Submitted:
15 December 2025
Posted:
15 December 2025
You are already at the latest version
Abstract
Marine triterpenoid saponins are structurally diverse metabolites with high pharmacological and nutraceutical potential, yet their characterization remains challenging due to extensive isomerism, aggregation phenomena, and the frequent co-extraction of lipids and other matrix components. In this work, we combine ATR–FTIR and high-resolution LC–MS to investigate the spectral and chromatographic behaviour of Cucumaria frondosa extracts and butanol-enriched fractions. FTIR spectra reveal a strong aliphatic signature, N–H-related features, and ester carbonyl bands consistent with the presence of co-extracted lipids and nitrogen-containing species such as ceramides or sphingolipids. LC–MS analysis of preparative fractions shows recurrent saponin-like ions— most prominently a feature at m/z ≈ 1347—reappearing across chromatographically distinct fractions, often accompanied by lipid-like ions in the 600–900 m/z range. These observations indicate that closely associated lipidic species can modulate the apparent chromatographic behaviour of saponin-containing fractions. Comparison with the Marine Animal Saponin Database (MASD v1.0) highlights both its value and its current MS1-centric limitations, including the lack of diagnostic MS/MS spectra and occasional inconsistencies between reported formulas and listed molecular weights. These findings underscore the need for integrated, multi-spectroscopic workflows and standardised spectral libraries to support confident annotation of marine saponins. Rather than proposing new structures, this study validates an analytical workflow that bridges early-stage MS screening with preparative fractionation and orthogonal spectroscopic assessment, offering a methodological reference to minimise misidentification and to guide future structural and biological investigations of marine triterpenoid saponins.
Keywords:
saponins
; thalassochemicals
; bioactive compounds
; mass spectrometry
; nutraceuticals
; marine biomaterials
1. Introduction
Marine triterpenoid saponins constitute a structurally rich and biologically potent family of secondary metabolites, particularly abundant in echinoderms such as sea cucumbers (Holothuroidea) [1,2]. Their amphiphilic architecture—comprising a hydrophobic triterpenoid aglycone linked to one or more hydrophilic glycosidic chains—underpins a broad palette of bioactivities, including cytotoxic [3,4,5], antiviral [6], immunomodulatory [7,8], and anti-inflammatory effects [9,10,11,12], making these thalassochemicals clear candidates in the evolution of next-generation nutraceuticals [13]. Within this group, Cucumaria frondosa has emerged as one of the most prolific sources of biologically active saponins, including the well-studied frondosides and numerous structural variants [14,15].
Despite their importance, the structural elucidation of marine saponins remains notoriously challenging. Several intrinsic factors contribute to this difficulty: (i) extensive structural isomerism arising from aglycone variation and complex glycosidic connectivity; (ii) sulfation of sugar moieties, which strongly influences ionisation efficiency and fragmentation pathways; (iii) the frequent co-extraction of lipids and other matrix components [16,17,18], which can mask or distort saponin-related signals; and (iv) the scarcity of purified standards for orthogonal validation via NMR or FTIR. Consequently, even high-resolution MS often yields ambiguous annotations when interpreted without additional spectroscopic context.
Efforts to consolidate available knowledge have led to the recent development of the Marine Animal Saponin Database (MASD v1.0) [19], a valuable resource compiling more than 900 reported saponins with associated formulas, molecular weights, taxonomic assignments, and literature references. MASD provides a much-needed reference framework, yet its MS1-centric architecture imposes inherent limitations for dereplication in complex extracts. Structural isomers cannot be distinguished on the basis of elemental formulas, and occasional inconsistencies between reported formulas and listed masses—likely inherited from heterogeneous primary sources—underscore the importance of complementary fragmentation evidence. The absence of MS/MS spectra further restricts the ability to confidently annotate saponins in mixtures enriched with lipids or other marine metabolites.
Matrix composition additionally exerts a profound influence on the extraction and chromatographic behaviour of saponins. Our Fourier-transform infrared (FTIR) data reveal that butanol-enriched C. frondosa fractions contain not only triterpenoid glycosides, but also lipids, sphingolipids, and ceramide-like species [20]. These co-extracted components alter both the vibrational fingerprint and chromatographic retention of the extract, and their association with saponin-like ions contributes to recurring MS features across fractions. Understanding how these components affect ionisation and retention is therefore essential for accurate interpretation of LC–MS data.
To address these analytical challenges [21,22], the present study integrates attenuated total reflection (ATR)–FTIR, and high-resolution LC–MS into a unified framework (see Figure 1) aimed at: (i) distinguishing saponin-consistent fragmentation pathways from non-specific fragmentation of co-eluting species; (ii) identifying lipid-associated ions that modulate chromatographic behaviour; and (iii) establishing practical decision criteria for dereplication in complex marine matrices. By articulating this reproducible and diagnostically rich workflow, we aim to support more rigorous annotation, minimise misidentification, and facilitate the transition from initial MS screening to preparative isolation and structural validation.
This study does not report the elucidation of new saponin structures. Instead, it validates an analytical strategy designed to reduce interpretative uncertainty and to clarify how lipidic co-extracts can distort MS-based analyses in marine metabolomics. The workflow presented here is intended as a practical tool for the community, offering guidance for future structural and functional studies of marine triterpenoid saponins.
2. Methods and Materials
2.1. Raw Extract Preparation
Dried specimens of Cucumaria frondosa were first ground into a fine powder using a mechanical grinder to maximize the surface area available for solvent penetration. A total of 100 grams of this powdered material was subjected to exhaustive solvent extraction to ensure maximal recovery of amphiphilic triterpenoid saponins.
The powdered tissue was transferred to a reflux apparatus and extracted with 500 mL of 70% ethanol (v/v) under reflux conditions for four hours. This hydroalcoholic mixture was selected to simultaneously solubilize both hydrophilic and hydrophobic domains of the target saponins, including sulfate-bearing moieties and lipophilic aglycones. Following the initial extraction, the solid residue was recovered via vacuum filtration using medium-pore Büchner funnel filters (20–25 µm pore size), and the process was repeated three additional times with 250 mL of fresh 70% ethanol per cycle. All filtrates were combined to form a crude ethanolic extract.
The pooled ethanolic extracts were then subjected to solvent removal under reduced pressure using a rotary evaporator at controlled temperature. This yielded an aqueous-enriched concentrate that contained the bulk of the water-soluble and amphiphilic compounds. No lyophilization was performed at this stage, in order to maintain solubility for the subsequent partitioning step.
To selectively enrich the extract in saponins and reduce polar impurities, the aqueous concentrate was subjected to liquid–liquid extraction using n-butanol (n-BuOH) as the organic solvent. The extract was mixed with n-BuOH in a separatory funnel, allowing amphiphilic compounds such as saponins to partition preferentially into the organic phase due to their moderate lipophilicity and surfactant-like character. After vigorous mixing and phase separation, the n-BuOH layer was carefully collected and retained for further analysis and purification.
This butanol fraction, enriched in triterpenoid saponins, was stored at 4 °C in amber glass vials and used directly for LC-MS/MS analysis and further fractionation steps. The protocol was designed to minimize thermal and oxidative degradation, preserving the structural integrity of the native saponin compounds throughout the process.
2.2. Instrumentation
Analytical HPLC–MS measurements were performed on a Shimadzu 2040 HPLC system coupled with a Shimadzu LCMS 2050 mass spectrometer, equipped with a heated DUIS (dual ion ESI/APCI source), an autosampler, degasser, quaternary low-pressure gradient pump, column oven, photodiode array detector, and a C18 column (Phenomenex Kinetex C18, particle size 2.6 m, pore size 100 Å, column size 2.1 mm × 100 mm) with a column oven temperature of 40 °C. The DUIS operated at a desolvation temperature of 450 °C with a desolvation line temperature of 200 °C. For ionization, the interface voltage was set at 1.5 kV for positive mode and kV for negative mode. Nitrogen, provided by the nitrogen generator NGA CASTORE XS iQ 18, was used as the nebulizer gas (nebulizing flow 2 L/min), the dry gas (drying gas flow 5 L/min), and the heating gas (heating gas flow 7 L/min). The mobile phase had a flow rate of 0.45 mL/min and a linear gradient consisting of A (H2O/MeCN/HCO2H, 950:50:1) and B (MeCN/H2O/HCO2H, 950:50:1) with the following percentages of B: 0.00–0.50 min, 40%; 0.50–6.00 min, 40%–100%; 6.00–12.00 min, 100%; 12.00–12.01 min, 100%–40%; 12.01–15.00 min, 40%.
Preparative HPLC separations were carried out at room temperature on a Shimadzu LC–20AP system equipped with an autosampler, a degasser (FLOM Gastorr PG32), two preparative HPLC pumps, a column oven, a UV–Vis detector, and a C18(2) column (Phenomenex Luna; particle size 5 m, pore size 100 Å, column dimensions 21.2 mm × 250 mm). An isocratic elution was used at a flow rate of 12 mL/min with UV detection at 210 nm. The mobile phase consisted of H2O/MeCN/HCO2H (14:86:0.1, v/v/v).
ATR–FTIR spectra were acquired using a single-reflection diamond ATR crystal with a Bruker IFS 66/S FTIR spectrometer. A liquid-nitrogen-cooled MCT detector was employed, and the interferometer mirror velocity was set to 150 kHz. The ATR unit was continuously purged with dry air to minimize atmospheric interference. Samples were dried on the crystal using dry air.
2.3. Interface-Voltage Modulation Experiments
To evaluate the propensity of marine triterpenoid saponins to undergo in-source fragmentation, the interface (capillary) voltage (IV) of the DUIS source was incrementally varied from to in steps while all other parameters were kept constant (see Section 2.2). Full-scan positive-ion spectra were recorded in cent-wave mode (m/z 100–1200, scan time 250 ). Five data files corresponding to the five IV settings were processed in LabSolutions (ver. 5.97) and exported as profile-mode centroid lists. The retention time window of interest was , where the putative molecular ion of the target saponin elutes.
2.4. Gaussian-Based Simulation of FTIR Spectra for Illustrative Purposes
In order to qualitatively illustrate how differences in concentration and vibrational extinction coefficients can account for the experimentally observed changes in FTIR spectra, synthetic spectra were generated as a superposition of Gaussian bands centered at characteristic wavenumbers. The simulated absorbance profile was constructed as
where denotes the absorbance in arbitrary units, is the central wavenumber of band i, and controls the band width. The amplitude of each contribution was taken to be proportional to the product of an effective molar extinction coefficient associated with the corresponding vibrational mode and the relative concentration c of the chemical species contributing to that band, consistent with a Beer–Lambert-type scaling under constant effective path length:
- Relative concentrations and choice of lipid excess.
For the “pure saponin” case, the saponin concentration was set to (arbitrary units), with a dominant contribution arising from the broad stretching band in the 3200–3600 cm−1 region and weaker contributions from stretching modes around 2850–2960 cm−1. For the “saponin with lipids” condition, additional Gaussian components associated with lipid vibrational modes were included, and the lipid concentration was increased relative to saponin according to
This value of X was chosen to reflect the fact that O–H stretching vibrations typically exhibit significantly larger effective extinction coefficients than C–H stretching modes of and groups, owing to their higher polarity and strong hydrogen-bonding contributions. As a consequence, even though , a sufficiently large excess of lipid leads to an increase in the term , resulting in an enhanced relative intensity of the bands in the simulated spectrum.
- Scope of the simulation.
These simulated spectra are not intended to provide a quantitative prediction of experimental FTIR intensities, but rather to serve as a conceptual and visual aid demonstrating how variations in relative concentrations and intrinsic extinction coefficients can qualitatively explain the spectral trends observed experimentally, particularly the apparent amplification of lipid-associated C–H stretching bands in mixed saponin–lipid systems.
3. Results and Discussion
3.1. FTIR Spectroscopy: Co-Extraction Hypothesis and Model Systems
The ATR–FTIR spectrum of the butanolic extract obtained from Cucumaria frondosa (Figure 4), measured as a film on a single reflexion diamond crystal after deposition from n-butanol and drying under dried air (trace solvent possibly remaining), displayed an unusually strong set of CH2/CH3 stretching bands centered near 2925 and 2850 cm−1, whose intensity markedly exceeded that of the OH stretching band typically found around 3400 cm−1. This observation deviates from the canonical spectral profile of saponins, which are characterized by rich hydroxylation patterns that dominate the IR spectrum via strong OH vibrations. The atypically high intensity of aliphatic bands raised the hypothesis that the extract contains a significant proportion of long hydrocarbon chains, possibly derived from co-extracted membrane lipids or lipid-like saponin architectures. Furthermore, a low-intensity but distinct C=O stretching band near 1735 cm−1 was observed, suggesting the presence of carbonyl functionalities; in our system these are most plausibly due to (i) ester carbonyls from co-extracted phospho-/glycolipids and (ii) a lactone (butenolide) associated with the triterpenoid aglycone, rather than a ketone, consistent with known marine triterpenoid motifs.
To further explore this hypothesis, a set of reference FTIR spectra were analyzed. These included simulated vibrational profiles under classical conditions for illustrative purposes (Figure 3) and experimental spectra of three structurally distinct commercial saponins—aescin, digitonin, and Quillaja saponins—measured in the solid state under the same ATR conditions (Figure 2). The simulations for a pure triterpenoid saponin (Figure 3a) show a broad OH band near 3400 cm−1, moderate CH2/CH3 stretches, and a dense fingerprint region between 1000–1100 cm−1 dominated by glycosidic C–O and C–O–C vibrations. Conversely, the simulation of a saponin–lipid mixture (Figure 3b) exhibits a spectral inversion, with dominant aliphatic stretching bands and a weakened. A narrower O–H stretching band—reflecting a change in the relative contribution of hydrogen-bonded versus non-hydrogen-bonded populations, rather than an intensity variation caused by the environment—suggests that the extract contains a larger proportion of less strongly hydrogen-bonded O–H groups compared with the pure saponin.
The experimental spectra of the purified saponins closely mirrored the simulated pure saponin pattern. All three compounds showed the expected broad OH band at 3200–3400 cm−1, moderate aliphatic stretches around 2925 and 2850 cm−1, and a glycosidic fingerprint rich in C–O and C–O–C bands (1200–1000 cm−1). No sulfate-associated signals were detected in these commercial samples, as expected from their known non-sulfated structures. Notably, the N–H stretching region (3300–3500 cm−1) was either absent or extremely weak in all purified samples, in agreement with the lack of free amino groups in the structures of aescin and related saponins.
In contrast, the FTIR spectrum of the crude C. frondosa extract (Figure 4) revealed a highly structured vibrational profile consistent with a mixture of triterpenoid saponins, lipids, sphingolipids, and ceramides. The aliphatic CH2/CH3 stretching bands at ∼2925 and ∼2850 cm−1 were particularly intense, markedly exceeding the OH stretching band near 3400 cm−1, and reinforcing the hypothesis of co-extracted or self-assembled lipidic domains. In comparison, pure aescin displayed only moderate aliphatic features, while the addition of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) to aescin (Figure 5a) replicated the spectral enhancement in the CH region—suggesting that the high intensity in the extract arises from similar components or interactions.
Beyond the aliphatic region, additional informative signals were observed. A sharp band at ∼1750 cm−1 and a shoulder near 1550 cm−1 were present, tentatively attributable to C=O stretching and N–H bending modes, respectively—likely arising from ester-containing lipids (phospho-/glycolipids) and amide-bearing lipids such as ceramides and sphingolipids. Consistent with this view, the C=O feature near 1735–1750 cm−1 becomes more prominent upon mixing with DMPC, supporting a lipidic origin for a significant fraction of this band, while a contribution from a triterpenoid lactone in the aglycone remains plausible. The typical glycosidic fingerprint between 1200–1000 cm−1 was also well defined, with features around 1225 and 1060 cm−1 that are compatible with the asymmetric/symmetric vibrations of sulfates (S=O) and/or phosphate () groups from phospholipids; we therefore retain both possibilities for these assignments. Interestingly, the OH stretching band in the extract was red-shifted (∼20 cm−1) compared to the purified samples, indicative of stronger hydrogen bonding or hydration—possibly related to micellar or supramolecular aggregation. Furthermore, the absence of a distinct, resolvable N–H stretching band (typically near 3250 cm−1). The pronounced absorption near 1650 cm−1 is consistent with the Amide I (C=O stretching) vibration of amide-bearing lipids, such as sphingolipids or ceramides. In this spectral region, C=C stretching modes would contribute only weakly due to their low extinction coefficients and would therefore be effectively obscured by the much stronger amide C=O signal. Together with the corresponding Amide II band, this provides strong evidence that the extract contains nitrogenous lipids not present in the aescin or aescin–DMPC model systems.
Two additional features reinforce the presence of nitrogenated compounds: a weak band near 1550 cm−1 tentatively assignable to amide II (N–H bending and C–N stretching), and a sharper band around 1700–1750 cm−1, consistent with C=O stretching from ester groups (and a possible lactone contribution in the triterpenoid aglycone). These signatures suggest the coexistence of triterpenoid saponins (rich in hydroxyl and glycosidic groups) with lipidic components bearing ester and amide functionalities. The fingerprint region (1000–1200 cm−1) remains rich in C–O and C–C vibrations from glycosidic linkages, supporting the dominance of saponin-type structures.
The annotated spectrum further helps disentangle overlapping contributions, and confirms that the extract contains a mixture of hydroxylated saponins, saturated lipid chains, possible amides, and glycosidic moieties. These observations are consistent with co-extraction of marine triterpenoids, phospholipids, and nitrogenous lipids such as ceramides or sphingomyelins, and are compatible with their association in supramolecular aggregates; however, we note that FTIR alone does not prove specific complexation.
In the 1550 cm−1 region, the expected Amide II band is partially obscured by overlapping contributions, but its presence is consistent with the pronounced Amide I absorption observed at ∼1650 cm−1. Given the much larger extinction coefficient of amide C=O stretching compared with C=C modes, the feature at 1650 cm−1 is most reasonably attributed to amide-bearing lipids such as ceramides or sphingolipids rather than to unsaturated lipid vibrations. This interpretation is further supported by the broad, structured envelope in the 3300–3500 cm−1 region, dominated by O–H stretching but likely containing a minor N–H component. Together, these spectral signatures reinforce the presence of nitrogenous lipids in the extract, in agreement with the known lipid-rich composition of echinoderms.
Crucially, the extract also revealed a broad and structured signal in the N–H/O–H region (3300–3400 cm−1), more complex than the spectra of the pure saponins. Combined with a deformation band near 1600 cm−1, this pattern is consistent with amine- and amide-containing compounds, including sphingolipids, ceramides, or even small peptides, all of which contain primary or secondary amines. This would explain both the enhanced breadth of the high-wavenumber region and the more complex bending patterns in the mid-IR, even though a distinct N–H stretching maximum is not resolved.
To experimentally test the co-extraction hypothesis and the potential role of lipid association in modifying the vibrational profile, binary mixtures of each saponin with DMPC were prepared and analyzed (Figure 5). In all three mixtures, the CH2/CH3 stretching bands increased substantially in intensity, accompanied by enhanced definition in the 1450–1375 cm−1 region, consistent with C–H bending and C–C skeletal modes typical of lipid chains. These enhancements occurred despite the lower molar extinction coefficients of hydrocarbon vibrations, suggesting a true compositional increase in lipidic content. The resulting spectra closely resembled that of the C. frondosa extract, supporting the presence of lipid-associated domains in the crude sample. Moreover, the C=O stretching band near 1735 cm−1 became more prominent in the DMPC mixtures, consistent with ester carbonyls from phospholipid backbones (with a possible superimposed contribution from a triterpenoid lactone). However, the N–H region remained weak, in agreement with the absence of free amines in both saponins and the DMPC headgroup, which contains only a quaternary ammonium.
Simulated FTIR spectra for mixed saponin–lipid systems (Figure 3b) reproduced this behavior, reinforcing the idea that vibrational profiles are strongly modulated by the local molecular environment. The combination of experimental and computational evidence suggests that naturally occurring saponins may exist in the extract as lipid-associated assemblies, possibly forming supramolecular aggregates through hydrophobic and hydrogen-bond interactions. Given the amphiphilic nature of marine saponins, such self-assembled states are plausible and have been previously reported in studies of membrane solubilization and adjuvancy.
The co-extraction or complexation with lipids may also have implications for chromatographic behavior. These lipid-enriched saponin assemblies are expected to exhibit greater hydrophobicity and thus longer retention times in reversed-phase HPLC, particularly under the high-acetonitrile conditions applied in the late gradient stages of our analysis. This would help explain the detection of late-eluting compounds with aliphatic-rich mass spectra and IR profiles.
In conclusion, FTIR spectroscopy—supported by simulations and model mixtures—suggests that the C. frondosa extract contains both sulfated triterpenoid saponins and co-extracted or self-assembled lipidic components. In addition, the presence of weak N–H-related features and associated bending modes is compatible with co-extracted amine-containing lipids, such as sphingolipids or ceramides. This highlights the importance of considering matrix effects and supramolecular associations when interpreting vibrational data from marine extracts. In the following section, we complement these findings with tandem mass spectrometry to further resolve the molecular composition and fragmentation behavior of the extract.
3.2. Preliminary HPLC-MS Profiling and Elution Behavior of the Extract
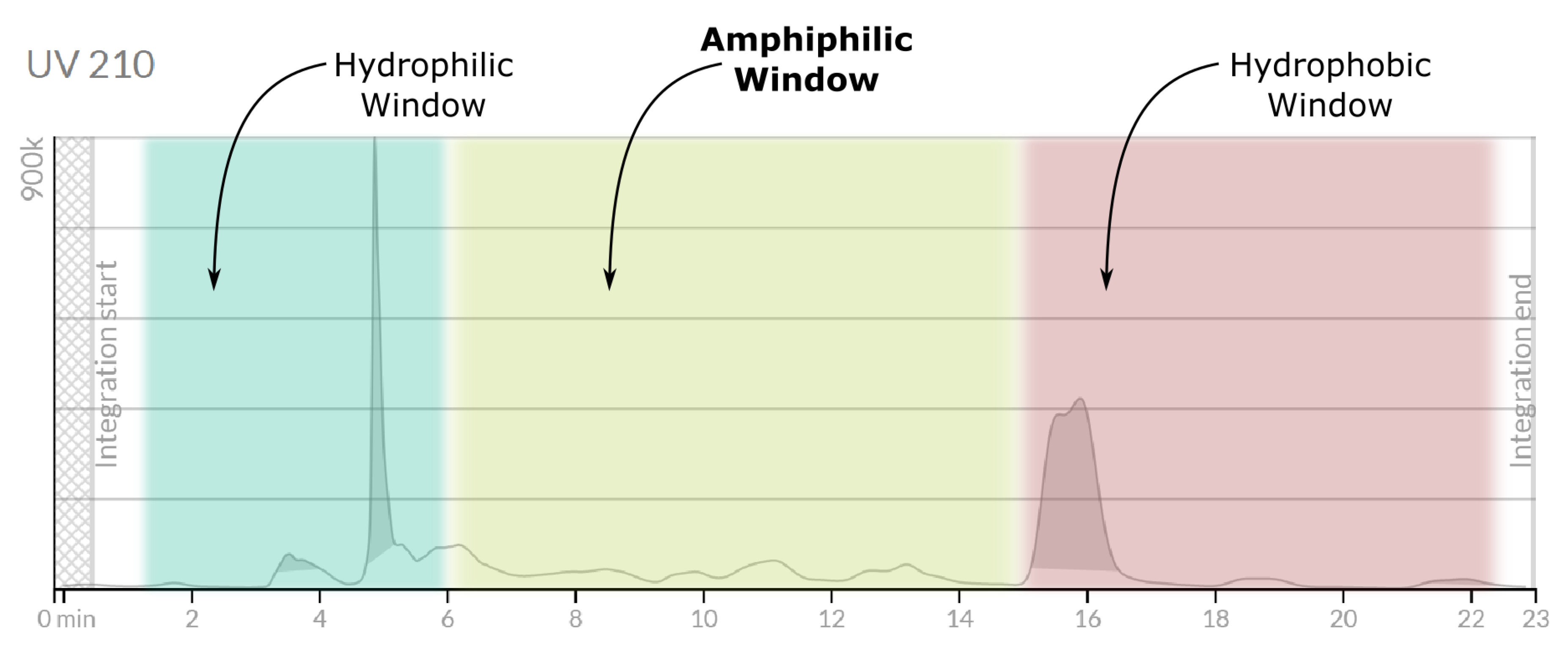
The butanolic extract of Cucumaria frondosa was subjected to reversed-phase high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS) for an initial compositional assessment. A C18 column was used under a gradient elution profile with 0.1% formic acid in water (A) and acetonitrile (B) as mobile phases. The gradient progressed from aqueous to highly organic conditions, favouring the elution of hydrophobic constituents during the later stages.
The UV chromatogram recorded at 214 nm (Figure 6) revealed three distinct elution domains:
- A prominent early peak (0–6 min), dominated by highly polar or low-molecular-weight species.
- An extended intermediate region (6–15 min) containing multiple, well-resolved signals.
- A late-eluting zone (after 15 min) composed of strongly hydrophobic components.
The mass spectra were acquired in both positive and negative modes employing a dual-ion ESI/APCI source showed minimal evidence of saponin-like ions in the early and late domains. In contrast, the intermediate region (6–15 min) displayed complex spectral signatures enriched in ions compatible with triterpenoid glycosides. This pattern aligns with FTIR observations indicating the presence of lipidic components associated with the saponin fraction, which may increase the effective hydrophobicity of saponin–lipid supramolecular assemblies and shift their retention times.
To more deeply interrogate this middle elution zone, a preparative-scale fractionation was carried out using the same C18 column and gradient. Twelve fractions (F1–F12) were collected across the 6–15 min window. Subsequent analytical HPLC-MS reanalysis revealed multiple chromatographic signals per fraction, with recurring mass features reproducibly detected across several fractions.
A particularly prominent ion at m/z= 1347 (positive mode) was consistently detected, especially in late-eluting preparative fractions. This ion is putatively assignable to a modified form of frondoside A. The 1347 m/z value may correspond to a frondoside adduct with a CH2 increment—potentially arising from methylene-bridge formylation via formic acid under acidic gradient conditions, a process previously described for polyol-containing molecules.
Strikingly, most fractions containing the 1347 m/z species also exhibited companion ions in the 600–900 m/z range, tentatively assignable to lipidic species (ceramides, phosphatidylcholine, sphingolipids). Their co-occurrence suggests the presence of stable or dynamically reorganising saponin–lipid aggregates, rather than isolated molecular entities.
Negative-mode ionization further revealed a robust and recurrent peak at m/z= 477, more intense in later fractions (e.g., F6, F12). This ion corresponds, according to LIPID MAPS [23], to trimethyl 1-((2,5,5,8a-tetramethyldecahydronaphthalen-1-yl)methoxy)propane-1,2,3-tricarboxylate, a molecule combining polar and aliphatic features. Its co-elution with saponin-like species and selective detection in negative mode indicate a potential role in modulating the hydrophilic–lipophilic balance of the saponin–lipid assemblies, possibly contributing to the observed retention-time shifts.
Consistent with FTIR data, the presence of nitrogen-containing lipid species (e.g., ceramides, sphingolipids) is supported by the NH-related stretching and bending signals detected in the extract. Since triterpenoid saponins themselves lack nitrogen atoms, these vibrational features must originate from lipidic co-extracts, reinforcing the interpretation of supramolecular association.
Figure 7.
LC-MS analysis of fractions F1, F2, F6 and F12 from the mid-elution zone (6–15 min). Each column (a–d) displays the total ion chromatogram (top), the average mass spectrum in positive mode (middle), and the average mass spectrum in negative mode (bottom). The recurring signal at m/z= 1347 appears in all fractions, accompanied by lipid-like ions in the 600–900 m/z range. The consistent negative-mode peak at m/z= 477 supports the presence of mixed saponin–lipid assemblies with modulated polarity.
Figure 7.
LC-MS analysis of fractions F1, F2, F6 and F12 from the mid-elution zone (6–15 min). Each column (a–d) displays the total ion chromatogram (top), the average mass spectrum in positive mode (middle), and the average mass spectrum in negative mode (bottom). The recurring signal at m/z= 1347 appears in all fractions, accompanied by lipid-like ions in the 600–900 m/z range. The consistent negative-mode peak at m/z= 477 supports the presence of mixed saponin–lipid assemblies with modulated polarity.
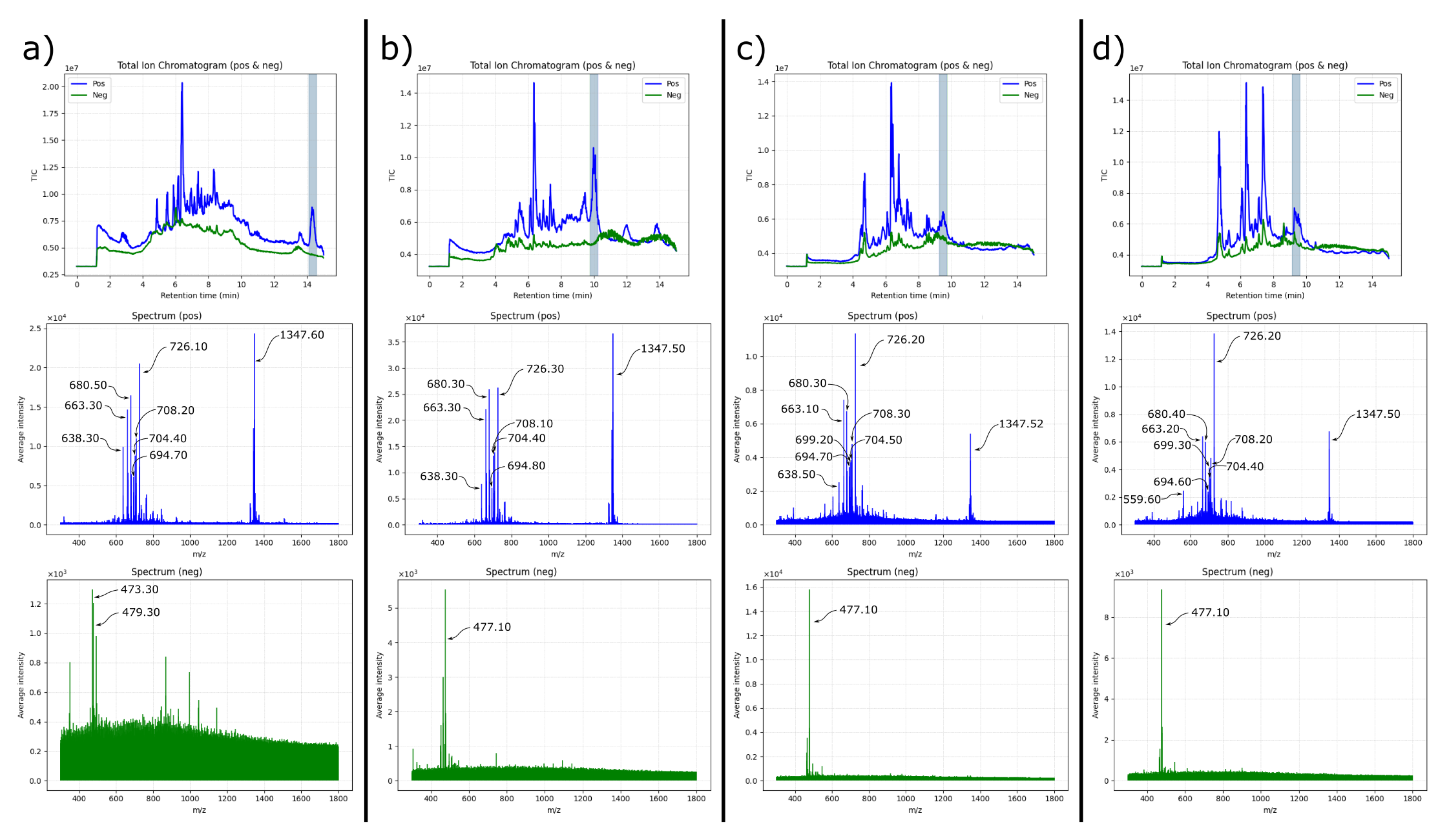
Interestingly, the retention time of the 1347 m/z species varied significantly among fractions. Early fractions (e.g., F1) showed the principal peak near 14 minutes, while later fractions (F6–F12) exhibited the same ion eluting earlier, around 9.5 minutes. Despite this mobility shift, the mass spectra remained remarkably consistent across fractions, strongly suggesting that the same molecular ensemble—rather than distinct chemical species—was present. We interpret this behaviour as evidence of dynamically reorganising saponin–lipid aggregates that redistribute into similar equilibrium assemblies after fraction collection and prior to analytical reinjection. This phenomenon explains the unexpected appearance of the target saponin and its lipidic companions across multiple, temporally distinct preparative fractions.
Collectively, these observations highlight the compositional and supramolecular complexity of the C. frondosa extract. The data indicate that the system does not consist of isolated molecules but of interconverting saponin–lipid aggregates whose chromatographic behaviour depends on their transient stoichiometry and microenvironment. Further purification and comprehensive structural analysis (see next section) are required to disentangle individual components and confirm molecular identities through NMR and targeted MS/MS fragmentation.
3.3. Critical Considerations for Structural Elucidation: Toward Comprehensive Spectral Validation
The release of MASD v1.0 marks an important milestone in the systematisation of marine triterpenoid saponins, providing unified molecular formulas, curated taxonomic information, and reference metadata for a large diversity of compounds. As the MASD authors clearly state, the first version of the database is deliberately centred on MS1-level information, with the explicit intention of establishing a platform that can later be expanded to include higher-order spectral data. This focus greatly increases accessibility and offers a consistent starting point for dereplication efforts across laboratories. At the same time, it naturally limits the specificity with which complex mixtures can be annotated, particularly those as chemically rich and structurally heterogeneous as Cucumaria frondosa extracts.
A long-recognised challenge in saponin research arises from the extensive structural diversity within this metabolite class. Numerous isomeric species—including aglycone variants, interchanged sugar sequences, and differences in sulfation—share identical elemental formulas. MASD v1.0 appropriately groups such structures under a common formula when MS1 data alone cannot distinguish them. Our results reflect this intrinsic limitation: several ions detected within the mid-elution window exhibit formulas consistent with saponin-like scaffolds, yet their annotation remains ambiguous without diagnostic fragmentation patterns. This behaviour is not a shortcoming of MASD itself, but rather an illustration of the fundamental inadequacy of MS1-only datasets to resolve isomeric complexity in marine secondary metabolites.
During the cross-examination of our experimental masses with the formulas listed in MASD, we observed a small subset of cases in which the reported molecular formula and the associated mass (labelled as “MWT” in the distributed dataset) did not fully coincide when recalculated as monoisotopic masses. Although the magnitude of these discrepancies is modest, they occasionally approach values consistent with CH2-equivalent shifts. With the utmost caution, we note that these deviations may arise from heterogeneity in the primary literature sources from which MASD compiles its structures, or from transcription or labelling inconsistencies inherited during data integration—challenges also acknowledged by the MASD authors in their discussion of curation limitations. Such instances are neither unexpected nor uncommon in natural product databases, particularly for structures established before the widespread implementation of high-resolution MS. Rather than detracting from the utility of MASD, these observations highlight the essential role of cross-validating formulas, exact masses, and fragmentation profiles when using the database for high-confidence annotation.
These considerations underscore the necessity of adopting an integrated analytical strategy for the structural elucidation of marine saponins. Tandem mass spectrometry (MS/MS) is indispensable for mapping fragmentation along both glycone and aglycone regions, enabling the discrimination of positional isomers, the identification of sugar linkages, and the confirmation of sulfation patterns. Complementarily, FTIR spectroscopy provides rapid and direct evidence of functional groups, as showcased by the characteristic S=O asymmetric stretching near 1225 cm−1 in our C. frondosa fractions. Together, MS/MS and FTIR greatly refine initial MS1-based annotations and help contextualise MASD entries within an experimentally validated framework.
Ultimately, however, full structural elucidation requires nuclear magnetic resonance (NMR) spectroscopy, provided that chromatographic purification yields fractions of adequate homogeneity. NMR remains the only technique capable of unambiguously assigning glycosidic sequences, aglycone stereochemistry, and substituent positions—elements that cannot be resolved by mass spectrometry alone. Such purification is likewise essential prior to biological testing: without molecularly defined samples, apparent bioactivities may reflect synergistic or antagonistic effects among co-extracted species, complicating structure–function interpretations.
In this light, MASD v1.0 serves as a valuable and much-needed foundation for the field, and our analysis highlights the complementary experimental efforts still required to achieve comprehensive structural resolution of marine saponins. The integration of MS/MS, FTIR, and NMR data into open-access repositories will not only enhance reproducibility, but will also enable future versions of MASD to incorporate richer, multi-dimensional spectral information. Such reciprocal development—where curated databases and experimental validation inform and strengthen one another—will be crucial for confidently identifying bioactive marine metabolites and for advancing their therapeutic potential.
Given these constraints, our analysis refrains from proposing new structures and instead focuses on demonstrating how multi-spectroscopic validation mitigates several sources of misannotation commonly encountered when relying solely on MS1-level information.
4. Conclusions
This study establishes and validates an integrated analytical workflow for the characterisation of triterpenoid saponins in complex marine extracts, using Cucumaria frondosa as a model system. By combining FTIR spectroscopy and high-resolution LC–MS, we reveal that butanol-enriched fractions contain not only triterpenoid glycosides but also a substantial proportion of co-extracted lipids and nitrogen-containing species. These components account for the pronounced aliphatic and carbonyl signatures detected by FTIR and must be considered when interpreting chromatographic and mass-spectrometric data from marine extracts.
LC–MS profiling of preparative fractions showed that saponin-like ions—most notably a feature at —recur across chromatographically distinct fractions, often accompanied by lipid-like ions in both positive and negative ionisation modes. This behaviour demonstrates how lipidic co-extracts can influence apparent retention times, ionisation patterns, and spectral complexity, underscoring the importance of contextualising MS signals within the broader chemical environment of the extract.
MS/MS could proved critical for discriminating canonical triterpenoid fragmentation pathways from non-specific fragmentation of co-eluting species. The resulting fragment patterns provided reliable structural constraints, enabling a more rigorous interpretation of ions tentatively associated with frondoside-like scaffolds and highlighting the limitations of relying solely on MS1 data for dereplication.
Comparison with the Marine Animal Saponin Database (MASD v1.0) reaffirmed its value as a reference resource while also illustrating the constraints of MS1-centric databases for annotation in complex matrices. Occasional inconsistencies between reported formulas and molecular weights, together with the absence of diagnostic MS/MS spectra, emphasise the need for community-wide deposition of validated spectral datasets and standardised fragmentation libraries.
Rather than reporting new saponin structures, this study provides a methodological framework designed to minimise misidentification and to guide the transition from broad MS-based screening to targeted isolation and structural validation. By integrating complementary spectroscopic tools into a coherent workflow, we contribute a practical resource for researchers working with structurally complex marine extracts. Such multidimensional approaches are essential not only for improving the accuracy of dereplication but also for advancing future structural, biological, and biotechnological investigations of echinoderm-derived saponins.
Author Contributions
Conceptualization, V.D-A.; methodology, V.D-A., M.Q., I.E., U.G., and A.M-P.; investigation, V.D-A., M.Q., I.E., U.G., A.M-P., L.G-Z., T.H. and L.T.A; writing—original draft preparation, V.D-A.; writing—review and editing, V.D-A., M.Q., I.E., U.G., A.M-P., L.G-Z., T.H. and L.T.A; visualization, V.D-A., M.Q., A.M-P., T.H. and L.T.A; supervision, T.H. and L.T.A.; funding acquisition, T.H. and L.T.A. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
Financial support by Xunta de Galicia through grant IN606B-2023/006 is gratefully acknowledged. Facilities provided by the Galician Supercomputing Centre (CESGA) are also acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dominguez-Arca, V.; Hellweg, T.; Antelo, L.T. Harnessing Thalassochemicals: Marine Saponins as Bioactive Agents in Nutraceuticals and Food Technologies. MARINE DRUGS 2025, 23. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Xue, C.H.; Zhang, T.T.; Wang, Y.M. Saponins from Sea Cucumber and Their Biological Activities. JOURNAL OF AGRICULTURAL AND FOOD CHEMISTRY 2018, 66, 7222–7237. [Google Scholar] [CrossRef]
- Podolak, I.; Grabowska, K.; Sobolewska, D.; Wrobel-Biedrawa, D.; Makowska-Was, J.; Galanty, A. Saponins as cytotoxic agents: an update (2010-2021). Part II-Triterpene saponins. PHYTOCHEMISTRY REVIEWS 2023, 22, 113–167. [Google Scholar] [CrossRef]
- Lu, Y.N.; Cui, P.; Tian, X.Q.; Lou, L.G.; Fan, C.Q. Unusual Cytotoxic Steroidal Saponins from the Gorgonian Astrogorgia dumbea. PLANTA MEDICA 2016, 82, 882–887. [Google Scholar] [CrossRef]
- Wang, W.; Hong, J.; Lee, C.; Im, K.; Choi, J.; Jung, J. Cytotoxic sterols and saponins from the starfish Certonardoa semiregularis. JOURNAL OF NATURAL PRODUCTS 2004, 67, 584–591. [Google Scholar] [CrossRef]
- Roccatagliata, A.; Maier, M.; Seldes, A.; Pujol, C.; Damonte, E. Antiviral sulfated steroids from the ophiuroid Ophioplocus januarii. JOURNAL OF NATURAL PRODUCTS 1996, 59, 887–889. [Google Scholar] [CrossRef]
- Cao, L.; Dong, P.; Liu, J.; Zhang, J.; Xie, H.; Yu, S.; Zhang, J. Advancements in saponin-based vaccine adjuvants. MEDICINAL CHEMISTRY RESEARCH 2025, 34, 1817–1832. [Google Scholar] [CrossRef]
- Sanina, N.; Chopenko, N.; Mazeika, A.; Kostetsky, E. Nanoparticulate Tubular Immunostimulating Complexes: Novel Formulation of Effective Adjuvants and Antigen Delivery Systems. BIOMED RESEARCH INTERNATIONAL 2017, 2017. [Google Scholar] [CrossRef]
- Wijesekara, T.; Luo, J.; Xu, B. Critical review on anti-inflammation effects of saponins and their molecular mechanisms. PHYTOTHERAPY RESEARCH 2024, 38, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Baharara, J.; Amini, E.; Salek, F. Anti-inflammatory properties of saponin fraction from (Spiny brittle star) Ophiocoma erinaceus (Muller and Troschel, 1842). IRANIAN JOURNAL OF FISHERIES SCIENCES 2020, 19, 638–652. [Google Scholar] [CrossRef]
- Chen, X.J.; Zhang, X.J.; Shui, Y.M.; Wan, J.B.; Gao, J.L. Anticancer Activities of Protopanaxadiol- and Protopanaxatriol-Type Ginsenosides and Their Metabolites. EVIDENCE-BASED COMPLEMENTARY AND ALTERNATIVE MEDICINE 2016, 2016. [Google Scholar] [CrossRef]
- RODRIGUEZ, J.; CASTRO, R.; RIGUERA, R. HOLOTHURINOSIDES - NEW ANTITUMOR NONSULFATED TRITERPENOID GLYCOSIDES FROM THE SEA-CUCUMBER HOLOTHURIA-FORSKALII. TETRAHEDRON 1991, 47, 4753–4762. [Google Scholar] [CrossRef]
- Hosseini, S.F.; Rezaei, M.; McClements, D.J. Bioactive functional ingredients from aquatic origin: a review of recent progress in marine-derived nutraceuticals. CRITICAL REVIEWS IN FOOD SCIENCE AND NUTRITION 2022, 62, 1242–1269. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Dave, D.; Shahidi, F. Northern Sea Cucumber (Cucumaria frondosa): A Potential Candidate for Functional Food, Nutraceutical, and Pharmaceutical Sector. MARINE DRUGS 2020, 18. [Google Scholar] [CrossRef]
- Fagbohun, O.F.; Rollins, A.; Mattern, L.; Cipollini, K.; Rupasinghe, H.P.V. Frondoside A of Cucumaria frondosa (Gennerus, 1767): Chemistry, biosynthesis, medicinal applications, and mechanism of actions. JOURNAL OF PHARMACY AND PHARMACOLOGY 2024, 77, 32–42. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, C.; Yu, D.; Hou, H. Methods of simultaneous preparation of various active substances from Stichopus chloronotus and functional evaluation of active substances. FOOD AND AGRICULTURAL IMMUNOLOGY 2022, 33, 563–574. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Avilov, S.A.; Andrijaschenko, V, P.; Popov, R.S.; Chingizova, E.A.; Grebnev, B.B.; Rasin, A.B.; Kalinin, I.V. The Isolation, Structure Elucidation and Bioactivity Study of Chilensosides A, A1, B, C, and D, Holostane Triterpene Di-, Tri- and Tetrasulfated Pentaosides from the Sea Cucumber Paracaudina chilensis (Caudinidae, Molpadida). MOLECULES 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, Y.; Zhang, W.; Franco, C.M.M. Distribution of Saponins in the Sea Cucumber Holothuria lessoni; the Body Wall Versus the Viscera, and Their Biological Activities. MARINE DRUGS 2018, 16. [Google Scholar] [CrossRef]
- Smith, S.J.; Cummins, S.F.; Motti, C.A.; Wang, T. A mass spectrometry database for the identification of marine animal saponin-related metabolites. ANALYTICAL AND BIOANALYTICAL CHEMISTRY 2024, 416, 6893–6907. [Google Scholar] [CrossRef]
- Hossain, A.; Dave, D.; Shahidi, F. Antioxidant Potential of Sea Cucumbers and Their Beneficial Effects on Human Health. MARINE DRUGS 2022, 20. [Google Scholar] [CrossRef]
- Bahrami, Y.; Zhang, W.; Chataway, T.; Franco, C. Structural Elucidation of Novel Saponins in the Sea Cucumber Holothuria lessoni. MARINE DRUGS 2014, 12, 4439–4473. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, Y.; Zhang, W.; Franco, C. Discovery of Novel Saponins from the Viscera of the Sea Cucumber Holothuria lessoni. MARINE DRUGS 2014, 12, 2633–2667. [Google Scholar] [CrossRef] [PubMed]
- Conroy, M.J.; Andrews, R.M.; Andrews, S.; Cockayne, L.; Dennis, E.A.; Fahy, E.; Gaud, C.; Griffiths, W.J.; Jukes, G.; Kolchin, M.; et al. LIPID MAPS: update to databases and tools for the lipidomics community. NUCLEIC ACIDS RESEARCH 2024, 52, D1677–D1682. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic overview of the analytical workflow validated in this study. The strategy integrates ATR–FTIR screening, LC–MS1 profiling, preparative C18 fractionation, and cross-comparison of recurring ions across fractions, followed by formula/mass matching against MASD v1.0. This workflow is intended as a practical decision-making tool for dereplication in complex marine extracts.
Figure 1.
Schematic overview of the analytical workflow validated in this study. The strategy integrates ATR–FTIR screening, LC–MS1 profiling, preparative C18 fractionation, and cross-comparison of recurring ions across fractions, followed by formula/mass matching against MASD v1.0. This workflow is intended as a practical decision-making tool for dereplication in complex marine extracts.
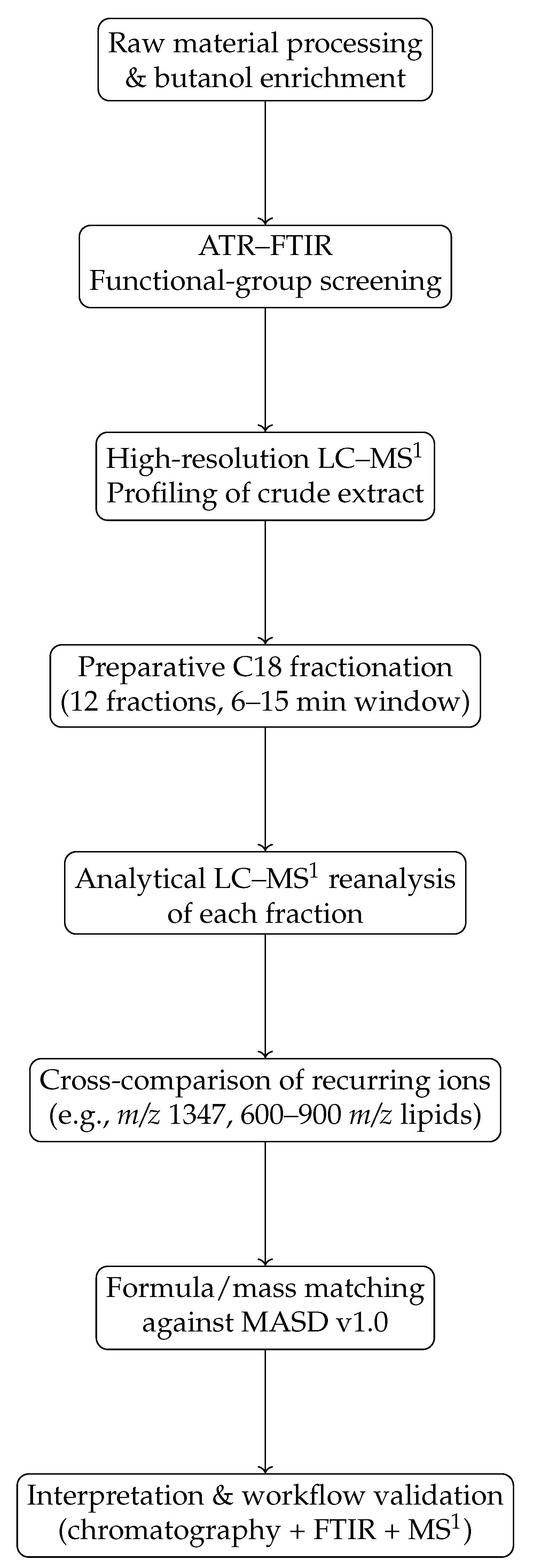
Figure 2.
Comparative FTIR spectra of triterpenoid saponins (ATR, films on diamond). (a) Aescin; (b) Digitonin; (c) Quillaja saponin. For reference, the spectra of the pure saponins with lipids (see Figure 5) and the Cucumaria frondosa extract (Figure 4) are overlaid in each panel. The increased intensity of CH2/CH3 stretching bands in the ternary mixtures mirrors the spectral profile of the crude extract, suggesting the presence of lipidic components or extended aliphatic domains in the natural matrix.
Figure 2.
Comparative FTIR spectra of triterpenoid saponins (ATR, films on diamond). (a) Aescin; (b) Digitonin; (c) Quillaja saponin. For reference, the spectra of the pure saponins with lipids (see Figure 5) and the Cucumaria frondosa extract (Figure 4) are overlaid in each panel. The increased intensity of CH2/CH3 stretching bands in the ternary mixtures mirrors the spectral profile of the crude extract, suggesting the presence of lipidic components or extended aliphatic domains in the natural matrix.
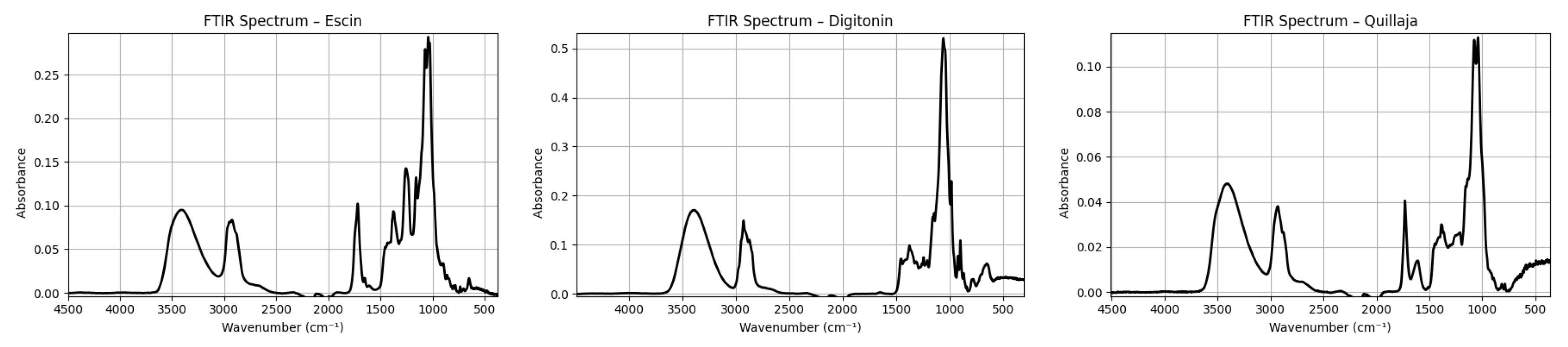
Figure 3.
Simulated FTIR spectra: (a) pure triterpenoid saponin, exhibiting a dominant OH stretching band and characteristic C–O signals from carbohydrate moieties; (b) mixed saponin–lipid system, where CH2/CH3 aliphatic stretching bands become predominant.
Figure 3.
Simulated FTIR spectra: (a) pure triterpenoid saponin, exhibiting a dominant OH stretching band and characteristic C–O signals from carbohydrate moieties; (b) mixed saponin–lipid system, where CH2/CH3 aliphatic stretching bands become predominant.
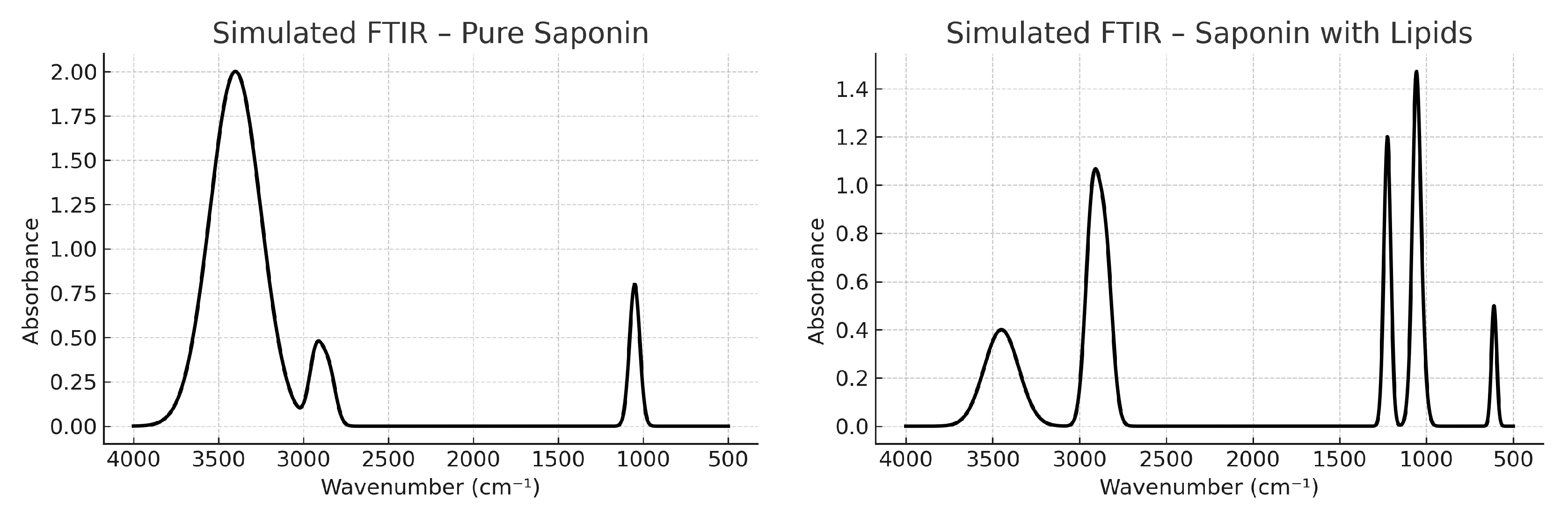
Figure 4.
Experimental ATR–FTIR spectrum of the Cucumaria frondosa extract measured as a film on the diamond crystal (deposit from n-butanol and dried with dry air; trace solvent possibly remaining). Band assignments highlight contributions from triterpenoid saponins (glycosidic fingerprint, OH stretching), lipids and DMPC (CH2/CH3 stretching), and nitrogenous lipids (amide-related N–H bending, C=O stretching).
Figure 4.
Experimental ATR–FTIR spectrum of the Cucumaria frondosa extract measured as a film on the diamond crystal (deposit from n-butanol and dried with dry air; trace solvent possibly remaining). Band assignments highlight contributions from triterpenoid saponins (glycosidic fingerprint, OH stretching), lipids and DMPC (CH2/CH3 stretching), and nitrogenous lipids (amide-related N–H bending, C=O stretching).

Figure 5.
Comparative FTIR spectra of triterpenoid saponins combined with DMPC. (a) Aescin + DMPC; (b) Digitonin + DMPC; (c) Quillaja saponin + DMPC. For reference, the spectra of the pure saponins (see Figure 2) and the Cucumaria frondosa extract (Figure 4) are overlaid in each panel. The increased intensity of CH2/CH3 stretching bands in the ternary mixtures mirrors the spectral profile of the crude extract, suggesting the presence of lipidic components or extended aliphatic domains in the natural matrix. The intensified band at ∼1735 cm−1 in the mixtures is consistent with ester carbonyls from the phospholipid backbone (with potential overlap from a triterpenoid lactone).
Figure 5.
Comparative FTIR spectra of triterpenoid saponins combined with DMPC. (a) Aescin + DMPC; (b) Digitonin + DMPC; (c) Quillaja saponin + DMPC. For reference, the spectra of the pure saponins (see Figure 2) and the Cucumaria frondosa extract (Figure 4) are overlaid in each panel. The increased intensity of CH2/CH3 stretching bands in the ternary mixtures mirrors the spectral profile of the crude extract, suggesting the presence of lipidic components or extended aliphatic domains in the natural matrix. The intensified band at ∼1735 cm−1 in the mixtures is consistent with ester carbonyls from the phospholipid backbone (with potential overlap from a triterpenoid lactone).
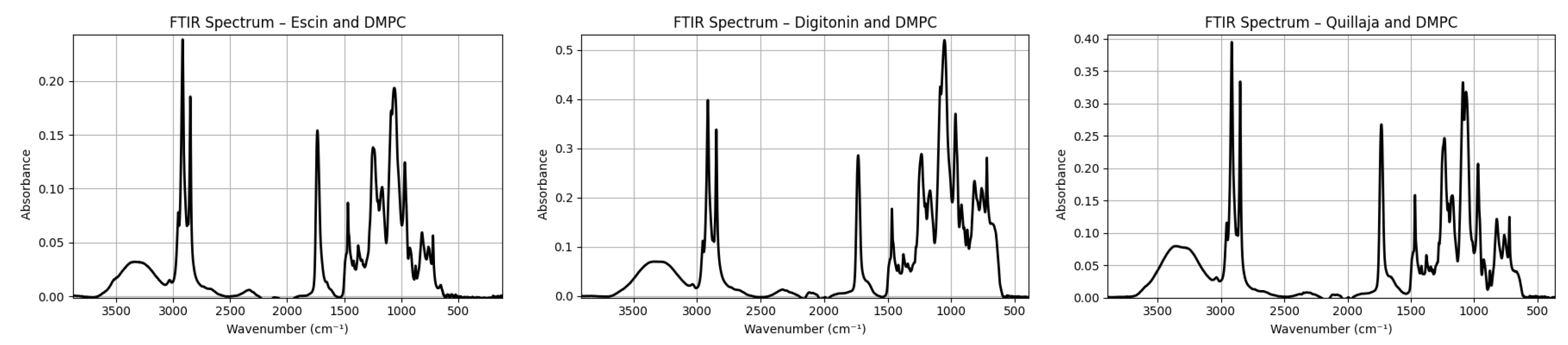
Figure 6.
UV chromatogram at 210 nm of the Cucumaria frondosa extract during the preparative C18 run. The mobile phase gradient is overlaid: blue for solvent B (acetonitrile) and cyan for solvent A (water). A clear elution window for moderately polar compounds is observed between 6 and 15 min.
Figure 6.
UV chromatogram at 210 nm of the Cucumaria frondosa extract during the preparative C18 run. The mobile phase gradient is overlaid: blue for solvent B (acetonitrile) and cyan for solvent A (water). A clear elution window for moderately polar compounds is observed between 6 and 15 min.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



