid. 17a Yield: 63.9 mg (0.168 mmol, 84%, two diastereomers in a ratio of 56:44 in DMSO-d6) of white solid; m.p. = 116–122 °C. EI-HRMS: m/z = 381.1641 (MH+); C18H25N2O7 requires: m/z = 381.1656 (MH+); νmax 3378, 2981, 1751, 1714, 1555, 1497, 1455, 1429, 1367, 1252, 1154, 1020, 974, 895, 858, 764, 699, 637 cm-1. 1H-NMR (500 MHz, DMSO-d6) for major diastereomer: δ 1.39 (s, 9H), 3.37 (s, 3H), 3.95 (dd, J=3.5, 5.8 Hz, 2H), 4.06 – 4.13 (m, 1H), 4.40 (d, J=10.7 Hz, 1H), 4.87 – 4.96 (m, 2H), 7.18 (t, J=5.9 Hz, 1H), 7.22 – 7.34 (m, 5H). 1H-NMR (500 MHz, DMSO-d6) for minor diastereomer: δ 1.33 (s, 9H), 3.70 (s, 3H), 3.50 (dd, J=5.9, 18.8 Hz, 1H), 3.80 (dd, J=5.8, 18.8 Hz, 1H), 4.46 (d, J=10.2 Hz, 1H), 4.75 (dd, J=4.3, 13.2 Hz, 1H), 4.85 (d, J=10.8 Hz, 1H), 7.06 (t, J=5.8 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) for both diastereomers: δ 28.10, 28.14, 42.35, 42.65, 50.18, 50.25, 52.45, 52.84, 57.28, 57.93, 59.78, 77.74, 78.22, 78.24, 78.44, 127.79, 127.85, 128.18, 128.25, 128.49, 128.64, 136.85, 136.92, 155.47, 155.81, 167.33, 170.36, 199.88, 200.62 (3 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/i-PrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm, T = 20°C. Diastereomer 1: tR = 9.24 minutes (minor); 17.62 minutes (major) – 91% ee. Diastereomer 2: tR = 14.12 minutes (major); 23.56 minutes (minor) – 93% ee.
Synthesis of methyl 5-((tert-butoxycarbonyl)amino)-2-((R)-2-nitro-1-phenylethyl)-3-oxopentanoate (17b) [22]
Following GP3 and GP4. Prepared from methyl 5-((tert-butoxycarbonyl)amino)-3-oxopentanoate (2b) (0.2 mmol, 49.1 mg) and trans-β-nitrostyrene (12) (0.3 mmol, 44.7 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:4). rac-17b Yield: 69.4 mg (0.176 mmol, 88%, two diastereomers in a ratio of 53:47 in DMSO-d6) of white solid. 17b Yield: 67.1 mg (0.170 mmol, 85%, two diastereomers in a ratio of 53:47 in DMSO-d6) of white solid; m.p. = 96.1–98.4 °C. EI-HRMS: m/z = 417.1618 (MNa+); C19H27N2NaO7 requires: m/z = 417.1632 (MNa+); νmax 3424, 2978, 1743, 1707, 1553, 1506, 1455, 1434, 1366, 1246, 1164, 1082, 966, 859, 756, 701 cm-1. 1H-NMR (500 MHz, DMSO-d6) for both diastereomers: δ 1.34 (s, 4.5H), 1.37 (s, 4.5H), 2.24 – 2.34 (m, 0.5H), 2.58 – 2.68 (m, 0.5H), 2.74 (t, J=6.8 Hz, 1H), 2.76 – 2.91 (m, 1H), 3.15 (q, J=6.4 Hz, 1H), 3.35 (s, 1.5H), 3.70 (s, 1.5H), 4.00 – 4.09 (m, 1H), 4.37 (dd, J=6.0, 10.5 Hz, 1H), 4.81 (d, J=7.5 Hz, 1H), 4.87 – 4.99 (m, 1H), 6.58 (t, J=5.7 Hz, 0.5H), 6.85 (t, J=5.7 Hz, 0.5H), 7.21 – 7.35 (m, 5H). 13C-NMR (126 MHz, DMSO-d6) for both diastereomers: δ 28.19, 28.22, 34.52, 34.79, 42.30, 42.36, 42.66, 42.89, 52.43, 52.83, 60.00, 60.84, 77.69, 77.78, 78.00, 78.15, 127.79, 127.86, 128.27, 128.46, 128.63, 136.80, 136.96, 155.33, 155.53, 166.88, 167.72, 201.83 (6 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/i-PrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm, T = 20°C. Diastereomer 1: tR = 7.22 minutes (minor); 8.98 minutes (major) – 95% ee. Diastereomer 2: tR = 12.53 minutes (minor); 20.94 minutes (major) – 95% ee.
Synthesis of methyl 2-(3-((tert-butoxycarbonyl)amino)propanoyl)-3-(nitromethyl)octadecanoate (rac-17c)
Following GP3. Prepared from methyl 5-((tert-butoxycarbonyl)amino)-3-oxopentanoate (2b) (0.3 mmol, 73.6 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst X, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:5). rac-17c Yield: 55.0 mg (0.104 mmol, 52%, two diastereomers in a ratio of 53:47 in CDCl3) of colorless oil. EI-HRMS: m/z = 429.3313 (MH+-Boc); C23H45N2O5 requires: m/z = 429.3323 (MH+-Boc); νmax 3413, 2923, 2853, 1743, 1712, 1552, 1505, 1436, 1366, 1248, 1168, 1084, 966, 911, 863, 781, 733 cm-1. 1H-NMR (600 MHz, CDCl3) for the major diastereomer: δ 0.88 (t, J=6.9 Hz, 3H), 1.19 – 1.40 (m, 28H), 1.43 (s, 9H), 2.71 – 2.79 (m, 1H), 2.82 – 2.96 (m, 2H), 3.30 – 3.45 (m, 2H), 3.76 (s, 3H), 3.73 – 3.83 (m, 1H), 4.60 (dd, J=5.0, 12.9 Hz, 1H), 4.65 (dd, J=4.5, 13.2 Hz, 1H), 4.86 – 4.96 (m, 1H). 1H-NMR (600 MHz, CDCl3) for the minor diastereomer: δ 3.76 (s, 3H), 4.52 (dd, J=5.9, 13.1 Hz, 2H). 13C-NMR (151 MHz, CDCl3) for both diastereomers: δ 14.27, 22.83, 26.82, 26.88, 28.50, 29.42, 29.43, 29.49, 29.50, 29.63, 29.65, 29.74, 29.79, 29.81, 29.83, 29.84, 30.28, 32.06, 35.17, 36.64, 36.66, 43.35, 43.56, 52.94, 53.08, 59.31, 59.76, 76.02, 76.47, 79.58, 79.63, 155.95, 168.46, 203.83 (18 signals missing due to overlapping).
Synthesis of methyl 4-((3-methylbut-2-en-1-yl)oxy)-2-((R)-2-nitro-1-phenylethyl)-3-oxobutanoate (17d)
Following GP3 and GP4. Prepared from methyl 4-((3-methylbut-2-en-1-yl)oxy)-3-oxobutanoate (2c) (0.2 mmol, 40.0 mg) and trans-β-nitrostyrene (12) (0.3 mmol, 44.7 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:5). rac-17d Yield: 54.5 mg (0.156 mmol, 78%, two diastereomers in a ratio of 60:40 in CDCl3) of colorless oil. 17d Yield: 62.2 mg (0.178 mmol, 89%, two diastereomers in a ratio of 60:40 in CDCl3) of colorless oil. EI-HRMS: m/z = 367.1858 (M+NH4+); C18H27N2O6 requires: m/z = 367.1864 (M+NH4+); νmax 3033, 2954, 2916, 1746, 1724, 1552, 1496, 1434, 1378, 1247, 1199, 1169, 1092, 1034, 981, 942, 893, 766, 700, 618 cm-1. 1H-NMR (500 MHz, CDCl3) for the major diastereomer: δ 1.61 (s, 3H), 1.74 (s, 3H), 3.73 (s, 3H), 3.76 – 3.90 (m, 2H), 4.21 – 4.32 (m, 3H), 4.79 – 4.95 (m, 3H), 5.20 – 5.24 (m, 1H), 7.18 – 7.24 (m, 2H), 7.25 – 7.33 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor diastereomer: δ 1.66 (s, 3H), 1.75 (s, 3H), 3.52 (s, 3H), 3.98 (d, J=7.0 Hz, 2H), 4.02 (d, J=17.5 Hz, 1H), 4.10 (d, J=17.4 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 18.12, 18.16, 25.90, 25.93, 42.13, 42.35, 52.80, 52.96, 56.60, 57.33, 67.75, 67.94, 74.72, 77.22, 77.77, 119.78, 119.80, 128.05, 128.22, 128.43, 128.50, 129.10, 129.22, 136.39, 136.41, 138.70, 138.84, 167.43, 167.90, 202.15, 202.77 (1 signal missing due to overlapping). HPLC: Chiralpak AS-H, n-Hexane/EtOH = 90:10, flow rate 1.0 mL/min, λ = 210 nm, T = 20 °C. Minor diastereomer: enantiomers: tR = 14.066 minutes (minor); 17.773 minutes (major) – 94% ee. Major diastereomer: enantiomers: tR = 17.164 minutes (major); 19.406 minutes (minor) – 91% ee.
Synthesis of methyl 2-(2-((3-methylbut-2-en-1-yl)oxy)acetyl)-3-(nitromethyl)octadecanoate (rac-17e)
Following GP3. Prepared from methyl 4-((3-methylbut-2-en-1-yl)oxy)-3-oxobutanoate (2c) (0.3 mmol, 60.1 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst X, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:5). rac-17e Yield: 52.2 mg (0.108 mmol, 54%, two diastereomers in a ratio of 51:49 in CDCl3) of colorless oil. EI-HRMS: m/z = 501.3887 (M+NH4+); C27H53N2O6 requires: m/z = 501.3898 (M+NH4+); νmax 2923, 2853, 1726, 1553, 1435, 1379, 1250, 1199, 1158, 1092, 1000, 780, 722 cm-1. 1H-NMR (600 MHz, CDCl3) for both diastereomers: δ 0.88 (t, J=7.0 Hz, 3H), 1.18 – 1.51 (m, 28H), 1.68 (dd, J=1.4, 4.8 Hz, 3H), 1.77 (dd, J=1.2, 4.1 Hz, 3H), 2.85 – 2.93 (m, 1H), 3.73 (s, 1.5H), 3.74 (s, 1.5H), 3.94 – 4.15 (m, 5H), 4.46 (dd, J=7.1, 13.3 Hz, 0.5H), 4.53 (dd, J=5.7, 13.2 Hz, 0.5H), 4.59 – 4.69 (m, 1H), 5.27 – 5.34 (m, 1H). 13C-NMR (151 MHz, CDCl3) for both diastereomers: δ 14.28, 18.19, 18.21, 22.84, 25.96, 26.88, 27.05, 29.38, 29.45, 29.51, 29.66, 29.75, 29.80, 29.83, 29.85, 30.29, 32.07, 36.10, 36.30, 52.67, 52.82, 54.92, 55.32, 67.99, 68.00, 74.63, 74.73, 76.40, 76.75, 119.82, 119.92, 138.71, 138.89, 168.55, 168.56, 203.55, 203.88 (17 signals missing due to overlapping).
Synthesis of methyl 2-((R)-2-nitro-1-phenylethyl)-3-oxooctadecanoate (17f)
Following GP3 and GP4. Prepared from methyl 3-oxooctadecanoate (2d) (0.3 mmol, 93.7 mg) and trans-β-nitrostyrene (12) (0.2 mmol, 29.8 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:10). rac-17f Yield: 34.2 mg (0.074 mmol, 37%, two diastereomers in a ratio of 56:44 in CDCl3) of white solid. 17f Yield: 55.4 mg (0.120 mmol, 60%, two diastereomers in a ratio of 50:50 in CDCl3) of white solid; m.p. = 60.0–61.2 °C. EI-HRMS: m/z = 462.3214 (MH+); C27H44NO5 requires: m/z = 462.3214 (MH+); νmax 2915, 2850, 1742, 1711, 1550, 1496, 1471, 1455, 1438, 1383, 1334, 1281, 1245, 1208, 1173, 1128, 1092, 1072, 1033, 1004, 983, 918, 892, 853, 765, 718, 700, 616 cm-1. 1H-NMR (500 MHz, CDCl3) for the major diastereomer: δ 0.88 (t, J=6.9 Hz, 3H), 0.94 – 1.04 (m, 2H), 1.06 – 1.39 (m, 22H), 1.51 – 1.60 (m, 2H), 2.13 (dt, J=7.2, 17.8, 1H), 2.38 – 2.50 (m, 1H), 3.76 (s, 3H), 4.03 (d, J=10.0 Hz, 1H), 4.19 – 4.28 (m, 1H), 4.75 – 4.89 (m, 2H), 7.16 – 7.22 (m, 2H), 7.24 – 7.34 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor diastereomer: δ 2.61 (dt, J=7.4, 17.7 Hz, 1H), 3.52 (s, 3H), 4.13 (d, J=9.5 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for both diastereomers: δ 14.27, 22.83, 23.05, 23.39, 28.74, 29.00, 29.33, 29.45, 29.48, 29.50, 29.56, 29.68, 29.73, 29.76, 29.78, 29.80, 29.83, 32.06, 42.53, 42.86, 43.50, 43.84, 52.87, 53.05, 60.97, 61.34, 77.67, 77.93, 127.94, 128.11, 128.42, 128.50, 129.15, 129.26, 136.44, 136.63, 167.62, 168.14, 202.76, 203.81 (10 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/EtOH = 95:5, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Minor diastereomer: enantiomers: tR = 7.287 minutes (minor); 22.511 minutes (major) – 95% ee. Major diastereomer: enantiomers: tR = 8.963 minutes (major); 13.215 minutes (minor) – 96% ee.
Synthesis of methyl 2-(1-nitroheptadecan-2-yl)-3-oxooctadecanoate (rac-17g)
Following GP3. Prepared from methyl 3-oxooctadecanoate (2d) (0.3 mmol, 93.7 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst X, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:10). rac-17g Yield: 78.7 mg (0.132 mmol, 66%, two diastereomers in a ratio of 68:32 in CDCl3) of white solid; m.p. = 40.0–40.9 °C. EI-HRMS: m/z = 594.5108 (M-H+)-; C36H68NO5 requires: m/z = 594.5103 (M-H+)-; νmax 2955, 2914, 2849, 1730, 1707, 1556, 1543, 1470, 1435, 1402, 1380, 1243, 1204, 1128, 1073, 1000, 863, 719 cm-1. 1H-NMR (600 MHz, CDCl3) for both diastereomers: δ 0.88 (t, J=7.0, 6H), 1.11 – 1.46 (m, 52H), 1.55 – 1.62 (m, 2H), 2.47 – 2.55 (m, 1H), 2.57 – 2.66 (m, 1H), 2.79 – 2.90 (m, 1H), 3.75 (s, 2.04H), 3.75 (s, 0.96H), 3.76 – 3.81 (m, 1H), 4.49 – 4.57 (m, 1H), 4.59 – 4.67 (m, 1H). 13C-NMR (151 MHz, CDCl3) for both diastereomers: δ 14.28, 22.85, 23.46, 23.50, 26.80, 26.91, 29.08, 29.10, 29.42, 29.45, 29.48, 29.50, 29.52, 29.60, 29.65, 29.67, 29.75, 29.81, 29.83, 29.85, 30.22, 32.08, 36.72, 36.77, 43.43, 43.60, 52.79, 52.91, 59.33, 59.72, 76.16, 76.57, 168.77, 168.81, 204.33, 204.46 (36 signals missing due to overlapping).
Synthesis of methyl 2-((R)-2-nitro-1-phenylethyl)-3-oxoicosanoate (17h)
Following GP3 and GP4. Prepared from methyl 3-oxoicosanoate (2e) (0.3 mmol, 102.2 mg) and trans-β-nitrostyrene (12) (0.2 mmol, 29.8 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:10). rac-17h Yield: 43.1 mg (0.088 mmol, 44%, two diastereomers in a ratio of 54:46 in CDCl3) of white solid. 17h Yield: 52.9 mg (0.108 mmol, 54%, two diastereomers in a ratio of 48:52 in CDCl3) of white solid; m.p. = 42.2–44.7 °C. EI-HRMS: m/z = 490.3524 (MH+); C29H48NO5 requires: m/z = 490.3527 (MH+); νmax 2914, 2849, 1737, 1712, 1555, 1496, 1471, 1455, 1433, 1404, 1378, 1271, 1198, 1168, 1113, 1082, 982, 891, 765, 716, 699 cm-1. 1H-NMR (500 MHz, CDCl3) for the major diastereomer: δ 0.88 (t, J=6.9 Hz, 3H), 1.06 – 1.37 (m, 30H), 2.38 – 2.50 (m, 1H), 2.61 (dt, J=7.4, 17.7 Hz, 1H), 3.52 (s, 3H), 4.13 (d, J=9.4 Hz, 1H), 4.19 – 4.28 (m, 1H), 4.74 – 4.89 (m, 2H), 7.16 – 7.22 (m, 2H), 7.24 – 7.33 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor diastereomer: δ 0.94 – 1.04 (m, 2H), 1.51 – 1.60 (m, 2H), 2.13 (dt, J=7.1, 17.7 Hz, 1H), 3.75 (s, 3H), 4.03 (d, J=10.0 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for both diastereomers: δ 14.25, 22.82, 23.03, 23.37, 28.72, 28.99, 29.32, 29.43, 29.47, 29.49, 29.55, 29.67, 29.72, 29.75, 29.77, 29.79, 29.82, 32.05, 42.51, 42.86, 43.48, 43.83, 52.84, 53.02, 60.94, 61.33, 77.65, 77.92, 127.93, 128.10, 128.39, 128.47, 129.13, 129.24, 136.44, 136.63, 167.61, 168.12, 202.74, 203.79 (14 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/EtOH = 95:5, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Major diastereomer: enantiomers: tR = 8.838 minutes (minor); 26.885 minutes (major) – 96% ee. Minor diastereomer: enantiomers: tR = 10.913 minutes (major); 15.371 minutes (minor) – 96% ee.
Synthesis of methyl 2-(1-nitroheptadecan-2-yl)-3-oxoicosanoate (rac-17i)
Following GP3. Prepared from methyl 3-oxoicosanoate (2e) (0.3 mmol, 102.2 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst X, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:10). rac-17i Yield: 52.4 mg (0.084 mmol, 42%, two diastereomers in a ratio of 50:50 in CDCl3) of white solid; m.p. = 42.0–43.9 °C. EI-HRMS: m/z = 622.5416 (M-H+)-; C38H73NO5 requires: m/z = 622.5421 (M-H+)-; νmax 2956, 2915, 2848, 1731, 1706, 1556, 1542, 1470, 1435, 1402, 1379, 1349, 1241, 1205, 1128, 1108, 1078, 1001, 719 cm-1. 1H-NMR (500 MHz, CDCl3) for both diastereomers: δ 0.87 (t, J=6.9 Hz, 6H), 1.08 – 1.44 (m, 56H), 1.53 – 1.61 (m, 2H), 2.45 – 2.55 (m, 1H), 2.56 – 2.66 (m, 1H), 2.78 – 2.89 (m, 1H), 3.74 (s, 1.5H), 3.74 (s, 1.5H), 3.75 (d, J=6.9 Hz, 0.5H), 3.79 (d, J=7.6 Hz, 0.5H), 4.45 – 4.55 (m, 1H), 4.62 (td, J=4.6, 13.5 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for both diastereomers: δ 14.25, 22.83, 23.43, 23.48, 26.77, 26.88, 29.06, 29.08, 29.40, 29.43, 29.46, 29.48, 29.50, 29.59, 29.64, 29.71, 29.74, 29.79, 29.82, 29.84, 30.20, 32.06, 36.68, 36.74, 43.38, 43.56, 52.74, 52.86, 59.30, 59.69, 76.13, 76.54, 168.74, 168.78, 204.28, 204.41 (40 signals missing due to overlapping).
Synthesis of tert-butyl 2-((R)-2-nitro-1-phenylethyl)-3-oxoicosanoate (17j)
Following GP3 and GP4. Prepared from tert-butyl 3-oxoicosanoate (2f) (0.3 mmol, 114.8 mg) and trans-β-nitrostyrene (12) (0.2 mmol, 29.8 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:10). rac-17j Yield: 42.5 mg (0.080 mmol, 40%, two diastereomers in a ratio of 61:39 in CDCl3) of white solid. 17j Yield: 50.0 mg (0.094 mmol, 47%, two diastereomers in a ratio of 36:64 in CDCl3) of white solid; m.p. = 54.0–56.4 °C. EI-HRMS: m/z = 549.4273 (M+NH4+); C32H57N2O5 requires: m/z = 549.4262 (M+NH4+); νmax 2916, 2849, 1731, 1712, 1553, 1496, 1468, 1434, 1394, 1378, 1284, 1250, 1148, 1126, 1092, 1062, 982, 914, 838, 770, 751, 720, 700 cm-1. 1H-NMR (500 MHz, CDCl3) for the major diastereomer: δ 0.88 (t, J=6.9 Hz, 3H), 0.98 – 1.07 (m, 2H), 1.09 – 1.39 (m, 28H), 1.46 (s, 9H), 1.55 – 1.63 (m, 1H), 2.14 (dt, J=7.1, 17.5 Hz, 1H), 2.38 – 2.52 (m, 1H), 3.91 (d, J=9.9 Hz, 1H), 4.12 – 4.23 (m, 1H), 4.65 – 4.76 (m, 1H), 7.16 – 7.34 (m, 5H). 1H-NMR (500 MHz, CDCl3) for the minor diastereomer: δ 2.62 (dt, J=7.4, 17.4 Hz, 1H), 4.01 (d, J=10.1 Hz, 1H), 4.77 – 4.90 (m, 2H). 13C-NMR (126 MHz, CDCl3) for both diastereomers: δ 14.26, 22.83, 23.18, 23.53, 27.52, 27.98, 28.90, 29.15, 29.38, 29.48, 29.50, 29.56, 29.69, 29.73, 29.76, 29.80, 29.83, 32.06, 42.54, 42.79, 42.87, 43.59, 62.16, 62.51, 78.06, 78.40, 82.95, 83.40, 128.18, 128.29, 128.32, 128.36, 128.95, 129.14, 136.81, 136.84, 166.04, 166.75, 203.13, 203.98 (16 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/EtOH = 95:5, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Minor diastereomer: enantiomers: tR = 6.051 minutes (minor); 8.241 minutes (major) – 93% ee. Major diastereomer: enantiomers: tR = 6.768 minutes (minor); 7.403 minutes (major) – 98% ee.
Synthesis of tert-butyl 2-(1-nitroheptadecan-2-yl)-3-oxoicosanoate (rac-17k)
Following GP3. Prepared from tert-butyl 3-oxoicosanoate (2f) (0.3 mmol, 114.8 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst X, 24 h; isolation by column chromatography (first CC: CH2Cl2/petroleum ether = 1:3; second CC: EtOAc/petroleum ether = 1:10). rac-17k Yield: 51.9 mg (0.079 mmol, 39%, two diastereomers in a ratio of 66:34 in CDCl3) of white solid; m.p. = 41.2–45.0 °C. EI-HRMS: m/z = 664.5894 (M-H+)-; C41H78NO5 requires: m/z = 664.5886 (M-H+)-; νmax 2916, 2848, 1726, 1703, 1556, 1545, 1466, 1370, 1256, 1210, 1157, 1047, 845, 720, 618 cm-1. 1H-NMR (500 MHz, CDCl3) for both diastereomers: δ 0.88 (t, J=7.0 Hz, 6H), 1.12 – 1.44 (m, 56H), 1.47 (s, 9H), 1.55 – 1.63 (m, 2H), 2.45 – 2.54 (m, 1H), 2.56 – 2.67 (m, 1H), 2.76 – 2.86 (m, 1H), 3.63 (d, J=6.9, 0.34H), 3.66 (d, J=7.5 Hz, 0.66H), 4.48 – 4.56 (m, 1H), 4.57 – 4.67 (m, 1H). 13C-NMR (126 MHz, CDCl3) for both diastereomers: δ 14.26, 22.84, 23.55, 23.60, 26.79, 26.88, 28.03, 28.07, 29.23, 29.49, 29.51, 29.54, 29.60, 29.67, 29.76, 29.81, 29.83, 29.85, 30.19, 32.08, 36.66, 36.77, 43.26, 43.47, 60.43, 60.98, 76.40, 76.77, 83.01, 83.07, 167.40, 167.43, 204.68, 204.83 (44 signals missing due to overlapping).
Synthesis of methyl (11Z,14Z)-2-((R)-2-nitro-1-phenylethyl)-3-oxoicosa-11,14-dienoate (17l)
Following GP3 and GP4. Prepared from methyl (11Z,14Z)-3-oxoicosa-11,14-dienoate (2g) (0.3 mmol, 101.0 mg) and trans-β-nitrostyrene (12) (0.2 mmol, 29.8 mg), organocatalyst VIII, 24 h; isolation by column chromatography (EtOAc/petroleum ether = 1:20). rac-17l Yield: 58 mg (0.120 mmol, 60%, two diastereomers in a ratio of 52:48 in CDCl3) of colorless oil. 17l Yield: 60.2 mg (0.124 mmol, 62%, two diastereomers in a ratio of 51:49 in CDCl3) of colorless oil. EI-HRMS: m/z = 508.3032 (MNa+); C29H43NO5Na requires: m/z = 508.3032 (MNa+); νmax 3009, 2925, 2855, 1744, 1717, 1554, 1496, 1455, 1434, 1377, 1243, 1168, 981, 914, 765, 699 cm-1. 1H-NMR (600 MHz, CDCl3) for both diastereomers: δ 0.89 (td, J=1.5, 7.0 Hz, 3H), 0.95 – 1.05 (m, 1H), 1.10 – 1.16 (m, 1H), 1.16 – 1.21 (m, 1H), 1.23 – 1.40 (m, 12H), 1.52 – 1.60 (m, 1H), 1.97 – 2.07 (m, 4H), 2.13 (dt, J=7.2, 17.8 Hz, 0.5H), 2.39 – 2.49 (m, 1H), 2.61 (dt, J=7.4, 17.7 Hz, 0.5H), 2.77 (q, J=6.5 Hz, 2H), 3.52 (s, 1.5H), 3.76 (s, 1.5H), 4.03 (d, J=10.0 Hz, 0.5H), 4.13 (d, J=9.4 Hz, 0.5H), 4.19 – 4.27 (m, 1H), 4.74 – 4.88 (m, 2H), 5.28 – 5.43 (m, 4H), 7.17 – 7.21 (m, 2H), 7.22 – 7.34 (m, 3H). 13C-NMR (151 MHz, CDCl3) for both diastereomers: δ 14.22, 22.71, 23.03, 23.37, 25.76, 27.30, 27.31, 27.34, 28.70, 28.97, 29.13, 29.20, 29.24, 29.36, 29.48, 29.67, 29.72, 31.66, 42.53, 42.87, 43.48, 43.83, 52.87, 53.05, 60.98, 61.35, 77.67, 77.93, 127.94, 128.00, 128.02, 128.12, 128.20, 128.23, 128.43, 128.51, 129.16, 129.26, 130.13, 130.38, 136.45, 136.64, 168.14, 202.70, 203.76 (9 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/EtOH = 95:5, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Minor diastereomer: enantiomers: tR = 7.439 minutes (minor); 19.360 minutes (major) – 96% ee. Major diastereomer: enantiomers: tR = 9.402 minutes (major); 12.992 minutes (minor) – 96% ee.
Synthesis of methyl (4S)-7-((tert-butoxycarbonyl)amino)-2-(2-nitro-1-phenylethyl)-3-oxo-4-stearamidoheptanoate (17m)
Following GP3. Prepared from methyl (S)-7-((tert-butoxycarbonyl)amino)-3-oxo-4-stearamidoheptanoate (2h) (0.2 mmol, 111.0 mg) and trans-β-nitrostyrene (12) (0.3 mmol, 44.7 mg), organocatalyst X, 48 h; isolation by column chromatography (1. EtOAc/petroleum ether = 1:2; 1. EtOAc/petroleum ether = 1:1). 17m Yield: 100.0 mg (0.142 mmol, 71%, 4 diastereomers in a ratio of 27:21:24:28 in CDCl3) of white semisolid. EI-HRMS: m/z = 704.4827 (MH+); C39H66N3O8 requires: m/z = 704.4844 (MH+); νmax 3375, 2920, 2851, 1739, 1720, 1687, 1648, 1551, 1518, 1455, 1436, 1365, 1247, 1214, 1168, 1089, 1005, 872, 764, 720, 701, 617 cm-1. 1H-NMR (500 MHz, CDCl3) for 4 diastereomers: δ 0.88 (t, J=6.9, 3H), 1.05 – 1.39 (m, 29H), 1.42 – 1.45 (m, 9H), 1.46 – 1.68 (m, 4H), 1.71 – 1.86 (m, 1H), 2.02 – 2.28 (m, 2H), 2.77 – 3.22 (m, 2H), 3.48, 3.49, 3.74, 3.77 (4 × s, 3H), 4.22 – 4.96 (m, 6H), 5.83 – 6.68 (m, 1H), 7.18 – 7.35 (m, 5H). 13C-NMR (126 MHz, CDCl3) for 4 diastereomers: δ 14.24, 22.80, 25.55, 25.62, 25.67, 25.90, 26.01, 26.20, 26.30, 26.90, 26.95, 27.05, 27.20, 28.48, 28.49, 29.38, 29.40, 29.43, 29.45, 29.47, 29.60, 29.61, 29.63, 29.73, 29.75, 29.77, 29.81, 32.03, 36.04, 36.40, 36.44, 36.50, 39.56, 39.71, 39.75, 42.54, 42.63, 43.10, 43.25, 52.84, 53.02, 53.11, 53.36, 57.89, 58.13, 58.32, 58.44, 58.62, 58.97, 59.18, 77.27, 77.36, 77.58, 77.70, 79.48, 79.52, 79.67, 79.70, 128.09, 128.14, 128.27, 128.36, 128.42, 128.50, 129.04, 129.18, 129.21, 136.21, 136.27, 136.50, 136.55, 156.26, 156.41, 156.55, 156.59, 166.95, 167.15, 167.30, 167.66, 173.10, 173.47, 173.85, 174.11, 200.93, 201.85, 202.18, 202.94 (69 signals missing due to overlapping).
Synthesis of methyl 2-((S)-5-((tert-butoxycarbonyl)amino)-2-stearamidopentanoyl)-3-(nitromethyl)octadecanoate (17n)
Following GP3 and GP4. Prepared from methyl (S)-7-((tert-butoxycarbonyl)amino)-3-oxo-4-stearamidoheptanoate (2h) (0.2 mmol, 111.0 mg) and (E)-1-nitroheptadec-1-ene (16) (0.3 mmol, 85.0 mg), organocatalyst X, 48 h; isolation by column chromatography (1. EtOAc/petroleum ether = 1:3; 1. EtOAc/petroleum ether = 1:2); 17n Yield: 129.1 mg (0.154 mmol, 77%, 4 diastereomers in a ratio of 26:28:25:21 in CDCl3) of white solid. Organocatalyst VIII, 48 h; isolation by column chromatography (1. EtOAc/petroleum ether = 1:3; 1. EtOAc/petroleum ether = 1:2); 17n Yield: 147.5 mg (0.176 mmol, 88%, 4 diastereomers in a ratio of 45:11:8:36 in CDCl3) of white solid. EI-HRMS: m/z = 838.6860 (MH+); C48H92N3O8 requires: m/z = 838.6879 (MH+); νmax 3361, 2917, 2850, 1741, 1718, 1687, 1644, 1553, 1524, 1467, 1366, 1250, 1222, 1171, 1039, 1011, 869, 721, 646 cm-1. 1H-NMR (500 MHz, CDCl3) for 4 diastereomers: δ 0.88 (t, J=6.9 Hz, 6H), 1.10 – 1.39 (m, 57H), 1.44 (s, 9H), 1.48 – 1.58 (m, 2H), 1.58 – 1.67 (m, 2H), 1.87 – 1.99 (m, 1H), 2.20 – 2.27 (m, 2H), 2.80 – 2.93 (m, 1H), 3.05 – 3.24 (m, 2H), 3.73, 3.74, 3.76, 3.77 (4 × s, 3H), 3.99 (d, J=6.3 Hz, 0.206H), 4.04 (d, J=6.9 Hz, 0.255H), 4.09 (d, J=5.2 Hz, 0.284H), 4.15 (d, J=7.0 Hz, 0.255H), 4.40 – 4.80 (m, 4H), 6.42 – 6.58 (m, 1H). 13C-NMR (126 MHz, CDCl3) for 4 diastereomers: δ 14.23, 22.80, 25.67, 25.70, 26.36, 26.74, 26.77, 26.83, 26.87, 27.08, 27.43, 28.48, 29.42, 29.44, 29.46, 29.48, 29.64, 29.66, 29.72, 29.73, 29.75, 29.78, 29.82, 30.43, 30.52, 32.04, 36.37, 36.43, 36.53, 36.60, 37.22, 37.42, 39.76, 39.79, 39.82, 39.83, 52.77, 52.92, 53.00, 53.15, 55.92, 56.15, 56.47, 58.05, 58.26, 58.39, 58.62, 76.25, 76.43, 76.48, 77.36, 79.56, 156.45, 168.14, 168.38, 168.43, 168.46, 173.58, 173.64, 173.71, 173.80, 203.24, 203.40, 203.67 (128 signals missing due to overlapping).
Synthesis of methyl (4S)-2-(2-nitro-1-phenylethyl)-3-oxo-4,7-distearamidoheptanoate (17o)
Following GP3. Prepared from methyl (S)-3-oxo-4,7-distearamidoheptanoate (2i) (0.2 mmol, 144.1 mg) and trans-β-nitrostyrene (12) (0.3 mmol, 44.7 mg), organocatalyst X, 48 h; isolation by column chromatography (first column chromatography: EtOAc/petroleum ether = 1:1; second column chromatography: CH2Cl2/MeOH = 75:1). 17o Yield: 50.5 mg (0.058 mmol, 29%, 4 diastereomers in a ratio of 24:22:30:24 in CDCl3) of light orange solid. EI-HRMS: m/z = 870.6936 (MH+); C52H92N3O7 requires: m/z = 870.6930 (MH+); νmax 3292, 2916, 2848, 1745, 1720, 1638, 1552, 1462, 1434, 1377, 1274, 1240, 1221, 1205, 1168, 1114, 755, 719, 700 cm-1. 1H-NMR (500 MHz, CDCl3) for 4 diastereomers: δ 0.88 (t, J=6.9 Hz, 6H), 0.99 – 1.42 (m, 56H), 1.42 – 1.68 (m, 6H), 1.69 – 1.86 (m, 1H), 2.05 – 2.29 (m, 4H), 2.91 – 3.35 (m, 3H), 3.47, 3.48, 3.74, 3.77 (4 × s, 3H), 4.22 – 4.65 (m, 3H), 4.72 – 4.95 (m, 2H), 5.57 – 5.85 (m, 1H), 6.14 (d, J=7.9 Hz, 0.244H), 6.56 (d, J=7.9 Hz, 0.296H), 6.85 (br s, 0.225H), 6.93 (d, J=7.1 Hz, 0.235H), 7.18 – 7.34 (m, 5H). 13C-NMR (126 MHz, CDCl3) for 4 diastereomers: δ 14.24, 22.81, 25.55, 25.60, 25.63, 25.67, 25.77, 25.91, 26.08, 26.27, 26.57, 26.77, 27.03, 27.27, 29.41, 29.43, 29.48, 29.50, 29.64, 29.66, 29.78, 29.80, 29.83, 32.04, 36.02, 36.38, 36.46, 36.89, 36.91, 38.52, 38.61, 38.68, 38.71, 42.54, 42.64, 43.09, 43.31, 52.83, 52.99, 53.14, 53.32, 58.08, 58.17, 58.19, 58.25, 58.27, 58.82, 58.95, 59.10, 77.29, 77.36, 77.56, 77.65, 77.69, 128.11, 128.16, 128.26, 128.36, 128.42, 128.49, 129.03, 129.17, 136.21, 136.33, 136.52, 136.60, 167.03, 167.21, 167.40, 167.69, 173.31, 173.74, 173.80, 174.07, 174.08, 174.21, 201.14, 201.89, 202.14, 203.10 (128 signals missing due to overlapping).
Synthesis of methyl 2-((S)-2,5-distearamidopentanoyl)-3-(nitromethyl)octadecanoate (17p)
Following GP3 and GP4. Prepared from methyl (S)-3-oxo-4,7-distearamidoheptanoate (2i) (0.2 mmol, 144.1 mg) and (E)-1-nitroheptadec-1-ene (16) (0.3 mmol, 85.0 mg), organocatalyst X, 48 h; isolation by column chromatography (1. EtOAc/petroleum ether = 1:3; 1. EtOAc/petroleum ether = 1:1); 17p Yield: 50.2 mg (0.05 mmol, 25%, 4 diastereomers in a ratio of 25:25:27:23 in CDCl3) of light orange solid. Organocatalyst VIII, 48 h; isolation by column chromatography (1. EtOAc/petroleum ether = 1:3; 1. EtOAc/petroleum ether = 1:1); 17p Yield: 46.2 mg (0.046 mmol, 23%, 4 diastereomers in a ratio of 39:11:12:38 in CDCl3) of light orange solid. EI-HRMS: m/z = 1004.8949 (MH+); C61H118N3O7 requires: m/z = 1004.8964 (MH+); νmax 3293, 2916, 2849, 1746, 1721, 1639, 1552, 1463, 1378, 1274, 1257, 1240, 1222, 1204, 719, 615 cm-1. 1H-NMR (500 MHz, CDCl3) for 4 diastereomers: δ 0.88 (t, J=6.9, 9H), 1.07 – 1.46 (m, 88H), 1.48 – 1.70 (m, 7H), 1.83 – 1.98 (m, 1H), 2.14 – 2.21 (m, 2H), 2.21 – 2.30 (m, 2H), 2.81 – 2.92 (m, 1H), 3.17 – 3.28 (m, 1H), 3.28 – 3.40 (m, 1H), 3.73, 3.74, 3.76, 3.77 (4 × s, 3H), 3.98 (d, J=5.9 Hz, 0.235H), 4.03 (d, J=7.0 Hz, 0.270H), 4.08 (d, J=5.2 Hz, 0.249H), 4.14 (d, J=7.0 Hz, 0.246H), 4.37 – 4.78 (m, 3H), 5.79 – 5.89 (m, 1H), [6.72 (d, J=7.6 Hz), 6.76 (d, J=7.7 Hz), 6.77 (d, J=7.8 Hz), 6.89 (d, J=7.6 Hz); 1H]. 13C-NMR (126 MHz, CDCl3) for 4 diastereomers: δ 22.81, 25.65, 25.68, 25.69, 25.91, 25.93, 26.35, 26.41, 26.45, 26.49, 26.75, 26.79, 26.85, 26.89, 27.11, 27.40, 27.57, 29.44, 29.45, 29.49, 29.52, 29.57, 29.66, 29.67, 29.69, 29.71, 29.74, 29.76, 29.79, 29.81, 29.83, 30.44, 30.59, 32.05, 36.34, 36.39, 36.46, 36.48, 36.56, 36.92, 37.19, 37.40, 38.78, 38.81, 52.80, 52.94, 52.97, 53.16, 55.83, 55.89, 56.43, 56.55, 58.28, 58.41, 58.46, 58.56, 76.25, 76.50, 76.58, 77.36, 168.25, 168.36, 168.52, 173.80, 173.90, 173.94, 173.95, 174.02, 203.12, 203.63, 203.70 (173 signals missing due to overlapping).
Organocatalyzed Michael addition of pyrrolones 11 to nitroalkene 16 – General procedure for the preparation of racemic mixtures – General procedure 5 (GP5)
To a solution/suspension of (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg; 1.0 equivalent) and the achiral organocatalyst X (0.04 mmol, 0.2 equivalents, 16.4 mg) in anhydrous toluene (1 mL) under argon at room temperature, pyrrolone 11 (0.3 mmol, 1.5 equivalents) was added and the resulting reaction mixture was stirred at room temperature for 48–72 hours. The volatiles were evaporated in vacuo and the residue was purified by column chromatography (Silica gel 60, mobile phase). The fractions containing the pure racemic product rac-18 were combined and the volatiles were evaporated in vacuo. The product rac-18 was fully characterized and analyzed by HPLC.
Organocatalyzed Michael addition of pyrrolones 11 to nitroalkene 16 – General procedure for the organocatalyzed asymmetric addition – General procedure 6 (GP6)
To a solution/suspension of (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg; 1.0 equivalent) and the chiral organocatalyst I (0.02 mmol, 0.1 equivalents, 10.9 mg) in anhydrous toluene (1 mL) under argon at room temperature, pyrrolone 11 (0.3 mmol, 1.5 equivalents) was added and the resulting reaction mixture was stirred at room temperature for 48–72 hours. The volatiles were evaporated in vacuo and the residue was purified by column chromatography (Silica gel 60, mobile phase). The fractions containing the pure chiral nonracemic product 18 were combined the volatiles were evaporated in vacuo. The product 18 was fully characterized and analyzed by HPLC.
Synthesis of 1-(tert-butyl) 3-methyl (S)-5-benzyl-5-((S)-1-nitroheptadecan-2-yl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (18a)
Following GP5 and GP6. Prepared from 1-(tert-butyl) 3-methyl 5-benzyl-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11a) (0.3 mmol, 99.4 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst I, 48 h; isolation by column chromatography (EtOAc/petroleum ether = 1:5). rac-18a Yield: 94.7 mg (0.154 mmol, 77%, diastereomer 1/diastereomer 2 = 97:3 in CDCl3) of colorless oil. 18a Yield: 84.8 mg (0.138 mmol, 69%, diastereomer 1/diastereomer 2 = 97:3 in CDCl3) of colorless oil. EI-HRMS: m/z = 615.3970 (MH+); C35H55N2O7 requires: m/z = 615.4004 (MH+); νmax 2923, 2853, 1732, 1713, 1581, 1554, 1497, 1456, 1438, 1370, 1295, 1224, 1143, 1089, 1063, 992, 914, 876, 843, 760, 725, 703, 628 cm-1. 1H-NMR (500 MHz, CDCl3) for diastereomer 1: δ 0.88 (t, J=6.9 Hz, 3H), 1.07 – 1.43 (m, 26H), 1.44 – 1.85 (m, 11H), 3.26 – 3.54 (m, 3H), 3.77 (s, 3H), 4.31 (dd, J=5.1, 13.7 Hz, 1H), 4.51 (br s, 1H), 6.95 – 7.03 (m, 2H), 7.11 – 7.21 (m, 3H), 8.60 (br s, 1H). 1H-NMR (500 MHz, CDCl3) for diastereomer 2: δ 3.71 (s, 3H). 13C-NMR (126 MHz, CDCl3) for diastereomer 1: δ 14.26, 22.83, 27.55, 27.74, 28.10, 29.50, 29.52, 29.61, 29.63, 29.73, 29.78, 29.79, 29.82, 29.83, 32.06, 39.94, 43.20, 51.92, 76.56, 85.81, 112.39, 127.69, 128.43, 129.63, 133.25, 147.60, 161.78, 164.91, 196.43 (2 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/iPrOH = 95:5, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Major diastereomer: enantiomers: tR = 9.350 minutes (major); 13.217 minutes (minor) – 82% ee.
Synthesis of 1-(tert-butyl) 3-methyl 5-(4-(((benzyloxy)carbonyl)amino)butyl)-5-(1-nitroheptadecan-2-yl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (18b)
Following GP5 and GP6. Prepared from 1-(tert-butyl) 3-methyl 5-(4-(((benzyloxy)carbonyl)amino)butyl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11b) (0.3 mmol, 134.0 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst I, 72 h; isolation by column chromatography (EtOAc/petroleum ether = 1:2). rac-18b Yield: 94.9 mg (0.130 mmol, 65%, diastereomer 1/diastereomer 2 = 90:10 in CDCl3) of colorless oil. 18b Yield: 26.3 mg (0.036 mmol, 18%, diastereomer 1/diastereomer 2 = 26:74 in CDCl3) of colorless oil. EI-HRMS: m/z = 730.4644 (MH+); C40H64N3O9 requires: m/z = 730.4637 (MH+); νmax 3675, 3369, 2923, 2854, 1711, 1582, 1553, 1455, 1438, 1394, 1371, 1280, 1226, 1140, 1067, 846, 763, 697 cm-1. 1H-NMR (500 MHz, CDCl3) for diastereomer 1: δ 0.88 (t, J=6.9 Hz, 3H), 0.91 – 1.03 (m, 2H), 1.06 – 1.34 (m, 27H), 1.35 – 1.49 (m, 4H), 1.56 (s, 9H), 1.96 – 2.07 (m, 1H), 2.10 – 2.22 (m, 1H), 3.03 – 3.17 (m, 3H), 3.84 (s, 3H), 4.22 (dd, J=5.3, 13.8 Hz, 1H), 4.30 – 4.41 (m, 1H), 4.71 (t, J=6.2 Hz, 1H), 5.06 (s, 2H), 7.27 – 7.39 (m, 5H). 1H-NMR (500 MHz, CDCl3) for diastereomer 2: δ 1.58 (s, 9H), 1.87 – 1.96 (m, 1H), 3.28 – 3.36 (m, 1H), 3.83 (s, 3H), 4.28 (dd, J=5.3, 14.6 Hz, 1H), 5.18 (dd, J=5.5, 14.7 Hz, 1H), 9.19 (s, 1H). 13C-NMR (126 MHz, CDCl3) for diastereomer 1: δ 14.26, 20.18, 22.82, 27.41, 27.49, 28.02, 29.49, 29.55, 29.62, 29.71, 29.77, 29.78, 29.81, 29.82, 32.05, 33.87, 40.61, 43.36, 52.03, 66.78, 76.35, 77.36, 86.22, 112.22, 128.24, 128.64, 136.59, 147.42, 156.40, 161.95, 165.24, 196.41 (4 signals missing due to overlapping). 13C-NMR (126 MHz, CDCl3) for diastereomer 1 and diastereomer 2 (diastereomer 1/diastereomer 2 = 26:74): δ 14.26, 20.19, 20.45, 22.82, 27.18, 27.50, 27.91, 28.03, 28.12, 29.44, 29.49, 29.50, 29.56, 29.63, 29.67, 29.72, 29.75, 29.79, 29.82, 29.83, 32.05, 33.89, 40.66, 41.98, 43.37, 52.03, 52.05, 66.79, 74.38, 76.11, 76.36, 77.40, 86.21, 112.23, 112.31, 128.25, 128.65, 136.59, 147.43, 156.39, 161.92, 165.29, 196.41, 197.67 (28 signals missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/iPrOH = 90:10, flow rate 1.0 mL/min, λ = 210 nm, T = 25 °C. Major diastereomer: enantiomers: tR = 24.088 minutes (minor); 31.095 minutes (major) – 57% ee. Minor diastereomer: enantiomers: tR = 26.634 minutes (minor); 30.099 minutes (major) – 11% ee.
Synthesis of 1-(tert-butyl) 3-methyl 5-(3-(benzyloxy)-3-oxopropyl)-5-(1-nitroheptadecan-2-yl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (18c)
Following GP5 and GP6. Prepared from 1-(tert-butyl) 3-methyl 5-(3-(benzyloxy)-3-oxopropyl)-4-oxo-4,5-dihydro-1H-pyrrole-1,3-dicarboxylate (11c) (0.3 mmol, 121.0 mg) and (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg), organocatalyst I, 48 h; isolation by column chromatography (EtOAc/petroleum ether = 1:5). rac-18c Yield: 92.0 mg (0.134 mmol, 67%, diastereomer 1/diastereomer 2 = 93:7 in CDCl3) of colorless oil. 18c Yield: 42.6 mg (0.062 mmol, 31%, diastereomer 1/diastereomer 2 = 31:69 in CDCl3) of colorless oil. EI-HRMS: m/z = 687.4224 (MH+); C38H59N2O9 requires: m/z = 687.4215 (MH+); νmax 3675, 2923, 2854, 1735, 1713, 1582, 1554, 1439, 1393, 1371, 1279, 1256, 1227, 1141, 1077, 846, 800, 752, 698 cm-1. 1H-NMR (600 MHz, CDCl3) for diastereomer 1: δ 0.88 (t, J=6.9 Hz, 3H), 1.17 – 1.34 (m, 27H), 1.35 – 1.46 (m, 1H), 1.56 (s, 9H), 1.98 – 2.06 (m, 1H), 2.09 – 2.17 (m, 1H), 2.37 – 2.45 (m, 1H), 2.48 – 2.55 (m, 1H), 3.13 – 3.20 (m, 1H), 3.83 (s, 3H), 4.25 (dd, J=5.6, 13.8 Hz, 1H), 4.30 – 4.39 (m, 1H), 5.07 (s, 2H), 7.28 – 7.39 (m, 5H), 9.08 (s, 1H). 1H-NMR (500 MHz, CDCl3) for diastereomer 2: δ 1.58 (s, 9H), 2.26 – 2.33 (m, 1H), 2.54 – 2.62 (m, 1H), 3.35 – 3.41 (m, 1H), 3.82 (s, 3H), 5.06 (s, 2H), 5.19 (dd, J=5.1, 14.7 Hz, 1H), 9.17 (s, 1H). 13C-NMR (151 MHz, CDCl3) for diastereomer 1: δ 14.26, 22.83, 27.48, 27.60, 28.01, 29.13, 29.49, 29.50, 29.54, 29.62, 29.72, 29.77, 29.79, 29.82, 29.84, 32.06, 43.15, 52.06, 66.88, 75.92, 76.42, 86.62, 112.16, 128.44, 128.47, 128.56, 128.75, 135.56, 147.29, 161.76, 165.19, 171.48, 195.61 (1 signal missing due to overlapping). 13C-NMR (126 MHz, CDCl3) for diastereomer 1 and diastereomer 2 (diastereomer 1/diastereomer 2 = 31:69): δ 14.28, 22.84, 27.17, 27.49, 28.02, 28.07, 28.11, 28.37, 29.28, 29.44, 29.50, 29.56, 29.64, 29.68, 29.73, 29.76, 29.80, 29.83, 29.85, 32.07, 41.85, 43.16, 52.06, 52.08, 66.89, 66.93, 74.46, 75.26, 75.93, 76.42, 77.39, 86.67, 112.15, 112.36, 128.45, 128.48, 128.58, 128.77, 135.51, 135.56, 147.29, 161.72, 161.77, 165.20, 165.34, 171.18, 171.49, 195.63, 196.88 (19 signal missing due to overlapping). HPLC: Chiralpak IA-3, n-Hexane/iPrOH = 90:10, flow rate 1.0 mL/min, λ = 280 nm, T = 25 °C. Major diastereomer: enantiomers: tR = 10.649 minutes (major); 12.074 minutes (minor) – 68% ee. Minor diastereomer: enantiomers: tR = 9.991 minutes (minor); 12.514 minutes (major) – 38% ee.
Synthesis of 4-hydroxy-3-(1-nitroheptadecan-2-yl)furan-2(5H)-one (20)
To a solution of (E)-1-nitroheptadec-1-ene (16) (0.2 mmol, 56.7 mg) and the achiral organocatalyst X (0.04 mmol, 0.2 equivalents, 16.4 mg) in anhydrous CH2Cl2 (1 mL) under argon at room temperature, tetronic acid (19) (0.3 mmol, 30.0 mg) was added and the resulting reaction mixture was stirred at room temperature for 72 hours. The volatiles were evaporated in vacuo and the residue was purified by column chromatography (Silica gel 60, EtOAc/petroleum ether = 1:1). The fractions containing the pure product 20 were combined and the volatiles were evaporated in vacuo. Yield: 33.8 mg (0.088 mmol, 44%) of white solid; m.p. = 86.8-88.2 °C. EI-HRMS: m/z = 384.2741 (MH+); C21H38NO5 requires: m/z = 384.2744 (MH+); νmax 2918, 2850, 1714, 1611, 1547, 1428, 1381, 1348, 1279, 1260, 1129, 1097, 1044, 971, 954, 780, 720, 682, 658 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (t, J=6.9 Hz, 3H), 1.09 – 1.35 (m, 26H), 1.50 – 1.60 (m, 1H), 1.65 – 1.77 (m, 1H), 3.37 (tt, J=5.6, 9.6 Hz, 1H), 4.53 (dd, J=5.6, 12.4 Hz, 1H), 4.71 – 4.81 (m, 3H), 10.97 (br s, 1H). 13C-NMR (126 MHz, CDCl3): δ 14.27, 22.84, 27.28, 29.44, 29.51, 29.62, 29.76, 29.79, 29.81, 29.84, 29.86, 29.99, 32.07, 32.87, 68.14, 76.88, 99.10, 177.35, 177.91 (2 signals missing due to overlapping).
X-ray Crystallography. Single-crystal X-ray diffraction data was collected on Agilent Technologies SuperNova Dual diffractometer with an Atlas detector using monochromated Mo-Kα radiation (λ = 0.71073 Å) at 150 K. The data was processed using CrysAlis PRO [67]. Using Olex2.1.2. [68], the structure was solved by direct methods implemented in SHELXS [69] or SHELXT [70] and refined by a full-matrix least-squares procedure based on F2 with SHELXT-2014/7 [71]. All nonhydrogen atoms were refined anisotropicallly. Hydrogen atoms were placed in geometrically calculated positions and were refined using a riding model. The drawing and the analysis of bond lengths, angles and intermolecular interactions were carried out using Mercury [72] and Platon [73]. Structural and other crystallographic details on data collection and refinement for compound 17a have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC Deposition Number 2513139. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk).
5. Conclusions
Glycine-, β-alanine-, lysine-, glycolic acid-, palmitic acid-, stearic acid-, and linoleic acid-derived β-keto esters 2a–i, as well as phenylalanine-, ornithine-, and glutamic acid-derived pyrrolones 11a–c and tetronic acid (19), were used as nucleophiles for the organocatalyzed addition to trans-β-nitrostyrene (12) and the palmitic acid-derived nitroalkene 16 electrophiles. All racemic products rac-17a–l, rac-18a–c, and 20 were prepared using the achiral organocatalyst X. For products with HPLC-separable stereomers, chiral nonracemic 1,4-adducts 17a,b,d,f,h,j,l and 18a–c were resynthesized using bifunctional noncovalent organocatalysts I and/or VIII. The doubly fatty acid-derived products rac-17g,i,k could not be separated on chiral HPLC columns. β-Keto ester adducts 17a,b,d,f,h,j,l were prepared with high enantioselectivity (91–98% ee) but low diastereoselectivity (dr up to 36:64) due to the epimerizable C-2 stereocenter. The (1S,2S)-absolute configuration of the stereoisomer of adduct 17a (prepared with organocatalyst I) was determined by single-crystal X-ray diffraction analysis. Based on this, we assigned the (1R)-absolute configuration to the major enantiomer of both diastereomers of products 17a, 17b, 17d, 17f, 17h, 17j, and 17l prepared with organocatalyst VIII. Products 17m–p, prepared from the addition of lysine-derived β-keto esters 2h and 2i, showed low diastereoselectivity with both achiral X and chiral organocatalyst VIII. In the pyrrolone series, the best diastereoselectivity (dr = 97:3) and enantioselectivity (82% ee for the major diastereomer) were obtained with phenylalanine-derived pyrrolone 11a, yielding adduct 18a. The (1'S,5S)-absolute configuration of the major diastereomer of product 18a was assigned based on our previous results in the series of 1,4-adducts of pyrrolones to nitroalkene acceptors [22]. Despite certain drawbacks, such as low diastereoselectivity and challenges in separating stereomers on chiral HPLC columns, this study demonstrates the ease of assembling readily available amino acid and fatty acid building blocks under ambient conditions for the synthesis of interesting classes of products.
Supplementary Materials
The following supporting information can be downloaded: copies of 1H- and 13C-NMR spectra; HPLC data, X-ray diffraction data.
Author Contributions
Conceptualization, L.C., U.G., J.S., and B.Š.; methodology, L.C. H.B., and U.G.; software, L.C., U.G., J.S., and B.Š.; validation, L.C., H.B., A.F., N.P., U.G., J.S., F.P., and B.Š.; formal analysis, U.G., H.B., and L.C.; investigation, A.F., L.C., and U.G.; resources, L.C., U.G., and J.S.; data curation, L.C., N.P., A.F., U.G., J.S., H.B., and B.Š.; writing—original draft preparation, L.C., U.G., J.S., and B.Š.; writing—review and editing, L.C., A.F., N.P., U.G., J.S., F.P., and B.Š.; visualization, L.C., U.G., B.Š., H.B., and J.S.; supervision, U.G.; project administration, U.G. and J.S.; funding acquisition, U.G. and J.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the support of the Centre for Research Infrastructure at the University of Ljubljana, Faculty of Chemistry and Chemical Technology, which is part of the Network of Research and Infrastructural Centres UL (MRIC UL) and is financially supported by the Slovenian Research Agency (Infrastructure program No. I0-0022). Eleven mass spectra were recorded by a commercial service at the Jožef Stefan Institute in Ljubljana, Slovenia.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reyes, E.; Prieto, L.; Milelli, A. Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists. Molecules 2023, 28, 271. [CrossRef]
- Pellissier, H., Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [CrossRef]
- Vicario, J. L.; Badia, D.; Carrillo, L., Organocatalytic enantioselective Michael and hetero-Michael reactions. Synthesis 2007, 2007, 2065–2092. [CrossRef]
- Almaşi, D.; Alonso, D. A.; Nájera, C., Organocatalytic asymmetric conjugate additions. Tetrahedron: Asymmetry 2007, 18, 299–365. [CrossRef]
- Tsogoeva, S. B., Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. [CrossRef]
- Thirumalaikumar, M., Enantioselective Michael Addition Reactions. Org. Prep. Proced. Int. 2011, 43, 67–129. [CrossRef]
- Wang, J.; Li, P.; Choy, P. Y.; Chan, A. S. C.; Kwong, F. Y., Advances and Applications in Organocatalytic Asymmetric aza-Michael Addition. ChemCatChem 2012, 4, 917–925. [CrossRef]
- Zhang, Y.; Wang, W., Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [CrossRef]
- Mondal, A.; Bhowmick, S.; Ghosh, A.; Chanda, T.; Bhowmick, K. C., Advances on asymmetric organocatalytic 1,4-conjugate addition reactions in aqueous and semi-aqueous media. Tetrahedron: Asymmetry 2017, 28, 849–875. [CrossRef]
- Das, T.; Mohapatra, S.; Mishra, N. P.; Nayak, S.; Raiguru, B. P., Recent Advances in Organocatalytic Asymmetric Michael Addition Reactions to α, β-Unsaturated Nitroolefins. ChemistrySelect 2021, 6, 3745–3781. [CrossRef]
- Shim, J. H.; Cheun, S. H.; Kim, H. S.; Ha, D.-C., Organocatalysis for the Asymmetric Michael Addition of Aldehydes and α,β-Unsaturated Nitroalkenes. Catalysts 2022, 12, 121. [CrossRef]
- Niu, C.; Du, D.-M., Recent Advances in Organocatalyzed Asymmetric Sulfa-Michael Addition Triggered Cascade Reactions. Chem. Rec. 2023, 23, e202200258. [CrossRef]
- Coutant, C.; De Bonfils, P.; Nun, P.; Coeffard, V., Asymmetric Organocatalyzed Intermolecular Functionalization of Cyclohexanone-Derived Dienones. Chem. Rec. 2023, 23, e202300042. [CrossRef]
- Wojaczyńska, E.; Steppeler, F.; Iwan, D.; Scherrmann, M.-C.; Marra, A. Synthesis and Applications of Carbohydrate-Based Organocatalysts. Molecules 2021, 26, 7291. [CrossRef]
- Kopyt, M.; Dudziński, J.; Barbasiewicz, M.; Kwiatkowski, P., Enantioselective Organocatalytic Addition of 1,3-Dicarbonyl Compounds to β-Arylvinyl Triflones. Org. Lett. 2025, 27, 2879–2884. [CrossRef]
- Luo, X.; Jiang, Z.; Yang, S.; Ren, X.; Wang, T., Organocatalyzed Asymmetric Conjugate Addition of Alcohols to β-Fluoroalkyl Vinylsulfones by Bifunctional Phosphonium Salt Catalyst. Chem. Eur. J. 2024, 30, e202401325. [CrossRef]
- Jin, Y.-H.; Wang, X.-P.; Ning, L.-W.; Wang, R.-J.; Li, Y.; Zhang, Y.; Chen, L.-Y., Organocatalyzed Asymmetric Michael Addition of 3-Fluorooxindole to Vinylidene Bisphosphonates. J. Org. Chem. 2024, 89, 8435–8446. [CrossRef]
- Goyal, P.; Dubey, A. K.; Chowdhury, R., Organocatalyzed Asymmetric Michael Addition of 4-Monosubstituted-pyrazol-5-ones to Enones: Construction of Vicinal Quaternary and Tertiary Stereocenters. Eur. J. Org. Chem. 2024, 27, e202400002. [CrossRef]
- Retini, M.; Bartolucci, S.; Bartoccini, F.; Piersanti, G., Asymmetric Alkylation of Cyclic Ketones with Dehydroalanine via H-Bond-Directing Enamine Catalysis: Straightforward Access to Enantiopure Unnatural α-Amino Acids. Chem. Eur. J. 2022, 28, e202201994. [CrossRef]
- Si, S.; Wang, Q.; Lv, Y.; Liao, H.; Chen, W.-W.; Liu, S.; Zhao, B., Direct Asymmetric α-C Addition of Glycinate to Nitroalkenes by Pyridoxal/Nd Dual Catalysis. Angew. Chem. Int. Ed. 2025, 64, e202506342. [CrossRef]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U., 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [CrossRef]
- Grošelj, U.; Žorž, M.; Golobič, A.; Stanovnik, B.; Svete, J., α-Amino acid derived enaminones and their application in the synthesis of N-protected methyl 5-substituted-4-hydroxypyrrole-3-carboxylates and other heterocycles. Tetrahedron 2013, 69, 11092–11108. [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, A.; Štefane, B.; Svete, J.; Grošelj, U., Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [CrossRef]
- Ričko, S.; Meden, A.; Ciber, L.; Štefane, B.; Požgan, F.; Svete, J.; Grošelj, U., Construction of Vicinal Tetrasubstituted Stereogenic Centers via a Mannich-Type Organocatalyzed Addition of Δ2-Pyrrolin-4-ones to Isatin Imines. Adv. Synth. Catal. 2018, 360, 1072–1076. [CrossRef]
- Dell’Amico, L.; Albrecht, Ł.; Naicker, T.; Poulsen, P. H.; Jørgensen, K. A., Beyond Classical Reactivity Patterns: Shifting from 1,4- to 1,6-Additions in Regio- and Enantioselective Organocatalyzed Vinylogous Reactions of Olefinic Lactones with Enals and 2,4-Dienals. J. Am. Chem. Soc. 2013, 135, 8063–8070. [CrossRef]
- Bojanowski, J.; Sieroń, L.; Albrecht, A. Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules 2019, 24, 2565. [CrossRef]
- Xie, L.; Dong, S.; Zhang, Q.; Feng, X.; Liu, X., Asymmetric construction of dihydrobenzofuran-2,5-dione derivatives via desymmetrization of p-quinols with azlactones. Chem. Commun. 2019, 55, 87–90. [CrossRef]
- Liu, Y.; Liu, J.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H., Asymmetric Michael addition of glycine-derived schiff bases to azadienes catalyzed by a quinidine-derived chiral catalyst. Tetrahedron 2026, 189, 134991. [CrossRef]
- Bernardi, L.; Fochi, M.; Carbone, R.; Martinelli, A.; Fox, M. E.; Cobley, C. J.; Kandagatla, B.; Oruganti, S.; Dahanukar, V. H.; Carlone, A., Organocatalytic Asymmetric Conjugate Additions to Cyclopent-1-enecarbaldehyde: A Critical Assessment of Organocatalytic Approaches towards the Telaprevir Bicyclic Core. Chem. Eur. J. 2015, 21, 19208–19222. [CrossRef]
- Guerrero-Corella, A.; Esteban, F.; Iniesta, M.; Martín-Somer, A.; Parra, M.; Díaz-Tendero, S.; Fraile, A.; Alemán, J., 2-Hydroxybenzophenone as a Chemical Auxiliary for the Activation of Ketiminoesters for Highly Enantioselective Addition to Nitroalkenes under Bifunctional Catalysis. Angew. Chem. Int. Ed. 2018, 57, 5350–5354. [CrossRef]
- Stockhammer, L.; Craik, R.; Monkowius, U.; Cordes, D. B.; Smith, A. D.; Waser, M., Isothiourea-Catalyzed Enantioselective Functionalisation of Glycine Schiff Base Aryl Esters via 1,6- and 1,4-Additions. ChemistryEurope 2023, 1, e202300015. [CrossRef]
- Breman, A. C.; Smits, J. M. M.; de Gelder, R.; van Maarseveen, J. H.; Ingemann, S.; Hiemstra, H., Enantioselective Organocatalytic Thiol Addition to α,β-Unsaturated α-Amino Acid Derivatives. Synlett 2012, 23, 2195–2200. [CrossRef]
- Buyck, T.; Wang, Q.; Zhu, J., Catalytic Enantioselective Michael Addition of α-Aryl-α-Isocyanoacetates to Vinyl Selenone: Synthesis of α,α-Disubstituted α-Amino Acids and (+)- and (−)-Trigonoliimine A. Angew. Chem. Int. Ed. 2013, 52, 12714–12718. [CrossRef]
- Uraguchi, D.; Yamada, K.; Ooi, T., Highly E-Selective and Enantioselective Michael Addition to Electron-Deficient Internal Alkynes Under Chiral Iminophosphorane Catalysis. Angew. Chem. Int. Ed. 2015, 54, 9954–9957. [CrossRef]
- Etxabe, J.; Izquierdo, J.; Landa, A.; Oiarbide, M.; Palomo, C., Catalytic Enantioselective Synthesis of N,Cα,Cα-Trisubstituted α-Amino Acid Derivatives Using 1H-Imidazol-4(5H)-ones as Key Templates. Angew. Chem. Int. Ed. 2015, 54, 6883–6886. [CrossRef]
- Yoshida, Y.; Mino, T.; Sakamoto, M., Organocatalytic Highly Regio- and Enantioselective Umpolung Michael Addition Reaction of α-Imino Esters. Chem. Eur. J. 2017, 23, 12749–12753. [CrossRef]
- Bourboula, A.; Limnios, D.; Kokotou, M.G.; Mountanea, O.G.; Kokotos, G. Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones. Molecules 2019, 24, 2081. [CrossRef]
- Mountanea, O. G.; Limnios, D.; Kokotou, M. G.; Bourboula, A.; Kokotos, G., Asymmetric Synthesis of Saturated Hydroxy Fatty Acids and Fatty Acid Esters of Hydroxy Fatty Acids. Eur. J. Org. Chem. 2019, 2019, 2010–2019. [CrossRef]
- Büttner, H.; Steinbauer, J.; Wulf, C.; Dindaroglu, M.; Schmalz, H.-G.; Werner, T., Organocatalyzed Synthesis of Oleochemical Carbonates from CO2 and Renewables. ChemSusChem 2017, 10, 1076–1079. [CrossRef]
- Shirshin, K. K.; Esipovich, A. L.; Kazantsev, O. A.; Rumyantsev, M.; Korotaev, M. S.; Rogozhin, A. E., Specific Organocatalysis in Amidation Reaction of Fatty Acid Methyl Esters with 3-(Dimethylamino)-1-propylamine. ChemistrySelect 2021, 6, 480–487. [CrossRef]
- Carolina, B. G.; Caroline, L. C.; Diego, C. C.; Marcelo, G. M. D.; Oca; Martha, R.; Tiago, C.; Lucielli, S.; Fabiana, K. S.; Diego, A., Organocatalytic Synthesis and Antitumor Activity of Novel 1,2,3-triazoles Derived from Fatty β-ketoesters. Med. Chem. 2022, 18, 463–472. [CrossRef]
- Mountanea, O.G.; Mantzourani, C.; Gkikas, D.; Politis, P.K.; Kokotos, G. Asymmetric Synthesis of Saturated and Unsaturated Hydroxy Fatty Acids (HFAs) and Study of Their Antiproliferative Activity. Biomolecules 2024, 14, 110. [CrossRef]
- Treptow, T. G. M.; Figueiró, F.; Jandrey, E. H. F.; Battastini, A. M. O.; Salbego, C. G.; Hoppe, J. B.; Taborda, P. S.; Rosa, S. B.; Piovesan, L. A.; Montes D'Oca, C. D. R.; Russowsky, D.; Montes D'Oca, M. G., Novel hybrid DHPM-fatty acids: Synthesis and activity against glioma cell growth in vitro. Eur. J. Med. Chem. 2015, 95, 552–562. [CrossRef]
- Wong, T.-Y.; Sharma, S.; Mehdiratta, K.; Bhosale, R. S.; Nimmakayala, K.; Wilharm, R. K.; Chakraborty, A.; Orimoloye, M.; Liu, Q.; Kamat, S. S.; Pierre, V. C.; Gokhale, R. S.; Aldrich, C. C., Kupyaphores─Self-Assembling Diisocyanolipopeptide ZnII Ionophores in Mycobacterium tuberculosis ZnII/CuI/II Homeostasis and Antibacterial Effects. J. Am. Chem. Soc. 2025, 147, 40652–40663. [CrossRef]
- Pavlíčková, T.; Bultel-Poncé, V.; Roy, J.; Reversat, G.; Vigor, C.; Durand, T.; Jahn, U.; Galano, J.-M.; Oger, C., Beyond Neuroprostanes: Total Synthesis and Epilipidomic Studies of a Novel Cyclopentenone Metabolite of Docosahexaenoic Acid. Chem. Eur. J. 2025, 31, e02310. [CrossRef]
- Koutoulogenis, G.S.; Kokotos, G. Nitro Fatty Acids (NO2-FAs): An Emerging Class of Bioactive Fatty Acids. Molecules 2021, 26, 7536. [CrossRef]
- Ni, H.; Tan, X.; Du, J.; Wang, Y., Nitro-fatty acids: mechanisms of action, roles in metabolic diseases, and therapeutics. Curr. Med. 2024, 3, 3. [CrossRef]
- Roos, J.; Manolikakes, G.; Schlomann, U.; Klinke, A.; Schopfer, F. J.; Neumann, C. A.; Maier, T. J., Nitro-fatty acids: promising agents for the development of new cancer therapeutics. Trends Pharmacol Sci 2024, 45, 1061–1080. [CrossRef]
- Chowdhury, F. A.; Colussi, N.; Sharma, M.; Wood, K. C.; Xu, J. Z.; Freeman, B. A.; Schopfer, F. J.; Straub, A. C., Fatty acid nitroalkenes – Multi-target agents for the treatment of sickle cell disease. Redox Biol. 2023, 68, 102941. [CrossRef]
- Rubbo, H.; Trostchansky, A., Nitro-fatty acid signaling: Therapeutic potential in inflammatory diseases. Redox Biochem Chem 2024, 8, 100027. [CrossRef]
- Schopfer, F. J.; Teng, L.; Sekandari, A.; Ekhator, E. S.; Kohan, A. B.; Freeman, B. A.; Fazzari, M., Fatty acid nitroalkenes regulate intestinal lipid absorption. J. Lipid Res. 2025, 66, 100855. [CrossRef]
- Khoo, N. K. H.; Li, L.; Salvatore, S. R.; Schopfer, F. J.; Freeman, B. A., Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-κB signaling:A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 2295. [CrossRef]
- Di Maio, R.; Keeney, M. T.; Cechova, V.; Mortimer, A.; Sekandari, A.; Rowart, P.; Greenamyre, J. T.; Freeman, B. A.; Fazzari, M., Neuroprotective actions of a fatty acid nitroalkene in Parkinson’s disease. npj Parkinson's Disease 2023, 9, 55. [CrossRef]
- Franchini, L.; Panza, L.; Kongmanas, K.; Tanphaichitr, N.; Faull, K. F.; Ronchetti, F., An efficient and convenient synthesis of deuterium-labelled seminolipid isotopomers and their ESI-MS characterization. Chem. Phys. Lipids 2008, 152, 78–85. [CrossRef]
- Bevan, T. W.; Francis-Taylor, J.; Wong, H.; Northcote, P. T.; Harvey, J. E., A colourful azulene-based protecting group for carboxylic acids. Tetrahedron 2018, 74, 2942–2955. [CrossRef]
- Jirošová, A.; Majer, P.; Jančařík, A.; Dolejšová, K.; Tykva, R.; Šobotník, J.; Jiroš, P.; Hanus, R., Sphinganine-Like Biogenesis of (E)-1-Nitropentadec-1-ene in Termite Soldiers of the Genus Prorhinotermes. ChemBioChem 2014, 15, 533–536. [CrossRef]
- Mailhol, D.; Duque, M. d. M. S.; Raimondi, W.; Bonne, D.; Constantieux, T.; Coquerel, Y.; Rodriguez, J., Enantioselective Organocatalytic Michael Addition of Cyclobutanones to Nitroalkenes. Adv. Synth. Catal. 2012, 354, 3523–3532. [CrossRef]
- Konishi, H.; Lam, T. Y.; Malerich, J. P.; Rawal, V. H., Enantioselective α-Amination of 1,3-Dicarbonyl Compounds Using Squaramide Derivatives as Hydrogen Bonding Catalysts. Org. Lett. 2010, 12, 2028–2031. [CrossRef]
- Badiola, E.; Fiser, B.; Gómez-Bengoa, E.; Mielgo, A.; Olaizola, I.; Urruzuno, I.; García, J. M.; Odriozola, J. M.; Razkin, J.; Oiarbide, M.; Palomo, C., Enantioselective Construction of Tetrasubstituted Stereogenic Carbons through Brønsted Base Catalyzed Michael Reactions: α′-Hydroxy Enones as Key Enoate Equivalent. J. Am. Chem. Soc. 2014, 136, 17869–17881. [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T., Highly Enantioselective Conjugate Addition of Nitromethane to Chalcones Using Bifunctional Cinchona Organocatalysts. Org. Lett. 2005, 7, 1967–1969. [CrossRef]
- Malerich, J. P.; Hagihara, K.; Rawal, V. H., Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [CrossRef]
- Yang, W.; Du, D.-M., Highly Enantioselective Michael Addition of Nitroalkanes to Chalcones Using Chiral Squaramides as Hydrogen Bonding Organocatalysts. Org. Lett. 2010, 12, 5450–5453. [CrossRef]
- Li, Y.-J.; Lee, P.-T.; Yang, C.-M.; Chang, Y.-K.; Weng, Y.-C.; Liu, Y.-H., [2,3]-Wittig rearrangement of methyl β-pyrrolidinyl-γ-allyloxy-(E)-2-butenoate. Expeditious synthesis of 5-alkenyl-4-pyrrolidin-1-yl-5H-furan-2-ones. Tetrahedron Lett. 2004, 45, 1865–1868. [CrossRef]
- Yatsuzuka, K.; Kawasaki, M.; Shirai, R., Enantioselective [2,3]-Wittig Rearrangement of Carboxylic Acid Derived Enolates by Tetradentate Chiral Lithium Amide. Synlett 2023, 34, 1727–1731. [CrossRef]
- Brinkerhoff, R. C.; Tarazona, H. F.; de Oliveira, P. M.; Flores, D. C.; Montes D'Oca, C. D. R.; Russowsky, D.; Montes D'Oca, M. G., Synthesis of β-ketoesters from renewable resources and Meldrum's acid. RSC Advances 2014, 4, 49556–49559. [CrossRef]
- Kuldová, J.; Hrdý, I.; Svatos, A., Defense Secretion of Prorhinotermes simplex: Toxicity to Insecticide Susceptible and Resistant House Fly. J. Chem. Ecol. 1999, 25, 657–662. [CrossRef]
- CrysAlis PRO; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cristallogr. 2009, 42, 339–341. [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. https://doi: 10.1107/S2053273314026370.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [CrossRef]
Scheme 1.
Synthesis of β-keto ester building blocks 2a–i.
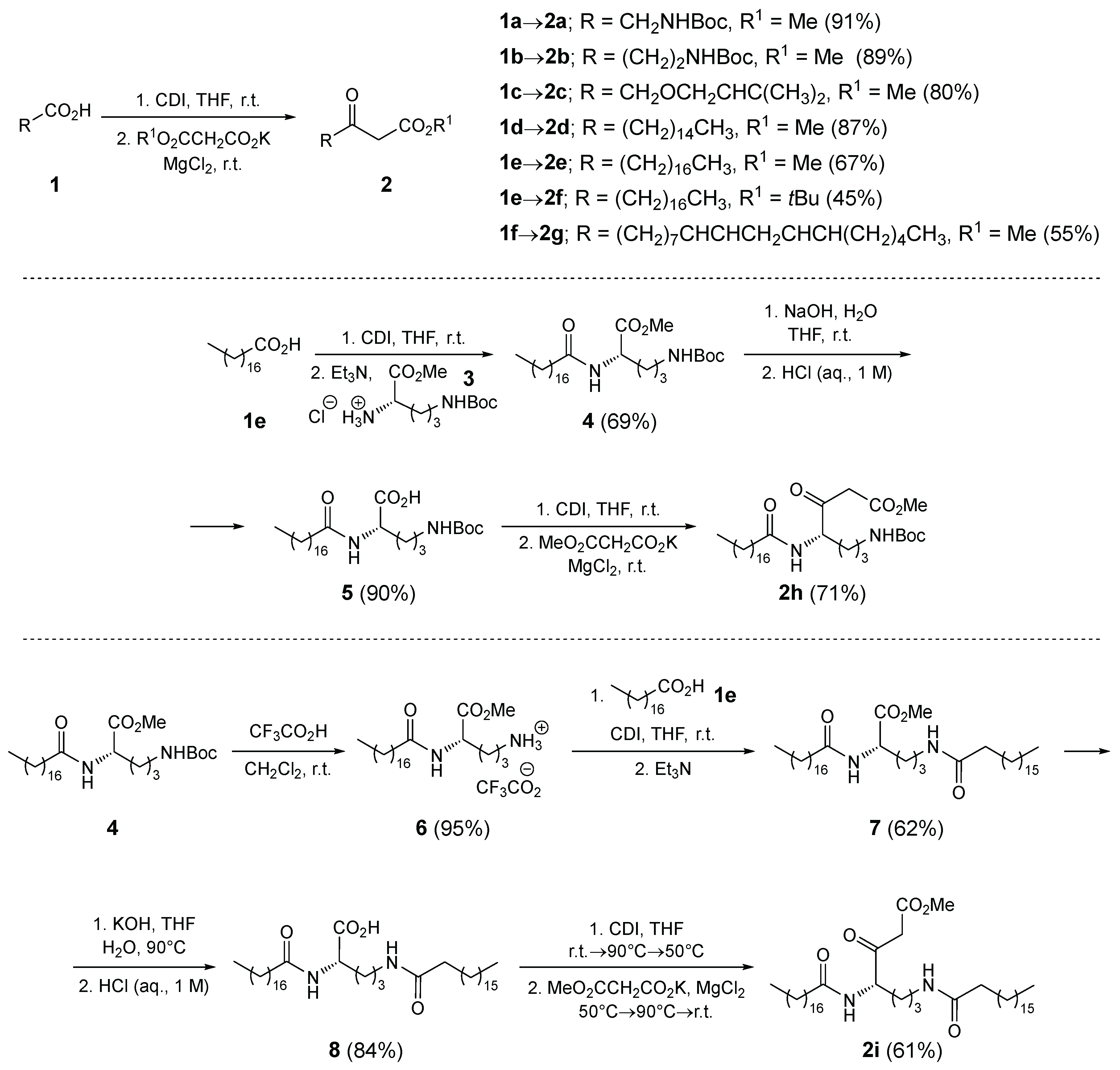
Scheme 2.
Synthesis of the starting building blocks: pyrrolones 11a–c and the fatty acid-derived nitroalkene 16.
Scheme 2.
Synthesis of the starting building blocks: pyrrolones 11a–c and the fatty acid-derived nitroalkene 16.
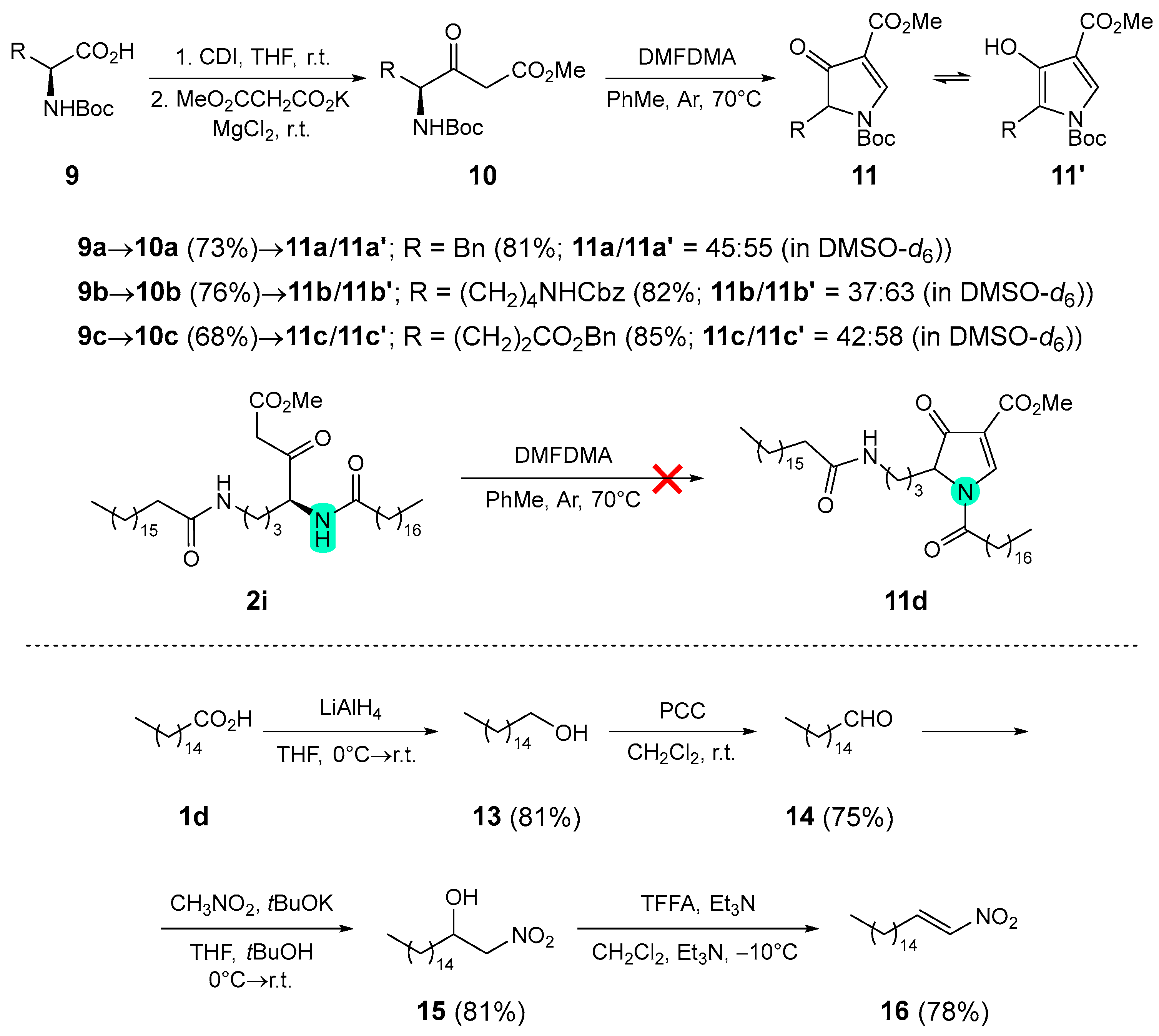
Scheme 3.
Organocatalyst and solvent optimization for the addition of Boc-β-alanine-derived β-keto ester 2b to trans-β-nitrostyrene (12). 2b (0.3 mmol), 12 (0.2 mmol), cat. I and VIII (10 mol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24 h. Catalyst X was used to prepare rac-17b.
Scheme 3.
Organocatalyst and solvent optimization for the addition of Boc-β-alanine-derived β-keto ester 2b to trans-β-nitrostyrene (12). 2b (0.3 mmol), 12 (0.2 mmol), cat. I and VIII (10 mol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24 h. Catalyst X was used to prepare rac-17b.
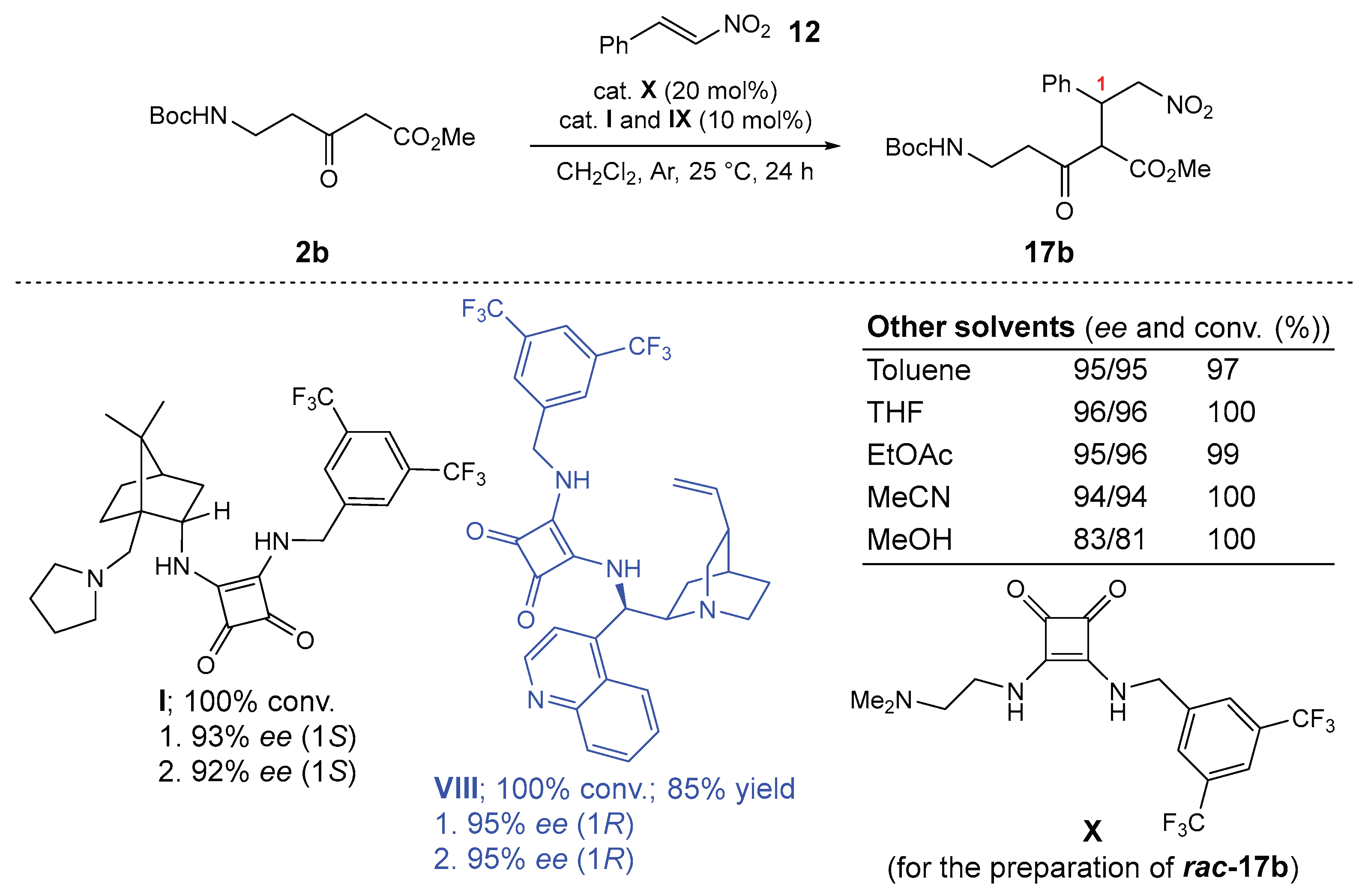
Scheme 4.
Scope for the addition of β-keto ester 2 to trans-β-nitrostyrene (12) and fatty acid-derived nitroalkene 16; if not stated otherwise, organocatalyst VIII was used, yielding (1R)-enantioselectivity. Catalyst X was used to prepare the racemic products rac-17a–l. a) 2 (0.3 mmol), 12 or 16 (0.2 mmol), cat. X (20 mol%), I (10 mol%), and VIII (10 mol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24–48 h. b) 2 (0.2 mmol), 12 or 16 (0.3 mmol), cat. X (20 mol%), I (10 mol%), and VIII (10 mmol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24–48 h. c) In DMSO-d6. d) In CDCl3.
Scheme 4.
Scope for the addition of β-keto ester 2 to trans-β-nitrostyrene (12) and fatty acid-derived nitroalkene 16; if not stated otherwise, organocatalyst VIII was used, yielding (1R)-enantioselectivity. Catalyst X was used to prepare the racemic products rac-17a–l. a) 2 (0.3 mmol), 12 or 16 (0.2 mmol), cat. X (20 mol%), I (10 mol%), and VIII (10 mol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24–48 h. b) 2 (0.2 mmol), 12 or 16 (0.3 mmol), cat. X (20 mol%), I (10 mol%), and VIII (10 mmol%), anhydrous CH2Cl2 (1.0 mL), 25 °C, Ar, 24–48 h. c) In DMSO-d6. d) In CDCl3.
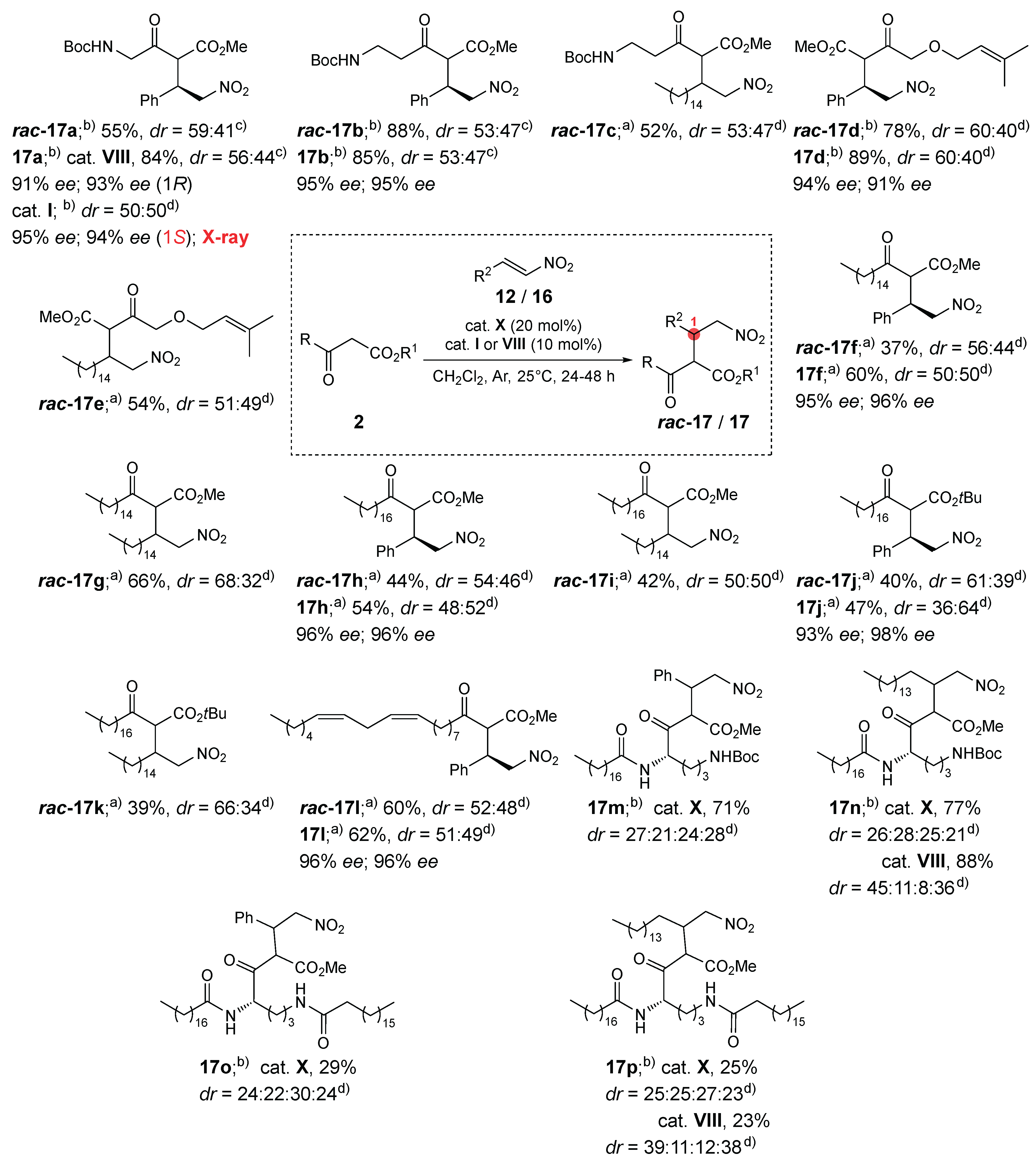
Scheme 5.
Scope for the addition of pyrrolones 11 and tetronic acid (19) to fatty acid-derived nitroalkene 16. a) 11 or 19 (0.3 mmol), 16 (0.2 mmol), cat. X (20 mol%) or I (10 mol%), anhydrous PhMe or CH2Cl2 (1.0 mL), 25 °C, Ar, 48–72 h. b) In CDCl3.
Scheme 5.
Scope for the addition of pyrrolones 11 and tetronic acid (19) to fatty acid-derived nitroalkene 16. a) 11 or 19 (0.3 mmol), 16 (0.2 mmol), cat. X (20 mol%) or I (10 mol%), anhydrous PhMe or CH2Cl2 (1.0 mL), 25 °C, Ar, 48–72 h. b) In CDCl3.
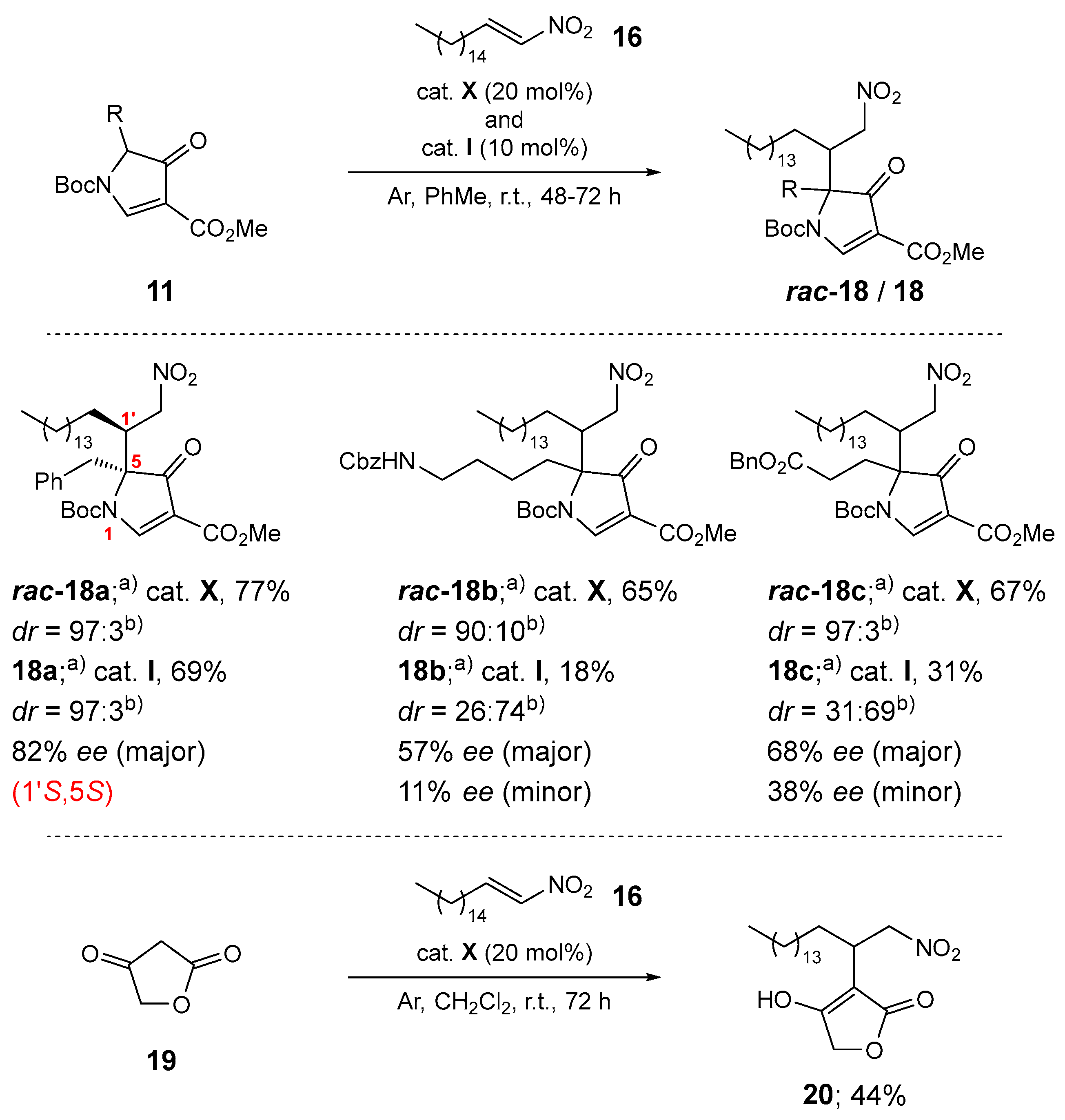
Figure 1.
Molecular structure of the (1S,2S)-stereoisomer of product 17a, synthesized using catalyst I. Thermal ellipsoids are shown at 50% probability.
Figure 1.
Molecular structure of the (1S,2S)-stereoisomer of product 17a, synthesized using catalyst I. Thermal ellipsoids are shown at 50% probability.
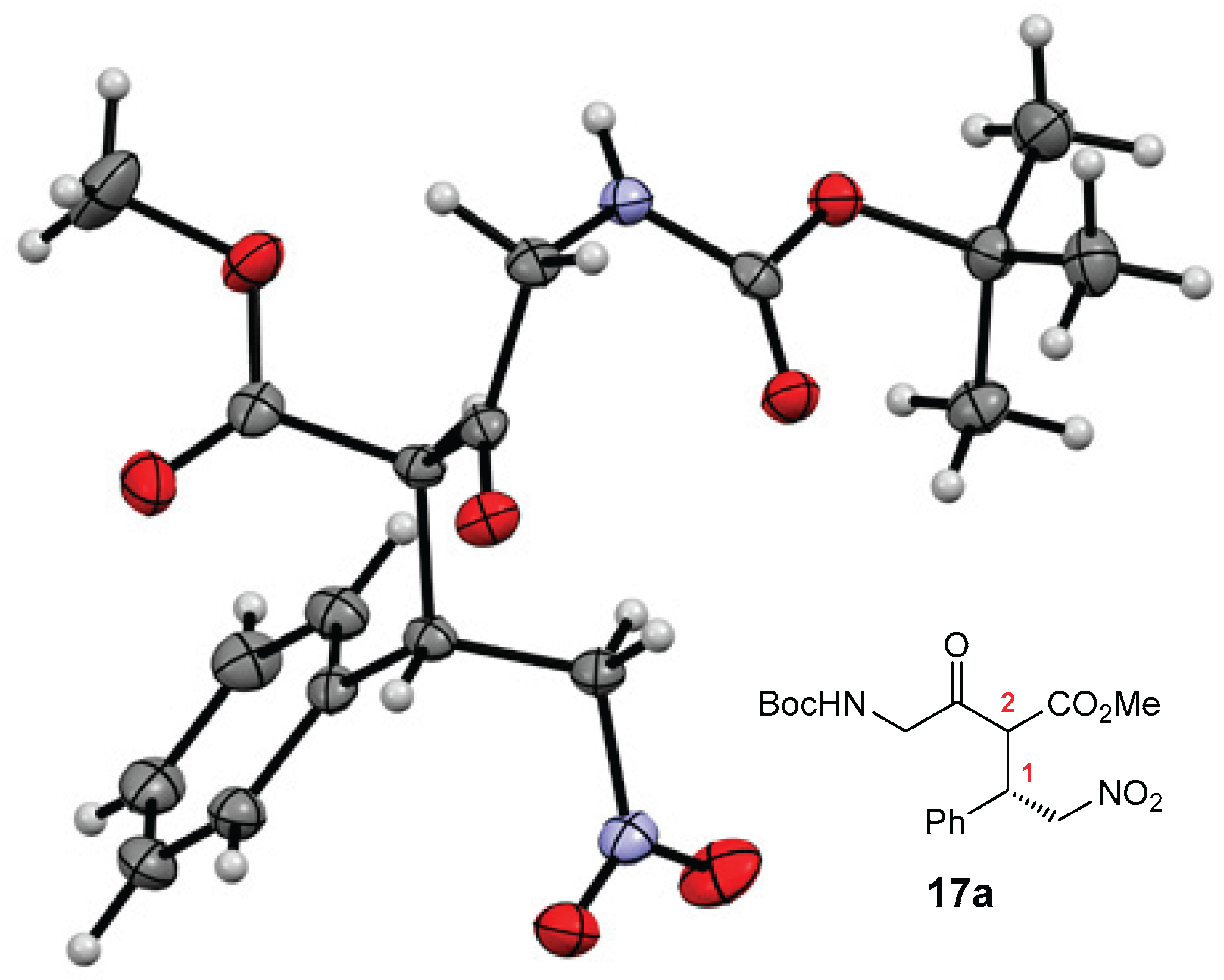
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



