Submitted:
24 December 2025
Posted:
25 December 2025
Read the latest preprint version here
Abstract
Background: Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is characterised by dysautonomia, unrefreshing sleep, and post-exertional malaise (PEM). Adverse childhood experiences (ACEs) are epidemiologically associated with ME/CFS, but mechanistic links remain unclear. Hypothesis: Repeated corticotropin-releasing factor (CRF)-driven stress from ACEs induces a maladaptive 'high-tonic/low-phasic' firing mode in the locus coeruleus (LC). This leads to mitochondrial strain, adenosine triphosphate (ATP) shortfall, and vesicular norepinephrine (NE) depletion—producing a 'wired-but-tired' state. An acute trigger, often infection, overwhelms this vulnerable system, resulting in persistent neuroinflammation and impaired glymphatic clearance. Rationale: (i) ACE-related autonomic and inflammatory signatures persist into adulthood; (ii) preliminary pharmacological observations suggest substrate rather than receptor limitation; (iii) LC neurons are metabolically vulnerable to chronic activation; (iv) glymphatic function depends on LC quiescence during sleep. Testable Predictions: (1) Lower LC neuromelanin MRI signal correlates with ACE scores and ME/CFS severity (a priori anticipated: r < −0.5). (2) Reduced norepinephrine transporter (NET) positron emission tomography (PET) binding in LC correlates with hypervigilance. (3) Paradoxically low cerebrospinal fluid (CSF) 3-methoxy-4-hydroxyphenylglycol (MHPG) despite autonomic symptoms. (4) Heart rate variability (HRV) and pupillometry track functional capacity. (5) Pharmacological probe studies differentiate substrate-limited from receptor-limited states. Limitations: No direct vesicular NE measurements exist; ACE-ME/CFS association is cross-sectional; pharmacological observations are anecdotal. Prospective validation required.
Keywords:
myalgic encephalomyelitis
; chronic fatigue syndrome
; locus coeruleus
; norepinephrine depletion
; adverse childhood experiences
; dysautonomia
; hypothesis
1. Introduction
1.1. The ME/CFS Enigma
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) represents one of the most challenging conditions in modern medicine. Despite affecting millions globally and causing profound disability, its pathophysiology remains incompletely understood. Recent research increasingly recognises ME/CFS as a systemic neuroimmunological disease (Komaroff and Lipkin, 2021, pp. 895–906). However, mechanistic models capable of explaining its core features—post-exertional malaise (PEM), unrefreshing sleep, cognitive impairment, orthostatic intolerance, and the paradoxical coexistence of hyperarousal and exhaustion—remain elusive.
1.2. The ACE Connection
Epidemiological studies have identified an association between adverse childhood experiences (ACEs) and ME/CFS risk, though large-scale prospective studies are lacking (Van Houdenhove et al., 2001, pp. 21–28). Clinical observation reveals patterns beyond classical acute trauma: chronic emotional neglect, excessive performance pressure, parentification, and conditional love based on achievement. These patients often exhibit self-critical perfectionism and high achievement orientation—patterns measurable via validated instruments such as the Childhood Trauma Questionnaire (CTQ) and consistent with long-term stress adaptation (Kempke et al., 2013, pp. 995–1002).
The ACE-ME/CFS connection has typically been discussed in psychological or behavioral terms. This paper proposes a specific neurobiological mechanism: chronic programming of the locus coeruleus–norepinephrine (LC–NE) system, leading to metabolic exhaustion that renders the system vulnerable to collapse after an acute trigger.
1.3. Preliminary Pharmacological Observations
Clinical reports describe unusual response patterns to stimulant medications in some ME/CFS patients. Randomised controlled trials have demonstrated modest, short-term improvements in fatigue and executive function in subgroups: Young (2013, pp. 127–133) reported significant improvement in executive functioning with lisdexamfetamine over eight weeks; Blockmans et al. (2006, pp. 1047–1053) found methylphenidate produced moderate symptom reduction in a subset of patients. Importantly, neither study documented rapid tolerance development.
Anecdotal clinical observations (not derived from controlled studies) suggest a different pattern in some patients: marked initial improvement followed by rapid tolerance requiring dose escalation. This sequence—if confirmed prospectively—would be consistent with substrate limitation (vesicular depletion) rather than receptor dysfunction. This observation is presented as a hypothesis-generating clinical impression requiring controlled validation, not as established evidence.
Mechanistically, such a pattern would be plausible given known pharmacology: amphetamines increase cytosolic monoamine concentration and promote vesicular release via vesicular monoamine transporter (VMAT) interaction and transporter reversal; vesicle filling requires vacuolar-type ATPase (V-ATPase)-dependent proton gradient formation and adequate ATP availability; the conversion of dopamine to NE via dopamine-β-hydroxylase (DBH) is energy- and cofactor-dependent (Eiden and Weihe, 2011, pp. 86–98; Edwards, 2007, pp. 835–849). If vesicular stores were chronically depleted, forced release would produce pronounced but unsustainable effects.
1.4. Scope and Structure
This paper presents a conceptual framework linking ACEs to ME/CFS through chronic locus coeruleus dysfunction. We: (1) review neurobiological foundations; (2) propose a specific mechanism; (3) integrate clinical phenomena; (4) generate specific, testable predictions with proposed study designs; (5) discuss alternative explanations and limitations. This is a hypothesis paper that synthesises existing data and proposes testable mechanisms. It makes no therapeutic claims.
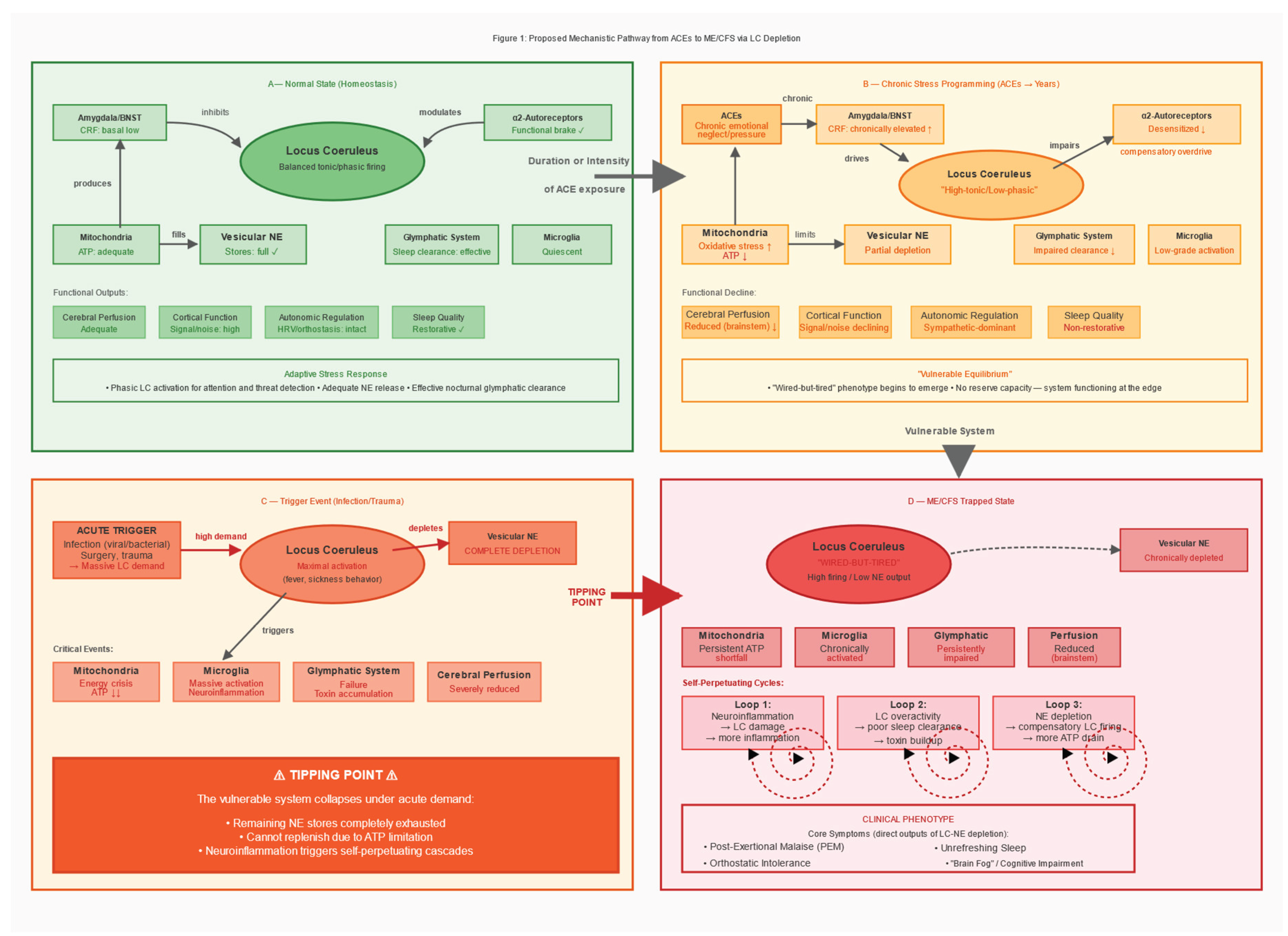
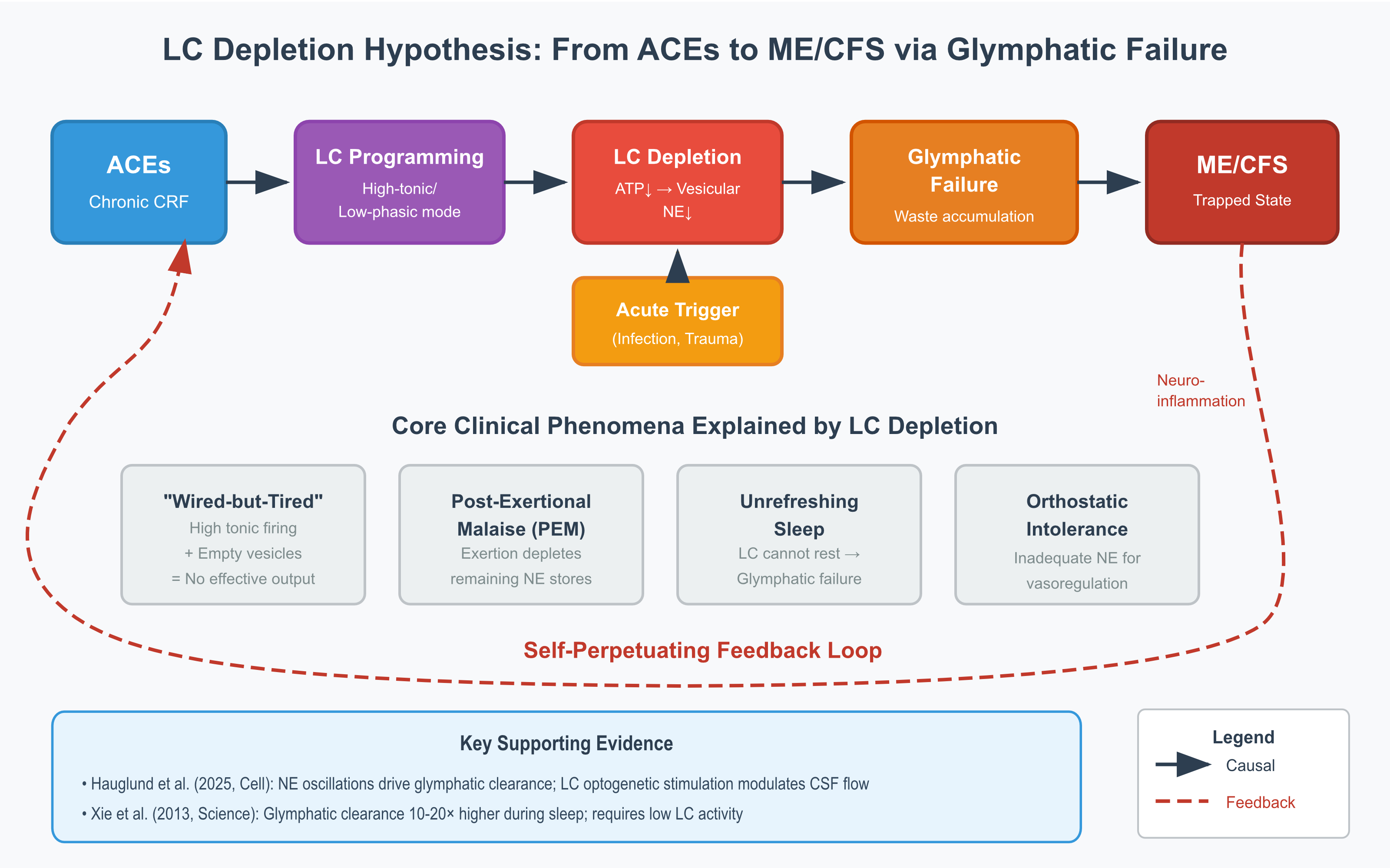
Figure 1.
Proposed Mechanistic Pathway from ACEs to ME/CFS via LC Depletion. Schematic representation of the proposed mechanism linking adverse childhood experiences (ACEs) to myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) through locus coeruleus (LC) dysfunction. Panel A shows normal LC function with balanced firing patterns and adequate norepinephrine (NE) levels. Panel B illustrates how chronic stress leads to high-tonic/low-phasic firing, mitochondrial exhaustion, and partial vesicle depletion. Panel C depicts an acute trigger (typically infection) overwhelming the vulnerable system. Panel D shows the trapped ME/CFS state characterised by self-perpetuating neuroinflammation, glymphatic dysfunction, and post-exertional malaise. Red arrows indicate positive feedback loops. Abbreviations: ATP, adenosine triphosphate; BNST, bed nucleus of the stria terminalis; CRF, corticotropin-releasing factor; NE, norepinephrine; PEM, post-exertional malaise.
Figure 1.
Proposed Mechanistic Pathway from ACEs to ME/CFS via LC Depletion. Schematic representation of the proposed mechanism linking adverse childhood experiences (ACEs) to myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) through locus coeruleus (LC) dysfunction. Panel A shows normal LC function with balanced firing patterns and adequate norepinephrine (NE) levels. Panel B illustrates how chronic stress leads to high-tonic/low-phasic firing, mitochondrial exhaustion, and partial vesicle depletion. Panel C depicts an acute trigger (typically infection) overwhelming the vulnerable system. Panel D shows the trapped ME/CFS state characterised by self-perpetuating neuroinflammation, glymphatic dysfunction, and post-exertional malaise. Red arrows indicate positive feedback loops. Abbreviations: ATP, adenosine triphosphate; BNST, bed nucleus of the stria terminalis; CRF, corticotropin-releasing factor; NE, norepinephrine; PEM, post-exertional malaise.
2. Neurobiological Foundations
2.1. The Locus Coeruleus: Anatomy and Function
The LC is a small, bilateral structure in the dorsal pons containing approximately 15,000–50,000 neurons per side (estimates vary across studies; Schwarz and Luo, 2015). Despite its modest size, the LC supplies approximately 70–80% of the brain’s norepinephrine through extensive projections to virtually all brain regions (Schwarz and Luo, 2015, pp. R1051–R1056). Functional roles include: arousal and attention modulation; stress response (as primary brain target of CRF); memory consolidation; pain modulation via descending pathways; cerebrovascular regulation; and immune modulation through sympathetic outflow.
The Yerkes-Dodson relationship demonstrates that LC activity follows an inverted-U function: too-low activity produces drowsiness; optimal activity enables alert focus; too-high activity causes hypervigilance and cognitive disorganisation (Aston-Jones and Cohen, 2005, pp. 403–450). This balance is maintained through two firing modes: tonic activity (steady background firing at 2–5 Hz) and phasic activity (burst firing >10 Hz in response to salient stimuli).
2.2. The CRF-LC Stress Axis
Under stress, the following cascade occurs: threat detection by the amygdala; CRF release from the central amygdala and bed nucleus of the stria terminalis (BNST); LC activation as CRF binds to CRFR1 receptors; NE surge increasing alertness and readiness; and parallel hypothalamic-pituitary-adrenal (HPA) axis activation (Valentino and Van Bockstaele, 2008, pp. 194–203). This system is highly adaptive to acute threats. The problem arises with chronic activation.
2.3. ACEs: Programming the Stress System
The landmark ACE Study (Felitti et al., 1998, pp. 245–258), including over 17,000 participants, revealed a dose-response relationship between childhood adversity and adult health outcomes. Each additional ACE increases risk of cardiovascular disease, autoimmune disorders, chronic pain syndromes, and neuroimmunological conditions. Crucially, these effects persist regardless of adult behavior, indicating biological programming.
Neurobiological consequences of ACEs include: HPA axis dysregulation with flattened diurnal cortisol rhythm (Bunea et al., 2017, p. 1274); chronic low-grade inflammation with elevated C-reactive protein, interleukin-6, and tumour necrosis factor-α (Baumeister et al., 2016, pp. 642–649); autonomic dysregulation with reduced heart rate variability; epigenetic modifications including methylation of the NR3C1 glucocorticoid receptor gene (McGowan et al., 2009, pp. 342–348; Perroud et al., 2011, p. e59); and brain structural changes including reduced hippocampal volume and increased amygdala reactivity.
Important caveats: The ACE-ME/CFS association is derived from cross-sectional data; prospective confirmation is lacking. Confounders including socioeconomic status, comorbidities, and retrospective reporting bias must be acknowledged. The model proposed here applies to a hypothesised subgroup with ACE histories; ME/CFS heterogeneity means other pathways likely exist.
2.4. Transgenerational Epigenetic Transmission: The ‘ACE-Negative’ Paradox
A significant proportion of ME/CFS patients-estimated at 25-35% in clinical samples-report no significant adverse childhood experiences on standardised instruments such as the Childhood Trauma Questionnaire (Kempke et al., 2013, pp. 995-1002). This ‘ACE-negative’ subgroup presents a conceptual challenge: if early-life stress programs the LC-NE system toward vulnerability, how do patients without reported ACEs develop ME/CFS?
Transgenerational epigenetic transmission offers a mechanistically plausible explanation. Three distinct pathways merit consideration:
Paternal preconceptional stress. Human spermatogenesis requires approximately 74 days (Amann, 2008, pp. 258-266). During this period, developing spermatocytes are susceptible to epigenetic modification. Animal studies demonstrate that paternal stress during the preconceptional period alters offspring HPA axis reactivity and anxiety-related behaviors through sperm RNA and DNA methylation changes (Rodgers et al., 2013, p. 1104; Dias and Ressler, 2014, pp. 89-96). While human evidence remains limited, the biological plausibility is established: chronic paternal stress in the 10 weeks preceding conception could program offspring stress reactivity without the offspring experiencing any direct adversity.
Periconceptional epigenetic reprogramming. Two critical windows of epigenetic vulnerability occur in the earliest stages of development. The first window (days 1-5 post-fertilisation) involves near-complete erasure of parental methylation marks during preimplantation development. The second window (approximately days 7-14) involves de novo methylation establishment following implantation (Reik et al., 2001, pp. 1089-1093). Maternal stress during these periods-typically before pregnancy recognition-can influence epigenetic reprogramming through altered oviductal and uterine environments (Burkusis et al., 2015, pp. 143-154). Importantly, these effects occur before any conscious maternal awareness of pregnancy, rendering them invisible to retrospective recall.
Grandmaternal germline exposure. The primordial germ cells that will become a woman’s oocytes develop during gestational weeks 6-12 of her own fetal development (Sasaki and Matsui, 2008, pp. 5-13). Consequently, stress experienced by the grandmother during this critical window directly exposes both the developing mother (F1) and her germline precursors (future F2). The Dutch Hunger Winter studies provide compelling epidemiological evidence: grandchildren of women pregnant during the 1944-45 famine exhibited increased adiposity and metabolic dysfunction, even when their mothers showed no such effects (Painter et al., 2008, pp. 467-473; Lumey et al., 2011, pp. 333-339). Rachel Yehuda’s Holocaust research similarly demonstrated transgenerational transmission of PTSD-related epigenetic signatures at the FKBP5 locus across three generations (Yehuda et al., 2016, p. 372; Yehuda and Lehrner, 2018, pp. 243-250).
Implications for the LC depletion hypothesis. These transgenerational mechanisms suggest that LC-NE system programming may occur through ancestral stress exposure rather than personal childhood adversity. The glucocorticoid receptor gene (NR3C1) methylation documented in ACE-exposed individuals (McGowan et al., 2009, pp. 342-348) may similarly be transmitted transgenerationally, as demonstrated in Holocaust offspring (Yehuda et al., 2016, p. 372). This would produce the same downstream effect-altered HPA axis reactivity and CRF-LC coupling-without any personally recalled trauma.
Critical caveat: Transgenerational epigenetic inheritance in humans remains an active and contested area of investigation. While animal evidence is robust, human studies face substantial methodological challenges including confounding by shared postnatal environments, retrospective ascertainment, and the difficulty of distinguishing biological from psychosocial transmission. The evidence in humans is therefore primarily associational rather than causal. The proposed mechanisms are biologically plausible but require prospective validation. Nonetheless, they offer a coherent explanation for why approximately one-third of ME/CFS patients report no personal ACE history yet present with the same neurobiological phenotype as ACE-positive patients.
2.5. LC Vulnerability to Chronic Stress
The LC has features making it particularly vulnerable to chronic stress. Metabolic fragility: High mitochondrial density reflects high energy demand; constant NE synthesis requires ATP via DBH; prolonged high firing rates generate oxidative stress. Autoreceptor desensitisation: α2-adrenergic autoreceptors normally inhibit NE release; with chronic CRF stimulation, these receptors desensitise, resulting in loss of negative feedback (Valentino and Van Bockstaele, 2008, pp. 194–203). The ‘high-tonic/low-phasic’ pattern: Chronic stress shifts the LC from responsive (phasic) to rigid (tonic) firing, documented in PTSD studies (Bangasser and Valentino, 2020, p. 601519), manifesting as hypervigilance with impaired attention shifting.
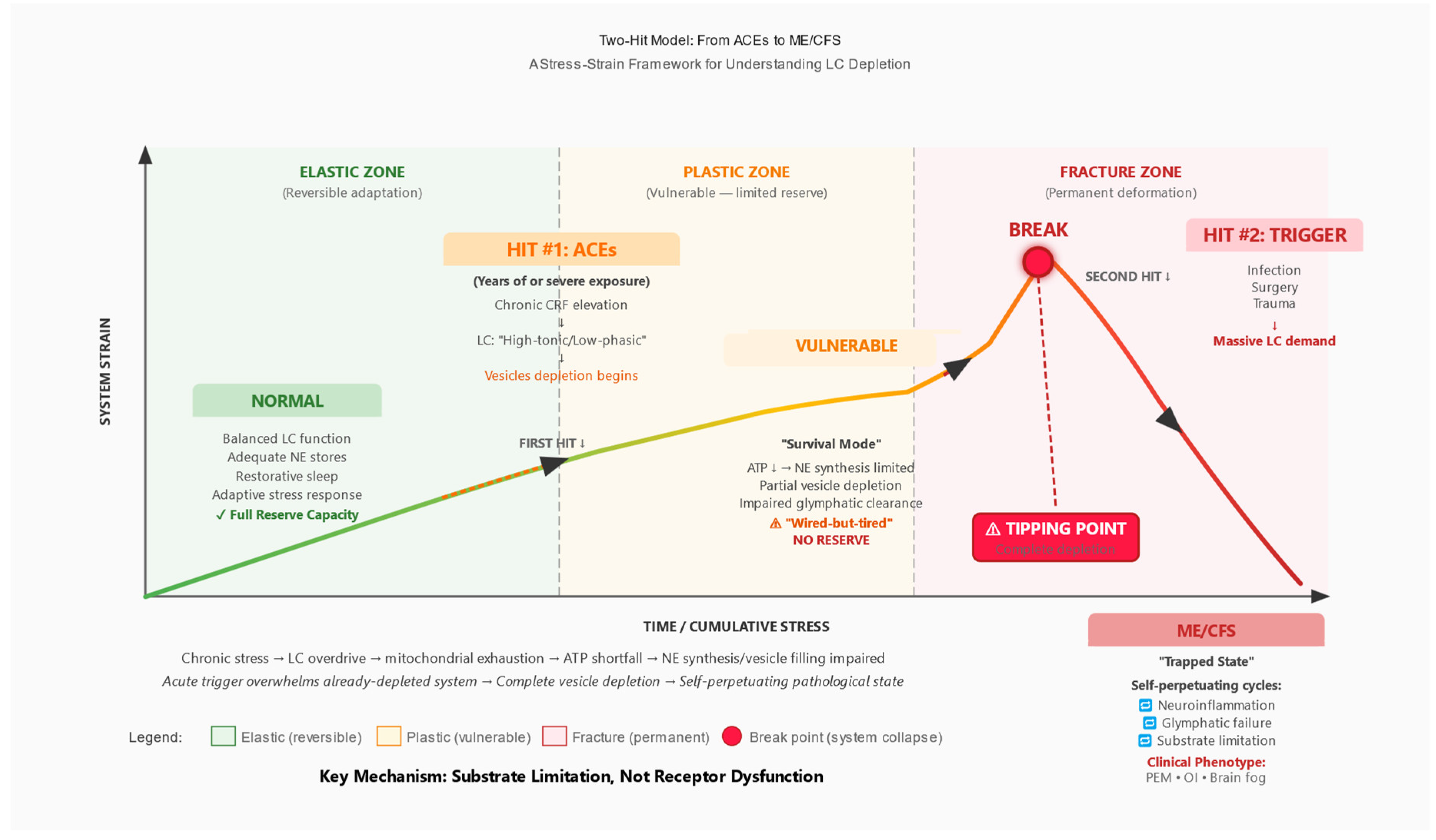
Figure 2.
Two-Hit Model: From ACEs to ME/CFS. A stress-strain framework illustrating the progression from normal state through vulnerable equilibrium to system collapse. Hit #1 (ACEs over years) produces chronic CRF elevation and LC programming into ‘high-tonic/low-phasic’ mode, creating a vulnerable state with depleted reserves. Hit #2 (acute trigger such as infection, surgery, or trauma) overwhelms the already-vulnerable system, crossing a tipping point into the trapped ME/CFS state with self-perpetuating pathological cycles. Key mechanism: substrate limitation (vesicular NE depletion), not receptor dysfunction. Abbreviations: ACEs, adverse childhood experiences; CRF, corticotropin-releasing factor; LC, locus coeruleus; NE, norepinephrine; OI, orthostatic intolerance; PEM, post-exertional malaise.
Figure 2.
Two-Hit Model: From ACEs to ME/CFS. A stress-strain framework illustrating the progression from normal state through vulnerable equilibrium to system collapse. Hit #1 (ACEs over years) produces chronic CRF elevation and LC programming into ‘high-tonic/low-phasic’ mode, creating a vulnerable state with depleted reserves. Hit #2 (acute trigger such as infection, surgery, or trauma) overwhelms the already-vulnerable system, crossing a tipping point into the trapped ME/CFS state with self-perpetuating pathological cycles. Key mechanism: substrate limitation (vesicular NE depletion), not receptor dysfunction. Abbreviations: ACEs, adverse childhood experiences; CRF, corticotropin-releasing factor; LC, locus coeruleus; NE, norepinephrine; OI, orthostatic intolerance; PEM, post-exertional malaise.
2.6. The Glymphatic Connection
The glymphatic system is the brain’s waste clearance mechanism (Nedergaard, 2013, pp. 1529–1530). During sleep, astrocytes shrink, expanding extracellular space by approximately 60%. Cerebrospinal fluid flows along perivascular spaces, washing out metabolic waste. Clearance is substantially enhanced during sleep compared to wakefulness (Xie et al., 2013, pp. 373–377).
Critical point: Effective glymphatic function requires reduced LC activity. Recent research has demonstrated that norepinephrine oscillations during non-rapid eye movement (NREM) sleep are the strongest predictors of glymphatic clearance; optogenetic stimulation of the LC directly modulates cerebrospinal fluid flow, while pharmacological suppression of NE oscillations (e.g., with certain hypnotics) may attenuate glymphatic clearance, though the magnitude requires further quantification (Hauglund et al., 2025, pp. 606–622). These findings establish a direct causal link between LC activity patterns and brain waste clearance. With chronic LC overactivation, the system cannot achieve the rhythmic NE fluctuations necessary for effective clearance; waste products accumulate, further damaging LC neurons in a vicious cycle. Clinical manifestation: ‘unrefreshing sleep.’
3. The Proposed Mechanism
3.1. Multi-Stage Model
We propose a multi-stage model from adaptation to exhaustion to collapse.
Stage 1 (Adaptive Response—Childhood/Adolescence): ACEs → chronic CRF elevation → sustained LC activation → hypervigilance pattern. From the child’s perspective, this is adaptive: in an unpredictable environment, constant vigilance is protective.
Stage 2 (Chronic Maladaptation—Years to Decades): Sustained high-tonic LC firing leads to progressive metabolic exhaustion: α2-autoreceptor desensitisation; mitochondrial dysfunction from chronic oxidative stress; ATP depletion; vesicular depletion (vesicle refilling is ATP-dependent via V-ATPase; DBH requires ascorbate and oxygen) and the ‘wired-but-tired’ state where neurons fire at high rate but release inadequate NE.
Stage 3 (Vulnerable Equilibrium—Pre-ME/CFS): The system maintains function but has no reserve capacity. Vesicles are chronically depleted, mitochondria stressed, glymphatic clearance impaired, neuroinflammation subclinical. One more stressor will cause system collapse.
Stage 4 (The Trigger): Typically viral infection (e.g., EBV, SARS-CoV-2, influenza), but may include major surgery, physical trauma, severe psychological stress, or pregnancy/childbirth. Massive LC activation occurs; complete vesicular depletion follows; post-infectious neuroinflammation persists; failed recovery ensues as the pre-damaged system cannot restore homeostasis.
Stage 5 (ME/CFS—Trapped State): The system becomes locked in pathological equilibrium: high neuronal activity with low NE output; persistent neuroinflammation; continued glymphatic dysfunction; progressive mitochondrial failure; PEM from any increased demand; orthostatic intolerance; cognitive impairment; unrefreshing sleep. Positive feedback loops trap the system.
3.2. Differentiation from Existing Two-Hit Models
Two-hit models exist in ME/CFS literature (Komaroff and Lipkin, 2021, pp. 895–906). The present model differs by specifying: (1) the precise neuroanatomical target (LC rather than diffuse ‘stress response’); (2) the cellular mechanism (vesicular NE depletion via ATP limitation rather than receptor dysfunction); (3) the link to glymphatic failure explaining unrefreshing sleep; and (4) testable predictions at cellular, circuit, and systems levels.
4. Clinical Phenomena Through the LC Depletion Lens
4.1. The ‘Wired-but-Tired’ Paradox
ME/CFS patients simultaneously exhibit hypervigilance, anxiety, difficulty ‘turning off,’ profound fatigue, inability to sustain activity, and disrupted sleep despite exhaustion. Mechanistic explanation: ‘Wired’ reflects high-tonic LC firing (desensitised autoreceptors). ‘Tired’ reflects empty vesicles with inadequate NE release despite high neuronal activity. The system runs at high RPMs but produces inadequate output.
4.2. Post-Exertional Malaise
Physical or cognitive exertion produces delayed (24–72 hours) severe symptom exacerbation, often lasting days to weeks. Mechanistic explanation: Exertion requires NE mobilisation; vesicles release remaining content; compromised mitochondria cannot rapidly resynthesise NE or refill vesicles; days are required for partial recovery (versus hours for healthy individuals).
4.3. Orthostatic Intolerance
Upon standing, many ME/CFS patients experience dizziness, tachycardia, cognitive impairment, and nausea. Mechanistic explanation: Vasoregulation requires NE to prevent blood pooling; with inadequate NE, compensation fails; heart rate increases to maintain cerebral perfusion. This reflects apparent sympathetic overactivity (tachycardia) with reduced effective central noradrenergic output. Comorbidities such as postural orthostatic tachycardia syndrome (POTS) and small fibre neuropathy (SFN) may act as moderator variables.
4.4. Unrefreshing Sleep
Sleep provides minimal restoration; patients often wake feeling as exhausted as when they went to bed. Mechanistic explanation: Glymphatic clearance requires rhythmic NE oscillations during NREM sleep (Hauglund et al., 2025, pp. 606–622); with chronic high-tonic LC firing, the system cannot achieve these oscillations; waste products accumulate; vesicles cannot refill (DBH requires ATP); morning cortisol surge activates a system with no reserve.
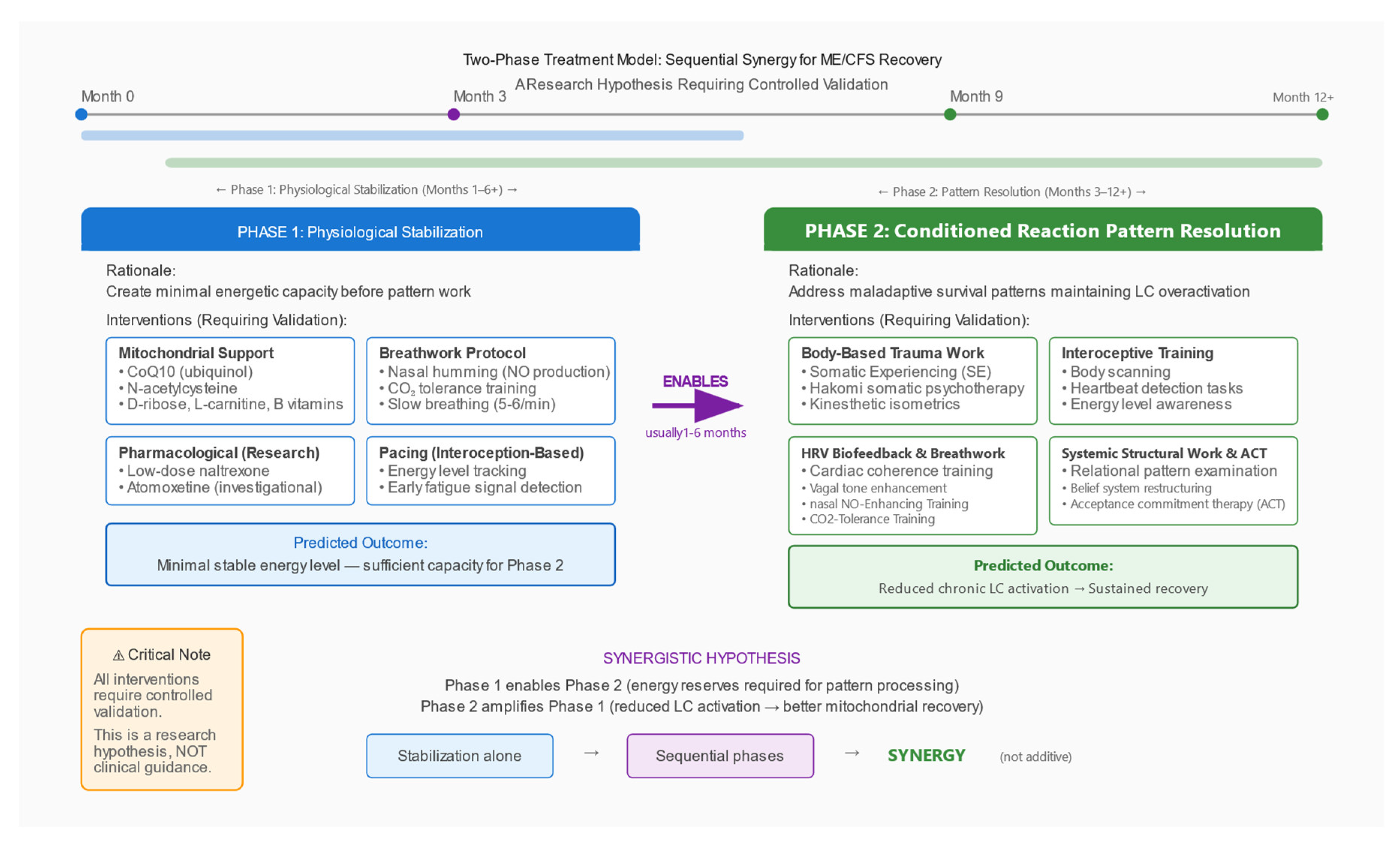
Figure 3.
Two-Phase Treatment Model: Sequential Synergy Hypothesis. Proposed therapeutic framework requiring controlled validation. Phase 1 (Physiological Stabilisation) creates minimal energetic capacity through mitochondrial support, pacing, breathwork, and pharmacological modulation. Phase 2 (Pattern Resolution) addresses maladaptive survival patterns through body-based trauma therapies, interoceptive training, and HRV biofeedback. Key insight: Phase 2 is only possible after Phase 1, but Phase 2 amplifies the effects of Phase 1 synergistically. This is a research hypothesis, not a treatment recommendation.
Figure 3.
Two-Phase Treatment Model: Sequential Synergy Hypothesis. Proposed therapeutic framework requiring controlled validation. Phase 1 (Physiological Stabilisation) creates minimal energetic capacity through mitochondrial support, pacing, breathwork, and pharmacological modulation. Phase 2 (Pattern Resolution) addresses maladaptive survival patterns through body-based trauma therapies, interoceptive training, and HRV biofeedback. Key insight: Phase 2 is only possible after Phase 1, but Phase 2 amplifies the effects of Phase 1 synergistically. This is a research hypothesis, not a treatment recommendation.
5. Supporting Evidence and Alternative Explanations
5.1. Evidence Supporting the Hypothesis
Biological plausibility: LC metabolic vulnerability is well-established; CRF-LC stress coupling is characterised; chronic stress leads to mitochondrial dysfunction; vesicular monoamine storage requires ATP; glymphatic clearance requires reduced LC activity.
Clinical phenomenology: The hypothesis explains the ‘wired-but-tired’ paradox, PEM, orthostatic intolerance, unrefreshing sleep, cognitive impairment, and coexisting hypervigilance and fatigue.
Dysautonomia profile: Reduced HRV is documented in ME/CFS (Nelson et al., 2019, p. e17600); orthostatic intolerance with reduced cerebral blood flow is documented (van Campen et al., 2020, p. 169); paradox of apparent sympathetic overactivity with ineffective central noradrenergic output is consistent with hyperactive neurons having inadequate neurotransmitter release.
Preliminary conference data (not peer-reviewed): Conference presentations at IACFS/ME 2025 reported findings consistent with this hypothesis, including low brain NE (Goldstein and Aregawi, 2025, conference abstract), reduced CRH-producing neurons in autopsy study (Da Silva et al., 2025, conference abstract), and sympathetic dysfunction meta-analysis (Hendrix et al., 2025, conference abstract). These presentations are hypothesis-generating and await peer review; they are not evidence on which the present model depends.
5.2. Alternative and Complementary Mechanisms
The LC depletion hypothesis is not mutually exclusive with other mechanisms: autoimmune mechanisms including β2-adrenergic receptor autoantibodies (Wirth and Scheibenbogen, 2020, p. 102527); small fibre neuropathy documented in significant subset; mast cell activation (histamine affects LC firing); cerebrovascular dysfunction with reduced cerebral blood flow; mitochondrial dysfunction which could be primary or secondary (Naviaux et al., 2016, pp. E5472–E5480); and immune-mediated LC damage from post-viral neuroinflammation (Nakatomi et al., 2014, pp. 945–950). The present hypothesis emphasises one pathway; ME/CFS is likely heterogeneous with multiple pathways converging on similar end states.
5.3. Limitations and Uncertainties
(1) No direct measurements of vesicular NE content in ME/CFS LC neurons exist.
(2) The ACE-ME/CFS link is associative from cross-sectional data; prospective studies are lacking; confounders (socioeconomic status, comorbidities, recall bias) are not fully controlled.
(3) The model’s reliance on ACE history presents a methodological limitation: ACE questionnaires cannot capture transgenerational epigenetic transmission. An estimated 25-35% of ME/CFS patients deny significant personal ACEs (Kempke et al., 2013, pp. 995-1002). These ‘ACE-negative’ patients may represent: (i) underreporting due to recall limitations; (ii) forms of adversity not captured by standard ACE instruments; (iii) transgenerational epigenetic programming from parental or grandparental stress; or (iv) alternative pathogenic pathways not involving early-life stress. The present model accommodates transgenerational transmission (Section 2.4) but acknowledges that ME/CFS is likely aetiologically heterogeneous.
(4) Pharmacological observations regarding tolerance are anecdotal, not from controlled studies.
(5) Conference data are preliminary.
(6) Causal direction is unclear and likely bidirectional.
(7) The model may be incomplete, focusing on LC while missing other critical nodes.
6. Testable Predictions and Research Agenda
To enable empirical evaluation of the model, we specify falsifiable predictions and corresponding study plans (primary endpoints, covariates, and power/analysis). MRI methods will conform to COBIDAS-MRI recommendations; PET acquisition, modeling, and quantification will follow SNMMI procedure standards and QIBA guidance; HRV analysis will follow the Task Force recommendations; and observational reporting will follow STROBE. Site-specific OSF preregistration and local IRB approval will precede any data collection.
6.1. Neuromelanin-Sensitive MRI of the LC
Prediction: LC contrast-to-noise ratio (CNR) will be reduced in ME/CFS patients compared to controls and will correlate inversely with ACE score (CTQ total; a priori anticipated: r < −0.5) and symptom severity.
Design: Cross-sectional comparison; ME/CFS (n ≥ 60) vs. healthy controls (n ≥ 60), stratified by ACE score tertiles.
Primary endpoint: LC CNR vs. dorsal pontine reference tissue. Neuromelanin MRI signal reflects multiple factors including NE synthesis history, iron content, and age-related changes; signal reductions cannot be directly equated with acute NE depletion. Age- and scanner-related partial volume effects will be modeled as covariates. Exploratory: Rostro-caudal LC profile analysis to detect regionally selective signal changes.
Covariates: Age (non-linear), sex, scanner/coil, signal-to-noise ratio, voxel size, head motion.
Power: Pilot (n ≈ 20/group) for variance estimation; main study powered for a medium effect size (d ≈ 0.5), α = 0.05 two-sided. Multiple comparison correction will follow a hierarchical testing strategy with family-wise error control (Bonferroni or FDR as appropriate). Bayesian analyses will supplement frequentist inference for pilot/N-of-1 designs.
Status: Neuromelanin-sensitive MRI is established for LC visualisation (Betts et al., 2019, pp. 2558–2571; Priovoulos et al., 2018, pp. 427–436); application to ME/CFS would be novel.
Acquisition, QC, preprocessing and reporting will conform to COBIDAS-MRI recommendations; protocol deviations will be documented a priori.
6.2. NET-PET Imaging
Prediction: NET binding potential (BPND) will be reduced in the LC region in ME/CFS and will correlate with hypervigilance symptoms and fatigue severity.
Protocol: Primary radioligand: [11C]MeNER; 90-minute dynamic acquisition; arterial input function or reference tissue model (occipital cortex); geometric transfer matrix partial volume correction for LC region-of-interest.
Covariates: Endogenous NE levels, medications (SNRIs, atomoxetine, TCAs, beta-blockers—with appropriate washout), smoking status, age, sex.
Interpretive note: Reduced NET binding can reflect multiple factors: transporter density, endogenous NE occupancy, or partial-volume effects (particularly relevant given the LC’s small size). Alternative or complementary PET ligands (e.g., [18F]-labeled tracers, if available) may provide converging evidence. Converging evidence from neuromelanin MRI, CSF markers, and physiology is essential (Pietrzak et al., 2013, pp. 1199–1205).
PET procedures (acquisition, kinetic modeling, PVC and reporting) will follow SNMMI procedure standards and QIBA guidance for neuroreceptor studies; ligand- and site-specific parameters will be preregistered (OSF).
6.3. CSF Metabolite Profiling
Prediction: Paradoxically low MHPG (principal NE metabolite) despite presumed high neuronal activity; elevated oxidative stress markers (GSH/GSSG ratio, 8-iso-PGF2α); elevated inflammatory markers; though interpretation requires caution given substantial inter-individual variability, posture effects, and diurnal fluctuations.
Design: ME/CFS (n ≥ 30) vs. controls, with subgroup analysis by ACE score.
Pre-analytics: Lumbar puncture 08:00–10:00 to control circadian variation; polypropylene collection tubes; immediate centrifugation; aliquoting and storage at −80 °C; haemolysis exclusion. Robust regression with adjustment for multiple comparisons (Shungu et al., 2012, pp. 1073–1087).
Pre-analytics and reporting will follow STROBE and BRISQ principles; site-level SOPs will be preregistered (OSF) before enrolment.
6.4. Physiological Measures (HRV, Pupillometry)
Prediction: HRV parameters (RMSSD, SDNN, LF/HF ratio) will show reduced parasympathetic indices; pupillometry will show reduced pupillary light reflex amplitude and altered spontaneous fluctuations (Murphy et al., 2014, pp. 4140–4154; Joshi et al., 2016, pp. 221–234). These will correlate with functional capacity and ACE score.
Protocol: HRV: 5-minute seated rest followed by 5-minute standing; ECG sampling ≥250 Hz; exclusion of significant arrhythmias; time-domain (RMSSD, SDNN) and frequency-domain (LF, HF, LF/HF) analysis. The LF/HF ratio as an index of sympathovagal balance remains controversial; notably, it becomes uninterpretable when the respiratory rate is ≤ 6 breaths per minute (as in coherent breathing), as the respiratory sinus arrhythmia then falls entirely within the LF band. Therefore, time-domain measures (RMSSD, SDNN) are prioritised as more robust markers of cardiac vagal tone. Pupillometry: infrared eye-tracking; baseline diameter, light reflex amplitude, latency, and return-to-baseline time; spontaneous pupil fluctuation index. Test-retest reliability reported as intraclass correlation coefficient (ICC). Primary: correlation with SF-36 and PEM intensity. Pre-registered primary endpoints: (1) Task-evoked pupil dilation (Δmm from baseline); (2) Pupillary light reflex amplitude (%); (3) RMSSD (ms) as primary vagal marker. Baseline pupil diameter (tonic LC activity proxy) and task-evoked pupil dilation (phasic LC activity proxy) will be analyzed separately. Constant room illumination (< 5 lux) and screen luminance will be documented.
HRV acquisition and analysis will follow the Task Force recommendations; pupillometry procedures and reporting will be standardised and preregistered (OSF).
6.5. Pharmacological Differentiation (N-of-1 Trials)
Rationale: To differentiate substrate-limited from receptor-limited states.
Design: Double-blind, placebo-controlled N-of-1 crossover with placebo run-in (2 weeks); comparing atomoxetine (blocks reuptake, does not force release), modafinil (DAT/NET blocker), and, if ethically justified, low-dose stimulant (forces release). Washout periods: minimum 5–7 half-lives between conditions.
Prediction: Atomoxetine/modafinil produce modest effect (substrate-limited); stimulant produces initial effect followed by tolerance (empties limited stores).
Safety: Data Safety Monitoring Board (DSMB) required; predefined stopping criteria (cardiovascular events, severe psychiatric symptoms); ECG, blood pressure, sleep quality, and anxiety monitoring at each visit; preregistration on OSF/ClinicalTrials.gov. Such studies require careful ethical review.
Reporting will follow the CONSORT Extension for N-of-1 Trials (CENT 2015); blinding, washouts and stopping rules will be preregistered (OSF) prior to enrolment.
6.6. Transcutaneous Auricular Vagus Nerve Stimulation (taVNS): A Therapeutic Window to the LC-NE System
The auricular branch of the vagus nerve (ABVN) provides a non-invasive access point to brainstem nuclei implicated in ME/CFS pathophysiology. Anatomical and neuroimaging studies have established that electrical stimulation of the cymba conchae - the ear region predominantly innervated by the ABVN - activates the nucleus tractus solitarius (NTS), which projects directly to the locus coeruleus (Frangos et al. 2015; Yakunina et al. 2017). This pathway offers a potential therapeutic target for restoring LC-NE function in ME/CFS.
6.6.1. Pupillary Responses as LC Activity Proxy
Pupil diameter is a well-validated non-invasive proxy of LC-NE activity under controlled luminance conditions, though it remains susceptible to confounding by light, cognition, and pharmacological factors (Joshi and Gold 2020). If taVNS activates the LC as proposed, measurable pupillary changes should occur. Recent evidence supports this mechanism:
Ludwig et al. (2024) demonstrated that phasic taVNS produces significantly greater pupil dilation compared to sham stimulation (0.18 +/- 0.03 vs. 0.09 +/- 0.03, p = 0.001). Higher stimulation intensity (5 mA vs. 3 mA) produced larger effects (p < 0.001), suggesting dose-dependent LC activation. Importantly, the influence of intensity on pupil dilation appears stronger than that of frequency, with 25 Hz showing no significant advantage over 10 Hz at matched intensities.
Villani et al. (2022) demonstrated that taVNS modulation of target-related pupil dilation depends on pre-stimulation pupil diameter - an index of baseline tonic LC-NE activity. Active taVNS was associated with differential pupil responses based on baseline state, consistent with the Adaptive Gain Theory prediction that LC-NE effects are state-dependent (Aston-Jones and Cohen 2005).
Critical caveat: Earlier studies reported null effects on pupillary measures (Keute et al. 2019; Warren et al. 2019). This heterogeneity likely reflects differences in stimulation protocols. Continuous stimulation paradigms show weaker effects than phasic (burst) protocols, and stimulation at perceptual threshold may be insufficient for reliable LC activation. For ME/CFS research applications, phasic protocols with intensity above sensory threshold (3-5 mA) appear most promising.
6.6.2. Orthostatic Intolerance: Direct Evidence from POTS
Orthostatic intolerance affects the majority of ME/CFS patients, with POTS (Postural Orthostatic Tachycardia Syndrome) representing a common comorbidity. Given the shared pathophysiology of autonomic dysregulation, evidence from POTS populations is directly relevant to ME/CFS.
A recent randomized, sham-controlled, double-blind trial (NCT05043051) provides promising preliminary evidence. After 2 months of daily taVNS at the cymba conchae, the active treatment group demonstrated a reduction in orthostatic tachycardia from 26.4 bpm (SD = 11.3) at baseline to 17.6 bpm (SD = 9.9) - a clinically meaningful reduction (preliminary data suggest approximately one-third improvement; final published effect sizes to be confirmed). No change occurred in the sham group. The active treatment group also showed HRV changes reflecting improved autonomic balance, including reduced LF/HF ratio during postural transition (Shiffer et al. 2025).
An earlier open-label study found that 4 hours of daily tVNS at the cymba conchae for 14 days significantly reduced orthostatic intolerance and gastrointestinal symptoms in POTS patients, with a tendency toward reduced standing heart rate. These findings align with mechanistic studies showing that tVNS increases HRV parameters (RMSSD, HF power) and decreases LF/HF ratio in heart failure patients, suggesting enhanced parasympathetic tone and attenuated sympathetic activity (Stavrakis et al. 2020).
Mechanistic plausibility for ME/CFS: The autonomic phenotype of ME/CFS - characterized by reduced HRV, exaggerated sympathetic responses to orthostasis, and impaired parasympathetic recovery - mirrors the profile responsive to taVNS in POTS. Both conditions show evidence of parasympathetic cardiovagal impairment alongside sympathetic hyperactivation. The reduction in anti-autonomic autoantibodies and inflammatory cytokines observed in the POTS trial suggests taVNS may address multiple pathophysiological pathways simultaneously.
6.6.3. Cognitive Function and Brain Fog
Cognitive dysfunction (brain fog) is a cardinal symptom of ME/CFS, encompassing deficits in attention, working memory, processing speed, and executive function. The LC-NE system is critically involved in all these domains (Sara 2009). If taVNS modulates LC function, cognitive improvements should follow.
A meta-analysis of 19 taVNS studies in healthy populations found a small but significant overall effect on cognition (Hedge’s g = 0.21, p < 0.0001), with the largest domain-specific effect for executive function (g = 0.66, 95% CI = 0.14-1.17, p = 0.02) (Ridgewell et al. 2021). Individual studies report improvements in cognitive flexibility (d = 0.49), working memory, and attentional processing (Borges et al. 2020; Jacobs et al. 2015; Ventura-Bort et al. 2018).
A Long COVID proof-of-concept trial - relevant given the overlap between Long COVID and ME/CFS - found that 4 weeks of daily taVNS reduced symptoms in the active group, while the sham group showed no change. Following the crossover, more than half of the participants no longer demonstrated any symptom prevalence (Badran et al. 2022).
Functional connectivity studies show that taVNS increases connectivity from hippocampus to prefrontal cortex and cingulate, while decreasing connectivity to anterior temporal regions - changes that may reflect normalization of networks disrupted in ME/CFS (Fang et al. 2016). The medial prefrontal cortex appears particularly responsive, with taVNS-induced connectivity changes correlating with working memory improvements in older adults.
6.6.4. Stimulation Laterality: Unilateral vs. Bilateral
Most taVNS research has employed unilateral left-ear stimulation, based on the historical convention from invasive VNS. However, transcutaneous auricular stimulation activates only afferent fibers, which are processed centrally before any efferent cardiac effects - mitigating the safety concerns that drove the left-only convention.
A systematic review and meta-analysis of 177 studies (n = 6,322 subjects) found that bilateral taVNS did not significantly increase the likelihood of cardiovascular adverse events compared to unilateral stimulation (Redgrave et al. 2022). Theoretical considerations suggest bilateral stimulation may enhance effects through increased afferent input to the brainstem. An ongoing multicenter RCT is investigating bilateral synchronous taVNS for disorders of consciousness using 200 microseconds pulse width at 20 Hz.
An fMRI study in 50 healthy controls directly compared left, right, and bilateral taVNS, finding distinct functional connectivity changes depending on stimulation laterality and frequency. Left stimulation preferentially altered connectivity in hippocampal and salience networks, while bilateral stimulation produced the most widespread effects.
6.6.5. Proposed Study Design for ME/CFS
Based on the converging evidence above, we propose a randomized, sham-controlled, crossover trial design. Washout period between conditions: ≥2 weeks to exclude carry-over effects.
Population: n ≥ 40 patients meeting ME-ICC (International Consensus Criteria, 2011) or CCC (Canadian Consensus Criteria, 2003) for ME/CFS, stratified by orthostatic intolerance severity
Intervention: Cymba conchae stimulation, 25 Hz, 200-500 microseconds pulse width, intensity 3-5 mA (above sensory threshold), 30-60 minutes daily for 8 weeks
Control: Earlobe (sham) stimulation with identical parameters
Primary endpoints: (1) HRV parameters (RMSSD, HF power, LF/HF ratio); (2) Pupillary light reflex and event-related pupil dilation (LC activity proxies); (3) Orthostatic heart rate response (tilt test)
Secondary endpoints: Fatigue severity (Chalder Fatigue Scale), cognitive function (executive function battery), orthostatic symptom burden, inflammatory markers (IL-6, TNF-alpha), post-exertional malaise frequency
Exploratory: Comparison of unilateral vs. bilateral stimulation in a subset; correlation of pupillary responses with clinical improvement as potential predictive biomarker
6.7. Prospective ACE-ME/CFS Cohort
Design: Large general population cohort (n ≥ 10,000); ACE assessment (CTQ) at baseline with biomarkers (HRV, inflammatory markers); long-term follow-up (10+ years); ME/CFS incidence documented per IOM/NICE criteria with infectious trigger documentation.
Prediction: Dose-response relationship—each additional ACE increases ME/CFS risk; mediation/moderation by HPA axis function and HRV; competing risks analysis.
Observational reporting will follow STROBE; analysis plans and amendments will be preregistered (OSF) before database lock.
7. Potential Disconfirming Evidence
The model should be abandoned or substantially revised if: (1) LC neuromelanin imaging shows no abnormalities in ME/CFS vs. controls; (2) CSF MHPG is normal or elevated rather than low; (3) HRV/pupillometry show no correlation with symptoms or ACE scores; (4) pharmacological probe trials show inconsistent patterns not matching substrate-limitation predictions; (5) mitochondrial support interventions show no benefit in appropriately powered trials; (6) prospective cohort shows no ACE dose-response relationship with ME/CFS incidence.
8. Discussion
8.1. Strengths
The proposed framework: (1) integrates disparate observations (ACE epidemiology, clinical phenomenology, autonomic dysfunction, sleep pathology); (2) is mechanistically specific, proposing a concrete pathway rather than vague ‘stress causes illness’; (3) generates testable predictions across imaging, CSF, physiology, and pharmacology; (4) explains heterogeneity through the two-hit model; (5) has translational potential if validated.
8.2. Weaknesses
(1) Evidence is indirect—no direct measurement of LC vesicular NE in humans exists. (2) ACE ascertainment involves retrospective reporting bias. (3) Pharmacological observations regarding tolerance are anecdotal. (4) Conference data are preliminary. (5) Causality is unclear and likely bidirectional. (6) The model may be incomplete.
8.3. Implications if Hypothesis Is Correct
For understanding ME/CFS: Shift from ‘mysterious post-viral syndrome’ to mechanistic model with identifiable vulnerability factors; explains why a subset of patients have ACE histories and high achievement orientation phenotypes; explains delayed onset and refractoriness. For research: Provides concrete targets, stratification strategies, and surrogate markers. For patients: Validates experiences (symptoms have neurobiological substrate); removes stigma. However: No immediate therapeutic implications; research must precede any clinical application.
9. Conclusion
We have proposed a specific, testable hypothesis: ME/CFS in a subset of patients results from chronic norepinephrine depletion of the locus coeruleus, programmed by adverse childhood experiences and triggered by acute systemic stressors. The hypothesis integrates ACE epidemiology, LC vulnerability to chronic stress, mitochondrial and vesicular mechanisms, glymphatic dysfunction, clinical phenomenology, and preliminary pharmacological observations.
It generates specific predictions testable through neuroimaging, CSF profiling, physiological surrogates, pharmacological probes, and prospective epidemiology. Most importantly, this is a hypothesis, not established fact. It requires rigorous testing. Until evidence exists, no therapeutic conclusions should be drawn. We present this framework to stimulate discussion and guide research.
Funding
No external funding.
Ethics
No human participants/data are included in this manuscript. The study plans are intended for institutional adoption; site-specific OSF preregistration and local IRB approval will precede any data collection.
Acknowledgments
The author thanks colleagues who shared clinical observations and ME/CFS patients who shared their experiences.
Data Availability
Not applicable (conceptual hypothesis paper). Preregistration of proposed studies is intended.
Conflicts of Interest
The author has clinical experience with body-based trauma therapies (Somatic Experiencing, Hakomi) and systemic work with ME/CFS patients, which informed this hypothesis. No proprietary methods are promoted. All therapeutic approaches discussed are well-established techniques requiring validation through controlled research. The author has no relationships with the pharmaceutical industry.
Abbreviations
ABVN, auricular branch of the vagus nerve; ACE, adverse childhood experience; ATP, adenosine triphosphate; BNST, bed nucleus of the stria terminalis; BPND, non-displaceable binding potential; BRISQ, Biospecimen Reporting for Improved Study Quality; CENT, CONSORT Extension for N-of-1 Trials; COBIDAS, Committee on Best Practices in Data Analysis and Sharing (MRI); CONSORT, Consolidated Standards of Reporting Trials; CNR, contrast-to-noise ratio; CRF, corticotropin-releasing factor; CSF, cerebrospinal fluid; CTQ, Childhood Trauma Questionnaire; DAT, dopamine transporter; DBH, dopamine-β-hydroxylase; DSMB, Data Safety Monitoring Board; EBV, Epstein–Barr virus; ECG, electrocardiogram; GTM, geometric transfer matrix; HF, high-frequency power; HPA, hypothalamic-pituitary-adrenal; HRV, heart rate variability; ICC, intraclass correlation coefficient; IOM, Institute of Medicine; IRB, Institutional Review Board; LC, locus coeruleus; LF, low-frequency power; ME/CFS, myalgic encephalomyelitis/chronic fatigue syndrome; MHPG, 3-methoxy-4-hydroxyphenylglycol; MRI, magnetic resonance imaging; NE, norepinephrine; NET, norepinephrine transporter; NICE, National Institute for Health and Care Excellence; NREM, non-rapid eye movement; NTS, nucleus tractus solitarius; OSF, Open Science Framework; PEM, post-exertional malaise; PET, positron emission tomography; OI, orthostatic intolerance; POTS, postural orthostatic tachycardia syndrome; PVC, partial volume correction; QIBA, Quantitative Imaging Biomarkers Alliance; RMSSD, root mean square of successive differences; ROI, region of interest; ROS, reactive oxygen species; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SDNN, standard deviation of normal-to-normal intervals; SFN, small fibre neuropathy; SNMMI, Society of Nuclear Medicine and Molecular Imaging; SNRI, serotonin-norepinephrine reuptake inhibitor; STROBE, Strengthening the Reporting of Observational Studies in Epidemiology; TCA, tricyclic antidepressant; V-ATPase, vacuolar-type ATPase; VMAT, vesicular monoamine transporter.
References
- Amann, Rupert P. 2008. “The Cycle of the Seminiferous Epithelium in Humans: A Need to Revisit?” Journal of Andrology 29 (5): 469-487. [CrossRef]
- Aston-Jones, Gary, and Jonathan D. Cohen. 2005. “An Integrative Theory of Locus Coeru-leus-Norepinephrine Function: Adaptive Gain and Optimal Performance.” Annual Review of Neuroscience 28: 403-450.
- Badran, Bashar W., Lowell T. Dowdle, Oliver J. Mithoefer, Nathan T. LaBate, Jeff Coatsworth, Jared C. Brown, William H. DeVries, Chris W. Austelle, Lisa M. McTea-gue, and Mark S. George. 2018. “Neurophysiologic Effects of Transcutaneous Auricular Vagus Nerve Stimulation (taVNS) via Electrical Stimulation of the Tragus: A Concur-rent taVNS/fMRI Study and Review.” Brain Stimulation 11 (3): 492-500. [CrossRef]
- Baig, Sheharyar S., Marharyta Kamarova, Ali Ali, Li Su, Jesse Dawson, Jessica N. Red-grave, and Arshad Majid. 2022. “Transcutaneous Vagus Nerve Stimulation (tVNS) in Stroke: The Evidence, Challenges and Future Directions.” Autonomic Neuroscience 237: 102909. [CrossRef]
- Bangasser, Debra A., and Rita J. Valentino. 2020. “The Locus Coeruleus-Norepinephrine System in Stress and Arousal: Unraveling Historical, Current, and Future Perspectives.” Frontiers in Psychiatry 11: 601519.
- Baumeister, David, Rabia Akhtar, Simona Ciufolini, Carmine M. Pariante, and Valeria Mondelli. 2016. “Childhood Trauma and Adulthood Inflammation: A Meta-Analysis.” Molecular Psychiatry 21 (5): 642-649.
- Betts, Matthew J., Evgeniya Kirilina, Maria C.G. Otaduy, et al. 2019. “Locus Coeruleus Imaging as a Biomarker for Noradrenergic Dysfunction in Neurodegenerative Diseases.” Brain 142 (9): 2558-2571.
- Blockmans, Daniel, Peter Persoons, Boudewijn Van Houdenhove, and Hugo Bobbaers. 2006. “Does Methylphenidate Reduce the Symptoms of Chronic Fatigue Syndrome?” The American Journal of Medicine 119 (12): 1047-1053.
- Borges, Uirassu, Leonie Knops, Sylvain Laborde, Stefanie Klatt, and Markus Raab. 2020. “Transcutaneous Vagus Nerve Stimulation May Enhance Only Specific Aspects of the Core Executive Functions. A Randomized Crossover Trial.” Frontiers in Neuro-science 14: 523. [CrossRef]
- Bremner, J. Douglas, Matthew T. Wittbrodt, and Nil Z. Gurel. 2024. “Transcutaneous Auri-cular Vagus Nerve Stimulation for Treating Emotional Dysregulation and Inflammation in Common Neuropsychiatric Disorders.” Brain Sciences 16 (1): 8. [CrossRef]
- Bunea, Ioana M., Aurora Szentagotai-Tatar, and Andrei C. Miu. 2017. “Early-Life Adversi-ty and Cortisol Response to Social Stress: A Meta-Analysis.” Translational Psychiatry 7 (12): 1274.
- Burger, Andreas M., Mathijs D’Agostini, Bart Verkuil, and Ilse Van Diest. 2020. “Moving Beyond Belief: A Narrative Review of Potential Biomarkers for Transcutaneous Vagus Ner-ve Stimulation.” Psychophysiology 57 (6): e13571. [CrossRef]
- Burkusis, Michal, Vladislav Plaks, Adi Michael, et al. 2015. “Environment and the Develo-ping Embryo.” In Principles of Developmental Genetics, 2nd ed., 143-154. Academic Press.
- D’Agostini, Mathijs, Cristina Ottaviani, Steven Verbeke, et al. 2022. “Transcutaneous Auri-cular Vagus Nerve Stimulation Cannot Modulate the P3b Event-Related Potential in Healthy Volunteers.” Clinical Neurophysiology 135: 22-29. [CrossRef]
- Da Silva, Joao P., et al. 2025. “Delineating Clinical Phenotypes and HPA-Axis Dysfunction in ME/CFS.” Presented at IACFS/ME 2025 Conference [conference abstract--not peer-reviewed].
- Dias, Brian G., and Kerry J. Ressler. 2014. “Parental Olfactory Experience Influences Beha-vior and Neural Structure in Subsequent Generations.” Nature Neuroscience 17 (1): 89-96. [CrossRef]
- Diedrich, André, Vasile Urechie, Dana Shiffer, Stefano Rigo, Maura Minonzio, Beatri-ce Cairo, Emily C. Smith, Luis E. Okamoto, Franca Barbic, Alessandro Bisoglio, Al-berto Porta, Italo Biaggioni, and Raffaello Furlan. 2021. “Transdermal Auricular Vagus Stimulation for the Treatment of Postural Tachycardia Syndrome.” Autonomic Neuro-science 236: 102886. [CrossRef]
- Edwards, Robert H. 2007. “The Neurotransmitter Cycle and Quantal Size.” Neuron 55 (6): 835-849.
- Eiden, Lee E., and Eberhard Weihe. 2011. “VMAT2: A Dynamic Regulator of Brain Mono-aminergic Neuronal Function Interacting with Drugs of Abuse.” Annals of the New York Academy of Sciences 1216: 86-98.
- Fang, Jiliang, Natalia Egorova, Peijing Rong, Jian Liu, Yue Hong, Yufeng Fan, Xian Wang, Hong Wang, Yazhuo Yu, Yuanpin Ma, Cong Xu, Shaoyuan Li, Jiping Zhao, Ming Luo, Bing Zhu, and Jian Kong. 2017. “Early Cortical Biomarkers of Longitudinal Transcutaneous Vagus Nerve Stimulation Treatment Success in Depression.” Neu-roImage: Clinical 14: 105-111. [CrossRef]
- Farmer, Adam D., Andreas Strzelczyk, Martina Finisguerra, et al. 2021. “International Con-sensus Based Review and Recommendations for Minimum Reporting Standards in Research on Transcutaneous Vagus Nerve Stimulation (Version 2020).” Frontiers in Human Neuro-science 14: 568051. [CrossRef]
- Felitti, Vincent J., Robert F. Anda, Dale Nordenberg, et al. 1998. “Relationship of Childhood Abuse and Household Dysfunction to Many of the Leading Causes of Death in Adults.” American Journal of Preventive Medicine 14 (4): 245-258.
- Frangos, Eleni, Jens Ellrich, and Barry R. Komisaruk. 2015. “Non-Invasive Access to the Vagus Nerve Central Projections via Electrical Stimulation of the External Ear: fMRI Evi-dence in Humans.” Brain Stimulation 8 (3): 624-636. [CrossRef]
- Goldstein, David S., and Dejene Aregawi. 2025. “Low Brain Norepinephrine in ME/CFS.” Presented at IACFS/ME 2025 Conference [conference abstract--not peer-reviewed].
- Hauglund, Natalie L., Mie Andersen, Kamilla Tokarska, et al. 2025. “Norepinephrine-Mediated Slow Vasomotion Drives Glymphatic Clearance During Sleep.” Cell 188 (3): 606-622. [CrossRef]
- Hendrix, Jolien, et al. 2025. “Sympathetic Dysfunction in ME/CFS: A Meta-Analysis.” Presented at IACFS/ME 2025 Conference [conference abstract--not peer-reviewed].
- Jacobs, Heidi I. L., Joost M. Riphagen, Carinne M. Razat, Sebastian Wiese, and Ale-xander T. Sack. 2015. “Transcutaneous Vagus Nerve Stimulation Boosts Associative Memory in Older Individuals.” Neurobiology of Aging 36 (5): 1860-1867. [CrossRef]
- Joshi, Siddhartha, Yin Li, Rishi M. Kalwani, and Joshua I. Gold. 2016. “Relationships Between Pupil Diameter and Neuronal Activity in the Locus Coeruleus, Colliculi, and Cin-gulate Cortex.” Neuron 89 (1): 221-234.
- Joshi, Siddhartha, and Joshua I. Gold. 2020. “Pupil Size as a Window on Neural Sub-strates of Cognition.” Trends in Cognitive Sciences 24 (6): 466-480. [CrossRef]
- Kempke, Stefan, Patrick Luyten, Steven Claes, et al. 2013. “Self-Critical Perfectionism and Fatigue and Pain in Chronic Fatigue Syndrome.” Psychological Medicine 43 (5): 995-1002.
- Keute, Marius, Mustafa Demirezen, Arnd Graf, Notger G. Mueller, and Tino Zaehle. 2019. “No Modulation of Pupil Size and Event-Related Pupil Response by Transcuta-neous Auricular Vagus Nerve Stimulation (taVNS).” Scientific Reports 9: 11452. [CrossRef]
- Komaroff, Anthony L., and W. Ian Lipkin. 2021. “Insights from Myalgic Encephalomyeli-tis/Chronic Fatigue Syndrome May Help Unravel the Pathogenesis of Postacute COVID-19 Syndrome.” Trends in Molecular Medicine 27 (9): 895-906.
- Ludwig, Mareike, Celine Wienke, Matthew J. Betts, Tino Zaehle, and Dorothea Hammerer. 2021. “Current Challenges in Reliably Targeting the Noradrenergic Locus Coeruleus Using Transcutaneous Auricular Vagus Nerve Stimulation (taVNS).” Autonomic Neuroscience 236: 102900. [CrossRef]
- Ludwig, Mareike, Celine Wienke, Matthew J. Betts, Tino Zaehle, and Dorothea Häm-merer. 2024. “Current and Future Directions in Transcutaneous Vagus Nerve Stimula-tion: Research, Application, and Effects on the Locus Coeruleus-Norepinephrine Sys-tem.” Neuroscience & Biobehavioral Reviews 159: 105607. [CrossRef]
- Lumey, L. H., Aryeh D. Stein, and Ezra Susser. 2011. “Prenatal Famine and Adult Health.” Annual Review of Public Health 32: 237-262. [CrossRef]
- McGowan, Patrick O., Aya Sasaki, Ana C. D’Alessio, et al. 2009. “Epigenetic Regulation of the Glucocorticoid Receptor in Human Brain Associates with Childhood Abuse.” Nature Neuroscience 12 (3): 342-348.
- Murphy, Peter R., Redmond G. O’Connell, Michael O’Sullivan, Ian H. Robertson, and Joshua H. Balsters. 2014. “Pupil Diameter Covaries with BOLD Activity in Human Locus Coeru-leus.” Human Brain Mapping 35 (8): 4140-4154.
- Nakatomi, Yasuhito, Kei Mizuno, Akira Ishii, et al. 2014. “Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An [11C-(R)]PK11195 PET Study.” Journal of Nuclear Medicine 55 (6): 945-950.
- Naviaux, Robert K., Jane C. Naviaux, Kefeng Li, et al. 2016. “Metabolic Features of Chronic Fatigue Syndrome.” Proceedings of the National Academy of Sciences USA 113 (37): E5472-E5480.
- Nedergaard, Maiken. 2013. “Garbage Truck of the Brain.” Science 340 (6140): 1529-1530.
- Nelson, Maximilian J., Joel S. Bahl, Jonathan D. Buckley, Ross L. Thomson, and Kade Davi-son. 2019. “Evidence of Altered Cardiac Autonomic Regulation in ME/CFS: A Systematic Review and Meta-Analysis.” Medicine 98 (43): e17600.
- Painter, Rebecca C., Clive Osmond, Peter Gluckman, et al. 2008. “Transgenerational Effects of Prenatal Exposure to the Dutch Famine on Neonatal Adiposity and Health in Later Life.” BJOG 115 (10): 1243-1249. [CrossRef]
- Perroud, Nader, Ariane Paoloni-Giacobino, Paco Prada, et al. 2011. “Increased Methylation of Glucocorticoid Receptor Gene (NR3C1) in Adults with a History of Childhood Maltreat-ment.” Translational Psychiatry 1 (12): e59.
- Pietrzak, Robert H., Jean-Dominique Gallezot, Yiyun S. Ding, et al. 2013. “Association of PTSD with Reduced In Vivo Norepinephrine Transporter Availability in the Locus Coeru-leus.” JAMA Psychiatry 70 (11): 1199-1205.
- Priovoulos, Nikos, Heidi I. L. Jacobs, Dimo Ivanov, Kamil Uludag, Frans R. J. Verhey, and Benedikt A. Poser. 2018. “High-Resolution In Vivo Imaging of Human Locus Coeruleus by Magnetization Transfer MRI at 3T and 7T.” NeuroImage 168: 427-436.
- Redgrave, Jessica N., Lyndsey Moore, Tobi Oyekunle, Mubarak Ebrahim, Konstanti-nos Falidas, Nicola Snowdon, Ali Ali, and Arshad Majid. 2018. “Transcutaneous Auri-cular Vagus Nerve Stimulation with Concurrent Upper Limb Repetitive Task Practice for Poststroke Motor Recovery: A Pilot Study.” Journal of Stroke and Cerebrovascular Diseases 27 (7): 1998-2005. [CrossRef]
- Reik, Wolf, Wendy Dean, and Jorn Walter. 2001. “Epigenetic Reprogramming in Mammali-an Development.” Science 293 (5532): 1089-1093. [CrossRef]
- Ridgewell, Christopher, Keith J. Heaton, Andrea Hildebrandt, Jeremy Couse, David Leber, and William H. Neumeier. 2021. “The Effects of Transcutaneous Auricular Va-gal Nerve Stimulation on Cognition in Healthy Individuals: A Meta-Analysis.” Neu-ropsychology 35 (4): 352-365. [CrossRef]
- Rodgers, Ali B., Christopher P. Morgan, Stefanie L. Bronson, et al. 2013. “Paternal Stress Exposure Alters Sperm MicroRNA Content and Reprograms Offspring HPA Stress Axis Regulation.” Journal of Neuroscience 33 (21): 9003-9012. [CrossRef]
- Sara, Susan J. 2009. “The Locus Coeruleus and Noradrenergic Modulation of Cogniti-on.” Nature Reviews Neuroscience 10 (3): 211-223. [CrossRef]
- Sasaki, Hiroyuki, and Yasuhisa Matsui. 2008. “Epigenetic Events in Mammalian Germ-Cell Development: Reprogramming and Beyond.” Nature Reviews Genetics 9 (2): 129-140. [CrossRef]
- Schwarz, Lindsay A., and Liqun Luo. 2015. “Organization of the Locus Coeruleus-Norepinephrine System.” Current Biology 25 (21): R1051-R1056.
- Shiffer, Dana, Stefano Rigo, Maura Minonzio, Deniz Timothy Yarsuvat, Eleonora To-baldini, Ludovico Furlan, Nicola Montano, Beatrice Cairo, Alberto Porta, Antonio Roberto Zamunér, Stefanos Bonovas, Vasile Urechie, Italo Biaggioni, André Diedrich, and Raffaello Furlan. 2025. “Short and Long Term Effects of a Two-Week Transcutane-ous Vagus Nerve Stimulation in Hyperadrenergic Postural Orthostatic Tachycardia Syndrome: A Proof-of-Concept Trial.” European Journal of Internal Medicine. [CrossRef]
- Shungu, Dikoma C., Natalie Weiduschat, James W. Murrough, et al. 2012. “Increased Ventricular Lactate in Chronic Fatigue Syndrome. III. Relationships to Cortical Glutathione and Clinical Symptoms.” NMR in Biomedicine 25 (9): 1073-1087.
- Skora, Lidia, Anna Marzecová, and Gerhard Jocham. 2024. “Tonic and Phasic Transcu-taneous Auricular Vagus Nerve Stimulation (taVNS) Both Evoke Rapid and Transient Pupil Dilation.” Brain Stimulation 17 (2): 233-244. [CrossRef]
- Stavrakis, Stavros, James A. Stoner, M. Beth Humphrey, Lynsie Morris, Andrea Fili-berti, John C. Reynolds, Khaled Elkholey, Imran Javed, Nik Twidale, Pavel Riha, Savita Varahan, Benjamin J. Scherlag, Warren M. Jackman, Tarun W. Dasari, and Sunny S. Po. 2020. “TREAT AF (Transcutaneous Electrical Vagus Nerve Stimulation to Suppress Atrial Fibrillation): A Randomized Clinical Trial.” JACC: Clinical Electro-physiology 6 (3): 282-291. [CrossRef]
- Stavrakis, Stavros, Praloy Chakraborty, Kassem Farhat, Seabrook Whyte, Lynsie Mor-ris, Zain Ul Abideen Asad, Brittany Karfonta, Juvaria Anjum, H. Greg Matlock, Xue Cai, and Xichun Yu. 2024. “Noninvasive Vagus Nerve Stimulation in Postural Ta-chycardia Syndrome: A Randomized Clinical Trial.” JACC: Clinical Electrophysiology 10 (2): 346-355. [CrossRef]
- Valentino, Rita J., and Elisabeth Van Bockstaele. 2008. “Convergent Regulation of Locus Coeruleus Activity as an Adaptive Response to Stress.” European Journal of Pharmacology 583 (2-3): 194-203.
- van Campen, C. Linda M. C., Peter C. Rowe, and Frans C. Visser. 2020. “Cerebral Blood Flow Is Reduced in Severe ME/CFS Patients During Mild Orthostatic Stress Testing.” Healthcare 8 (2): 169.
- Van Houdenhove, Boudewijn, Etienne Neerinckx, Romain Lysens, et al. 2001. “Victimizati-on in Chronic Fatigue Syndrome and Fibromyalgia in Tertiary Care.” Psychosomatics 42 (1): 21-28.
- Ventura-Bort, Carlos, Iria Garcia de Gurtubay, Pablo Bermejo, et al. 2023. “Immediate Effects and Duration of a Short and Single Application of Transcutaneous Auricular Vagus Nerve Stimulation on P300 Event Related Potential.” Frontiers in Neuroscience 17: 1096865. [CrossRef]
- Villani, Valentina, Manos Tsakiris, and Ruben T. Azevedo. 2022. “Transcutaneous Vagus Nerve Stimulation Modulates Pupil Size and Interoceptive Attention.” Psycho-physiology 59 (4): e13983. [CrossRef]
- Warren, Christopher M., Kim D. Tona, Lucia Ber, Ruud L. van den Brink, Loes van Nieuwenhoven, Eco J. C. de Geus, and Sander Nieuwenhuis. 2019. “The Neuromodu-latory and Hormonal Effects of Transcutaneous Vagus Nerve Stimulation as Evidenced by Salivary Alpha Amylase, Salivary Cortisol, Pupil Diameter, and the P3 Event-Related Potential.” Brain Stimulation 12 (3): 635-642. [CrossRef]
- Wienke, Celine, Marcus Grueschow, Aiden Haghikia, and Tino Zaehle. 2023. “Phasic, Event-Related Transcutaneous Auricular Vagus Nerve Stimulation Modifies Behavio-ral, Pupillary, and Low-Frequency Oscillatory Power Responses.” Journal of Neuro-science 43 (36): 6306-6319. [CrossRef]
- Wirth, Klaus, and Carmen Scheibenbogen. 2020. “A Unifying Hypothesis of the Pathophy-siology of ME/CFS: Insights from Autoantibodies Against Beta2-Adrenergic Receptors.” Autoimmunity Reviews 19 (6): 102527.
- Xie, Lulu, Hongyi Kang, Qiwu Xu, et al. 2013. “Sleep Drives Metabolite Clearance from the Adult Brain.” Science 342 (6156): 373-377. [CrossRef]
- Yakunina, Natalia, Sam Soo Kim, and Eui-Cheol Nam. 2017. “Optimization of Transcuta-neous Vagus Nerve Stimulation Using Functional MRI.” Neuromodulation: Technology at the Neural Interface 20 (3): 290-300. [CrossRef]
- Yehuda, Rachel, Nikolaos P. Daskalakis, Linda M. Bierer, et al. 2016. “Holocaust Exposure Induced Intergenerational Effects on FKBP5 Methylation.” Biological Psychiatry 80 (5): 372-380. [CrossRef]
- Yehuda, Rachel, and Amy Lehrner. 2018. “Intergenerational Transmission of Trauma Effects: Putative Role of Epigenetic Mechanisms.” World Psychiatry 17 (3): 243-257. [CrossRef]
- Young, Joel L. 2013. “Use of Lisdexamfetamine Dimesylate in Treatment of Executive Functioning Deficits and Chronic Fatigue Syndrome: A Double-Blind, Placebo-Controlled Study.” Psychiatry Research 207 (1-2): 127-133.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



