
Submitted:
15 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
Neurodevelopmental disorders (NDDs) are a class of childhood-onset conditions that affect brain development and function. NDDs have a heterogeneous etiology, a wide genetic and clinical variability and generally lead to impaired cognition, communication, psychomotor skills, and adaptive behavior. These disorders include intellectual disability (ID), autism spectrum disorder (ASD), and developmental and epileptic encephalopathies that manifest during childhood. Over the past 2 decades, genetic research has discovered more than 1,500 genes in different signaling pathways that are involved in NDDs, including many transcriptional regulators such as DNA/histone modifiers and chromatin-regulatory protein complexes. These same investigations have led to the accessibility and availability of next-generation sequencing in the assessment of children with NDDs in the clinical setting. The advances have dramatically altered the approach to the genetic diagnostic assessment of the child with NDDs and have increased the diagnostic yield of genetic testing in the pediatric setting. The purpose of this review is to provide the historical background to the rational assessment of child with an NDD and present a perspective on the current evaluation given the modern repertoire available to the pediatric practitioner facing this challenge in the clinical setting.
Keywords:
global developmental delay
; intellectual disability
; autism spectrum disorder
; phenotype
; genotype
; next generation sequencing
; genomic medicine
One of the most common presentations to the primary care pediatric setting is the individual with a neurodevelopmental disorder (NDD). The patient usually has a global developmental delay (GDD), intellectual disability (ID), and/or autism spectrum disorder (ASD). The question of a genetic cause or syndrome arises because of parents’ questions about prognosis and risk for the condition to reoccur in future pregnancies. The recognition of phenotypic signs leads to a referral to genetics or child neuropsychiatry. The reason for the specialist referral is to establish an etiologic diagnosis to inform management. Currently, there exists a lack of clarity among health care professionals regarding the necessity of genetic testing [1]. The purpose of this review is to provide the historical background to the rational assessment of child with NDDs and to present a perspective on the current optimal evaluation given the modern repertoire available to the pediatric practitioner facing this challenge in the clinical setting. As genetic testing options rapidly expand, the clinical community needs to be mindful of their individual strengths and limitations in order to select the most appropriate diagnostic pathway. While the term NDDs can often include other neurologic and behavioral conditions, this review will focus on the assessment of children with GDD/ID/ASD.
In Diagnostic and Statistical Manual of Mental Disorders (DSM-5) the designation GDD is used to explain developmental disability in children younger than age five [2,3]. About 5%-10% of the pediatric population experiences GDD [2]. ID is a term used for children older than 5 years of age when proper standardized measurements of developmental skills can be performed [4]. It is a variable, heterogeneous manifestation of central nervous system dysfunction with a prevalence of about 3% of the global population [4,5,6]. According to the DSM-5 [3], ID is defined as a defect in intellectual functioning and adaptive behavior starting before age 22 years, influencing the conceptual, social, and practical domains in daily life. ASD is a complex developmental disorder with early onset, manifesting as deficits in social communication and interaction and the presence of restrictive/repetitive behaviors and interests and/or sensory disorders interfering with daily functioning. Having a child with a NDD has a major impact on families regarding many aspects of daily life, and the financial burden on society is huge [7]. ID/ASD are both clinically and genetically heterogeneous. Clinical heterogeneity (diversity) is reflected by a wide range of different ID/ASD conditions due to variants of a large number of genes and by the broad clinical spectrum caused by mutations in the same gene [8].
ID/ASD are often one component of a broader condition including disorders of known etiology such as Down and fragile X syndromes. Associated neurodevelopmental comorbidities, such as epilepsy, attention deficit hyperactivity disorder (ADHD), are common, and specific characterization of these comorbidities is important in management. We will discuss a phenotypic classification of presentations of ID/ASD below. The genetic heterogeneity of NDDs is reflected by the large number of genes known and estimated to be involved in ID (about 2,000) and ASD (about 1,000), which has led to the latest ID/ASD gene panels used in diagnostic laboratories containing about 1,500 genes [9,10]. A significant genetic overlap between ID, epilepsy, and ASD has also been shown [11,12].
The Importance of Diagnosis
The clinical and genetic heterogeneity complicate the process of obtaining a precise etiological diagnosis, which is of paramount importance for the affected individual, the family, and the care providers. Although the labeling process might seem stigmatizing, diagnosis provides prediction, and it is usually sought by the family members [13], who want to know why and how it happened and if it will happen again. Knowing the etiology can be liberating, providing relief following years of uncertainty. Precise diagnosis may help establish an accurate recurrence risk, predict the prognosis with relative certainty, and organize appropriate laboratory testing, avoiding a diagnostic evaluation of unnecessary expense and invasiveness. The diagnosis and the knowledge of the natural history together make it possible to plan specific management and treatment, and may help the family in coping with the potential serious manifestations and/or developmental disabilities [14,15] (see Figure 1). Referral to support groups, the strong stimulus to initiate a new parent support group, or arrangement of a meeting with other parents and children with the same disorder are examples of how much can be accomplished in the care setting with an established diagnosis [16]. As clinicians we have experience with other individuals having the same disorder, access to management programs, knowledge of the prognosis, awareness of research on the disease, and many other elements that when shared with the parents will give them a sense that some control is possible [17,18].
In addition, continuous scientific advancements and ongoing studies are making feasible the specific treatment of a number of genetic disorders. The treatment is usually directed against the mechanism of the disease and thereby alters its natural history [19,20,21,22] (See illustrative examples in the Appendix). In our experience, the communication of diagnosis has been crucial for the therapeutic alliance and is the first step toward the study of behavioral phenotype [25].
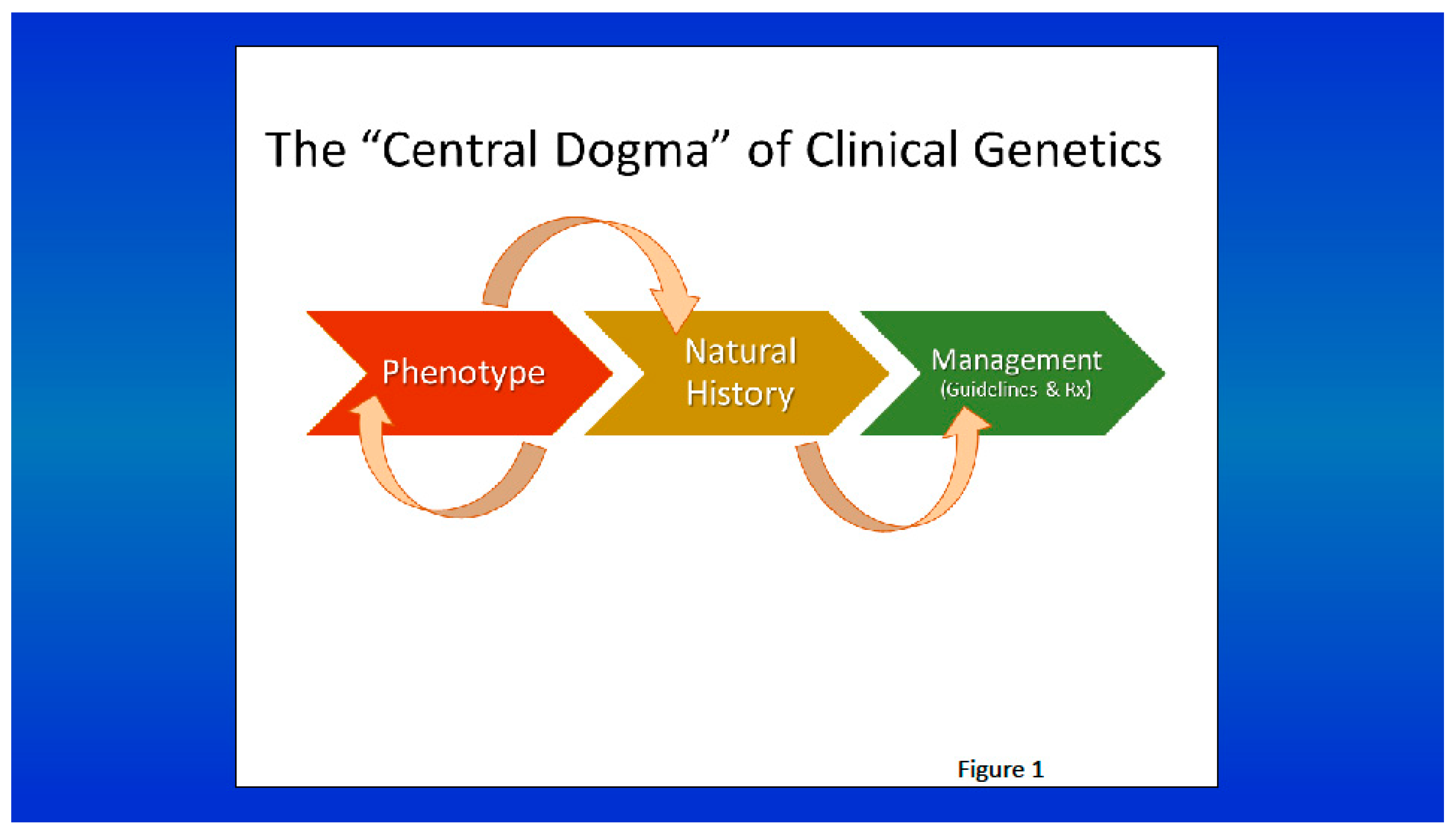
Figure 1.
The central dogma of medical genetics, like the central dogma of molecular biology, progresses from the description of the phenotype of a genetic condition to the characterization of its natural history, which is vital to the establishment of management guidelines and treatment (from Cassidy & Allanson’s Management of Genetic Syndromes, eds, Carey JC, Battaglia A, Viskochil D, and Cassidy SB. Hoboken, NJ, John Wiley & Sons, 2021).
Figure 1.
The central dogma of medical genetics, like the central dogma of molecular biology, progresses from the description of the phenotype of a genetic condition to the characterization of its natural history, which is vital to the establishment of management guidelines and treatment (from Cassidy & Allanson’s Management of Genetic Syndromes, eds, Carey JC, Battaglia A, Viskochil D, and Cassidy SB. Hoboken, NJ, John Wiley & Sons, 2021).
When GDD/ID/ASD are identified in a child, there is a shared sense of urgency to determine the causative factors. Given the potential impact of such a diagnosis and the hundreds of conditions known to cause GDD/ID/ASD, hypothetically one could perform a number of investigations. Powerful recent advances in technologies to analyze the genome have had a profound impact on the practice of medical genetics, both in the laboratory and in the clinic. Numerous techniques are now available to diagnose a particular phenotype, and while traditional techniques remain efficient tools in certain situations, higher-throughput technologies have become the de facto laboratory tool for diagnosis of most conditions. Maintaining a clear understanding of the rapidly evolving landscape of diagnostic tests, and their limitations, presents a challenge for non-genetics professionals. Therefore, selecting the right technology is challenging, and the wrong choice may lead to prolonged time to diagnosis, high costs, or even a missed diagnosis. Literature review, coupled with our own experience, highlights the need to further educate primary care clinicians in the uses and limitations of genetic testing for NDDs [26].
A rational approach to a diagnostic evaluation promises to provide significant benefits to the individual, family, and practitioner. For reaching a diagnosis, two main approaches are recognized historically: “phenotype-first” and “genotype-first”. Phenotype-first refers to the traditional approach to the recognition and delineation of a diagnostic entity. All of the conditions with eponyms exemplify this: a set of patients in the clinical setting are recognized to have overlapping manifestations and are documented, suggesting that the patients represent a diagnostic entity (e.g., Coffin-Siris syndrome). The phenotypic spectrum, the natural history, and more recently the etiology are characterized in later publications. Genotype-first refers to the early genotyping of patients with NDD (or other clinical presentations) whenever a given condition is not diagnosed clinically, and subsequent establishment of a causative diagnosis with ES/GS testing, which has become more readily available and expansive. The earliest genotype-first approach is represented by the broad use of karyotyping in individuals with GDD/ID and congenital abnormalities, not clinically recognizable, which explained 10-15% of ID individuals [27,28]. Due to the ongoing advances in comprehensive phenotype analysis and cytogenomic technology, the “genotype-first” approach has increasingly become the first step within the diagnostic process in NDDs.
Methods
The authors completed a narrative review of the literature on the rational assessment of the child with NDDs by collecting the many historical articles on the etiology of GDD/ID from prior work [5,29,30,31,32,33,34]. This search provided the historical framework. This was followed by a comprehensive review using PubMed, Medline, and Google Scholar databases led by AB to detect current papers on the diagnostic yield of next generation sequencing in individuals presenting with NDDs. Manuscripts, consisting of original papers and reviews, were reviewed for titles, abstracts, and entire texts.
The “Phenotype-First” Era Prebanding-Banding Period/FISH-Subtelomeric FISH Period
With the description of many of the now well known common congenital anomaly syndromes during the 1960s-1980s era (the “Phenotype-First” approach) researchers in the field of medical genetics developed approaches to the diagnosis of the child with these neurodevelopmental differences. The articles by Smith and Simons and Opitz et al. launched this approach and are considered classics [29,30]. From the 1970s on, research studies of individuals with NDDs helped establish a rational approach to the diagnosis of persons with these presentations [29,30,31,32,33,34]. Various investigations of individuals with GDD/ID (and more recently ASD) focused on determining a cause for the condition. These studies showed that the patients fell into six clinically-based categories [29,30,31,32,33,34,35]. This framework led to what was considered a more rational approach to the diagnostic assessment rather than performing all available testing. In the last three decades, working groups created by the AAP/AAN/ACMG reviewed available data and suggested focused consensus approaches to the assessment of patients [36,37,38]. Such consensus guidelines recommended appropriate laboratory testing and imaging based on the clinical presentation. A rational approach to a diagnostic evaluation promises to provide significant benefits to the patient, family, and practitioner. Usually the primary care practitioner weighs a variety of factors when deciding which diagnostic or screening tests to pursue on a given individual. Evaluation of the seriousness of the condition, acceptability of the test, and the importance of making a diagnosis are generally considered before proceeding with the tests. Rational evaluation is an expansion and extension of this approach specific to GDD/ID/ASD.
A thorough clinical history is of the utmost importance. A careful, head-to-toe physical examination, including a neurological assessment, and a search for skin changes should be performed. Documentation of abnormal findings, together with anthropometric measurements, is critical. Photographs, especially of the face, and video can prove very useful. Videos are an invaluable tool, documenting gait, posture, behavior characteristics, and movement disorders. The information gained from the physical examination alone can help in determining a diagnosis in about 20% of individuals or in postulating a provisional diagnosis for appropriate testing [32,33,34,35].
In the years 1960-1980, karyotyping was performed in individuals suspected of having a distinct condition, such as trisomy 21 or 18. For decades, a clinical assessment followed by standard cytogenetics of the times was the only option for several conditions as molecular testing was not available (examples in Appendix). Later, karyotyping was used more broadly in individuals with GDD/ID and multiple congenital abnormalities (the earliest “genotype-first” approach), and was successful in detecting chromosomal rearrangements larger than 5Mb, with an overall diagnostic yield of 10-15%. The identification of the first individuals with the fragile site at the bottom end of the X chromosome in 1969 [44], followed by recognition of the disorder in several other individuals throughout 1970s and 1980s with the use of cytogenetic studies, provided an extra diagnostic yield, with 1% of the GDD/ID males being affected [45]. Subsequently FISH probes, developed in 1980s-1990s, enabled the detection of smaller copy number variations (CNVs) than karyotyping and led to the recognition of many of the now classic chromosome disorders such as deletion 1p36, in the 1990s [46,47,48,49,50]; and to the boundaries of what is now known as the critical region of a number of disorders, such as Wolf-Hirschhorn syndrome [51]. Thereafter, the multiprobe FISH was shown to be highly suitable for detecting subtelomeric deletions and duplications accounting for an additional 2.5-5% of the diagnostic yield in individuals with NDDs [52,53,54,55]. To assist in this diagnostic pathway authors of previously cited articles on the rational assessment suggested algorithms [32,33,34].
Comprehensive phenotype analysis, including the understanding of the natural history of the condition, logically informs the development of clinical guidelines and treatment modalities. This progression from phenotype to natural history to management can be considered the central dogma of medical genetics (following the theme of the central dogma of biology, DNA→RNA→protein) [56] (Figure 1).
Even after the identification of the molecular defect, obtaining a diagnosis still depended on the clinician’s expertise and ability to recognize the many hundreds of clinically recognizable disorders. Moreover, the approach could fail when the patient’s characteristics did not entirely fit a known disorder, the syndrome was genetically heterogeneous, or the genetic defect was unknown. In these latter scenarios, the clinician simply did not know which gene to test, and a clinical diagnosis could not be confirmed by any molecular testing. As a consequence, the only way to identify a causative gene mutation was using Sanger sequencing [57], which, however, only allowed for the testing of one gene at a time. Thereafter, sequential single-by-gene testing was performed until a causative mutation was found based on clinical suspicion. Such an expensive diagnostic odyssey often took years, being a burden to the patients and families involved [58]. Moreover, for diagnosing patients with more than one Mendelian disorder, the phenotype-first approach would not work, and in only 7% of patients could the gene mutation causing the phenotype be identified [59]. This low diagnostic yield spawned a search for a genome-wide screening test without the a priori knowledge of the genomic locus and faster than Sanger sequencing.
The “Genotype-First” Era Cytogenomic Microarray Period
In the early 2000s, the introduction of multiplex ligation-dependent probe amplification (MLPA) and cytogenomic hybridization microarray (CMA) in both research and clinics spurred both improved diagnosis of patients with known MCA syndromes and documentation of CNVs of less than 5Mb or even smaller (<1Mb), helping both to characterize the critical regions for specific component phenotypes (seizures, face), and increasing the diagnostic yield of NDDs by another 15% [10,60]. CMA was also helpful in demonstrating the high prevalence of translocations, even cryptic, not detected by a previous karyotype combined with a given syndrome-specific FISH [61]. CMA can determine if a deletion is pure or part of a more complex imbalance, more accurately than either FISH or standard cytogenetics alone.
These new laboratory techniques also led to the identification of novel GDD/ID syndromes, such as 1q21.1 and 17q21.3 microdeletion syndromes [62,63,64]. All are clear-cut examples of the genotype-first approach: wide cytogenomic screening leading to a clinical characterization of a novel entity and even to a causative single gene.
Next Generation Sequencing (NGS): a Shift in the Genetic Testing Paradigm
A quantum leap in genetic testing occurred with the development of NGS whereby DNA from individuals could be screened for sequence variants in days to weeks rather than months to years [65]. Not only did this technique increase sensitivity for detection of pathogenic mutations in single genes, it enabled laboratories to develop panels to screen many genes for a given phenotype. Genetic syndromes with overlapping manifestations could be easily screened simultaneously. The convenience of NGS led to the development of wider and wider testing panels, ultimately making exome sequence (ES) analysis a reality in clinical care, considered by many to be the first tier test in the genetic assessment of individuals with NDDs [66,67,68,69]. These advances represent a paradigm shift in the assessment of NDD.
Exome Sequencing Period
The availability of ES at lower costs and increased availability spawned the emergence of a “genotype first” approach where a genetic syndrome is diagnosed by molecular testing and not clinically. This is particularly the case when studying individuals with milder manifestations that may not be easily recognized. In addition, a broader molecular screening approach may lead to the diagnosis of individuals on the milder end of a given syndrome spectrum. This application of “genotype first” gave rise to the recognition of phenotype expansion for some of the disorders in clinical genetics, which may provide a better understanding of specific management issues on a genotype–phenotype basis [69]. The methodologies for genetic testing have been optimized for many of the syndromes. Over recent years, ES has been strongly recommended and introduced, in most developed countries, within the diagnostic setting as a first or second-tier test for individuals with MCA/GDD/ID [70,71,72,73]. Moreover, ES has been shown to be successful also in persons with ASD and epilepsy [74,75,76]. Overall, the use of ES in individuals with NDDs has improved the diagnostic yield up to 30-55% [10,77,78,79,80,81,82,83,84], depending on the clinical features of the population tested, year of testing, and analytical strategy [79,85,86,87,88,89], leaving 45% of individuals without an etiology.
As the clinical experience illustrates, the approach and yield of diagnostic testing has changed dramatically with the application of NGS from the research laboratory to the clinical setting during the decade beginning in 2010. By the end of the decade, a number of scientific journals were publishing reports of series of individuals, with similar pathogenic variants of previously known genes leading to the description of novel disease entities on a regular basis. Other studies showed that performing NGS early in the diagnostic process provided changes to the individual’s management and was cost-effective, ending the diagnostic odyssey [71].
First Tier Testing for NDDs
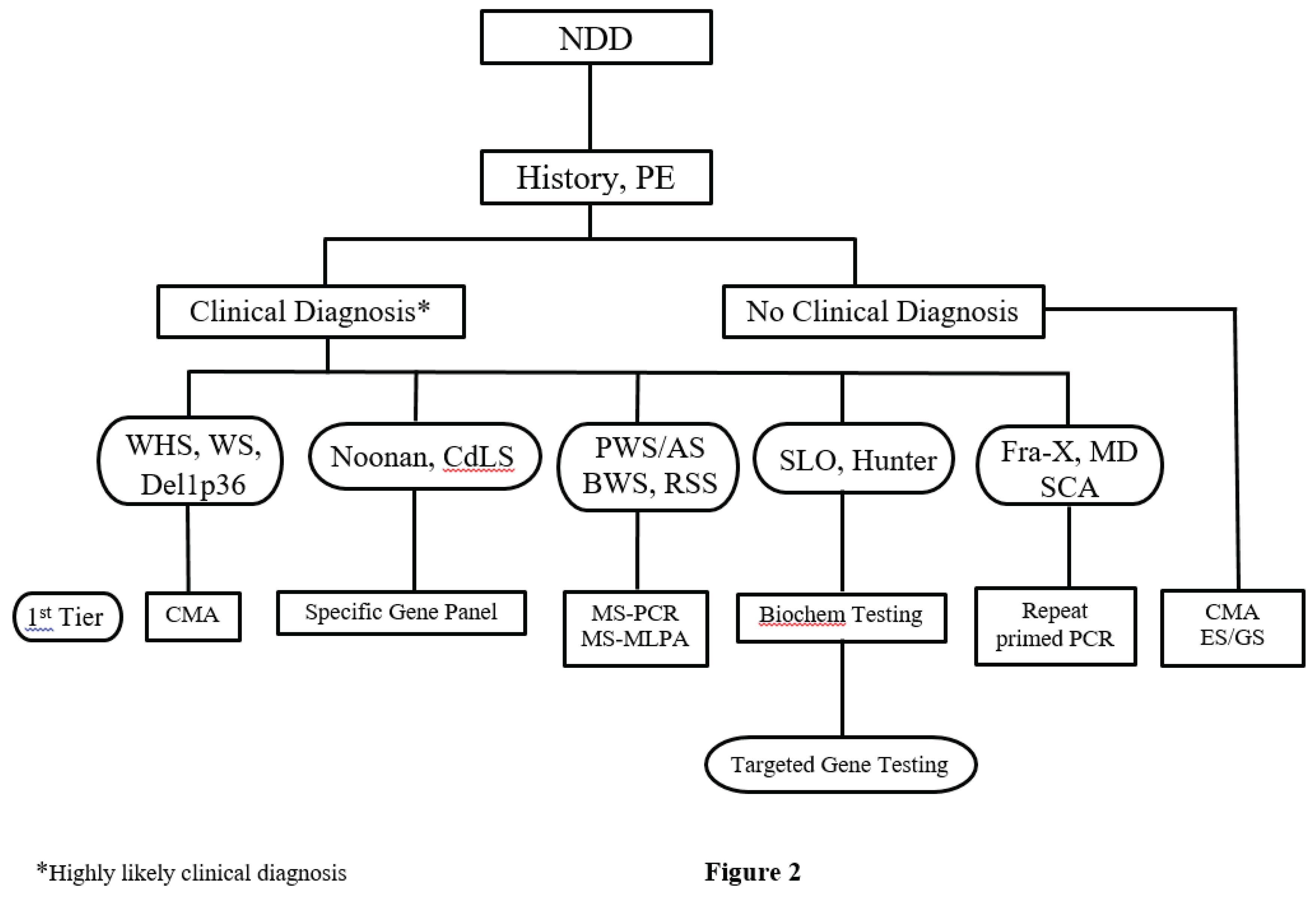
Based on scientific data [90] and on our own experience, we propose to consider ES as a first tier test for NDDs [91]. But for the individuals who likely have a diagnosis based on the clinical presentation, such as those children with WHS or WS, the first tier test would necessarily be CMA. Other conditions in which ES should not be the first tier test include those disorders caused by a number of different genetic and epigenetic alterations (PWS/AS, RSS), in which the first tier test should be MS-PCR and MS-MLPA; and repeat-expansion diseases (fragile X syndrome, myotonic dystrophy) in which the first tier test should be repeat-primed PCR. Fragile-X syndrome remains a common cause of ID/ASD, and should be offered in addition to ES analysis to those individuals with a positive family history of ID and ASD (without microcephaly), and to those with a clinical-behavioral phenotype reminiscent of the syndrome. Lastly, targeted gene panels would be ideal for analyzing specific variants of genes that have suspected associations with disease (CdLS, Noonan syndromes). In summary, ES is a better choice when one is uncertain what genes need to be tested. See Figure 2, an updated algorithm summarizing this thought process and diagnostic pathway. An example illustrating the approach is in the Appendix.
In the upcoming years, WGS will likely supplant ES as the most comprehensive clinical genomic diagnostic test. However, the interpretation of and counseling for the many variants of uncertain significance (VUS) that can be found when performing ES/GS constitutes an enormous challenge.
This paradigm shift in genetic testing has created a renewed interest in phenotype, and the notion of “deep phenotyping” has emerged, and has highlighted the paramount importance of the partnership between clinicians and laboratory geneticists [94]. The techniques for exome capture and sequencing have greatly improved over time, and are relatively stable in most laboratories, but the most significant challenges are in the complexity of data analysis and interpretation of the functional impact of the detected variants and identification of the underlying affected pathway. The large amount of sequence data generated by NGS platforms has fueled the addition of bioinformatics teams into clinical diagnostic laboratories to develop data handling and analysis pipelines for such complex tests. Variants that are rare in the population and are predicted to have a functional impact on the gene, by altering the gene’s protein coding sequence, are prioritized for analysis and human interpretation. Numerous efforts have been carried out to develop computational tools to functionally interpret both coding and non-coding genomic elements and to estimate the variants pathogenicity [95,96,97,98]. Progress made with the introduction of WGS [70] has allowed for the identification of non-coding variants and also provided more uniform coverage throughout the genome which is useful for the identification of structural variation at base-pair resolution. Overall, it seems that pathogenic de novo non-coding mutations probably account for less than 5% of exome-negative NDDs individuals [100,101,102,103].
Recent studies show that both re-analysis of existing ES/GS data as well as resequencing strategies increase the diagnostic yield by 6-13% in undiagnosed NDDs individuals [103,104,105,106], and the suggested cadence of re-analysis was no more than 3 years [107,108,109]. A recent case report illustrates this point (see Appendix).
Notably, functional validation in specialist laboratories is essential to support the causal link between the genetic variant and the individual’s phenotype, providing evidence for the variant to be considered a pathogenic variant according to the ACMG guidelines [110].
Updating clinical information before revisiting genetic data is essential. As children may not yet display the full characteristic phenotype of a given disorder, systematic reassessment of the child clinical and behavioral phenotype might show additional features implicative of a specific condition. Such evaluations may be crucial for genetic re-analysis. Assessment of the parental phenotypes is also of paramount importance, as, for instance, by assuming full penetrance of variants and apparently unaffected parents, variants in the index may be disregarded during interpretation [111]. It has similarly been found that incomplete penetrance or variable expressivity makes it hard the discovery of novel genes underlying NDDs [112].
It is reasonable to expect that other diagnostic techniques, such as methylation profiling [113], optical mapping [114], long read sequencing, or RNA sequencing [115,116], will increase diagnostic yield (for in-depth description see Appendix). Clinical utility of combining trio-ES sequencing with copy number variation sequencing in the etiologic diagnosis and clinical management of GDD is proven [134]. The feasibility of implementing such automated systems for re-analysis depends, however, on available budget, local infrastructure, and bioinformatic support. The budget constitutes the main obstacle both in low and middle income countries and in those countries with a national health scheme [135,136,137]. With not all individuals returning to the clinic for systematic follow-up, it is important to request consent from families to allow for re-analysis, also including the use of new technologies, after the initial negative or uncertain diagnostic analysis, to maximize benefits.
Due to these advances in NGS other models of care have emerged in recent years: the establishment of Undiagnosed Disease Clinics and a US clinics network [138,139,140] and the strategy of WGS as a first-tier diagnostic test for sick infants admitted to the NICU [86,141,143]. (see Appendix for more discussion of these approaches).
Most healthcare professionals have progressively acknowledged the benefit of NGS since it helps mitigate the “fast and furious” pace of discovery in genomics, considering the estimated 250 new gene-phenotype associations being unveiled each year [83,154]. A recent study has shown how re-analysis of trio GS data doubles the yield in NDDs, and the genetic data obtained were actionable in terms of altering management in most cases (up to 74%) [104]. Most importantly, two other recent studies showed the potential to use a single technology (long-read genomes) to accurately identify all types of clinically relevant variants, potentially qualifying long-read sequencing in the future as a first-tier test for all rare diseases [155,156], also allowing for the detection of dual pathology.
ES/GS are feasible in clinical practice and identify causal variants in a substantial proportion of persons with NDDs. But neither test can diagnose a genetic disease in a patient [94]. Clinical laboratories report variant classifications, not patient diagnoses. The ACMG guidelines recommend against making diagnostic claims or basing clinical management on genetic test result alone [157]. Diagnosis must be performed by a clinician who can assess genetic variants in the context of all of a patient’s clinical features and their consistency with the full spectrum of clinical manifestations that occur in the disease being considered.
Summary and Conclusions
We have provided the historical framework and an updated approach to the rational assessment of the person with NDDs. In the child with a strong suspicion of a specific condition, the appropriate first tier testing depends on the diagnosis in question (Figure 2). The history and physical examination performed by an experienced clinician drives the choice of the most appropriate genetic testing in that situation.. In the child without a clinical diagnosis we recommend ES/GS as the first step (see Appendix for further due considerations).
Author Contributions
Dr. Agatino Battaglia: conception of idea, methods/review, analysis of results, composed draft, completed the final. Dr. John C Carey: conception of idea, analysis of results, reviewed draft and read the final.
Funding
The study was funded by the Italian Ministry of Health under the "Ricerca Corrente" program, which supports institutional research activities without direct involvement in specific study procedures or publications.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We thank the patients and families we have cared for over the years who spurred our deep interest in the topic, and our postgraduate medical students, residents, and fellows who by asking so many probing questions, taught us in the process.We thank Feliz Martinez for technical assistance with the algorithm. We dedicate this work to our mentor, teacher, and colleague, Dr. John M. Opitz.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
PE: physical examination; WHS: Wolf-Hirschhorn syndrome; WS: Williams syndrome; CdLS: Cornelia de Lange syndrome; PWS: Prader-Willi syndrome; AS: Angelman syndrome; BWS: Beckwith-Wiedemann syndrome; RSS: Russel-Silver syndrome; SLOS: Smith-Lemli-Opitz syndrome; MD: myotonic dystrophy; SCA: spino-cerebellar ataxia; CMA: cytogenomic microarray; MS-PCR/MS-MLPA: Methylation-Specific Polymerase Chain Reaction/Methylation-Specific Multiplex Ligation-dependent Probe Amplification; ES: exome sequencing; WGS: whole genome sequencing
References
- Callahan, K.P.; Feudtner, C. Genetic Testing Is Messier in Practice than in Theory: Lessons from Neonatology. Am. J. Bioeth. 2022, 22, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Thomaidis, L.; Zantopoulos, G.Z.; Fouzas, S.; Mantagou, L.; Bakoula, C.; Konstantopoulos, A. Predictors of severity and outcome of global developmental delay without definitive etiologic yield: a prospective observational study. BMC Pediatr. 2014, 14, 40–40. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, 2013. [Google Scholar]
- Moeschler, J.B.; Shevell, M.; Saul, R.A.; Chen, E.; Freedenberg, D.L.; Hamid, R.; Jones, M.C.; Stoler, J.M.; Tarini, B.A.; COMMITTEE ON GENETICS. Comprehensive Evaluation of the Child With Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef]
- Majnemer, A.; Shevell, M.I. Diagnostic yield of the neurologic assessment of the developmentally delayed child. J. Pediatr. 1995, 127, 193–199. [Google Scholar] [CrossRef]
- Maulik, P.K.; Mascarenhas, M.N.; Mathers, C.D.; Dua, T.; Saxena, S. Prevalence of intellectual disability: A meta-analysis of population-based studies. Res. Dev. Disabil.;Correction to Res. Dev. Disabil. 2011, 32 34, 419–436 729. [Google Scholar] [CrossRef]
- Xie, D.; Duan, R.; Li, C.; Xie, Z.; Wang, A.; Xiong, L.; Wei, J.; Xi, H.; Fang, J.; Yan, H.; et al. Study on the Economic Burden of Neurodevelopmental Diseases on Patients With Genetic Diagnosis. Front. Public Heal. 2022, 10, 887796. [Google Scholar] [CrossRef]
- Sadleir, L.G; Mountier, EI; Gill, D; Davis, S; Joshi, C; DeVile, C; et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology 2017, 89, 1035–1042. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Pirozzi, F. Advances in understanding–genetic basis of intellectual disability. F1000Research 2016, 5, 599. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2015, 17, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al.; The Brainstorm Consortium Analysis of shared heritability in common disorders of the brain. Science 2018, 360. [Google Scholar] [CrossRef]
- Jamet, E. An eye-tracking study of cueing effects in multimedia learning. Comput. Human Behav. 2014, 32, 47–53. [Google Scholar] [CrossRef]
- Battaglia, A.; Lortz, A.; Carey, J.C. Natural history study of adults with Wolf–Hirschhorn syndrome 1: Case series of personally observed 35 individuals. Am. J. Med Genet. Part A 2021, 185, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.C.; Lortz, A.; Mendel, A.; Battaglia, A. Natural history study of adults with Wolf–Hirschhorn syndrome 2: Patient-reported outcomes study. Am. J. Med Genet. Part A 2021, 185, 2065–2069. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A. Deletion 4p: Wolf-Hirschhorn syndrome. In “Cassidy’s and Allansons’ Management of Genetic Syndromes”, 4th edition; Carey, JC., Battaglia, A., Viskochil, D., Cassidy, SB., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA and Chichester, West Sussex, UK, 2021; Volume chapter 19, pp. pages 265–280. [Google Scholar]
- Rosenbaum, P.L. Prevention of psychosocial problems in children with chronic illness. 1988, 139, 293–5. [Google Scholar] [PubMed]
- Dingemans, A.J.M.; Stremmelaar, D.E.; Vissers, L.E.L.M.; Jansen, S.; Sá, M.J.N.; van Remortele, A.; Jonis, N.; Truijen, K.; van de Ven, S.; Ewals, J.; et al. Human disease genes website series: An international, open and dynamic library for up-to-date clinical information. Am. J. Med Genet. Part A 2021, 185, 1039–1046. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Yin, J.; Bajic, A.; Zhang, P.; Cummock, S.; Kim, J.J.; Schaaf, C.P. Functional Consequences of CHRNA7 Copy-Number Alterations in Induced Pluripotent Stem Cells and Neural Progenitor Cells. Am. J. Hum. Genet. 2017, 101, 874–887. [Google Scholar] [CrossRef]
- Bick, D.; Bick, S.L.; Dimmock, D.P.; Fowler, T.A.; Caulfield, M.J.; Scott, R.H. An online compendium of treatable genetic disorders. Am. J. Med Genet. Part C: Semin. Med Genet. 2020, 187, 48–54. [Google Scholar] [CrossRef]
- Ioannidis, V.; Pandey, R.; Bauer, H.F.; Schön, M.; Bockmann, J.; Boeckers, T.M.; Lutz, A.-K. Disrupted extracellular matrix and cell cycle genes in autism-associated Shank3 deficiency are targeted by lithium. Mol. Psychiatry 2023, 29, 704–717. [Google Scholar] [CrossRef]
- Bupp, C.P.; Schultz, C.R.; Uhl, K.L.; Rajasekaran, S.; Bachmann, A.S. Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features. Am. J. Med Genet. Part A 2018, 176, 2548–2553. [Google Scholar] [CrossRef]
- Gong, M.; Li, J.; Qin, Z.; Wilke, M.V.M.B.; Liu, Y.; Li, Q.; Liu, H.; Liang, C.; Morales-Rosado, J.A.; Cohen, A.S.; et al. MARK2 variants cause autism spectrum disorder via the downregulation of WNT/β-catenin signaling pathway. Am. J. Hum. Genet. 2024, 111, 2392–2410. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Battaglia, A.; Parrini, B.; Tancredi, R. The behavioral phenotype of the idic(15) syndrome. Am. J. Med Genet. Part C: Semin. Med Genet. 2010, 154C, 448–455. [Google Scholar] [CrossRef]
- Wang, A.; Little, I.D.; Carter, D.; Pham, S.; Piper, M.; Ramírez-Renta, G.M.; Telaak, S.; Gunter, C. Provider-reported experiences, barriers, and perspectives on genetic testing as part of autism diagnosis. PLOS ONE 2024, 19, e0296942. [Google Scholar] [CrossRef]
- O’Connor, C. Chromosomes and cytogenetics. 2014. Available online: https://www.nature.com.
- Warkany, J.; Weinstein, E.D.; Soukup, S.W.; Rubinstein, J.H.; Curless, M.C. CHROMOSOME ANALYSES IN A CHILDREN'S HOSPITAL. Pediatrics 1964, 33, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Smith, DW; Simons, ER. Rational diagnostic evaluation of the child with mental deficiency. Am J Dis Child 1975, 129, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Opitz, J.M.; Kaveggia, E.G.; Durkin-Stamm, M.V.; Pendleton, E. Diagnostic/genetic studies in severe mental retardation. 1978, 14, 1–38. [Google Scholar] [PubMed]
- McLaren, J.; E Bryson, S. Review of recent epidemiological studies of mental retardation: prevalence, associated disorders, and etiology. 1987, 92, 243–54. [Google Scholar]
- Battaglia, A; Bianchini, E; Carey, JC. Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet 1999, 82, 60–66. [Google Scholar] [CrossRef]
- Battaglia, A. Genetics of mental retardation. Am J Med Genet Part C, Semin Med Genet 2003, 117C, 1–2. [Google Scholar] [CrossRef]
- Battaglia, A.; Carey, J.C. Diagnostic evaluation of developmental delay/mental retardation: An overview. Am. J. Med Genet. Part C: Semin. Med Genet. 2003, 117C, 3–14. [Google Scholar] [CrossRef]
- Van Karnebeek, CDM; Jansweijer, MCE; Leenders, AGE; Offringa, M; Hennekam, RCM. Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet 2005, 13, 6–25. [Google Scholar] [CrossRef]
- Curry, C.J.; Stevenson, R.E.; Aughton, D.; Byrne, J.; Carey, J.C.; Cassidy, S.; Cunniff, C.; Graham, J.M.; Jones, M.C.; Kaback, M.M.; et al. Evaluation of mental retardation: Recommendations of a consensus conference. Am. J. Med Genet. 1997, 72, 468–477. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Moeschler, J.B.; Shevell, M.; Saul, R.A.; Chen, E.; Freedenberg, D.L.; Hamid, R.; Jones, M.C.; Stoler, J.M.; Tarini, B.A.; COMMITTEE ON GENETICS. Comprehensive Evaluation of the Child With Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef]
- Angelman, H. “Puppet children”. A report on three cases. Dev Med Child Neurol 1965, 7, 681–688. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.W.; Lemli, L.; Opitz, J.M. A newly recognized syndromeof multiple congenital anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Maslen, C.; Kachilele-Linjewile, S.; Lin, D.; Linck, L.M.; Connor, W.E.; Steiner, R.D.; Porter, F.D. Mutations in the Human Sterol Δ7-Reductase Gene at 11q12-13 Cause Smith-Lemli-Opitz Syndrome. Am. J. Hum. Genet. 1998, 63, 55–62. [Google Scholar] [CrossRef]
- Waterham, H.R.; Wijburg, F.A.; Hennekam, R.C.; Vreken, P.; Poll-The, B.T.; Dorland, L.; Duran, M.; Jira, P.E.; Smeitink, J.A.; Wevers, R.A.; et al. Smith-Lemli-Opitz Syndrome Is Caused by Mutations in the 7-Dehydrocholesterol Reductase Gene. Am. J. Hum. Genet. 1998, 63, 329–338. [Google Scholar] [CrossRef]
- A Lubs, H. A marker X chromosome. 1969, 21, 231–44. [Google Scholar] [PubMed]
- de Vries, B.B.; Ouweland, A.M.v.D.; Mohkamsing, S.; Duivenvoorden, H.J.; Mol, E.; Gelsema, K.; van Rijn, M.; Halley, D.J.; Sandkuijl, L.A.; Oostra, B.A.; et al. Screening and Diagnosis for the Fragile X Syndrome among the Mentally Retarded: An Epidemiological and Psychological Survey. Am. J. Hum. Genet. 1997, 61, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Ewart, A.K.; A Morris, C.; Ensing, G.J.; Loker, J.; Moore, C.; Leppert, M.; Keating, M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc. Natl. Acad. Sci. 1993, 90, 3226–3230. [Google Scholar] [CrossRef]
- Ewart, A.K.; Morris, C.A.; Atkinson, D.; Jin, W.; Sternes, K.; Spallone, P.; Stock, A.D.; Leppert, M.; Keating, M.T. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 1993, 5, 11–16. [Google Scholar] [CrossRef]
- Breuning, M.H.; Dauwerse, H.G.; Fugazza, G.; Saris, J.J.; Spruit, L.; Wijnen, H.; Tommerup, N.; Van Der Hagen, C.B.; Imaizumi, K.; Kuroki, Y.; et al. Rubinstein-Taybi syndrome caused by submicroscopic deletions within 16p13.3. 1993, 52, 249–54. [Google Scholar]
- Battaglia, A; Gurrieri, F; Bertini, E; Bellacosa, A; Pomponi, MG; Paravatou-Petsotas, M; et al. The inv dup(15) syndrome: a clinically recognizable syndrome with altered behavior, mental retardation and epilepsy. Neurology 1997, 48, 1081–1086. [Google Scholar] [CrossRef]
- Shapira, S.K.; McCaskill, C.; Northrup, H.; Spikes, A.S.; Elder, F.; Sutton, V.R.; Korenberg, J.R.; Greenberg, F.; Shaffer, L.G. Chromosome 1p36 Deletions: The Clinical Phenotype and Molecular Characterization of a Common Newly Delineated Syndrome. Am. J. Hum. Genet. 1997, 61, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Estabrooks, L.L.; Rao, K.W.; Driscoll, D.A.; Crandall, B.F.; Dean, J.C.S.; Ikonen, E.; Korf, B.; Aylsworth, A.S. Preliminary phenotypic map of chromosome 4p16 based on 4p deletions. Am. J. Med Genet. 1995, 57, 581–586. [Google Scholar] [CrossRef]
- Flint, J.; Wilkie, A.O.; Buckle, V.J.; Winter, R.M.; Holland, A.J.; McDermid, H.E. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat. Genet. 1995, 9, 132–140. [Google Scholar] [CrossRef]
- Knight, S.J.; Regan, R.; Nicod, A.; Horsley, S.W.; Kearney, L.; Homfray, T.; Winter, R.M.; Bolton, P.; Flint, J. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet 1999, 354, 1676–1681. [Google Scholar] [CrossRef]
- A De Vries, B.B.; Winter, R.; Schinzel, A.; van Ravenswaaij-Arts, C. Telomeres: a diagnosis at the end of the chromosomes. J. Med Genet. 2003, 40, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Ravnan, J.B.; Tepperberg, J.H.; Papenhausen, P.; Lamb, A.N.; Hedrick, J.; Eash, D.; Ledbetter, D.H.; Martin, C.L. Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med Genet. 2006, 43, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Carey, JC; Battaglia, A; Viskochil, D; Cassidy, SB. Introduction. In “Cassidy’s and Allansons’ Management of Genetic Syndromes”, 4th ed.; Carey, JC., Battaglia, A., Viskochil, D., Cassidy, SB., Eds.; Wiley Blackwell: Hoboken, NJ, USA, 2021; pp. XXVII–XXXV. [Google Scholar]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage φX174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Mefford, HC. Genotype to phenotype-discovery and characterization of novel genomic disorders in a “genotype-first” era. Genet Med 2009, 11, 836–842. [Google Scholar]
- Jansen, S.; Vissers, L.E.L.M.; de Vries, B.B.A. The Genetics of Intellectual Disability. Brain Sci. 2023, 13, 231. [Google Scholar] [CrossRef]
- de Vries, B.B.; Pfundt, R.; Leisink, M.; Koolen, D.A.; Vissers, L.E.; Janssen, I.M.; van Reijmersdal, S.; Nillesen, W.M.; Huys, E.H.; de Leeuw, N.; et al. Diagnostic Genome Profiling in Mental Retardation. Am. J. Hum. Genet. 2005, 77, 606–616. [Google Scholar] [CrossRef]
- South, S.T.; Whitby, H.; Battaglia, A.; Carey, J.C.; Brothman, A.R. Comprehensive analysis of Wolf–Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur. J. Hum. Genet. 2007, 16, 45–52. [Google Scholar] [CrossRef]
- A Koolen, D.; Vissers, L.E.L.M.; Pfundt, R.; de Leeuw, N.; Knight, S.J.; Regan, R.; Kooy, R.F.; Reyniers, E.; Romano, C.; Fichera, M.; et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006, 38, 999–1001. [Google Scholar] [CrossRef]
- Sharp, A.J.; Selzer, R.R.; Veltman, J.A.; Gimelli, S.; Gimelli, G.; Striano, P.; Coppola, A.; Regan, R.; Price, S.M.; Knoers, N.V.; et al. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet. 2007, 16, 567–572. [Google Scholar] [CrossRef]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent Rearrangements of Chromosome 1q21.1 and Variable Pediatric Phenotypes. New Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Anesthesia Analg. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Glusman, G.; Smit, A.F.A.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.-I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Anesthesia Analg. 2019, 21, 2413–2421. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.; Thung, D.; Van De Vorst, M.; Van Bon, B.; Willemsen, M.; Kwint, M.; Janssen, I.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2021, 23, 2029–2037. [Google Scholar] [CrossRef]
- Stark, Z.; Schofield, D.; Martyn, M.; Rynehart, L.; Shrestha, R.; Alam, K.; Lunke, S.; Tan, T.Y.; Gaff, C.L.; White, S.M. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Anesthesia Analg. 2019, 21, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; van Nimwegen, K.J.; Schieving, J.H.; Kamsteeg, E.-J.; Kleefstra, T.; Yntema, H.G.; Pfundt, R.; van der Wilt, G.J.; Krabbenborg, L.; Brunner, H.G.; et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Anesthesia Analg. 2017, 19, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Kim, G.E.; Pagnamenta, A.T.; Murakami, Y.; Carvill, G.L.; Meyer, E.; Copley, R.R.; Rimmer, A.; Barcia, G.; Fleming, M.R.; et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 2014, 23, 3200–3211. [Google Scholar] [CrossRef]
- Turner, T.N.; Hormozdiari, F.; Duyzend, M.H.; McClymont, S.A.; Hook, P.W.; Iossifov, I.; Raja, A.; Baker, C.; Hoekzema, K.; Stessman, H.A.; et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet. 2016, 98, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Allen, AS; Berkovic, SF; Cossette, P; Delanty, N; Dlugos, D; Eichler, EE.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef]
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. npj Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef]
- De Ligt, J.; Willemsen, M.H.; Van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; De Vries, P.; Gilissen, C.; et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De Novo Mutations in Moderate or Severe Intellectual Disability. PLOS Genet. 2014, 10, e1004772–e1004772. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.M.; Hildreth, A.; Batalov, S.; Ding, Y.; Chowdhury, S.; Watkins, K.; Ellsworth, K.; Camp, B.; Kint, C.I.; Yacoubian, C.; et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Farnaes, L.; Hildreth, A.; Sweeney, N.M.; Clark, M.M.; Chowdhury, S.; Nahas, S.; Cakici, J.A.; Benson, W.; Kaplan, R.H.; Kronick, R.; et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. npj Genom. Med. 2018, 3, 1–8. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; A Nickerson, D.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2009, 42, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; I Gildersleeve, H.; E Beck, A.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. New Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Testard, Q.; Vanhoye, X.; Yauy, K.; Naud, M.-E.; Vieville, G.; Rousseau, F.; Dauriat, B.; Marquet, V.; Bourthoumieu, S.; Geneviève, D.; et al. Exome sequencing as a first-tier test for copy number variant detection: retrospective evaluation and prospective screening in 2418 cases. J. Med Genet. 2022, 59, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Mariano, V.; Kanellopoulos, A.K.; Ricci, C.; Di Marino, D.; Borrie, S.C.; Dupraz, S.; Bradke, F.; Achsel, T.; Legius, E.; Odent, S.; et al. Intellectual Disability and Behavioral Deficits Linked to CYFIP1 Missense Variants Disrupting Actin Polymerization. Biol. Psychiatry 2023, 95, 161–174. [Google Scholar] [CrossRef]
- Kim, J.-H.; Shinde, D.N.; Reijnders, M.R.; Hauser, N.S.; Belmonte, R.L.; Wilson, G.R.; Bosch, D.G.; Bubulya, P.A.; Shashi, V.; Petrovski, S.; et al. De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am. J. Hum. Genet. 2016, 99, 711–719. [Google Scholar] [CrossRef]
- Tokita, M.J.; Braxton, A.A.; Shao, Y.; Lewis, A.M.; Vincent, M.; Küry, S.; Besnard, T.; Isidor, B.; Latypova, X.; Bézieau, S.; et al. De Novo Truncating Variants in SON Cause Intellectual Disability, Congenital Malformations, and Failure to Thrive. Am. J. Hum. Genet. 2016, 99, 720–727. [Google Scholar] [CrossRef]
- Friedman, J.M.; Jones, K.L.; Carey, J.C. Exome Sequencing and Clinical Diagnosis. JAMA 2020, 324, 627–628. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- I Davydov, I.; Salamin, N.; Robinson-Rechavi, M. Large-Scale Comparative Analysis of Codon Models Accounting for Protein and Nucleotide Selection. Mol. Biol. Evol. 2019, 36, 1316–1332. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting the Effects of Amino Acid Substitutions on Protein Function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Rivière, J.-B.; Fombonne, E.; O’rOak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; FitzPatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Takata, A. Estimating contribution of rare non-coding variants to neuropsychiatric disorders. Psychiatry Clin. Neurosci. 2018, 73, 2–10. [Google Scholar] [CrossRef]
- Turner, T.N.; Coe, B.P.; Dickel, D.E.; Hoekzema, K.; Nelson, B.J.; Zody, M.C.; Kronenberg, Z.N.; Hormozdiari, F.; Raja, A.; Pennacchio, L.A.; et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 2017, 171, 710–722.e12. [Google Scholar] [CrossRef] [PubMed]
- Bar, O.; Vahey, E.; Mintz, M.; Frye, R.E.; Boles, R.G. Reanalysis of Trio Whole-Genome Sequencing Data Doubles the Yield in Autism Spectrum Disorder: De Novo Variants Present in Half. Int. J. Mol. Sci. 2024, 25, 1192. [Google Scholar] [CrossRef]
- Miyake, N.; Tsurusaki, Y.; Fukai, R.; Kushima, I.; Okamoto, N.; Ohashi, K.; Hashimoto, R.; Hiraki, Y.; Son, S.; Kato, M.; et al. Molecular diagnosis of 405 individuals with autism spectrum disorder. Eur. J. Hum. Genet. 2023, 32, 1551–1558. [Google Scholar] [CrossRef]
- Laurie, S.; Steyaert, W.; de Boer, E.; Polavarapu, K.; Schuermans, N.; Sommer, A.K.; Demidov, G.; Ellwanger, K.; Paramonov, I.; Thomas, C.; et al. Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses. Nat. Med. 2025, 31, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Innella, G.; Ferrari, S.; Miccoli, S.; Luppi, E.; Fortuno, C.; Parsons, M.T.; Spurdle, A.B.; Turchetti, D. Clinical implications of VUS reclassification in a single-centre series from application of ACMG/AMP classification rules specified forBRCA1/2. J. Med Genet. 2023, 61, 483–489. [Google Scholar] [CrossRef]
- Jamet, E. An eye-tracking study of cueing effects in multimedia learning. Comput. Human Behav. 2014, 32, 47–53. [Google Scholar] [CrossRef]
- Schobers, G.; Schieving, J.H.; Yntema, H.G.; Pennings, M.; Pfundt, R.; Derks, R.; Hofste, T.; de Wijs, I.; Wieskamp, N.; Heuvel, S.v.D.; et al. Reanalysis of exome negative patients with rare disease: a pragmatic workflow for diagnostic applications. Genome Med. 2022, 14, 1–10. [Google Scholar] [CrossRef]
- Farris, J.; Khanna, C.; Smadbeck, J.B.; Johnson, S.H.; Bothun, E.; Kaplan, T.; Hoffman, F.; Polonis, K.; Oliver, G.; Reis, L.M.; et al. Complex balanced intrachromosomal rearrangement involving PITX2 identified as a cause of Axenfeld-Rieger Syndrome. Am. J. Med Genet. Part A 2024, 194. [Google Scholar] [CrossRef]
- Farley, K.O.; Forbes, C.A.; Shaw, N.C.; Kuzminski, E.; Ward, M.; Baynam, G.; Lassmann, T.; Fear, V.S. CRISPR-Cas9-generated PTCHD1. Hum. Genet. Genom. Adv. 2023, 5, 100257. [Google Scholar] [CrossRef]
- Salfati, E.L.; Spencer, E.G.; Topol, S.E.; Muse, E.D.; Rueda, M.; Lucas, J.R.; Wagner, G.N.; Campman, S.; Topol, E.J.; Torkamani, A. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med. 2019, 11, 1–8. [Google Scholar] [CrossRef]
- Kaplanis, J.; Samocha, K.E.; Wiel, L.; Zhang, Z.; Arvai, K.J.; Eberhardt, R.Y.; Gallone, G.; Lelieveld, S.H.; Martin, H.C.; McRae, J.F.; et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020, 586, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Marshall, A.E.; Lemire, G.; Liang, Y.; Davila, J.; Couse, M.; Boycott, K.M.; Kernohan, K.D. RNA sequencing reveals deep intronic CEP120 variant: A report of the diagnostic odyssey for two siblings with Joubert syndrome type 31. Am. J. Med Genet. Part A 2023, 194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Longo, N.; Lewis, R.G.; Nicholas, T.J.; Boyden, S.E.; Andrews, A.; Larson, A.; Network, U.D.; Bayrak-Toydemir, P.; Botto, L.D.; et al. Novel molecular mechanism in Malan syndrome uncovered through genome sequencing reanalysis, exon-level Array, and RNA sequencing. Am. J. Med Genet. Part A 2024, 194. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Bourque, D.K.; Kerkhof, J.; Carere, D.A.; Ainsworth, P.; Sadikovic, B.; Armour, C.M.; Lin, H. Genome-wide DNA methylation and RNA analyses enable reclassification of two variants of uncertain significance in a patient with clinical Kabuki syndrome. Hum. Mutat. 2019, 40, 1684–1689. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Kerkhof, J.; Rastin, C.; Levy, M.A.; Relator, R.; McConkey, H.; Demain, L.; Dominguez-Garrido, E.; Kaat, L.D.; Houge, S.D.; DuPont, B.R.; et al. Diagnostic utility and reporting recommendations for clinical DNA methylation episignature testing in genetically undiagnosed rare diseases. Anesthesia Analg. 2024, 26, 101075. [Google Scholar] [CrossRef]
- Andre, G.; Kulakauskas, S.; Chapot-Chartier, M.-P.; Navet, B.; Deghorain, M.; Bernard, E.; Hols, P.; Dufrêne, Y.F. Imaging the nanoscale organization of peptidoglycan in living Lactococcus lactis cells. Nat. Commun. 2010, 1, 27–8. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Schwartz, C.; Skinner, C.; Rodenhiser, D.I.; Ainsworth, P.J.; Pare, G.; Sadikovic, B. Clinical Validation of Fragile X Syndrome Screening by DNA Methylation Array. J. Mol. Diagn. 2016, 18, 834–841. [Google Scholar] [CrossRef]
- Wojcik, M.H.; Reuter, C.M.; Marwaha, S.; Mahmoud, M.; Duyzend, M.H.; Barseghyan, H.; Yuan, B.; Boone, P.M.; E Groopman, E.; Délot, E.C. [PubMed]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Cope, H; Barseghyan, H; Bhattacharya, S; Fu, Y; Hoppman, N; Marcou, C.
- Detection of a mosaic CDKL5 deletion and inversion by optical genome mapping ends an exhaustive diagnostic odyssey. Mol Genet Genom Med 2021, 9, e1665. [CrossRef]
- Schnause, A.C.; Komlosi, K.; Herr, B.; Neesen, J.; Dremsek, P.; Schwarz, T.; Tzschach, A.; Jägle, S.; Lausch, E.; Fischer, J.; et al. Marfan Syndrome Caused by Disruption of the FBN1 Gene due to A Reciprocal Chromosome Translocation. Genes 2021, 12, 1836. [Google Scholar] [CrossRef] [PubMed]
- Sabatella, M.; Mantere, T.; Waanders, E.; Neveling, K.; Mensenkamp, A.R.; van Dijk, F.; Hehir-Kwa, J.Y.; Derks, R.; Kwint, M.; O'GOrman, L.; et al. Optical genome mapping identifies a germline retrotransposon insertion in SMARCB1 in two siblings with atypical teratoid rhabdoid tumors. J. Pathol. 2021, 255, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, M.; Qian, Y.; Yang, Y.; Sun, Y.; Liu, B.; Wang, L.; Dong, M. Identification of a likely pathogenic structural variation in the LAMA1 gene by Bionano optical mapping. npj Genom. Med. 2020, 5, 1–6. [Google Scholar] [CrossRef]
- Fahiminiya, S.; Rivard, G.; Scott, P.; Montpetit, A.; Bacot, F.; St-Louis, J.; Mitchell, G.A.; Foulkes, W.D.; Soucy, J.; Gauthier, J. A full molecular picture of F8 intron 1 inversion created with optical genome mapping. Haemophilia 2021, 27, E638–E640. [Google Scholar] [CrossRef]
- Mackie, S.L.; Koduri, G.; Hill, C.L.; Wakefield, R.J.; Hutchings, R.; Loy, C.; Dasgupta, B.; Wyatt, J.C. Accuracy of musculoskeletal imaging for the diagnosis of polymyalgia rheumatica: Systematic review. RMD Open 2015, 1, e000100. [Google Scholar] [CrossRef]
- Macke, E.L.; Miller, A.R.; Colwell, C.M.; Gonzalez, M.H.; Hunter, J.; Venkata, L.P.R.; Walker, L.; Wheeler, G.; Wilson, R.K.; Mardis, E.R.; et al. Optical Genome Mapping (OGM) Identifies Multiple Structural Variants in a Case With Atypical Phelan-McDermid Syndrome. Am. J. Med Genet. Part A 2024, 197, e63929. [Google Scholar] [CrossRef]
- Dremsek, P.; Schachner, A.; Reischer, T.; Krampl-Bettelheim, E.; Bettelheim, D.; Vrabel, S.; Delissen, Z.; Pfeifer, M.; Weil, B.; Bajtela, R.; et al. Retrospective study on the utility of optical genome mapping as a follow-up method in genetic diagnostics. J. Med Genet. 2024, 62, 89–96. [Google Scholar] [CrossRef]
- Tahim, A.S.; Bryant, C.; Greaney, L.; Rashid, A.; Fan, K. Improving documentation of visual acuity in patients suffering facial fractures. Emerg. Med. J. 2013, 30, 949–950. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.E.; Lemire, G.; Liang, Y.; Davila, J.; Couse, M.; Boycott, K.M.; Kernohan, K.D. RNA sequencing reveals deep intronic CEP120 variant: A report of the diagnostic odyssey for two siblings with Joubert syndrome type 31. Am. J. Med Genet. Part A 2023, 194. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.; Liu, Y.; Yue, L.; Jin, H.; Chen, Y.; Wang, D.; Wang, M.; Chen, G.; Yang, L.; et al. Genetic Testing for Global Developmental Delay in Early Childhood. JAMA Netw. Open 2024, 7, e2415084–e2415084. [Google Scholar] [CrossRef] [PubMed]
- Chand, R.P.; Vinit, W.; Vaidya, V.; Iyer, A.S.; Shelke, M.; Aggarwal, S.; Magar, S.; Danda, S.; Moirangthem, A.; Phadke, S.R.; et al. Proband only exome sequencing in 403 Indian children with neurodevelopmental disorders: Diagnostic yield, utility and challenges in a resource-limited setting. Eur. J. Med Genet. 2023, 66, 104730. [Google Scholar] [CrossRef]
- Gorukmez, O; Gorukmez, O; Topak, A. Clinical exome sequencing findings in 1589 patients. Am J Med Genet 2023, 191A, 1557–1564. [Google Scholar] [CrossRef]
- Migliavacca, M.P.; Sobreira, J.; Bermeo, D.; Gomes, M.; Alencar, D.; Sussuchi, L.; Souza, C.A.; Silva, J.S.; Kroll, J.E.; Burger, M.; et al. Whole genome sequencing as a first-tier diagnostic test for infants in neonatal intensive care units: A pilot study in Brazil. Am. J. Med Genet. Part A 2024, 194. [Google Scholar] [CrossRef]
- Gahl, W.A.; Markello, T.C.; Toro, C.; Fajardo, K.F.; Sincan, M.; Gill, F.; Carlson-Donohoe, H.; Gropman, A.; Pierson, T.M.; Golas, G.; et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Anesthesia Analg. 2011, 14, 51–59. [Google Scholar] [CrossRef]
- Ramoni, R.B.; Mulvihill, J.J.; Adams, D.R.; Allard, P.; Ashley, E.A.; Bernstein, J.A.; Gahl, W.A.; Hamid, R.; Loscalzo, J.; McCray, A.T.; et al. The Undiagnosed Diseases Network: Accelerating Discovery about Health and Disease. Am. J. Hum. Genet. 2017, 100, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Curic, E.; Ewans, L.; Pysar, R.; Taylan, F.; Botto, L.D.; Nordgren, A.; Gahl, W.; Palmer, E.E. International Undiagnosed Diseases Programs (UDPs): components and outcomes. Orphanet J. Rare Dis. 2023, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bowling, K.M.; Thompson, M.L.; Finnila, C.R.; Hiatt, S.M.; Latner, D.R.; Amaral, M.D.; Lawlor, J.M.; East, K.M.; Cochran, M.E.; Greve, V.; et al. Genome sequencing as a first-line diagnostic test for hospitalized infants. Anesthesia Analg. 2021, 24, 851–861. [Google Scholar] [CrossRef]
- Denommé-Pichon, A.-S.; Vitobello, A.; Olaso, R.; Ziegler, A.; Jeanne, M.; Mau-Them, F.T.; Couturier, V.; Racine, C.; Isidor, B.; Poë, C.; et al. Accelerated genome sequencing with controlled costs for infants in intensive care units: a feasibility study in a French hospital network. Eur. J. Hum. Genet. 2021, 30, 567–576. [Google Scholar] [CrossRef]
- Disease, N.B.; French, C.E.; Project, N.G.C.; Delon, I.; Dolling, H.; Sanchis-Juan, A.; Shamardina, O.; Mégy, K.; Abbs, S.; Austin, T.; et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensiv. Care Med. 2019, 45, 627–636. [Google Scholar] [CrossRef]
- Hayeems, R.Z.; Bhawra, J.; Tsiplova, K.; Meyn, M.S.; Monfared, N.; Bowdin, S.; Stavropoulos, D.J.; Marshall, C.R.; Basran, R.; Shuman, C.; et al. Care and cost consequences of pediatric whole genome sequencing compared to chromosome microarray. Eur. J. Hum. Genet. 2017, 25, 1303–1312. [Google Scholar] [CrossRef]
- Sanford, E.F.; Clark, M.M.; Farnaes, L.; Williams, M.R.; Perry, J.C.; Ingulli, E.G.; Sweeney, N.M.; Doshi, A.; Gold, J.J.; Briggs, B.; et al. Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU*. Pediatr. Crit. Care Med. 2019, 20, 1007–1020. [Google Scholar] [CrossRef]
- Hutchings, A.; Hollywood, J.; Lamping, D.L.; Pease, C.T.; Chakravarty, K.; Silverman, B.; Choy, E.H.S.; Scott, D.G.; Hazleman, B.L.; Bourke, B.; et al. Clinical outcomes, quality of life, and diagnostic uncertainty in the first year of polymyalgia rheumatica. Arthritis Rheum. 2007, 57, 803–809. [Google Scholar] [CrossRef]
- Aekka, A.; Weisman, A.G.; Papadakis, J.; Yerkes, E.; Baker, J.; Keswani, M.; Weinstein, J.; Finlayson, C. Clinical utility of early rapid genome sequencing in the evaluation of patients with differences of sex development. Am. J. Med Genet. Part A 2023, 194, 351–357. [Google Scholar] [CrossRef]
- Chung, C.C.; Leung, G.K.; Mak, C.C.; Fung, J.L.; Lee, M.; Pei, S.L.; Yu, M.H.; Hui, V.C.; Chan, J.C.; Chau, J.F.; et al. Rapid whole-exome sequencing facilitates precision medicine in paediatric rare disease patients and reduces healthcare costs. Lancet Reg. Heal. - West. Pac. 2020, 1, 100001. [Google Scholar] [CrossRef] [PubMed]
- Maron, JL; Kingsmore, SF; Wigby, K; Chowdhury, S; Dimmock, D; Poindexter, B; et al. Novel Variant Findings and Challenges Associated With the Clinical Integration of Genomic Testing: An Interim Report of the Genomic Medicine for Ill Neonates and Infants (GEMINI) Study. JAMA Pediatr 2021, 175, e205906. [Google Scholar] [CrossRef]
- Nomakuchi, T.T.; Teferedegn, E.Y.; Li, D.; Muirhead, K.J.; Dubbs, H.; Leonard, J.; Muraresku, C.; Sergio, E.; Arnold, K.; Pizzino, A.; et al. Utility of genome sequencing in exome-negative pediatric patients with neurodevelopmental phenotypes. Am. J. Med Genet. Part A 2024, 194, e63817. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Smith, L.D.; Kunard, C.M.; Bainbridge, M.; Batalov, S.; Benson, W.; Blincow, E.; Caylor, S.; Chambers, C.; Del Angel, G.; et al. A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. Am. J. Hum. Genet. 2022, 109, 1605–1619. [Google Scholar] [CrossRef]
- Guo, F.; Liu, R.; Pan, Y.; Collins, C.; Bean, L.; Ma, Z.; Mathur, A.; Da Silva, C.; Nallamilli, B.; Guruju, N.; et al. Evidence from 2100 index cases supports genome sequencing as a first-tier genetic test. Anesthesia Analg. 2023, 26, 100995. [Google Scholar] [CrossRef]
- Goldin, M.R.; Ruderfer, D.M.; Bick, A.; Roden, D.M.; Schuler, B.A.; Robinson, J.R. Benefits and barriers to broad implementation of genomic sequencing in the NICU. Am. J. Hum. Genet. 2025, 112, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, MJ; Nickerson; Chong, JX. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet 2019, 105(3), 448–455. [Google Scholar] [CrossRef]
- Damaraju, N.; Miller, A.L.; E Miller, D. Long-Read DNA and RNA Sequencing to Streamline Clinical Genetic Testing and Reduce Barriers to Comprehensive Genetic Testing. J. Appl. Lab. Med. 2024, 9, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Höps, W.; Weiss, M.M.; Derks, R.; Galbany, J.C.; Ouden, A.D.; Heuvel, S.v.D.; Timmermans, R.; Smits, J.; Mokveld, T.; Dolzhenko, E.; et al. HiFi long-read genomes for difficult-to-detect, clinically relevant variants. Am. J. Hum. Genet. 2025, 112, 450–456. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Azzariti, D.R.; Hamosh, A. Genomic Data Sharing for Novel Mendelian Disease Gene Discovery: The Matchmaker Exchange. Annu. Rev. Genom. Hum. Genet. 2020, 21, 305–326. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Sobreira, N.L.M.; Arachchi, H.; Buske, O.J.; Chong, J.X.; Hutton, B.; Foreman, J.; Schiettecatte, F.; Groza, T.; Jacobsen, J.O.; Haendel, M.A.; et al. Matchmaker Exchange. Curr. Protoc. Hum. Genet. 2017, 95, 9.31.1–9.31.15. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; DiStefano, M.T.; Riggs, E.R.; Josephs, K.S.; Alkuraya, F.S.; Amberger, J.; Amin, M.; Berg, J.S.; Cunningham, F.; Eilbeck, K.; et al. Toward robust clinical genome interpretation: Developing a consistent terminology to characterize Mendelian disease-gene relationships—allelic requirement, inheritance modes, and disease mechanisms. Anesthesia Analg. 2023, 26, 101029–101029. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Aylsworth, A.S.; Allanson, J.; Battaglia, A.; Carey, J.C.; Curry, C.J.; Davies, K.E.; Eichler, E.E.; Graham, J.M.; Hall, B.; et al. Personal journeys to and in human genetics and dysmorphology. Am. J. Med Genet. Part A 2024, 194, e63514. [Google Scholar] [CrossRef]
- Horowitz, K.; Fotopoulos, N.H.; Mistry, A.J.; Simo, J.; Medeiros, M.; Bucco, I.D.; Ginsberg, M.; Dwosh, E.; La Piana, R.; A Rouleau, G.; et al. Enhancing variant of uncertain significance (VUS) interpretation in neurogenetics: collaborative experiences from a tertiary care centre. J. Med Genet. 2024, 62, 37–45. [Google Scholar] [CrossRef]
- Carey, J.C.; Curry, C.J.; Grix, A.W.; Golabi, M.; Graham, J.M.; Buehler, B.A. A tribute to Bryan D. Hall: Festschrift 2003. Am. J. Med Genet. Part A 2003, 123A, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, E.; Williams, T.; Shaw, C.; Chekalin, E.; Ortega, J.; Robinson, K.; Button, J.; Jones, M.C.; del Campo, M.; Basel, D.; et al. The impact of clinical genome sequencing in a global population with suspected rare genetic disease. Am. J. Hum. Genet. 2024, 111, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Grandinette, S.A.; Wang, X.; Hudson, T.R.; Briseno, K.; Berry, A.M.; Hacker, J.L.; Hsu, A.; Silverstein, R.A.; Hille, L.T.; et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. New Engl. J. Med. 2025, 392, 2235–2243. [Google Scholar] [CrossRef]
- Miga, K.H.; Eichler, E.E. Envisioning a new era: Complete genetic information from routine, telomere-to-telomere genomes. Am. J. Hum. Genet. 2023, 110, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Ricordi, C. Il codice della longevità sana; Mondadori libri SpA.: Milano, 2022. [Google Scholar]
Figure 2.
The algorithm depicts the current assessment of the child with a NDD. See text.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



