Submitted:
26 November 2025
Posted:
28 November 2025
You are already at the latest version
Abstract
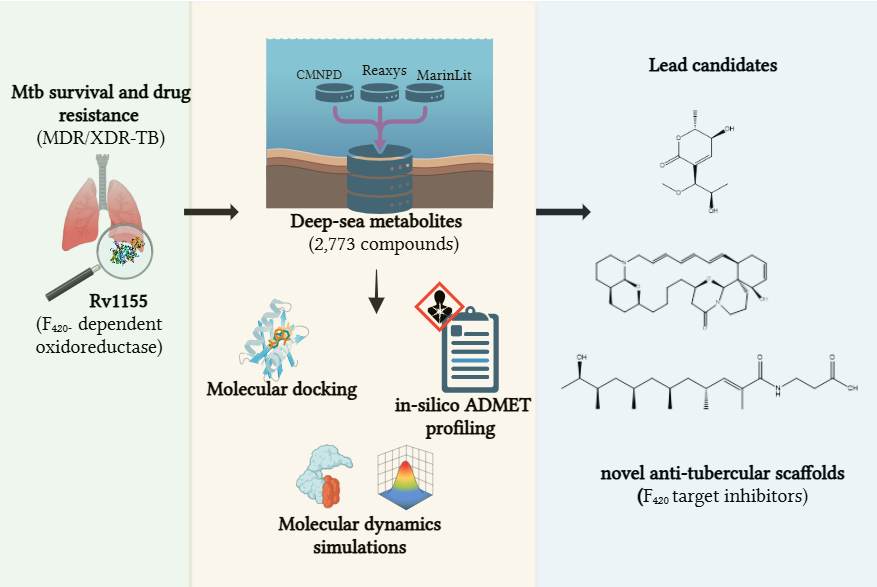
Tuberculosis, caused by Mycobacterium tuberculosis (M. tb), remains a leading global threat, escalated now by the rise of multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains. In search of a novel anti-tubercular agent with a distinct mechanism of action, this study explores deep-sea marine metabolites as potential inhibitors of the F₄₂₀-dependent oxidoreductase Rv1155, a redox enzyme essential for M. tb survival. A total of 2,773 marine-derived compounds curated from the CMNPD, Reaxys, and MarinLit databases were screened using an integrated CADD workflow combining molecular docking, in-silico ADMET profiling, and molecular dynamics (MD) simulations. Docking results revealed several metabolites with high affinity for the Rv1155 binding pocket, and three compounds: Upenamide (CMNPD_22964), Aspyronol (Compound_1749), and Fiscpropionate F (Compound_1796) as hit candidates. Among these, Upenamide displayed the strongest binding and stable protein-ligand dynamics, while Aspyronol demonstrated a promising ADMET profile comparable to that of the native cofactor F₄₂₀₂. These findings highlight the potential of deep-sea marine metabolites as a valuable source of anti-tubercular scaffolds and establish a computationally driven, cost-effective framework for discovering inhibitors targeting F₄₂₀-dependent enzymes. This approach provides a foundation for future experimental validation and expansion to additional F₄₂₀-related drug targets in M. tb.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Source of Compounds
2.2. Molecular Docking
2.3. ADMET Evaluation
2.4. Molecular Dynamics (MD) Simulations
2.5. MM-GBSA Calculations
3. Results and Discussion
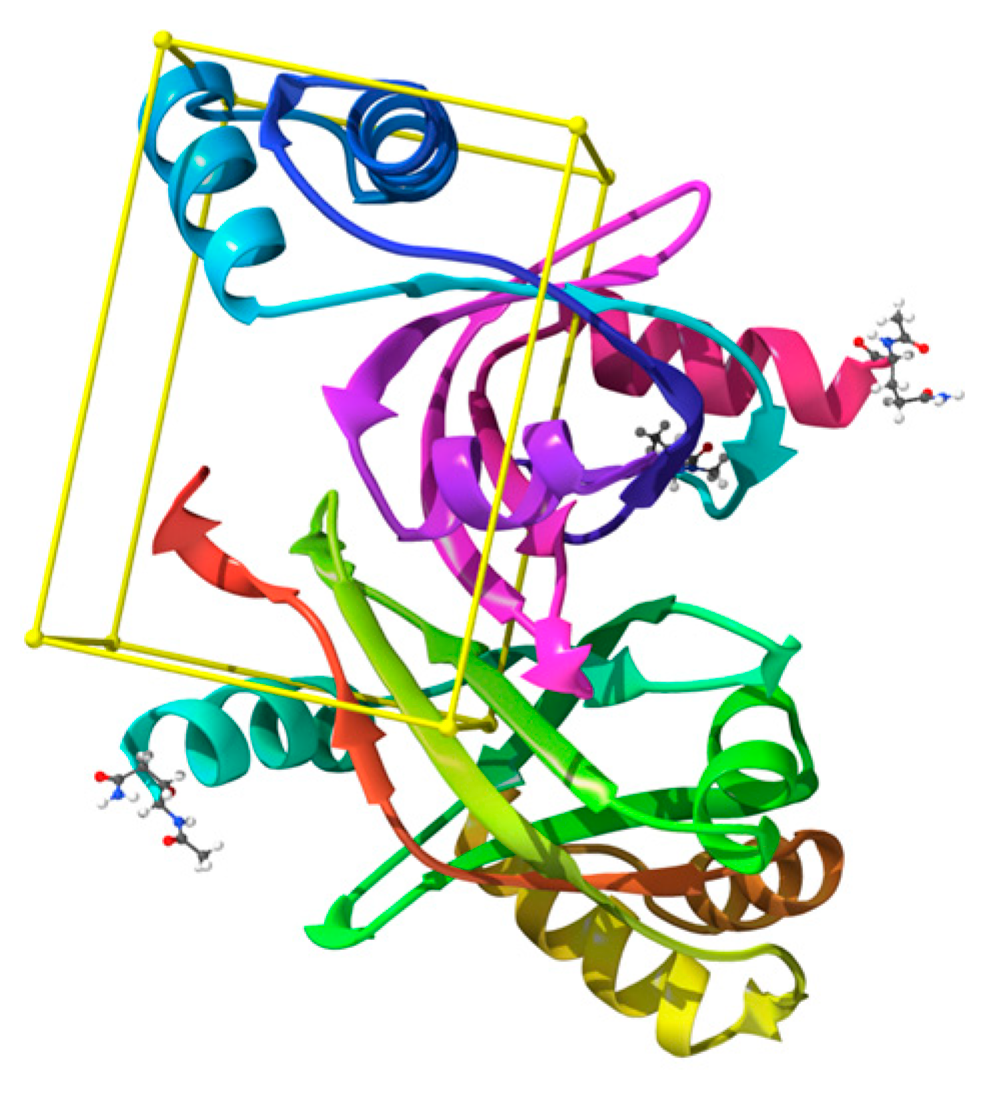
3.1. Molecular Docking Analysis
3.1.1. Validation of Docking Methodology
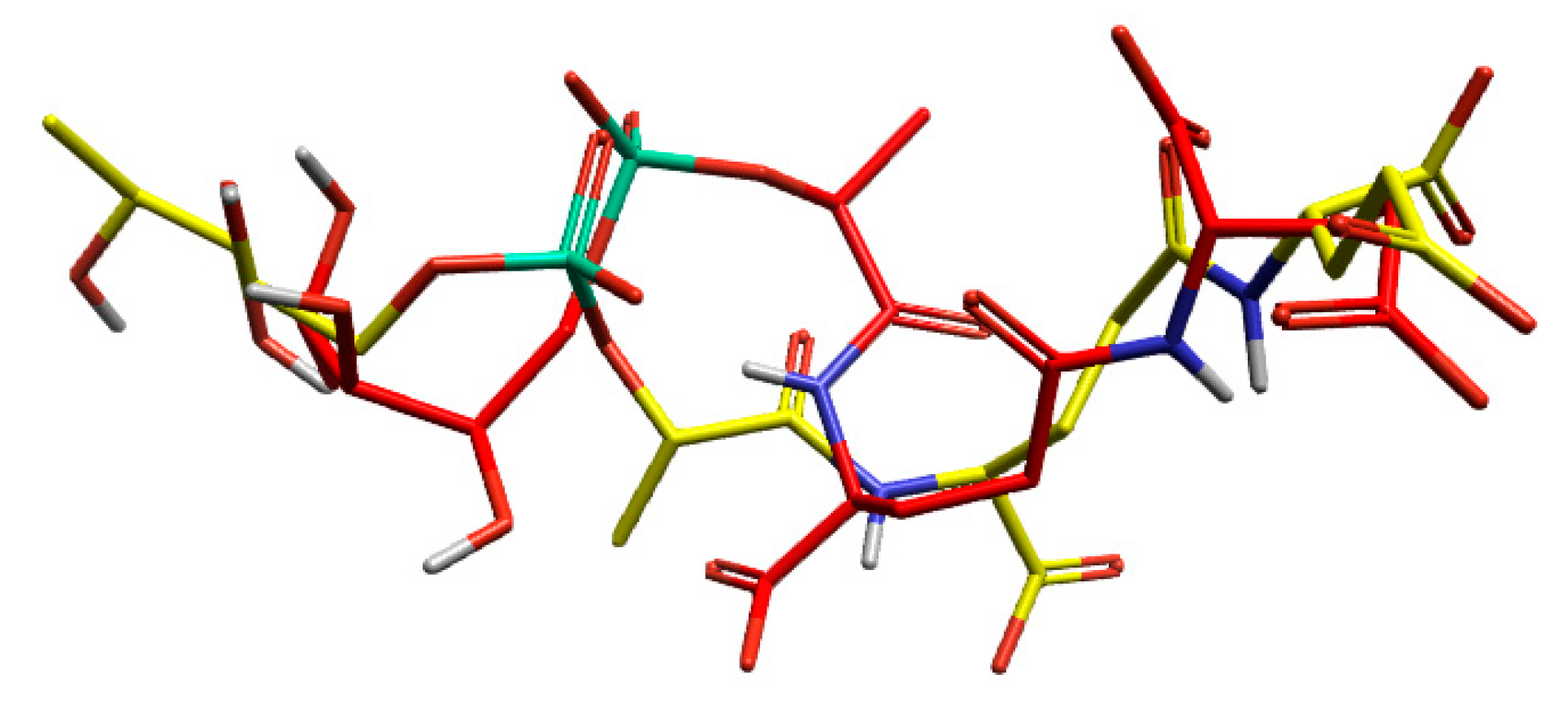
3.2. ADMET Evaluation
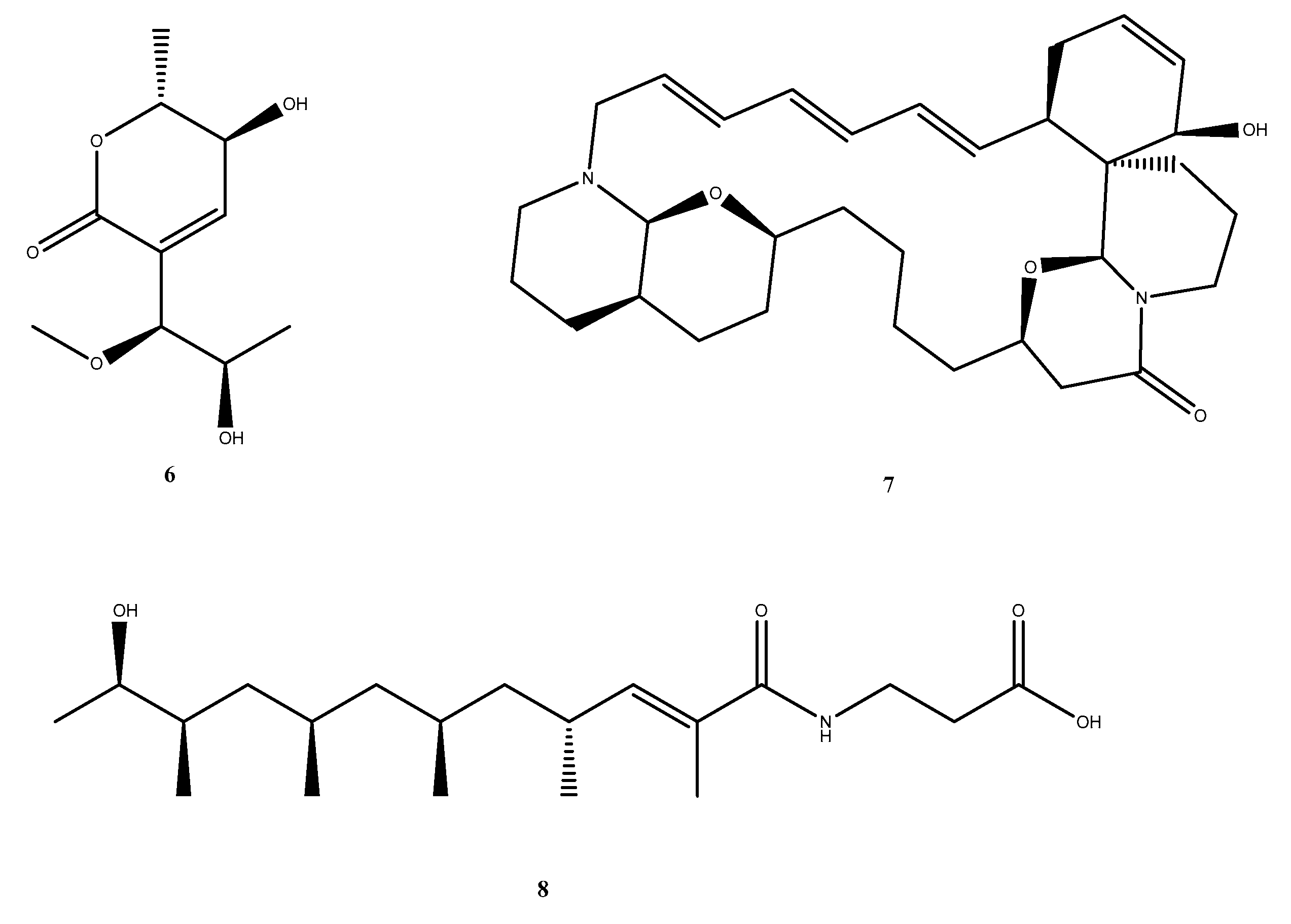
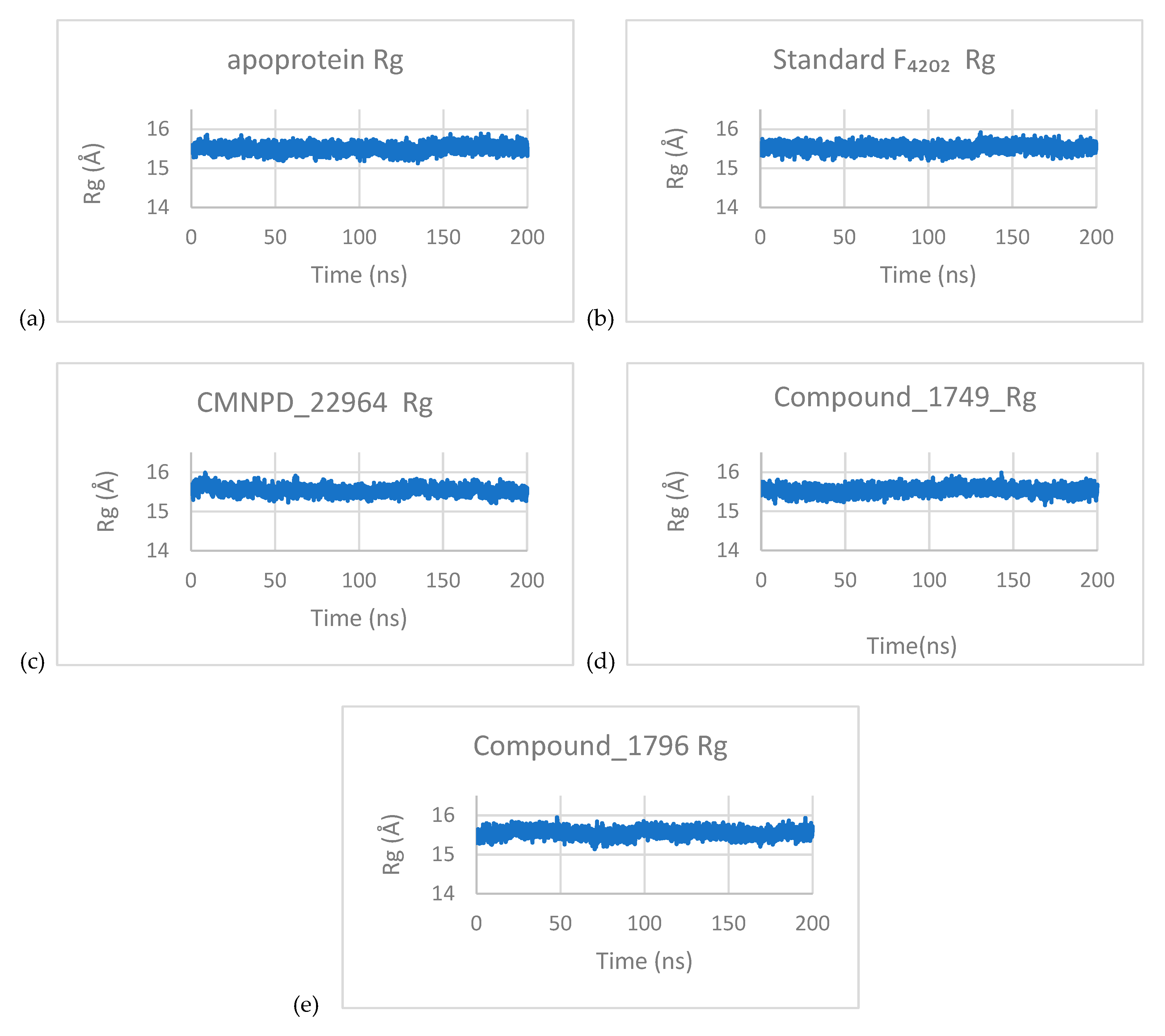
3.3. Molecular Dynamics (MD) Simulation
3.3.1. Root Mean Square Deviation (RMSD)
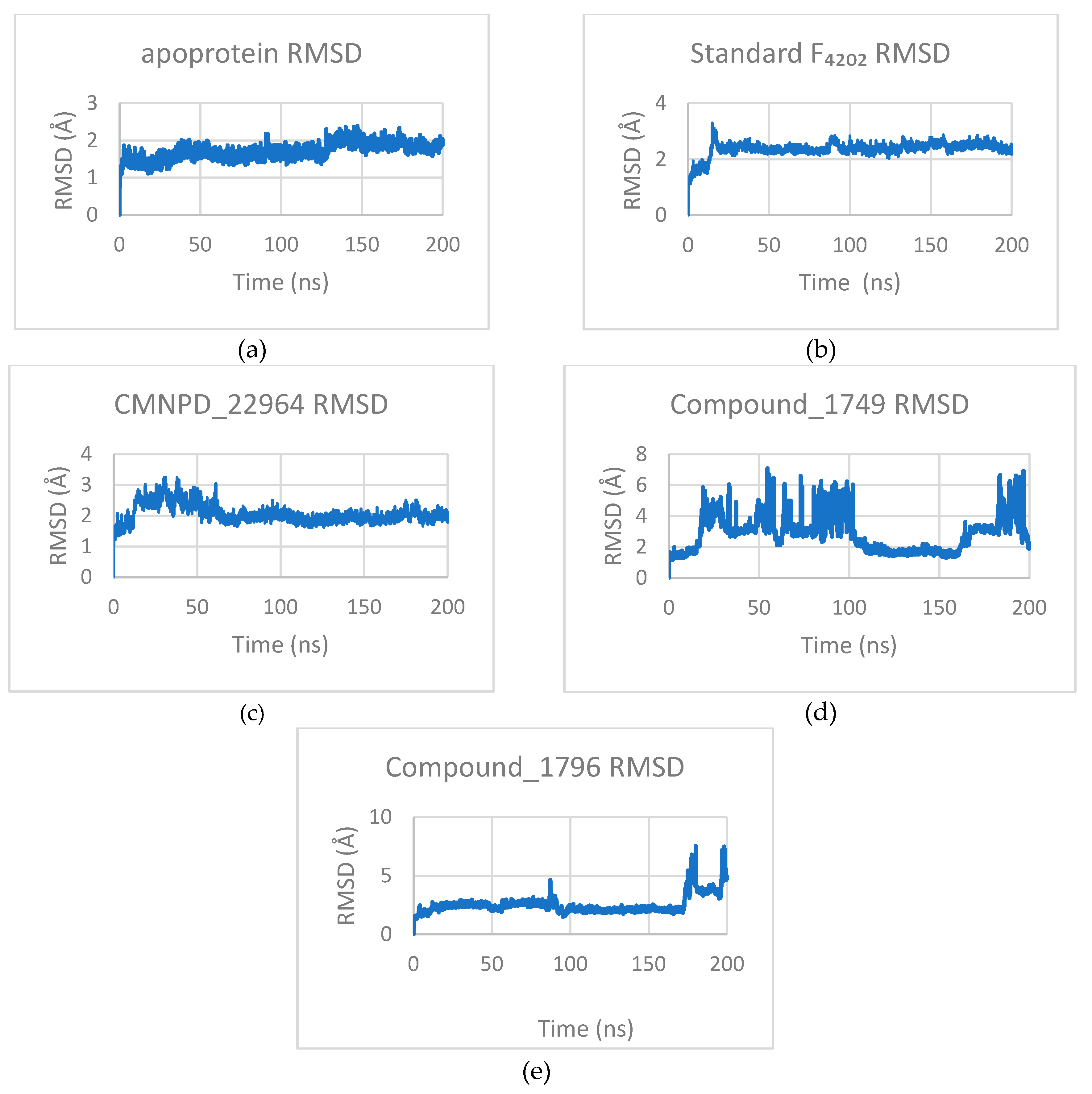
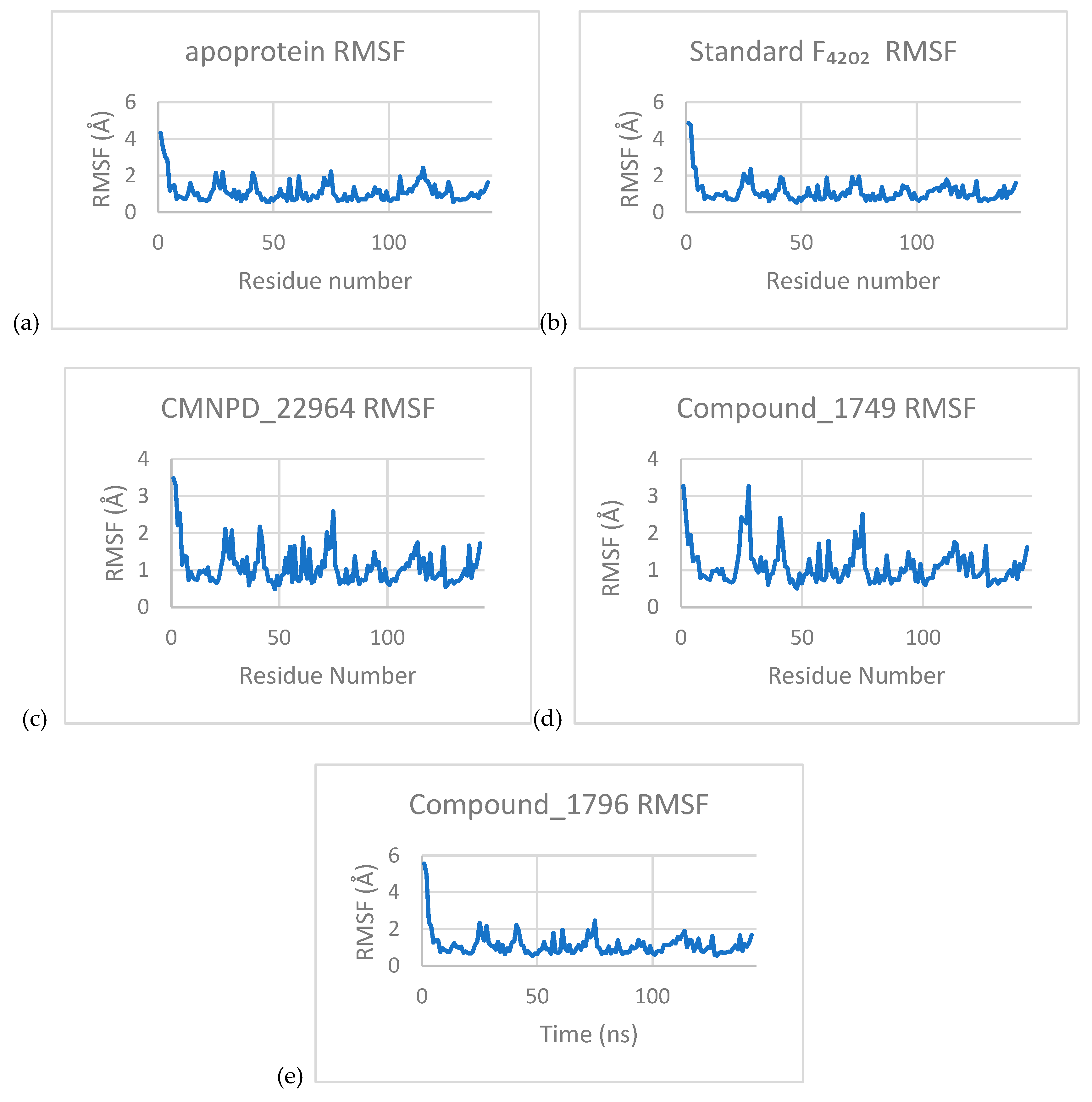
3.3.2. Root Mean Square Fluctuation (RMSF)
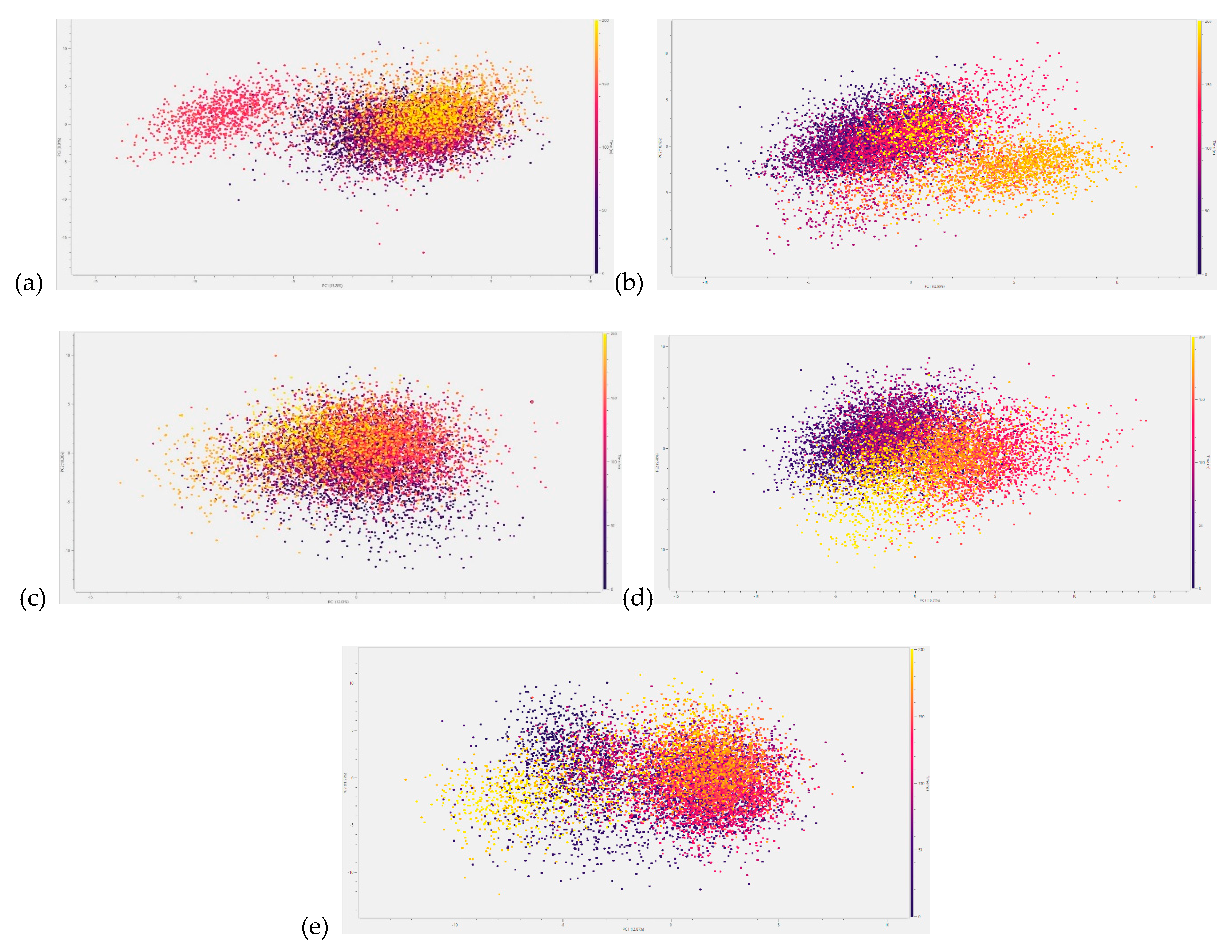
3.3.3. Principal Component Analysis (PCA)
3.4. Hydrogen Bond Contacts
3.5. MM-GBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2025. Geneva: World Health Organization; 2025. Licence: CC BY-NC-SA 3.0 IGO.
- Clinical Overview of Drug-Resistant Tuberculosis Disease (https://www.cdc.gov/tb/hcp/clinical-overview/index.html).
- D. J. Newman and G. M. Cragg, “Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019,” J Nat Prod, vol. 83, no. 3, pp. 770–803, Mar. 2020. [CrossRef]
- C. N. Aguilar, N. P. Meléndez-Renteria, and K. N. Ramirez-Guzman, Exploration and Valorization of Natural Resources from Arid Zones. New York: Apple Academic Press, 2024. [CrossRef]
- K. V. Rao, N. Kasanah, S. Wahyuono, B. L. Tekwani, R. F. Schinazi, and M. T. Hamann, “Three New Manzamine Alkaloids from a Common Indonesian Sponge and Their Activity against Infectious and Tropical Parasitic Diseases,” J Nat Prod, vol. 67, no. 8, pp. 1314–1318, Aug. 2004. [CrossRef]
- K. V. Rao et al., “Manzamine B and E and Ircinal A Related Alkaloids from an Indonesian Acanthostrongylophora Sponge and Their Activity against Infectious, Tropical Parasitic, and Alzheimer’s Diseases,” J Nat Prod, vol. 69, no. 7, pp. 1034–1040, Jul. 2006. [CrossRef]
- M. Yousaf et al., “New Manzamine Alkaloids from an Indo-Pacific Sponge. Pharmacokinetics, Oral Availability, and the Significant Activity of Several Manzamines against HIV-I, AIDS Opportunistic Infections, and Inflammatory Diseases,” J Med Chem, vol. 47, no. 14, pp. 3512–3517, Jul. 2004. [CrossRef]
- P. Pruksakorn et al., “Trichoderins, novel aminolipopeptides from a marine sponge-derived Trichoderma sp., are active against dormant mycobacteria,” Bioorg Med Chem Lett, vol. 20, no. 12, pp. 3658–3663, Jun. 2010. [CrossRef]
- H.-M. Hua et al., “Batzelladine alkaloids from the caribbean sponge Monanchora unguifera and the significant activities against HIV-1 and AIDS opportunistic infectious pathogens,” Tetrahedron, vol. 63, no. 45, pp. 11179–11188, Nov. 2007. [CrossRef]
- N. Z. Abd Rani, Y. K. Lee, S. Ahmad, R. Meesala, and I. Abdullah, “Fused Tricyclic Guanidine Alkaloids: Insights into Their Structure, Synthesis and Bioactivity,” Mar Drugs, vol. 20, no. 9, p. 579, Sep. 2022. [CrossRef]
- V. Kumar, S. Parate, S. Yoon, G. Lee, and K. W. Lee, “Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2,” Front Microbiol, vol. 12, Apr. 2021. [CrossRef]
- M. Jayaraman, V. Gosu, R. Kumar, and J. Jeyaraman, “Computational insights into potential marine natural products as selective inhibitors of Mycobacterium tuberculosis InhA: A structure-based virtual screening study,” Comput Biol Chem, vol. 108, p. 107991, Feb. 2024. [CrossRef]
- M. Azhari, N. Merliani, M. Singgih, M. Arai, and E. Julianti, “Insights into Natural Products from Marine-Derived Fungi with Antimycobacterial Properties: Opportunities and Challenges,” Mar Drugs, vol. 23, no. 7, p. 279, Jul. 2025. [CrossRef]
- H. Zandi and R. Safari, “Computational Approaches in Natural Product Research: Advances, Challenges, and Future Directions,” Advances in Energy and Materials Research, vol. 1, no. 2, pp. 10–15, 2024. [CrossRef]
- S.-Y. Huang and X. Zou, “Advances and Challenges in Protein-Ligand Docking,” Int J Mol Sci, vol. 11, no. 8, pp. 3016–3034, Aug. 2010. [CrossRef]
- A. Serghini, S. Portelli, and D. B. Ascher, “AI-Driven Enhancements in Drug Screening and Optimization,” 2024, pp. 269–294. [CrossRef]
- L. Harris et al., “Global distribution of deep-sea natural products shows environmental and phylogenetic undersampling and potential for biodiscovery,” Jul. 03, 2025. (in press). [CrossRef]
- “MarinLit a database of the marine natural products literature (https://marinlit.rsc.org/).”.
- “Reaxys (http://www.reaxys.com/).”.
- C. Lyu et al., “CMNPD: a comprehensive marine natural products database towards facilitating drug discovery from the ocean,” Nucleic Acids Res, vol. 49, no. D1, pp. D509–D515, Jan. 2021. [CrossRef]
- J. Eberhardt, D. Santos-Martins, A. F. Tillack, and S. Forli, “AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings,” J Chem Inf Model, vol. 61, no. 8, pp. 3891–3898, Aug. 2021. [CrossRef]
- O. Trott and A. J. Olson, “AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading,” J Comput Chem, vol. 31, no. 2, pp. 455–461, Jan. 2010. [CrossRef]
- G. Wolber and T. Langer, “LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters,” J Chem Inf Model, vol. 45, no. 1, pp. 160–169, Jan. 2005. [CrossRef]
- L. Fu et al., “ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support,” Nucleic Acids Res, vol. 52, no. W1, pp. W422–W431, Jul. 2024. [CrossRef]
- P. Eastman et al., “OpenMM 7: Rapid development of high performance algorithms for molecular dynamics,” PLoS Comput Biol, vol. 13, no. 7, p. e1005659, Jul. 2017. [CrossRef]
- S. Boothroyd et al., “Development and Benchmarking of Open Force Field 2.0.0: The Sage Small Molecule Force Field,” J Chem Theory Comput, vol. 19, no. 11, pp. 3251–3275, Jun. 2023. [CrossRef]
- D. Sitkoff, K. A. Sharp, and B. Honig, “Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models,” J Phys Chem, vol. 98, no. 7, pp. 1978–1988, Feb. 1994. [CrossRef]
- “Molecular docking and its application in search of antisickling agent from Carica papaya,” J Appl Biol Biotechnol, vol. 8, no. 1, pp. 105–116, Jan. 2020. [CrossRef]
- E. H. Mashalidis, A. G. Gittis, A. Tomczak, C. Abell, C. E. Barry, and D. N. Garboczi, “Molecular insights into the binding of coenzyme <scp> F 420 </scp> to the conserved protein <scp>R</scp> v1155 from <scp> M </scp> ycobacterium tuberculosis,” Protein Science, vol. 24, no. 5, pp. 729–740, May 2015. [CrossRef]
- C. Greening et al., “Physiology, Biochemistry, and Applications of F 420 - and F o -Dependent Redox Reactions,” Microbiology and Molecular Biology Reviews, vol. 80, no. 2, pp. 451–493, Jun. 2016. [CrossRef]
- B. M. Lee, “The Role of F 420-dependent Enzymes in Mycobacteria Declaration,” 2017.
- G. Bashiri et al., “A revised biosynthetic pathway for the cofactor F420 in prokaryotes,” Nat Commun, vol. 10, no. 1, p. 1558, Apr. 2019. [CrossRef]
- P. C. Agu et al., “Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management,” Sci Rep, vol. 13, no. 1, p. 13398, Aug. 2023. [CrossRef]
- K. A. Johnson, “Role of Induced Fit in Enzyme Specificity: A Molecular Forward/Reverse Switch,” Journal of Biological Chemistry, vol. 283, no. 39, pp. 26297–26301, Sep. 2008. [CrossRef]
- S. Mhambi, D. Fisher, M. B. T. Tchokonte, and A. Dube, “Permeation Challenges of Drugs for Treatment of Neurological Tuberculosis and HIV and the Application of Magneto-Electric Nanoparticle Drug Delivery Systems,” Pharmaceutics, vol. 13, no. 9, p. 1479, Sep. 2021. [CrossRef]
- C. H. C. Litjens, R. E. Aarnoutse, and L. H. M. te Brake, “Preclinical models to optimize treatment of tuberculous meningitis – A systematic review,” Tuberculosis, vol. 122, p. 101924, May 2020. [CrossRef]
- J. I. Jiménez et al., “‘Upenamide: An Unprecedented Macrocyclic Alkaloid from the Indonesian Sponge Echinochalina sp.,” J Org Chem, vol. 65, no. 25, pp. 8465–8469, Dec. 2000. [CrossRef]
- X.-W. Chen, C.-W. Li, C.-B. Cui, W. Hua, T.-J. Zhu, and Q.-Q. Gu, “Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1,” Mar Drugs, vol. 12, no. 6, pp. 3116–3137, May 2014. [CrossRef]
- Z. Liu et al., “Polypropionate Derivatives with Mycobacterium tuberculosis Protein Tyrosine Phosphatase B Inhibitory Activities from the Deep-Sea-Derived Fungus Aspergillus fischeri FS452,” J Nat Prod, vol. 82, no. 12, pp. 3440–3449, Dec. 2019. [CrossRef]
- J. D. Selengut and D. H. Haft, “Unexpected Abundance of Coenzyme F 420 -Dependent Enzymes in Mycobacterium tuberculosis and Other Actinobacteria,” J Bacteriol, vol. 192, no. 21, pp. 5788–5798, Nov. 2010. [CrossRef]
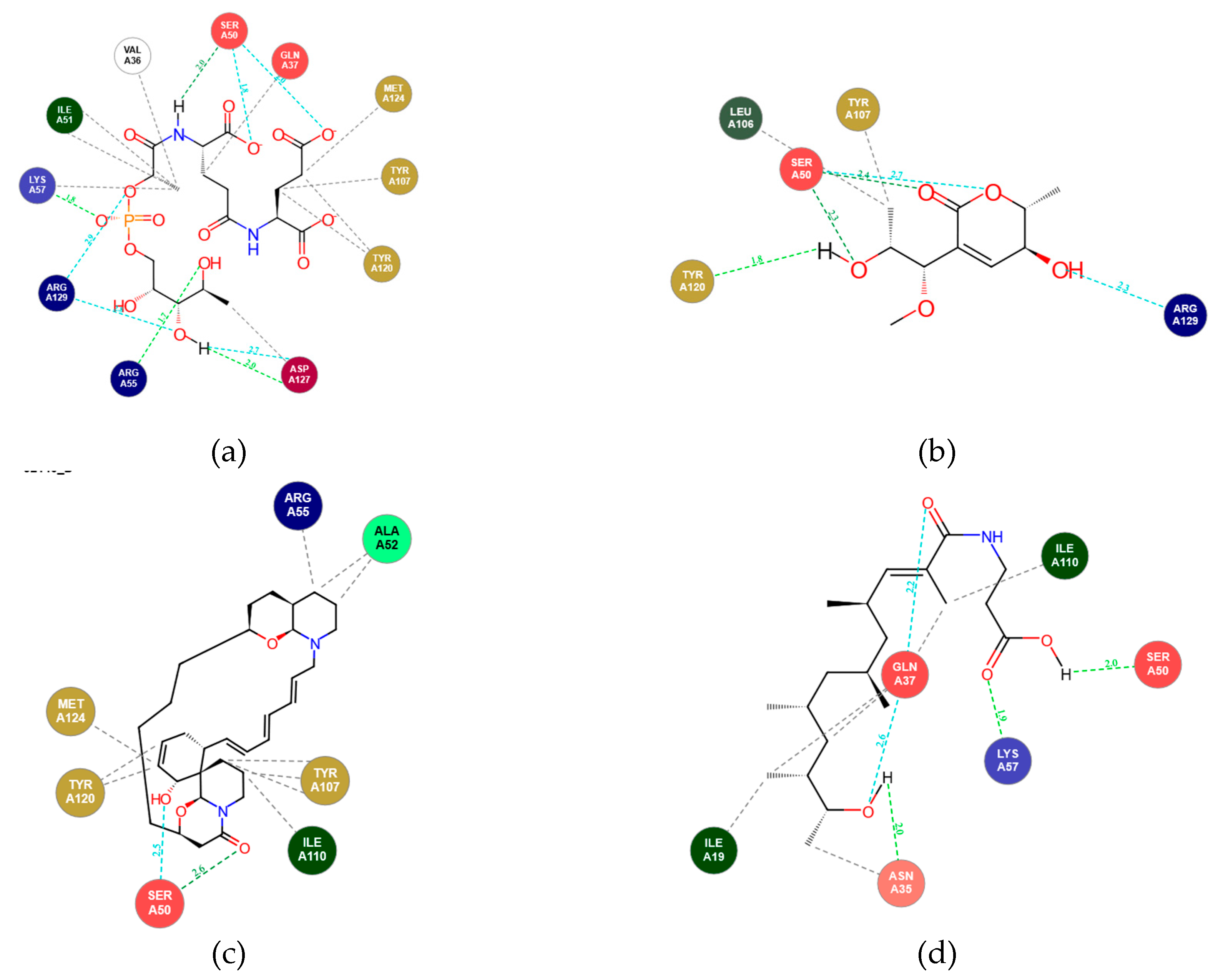
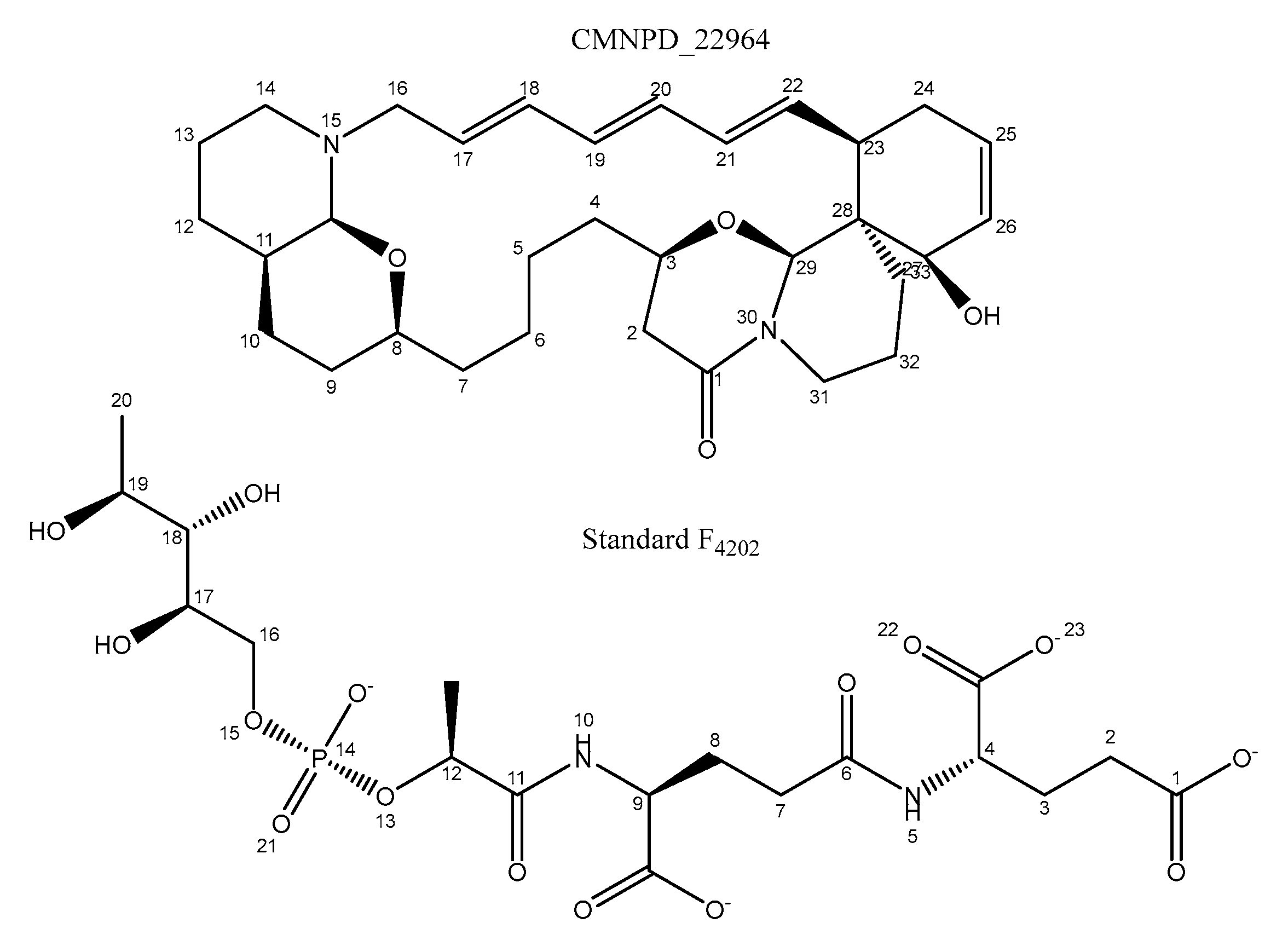

| Atom number in standard | Proton in Arg129(A) | Proportion of frame (%) |
|---|---|---|
| O22 O23 |
Imine guanidine proton | 58.1 54.8 |
| O22 O23 |
Terminal guanidine proton | 56.4 54.2 |
| Protein complexed with | ΔMM − GBSA (kcal/mol) |
Non-polar solvation energy of ΔMM − GBSA (kcal/mol/Å2) |
ΔSASA (Å2) |
|---|---|---|---|
| Standard_F4202 | -6.33 | -39.29 | -8030 |
| Aspyronol (Compound_1749) |
-24.77 |
-30.45 |
-6262 |
| Upenamide (CMNPD _22964) |
-28.56 |
-40.06 |
-8184 |
| Fiscpropionate F (Compound_1796) |
-34.07 |
-30.50 |
-6272 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



