Submitted:
11 December 2025
Posted:
15 December 2025
You are already at the latest version
Abstract
From a biochemical perspective, the recently introduced collective term luciferase-like monooxygenase family (LLM family) is notable for grouping together bacterial enzymes with fundamentally different functional characteristics. Thus not only does the family include non-bioluminescent and well as bioluminescent enzymes, but additionally both anoxybiontic and oxybiontic enzymes. By reviewing both the relatively short history of the LLM family itself, and the more protracted development of our present understanding of a number of the biochemically disparate composite enzyme groups, alternative representational descriptors can be identified that better serve both to succinctly delineate and functionally characterise the discrete groups currently corralled into the LLM family.
Keywords:
1. Introduction
2. The Luciferase-like Monooxygenase Family (= Bac_Luciferases)—Issues of Perspective
3. Bacterial Luciferases—A Historical Overview
4. De Facto to In Silico Progression—The Advance of the LLM Family
5. The Emergence of a Functional Understanding of the Bacterial Luciferases
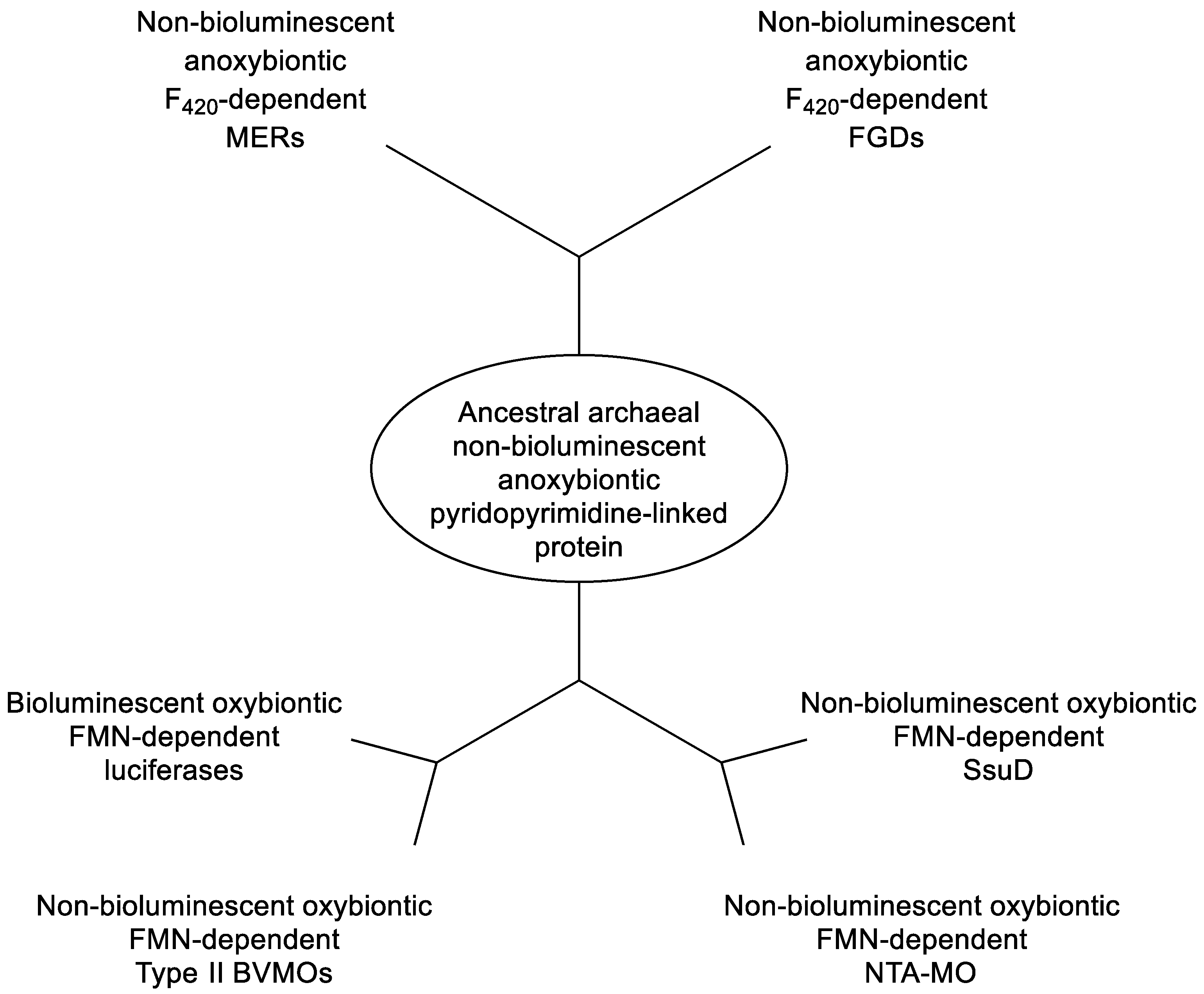
6. Beyond Bacterial Luciferases—The Discovery of Functionally Related Non-Bioluminescent Monooxygenases
7. What’s in a Name? The Established Value of FD-TCMO as a Moniker, and Its Relevance to Further More Recent Studies
Funding
Data availability Statement
Acknowledgments
Conflicts of interest
References
- Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. Enzyme Nomenclature. Academic Press: New York, NY, USA, 1992.
- Orengo, C.A.;Thornton, J.M. Protein families and their evolution - a structural perspective. Ann. Rev. Biochem. 2005, 74, 867-900. [CrossRef]
- Mascotti, M.L.; Kumar, H.; Nguyen, Q-T.; Ayub, M.J.; Fraaije,M.W. Reconstituting the evolutionary history of F420-dependent dehydrogenases. Scientific Reports, 2018, 8, 17571. [CrossRef]
- Harms, M.J.; Thornton, J.W. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev, Genet., 2013,14, 559-571. [CrossRef]
- Selengut, J.D.; Haft, D.H. Unexpected abundance of coenzyme F420-dependent enzymes in Mycobacterium tuberculosis and other Actinobacteria. J. Bacteriol., 2010, 192, 5788-5798. [CrossRef]
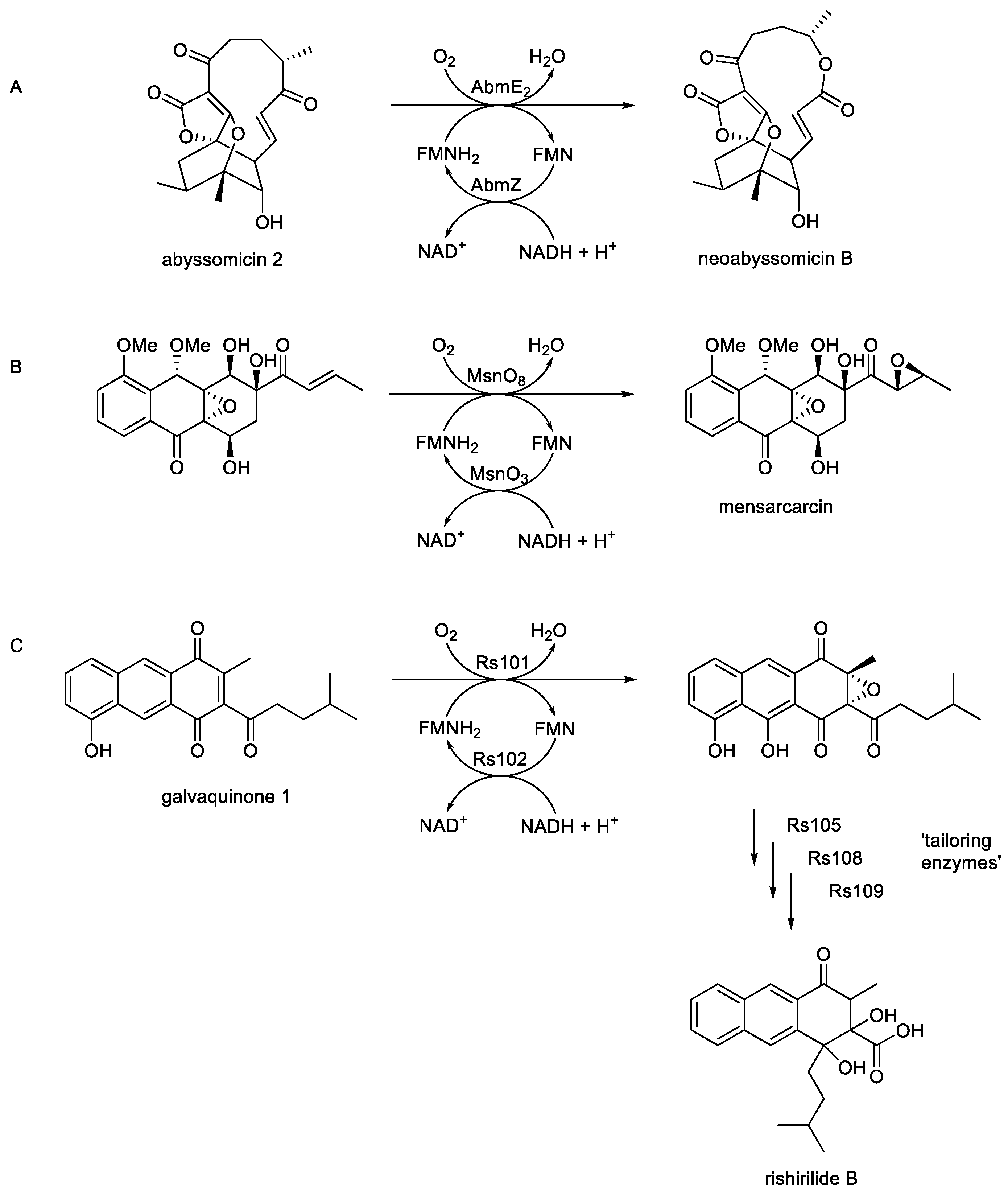
- Song, Y.; Li, Q.; Qiu, F.; Sun, C.; Liang, H; Wei, X.; Wong, N.K.; Yi, L.; Zhang, Y.; Shao, M.; Ju, I. Neoabyssomicins A-C, polycyclic macrolactones from deep-sea derived Streptomyces koyangensis SCS 10 5802. Tetrahedron, 2017, 73, 5366-5372.
- Maier, S.; Heitzler, T.; Asmus, K.; Brotz, E.; Hardter, U.; Hesselbach, K.; Paululat, T.; Bechthold, A. Functional characterization of different ORFs including luciferase-like monooxygenase genes from the mensacarcin gene cluster. ChemBioChem., 2015, 16, 1175-1182. [CrossRef]
- Alali, A.; Zhang, L.; Li, J.; Zuo, C.; Wassouf, D.; Yan, X.; Schwarzer, P.; Gunther, B.; Rinsle, O.; Bechthold, A. Biosynthesis of the tricyclic aromatic Type II polyketide rishirilide: new potential third ring oxygenation after three cyclisation steps. Mol. Biotechnol., 2021, 63, 502-514. [CrossRef]
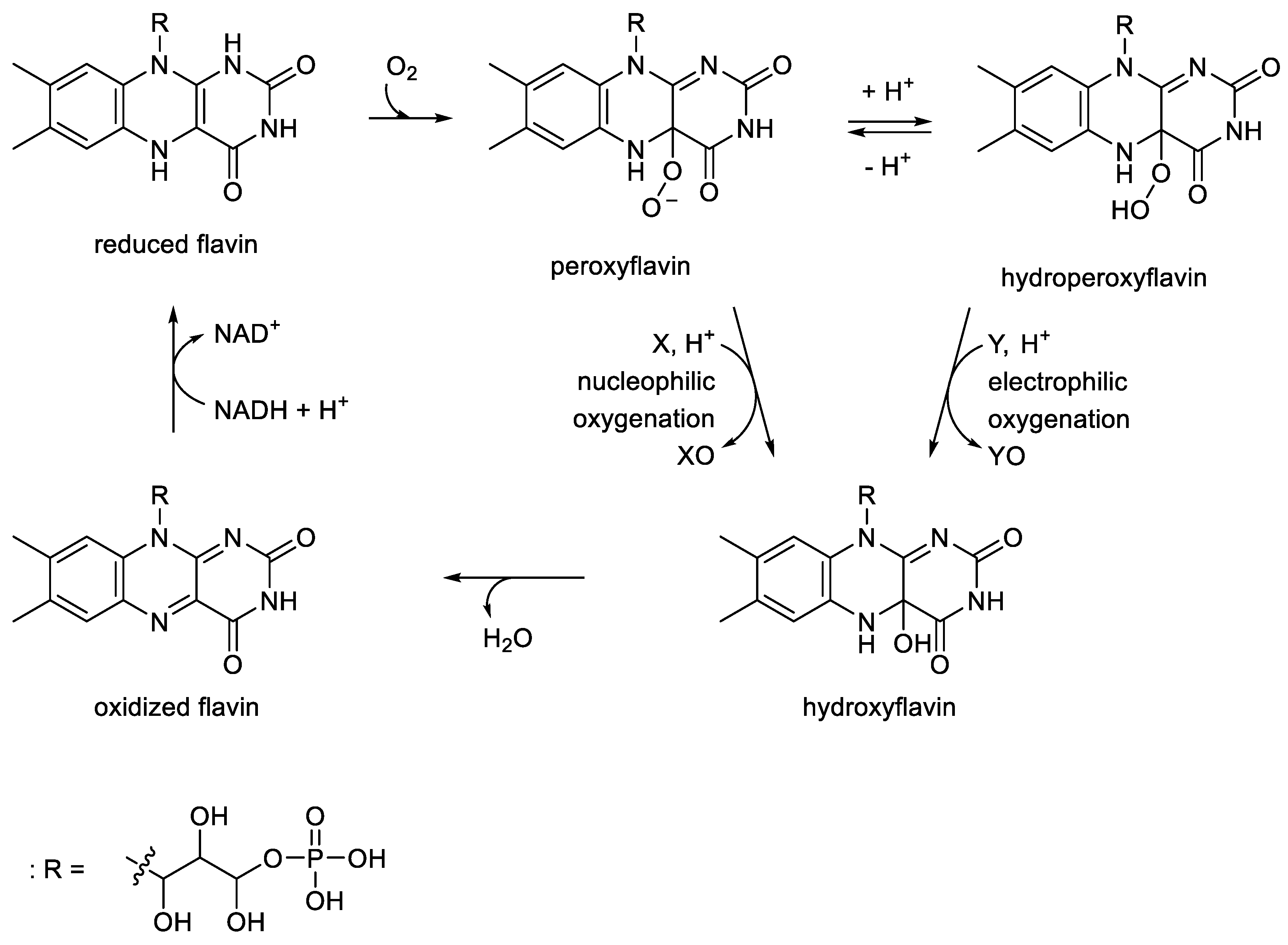
- Ghisla, S.; Massey, V. Mechanisms of flavoprotein-catalysed reactions. Eur. J. Biochem., 1989, 181, 1-17.
- Hastings, J.W.; Potrikus, C.J.; Gupta, S.C.; Kurfurst, M.; Makemson, J.C. Biochemistry and physiology of bioluminescent bacteria. Adv. Microb. Physiol., 1985, 26, 236-291.
- Greening, C.; Ahmed, F.H.; Mohamed, A.E.; Lee, B.M.; Pandey, G.; Warden, A.C.; Scott, C.; Oakeshott, J.G.; Taylor, M.C.; Jackson, C.J. Physiology, biochemistry, and applications of F420-dependent redox reactions. Microbiol. Mol. Biol. 2016, 80, 451-493.
- Lavoisier, A. On the combustion of candles in atmospheric air and dephlogistated air. Communicated to the Academie des Sciences, Paris, 3rd May 1777.
- Payen, A.; Persoz, J.F. Memoire sur la diastase, les principaux produits de ses reaction, et leurs applications aux arts industrials. Ann. Chim. Phys., 1833, 53, 73-92.
- Dubois, R. Sur le mechanisme des fonctions photodermatique et photogenique dans le siphon du Pholas dactylus. Compt. Rend. Soc. Biol., 1887, 3, 564-568.
- Hastings, J.W.; Presswood, R.P. Bacterial luciferase: FMNH2-aldehyde oxidase. Meth. Enzymol., 1978, 53, 558-570.
- Delroisse, J.; Duchatelet, L.; Flammang, P.; Mallefet, J. Leaving the dark side? Insights into the evolution of luciferases. Front. Mar. Sci., 2021, 8, 673620. [CrossRef]
- Haddock, S.H.D.; Moline, M.A.; Case, J.F. Bioluminescence in the sea. Ann. Rev. Mar. Sci., 2010, 2, 443-493. [CrossRef]
- Meighen, E.A. Molecular biology of bacterial bioluminescence. Microbiol. Rev. 1991, 55, 123-142. [CrossRef]
- Fisher, A.J.; Thompson, T.B.; Thoden, J.B.; Baldwin, T.O.; Rayment, I. The 1.5- Å crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem., 1986, 271, 21956-21968.
- Campbell, Z.T.; Weichsel, A.; Montfort, W.R.; Baldwin, T.O. Crystal . structure of the bacterial luciferase/flavin complex provides insights into the function of the β-subunit Biochem., 2009, 48, 6085-6094.
- Deeva, A.A.; Temlyakova, E.A.; Sorokin, A.A.; Nemtseva, E.V.; Kratasyuk, V.A. Structural distinctions of the fast and slow bacterial luciferases revealed by phylogenetic analysis. Bioinform., 2016, 32, 3053-3057. [CrossRef]
- Wierenga, R.K. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett., 2001, 492, 193-198. [CrossRef]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One fold with many functions: the evolutionary relationships between TIM-barrel families based on their sequences, structures, and functions. J. Mol. Biol., 2002, 321, 741-765. [CrossRef]
- Aufhammer, S.W.; Warkentin, E.; Berk, H.; Shima, S,; Thauer, R.K.; Ermier, U. Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure, 2004, 12, 361-370. [CrossRef]
- Nolling, J.; Ishii, M.; Koch, J.; Pihl, T.D.; Reeve, J.N.; Thauer,R.K.; Hedderich, R. Characterization of a 45-kDa flavoprotein and evidence for a rubredoxin, two proteins that could participate in electron transport from H2 to CO2 in methanogenesis in Methanobacterium thermoautotrophicum. Eur. J. Biochem., 1995, 231, 628-638. [CrossRef]
- Aufhammer, S.W.; Warkentin, E.; Ermler, U.; Hagemeier, C.H.; Thauer, R.K.; Shima, S. Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: archetecture of the F420/FMN binding site of the enzymes within the nonpropyl cis-peptide containing bacterial luciferase family. Protein Sci., 2005, 14, 1840-1849. [CrossRef]
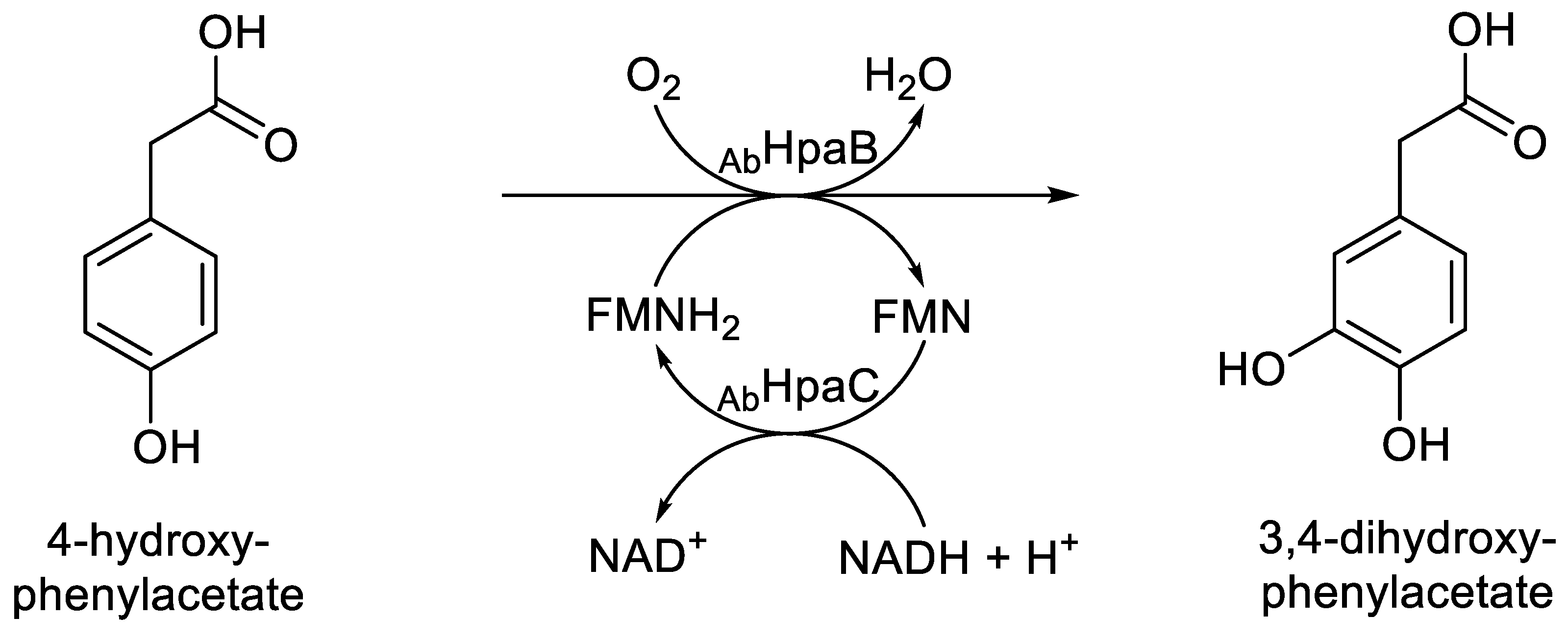
- Sucharitakul, J.; Chaiyen, P.; Entsch, B.; Ballou, D.P. The reductase of p-hydroxyphenylacetate 3-hydroxylase requires p-hydroxyphenylacetate for effective catalysis. Biochem., 2005, 44, 10434-10442.
- Eichhorn, E.; van der Ploeg, J.R.; Leisinger, T. Characterization of a two-component alkanesulfonate monooxygenase from Escherichia coli. J. Biol. Chem., 1999, 274, 26639-26646. [CrossRef]
- Knoebel, H.R.; Egli, T.; van der Meer, J. Cloning and characterization of the genes encoding nitrilotriacetate monooxygenase of Chelobacter heinzii ATCC 29600. J. Bacteriol., 1996, 178, 6123-6132.
- Grinter, R.; Greening, C. Cofactor F420: an expended view of its distribution, biosynthesis, and roles in bacteria and archaea. FEBS Microbiol. Rev., 2021, 45, 1-46. [CrossRef]
- Mascotti, M.L.; Ayub, M.J.; Fraaije, M.W. On the diversity of .F420-dependent oxidoreductases: a sequence- and structure-based classification. Proteus, 2021, 89, 1497-1507.
- Fischer, W.W.; Hemp, J.; Valentine, J.S. How did life survive Earth’s great oxygenation?. Curr. Opin. Chem. Biol., 2016, 166-178. [CrossRef]
- Muller, H. Reversibility in evolution considered from the standpoint of genetics. Biol. Rev. Camb. Phil. Soc., 1939, 14, 261-280. [CrossRef]
- Riedl, R. A systems-analytical approach to macro-evolutionary . phenomenon. Q. Rev. Biol., 1977, 52, 351-370. [CrossRef]
- Shah, P.; McCandlish, D.M.; Plotkin, J.B. Contingency and entrenchment protein evolution under purifying selection. Proc. Natl. Acad. Sci. USA, 2015, E3226-E3225. [CrossRef]
- McCandlish, D.M.; Shah, P.; Plotkin, J.B. Epistasis and the dynamics of recession in molecular evolution. Genetics, 2016, 203, 1335-1351. [CrossRef]
- Harvey, E.N. A history of bioluminescence: from the earliest times until 1900. American Philosophical Society, Philadelphia, Pennsylvania, USA, 1957.
- Harvey, E.N. Oxygen and bioluminescence. Biol. Bull., 1926, 51, 89-97.
- Strehler, B.L. Luminescence in cell-free extracts of luminous bacteria. and its activation by DPNH. J. Amer. Chem. Soc., 1953, 75, 1264. [CrossRef]
- McElroy, W.D.; Hastings, J.W.; Sonnenfeld, V.; Coulombre, J. The requirement of riboflavin phosphate for bacterial luminescence. . Science, 1953, 118, 385-386. [CrossRef]
- Strehler, B.L.; Cormier, M.J. Factors affecting the luminescence of cell-free extracts of the luminous bacterium Achromobacter fischeri. Arch. Biochem. Biophys., 1953, 47, 16-33. [CrossRef]
- Cormier, M.J.; Strehler, B.L. The identification of KCF: requirement of long-chain aldehyde for bacterial extract luminescence. J. Amer. Chem. Soc., 1953, 75, 4864-4865. [CrossRef]
- Rogers, P.; McElroy, W.D. Enzymatic determination of aldehyde . permeability in luminous bacteria. I. Effect of chain length on light. emission and penetration. Arch. Biochem. Biophys., 1958, 75, 87-105.
- Totter, J.R.; Cormier, M.J. The relation of bacterial luciferase to alternative pathways of dihydroflavin mononucleotide oxidation..J. Biol. Chem., 1955, 216, 801-911. [CrossRef]
- Mason, H.S.; Fowlks, W.L.; Peterson, E. Oxygen transfer and electron , transport by the phenolase complex. J. Amer. Chem. Soc., 1955, 77, 2914-2915. [CrossRef]
- Hastings, J.W.; Riley, W.H.; Massa, J. The purification, properties, and chemiluminescent quantum yield of bacterial luciferase. J. Biol. Chem., 1965, 240, 1473-1481. [CrossRef]
- Hayaishi, O. Biological oxidation. Ann. Rev. Biochem., 1963, 31, 25-46.
- Hastings, J.W.; Gibson, Q.H.; Greenwood, C. On the molecular mechanism of bioluminescence. I. The role of long-chain aldehyde. Proc. Natl. Acad. Sci. USA, 1964, 52, 1529-1535. [CrossRef]
- Meighan, E.A.; Hastings, J.W. Binding site determination from kinetic data: reduced flavin mononucleotide binding to bacterial luciferase. J. Biol. Chem., 1971, 246, 766-767.
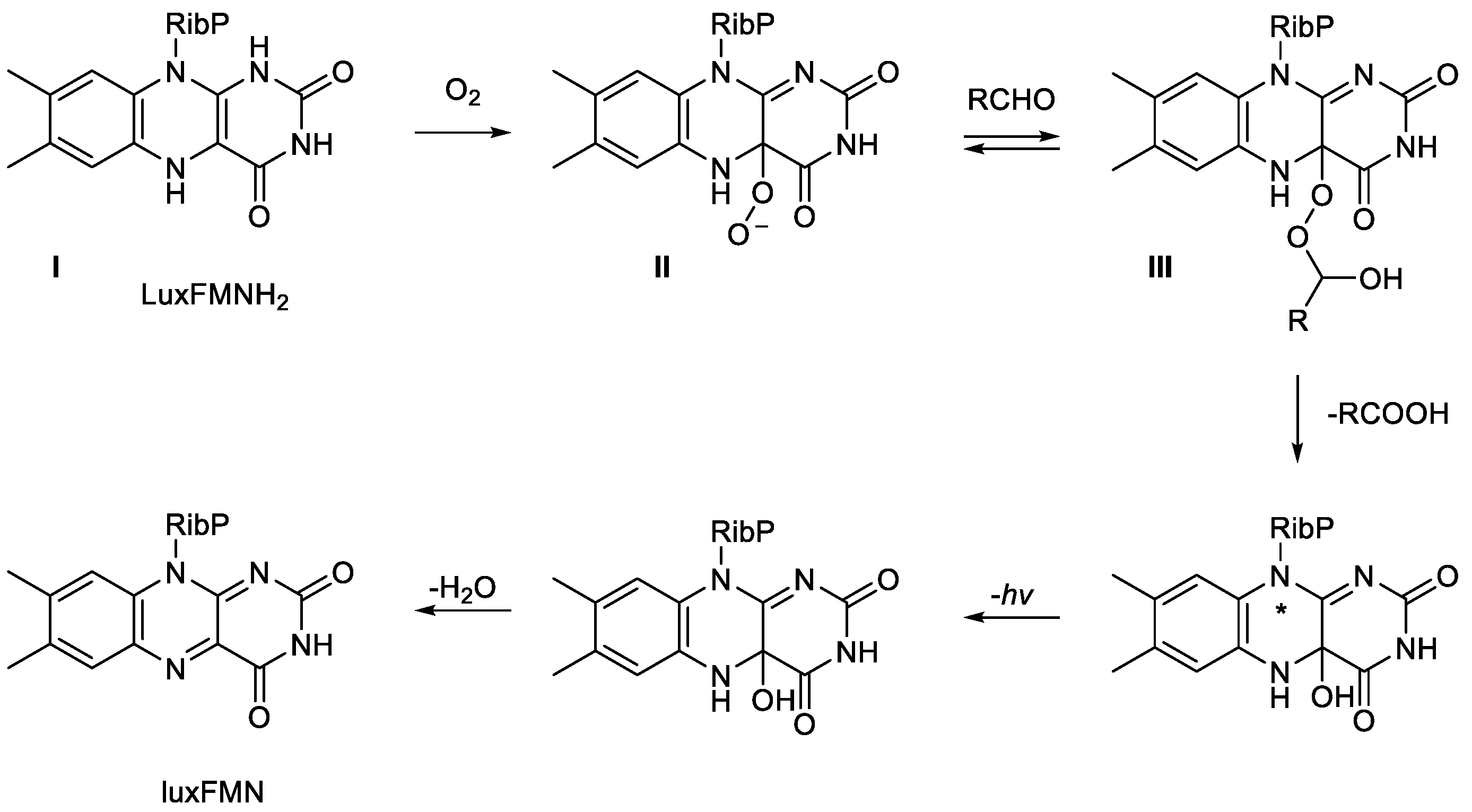
- Eberhard, A.; Hastings, J.W. A postulated mechanism for the b . . bioluminescent oxidation of reduced flavin mononucleotide. Biochem. Biophys. Res. Commun., 1972, 47, 348-353. [CrossRef]
- Romero, E.; Gomez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same substrate, many reactions: oxygen activation in flavoenzymes. Chem. Rev., 2018, 118, 1742-1769. [CrossRef]
- Criegee, R. Die umlangerung de dekalin peroxydester als folge von . . kationischem sauerstoff. Liebigs Ann. Chem., 1948, 560, 127-135.
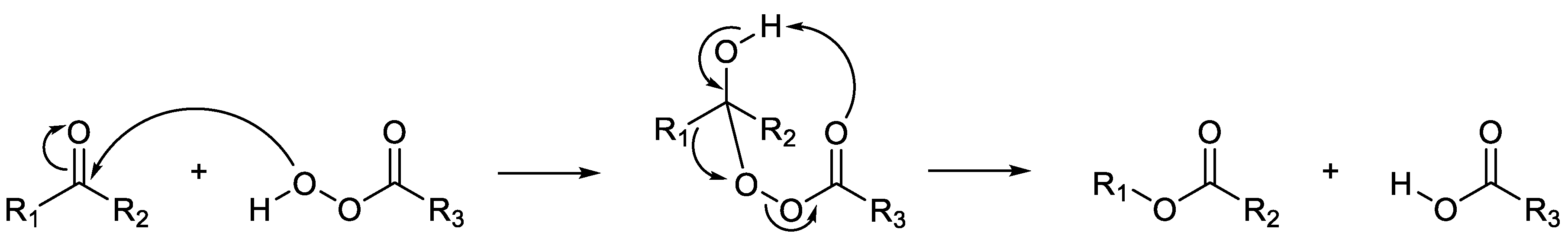
- Baeyer, A.; Villiger, V. Einwirkung des caro’schen reagens auf ketone. Ber. Dtsch. Chem.,1899, 32, 3625-3633. [CrossRef]
- Gunsalus, I.C. The biology, chemistry, and physics of oxygen reduction. Fed. Proc., 1976, 35, 1345-1346.
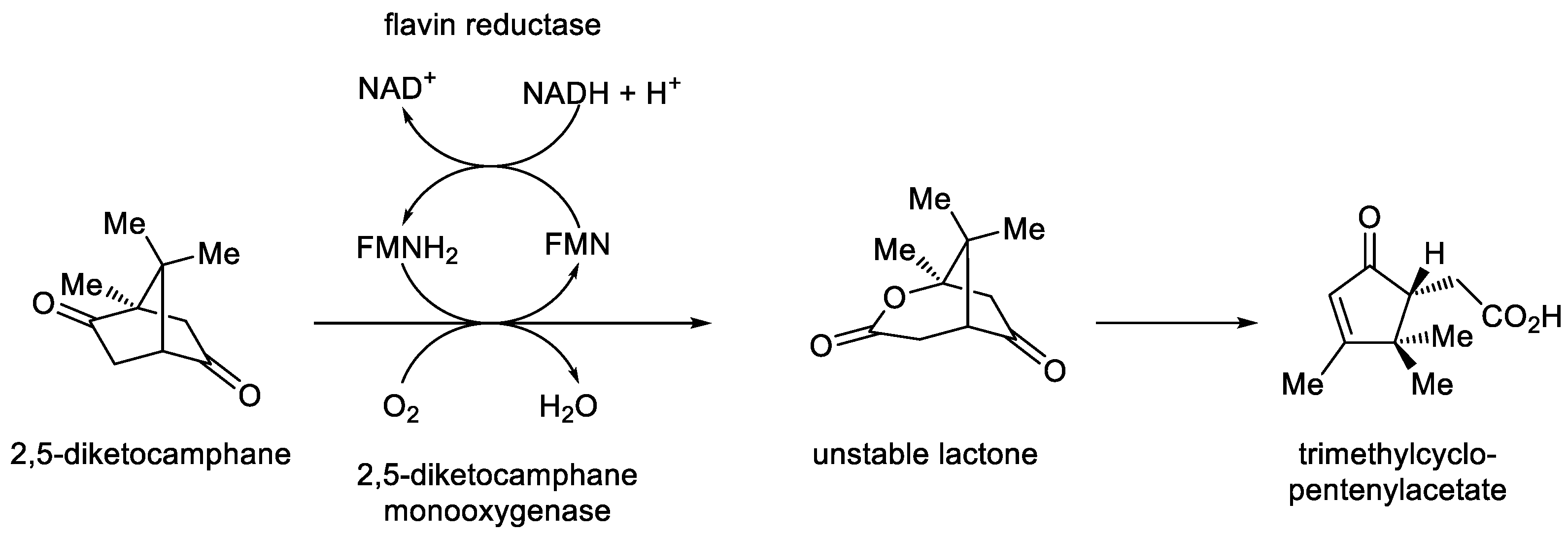
- Conrad, H.E.; Lieb, K.; Gunsalus, I.C. An enzyme system for cyclic . . ketolactonization. Biochem. Biophys. Res. Commun., 1961, 6, 293-297. [CrossRef]
- Conrad, H.E.; Lieb, K.; Gunsalus, I.C. Mixed function oxidation. III. An electron transport complex in camphor lactonization. J. Biol. Chem., 1965, 240, 4029-4037.
- Hastings, J.W.; Nealson, K.H. Bacterial bioluminescence. Ann. Rev. Microbiol., 1977, 31, 549-595.
- Eckstein, J.W.; Hastings, J.W.; Ghisla, S. Mechanism of bacterial bioluminescence: 4a,5-dihydroflavin analogues as models for luciferase hydroperoxide intermediates and the effect of substituents at the 8-position of flavin on luciferase kinetics. Biochem., 1993, 32, 404-411. [CrossRef]
- Suzuki, K.; Kaidoh, T.; Katagiri, M.; Tsuchiya, T. O2 incorporation into a long-chain fatty acid during bacterial bioluminescence. Biochem. Biophys. Acta, 1983, 722, 297-301. [CrossRef]
- Mason, H.S.; Fowlkes, W.L.; Peterson, E. Oxygen transfer and electron transport by a phenolase complex. J. Amer. Chem. Soc., 1955, 77, 2914-2915.
- Grogan, G.; Roberts, S.M.; Willetts, A. Camphor-grown Pseudomonas putida, a multifunctional biocatalyst. Biotechnol. Lett., 1993, 15, 913-918. [CrossRef]
- Willetts, A. Structural studies and synthetic applications of Baeyer-Villiger monooxygenases. Trends Biotechnol., 1998, 15, 55-62. [CrossRef]
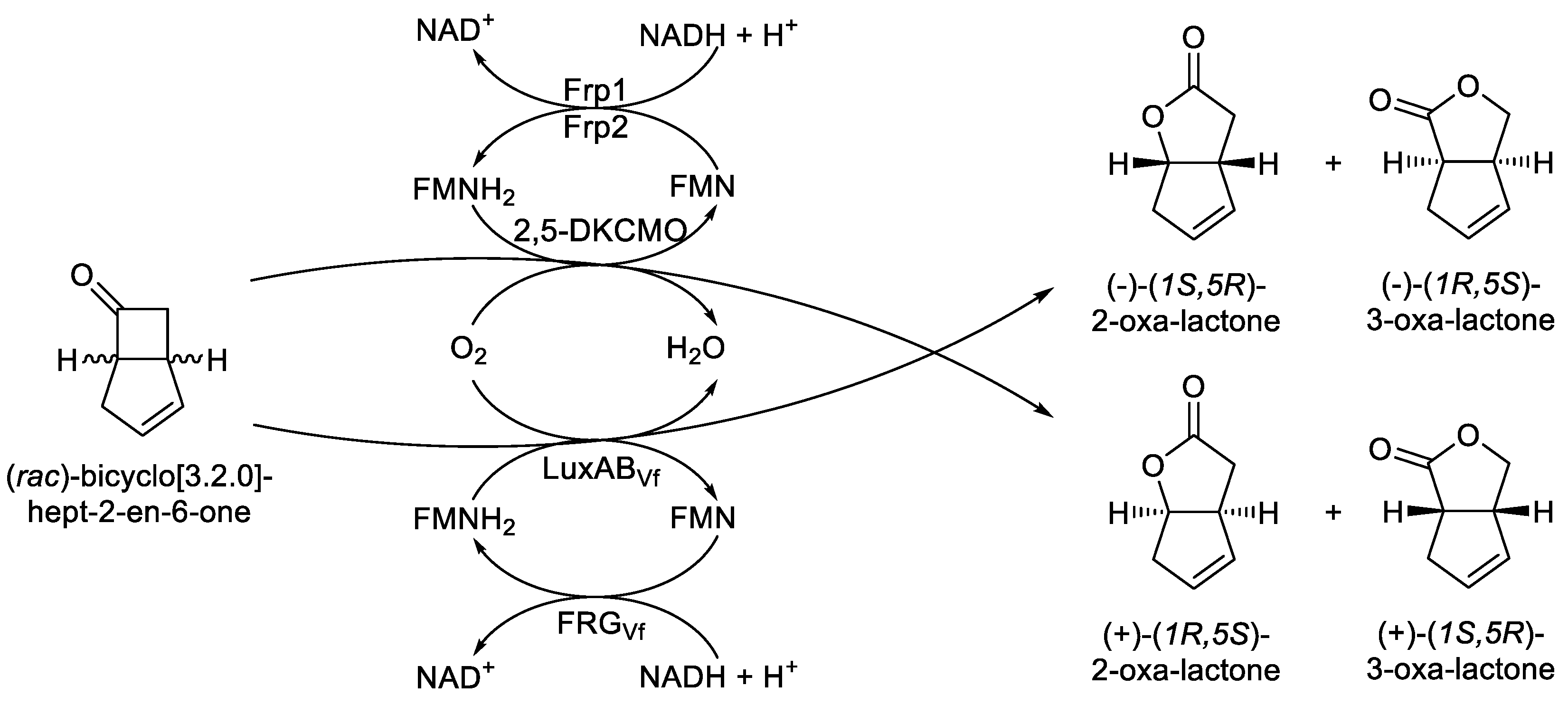
- Willetts, A.; Kelly, D.R. Flavin-dependent redox transfers by two-component diketocamphane monooxygenases of camphor-grown Pseudomonas putida NCIMB 10007. Microorganisms, 2016, 4, 38. [CrossRef]
- Villa, R.; Willetts, A. Oxidation by microbial NADH plus FMN-dependent luciferases from Photobacterium phosphoreum and Vibrio fischeri. J. Molec. Catal. B: Enzymatic, 1997, 2, 193-197. [CrossRef]
- Trudgill, P.W. Bacteria and the challenge of acyclic molecules. In Experiences in Biochemical Perception; Ornston, L.N.; Sligar, S.G., Eds,; Academic Press: New York, NY, USA, 1982; pp59-73.
- Xun, L.; Reeder, R.B.; Plymale, A.E.; Girvin, D.C.; Bolton, H. Degradation of metal-trinitriloacetate complexes by nitrilotriacetate monooxygenase. Environ. Sci. Technol., 1996, 30, 1752-1755. [CrossRef]
- Xu, Y.; Mortimer, M.W.; Fisher, T.S.; Kahn, M.L.; Brockman, F.J.; Xun, L. Cloning, sequencing, and analysis of a gene cluster from Chelatobacter heintzii ATCC 29600 encoding nitrilotriacetate monooxygenase and NADH:flavin mononucleotide oxidoreductase. J. Bacteriol., 1997, 179, 1112-1116. [CrossRef]
- Egli, T. Biodegradation of metal-complexing aminopolycarboxylic acids. J. Biosci. Bioeng., 2001, 92, 89-97.
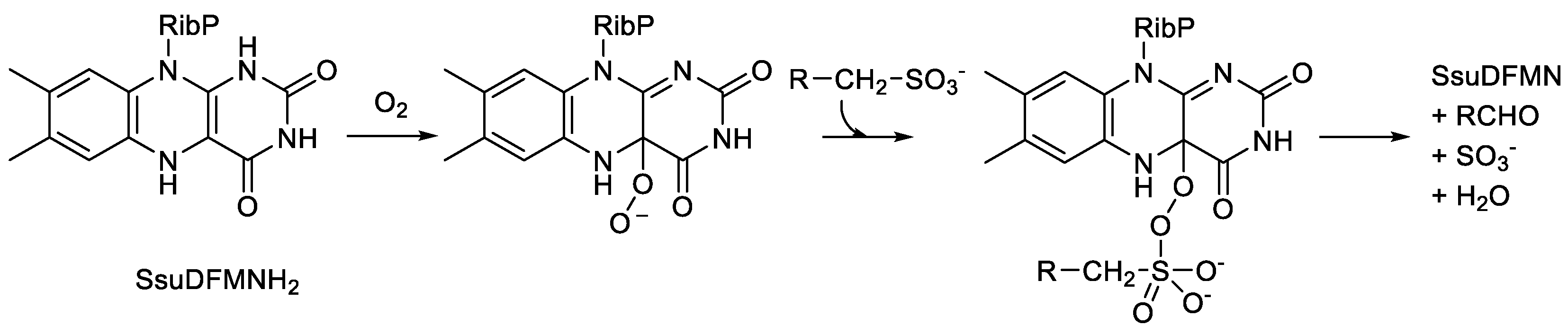
- van der Ploeg, J.R.; Ivanicka-Nowicka, R.; Bykowski, T.; Hryniewicz, M.M.; Leisinger, T. The Escherichia coli ssuEACB gene cluster is required for the utilisation of sulfur from aliphatic sulfonates and is regulated by the transcriptional factor cb1. J. Biol. Chem., 1999, 274, 29358-29365.
- Eichhorn, E.; van der Ploeg, J.R.; Leisinger, T. Deletion analysis of the Escherichia coli alkanesulfonate transport system. J. Bacteriol., 2000, 182, 2687-2695.
- van der Ploeg, J.R.; Eichhorn, E.; Leisinger, T. Sulfonate-sulfur metabolism and its regulation in Escherchia coli. Arch. Microbiol., 2001, 176, 1-8. [CrossRef]
- Payne, J.W.; Bolton, H.J.; Campbell, J.A.; Xun, L. Purification and characterization of EDTA monooxygenase from the EDTA-degrading bacterium BNS1. J. Bacteriol., 1998, 180, 3823-3827.
- Zhan, X.; Carpenter, R.A.; Ellis, H.R. Catalytic importance of the substrate binding order for the FMNH2-dependent alkanesulfonate monooxygenase enzyme. Biochem., 2008, 47, 2221-2230. [CrossRef]
- Kertesz, M.A.; Schmidt-Larbig, K.; Wuest, T. A novel reduced flavin mononucleotide-dependent methanesulfonate sulfonatase encoded by the sulfur-regulated msu operon of Pseudomonas aeruginosa. J. Bacteriol., 1999, 181, 1464-1473. [CrossRef]
- Liew, J.J.M.; El Saudi, I.M.; Nguyen, S.V.; Wicht, D.K.; Douling, D.P. Studies of the alkanesulfonate monooxygenase MsuD provide insights into C-S bond cleavage, substrate scope, and unexpected role for the tetramer. J. Biol. Chem., 2021, 297, 100823. [CrossRef]
- Prieto, M.A.; Garcia, J.L. Molecular characterization of 4-hydroxy- phenylacetate 3-hydroxylase of Escherichia coli: a two-protein component system. J. Biol. Chem., 1994, 269, 22823-22829.
- Prieto, M.A.; Diaz, E.; Garcia, J.L. Molecular characterization of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli W: engineering a mobile aromatic degradative cluster. J. Bacteriol., 1994, 176, 111-120. [CrossRef]
- Galan, B.; Diaz, E.; Prieto, M.A.; Garcia, J.L. Functional analysis of the small component of the 4-hydroxyphenylacetate 3-monooxygenase of Escherichia coli W: a prototype of a new flavin:NAD(P)H reductase family. J. Bacteriol., 2000, 182, 627-636. [CrossRef]
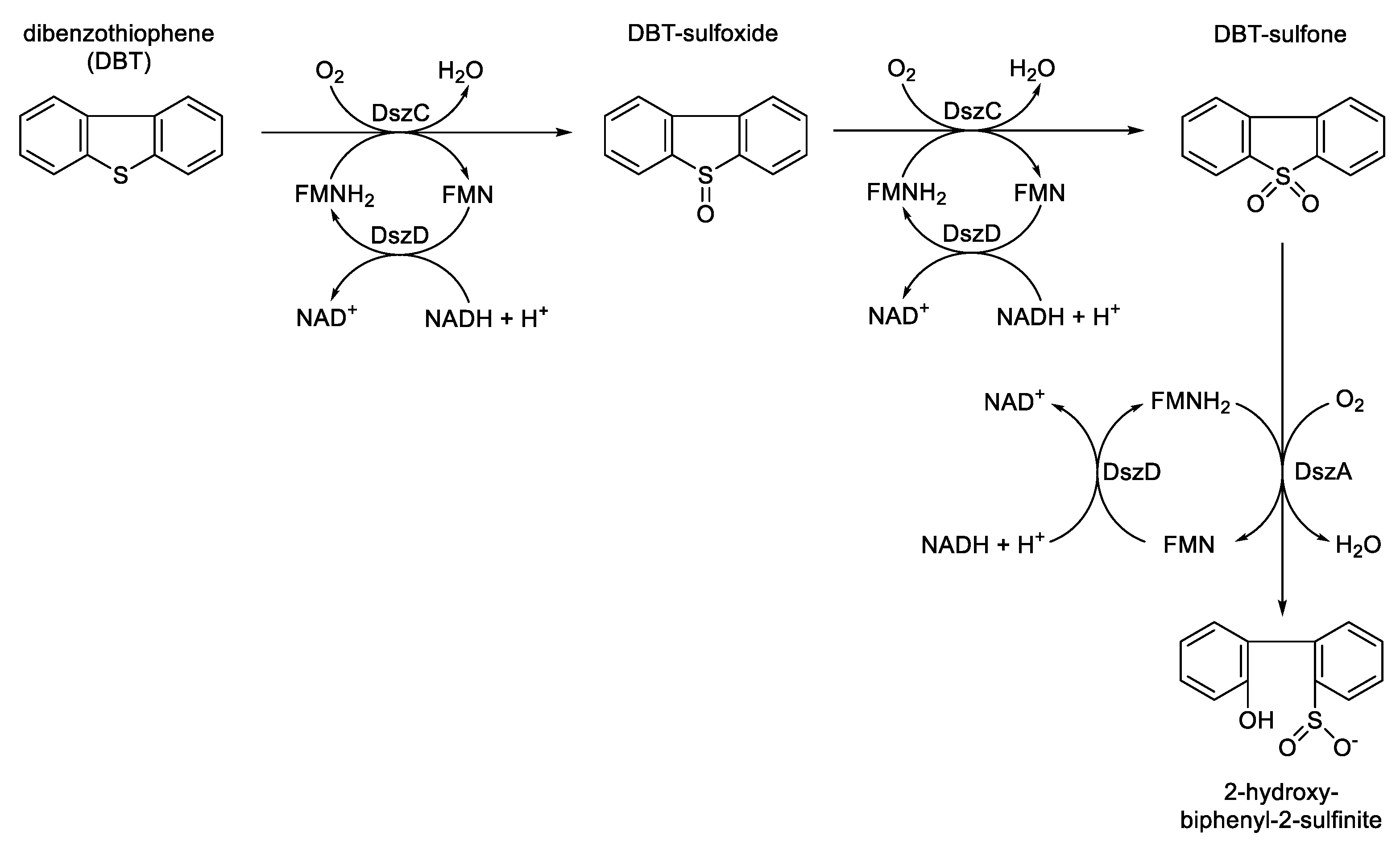
- Oldfield, C.; Pogrebinsky, O.; Simmonds, J.; Olsen, E.S.; Kulpa, C.F. Elucidation of the metabolic pathway for dibenzothiophene desulfurization by Rhodococcus sp. strain IGTS8 (ATCC 53968). Microbiology, 1997, 143, 2961-2973.
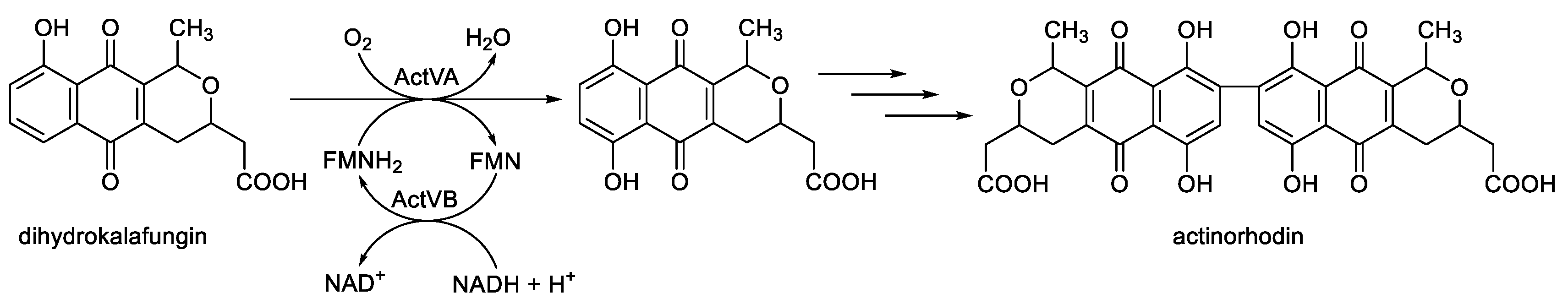
- Kendrew, S.G.; Hopwood, D.A.; Marsh, E.N.G. Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in the biosynthesis of actinorhodin: purification and characterization of the recombinant enzyme. J. Bacteriol., 1997, 179, 4305-4310. [CrossRef]
- Sciara, G.; Kendrew, S.G.; Miele, A.E.; Marsh, E.N.G.; Federici, L.; Malatesta, F.; Schimperna, G.; Savino, C.; Valione, B. The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin synthesis. EMBO J., 2003, 22, 205-215.
- Valton, J.; Filisetti, L.; Fontecave, M.; Niviere, V. A two-component flavin-dependent monooxygenase involved in actinorhodin biosynthesis in Streptomyces coelicolor. J. Biol. Chem., 2004, 279, 44362-44369. [CrossRef]
- Filisetti, L.; Valton, J,; Fontecave, M.; Niviere, V. The flavin reductase ActVB from Streptomyces coelicolor: characterization of the electron transfer activity of the flavoprotein form. FEBS Lett., 2005, 579, 2817-2820. [CrossRef]
- Valton, J.; Fontecave, M.; Douki, T. Kendrew, S.C.; Niviere, V. An aromatic hydroxylation reaction catalysed by a two-component FMN-dependent monooxygenase. The ActVA-ActVB system from Streptomyces coelicolor. J. Biol. Chem., 2006, 281, 27-35. [CrossRef]
- Valton, J.; Mathevon, C.; Fontecave, M.; Niviere, V.; Ballou, D.P. Mechanism and regulation of the two-component FMN-dependent monooxygenase ActVA-ActVB from Streptomyces coelicolor. J. Biol. Chem., 2008, 283, 10287-10296. [CrossRef]
- Tang, M-C.; Zou, Y.; Watanabe, K.; Walsh, C.T.; Tang, Y. Oxidative cyclization in natural product biosynthesis. Chem. Rev., 2017, 117, 5226-5333. [CrossRef]
- Tolmie, C.; Smit, M.S.; Opperman, J. Native roles of Baeyer-Villiger monooxygenases in microbial metabolism of natural products. Nat. Prod. Rev., 2017, 36, 326-353.
- Ichinose, K.; Bedford, D.J.; Tornus, D.; Bachthold, A.; Bibb, M.J.; Revill, W.P.; Floss, H.G.; Hopwood, D.A. The granaticin biosynthetic gene cluster of Streptomyces violaceoruber Tu22: sequence analysis and expression in a heterologous host. Chem. Bio., 1998, 6, 647-659. [CrossRef]
- Van Berkel, W.J.H.; Kamerbeck, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol., 2006, 4, 670-689. [CrossRef]
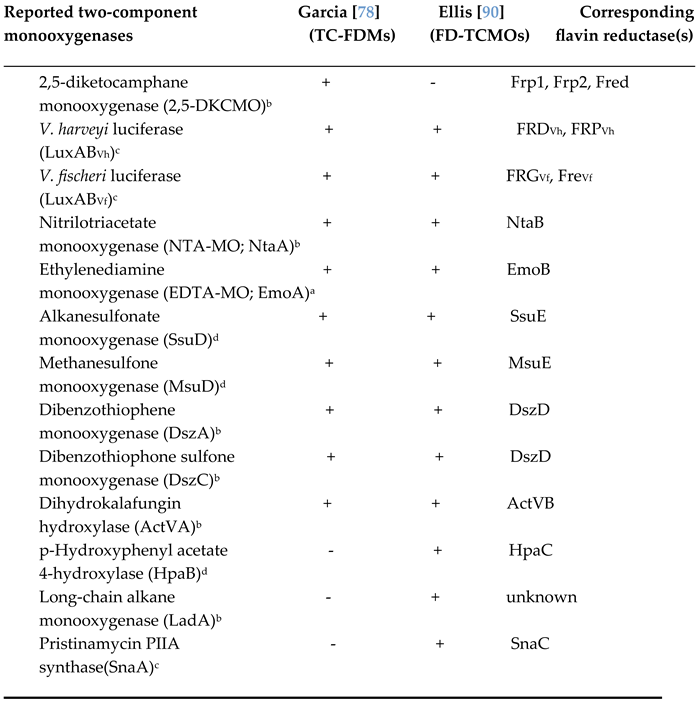
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys., 2010, 497, 1-12. [CrossRef]
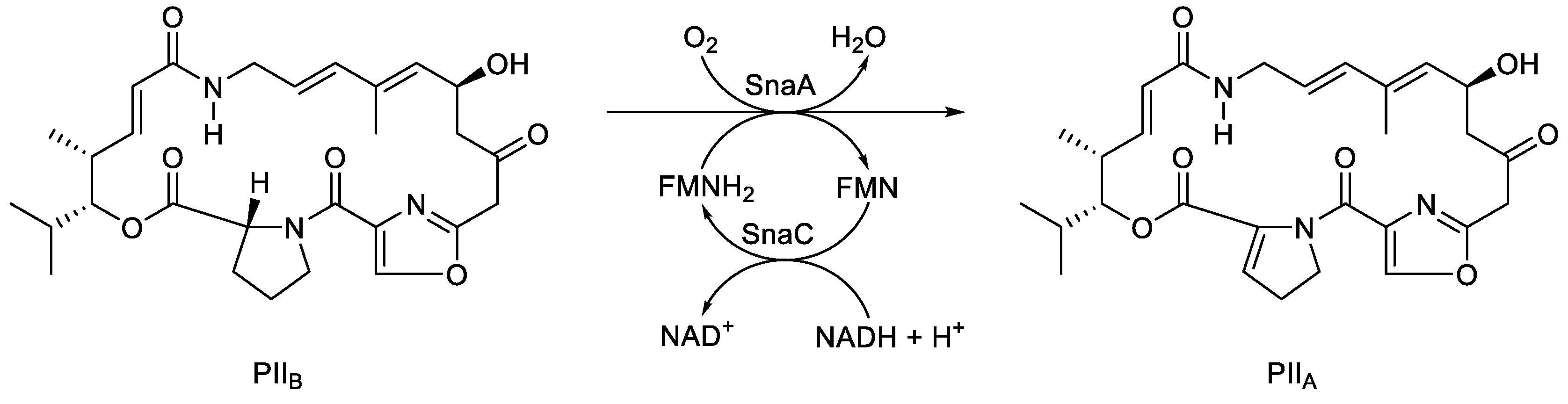
- Thibaut, D.; Ratet, N.; Bisch, D.; Faucher, D.; Debussche, L.; Blanche, F. Purification of the two-enzyme system catalysing the oxidation of the D-proline residue of pristinamycin IIB during the last step of pristinamycin IIA biosynthesis. J. Bacteriol., 1995, 177, 5199-5205. [CrossRef]
- Blanc, V.; Lagneaux, D.; Didier, P.; Gill, P.; Lacroix, P.; Crouzet, J. Cloning and analysis of structural genes from Streptomyces pristinaespirilis encoding enzymes involved in the conversion of pritinamycinIIB to pristinamycin IIA(PIIA): PIIA synthase and NADH: riboflavin 5’-phosphate oxidoreductase. J. Bacteriol., 1995, 177, 5206-5214. [CrossRef]
- Chaiyen, P.; Suadee, C.; Wilairat, P. A novel two-component flavoprotein hydroxylase p-hydroxyphenylacetate hydroxylase from Acinetobacter baumanii. Eur. J. Biochem., 2001, 268, 5550-5561. [CrossRef]
- Feng, L.; Wang, W.; Cheng, I.; Wang, L. Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans N980-2 isolated from a deep-subsurface oil reservoir. Proc. Natl. Acad. Sci. USA, 2007, 104, 5602-5607. [CrossRef]
- Li, L.; Liu, X.; Yang, W.; Xu, F.; Wang, W.; Feng, L.; Bartlam, L.; Wang, L.; Rao, Z. Crystal structure of long-chain alkane monooxygenase (LadA) in complex with coenzyme FMN: unveiling the long-chain alkane hydroxylase. J. Mol. Biol., 2008, 376, 453-465. [CrossRef]
- Abbasian, F.; Lockington, R.; Mallavarapu, M.; Naidu, R. A comprehensive review of aliphatic hydrocarbon biodegradability by bacteria. Appl. Biochem. Biotechnol., 2015, 176, 670-699. [CrossRef]
- Toplak, M.; Teufel, R. Three rings to rule them all: how versatile flavoproteins orchestrate the structural diversification of natural products. Biochem., 2022, 61, 47-56. [CrossRef]
- Doraghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K-S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A road map for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol., 2014, 10, 963-968.
- Hoskisson, P.A.; Seipke, R.A. Cryptic or silent? The known unknowns, unknown knowns, and unknown unknowns of secondary metabolism. Microbial Interactions, 2020, 11, 5. [CrossRef]
- Zapf, C.W.; Harrison, B.D.; Drahl, C.; Sorensen, E.J. A Diels-Alder macrocyclization enables an efficient asymmetric synthesis of the antibacterial natural product abyssomicin C. Angew. Chem. Int, Ed., 2005, 44, 6533-6537. [CrossRef]
- Keller, S.; Nicholson, G.; Drahl, C.; Sorensen, E.J.; Fiedler, H.P.; Sussmuth, R.D. Abyssomicins G and H and atrop-abyssomicin C from marine Verrucosispora strain Ass-18-032. J. Antibiot., 2007, 60, 391-394. [CrossRef]
- Maier, S.; Heitzler, T.; Asmus, K.; Brotz, K.; Hardter, U.; Hesselbach, K.; Paululat, T.; Bechthold, A. Functional characterization of different ORFs including luciferase-like monoxygenase genes from the mensacarcin gene cluster. ChemBioChem., 2015, 16, 1175-1182. [CrossRef]
- Colonna, S.; Gaggero, N.; Carrea, G.; Ottolina, G.; Pasta, P.; Zambianchi, F. First asymmetric epoxidation catalysed by cyclohexanone monooxygenase. Tet. Lett., 2002, 43, 1797-1799. [CrossRef]
- Mendelovici, M.; Glotter, E. Epoxidation and Baeyer-Villiger oxidation of γ-hydroxy-α,β-unsaturated ketones on exposure to m-chloroperbenzoic acid. J. Chem. Soc, Perkin Trans I, 1992, 1735-1749.
- Osumak, A.; Magolan, J.; Waynant, K.V. One-pot carbonyl reduction and carbonate formation using sodium borohydride in dialkyl carbonate solvents. Tet Lett., 2019, 60, 151203. [CrossRef]
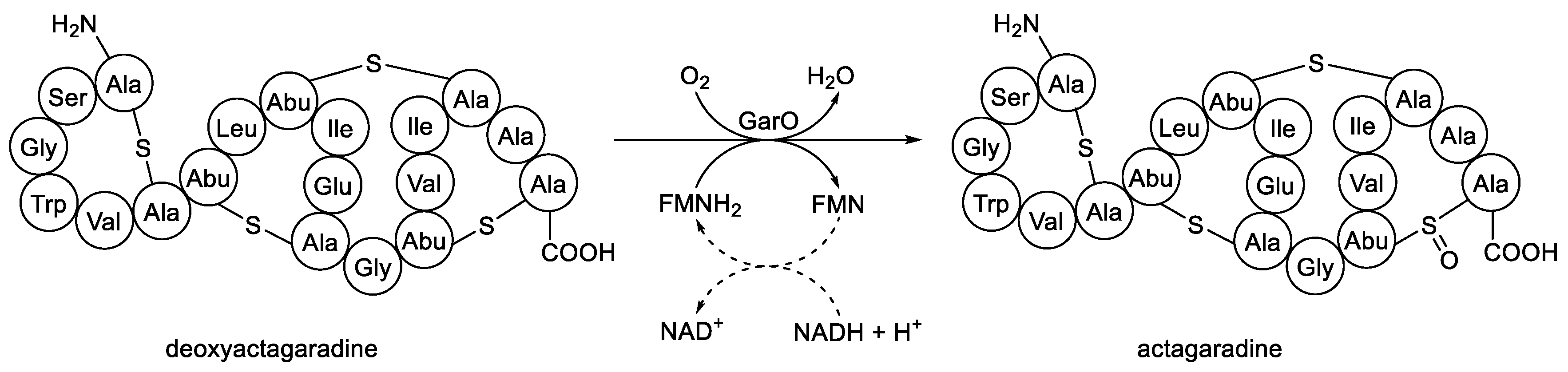
- Shi, Y.; Bueno, A.; van der Donk, W.A. Heterologous production of the lantibiotic Ala(0)actagardine in Escherichia coli. Chem. Commun., 2012, 48, 10966-10968. [CrossRef]
- Zimmermann, N.; Metzger, J.W.; Jung, G. The tetracyclic lantibiotic actagaradine 1H-NMR and 13C-NMR assignments and revised primary structure. Eur. J. Biochem., 1995, 228, 786-797.
- Dawson, M.W.; Appleyard, N.; Cortes-Bargallo, J.; Wadman, S.N. Actagaradine derivatives, and pharmaceutical uses thereof. 2011. WO/2011/095769.
- Shi, Y.; Yang, X.; Garg, N.; van der Donk, W.A. Production of lantipeptides in Escherichia coli. J. Amer. Chem. Soc., 2011, 133, 2338-2341.
- Okesli, A.; Cooper, L.E.; Fogle, E.J.; van der Donk, W.A. Nine post-translational modifications during biosynthesis of cinnamycin. J. Amer. Chem. Soc., 2011, 133, 13753-13760. [CrossRef]
- Garg, N.; Tang, W.; Goto, Y.; van der Donk, W.A. Lantibiotics from Geobacillus thermodenitrificans. Proc. Natl. Acad. Sci. USA, 2012, 109, 5241-5246. [CrossRef]
- Tang, W.; van der Donk, W.A. Structural characterization of four prochlorosins: a novel class of lantipeptides produced by planktonic marine cyanobacteria. Biochem., 2012, 51, 4271-4279.
- Campbell, Z.; Baldwin, T.O. Fre is the major flavin reductase supporting bioluminescence from Vibrio harveyi luciferase in Escherichia coli. J. Biol. Chem., 2009, 284, 8322-8328. [CrossRef]
- Kadow, M.; Sass, S.; Schmidt, M.; Bornscheuer, U.T. Recombinant expression and purification of the 2,5-diketocamphane 1,2-monooxygenase from the camphor-metabolising Pseudomonas strain NCIMB 10007. AMB Express, 2011, 1, 13. [CrossRef]
- Willetts, A. Inter-species redox coupling by flavin reductases and FMN-dependent two-componenent monooxygenases undertaking nucleophilic Baeyer-Villiger bioxygenations. Microorganisms, 2023, 11, 71. [CrossRef]
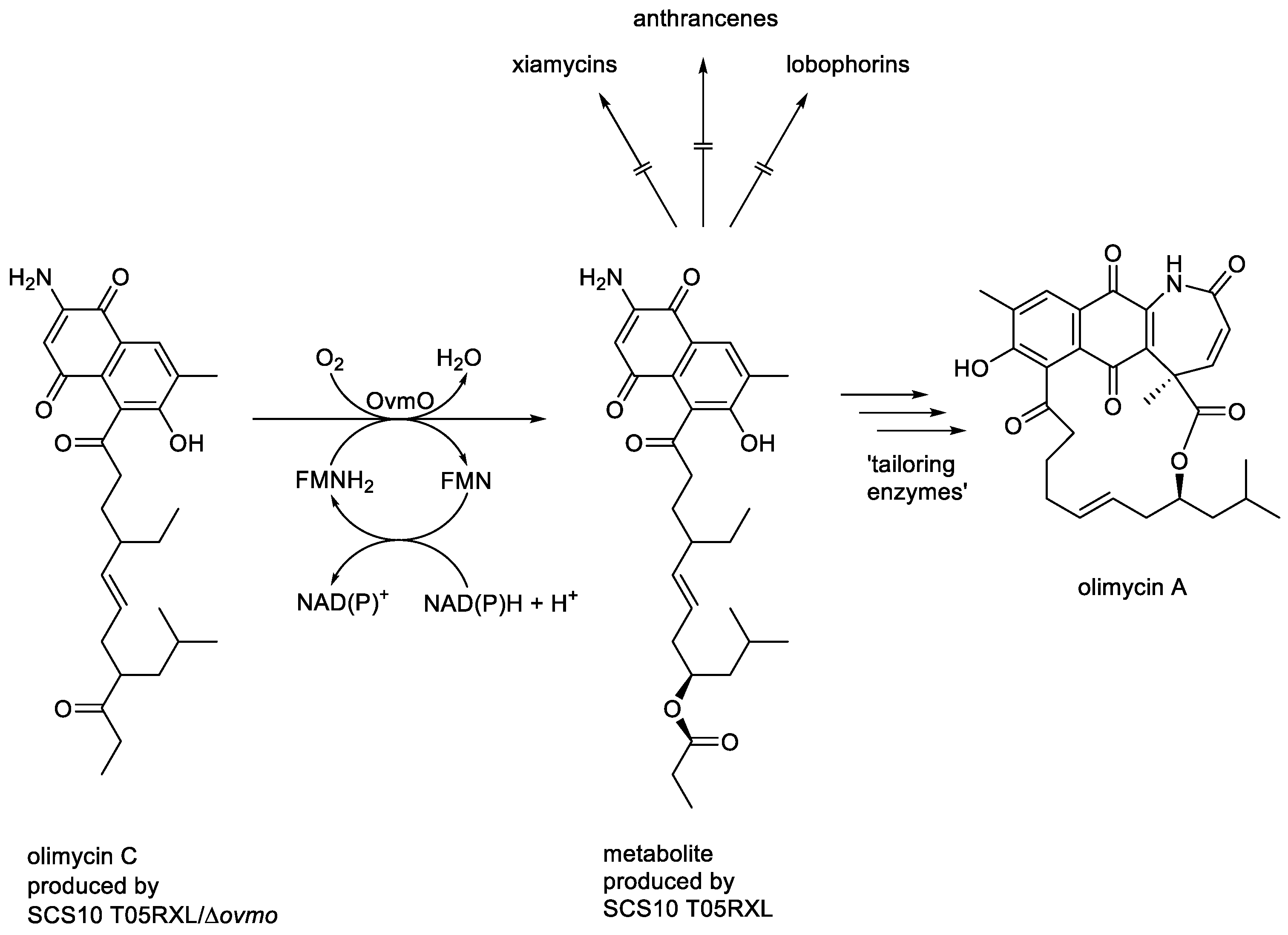
- Zhang, C.; Sun, C.; Huang, H.; Gui, C.; Wang, L.; Li, Q.; Ju, J. Biosynthetic Baeyer-Villiger chemistry enables access to two anthracene scaffolds from a single gene cluster in deep-sea-derived Streptomyces olivaceus SCS10 T05. J. Nat. Prod., 2018, 81, 1570-15 . [CrossRef]
- Zhang, C.; Zhang, H.; Ju, J. On-PKS Baeyer-Villiger-type O-atom insertion catalysed by luciferase-like monooxygenase Ovmo during olimycin biosynthesis. Org. Lett., 2022, 22, 1780-1784. [CrossRef]
- Dong, Y.; Enjing, J.; Wang, R.; Bao, Y.; Li, H. New olimycins from cold-seep-derived Streptomyces olivaceus. Chem. Biodiversity, 2023, 20. [CrossRef]
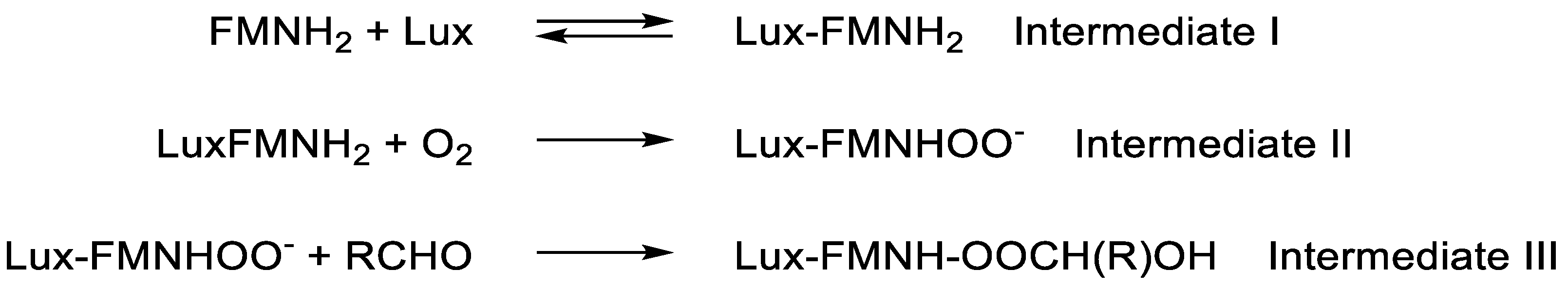


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



