
Submitted:
28 November 2025
Posted:
28 November 2025
You are already at the latest version
Abstract
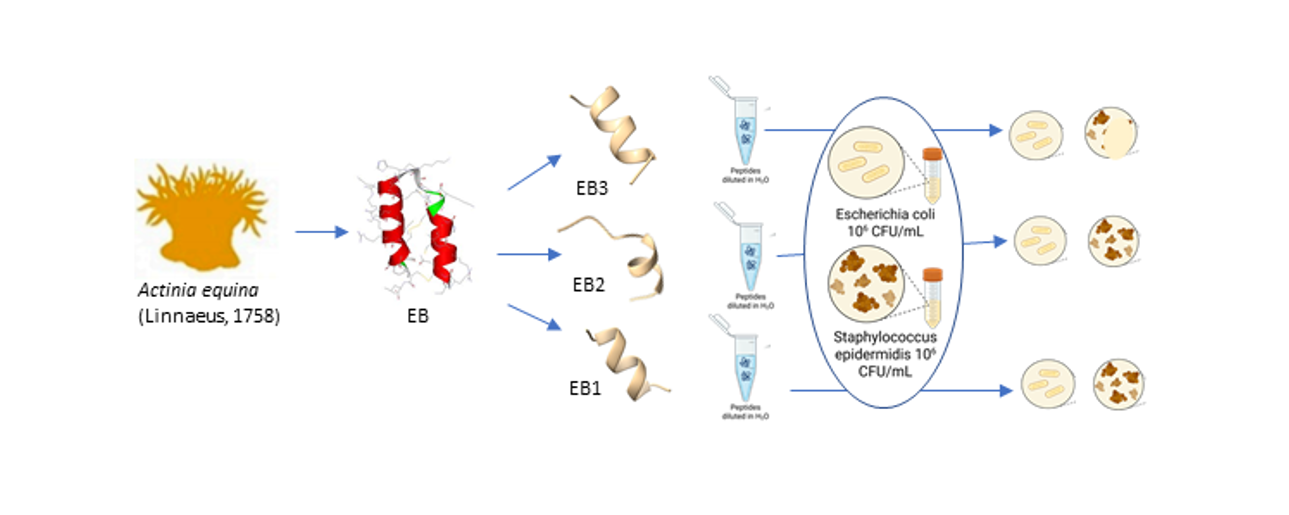
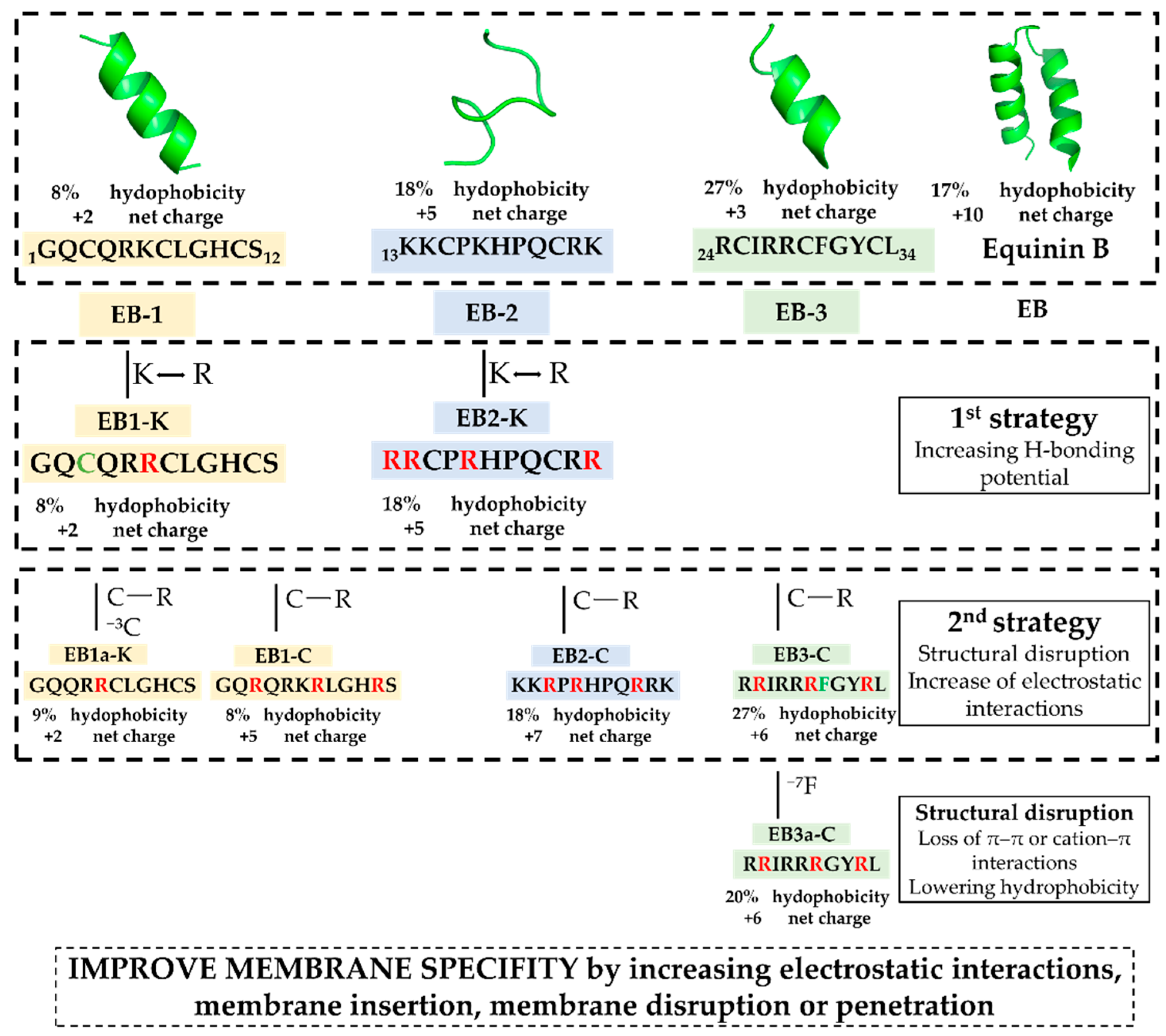
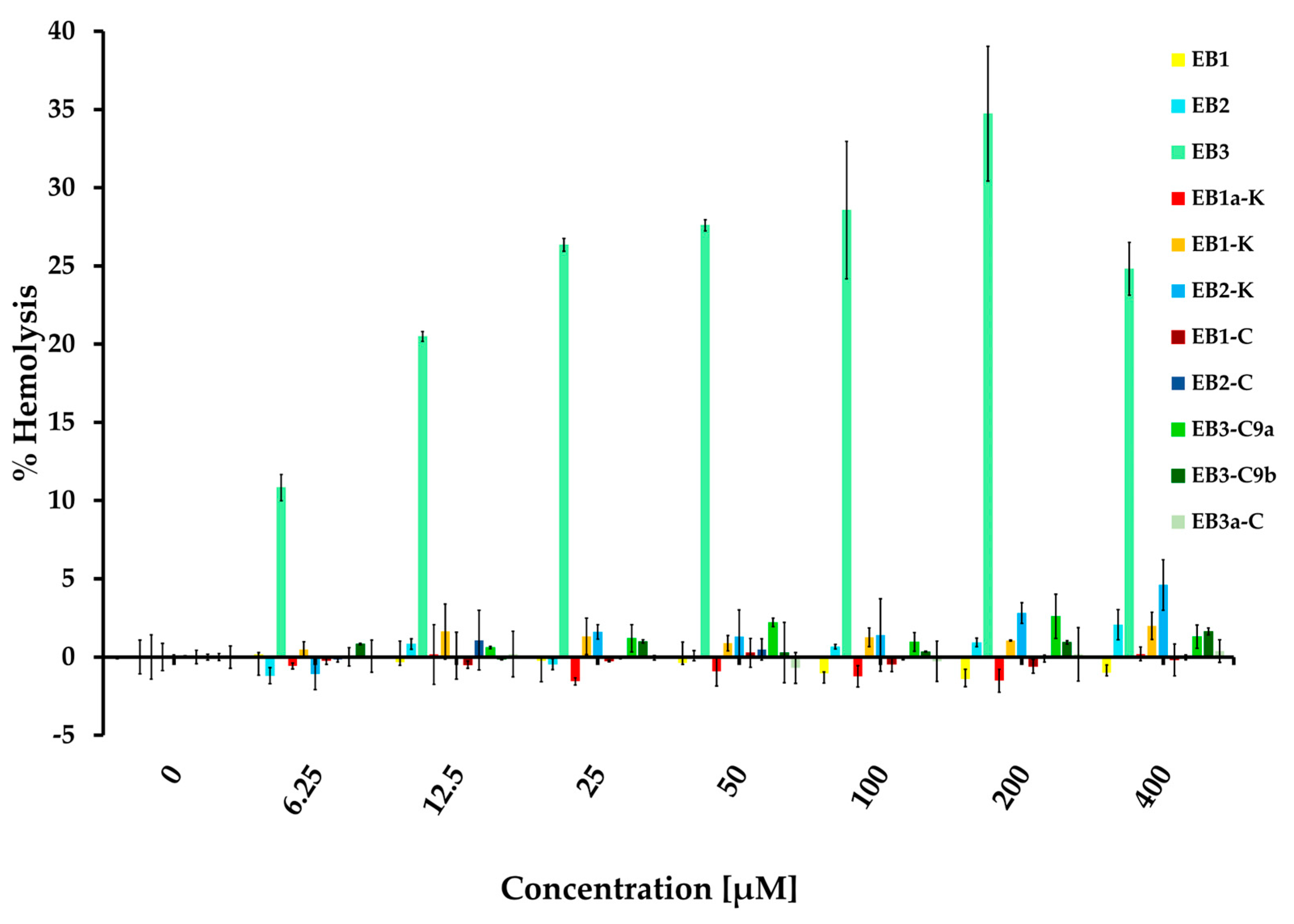
Equinin B (GQCQRKCLGHCSKKCPKHPQCRKRCIRRCFGYCL), a marine peptide from Actinia equina exhibits antibacterial activity against both Gram-positive and Gram-negative bacteria. To identify a smaller active region, the peptide was cleaved into three fragments: GQCQRKCLGHCS (EB-1), KKCPKHPQCRK (EB-2) and RCIRRCFGYCL (EB-3). Only the 11-residue C-terminal fragment showed selective activity against Gram-positive bacteria, including Staphylococcus epidermidis, Bacillus subtilis, and Enterococcus hirae, while remaining inactive against Escherichia coli. Peptide modifications, achieved by replacing cysteine residues with arginine, generally did not enhance activity, but in the C-terminal fragment they reduced hemolytic activity and increased bacterial specificity. Membrane depolarization assays confirmed that the unmodified fragment strongly disrupts bacterial membranes, whereas the modified variant showed minimal depolarization, highlighting its markedly reduced membrane-disruptive potential. In silico modelling revealed that the unmodified fragment (EB-3) can adopt multiple membrane-disruption modes, from transient shallow pores to carpet-like mechanisms, while the cysteine-to-arginine variant interacts mainly via partial insertion anchored by arginine residues. Phenylalanine appears to interact with the membrane, and reducing hydrophobicity by its removal abolished antibacterial activity. These findings highlight the 11-residue C-terminal fragment as a tunable, membrane-targeting motif with mechanistic novelty, offering a blueprint for developing safer, selective antimicrobial peptides with reduced cytotoxicity.
Keywords:
1. Introduction
2. Results and Discussion
2.1. Structure-Function Guided Design of the Peptide Library
2.2. Evaluation of Peptide Specificity
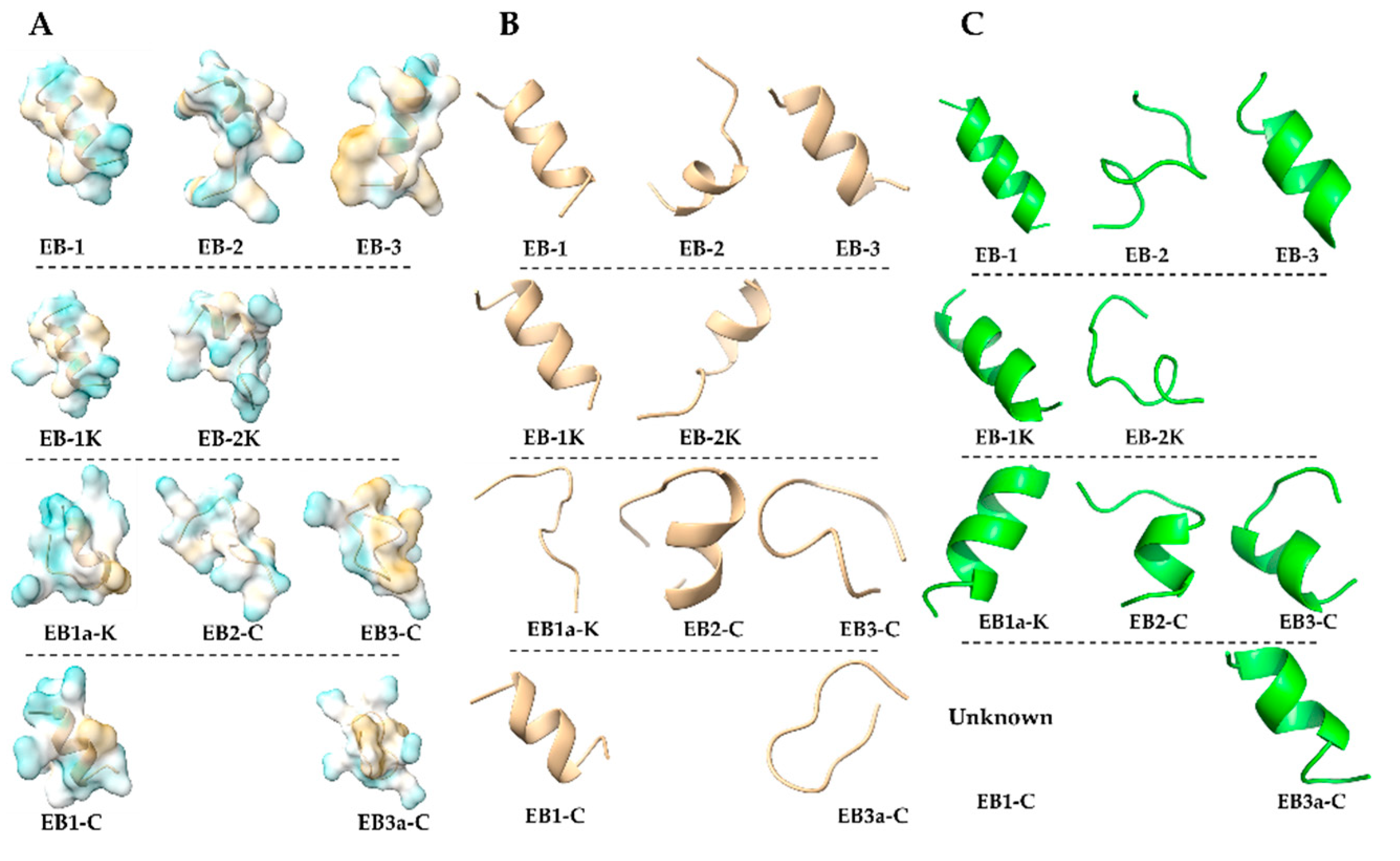
2.3. In Silico Structural Profiling
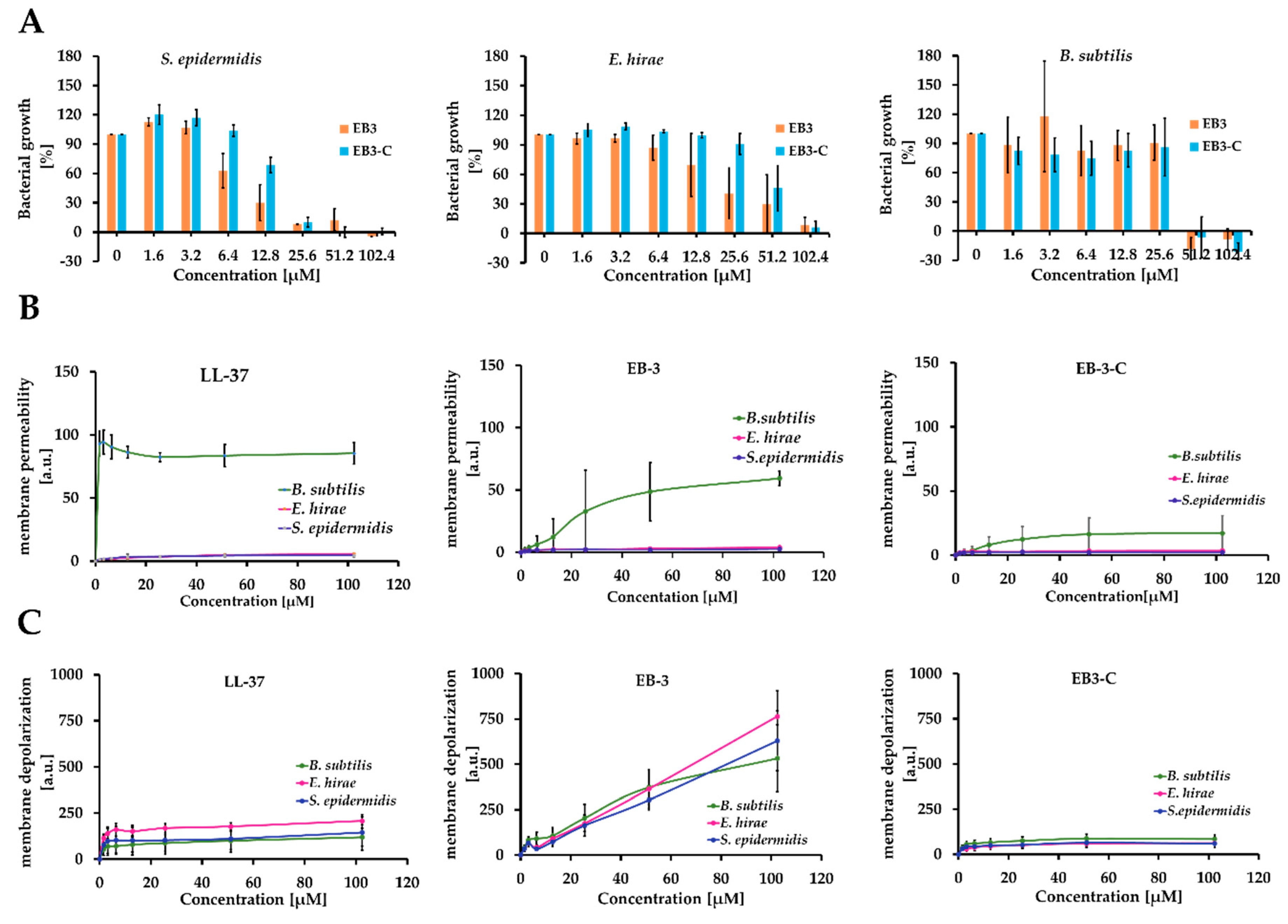
2.4. Evaluation of Membrane Permeability and Activity
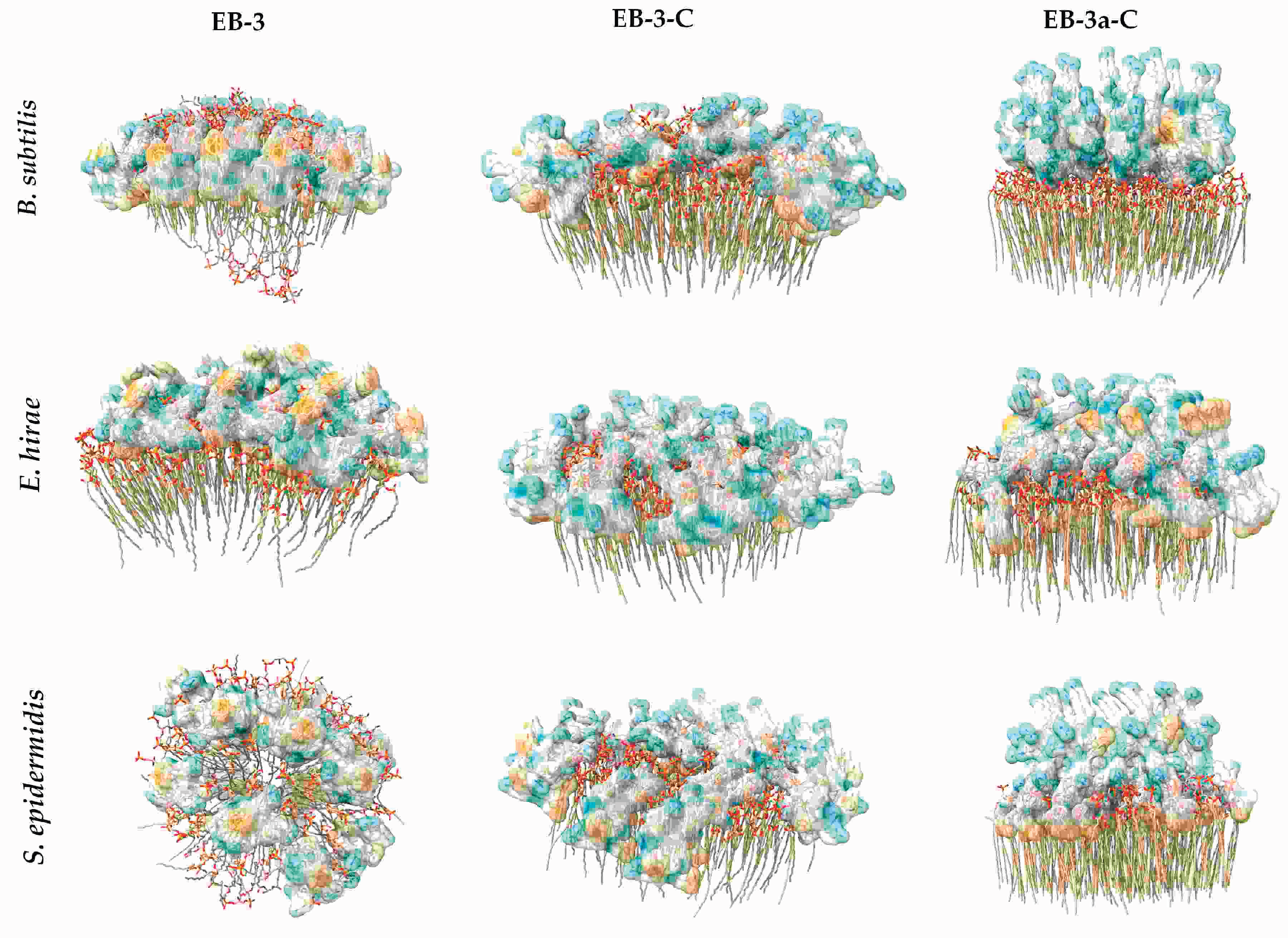
2.5. In Silico Evaluation of Peptide-Membrane Interactions
3. Materials and Methods
3.1. General
3.2. Peptide Synthesis
3.2. Biological Activity
3.3.1. Minimum Inhibitory Concentration (MIC) Assay
3.3.2. Hemolysis Assay
3.3.3. In Silico Analysis of Peptides Structural Properties
3.3.4. Membrane Permeability Assay
3.3.5. Membrane Depolarization Assay
3.3.6. In Silico Peptide-Membrane Interactions
3.4. Nano-Ultra-Performance Liquid Chromatography-Electrospray Ionization-Quadrupole Mass Spectrometry Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Antibiotic resistance |
| AMPs | Antimicrobial peptides |
| HBTU | N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate |
| NMM | N-methylmorpholine |
| TFA | Trifluoroacetic acid |
| TIS | Triisopropylsylane |
| SPE | Solid Phase Extraction |
| SPPS | Solid Phase Peptide Synthesis |
| MIC | Minimum Inhibitory Concentration |
| PI | Propidium iodide |
| MHB | Mueller-Hinton Broth |
| DMSO | Dimethyl sulfoxide |
| DMF | Dimethylformamide |
Appendix A

| Peptide ID | Peptide sequence | Precursor m/z | Rt/min | |
|---|---|---|---|---|
| 1 | EB-1 | GQCQRKCLGHCS | [660.2925+2H]2+ | 11.53 |
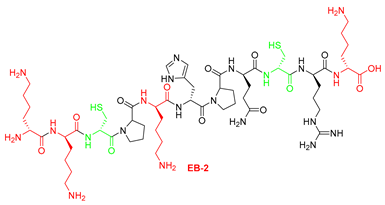
| 2 | EB-2 | KKCPKHPQCRK | [676.8743+2H]2+ | 09.17 |
| 3 | EB-3 | RCIRRCFGYCL | [695.3392+2H]2+ | 18.06 |
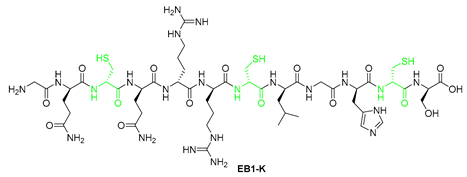
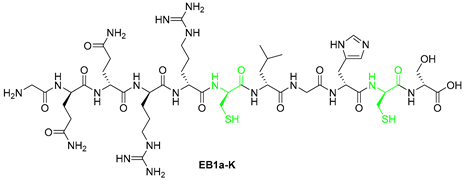
| 4 | EB1a-K | GQQRRCLGHCS | [622.7909+2H]2+ | 10.17 |
| 5 | EB1-K | GQCQRRCLGHCS | [674.2955+2H]2+ | 10.77 |
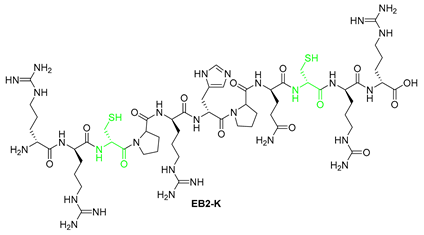
| 6 | EB2-K | RRCPRHPQCRR | [732.8866+2H]2+ | 09.17 |
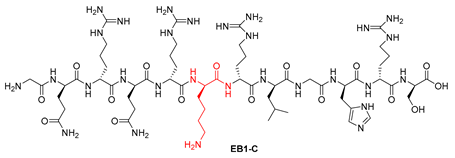
| 7 | EB1-C | GQRQRKRLGHRS | [739.9304+2H]2+ | 09.76 |
| 8 | EB2-K | KKRPKHPQRRK | [729.9662+2H]2+ | 08.76 |
| 9 | EB3-C | RRIRRRFGYRL | [774.9771+2H]2+ | 09.07 |
| 10 | EB3a-C | RRIRRRGYRL | [701.4429+2H]2+ | 09.23 |
References
- Boman, H.G. Antibacterial Peptides: Basic Facts and Emerging Concepts. Journal of Internal Medicine 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and Specificity of Two Antibacterial Proteins Involved in Insect Immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.; Strom, M.; M. Svendsen, J. The Medicinal Chemistry of Short Lactoferricin-Based Antibacterial Peptides. CMC 2007, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From Antimicrobial to Anticancer Peptides. A Review. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Brogden, N.K.; Brogden, K.A. Will New Generations of Modified Antimicrobial Peptides Improve Their Potential as Pharmaceuticals? International Journal of Antimicrobial Agents 2011, S0924857911002342. [Google Scholar] [CrossRef]
- Bocchinfuso, G.; Palleschi, A.; Orioni, B.; Grande, G.; Formaggio, F.; Toniolo, C.; Park, Y.; Hahm, K.; Stella, L. Different Mechanisms of Action of Antimicrobial Peptides: Insights from Fluorescence Spectroscopy Experiments and Molecular Dynamics Simulations. Journal of Peptide Science 2009, 15, 550–558. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the Past and Charting the Future of Marine Natural Products Drug Discovery and Chemical Biology. Chemistry & Biology 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Meng, L.-H.; Wang, B.-G. Progress in Research on Bioactive Secondary Metabolites from Deep-Sea Derived Microorganisms. Marine Drugs 2020, 18, 614. [Google Scholar] [CrossRef]
- Satpute, S.K.; Banat, I.M.; Dhakephalkar, P.K.; Banpurkar, A.G.; Chopade, B.A. Biosurfactants, Bioemulsifiers and Exopolysaccharides from Marine Microorganisms. Biotechnology Advances 2010, 28, 436–450. [Google Scholar] [CrossRef]
- Laport, M.; Santos, O.; Muricy, G. Marine Sponges: Potential Sources of New Antimicrobial Drugs. CPB 2009, 10, 86–105. [Google Scholar] [CrossRef]
- Macedo, M.W.F.S.; Cunha, N.B.D.; Carneiro, J.A.; Costa, R.A.D.; Alencar, S.A.D.; Cardoso, M.H.; Franco, O.L.; Dias, S.C. Marine Organisms as a Rich Source of Biologically Active Peptides. Front. Mar. Sci. 2021, 8, 667764. [Google Scholar] [CrossRef]
- La Corte, C.; Catania, V.; Dara, M.; Parrinello, D.; Staropoli, M.; Trapani, M.R.; Cammarata, M.; Parisi, M.G. Equinins as Novel Broad-Spectrum Antimicrobial Peptides Isolated from the Cnidarian Actinia Equina (Linnaeus, 1758). Marine Drugs 2024, 22, 172. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, T.V.; Balandin, S.V.; Aleshina, G.M.; Tagaev, A.A.; Leonova, Y.F.; Krasnodembsky, E.D.; Men’shenin, A.V.; Kokryakov, V.N. Aurelin, a Novel Antimicrobial Peptide from Jellyfish Aurelia Aurita with Structural Features of Defensins and Channel-Blocking Toxins. Biochemical and Biophysical Research Communications 2006, 348, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Shenkarev, Z.O.; Panteleev, P.V.; Balandin, S.V.; Gizatullina, A.K.; Altukhov, D.A.; Finkina, E.I.; Kokryakov, V.N.; Arseniev, A.S.; Ovchinnikova, T.V. Recombinant Expression and Solution Structure of Antimicrobial Peptide Aurelin from Jellyfish Aurelia Aurita. Biochemical and Biophysical Research Communications 2012, 429, 63–69. [Google Scholar] [CrossRef]
- Huertas, N.; Monroy, Z.; Medina, R.; Castañeda, J. Antimicrobial Activity of Truncated and Polyvalent Peptides Derived from the FKCRRQWQWRMKKGLA Sequence against Escherichia Coli ATCC 25922 and Staphylococcus Aureus ATCC 25923. Molecules 2017, 22, 987. [Google Scholar] [CrossRef]
- Wang, G. The Antimicrobial Peptide Database Is 20 Years Old: Recent Developments and Future Directions. Protein Science 2023, 32, e4778. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Guzmán, F.; Aróstica, M.; Román, T.; Beltrán, D.; Gauna, A.; Albericio, F.; Cárdenas, C. Peptides, Solid-Phase Synthesis and Characterization: Tailor-Made Methodologies. Electronic Journal of Biotechnology 2023, 64, 27–33. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Gram-Positive Bacterial Cell Envelopes: The Impact on the Activity of Antimicrobial Peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 2016, 1858, 936–946. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Piller, P.; Wolinski, H.; Cordfunke, R.A.; Drijfhout, J.W.; Keller, S.; Lohner, K.; Malanovic, N. Membrane Activity of LL-37 Derived Antimicrobial Peptides against Enterococcus Hirae: Superiority of SAAP-148 over OP-145. Biomolecules 2022, 12, 523. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX : Tools for Structure Building and Analysis. Protein Science 2023, 32, e4792. [Google Scholar] [CrossRef]
- Thevenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An Updated de Novo Structure Prediction Server for Both Linear and Disulfide Bonded Cyclic Peptides. Nucleic Acids Research 2012, 40, W288–W293. [Google Scholar] [CrossRef]
- Beaufays, J.; Lins, L.; Thomas, A.; Brasseur, R. In Silico Predictions of 3D Structures of Linear and Cyclic Peptides with Natural and Non-proteinogenic Residues. Journal of Peptide Science 2012, 18, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Snider, C.; Jayasinghe, S.; Hristova, K.; White, S.H. MPEx: A Tool for Exploring Membrane Proteins. Protein Science 2009, 18, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. MEMBRANE PROTEIN FOLDING AND STABILITY: Physical Principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Marx, L.; Blondelle, S.E.; Pabst, G.; Semeraro, E.F. Experimental Concepts for Linking the Biological Activities of Antimicrobial Peptides to Their Molecular Modes of Action. Biochimica et Biophysica Acta (BBA) - Biomembranes 2020, 1862, 183275. [Google Scholar] [CrossRef]
| Compound | Sequence | Escherichia coli | Staphylococcus epidermidis |
|---|---|---|---|
| MIC90/µgmL-1 | MIC90/µgmL-1 | ||
| EB-1 | GQCQRKCLGHCS | >100 | >100 |
| EB-2 | KKCPKHPQCRK | >100 | >100 |
| EB-3 | RCIRRCFGYCL | 100 | 25 |
| EB1a-K | GQQRRCLGHCS | >100 | >100 |
| EB-1K | GQCQRRCLGHCS | >100 | >100 |
| EB-2K | RRCPRHPQCRR | >100 | >100 |
| EB1-C | GQRQRKRLGHRS | >100 | >100 |
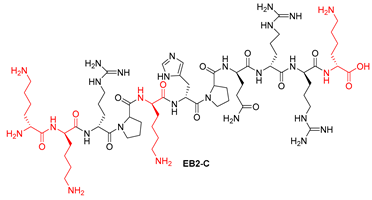
| EB2-C | KKRPKHPQRRK | >100 | >100 |
| EB3a1-C | RRIRRRFGYRL | >100 | >100 |
| EB3-C | RRIRRRFGYRL | >100 | 25-50 |
| EB3a-C | RRIRRRGYRL | >100 | >100 |
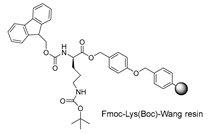
| Starting resin for SPPS | Final peptide* |
|---|---|
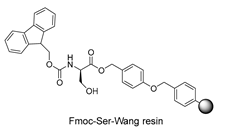 |
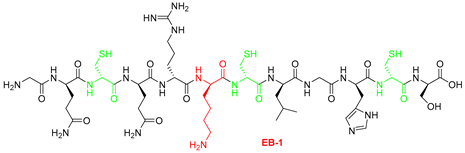 |
 | |
 | |
 | |
 |
 |
 | |
 |
 |
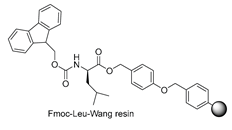 |
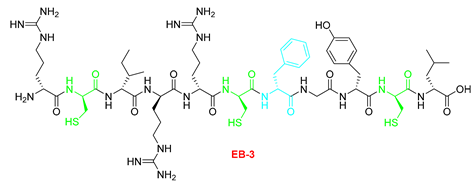 |
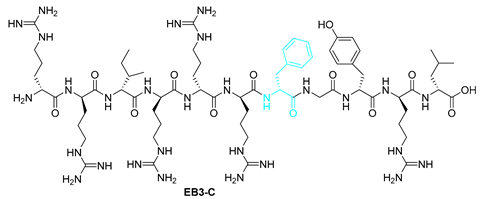 | |
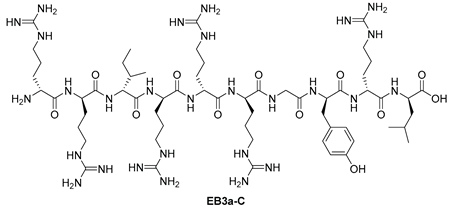 |
| Compound | ΔG | Total Hydr. Moment |
|---|---|---|
| EB-1 | 9.93 | 5.3 |
| EB-2 | 16.35 | 4.48 |
| EB-3 | 1.73 | 6.97 |
| EB1a-K | 16.86 | 5.47 |
| EB1-K | 16.84 | 4.42 |
| EB2-K | 20.29 | 2.17 |
| EB1-C | 15.42 | 4.59 |
| EB2-C | 20.01 | 4.01 |
| EB3-C | 7.22 | 5.58 |
| EB3a-C | 8.93 | 5.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



