
Submitted:
16 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) and next-generation al-logeneic T cell therapies are curative options for hematologic malignancies, but their effi-cacy is often limited by graft-versus-host disease (GvHD), a complex syndrome resulting from dysregulated donor–host immune interactions. GvHD is mediated by interconnected signaling pathways that regulate T cell activation, metabolic and epigenetic program-ming, tissue-specific migration, stromal-immune interactions, and fibrotic remodeling. Therapeutic agents targeting key pathways, including ruxolitinib (JAK1/2), ibrutinib (BTK/ITK), and belumosudil (ROCK2), can modulate inflammatory responses while pre-serving graft-versus-tumor (GvT) activity. Emerging technologies, such as CRISPR-based genome editing, single-cell and spatial multi-omics, and AI-driven network modeling, enable patient-specific mapping of signaling hierarchies, prediction of disease trajectories, and identification of actionable targets. Integration of these approaches supports precise modulation of immune circuits, offering alternatives to broad immunosuppression. These insights provide a framework for next-generation, individualized interventions that pro-mote durable immune tolerance without compromising anti-tumor immunity and high-light rational combination strategies to improve outcomes in allo-HSCT and allogeneic T cell therapies.
Keywords:
allogeneic hematopoietic stem cell transplantation
; allogeneic T cell therapy
; graft-versus-host disease
; signaling pathways
; CRISPR
; multi-omics
1. Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains a curative strategy for hematologic malignancies, selected solid tumors, and non-malignant disorders, primarily via the graft-versus-leukemia/tumor (GvL/GvT) effect mediated by donor-derived immune cells. However, its benefit is frequently offset by graft-versus-host disease (GvHD), a major cause of morbidity and mortality, affecting up to 50% of patients and accounting for 15%–30% of transplant-related deaths (1–5). GvHD also arises in donor lymphocyte infusion (DLI) and engineered allogeneic T cell therapies, where donor T cells recognize host alloantigens and trigger immunotoxicity [6,7,8].
Clinically, GvHD manifests as acute (aGvHD) or chronic (cGvHD). aGvHD develops within 3–12 weeks post-transplant and primarily affects the skin, liver, gastrointestinal (GI) tract, and occasionally lungs or CNS. cGvHD emerges after day 100, resembling systemic autoimmune disease with fibrosis, tissue remodeling, and multi-organ dysfunction [1,9,10]. Despite divergent phenotypes, aGvHD and cGvHD share overlapping immunopathological mechanisms [11,12,13].
GvHD pathogenesis occurs in three interconnected phases. Conditioning-induced tissue damage releases danger-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and alarmins (e.g., Interleukin [IL]-1α, IL-33, and high mobility group box 1 [HMGB1]) with pro-inflammatory cytokines (tumor necrosis factor alpha [TNF-α], IL-1, type I interferons [IFNs], and IL-6) and chemokines (CCL1–5, CCL17, CCL25, CXCL9–11), activating donor and host antigen-presenting cells (APCs) [14,15,16,17]. APCs present host alloantigens via MHC I/II to CD4⁺/CD8⁺ T cells, driving effector differentiation (T helper 1 [Th1], Th17) and tissue injury through cytokines (IFN-γ, IL-2, IL-17, TNF-α) and cytotoxic mechanisms (FasL, perforin, granzymes). cGvHD is dominated by persistent T/B cell activation, B cell activating factor (BAFF)-driven B cell dysregulation, and progressive fibrosis [18,19,20,21].
Mechanistically, GvHD arises from dysregulation of kinase-driven cascades, including Janus kinase/Signal Transducer and Activator of Transcription (JAK/STAT), Bruton’s tyrosine kinase/interleukin-2–inducible T cell kinase (BTK/ITK), phosphoinositide 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR), lck/yes-related novel tyrosine kinase/spleen tyrosine kinase (LYN/SYK) tyrosine kinase, mitogen-activated protein kinase (MAPK), and downstream axes such as NFAT, and NF-κB, which collectively govern activation, differentiation, migration, and tissue remodeling of alloreactive immune cells [22,23].
Current therapies, such as corticosteroids and calcineurin inhibitors, remain standard but are limited by steroid-refractory disease (SR-GvHD)[24,25,26]. Targeted therapies, including ruxolitinib (JAK1/2), ibrutinib (BTK/ITK), belumosudil (rho-associated coiled-coil protein kinases2 [ROCK2]), and remestemcel-L (allogeneic mesenchymal stromal cell [MSC] product), highlight the potential of precision-based, pathway-specific interventions [27,28,29,30].
Despite these advances, outcomes remain suboptimal, emphasizing the need for deeper mechanistic insight. Building on our prior mapping of extracellular immunoregulatory networks [21], this review focuses on intracellular signaling pathways that integrate upstream cues and dictate effector differentiation, inflammatory amplification, tissue injury, and fibrosis. Table 1 summarizes clinical trials targeting these pathways, illustrating rapid translational progress toward signaling-directed therapy. This signaling-centric framework sets the stage for the Future Therapeutic Directions section, where multi-omics, AI-guided modeling, and CRISPR-based engineering are discussed as strategies to recalibrate dysregulated signaling in a patient-specific manner.

Table 1.
Selected clinical trials of immune signaling molecules in acute and chronic GvHD.
| Target | Reagent | Clinical trial description | Trial number | Reference |
|---|---|---|---|---|
|
JAK/STAT |
Ruxolitinib Ruxolitinib Ruxolitinib Tacrolimus + Ruxolitinib + Methotrexate Itacitinib Itacitinib + PTCy Baricitinib Belumosudil Belumosudil |
Phase 2 single-arm trial in SR-aGvHD; 55% ORR at day 28, 73% best ORR, durable responses, manageable safety profile. Phase III trial in SR-aGvHD; higher day-28 ORR with ruxolitinib vs BAT (62% vs 39%) and durable responses. Phase III randomized trial in SR-cGvHD; superior week-24 ORR vs BAT (50% vs 26%), durable responses, manageable safety. Phase III, randomized trial in allo-PBSC transplant; 24-month GVHD-free survival, aiming to assess whether addition of ruxolitinib to standard prophylaxis improves long-term GVHD control. Phase 1, open-label trial in steroid-naïve and SR aGVHD; Day-28 ORR 79% (200 mg) and 67% (300 mg); manageable safety profile with diarrhea and anemia. Open-label Phase 1 study; low-grade CRS (0–1), no ≥grade 2 CRS; 1-year OS ~80%, NRM ~8%, 1-year moderate/severe cGvHD ~5%. Phase 1/2 study in SR cGvHD; manageable neutropenia/infections, 6-month ORR ~63%, best overall response ~90%, durable at 12 months, steroid-sparing, preserves GvL activity. Phase IIa, open-label dose-finding trial in cGvHD; ORR 65–69%, QoL improvement, corticosteroid reduction, mild cytopenias. Phase II randomized trial in heavily pretreated cGvHD; ORR ~74% (QD) and ~77% (BID) with durable and well-tolerated responses. |
NCT02953678 (REACH1) NCT02913261 (REACH2) NCT03112603 (REACH3) NCT06615050 (BMT CTN 2203) NCT02614612 NCT03755414 NCT02759731 NCT02841995 NCT03640481 |
[48] [28] [47] [52] [155] [51] [38] [30] |
|
BTK/ITK |
Ibrutinib | Multi-center, single-arm study in SR- cGvHD, ORR 67%, durable responses; improved quality of life; led to FDA approval |
NCT02195869 | [29,67] |
|
LYN/SYK |
Fostamatinib (R788) | Phase I study for prophylaxis and treatment of cGvHD after allo-HCT, ORR ≈ 77 %; median duration ~19.3 months; steroid-dose reduced by ~80 % |
NCT02611063 | [85] |
|
PI3K/AKT/mTOR |
Sirolimus+ Tacrolimus + Methotrexate Sirolimus + Tacrolimus ± Methotrexate |
Phase II RCT, RIC allo-HCT in lymphoma; reduced grade II–IV aGvHD 9% vs 25%, no increased overall toxicity Phase II RCT, RIC allo-HSCT with HLA-mismatched; cumulative incidence of grade II–IV aGvHD 36% vs historical ~70%, preserved GvL, no added toxicity |
NCT00928018 NCT01251575 |
[78] [77] |
|
NFAT |
Tacrolimus (FK506) + Methotrexate Cyclosporine A + Methotrexate |
Phase III RCT for aGvHD prophylaxis after allo-HSCT; Reduced grade II–IV aGvHD (17.5%) vs cyclosporine (48%) Phase III trial for aGvHD prophylaxis after allo-BMT; with long-term follow-up demonstrating sustained survival and cGvHD incidence over several years |
|
[100] [99] |
|
NF-κB |
Bortezomib + prednisone Bortezomib |
Single-arm Phase II in SR-cGvHD; ORR ≈ ~80% at 15 wks; steroid-sparing; responses in multiple organ sites; tolerable/expected toxicity profile. Phase I/II, prospective treatment in SR-cGvHD; manageable safety and tolerability; evidence of clinical activity |
NCT00815919 NCT01672229 |
[108] [106] |
| BAT, best available therapy; CR, complete response; GFS, GvHD-free survival; GRFS, GvHD/relapse-free survival; MMF, mycophenolic acid; NRM, non-relapse mortality; OS, overall survival; ORR, overall response rate; ROM, range of motion; MMUD, mismatched unrelated donor; Tac/MTX/Rux, tacrolimus/ methotrexate/ ruxolitinib; PTCy/Tac/MMF, post-transplant cyclophosphamide/ tacrolimus/ mycophenolate mofetil; CRS, cytokine release syndrome; QoL, quality of life; RCT, Randomized Controlled Trial; RIC, reduced intensity conditioning. | ||||
2. Methodology of Literature Selection
A structured literature search was performed across PubMed, Scopus, Web of Science, and Google Scholar (1989–2025) to integrate foundational GvHD biology with emerging mechanistic and technological advances. Search strategies combined major alloimmune signaling pathways (JAK/STAT, LYN/SYK, BTK/ITK, PI3K/AKT/mTOR, NFAT, NF-κB, Notch) with contemporary platforms including CRISPR-based gene editing, single-cell multi-omics, and AI-driven modeling. Studies underwent two-stage screening (title/abstract followed by full-text review), and inclusion was restricted to English-language preclinical, translational, and clinical investigations with mechanistic relevance. This approach enabled a cross-disciplinary synthesis of pathway-level insights, forming a rigorous foundation for the integrative framework presented in this review.
3. Mechanistic Signaling Pathways Underlying GvHD
GvHD arises from a highly orchestrated network of intracellular signaling pathways that coordinate immune cell activation, differentiation, cytokine production, and tissue remodeling. Although acute and chronic GvHD share overlapping networks, discrete signaling nodes integrate upstream inflammatory cues to drive context-specific pathogenic T and B cell responses [22,31,32].
Central to alloimmune regulation are interconnected signaling modules, including JAK/STAT, BTK/ITK, LYN/SYK, PI3K/AKT/mTOR, MAPK, NF-κB, NFAT, and Notch. Each function as a critical integrator, converting cytokine and antigen-receptor signals into coordinated effector programs. For instance, STAT3 downstream of IL-6R and platelet-derived growth factor receptor (PDGF-R) amplifies pro-inflammatory cytokine cascades and drives fibro-inflammatory tissue injury. Antigen receptor–associated kinases (BTK, ITK, SYK, PI3K) reinforce TCR/BCR signaling, sustaining alloreactive responses underlying tissue damage and chronic inflammation [23,32,33].
Mechanistic insights into these pathways have guided therapeutic strategies. JAK inhibitors (ruxolitinib, itacitinib, baricitinib) mitigate cytokine-driven hyperactivation and are effective in SR-GvHD [34,35]. BTK/ITK blockade with ibrutinib disrupts T and B cell activation, forming a cornerstone of cGvHD management [36,37]. ROCK2 inhibition via belumosudil modulates both inflammatory signaling and fibrosis [30,38]. Emerging approaches targeting Notch, NFAT, NF-κB, and PI3K/AKT/mTOR pathways aim to selectively temper alloimmunity while preserving GvL activity [39,40,41,42].
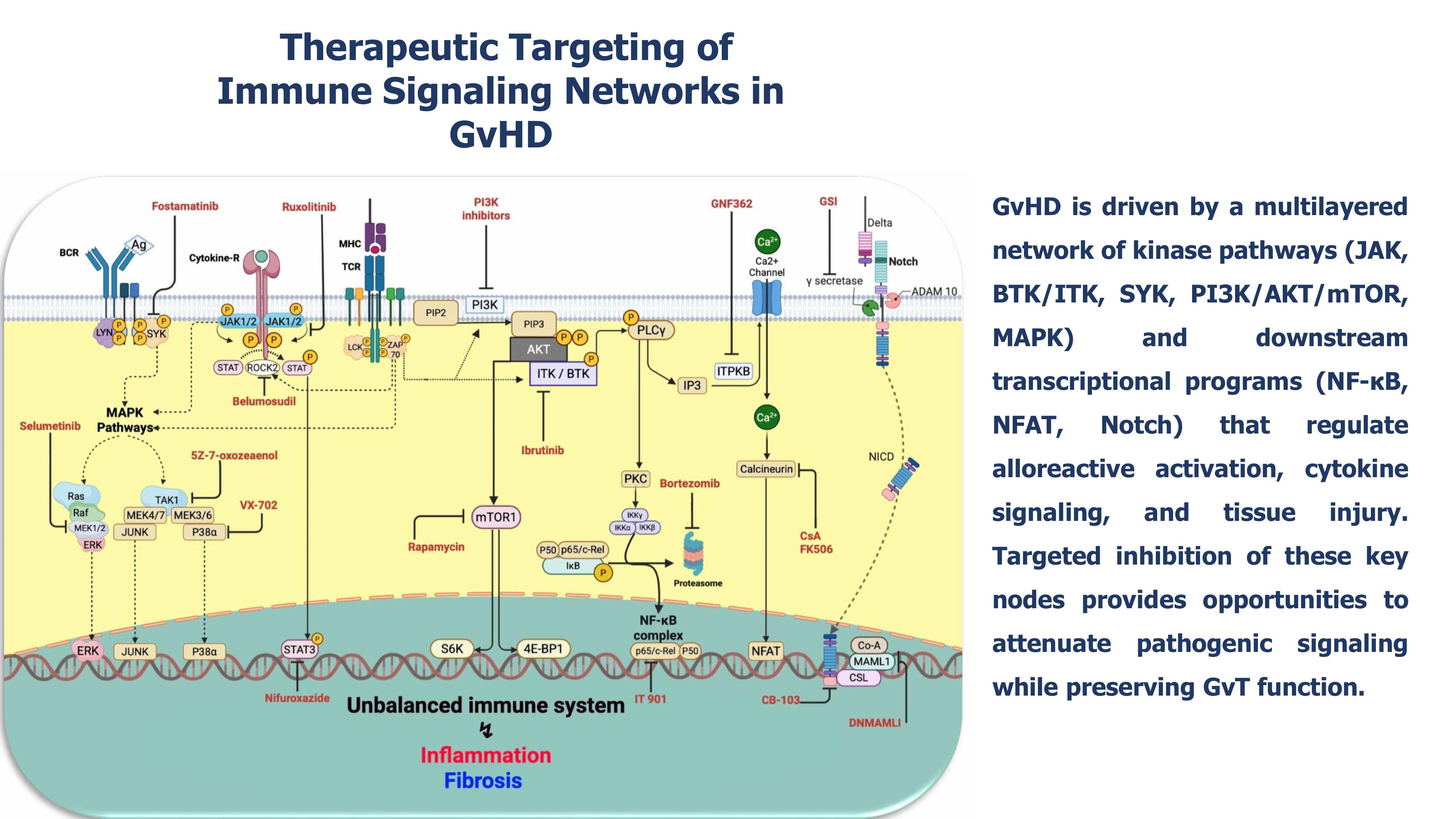
Together, these findings define a conceptual framework in which precise, pathway-specific modulation of intracellular networks recalibrates alloimmune responses, attenuates GvHD, and sustains GvL function. Figure 1 schematically integrates these pivotal signaling circuits, highlighting key molecular nodes and pharmacologic inhibitors, providing a coherent visual blueprint for translational and therapeutic targeting.
Figure 1.
Therapeutic targeting of immune signaling networks in GvHD.
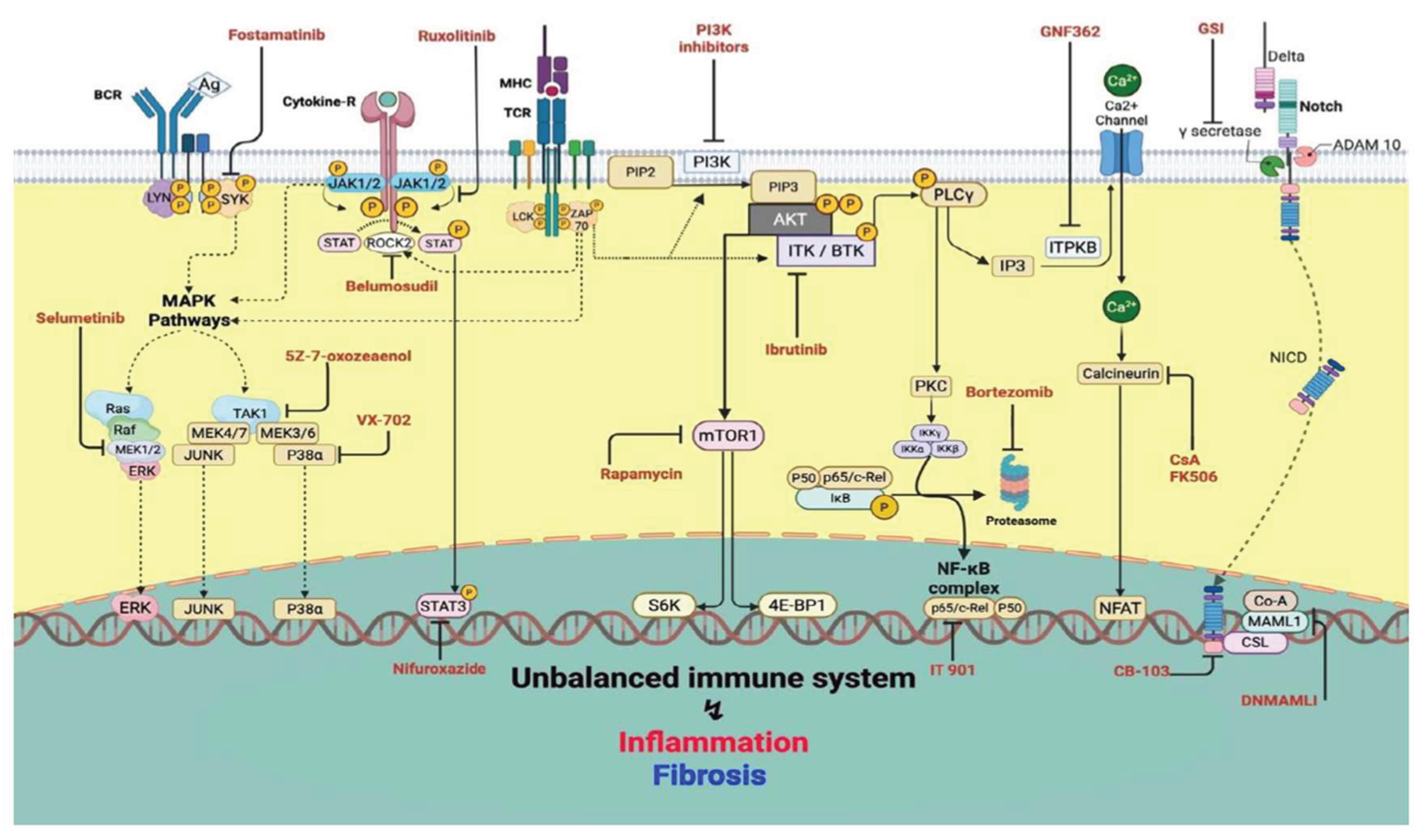
GvHD is sustained by a multilayered signaling architecture integrating kinase-driven cascades and downstream transcriptional programs that collectively regulate alloreactive activation, differentiation, cytokine output, and tissue injury. Key kinase hubs, including JAK, BTK, ITK, and SYK, alongside the PI3K/AKT/mTOR and MAPK (MEK1/2, p38, TAK1) pathways, interface with transcriptional effectors such as NF-κB, Notch, and NFAT to shape pathogenic immune circuits. Therapeutic interventions exploit these nodal vulnerabilities: JAK inhibitors (ruxolitinib, itacitinib, baricitinib), ROCK2 blockade (belumosudil), and selective STAT3 inhibition (nifuroxazide) attenuate cytokine-driven signaling; BTK/ITK (ibrutinib) and SYK inhibitors (fostamatinib) reduce lymphocyte activation; PI3K/mTOR inhibitors (rapamycin and selective PI3K inhibitors) restrict metabolic support and survival; MEK1/2 inhibitors (selumetinib, trametinib), p38α blockade (VX-702), and TAK1 inhibition (5Z-7-oxozeanol) dampen inflammatory amplification and tissue damage. Notch pathway modulation via dominant-negative MAML1, CSL-NICD inhibitors (CB-103), and γ-secretase inhibitors (GSIs); suppression of NF-κB through c-Rel inhibition (IT-901) or proteasome targeting (bortezomib), and NFAT inhibition via calcineurin blockers (CsA, FK506) or ITPKB targeting (GNF362) further disrupt downstream transcriptional programs. This schematic highlights the hierarchical structure of GvHD signaling and the spectrum of opportunities for precise therapeutic intervention across interconnected regulatory layers.
3.1. Therapeutic Targeting of Central Signaling Pathways in GvHD
3.1.1. JAK/STAT
The JAK/STAT pathway is a central intracellular signaling cascade in GvHD, orchestrating immune cell activation, differentiation, and inflammatory amplification. It comprises four JAK kinases (JAK1, JAK2, JAK3, TYK2) and seven STATs (STAT1–4, STAT5a/b, STAT6). Engagement of cytokine or antigen receptors triggers JAK1/2-mediated phosphorylation of STATs, notably STAT1 and STAT3, which dimerize and translocate to the nucleus to regulate gene transcription [22,43,44]. The JAK1/2–STAT1/3 axis drives T cell activation and polarization toward pro-inflammatory Th1/Th17 subsets, supports B cell proliferation and maturation, and enhances APC function via dendritic cells (DCs) activation, maturation, and trafficking. Together, these events amplify tissue inflammation and promote both acute and chronic GvHD [30,31].
Pharmacologic JAK1/2 inhibition with ruxolitinib effectively modulates immune responses in GvHD by dampening inflammatory cytokine signaling and reducing alloreactive T cell trafficking to target tissues [45,46]. This underlies its proven efficacy in SR-aGvHD and emerging use in SR-cGvHD (NCT02953678, NCT02913261, NCT03112603) [28,47,48]. Hematologic toxicities associated with broader JAK2 inhibition have spurred the development of next-generation agents [23,49,50]. Selective JAK1 inhibition with itacitinib and dual JAK1/2 inhibition with baricitinib show promising activity in both SR-aGvHD and SR-cGvHD, with improved tolerability in early-phase trials (NCT02614612, NCT02759731) [51,52].
Accessory modulatory kinases fine-tune STAT3/5 phosphorylation and regulate the Th17/ regulatory T cell (Treg) balance. ROCK2 has emerged as a critical upstream kinase controlling pathogenic Th17 responses, Treg homeostasis, and downstream processes such as tissue fibrosis and inflammation in cGvHD. Pharmacologic ROCK2 inhibition with belumosudil (KD025) suppresses STAT3 activation, restores Th17/Treg equilibrium, and mitigates tissue injury [53,54,55]. These effects were demonstrated in phase II randomized trials (NCT02841995, NCT03640481) and led to FDA approval for SR-cGvHD [30,38].
Beyond the canonical JAK1/2–STAT1/3 axis, additional regulatory checkpoints within STAT3 provide potential therapeutic targets. The endogenous inhibitor PIAS3 restrains effector T and B cell activation, attenuating aGvHD [56], while pharmacologic STAT3 blockade with nifuroxazide similarly reduces inflammation [57]. Parallel strategies targeting JAK3, including baricitinib and CS12192, promote Treg expansion and facilitate intestinal tissue repair, alleviating both acute and chronic GvHD [58,59]. These findings underscore the potential of precise, pathway-specific modulation of the JAK/STAT network to recalibrate alloimmunity while preserving beneficial immune function.
3.1.2. BTK/ITK
BTK and ITK pathways act as central hubs downstream of antigen receptor signaling in B and T cells. Upon TCR and BCR engagement, proximal kinases including LYN and LCK, followed by SYK and ZAP-70, activate BTK and ITK. These kinases phosphorylate and activate phospholipase Cγ2 (PLCγ2), which induces calcium mobilization and triggers NF-κB, NFAT, and MAPK activation. This coordinated signaling regulates lymphocyte survival, differentiation, metabolism, migration, and inflammatory cytokine production [60,61,62,63].
Preclinical studies show that targeted inhibition of BTK/ITK reshapes alloreactive immunity. Blockade with ibrutinib or selective ITK inhibitors reduces effector T cell expansion, limits B cell activation and antigen presentation, and diminishes tissue-damaging inflammatory mediators, while partially preserving GvL activity in murine models of both acute and chronic GvHD [64,65].
Mechanistic insights have translated into clinical application. Ibrutinib, a first-in-class dual BTK/ITK inhibitor, was tested in a multicenter phase 1b/2 study for patients with SR-cGvHD (NCT02195869). The trial demonstrated durable clinical responses across multiple organs with an acceptable safety profile [29,66,67]. These results led to FDA approval, establishing BTK/ITK blockade as the first clinically validated, pathway-specific therapy for cGvHD.
3.1.3. PI3K/AKT/mTOR
The PI3K/AKT/mTOR pathway is a central regulator of T and B cell survival, proliferation, differentiation, metabolic programming, and migration. Activation of PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), recruiting AKT and activating mTORC1/2 complexes. These complexes orchestrate effector T cell differentiation, memory formation, and cytokine production [68,69]. Dysregulation of this pathway drives GvHD by promoting expansion of alloreactive T cells and excessive inflammatory cytokine release [39,70].
Preclinical studies demonstrate that pharmacologic inhibition of PI3K, AKT, or mTOR mitigates GvHD severity. Sirolimus, an mTOR inhibitor, limits donor T cell proliferation, suppresses APC activation, and preserves Treg function [39,70]. Dual PI3K/mTOR inhibitors, such as BEZ235 and dactolisib, reduce pathogenic cytokine production, T cell infiltration, and tissue injury while maintaining GvL activity [71,72]. Isoform-selective PI3Kδ inhibitors, including idelalisib, specifically dampen effector T cell responses and B cell–mediated antibody production, offering targeted immunomodulation with improved tolerability [73].
Clinically, sirolimus, administered alone or with calcineurin inhibitors, has effectively reduced both the incidence and severity of aGvHD while preserving robust GvL responses [74,75,76]. Phase II trials (NCT00928018, NCT01251575) confirmed these results, showing significant reductions in grade II–IV aGvHD without increased overall toxicity [77,78]. Careful monitoring is required due to potential adverse effects [74,76]. Collectively, preclinical and early translational studies highlight the potential of dual PI3K/mTOR and PI3Kδ-selective inhibition as a precise prophylactic and therapeutic strategy in allogeneic transplantation [71,73].
3.1.4. LYN/SYK
The LYN/SYK axis is a central regulator of lymphocyte and myeloid cell function, controlling activation, differentiation, and cytokine production. Upon BCR or TCR engagement, LYN phosphorylates immunoreceptor tyrosine-based activation motifs (ITAMs) motifs on receptor subunits, recruiting SYK. SYK then activates downstream PLCγ, PI3K/AKT, and MAPK pathways to drive proliferation, effector differentiation, and inflammatory responses. Simultaneously, LYN transmits inhibitory signals via ITIM-containing receptors (e.g., CD22, FcγRIIB), recruiting phosphatases such as SH2-containing tyrosine phosphatase-1 (SHP-1) and inositol phosphatase (SHIP-1), providing essential negative feedback to prevent excessive activation [79,80,81,82].
Dysregulation of LYN/SYK promotes both acute and chronic GvHD. SYK-dependent B cell activation enhances germinal center responses, activates dendritic cells, and triggers fibrosis through pathogenic cytokine production [79,83]. Preclinical murine studies show that SYK inhibition with fostamatinib (R788) reduces disease severity by depleting plasmablast-like B cells (IgD⁻CD38high) and inducing apoptosis in human cGvHD B cells in vitro. In sclerodermatous cGvHD (Scl-cGvHD) models, SYK blockade mitigates fibrosis and improves skin pathology [79,83,84].
Building on these findings, fostamatinib was evaluated in a phase I trial (NCT02611063) in cGvHD patients post-allo-HSCT. It demonstrated meaningful clinical activity with an acceptable safety and tolerability profile. Correlative immune profiling revealed that SYK inhibition modulates activated B cell and myeloid subsets while preserving overall B cell reconstitution [85]. Second-generation selective SYK inhibitors, such as entospletinib, showed additional promise in preclinical ocular and cutaneous cGvHD models, reducing pathogenic immune infiltration and improving disease outcomes while maintaining immune recovery [86]. These data support SYK-targeted therapy as a biologically rational and mechanistically precise strategy for managing cGvHD.
3.1.5. MAPK
MAPK pathway is a highly conserved signaling cascade that regulates immune cell proliferation, survival, differentiation, and cytokine production. It consists of three primary branches: ERK1/2, JNK1/2/3, and p38 MAPKs, which respond to growth factors, cellular stress, and antigen receptor activation. In both lymphoid and myeloid cells, MAPK pathways integrate signals from TCR, BCR, Toll-like receptors (TLR), and cytokine receptors, orchestrating effector functions, inflammatory responses, and apoptosis. Dysregulation of these pathways contributes to aberrant immune activation and has been implicated in autoimmune and alloimmune disorders, including GvHD [87,88,89].
In murine models of allo-HSCT, pharmacologic inhibition of MAPK pathway components has been shown to modulate GvHD pathology. MEK1/2 inhibitors, such as selumetinib and trametinib, suppress donor T cell proliferation, reduce inflammatory cytokine production, and limit tissue injury, while preserving regulatory T cells and GvT effects [87,90,91]. Similarly, p38α MAPK inhibitors (e.g., VX-702) and TAK1 inhibitors (e.g., 5Z-7-oxozeanol) mitigate fibrosis, inflammation, and organ damage in both acute and chronic preclinical GvHD models [92,93].
Collectively, these studies highlight the therapeutic potential of targeting multiple nodes within the MAPK cascade for both prophylaxis and treatment of GvHD. Although no MAPK inhibitors have yet reached late-phase clinical trials for GvHD, robust preclinical and translational evidence supports ongoing exploration of this pathway as a mechanistically rational target.
3.1.6. NFAT
The NFAT pathway serves as a central Ca²⁺/calcineurin-dependent axis controlling donor T cell activation and effector differentiation. Upon TCR engagement, inositol 1,4,5-trisphosphate (IP₃)-mediated Ca²⁺ influx activates calcineurin, leading to NFAT1/2 nuclear translocation; sustained activation promotes pathogenic alloreactivity in GvHD [94,95].
In experimental models, selective deletion of NFAT1 or NFAT2 in donor T cells limits proliferation and inflammatory cytokine output while preserving GvL activity [41]. Upstream regulation is mediated by inositol 1,4,5-trisphosphate 3-kinase B (Itpkb), which converts IP₃ to IP₄ and constrains Ca²⁺ flux and NFAT activation. Pharmacologic inhibition using selective Itpkb inhibitors (e.g., GNF362) or genetic ablation of Itpkb markedly attenuates both acute and chronic GvHD without compromising anti-tumor immunity [23,97,98], positioning the IP₃/IP₄–NFAT axis as a promising target for selective alloimmune modulation.
Clinically, calcineurin inhibitors such as cyclosporine A and tacrolimus remain the standard of care for GvHD prophylaxis by broadly suppressing NFAT signaling. Although effective in reducing aGvHD incidence and severity, such global inhibition may impair GvL responses [24,99,100]. Overall, NFAT and its upstream regulators, particularly Itpkb, represent mechanistically tractable nodes integrating calcium signaling with T cell–mediated alloimmunity, offering promising opportunities to develop next-generation therapies that mitigate GvHD while preserving GvT effects.
3.1.7. NF-κB
The NF-κB family of transcription factors, including p50 (NF-κB1), p52 (NF-κB2), p65/RelA, RelB, and c-Rel, plays a pivotal role in immune regulation by controlling lymphocyte activation, cytokine production, survival, and inflammatory responses. Dysregulated NF-κB signaling contributes to pathogenic inflammation in allo-HCT, where donor T cell activation and differentiation during GvHD rely heavily on this pathway [101,102,103].
Preclinical evidence indicates that selective blockade of the c-Rel subunit with small-molecule inhibitors such as IT-901 suppresses alloreactive T cell activation while preserving regulatory T cells and maintaining GvT responses. Proteasome inhibition with bortezomib also indirectly suppresses NF-κB by preventing IκBα degradation, thereby limiting alloreactive T cell and APC expansion, reducing inflammatory cytokine release, and promoting Treg enrichment [102,104,105].
Early clinical studies have positioned bortezomib as a mechanistically guided therapy for SR-cGvHD. In a Phase I/II trial (NCT01672229), bortezomib demonstrated acceptable safety and manageable tolerability, with signals of clinical activity across multiple organ systems [106]. Additional Phase I/II trials, including monotherapy (NCT00815919) and combination approaches, have likewise shown promising efficacy with controlled toxicity [107,108]. Although current clinical findings are encouraging, larger randomized trials are needed to determine the long-term efficacy and safety of NF-κB–directed therapies in GvHD.
3.1.8. Notch
The Notch pathway is a conserved, contact-dependent signaling cascade that links ligand engagement to rapid transcriptional reprogramming. Upon interaction with Delta-like (DLL1, DLL3, DLL4) or Jagged (JAG1, JAG2) ligands, NOTCH1–4 receptors undergo proteolytic cleavage to release the Notch intracellular domain (NICD), which translocates to the nucleus and complexes with RBP-Jκ and MAML co-activators to induce target gene expression. Through these mechanisms, Notch signaling shapes T cell differentiation, cytokine production, and effector programming [40,109,110,111,112].
Extensive preclinical evidence identifies canonical Notch signaling as a central driver of GvHD. Disruption of Notch-dependent transcription using dominant-negative MAML1 expressed in donor T cells markedly reduces acute GvHD in murine models by expanding Tregs and suppressing inflammatory cytokines, including TNF-α, IFN-γ, and IL-2 while preserving GvL activity [40,113]. Antibody-mediated blockade of DLL1 and DLL4 similarly limits intestinal T cell trafficking and improves survival in mouse and non-human primate models without compromising GvL responses [114,115,116].
Therapeutic targeting of Notch signaling remains challenging due to toxicity concerns. γ-secretase inhibitors (GSIs), which prevent NICD release, effectively suppress Notch activation but induce severe intestinal epithelial injury in preclinical GvHD systems, restricting their clinical applicability [40,114]. To overcome these limitations, more selective approaches are being developed. These include CSL–NICD inhibitors such as CB-103 and ligand- or receptor-specific monoclonal antibodies. Early clinical studies of CB-103 in NOTCH-driven T cell acute lymphoblastic leukemia (T-ALL) have demonstrated biological activity and a favorable safety profile, supporting its potential translation to GvHD therapeutics [40,117].
3.2. Other Signaling Pathways
In addition to the major signaling axes driving GvHD, several auxiliary pathways shape alloimmune activation, tissue injury, and repair. These include the Wnt/β-catenin network, endoplasmic reticulum (ER) stress–responsive IRE1α/XBP-1 axis, β-adrenergic receptor (β-AR)–mediated cyclic adenosine monophosphate/Protein kinase A (cAMP/PKA) signaling, PKC-dependent nodes embedded within the TCR pathway, and Ikaros regulatory axis.
The Wnt/β-catenin pathway orchestrates cell proliferation, differentiation, and tissue regeneration, with dysregulation in donor T cells, APCs, intestinal epithelium, and MSCs disrupting homeostasis, driving inflammation, and skewing MSC fate toward adipogenesis at the expense of osteogenesis [118,119,120]. LGR5, a receptor for R-spondin ligands, sustains Wnt signaling in intestinal stem cells vulnerable to conditioning-induced injuries. Therapeutic interventions, such as R-spondin1 administration or LGR5⁺ organoid transplantation, enhance epithelial repair, reduce inflammation, and preserve GvL, whereas selective Wnt inhibition can mitigate fibrosis in cGvHD, highlighting its context-dependent effects [118,119,120,121,122].
IRE1α/XBP-1 signaling axis constitutes a critical molecular link between ER stress and the immunofibrotic responses characteristic of cGvHD. Pharmacological inhibition of IRE1α attenuates pro-inflammatory cytokine production, suppresses tissue fibrosis, and mitigates multiorgan injury in murine models, while retaining protective GvL activity [123]. Downstream, the spliced transcription factor XBP-1s reinforces B cell–driven pathology by constraining regulated IRE1α-dependent decay (RIDD), thereby promoting the survival and persistence of pathogenic B cell subsets [124]. Together, these findings establish the IRE1α/XBP-1 pathway as a mechanistically actionable and clinically relevant target for therapeutic intervention in cGvHD.
β-AR signaling, predominantly acting through cAMP/PKA activation, emerges as a critical modulatory pathway shaping alloimmune responses in GvHD. Engagement of β₂-ARs on donor T cells skews differentiation toward regulatory and less inflammatory phenotypes, thereby attenuating aGvHD without impairing the beneficial GvL response [125,126,127]. Beyond donor immunity, physiological adrenergic stressors, such as cold-induced sympathetic activation, similarly suppress GvHD through host β₂-AR–dependent mechanisms, highlighting the broader neuroimmune interface in transplant biology [128]. At the mechanistic level, β-AR–driven PKA signaling negatively regulates innate antiviral pathways and dampens inflammatory activation [126], collectively positioning the β-AR/PKA axis as a promising and underexplored therapeutic target for selective immune modulation in allo-HCT.
PKC isoforms, particularly PKCθ and PKCα, represent a critical signaling axis embedded within the broader TCR pathway and function as key regulators of downstream effector activation. Genetic ablation or pharmacological inhibition of these isoforms markedly suppresses donor T cell–driven GvHD without compromising GvL activity or anti-microbial immunity [129,130]. Positioned as a discrete yet functionally central signaling node, PKC offers a mechanistically precise target for selective immune modulation aimed at uncoupling pathogenic alloresponses from beneficial antitumor and anti-infectious immunity.
Ikaros (IKZF1), a zinc-finger transcription factor indispensable for lymphocyte differentiation, has emerged as a key regulator of alloimmune activation in GvHD. In murine models, loss of Ikaros within host APCs amplifies donor T cell priming, leading to a more severe disease and increased mortality [131,132]. Clinically, elevated Ikaros expression correlates with a higher incidence of moderate to severe cGvHD, underscoring its relevance beyond experimental systems [133]. Together, these findings position Ikaros as both a biomarker of pathogenic alloimmunity and a mechanistic node within the Ikaros–Notch axis, highlighting its potential as a therapeutic target for selective immune modulation in GvHD.
Collectively, these auxiliary pathways, including Wnt/β-catenin, ER stress–responsive IRE1α/XBP-1, β-AR–driven cAMP/PKA signaling, PKC nodes within TCR signaling, and the Ikaros regulatory network, form key layers of alloimmune regulation. Although largely preclinical, their mechanistic relevance underscores their potential as therapeutic targets in GvHD.
4. Future Therapeutic Directions
Rapid progress in single-cell and spatial multi-omics, AI-based immune network modeling, and CRISPR genome engineering is reshaping next-generation therapeutic strategies for GvHD. Together, these technologies now establish an integrated translational workflow that enables: (i) systematic discovery of cell- and tissue-specific signaling hubs, (ii) computational prioritization of targets using patient-tailored immune network models, and (iii) functional pathway modulation through genome engineering or rational pharmacology, validated within iterative, closed-loop experimental systems.
4.1. Data-driven discovery of actionable signaling nodes
Single-cell and spatial multi-omics profiling of donor grafts has revealed substantial heterogeneity among donor-derived immune subsets, including mucosa-associated invariant T (MAIT) cells and tissue-resident T and B lymphocytes, providing mechanistic insight into organ-specific GvHD [134,135,136]. Integrated regulatory analyses highlight checkpoints such as SOCS1, which constrain T cell activation and correlate with reduced GvHD severity in preclinical models [136]. Complementary metabolomic studies further identify glycerophospholipid signatures predictive of aGvHD risk, offering quantitative biomarkers for patient stratification [134]. Collectively, these transcriptional, regulatory, and metabolic layers converge on biologically coherent and potentially druggable signaling nodes suitable for precision intervention.
4.2. Computational prioritization using AI-driven modeling and immune digital twins
Regulatory network inference tools such as SCENIC reconstruct cell type-specific regulons and support in silico simulation of gene or pathway perturbations [137,138]. When integrated with multi-omics datasets and patient metadata, immune digital twin models capture alloimmune dynamics, forecast disease trajectories, and quantitatively rank high-value signaling pathways for therapeutic targeting [139,140,141]. These computational pipelines function as mechanistically informed triage systems that guide CRISPR perturbation screens and rational combinatorial therapy design, while substantially reducing experimental burden.
4.3. Precision genome engineering for pathway-level modulation
CRISPR/Cas platforms enable targeted modulation of dysregulated signaling pathways in allogeneic T cells [142]. Deletion of MIR155HG reduces aGvHD severity in xenogeneic systems by dampening pathogenic inflammatory signaling [143]. One-step TCRα/β knockout eliminates alloreactivity without compromising T cell fitness, whereas emerging knock-in and epigenetic stabilization strategies enhance Treg lineage stability independently of FOXP3 [144,145,146]. Integrated analyses suggest that combining TCR deletion, antigen-specific redirection, and reinforcement of tolerogenic programs provides a scalable blueprint for next-generation pathway-focused cellular therapies that maintain GvL activity [146,147].
4.4. Closed-loop experimental validation using single-cell perturbation platforms
High-throughput pooled CRISPR screens coupled with single-cell readouts, such as Perturb-seq and compressed Perturb-seq, enable parallel, high-resolution assessment of thousands of genetic perturbations [149,150,151]. Integration of these platforms with humanized xenograft or organoid co-culture systems creates a closed-loop workflow that links computational target nomination to functional testing, iterative regulatory network refinement, and systematic evaluation of how pathway-targeted interventions modulate both GvHD pathology and GvL responses.
4.5. Translational roadmap: integrating safety, specificity, and immunological fidelity
Clinical translation of genome-engineered, pathway-targeted therapies requires comprehensive evaluation of specificity and genomic integrity. gRNA design tools such as GuideScan2, alongside off-target detection platforms including GUIDE-seq and CIRCLE-seq, provide genome-wide maps of unintended Cas9 activity [152,153]. Long-read sequencing technologies further detect structural variants that may escape short-read approaches [154].
In parallel, maintaining GvL/GvT activity while suppressing GvHD remains essential. Multi-omics biomarkers and patient-specific immune digital twins support refined patient selection, optimized dosing, and precise temporal coordination of pathway-focused therapeutic interventions. Collectively, the convergence of multi-omics profiling, AI-driven immune network modeling, and CRISPR-enabled genome engineering, anchored in robust safety frameworks, establishes a precision, signaling-centric roadmap for next-generation immunoregulation in allogeneic transplantation and cellular therapies
5. Conclusions
GvHD remains a major barrier to the curative potential of allo-HSCT and emerging allogeneic T cell therapies, arising from integrated immune signaling networks rather than isolated cytokines or cell types. Mapping core pathways, including JAK/STAT, PI3K/AKT/mTOR, NFAT, NF-κB, and MAPK, has shifted understanding toward a mechanistic framework in which dysregulated signaling hierarchies collectively drive alloreactivity, tissue injury, and fibrosis.
Pathway-targeted therapies, including JAK1/2 inhibitors, ROCK2 blockers, BTK/ITK modulators, and mTOR-directed agents, have demonstrated clinically meaningful efficacy, underscoring the translational potential of signaling-centric approaches that control pathological immune activation while preserving GvT/GvL effects.
Looking forward, the integration of multi-omics profiling, CRISPR-based functional studies, and AI-enabled network modeling provides patient-specific insights into signaling dynamics. These tools support rational design of individualized interventions capable of precisely recalibrating immune circuits without broad immunosuppression.
Together, this evolving framework positions GvHD management within the era of precision immunotherapy. Future strategies that combine pathway-specific therapeutics with genome- and data-driven modalities may deliver safer, more durable, and personalized outcomes while maintaining the critical balance between immune tolerance and anti-tumor immunity.
Supplementary Materials
Not applicable. No supplementary materials are associated with this review article.
Author Contributions
Conceptualization, A.R.; methodology, A.R.; investigation, A.R., F.M., P.P., A.A., and M.K.; formal analysis, A.R., F.M., and M.K.; data curation, A.R.; visualization, A.R. and P.P.; writing—original draft preparation, A.R.; writing—review and editing, A.R., F.M., and A.A.; supervision, A.R.; project administration, A.R.; resources, A.R.; funding acquisition, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable. This review article did not involve any studies with human participants or animals and therefore did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. No new data were generated or analyzed in this review.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| allo-HSCT | allogeneic hematopoietic stem cell transplantation |
| GvHD | graft-versus-host disease |
| GvT | graft-versus-tumor |
| JAK | Janus kinase |
| STAT | signal transducer and activator of transcription |
| BTK | Bruton’s tyrosine kinase |
| ITK | interleukin-2–inducible T-cell kinase |
| ROCK2 | Rho-associated coiled-coil-containing protein kinase 2 |
| NF-κB | nuclear factor kappa B |
| MAPK | mitogen-activated protein kinase |
| CRISPR | clustered regularly interspaced short palindromic repeats |
References
- Pidala, J; Onstad, L; Carpenter, P; Hamilton, BK; Kitko, CL; Juckett, M; et al. Longitudinal study of late acute and chronic GVHD after allogeneic hematopoietic cell transplantation: A long-term follow up study from the Chronic GVHD Consortium. Transplant Cell Ther [Internet]. 2025. Available online: https://www.sciencedirect.com/science/article/pii/S2666636725015313.
- Arai, S; Arora, M; Wang, T; Spellman, SR; He, W; Couriel, DR; et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015, 21(2), 266–74. [Google Scholar] [CrossRef] [PubMed]
- Hill, GR; Betts, BC; Tkachev, V; Kean, LS; Blazar, BR. Current concepts and advances in graft-versus-host disease immunology. Annu Rev Immunol. 2021, 39(1), 19–49. [Google Scholar] [CrossRef]
- Malard, F; Holler, E; Sandmaier, BM; Huang, H; Mohty, M. Acute graft-versus-host disease. Nat Rev Dis Primer 2023, 9(1), 27. [Google Scholar] [CrossRef]
- Zeiser, R; Blazar, BR. Acute graft-versus-host disease—biologic process, prevention, and therapy. N Engl J Med. 2017, 377(22), 2167–79. [Google Scholar] [CrossRef] [PubMed]
- Manduzio, P. Transfusion-associated graft-versus-host disease: A concise review. Hematol Rep. 2018, 10(4), 7724. [Google Scholar] [CrossRef]
- Frey, NV; Porter, DL. Graft-versus-host disease after donor leukocyte infusions: presentation and management. Best Pract Res Clin Haematol. 2008, 21(2), 205–22. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z; Niu, S; Fang, Y; Chen, Y; Li, YR; Yang, L. Addressing graft-versus-host disease in allogeneic cell-based immunotherapy for cancer. Exp Hematol Oncol. 2025, 14(1), 66. [Google Scholar] [CrossRef]
- Baumrin, E; Loren, AW; Falk, SJ; Mays, JW; Cowen, EW. Chronic graft-versus-host disease. Part I: Epidemiology, pathogenesis, and clinical manifestations. J Am Acad Dermatol 2024, 90(1), 1–16. [Google Scholar] [CrossRef]
- Flowers, ME; Inamoto, Y; Carpenter, PA; Lee, SJ; Kiem, HP; Petersdorf, EW; et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood J Am Soc Hematol. 2011, 117(11), 3214–9. [Google Scholar] [CrossRef]
- Zeiser, R; Blazar, BR. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N Engl J Med. 2017, 377(26), 2565–79. [Google Scholar] [CrossRef]
- Socié, G; Ritz, J. Current issues in chronic graft-versus-host disease. Blood J Am Soc Hematol. 2014, 124(3), 374–84. [Google Scholar] [CrossRef] [PubMed]
- Holtan, SG; Pasquini, M; Weisdorf, DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood J Am Soc Hematol. 2014, 124(3), 363–73. [Google Scholar] [CrossRef]
- Castor, MGM; Pinho, V; Teixeira, MM. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front Pharmacol. 2012, 3, 23. [Google Scholar] [CrossRef]
- Ara, T; Hashimoto, D. Novel Insights Into the Mechanism of GVHD-Induced Tissue Damage. Front Immunol. 2021, 12, 713631. [Google Scholar] [CrossRef]
- Gong, T; Liu, L; Jiang, W; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020, 20(2), 95–112. [Google Scholar] [CrossRef]
- Hill, GR; Koyama, M. Cytokines and costimulation in acute graft-versus-host disease. Blood J Am Soc Hematol. 2020, 136(4), 418–28. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M; Flynn, R; Price, A; Ranger, A; Browning, JL; Taylor, PA; et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood J Am Soc Hematol. 2012, 119(6), 1570–80. [Google Scholar] [CrossRef]
- Kumar, S; Leigh, ND; Cao, X. The role of co-stimulatory/co-inhibitory signals in graft-vs.-host disease. Front Immunol. 2018, 9, 3003. [Google Scholar] [CrossRef]
- Jiang, H; Fu, D; Bidgoli, A; Paczesny, S. T cell subsets in graft versus host disease and graft versus tumor. Front Immunol. 2021, 12, 761448. [Google Scholar] [CrossRef]
- Rastegari, A; Mohebbi, F. Immune mediators as therapeutic targets in GvHD; cytokines, growth factors, chemokines, and co-stimulation /co-inhibition. Transpl Immunol. 2025, 93, 102328. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C; Levy, DE; Decker, T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007, 282(28), 20059–63. [Google Scholar] [CrossRef]
- Braun, LM; Zeiser, R. Kinase Inhibition as Treatment for Acute and Chronic Graft-Versus-Host Disease. Front Immunol. 2021, 12, 760199. [Google Scholar] [CrossRef]
- Jaglowski, SM; Devine, SM. Graft-versus-host disease: why have we not made more progress? Curr Opin Hematol 2014, 21(2), 141–7. [Google Scholar] [CrossRef] [PubMed]
- Kurya, AU; Aliyu, U; Tudu, AI; Usman, AG; Yusuf, M; Gupta, S; et al. Graft-versus-host disease: Therapeutic prospects of improving the long-term post-transplant outcomes. Transplant Rep. 2022, 7(4), 100107. [Google Scholar] [CrossRef]
- Blazar, BR; Murphy, WJ; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012, 12(6), 443–58. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K; Dazzi, F; English, K; Farge, D; Galipeau, J; Horwitz, EM; et al. ISCT MSC committee statement on the US FDA approval of allogenic bone-marrow mesenchymal stromal cells. In Cytotherapy; Elsevier, 2025. [Google Scholar]
- Zeiser, R; von Bubnoff, N; Butler, J; Mohty, M; Niederwieser, D; Or, R; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N Engl J Med. 2020, 382(19), 1800–10. [Google Scholar] [CrossRef]
- Miklos, D; Cutler, CS; Arora, M; Waller, EK; Jagasia, M; Pusic, I; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood J Am Soc Hematol. 2017, 130(21), 2243–50. [Google Scholar] [CrossRef]
- Cutler, C; Lee, SJ; Arai, S; Rotta, M; Zoghi, B; Lazaryan, A; et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study. Blood 2021, 138(22), 2278–89. [Google Scholar] [CrossRef]
- Abboud, R; Choi, J; Ruminski, P; Schroeder, MA; Kim, S; Abboud, CN; et al. Insights into the role of the JAK/STAT signaling pathway in graft-versus-host disease. Ther Adv Hematol. 2020, 11, 2040620720914489. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, KP; Blazar, BR; Hill, GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest. 2017, 127(7), 2452–63. [Google Scholar] [CrossRef]
- Schroeder, MA; Choi, J; Staser, K; DiPersio, JF. The role of Janus kinase signaling in graft-versus-host disease and graft versus leukemia. Biol Blood Marrow Transplant. 2018, 24(6), 1125–34. [Google Scholar] [CrossRef] [PubMed]
- De Togni, E; Cole, O; Abboud, R. Janus kinase inhibition in the treatment and prevention of graft-versus-host disease. Front Immunol 2024, 15, 1304065. [Google Scholar] [CrossRef]
- Dou, L; Zhao, Y; Yang, J; Deng, L; Wang, N; Zhang, X; et al. Ruxolitinib plus steroids for acute graft versus host disease: a multicenter, randomized, phase 3 trial. Signal Transduct Target Ther 2024, 9(1), 288. [Google Scholar] [CrossRef] [PubMed]
- Santosa, D; Rizky, D; Tandarto, K; Kartiyani, I; Yunarvika, V; Ardini, DNE; et al. Efficacy and Safety of Ibrutinib for Chronic Graft-Versus-Host Disease: A Systematic Review. Asian Pac J Cancer Prev APJCP 2023, 24(12), 4025–33. [Google Scholar] [CrossRef]
- Zhu, S; Jung, J; Victor, E; Arceo, J; Gokhale, S; Xie, P. Clinical Trials of the BTK Inhibitors Ibrutinib and Acalabrutinib in Human Diseases Beyond B Cell Malignancies. Front Oncol. 2021, 11, 737943. [Google Scholar] [CrossRef]
- Jagasia, M; Lazaryan, A; Bachier, CR; Salhotra, A; Weisdorf, DJ; Zoghi, B; et al. ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol Off J Am Soc Clin Oncol 2021, 39(17), 1888–98. [Google Scholar] [CrossRef]
- Herrero-Sánchez, MC; Rodríguez-Serrano, C; Almeida, J; San Segundo, L; Inogés, S; Santos-Briz, Á; et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol OncolJ Hematol Oncol 2016, 9(1), 113. [Google Scholar] [CrossRef]
- Di Ianni, M; Del Papa, B; Baldoni, S; Di Tommaso, A; Fabi, B; Rosati, E; et al. NOTCH and graft-versus-host disease. Front Immunol. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Vaeth, M; Bäuerlein, CA; Pusch, T; Findeis, J; Chopra, M; Mottok, A; et al. Selective NFAT targeting in T cells ameliorates GvHD while maintaining antitumor activity. Proc Natl Acad Sci U S A 2015, 112(4), 1125–30. [Google Scholar] [CrossRef]
- Sánchez-Valdepeñas, C; Casanova, L; Colmenero, I; Arriero, M; González, A; Lozano, N; et al. Nuclear factor-kappaB inducing kinase is required for graft-versus-host disease. Haematologica 2010, 95(12), 2111–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, HH; Ziegler, J; Li, C; Sepulveda, A; Bedeir, A; Grandis, J; et al. Sequential activation of inflammatory signaling pathways during graft-versus-host disease (GVHD): early role for STAT1 and STAT3. Cell Immunol. 2011, 268(1), 37–46. [Google Scholar] [CrossRef]
- Heine, A; Held, SAE; Daecke, SN; Wallner, S; Yajnanarayana, SP; Kurts, C; et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122(7), 1192–202. [Google Scholar] [CrossRef]
- Spoerl, S; Mathew, NR; Bscheider, M; Schmitt-Graeff, A; Chen, S; Mueller, T; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123(24), 3832–42. [Google Scholar] [CrossRef]
- Ryu, DB; Lim, JY; Kim, TW; Shin, S; Lee, SE; Park, G; et al. Preclinical evaluation of JAK1/2 inhibition by ruxolitinib in a murine model of chronic graft-versus-host disease. Exp Hematol 2021, 98, 36–46.e2. [Google Scholar] [CrossRef]
- Zeiser, R; Russo, D; Ram, R; Hashmi, SK; Chakraverty, R; Middeke, JM; et al. Ruxolitinib in Patients With Corticosteroid-Refractory or Corticosteroid-Dependent Chronic Graft-Versus-Host Disease: 3-Year Final Analysis of the Phase III REACH3 Study. J Clin Oncol Off J Am Soc Clin Oncol 2025, 43(23), 2566–71. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, M; Perales, MA; Schroeder, MA; Ali, H; Shah, NN; Chen, YB; et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood J Am Soc Hematol. 2020, 135(20), 1739–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, D; Liu, Y; Lai, X; Chen, J; Cheng, Q; Ma, X; et al. Efficiency and Toxicity of Ruxolitinib as a Salvage Treatment for Steroid-Refractory Chronic Graft-Versus-Host Disease. Front Immunol. 2021, 12, 673636. [Google Scholar] [CrossRef] [PubMed]
- Redondo, S; Esquirol, A; Novelli, S; Caballero, AC; Garrido, A; Oñate, G; et al. Efficacy and Safety of Ruxolitinib in Steroid-Refractory/Dependent Chronic Graft-versus-Host Disease: Real-World Data and Challenges. Transplant Cell Ther 2022, 28(1), 43.e1–43.e5. [Google Scholar] [CrossRef]
- Holtzman, NG; Im, A; Ostojic, A; Curtis, LM; Parsons-Wandell, L; Nashed, J; et al. Efficacy and Safety of Baricitinib in Refractory Chronic Graft-Versus-Host Disease (cGVHD): Preliminary Analysis Results of a Phase 1/2 Study. Blood 2020, 136, 1. [Google Scholar] [CrossRef]
- Schroeder, MA; Khoury, HJ; Jagasia, M; Ali, H; Schiller, GJ; Staser, K; et al. A phase 1 trial of itacitinib, a selective JAK1 inhibitor, in patients with acute graft-versus-host disease. Blood Adv. 2020, 4(8), 1656–69. [Google Scholar] [CrossRef]
- Chen, W; Nyuydzefe, MS; Weiss, JM; Zhang, J; Waksal, SD; Zanin-Zhorov, A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci Rep. 2018, 8(1), 16636. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A; Weiss, JM; Nyuydzefe, MS; Chen, W; Scher, JU; Mo, R; et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A 2014, 111(47), 16814–9. [Google Scholar] [CrossRef]
- Flynn, R; Paz, K; Du, J; Reichenbach, DK; Taylor, PA; Panoskaltsis-Mortari, A; et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016, 127(17), 2144–54. [Google Scholar] [CrossRef]
- Lee, SH; Moon, SJ; Park, MJ; Kim, EK; Moon, YM; Cho, ML. PIAS3 suppresses acute graft-versus-host disease by modulating effector T and B cell subsets through inhibition of STAT3 activation. Immunol Lett 2014, 160(1), 79–88. [Google Scholar] [CrossRef]
- Jia, H; Cui, J; Jia, X; Zhao, J; Feng, Y; Zhao, P; et al. Therapeutic effects of STAT3 inhibition by nifuroxazide on murine acute graft graft-vs.-host disease: Old drug, new use. Mol Med Rep. 2017, 16(6), 9480–6. [Google Scholar] [CrossRef]
- Huang, S; Yang, Q; Zhou, Y; Li, L; Shan, S. CS12192: A novel selective and potent JAK3 inhibitor mitigates acute graft-versus-host disease in bone marrow transplantation. Transpl Immunol 2024, 85, 102075. [Google Scholar] [CrossRef]
- Kim, S; Ashami, K; Lim, S; Staser, K; Vij, K; Santhanam, S; et al. Baricitinib prevents GvHD by increasing Tregs via JAK3 and treats established GvHD by promoting intestinal tissue repair via EGFR. Leukemia 2022, 36(1), 292–5. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Rodriguez, J; Kraus, ZJ; Schwartzberg, PL. Tec family kinases Itk and Rlk / Txk in T lymphocytes: cross-regulation of cytokine production and T-cell fates. FEBS J 2011, 278(12), 1980–9. [Google Scholar] [CrossRef]
- Allen, JL; Tata, PV; Fore, MS; Wooten, J; Rudra, S; Deal, AM; et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood 2014, 123(13), 2108–15. [Google Scholar] [CrossRef] [PubMed]
- Miller, AT; Wilcox, HM; Lai, Z; Berg, LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity 2004, 21(1), 67–80. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, AJ; Yu, L; Bäckesjö, CM; Vargas, L; Faryal, R; Aints, A; et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009, 228(1), 58–73. [Google Scholar] [CrossRef]
- Schutt, SD; Fu, J; Nguyen, H; Bastian, D; Heinrichs, J; Wu, Y; et al. Inhibition of BTK and ITK with Ibrutinib Is Effective in the Prevention of Chronic Graft-versus-Host Disease in Mice. PloS One 2015, 10(9), e0137641. [Google Scholar] [CrossRef]
- Dubovsky, JA; Flynn, R; Du, J; Harrington, BK; Zhong, Y; Kaffenberger, B; et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014, 124(11), 4867–76. [Google Scholar] [CrossRef]
- Rahmat, LT; Logan, AC. Ibrutinib for the treatment of patients with chronic graft-versus-host disease after failure of one or more lines of systemic therapy. Drugs Today Barc Spain 1998 2018, 54(5), 305–13. [Google Scholar]
- Waller, EK; Miklos, D; Cutler, C; Arora, M; Jagasia, MH; Pusic, I; et al. Ibrutinib for Chronic Graft-versus-Host Disease After Failure of Prior Therapy: 1-Year Update of a Phase 1b/2 Study. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 2019, 25(10), 2002–7. [Google Scholar] [CrossRef]
- Weichhart, T; Hengstschläger, M; Linke, M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 2015, 15(10), 599–614. [Google Scholar] [CrossRef] [PubMed]
- Fruman, DA; Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014, 13(2), 140–56. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Sánchez, MC; Rodríguez-Serrano, C; Almeida, J; San-Segundo, L; Inogés, S; Santos-Briz, Á; et al. Effect of mTORC1/mTORC2 inhibition on T cell function: potential role in graft-versus-host disease control. Br J Haematol 2016, 173(5), 754–68. [Google Scholar] [CrossRef]
- Wang, S; Passang, T; Li, Y; Zhang, H; Das, PK; Chen, K; et al. Dual Inhibition of PI3K Delta and PI3K Gamma Isoforms to Prevent Graft-Versus-Host Disease By Limiting T Cell Expansion In Vivo. Blood 2022, 140 Supplement 1, 10219–20. [Google Scholar] [CrossRef]
- Castor, MGM; Rezende, BM; Bernardes, PTT; Vieira, AT; Vieira, ELM; Arantes, RME; et al. PI3Kγ controls leukocyte recruitment, tissue injury, and lethality in a model of graft-versus-host disease in mice. J Leukoc Biol. 2011, 89(6), 955–64. [Google Scholar] [CrossRef] [PubMed]
- Paz, K; Flynn, R; Du, J; Tannheimer, S; Johnson, AJ; Dong, S; et al. Targeting PI3Kδ function for amelioration of murine chronic graft-versus-host disease. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg 2019, 19(6), 1820–30. [Google Scholar] [CrossRef]
- Wang, L; Gu, Z; Zhai, R; Li, D; Zhao, S; Luo, L; et al. The efficacy and safety of sirolimus-based graft-versus-host disease prophylaxis in patients undergoing allogeneic hematopoietic stem cell transplantation: a meta-analysis of randomized controlled trials. Transfusion (Paris) 2015, 55(9), 2134–41. [Google Scholar] [CrossRef]
- Cutler, C; Kim, HT; Hochberg, E; Ho, V; Alyea, E; Lee, SJ; et al. Sirolimus and tacrolimus without methotrexate as graft-versus-host disease prophylaxis after matched related donor peripheral blood stem cell transplantation. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 2004, 10(5), 328–36. [Google Scholar] [CrossRef] [PubMed]
- Ho, VT; Aldridge, J; Kim, HT; Cutler, C; Koreth, J; Armand, P; et al. Comparison of Tacrolimus and Sirolimus (Tac/Sir) versus Tacrolimus, Sirolimus, and mini-methotrexate (Tac/Sir/MTX) as acute graft-versus-host disease prophylaxis after reduced-intensity conditioning allogeneic peripheral blood stem cell transplantation. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 2009, 15(7), 844–50. [Google Scholar] [CrossRef]
- Kornblit, B; Storer, BE; Andersen, NS; Maris, MB; Chauncey, TR; Petersdorf, EW; et al. Sirolimus with CSP and MMF as GVHD prophylaxis for allogeneic transplantation with HLA antigen-mismatched donors. Blood 2020, 136(13), 1499–506. [Google Scholar] [CrossRef] [PubMed]
- Armand, P; Kim, HT; Sainvil, MM; Lange, PB; Giardino, AA; Bachanova, V; et al. The addition of sirolimus to the graft-versus-host disease prophylaxis regimen in reduced intensity allogeneic stem cell transplantation for lymphoma: a multicentre randomized trial. Br J Haematol 2016, 173(1), 96–104. [Google Scholar] [CrossRef]
- Leonhardt, F; Zirlik, K; Buchner, M; Prinz, G; Hechinger, AK; Gerlach, UV; et al. Spleen tyrosine kinase (Syk) is a potent target for GvHD prevention at different cellular levels. Leukemia 2012, 26(7), 1617–29. [Google Scholar] [CrossRef]
- Brodie, EJ; Infantino, S; Low, MSY; Tarlinton, DM. Lyn, Lupus, and (B) Lymphocytes, a Lesson on the Critical Balance of Kinase Signaling in Immunity. Front Immunol. 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Li, HL; Davis, WW; Whiteman, EL; Birnbaum, MJ; Puré, E. The tyrosine kinases Syk and Lyn exert opposing effects on the activation of protein kinase Akt/PKB in B lymphocytes. Proc Natl Acad Sci U S A 1999, 96(12), 6890–5. [Google Scholar] [CrossRef]
- Takata, M; Sabe, H; Hata, A; Inazu, T; Homma, Y; Nukada, T; et al. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J 1994, 13(6), 1341–9. [Google Scholar] [CrossRef]
- Flynn, R; Allen, JL; Luznik, L; MacDonald, KP; Paz, K; Alexander, KA; et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood 2015, 125(26), 4085–94. [Google Scholar] [CrossRef]
- Le Huu, D; Kimura, H; Date, M; Hamaguchi, Y; Hasegawa, M; Hau, KT; et al. Blockade of Syk ameliorates the development of murine sclerodermatous chronic graft-versus-host disease. J Dermatol Sci 2014, 74(3), 214–21. [Google Scholar] [CrossRef]
- Lin, C; DiCioccio, RA; Haykal, T; McManigle, WC; Li, Z; Anand, SM; et al. A Phase I Trial of SYK Inhibition with Fostamatinib in the Prevention and Treatment of Chronic Graft-Versus-Host Disease. Transplant Cell Ther. 2023, 29(3), 179.e1–179.e10. [Google Scholar] [CrossRef]
- Poe, JC; Jia, W; Di Paolo, JA; Reyes, NJ; Kim, JY; Su, H; et al. SYK inhibitor entospletinib prevents ocular and skin GVHD in mice. JCI Insight 2018, 3(19), 122430. [Google Scholar] [CrossRef]
- Shindo, T; Kim, TK; Benjamin, CL; Wieder, ED; Levy, RB; Komanduri, KV. MEK inhibitors selectively suppress alloreactivity and graft-versus-host disease in a memory stage-dependent manner. Blood 2013, 121(23), 4617–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, S; Rauch, J; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int J Mol Sci 2020, 21(3). [Google Scholar] [CrossRef]
- Johnson, GL; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298(5600), 1911–2. [Google Scholar] [CrossRef]
- Itamura, H; Shindo, T; Tawara, I; Kubota, Y; Kariya, R; Okada, S; et al. The MEK inhibitor trametinib separates murine graft-versus-host disease from graft-versus-tumor effects. JCI Insight 2016, 1(10), e86331. [Google Scholar] [CrossRef]
- Itamura, H; Shindo, T; Muranushi, H; Kitaura, K; Okada, S; Shin-I, T; et al. Pharmacological MEK inhibition promotes polyclonal T-cell reconstitution and suppresses xenogeneic GVHD. Cell Immunol 2021, 367, 104410. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A; Kobayashi, S; Miyai, K; Osawa, Y; Horiuchi, T; Kato, S; et al. TAK1 inhibition ameliorates survival from graft-versus-host disease in an allogeneic murine marrow transplantation model. Int J Hematol 2018, 107(2), 222–9. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T; Date, M; Kano, M; Mizumaki, K; Tennichi, M; Kobayashi, T; et al. Blockade of p38 Mitogen-Activated Protein Kinase Inhibits Murine Sclerodermatous Chronic Graft-versus-Host Disease. Am J Pathol 2017, 187(4), 841–50. [Google Scholar] [CrossRef]
- Lin, Y; Song, Y; Zhang, Y; Shi, M; Hou, A; Han, S. NFAT signaling dysregulation in cancer: Emerging roles in cancer stem cells. Biomed Pharmacother Biomedecine Pharmacother 2023, 165, 115167. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 2007, 7(9), 690–702. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, G; Du, J; Paz, KG; Loschi, M; Zaiken, MC; Flynn, R; et al. Inhibition of inositol kinase B controls acute and chronic graft-versus-host disease. Blood 2020, 135(1), 28–40. [Google Scholar] [CrossRef] [PubMed]
- Miller, AT; Dahlberg, C; Sandberg, ML; Wen, BG; Beisner, DR; Hoerter, JAH; et al. Inhibition of the Inositol Kinase Itpkb Augments Calcium Signaling in Lymphocytes and Reveals a Novel Strategy to Treat Autoimmune Disease. PloS One 2015, 10(6), e0131071. [Google Scholar] [CrossRef]
- Storb, R; Deeg, HJ; Pepe, M; Appelbaum, F; Anasetti, C; Beatty, P; et al. Methotrexate and cyclosporine versus cyclosporine alone for prophylaxis of graft-versus-host disease in patients given HLA-identical marrow grafts for leukemia: long-term follow-up of a controlled trial. Blood 1989, 73(6), 1729–34. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, A; Ohashi, Y; Okamoto, S; Moriyama, Y; Nagao, T; Kodera, Y; et al. Phase III study comparing tacrolimus (FK506) with cyclosporine for graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation. Bone Marrow Transplant 2001, 28(2), 181–5. [Google Scholar] [CrossRef]
- Oeckinghaus, A; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009, 1(4), a000034. [Google Scholar] [CrossRef]
- Sun, K; Welniak, LA; Panoskaltsis-Mortari, A; O’Shaughnessy, MJ; Liu, H; Barao, I; et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U S A 2004, 101(21), 8120–5. [Google Scholar] [CrossRef]
- Liu, T; Zhang, L; Joo, D; Sun, SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Vodanovic-Jankovic, S; Hari, P; Jacobs, P; Komorowski, R; Drobyski, WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood 2006, 107(2), 827–34. [Google Scholar] [CrossRef]
- Shono, Y; Tuckett, AZ; Ouk, S; Liou, HC; Altan-Bonnet, G; Tsai, JJ; et al. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising antitumor activity. Cancer Discov. 2014, 4(5), 578–91. [Google Scholar] [CrossRef]
- Pai, CCS; Chen, M; Mirsoian, A; Grossenbacher, SK; Tellez, J; Ames, E; et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood 2014, 124(10), 1677–88. [Google Scholar] [CrossRef] [PubMed]
- Al-Homsi, AS; Feng, Y; Duffner, U; Al Malki, MM; Goodyke, A; Cole, K; et al. Bortezomib for the prevention and treatment of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Exp Hematol 2016, 44(9), 771–7. [Google Scholar] [CrossRef]
- Herrera, AF; Kim, HT; Bindra, B; Jones, KT; Alyea, EP, 3rd; Armand, P; et al. A phase II study of bortezomib plus prednisone for initial therapy of chronic graft-versus-host disease. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 2014, 20(11), 1737–43. [Google Scholar] [CrossRef]
- Kitagawa, M. Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J Biochem (Tokyo) 2016, 159(3), 287–94. [Google Scholar] [CrossRef]
- Chung, J; Ebens, CL; Perkey, E; Radojcic, V; Koch, U; Scarpellino, L; et al. Fibroblastic niches prime T cell alloimmunity through Delta-like Notch ligands. J Clin Invest. 2017, 127(4), 1574–88. [Google Scholar] [CrossRef]
- Radtke, F; MacDonald, HR; Tacchini-Cottier, F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol 2013, 13(6), 427–37. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, P; Felli, MP; Screpanti, I; Campese, AF. The mazy case of Notch and immunoregulatory cells. J Leukoc Biol. 2017, 102(2), 361–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y; Sandy, AR; Wang, J; Radojcic, V; Shan, GT; Tran, IT; et al. Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood 2011, 117(1), 299–308. [Google Scholar] [CrossRef]
- Tran, IT; Sandy, AR; Carulli, AJ; Ebens, C; Chung, J; Shan, GT; et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J Clin Invest. 2013, 123(4), 1590–604. [Google Scholar] [CrossRef]
- Tkachev, V; Vanderbeck, A; Perkey, E; Furlan, SN; McGuckin, C; Gómez Atria, D; et al. Notch signaling drives intestinal graft-versus-host disease in mice and nonhuman primates. Sci Transl Med. 2023, 15(702), eadd1175. [Google Scholar] [CrossRef]
- Radojcic, V; Paz, K; Chung, J; Du, J; Perkey, ET; Flynn, R; et al. Notch signaling mediated by Delta-like ligands 1 and 4 controls the pathogenesis of chronic GVHD in mice. Blood 2018, 132(20), 2188–200. [Google Scholar] [CrossRef]
- Medinger, M; Junker, T; Heim, D; Tzankov, A; Jermann, PM; Bobadilla, M; et al. CB-103: A novel CSL-NICD inhibitor for the treatment of NOTCH-driven T-cell acute lymphoblastic leukemia: A case report of complete clinical response in a patient with relapsed and refractory T-ALL. EJHaem 2022, 3(3), 1009–12. [Google Scholar] [CrossRef]
- Zhang, Y; Shen, L; Dreißigacker, K; Zhu, H; Trinh-Minh, T; Meng, X; et al. Targeting of canonical WNT signaling ameliorates experimental sclerodermatous chronic graft-versus-host disease. Blood 2021, 137(17), 2403–16. [Google Scholar] [CrossRef]
- Mammadli, M; Harris, R; Mahmudlu, S; Verma, A; May, A; Dhawan, R; et al. Human Wnt/β-Catenin Regulates Alloimmune Signaling during Allogeneic Transplantation. Cancers 2021, 13(15). [Google Scholar] [CrossRef] [PubMed]
- Qi, HZ; Ye, YL; Suo, Y; Qu, H; Zhang, HY; Yang, KB; et al. Wnt/β-catenin signaling mediates the abnormal osteogenic and adipogenic capabilities of bone marrow mesenchymal stem cells from chronic graft-versus-host disease patients. Cell Death Dis. 2021, 12(4), 308. [Google Scholar] [CrossRef]
- Hayase, E; Ara, T; Saito, Y; Takahashi, S; Yoshioka, K; Ohigashi, H; et al. R-Spondin1 protects gastric stem cells and mitigates gastric GVHD in allogeneic hematopoietic stem cell transplantation. Blood Adv. 2024, 8(3), 725–31. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S; Kadowaki, M; Aoyama, K; Koyama, M; Oshima, T; Tomizuka, K; et al. The Wnt agonist R-spondin1 regulates systemic graft-versus-host disease by protecting intestinal stem cells. J Exp Med. 2011, 208(2), 285–94. [Google Scholar] [CrossRef] [PubMed]
- Schutt, SD; Wu, Y; Tang, CHA; Bastian, D; Nguyen, H; Sofi, MH; et al. Inhibition of the IRE-1α/XBP-1 pathway prevents chronic GVHD and preserves the GVL effect in mice. Blood Adv. 2018, 2(4), 414–27. [Google Scholar] [CrossRef]
- Choi, HJ; Tang, CHA; Tian, L; Wu, Y; Sofi, MH; Ticer, T; et al. XBP-1s Promotes B Cell Pathogenicity in Chronic GVHD by Restraining the Activity of Regulated IRE-1α-Dependent Decay. Front Immunol. 2021, 12, 705484. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D; Bellinger, DL. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int J Mol Sci. 2015, 16(3), 5635–65. [Google Scholar] [CrossRef]
- Guo, Y; Zhang, XN; Su, S; Ruan, ZL; Hu, MM; Shu, HB. β-adrenoreceptor-triggered PKA activation negatively regulates the innate antiviral response. Cell Mol Immunol. 2023, 20(2), 175–88. [Google Scholar] [CrossRef] [PubMed]
- Gazova, S; Klena, L; Galvankova, K; Babula, P; Krizanova, O. Role of adrenergic receptors and their blocking in cancer research. Biomed Pharmacother Biomedecine Pharmacother 2025, 192, 118637. [Google Scholar] [CrossRef] [PubMed]
- Leigh, ND; Kokolus, KM; O’Neill, RE; Du, W; Eng, JWL; Qiu, J; et al. Housing Temperature-Induced Stress Is Suppressing Murine Graft-versus-Host Disease through β2-Adrenergic Receptor Signaling. J Immunol Baltim Md 1950 2015, 195(10), 5045–54. [Google Scholar] [CrossRef]
- Haarberg, KMK; Li, J; Heinrichs, J; Wang, D; Liu, C; Bronk, CC; et al. Pharmacologic inhibition of PKCα and PKCθ prevents GVHD while preserving GVL activity in mice. Blood 2013, 122(14), 2500–11. [Google Scholar] [CrossRef]
- Valenzuela, JO; Iclozan, C; Hossain, MS; Prlic, M; Hopewell, E; Bronk, CC; et al. PKCtheta is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. J Clin Invest. 2009, 119(12), 3774–86. [Google Scholar] [CrossRef]
- Toubai, T; Sun, Y; Tawara, I; Friedman, A; Liu, C; Evers, R; et al. Ikaros-Notch axis in host hematopoietic cells regulates experimental graft-versus-host disease. Blood 2011, 118(1), 192–204. [Google Scholar] [CrossRef]
- Heizmann, B; Kastner, P; Chan, S. The Ikaros family in lymphocyte development. Curr Opin Immunol 2018, 51, 14–23. [Google Scholar] [CrossRef]
- Pereira, AD; de Molla, VC; Fonseca, ARBM; Tucunduva, L; Novis, Y; Pires, MS; et al. Ikaros expression is associated with an increased risk of chronic graft-versus-host disease. Sci Rep. 2023, 13(1), 8458. [Google Scholar] [CrossRef]
- Liu, Y; Huang, A; Chen, Q; Chen, X; Fei, Y; Zhao, X; et al. A distinct glycerophospholipid metabolism signature of acute graft versus host disease with predictive value. JCI Insight 2019, 5(16), 129494. [Google Scholar] [CrossRef]
- Gao, M; Zhang, K; Li, S; Guo, H; Sun, Y; Kong, J; et al. Single-cell RNA sequencing reveals heterogeneity of mucosa-associated invariant T cells in donor grafts and its diagnostic relevance in gastrointestinal graft-versus-host disease. Haematologica 2025. [Google Scholar] [CrossRef]
- Guo, H; Li, R; Wang, M; Hou, Y; Liu, S; Peng, T; et al. Multiomics Analysis Identifies SOCS1 as Restraining T Cell Activation and Preventing Graft-Versus-Host Disease. Adv Sci Weinh Baden-Wurtt Ger 2022, 9(21), e2200978. [Google Scholar] [CrossRef] [PubMed]
- Kamimoto, K; Stringa, B; Hoffmann, CM; Jindal, K; Solnica-Krezel, L; Morris, SA. Dissecting cell identity via network inference and in silico gene perturbation. Nature 2023, 614(7949), 742–51. [Google Scholar] [CrossRef]
- Aibar, S; González-Blas, CB; Moerman, T; Huynh-Thu, VA; Imrichova, H; Hulselmans, G; et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017, 14(11), 1083–6. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J; Clark, D; Czodrowski, P; Dunham, I; Ferran, E; Lee, G; et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019, 18(6), 463–77. [Google Scholar] [CrossRef]
- Aghamiri, SS; Amin, R. The Potential Use of Digital Twin Technology for Advancing CAR-T Cell Therapy. Curr Issues Mol Biol. 2025, 47(5). [Google Scholar] [CrossRef]
- Niarakis, A; Laubenbacher, R; An, G; Ilan, Y; Fisher, J; Flobak, Å; et al. Immune digital twins for complex human pathologies: applications, limitations, and challenges. NPJ Syst Biol Appl. 2024, 10(1), 141. [Google Scholar] [CrossRef]
- Pandey, V; Sharma, S; Pokharel, YR. Exploring CRISPR-Cas: The transformative impact of gene editing in molecular biology. Mol Ther Nucleic Acids 2025, 36(4), 102717. [Google Scholar] [CrossRef] [PubMed]
- Neidemire-Colley, L; Khanal, S; Braunreiter, KM; Gao, Y; Kumar, R; Snyder, KJ; et al. CRISPR/Cas9 deletion of MIR155HG in human T cells reduces incidence and severity of acute GVHD in a xenogeneic model. Blood Adv. 2024, 8(4), 947–58. [Google Scholar] [CrossRef]
- Lam, AJ; Lin, DTS; Gillies, JK; Uday, P; Pesenacker, AM; Kobor, MS; et al. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human Treg identity. Cell Rep. 2021, 36(5), 109494. [Google Scholar] [CrossRef]
- Lo Presti, V; Meringa, A; Dunnebach, E; van Velzen, A; Moreira, AV; Stam, RW; et al. Combining CRISPR-Cas9 and TCR exchange to generate a safe and efficient cord blood-derived T cell product for pediatric relapsed AML. J Immunother Cancer 2024, 12(4). [Google Scholar] [CrossRef]
- Flumens, D; Campillo-Davo, D; Janssens, I; Roex, G; De Waele, J; Anguille, S; et al. One-step CRISPR-Cas9-mediated knockout of native TCRαβ genes in human T cells using RNA electroporation. STAR Protoc. 2023, 4(1), 102112. [Google Scholar] [CrossRef]
- Lei, T; Wang, Y; Zhang, Y; Yang, Y; Cao, J; Huang, J; et al. Leveraging CRISPR gene editing technology to optimize the efficacy, safety and accessibility of CAR T-cell therapy. Leukemia 2024, 38(12), 2517–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, X; Zhong, S; Zhan, Y; Zhang, X. CRISPR-Cas9 applications in T cells and adoptive T cell therapies. Cell Mol Biol Lett. 2024, 29(1), 52. [Google Scholar] [CrossRef] [PubMed]
- Yao, D; Binan, L; Bezney, J; Simonton, B; Freedman, J; Frangieh, CJ; et al. Scalable genetic screening for regulatory circuits using compressed Perturb-seq. Nat Biotechnol. 2024, 42(8), 1282–95. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A; Parnas, O; Li, B; Chen, J; Fulco, CP; Jerby-Arnon, L; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167(7), 1853–1866.e17. [Google Scholar] [CrossRef]
- Schraivogel, D; Steinmetz, LM; Parts, L. Pooled Genome-Scale CRISPR Screens in Single Cells. Annu Rev Genet. 2023, 57, 223–44. [Google Scholar] [CrossRef]
- Tsai, SQ; Zheng, Z; Nguyen, NT; Liebers, M; Topkar, VV; Thapar, V; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015, 33(2), 187–97. [Google Scholar] [CrossRef]
- Schmidt, H; Zhang, M; Chakarov, D; Bansal, V; Mourelatos, H; Sánchez-Rivera, FJ; et al. Genome-wide CRISPR guide RNA design and specificity analysis with GuideScan2. Genome Biol. 2025, 26(1), 41. [Google Scholar] [CrossRef]
- Höijer, I; Johansson, J; Gudmundsson, S; Chin, CS; Bunikis, I; Häggqvist, S; et al. Amplification-free long-read sequencing reveals unforeseen CRISPR-Cas9 off-target activity. Genome Biol. 2020, 21(1), 290. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R; Schroeder, MA; Rettig, MP; Jayasinghe, RG; Gao, F; Eisele, J; et al. Itacitinib for prevention of graft-versus-host disease and cytokine release syndrome in haploidentical transplantation. Blood 2025, 145(13), 1382–94. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



