Submitted:
30 November 2025
Posted:
02 December 2025
Read the latest preprint version here
Abstract
Glycosylphosphatidylinositol-anchored proteins (GPI-APs) represent a distinct class of eukaryotic, membrane-associated cell surface proteins, characterized by the presence of a glycolipid anchor at their C-terminal end. These proteins exhibit unique biophysical properties and play a crucial role in several human diseases, such as transmissible spongiform encephalopathies (TSEs), malaria, and sleeping sickness, as well as in rare disorders collectively known as Inherited GPI Deficiency (IGD)-related diseases. Due to their broad clinical significance, GPI-APs have become a focus of extensive research, including their intracellular quality control (QC) mechanisms. Findings from various model organisms have revealed remarkable interspecies variations in these QC pathways, as well as differences when compared to other proteins within the secretory pathway. In this review, we summarize recent advances in understanding the cellular QC of GPI-APs and the degradation of their misfolded variants.
Keywords:
endoplasmic reticulum (ER)
; glycosylphosphatidylinositol (GPI) anchor
; GPI anchor remodeling
; post-ER quality control
; vacuole/lysosome
; microautophagy
1. Biogenesis of GPI-Anchored Proteins
Approximately 1% of all encoded proteins in protozoa, fungi, plants, and mammals undergo post-translational attachment to a glycosylphosphatidylinositol (GPI) anchor within the endoplasmic reticulum (ER) [1,2]. The biogenesis of a GPI-AP is a complex, multi-step process preceded by the biosynthesis of the GPI anchor, which is preassembled by an orchestrated series of reactions at the ER membrane. The fully assembled GPI anchor consists of an inositol phospholipid and several sugar moieties (Figure 1). Both the biosynthesis of the GPI anchor and the subsequent protein attachment are well understood and have been extensively reviewed [3,4]. In brief, the generation of the GPI anchor starts with the enzymatic transfer of N-acetylglucosamine (GlcNAc) to phosphatidylinositol (PI) at the cytosolic leaflet of the lipid bilayer, generating GlcNAc-PI [5]. Afterwards, GlcNAc is deacetylated and the product GlcN-PI is flipped to the luminal leaflet of the ER membrane [6,7]. On the luminal side, inositol is acylated, generating GlcN-(acyl)PI and in mammals, but not in fungi and plants, the diacylglycerol portion is exchanged to form a 1-alkyl, 2-acyl glycerol backbone [8,9]. Next, three mannoses are added to the GlcN in a linear fashion but with different linkage types, followed by further modification of each mannose with a phospho-ethanolamine (PEtN). At this stage, a client protein in the ER lumen is coupled to the GPI anchor precursor. In a series of reactions coordinated by GPI transamidase, a large membrane protein complex, the client protein’s C-terminal transmembrane (TM) domain is removed, and the newly generated terminal carboxyl group is coupled to the amino group of the PEtN on the third mannose of the GPI anchor [10,11]. This transamination reaction is also referred to as “attachment” [12,13,14]. Whereas the mechanism of attachment appears well conserved among species, some variations in GPI anchor precursor structures have been observed. For instance, in yeast, the GPI anchor is extended with a fourth α1,2-linked mannose prior to attachment [15]. This reaction precedes the modification of the third mannose with PEtN that is important for attachment. Thus, whereas most (if not all) attached GPI anchors in yeast contain four mannoses, this GPI structure has so far only been detected in few mammalian cell types, mostly in brain cells [15,16]. In addition, work with mutant mammalian cell lines recently showed that some proteins are preferentially attached to the PEtN of the second mannose of a GPI precursor, indicating that attachment to a GPI anchor can occur in more than one way [17].
2. Physiological and Clinical Significance of GPI-Anchored Proteins
In contrast to other lipid anchors such as single myristoyl, palmitoyl, and prenyl groups, the biophysical properties of the GPI anchor are thought to provide a stable membrane association for the attached protein without the need for additional protein-protein or protein-membrane interactions [18,19]. However, the complexity of the GPI anchor structure suggests cellular functions that go beyond simple membrane association. It has long been known that GPI-APs tend to concentrate on specific lipid microdomains, or rafts, which affect protein trafficking and signaling [20,21]. Consequently, the biogenesis of GPI anchors and GPI-APs is tightly intertwined with membrane homeostasis and membrane traffic [22].
Other features of GPI-APs include the regulated transfer and release of these proteins once they are at the cell surface. In certain unicellular organisms, such as yeast, many GPI-APs become cross-linked to the cell wall, where they are covalently bound to glycans. In a transglycosylation reaction, the GPI anchor is first enzymatically cleaved between GlcN and the first mannose of the GPI anchor core structure and the liberated mannose-protein is then glycosidically linked to the nonreducing ends of cell wall glycan chains [23]. In certain protozoa, coordinated enzyme-mediated release (shedding) of GPI-APs from the cell surface is coupled to the changes of life cycle stages and occurs during host switching in parasites like Trypanosoma brucei [24]. In mammals, the cell type-specific shedding of GPI-APs from the plasma membrane is important for development, immunity and neurogenesis, and is regulated by a variety of enzymes that are capable to cleave the GPI anchor at distinct sites [25,26,27,28,29].
Beyond their roles in membrane homeostasis and protein traffic, GPI-APs have significant clinical relevance and are directly or indirectly implicated in several major human diseases. A prominent example is the human cellular prion protein (PrP), a GPI-AP whose physiological function remains incompletely defined, although recent studies suggest roles in the peripheral and central nervous systems (CNS), as a receptor mediating the internalization of α-synuclein amyloid fibrils, and promoting cancer [30,31,32,33,34]. In neurons, the induced or sporadic conversion of its normal fold (PrP(C)) into the pathogenic version (PrP(Sc)) causes the formation of amyloids and onset of Creutzfeld-Jakob and related neurodegenerative diseases [35]. The presence of the GPI anchor in PrP affects the clinical symptoms and disease progression but the underlying mechanisms remain unclear [36,37,38]. Germline mutations in GPI-related genes in humans cause various Inherited GPI Deficiency (IGD)-related diseases, as summarized by [39]. For example, defects in the addition of ethanolamine phosphate (EtNP) to the third mannose of the GPI anchor precursor have been associated with global developmental and intellectual delay, seizures, polymicrogyria, and peripheral neuropathy in multiple unrelated families with bi-allelic PIG-B mutations [40]. Comprehensive clinical reports and surveys on pathogenic variants of GPI-related genes highlight developmental delay, intellectual disability, and hyperphosphatasia, resulting from the release of cellular alkaline phosphatase, as frequent phenotypes of IGDs [41,42,43]. Paroxysmal nocturnal haemoglobinuria (PNH), a well-characterized human disease, is caused by a defect in PIG-A, which is essential for the initial step in GPI precursor synthesis. Dysfunctional PIG-A primarily affects blood cells, leading to the loss of CD55 and CD59, two GPI-anchored proteins crucial for complement regulation [44]. Additionally, impaired GPI-AP biosynthesis has been linked to several types of cancer [45].
Apart from IGDs, GPI-APs have garnered enormous attention due to their critical role in host infection by human parasites. This has fueled research aimed at understanding the underlying mechanisms of intracellular trafficking of GPI-AP to develop effective strategies against these diseases. The major surface proteins of Plasmodium falciparum, the causative agent of malaria, are GPI-APs that undergo frequent exchange during the parasite’s life cycle to evade immune recognition [46,47,48]. Similarly, trypanosomes, responsible for Chagas disease (Trypanosoma cruzi) and sleeping sickness (Trypanosoma brucei), feature a dense layer of GPI-APs on their cell surface. The dependence of these parasites on their hosts, coupled with the pressures of immune system counteraction, has driven the evolution of intricate microbial life cycles and specific cellular strategies for the biogenesis and QC of GPI-AP. Consequently, trypanosomes have become pioneering model organisms for studying GPI-APs [49,50]. Recent findings indicate that certain Trypanosoma exhibit distinct mechanisms for the degradation of misfolded GPI-APs, differing in key steps from those observed in other model organisms and illustrating the existence of various uncharacterized cellular mechanisms directly linked to protein QC [51].
3. Protein Quality Control in the Secretory Pathway
Whereas protein quality control (QC) in the secretory pathway was originally thought to be restricted to its point of entry, the ER, it has become clear that the eukaryotic cell harbors a variety of distinct surveillance systems in many, if not all organelles of the secretory pathway. As will be discussed later, QC systems that operate outside the ER have specific relevance for GPI-APs.
The early detection and retention of misfolded proteins inside the ER, also known as ER quality control (ERQC), includes the unfolded protein response (UPR) and has been extensively studied and discussed [52,53,54]. In addition, it became clear that most misfolded ER proteins are eliminated by ER-associated degradation (ERAD), which involves retrotranslocation into the cytosol followed by proteasomal degradation and has been the subject of excellent recent reviews [55,56]. Certain misfolded secretory proteins that are detected inside the ER are refractory to ERAD, for instance due to the formation of mutation-specific aggregates that impede their retrotranslocation. This led to the discovery and initial characterization of several specific trafficking pathways that are used for the delivery of these proteins to the vacuole/lysosome for degradation, termed collectively ER-to-lysosomes-associated degradation (ERLAD) pathways, for review see [57].
Protein misfolding can also be detected in the Golgi, in endosomes and in the vacuolar membrane, often linked to the homeostasis of individual organelles. These systems can be referred to as post-ER quality control (post-ERQC), for review see [58]. Work from yeast early on suggested the existence of a Golgi-localized QC [59,60]. As in the ER, misfolded proteins can be eliminated via several cellular routes from the Golgi. Pathways named EGAD (yeast) and GARD (mammals) promote the retrotranslocation and proteasomal degradation of misfolded and regulatory proteins from the Golgi and from endosomes [61,62]. The detection and degradation of regulators might involve their transient misfolding due to a particular kind of stress or metabolic situation, in analogy to the situation in the ER where key enzymes of lipid metabolism are degraded in a regulated fashion by ERAD [63,64,65]. Alternatively, misfolded proteins or those that lack binding partners inside the Golgi are either retrieved back to the ER by receptor-mediated incorporation into transport vesicles for ultimate degradation through ERAD or are trafficked to endosomes for subsequent delivery to the vacuole/lysosome for degradation [66,67]. TM proteins are marked for degradation in the vacuole/lysosome through ubiquitination on their cytosolic tail followed by endosomal sorting complex required for transport (ESCRT)-mediated internalization into multivesicular bodies (MVBs), for review see [68]. Misfolded soluble proteins destined for vacuolar/lysosomal degradation are sorted from the Golgi to endosomes by the conserved cycling receptor Vps10/sortilin. It has been shown to bind unfolded proteins through its luminal 1 domain [69,70,71]. Work from yeast has demonstrated that this pathway has relevance for the QC and degradation of misfolded GPI-APs [72].
4. GPI Anchor Remodeling and Implications for GPI-AP QC
The lack of a TM domain makes GPI-APs unique membrane-attached cell surface proteins with no exposure to the cytosol. All intracellular trafficking of GPI-APs therefore depends on TM adaptor proteins that connect the luminal or extracellular protein with the cytosolic machinery that regulates membrane traffic. For instance, the internalization of GPI-APs from the plasma membrane into endosomes for their regulated turnover by vacuolar/lysosomal degradation depends on cycling Cos proteins, which constitute a conserved class of TM adaptor proteins [73]. Similarly, the initial export from the ER depends on adaptor proteins classified as p24 proteins, which form heteromeric complexes [74,75,76,77]. p24 proteins bind to GPI-APs on the luminal side of the ER membrane, while their cytosolic tails interact with the COPII machinery to facilitate the concentration of GPI-APs at ER exit sites (ERES) [78].
The binding of GPI-APs to p24 proteins is regulated by GPI anchor remodeling, a process that includes modifications on both sugar and lipid moieties of the GPI anchor. Remodeling starts usually after the attachment of proteins to the preassembled GPI anchor precursor, although exceptions exist [79]. Soon after attachment, the acyl chain linked to inositol is removed by a conserved ER resident deacylase, Bst1 (yeast), PGAP1 (mammals), or PtPGAP1 (plants) [80,81], with few exceptions reported, such as human erythrocytes [82]. In yeast, either the acyl chain at the sn-2 position of the diacyl glycerol is then exchanged for a long saturated C-26 fatty acid by the ER resident enzymes Per1 and Gup1 or, alternatively, the diacylglycerol moiety is exchanged entirely for a ceramide by the ER resident remodelase Cwh43 [83,84,85]. In mammalian cells, lipid remodeling on the 1-alkyl, 2-acyl glycerol backbone occurs only after ER export, in the Golgi, for review see [86]. Prior to ER export, the PEtN on the second mannose of the GPI anchor is removed by a conserved ER resident deacetylase, Ted1 (yeast) or PGAP5 (mammals) [87,88]. In addition to enhancing the binding of GPI-APs to p24 proteins, GPI remodeling also promotes their concentration within sphingolipid-enriched microdomains prior to export. Together, these processes coordinate the efficient export of GPI-APs from the ER [87,89,90,91,92].
Protein folding and GPI anchor remodeling occur concurrently within the ER, raising a key question: to what extent are these complex processes functionally interconnected and co-regulated? For example, does terminal GPI anchor remodeling take place only after proteins have successfully folded and passed ERQC? Early insights from studies in yeast and mammalian cells revealed that misfolded GPI-APs are predominantly, though not exclusively, degraded via vacuolar or lysosomal pathways, rather than through ERAD [93,94,95,96]. Interestingly, in yeast mutants that block ER protein export, ERAD became the predominant pathway for the degradation of misfolded GPI-APs [96]. These findings demonstrated that misfolded GPI-APs are generally ERAD-competent, contrary to the notion that the GPI anchor might present a steric barrier to retrotranslocation [97]. The observation that, under normal conditions, these proteins are frequently exported from the ER and subsequently degraded in vacuoles/lysosomes supports a model in which GPI anchor remodeling, rather than protein folding, determines the kinetics of ER export. Accordingly, at least in yeast, protein folding and GPI anchor remodeling appear to operate independently, with misfolded GPI-APs relying predominantly on post-ERQC mechanisms for their disposal [72,96,98].
These mechanisms, however, do not appear to be universal. Comparative analyses of yeast data alongside both established and emerging findings from other model organisms reveal shared features as well as striking differences in the strategies used by the eukaryotic cell to handle misfolded GPI-APs. In the sections that follow, we summarize and discuss the current understanding of GPI-AP QC across yeast, mammals, plants, and protozoa.
5. Mechanisms of GPI-AP QC in Different Model Systems
5.1. Yeast
A prominent model misfolded GPI-AP for studying its QC in the yeast S. cerevisiae is Gas1*, the G291R mutant of the β-1,3-glucanosyltransferase Gas1 involved in cell wall synthesis [99]. Gas1* is degraded with a half-life of 45-60min [96,99]. In wild type cells, Gas1* is degraded mainly inside the vacuole, whereas a minor fraction is routed to Hrd1-dependent ERAD [96]. Like Gas1*, other chimeric or non-related misfolded GPI-APs, such as CPY*GPI or Cwp2*, are routed predominantly to the vacuole for degradation [72,96]. Interestingly, the fraction of Gas1* that is targeted to ERAD depends in part on protein O-mannosylation, a modification of serine and threonine side chains with single mannose moieties inside the ER that was previously known to have functional importance for ERQC and ERAD [98,99,100,101,102]. Gas1*, but not Gas1, becomes O-mannosylated by ER-resident protein mannosyltransferases Pmt1 and Pmt2, which form a heterodimeric complex and also have a chaperone function in addition to their mannosyltransferase activity [98]. The Pmt1/2 complex interacts physically with components of the ER export machinery for GPI-APs as well as with the Hrd1 ERAD machinery, and its deletion shifts Gas1* degradation entirely to vacuolar degradation [93,98]. These findings suggest that the Pmt1/2 complex retains Gas1* inside the ER to facilitate folding and can shuttle a subpopulation of terminally misfolded Gas1* to ERAD.
The fact that most cellular Gas1* is degraded inside the vacuole rather than by ERAD does not result from the strong endogenous expression level of Gas1* and/or saturation of the ERAD pathway because “rerouting” to ERAD can be observed when its ER export is genetically blocked [96]. In addition, Gas1* is degraded almost entirely by ERAD in wild type cells if its attachment to a GPI anchor is prevented by a single point mutation that abolishes recognition by the transamidase complex [96]. Collectively these results show that the presence of the GPI anchor is the cause for ER export and predominant vacuolar degradation of Gas1*. It also argues that the GPI anchor itself is not a structural obstacle for protein retrotranslocation and ERAD. How ERAD processes a GPI-AP and what happens to its GPI anchor are poorly understood mechanistically, though studies in protozoa might yield relevant information (see 5.4 below).
Why then is Gas1* exported in large quantities from the ER rather than being retained and routed to ERAD? It was found that Gas1 and Gas1* bind with the same efficiency to the p24 family member Emp24, the adaptor for connecting GPI-APs with the cytosolic COPII machinery for ER export [96]. At the same time, binding was equally abrogated in mutant cells that prevent GPI anchor remodeling [96]. These data showed that the GPI anchors of correctly folded Gas1 and misfolded Gas1* are remodeled with indistinguishable efficiencies in vivo and support the notion that GPI anchor remodeling is not functionally connected to protein folding in yeast. From the model systems that have been studied so far, yeast appears to provide the most limited ERQC for GPI-APs (Figure 2). Possible reasons for this will be discussed in the last section.
Gas1* fails to internalize from the endosomal membrane, probably because of the lack of adaptors that can be ubiquitinated by the cytosolic ubiquitination machinery and provide the crucial link for recruiting the ESCRT machinery for membrane invagination and formation of Gas1*-containing multi-vesicular bodies [68]. Therefore, after fusion of endosomes with the vacuole, Gas1* is transiently present at the vacuolar membrane, facing the lumen of the vacuole. From there it is internalized into intravacuolar vesicles in a yet poorly defined process that is regulated by members of the vacuolar transport chaperone (VTC) family [72]. VTCs are multispanning vacuolar membrane proteins with previously described functions in microautophagy [103]. Interestingly, internalization of Gas1* is inhibited in a Δpep4 mutant, a phenotype that mimics the one observed for the vacuolar uptake of lipid droplets and correlates with the inhibition of the formation of specialized lipid microdomains in the vacuolar membrane [104,105]. This may indicate indicate that cargo sequestering into vacuolar membrane microdomains with raft-like properties and with affinity to GPI anchors is part of the mechanism that mediates Gas1* internalization from the vacuolar membrane, but precisely how it works remains an important open question [106]. Although the protein part of Gas1* could be degraded directly from the vacuolar membrane by vacuolar proteases, internalization ensures that the GPI-AP is degraded in its entirety, including its remodeled GPI anchor, which contains long chain fatty acids or ceramide. Thus, internalization of most (if not all) Gas1* into intravacuolar vesicles prior to degradation might also be required to maintain vacuolar membrane homeostasis.
Figure 3.
The post-ER degradation pathway for misfolded GPI-APs in yeast. Misfolded GPI-APs are rapidly exported from the ER via the p24-complex and transported to the Golgi. There, they are diverted from the early secretory pathway and routed to endosomes in a post-ER quality control (post-ERQC) step that requires Vps10. At endosomes, misfolded GPI-APs are not internalized into intraluminal vesicles but instead remain on the limiting membrane of multivesicular bodies (MVBs). Upon fusion of MVBs with the vacuole, these proteins are delivered to the vacuolar membrane and become exposed to the vacuolar lumen, a transient intermediate that accumulates in Δpep4 cells. From the vacuolar membrane, misfolded GPI-APs are internalized through a process resembling microautophagy and dependent on the vacuolar transporter chaperone (VTC) complex. The resulting intravacuolar vesicles (IVVs) containing misfolded GPI-APs are ultimately degraded in an Atg15-dependent manner. The regulation of vacuolar membrane invagination during this internalization process, and the specific mechanistic role of the VTC complex, remain open questions.
Figure 3.
The post-ER degradation pathway for misfolded GPI-APs in yeast. Misfolded GPI-APs are rapidly exported from the ER via the p24-complex and transported to the Golgi. There, they are diverted from the early secretory pathway and routed to endosomes in a post-ER quality control (post-ERQC) step that requires Vps10. At endosomes, misfolded GPI-APs are not internalized into intraluminal vesicles but instead remain on the limiting membrane of multivesicular bodies (MVBs). Upon fusion of MVBs with the vacuole, these proteins are delivered to the vacuolar membrane and become exposed to the vacuolar lumen, a transient intermediate that accumulates in Δpep4 cells. From the vacuolar membrane, misfolded GPI-APs are internalized through a process resembling microautophagy and dependent on the vacuolar transporter chaperone (VTC) complex. The resulting intravacuolar vesicles (IVVs) containing misfolded GPI-APs are ultimately degraded in an Atg15-dependent manner. The regulation of vacuolar membrane invagination during this internalization process, and the specific mechanistic role of the VTC complex, remain open questions.
5.2. Mammalian Cells
Research on the QC of GPI-APs and the degradation pathways of misfolded versions in mammalian cells has focused on human disease-related species, such as the human prion protein (PrP), which is associated with neuronal death and neurodegeneration if it converts into its pathological isoform, and on CD55 and CD59, which are complement regulatory proteins and cause paroxysmal nocturnal hemoglobinuria when defective [107,108,109]. Several studies showed that pharmacological inhibition of the proteasome affected the turnover of mutant GPI-APs in mammalian cells, initially implying ERAD as their major degradation pathway [110,111,112,113]. However, this notion was challenged when it became clear that overexpression of misfolded versions of PrP (PrP*) in transfected cells can lead to inefficient translocation of PrP* into the ER and to its accumulation in the cytosol where its proteasomal degradation would occur unconnected to ERAD [114]. In fact, detailed analysis of the cellular itinerary of PrPs* showed that they are poor substrates for ERAD and instead are routed to the lysosome for degradation, with the GPI anchor being the cause for ER export and for preventing ERAD [94,97]. These findings suggested that similar cellular strategies exist in yeast and mammalian cells for the degradation of misfolded GPI-APs.
However, there are also notable differences. Prolonged ER retention of misfolded GPI-APs dependent on ER lectins has been observed. For instance, simultaneous deletion of Calnexin and Calreticulin led to a faster ER exit of CD59* (C94S) compared to control cells and resulted in inefficient deacetylation of inositol on its GPI anchor by PGAP1 [115,116]. In addition, Calnexin was found to physically interact with PGAP1 [115]. These data prompted the hypothesis that N-glycan-dependent ER retention through Calnexin and Calreticulin prevents premature GPI anchor remodeling and ER export [115,116,117,118]. This differs from the scenario described in yeast and suggests that mammalian cells do possess a certain degree of physical and functional connection between protein folding and GPI anchor remodeling (Figure 4). This would allow for an increased time window for the ERQC of GPI-APs.
Detailed work on PrPs* revealed a surprising connection between GPI-AP QC and ER homeostasis. The ER retention of misfolded GPI-APs was no longer observed when global ER protein folding stress was induced [95]. Under these conditions, misfolded GPI-APs were rapidly exported from the ER and routed to lysosomes via the plasma membrane a mechanism termed “rapid ER stress-induced export” (RESET) [119]. Because rapid ER export of misfolded GPI-APs preceded the transcriptional activation of the UPR, it was suggested to be a fast cellular mechanism for contributing to ER homeostasis under acute stress [95]. This in turn suggests that the accumulation of numerous GPI anchors in case of ER retention of misfolded GPI-APs produces a basal ER membrane stress that becomes overwhelming in presence of conditions that cause global protein misfolding. Consequently, rapid ER export of GPI-APs under these conditions will alleviate ER membrane stress or, alternatively, might be required to facilitate reorganization of the ER membrane upon stress adaptation [120]. Support for such a scenario comes from findings indicating that the accumulation of non-attached GPI anchors in the ER membrane triggers suppression of de novo GPI anchor synthesis via degradation of its biosynthetic machinery [121]. Prolonged basal ER stress caused by the retention of misfolded GPI-anchored proteins may still be deleterious over time. For example, neuronal death in prion diseases has been partly attributed to chronic ER stress induced by misfolded PrP mutants [122].
Work that addressed the trafficking itinerary of misfolded GPI-APs under stress conditions revealed additional complexities and cellular mechanisms specific for this class of proteins. RESET of PrP* variants was mediated by the p24 family member TMED9 [123]. Interestingly though, the population of exiting PrP* showed an invariant interactome and often stayed bound to the ER chaperones BiP and Calnexin when trafficked to the plasma membrane [124]. Exposure of the misfolded GPI-AP together with the chaperones at the plasma membrane was required for subsequent endocytosis prior to lysosomal degradation, suggesting that this mode of surface presentation provides critical cues for internalization [124]. How misfolded GPI-APs are recognized at the plasma membrane for endocytosis in presence or absence of binding partners is still unknown.
5.3. Plants
Almost all genes involved in GPI-anchor biogenesis and protein attachment in mammalian cells have homologs in the plant model Arabidopsis thaliana, and initial studies indicate that they perform comparable functions [125,126]. Likewise, the role of GPI lipid remodeling and p24 family proteins in the ER export of GPI-APs is conserved in plants [78,81]. Recent work with the model organisms Nicotiana benthamiana and Arabidopsis thaliana has started to provide insight into ERQC of GPI-APs in the plant kingdom [127]. The expression of misfolding mutants of the GPI-anchored lipid transfer protein LTPG1 led to their routing to the vacuole for degradation. Additional mutations that prevented the attachment to the GPI anchor resulted in targeting the same protein to ERAD. Overall, the results are in good agreement with those obtained with yeast and mammalian cells: a terminally misfolded GPI-AP is predominantly routed to the vacuole/lysosome for degradation.
Interesting insights into additional QC mechanisms for GPI-APs came from studying chimeric proteins where a C-terminal domain that contained the site for GPI anchor attachment was fused with different types of misfolding N-terminal domains. Whereas most mutants were routed to the vacuole for degradation, a mutant containing the misfolding domain of STRUBBELIG (SUBEX-C57S) was routed to ERAD instead [127]. Detailed analysis revealed that this was due to failure of the chimeric protein to become attached to a GPI anchor, explaining their routing to ERAD. Of note, removing the misfolded domain from the fusion proteins restored GPI anchoring. These results were used to suggest that ERQC precedes protein attachment to a GPI anchor and can prevent attachment in case of certain types of misfolding [127,128], (Figure 5). It was further suggested that the degree or “severity” of misfolding was critical for allowing or preventing attachment. Although failure of attachment could be caused by a variety of factors unrelated to protein QC mechanisms, these data provide useful hypotheses for further testing. It appears plausible that rapid protein aggregation inside the ER or failure to attract chaperones that promote folding could inhibit attachment to a GPI anchor. Likely, early detection and degradation of a terminally misfolded protein via ERAD would be more energy-efficient than degrading the same protein after attachment of a metabolically expensive GPI anchor and subsequent trafficking to the vacuole. Why only certain types of misfolded proteins abort attachment to a GPI anchor, and what distinguishes these misfolding variants, remain important open questions.
5.4. Protozoa
One of the most relevant human parasites, the malaria-causing Plasmodium falciparum, has most of its cell surface covered with GPI-APs, which are intensely studied for their roles in virulence and immune defense [46]. Most insights into the QC of GPI-APs in protozoa comes from studies related to Trypanosoma brucei, which causes sleeping sickness in humans. Research on this parasite has been paramount for the discovery and elucidation of the GPI structure [49]. The major protein on the cell surface of the parasite in the bloodstream form is the GPI-AP VSG (variant surface glycoprotein). During each new life cycle, VSG is synthesized as a variant due to expression switching between up to 2000 different VSG-encoding genes that are clustered at distinct expression sites as well as due to gene conversion during recombination events. Both mechanisms contribute to virulence because VSG variants protect the parasite from the immune system of the host [24]. Due to the constant and massive synthesis of VSG, failure to rapidly detect and eliminate misfolded or aggregation-prone VSG variants by efficient ERQC or post-ERQC systems was predicted to be lethal to the parasite. This sparked an interest in studying mechanisms of QC of GPI-APs in T. brucei. Initial studies showed that the ER exit of VSG and trans-sialidase, another GPI-AP in T. brucei, was significantly slowed when GPI anchoring failed [129]. Conversely, the ER exit was accelerated when an otherwise slowly secreted soluble protein was attached to a GPI anchor [130]. These data agree with scenarios found in other eukaryotes and confirm that GPI anchors are bona-fide ER export signals.
Recently, the QC and degradation mechanisms of the VSG-related high-affinity transferrin receptor (TfR) in Trypanosoma brucei have been investigated in detail [131]. The TfR is formed by heterodimerization of two subunits within the ER: ESAG6, a GPI-anchored protein, and ESAG7, a TM protein. When a misfolded version of ESAG6 (HA:E6) was expressed, it accumulated as a cytosolic intermediate upon proteasome inhibition. Interestingly, this intermediate was recognized by an antibody specific for GPI anchors lacking lipid moieties [131]. This suggests that the HA:E6 was fully translocated into the ER and has been attached to the GPI anchor, before it was recognized by ERQC and retrotranslocated back into the cytosol as part of ERAD. Whereas experiments with yeast suggested that misfolded GPI-APs can be targeted to ERAD, it was unclear if and how the GPI anchor is removed during this process. The experiments with HA:A6 suggest that the removal of GPI lipids might be linked to ERAD of GPI-APs. Where and how the lipids of the GPI anchor are removed is currently unclear and poses relevant mechanistic and topological questions. Interestingly, T. brucei strongly expresses a cytosolic GPI-specific phospholipase C (GPI-PLC). It has long been thought to have its major function in releasing (shedding) VSG from the cell surface of the bloodstream form of the parasite upon host switching, although the topological paradox (cytosolic GPI-PLC versus extracellular VSG) has been difficult to reconcile with the proposed mechanism. Results from work addressing this issue suggested that the process involves the translocation of GPI-PLC into the extracellular space mediated by reversible acylation or by secretion in extracellular vesicles but the contribution of either pathway to VSG shedding is not clear [132,133]. In addition, a recent study suggests that GPI-PLC has only a minor role in VSG shedding and is more important for mediating VSG endocytosis [134]. The data linked to QC of GPI-APs now suggest that GPI-PLC might have an important cellular function in removing GPI lipids of retrotranslocated GPI-APs prior to proteasomal degradation [131]. The authors propose that retrotranslocated HA:A6 has to be completely extracted from the ER membrane together with its GPI anchor for the reaction to occur [131]. In an alternative scenario, GPI-PLC-mediated cleavage of the GPI anchor could occur on a retrotranslocated HA:A6 with its GPI anchor flipped to the cytosolic side of the ER membrane. This would avoid exposure or shielding of lipids to the cytosol. To resolve these questions, further experiments are needed.
In summary, host-switching parasites, such as T. brucei, might have evolved a highly specialized cellular machinery for the rapid and efficient detection and elimination of misfolded GPI-APs (Figure 6). In T. brucei, such a need could be based on the fact that the parasite produces up to an estimated 30.000 VSG molecules/min [135]. A highly abundant GPI-specific PLC might be part of an ERAD machinery with a specific branch for GPI-APs.
6. Summary and Outlook
The biophysical properties of the GPI anchor have long been known to cause the concentration of GPI-APs in specific membrane domains, or rafts. This feature, together with an entirely luminal topology, makes GPI-APs special in that they depend on lipid-based sorting and adaptor-dependent trafficking inside the cell. The studies presented and discussed in this review showcase that the QC of GPI-APs is equally special. We propose that the described organism-specific variations in QC reflect different evolutionary strategies and adaptations to the intracellular locations of GPI anchor remodeling in order to maintain protein and membrane homeostasis inside the ER, which are both affected by GPI-APs.
In yeast, GPI anchor lipid remodeling is exclusively occurring inside the ER, including the addition of long chain fatty acids or ceramide. Once remodeled, rapid ER export of all GPI-APs, regardless of their folding state, would prevent ER membrane stress that would otherwise result from the accumulation of long-chain fatty acids or ceramides when misfolded species are retained [136,137]. In support of such a scenario, deletion of p24 family members dedicated to the ER export of GPI-APs results in profound ER stress and robust inductions of the UPR [138]. In addition, GPI-APs containing ceramides are very efficiently incorporated into specific vesicles for rapid ER export [91]. Maintaining ER membrane homeostasis likely explains why GPI-APs are largely shifted to post-ERQC, i.e., why they are rapidly exported from the ER, even when misfolded.
The situation is different in mammalian cells where parts of GPI anchor lipid remodeling, such as modification with long-chain saturated fatty acids, are occurring in the Golgi. Retention of misfolded GPI-APs inside the ER is observed and a certain degree of accumulation of misfolded species in the ER therefore likely poses tolerable or no ER stress. Only if ER stress is further enhanced, i.e., by drugs such as tunicamycin, rapid and comprehensive export of ER-retained misfolded GPI-APs occurs, followed by lysosomal degradation [95]. Thus, under physiological conditions, GPI anchor remodeling in the Golgi in mammalian cells would allow for a generally larger time window for the ERQC of GPI-APs.
The data from plants can be explained as yet another evolutionary strategy to combine maintenance of ER protein and membrane homeostasis. Rapid and rigorous ERQC that effectively prevents attachment of terminally misfolded species to GPI anchors, as observed in some cases, eliminates potential ER membrane stress and therefore the need for ER export.
It is interesting to observe that the host switching parasite T. brucei employs an entirely different modus operandi for the ERQC of GPI-APs. The tight connection of this process to ERAD might be linked to the unique biology of this and related parasites, which involves the frequent generation of new variants of VSG, the most abundant GPI-AP that makes up almost all protein on the cell surface and is paramount for the evasion of the host immune system and thus for cellular survival. One plausible scenario for the preferred routing of misfolded VSG variants to ERAD might be that massive expression of misfolded species only to be routed along the secretory pathway to the lysosomes for degradation might prove too costly. Although questions concerning the mechanism for the ERAD of misfolded VSG, such as topologies of intermediates, remain open, the contribution of the specific lipase GPI-PLC in this process serves as a valuable working hypothesis and should be rigorously investigated further. Another scenario might be linked to the GPI anchor itself. The lipid moieties of the VSG GPI anchor consist exclusively of myristic acid and are therefore unusually short compared to anchors in other cells [139]. This might reduce the ER membrane stress upon ER retention and accumulation of misfolded species followed by subsequent ERAD.
The observed variations in ERQC and degradation of misfolded GPI-APs might not be exclusive to certain organisms. The use of only a few model proteins in each biological system might generate a misleading bias. It is possible that all modes of ERQC and degradation of GPI-APs are present in all eukaryotic cells and are utilized to a certain degree, with each system having preferred mechanisms and pathways in place in adaptation to its cellular biology.
Supporting such a scenario, yeast can route misfolded GPI-APs to ERAD when ER exit is genetically blocked, and in mammalian cells ER retention of misfolded GPI-APs is rapidly lost upon ER stress, leading to their export from the ER. Together, these observations show that both systems retain flexibility and can mount rapid responses to changing environmental conditions In fact, the data from parasites, where ERAD is preferentially used for the degradation of misfolded GPI-APs, is arguably an excellent starting point for investigating mechanisms of ERAD of GPI-APs in other model systems, including addressing important mechanistic questions, such as the fate of the GPI anchor before or after retrotranslocation and extraction of misfolded GPI-APs from the membrane.
Acknowledgments
We thank Thomas Williams for critical reading of the manuscript and for many helpful comments. L.L. and V.G. were supported by a grant of the Agencia Estatal de Investigación (AEI/10.13039/501100011033/PID2022-136665NB-I00). L.L. was supported by the “Talento Doctor” Fellowship co-funded by the European Regional Development Fund (ERDF) and the Junta de Andalucía, Spain, and by the EMBO Scientific Exchange Grant (11267) for supporting a visiting researcher position in the Pedro Carvalho lab at the Dunn School of Pathology, University of Oxford, UK.
Conflicts of Interest
The authors declare no competing interests.
References
- O. Nosjean, A. Briolay, and B. Roux, ‘Mammalian GPI proteins: sorting, membrane residence and functions’, Biochim Biophys Acta, vol. 1331, no. 2, pp. 153–186, Sep. 1997. [CrossRef]
- P. Orlean and A. K. Menon, ‘Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids’, J Lipid Res, vol. 48, no. 5, pp. 993–1011, May 2007. [CrossRef]
- T. Kinoshita, ‘Biosynthesis and biology of mammalian GPI-anchored proteins’, vol. 10, no. 3, Mar. 2020. [CrossRef]
- T. Kinoshita, ‘Towards a thorough understanding of mammalian glycosylphosphatidylinositol-anchored protein biosynthesis’, Glycobiology, vol. 34, no. 11, Sep. 2024. [CrossRef]
- R. Watanabe et al., ‘Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2’, EMBO J, vol. 19, no. 16, pp. 4402–4411, Aug. 2000. [CrossRef]
- R. Watanabe, K. Ohishi, Y. Maeda, N. Nakamura, and T. Kinoshita, ‘Mammalian PIG-L and its yeast homologue Gpi12p are N-acetylglucosaminylphosphatidylinositol de-N-acetylases essential in glycosylphosphatidylinositol biosynthesis’, Biochem J, vol. 339 ( Pt 1), no. Pt 1, pp. 185–192, Apr. 1999. [CrossRef]
- Y. Wang et al., ‘Genome-wide CRISPR screen reveals CLPTM1L as a lipid scramblase required for efficient glycosylphosphatidylinositol biosynthesis’, Proc Natl Acad Sci U S A, vol. 119, no. 14, Apr. 2022. [CrossRef]
- Y. Murakami et al., ‘PIG-W is critical for inositol acylation but not for flipping of glycosylphosphatidylinositol-anchor’, Mol Biol Cell, vol. 14, no. 10, pp. 4285–4295, Oct. 2003. [CrossRef]
- N. Jiménez-Rojo and H. Riezman, ‘On the road to unraveling the molecular functions of ether lipids’, FEBS Lett, vol. 593, no. 17, pp. 2378–2389, Sep. 2019. [CrossRef]
- Y. Xu et al., ‘Structures of liganded glycosylphosphatidylinositol transamidase illuminate GPI-AP biogenesis’, Nature Communications 2023 14:1, vol. 14, no. 1, pp. 1–17, Sep. 2023. [CrossRef]
- D. Li, ‘Structure and Function of the Glycosylphosphatidylinositol Transamidase, a Transmembrane Complex Catalyzing GPI Anchoring of Proteins’, Subcell Biochem, vol. 104, pp. 425–458, 2024. [CrossRef]
- S. Mayor, A. K. Menon, and G. A. M. Cross, ‘Transfer of glycosyl-phosphatidylinositol membrane anchors to polypeptide acceptors in a cell-free system’, J Cell Biol, vol. 114, no. 1, pp. 61–71, 1991. [CrossRef]
- S. E. Maxwell, S. Ramalingam, L. D. Gerber, L. Brink, and S. Udenfriend, ‘An active carbonyl formed during glycosylphosphatidylinositol addition to a protein is evidence of catalysis by a transamidase’, J Biol Chem, vol. 270, no. 33, pp. 19576–19582, 1995. [CrossRef]
- D. K. Sharma, J. Vidugiriene, J. D. Bangs, and A. K. Menon, ‘A cell-free assay for glycosylphosphatidylinositol anchoring in African trypanosomes. Demonstration of a transamidation reaction mechanism’, J Biol Chem, vol. 274, no. 23, pp. 16479–16486, Jun. 1999. [CrossRef]
- S. J. Grimme, B. A. Westfall, J. M. Wiedman, C. H. Taron, and P. Orlean, ‘The essential Smp3 protein is required for addition of the side-branching fourth mannose during assembly of yeast glycosylphosphatidylinositols’, J Biol Chem, vol. 276, no. 29, pp. 27731–27739, Jul. 2001. [CrossRef]
- N. Stahl, M. A. Baldwin, R. Hecker, K. M. Pan, A. L. Burlingame, and S. B. Prusiner, ‘Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid’, Biochemistry, vol. 31, no. 21, pp. 5043–5053, Feb. 1992. [CrossRef]
- M. Ishida et al., ‘Ethanolamine-phosphate on the second mannose is a preferential bridge for some GPI-anchored proteins’, EMBO Rep, vol. 23, no. 7, Jul. 2022. [CrossRef]
- M. D. Resh, ‘Trafficking and signaling by fatty-acylated and prenylated proteins’, Nat Chem Biol, vol. 2, no. 11, pp. 584–590, 2006. [CrossRef]
- M. E. Linder and R. J. Deschenes, ‘Palmitoylation: policing protein stability and traffic’, Nat Rev Mol Cell Biol, vol. 8, no. 1, pp. 74–84, Jan. 2007. [CrossRef]
- K. Simons and D. Toomre, ‘Lipid rafts and signal transduction’, Nat Rev Mol Cell Biol, vol. 1, no. 1, pp. 31–39, 2000. [CrossRef]
- J. B. Helms and C. Zurzolo, ‘Lipids as targeting signals: lipid rafts and intracellular trafficking’, Traffic, vol. 5, no. 4, pp. 247–254, Apr. 2004. [CrossRef]
- S. Sasaki et al., ‘Protein sorting upon exit from the endoplasmic reticulum dominates Golgi biogenesis in budding yeast’, FEBS Lett, vol. 598, no. 5, pp. 548–555, Mar. 2024. [CrossRef]
- M. S. Vogt, G. F. Schmitz, D. V. Silva, H. U. Mösch, and L. O. Essen, ‘Structural base for the transfer of GPI-anchored glycoproteins into fungal cell walls’, Proc Natl Acad Sci U S A, vol. 117, no. 36, pp. 22061–22067, Sep. 2020. [CrossRef]
- A. Schwede and M. Carrington, ‘Bloodstream form Trypanosome plasma membrane proteins: antigenic variation and invariant antigens’, Parasitology, vol. 137, no. 14, pp. 2029–2039, Dec. 2010. [CrossRef]
- M. A. Davitz, D. Hereld, S. Shak, J. Krakow, P. T. Englund, and V. Nussenzweig, ‘A glycan-phosphatidylinositol-specific phospholipase D in human serum’, Science, vol. 238, no. 4823, pp. 81–84, 1987. [CrossRef]
- G. Kondoh et al., ‘Angiotensin-converting enzyme is a GPI-anchored protein releasing factor crucial for fertilization’, Nat Med, vol. 11, no. 2, pp. 160–166, Feb. 2005. [CrossRef]
- K. Watanabe et al., ‘Growth factor induction of Cripto-1 shedding by glycosylphosphatidylinositol-phospholipase D and enhancement of endothelial cell migration’, J Biol Chem, vol. 282, no. 43, pp. 31643–31655, Oct. 2007. [CrossRef]
- D. Corda, M. G. Mosca, N. Ohshima, L. Grauso, N. Yanaka, and S. Mariggiò, ‘The emerging physiological roles of the glycerophosphodiesterase family’, FEBS J, vol. 281, no. 4, pp. 998–1016, Feb. 2014. [CrossRef]
- Y. Fujihara and M. Ikawa, ‘GPI-AP release in cellular, developmental, and reproductive biology’, J Lipid Res, vol. 57, no. 4, pp. 538–545, Apr. 2016. [CrossRef]
- M. A. Wulf, A. Senatore, and A. Aguzzi, ‘The biological function of the cellular prion protein: an update’, BMC Biology 2017 15:1, vol. 15, no. 1, pp. 34-, May 2017. [CrossRef]
- E. De Cecco and G. Legname, ‘The role of the prion protein in the internalization of α-synuclein amyloids’, Prion, vol. 12, no. 1, pp. 23–27, Jan. 2018. [CrossRef]
- A. R. Castle and D. Westaway, ‘Prion Protein Endoproteolysis: Cleavage Sites, Mechanisms and Connections to Prion Disease’, J Neurochem, vol. 169, no. 1, p. e16310, Jan. 2025. [CrossRef]
- B. Caughey et al., ‘Prions and protein aggregates as pathogens, self-propagating structures, biomarkers, and therapeutic targets’, Microbiology and Molecular Biology Reviews, Sep. 2025. [CrossRef]
- M. Zahra, A. Idris, M. Q. Wei, N. A. J. McMillan, and A. L. Munn, ‘Unmasking the tumorigenic potential of cellular prion protein in cancer progression’, Biochim Biophys Acta Mol Basis Dis, vol. 1872, no. 1, Jan. 2026. [CrossRef]
- S. B. Prusiner, ‘Molecular biology of prions causing infectious and genetic encephalopathies of humans as well as scrapie of sheep and BSE of cattle.’, Dev Biol Stand, vol. 75, pp. 55–74, 1991.
- E. Dvorakova et al., ‘Detection of the GPI-anchorless prion protein fragment PrP226* in human brain’, BMC Neurol, vol. 13, Sep. 2013. [CrossRef]
- G. Zanusso et al., ‘Gerstmann-sträussler-scheinker disease and anchorless prion protein mice share prion conformational properties diverging from sporadic creutzfeldt-jakob disease’, Journal of Biological Chemistry, vol. 289, no. 8, pp. 4870–4881, Feb. 2014. [CrossRef]
- P. Shen et al., ‘Characterization of Anchorless Human PrP With Q227X Stop Mutation Linked to Gerstmann-Sträussler-Scheinker Syndrome In Vivo and In Vitro’, Mol Neurobiol, vol. 58, no. 1, pp. 21–33, Jan. 2021. [CrossRef]
- S. Li, Q. Tang, Y. Jiang, and X. Chen, ‘Inherited glycosylphosphatidylinositol deficiency: a review from molecular and clinical perspectives’, Acta Biochim Biophys Sin (Shanghai), vol. 56, no. 8, pp. 1234–1243, Aug. 2024. [CrossRef]
- Y. Murakami et al., ‘Mutations in PIGB Cause an Inherited GPI Biosynthesis Defect with an Axonal Neuropathy and Metabolic Abnormality in Severe Cases’, Am J Hum Genet, vol. 105, no. 2, pp. 384–394, Aug. 2019. [CrossRef]
- Y. Murakami and T. Kinoshita, ‘Inherited GPI deficiencies: A new disease with intellectual disability and epilepsy’, No To Hattatsu, vol. 47, no. 1, pp. 5–13, Jan. 2015.
- K. Bellai-Dussault, T. T. M. Nguyen, N. V. Baratang, D. A. Jimenez-Cruz, and P. M. Campeau, ‘Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders’, Clin Genet, vol. 95, no. 1, pp. 112–121, Jan. 2019. [CrossRef]
- J. Sidpra et al., ‘The clinical and genetic spectrum of inherited glycosylphosphatidylinositol deficiency disorders’, Brain, vol. 147, no. 8, pp. 2775–2790, Aug. 2024. [CrossRef]
- A. Hill, A. E. Dezern, T. Kinoshita, and R. A. Brodsky, ‘Paroxysmal nocturnal haemoglobinuria’, Nat Rev Dis Primers, vol. 3, May 2017. [CrossRef]
- D. G. Gamage and T. L. Hendrickson, ‘Gpi transamidase and gpi anchored proteins: Oncogenes and biomarkers for cancer’, Crit Rev Biochem Mol Biol, vol. 48, no. 5, pp. 446–464, Sep. 2013. [CrossRef]
- P. R. Gilson et al., ‘Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum’, Mol Cell Proteomics, vol. 5, no. 7, pp. 1286–1299, 2006. [CrossRef]
- F. Kamena et al., ‘Synthetic GPI array to study antitoxic malaria response’, Nat Chem Biol, vol. 4, no. 4, pp. 238–240, 2008.
- R. Nagar et al., ‘The major surface protein of malaria sporozoites is GPI-anchored to the plasma membrane’, Journal of Biological Chemistry, vol. 300, no. 8, Aug. 2024. [CrossRef]
- M. A. J. Ferguson, ‘The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research’, J Cell Sci, vol. 112, no. 17, pp. 2799–2809, 1999. [CrossRef]
- J. Lukeš, D. Speijer, A. Zíková, J. D. Alfonzo, H. Hashimi, and M. C. Field, ‘Trypanosomes as a magnifying glass for cell and molecular biology’, Trends Parasitol, vol. 39, no. 11, pp. 902–912, Nov. 2023. [CrossRef]
- L. Lemus, R. S. Hegde, and V. Goder, ‘New frontiers in quality control: the case of GPI-anchored proteins’, Nat Rev Mol Cell Biol, May 2023. [CrossRef]
- L. Ellgaard and A. Helenius, ‘Quality control in the endoplasmic reticulum’, Nat Rev Mol Cell Biol, vol. 4, no. 3, pp. 181–191, 2003.
- R. Strasser, ‘Protein Quality Control in the Endoplasmic Reticulum of Plants’, Annu Rev Plant Biol, vol. 69, pp. 147–172, Apr. 2018. [CrossRef]
- R. L. Wiseman, J. S. Mesgarzadeh, and L. M. Hendershot, ‘Reshaping endoplasmic reticulum quality control through the unfolded protein response’, Mol Cell, vol. 82, no. 8, pp. 1477–1491, Apr. 2022. [CrossRef]
- L. Krshnan, M. L. van de Weijer, and P. Carvalho, ‘Endoplasmic Reticulum-Associated Protein Degradation’, Cold Spring Harb Perspect Biol, vol. 14, no. 12, 2022. [CrossRef]
- J. C. Christianson, E. Jarosch, and T. Sommer, ‘Mechanisms of substrate processing during ER-associated protein degradation’, Nat Rev Mol Cell Biol, vol. 24, no. 11, pp. 777–796, Nov. 2023. [CrossRef]
- I. Fregno and M. Molinari, ‘Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways’, Crit Rev Biochem Mol Biol, vol. 54, no. 2, pp. 153–163, Mar. 2019. [CrossRef]
- S. Schwabl and D. Teis, ‘Protein quality control at the Golgi’, Curr Opin Cell Biol, vol. 75, Apr. 2022. [CrossRef]
- E. Hong, A. R. Davidson, and C. A. Kaiser, ‘A pathway for targeting soluble misfolded proteins to the yeast vacuole’, J Cell Biol, vol. 135, no. 3, pp. 623–633, 1996.
- S. Wang and D. T. Ng, ‘Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control’, Mol Biol Cell, vol. 21, no. 7, pp. 1153–1165, 2010.
- O. Schmidt et al., ‘ Endosome and Golgi-associated degradation ( EGAD ) of membrane proteins regulates sphingolipid metabolism ’, EMBO J, vol. 38, no. 15, Aug. 2019. [CrossRef]
- A. Eisenberg-Lerner et al., ‘Golgi organization is regulated by proteasomal degradation’, Nat Commun, vol. 11, no. 1, Dec. 2020. [CrossRef]
- A. Ruggiano, O. Foresti, and P. Carvalho, ‘ER-associated degradation: Protein quality control and beyond’, 2014. [CrossRef]
- V. Goder, E. Alanis-Dominguez, and M. Bustamante-Sequeiros, ‘Lipids and their (un)known effects on ER-associated protein degradation (ERAD)’, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, Jun. 2019. [CrossRef]
- D. Kumari and J. L. Brodsky, ‘The targeting of native proteins to the endoplasmic reticulum-associated degradation (Erad) pathway: An expanding repertoire of regulated substrates’, Biomolecules, vol. 11, no. 8, Aug. 2021. [CrossRef]
- S. Vashist, W. Kim, W. J. Belden, E. D. Spear, C. Barlowe, and D. T. Ng, ‘Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding’, J Cell Biol, vol. 155, no. 3, pp. 355–368, 2001.
- D. Hellerschmied, Y. V. Serebrenik, L. Shao, G. M. Burslem, and C. M. Crews, ‘Protein folding state-dependent sorting at the Golgi apparatus’, Mol Biol Cell, vol. 30, no. 17, pp. 2296–2308, Aug. 2019. [CrossRef]
- W. M. Henne, N. J. Buchkovich, and S. D. Emr, ‘The ESCRT pathway’, Dev Cell, vol. 21, no. 1, pp. 77–91, Jul. 2011. [CrossRef]
- M. U. Jørgensen, S. D. Emr, and J. R. Winther, ‘Ligand recognition and domain structure of Vps10p, a vacuolar protein sorting receptor in Saccharomyces cerevisiae’, Eur J Biochem, vol. 260, no. 2, pp. 461–469, Mar. 1999. [CrossRef]
- C. L. Gelling, I. W. Dawes, D. H. Perlmutter, E. A. Fisher, and J. L. Brodsky, ‘The Endosomal protein-sorting receptor sortilin has a role in trafficking α-1 antitrypsin’, Genetics, vol. 192, no. 3, pp. 889–903, Nov. 2012. [CrossRef]
- I. Fitzgerald and B. S. Glick, ‘Secretion of a foreign protein from budding yeasts is enhanced by cotranslational translocation and by suppression of vacuolar targeting’, Microb Cell Fact, vol. 13, no. 1, Aug. 2014. [CrossRef]
- L. Lemus, Z. Matić, L. Gal, A. Fadel, M. Schuldiner, and V. Goder, ‘Post-ER degradation of misfolded GPI-anchored proteins is linked with microautophagy’, Curr Biol, vol. 31, no. 18, pp. 4025-4037.e5, Sep. 2021. [CrossRef]
- C. MacDonald, J. A. Payne, M. Aboian, W. Smith, D. J. Katzmann, and R. C. Piper, ‘A Family of Tetraspans Organizes Cargo for Sorting into Multivesicular Bodies’, Dev Cell, vol. 33, no. 3, pp. 328–342, May 2015. [CrossRef]
- F. Schimmoller, B. Singer-Kruger, S. Schroder, U. Kruger, C. Barlowe, and H. Riezman, ‘The absence of Emp24p, a component of ER-derived COPII-coated vesicles, causes a defect in transport of selected proteins to the Golgi’, Embo J, vol. 14, no. 7, pp. 1329–1339, 1995.
- M. Muniz, C. Nuoffer, H. P. Hauri, and H. Riezman, ‘The Emp24 complex recruits a specific cargo molecule into endoplasmic reticulum-derived vesicles’, J Cell Biol, vol. 148, no. 5, pp. 925–930, 2000.
- S. Takida, Y. Maeda, and T. Kinoshita, ‘Mammalian GPI-anchored proteins require p24 proteins for their efficient transport from the ER to the plasma membrane’, Biochemical Journal, vol. 409, no. 2, pp. 555–562, Jan. 2008. [CrossRef]
- S. Lopez et al., ‘Dual independent roles of the p24 complex in selectivity of secretory cargo export from the endoplasmic reticulum’, Cells, vol. 9, no. 5, May 2020. [CrossRef]
- C. Bernat-Silvestre et al., ‘P24 Family proteins are involved in transport to the plasma membrane of gpi-anchored proteins in plants’, Plant Physiol, vol. 184, no. 3, pp. 1333–1347, Nov. 2020. [CrossRef]
- Z. Ji, R. Nagar, S. M. Duncan, M. L. Sampaio Guther, and M. A. J. Ferguson, ‘Identification of the glycosylphosphatidylinositol-specific phospholipase A2 (GPI-PLA2) that mediates GPI fatty acid remodeling in Trypanosoma brucei’, Journal of Biological Chemistry, vol. 299, no. 8, Aug. 2023. [CrossRef]
- S. Tanaka, Y. Maeda, Y. Tashima, and T. Kinoshita, ‘Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p’, J Biol Chem, vol. 279, no. 14, pp. 14256–14263, Apr. 2004. [CrossRef]
- C. Bernat-Silvestre et al., ‘AtPGAP1 functions as a GPI inositol-deacylase required for efficient transport of GPI-anchored proteins’, Plant Physiol, vol. 187, no. 4, pp. 2156–2173, Dec. 2021. [CrossRef]
- W. L. Roberts, J. J. Myher, A. Kuksis, M. G. Low, and T. L. Rosenberry, ‘Lipid analysis of the glycoinositol phospholipid membrane anchor of human erythrocyte acetylcholinesterase. Palmitoylation of inositol results in resistance to phosphatidylinositol-specific phospholipase C’, J Biol Chem, vol. 263, no. 35, pp. 18766–18775, 1988. [CrossRef]
- M. Fujita, M. Umemura, T. Yoko-o, and Y. Jigami, ‘PER1 Is Required for GPI-Phospholipase A(2) Activity and Involved in Lipid Remodeling of GPI-anchored Proteins’, Mol Biol Cell, vol. 17, no. 12, pp. 5253–5264, 2006.
- R. Bosson, M. Jaquenoud, and A. Conzelmann, ‘GUP1 of Saccharomyces cerevisiae Encodes an O-Acyltransferase Involved in Remodeling of the GPI Anchor’, Mol Biol Cell, vol. 17, no. 6, pp. 2636–2645, 2006.
- M. Umemura, M. Fujita, O. T. Yoko, A. Fukamizu, and Y. Jigami, ‘Saccharomyces cerevisiae CWH43 is involved in the remodeling of the lipid moiety of GPI anchors to ceramides’, Mol Biol Cell, vol. 18, no. 11, pp. 4304–4316, 2007.
- T. Kinoshita and M. Fujita, ‘Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling’, J Lipid Res, vol. 57, no. 1, pp. 6–24, Jan. 2016. [CrossRef]
- M. Fujita, Y. Maeda, M. Ra, Y. Yamaguchi, R. Taguchi, and T. Kinoshita, ‘GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi’, Cell, vol. 139, no. 2, pp. 352–365, 2009.
- J. Manzano-Lopez et al., ‘COPII Coat Composition Is Actively Regulated by Luminal Cargo Maturation’, Curr Biol, vol. 25, no. 2, pp. 152–162, 2015.
- M. Fujita et al., ‘Sorting of GPI-anchored proteins into ER exit sites by p24 proteins is dependent on remodeled GPI’, J Cell Biol, vol. 194, no. 1, pp. 61–75, 2011.
- G. A. Castillon et al., ‘The yeast p24 complex regulates GPI-anchored protein transport and quality control by monitoring anchor remodeling’, Mol Biol Cell, vol. 22, no. 16, pp. 2924–2936, 2011.
- S. Rodriguez-Gallardo et al., ‘Ceramide chain length-dependent protein sorting into selective endoplasmic reticulum exit sites.’, Sci Adv, vol. 6, no. 50, pp. 8237–8248, Dec. 2020. [CrossRef]
- S. Rodriguez-Gallardo et al., ‘Quality-controlled ceramide-based GPI-anchored protein sorting into selective ER exit sites’, Cell Rep, vol. 39, no. 5, May 2022. [CrossRef]
- H. Hirayama, M. Fujita, T. Yoko-o, and Y. Jigami, ‘O-mannosylation is required for degradation of the endoplasmic reticulum-associated degradation substrate Gas1*p via the ubiquitin/proteasome pathway in Saccharomyces cerevisiae’, J Biochem, vol. 143, no. 4, pp. 555–567, 2008.
- A. Ashok and R. S. Hegde, ‘Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation’, Mol Biol Cell, vol. 19, no. 8, pp. 3463–3476, 2008.
- P. Satpute-Krishnan, M. Ajinkya, S. Bhat, E. Itakura, R. S. Hegde, and J. Lippincott-Schwartz, ‘ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway’, Cell, vol. 158, no. 3, pp. 522–533, 2014.
- N. Sikorska et al., ‘Limited ER quality control for GPI-anchored proteins’, J Cell Biol, vol. 213, no. 6, pp. 693–704, 2016.
- A. Ashok and R. S. Hegde, ‘Selective Processing and Metabolism of Disease-Causing Mutant Prion Proteins’, PLoS Pathog, vol. 5, no. 6, p. e1000479, Jun. 2009. [CrossRef]
- V. Goder and A. Melero, ‘Protein O-mannosyltransferases participate in ER protein quality control’, J Cell Sci, vol. 124, no. Pt 1, pp. 144–153, 2011.
- M. Fujita, O. T. Yoko, and Y. Jigami, ‘Inositol deacylation by Bst1p is required for the quality control of glycosylphosphatidylinositol-anchored proteins’, Mol Biol Cell, vol. 17, no. 2, pp. 834–850, 2006.
- C. Harty, S. Strahl, and K. Romisch, ‘O-mannosylation protects mutant alpha-factor precursor from endoplasmic reticulum-associated degradation’, Mol Biol Cell, vol. 12, no. 4, pp. 1093–1101, 2001.
- C. Xu, S. Wang, G. Thibault, and D. T. W. Ng, ‘Futile Protein Folding Cycles in the ER Are Terminated by the Unfolded Protein O-Mannosylation Pathway’, Science (1979), vol. 340, no. 6135, pp. 978 LP-- 981, 2013.
- L. Lemus, H. Meyer, A. I. Rodríguez-Rosado, M. Schuldiner, and V. Goder, ‘O-mannosylation of misfolded ER proteins promotes ERAD’, EMBO J, vol. in press, Nov. 2025. [CrossRef]
- A. Uttenweiler, H. Schwarz, H. Neumann, and A. Mayer, ‘The Vacuolar Transporter Chaperone (VTC) Complex Is Required for Microautophagy’, Mol Biol Cell, vol. 18, no. 1, pp. 166–175, Jan. 2007. [CrossRef]
- C. W. Wang, Y. H. Miao, and Y. S. Chang, ‘A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast’, Journal of Cell Biology, vol. 206, no. 3, pp. 357–366, 2014. [CrossRef]
- T. Tsuji et al., ‘Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole’, Elife, vol. 6, Jun. 2017. [CrossRef]
- L. Lemus and V. Goder, ‘Pep4-dependent microautophagy is required for post-ER degradation of GPI-anchored proteins’, Autophagy, vol. 18, no. 1, pp. 223–225, 2022. [CrossRef]
- G. G. Kovács, G. Trabattoni, J. A. Hainfellner, J. W. Ironside, R. S. G. Knight, and H. Budka, ‘Mutations of the prion protein gene phenotypic spectrum’, J Neurol, vol. 249, no. 11, pp. 1567–1582, 2002. [CrossRef]
- A. Ruiz-Argüelles and L. Llorente, ‘The role of complement regulatory proteins (CD55 and CD59) in the pathogenesis of autoimmune hemocytopenias’, Autoimmun Rev, vol. 6, no. 3, pp. 155–161, Jan. 2007. [CrossRef]
- B. Puig, H. Altmeppen, and M. Glatzel, ‘The GPI-anchoring of PrP: implications in sorting and pathogenesis’, Prion, vol. 8, no. 1, pp. 11–18, 2014.
- J. Ma and S. Lindquist, ‘Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation’, Proc Natl Acad Sci U S A, vol. 98, no. 26, pp. 14955–14960, 2001.
- Y. Yedidia, L. Horonchik, S. Tzaban, A. Yanai, and A. Taraboulos, ‘Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein’, Embo J, vol. 20, no. 19, pp. 5383–5391, 2001.
- G. Petris et al., ‘CD4 and BST-2/tetherin proteins retro-translocate from endoplasmic reticulum to cytosol as partially folded and multimeric molecules’, J Biol Chem, vol. 289, no. 1, pp. 1–12, 2014.
- Y. J. Wang et al., ‘The association of the vanin-1 N131S variant with blood pressure is mediated by endoplasmic reticulum-associated degradation and loss of function’, PLoS Genet, vol. 10, no. 9, p. e1004641, 2014.
- B. Drisaldi et al., ‘Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation’, J Biol Chem, vol. 278, no. 24, pp. 21732–21743, 2003.
- Y.-S. S. Liu et al., ‘N -Glycan–dependent protein folding and endoplasmic reticulum retention regulate GPI-anchor processing’, J Cell Biol, vol. 217, no. 2, pp. 585–599, Feb. 2018. [CrossRef]
- X. Y. Guo, Y. S. Liu, X. D. Gao, T. Kinoshita, and M. Fujita, ‘Calnexin mediates the maturation of GPI-anchors through ER retention’, vol. 295, no. 48, pp. 16393–16410, Nov. 2020.
- A. M. Cheatham, N. R. Sharma, and P. Satpute-Krishnan, ‘Competition for calnexin binding regulates secretion and turnover of misfolded GPI-anchored proteins’, Journal of Cell Biology, vol. 222, no. 10, Oct. 2023. [CrossRef]
- J. Hong et al., ‘Molecular basis of the inositol deacylase PGAP1 involved in quality control of GPI-AP biogenesis’, Nat Commun, vol. 15, no. 1, Dec. 2024. [CrossRef]
- P. Satpute-Krishnan, M. Ajinkya, S. Bhat, E. Itakura, R. S. Hegde, and J. Lippincott-Schwartz, ‘ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway’, Cell, vol. 158, no. 3, pp. 522–533, 2014, [Online]. Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=25083867.
- C. Stordeur, K. Puth, J. P. Saenz, and R. Ernst, ‘Crosstalk of lipid and protein homeostasis to maintain membrane function’, Biol Chem, vol. 395, no. 3, pp. 313–326, 2014.
- Y. Wang et al., ‘Cross-talks of glycosylphosphatidylinositol biosynthesis with glycosphingolipid biosynthesis and ER-associated degradation’, Nat Commun, vol. 11, no. 1, Dec. 2020. [CrossRef]
- C. Hetz and C. Soto, ‘Stressing out the ER: a role of the unfolded protein response in prion-related disorders’, Curr Mol Med, vol. 6, no. 1, pp. 37–43, Feb. 2006. [CrossRef]
- E. Ronzier and P. Satpute-Krishnan, ‘TMED9 coordinates the clearance of misfolded GPI-anchored proteins out of the ER and into the Golgi’, PLoS Biol, vol. 23, no. 4, Apr. 2025. [CrossRef]
- E. Zavodszky and R. S. Hegde, ‘Misfolded GPI-anchored proteins are escorted through the secretory pathway by ER-derived factors’, Elife, vol. 8, May 2019. [CrossRef]
- N. Desnoyer and R. Palanivelu, ‘Bridging the GAPs in plant reproduction: a comparison of plant and animal GPI-anchored proteins’, Plant Reprod, vol. 33, no. 3–4, pp. 129–142, Dec. 2020. [CrossRef]
- Z. Xu et al., ‘Glycosylphosphatidylinositol anchor lipid remodeling directs proteins to the plasma membrane and governs cell wall mechanics’, Aug. 2022. [CrossRef]
- Y. J. Shin, U. Vavra, and R. Strasser, ‘Proper protein folding in the endoplasmic reticulum is required for attachment of a glycosylphosphatidylinositol anchor in plants’, Plant Physiol, vol. 186, no. 4, pp. 1878–1892, Aug. 2021. [CrossRef]
- M. J. Skelly, ‘Dropping anchor: Stringent quality control prevents GPI anchoring of severely misfolded proteins in plants’, Aug. 01, 2021, Plant Physiol. [CrossRef]
- M. A. McDowell, D. M. Ransom, and J. D. Bangs, ‘Glycosylphosphatidylinositol-dependent secretory transport in Trypanosoma brucei’, Biochem J, vol. 335 ( Pt 3), no. Pt 3, pp. 681–689, Nov. 1998. [CrossRef]
- J. D. Bangs, D. M. Ransom, M. A. McDowell, and E. M. Brouch, ‘Expression of bloodstream variant surface glycoproteins in procyclic stage Trypanosoma brucei: role of GPI anchors in secretion’, EMBO J, vol. 16, no. 14, pp. 4285–4294, Jul. 1997. [CrossRef]
- C. Tiengwe, C. M. Koeller, and J. D. Bangs, ‘Endoplasmic reticulum-associated degradation and disposal of misfolded GPI-anchored proteins in Trypanosoma brucei’, Mol Biol Cell, vol. 29, no. 20, pp. 2397–2409, Oct. 2018. [CrossRef]
- F. Paturiaux-Hanocq et al., ‘A role for the dynamic acylation of a cluster of cysteine residues in regulating the activity of the glycosylphosphatidylinositol-specific phospholipase C of Trypanosoma brucei’, J Biol Chem, vol. 275, no. 16, pp. 12147–12155, Apr. 2000. [CrossRef]
- A. J. Szempruch et al., ‘Extracellular Vesicles from Trypanosoma brucei Mediate Virulence Factor Transfer and Cause Host Anemia’, Cell, vol. 164, no. 1–2, pp. 246–257, Jan. 2016. [CrossRef]
- P. Garrison et al., ‘Turnover of Variant Surface Glycoprotein in Trypanosoma brucei Is a Bimodal Process’, mBio, vol. 12, no. 4, pp. 1725–1746, Aug. 2021. [CrossRef]
- J. D. Bangs, ‘Evolution of Antigenic Variation in African Trypanosomes: Variant Surface Glycoprotein Expression, Structure, and Function’, Bioessays, vol. 40, no. 12, Dec. 2018. [CrossRef]
- K. Halbleib et al., ‘Activation of the Unfolded Protein Response by Lipid Bilayer Stress’, Mol Cell, vol. 67, no. 4, pp. 673-684.e8, Aug. 2017. [CrossRef]
- R. Covino, G. Hummer, and R. Ernst, ‘Integrated Functions of Membrane Property Sensors and a Hidden Side of the Unfolded Protein Response’, Mol Cell, vol. 71, no. 3, pp. 458–467, Aug. 2018. [CrossRef]
- M. C. Jonikas et al., ‘Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum’, Science (1979), vol. 323, no. 5922, pp. 1693–1697, 2009.
- W. J. Masterson, J. Raper, T. L. Doering, G. W. Hart, and P. T. Englund, ‘Fatty acid remodeling: a novel reaction sequence in the biosynthesis of trypanosome glycosyl phosphatidylinositol membrane anchors’, Cell, vol. 62, no. 1, pp. 73–80, Jul. 1990. [CrossRef]
Figure 1.
Scheme of the preassembled GPI anchor in the yeast ER. The core GPI structure Manα1-4GlcNα1-6myo-inositol-1-P-lipid is conserved across eukaryotes. Yeast GPI anchors typically contain four mannose residues, as shown, whereas most mammalian GPIs contain three. Proteins are usually linked to the GPI anchor via an ethanolamine phosphate bridge connecting the C6 hydroxyl of the third mannose to the α-carboxyl group of the protein’s C-terminal amino acid (arrow). Beyond the conserved core, GPI anchors can carry additional linear or branched glycan modifications of largely unknown function; these vary depending on the attached protein and the organism in which the anchor is synthesized. Differences also exist in the phosphatidylinositol lipid moieties, which may be modified during GPI precursor assembly (in mammals) or more generally during remodeling after protein attachment. Variations include diacylglycerol as well as lysoacyl-, alkylacyl-, and alkenylacyl-phosphatidylinositols, and in yeast many remodeled GPIs contain phosphoceramide. Furthermore, the C2 hydroxyl of inositol is initially esterified to a fatty acid, which is typically removed during the remodeling process.
Figure 1.
Scheme of the preassembled GPI anchor in the yeast ER. The core GPI structure Manα1-4GlcNα1-6myo-inositol-1-P-lipid is conserved across eukaryotes. Yeast GPI anchors typically contain four mannose residues, as shown, whereas most mammalian GPIs contain three. Proteins are usually linked to the GPI anchor via an ethanolamine phosphate bridge connecting the C6 hydroxyl of the third mannose to the α-carboxyl group of the protein’s C-terminal amino acid (arrow). Beyond the conserved core, GPI anchors can carry additional linear or branched glycan modifications of largely unknown function; these vary depending on the attached protein and the organism in which the anchor is synthesized. Differences also exist in the phosphatidylinositol lipid moieties, which may be modified during GPI precursor assembly (in mammals) or more generally during remodeling after protein attachment. Variations include diacylglycerol as well as lysoacyl-, alkylacyl-, and alkenylacyl-phosphatidylinositols, and in yeast many remodeled GPIs contain phosphoceramide. Furthermore, the C2 hydroxyl of inositol is initially esterified to a fatty acid, which is typically removed during the remodeling process.
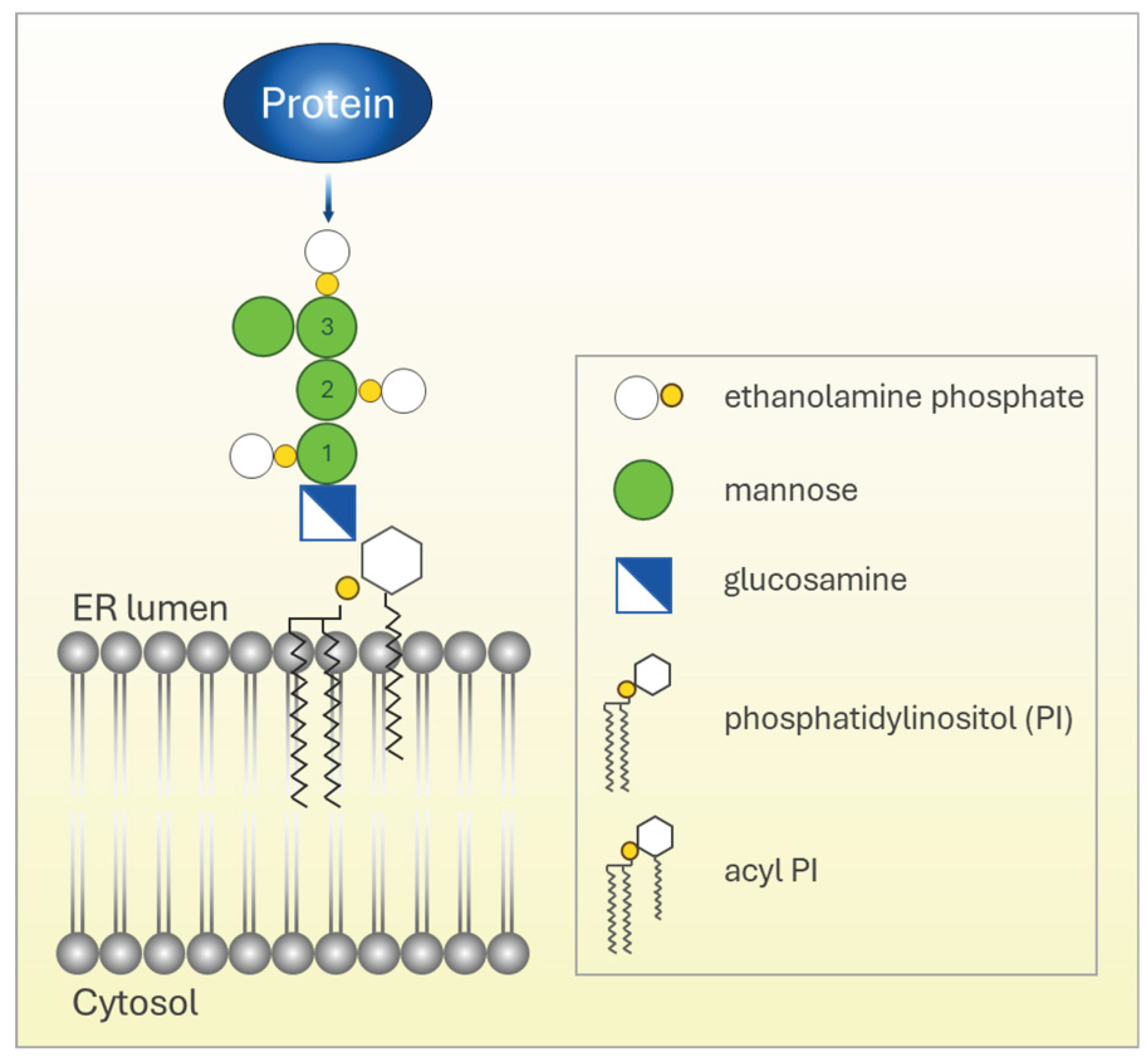
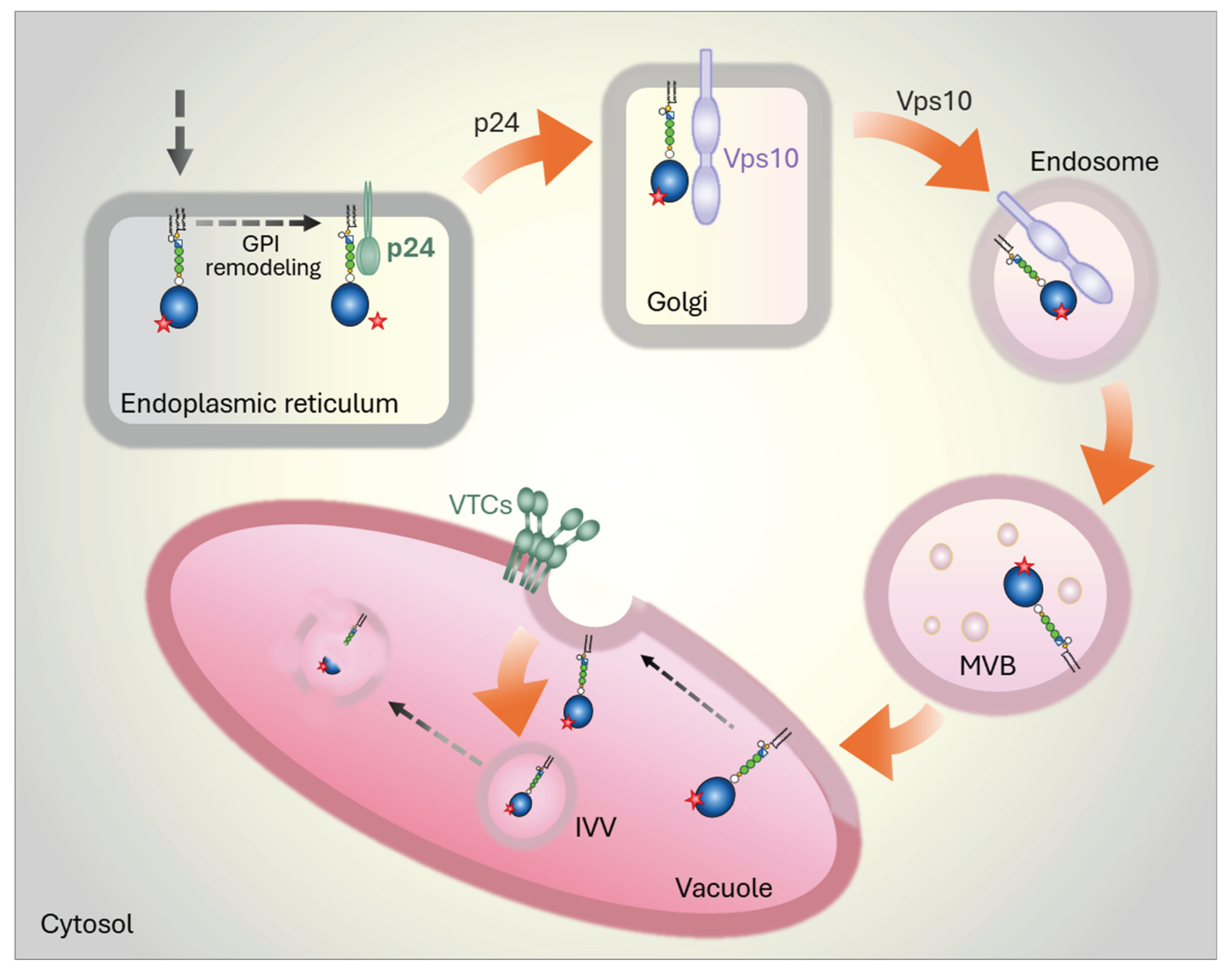
Figure 2.
Degradation pathways for misfolded GPI-APs in yeast. After protein synthesis and translocation into the ER, the C-terminal TM domain (not shown) is removed, and the protein is attached to a preassembled GPI anchor. Two degradation pathways are known in yeast and are used to different extents, as illustrated by the arrow thicknesses. Top half: a variable but generally minor fraction of misfolded GPI-APs (red star indicates misfolding) is routed to ERAD. It remains unknown what happens to the GPI anchor during retrotranslocation of GPI-APs via the Hrd1-complex. Bottom half: Following attachment, the GPI anchor undergoes remodeling, a process that is independent of protein folding and occurs even on misfolded GPI-APs. The remodeled GPI anchor acts as a strong ER export signal and promotes p24-dependent vesicular export of misfolded GPI-APs. Once exported, these proteins are subject to post-ER QC and are ultimately directed to the vacuole for degradation. Major components and mechanisms governing this post-ER degradation pathway have recently been elucidated.
Figure 2.
Degradation pathways for misfolded GPI-APs in yeast. After protein synthesis and translocation into the ER, the C-terminal TM domain (not shown) is removed, and the protein is attached to a preassembled GPI anchor. Two degradation pathways are known in yeast and are used to different extents, as illustrated by the arrow thicknesses. Top half: a variable but generally minor fraction of misfolded GPI-APs (red star indicates misfolding) is routed to ERAD. It remains unknown what happens to the GPI anchor during retrotranslocation of GPI-APs via the Hrd1-complex. Bottom half: Following attachment, the GPI anchor undergoes remodeling, a process that is independent of protein folding and occurs even on misfolded GPI-APs. The remodeled GPI anchor acts as a strong ER export signal and promotes p24-dependent vesicular export of misfolded GPI-APs. Once exported, these proteins are subject to post-ER QC and are ultimately directed to the vacuole for degradation. Major components and mechanisms governing this post-ER degradation pathway have recently been elucidated.
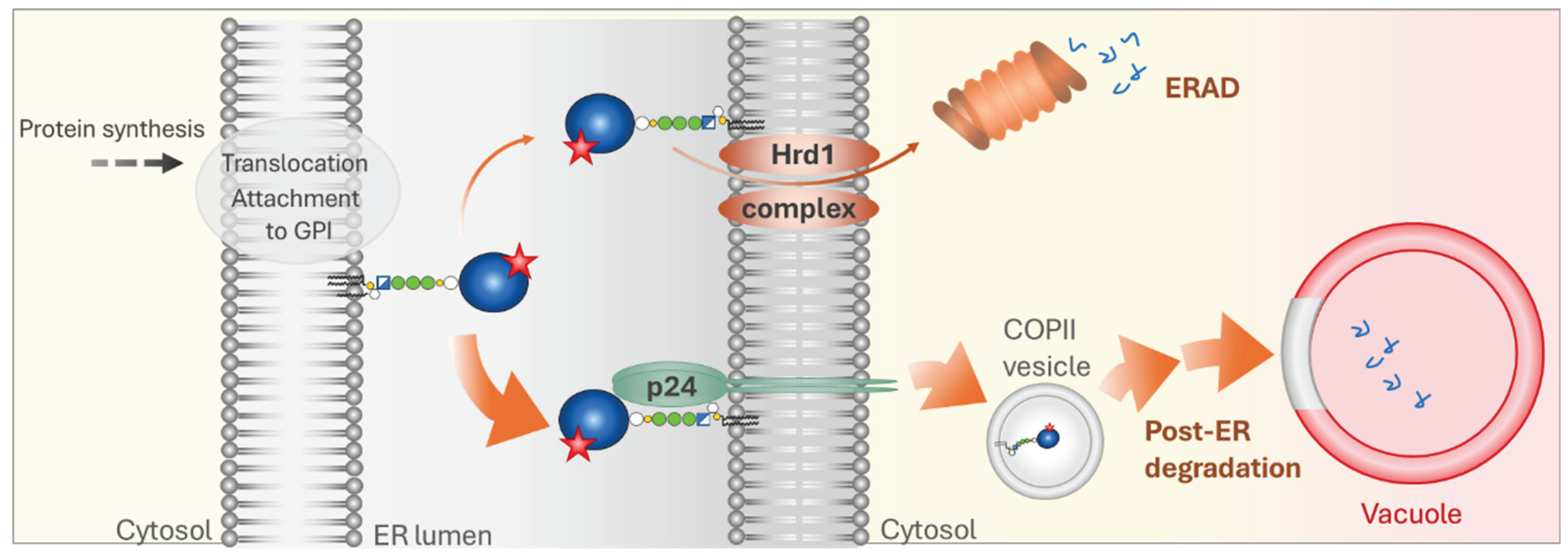
Figure 4.
Degradation pathways for misfolded GPI-APs in mammalian cells. Several studies have demonstrated a specific role for Calnexin/Calreticulin-dependent ERQC in retaining misfolded GPI-APs in the ER. GPI anchor remodeling has also been linked to Calnexin/Calreticulin, providing a physical connection between anchor remodeling and protein folding. Under conditions of ER stress, retention of misfolded GPI-APs is markedly reduced, leading to their rapid p24-dependent export from the ER and subsequent routing to lysosomes for post-ER degradation. In contrast to yeast, misfolded GPI-APs in mammalian cells are first transported to the plasma membrane in a complex with Calnexin before being delivered to lysosomes. Surface exposure of Calnexin is required to trigger endocytosis of these complexes. The precise trafficking itinerary from the cell surface to lysosomes remains unclear. Similar to yeast, little to no degradation of misfolded GPI-APs occurs via ERAD.
Figure 4.
Degradation pathways for misfolded GPI-APs in mammalian cells. Several studies have demonstrated a specific role for Calnexin/Calreticulin-dependent ERQC in retaining misfolded GPI-APs in the ER. GPI anchor remodeling has also been linked to Calnexin/Calreticulin, providing a physical connection between anchor remodeling and protein folding. Under conditions of ER stress, retention of misfolded GPI-APs is markedly reduced, leading to their rapid p24-dependent export from the ER and subsequent routing to lysosomes for post-ER degradation. In contrast to yeast, misfolded GPI-APs in mammalian cells are first transported to the plasma membrane in a complex with Calnexin before being delivered to lysosomes. Surface exposure of Calnexin is required to trigger endocytosis of these complexes. The precise trafficking itinerary from the cell surface to lysosomes remains unclear. Similar to yeast, little to no degradation of misfolded GPI-APs occurs via ERAD.
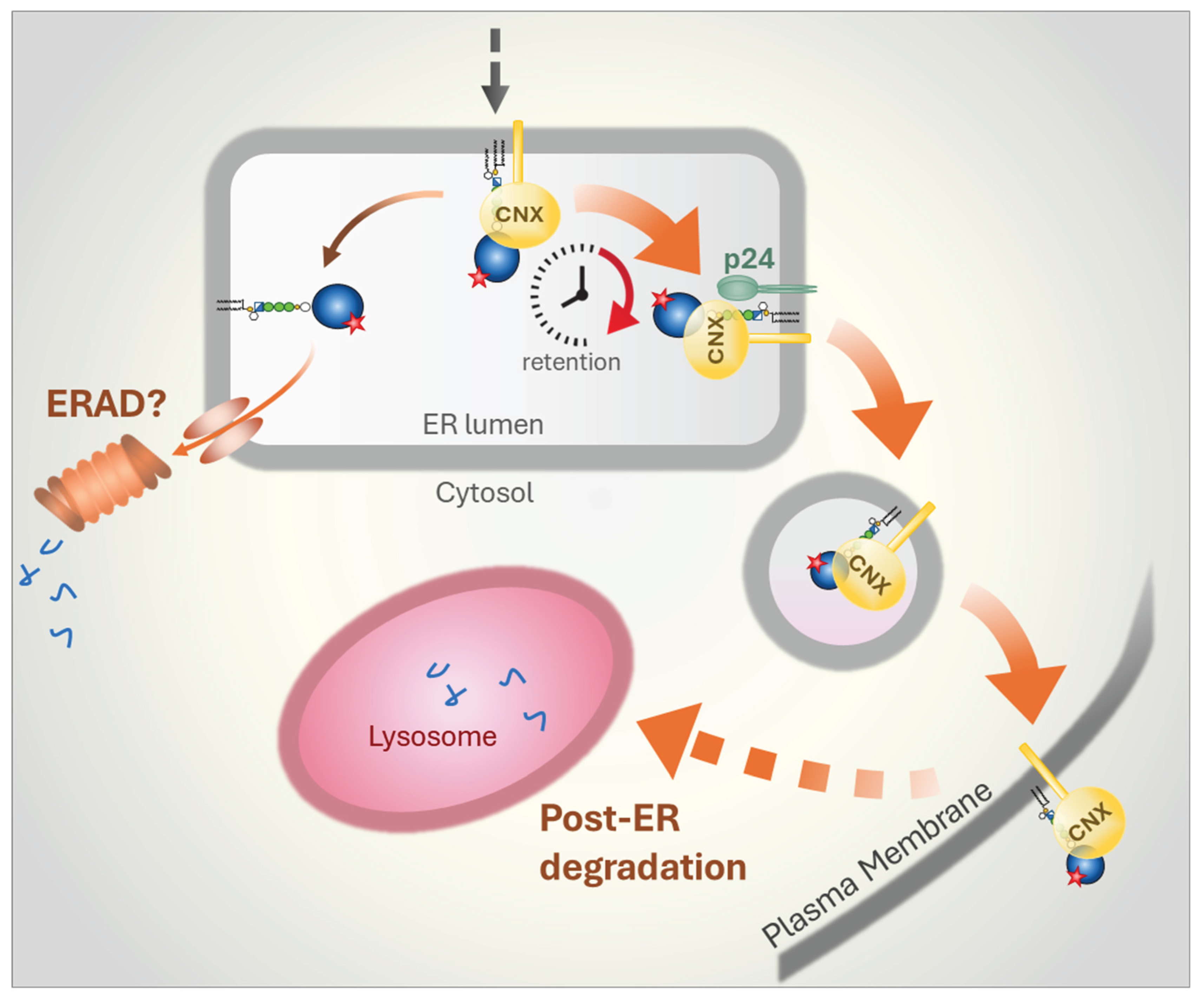
Figure 5.
Degradation pathways for misfolded GPI-APs in plants. Current evidence suggests that, as in yeast, most misfolded GPI-APs in plant cells are routed to the vacuole for degradation through post-ER QC pathways that remain largely unexplored. The available data further indicate that proteins containing severely misfolded domains (yellow star) are prevented from being attached to preassembled GPI anchors within the ER. This preemptive form of ERQC preserves the proteins’ original C-terminal TM domain and enables efficient ERAD of these species. The degree to which this pathway is used is not known. In addition, whether such a preemptive ERQC mechanism for GPI-APs operates in yeast or mammalian cells remains unknown.
Figure 5.
Degradation pathways for misfolded GPI-APs in plants. Current evidence suggests that, as in yeast, most misfolded GPI-APs in plant cells are routed to the vacuole for degradation through post-ER QC pathways that remain largely unexplored. The available data further indicate that proteins containing severely misfolded domains (yellow star) are prevented from being attached to preassembled GPI anchors within the ER. This preemptive form of ERQC preserves the proteins’ original C-terminal TM domain and enables efficient ERAD of these species. The degree to which this pathway is used is not known. In addition, whether such a preemptive ERQC mechanism for GPI-APs operates in yeast or mammalian cells remains unknown.
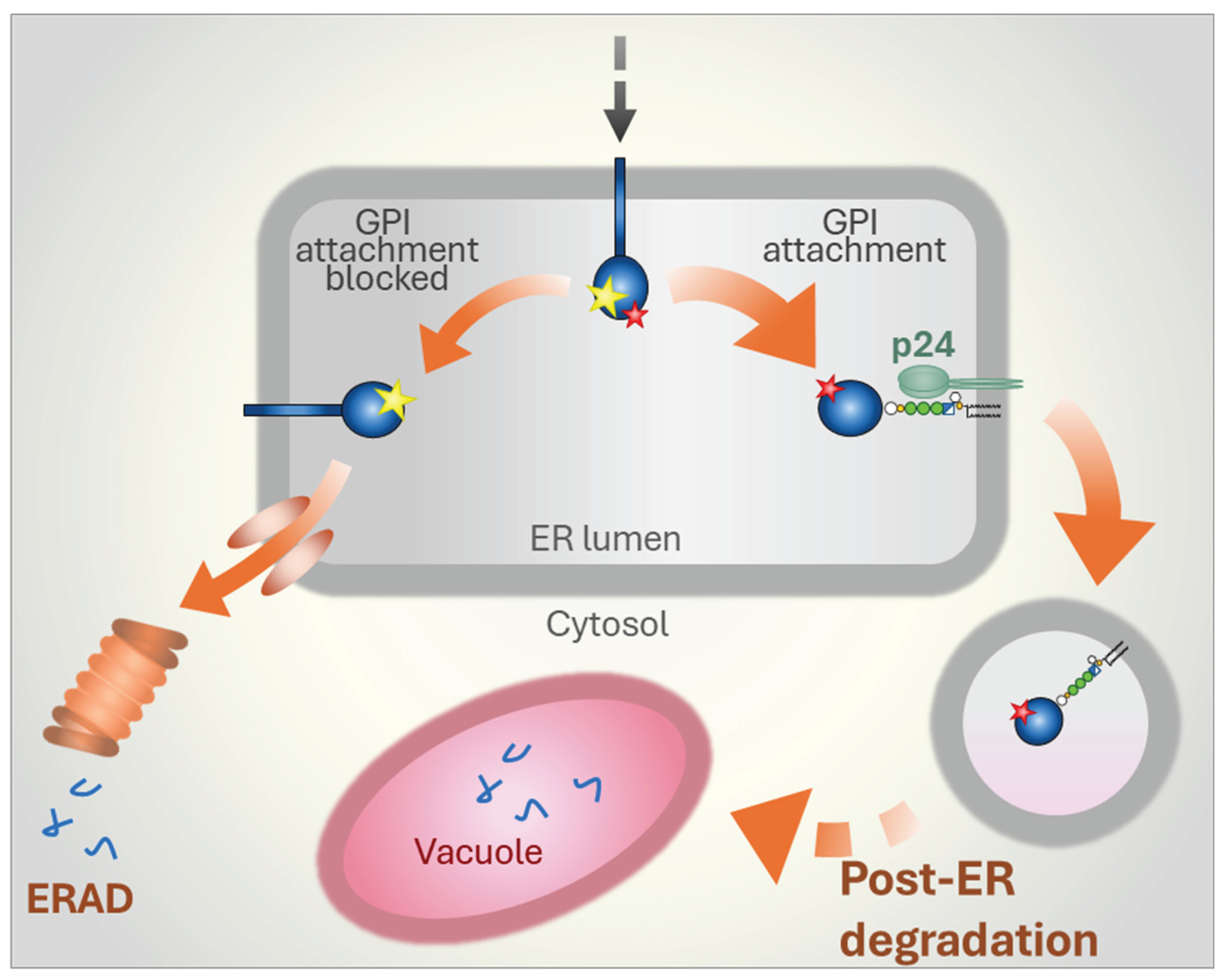
Figure 6.
Degradation pathways for misfolded GPI-APs in protozoa. Data from the parasite Trypanosoma brucei indicate that misfolded GPI-APs are efficiently routed to ERAD, with little to no protein directed to lysosomes for post-ER degradation. The requirement to synthesize large quantities of GPI-APs at specific life cycle stages, combined with the need for stringent QC, may have driven the evolution of a dedicated ERAD branch for disposing of misfolded GPI-APs. A key component of this pathway appears to be a highly abundant, GPI-specific phospholipase C (GPI-PLC) present in the cytosol of T. brucei. The presence of similarly abundant GPI-APs on the surface of other parasites suggests that this efficient ERAD mechanism may be common among certain protozoa. Whether ERAD of misfolded GPI-APs in other organisms, such as yeast or mammalian cells, also relies on specific lipases or instead employs distinct mechanisms remains an open question.
Figure 6.
Degradation pathways for misfolded GPI-APs in protozoa. Data from the parasite Trypanosoma brucei indicate that misfolded GPI-APs are efficiently routed to ERAD, with little to no protein directed to lysosomes for post-ER degradation. The requirement to synthesize large quantities of GPI-APs at specific life cycle stages, combined with the need for stringent QC, may have driven the evolution of a dedicated ERAD branch for disposing of misfolded GPI-APs. A key component of this pathway appears to be a highly abundant, GPI-specific phospholipase C (GPI-PLC) present in the cytosol of T. brucei. The presence of similarly abundant GPI-APs on the surface of other parasites suggests that this efficient ERAD mechanism may be common among certain protozoa. Whether ERAD of misfolded GPI-APs in other organisms, such as yeast or mammalian cells, also relies on specific lipases or instead employs distinct mechanisms remains an open question.
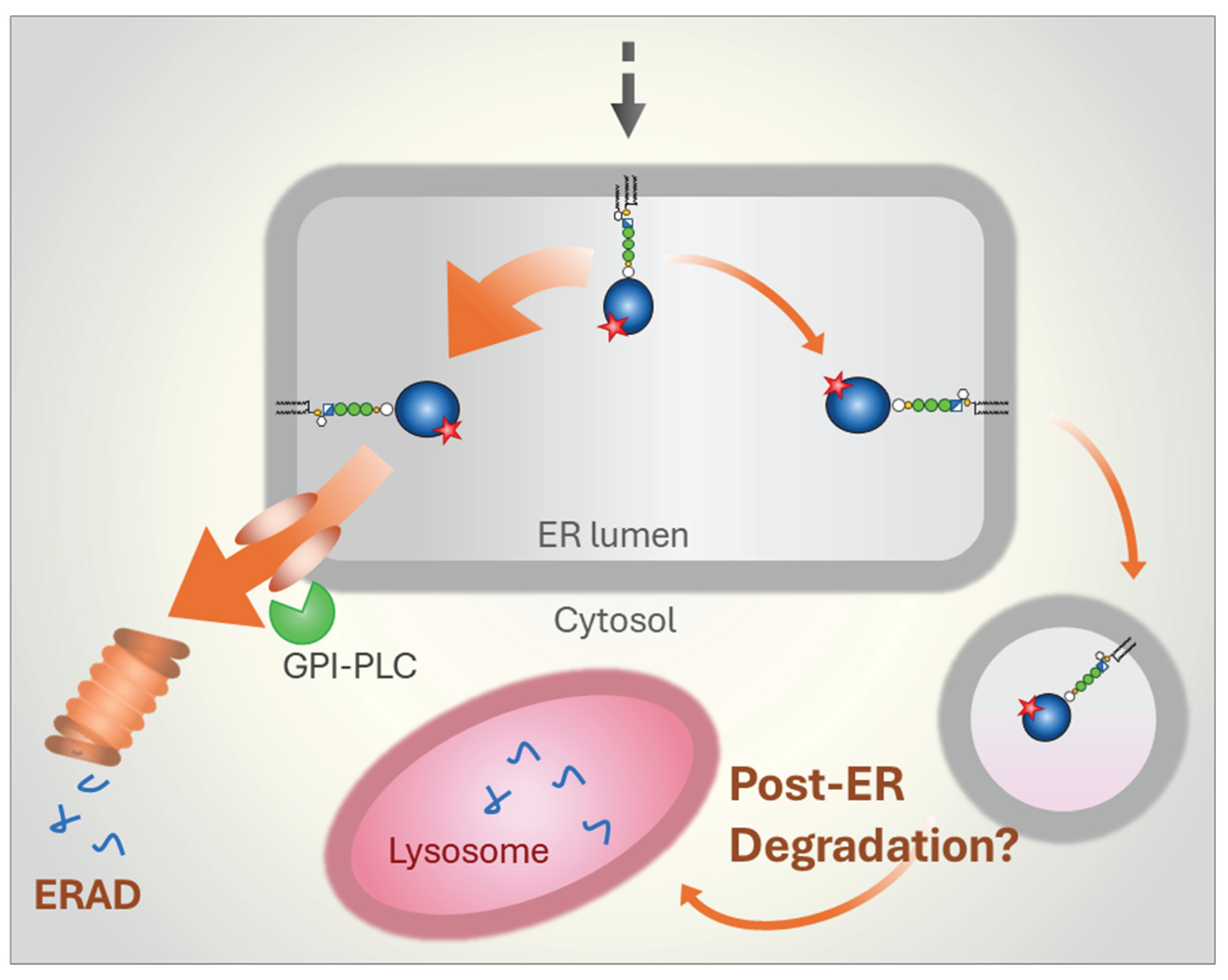
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



