1. Introduction
The genus
Staphylococcus (the
Micrococcaceae family) contains facultative anaerobic, Gram-positive, non-spore-forming bacteria that are widespread in soil and aquatic environments and are also components of the microbiota of the skin and mucous surfaces of humans and animals [
1]. Some of staphylococci can cause opportunistic infections in individuals with normal immune status and are a serious threat to immunocompromised patients [
2,
3]. One of the signs of staphylococcal pathogenicity is the ability to coagulate blood plasma. Depending on this ability, coagulase-positive staphylococci (CoPS) and coagulase-negative staphylococci (CoNS) are distinguished [
4].
To date, 70 species of the
Staphylococcus genus are known (
https://lpsn.dsmz.de/genus/staphylococcus, accessed on 21 September 2025), of which only 9 species (
S. aureus,
S. intermedius,
S. pseudintermedius,
S. delphini,
S. hyicus,
S. schleiferi subsp.
coagulans,
S. lutrae,
S. agnetis and
S. cornubiensis) belong to CoPS [
5]. Among CoNS, the species
S. epidermidis,
S. lugdunensis,
S. capitis,
S. caprae, and
S. saprophyticus are the most common pathogens causing nosocomial infections. Notably,
S. epidermidis,
S. capitis and
S. caprae belong to the same genetic cluster [
6] and have similar virulence factors involved in biofilm formation. These factors include the genes encoding teichoic acid biosynthesis, capsule genes, and other genes responsible for nonprotein adhesins biosynthesis [
7].
The bacterium
S. capitis is a commensal of human skin, primarily the skin of head. However, in neonatal intensive care units,
S. capitis is a serious clinical problem causing bacteremia and sepsis.
S. capitis can also cause catheter-associated bloodstream infections and endocarditis [
8,
9,
10]. Wang et al. (2022) showed that the
S. capitis population consists of five clones, among which a widespread multidrug-resistant clone has been identified [
11].
S. caprae, first isolated from goat milk, is able to colonize healthy skin and nasal mucosa of humans. Over the past decade, it has been shown that
S. caprae is capable of causing nosocomial infections, as most cases of
S. caprae infection were healthcare-associated and occurred in hospitals. [
7]. Reported cases of
S. caprae infection include bacteremia, bone and joint infections, urinary tract infections, endocarditis, and otitis [
12,
13,
14,
15]. A case of circulating methicillin-resistant
S. caprae in a neonatal intensive care unit has been described [
12].
Due to the growing number of drug-resistant bacterial strains, the amount of studies aimed at phage isolation and characterization has increased. Currently, 764 complete genomes of staphylococcal phages can be found in the NCBI database (
https://www.ncbi.nlm.nih.gov/nuccore, accessed on 10 September 2025). All of them belong to the class
Caudoviricetes and staphylococcal phages are divided into three groups: myovirus phages of the
Herelleviridae family (six genera), podovirus phages of the
Rountreeviridae family (two genera), and siphovirus phages (two subfamilies and 12 genera) [
16]. In addition, 92 staphylococcal phages (12%) are unclassified, among them several myovirus jumbo phages have been described [
17,
18]. Phages capable of infecting 23 distinct species within the
Staphylococcus genus have been identified. Among them, only six phages can infect
S. capitis and one phage IME1323_01 is specific to
S. caprae [
19].
In this study, a novel staphylococcal phage StaphC_127, capable of infecting both S. capitis and S. caprae strains, was isolated and characterized. Based on its genomic characteristics, this phage was classified as temperate siphovirus. Comparative analysis of the StaphC_127 genome allowed us to propose a new genus "Staphcevirus" and a new subfamily "Estebevirinae", which is closely related to several staphylococcal phages.
2. Materials and Methods
2.1. Bacterial Host Strain Identification
S. caprae strain CEMTC 1849 was isolated from the irritated surface of the ear skin. To isolate the strain, tenfold dilutions of the test sample were prepared in sterile phosphate-buffered saline (PBS) at pH 7.5. Then, 100 µL aliquots of the cell suspensions were spread on the surface of Mannitol salt agar (MSA) (Condalab, Madrid, Spain). Bacterial colonies of interest were transferred to nutrient agar (NA, Microgen, Makhachkala, Russia) and dispersed to obtain individual colonies. One of the colonies was subsequently reseeded onto new NA. This procedure was repeated three times to obtain a pure culture. To identify the bacterial species, sequencing of a 1300 base pair (bp) fragment of the 16S rRNA gene and a 751 bp fragment of the
rpoB gene was performed. Primers 16s-8-f-B 5′-AGRGTTTGATCCTGGCTCA-3′ and 16s-1350-r-B 5′-GACGGGGGGCGGTGTGTGTGTGTACAAG-3′, as well as 2491F 5’-AACCAATTCCGTATIGGTTT-3’ and 3241R 5’-GCIACITGITCCATACCTGT-3’, respectively, were used for PCR and sequencing, as described previously [
20,
21]. Sequencing reactions were carried out using BigDye Terminator v.3.1 (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Capillary electrophoresis of the reaction products was performed using an ABI 3500 genetic analyzer (Applied Biosystems, Foster City, CA, USA) and the obtained nucleotide sequences were compared to the appropriate sequences from the NCBI GenBank database (
https://www.ncbi.nlm.nih.gov, accessed on 28 April 2025). The bacterial strain was deposited as
S. caprae CEMTC 1849 in the Collection of Extremophilic Microorganisms and Type Cultures of the Institute of Chemical Biology and Fundamental Medicine of the Siberian Branch of the Russian Academy of Sciences (CEMTC, ICBFM SB RAS), Novosibirsk, Russia.
2.2. Phage Isolation and Propagation
Bacteriophage StaphC_127 was isolated from a sample obtained by washing the surface of a spoiled tomato. The swab sample was suspended in 1 mL PBS (pH 7.5) and then filtered through a 0.22 μm membrane filter (Wuxi Nest Biotechnology, Wuxi, China). Phage plaques were detected by applying a 10 μL aliquot of the filtrate to a freshly prepared lawn of S. caprae strain CEMTC 1849 grown in top agar (Becton, Dickinson and Company, Sparks, Difco, Laboratories, Franklin Lakes, NJ, USA). Plates containing the culture were incubated for 24 hours at 37 °C. Agar fragments containing phage plaques were excised, suspended in sterile PBS, and incubated overnight to eluate phage particles. The next day, tenfold dilutions of the phage-containing eluate were applied to a fresh layer of S. caprae CEMTC 1849 culture to obtain individual plaques. These plaques were used for subsequent phage isolation and the dilution and extraction cycle was repeated three times.
To propagate the phage,
S. caprae CEMTC 1849 was grown in 20 mL of lysogenic broth (LB) (BD Difco, Franklin Lakes, NJ, USA) to an optical density (OD
595) of 0.4. Next, phage particles were added to the exponentially growing
S. caprae CEMTC 1849 in an amount corresponding to a multiplicity of infection (MOI) of 0.1, and the infected culture was incubated with shaking at a temperature of 37 °C until the bacteria were lysed. The resulting bacterial lysate was centrifuged at 6000×
g for 30 minutes. Phage particles were precipitated from the lysate by adding 0.4 volume of solution containing 30% polyethylene glycol 6000 (AppliChem, Darmstadt, Germany) and 2.5 M sodium chloride. The procedure was performed as described previously [
22]. The resulting pellet containing phage particles was resuspended in 500 µL sterile PBS. The obtained phage was deposited in the CEMTC, ICBFM SB RAS.
2.3. Phage Plaques and Phage Particles Morphology
To determine the morphology of StaphC_127 plaques, the standard method on double-layer agar was used [
23]. Plaques of the phage were obtained after overnight incubation at 37 °C of plates with a lawn of the host strain
S. caprae CEMTC 1849 infected with StaphC_127. In addition, sterile filtrate with a phage titer of at least 10
8 plaque-forming units per mL (pfu/mL) was prepared for electron microscopy. Phage particle morphology was determined using transmission electron microscopy on a JEM 1400 microscope (JEOL, Japan) with preliminary negative staining with 1% uranyl acetate; digital images were collected using a side-mounted Veleta digital camera (Olympus Soft Imaging Solutions GmbH, Germany).
2.4. Host Range Analysis of the Phage StaphC_127
The host range was tested on 294 strains of
Staphylococcus spp., including 22 CoNS species:
S. arlettae (1 strain),
S. auricularis (7 str.),
S. borealis (15 str.),
S. capitis (6 str.),
S. caprae (5 str.),
S. carnosus (2 str.),
S. casei (2 str.),
S. coagulans (8 str.),
S. cohnii (8 str.),
S. devriesei (6 str.),
S. epidermidis (32 str.),
S. equorum (13 str.),
S. felis (6 str.),
S. haemolyticus (46 str.),
S. hominis (30 str.),
S. lugdunensis (4 str.),
S. pasteuri (10 str.),
S. saprophyticus (5 str.),
S. simulans (9 str.),
S. succinus (4 str.),
S. taiwanensis (1 str.),
S. warneri (30 str.), and
S. xylosus (2 str.), as well as two species of CoPS –
S. aureus (6 str.) and
S. pseudintermedius (36 str.). All strains were obtained from the CEMTC, ICBFM SB RAS and cultivated in Nutrient broth (Thermo Fisher Scientific, Waltham, MA, USA) and plated onto NA (Microgen, Obolensk, Russia). Notably, all the strains of a particular species used were isolated from different sources at different times. The susceptibility of staphylococcal strains to antibiotics was tested using the disk diffusion method (OXOID, Basingstoke, UK) in accordance with the EUCAST recommendations (
https://www.eucast.org, accessed: 25 June 2025). The following antibiotics were used (abbreviations and the amount in the disk, μg, are given in brackets): amoxicillin/clavulanic acid (AMC, 20/10); cefoxitin (FOX, 30); benzylpenicillin (P0, 6); oxacillin (OX, 1); ampicillin (AM, 10); levofloxacin (LEV, 5); erythromycin (E, 15); clindamycin (DA, 2); linezolid (LZD, 10); tetracycline (TE, 30); gentamicin (CN, 10); ciprofloxacin (CIP, 5); tobramycin (TOB, 10).
The spot test described previously was used [
24]. Fresh bacterial lawns were prepared by mixing 100 µL of the bacterial culture with top agar. Tenfold dilutions of the phage preparation were then applied to the lawn culture and allowed to dry. After overnight incubation at 37 °C, the plates were recorded for the presence/absence of plaques. The sensitivity of bacterial strains to phages was assessed by the relative efficiency of plating (EOP). The EOP value was calculated as the ratio of the phage titer on the test strain to the phage titer on the host strain. EOP values >0.5 were ranked as ‘high’ efficiency; 0.2–0.5 as ‘medium’ efficiency; 0.001–0.2 as ‘low’ efficiency [
25].
2.5. Induction of Prophages
To induce prophages in the host strain and susceptible strains, an exponentially growing culture of Staphylococcus spp. (the OD600 value of 0.4) was treated with mitomycin C, 0.5 μg/mL (Sigma-Aldrich, St. Louis, MO, USA) and incubated with shaking overnight at 37 °C. The OD600 value was measured every hour to record growth or possible lysis. In addition, fresh layers of S. caprae CEMTC 1849 and susceptible strains grown on top agar were exposed to ultraviolet irradiation for 5, 10, and 15 s. Then, plates were incubated for 42 h at 37 °C and the appearance of plaques was regularly checked. The assays were carried out with three replicates.
To estimate whether StaphC_127 DNA can integrate into the genome of susceptible
S. caprae and
S. capitis strains, phage particles and bacterial cells were co-cultured to generate bacteriophage insensitive mutants (BIMs). Experiments were performed as described previously, with some modifications [
26]. Briefly, 5 mL of certain bacterial culture (10
8 cfu/mL) was mixed with the corresponding volume of phage lysate at an MOI of 1. After 24 hours of incubation at 37 °C, 10 µL of the mixture was plated on MSA plates. All grown bacterial colonies were tested for sensitivity to StaphC_127. To purify the BIM from residual phage particles, they were passaged five times on MSA plates and then used to detect the genomic DNA of StaphC_127.
After sequencing of the complete genome, phage DNA screening was performed using the primers StaphC_127_F 5′-GAACCTATTCACGTTATGGGC-3′ and StaphC_127_R 5′-GGATTGGTTAACTTCGATTCCG-3′ (amplify a 560 bp fragment of the ORF38). PCR protocol consisted of 30 cycles of amplification, with each cycle including denaturation (95 °C, 30 s), annealing (55 °C, 30 s), and elongation (72 °C, 1 min). DNA of StaphC_127 was used as positive control.
2.6. Biological Properties of StaphC_127
All experiments were carried out in three biological replicates with three repeats per each experiment. Statistical data processing and graphing were performed using GraphPad Prism 8.0.1 software. To generate graphs, means and standard deviations were calculated. All experiments were conducted as described previously [
23] with minor modifications.
To evaluate phage adsorption, exponentially growing host strain cells were infected with the phage (105 pfu/mL) and the infected culture was incubated with shaking at 37 °C for 30 min. Aliquots were taken every minute to determine the titer of free phages on the host strain. The obtained plates were incubated for 24 hours at 37 °C. After 24 hours, plaques were counted at each time point.
To determine the latent period and burst size, 10 mL of exponentially growing bacteria were centrifuged at 6000× g for 5 minutes. The supernatant was then removed, and the pellet was resuspended in 0.5 mL of LB, followed by the addition of StaphC_127 at an MOI of 0.001. The infected bacterial culture was incubated with shaking at 37 °C for 60 min; aliquots were taken every 3 min and used to determine the phage titer. The latent period was defined as the time interval between phage adsorption to the host cell surface and the release of new viral particles. Phage yield was calculated as the ratio of the titer of released phage particles to the residual phage titer during the latent period.
The lytic properties of bacteriophages were observed by adding the phage to an exponentially growing bacterial culture at an MOI of 1. The infected culture was incubated at 37 °C at 200 rpm, and aliquots were taken every 30 min for 6 hours to determine the bacterial titer. The next day, bacterial colonies were counted and a multi-step culture lysis curve was constructed based on the data obtained.
2.7. Phage DNA Purification and Complete Genome Sequencing
To isolate phage DNA, 0.1 volume of 10×DNase buffer, DNase, and RNase (Thermo Fisher Scientific, Waltham, MA, USA) were added to phage suspensions (400 μL) to a final concentration of 5 μg/mL. The mixture was incubated for 30 min at 37 °C. Then, 1/25 volume of EDTA (pH 8.0) and 1/20 volume of 10% SDS were added to the suspension, as well as proteinase K (Thermo Fisher Scientific, Waltham, MA, USA to a final concentration of 150 μg/mL. The solution was incubated for three hours at 55 °C.
Phage DNA was purified using protein extraction with phenol and chloroform. An equal volume of phenol was added to the suspension, mixed, and centrifuged at 10,000× g for 5 minutes. The upper liquid fraction was collected in a fresh tube, an equal volume of chloroform was added, mixed, and centrifuged at 10,000× g for 5 minutes. Phage DNA was precipitated using 95% ethanol supplemented with 1/30 volume of 3 M sodium acetate (pH 5.1). The mixture was kept at –20 °C for 24 hours and then centrifuged at 14,000× g for 20 minutes, the supernatant was removed, and the precipitate was dried and then dissolved in 50 µL of distilled water.
A paired-end sequencing library was prepared using NEBNext Ultra II DNA Library Prep Kit for Illumina and Multiplex Oligos for Illumina Dual Index Primers Set 1 (New England Biolabs, Inc., Ipswich, MA, USA) for further sequencing on the MiSeq sequencer with a MiSeq reagent kit v.2 (500 cycles) (Illumina, San Diego, CA, USA). To check the quality of the obtained sequencing data and remove adapter sequences, the Trimmomatic 0.32 tool was used (parameters: SLIDINGWINDOW:10:20, ILLUMINACLIP:2:30:10, TRAILING:20, LEADING:20) [
27]. The phage genome de novo assembly was performed using SPAdes genome assembler v.3.15.2 (
http://cab.spbu.ru/software/spades, accessed on 20 August 2025) with standard parameters [
28]. The StaphC_127 genome sequence was deposited in the NCBI GenBank database (accession number PX570736). The complete StaphC_127 genome is applied (Data S1).
2.8. Annotation and Characterization of Phage Genomes
The Rapid Automated Annotation Service (RAST) v.2.0 program (
https://rast.nmpdr.org, accessed on 25 September 2025) was used to search for open reading frames (ORFs) [
29]. The annotation was then manually verified using BLASTX searches against sequences deposited in the NCBI GenBank database (
https://ncbi.nlm.nih.gov, accessed on 25 September 2025). In addition, InterProScan and HHpred tools were used to identify protein functions [
30,
31]. The search for virulence factors and antibiotic resistance genes was performed using the Virulence Factor database (
http://www.mgc.ac.cn/VFs, accessed on 30 September 2025) [
32] and Antibiotic Resistance Gene database (
https://card.mcmaster.ca/analyze/rgi, accessed on 30 September 2025) [
33], respectively. Phage genome termini and DNA packaging strategy were determined using the PhageTerm v.1.0.12 tool [
34]. SnapGene Viewer (GSL Biotech; available at
https://www.snapgene.com/; accessed on 01 October 2025) was used to generate a genomic map. Phage lifestyle analysis was performed using the PhageAI web service (
https://app.phage.ai/, accessed on 02 October 2025).
2.9. Comparative Analysis of Phage Genomes and Phage Proteins
Nucleotide identity between the complete genomes was calculated in the BioEdit 7.2.5 program [
35] based on the alignment, which was performed in the MAFFT [
36] program (
https://mafft.cbrc.jp accessed on 03 September 2025). The starting points of the analyzed sequences were manually changed in accordance with the StaphC_127 sequence based on dot-plot analysis in the UGENE program [
37].
To assess phage taxonomy, a comparative proteomic phylogenetic analysis was performed using the Viral Proteome Tree Server (ViPTree) (
https://www.genome.jp/viptree, accessed on 3 October 2025) [
38]. Intergenomic similarity (SG) was calculated using the Virus Intergenomic Distance Calculator (VIRIDIC) [
39]. To search for the core genes, the CoreGenes5.0 web server (
https://coregenes.ngrok.io/; accessed 04 October 2025) [
40] was used with the Bidirectional Best Hit option with an E value of 1e-5. The amino acid sequences of interest were searched against the NCBI GenBank database using BLASTP and BLASTX (
https://blast.ncbi.nlm.nih.gov/, accessed on 04 October 2025) and retrieved for further analysis. Protein sequences were aligned and phylogenetic analysis was performed using the MEGA XII program [
41].
3. Results
3.1. Identification of the Host
The host strain S. caprae CEMTC 1849 was isolated from the surface of the ear skin with irritation and it was the only bacterial strain growing under aerobic conditions from this isolation sample. This strain was checked for the presence of the lysogenic phages using mitomycin C and ultraviolet irradiation. In both cases, plaques were not observed indicating the absence of functional prophages in the S. caprae CEMTC 1849 genome.
3.2. Phage StaphC_127 Plaque and Virion Morphology
Phage StaphC_127 formed translucent plaques with a diameter of 1 mm on a lawn of the host strain
S. caprae CEMTC 1849 (
Figure 1A). Electron microscopy revealed a capsid 60,7 ± 0.9 nm in diameter, connected to a long, non-contractile tail 252.3 ± 13.4 nm long. The morphology of phage particles corresponded to that of siphovirus (
Figure 1B).
3.3. Host Range of the Phage StaphC_127
To determine the host range of StaphC_127, the phage was tested on 294
Staphylococcus spp. strains, including two species of CoPS (
S. aureus and
S. pseudintermedius, n = 42 in total) and 22 species of CoNS (n = 252 str.). It was shown that StaphC_127 was able to infect three of six tested strains of
S. capitis and four of five tested of
S. caprae strains, including the host strain (
Table 1). All susceptible strains of both
S. capitis and
S. caprae were isolated from clinical samples collected from the skin surface. One them,
S. capitis CEMTC 10051, exhibited resistance to beta-lactam antibiotics. According to the relative efficiency of plating (EOP), which indicates the ratio of the phage titer on the tested bacterial strain to the phage titer on the host strain, StaphC_127 was able to effectively infect strains of both
S. capitis and
S. caprae. Other CoNS strains, including all tested
S. epidermidis, were not sensitive to this phage.
To check whether the genome of these strains contain functional prophages, ultraviolet irradiation and mitomycin C were used. Prophages were not induced as plaques were not observed in all strains.
3.4. Biological Properties of StaphC_127
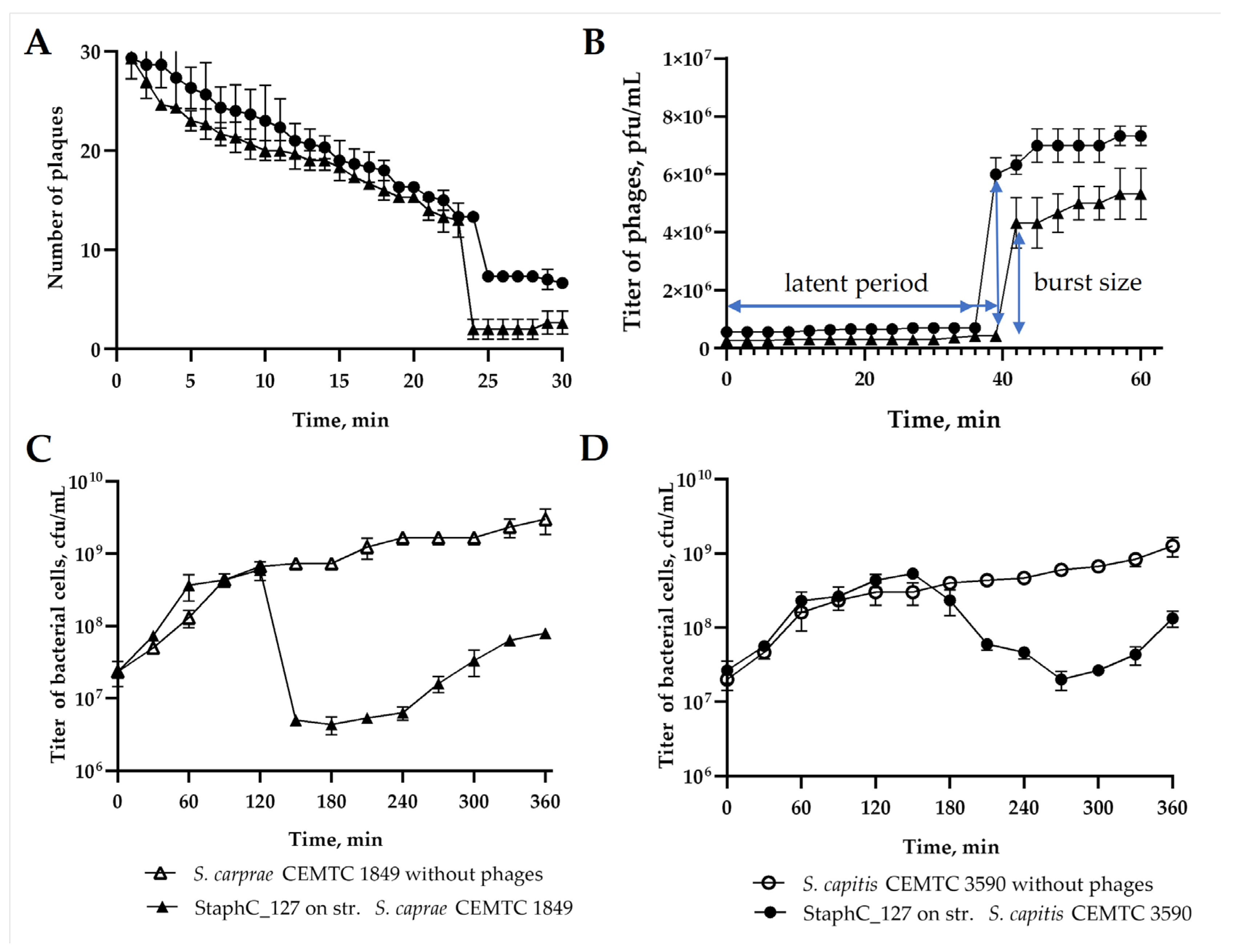
The biological properties of StaphC_127 were investigated using the
S. caprae CEMTC 1849 and
S. capitis CEMTC 3590 strains. The maximum attachment of phage particles was recorded at 24 and 26 minutes in the adsorption experiments, respectively (
Figure 2A). The adsorption rate constant was calculated according to Kropinsky [
23] and it was found as 1.3 × 10
-8 mL/min and 1.4 × 10
-9 mL/min for
S. caprae CEMTC 1849 and
S. capitis CEMTC 3590, respectively. The latent period of the phage was 36 and 39 minutes, respectively. After the latent period, phage particle release from host cells was observed, and the burst size was approximately 15 phage particles per infected cell for
S. caprae CEMTC 1849 and 44 phage particles per infected cell for
S. capitis CEMTC 3590 (
Figure 2B). The lytic activity experiment was performed by adding StaphC_127 to cells with an MOI of 1. When infected with
S. caprae CEMTC 1849, the number of viable host cells decreased by two orders of magnitude, starting at 150 minutes (
Figure 2C). After infection of
S. capitis CEMTC 3590 with the StaphC_127 phage, a decrease in bacterial cell titer was observed after 180 minutes of the experiment (
Figure 2D). At 240 minutes for
S. caprae CEMTC 1849 and 300 minutes for
S. capitis CEMTC 3590, phage-resistant bacterial cells were identified, which continued to accumulate until the end of the experiment. The established biological characteristics indicated low lytic activity of the StaphC_127 phage.
3.5. Annotation and Characterization of the StaphC_127 Genome
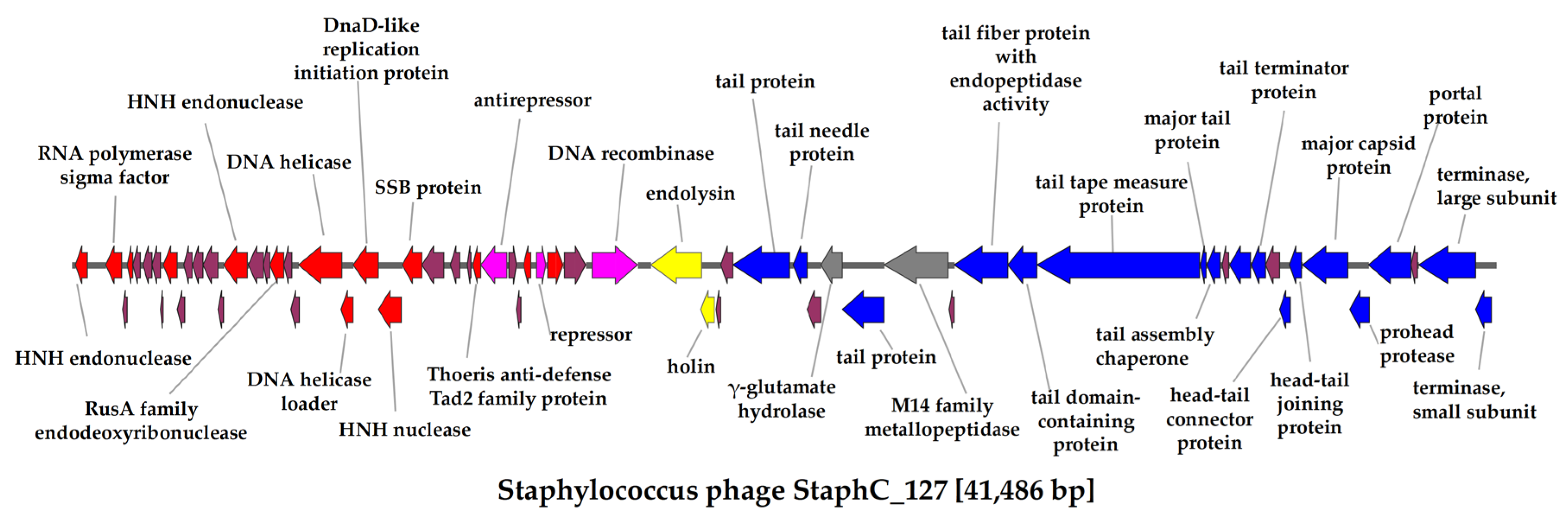
The length of the StaphC_127 complete genome was 41,486 bp with a GC content of 33.92%. The StaphC_127 genome contained 65 ORFs; of them, 38 ORFs encoded proteins with predicted functions, and the remaining 27 ORFs had unspecified functions (
Figure 3). The list of annotated ORFs with functions verified using BLASTX InterProScan, and HHpred tools is presented in
Table S1).
The StaphC_127 genome has 14 ORFs encoding proteins involved in nucleic acid metabolism (
Figure 3). No genes of DNA- and RNA polymerases or DNA primase were identified, indicating that this phage uses host enzymes for replication and transcription. Like other temperate phages [
42], the StaphC_127 genome contains the genes encoding the antirepressor (gp30) and repressor (gp34) of transcription, which play a key role in switching the life cycle of phages from lysogenic to lytic. In addition, this genome encodes the site-specific DNA recombinase (gp37) and does not contain the tRNA genes.
Seventeen ORFs in the StaphC_127 genome encodes proteins responsible for virion assembly and DNA packaging (
Figure 3). Among them, the tape measure protein (gp51), a tail protein with endopeptidase activity (gp49), major capsid protein (gp60), portal protein (gp62), large and small subunits of terminase (gp64 and gp65) were identified (
Figure 3). Two lytic cassette proteins are encoded by the StaphC_127 genome: the endolysin with N-acetylmuramoyl-L-alanine amidase activity (gp38) and holin (gp39).
Notably, the gene encoding the Tad2 protein (gp29), which inhibits the Thoeris antiphage defense system, was found in the StaphC_127 genome. BLAST search indicated that the amino acid sequence of gp29 was similar (the identity ranged from 35% to 94%) to the Tad2-family proteins encoded by the Staphylococcus genomes. The genes responsible for the production of toxins or conferring resistance to antibiotics were not identified in the StaphC_127 genome.
Using the PhageTerm tool, it was determined that the ends of the StaphC_127 genome are cohesive terminal sites (cos) 16 bp long. In the cos-type of genome packaging, segments of the phage genome in concatemers are separated by a specific nucleotide sequence called cos. Usually, phage terminase recognizes the first of these cos sequences and binds to it, packaging the viral DNA into an empty capsid until the next cos sequence is encountered. At this point, it cuts the DNA molecule, triggering the process of closing the capsid filled with a single segment of the phage genome [
43].
3.6. Lifestyle Determination of StaphC_127
The absence of prophages relative to StaphC_127 within the genomes of the host and other susceptible strains was confirmed using PCR with primers specific to the ORF38 of the phage. Then, it was determined whether the StaphC_127 genome can integrate into the genome of the sensitive bacterium. DNA of the obtained bacteriophage-insensitive mutants (BIMs) (ten clones from each susceptible strain) were checked for the presence of the StaphC_127 genome. The obtained data indicated that integration of the StaphC_127 genome was not detected in the host strain S. caprae CEMTC 1849 and four other susceptible strains. However, DNA of BIMs obtained from S. caprae CEMTC 2739 and S. caprae CEMTC 3411 contained the nucleotide sequence of the StaphC_127 ORF38. Presumably, the StaphC_127 genome integrated into the bacterial chromosome. As for the host strain S. caprae CEMTC 1849 and four other susceptible strains, the antiphage mechanisms of these five strains probably prevent the phage from entering the lysogenic life cycle. Therefore, it is possible to assume that StaphC_127 is a temperate phage that was additionally confirmed by the results of a bioinformatic analysis using the PhageAI program, which predicted the temperate life cycle of StaphC_127 with a probability of 98.48%.
3.7. Comparative Analysis of StaphC_127 and Estimated Taxonomy
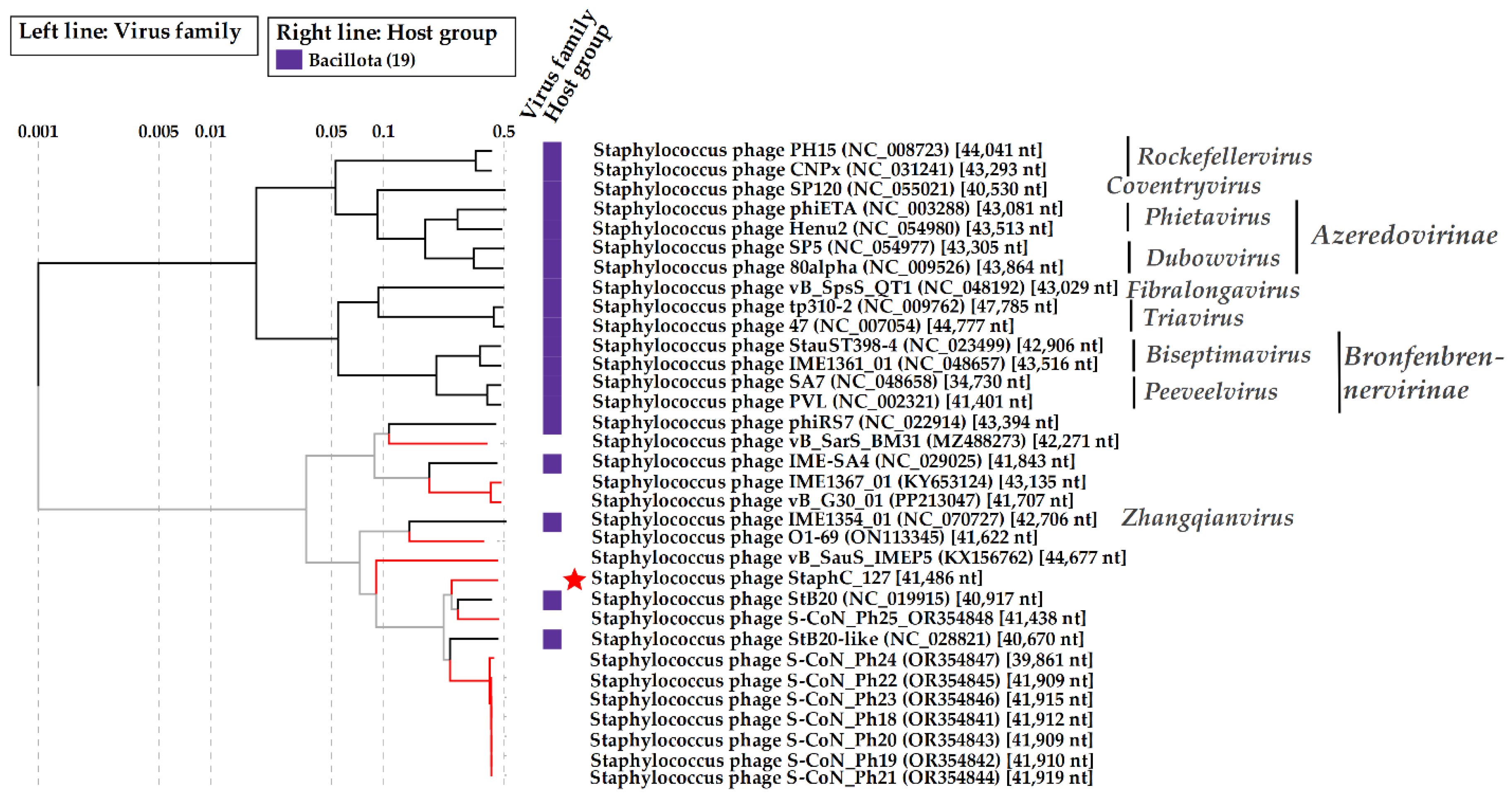
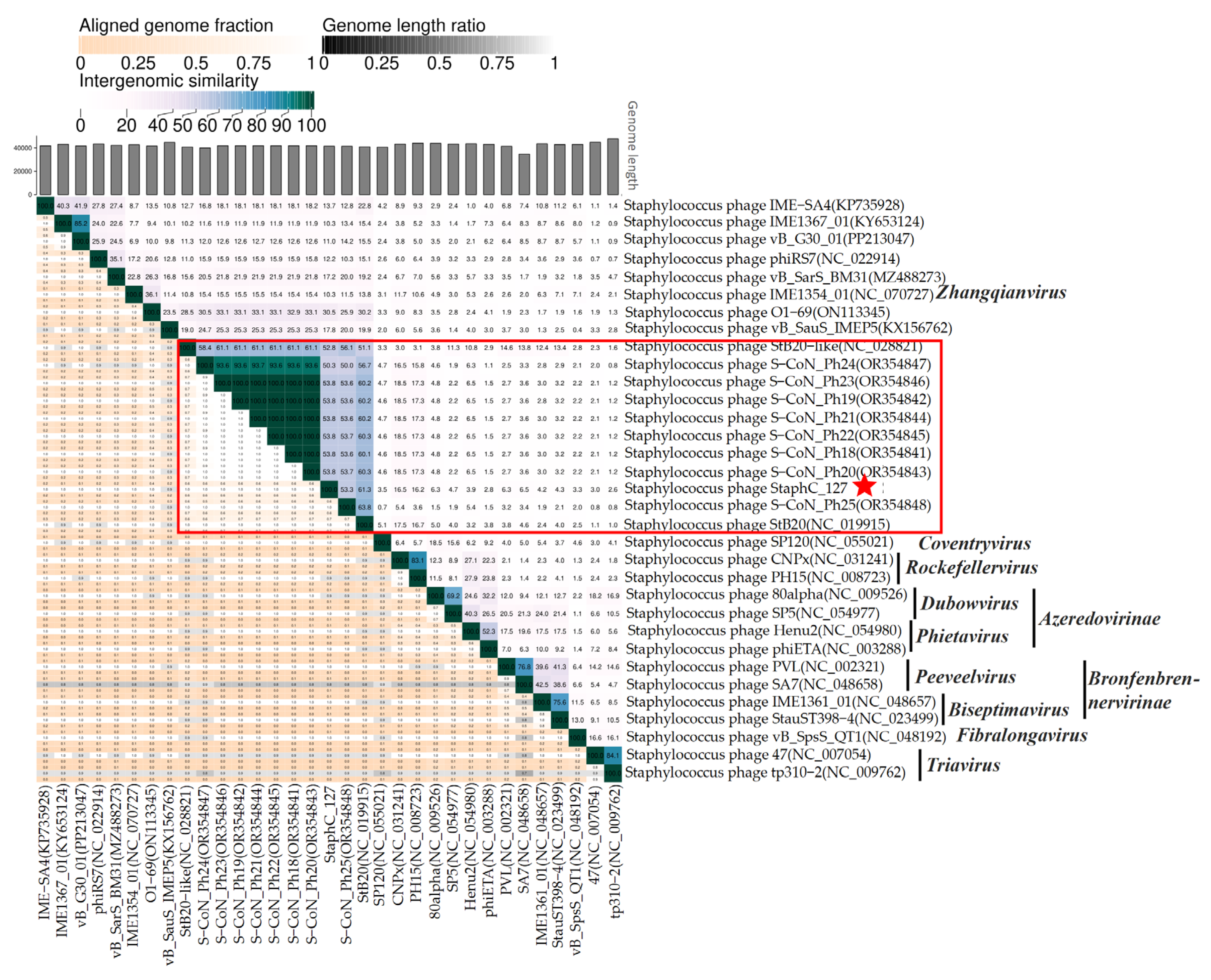
The StaphC_127 genome was compared with available phage sequences extracted from the NCBI GenBank database using BLASTN search. StaphC_127 exhibited some similarity to prophage sequences in the
S. epidermidis and
S. capitis genomes. Among the phage sequences, 18 similar genomes were found using the BLASTN search. Comparative ViPTree analysis indicated that StaphC_127, along with 18 staphylococcal phages, formed a monophyletic group containing two branches and distinct from other
Staphylococcus phages (
Figure 4).
Of the phages from this monophyletic group, only the
Staphylococcus phage IME1354_01 (NC_070727) was previously classified as a member of the
Zhangqianvirus genus [
44]; the remaining 17 phages had no definite taxonomic position. The phage IME1354_01 was in the branch with StaphC_127. The calculated nucleotide identity (NI) of the StaphC_127 genome with those of the closest phages (
Staphylococcus phages StB20 and S-CoN_Ph25) was 68.8% and 60.7%, respectively. NI of the StaphC_127 genome with other genomes from the branch containing StaphC_127 was lower and varied from 40.7% to 57.9% (
Staphylococcus phages IME1354_01 and StB20-like, respectively).
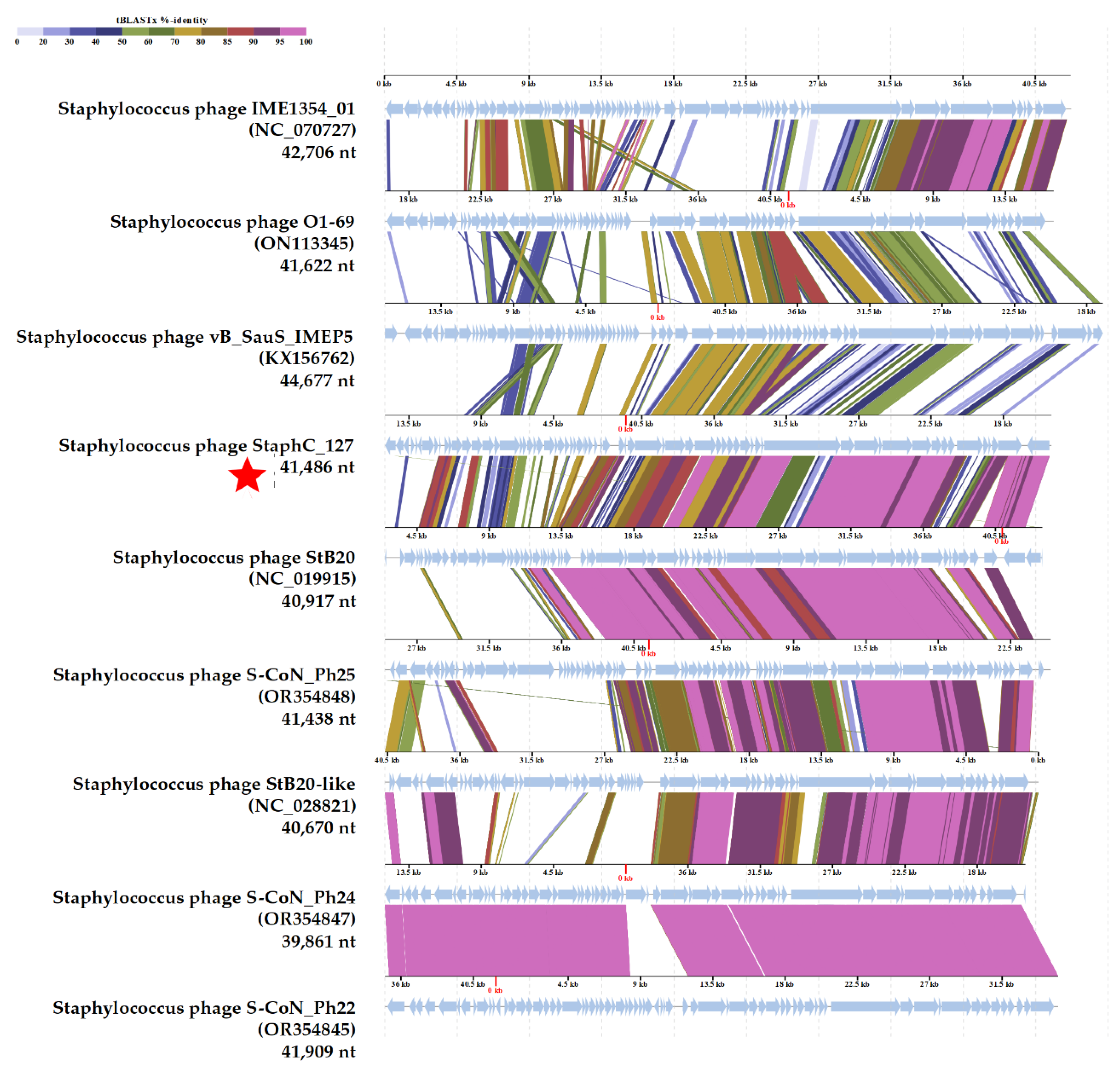
Pairwise comparisons of the StaphC_127 genome with nine genomes of phages from this branch (
Staphylococcus phages S-CoN_Ph24 and S-CoN_Ph22 represented a group of closely related phages) was carried out using the VipTree program (
Figure 5). Obviously, the StaphC_127 genome showed greater similarity to the genomes of the
Staphylococcus phages StB20, S-CoN_Ph25, StB-like, S-CoN_Ph24, and S-CoN_Ph22 than with other phages from this branch. The identity levels of the structural genes were on average higher than those of the genes responsible for the metabolism of nucleic sequences (
Figure 5).
In addition, the intergenomic similarity (SG) levels between StaphC_127 and
Staphylococcus siphoviruses shown in
Figure 4 were calculated using VIRIDIC (
Figure 6). Notably, StaphC_127 and a group of the above relative phages (
Staphylococcus phages StB20, S-CoN_Ph25, StB20-like, S-CoN_Ph24, and S-CoN_Ph22) showed SG levels of more than 50%, which was substantially more than with other tested
Staphylococcus siphoviruses. Moreover, the staphylococcal phages S-CoN_Ph18 – S-CoN_Ph23 probably belong to the same species.
Based on the obtained data and considering previous recommendations [
45], it can be assumed that StaphC_127 represent a new putative genus, which is proposed to name "
Staphcevirus". Furthermore, StaphC_127 and a number of relative phages might be combined into a new putative subfamily "
Estebevirinae" after the first described phage from this group,
Staphylococcus phages StB20 [
46]. Importantly, CoreGenes 5.0 identified 23 core proteins for 11 phages belonging to the proposed subfamily. Of these 23 core proteins, 14 are structural proteins and DNA packaging proteins, two lytic cassette proteins (holin and endolysin), the repressor, poly-gamma-glutamate hydrolase, and five hypothetical proteins. In an attempt to identify core proteins for the monophyletic group of 19 staphylococcal phages, including StaphC_127 and IME1354_01 (the
Zhangqianvirus genus) [
44] only two proteins were revealed: the terminase large subunit and the tail length tape measure protein.
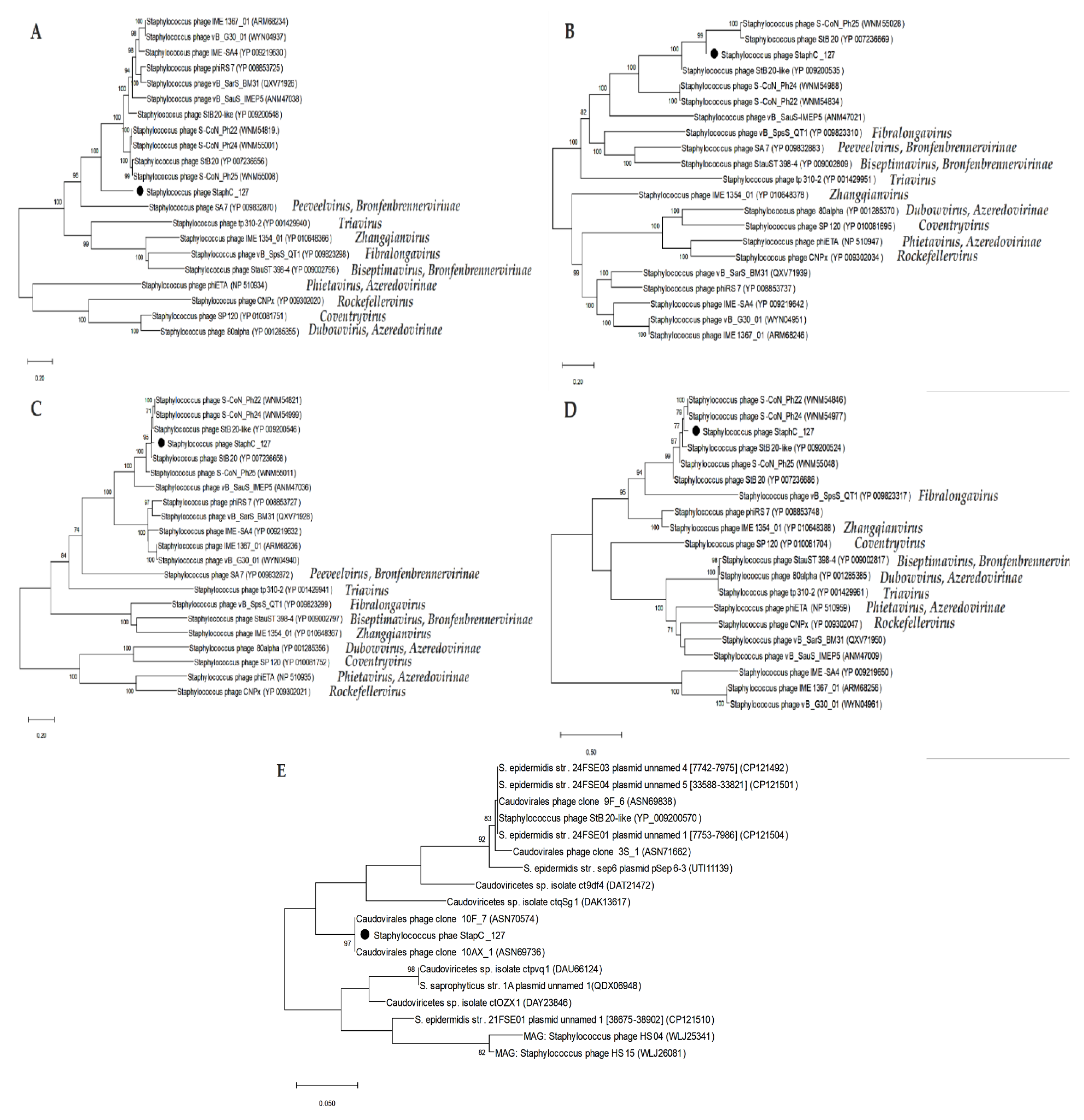
3.8. Phylogenetic Analysis of StaphC_127 Proteins
Based on the CoreGenes results, the amino acid sequences of four signature proteins of the putative subfamily
Estebevirinae (terminase large subunit, portal protein, tail length tape measure protein, and endolysin) were used for phylogenetic analysis (
Figure 7). Notably, the topology of the constructed phylogenetic trees does not demonstrate good coincidence with the ViPTree topology indicating the evidence of mosaicism inherent in staphylococcal siphoviruses. However, the appropriate sequences of all phages from the putative
Estebevirinae subfamily formed monophyletic clades in all constructed trees (
Figure 7).
As for the Tad2 protein sequences, 17 similar ones were found among the viral sequences of both individual and metagenome-associated phage genomes (
Figure 7E). Notably, two identical sequences were extracted from the metagenome-associated genomes (MAGs), whereas only one genome from the propose
Estebevirinae subfamily (
Staphylococcus phages StB20-like) encodes relative Tad2 protein.
4. Discussion
StaphC_127 is currently the second described phage that infect
S. caprae. This phage, like the first described phage against
S. caprae, IME1323_01 [
19], belongs to the siphoviruses; however, IME1323_01 is a member of the
Azeredovirinae subfamily [
19], whereas StaphC_127 is distant from IME1323_01 and probably represents a new putative genus
Staphcevirus within the new undefined subfamily that might be named
Estebevirinae after one of the first described phage from this branch,
Staphylococcus phages StB20 [
46].
Previously, a group of
Staphylococcus siphoviruses infecting only CoNS was identified: StB20 was specific to
S. capitis [
46], S-CoN_Ph25 was able to infect
S. capitis,
S. epidermidis,
S. haemolyticus,
S. hominis,
S. lugdunensis, and
S. warneri [
47], while each of the eight closely related phages S-CoN_Ph18 – S-CoN_Ph24 were active able to infect several different species of bacteria listed above [
47]. The studied phage StaphC_127, the first one active against both
S. caprae and
S. capitis, is a member of this group. However,
Staphylococcus phages IME1354_1 [
44] and vB_SauS [
48] that are specific to CoPS
S aureus form the monophyletic branch with the aforesaid phages infecting only CoNS. Given that the GC content of the StaphC_127 genome (33.9%) is close to that of
S. caprae (33,6%) and
S. capitis (33%) [
7,
49], it is possible to assume a long coevolution of the phage with sensitive CoNS strains.
Various studies of staphylococcal phages indicated that the phages infecting these bacteria and having the siphovirus morphotype are temperate and belong to different taxonomic groups –
Azeredovirinae,
Bronfenbrennervirinae,
Coventryvirus,
Fibralongavirus,
Rockefellervirus,
Sextaecvirus,
Triavirus [
50,
51,
52,
53,
54,
55]. It has been shown that many staphylococcal siphoviruses are able to integrate into the bacterial genome and carry the toxin and virulence genes [
56,
57,
58,
59]. All staphylococcal siphoviruses exhibit a high degree of mosaicism; comparison at the amino acid sequence level allows the identification of homologous proteins found in distantly related phages [
46]. This fact indicates that all staphylococcal siphoviruses probably originated from a common ancestor and over time diverged to such an extent that similarity at the nucleotide sequence level has become low. Such mosaicism is inherent in tailed phages and genetic exchange can involve both individual genes and entire gene clusters resulting in the formation of fully functional phages that differ significantly in genome organization and biological properties from their putative parental phages [
60]. Apparently, "local" phage groups frequently exchange genes with other members of the group that can explain, among other things, the low level of identity of the nucleic acid metabolism genes in phages included in the putative
Estebevirinae subfamily.
One of the features of the StaphC_127 phage is the presence of a gene encoding the Tad2 protein, which is a product of the anti-Thoeris phage genome system. In bacteria, the Thoeris defense system triggers an abortive infection (Abi) upon detection of a phage. The Thoeris system was found in more than 2000 bacterial and archaeal genomes and consists of one copy of the
thsA gene and one or several copies of the
thsB genes. The
thsA genes encodes an effector protein that triggers Abi, whereas
thsB encode proteins responsible for phage detection. In turn, Tad2 acts as a "sponge" that binds immune signaling molecules produced by the
thsB gene [
61]. Notably, only 17 Tad2 sequences similar to Tad2 of StaphC_127 were found to encode by viral genomes, both phage and MAGs, and only one sequence belonged to the
Staphylococcus phages StB20, a member of the putative
Estebevirinae subfamily. In addition, two sequences identical to the Tad2 protein of StaphC_127 were identified in two MAGs. The SG matrix calculated for phage genomes relative similar Tad2 (
Figure S1) showed that SGs between the StaphC_127 genome and two MAGs with the identical Tad2-genes are 75.6% and 79.5% that clearly indicated that the MAGs, like StaphC_127, are probable members of the proposed
Staphcevirus genus.
StaphC_127 is possibly a temperate phage. This assumption is supported by the presence of the genes encoding the repressor, antirepressor, and site-specific DNA recombinase in its genomes. In addition, the StaphC_127 genome was detected in BIMs obtained from two
S. caprae strains after StaphC_127 infection. Certainly, further genome sequencing of these BIMs is required to confirm the assumption and indicate the location of the prophages in the BIM genomes. In addition, genome sequencing of
S. caprae CEMTC 2739 and
S. caprae CEMTC 3411 strains could indicate the differences in their anti-phage defense systems. Previously, integration of phage genomes into the bacterial genome has been proven for several closely related phages [
44,
62,
63].
Since the significant increase in antimicrobial resistance, phage therapy has attracted increasing attention. Phages are an affordable and effective treatment option for infections. However, the use of temperate phages or phages with integrase/recombinase in phage therapy is limited due to complications caused by lysogeny that can be accompanied by uncontrolled transduction of virulence genes or host antibiotic resistance. Direct use of lytic cassette proteins may become an alternative to the use of whole bacteriophages. Given that StaphC_127 is capable of lysing both
S. caprae and
S. capitis, its endolysin could have extended specificity, which certainly requires verification. In addition, options have been considered recently for using temperate phages together with stressors (antibiotics, mitomycin C), which promote the switching of phages to the lytic cycle, thereby causing a synergistic effect of the phage-stressor action on bacterial cells [
64,
65].
5. Conclusion
In this study, we characterized a novel Staphylococcus phage StaphC_127 capable of infecting both S. caprae and S. capitis. This phage exhibited low lytic activity against its host strain S. caprae CEMTC 1849 and another strain S. capitis CEMTC 3590, moderate host range against the tested S. caprae and S. capitis strains, and was possibly able to integrate its genome into the chromosome of two S. caprae strains (among seven susceptible CoNS strains). Taking into consideration the obtained results of the genome analysis, we can assume that StaphC_127 is the first member of a new supposed Staphcevirus genus that, in turn, is part of a new putative subfamily Estebevirinae.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Data S1: The complete nucleotide sequence of StaphC_127 in fasta format; Figure S1: SG matrix for the StaphC_127 phage Tad2-related genomes prepared using VIRIDIC; Table S1: StaphC_127 genome annotation prepared using RAST tool.
Author Contributions
Conceptualization, V.I.Y. and J.I.M.; methodology, V.I.Y. and I.V.B.; validation, A.Y.T. and V.A.F.; formal analysis, I.V.B.; investigation, Y.N.K. and A.V.B.; resources, N.V.T.; data curation, E.V.Z.; writing—original draft preparation, V.I.Y.; writing—review and editing, V.V.M and N.V.T.; visualization, V.I.Y.; supervision, N.V.T.; project administration, N.V.T.; funding acquisition, N.V.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (project 25-64-00030). This study was supported by the Russian state-funded project for ICBFM SB RAS, grant No. 125012300671-8.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The StaphC_127 genome sequence was deposited in the ICBI GenBank database accession number PX570736.
Acknowledgments
The authors express their gratitude to technical specialist Kaverina Galina for her helpful assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bello, S.; Mudassir, S.H.; Rudra, B.; Gupta, R.S. Phylogenomic and molecular markers based studies on Staphylococcaceae and Gemella species. Proposals for an emended family Staphylococcaceae and three new families (Abyssicoccaceae fam. nov., Salinicoccaceae fam. nov. and Gemellaceae fam. nov.) harboring four new genera, Lacicoccus gen. nov., Macrococcoides gen. nov., Gemelliphila gen. nov., and Phocicoccus gen. nov. Antonie van Leeuwenhoek 2023, 116, 937–973. [Google Scholar] [CrossRef] [PubMed]
- Noshak, M.A.; Rezaee, M.A.; Hasani, A.; Mirzaii, M. The Role of the Coagulase-negative Staphylococci (CoNS) in Infective Endocarditis; A Narrative Review from 2000 to 2020. Current pharmaceutical biotechnology 2020, 21, 1140–1153. [Google Scholar] [CrossRef]
- Ye, Y.; Tian, Y.; Kong, Y.; Ma, J.; Shi, G. Trends of Antimicrobial Susceptibility in Clinically Significant Coagulase-Negative Staphylococci Isolated from Cerebrospinal Fluid Cultures in Neurosurgical Adults: a Nine-Year Analysis. Microbiology spectrum 2022, 10, e0146221. [Google Scholar] [CrossRef]
- Argemi, X; Hansmann, Y; Prola, K; Prevost, G. Coagulase-Negative Staphylococci Pathogenomics. International journal of molecular sciences 2019, 20, 1215. [Google Scholar] [CrossRef]
- Gonzalez-Martin, M.; Corbera, J.A.; Suarez-Bonnet, A.; Tejedor-Junco, M.T. Virulence factors in coagulase-positive staphylococci of veterinary interest other than Staphylococcus aureus. The veterinary quarterly 2020, 40, 118–131. [Google Scholar] [CrossRef]
- Lamers, R.P.; Muthukrishnan, G.; Castoe, T.A.; Tafur, S.; Cole, A.M.; Parkinson, C.L. Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC evolutionary biology 2012, 12, 171. [Google Scholar] [CrossRef]
- Watanabe, S.; Aiba, Y.; Tan, X.E.; Li, F.Y.; Boonsiri, T.; Thitiananpakorn, K.; Cui, B.; Sato'o, Y.; Kiga, K.; Sasahara, T.; Cui, L. Complete genome sequencing of three human clinical isolates of Staphylococcus caprae reveals virulence factors similar to those of S. epidermidis and S. capitis. BMC Genomics 2018, 19, 810. [Google Scholar] [CrossRef]
- Decalonne, M.; Dos Santos, S.; Gimenes, R.; Goube, F.; Abadie, G.; Aberrane, S.; Ambrogi, V.; Baron, R.; Barthelemy, P.; Bauvin, I.; et al. Staphylococcus capitis isolated from bloodstream infections: a nationwide 3-month survey in 38 neonatal intensive care units. European journal of clinical microbiology and infectious diseases: official publication of the European Society of Clinical Microbiology 2020, 39, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Al Hennawi, H.E.T.; Mahdi, E.M.; Memish, Z.A. Native valve Staphylococcus capitis infective endocarditis: a mini review. Infection 2020, 48, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Pivetti, V.; Vittorini, A.; Viggiano, C.; Castoldi, F.; Fabiano, V.; Lista, G.; Cavigioli, F. Staphylococcus capitis Central-Line-Associated Bloodstream Infections in the Neonatal Intensive Care Unit: A Single-Center, Four-Year Experience in Central-Line Management during Sepsis Treatment. Pathogens 2024, 13, 234. [Google Scholar] [CrossRef]
- Wang, Z.; Gu, C.; Sun, L.; Zhao, F.; Fu, Y.; Di, L.; Zhang, J.; Zhuang, H.; Jiang, S.; Wang, H.; Zhu, F.; Chen, Y.; Chen, M.; Ling, X.; Chen, Y.; Yu, Y. Development of a novel core genome MLST scheme for tracing multidrug resistant Staphylococcus capitis. Nature communications 2022, 13, 4254. [Google Scholar] [CrossRef]
- Ross, T.L.; Fuss, E.P.; Harrington, S.M.; Cai, M.; Perl, T.M.; Merz, W.G. Methicillin-resistant Staphylococcus caprae in a neonatal intensive care unit. Journal of clinical microbiology 2005, 43, 363–7. [Google Scholar] [CrossRef]
- Seng, P.; Barbe, M.; Pinelli, P.O.; Gouriet, F.; Drancourt, M.; Minebois, A.; Cellier, N.; Lechiche, C.; Asencio, G.; Lavigne, J.P.; Sotto, A.; Stein, A. Staphylococcus caprae bone and joint infections: a re-emerging infection? Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 2014, 20, O1052–O1058. [Google Scholar] [CrossRef] [PubMed]
- Gowda, A.; Pensiero, A.L.; Packer, C.D. Staphylococcus caprae: A Skin Commensal with Pathogenic Potential. Cureus 2018, 10, e3485. [Google Scholar] [CrossRef] [PubMed]
- Diez de Los Rios, J.; Hernandez-Meneses, M.; Navarro, M.; Montserrat, S.; Perissinotti, A.; Miro, J.M. Staphylococcus caprae: an emerging pathogen related to infective endocarditis. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 2023, 29, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.S.; Silva, M.D.; Azeredo, J.; Melo, L.D. Coagulase-Negative Staphylococci phages panorama: Genomic diversity and in vitro studies for a therapeutic use. Microbiological research 2025, 290, 127944. [Google Scholar] [CrossRef]
- Uchiyama, J.; Takemura-Uchiyama, I.; Sakaguchi, Y.; Gamoh, K.; Kato, S.; Daibata, M.; Ujihara, T.; Misawa, N.; Matsuzaki, S. Intragenus generalized transduction in Staphylococcus spp. by a novel giant phage. The ISME journal 2014, 8, 1949–1952. [Google Scholar] [CrossRef]
- da Silva Duarte, V.; Treu, L.; Sartori, C.; Dias, R.S.; da Silva Paes, I.; Vieira, M.S.; Santana, G.R.; Marcondes, M.I.; Giacomini, A.; Corich, V.; Campanaro, S.; da Silva, C.C.; de Paula, S.O. Milk microbial composition of Brazilian dairy cows entering the dry period and genomic comparison between Staphylococcus aureus strains susceptible to the bacteriophage vB_SauM-UFV_DC4. Scientific reports 2020, 10, 5520. [Google Scholar] [CrossRef]
- Tian, F.; Li, J.; Li, F.; Tong, Y. Characteristics and genome analysis of a novel bacteriophage IME1323_01, the first temperate bacteriophage induced from Staphylococcus caprae. Virus research 2021, 305, 198569. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef]
- Drancourt, M.; Raoult, D. rpoB gene sequence-based identification of Staphylococcus species. Journal of clinical microbiology 2002, 40, 1333–8. [Google Scholar] [CrossRef]
- Bacteriophage λ and its vectors. In Molecular cloning; Sambrook, J, Russell, D, Eds.; Cold Spring Harbour Laboratory Press, 2001; Volume 1, pp. 187–303. [Google Scholar]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods in molecular biology 2009, 501, 69–76. [Google Scholar] [CrossRef]
- Kutter, E.; Clokie, M.R.; Kropinski, A.M. Phage host range and efficiency of plating. In Bacteriophages. Methods in Molecular Biology; Humana Press: Totowa, New Jersey, USA, 2009; Volume 501, pp. 141–149. [Google Scholar] [CrossRef]
- Fanaei, P.R.; Wagemans, J.; Kunisch, F.; Lavigne, R.; Trampuz, A.; Gonzalez, M.M. Novel Stenotrophomonas maltophilia bacteriophage as potential therapeutic agent. Pharmaceutics 2022, 14, 2216. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Vandenheuvel, D.; Kropinski, A.M.; Mast, J.; De Vos, D.; Verbeken, G.; Noben, J.P.; Lavigne, R.; Vaneechoutte, M.; Pirnay, J.P. Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS ONE 2014, 9, e104853. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Current protocols in bioinformatics 2020, 70, e102. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Xia, F.; Stevens, R. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic acids research 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic acids research 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids research 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A Database for Classification of Antimicrobial Drug, Biocide and Metal Resistance Determinants in Metagenomic Sequence Data. Nucleic acids research 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a New Bioinformatic Tool To Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrobial agents and chemotherapy 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Scientific reports 2017, 7, 8292. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in bioinformatics 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Davis, P.; Seto, D.; Mahadevan, P. CoreGenes5.0: An Updated User-Friendly Webserver for the Determination of Core Genes from Sets of Viral and Bacterial Genomes. Viruses 2022, 14, 2534. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Molecular biology and evolution 2024, 41, msae263. [Google Scholar] [CrossRef]
- Węgrzyn, G.; Licznerska, K.; Węgrzyn, A. Phage λ--new insights into regulatory circuits. Advances in virus research 2012, 82, 155–178. [Google Scholar] [CrossRef]
- Marcelli, B.; Karsens, H.; Nijland, M.; Oudshoorn, R.; Kuipers, O.P.; Kok, J. Employing lytic phage-mediated horizontal gene transfer in Lactococcus lactis. PLoS ONE 2020, 15, e0238988. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Li, J.; Li, L.; Li, F.; Tong, Y. Molecular dissection of the first Staphylococcus cohnii temperate phage IME1354_01. Virus research 2022, 318, 198812. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Deghorain, M.; Bobay, L.M.; Smeesters, P.R.; Bousbata, S.; Vermeersch, M.; Perez-Morga, D.; Dreze, P.A.; Rocha, E.P.; Touchon, M.; Van Melderen, L. Characterization of novel phages isolated in coagulase-negative staphylococci reveals evolutionary relationships with Staphylococcus aureus phages. Journal of bacteriology 2012, 194, 5829–39. [Google Scholar] [CrossRef]
- Alsaadi, S.E.; Lu, H.; Zhang, M.; Dykes, G.F.; Allison, H.E.; Horsburgh, M.J. Bacteriophages from human skin infecting coagulase-negative Staphylococcus: diversity, novelty and host resistance. Scientific reports 2024, 14, 8245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xing, S.; Sun, Q.; Pei, G.; Cheng, S.; Liu, Y.; An, X.; Zhang, X.; Qu, Y.; Tong, Y. Characterization and complete genome sequence analysis of a novel virulent Siphoviridae phage against Staphylococcus aureus isolated from bovine mastitis in Xinjiang, China. Virus genes 2017, 53, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Pike, R.; Harley, A.; Mumin, Z.; Potterill, I.; Meunier, D.; Ganner, M.; Getino, M.; Coelho, J.; Jauneikaite, E.; Moganeradj, K.; Brown, C.; Holmes, A.; Demirjian, A.; Hopkins, K.; Pichon, B. Complete genome assemblies and antibiograms of 22 Staphylococcus capitis isolates. BMC Genom Data 2025, 26, 12. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 5174–5179. [Google Scholar] [CrossRef]
- Daniel, A.; Bonnen, P.E.; Fischetti, V.A. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. Journal of bacteriology 2007, 189, 2086–2100. [Google Scholar] [CrossRef]
- Yoon, H.; Yun, J.; Lim, J.A.; Roh, E.; Jung, K.S.; Chang, Y.; Ryu, S.; Heu, S. Characterization and genomic analysis of two Staphylococcus aureus bacteriophages isolated from poultry/livestock farms. The Journal of general virology 2013, 94, 2569–2576. [Google Scholar] [CrossRef]
- Melo, L. D.; Sillankorva, S.; Ackermann, H.W.; Kropinski, A.M.; Azeredo, J.; Cerca, N. Characterization of Staphylococcus epidermidis phage vB_SepS_SEP9 - a unique member of the Siphoviridae family. Research in microbiology 2014, 165, 679–685. [Google Scholar] [CrossRef]
- Zeman, M.; Bardy, P.; Vrbovska, V.; Roudnicky, P.; Zdrahal, Z.; Ruzickova, V.; Doskar, J.; Pantucek, R. New Genus Fibralongavirus in Siphoviridae Phages of Staphylococcus pseudintermedius. Viruses 2019, 11, 1143. [Google Scholar] [CrossRef] [PubMed]
- Tetens, J.; Sprotte, S.; Thimm, G.; Wagner, N.; Brinks, E.; Neve, H.; Holzel, C. S.; Franz, C.M. First Molecular Characterization of Siphoviridae-Like Bacteriophages Infecting Staphylococcus hyicus in a Case of Exudative Epidermitis. Frontiers in microbiology 2021, 12, 653501. [Google Scholar] [CrossRef]
- Iandolo, J.J.; Worrell, V.; Groicher, K.H.; Qian, Y.; Tian, R.; Kenton, S.; Dorman, A.; Ji, H.; Lin, S.; Loh, P.; Qi, S.; Zhu, H.; Roe, B.A. Comparative analysis of the genomes of the temperate bacteriophage’s phi 11, phi 12 and phi 13 of Staphylococcus aureus 8325. Gene 2002, 289(1-2), 109–118. [Google Scholar] [CrossRef] [PubMed]
- Christie, G.E.; Matthews, A.M.; King, D.G.; Lane, K.D.; Olivarez, N.P.; Tallent, S.M.; Gill, S.R.; Novick, R. P. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α--implications for the specificity of SaPI mobilization. Virology 2010, 407, 381–390. [Google Scholar] [CrossRef]
- van der Mee-Marquet, N.; Corvaglia, A.R.; Valentin, A.S.; Hernandez, D.; Bertrand, X.; Girard, M.; Kluytmans, J.; Donnio, P.Y.; Quentin, R.; Francois, P. Analysis of prophages harbored by the human-adapted subpopulation of Staphylococcus aureus CC398. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 2013, 18, 299–308. [Google Scholar] [CrossRef]
- Coombs, G.W.; Baines, S.L.; Howden, B.P.; Swenson, K.M.; O'Brien, F.G. Diversity of bacteriophages encoding Panton-Valentine leukocidin in temporally and geographically related Staphylococcus aureus. PloS ONE 2020, 15, e0228676. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophage genomics. Current opinion in microbiology 2003, 6, 506–511. [Google Scholar] [CrossRef]
- Shi, Y.; Masic, V.; Mosaiab, T.; Rajaratman, P.; Hartley-Tassell, L.; Sorbello, M.; Goulart, C.C.; Vasquez, E.; Mishra, B.P.; Holt, S.; Gu, W.; Kobe, B.; Ve, T. Structural characterization of macro domain-containing Thoeris antiphage defense systems. Science advances 2024, 10, eadn3310. [Google Scholar] [CrossRef]
- Han, G.; Zhang, J.; Luo, Z.; Lu, B.; Zhang, P.; Yong, K.; Wang, Y.; Luo, Y.; Yang, Z.; Ren, M.; Cao, S.; Yao, X. Characteristics of a novel temperate bacteriophage against Staphylococcus arlettae (vB_SarS_BM31). International microbiology: the official journal of the Spanish Society for Microbiology 2023, 26, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Pu, F.; Zhang, N.; Pang, J.; Zeng, N.; Baloch, F.B.; Li, Z.; Li, B. Deciphering the Genetic Architecture of Staphylococcus warneri Prophage vB_G30_01: A Comprehensive Molecular Analysis. Viruses 2024, 16, 1631. [Google Scholar] [CrossRef] [PubMed]
- Al-Anany, A.M.; Fatima, R.; Hynes, A.P. Temperate phage-antibiotic synergy eradicates bacteria through depletion of lysogens. Cell reports 2021, 35, 109172. [Google Scholar] [CrossRef] [PubMed]
- Al-Anany, A.M.; Fatima, R.; Nair, G.; Mayol, J.T.; Hynes, A.P. Temperate phage-antibiotic synergy across antibiotic classes reveals new mechanism for preventing lysogeny. mBio 2024, 15, e0050424. [Google Scholar] [CrossRef] [PubMed]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).