
Submitted:
09 December 2025
Posted:
11 December 2025
You are already at the latest version
Abstract
Peters-Plus syndrome is a rare autosomal recessive disorder characterized by multisystem involvement, with a primary manifestation in the anterior segment of the eye. The hallmark feature, Peters anomaly, presents as central corneal opacity with iridocorneal adhesions. Clinically, patients often exhibit the classic triad of anterior chamber defects, short stature, and brachydactyly, accompanied by craniofacial dysmorphisms such as cleft lip and/or palate, hypertelorism, and short palpebral fissures, as well as variable intellectual disability. Skeletal abnormalities include rhizomelic limb shortening, clinodactyly, and restricted growth, frequently responsive to growth hormone therapy. Additional manifestations may involve congenital heart defects, genitourinary anomalies, and endocrine disturbances such as hypothyroidism. Prenatal growth restriction is common, and structural brain anomalies can occasionally be present, though they do not consistently correlate with neurodevelopmental outcomes. Diagnosis is suspected based on characteristic clinical findings and confirmed through the identification of biallelic pathogenic variants in the B3GLCT gene. Genetic counseling is essential due to the autosomal recessive inheritance pattern. Management is individualized, including early corneal transplantation between 3–6 months to prevent amblyopia, treatment of glaucoma or cataracts, and multidisciplinary follow-up addressing ophthalmologic, endocrine, neurologic and developmental needs. Prognosis varies widely depending on the severity of ocular and systemic involvement. This overview underscores the importance of early recognition, genetic confirmation, and comprehensive, patient-centered care in optimizing outcomes for individuals with Peters-Plus syndrome.
Keywords:
Peters-plus syndrome
; Peters anomaly
; anterior chamber reye defects
; B3GLCT gene
; brachydactyly
; corneal opacity
1. Introduction
Peters-Plus syndrome is a rare polymalformative condition of unknown current prevalence, with equal sex distribution and a high incidence in consanguineous families. Approximately 100 cases have been reported in the literature, highlighting its exceptional rarity. It is characterized by ocular abnormalities of the anterior chamber—most notably Peters anomaly—along with additional findings such as limb shortening, cleft lip, growth impairment, and variable intellectual disability. Following clinical suspicion, definitive diagnosis is achieved genetically by identifying biallelic pathogenic variants in B3GLCT. This autosomal recessive disorder implies that parents of an affected child are asymptomatic carriers. Management focuses on the individual's clinical manifestations, primarily corneal transplantation in cases of severe corneal opacification, typically performed between 3–6 months of age to prevent amblyopia (1–5).
The study was approved by the Institutional Research Ethics Committee. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the CEI de Provincial Centre of Malaga, Spain, protocol code SICEIA-2024-002941and date of approval date 2 December 2024.
2. Case Description
A case from 2024 is presented involving a 33-year-old primigravida with a spontaneous monochorionic diamniotic twin pregnancy. The patient had no relevant medical history. The first-trimester ultrasound was satisfactory, showing no morphological abnormalities in either fetus, with findings consistent with gestational age and a low-risk combined aneuploidy screening.
The morphological ultrasound at 19+4 weeks revealed the following findings:
The first fetus, located in the left hemiabdomen in cephalic presentation, showed biometry consistent with 18+1 weeks, with an estimated fetal weight of 235 g and a three-vessel umbilical cord with normal umbilical Doppler. Intracranially, visible lateral ventricles were noted in both anterior horns (8.5 mm) without criteria for ventriculomegaly, a normal posterior fossa, and a corpus callosum of normal morphology.
The second fetus, located in the right hemiabdomen in transverse lie, showed biometry consistent with 17+4 weeks, with an estimated fetal weight of 209 g, a single umbilical artery, and normal umbilical Doppler. Intracranial assessment demonstrated bilateral ventriculomegaly (Figure 1), predominantly posterior (anterior horns 6 mm; posterior horns 11–13 mm), and a dilated third ventricle measuring 7×3 mm (Figure 2), with a normal-appearing posterior fossa and corpus callosum. The intertwin discordance in estimated fetal weight was 11%.
The main findings included biometry not concordant with gestational age in both fetuses and a central nervous system malformation in the second fetus, characterized by bilateral ventriculomegaly, whereas the first fetus demonstrated prominent ventricles without fulfilling criteria for ventriculomegaly.
Based on these findings, etiological evaluation was recommended, and an amniocentesis was planned for genetic and infectious studies.
Amniocentesis was performed without complications at 20+2 weeks, and sequential prenatal genetic tests were requested, including QF-PCR, karyotype, microarray (CGH/SNP), and targeted clinical exome sequencing.
Initial QF-PCR analysis demonstrated a normal chromosomal complement, with no aneuploidies of chromosomes 13, 18, or 21, and male sex (XY) in both fetuses. Subsequent high-resolution microarray confirmed the absence of pathogenic or uncertain copy-number variants, with no findings to explain the ventriculomegaly. Cytomegalovirus PCR in amniotic fluid was negative in both fetuses.
Biweekly ultrasound evaluations were performed to monitor fetal development.
At 23+5 weeks, the first fetus, with positive cardiac activity and fetal movements and located in the left hemiabdomen in cephalic presentation, showed biometry consistent with 22+5 weeks, a three-vessel umbilical cord, and normal umbilical artery Doppler. Middle cerebral artery Doppler indicated absence of fetal anemia. Intracranial evaluation revealed normal lateral ventricles and supratentorial structures, with a cephalic index of 87%, compatible with brachycephaly (Figure 3).
The second fetus, with positive cardiac activity and fetal movements and located in the right hemiabdomen in cephalic presentation, showed biometry consistent with 21+2 weeks, a single umbilical artery, and umbilical Doppler with preserved diastolic flow. Middle cerebral artery Doppler showed no evidence of anemia. Intracranially, mild bilateral ventriculomegaly was observed (anterior horns 5 mm; posterior horns 9.7–10.6 mm) along with a 5.4×2.8 mm interhemispheric cyst compatible with an interhemispheric cyst. The cerebral sulci and corpus callosum appeared normal. Intertwin estimated fetal weight discordance was 22%. These findings indicated globally favorable evolution in the first fetus and persistent neurodevelopmental abnormalities in the second fetus.
Given the normal prior genetic studies, expanded analysis via next-generation sequencing of the clinical exome was performed. A homozygous c.660+1G>A variant in the B3GLCT gene, located at the canonical splice site of exon 8, was identified in both fetuses. This variant is described as pathogenic in international databases and is associated with Peters-Plus syndrome.
Upon receiving the genetic results for both fetuses, consistent with Peters-Plus syndrome, a consultation at 27+2 weeks was conducted to inform the parents about the genetic nature, clinical prognosis, and functional implications of the disorder, including possible ocular, neurological, skeletal, and systemic complications, as well as future reproductive options. A detailed neurosonographic assessment of both twins was also performed, revealing stage I intrauterine growth restriction in both fetuses associated with marked rhizomelia (Figure 4 and Figure 5).
The first fetus showed biometry corresponding to 25+2 weeks with an estimated fetal weight of 833 g (3rd percentile), normal Doppler, a three-vessel umbilical cord, and a general morphological exam without major structural anomalies. Neurosonography showed lateral ventricles within normal limits, a corpus callosum of adequate morphology, and preserved cortical development. No ventriculomegaly or ocular abnormalities were identified, except for mild midfacial hypoplasia compatible with the syndrome.
The second fetus showed biometry corresponding to 24 weeks with an estimated fetal weight of 686 g (0.23rd percentile), a single umbilical artery, and umbilical Doppler with preserved diastolic flow. Intracranially, mild-to-moderate bilateral ventriculomegaly was observed (Figure 6), along with third ventricle dilation, a 5×3 mm interhemispheric cyst, and a thin but present corpus callosum. The remaining supratentorial and posterior fossa structures showed maturation appropriate for gestational age.
Both fetuses demonstrated marked rhizomelia without additional evident skeletal anomalies. Intertwin estimated fetal weight discordance was 17.6%.
Prenatal genetic studies (QF-PCR, 750K microarray) confirmed a normal male karyotype in both fetuses without relevant numerical or structural chromosomal abnormalities. The extended prenatal exome identified the pathogenic c.660+1G>A B3GLCT variant in both fetuses, consistent with autosomal recessive Peters-Plus syndrome. Cytomegalovirus PCR in amniotic fluid was negative. The ultrasound findings—rhizomelia, mild-to-moderate brain anomalies, subtle dysmorphic features, and additional structural abnormalities—were compatible with the phenotypic spectrum of the syndrome.
Given these imaging and genetic findings, the patient expressed her desire to proceed with pregnancy termination under legal provisions for severe and incurable fetal disease. Fetal MRI was requested for anatomical characterization, parental segregation studies were initiated, and the case was submitted for evaluation by the hospital’s Clinical Committee for Pregnancy Termination.
Fetal MRI at 27 weeks identified the cavum septi pellucidi and velum interpositum in both fetuses, with normal configuration of the third and fourth ventricles, cerebellum, brainstem, and craniocervical junction. The first fetus, in cephalic presentation, showed mild bilateral ventriculomegaly with atrial diameter of approximately 12 mm. Cortical maturation assessment revealed delayed sulcation, with a shallow cingulate sulcus and a Sylvian fissure symmetric but underdeveloped for gestational age. Calcarine, parieto-occipital, and convexity sulci were identifiable and preserved. The second fetus, in transverse lie, showed lateral ventricles at the upper limit of normal (10–11 mm) without definitive criteria for ventriculomegaly. Partial delay in sulcation was also noted, with unilateral shallow cingulate sulcus and less-developed convexity sulci. The Sylvian fissure, calcarine sulci, and parieto-occipital sulci appeared age appropriate. In conclusion, MRI confirmed mild ventriculomegaly in the first fetus and, in both fetuses, partial and non-uniform delayed sulcation compatible with mild-to-moderate cortical developmental abnormalities.
The Clinical Committee for Pregnancy Termination, based on imaging findings, genetic results, and current literature regarding functional implications and clinical prognosis of Peters-Plus syndrome, determined that the case met criteria for legal termination due to serious and incurable fetal disease.
Fetocide was performed at an external facility without complications, and the patient was admitted to our center for completion of the termination. Cervical ripening was induced with oral mifepristone followed by vaginal prostaglandins. Fourteen hours after admission, vaginal delivery occurred: the first fetus weighed 1200 g (Apgar 0/0) and the second weighed 990 g (Apgar 0/0). The patient had an uncomplicated postoperative course and was discharged after 24 hours.
Parental segregation studies confirmed that both parents were heterozygous carriers of the variant, while both fetuses were homozygous affected, establishing a 25% recurrence risk in future pregnancies. These findings confirmed the genetic etiology of the ultrasound abnormalities, representing the first prenatal diagnosis in this family, and preimplantation genetic testing (PGT) was recommended for future reproductive planning.
In July 2025, the patient reported a new spontaneous pregnancy. An early ultrasound was scheduled to assess viability, and chorionic villus sampling was recommended given the genetic status of both parents and prior obstetric history.
At 9 weeks, ultrasound identified a viable single embryo consistent with gestational age, accompanied by a retroplacental hematoma and a small subserosal fibroid. During the first-trimester screening ultrasound at 12+3 weeks, major fetal malformations were identified, including an occipital meningocele with indirect signs of neural tube defect (crash sign, “dry brain,” and increased intracranial translucency), and unilateral cleft lip. Additional soft markers for aneuploidy were observed (increased nuchal translucency, tricuspid regurgitation, and single umbilical artery), whereas kidneys and limbs appeared normal.
Transcervical chorionic villus sampling was performed without complications, with subsequent confirmation of fetal viability. Rapid QF-PCR demonstrated diploid complement for chromosomes 13, 18, 21, and XY. Later molecular analysis confirmed fetal homozygosity for the c.660+1G>A variant, inherited biallelically, consistent with the expected autosomal recessive disorder.
After multidisciplinary counseling regarding the perinatal prognosis—considered severely compromised due to the malformations and molecular diagnosis—the patient opted for legal pregnancy termination, which proceeded uneventfully.
She was again counseled regarding the 25% recurrence risk in future pregnancies and referred for specialized reproductive counseling.
3. Discussion
3.1. Clinical Characteristics
Patients with Peters-Plus syndrome may present with multiple physical anomalies characteristic of this condition. The typical triad includes anterior chamber defects, short stature, and brachydactyly (1,4,6).
The most common defect affects the anterior segment of the eye and is defined as Peters anomaly, consisting of central corneal opacity associated with thinning or absence of Descemet membrane and posterior corneal thinning with iridocorneal adhesions. Two types exist: type I, a milder form involving only the cornea and often unilateral; and type II, a more severe variant with lens abnormalities and poorer visual prognosis. Common ocular complications include cataracts and glaucoma, with the latter present at birth in 50% of patients. Although Peters anomaly is the most frequent ocular defect, other findings such as mild mesenchymal dysgenesis or iris coloboma may also occur (1,3,5–8).
Skeletal alterations include growth restriction, more pronounced in height than weight, with rhizomelic limb shortening and characteristic brachydactyly. Clinodactyly of the fifth finger may be prominent. Limited elbow range of motion or hypermobility in other joints may be observed. Growth hormone deficiency is common and typically responds well to replacement therapy. Growth restriction begins prenatally, although not always detected. Adult height ranges from 1.28–1.51 m in females and 1.41–1.55 m in males. Although no specific imaging findings have been described, typical early-onset arthritis and thoracic hemivertebrae may be identified (1–3,6,9).
Intellectual disability occurs in 78–83% of patients and ranges from mild to severe forms; severe impairment is seen in up to 26% of affected individuals. CNS anomalies may include microcephaly, neural tube defects, and corpus callosum agenesis, although structural abnormalities may be absent even in patients with intellectual disability, indicating poor correlation between imaging findings and neurodevelopmental outcomes. Epilepsy is another possible manifestation (1–4,6).
Craniofacial findings include short palpebral fissures, prominent forehead, hypertelorism, exaggerated cupid’s bow, and long philtrum. Cleft lip occurs in 45% of cases and cleft palate in 33%. Less common features include micrognathia, ptosis, and small dysmorphic ears. Facial hirsutism has also been described (1–3,5).
Other associated anomalies include congenital heart defects such as atrial or ventricular septal defects, pulmonary or aortic stenosis, hypoplastic left heart syndrome, or bicuspid pulmonary valve, occurring in fewer than one-third of patients. Genitourinary anomalies affect fewer than 20% and include hydronephrosis, renal or ureteral duplication, and renal agenesis. Congenital hypothyroidism has also been reported. CNS anomalies may include corpus callosum hypoplasia or agenesis, hydrocephalus, encephalocele, or Dandy-Walker malformation (1,5,6).
Prenatally, increased rates of miscarriage and intrauterine fetal demise in the second and third trimesters have been noted (1–3).
A summary table of prenatal and clinical findings of Peters-Plus syndrome is shown below (Table 1).
Es importante recalcar que se trata de una entidad que se presenta por medio de alteraciones variables y no específicas de dicha patología, por lo que el diagnóstico prenatal basado exclusivamente en los hallazgos ecográficos puede resultar muy complicado (5).
3.2. Diagnosis and Genetic Counseling
Initial suspicion arises in patients with bilateral anterior chamber anomalies, although unilateral cases occur, especially when associated with skeletal dysplasia (short limbs with broad distal segments), characteristic facial features (cleft lip/palate, exaggerated cupid’s bow, short palpebral fissures, ear anomalies), and variable intellectual disability. Family history with similar findings strengthens suspicion (1,5,6).
Confirmatory diagnosis requires genetic testing demonstrating biallelic pathogenic variants in B3GLCT (21 pathogenic variants identified to date), located at 13q12.3. The gene encodes β-1,3-glucosyltransferase, involved in glycosylation of thrombospondin type-1 repeats via O-fucosyltransferase-mediated mechanisms. Loss-of-function mutations impair protein glycosylation. The splice-site mutation c.660+1G>A in intron 8 is the most frequently reported variant (1,6,7,10).
Inheritance is autosomal recessive; thus, parents are asymptomatic heterozygous carriers. Each sibling has a 25% chance of being affected, 50% chance of being a carrier, and 25% chance of being unaffected. Carrier testing and preconception counseling are recommended, including discussion of prenatal or preimplantation genetic testing. Syndromes with similar findings but without B3GLCT mutations are classified as Peters-Plus-like (1–4,9).
3.3. Differential Diagnosis
Differential diagnosis includes syndromes involving short limbs and short stature: Cornelia de Lange, Smith-Lemli-Opitz, Robinow, Abruzzo-Erickson, Weill-Marchesani, fetal alcohol syndrome, and SHORT syndrome. No clear association has been established between Peters-Plus syndrome and other anterior chamber disorders. Peters anomaly may occur in isolation or be associated with mutations in PAX6, PITX2, PITX3, CYP1B1, and FKHL7. Other conditions to consider include Peters anomaly with intellectual disability, such as corpus callosum agenesis (1–4).
3.4. Management
Once diagnosed, evaluation must determine disease extent and individual needs. Assessments include complete ophthalmologic evaluation, growth hormone stimulation testing, early intervention programs, echocardiography, renal ultrasound, and neuroimaging when indicated. Hearing assessment and thyroid function tests are recommended. Genetic counseling is essential for family planning (1,6).
Treatment targets individual manifestations. Penetrating keratoplasty is indicated for severe bilateral corneal opacification and ideally performed at 3–6 months to prevent amblyopia. Keratoprosthesis is considered after repeated graft failures. In milder cases, release of iridocorneal adhesions or medical/surgical glaucoma treatment may be performed. Congenital glaucoma associated with Peters anomaly is more difficult to treat and has poorer outcomes compared to primary congenital glaucoma; adequate intraocular pressure is achieved in only 32% of cases, and long-term visual outcomes are poor due to underlying ocular pathology (1,6,8).
3.5. Prognosis
Prognosis depends on the severity of ocular disease, associated systemic anomalies, and age at presentation. Penetrating keratoplasty may prevent amblyopia but carries high graft-failure risk. Close ophthalmologic monitoring every three months during childhood is recommended to manage glaucoma and amblyopia risk. A multidisciplinary team is essential to address developmental and systemic needs (1,8).
4. Conclusions
Peters-Plus syndrome is a rare, complex autosomal recessive disorder requiring high clinical suspicion and molecular confirmation. Identification of biallelic pathogenic B3GLCT variants establishes diagnosis and clarifies the pathophysiologic mechanism involving impaired glycosylation of thrombospondin-type proteins.
The present case and literature review demonstrate the marked phenotypic variability of this syndrome, with ocular anomalies—particularly Peters anomaly—representing the core manifestation, often associated with craniofacial, skeletal, genitourinary, and CNS abnormalities. Clinical severity and visual prognosis depend on corneal involvement and complications such as glaucoma and cataracts.
Prenatal ultrasound findings—including growth restriction, craniofacial anomalies, limb shortening, and CNS defects—should raise suspicion of a genetic syndrome, warranting invasive testing for genetic confirmation and appropriate parental counseling.
Identifying heterozygous carrier status in both parents enables tailored genetic counseling and reproductive options such as preimplantation genetic testing to reduce recurrence risk.
Clinical management must be multidisciplinary, integrating ophthalmologic, endocrine, neurologic, cardiac, and genetic care. Early corneal transplantation in severe cases, growth hormone therapy, and early developmental support are fundamental pillars to optimize patient outcomes.
Author Contributions
F.A.M., M.R.S. drafted and designed the article; F.A.M. and M.R.S. reviewed the clinical case and the current literature; F.A.M., M.R.S. and G.M.C., writing—original draft preparation; J.L.J.S., N.A.I., A.H.J.R. and B.A.M., writing—review and editing; J.L.J.S., N.A.I., M.R.S., B.A.M. and G.M.C. reviewed the article critically; M.R.S. and M.R.S. continued the patient follow-ups. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by the Institutional Research Ethics Committee. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the CEI de Provincial Centre of Malaga, Spain, protocol code SICEIA-2024-002941and date of approval date 2 December 2024.
Informed Consent Statement
Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Oberstein, S.A.L.; Ruivenkamp, C.A.; Hennekam, R.C. Peters Plus Syndrome. GeneReviews® [Internet]. 2025.
- de Buy Wenniger, L.J.M.; Hennekam, R.C. The Peters' plus syndrome: a review. Ann Genet. 2002, 45, 97–103. [Google Scholar]
- Boog, G.; Le Vaillant, C.; Joubert, M. Prenatal sonographic findings in Peters-plus syndrome. Ultrasound Obstet Gynecol. 2005, 25, 602–6. [Google Scholar] [CrossRef] [PubMed]
- Canda, M.T.; Çağlayan, L.D.; Demir, A.B.; Demir, N. Prenatal detection of Peters plus-like syndrome. Turk J Obstet Gynecol. 2019, 15, 273–7. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Kaul, A.; Kabra, M. Prenatal diagnosis of fetal Peters' plus syndrome: a case report. Case Rep Genet. 2013, 2013, 364529. [Google Scholar] [CrossRef] [PubMed]
- Demir, G.Ü.; Lafcı, N.G.; Doğan, Ö.A.; Şimşek-Kiper, P.Ö.; Utine, G.E. Peters Plus syndrome: a recognizable clinical entity. Turk J Pediatr. 2020, 62, 136–40. [Google Scholar] [CrossRef] [PubMed]
- Schoner, K.; Kohlhase, J.; Müller, A.M.; Schramm, T.; Plassmann, M.; Schmitz, R.; et al. Hydrocephalus, agenesis of the corpus callosum, and cleft lip/palate in fetuses with Peters' plus syndrome and B3GALTL mutations: expanded PPS phenotypes. Prenat Diagn 2013, 33, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Jat, N.S.; Tripathy, K. Peters Anomaly. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. Updated 2023 Aug 25. Available from: https://www.ncbi.nlm.nih.gov/books/NBK58054.
- Kapoor, S.; Mukherjee, S.B.; Arora, R.; Shroff, D. Peters plus syndrome. Indian J Pediatr. 2008, 75, 635–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Venkat, A.; Taujale, R.; Mull, J.L.; Ito, A.; Kannan, N.; Haltiwanger, R.S. Peters plus syndrome mutations affect function and stability of human β1,3-glucosyltransferase. J Biol Chem. 2021, 297. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Ventriculomegaly in the second fetus at 19+4 weeks.
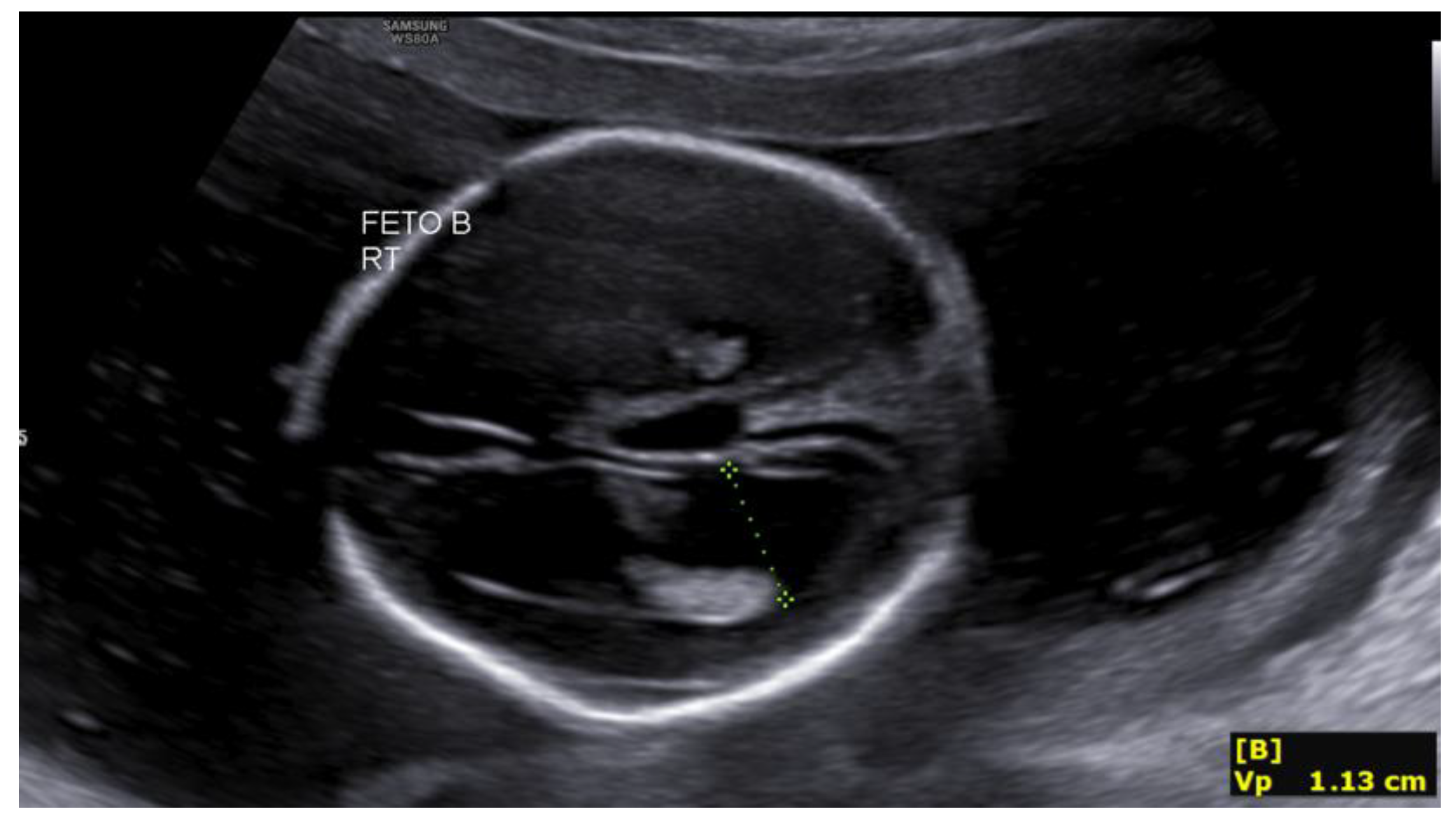
Figure 2.
Third ventricle dilation in the second fetus at 19+4 weeks.
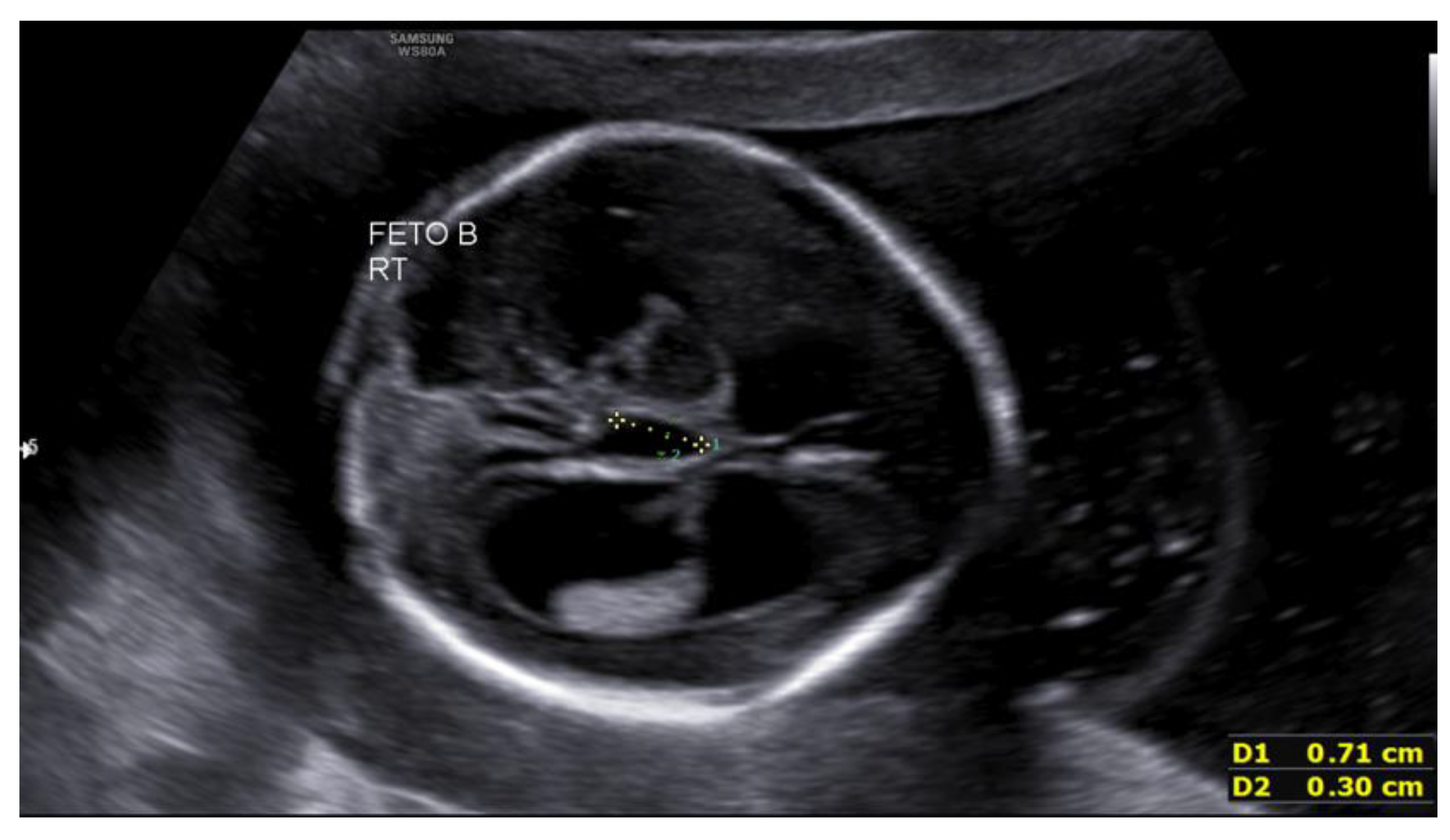
Figure 3.
Cephalic index >85% (brachycephaly) in the first fetus at 23+5 weeks.
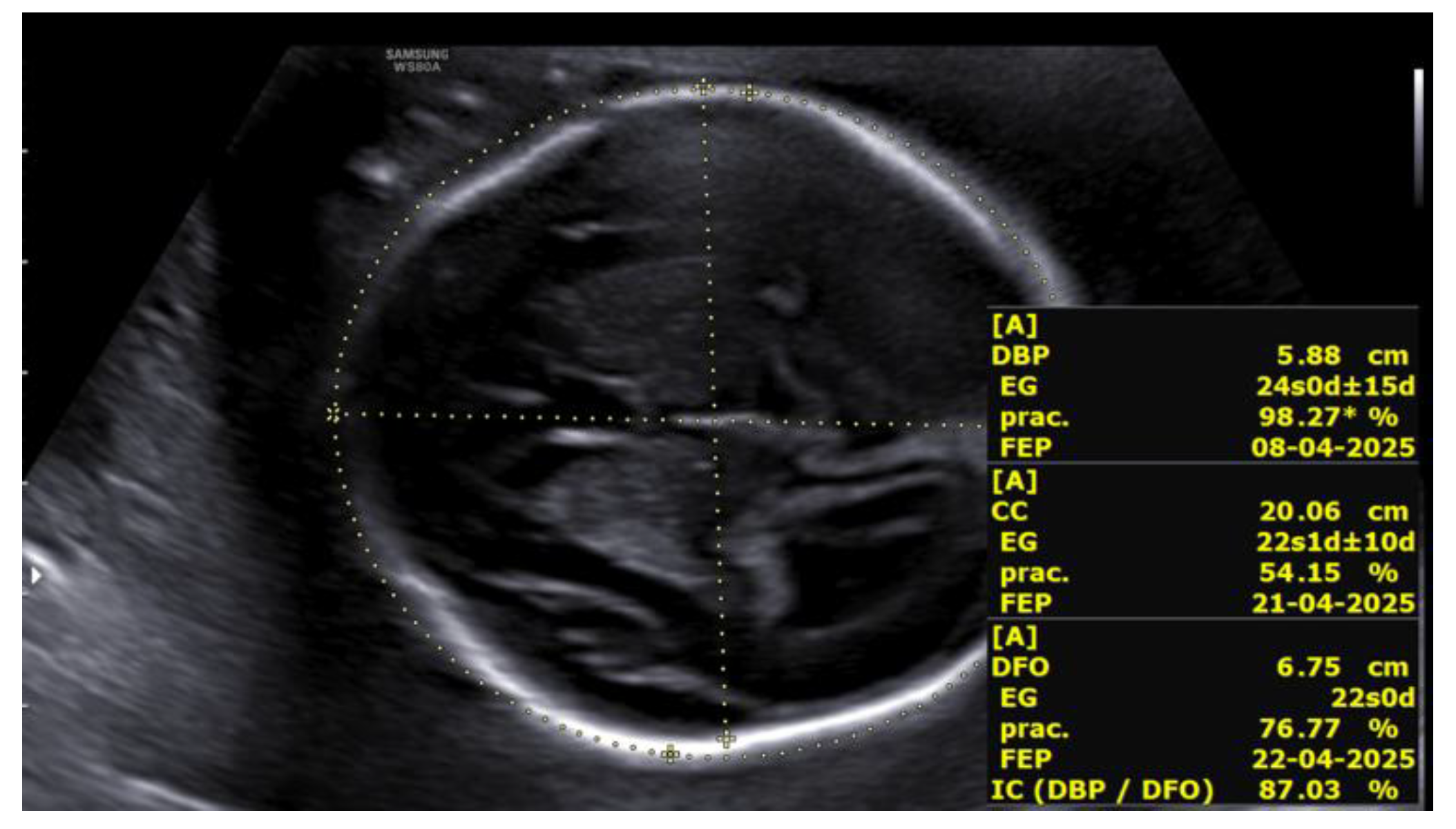
Figure 4.
Rhizomelic shortening of the lower limb in the first fetus at 27+2 weeks.
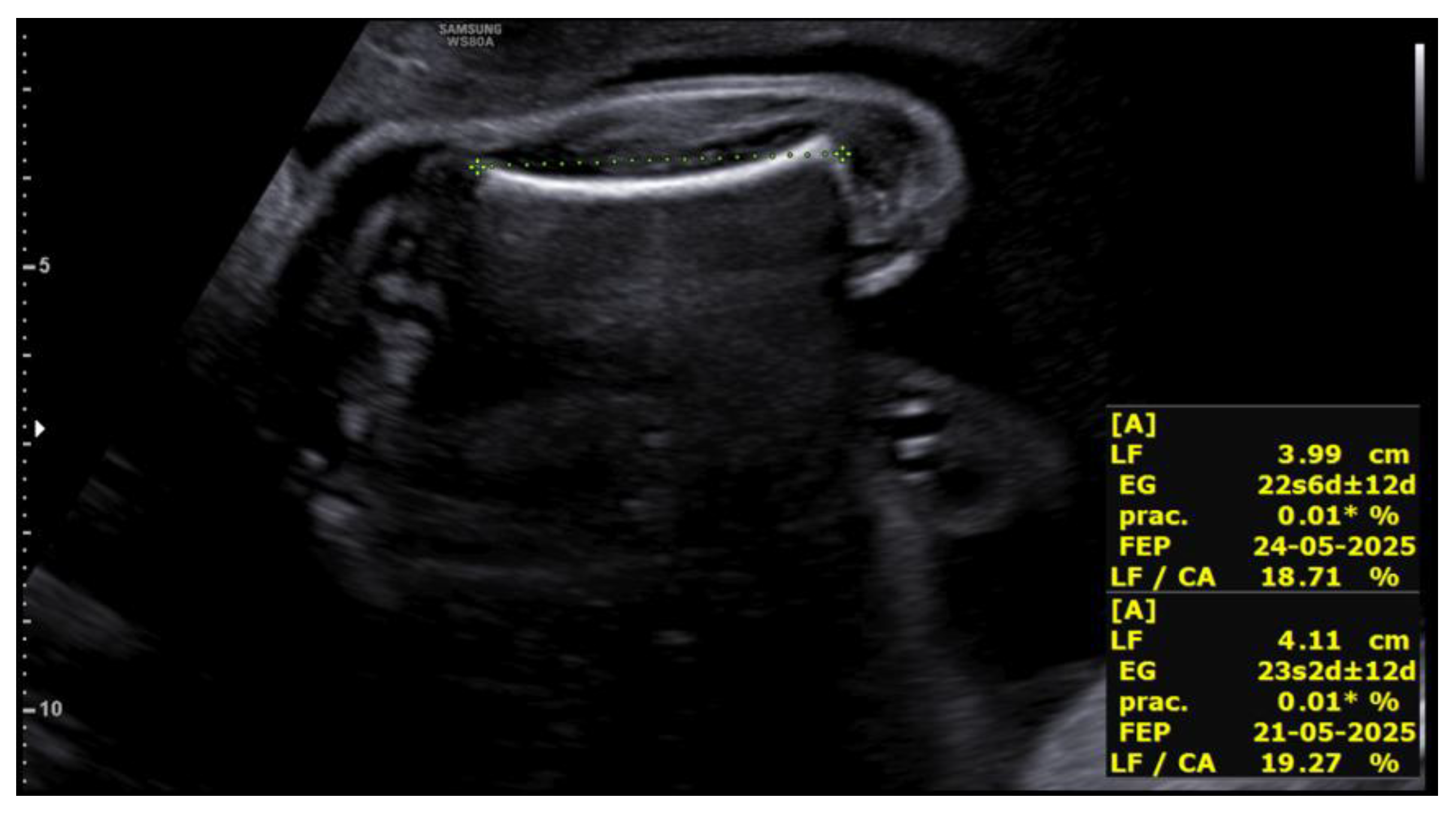
Figure 5.
Rhizomelic shortening of the upper limb in the second fetus at 27+2 weeks.
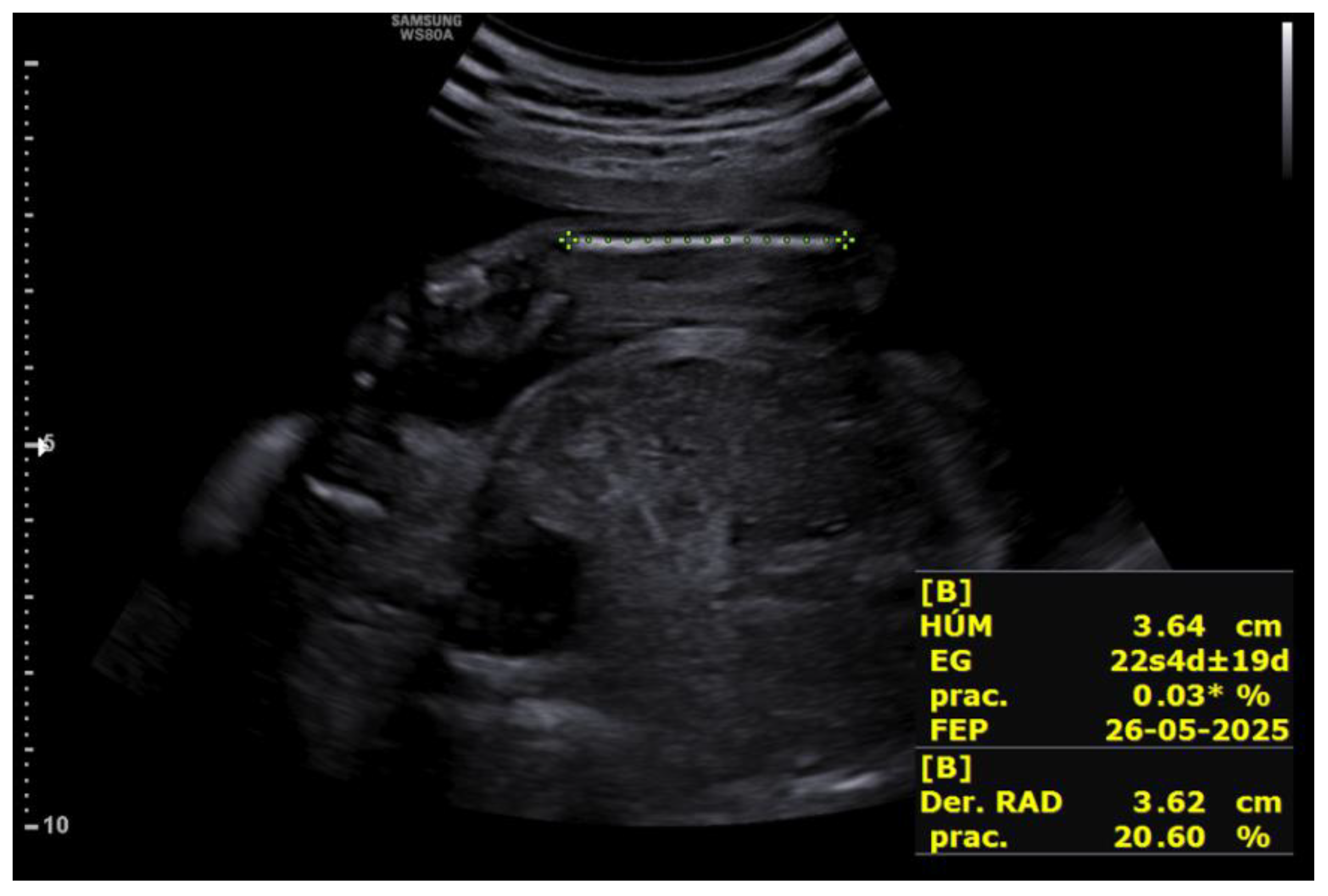
Figure 6.
Mild bilateral ventriculomegaly in the second fetus at 27+2 weeks.
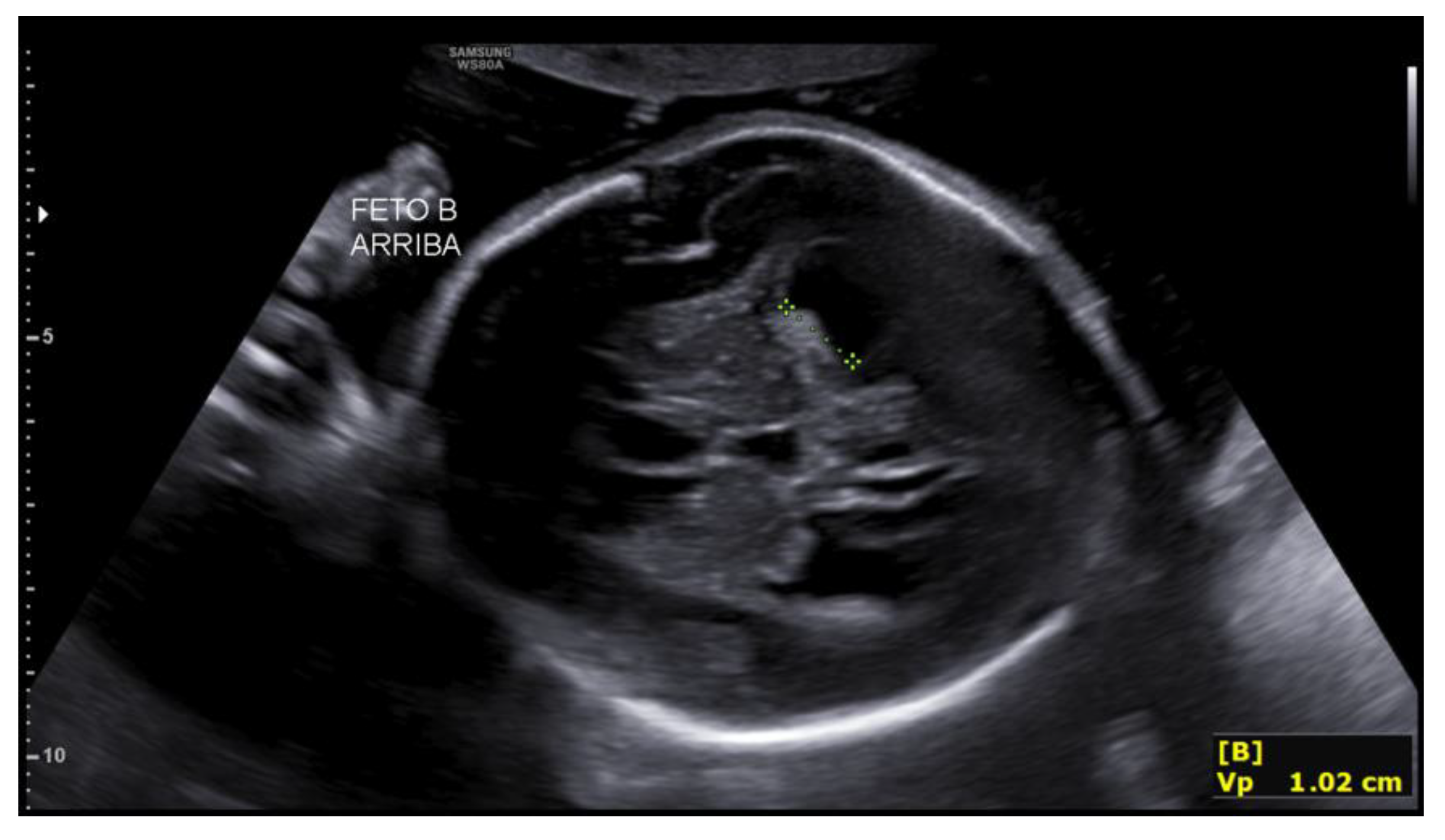

Table 1.
Main Prenatal and Clinical Findings of Peters-Plus Syndrome.
| System / Region Affected | Typical Finding | Description |
|---|---|---|
| Fetal Growth | Intrauterine growth restriction (IUGR) | Prenatal onset in many cases; may progress during late gestation. |
| Craniofacial | Anterior chamber anomalies (Peters anomaly) | Corneal opacity, iridocorneal adhesions, increased anterior segment echogenicity; absence of the lens in severe cases. |
| Microphthalmia / partial anophthalmia | Underdeveloped or small ocular globes. | |
| Micrognathia | Small mandible with retruded facial profile. | |
| Elongated philtrum | Characteristic feature with prominent upper lip groove. | |
| Cleft lip and/or palate | Detectable on 3D facial ultrasound or coronal planes. | |
| Skeleton / Limbs | Rhizomelic shortening | Disproportionately shortened proximal limb segments. |
| Brachydactyly | Short digits; broad hands and feet. | |
| Fifth-finger clinodactyly | Curvature of the little finger toward the fourth finger. | |
| Single palmar crease | Transverse palmar crease (typically identified postnatally). | |
| Genitourinary System | Hydronephrosis | Pelvicalyceal dilatation. |
| Ureteral duplication | Double ureteral system. | |
| Renal hypoplasia | Small or underdeveloped kidneys. | |
| Hypospadias | Urethral meatus located on the ventral aspect of the penis. | |
| Cryptorchidism | Undescended testes. | |
| Rudimentary uterus/vagina | Underdeveloped or absent Müllerian structures. | |
| Cardiac | Septal defects (atrial or ventricular) | Congenital structural heart defects. |
| Subvalvular aortic stenosis | Obstruction below the aortic valve. | |
| Hypoplastic left heart | Underdevelopment of left-sided cardiac structures. | |
| Central Nervous System | Intellectual disability (postnatal; not detectable prenatally) | May occur with or without structural CNS abnormalities. |
| Mild structural abnormalities | Partial agenesis or hypoplasia of the corpus callosum may occur. | |
| Other Clinical Features | Short stature, coarse facial features, short neck, inguinal hernias, rectus diastasis | Multisystem involvement characteristic of the syndrome. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



