1. Introduction
Periodontitis is an inflammatory condition affecting the supporting structures of the teeth and represents one of the most prevalent chronic infectious diseases in humans [
1]. This disease results in the destruction of periodontal connective tissue and resorption of the alveolar bone, constituting the primary cause of tooth loss in adults. Inflammation of the periodontal tissues can lead to localized tissue damage, ulceration, and bleeding within the lining of periodontal pockets [
1,
2]. In cases of severe, extensive periodontitis, the total ulcerated area on the inner walls of periodontal pockets may reach approximately 72 cm², which is comparable to the surface area of an adult human palm. This extensive ulceration facilitates transient but recurrent bacteremia, which can be induced by routine oral hygiene practices, periodontal treatments, and even mastication [
3,
4]. Consequently, periodontal pathogens such as
Porphyromonas gingivalis (
P. gingivalis) and their virulence factors can enter the bloodstream, potentially compromising the integrity and function of endothelial cells and exerting effects on vascular smooth muscle cells [
2].
Numerous ex vivo and in vivo studies have demonstrated that infection with
P. gingivalis induces the transformation of mouse macrophages into foam cells. Repeated bacteremia caused by
P. gingivalis has been shown to induce or exacerbate atherosclerosis (As)-like lesions in the aorta and coronary arteries of experimental pigs [
4,
5]. Furthermore,
P. gingivalis oral infection accelerates the progression of early atherosclerotic lesions in apolipoprotein E knockout (ApoE-/-) mice [
4]. However, despite numerous studies suggesting a link between
P. gingivalis infection and the development of As and the frequent detection of bacterial biomolecules such as nucleic acids within atherosclerotic plaques, live bacteria are rarely isolated from these lesions [
6]. Moreover, some clinical studies have reported that antibiotic treatment does not significantly reduce vascular wall inflammation or subsequent adverse cardiovascular events in patients with periodontitis and cardiovascular disease (CVD) [
7,
8]. This paradoxical observation raises important questions regarding the hypothesis that
P. gingivalis influences the progression of As and the association between periodontitis and As.
Gram-negative bacteria continuously produce and secrete extracellular vesicles (EVs) during their growth [
9]. Among the principal periodontal pathogens,
P. gingivalis is recognized as a particularly prolific producer of EVs. These EVs are double-membrane vesicles ranging from approximately 20 to 250 nm in diameter and are enriched with key virulence factors, including lipopolysaccharide (LPS) and gingipains. Notably,
P. gingivalis EVs exhibit greater stability and pathogenic potential compared to their parent bacterial cells [
9]. Functionally,
P. gingivalis EVs contribute to adhesion and colonization, host cell invasion, biofilm formation, host cell damage, and modulation of host immune responses, often exerting effects comparable to or exceeding those of the parental bacteria. Furthermore, studies have demonstrated that
P. gingivalis EVs can disseminate via the bloodstream to distant organs such as the heart, liver, lungs, and spleen, where they are implicated in the pathogenesis of various systemic diseases [
10].
Although the biogenesis of
P. gingivalis EVs has not been fully elucidated, various factors have been demonstrated to influence their production and yield [
9,
10]. Recent studies have shown that antibiotic-induced stress not only enhances the secretion of bacterial EVs, but also significantly modifies their pathogenic characteristics. For instance, Bauwens et al. [
11] reported that ciprofloxacin treatment increased the production of enterohemorrhagic
Escherichia coli (
E. coli) EVs and concurrently upregulated the levels of its principal virulence factor, Shiga toxin 2a, potentially exacerbating the severity of clinically relevant infections.
In summary, the pathogenic effects and mechanisms of bacterial EVs produced under antibiotic-induced stress conditions may differ from those generated under conventional conditions. To date, the influence of antibiotics—particularly those commonly employed in periodontal therapy—on the biogenesis of P. gingivalis EVs and their pathogenic characteristics remains unclear. Therefore, the present study aims to investigate the impact of sub-minimum inhibitory concentration (sub-MIC) metronidazole on the biogenesis of P. gingivalis EVs and to evaluate whether these EVs enhance cytotoxicity, induce oxidative stress, and exacerbate inflammatory responses in endothelial cells. This investigation seeks to provide a foundation for elucidating the underlying mechanisms contributing to the limited clinical efficacy of antibiotics in the treatment of periodontitis-associated CVD.
2. Materials and Methods
2.1. Bacterial Culture
P. gingivalis (strain ATCC 33277) was purchased from BeiNa Biological Company (BNCC, Beijing, China) and initially cultured on blood agar plates at 37 °C under anaerobic conditions (80% N₂, 10% CO₂, 10% H₂) for 6–8 days to reactivate the strain. Subsequently, the activated bacteria were inoculated at 5% (v/v) into TSB (Cat No: BNCC368224, BNCC) supplemented with 0.5% hemoglobin chloride (Cat No: H8132, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) and 0.1% vitamin K₁ (Cat No: V8151, Solarbio). Cultures were incubated anaerobically at 37 °C until reaching the logarithmic growth phase, with subculturing performed every 54 hours to maintain bacterial viability. After three successive centrifugation steps (1000 × g, 5 min), the bacterial pellet was collected, resuspended in fresh medium, and adjusted to a final concentration of 1 × 10⁶ CFU/mL for subsequent use.
2.2. Transmission Electron Microscopy Observations
P. gingivalis bacteria, with or without sub-MIC metronidazole treatment for 30 minutes, were fixed with 2.5% glutaraldehyde at 4 °C for 24 hours. Subsequently, the bacterial pellets were collected by centrifugation at 3,000 × g for 20 minutes and fixed again with fresh 2.5% glutaraldehyde for another 24 hours. After post-fixation with 2% osmium tetroxide for 2 hours, the samples were dehydrated through a graded ethanol series (30%, 50%, 70%, 85%, 95%, 100%, and 100%), dried in acetone (three changes, 15 minutes each), and embedded in resin blocks. The blocks were sectioned into ultrathin (70 nm) slices, which were stained with uranyl acetate and lead citrate [
14]. The prepared samples were observed using a JEM-1400FLASH transmission electron microscopy (TEM; JEOL, Japan).
2.3. Scanning Electron Microscope Observation
Sterile glass coverslips (φ = 24 mm) were placed in 6-well plates. Activated bacterial suspension (as described in
Section 2.1) at 1×10⁶ CFU/mL was added at 2 mL per well and incubated anaerobically for 2 hours. Metronidazole solution dissolved in DMSO was then added to each well to achieve a final concentration of sub-MIC, with an equal volume of PBS used as a blank control. After 30 minutes of anaerobic incubation at 37 °C, the bacteria were fixed with 2.5% glutaraldehyde at 4 °C for 24 hours. Alternatively, when
P. gingivalis cultures reached the late logarithmic growth phase under anaerobic conditions at 37 °C, 200 μL of the culture medium (untreated or treated with sub-MIC metronidazole) was transferred onto sterile glass coverslips (φ= 24 mm) in 6-well plates, allowed to adhere at room temperature for 1 hour, and then fixed with 2.5% glutaraldehyde at 4 °C for 24 hours. For all samples, the fixative was carefully aspirated, and the coverslips were washed by immersion in PBS buffer for 10 minutes (repeated three times). After PBS removal, dehydration was performed sequentially using 50%, 70%, and 90% ethanol (10 minutes each), followed by two changes of absolute ethanol (10 minutes each). The samples were then treated with an ethanol-pure tert-butanol mixture (1:1, V/V) for 15 minutes, followed by two changes of pure tert-butanol (15 minutes each). After tert-butanol removal, the samples were freeze-dried and sputter-coated with gold [
15]. Observation and imaging were performed using an SU8010 scanning electron microscope (SEM; Hitachi, Japan).
2.4. Isolation and Identification of P. gingivalis EVs
The protocol for extracting
P. gingivalis EVs was modified from a previously described method [
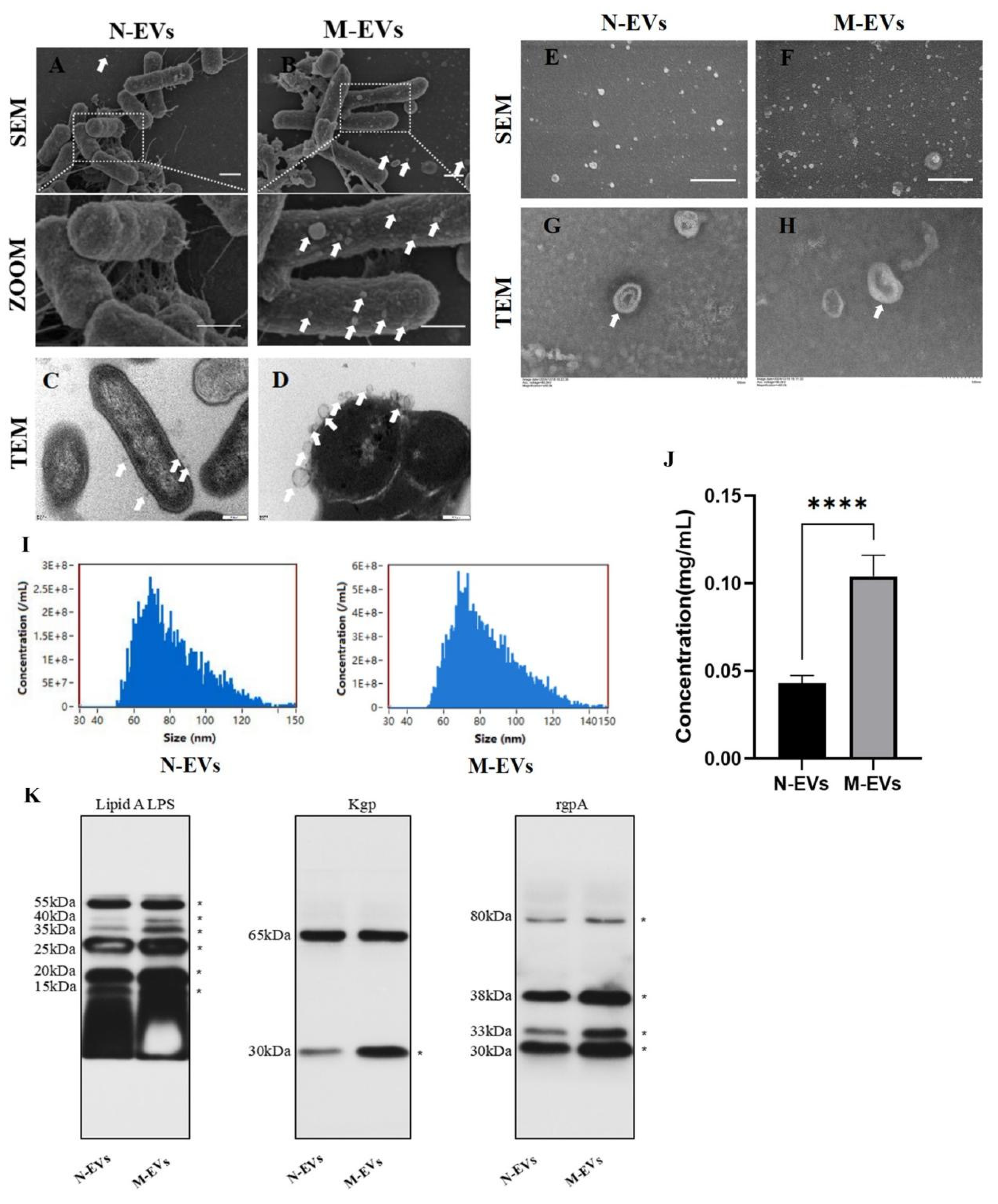
16]. Culture media from untreated or sub-MIC metronidazole-treated bacteria at the late logarithmic growth phase were centrifuged at 4 °C, 8,000 × g for 30 minutes to remove the bulk of bacterial cells. The resulting supernatant was collected and filtered through a 0.22-μm sterile filter to remove residual bacteria and debris. The filtrate was then concentrated using a 100 kDa ultrafiltration unit centrifuged at 4 °C, 2,500 × g for 30 minutes. The concentrate was transferred to a 30 mL ultracentrifuge tube and subjected to ultracentrifugation at 4 °C, 100,000 × g for 80 minutes. The pellet was resuspended in a small volume of sterile phosphate-buffered saline (PBS: Cat No: P1020, pH 7.2-7.4, Solarbio) and filtered again through a 0.22-μm filter to remove larger vesicles and residual debris. A second round of ultracentrifugation was performed under the same conditions (4 °C, 100,000 × g, 80 minutes). The final pellet was resuspended in 1.5 mL of sterile PBS, yielding naturally derived EVs (N-EVs) and sub-MIC metronidazole-treated derived EVs (M-EVs), which were stored at -80 °C for subsequent use.
For TEM characterization, 10 μL of N/M-EVs sample was applied onto a copper grid, allowed to settle for 1 minute, and the excess liquid was blotted away with filter paper. Then, 10 μL of uranyl acetate (Cat No: 1722586, Beijing Zhongjing Keqi Technology Co., Ltd., Beijing, China) was applied to the grid for 1 minute, and the excess was blotted off. After air-drying at room temperature for several minutes, the samples were imaged using an HT7700 TEM (Hitachi, Japan). For nanoparticle characterization, 10 μL of N/ M-EVs were each diluted to an appropriate concentration and analyzed using an N30E Nanoflow cytometer (NanoFCM, China). The instrument acquired and analyzed scattered light signals from individual particles to determine the EVs’ size distribution, average particle size, and concentration.
2.5. Protein Concentration Measurement
The protein concentration of the extracted P. gingivalis EVs was quantified using a bicinchoninic acid (BCA) protein assay kit (Cat No: PC0021, Solarbio) according to the manufacturer’s instructions.
2.6. Immunoblotting
EV samples were lysed using 80 μL of exosomal protein lysis buffer (Cat No: IL9020, Solarbio). Protein concentration was determined using the BCA protein assay kit as per the manufacturer’s protocol. For Western blotting, equal amounts of protein were loaded onto SDS-PAGE gels and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes (Cat No: IPVH00010, Millipore, Darmstadt, Germany). The membranes were blocked with 5% BSA for 30 minutes at room temperature. They were then incubated overnight at 4 °C with the following primary antibodies: lipid A LPS (Cat No: NB100-64484, Novus Biologicals, Littleton, CO, USA), Kgp (Cat No: abx338983, Abbexa, Cambridge, UK), and RgpA (Cat No: LS-C371047, LSBio, Seattle, WA, USA). Following primary antibody incubation, the membranes were incubated with corresponding species-specific secondary antibodies for 30 minutes at room temperature with shaking. Enhanced chemiluminescence (ECL) reagent was prepared by mixing the two components in a 1:1 ratio. The PVDF membranes were exposed to the ECL reagent, and images were automatically captured using a protein imaging system.
2.7. Cell Culture
HUVECs (Cat No: DFSC-EC-01, Shanghai ZQXZBIO Biotechnology Co., Ltd., Shanghai, China) were cultured in a specialized medium (Cat No:PCM-H-040, ZQXZBIO) according to the manufacturer’s instructions. The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. Upon reaching approximately 80% confluence, HUVECs were passaged and employed for experimental procedures between the third and fifth passages.
2.8. Endocytosis Analysis
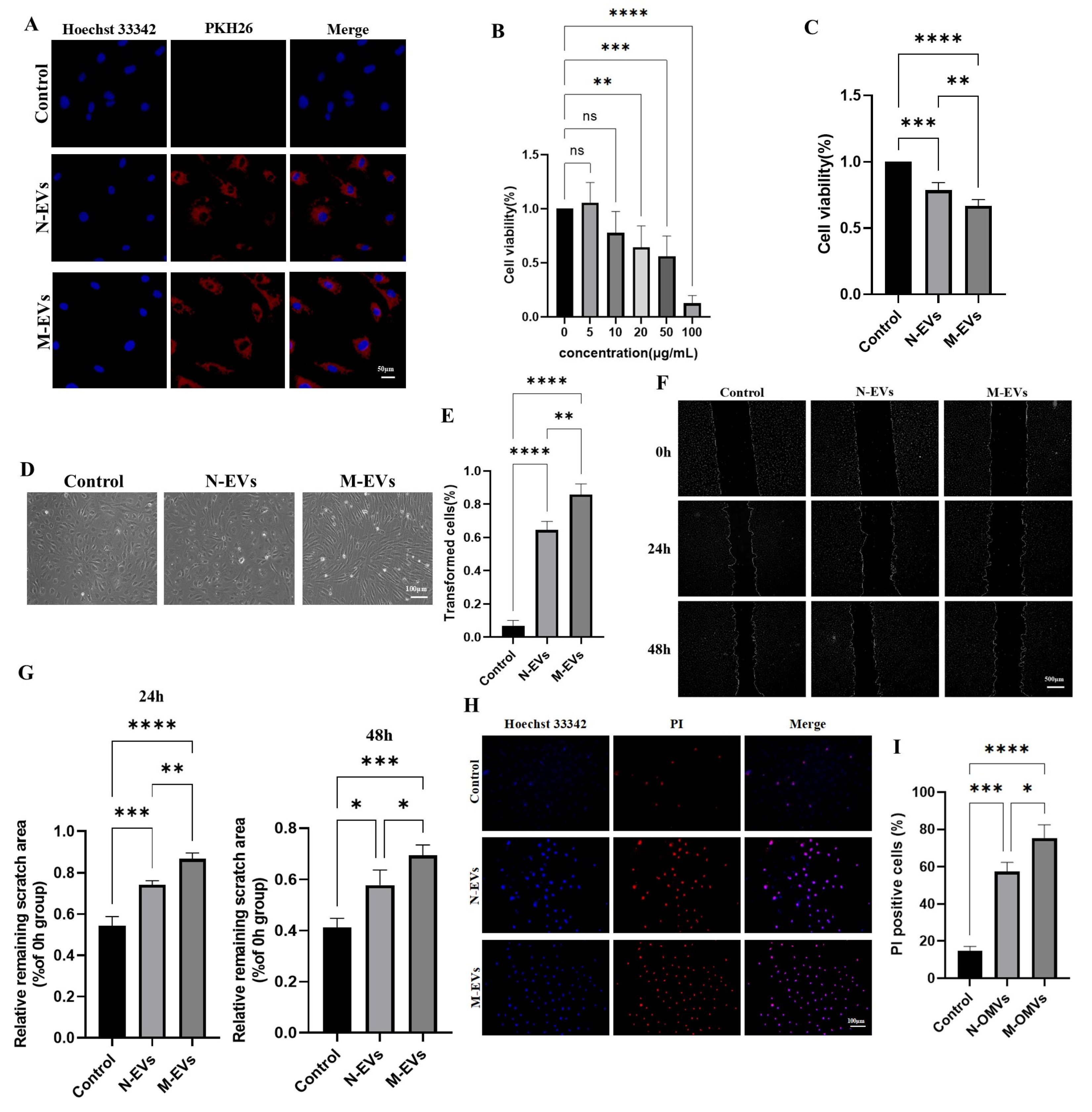
The lipid membranes of P. gingivalis EVs were labeled using the PKH26 red fluorescent cell linker kit (Cat No: UR52302, Umibio (Shanghai) Co., Ltd., Shanghai, China). Briefly, the extracted N-EVs and M-EVs were incubated with 1 μL PKH26 dye and 9 μL Diluent C in 100 μL PBS for 15 minutes (PBS buffer alone served as the control). Subsequently, 1 mL of PBS was added, and the mixture was centrifuged at 4 °C, 100,000 × g for 2 hours. The supernatant was discarded, and the pellet was resuspended in sterile PBS. HUVECs were then incubated with the PKH26-labeled N-EVs or M-EVs at 37 °C for 24 hours. After incubation, the cells were fixed with 4% paraformaldehyde for 15 minutes. Images were acquired using a Ti-s inverted fluorescence microscope (Nikon, Japan).
2.9. Cell Viability Analysis
Cell viability of HUVECs was assessed using the Cell Counting Kit-8 (CCK-8; Cat No: K1018, APExBIO, Houston, TX, USA). HUVECs were seeded in 96-well plates at a density of 1 × 10⁴ cells per well. After treatment with P. gingivalis EVs at various concentrations (0, 5, 10, 20, 50, and 100 μg/mL) for 24 hours, 10 μL of CCK-8 solution was added to each well. The plate was incubated for 24 hours, and the optical density (OD) at 450 nm was measured. Cell viability (%) was calculated as follows: [(OD experiment - OD control) / (OD control - OD blank)] × 100%.
2.10. Cell Morphology Analysis
HUVECs were seeded directly into 12-well plates at a density of 1 × 10⁵ cells per well and incubated overnight at 37 °C with 5% CO₂. The cells were then treated with 20 μg/mL of either N-EVs or M-EVs for 24 hours. After treatment, the old medium was removed, and the cells were gently washed three times with PBS. Fresh medium (1 mL) was added to each well, and changes in cell morphology were observed using an inverted optical microscope.
2.11. Cell Migration Capacity Analysis
HUVECs were seeded in 6-well plates at a density of 2 × 10⁵ cells per well. When the cells reached 80-90% confluence, a straight scratch was created using a 1000 μL pipette tip. The dislodged cells were washed away with PBS. An appropriate amount of Hoechst 33342 (Cat No: C1025, Shanghai Beyotime Biotechnology Co., Ltd., Shanghai, China) was added to stain the nuclei, incubating for 5 minutes. Images of the scratch center were captured at 0 hours using an inverted fluorescence microscope. After treatment with 20 μg/mL of N-EVs or M-EVs for 24 and 48 hours, respectively, the same staining and imaging procedures were repeated to capture images at the respective time points. The scratch area was measured and analyzed using ImageJ software.
2.12. Propidium Iodide Staining
The number of late apoptotic cells was detected using the Propidium Iodide (PI) Apoptosis Detection Kit (Cat No: C1352S, Beyotime) according to the manufacturer’s instructions. HUVECs were seeded on coverslips (1 × 1.0 cm) placed in 12-well plates at a density of 1 × 10⁵ cells per well and incubated overnight at 37 °C with 5% CO₂. After treatment with 20 μg/mL of N-EVs or M-EVs for 24 hours, the old medium was carefully aspirated. Without washing with PBS, 400 μL of binding buffer and 10 μL of PI staining solution were directly added to each well, followed by incubation in the dark at room temperature for 10 minutes. Subsequently, an appropriate amount of Hoechst 33342 was added and incubated for 5 minutes to stain all nuclei. Images were acquired directly using an inverted fluorescence microscope.
2.13. Reactive Oxygen Species Analysis
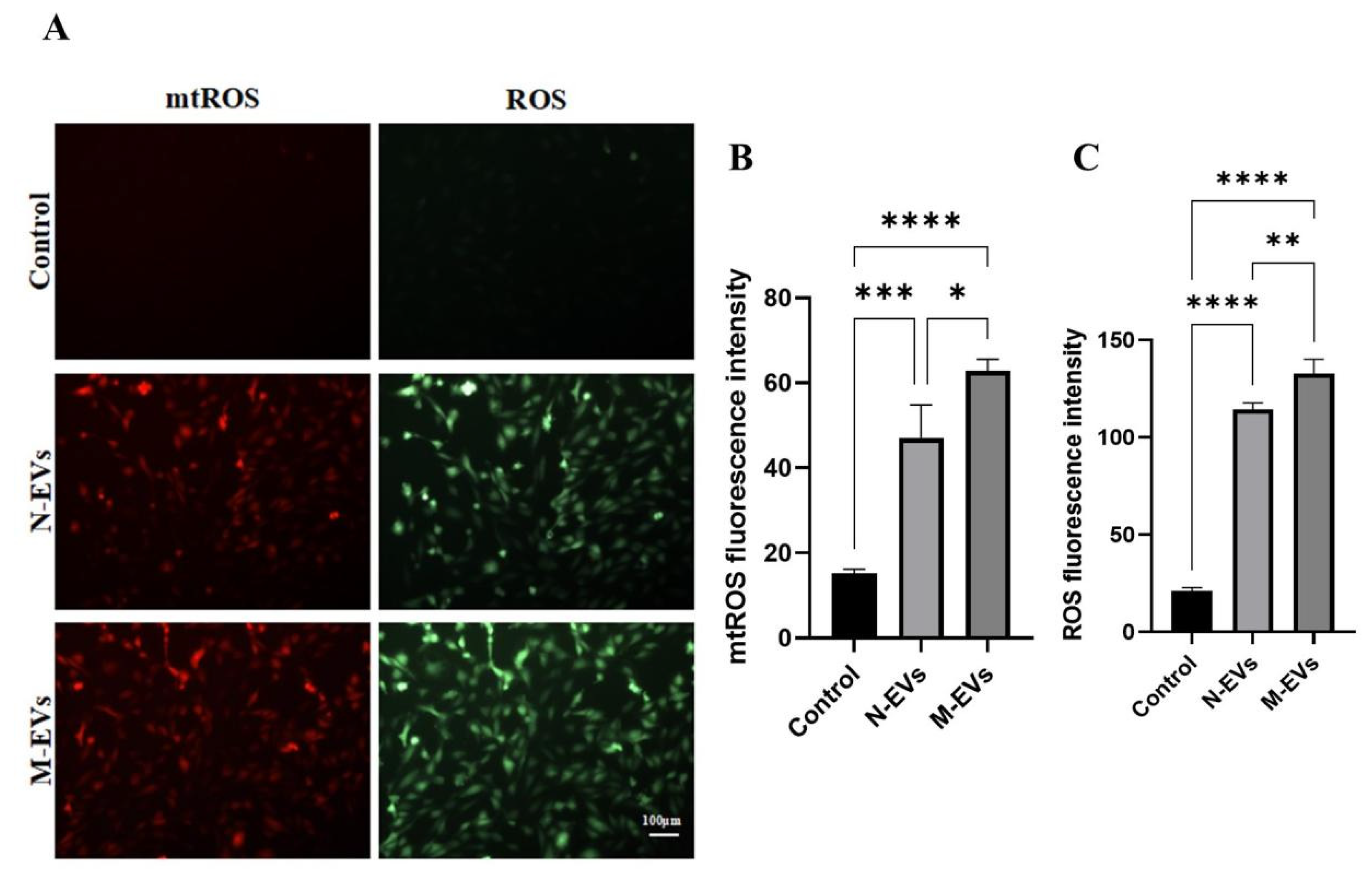
The levels of mitochondrial reactive oxygen species (mtROS) and total cellular reactive oxygen species (ROS) in HUVECs treated with 20 μg/mL of N-EVs or M-EVs for 24 hours were detected using Mito-SOX Red mitochondrial superoxide indicator (Cat No: BB25021, BestBio) and 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA; Cat No: S0033S, Beyotime) probes, respectively, following the manufacturers’ protocols. HUVECs were seeded in 12-well plates at a density of 1 × 10⁵ cells per well and incubated overnight at 37 °C with 5% CO₂. After respective treatments for 24 hours, the old medium was removed, and the cells were washed with PBS. Pre-warmed (37 °C) Mito-SOX Red working solution (prepared at a ratio of Mito-SOX Red fluorescent probe: probe diluent: medium = 1:10:200) was added, and the cells were incubated under growth conditions in the dark for 15 minutes. The cells were then washed with PBS. Subsequently, an appropriate amount of DCFH-DA probe was added and incubated in the dark for 30 minutes. After three washes with PBS, Hoechst 33342 was added and incubated for 5 minutes to stain nuclei. Following three additional PBS washes, 2 mL of fresh, pre-warmed (37 °C) medium was added. Images were captured using an inverted fluorescence microscope, and fluorescence intensity was analyzed using ImageJ V1.54g.
2.14. Immunofluorescence Staining
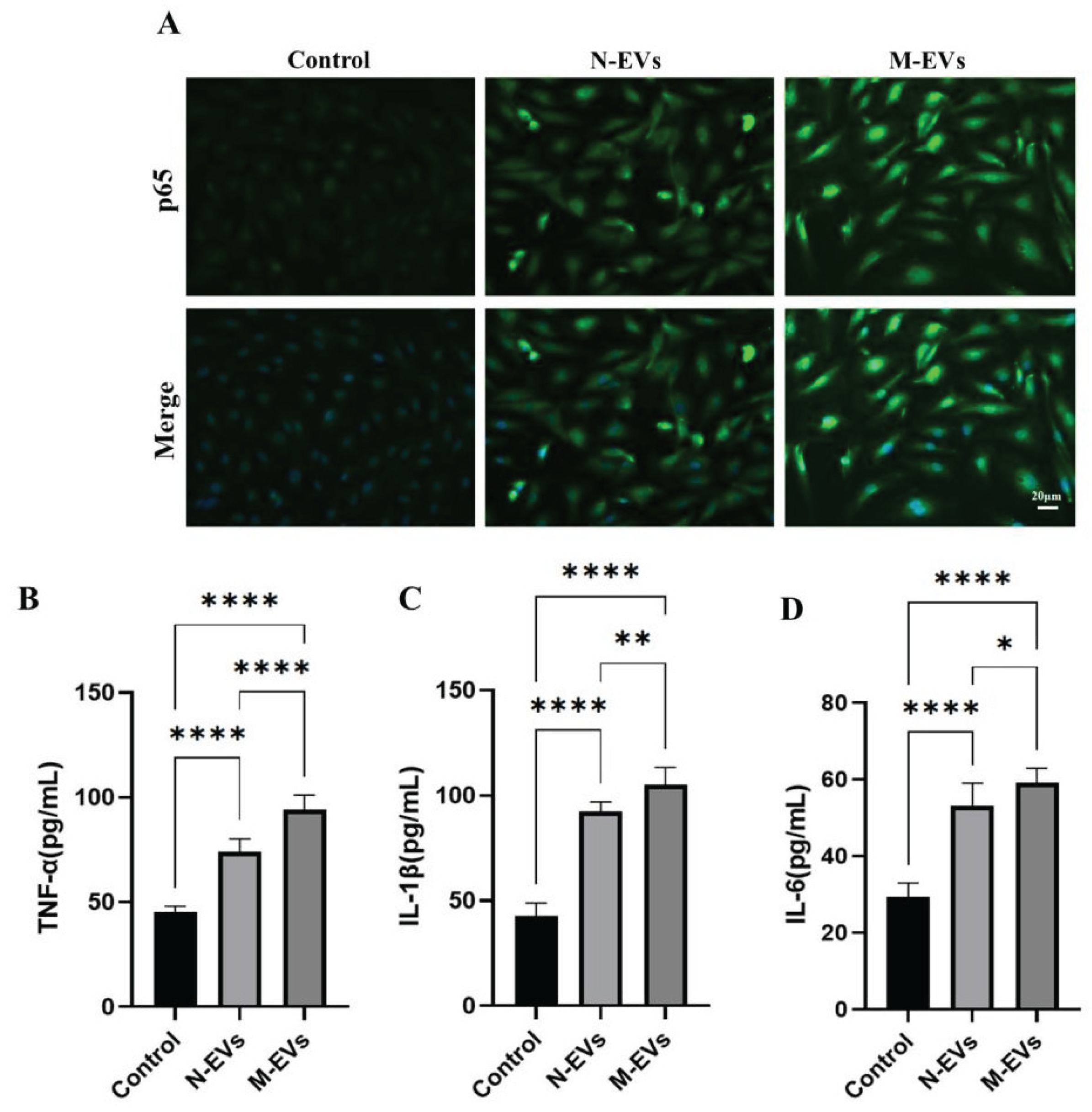
HUVECs were seeded at a density of 2 × 104 cells per well in 48-well plates. Following 24 hours of the designated treatment, the cells were fixed with 4% paraformaldehyde for 10 minutes, washed three times with PBS, and subsequently permeabilized with immunostaining permeabilizing solution containing Triton X-100 (Cat No: P0096, Beyotime) for 10 minutes at room temperature. The cells were then blocked with 5% bovine serum albumin (BSA) blocking solution (Cat No: SW3015, Solarbio) for 1 hour at room temperature. Primary antibodies against nuclear factor-kappa B (NF-κB) p65 (1:100, Cat No: AB2020, Beyotime, China) was applied and incubated overnight at 4 °C. After removal of the primary antibodies and three washes with PBS, the cells were incubated with the secondary antibody (1:1000, Cat No: AF350-labeled goat anti-rabbit IgG, Beyotime) for 1 hour at room temperature. Following three additional PBS washes, cell nuclei were counterstained with Hoechst 33342. IF images were captured using an inverted fluorescence microscope, and fluorescence intensity was quantified using ImageJ software.
2.15. Enzyme-Linked Immunosorbent Assay Detection
The cell culture supernatants from each group were collected into 1.5 mL microcentrifuge tubes and centrifuged at 350 × g for 20 minutes at 2-8 °C to remove cell debris and impurities. The resulting clarified supernatants were collected. The expression levels of tumor necrosis factor-alpha (TNF-α; Cat No: HTQ-ES-00090, Changzhou Haotianqi Biotechnology Co., Ltd. (Mostcell), Changzhou, China), interleukin-1 beta (IL-1β; Cat No: HTQ-ES-00140, Mostcell), and interleukin-6 (IL-6; Cat No: HTQ-ES-00035, Mostcell) were measured using respective commercial ELISA kits according to the manufacturers’ instructions.
2.16. Statistical Analysis
All experiments were conducted in triplicate or more, and the resulting data were analyzed and visualized using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Statistical significance was assessed using unpaired t-tests or ANOVA. The results are presented as the mean ± standard deviation (SD). A P value < 0.05 was considered significant.
4. Discussion
Previous research has demonstrated that multiple factors influence the production and yield of
P. gingivalis EVs, including the structural characteristics and modification status of lipid A, gingipains, peptidyl arginine deiminase (PPAD), autolysin, and the fimbrial subunit A (FimA) [
9,
17,
20,
21]. More recently, environmental conditions affecting bacterial growth—such as antibiotic exposure, pH, temperature, nutrient availability, and oxidative stress—have also been implicated in the regulation of EVs formation [
22]. Among these, antibiotics, particularly those targeting Gram-negative bacteria, have garnered increasing attention for their role in modulating EVs biogenesis. For instance, Kadurugamuwa et al. [
23] reported that gentamicin at four times the MIC, destabilized the outer membrane of
Pseudomonas aeruginosa (
P. aeruginosa), resulting in a 3 to 5 fold increase in EVs release. Metronidazole, a nitroimidazole antibiotic widely employed in clinical settings due to its efficacy against anaerobic bacteria, has similarly been shown to influence EVs secretion. Ribeiro de Freitas et al. [
24] demonstrated that sub-MIC levels of metronidazole significantly enhanced EVs secretion in
Bacteroides fragilis (
B. fragilis). In the present study, we provide the first evidence that sub-MIC metronidazole induces an approximately 2.4-fold increase in EVs release from
P. gingivalis. Although this phenomenon has been observed previously and herein, the precise molecular mechanisms underlying metronidazole-induced EVs secretion in
P. gingivalis remain unclear. Notably, Ribeiro de Freitas et al. [
24] highlighted a strong association between increased
B. fragilis EVs secretion and bacterial stress responses. Furthermore, our findings indicate that sub-MIC metronidazole exposure leads to a reduction in
P. gingivalis cell size and a morphological shift from elongated rods to elliptical forms. This morphological alteration likely represents a direct response to metronidazole-induced stress, potentially involving the SOS response and oxidative stress pathways [
25]. Such changes may facilitate EVs biogenesis by decreasing outer membrane stability and modifying its composition, thereby promoting EVs outgrowth and secretion.
Lipid A of
P. gingivalis constitutes a principal toxic component of its LPS and plays a critical role in the pathogenesis of periodontal disease and its associated systemic disorders [
17]. Lipid A activates the NF-κB signaling pathway through binding to Toll-like receptors (TLRs) on host cell surfaces, thereby inducing excessive release of pro-inflammatory cytokines and chemokines, which leads to persistent localized periodontal inflammation [
27]. Upon translocation into the circulation via disrupted periodontal epithelium, lipid A can provoke systemic low-grade inflammation, which is closely linked to As, diabetes, and Alzheimer’s disease [
17,
27]. The gingipains of
P. gingivalis, primarily RgpA/B and Kgp, are key virulence factors belonging to the cysteine protease family; they play a central role in host tissue destruction, immune evasion, and facilitation of bacterial colonization and biofilm formation [
9,
10]. In the present study,
P. gingivalis EVs were found to be enriched in lipid A, RgpA, and Kgp. Notably, EVs produced under sub-MIC metronidazole exposure exhibited increased abundance of lipid A, Kgp, and RgpA, potentially enhancing their invasive capacity against host cells. Kim et al. [
28] demonstrated that EVs induced by 1/4 MIC ceftazidime from
Burkholderia cepacia (
B. cepacia) augmented host cytotoxicity and pro-inflammatory responses. Similarly, Ribeiro de Freitas et al. [
24] reported that sub-MIC metronidazole significantly increased secretion of
B. fragilis EVs, which exacerbated inflammatory responses by activating host immune cells and markedly promoting TNF-α and IL-6 secretion. Consistent with these findings, the in vitro experiments conducted in this study provide the first evidence that sub-MIC metronidazole-induced
P. gingivalis EVs exhibit significantly enhanced cytotoxicity toward HUVECs. Mechanistically, compared to naturally derived
P. gingivalis EVs, those induced by sub-MIC metronidazole elicited significantly elevated levels of mtROS and total cellular ROS generation. It is well established that ROS can activate IκB kinase (IKK), resulting in the phosphorylation and ubiquitin-mediated degradation of the inhibitory protein IκBα. This degradation releases NF-κB, which is otherwise sequestered in the cytoplasm in an inactive complex with IκBα, thereby permitting its translocation into the nucleus. Once in the nucleus, NF-κB promotes a marked upregulation in the expression of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 [
29,
30]. This study further demonstrated that sub-MIC metronidazole-induced
P. gingivalis EVs robustly induced nuclear translocation of the NF-κB p65 subunit and increased expression of its downstream pro-inflammatory cytokines TNF-α, IL-6, and IL-1β. The overexpression of these cytokines may contribute to atherosclerotic plaque formation and instability by compromising vascular endothelial barrier integrity, promoting lipid deposition, and facilitating platelet aggregation, thereby elevating the risk of CVD [
30,
31]. Consequently,
P. gingivalis EVs induce endothelial dysfunction via the ROS/NF-κB signaling pathway, with sub-MIC metronidazole-induced EVs exacerbating endothelial damage through this mechanism. This critical finding provides a novel mechanistic explanation for the suboptimal therapeutic efficacy of antibiotics observed clinically in periodontitis patients with comorbid CVD. We hypothesize that under the selective pressure of sub-MIC antibiotic concentrations, periodontal pathogens secrete EVs with enhanced pathogenicity. These EVs persistently disrupt the vascular endothelial barrier and, by amplifying systemic inflammation, accelerate the progression of vascular pathology.