Submitted:
28 November 2025
Posted:
28 November 2025
You are already at the latest version
Abstract
Natural products have and continue to be a remarkable resource, rich in structural di-versity, and endowed with valuable chemical and biological properties that have ad-vanced both science and society. Some natural products, especially those from marine organisms, are chemically reactive, and during extraction and handling can partially or totally transform into artifacts. All too often overlooked or mischaracterised as natural products, artifacts can be invaluable indicators of a uniquely evolved and primed chemical space, with enhanced chemical and biological properties highly prized for drug discovery. To demonstrate this potential, we review a wide selection of marine and mi-crobial case studies, revealing the factors that initiate artifact formation (e.g. solvents, heat, pH, light and air oxidation) and commenting on the mechanisms behind artifact for-mation. We conclude with reflections on how to recognise and control artifact formation, and how to exploit knowledge of artifacts as a window into unique regions of natural product chemical space — to better inform the development of future marine bioproducts.

Keywords:
marine natural products
; marine bioproducts
; artifacts
; chemical reactivity
; chemical stability
; drug discovery
1. Introduction
Since the emergence of the first single cell organisms, natural products have co-evolved with all kingdoms of life to deliver a multitude of survival advantages – defending against predators/competitors, immobilising/killing prey, facilitating reproduction, and articulating intra/inter species communication and ecological behaviour. With the advent of modern science, knowledge of the molecular structures and properties of natural products has provided valuable insights into what is often synthetically challenging/inaccessible regions of chemical space, populated by diverse structures featuring complex carbo/heterocyclic scaffolds, functionality and chirality. Many natural products possess potent and selective ecological, chemical and biological properties, knowledge of which has informed our understanding of living systems, inspiring many of the world’s most successful drugs, agrochemicals and biomaterials, and fuelling a revolution in industry, commerce, healthcare and agriculture. Notwithstanding the extraordinary impact that natural products have had on science and society, the innate chemical reactivity of some natural products both defines their uniqueness and potential, while simultaneously presenting a technical challenge. For example, some natural products partially or completely transform into artifacts during extraction, isolation, handling and/or storage. When these transformations go unnoticed, it represents a lost opportunity, as we forgo insights into molecular structures and chemical reactivity that might otherwise inform biosynthetic investigations, biomimetic syntheses, structure activity relationships (SAR), mechanisms of action, and ultimately advance drug discovery and development.
Recent reviews in the area of natural product artifacts have been predominantly focused on analytical chemistry, and for the most part do not address the chemical reactivity of natural products at a mechanistic or molecular level.[1,2,3,4,5,6] An exception to this trend (2020, Capon)[7] presents the case for extracting value from mechanistic insights into the formation of natural products artifacts, using selected marine natural product case studies. This current review returns to and expands on that earlier value proposition, to address the inter-connected concepts of natural product artifacts and innate chemical reactivity — as illustrated by a new set of marine and microbial natural product case studies, with particular attention paid to the factors that initiate, and the underlying mechanisms behind, artifact formation (Table 1). For example, given the critical and nearly ubiquitous role of solvents in natural products science, we survey the risks posed by different solvents in artifact formation. We also explore the influence of heat, pH, light and air, and the propensity of certain classes of natural products to undergo structural diversification through the likes of acetal equilibration, trans-esterification and epimerisation. Another noteworthy discussion point is the specialised subset of cryptic natural products, those endowed with levels of chemical reactivity so high as to preclude detection and isolation. The “unknown unknowns” of the natural product world, such natural products are often only hinted at by the artifacts they leave behind. For the observant researcher, however, perseverance can be rewarded by a hidden cache of knowledge. Finally, we draw attention to a phenomenon where natural products applied to cell-based bioassays undergo in situ biotransformation. Reports on this phenomenon are not prominent in the natural products literature. This is unfortunate, as a failure to acknowledge biotransformation risks can compromise bioassay data analysis and distort conclusions related to potency and selectivity, SAR, pharmacophores and mechanisms-of-action.
In the concluding remarks, the authors integrate the case studies presented and their own experiences to outline best practices for detecting and mitigating artifact formation, and, equally importantly, for recognising it and leveraging the insights it can provide.

Table 1.
Case Studies in Marine and Microbial Natural Products.
| 2 Solvent | cavoxin/cavoxone | photopiperazines |
| 2.1 DMSO | pyrrolizin-3-ones | aspochracin/sclerotiolides |
| migrastatins/dorrigocins | 2.7 dichloromethane | clavosines/calyculins |
| discorhabdins | bromotyramines | 5.2 Photooxidation |
| dendrillic acids | 2.8 benzene | cadinanes |
| methylsulfonated polyketides | theonellastrols | 5.3 Photoreactive |
| cerulenin | 2.9 ethyl acetate | chetomins |
| bisanthraquinones | sorbicillinol/sorbivetone | talaromycins/purpactins |
| glyclauxins | 2.10 aprotic vs protic | 6 Air oxidation |
| aculeaxanthones/chrysoxanthones | oxandrastins | ketidocillinones |
| versixanthones | alaeolide | pseudopyronines |
| secalonic acid/parnafungins | pratensilins | norpectinatone |
| 2.2 pyridine | 3 Heat | linfuranones |
| acremoxanthone/acremonidins | psammaplins/bastadins | hyafurones/aurafurones |
| 2.3 methanol | creolophins | avermectins |
| brevianamides | neobulgarones | penilumamides |
| talaronins | 4 pH | 7 Acetal/ketal equilibration |
| pyrasplorins | 4.1 basic | okichromanone |
| penicipyridones | pestalone/pestalachloride | sphydrofuran |
| varacins | neoenterocins | 8 Trans-esterification |
| epithiodiketopiperazines | asperazepanones | kipukasins |
| eleutherobins/caribaeoranes | hydroxybrevianamides | glenthmycins |
| 2.4 acetone | salinosporamides | amaurones |
| kutzneridines | 4.2 acidic | 9 Epimerization |
| enamidonins/K97-0239A and B | enterocins | aspergillazines |
| autucedines | serratiochelin | quinolactacins |
| madurastatins | franklinolides | 10 Cryptic natural products |
| drimanes | oxanthromicins/eurotones | N-amino-l-proline methyl ester |
| duclauxin/verruculosins | 4.3 silica gel | prolinimines |
| 2.5 acetonitrile | sphydrofurans | N-amino-anthranilic acid |
| talcarpones | duclauxin/bacillisporins | penipacids |
| 2.6 chloroform | xenoclauxin/talaromycesone B | elansolids |
| greensporones | xanthepinone | 11 Bioassay biotransformation |
| alkyl resorcinols | daldinones | abyssomicins |
| azodyrecins | 5 Light | roseopurpurins |
| schipenindolenes | 5.1 Photoisomerization | kendomycin/goondomycins |
| shearinines | pyranpolyenolides |
2. Solvents
To isolate, identify and study natural products first requires extraction from the producing organism (i.e. a microbial fermentation, or a macro marine organism such as a sponge, alga, tunicate etc…), followed by fractionation, spectroscopic, chemical and biological data acquisition and analysis, as well as handling and storage. All these processes require exposure to solvents, leaving open the possibility of forming artifacts. As is evident from the case studies outlined below, while all solvents bring with them some risk of forming artifacts, some solvents clearly present higher risk than others.
2.1. DMSO
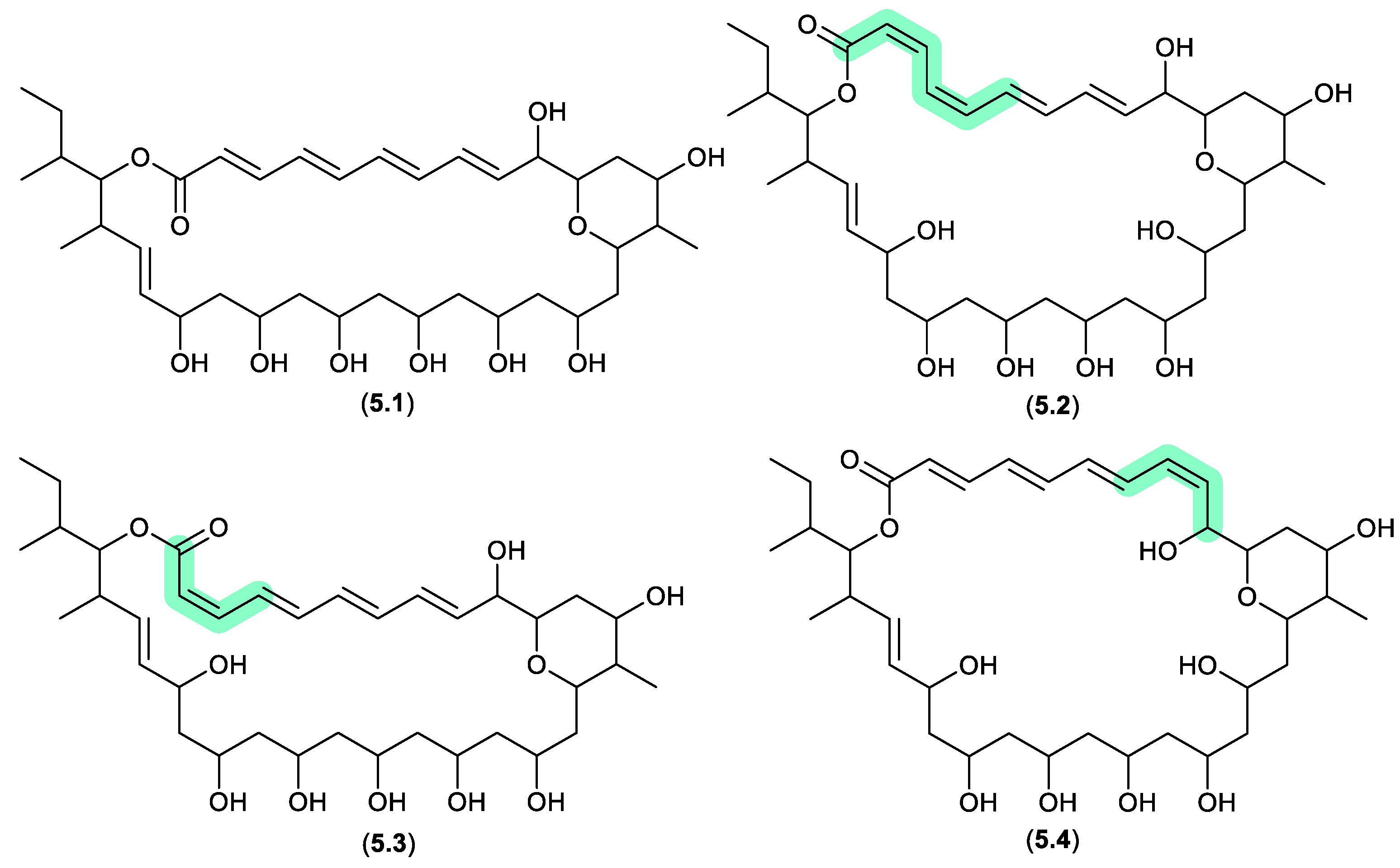
migrastatins/dorrigocins (Figure 2.1.1)
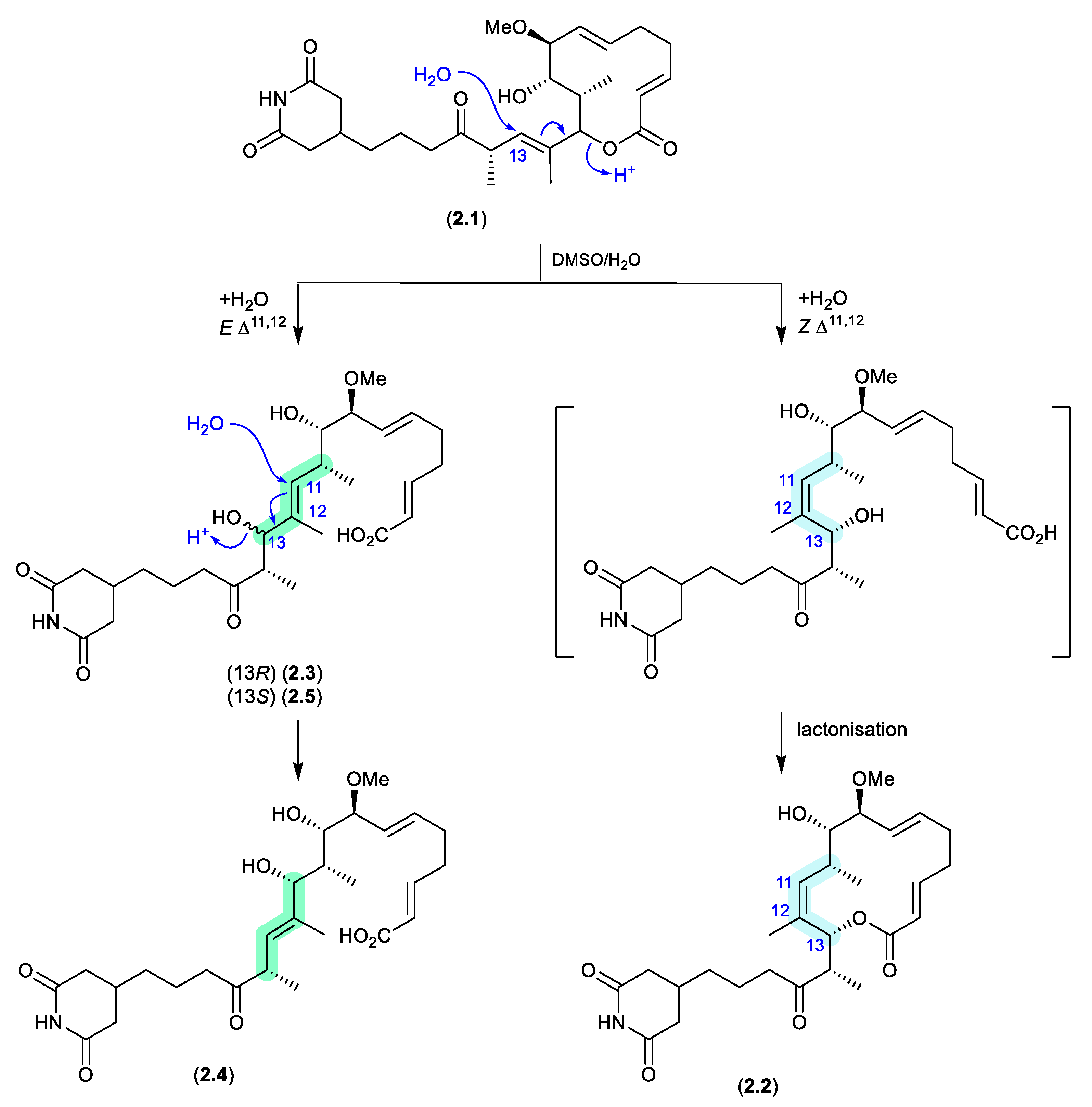
The glutarimide polyketides, iso-migrastatin (2.1), migrastatin (2.2), and dorrigocins A (2.3) and B (2.4), were initially isolated as co-metabolites of Streptomyces platensis (NRRL 18993).[8,9] Subsequent studies identified an additional analogue, 13-epi-dorrigocin A (2.5), and revealed that 2.1 alone was a natural product, with 2.2 to 2.5 produced when 2.1 was stored in DMSO-H2O.[10] These latter transformations can be rationalised as H2O addition to C-13 with concomitant double bond migration and opening of the macrolactone to generate E and Z D11,12 isomers: the E isomer leads to dorrigocin A (2.3), 13-epi-dorrigocin A (2.5) and dorrigocin B (2.4) and the Z isomer undergoes relactonisation to yield migrastatin (2.2). Knowledge of this chemical reactivity was later exploited to produce a library of iso-migrastatin congeners,[11] while further studies demonstrated a direct thermally induced [3,3]-sigmatropic rearrangement of 2.1 to 2.2 paving the way for new analogues.[12]
Figure 2.
1.1. discorhabdins (Figure 2.1.2).
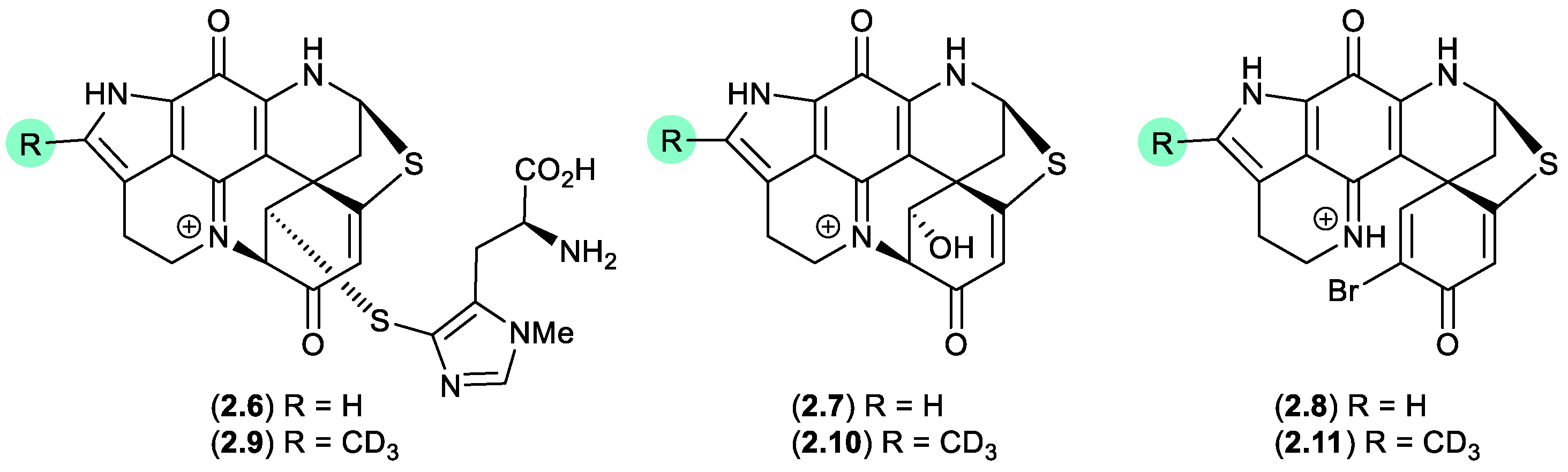
On handling in DMSO-d6, the three pyrroloiminoquinone discorhabdins H (2.6), L (2.7) and B (2.8), isolated from the New Zealand marine sponge Latrunculia kaakaariki, transformed to the trideuteromethyl artifacts 2.9–2.11.[13] Investigations into the mechanism behind this transformation suggested it occurred during recovery of NMR (DMSO-d6) samples in the presence of MeOH, H2O, and trifluoroacetic acid (TFA) (with all three of these solvents being essential for efficient incorporation of the CD3 moiety). ICP-MS revealed trace levels of iron present in both the DMSO-d6 and TFA, presumably initiating OH radicals via a Fenton reaction.
Figure 2.
1.2 dendrillic acids (Figure 2.1.3).
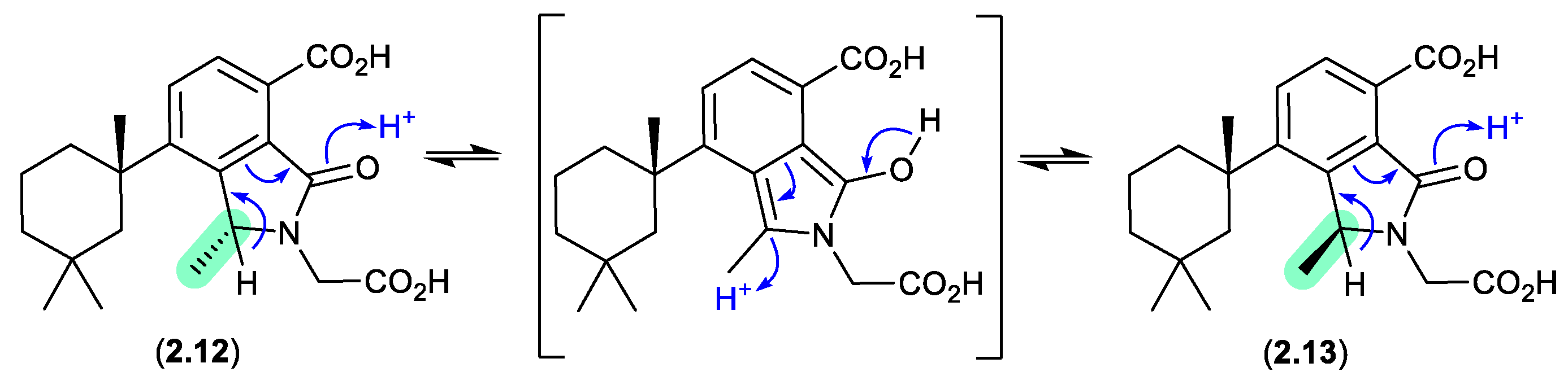
On handling in DMSO-d6, the spongian norditerpenoid dendrillic acids A (2.12) and B (2.13), isolated from the Western Australian marine sponge Dendrilla sp., readily equilibrated at room temperature (r.t.) to an epimeric mixture (2.12/2.13).[14] This epimerisation likely proceeds via enolization of the lactam to an achiral pyrrolo intermediate, as supported by partial incorporation of deuterium into the γ-lactam methine when 5% D2O was added to the DMSO-d6 solution.
Figure 2.
1.3 methylsulfonated polyketides (Figure 2.1.4).
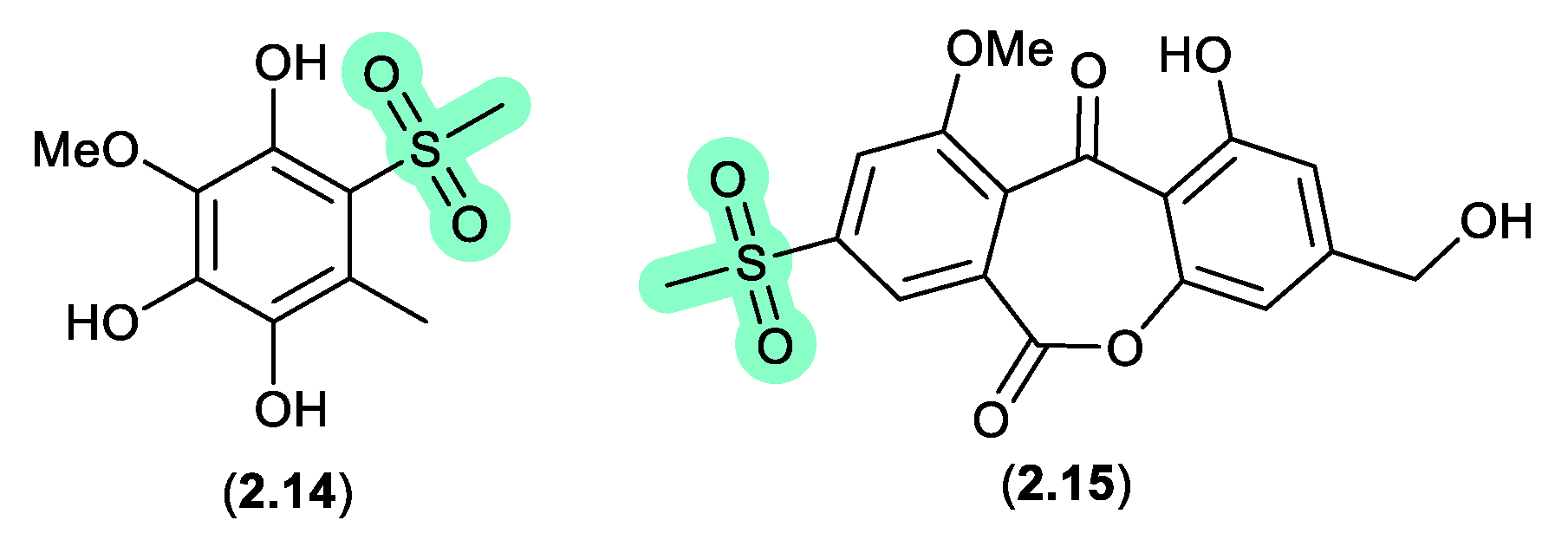
Supplementation of a fermentation of a mangrove-derived fungus Neosartorya udagawae HDN13-313 with a DMSO solution of the DNA methyltransferase inhibitor 5-azacytidine yielded the methylsulfonylated artifacts 2.14 and neosartoryone A (2.15).[15] Investigating this transformation revealed that a fermentation supplemented with DMSO alone also produced 2.14, which suggested that the methylsulfonyl moiety was derived from DMSO. In support of this hypothesis, fermentation in the presence of DMSO-d6 returned the corresponding deuterated analogues of both 2.14 and 2.15.
Figure 2.
1.4 cerulenin (Figure 2.1.5).
Cerulenin (2.16), originally reported mid last century from the fungus Cephalosporium caerulens KF-140, was the first reported natural fatty acid synthase inhibitor, with anticancer, antifungal and anti-obesity potential. In protic solvents 2.16 undergoes intramolecular 5-exo-trig cyclization to generate cerulenin hydroxylactams 2.17 and 2.18. For example, a recent report confirmed that an NMR (DMSO-d6) sample exists almost entirely in the acyclic form (2.16), whereas LC-MS in MeOH/H2O/HCO2H revealed a three-component mixture (2.16–2.18).[16]
Figure 2.
1.5 bisanthraquinones (Figure 2.1.6).
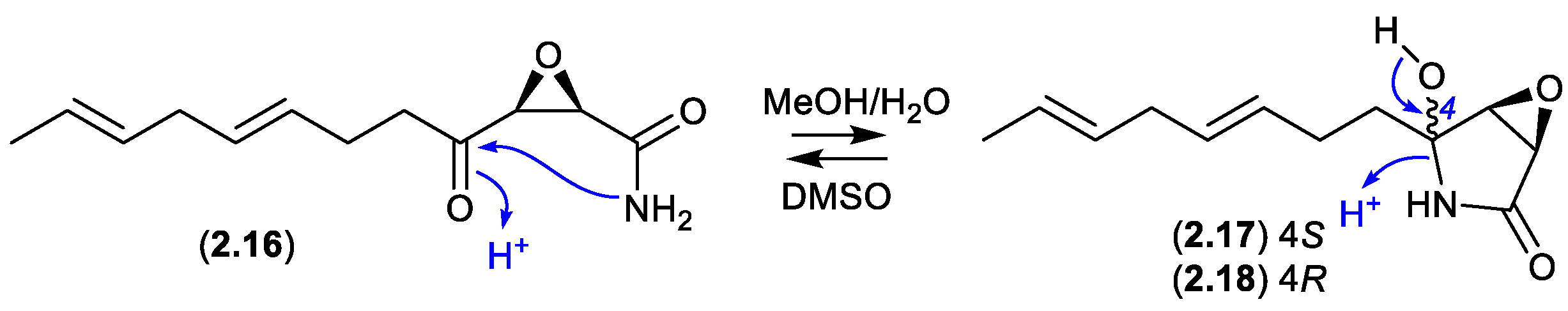
An investigation into a Streptomyces sp. isolated from a cyanobacterium associated with the Puerto Rican tunicate Ecteinascidia turbinata yielded two antibacterial bisanthraquinones, 2.19 and 2.20.[17] During long NMR (DMSO-d6) acquisitions both these metabolites degraded to a single common dehydration artifact 2.21. Partial conversion of 2.19 to 2.21 was also achieved by allowing 2.19 to stand at r.t. in DMSO for several days. Of note, the artifact 2.21 was 220-fold more active against methicillin-resistant Staphylococcus aureus (MRSA) than the parent natural product 2.19.
Figure 2.
1.6 glyclauxins (Figure 2.1.7).
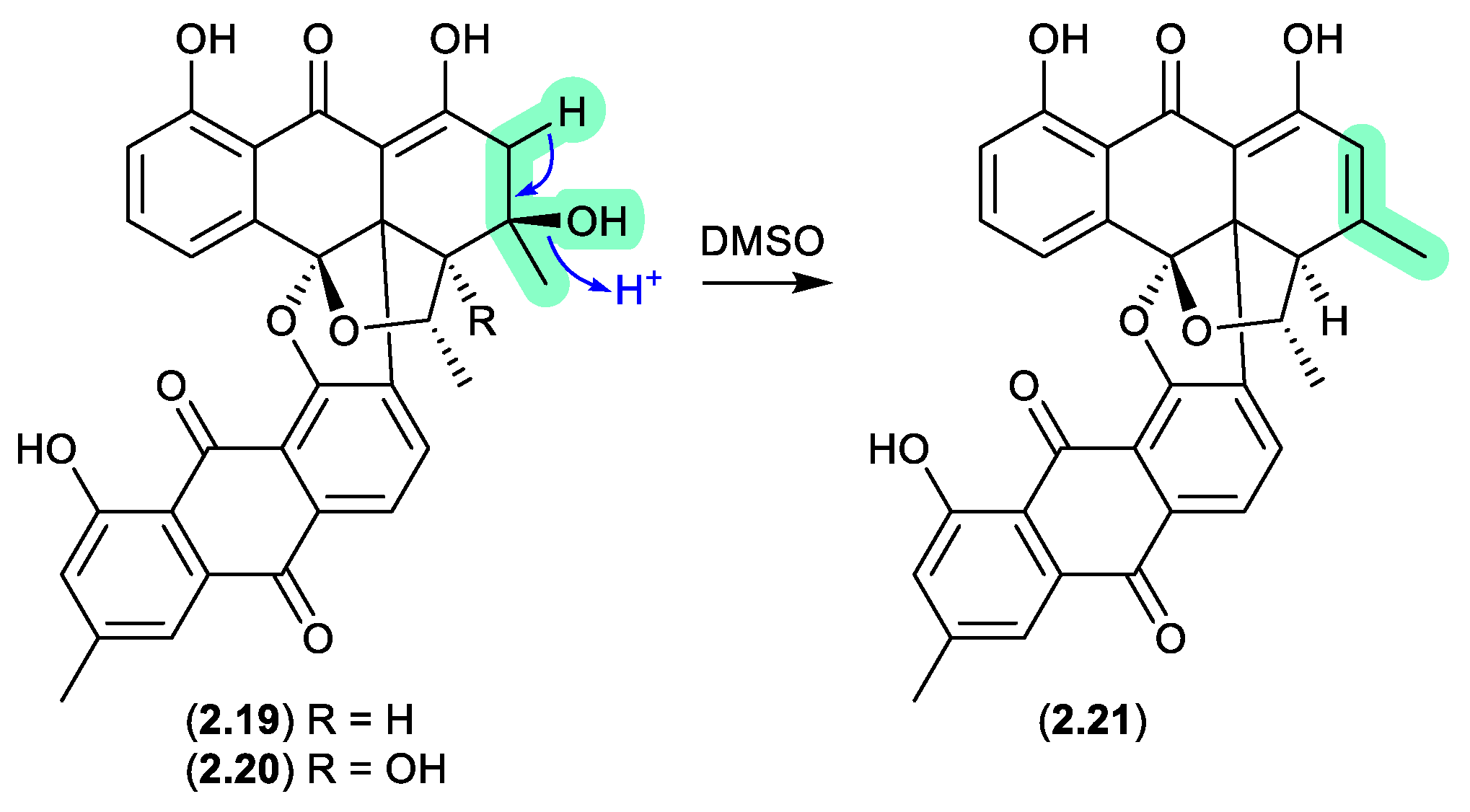
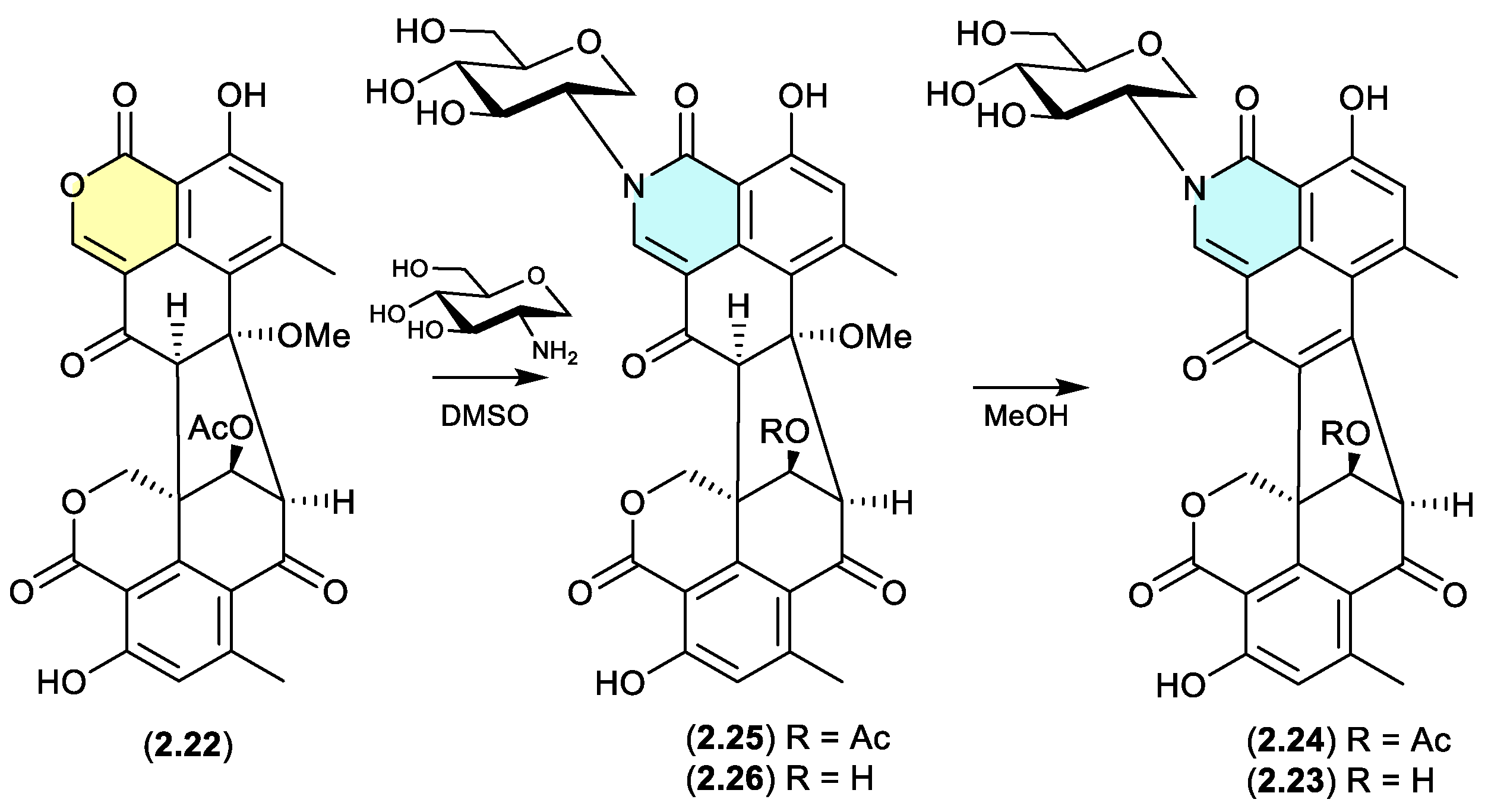
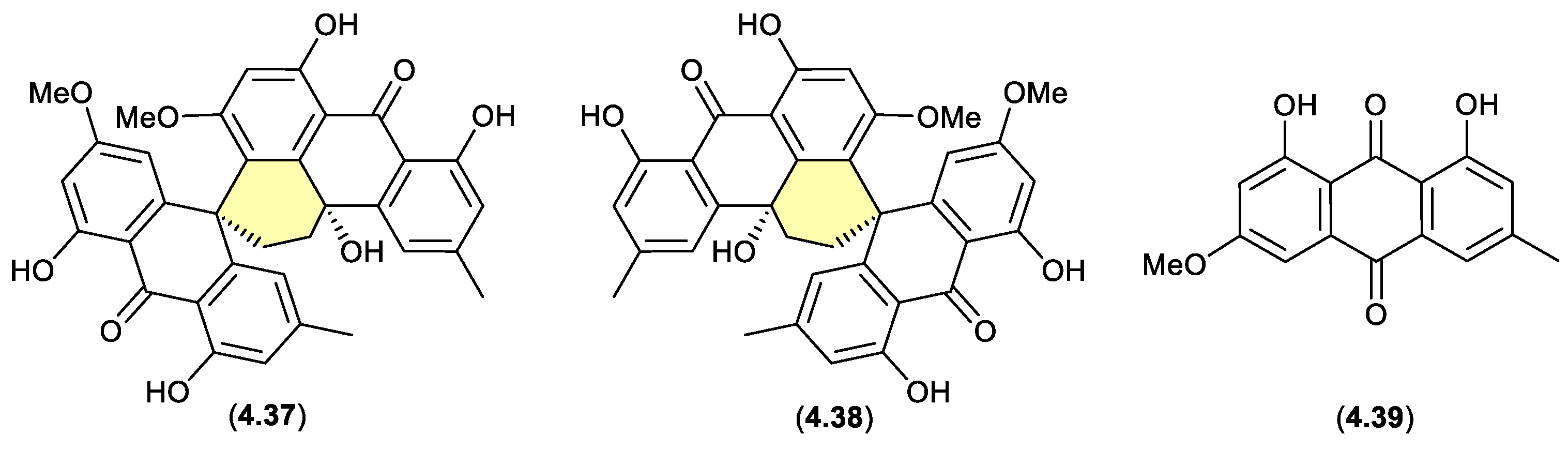
The Australian wasp nest-derived fungus Talaromyces sp. CMB-MW102 yielded the known duclauxin (2.22) (see Section 4.3) and a series of 1-deoxy-d-glucosamine adducts, glyclauxins A–E.[18] In an effort to understand the biosynthetic relationship between duclauxin and glyclauxins, and implement a biomimetic synthesis, a r.t. DMSO solution of 2.22 and synthetic 1-deoxy-d-glucosamine underwent quantitative conversion to glyclauxin B (2.24), while MeOH solutions of glyclauxins C (2.25) and D (2.26) underwent rapid and quantitative transformation to glyclauxins B (2.24) and A (2.23), respectively. Despite the ease of these latter transformations, both 2.23 and 2.24 were detected in fresh unfractionated culture extracts — attesting to their dual status as both natural products and artifacts (see Section 12).
Figure 2.
1.7 aculeaxanthones/chrysoxanthones (Figure 2.1.8).
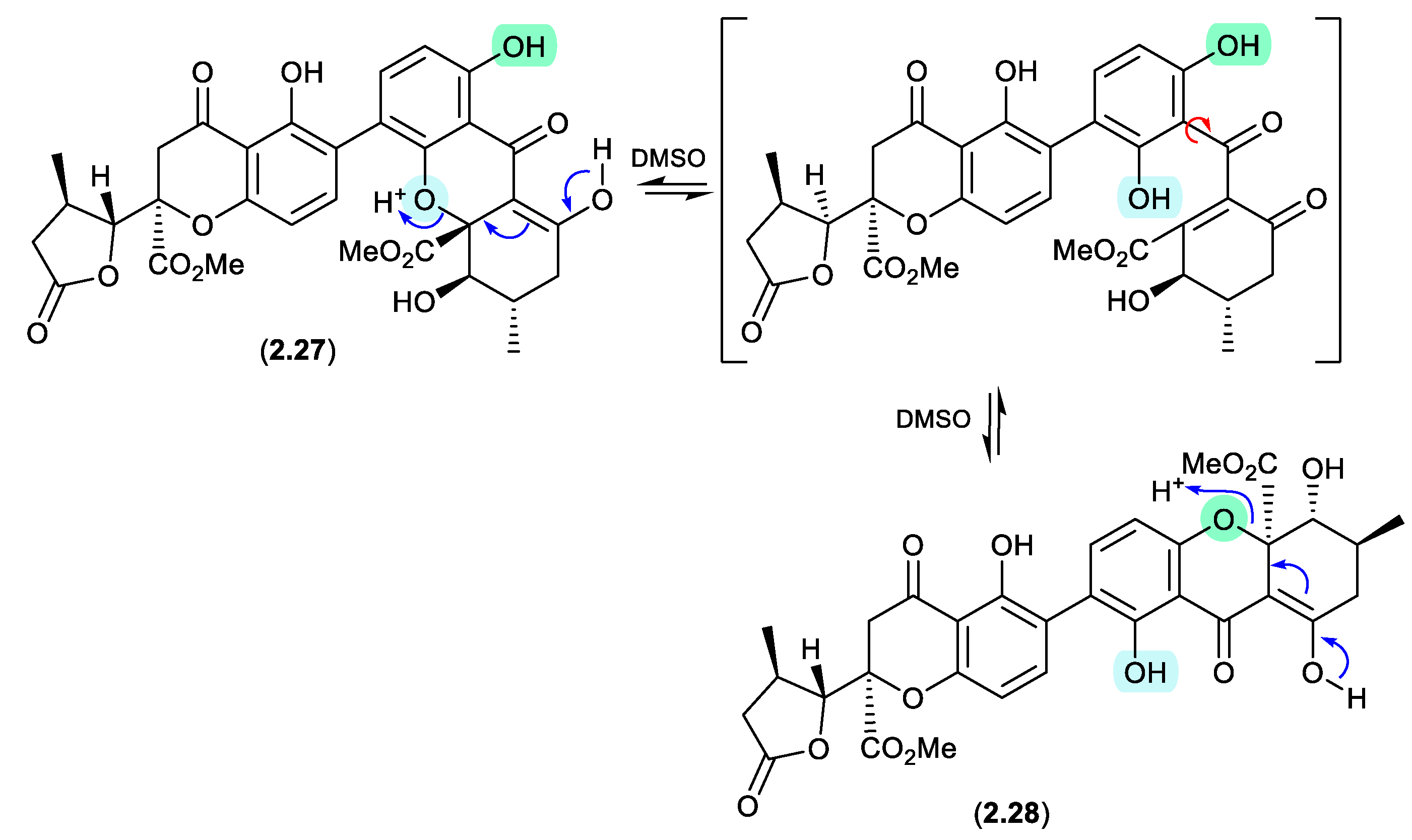
Aculeaxanthone B (2.27), from the marine-derived fungus Aspergillus aculeatinus WHUF0198, undergoes a retro-oxa-Michael equilibration in DMSO-d6 to the regioisomer, chrysoaxanthone B (2.28).[19] As 2.28 was itself reported as a natural product from the sponge-derived Penicillium chrysogenum HLS111,[20] this raises an interesting question — “Is one regioisomer an artifact, and if so, which one?”
Figure 2.
1.8 versixanthones (Figure 2.1.9).
Similar DMSO mediated retro-oxa-Michael equilibrations were reported in 2015 between versixanthones A (2.29) and D (2.30), and versixanthones B (2.31) and C (2.32), which were isolated from the marine-derived fungus Aspergillus versicolor HDN100.9.[21]
Figure 2.
1.9 secalonic acid/parnafungins (Figure 2.1.10 and 2.1.11).
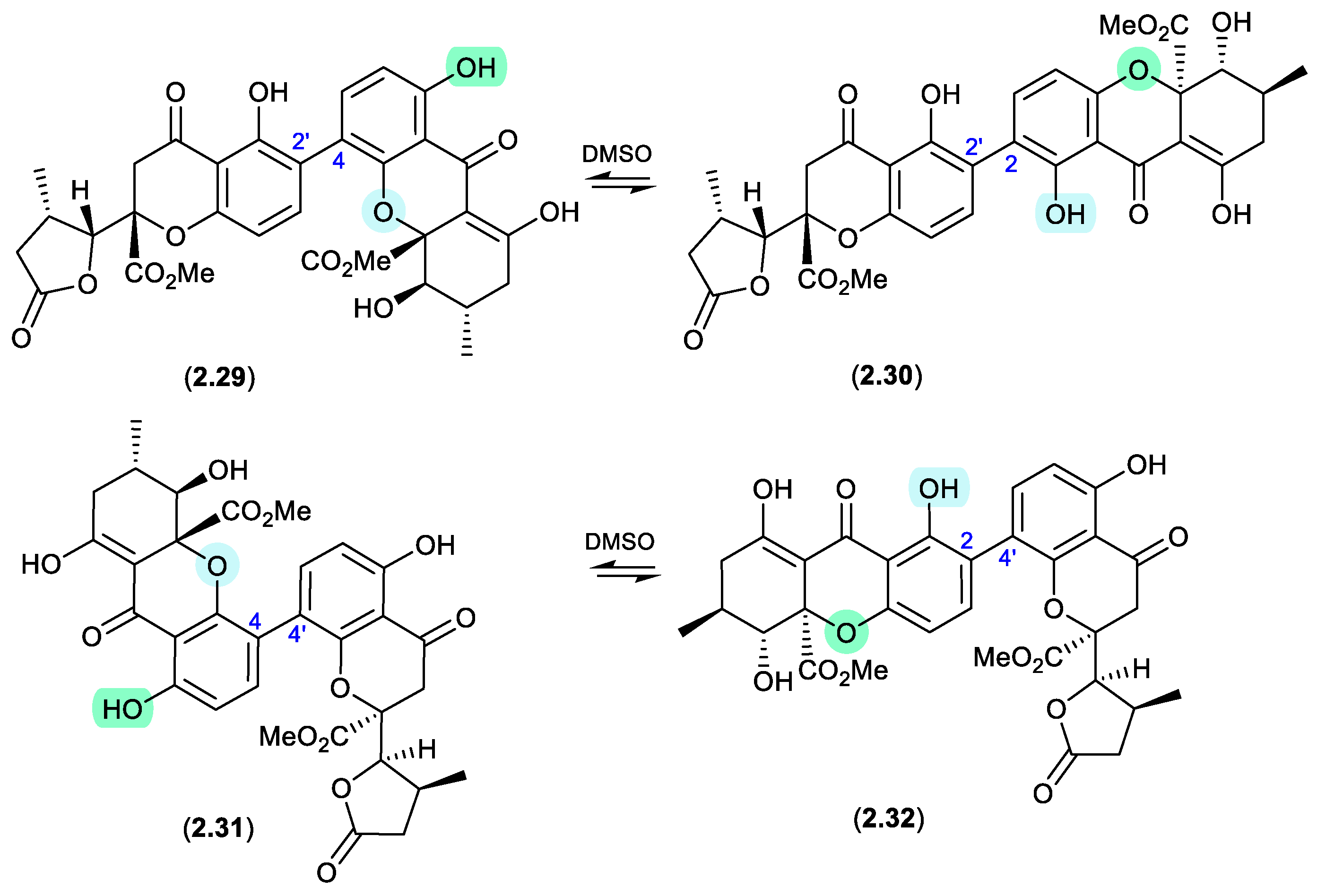
This solvent mediated intramolecular retro-oxa-Michael equilibration was further demonstrated in studies of secalonic acid A (2.33) and its associated regioisomers,[22,23] as well as in the relationship between parnafungin A, which is a mixture of the C-15a epimers parnafungins A1 (2.34) and A2 (2.35), and parnafungin B, a mixture of the C-15a epimers parnafungins B1 (2.36) and B2 (2.37).[24]
Figure 2.
1.10.
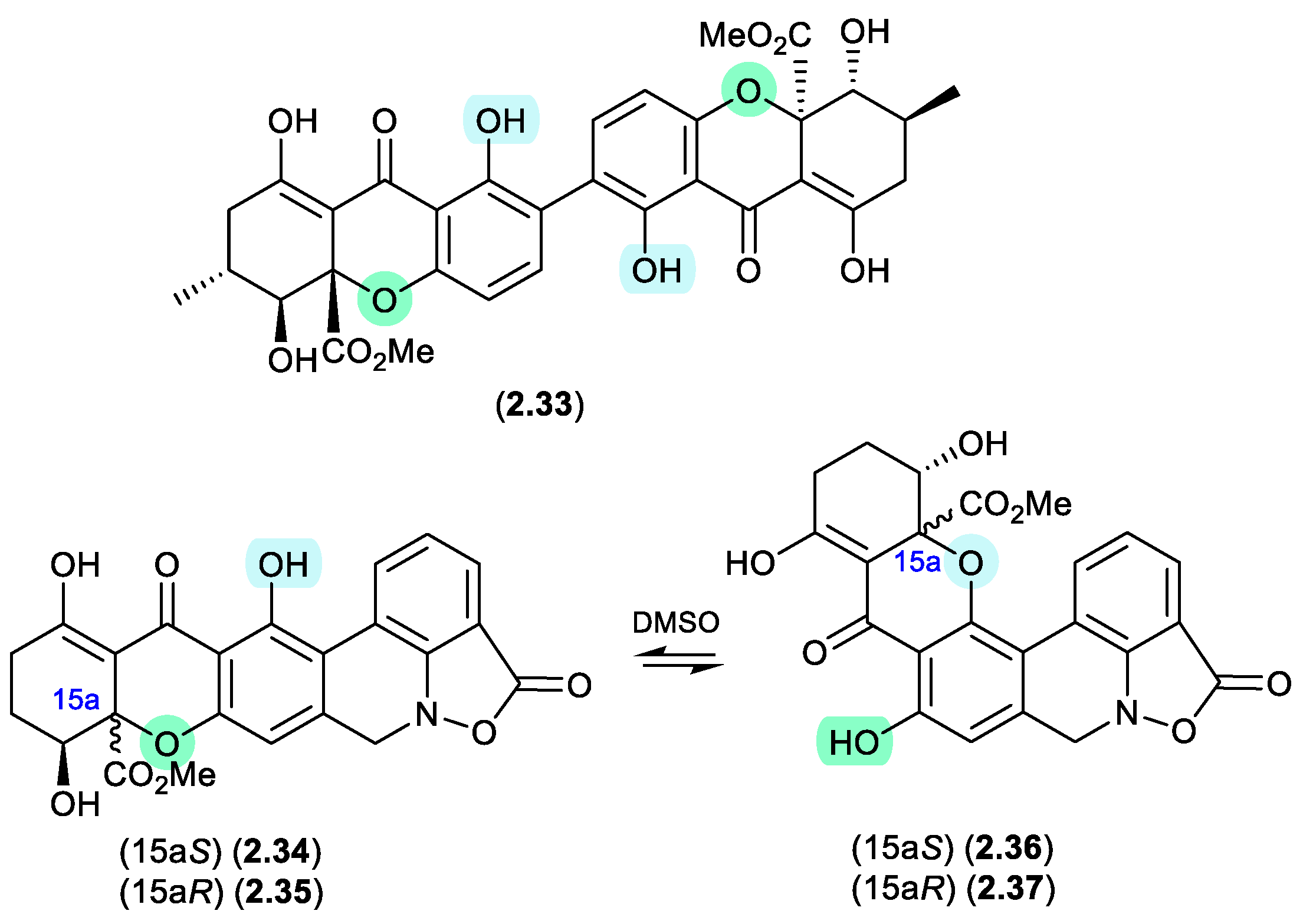
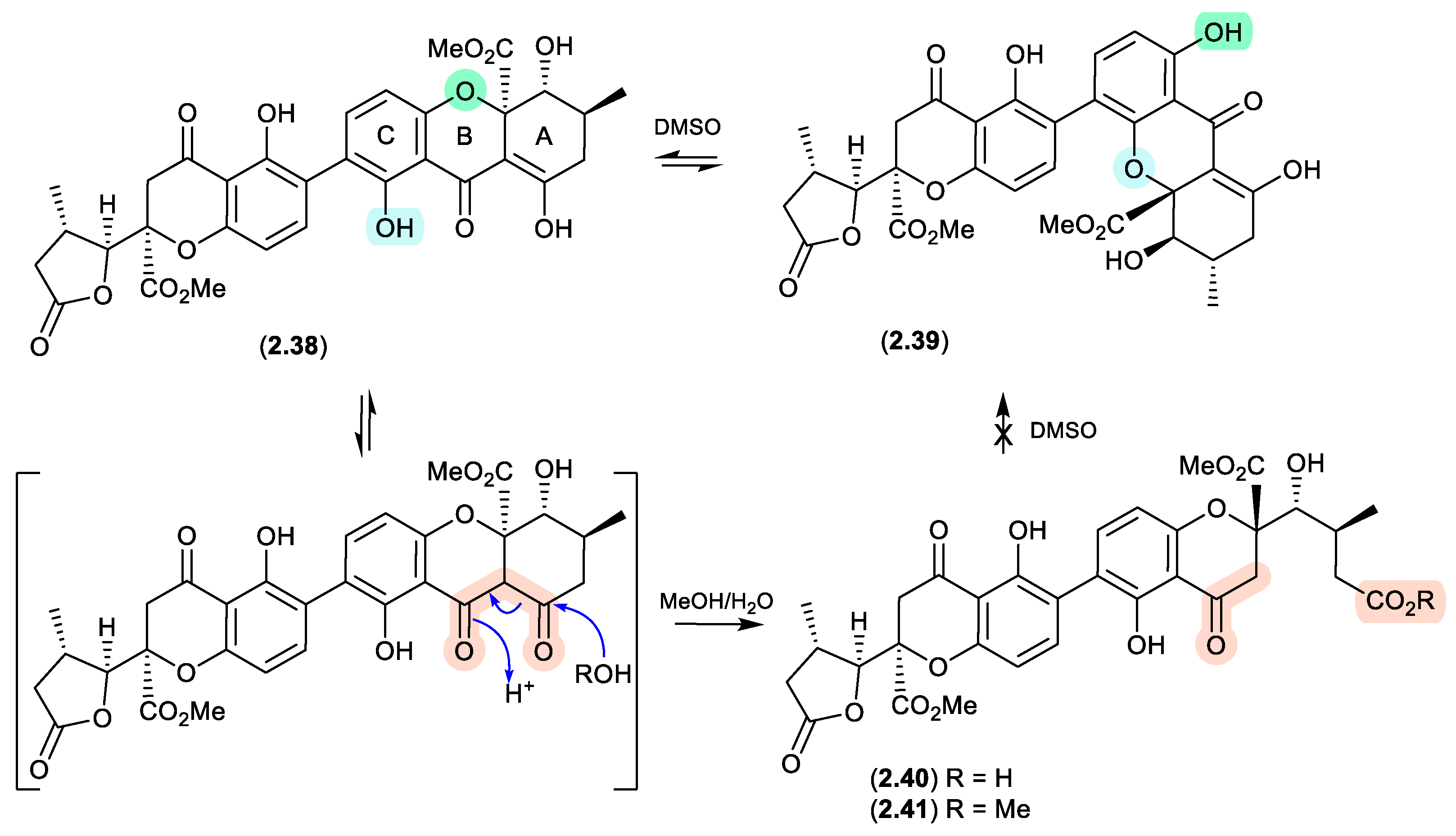
Of note, the chemical reactivity of the tetrahydroxanthone-chromanone core common across 2.33–2.37 is not restricted to the intramolecular retro-oxa-Michael equilibrations outlined above. For example, while DMSO mediated an intra-molecular retro-oxa-Michael equilibration between chrysoxanthone E (2.38) and G (2.39), exposure of 2.38 to H2O facilitated hydrolysis of ring A to chrysoxanthone O (2.40), while addition of MeOH extended this to include the methanolysis product chrysoxanthone P (2.41).[25] Unsurprisingly, the opening of ring A removed the ability to undertake retro-oxa-Michael equilibration. With DMSO, MeOH and H2O being common extraction, purification and handling solvents, the ease with which these r.t. transformations take place raises the likelihood that handling artifacts have been misidentified as natural products. It also challenges the veracity of bioactivity data and the ability to assign SAR, where analytes can transformation during bioassays (see Section 11).
Figure 2.
1.11.
2.2. Pyridine
acremoxanthones/acremonidins (Figure 2.2.1)
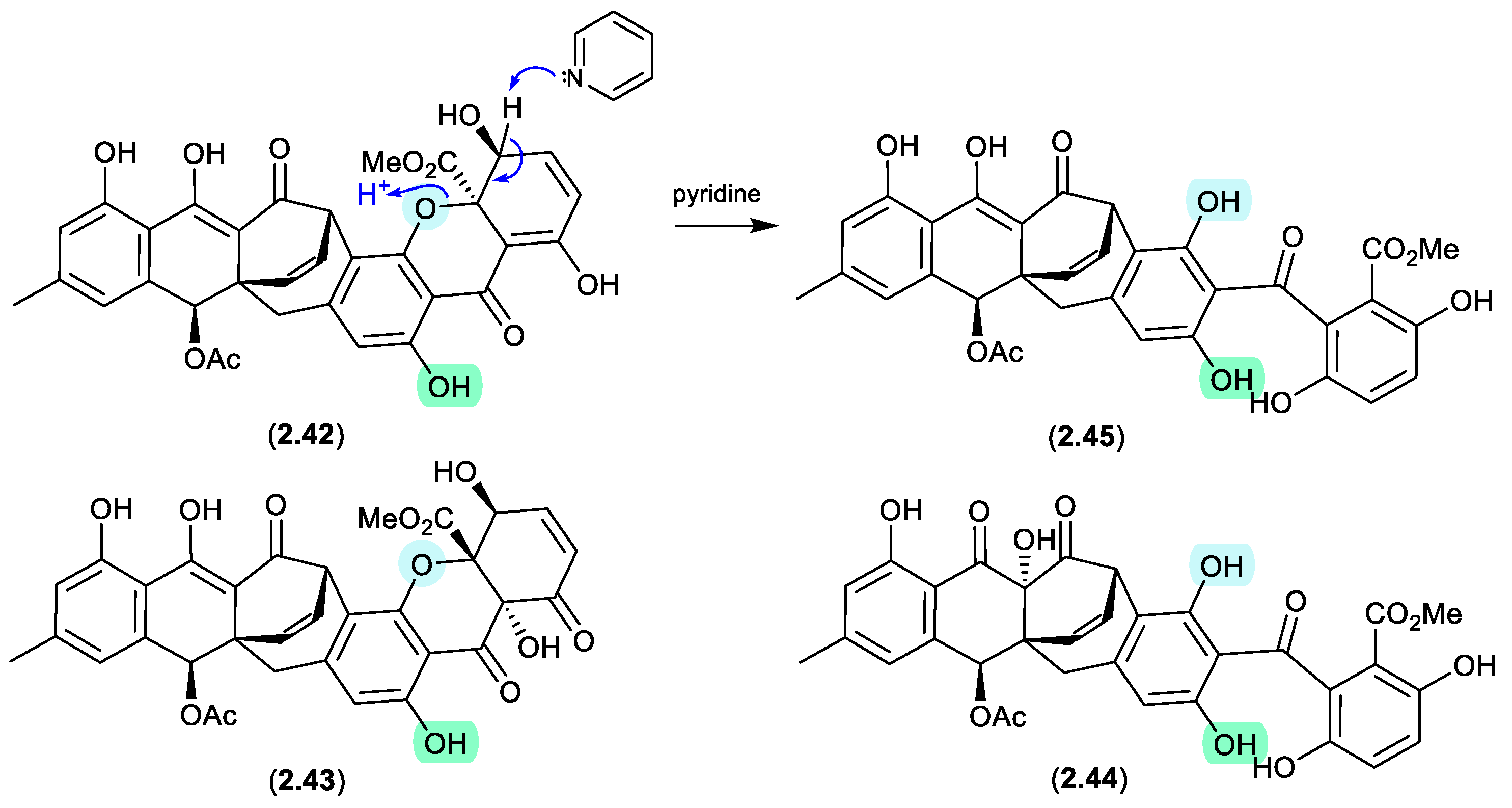
An unidentified fungus of the order Hypocreales (MSX 17022) yielded two new xanthone-anthraquinone heterodimers, acremoxanthones C (2.42) and D (2.43), and the closely related known metabolites acremonidins C (2.44) and A (2.45).[26] Interestingly, attempts at preparing Mosher esters revealed that 2.43 was unstable to pyridine. Indeed, when exposed to pyridine alone at r.t. for 4.5 h, 2.43 underwent complete conversion to 2.45. As noted above (Section 2.1) such a transformation would be expected to occur under neutral conditions in DMSO.
Figure 2.
2.1.
2.3. Methanol
brevianamides (Figure 2.3.1 and 2.3.2)
The Lake Michigan deep-water sediment-derived fungus Penicillium sp. 5-PBA-2 yielded two diketopiperazines, brevianamides E1 (2.46) and E2 (2.47), along with known analogues, including brevianamide E (2.48).[27] Of note, 2.46 and 2.47 rapidly equilibrated (1:9 ratio) in acidic HPLC solvents (MeCN/H2O with either 0.1% formic acid or 0.1% TFA), while prolonged exposure of the equilibrating mixture to acidic MeOH (0.1% TFA) yielded the methyl ether 2.49, as well as the two rearranged products 2.50 and 2.51. It is proposed that the latter are formed by reaction with formaldehyde present as an impurity in commercial MeOH — confirmed by addition of 2,4-dinitrophenylhydrazine to commercial MeOH and the detection of formaldehyde 2,4-dinitrophenylhydrazone.
Figure 2.
3.1.
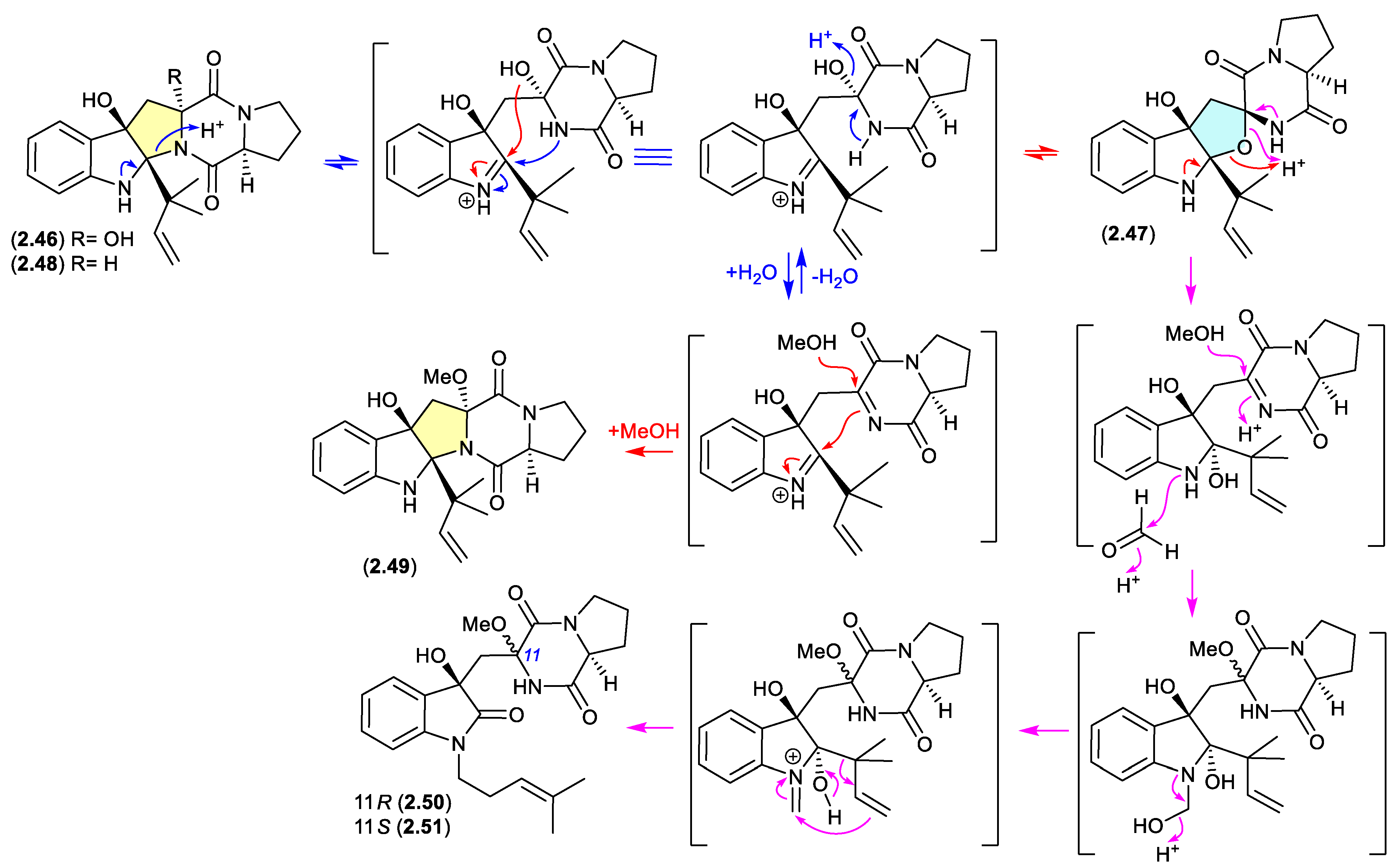
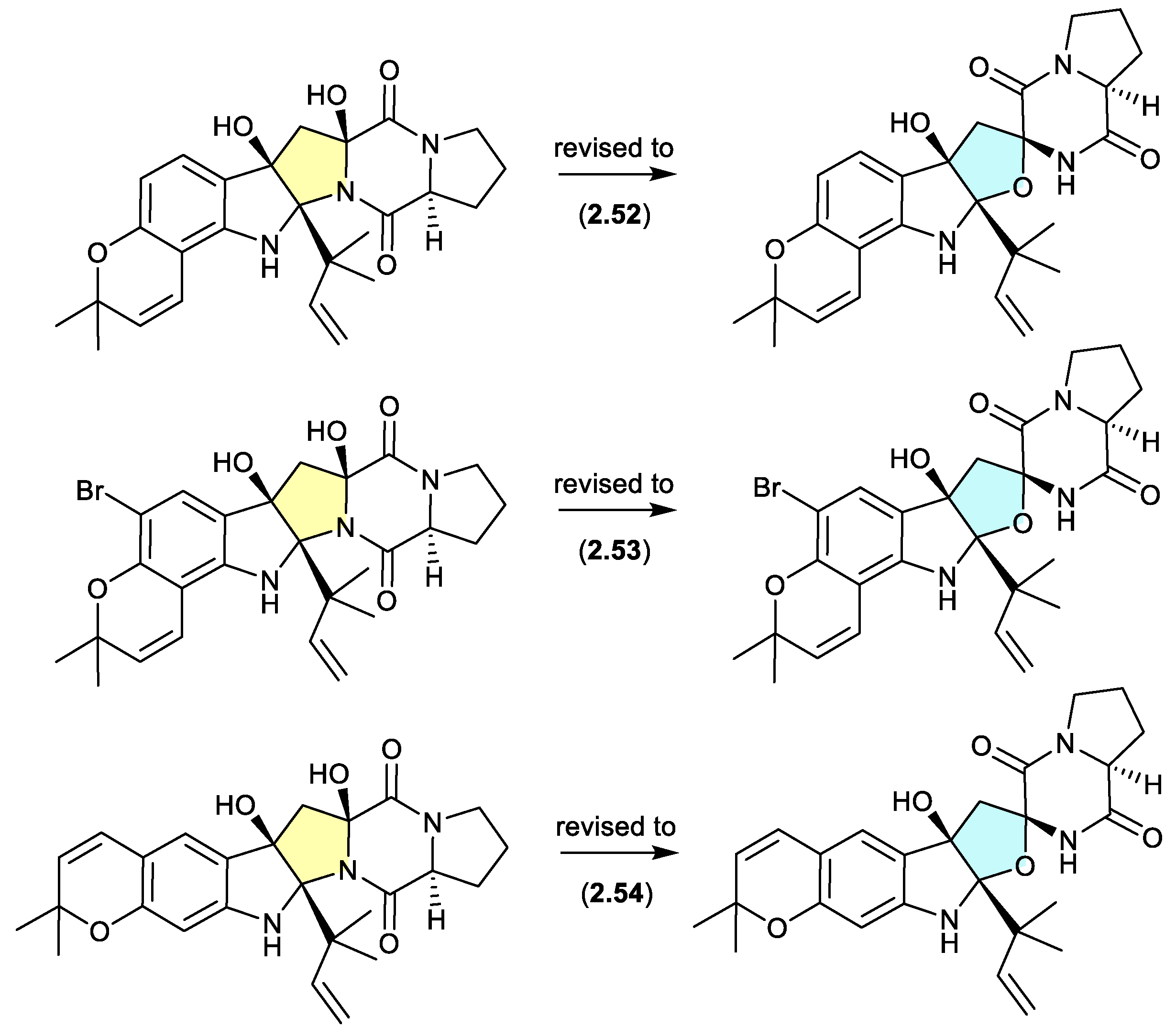
A comparison of experimental and calculated ECD spectra (with 2.47) led to structure revisions for known diketopiperazines featuring a rare 11-oxy moiety, including notoamides K (2.52) and P (2.53), and asperversiamide L (2.54).[27]
Figure 2.
3.2. talaronins (Figure 2.3.3).
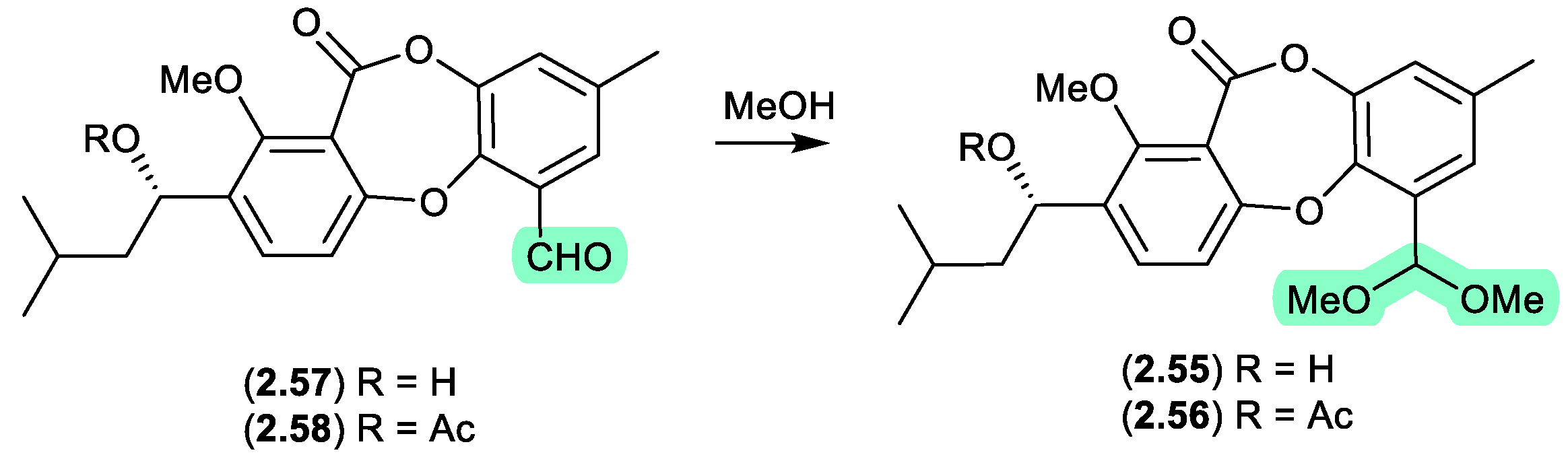
The Chinese mangrove derived fungus Talaromyces sp. WHUF0362 yielded a range of polyketides, including the new depsidone dimethylacetals, talaronins A (2.55) and B (2.56), the new depsidone benzaldehyde talaronin D (2.57), and the known purpactin C′ (2.58).[28] Although the dimethylacetals 2.55 and 2.56 are designated as natural products, the extensive use of MeOH eluant across Sephadex LH-20 and HPLC chromatography makes it far more probable that the dimethylacetals are methanolysis artifacts of 2.57 and 2.58, respectively. Other examples of natural product aldehydes transforming in MeOH to dimethyl acetals include the colletotrichalactones from the Korean leaf-derived endophytic fungus Colletotrichum sp. JS-0361,[29] cladosporisteroids from the Chinese marine sponge-derived fungus Cladosporium sp. SCSIO41007,[30] and the talaromycins from the South China Sea gorgonian-derived fungus Talaromyces sp. (see Section 5.3).[31]
Figure 2.
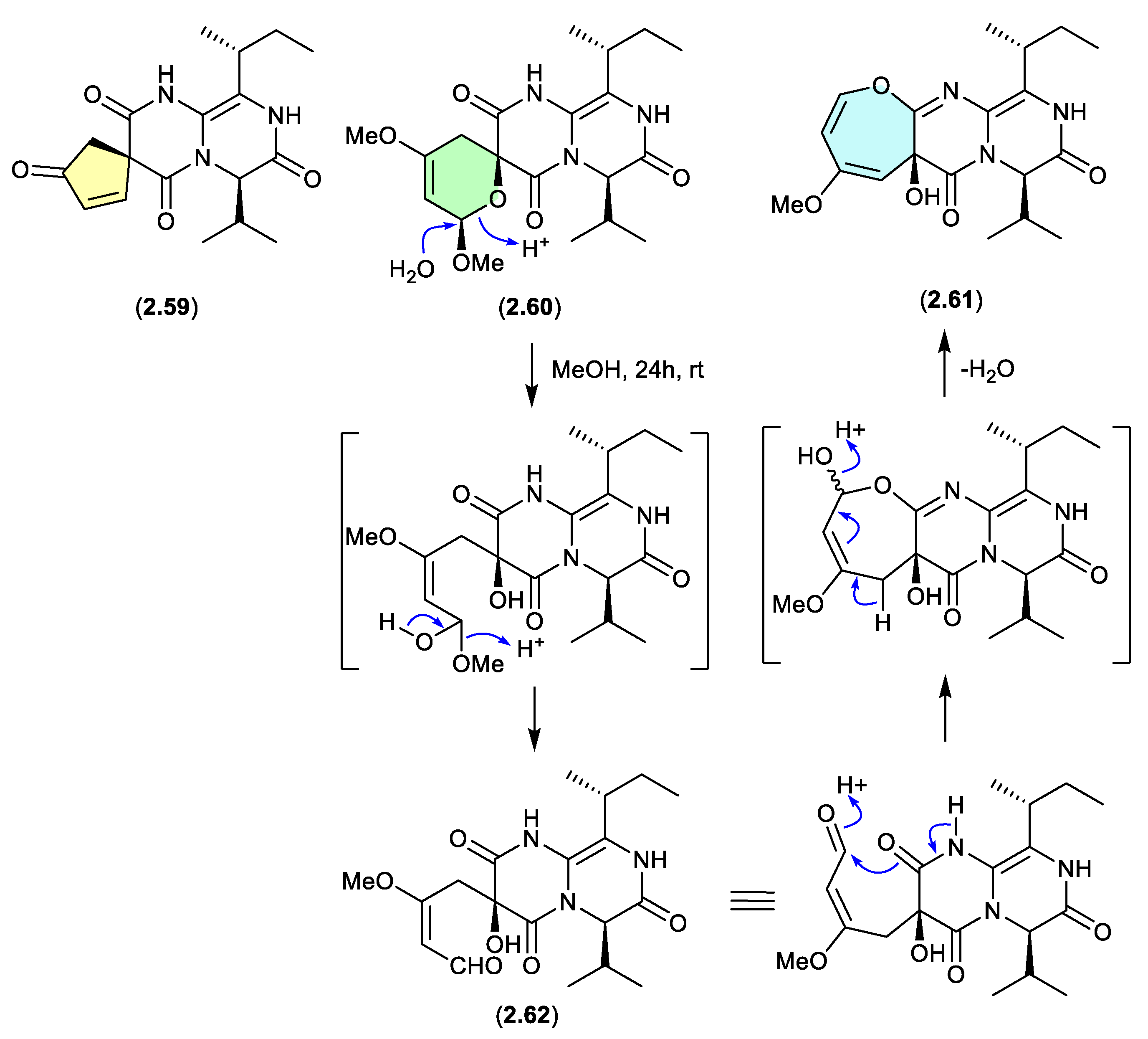
3.3 pyrasplorins (Figure 2.3.4).
The mangrove-derived fungus Aspergillus versicolor yielded an array of pyrazinopyrimidine alkaloids, pyrasplorins A–C (2.59–2.61).[32] On handling in MeOH, 2.60 underwent ring opening to the artifact 2.62; however, it’s likely that the ring opening was mediated by H2O present in the MeOH). Although the authors make no mention of a plausible dehydration pathway from the artifact 2.62 to 2.61, it raises the prospect that the latter is also an artifact.
Figure 2.
3.4. penicipyridones (Figure 2.3.5).
The marine mangrove plant-derived fungus Penicillium oxalicum QDU1 yielded 11 new pyridine alkaloids, exemplified by penicipyridones A–C (2.63–2.65).[33] Indicative of their chemical reactivity, over 3 d acidic solutions of 2.63 (MeCN/H2O or MeOH/H2O, with 0.1% formic acid) effected a transformation to 2.64, with trace amounts of 2.65. After 14 d, acidic MeOH solutions of any one of 2.63–2.65 equilibrated to an 8:7:1 mixture. This reactivity reveals a biomimetic path to adding unnatural nucleophiles at C-4.
Figure 2.
3.5. variacins (Figure 2.3.6).
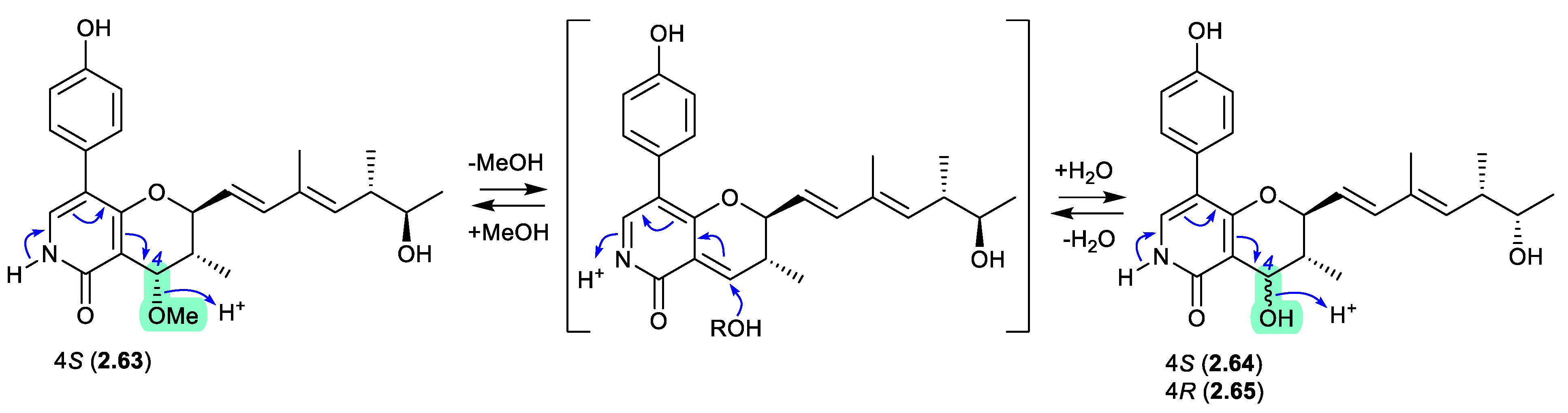
The benzopentathiepin varacin (2.66), isolated from a Fijian sample of the marine ascidian Lissoclinum vareau, exhibited potent cytotoxic properties against human colon carcinoma cells (HCT 116).[34] Subsequently, 2.66 was reported along with three analogues, varacins A–C (2.67–2.69), from a Sea of Japan ascidian Polycitor sp.[35] Interestingly, solutions of 2.66 or 2.67 (in MeOH, CH2Cl2 or pyridine) equilibrate to mixtures of 2.66, 2.67 and S8 (2.70).
Figure 2.
3.6. epithiodiketopiperazines (Figure 2.3.7 and 2.3.8).
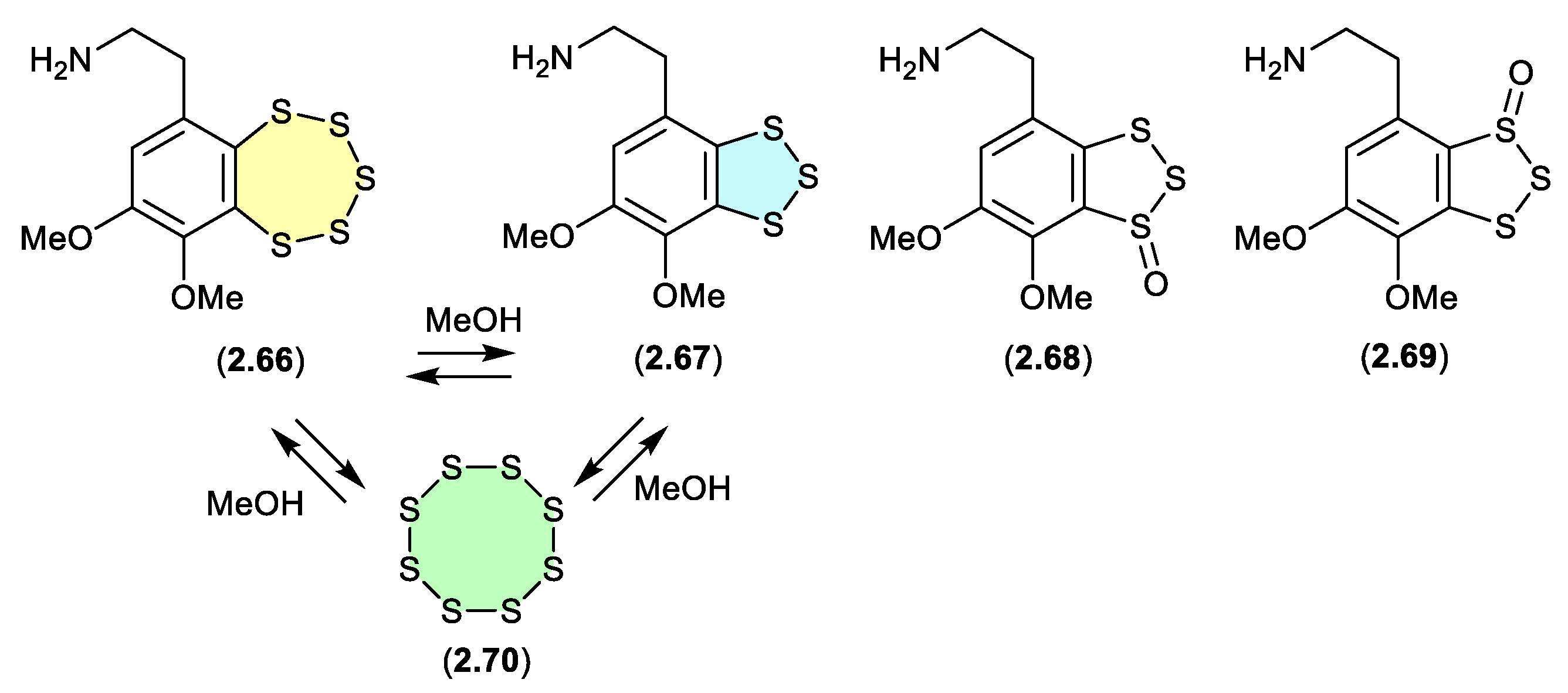
The study of a fungal strain Penicillium sp. YE isolated from a Florida collection of a coral Pseudodiploria strigosa infected with coral black band disease, yielded 4 new epithiodiketopiperazines, penigainamides A (2.71), B (2.72), C (2.73) and D (2.74), and five known analogues, adametizine A (2.75), FA2097 (2.76), outovirins A (2.77) and C (2.78) and pretrichodermamide C (2.79).[36] Some of these proved unstable to handling in a range of solvents, leading to either contraction/expansion of the di, tri and tetra thioether ring, and/or transformation between chlorohydrin and epoxide moieties. For example, exposure of (i) 2.75 to MeOH (1 wk) lead to partial conversion to 2.71–2.73 and 2.76, and H2O (1 wk) to 2.72, 2.76, 2.78 and 2.79; (ii) 2.71 to CDCl3 (1 h) lead to partial conversion to 2.74; (iii) 2.72 to methanol-d4 (30 min) to 2.73; and (iv) 2.78 to H2O (1 wk) to 2.77, to acetone-d6 (30 min) to 2.74, and to artificial sea water culture media (PDB) to 2.75.
Figure 2.
3.7.
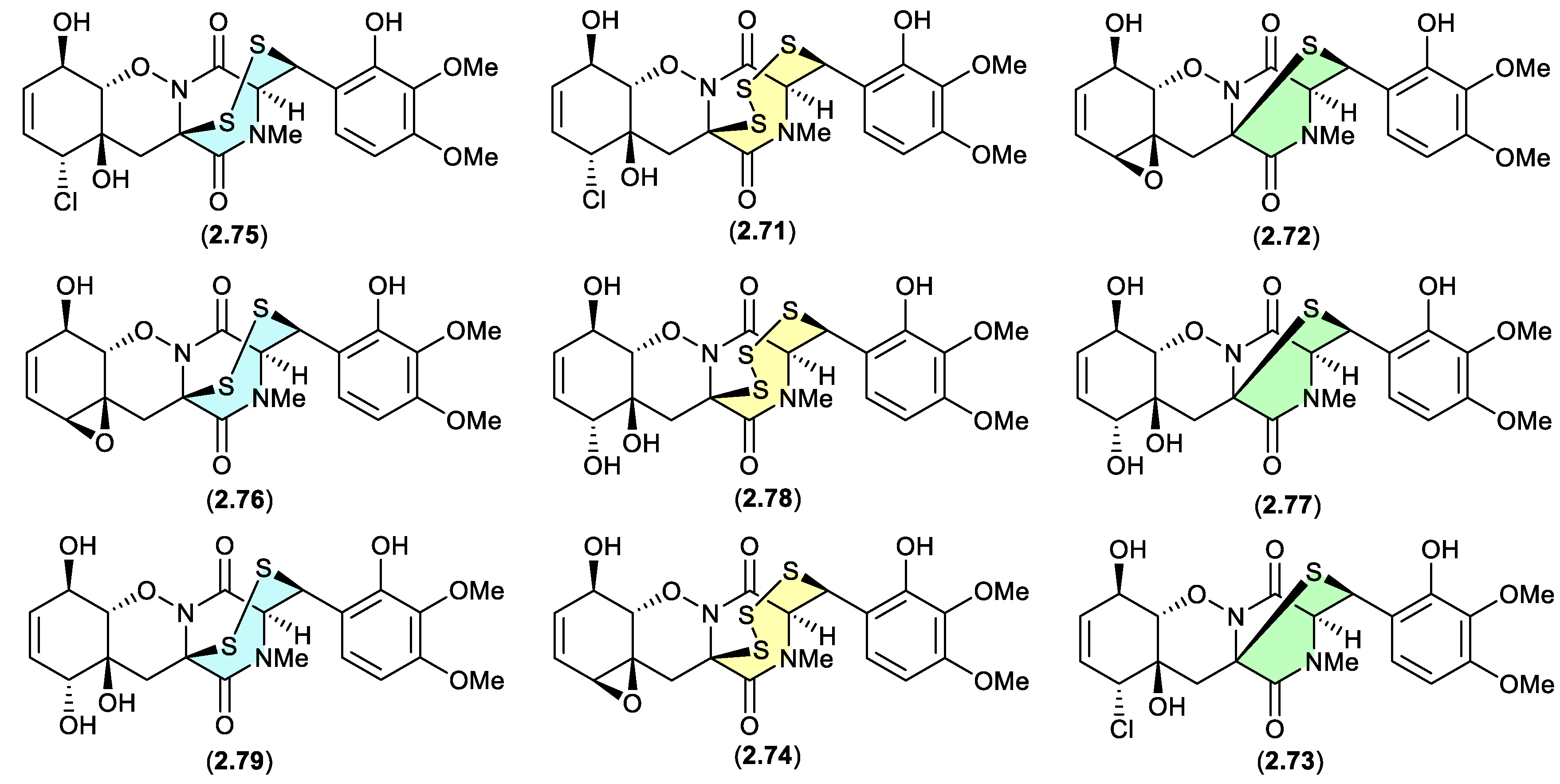
Figure 2.
3.8.
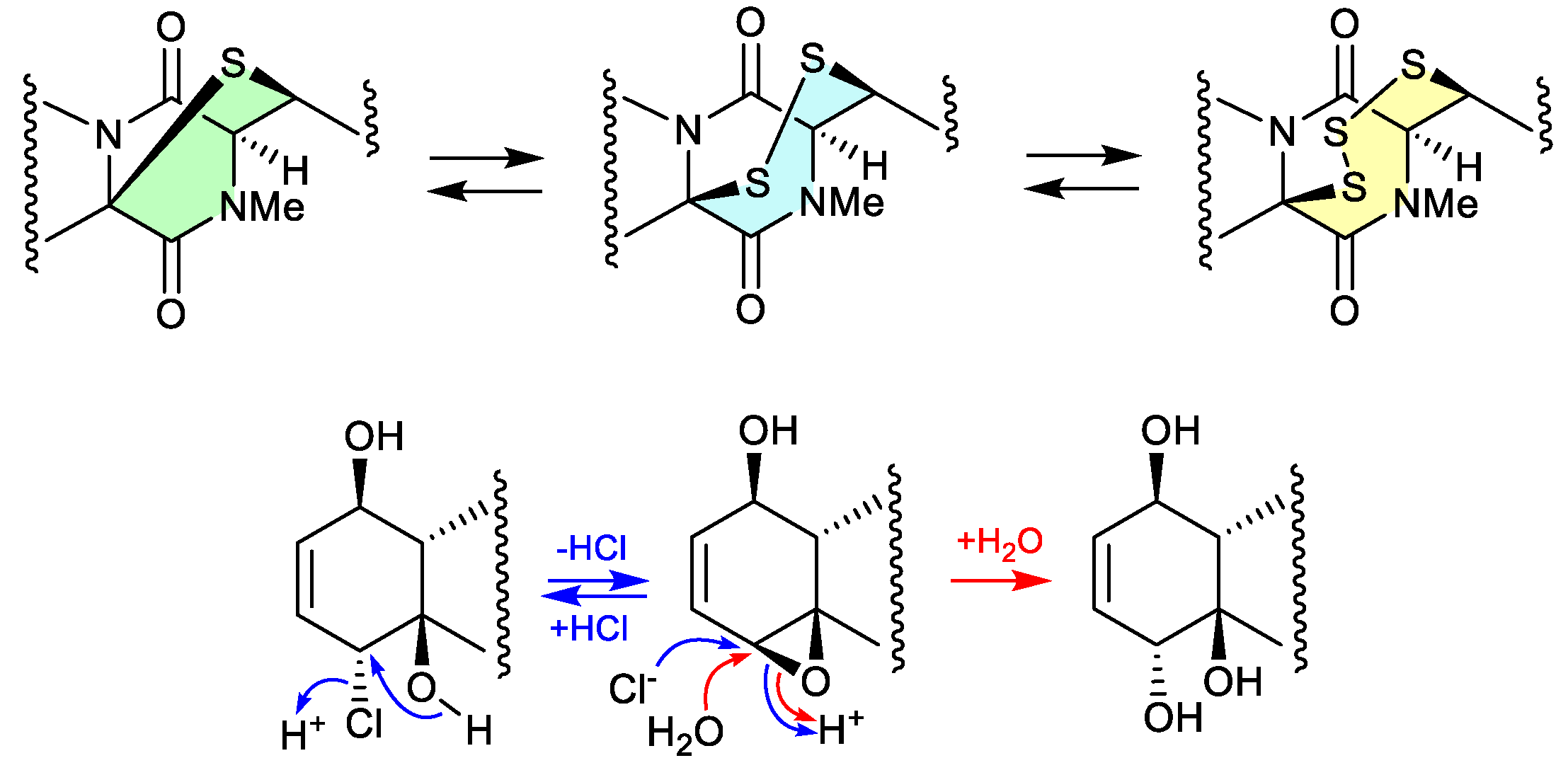
eleutherobins/caribaeoranes (Figure 2.3.9)
Eleutherobin (2.80), first reported in 1997 by Fenical et al from a Western Australian sample of soft coral Elutherobia sp., exhibited exceptionally activity as a microtubule-stabilising antimitotic agent that was comparable to paclitaxel.[37] Subsequently, Andersen et al reported the re-isolation of 2.80 in 2000, along with the six new analogues, desmethyleleutherobin (2.81), desacetyleleutherobin (2.82), isoeleutherobin A (2.83), Z-eleutherobin (2.84), caribaeoside (2.85) and caribaeolin (2.86), from southern Caribbean samples of the octocoral Erythropodium caribaeorum.[38] In both studies, fractionation involved the use of silica gel (MeOH) chromatography, raising the possibility that the 4-methylacetals were handling artifacts. In a follow-up 2001 report, Andersen et al described alternate solvent extractions of fresh collections of E. caribaeorum, with MeOH extraction returning the previously reported 4-methylacetals, and EtOH extraction returning the corresponding 4-ethylketals — revealing the true natural products in this class to be 4-hemiketals.[39]
Figure 2.
3.9.
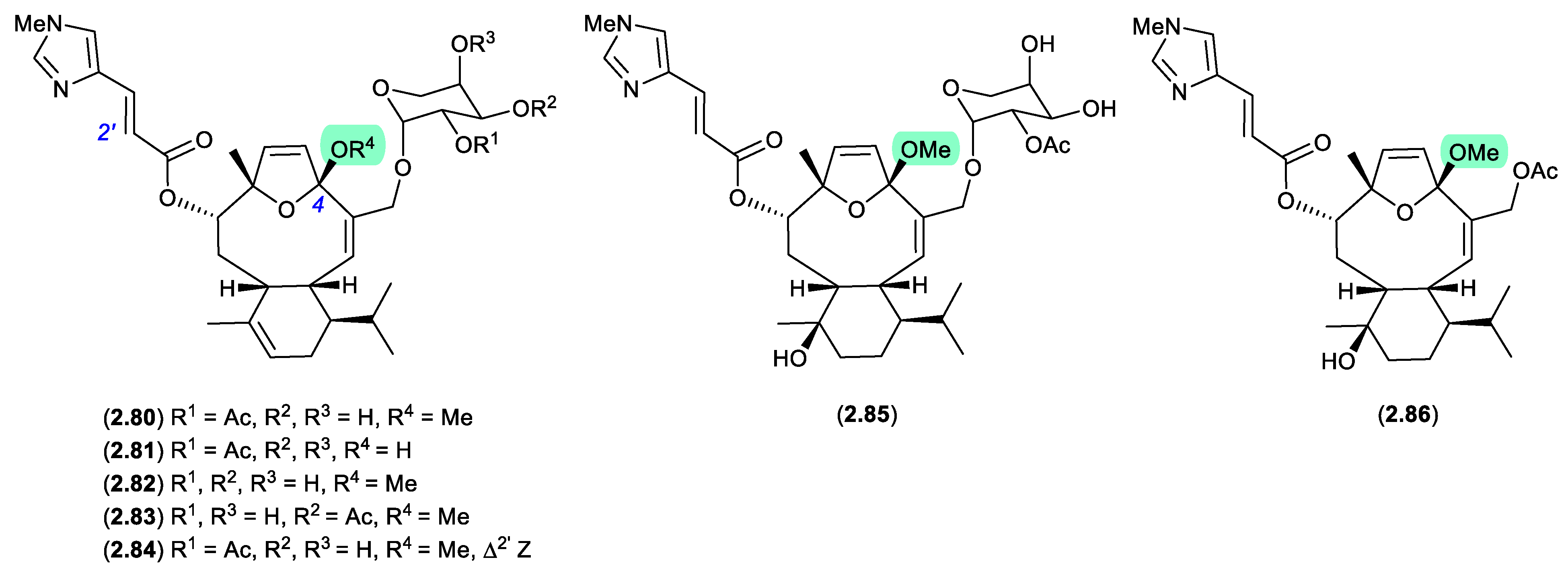
2.4. Acetone
kutzneridines (Figure 2.4.1)
Genomic analysis of the Panama soil-derived actinobacterium Kutzneria sp. CA-103260 revealed a biosynthetic gene cluster (BGC) encoding for a putative lipopeptide — with heterologous expression in Streptomyces coelicolor M1152 yielding kutzneridine A (2.87), bearing an exotic N,N-acetonide moiety.[40] However, as acetone was used in the extraction process, it was determined that 2.87 was an artifact of a cryptic natural product 2.88 (detected but not isolated/characterised – see Section 10).
Figure 2.
4.1.
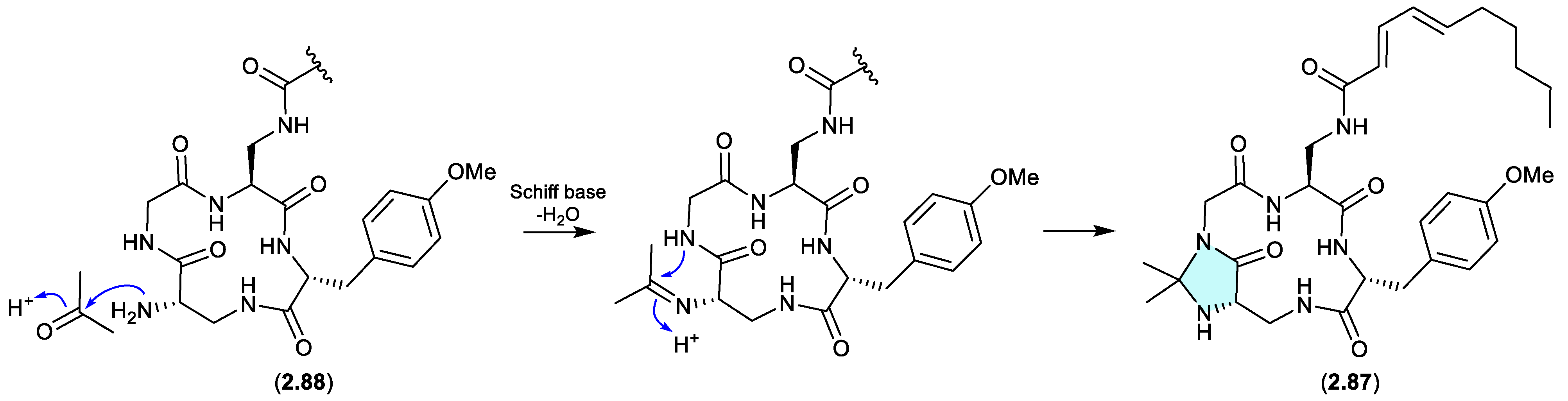
enamidonins/ K97-0239A and B (Figure 2.4.2)
The N,N-acetonide containing lipopeptide enamidonin (2.89) was first reported in 1995 from the soil-derived Streptomyces sp. 91-75,[41] and later in 2018 along with two new analogues, enamidonins B (2.90) and C (2.91) from a Korean soil-derived Streptomyces sp. KCB14A132.[42] Likewise, the Japanese soil-derived Streptomyces sp. K97-0239 yielded the N,N-acetonide lipopeptides K97-0239A (2.92) and K97-0239AB (2.93).[43] As all of the N,N-acetonides 2.89–2.93 were recovered by acetone extraction from their respective fermentations, it is likely all are handling artifacts.
Figure 2.
4.2.
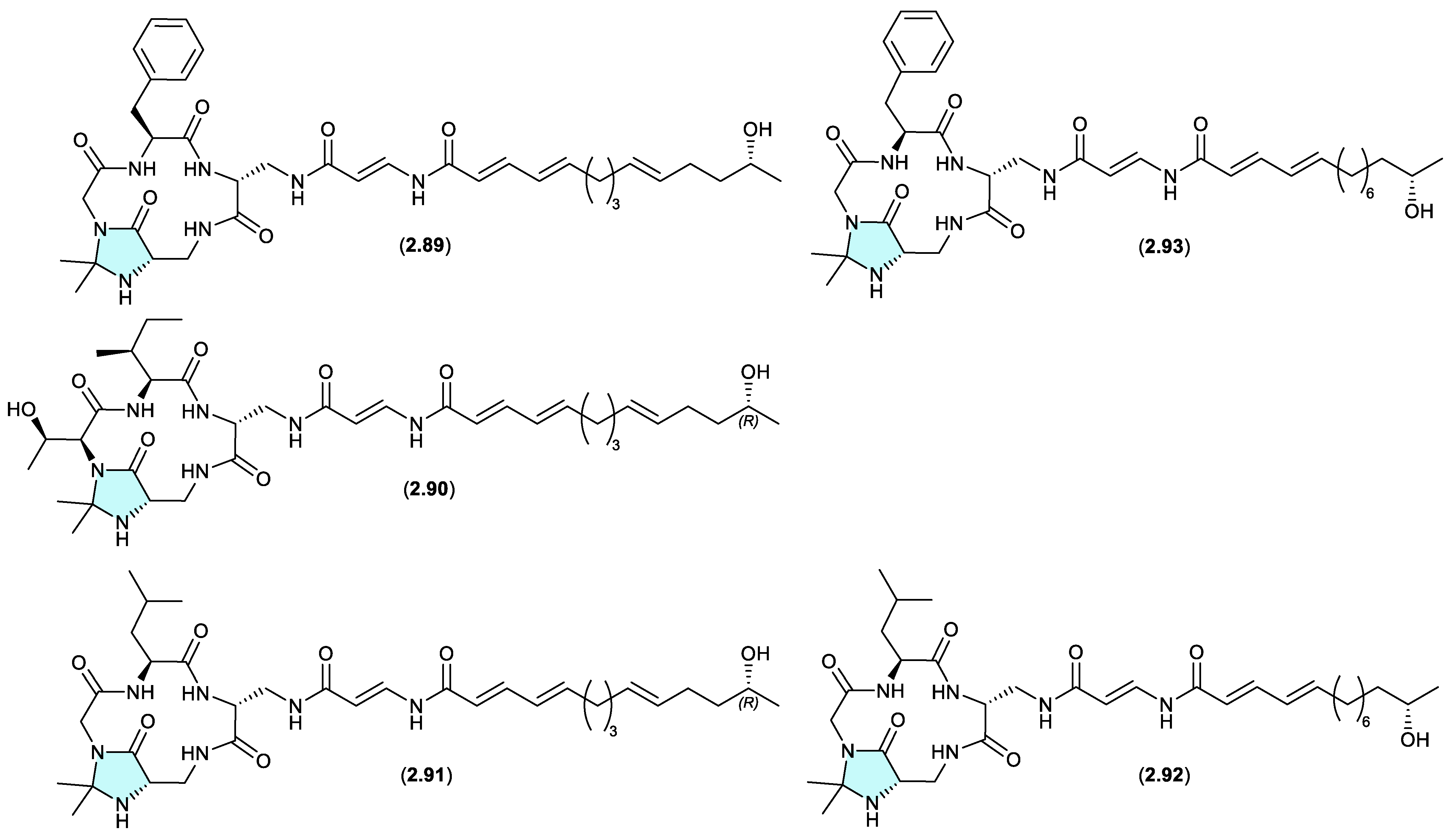
autucedines (Figure 2.4.3)
Genome mining of the deep-sea derived Streptomyces olivaceus SCSIO T05 led to the discovery of five new enamidonin (2.89) related lipopeptides.[44] Fermentations extracted with acetone produced three N,N-acetonide adducts autucedines A (2.94), D (2.95) and E (2.96), while extraction with 2-butanone produced the analogous N,N-ethyl,methyl adducts autucedines B (2.97) and C (2.98). This study has taken advantage of this handling artifact reaction to produce new analogues.
Figure 2.
4.3.
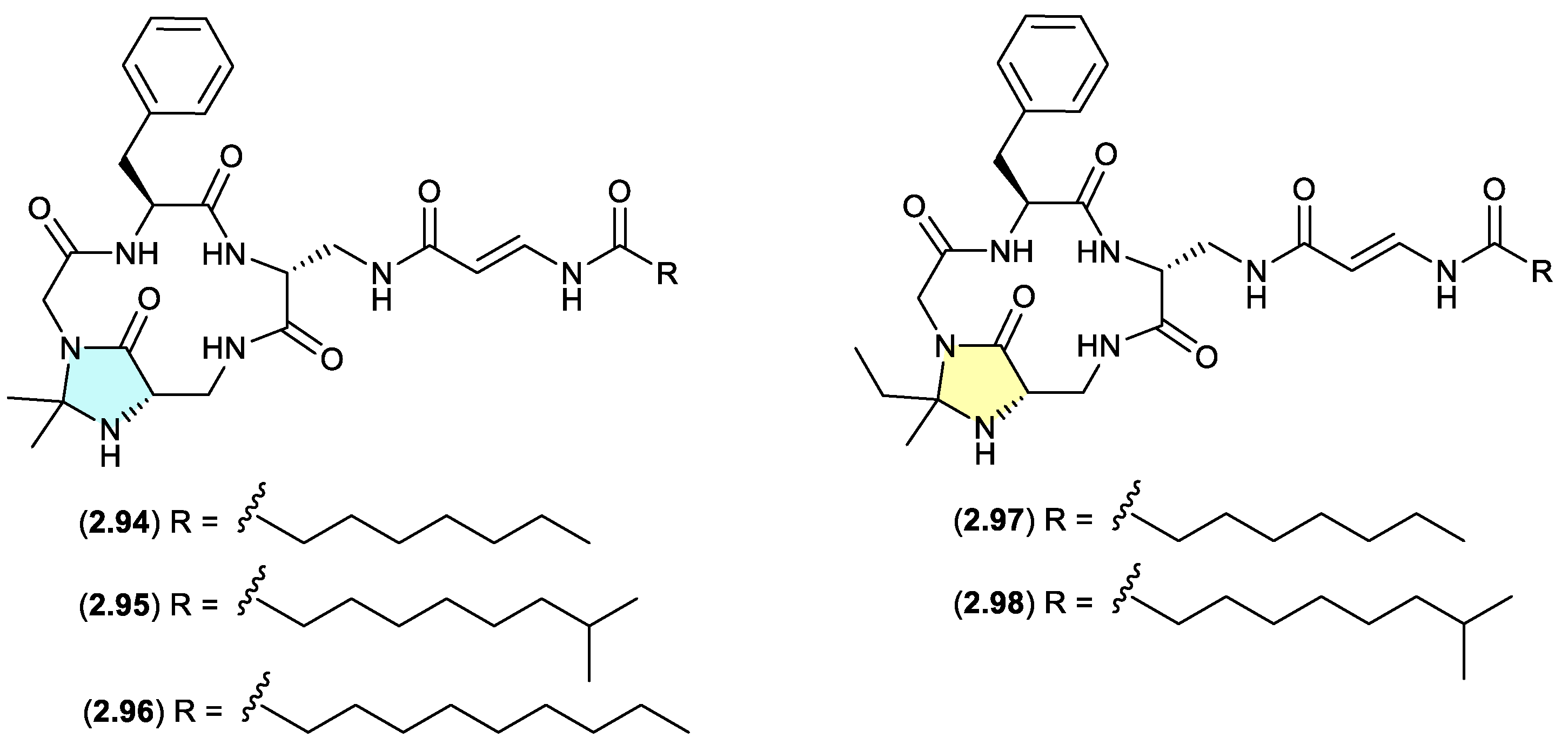
madurastatins (Figure 2.4.4)
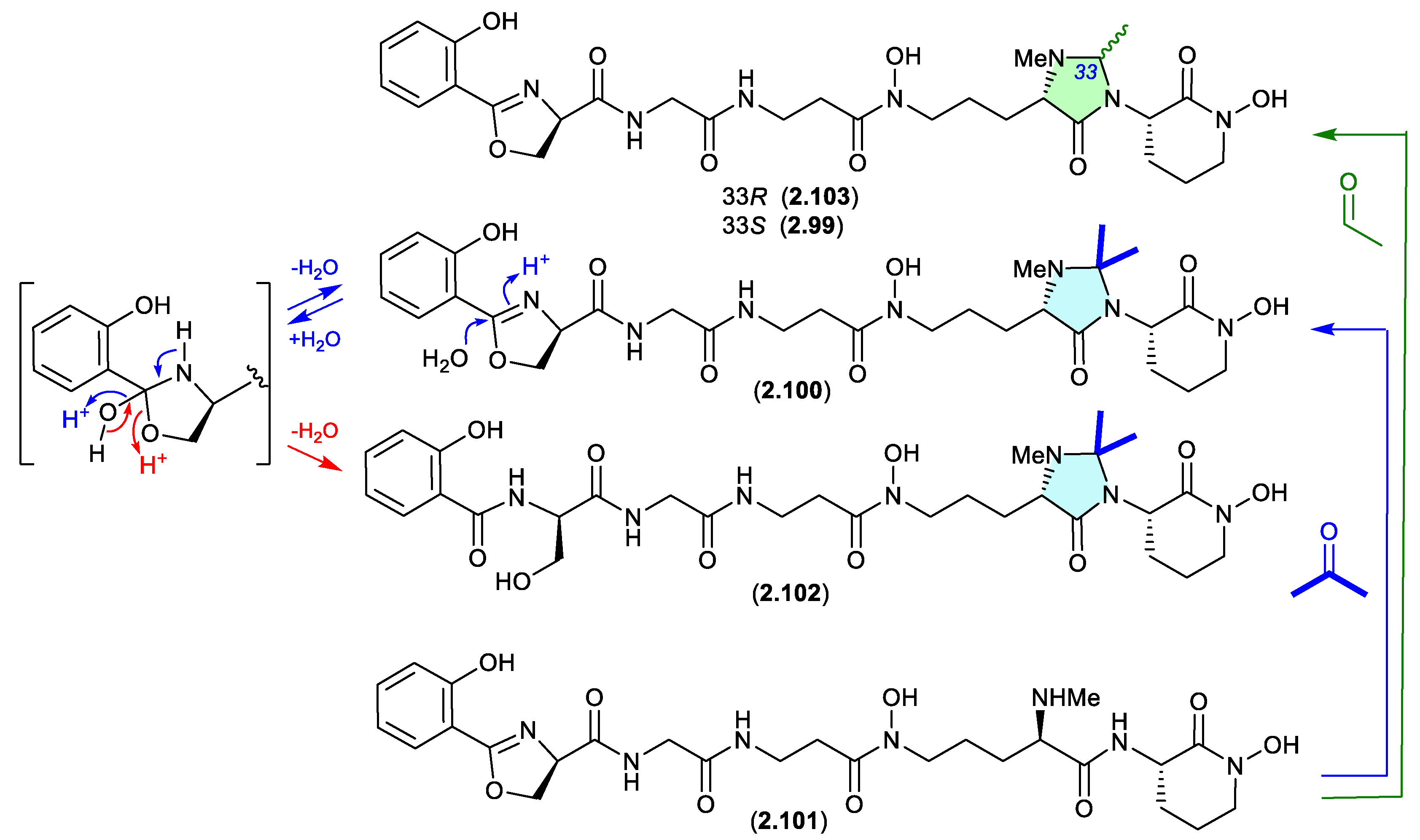
The marine sponge-derived Actinomadura sp. WMMA-1423 yielded two new N,N-acetal siderophores, madurastatins D1 (2.99) and D2 (2.100), along with madurastatin C1 (2.101).[45] As the fermentation was extracted with acetone, its likely 2.101 reacts with acetone to yield the N,N-acetonide artifact 2.100, and with acetaldehyde — a common contaminant in commercial (especially recycled) acetone — to yield the artifact 2.99. Presumably N-methylation diminishes chemical reactivity, which is why on this occasion the precursor 2.101 survives extraction — unlike the case with 2.89–2.98.
A 2024 study of the acetone extracts of a South African plant rhizosphere-derived Actinomadura sp. CA-135719, identified madurastatins H2 (2.102) and 33-epi-D (2.103) and reassigned absolute configurations across 2.99–2.103.[46] Significantly, these authors concluded that the madurastatins 2.99, 2.100, 2.102 and 2.103 are in all probability acetone handling artifacts. Although there could be some contribution from low levels of endogenous aldehydes. Of note, 2.102 is also likely an acid-mediated hydrolysis analogue (artifact) of 2.100 where the oxazoline ring is opened to a serine residue (see Section 4.2, serratiochelin).
Figure 2.
4.4.
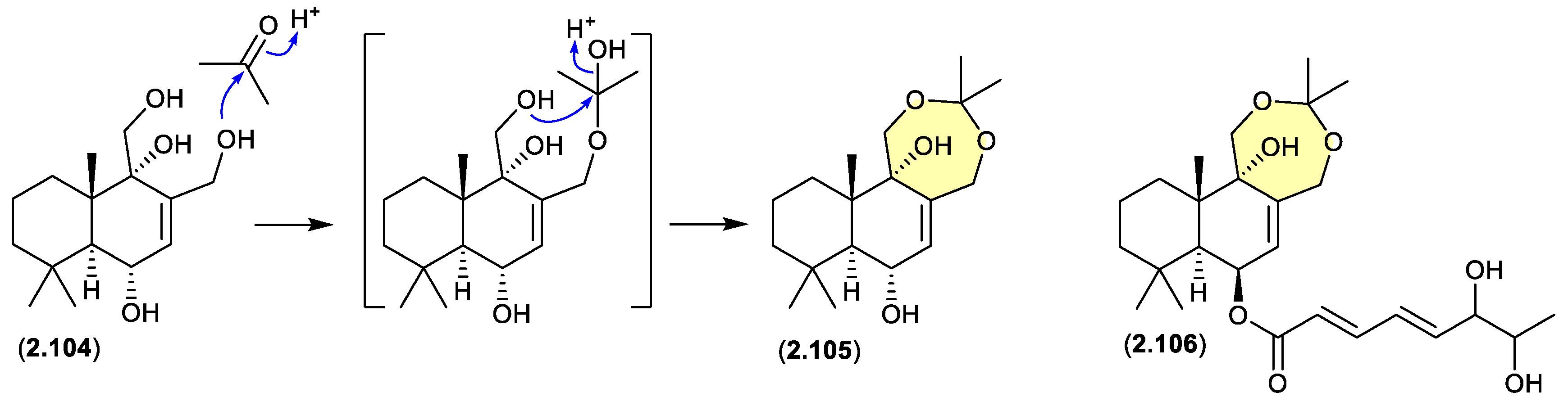
drimanes (Figure 2.4.5)
The Okhotsk Sea sediment-derived fungus Aspergillus ustus KMM 4664 yielded a range of metabolites including the known drimane 12-hydroxyalbrassitriol (2.104) and the new acetonides 2.105 and 2.106.[47] As the fungal fermentation was extracted with acetone (and chromatographed on silica gel) the acetonides are likely acetone artifacts.
Figure 2.
4.5.
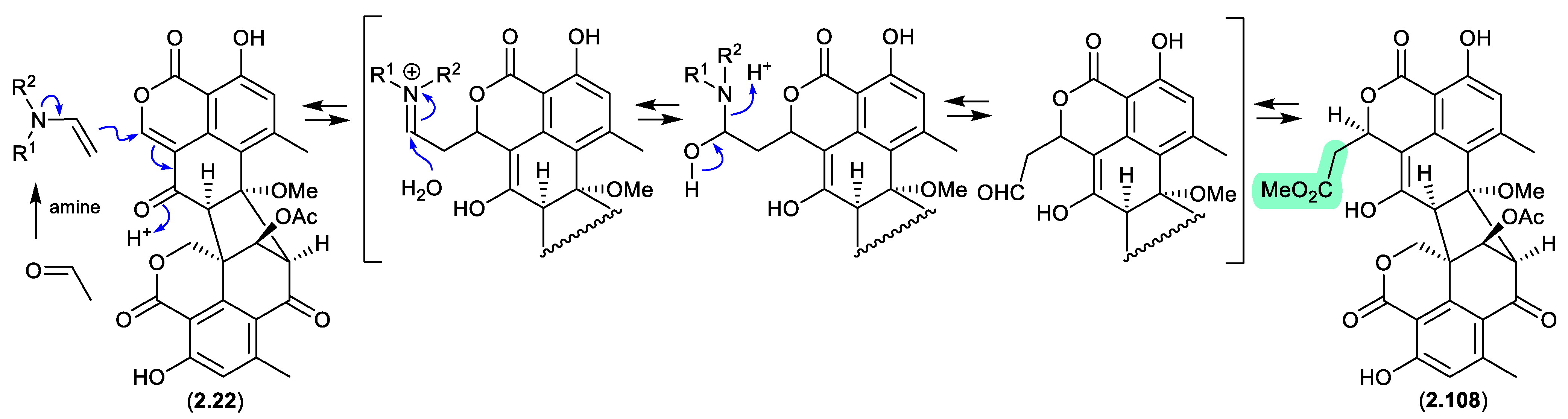
duclauxin/verruculosins (Figure 2.4.6–2.4.7)
A 2024 report by Rivera-Chávez et al on the chemical reactivity of the fungal natural product duclauxin (2.22) (see Section 4.3) established that exposure to acetone led to the formation of verruculosin A (2.107).[48] First reported from the acetone extract of the South China Sea soft coral-derived fungus Talaromyces verruculosus along with verruculosin B (2.108), 2.107 was originally designated as the first duclauxin-like natural product to possess an octacyclic skeleton.[49] Unlike the acetonide adducts described above (e.g. enamidonins, madurastatins), Rivera-Chávez et al proposed that the transformation of 2.22 to 2.107 proceeds via an enamine activated adduct of acetone (with a biogenetic amine present in the acetone extract). This hypothesis was confirmed by transformation of 2.22 to 2.107 in acetone supplemented with morpholine.
Building on this enamine-mediated mechanism, it’s reasonable to propose a comparable transformation utilising acetaldehyde, a known contaminant in commercial acetone (see Section 2.4, madurastatins), followed by oxidation and methylation during silica gel (CH2Cl2/MeOH) chromatography (see Section 4.3), would see verruculosin B (2.108) also designated as an artifact of duclauxin (2.22).
Figure 2.
4.6.
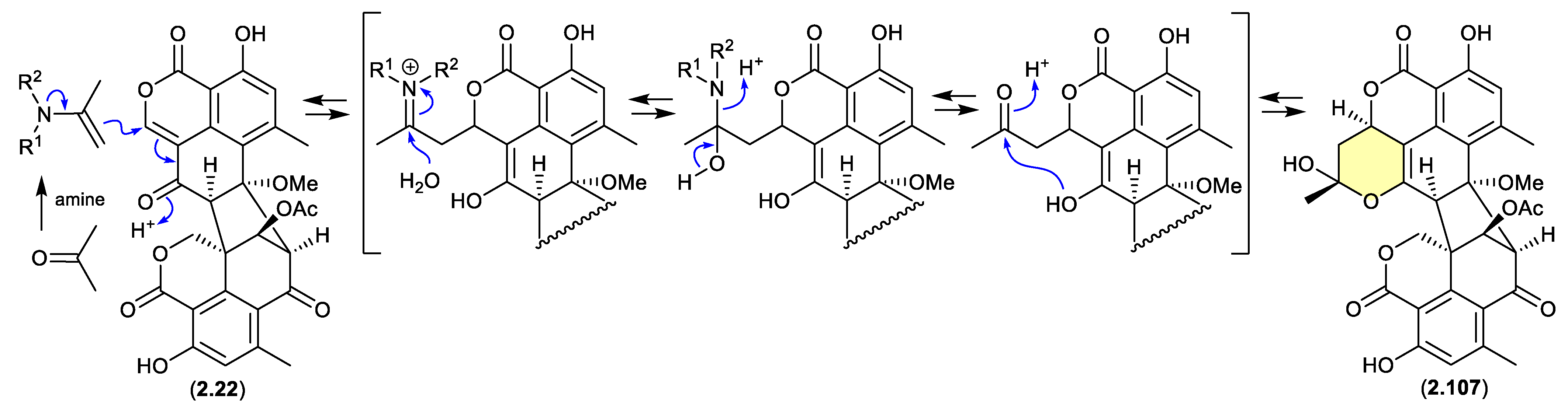
Figure 2.
4.7.
2.5. Acetonitrile
talcarpones (Figure 2.5.1)
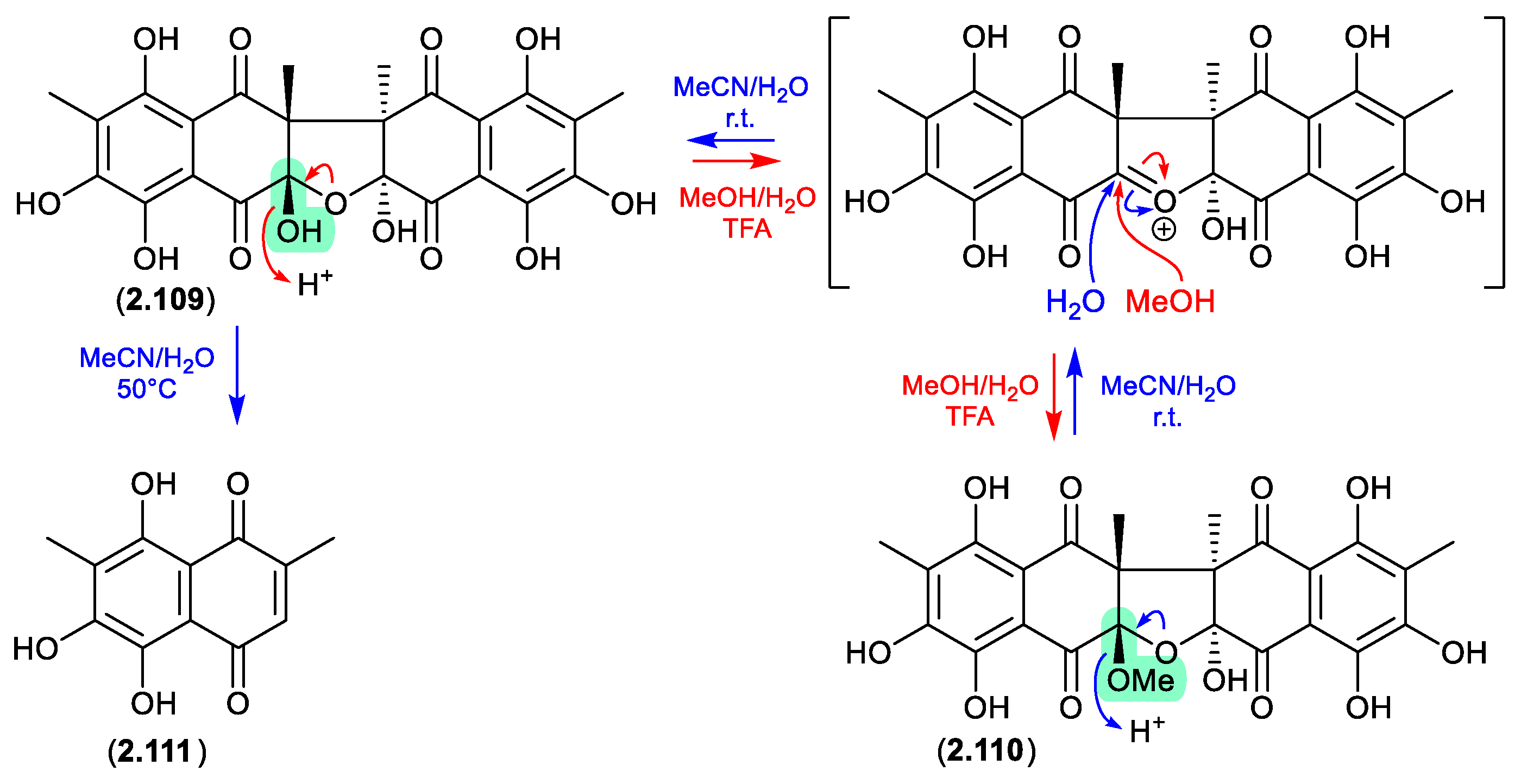
The Australian soil-derived fungus Talaromyces johnpittii MST-FP2594 yielded the new binaphthazarin talcarpones A (2.109) and B (2.110), along with the known monomeric naphthoquinone aureoquinone (2.111).[50] When stored in aqueous MeCN at r.t. 2.110 transformed to 2.109 (presumably via H2O addition to an oxonium intermediate), and on heating (50 °C) further transformed to 2.111. Conversely, at r.t. an acidic MeOH (5% TFA) solution of 2.109 transformed to 2.110 (presumably via MeOH addition to an oxonium intermediate). As the acetone extract of T. johnpittii was subjected to silica gel (CH2Cl2/MeOH) chromatography (see Section 4.3), it seems likely 2.110 is a methanolysis artifact of 2.109.
Figure 2.
5.1.
2.6. Chloroform
greensporones (Figure 2.6.1)
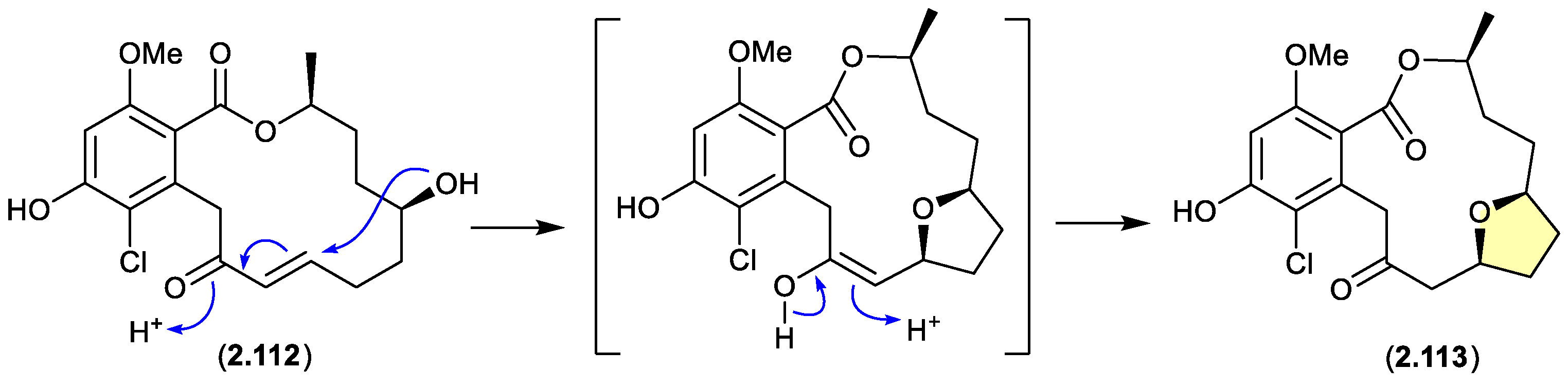
A CDCl3 solution of the resorcylic acid lactone greensporone D (2.112), isolated from the aquatic fungus Halenospora sp. G87, underwent conversion via an intramolecular Michael addition to the tetrahydrofuran greensporone F (2.113).[51]
Figure 2.
6.1.
alkyl resorcinols (Figure 2.6.2)
Alkyl resorcinols isolated from the Chinese soil-derived Pseudomonas aurantiaca YM03-Y3, underwent oxidative transformation a r.t. in CDCl3 solution, with 2.114 yielding the quinone 2.115, dimer 2.116, ring contracted butanolide 2.117 (the latter likely through the oxidative decarboxylation), and a range of minor products.[52]
Figure 2.
6.2.
azodyrecins (Figure 2.6.3)
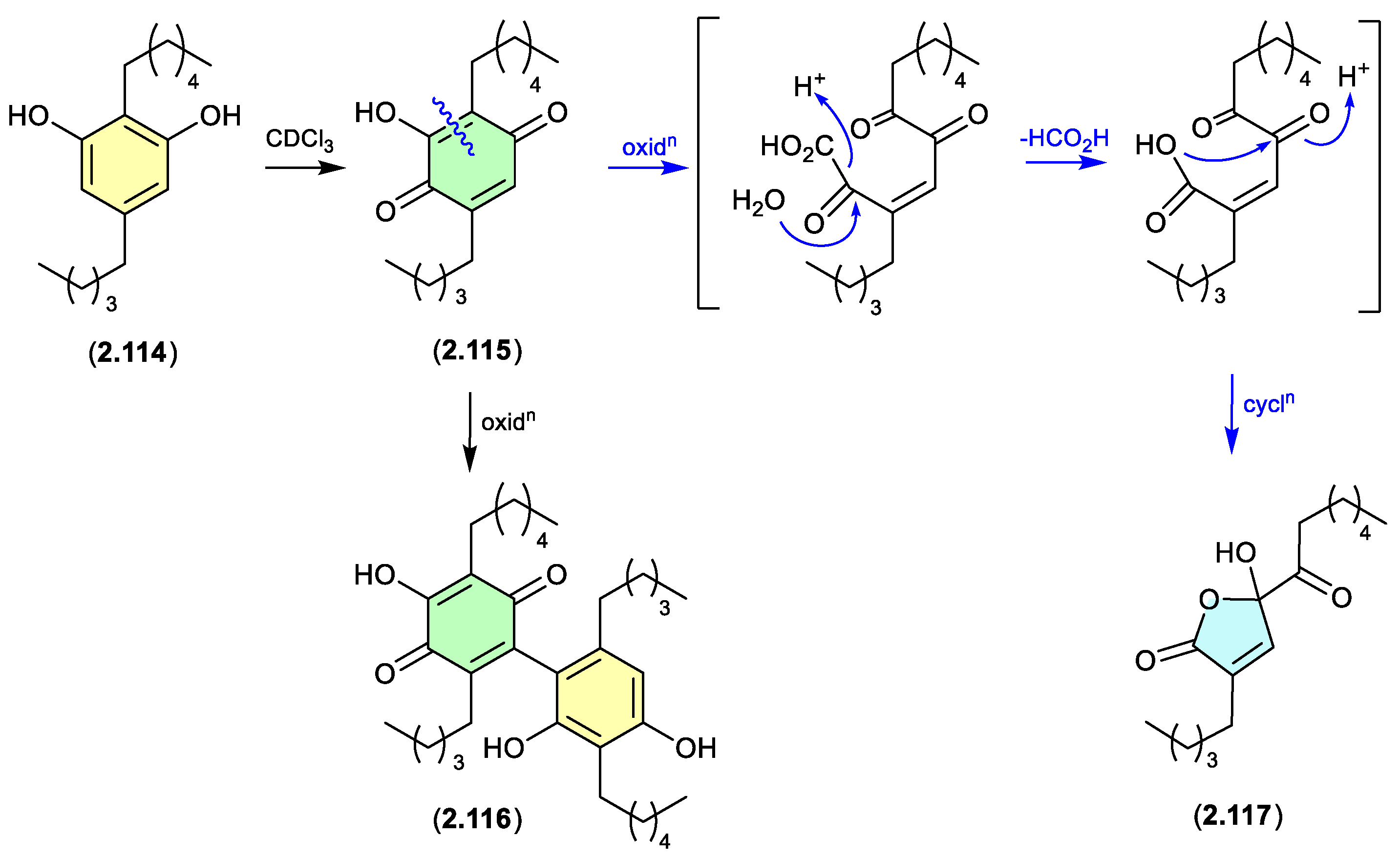
On prolonged storage (30 d) at r.t. a CDCl3 solution of the soil-derived Streptomyces sp. P8-A2 azoxy compounds, azodyrecins A–C (2.118–2.120), underwent quantitative double bond isomerization to the E-isomers, 1’-trans-azodyrecins A–C (2.121–2.123).[53]
Figure 2.
6.3.
schipenindolenes (Figure 2.6.4)
The Chinese fungal endophyte Penicillium sp. DG23 yielded indole-diterpenes with HMG-CoA reductase degrading activity, with schipenindolenes E (2.124) and I (2.125) undergoing an unexpected equilibration when stored for 2 days at r.t. in CDCl3.[54]
Figure 2.
6.4.
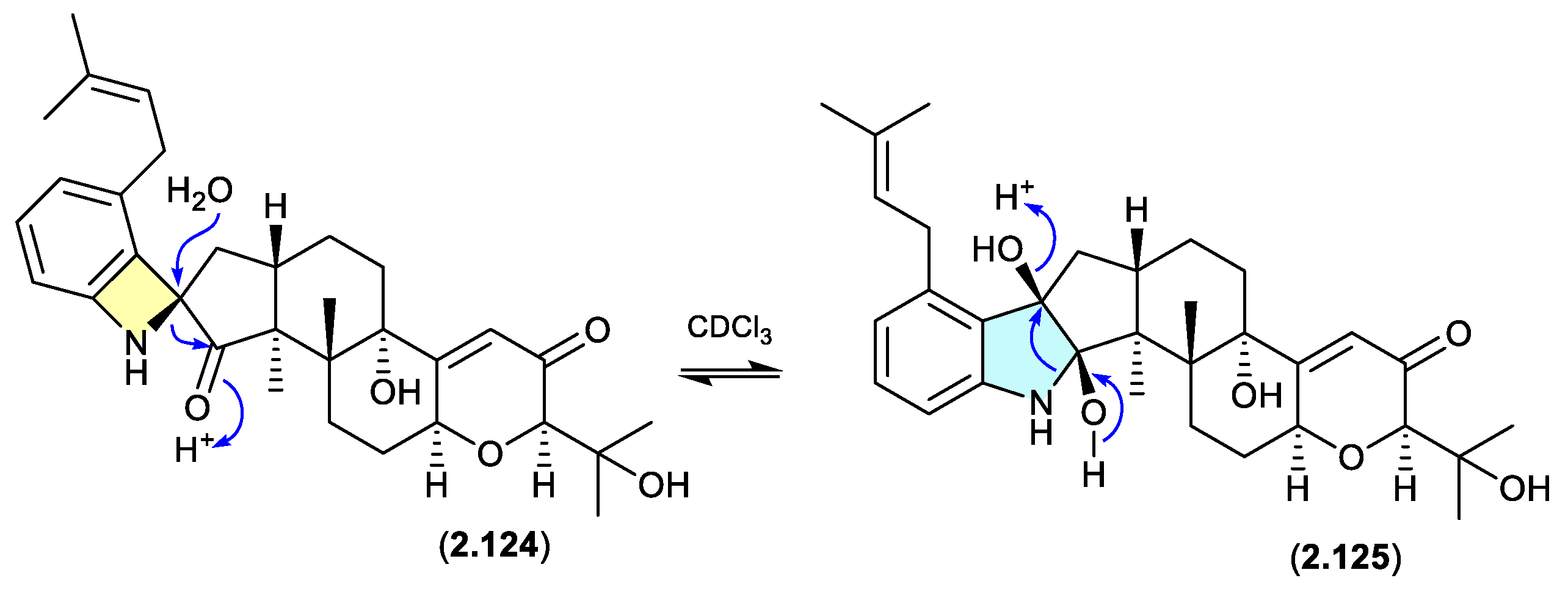
shearinines (Figures 2.6.5 and 2.6.6)
A Chinese mangrove-derived Penicillium sp. yielded an array of new indolo-terpenes, of which noteworthy examples (relevant to this review) include shearinines A (2.126), F (2.127), I (2.128), H (2.129), J (2.130) and K (2.131).[55] Significantly, in CDCl3 at r.t. shearinine K (2.131) partially transformed to shearinine J (2.130). As fractionation of the fermentation involved silica gel (CH2Cl2/MeOH) chromatography, it is likely 2.130 is an artifact of 2.131. Extrapolating on this, it’s also likely that shearinines I (2.128) and H (2.129) are comparable artifacts of shearinines A (2.126) and F (2.127), respectively.
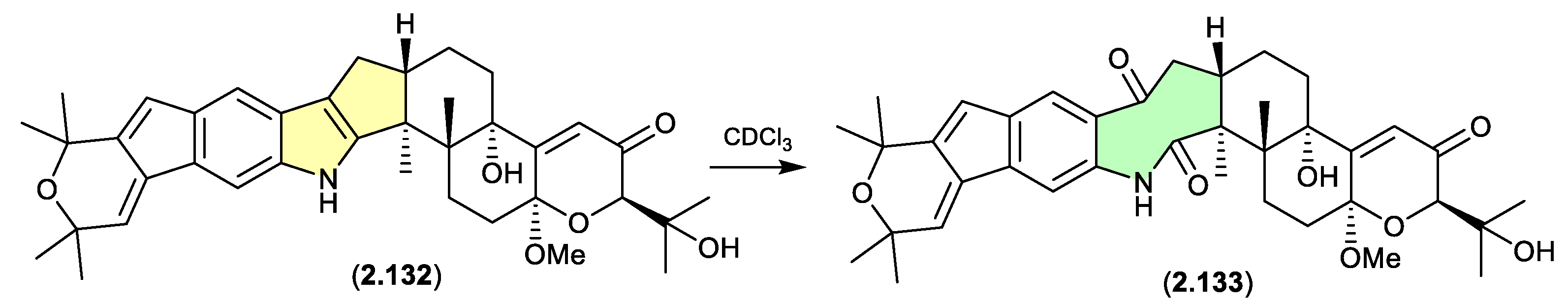
The Chinese mangrove rhizosphere soil-derived fungus Penicillium sp. N4-3 yielded further examples of the shearinine structure class, including shearinine S (2.132), which in CDCl3 transformed to the artifact shearinine T (2.133).[56] It has been proposed that this oxidative ring opening is initiated by autoxidation.[55,56]
Figure 2.
6.5.
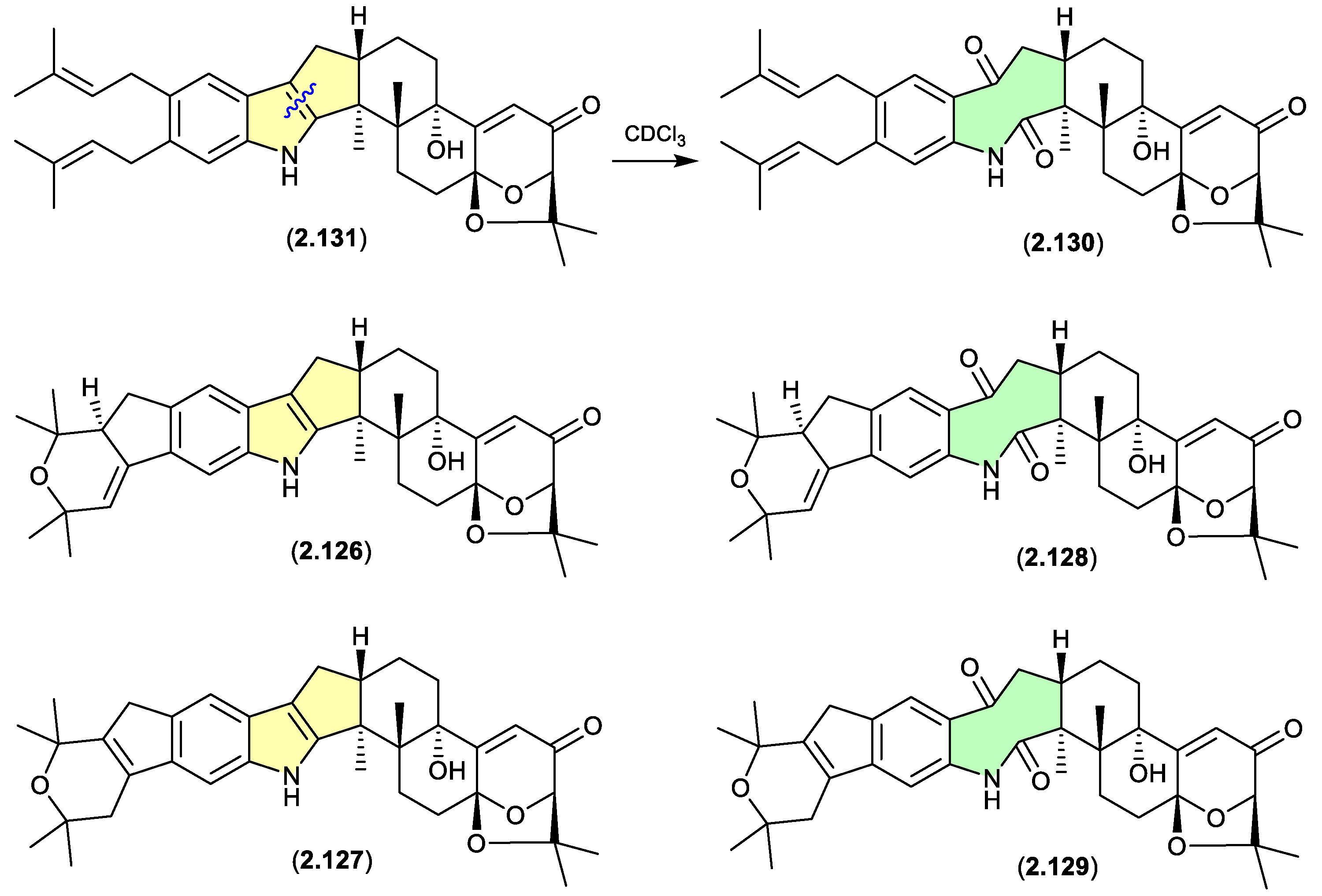
Figure 2.
6.6.
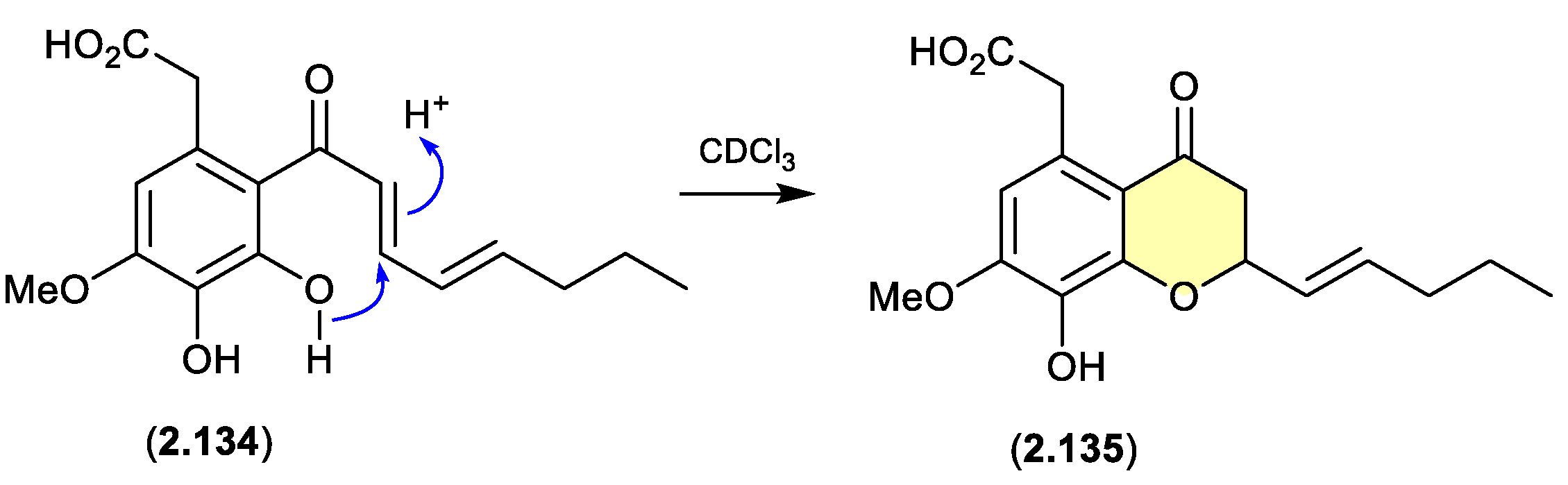
cavoxin/cavoxone (Figure 2.6.7)
A revision of the structure of cavoxin (2.134) (also known as aposphaerin A) produced by the fungus Phoma cava, reaffirmed that exposure to CDCl3 initiates an intramolecular acid-mediated cyclisation to yield the artifact cavoxone (2.135).[57]
Figure 2.
6.7.
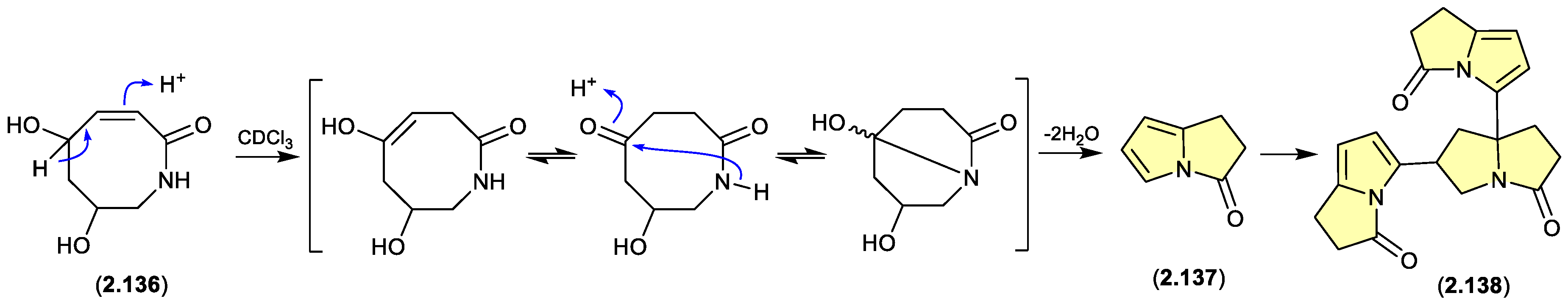
pyrrolizin-3-ones (Figure 2.6.8)
The marine-derived Streptomyces sp. QD518 yielded the azocin-2-one 2.136, which during NMR (CDCl3) data acquisition underwent complete transformation to 2.137 and 2.138.[58] A plausible ring rearrangement mechanism is proposed below.
Figure 2.
6.8.
2.7. Dichloromethane
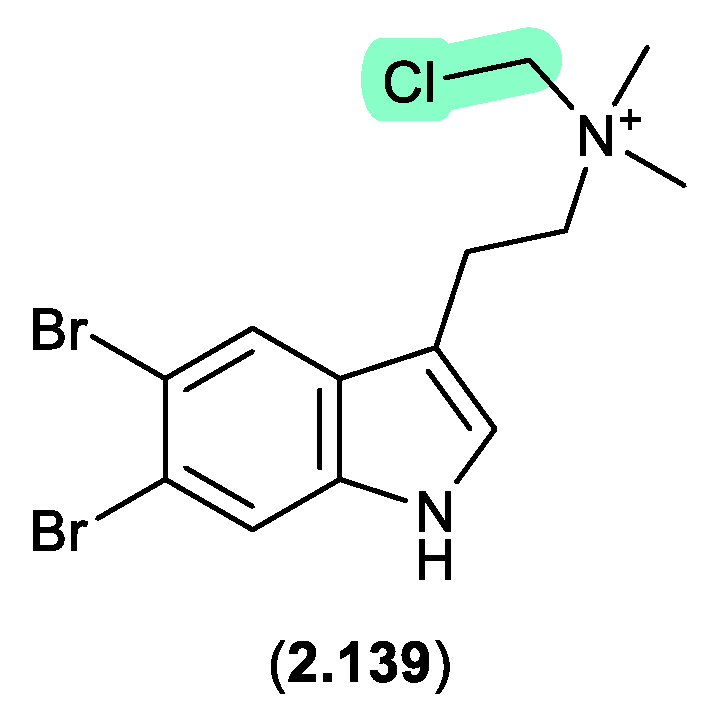
bromotyramines (Figure 2.7.1)
Bromotryptamines recovered from a tropical southwestern Pacific Ocean collection of the marine sponge Narrabeena nigra included the N-chloromethyl 2.139, which would have been formed as an artifact during exposure to CH2Cl2.[59]
Figure 2.
7.1.
2.8. Benzene
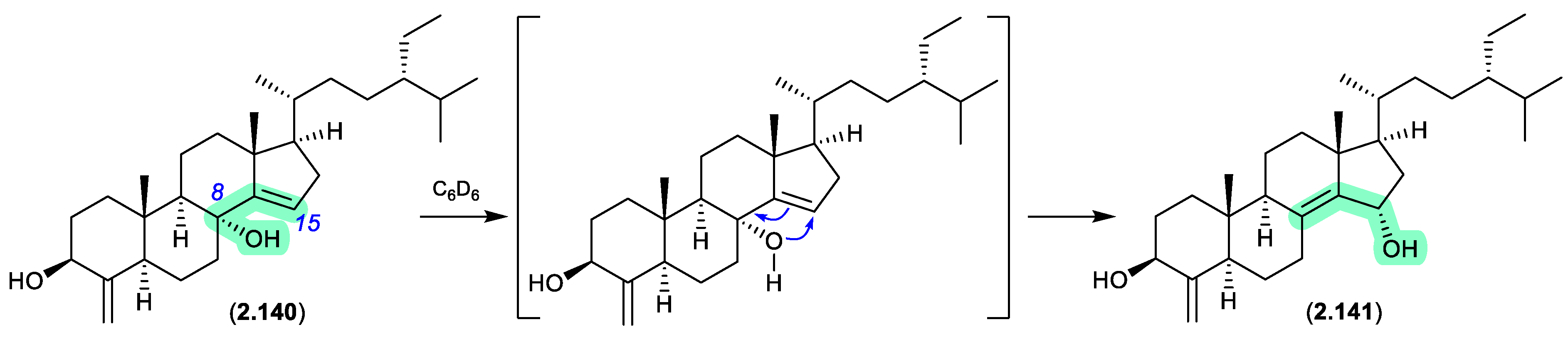
theonellastrols (Figure 2.8.1)
Oxygenated sterols from a Philippines marine sponge Theonella swinhoei included 8α -hydroxytheonellasterol (2.140), which in benzene-d6 at r.t. underwent spontaneous allylic hydroxy migration to the artifact 15α-hydroxytheonellasterol (2.141).[60]
Figure 2.
8.1.
2.9. Ethyl Acetate
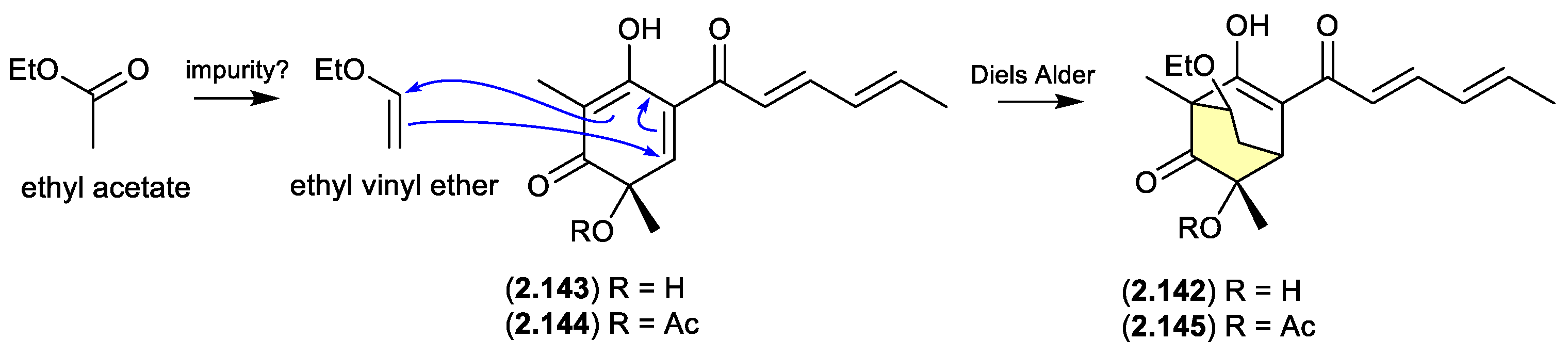
sorbicillinol/sorbivetone (Figure 2.9.1)
The new sorbicillinoid sorbivinetone (2.142) (also known as rezishanone C) isolated from the sponge-derived fungus Penicillium chrysogenum DSM 16137 featured a biosynthetically unusual ethoxy moiety.[61] This prompted speculation that 2.142 could be a Diels Alder cycloaddition artifact of ethyl vinyl ether (potentially an impurity in EtOAc) and sorbicillinol (2.143) — the latter a highly reactive natural product and precursor of many sorbicillin-derived fungal metabolites, known to readily undergo Diels Alder reactions.[62] Supportive of this hypothesis, [13C2]-acetate incorporation studies revealed that neither the ethoxy moiety, or the two bridging C-atoms were labelled. Furthermore, a dilute aqueous MeCN solution of ethyl vinyl ethyl and the more stable O-acetylsorbicillinol (2.144), yielded the predicted Diels Alder adduct, O-acetylsorbivinetone (2.145).
Figure 2.
9.1.
2.10. Aprotic vs Protic
While not strictly artifacts, the following examples reveal how natural products can change structure in response to the solvent they are dissolved in — in particular between polar protic, versus less polar aprotic solvents. This has particular relevance when exploring SAR and in attempting molecular docking analyses of binding interactions with putative molecular targets.
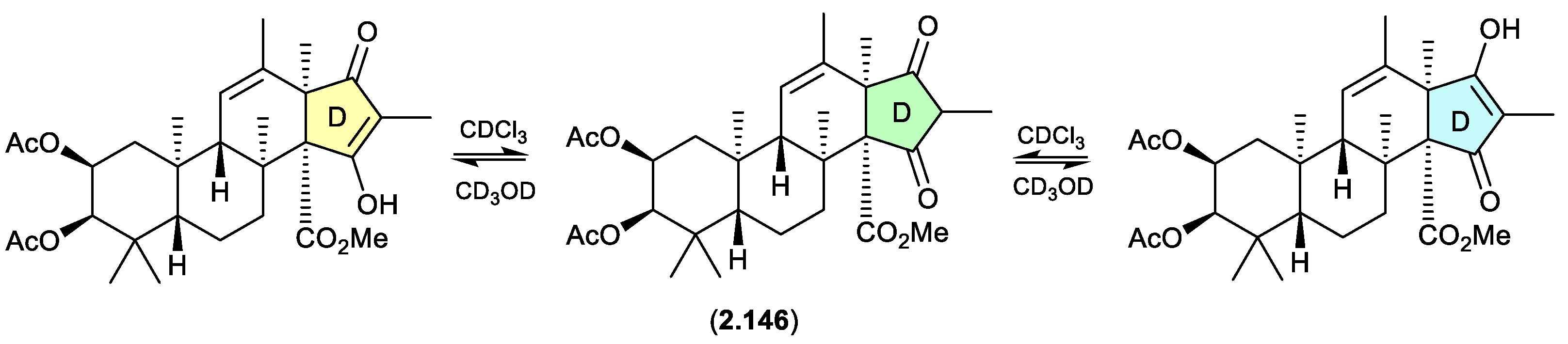
oxandrastins (Figure 2.10.1)
The Australian fungus Penicillium sp. CMB-MD14 yielded an array of meroterpenes, exemplified by oxandrastin A (2.146), where ring D exists in two solvent dependent tautomers, as evident in the NMR (CDCl3) and NMR (methanol-d4) spectra.[63]
Figure 2.
10.1.
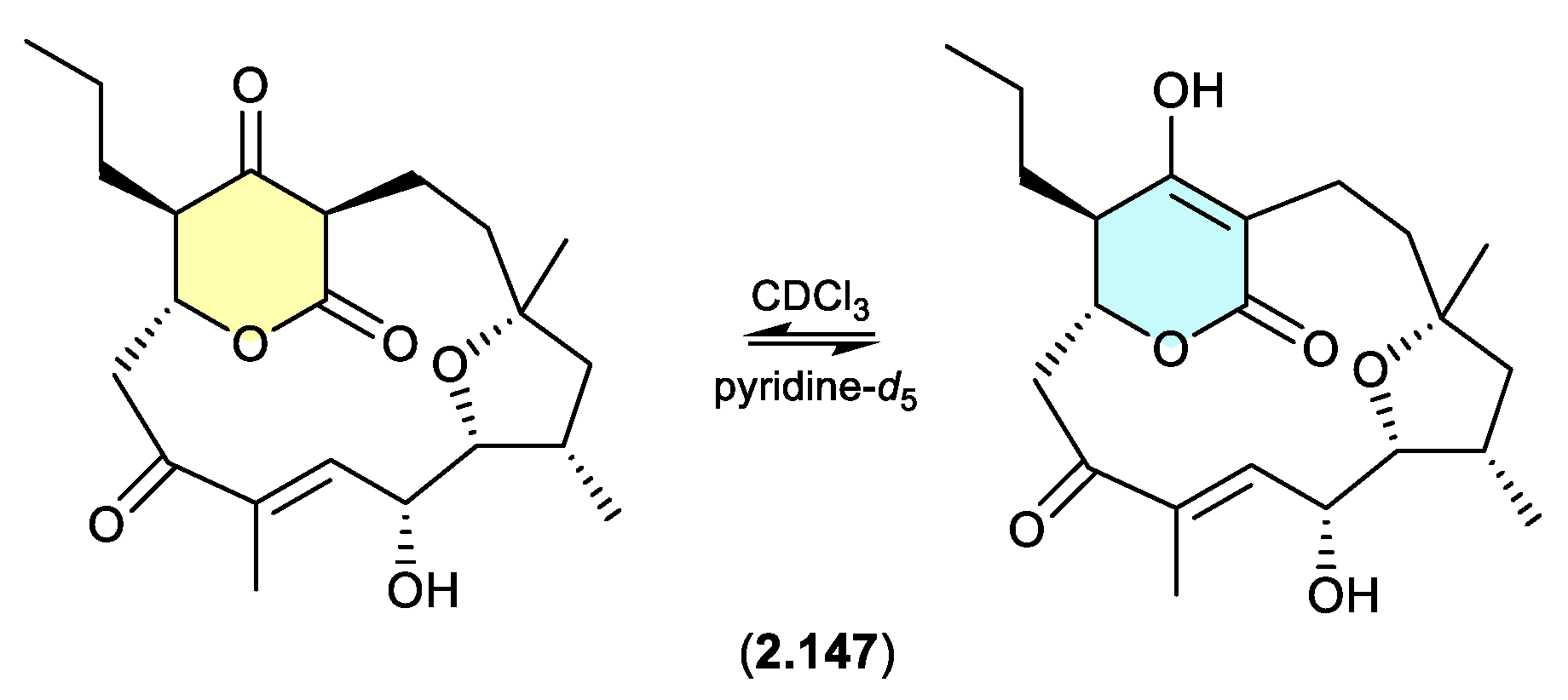
alaeolide (Figure 2.10.2)
The Japanese marine sediment-derived Streptomyces sp. NPS554 yielded the polycyclic polyketide akaeolide (2.147), which exists as two solvent dependent tautomers — as evident in the NMR (CDCl3) and NMR (pyridine-d5) spectra.[64]
Figure 2.
10.2.
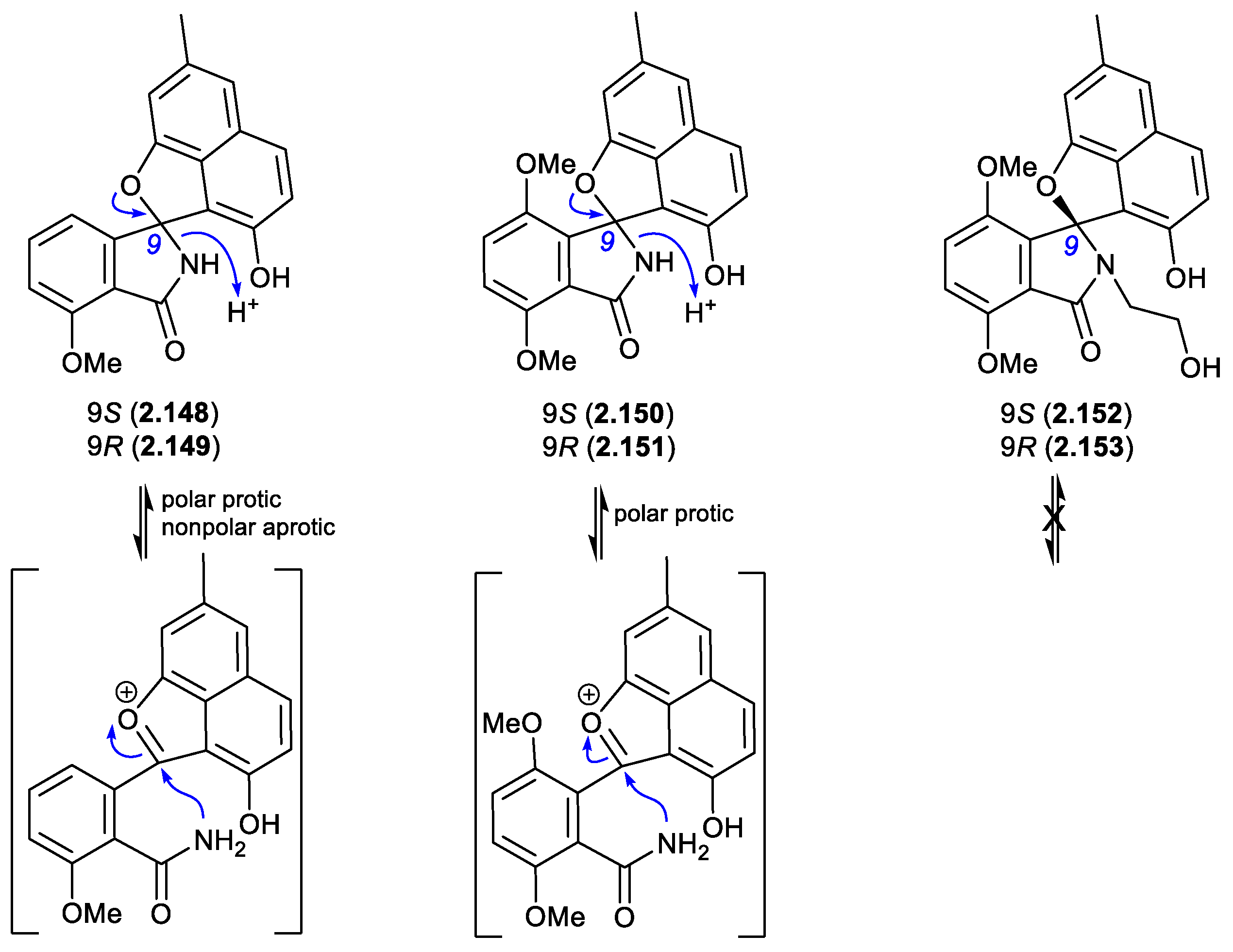
pratensilins/pratenone A (Figures 2.10.3 and 2.10.4)
A 2017 account of the Chinese marine sediment-derived Streptomyces sp. KCB-132 reported three spiro indolinone-naphthofuran enantiomeric pairs, (+)-(S)-pratensilin A (2.148) and (–)-(R)-pratensilin A (2.149); (+)-(S)-pratensilin B (2.150) and (–)-(R)-pratensilin B (2.151); and (+)-(S)-pratensilin C (2.152) and (–)-(R)-pratensilin C (2.153).[65] Following resolution by chiral HPLC, the A pair (2.148/2.149) equilibrated in both MeOH and CH2Cl2, whereas the B pair (2.150/2.151) only equilibrated in MeOH, and the C pair (2.152/2.153) did not equilibrate in either solvent. While the authors propose two mechanisms, both require the loss of naphthalene aromaticity. An alternate mechanism proceeds via collapse of the embedded aminol to an achiral oxonium intermediate, followed by recyclization with scrambling of chirality. The increased level of substitution on the spiro lactam nitrogen could explain the lack of reactivity of 2.152/2.153. In addition to retaining naphthalene aromaticity, an oxonium pathway would also benefit from stabilisation by polar solvents and the adjacent electron rich naphthalene system. As the isolation of the pratensilins involved MeOH extraction followed by silica gel and HPLC with a MeOH eluant, it’s possible one of the enantiomers in each pair is an artifact.
Figure 2.
10.3.
In a follow-up 2020 study, Streptomyces sp. KCB-132 was also reported to yield another enantiomeric pair, (–)-(S)-pratenone (2.154) and (+)-(R)-pratenone (2.155).[66] Following resolution by chiral HPLC, both enantiomers rapidly racemised (~10 min) in the polar solvents MeOH and THF, but were less prone to racemization in CH2Cl2 and MeCN. A proposed equilibration mechanism that proceeds via a carbocation (and retains naphthalene aromaticity) would be stabilised by polar over non-polar solvents, and by the electron rich naphthalene system. As with the pratensilins, the fermentation was initially extracted with MeOH, before being subjected to both silica gel and HPLC with a MeOH eluant, leaving open the possibility that one of the enantiomers is a handling artifact.
Figure 2.
10.4.
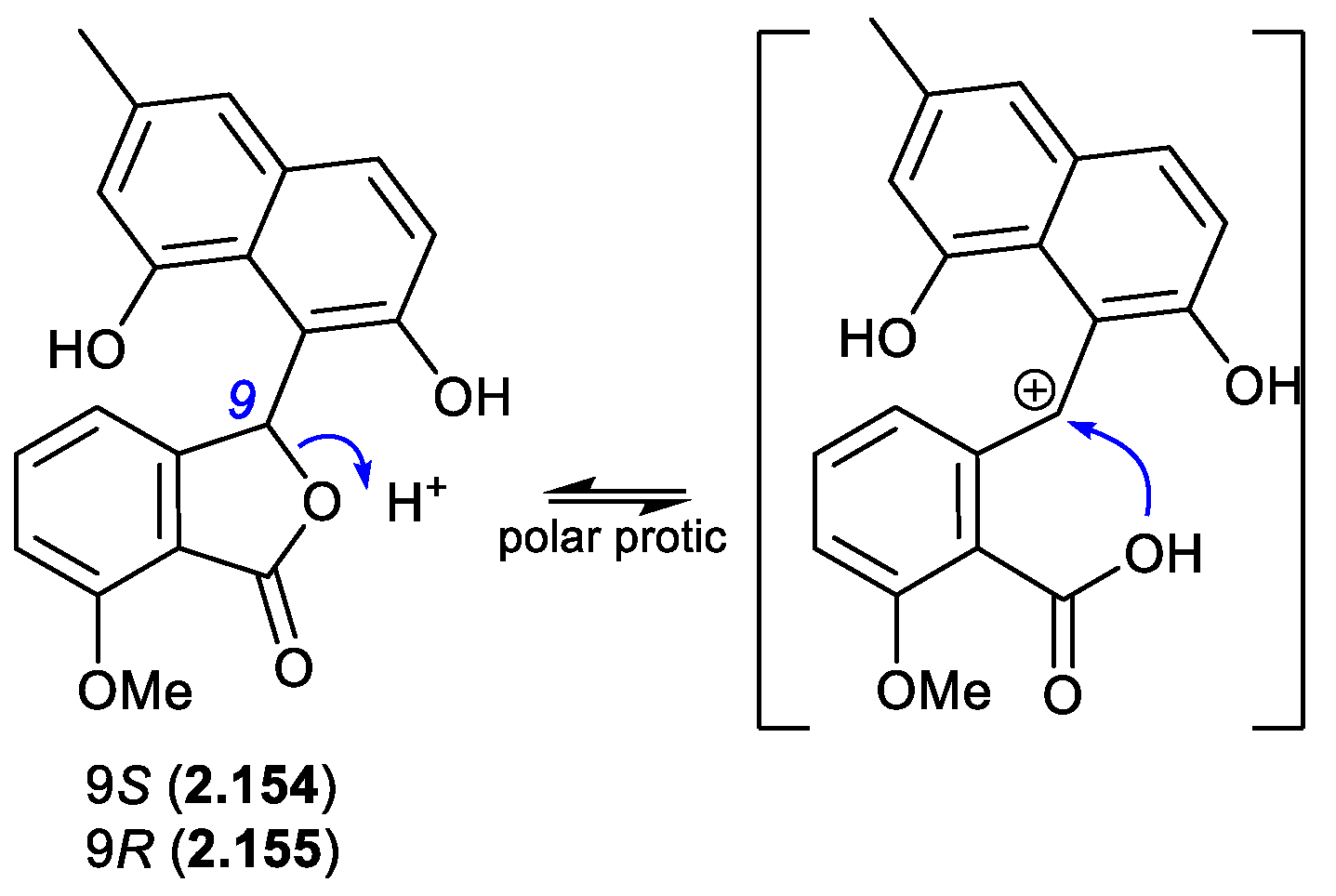
3. Heat
If subjected to elevated temperatures, such as during in vacuo solvent removal from extracts, fractions or pure compounds on a rotary evaporator with a water bath heated to >50 °C, many natural products will decompose (partially or fully), and yet others will transform to artifacts. Consider the following examples.
psammaplins and bastadins (Figure 3.1)
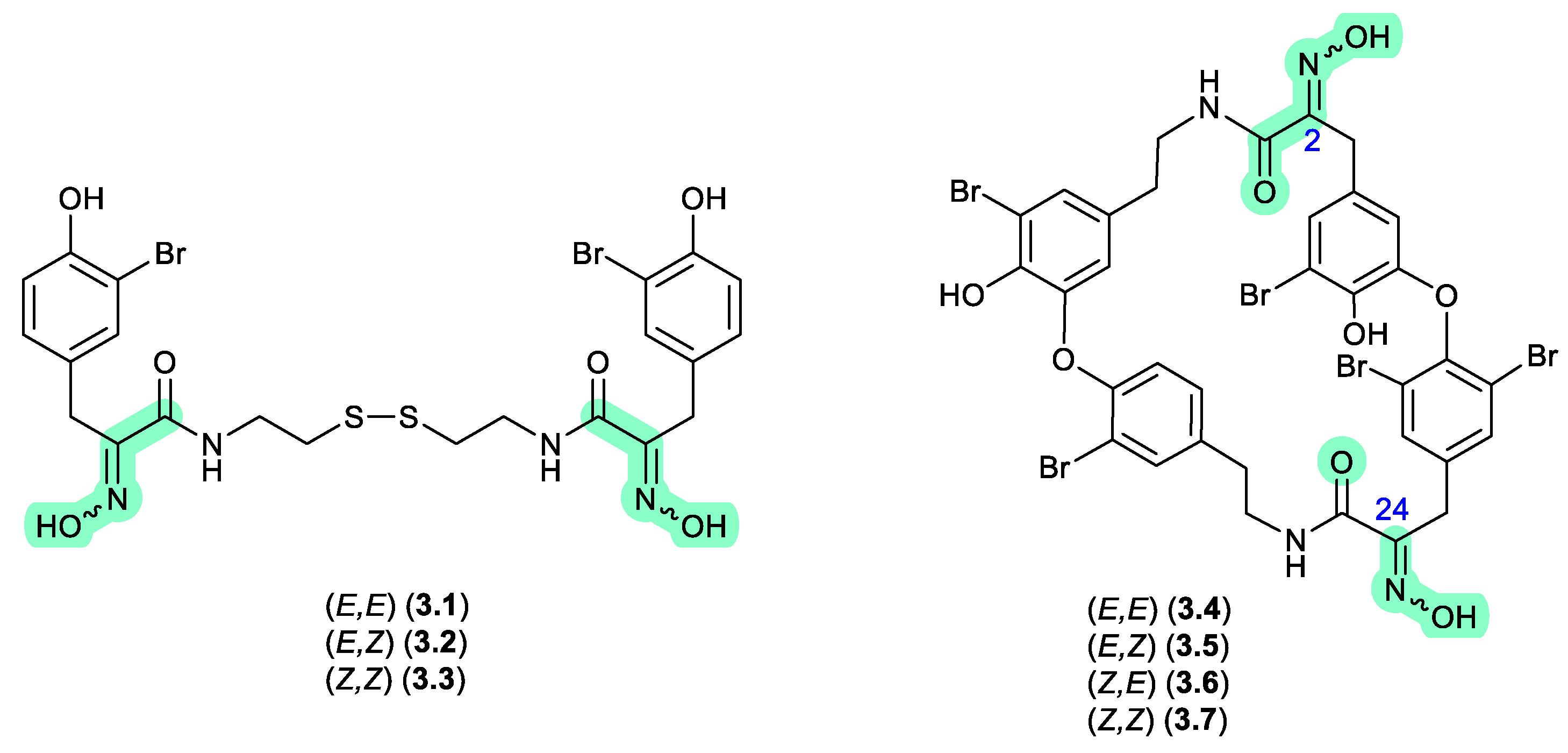
In 1987, Schmitz et al reported the isomeric oximes, (E,E)-psammaplin A (3.1) and (E,Z)-psammaplin A (3.2) from an unidentified verongiid sponge.[67] This study revealed that on recovery from NMR solvents with mild heating, and subsequent storage over a couple of weeks, 3.2 underwent quantitative transformation to 3.1 — prompting speculation that the true natural product may be the chemically reactive Z,Z isomer (3.3) (see Section 10). The next three decades saw a wealth of reports on structural diverse tyrosine-oxime sponge metabolites — perhaps best exemplified by the cyclic tetrapeptide bastadins common to the genera Ianthella. With the literature largely dominated by E oxime configurations, in 2010 Crews et al reported the known (E,E)-bastadin 19 (3.4) along with the new isomer (E,Z)-bastadin 19 (3.5), from a Papua New Guinea collection of Ianthella cf. reticulata.[68] Of particular note, a 1 h photolysis of (E,E)-bastadin 19 (3.4) in MeCN/MeOH with acetophenone as a sensitiser yielded all possible oxime isomers, namely (E,E) (3.4), (E,Z) (3.5), (Z,E) (3.6) and (Z,Z) (3.7). Likewise, a 30 min sonication (warming) of a DMSO solution of synthetic (Z,Z) (3.7) returned a mixture comprising the full suite of oximes isomers, 3.4–3.7, which on standing in DMSO at r.t. for a further 720 h underwent quantitative conversion to (E,E) (3.4). These observations prompted Crews et al to speculate that “…the bastadins and psammaplins initially contain the (Z)-oxime configuration and subsequently isomerize to the more thermodynamically stable E isomer in solution during extraction and/or workup.” — opening up the prospect that a great many sponge oximes reported in the scientific literature were in fact isolation and/or handling artifacts, and that the true (cryptic) natural products have gone unnoticed.
Figure 3.
1.
creolophins (Figure 3.2)
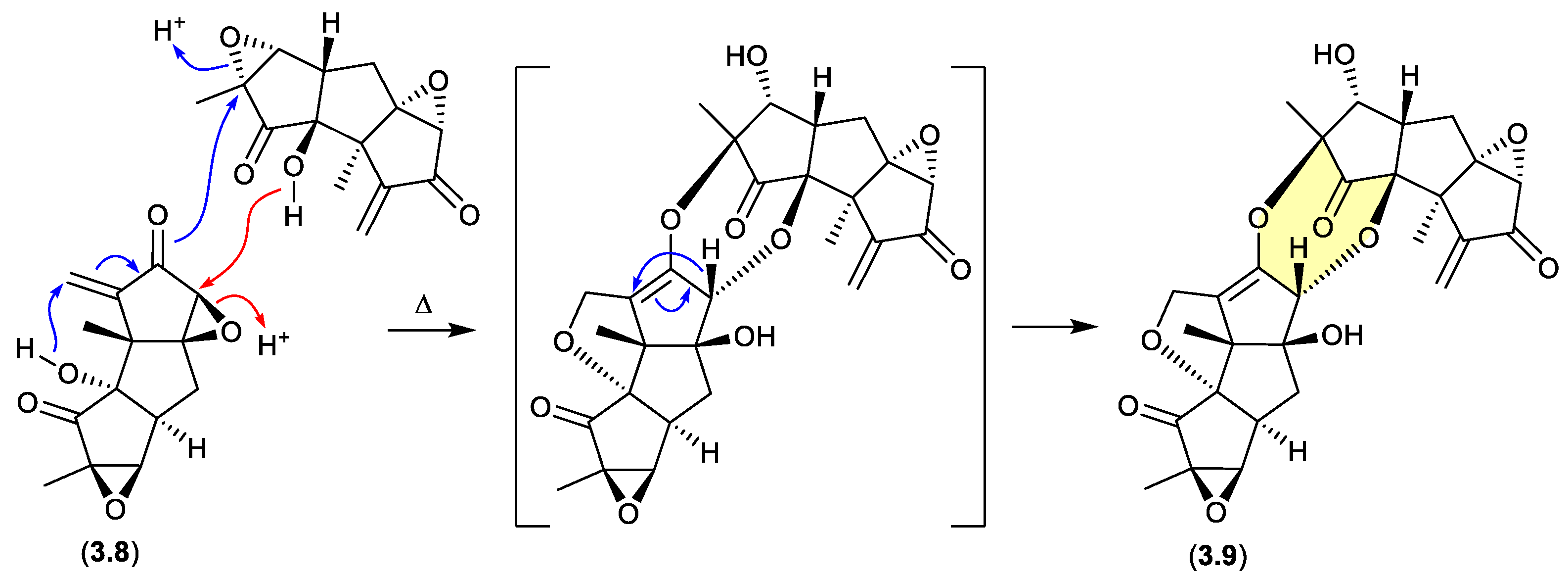
During isolation of the norhirsutane metabolites from culture extracts of the fungus Creolophus cirrhatus, creolophin E (3.8) was found to be thermally unstable, and on mild heating to 50 °C in vacuo yielded the dimer neocreolophin (3.9) through a cascading series of nucleophilic additions and a 1,3-sigmatropic hydride shift.[69]
Figure 3.
2.
neobulgarones (Figure 3.3)
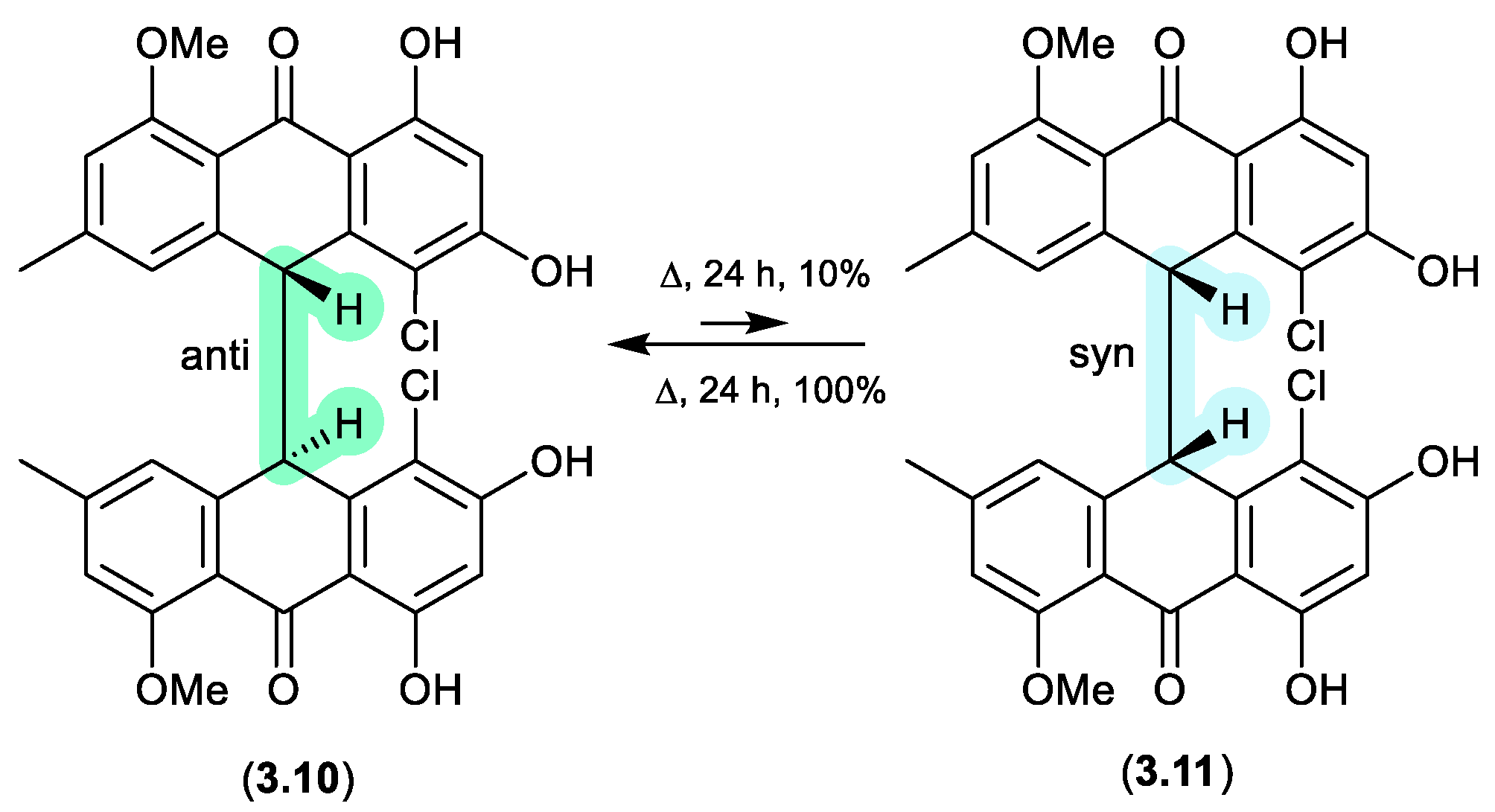
The Australian fungus Penicillium sp. CMB-MD22 yielded a large array of bianthrones as pairs of selectively heat labile anti and syn diastereomers, exemplified by (±)-neobulgarone E (3.10) and neobulgarone F (3.11).[70] For example, while heating of a MeOH solution of anti 3.10 (24 h, 65 °C) led to only 10% conversion to syn 3.11, similar treatment of 3.11 led to complete conversion to 3.10. Notwithstanding, careful analysis of a fresh fermentation prior to heating or fractionation detected an ~1:1 ratio of all anti and syn neobulgarones, confirming their status as natural products.
Figure 3.
3.
4. pH
Many natural products are susceptible to transformation under basic and/or acidic conditions. Variations in pH that occur naturally in fermentation broths, as well as in extracts rich in phenolics or alkaloids, can carry over to organic partitions and be further amplified during in vacuo concentration – particularly as pH modified aqueous residues are the last to evaporate. Changes in pH can also be introduced either inadvertently or deliberately during fractionation, for example, using acidic media (e.g. silica gel) and/or solvents (e.g. CHCl3 or CH2Cl2), or through the addition of eluant modifiers (e.g. TFA, formic acid, triethylamine (TEA), or various buffers). Variations in pH can also occur during long term storage in solvents that may become acidic over time (e.g. DMSO, CH2Cl2). Furthermore, as many natural products incorporate acidic or basic functional groups (e.g. phenols, carboxylic acids, amines, guanidines), concentrating/drying enriched fractions and/or pure samples can facilitate pH mediated transformations.
4.1. Basic
pestalone/pestalachloride (Figure 4.1.1)
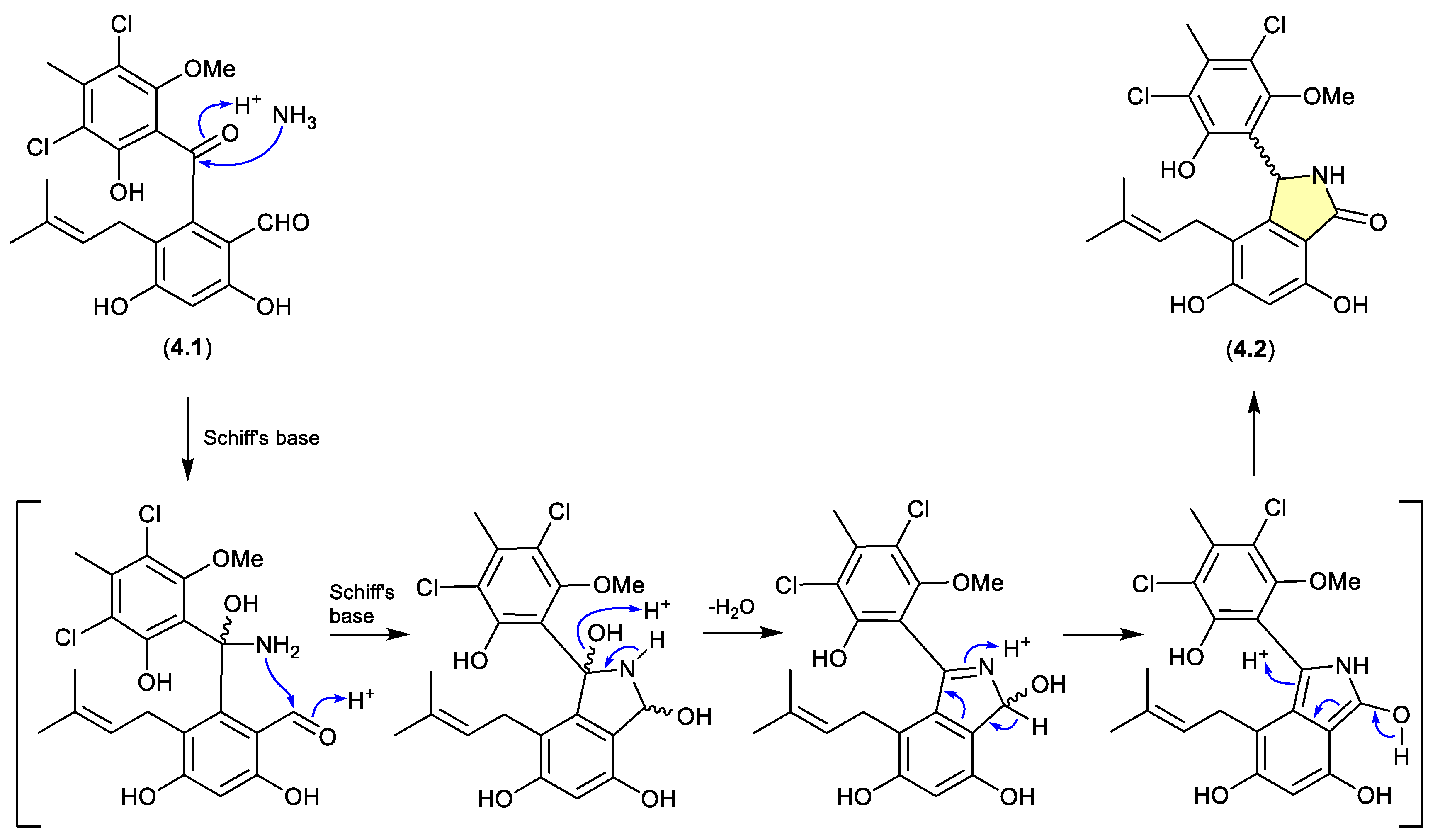
The benzophenone pestalone (4.1) was first reported in 2001 from the marine brown algae-derived fungus Pestalotia sp. CNL-365 (now classified under the genus Pestalotiopsis),[71] with a subsequent 2008 report describing the racemic pestalachloride A (4.2) from the plant endophytic fungus Pestalotiopsis adusta L416.[72] While racemic natural products are well known, they can nevertheless be indicators of inherent chemical reactivity that enable non-stereo controlled non-enzyme mediated transformations — much as is the case for artifacts. Interestingly, a 2010 total synthesis revealed that under mild conditions (NH3/NH4Cl/H2O, r.t., 80 min) 4.1 underwent high yield conversion to 4.2.[73] Of note, literature inference that 4.1 and 4.2 exist as enantiomeric mixtures of “stable” atropisomers does not seem plausible and lacks experimental validation. It’s more probable that 4.1 is achiral, and 4.2 is a racemic mixture of epimers.
Figure 4.
1.1.
neoenterocins (Figure 4.1.2)
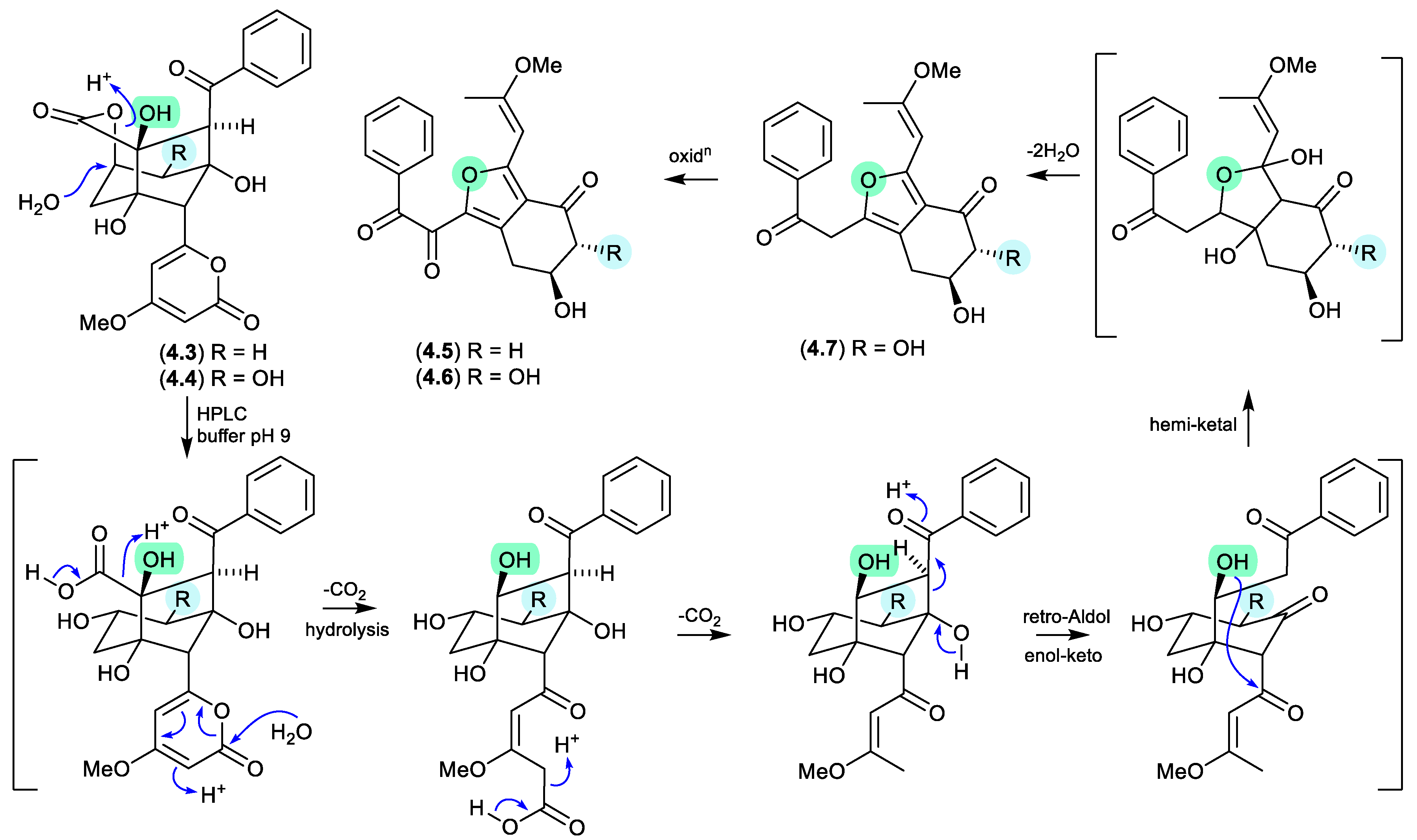
Modified M-AM3-D media cultivations of the South China Sea sediment-derived Streptomyces sp. SCSIO 11863 yielded the known polyketides 5-deoxyenterocin (4.3) and enterocin (4.4), and the new neoenterocins A (4.5) and B (4.6).[74] Significantly, HPLC analysis of enterocin (4.4) in 20 mM PBS buffer (pH 9) yielded 4.6, and the putative precursor 4.7, revealing a non-enzymatic pathway from enterocins to neoenterocins.
Figure 4.
1.2.
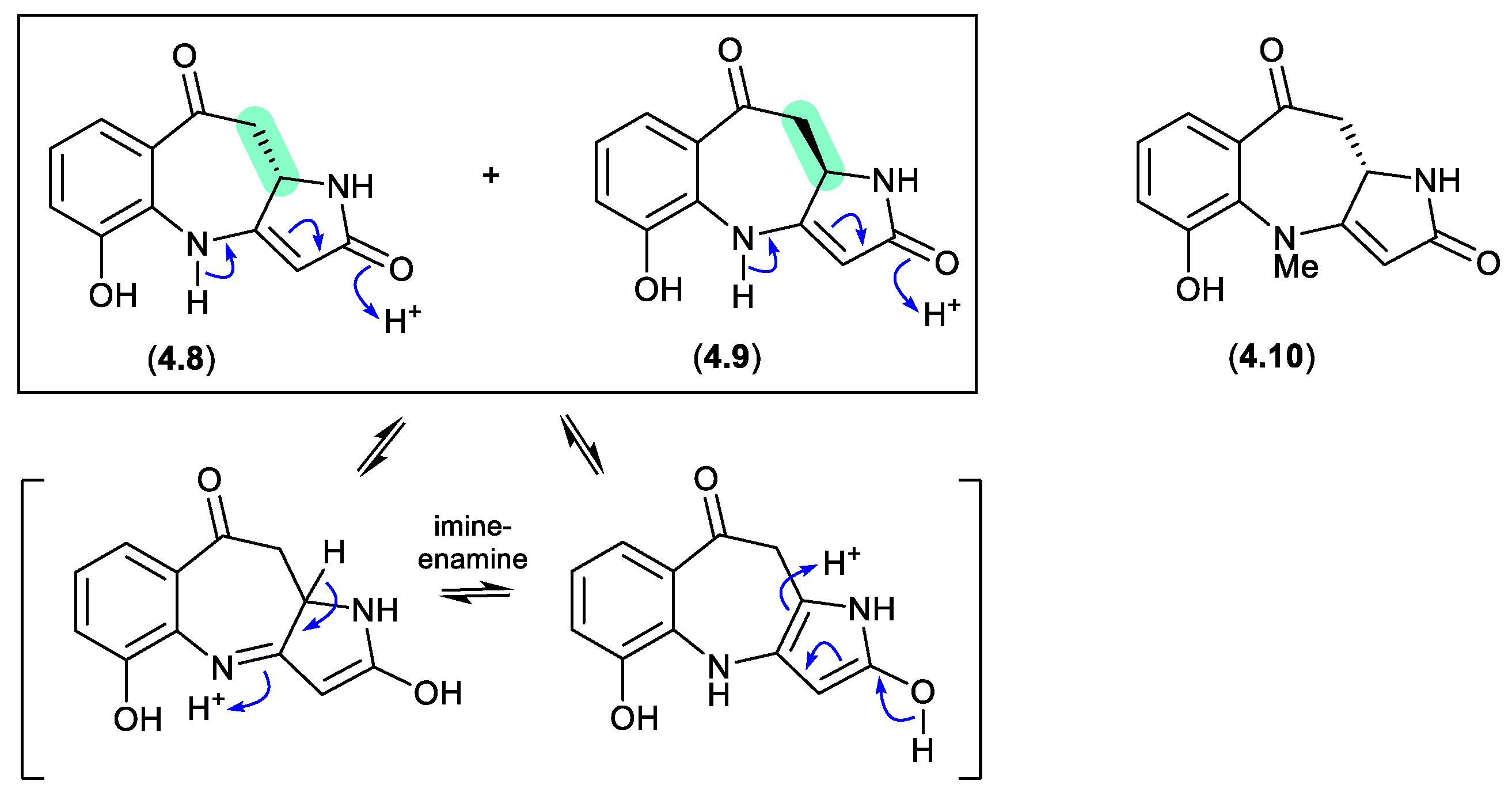
asperazepanones (Figure 4.1.3)
The gorgonian coral-derived fungus Aspergillus candidus CHNSCLM-0393 yielded the benzoazepine alkaloids, (+)-asperazepanone A (4.8), (–)-asperazepanone A (4.9) and (+)-asperazepanone B (4.10).[75] While the enantiomers 4.8 and 4.9 could be resolved by chiral HPLC, under mildly basic conditions they rapidly equilibrated to a racemic mixture. The proposed enamine-imino tautomerism mechanism is not available to the N-methylated analogue 4.10, which was isolated as the single (+) enantiomer, suggesting that 4.9 is an artifact.
Figure 4.
1.3.
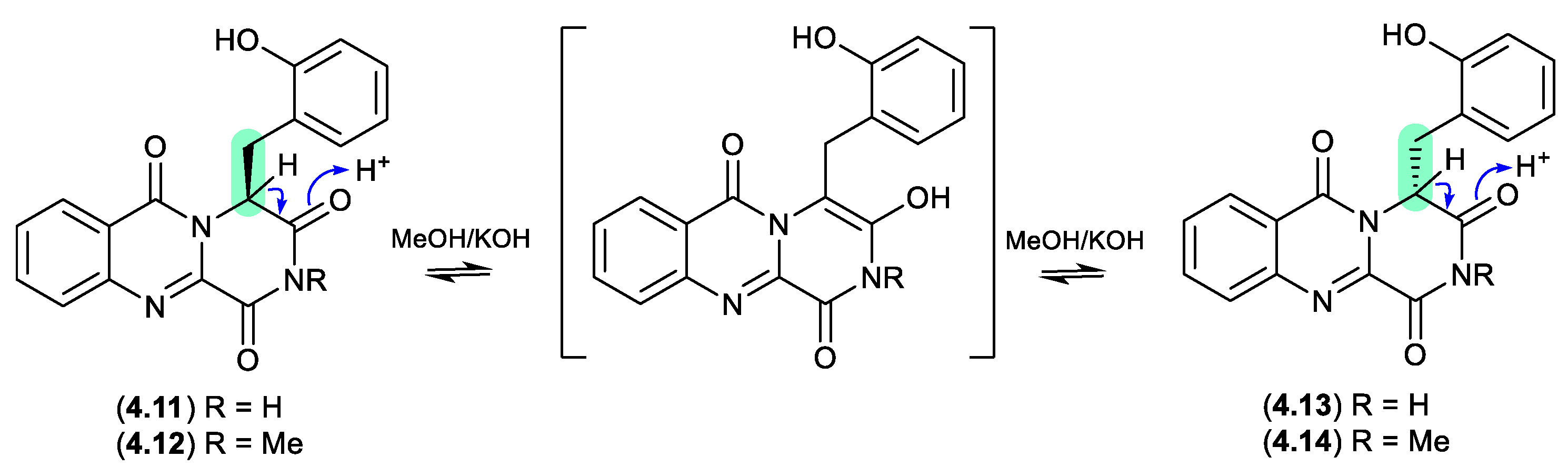
hydroxybrevianamides (Figure 4.1.4)
The South China Sea soft coral-derived fungus Aspergillus sp. CHNSCLM-0151 yielded two natural products, (+)-17-hydroxybrevianamide N (4.11) and (+)-N1-methyl-17-hydroxybrevianamide N (4.12), each featuring a rare o-hydroxyphenylalanine residue.[76] On exposure to mild base, 4.11 and 4.12 formed a racemic mixture with the corresponding epimers 4.13 and 4.14, respectively.
Figure 4.
1.4.
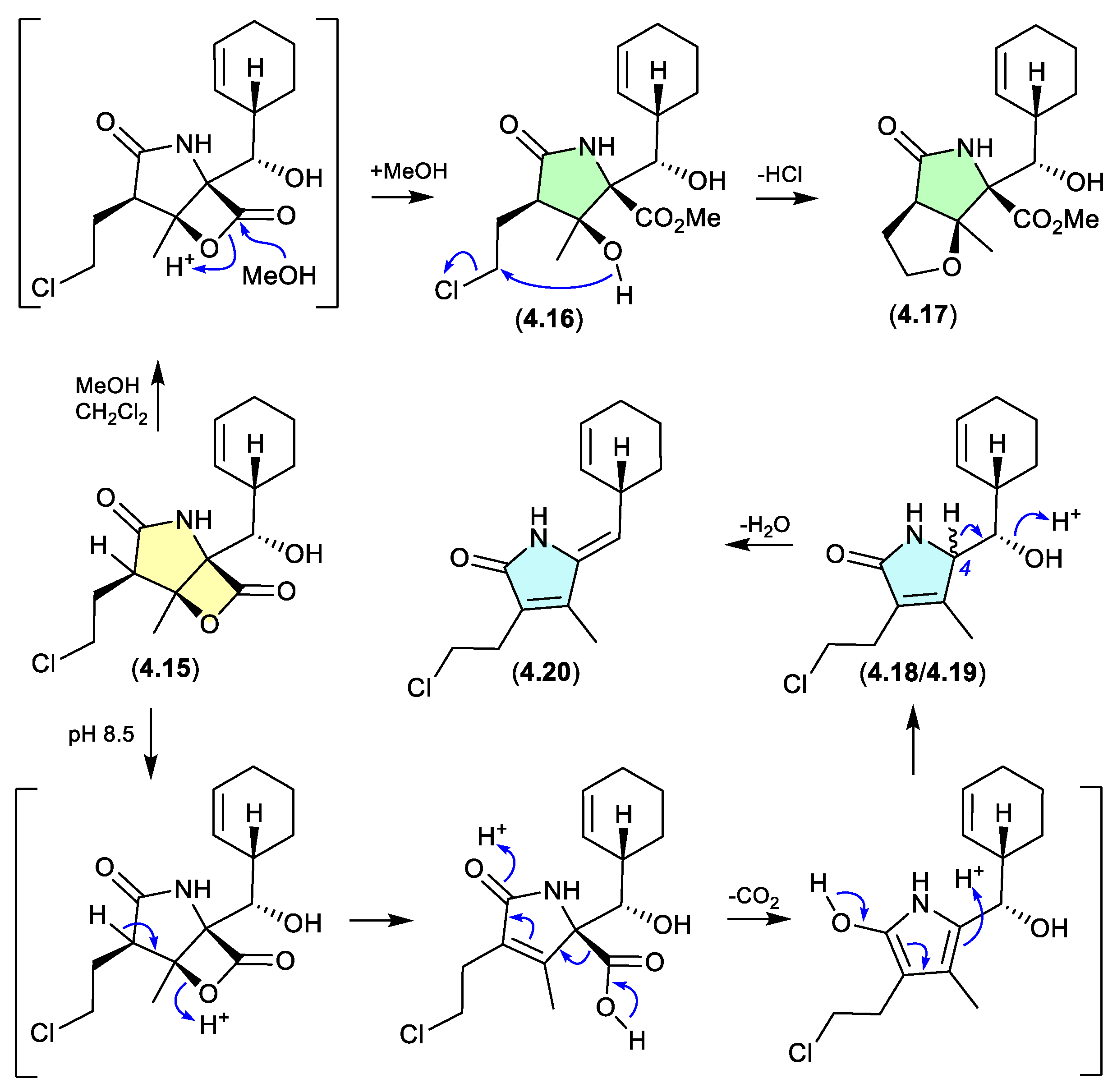
salinosporamides (Figure 4.1.5)
The marine obligate actinomycete Salinispora tropica CNB-392 produced the potent proteasome inhibitor salinosporamide A (4.15), along with five handling artifacts.[77] These artifacts included the methanolysis products 4.16 and 4.17 (mimicking isolation conditions), and the base-mediated seco-decarboxy epimers 4.18/4.19, and its dehydration product 4.20 (mimicking the pH of the fermentation broth).
Figure 4.
1.5.
4.2. Acidic
enterocins (Figure 4.2.1)
The polyketide enterocin (4.4) has been reported along with related analogues from a number of sources, including a Western Australian ascidian of the genus Didemnum,[78] the marine green alga-derived Streptomyces sp. OUCMDZ-3434,[79] and the shallow water marine sediment-derived Streptomyces sp. BD-26T.[80] A re-isolation of 4.4 from an Australian soil-derived Streptomyces sp. CMB-MRB492 prompted the first detailed assessment of its chemical reactivity.[81] When dried under nitrogen at r.t., extracts containing 4.4 proved stable, however, drying with warming to 40 °C resulted in partial conversion of 4.4 to a mixture of enterocins B (4.21) and C (4.22). Curiously, contrary to the more common role of acid activation of artifact formation, addition of trace amounts of TFA suppressed this thermal transformation. The same could not be said for 4.21 and 4.22, both of which when stored at r.t. for 24 h in MeCN/H2O with 0.01% TFA underwent quantitative conversion to enterocin D (4.23). While 4.23 proved stable to long term storage at low pH, when stored at r.t. for 24 h in MeCN/H2O at pH 9 it underwent partial transformation to enterocin E (4.24). The pH sensitivity of enterocin (4.4) highlights the challenges faced in exploring SAR using cell-based bioassays, where the pH of cultures may vary, even during the course of the bioassay.
Figure 4.
2.1.
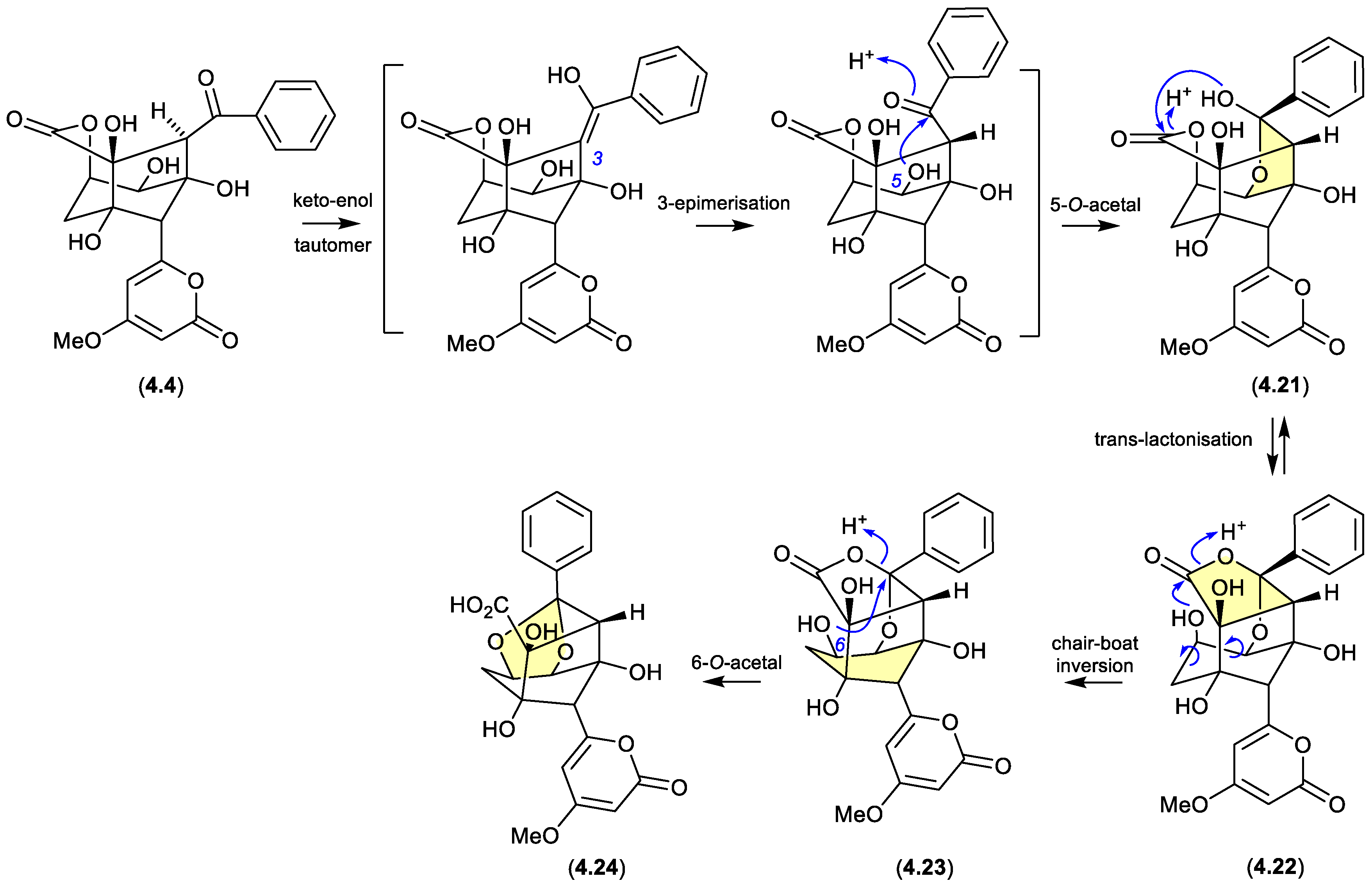
serratiochelin (Figure 4.2.2)
A co-culture of a Shewanella sp. and a Serratia sp., both sourced as a mixed culture from the intestine/stomach of an Atlantic hagfish (Myxine glutinosa) collected by benthic trawl in Hadselfjorden (Norwegian Sea), yielded the known siderophore serratiochelin A (4.25) and its formic acid-mediated hydrolysis artifact serratiochelin C (4.26).[82]
Figure 4.
2.2.
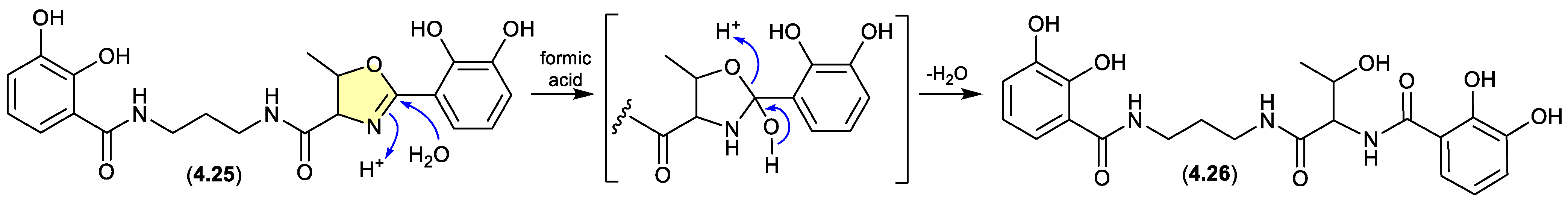
franklinolides (Figure 4.2.3)
An Australian marine sponge complex comprising a massive Geodia sp. thinly encrusted with a Halichondria sp. yielded the exceptionally cytotoxic franklinolide A (4.27).[83] A sample of 4.27 stored at r.t. in methanol-d4 for several days yielded the deutero-methyl ester 4.28 and the hydrolysed artifact, bitungolide A (4.29). The ease of transformation from 4.28 to 4.29 is noteworthy, given that 4.29 was first reported in 2002 from an Indonesian marine sponge, along with an array of geometric isomers, following extensive extraction with MeOH, silica gel chromatography (MeOH/CH2Cl2), and HPLC chromatography (MeOH/H2O).[84]
Figure 4.
2.3.
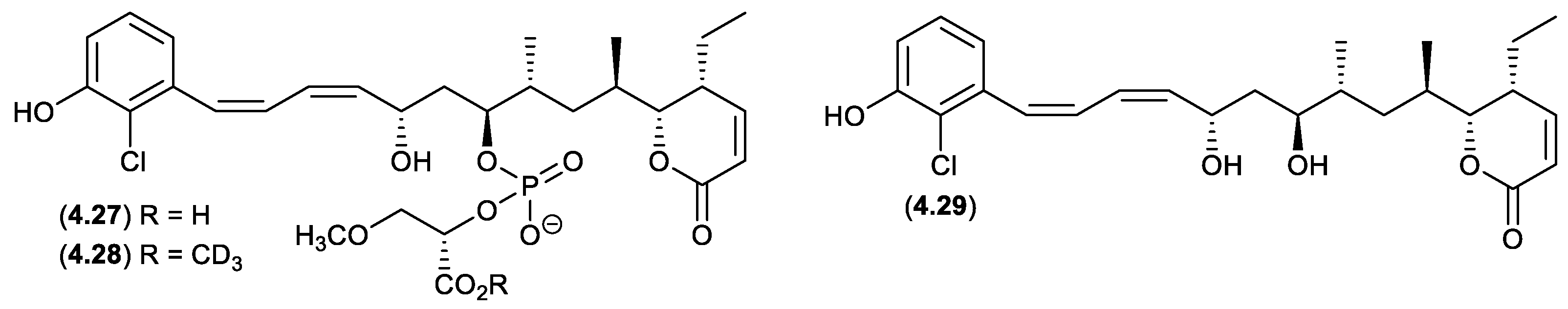
oxanthromicins/eurotones (Figure 4.2.4 and 4.2.5)
In 2014, the racemic polyketide (±)-hemi-oxanthromicin A (4.30), isolated from the soil-derived Streptomyces sp. MST-134270, was shown to be unstable to acid chromatography (MeOH/H2O with 0.1% TFA).[85] During handling 4.30 transformed to the methanolysis adduct (±)-hemi-oxanthromicin A (4.31) and the dimeric (±)-spiro-oxanthromicin A (4.32). Formation of latter likely proceeds via the co-isolated (±)-spiro-oxanthromicins B1/B2 (4.33/4.34) and C1/C2 (4.35/4.36), with B1/B2 (4.33/4.34) transforming during purification (MeOH/H2O with 0.1% TFA) to 4.32.
Figure 4.
2.4.
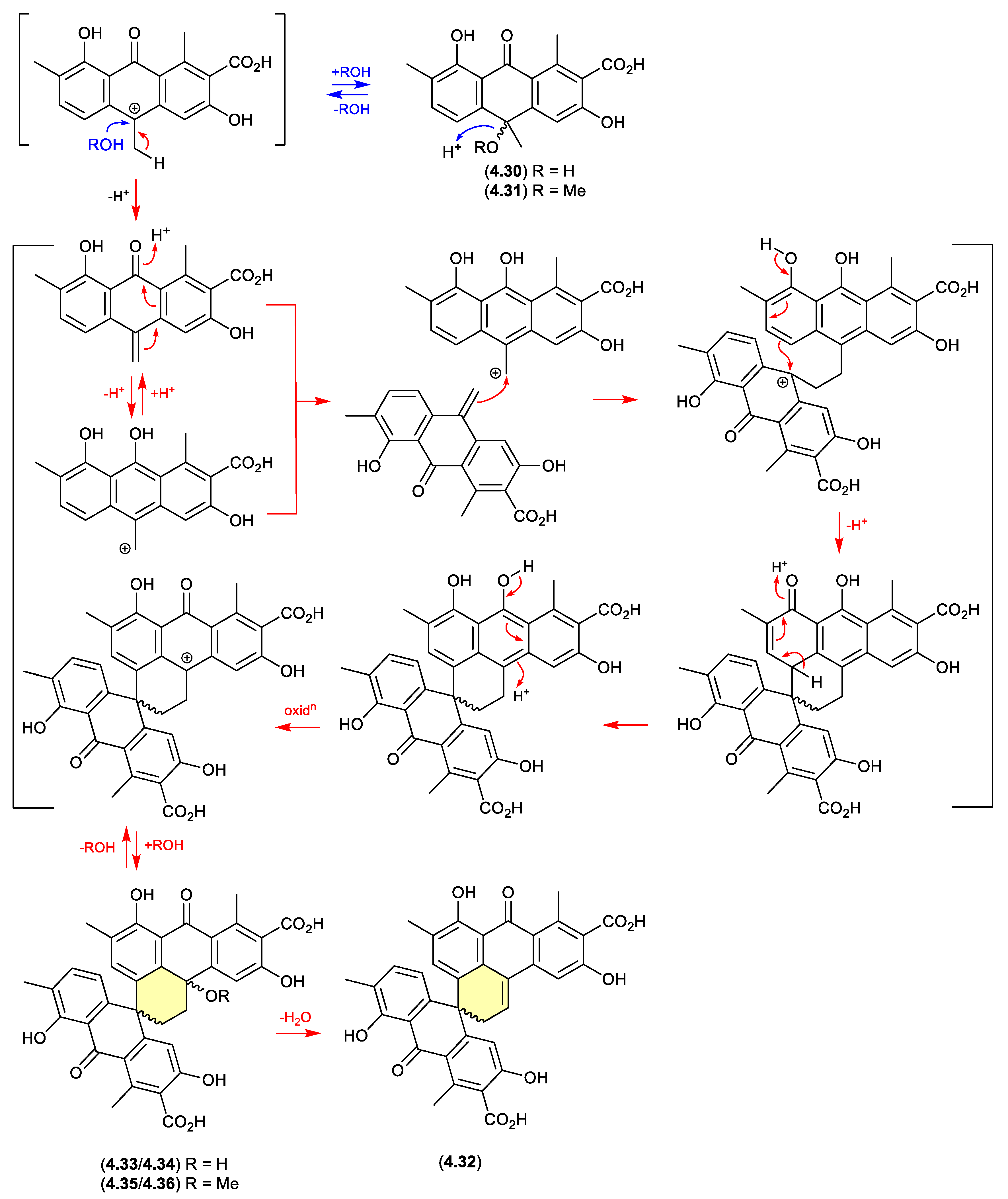
Curiously, in 2019 the enantiomeric (+)-eurotone A (4.37) and (–)-eurotone A (4.38) and related monomer, physcion (4.39), were reported following silica gel (CHCl3/MeOH) fractionation of an extract prepared from the marine-derived fungus Eurotium sp. SCSIO F452[86] — where 4.37/4.38 may be indicative of a chemically reactive and cryptic hemi-quinone co-metabolite akin to 4.33/4.34.
Figure 4.
2.5.
4.3. Silica Gel
sphydrofurans (Figure 4.3.1)
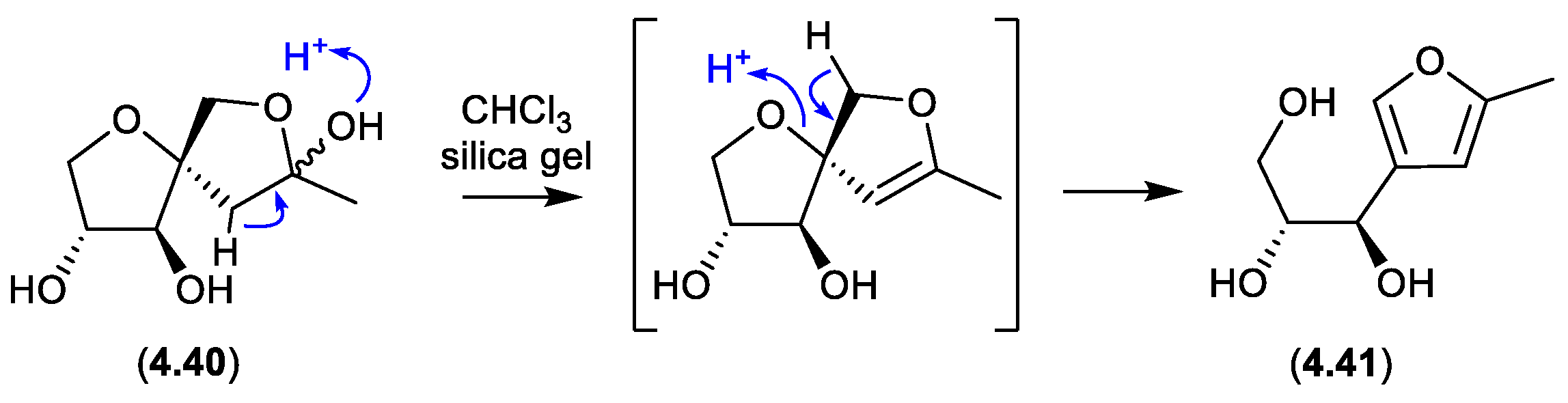
On exposure to silica gel chromatography (CHCl3/MeOH), the Streptomyces metabolite sphydrofuran (4.40) transforms to the furan artifact (4.41).[87]
Figure 4.
3.1.
duclauxin/bacillisporins/xenoclauxin/talaromycesone B (Figure 4.3.2 and 4.3.3)
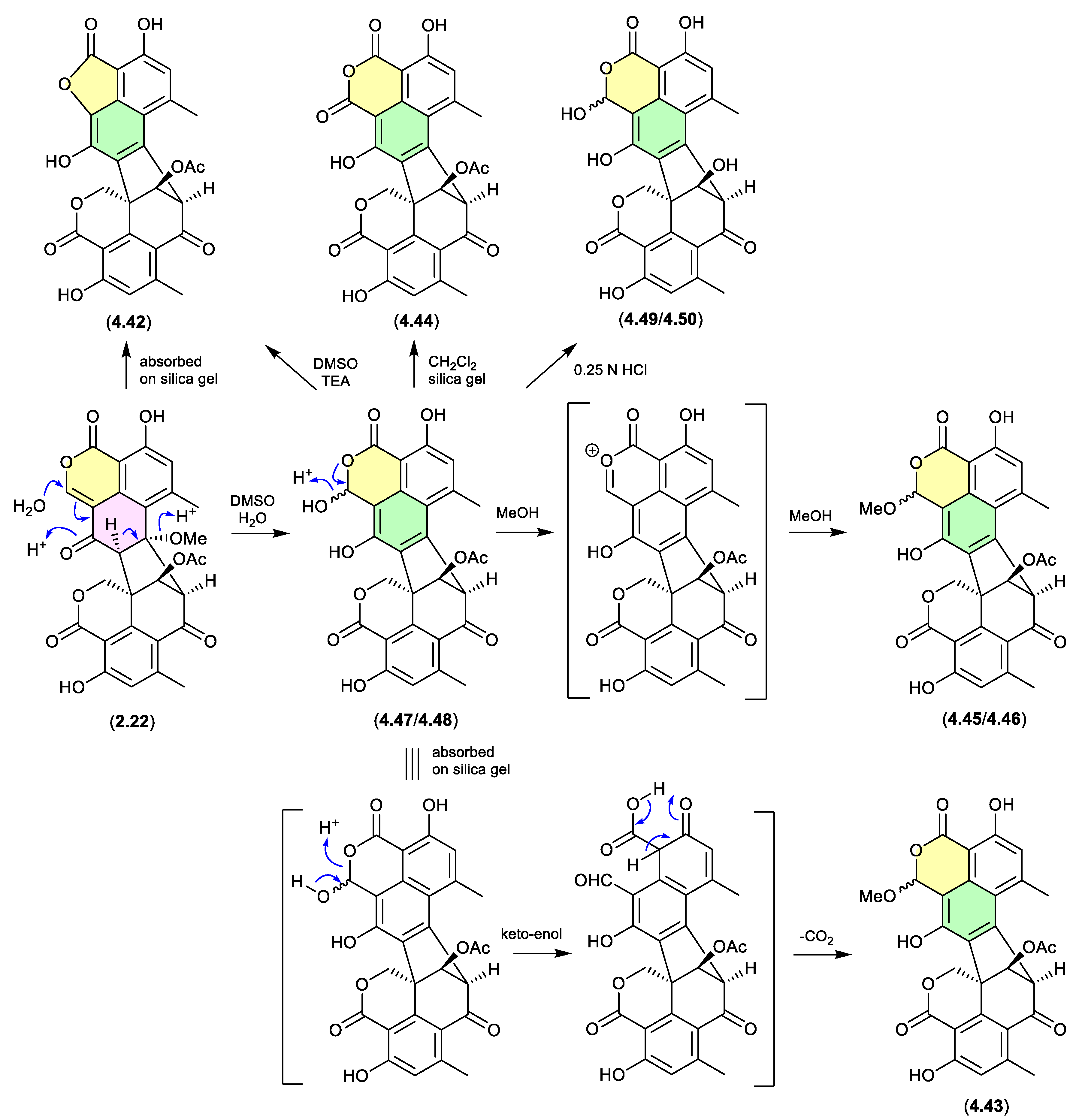
Since the polyketide duclauxin (2.22) was first reported in 1965 from Penicillium duclauxii,[88] in excess of 50 analogues have been reported as natural products from both terrestrial and marine fungi of the genera Penicillium and Talaromyces — many with a wide range of promising biological properties. Over the course of these studies it has been noted/speculated that some duclauxin-like natural products (co-metabolites of 2.22) may be handling artifacts. Recent investigations into the Mexican soil-derived Talaromyces sp. IQ-313,[48] examined duclauxin (2.22) and the artifact status of the known natural products, talaromycesone B (4.42), bacillisporin G (4.43), xenoclauxin (4.44), and bacillisporins F (4.45/4.46), J (4.47/4.48) and I (4.49/4.50). For example, a solution of 2.22 in DMSO/H2O transformed over 24 h to the acetal epimer bacillisporins J (4.47/4.48), which on dissolution in MeOH underwent spontaneous transformation to bacillisporins F (4.45/4.46). Similarly, 2.22 adsorbed on silica gel transformed to talaromycesone B (4.42); a mixture of 2.22 and 4.47/4.48 adsorbed on silica gel transformed to bacillisporin G (4.43); a CH2Cl2 suspension of bacillisporins J (4.47/4.48) on silica gel generated xenoclauxin (4.44); and exposure of 4.47/4.48 to mild acid (0.25 N HCl) effected hydrolysis of the acetate moiety to yield bacillisporins I (4.49/4.50). These studies supported the view that other members of the duclauxin family of natural product are also handling artifacts, including the marine-derived fungal verruculosins (see Section 2.5, duclauxin/verruculosins)[49] and adpressins B–F,[89] and terrestrial-derived fungal talaroketals A and B,[90] talaroclauxins A and B,[91] macrosporusones A–C,[92] and bacillisporones A, B, D and E.[93]
Figure 4.
3.2.
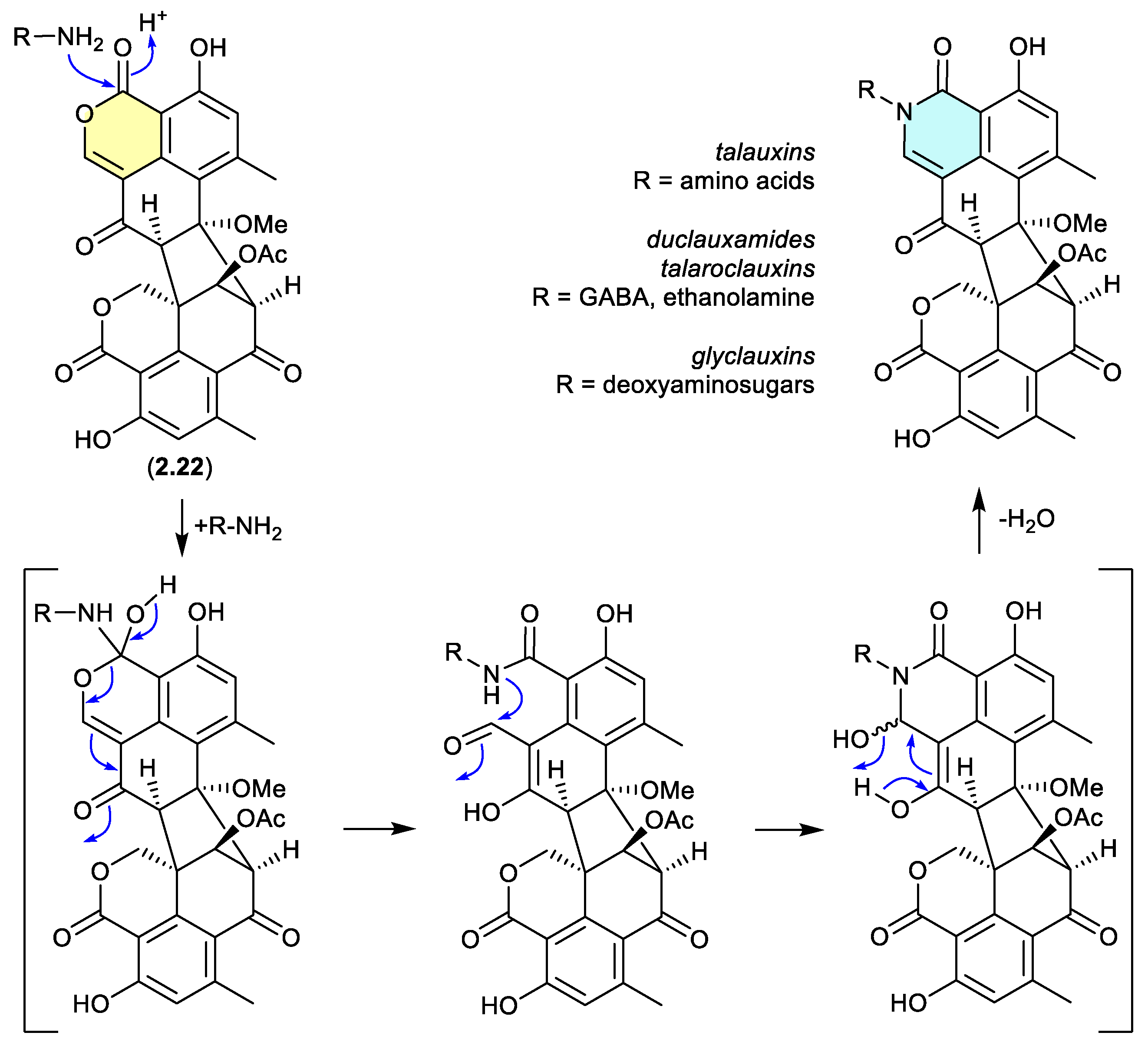
Duclauxin (2.22) has also been shown to react rapidly (in situ during cultivation, or extraction – see comment below) with various biogenetically available amines to produce lactams, including with, (i) amino acids to yield the talauxins,[94] (ii) ethanolamine and g-aminobutyric acids to yield duclauxamides[95,96] and talaroclauxins,[91] and (iii) deoxyaminosugars to yield glyclauxins.[18] While there is no evidence to suggest that these lactams are artifacts, their natural products status does raise an interesting possibility. If the fungus only produces duclauxin (2.22), and it is only during solvent extraction that 2.22 is released from the mycelia and comes into contact with biogenic amines present in the culture media — the resulting lactams could be better described as artifacts, as they only come into existence at the point of solvent extraction. This very scenario was encountered in the case of the prolinimines (see Section 10.1).
Figure 4.
3.3.
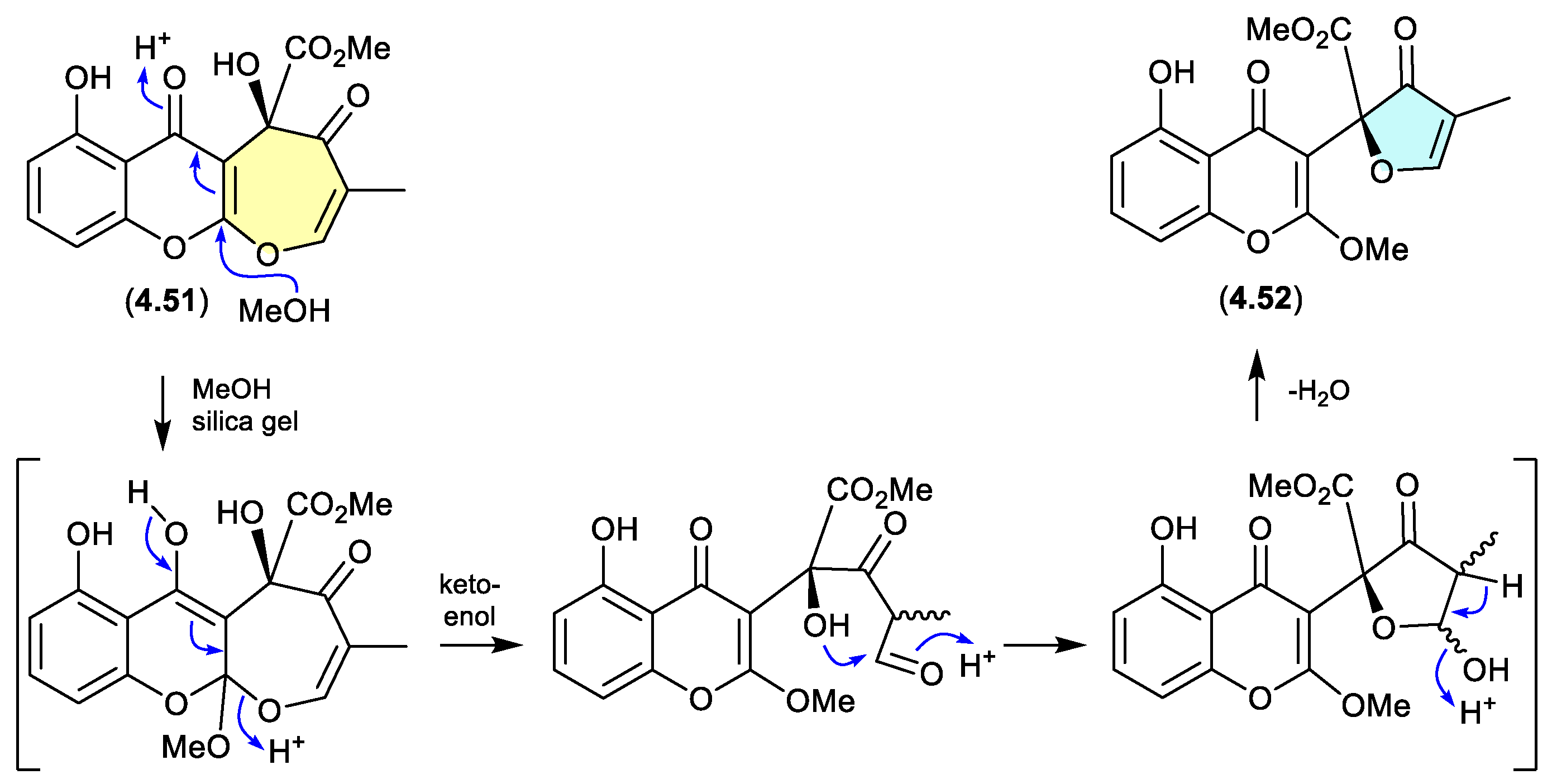
xanthepinone (Figure 4.3.4)
The chromone 4.51 recovered from the wood-decay fungus Rhizina sp. BCC 12292 was found to be a ring-contracted artifact, induced by silica gel chromatography (MeOH/CH2Cl2) of the known co-metabolite xanthepinone (4.52).[97]
Figure 4.
3.4.
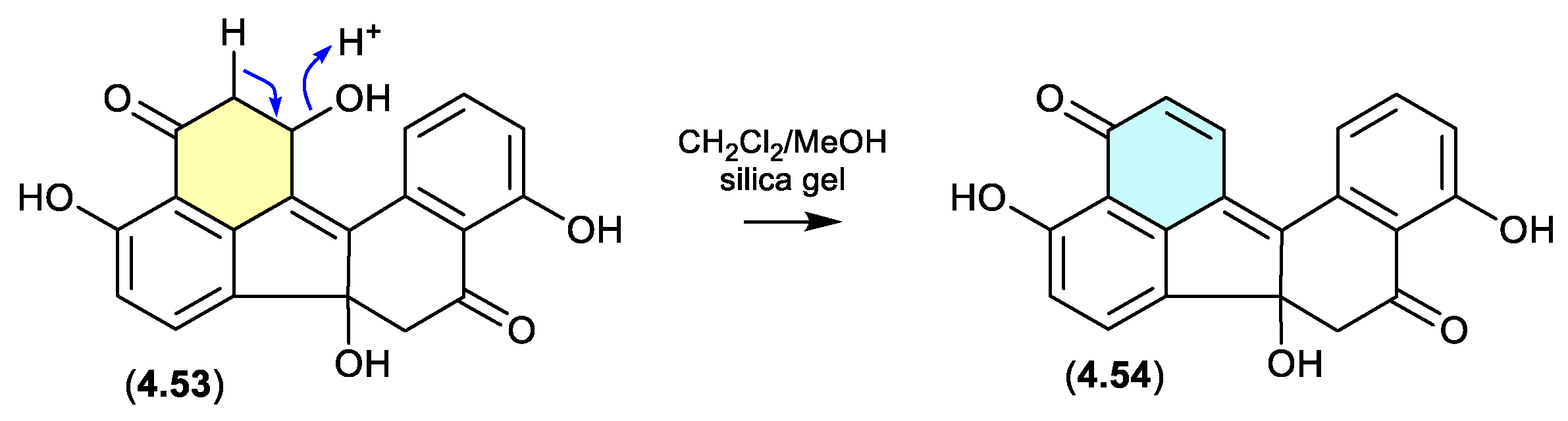
daldinones (Figure 4.3.5)
Daldinone H (4.53) isolated from the Cameroon mangrove plant-derived fungus Annulohypoxylon sp., underwent rapid dehydration on exposure to silica gel chromatography (CH2Cl2/MeOH) to the artifact daldinone I (4.54).[98]
Figure 4.
3.5.
5. Light
Many natural products are sensitive to sunlight, and it is advisable to minimise exposure to direct sunlight and store them longer term in the dark.
5.1. Photoisomerization
pyranpolyenolides (Figure 5.1.1)
The South China Sea sediment-derived Streptomyces sp. MS110128 yielded two new polyene macrolides that were prone to photoisomerization. For example, after only 5 h exposure to natural light, pyranpolyenolide B (5.1) underwent double bond isomerisation to pyranpolyenolides D (5.2), E (5.3) and F (5.4).[99]
Figure 5.
1.1.
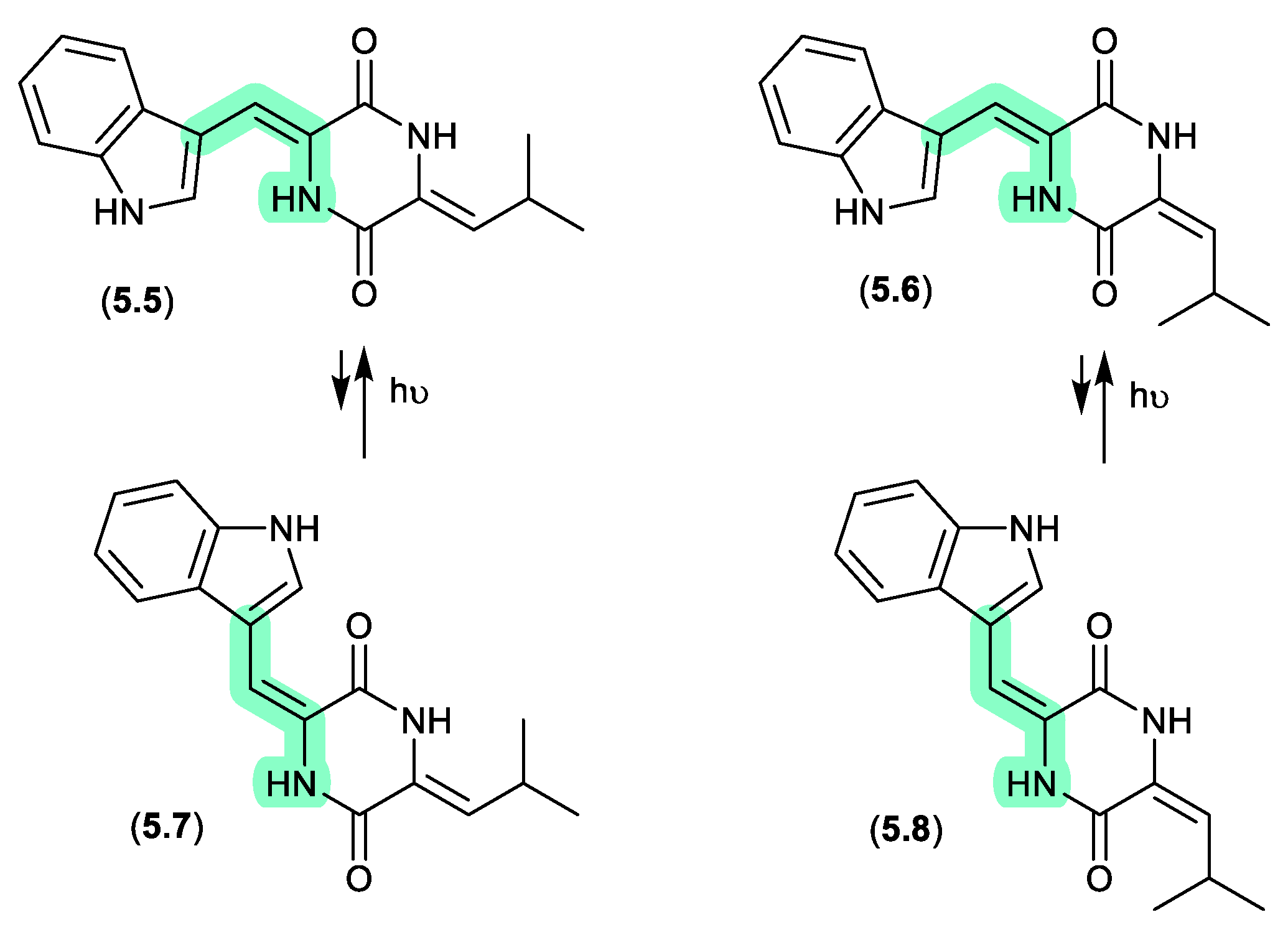
photopiperazines (Figure 5.1.2)
A Californian marine sponge-derived actinomycete (strain AJS-327) yielded four isomeric diketopiperazines, photopiperazines A–D (5.5–5.8), that were prone to photoisomerization.[100] For example, following HPLC purification, and even where care was taken to handle samples under dim light conditions, 5.5 equilibrated to a 1:0.2 ratio with 5.7. The same was also observed for 5.6 and 5.8. On exposure to long wavelength UV (365 nm) for 2 h this ratio adjusted to 1:1.3; however, when stored under room lighting for 6 h the mixture re-equilibrated to the original ratio. Significantly, photoisomerization was restricted to the double bond associated with the Trp, but not the Leu moiety.
Figure 5.
1.2.
aspochracins/sclerotiolides (Figure 5.1.3)
The halotolerant fungus, Aspergillus sclerotiorum PT06-1 isolated from salt sediments collected from the Putian Sea Salt Field, Fujian, China, yielded an array of cyclic tripeptides prone to photoisomerism and oxidation.[101] For example, when exposed to daylight and air for 1 d, a MeOH/H2O solution of the known cyclic tripeptide aspochracin (5.9) underwent isomerisation to sclerotiolide E (5.10), and when extended to 10 d, yielded sclerotiolides F–J (5.11–5.16).
Figure 5.
1.3.
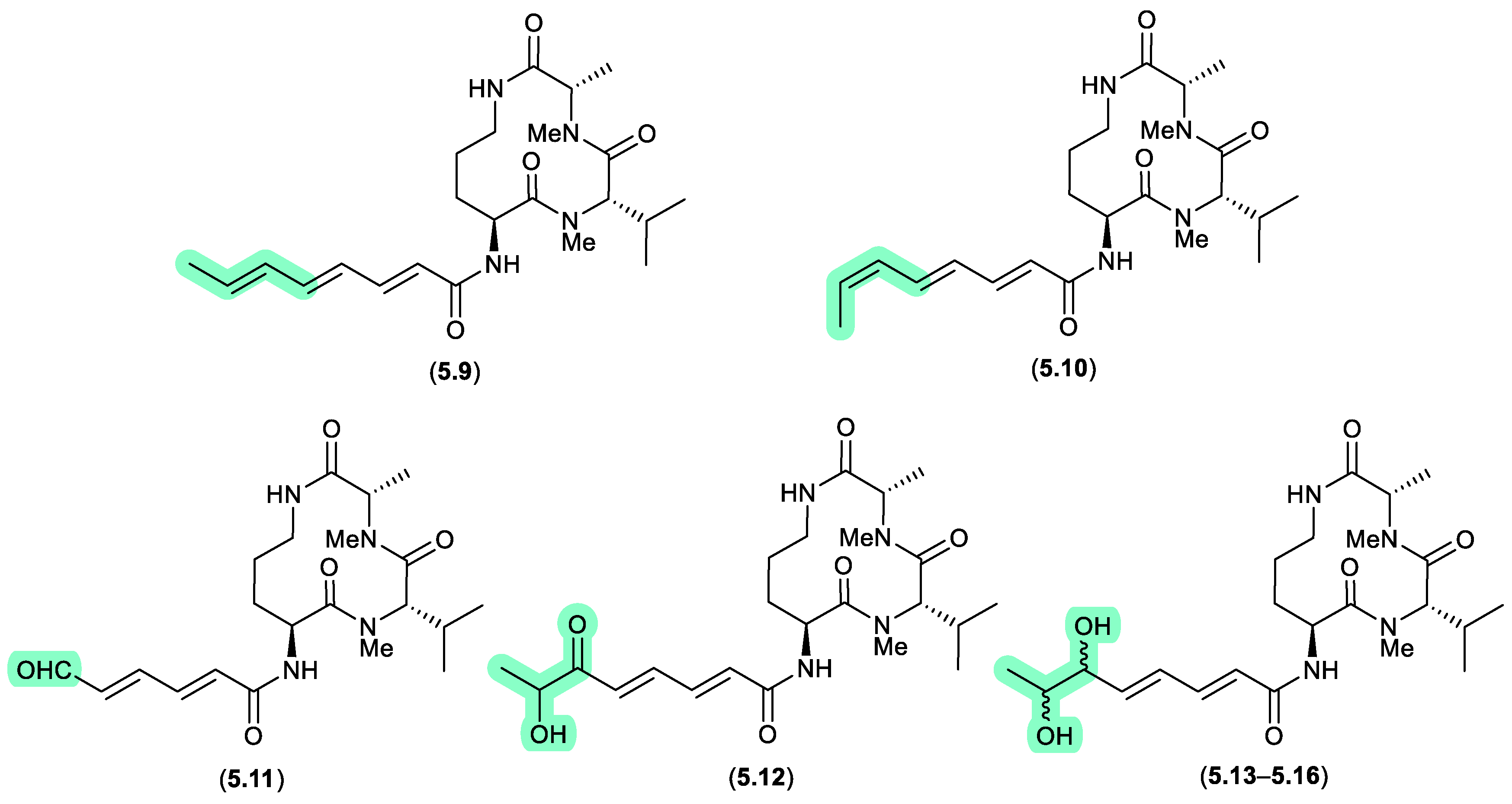
clavosines/calyculins (Figure 5.1.4)
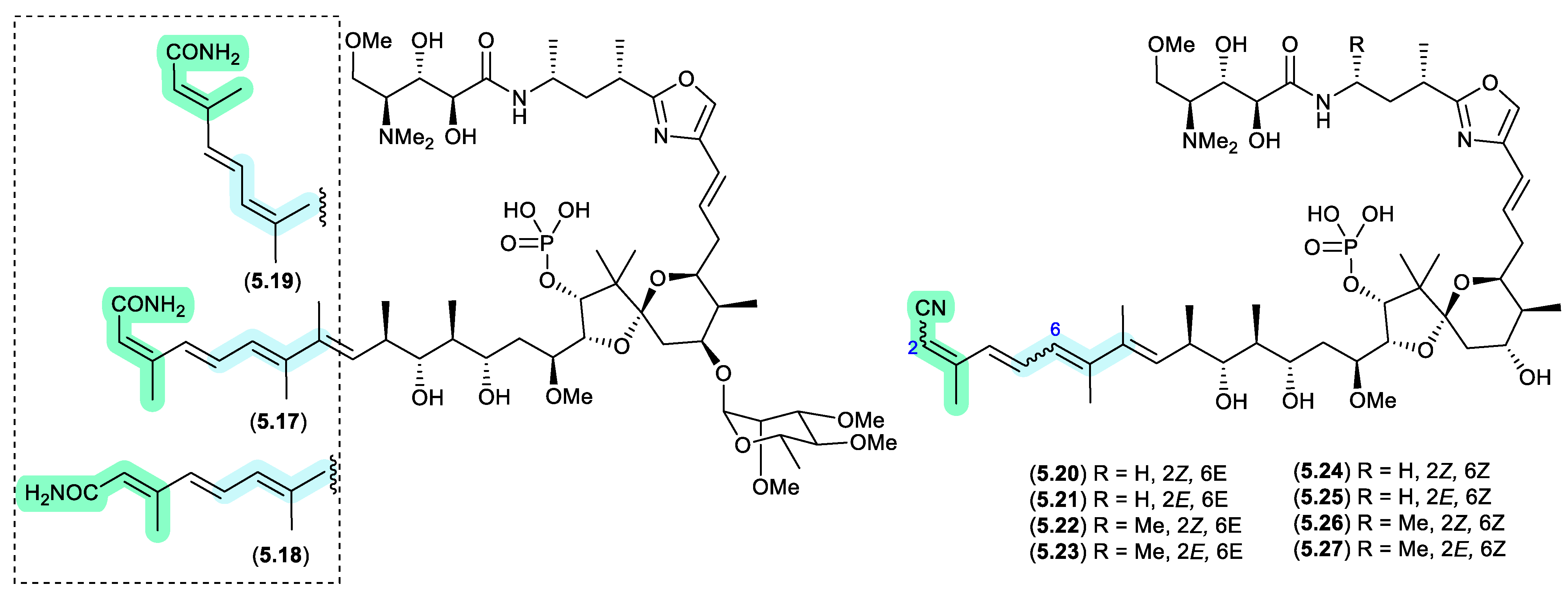
A sample of marine sponge Myriastra clavosa collected from Chuuk, Federated States of Micronesia, yielded clavosines A–C (5.17–5.19) as potent cytotoxins and inhibitors of Protein Phosphatase 1 and 2A, of which clavosine C (5.19) is a photoisomerization artifact of clavosine B (5.18).[102] Similar issues arose in the case of the structurally related calyculins A–H (5.20–5.27), where it has been suggested all but calyculins A and C (5.20 and 5.22) may be photoisomerization artifacts.[103]
Figure 5.
1.4.
5.2. Photooxidation
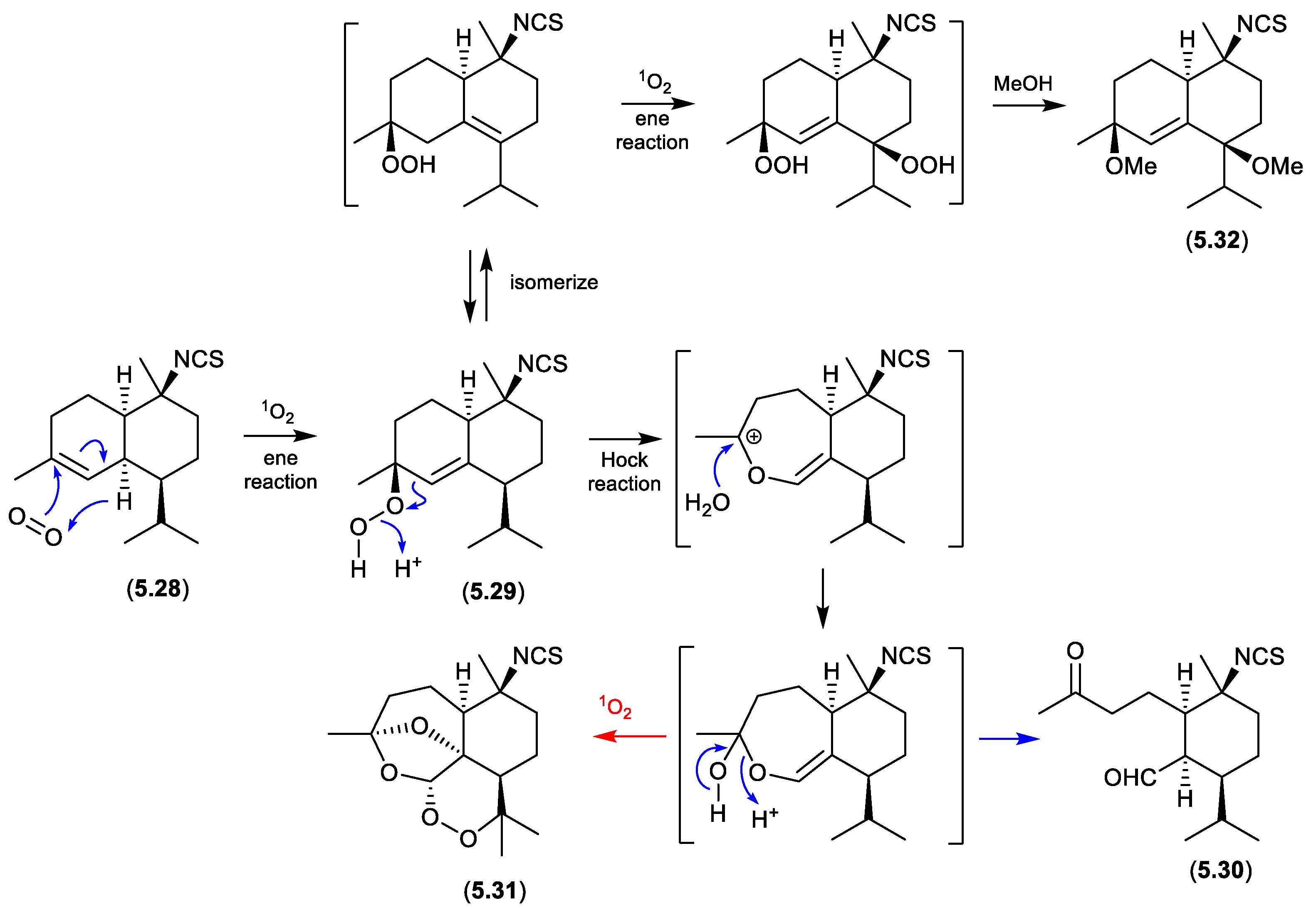
cadinanes (Figure 5.2.1)
Marine and microbial extracts can be rich in pigments, which could in principle act as photosensitisers, leading to photooxidation artifacts. In an interesting study, Tanaka et al noted “We have observed that a neighboring natural products laboratory often isolated molecules with a hydroperoxide moiety, while we have rarely isolated molecules with this functionality. One difference between the two labs is their orientation, with our lab facing north and the neighboring lab facing south. It was therefore suspected that isolation of hydroperoxide molecules could be affected by sunlight coming through windows during laboratory procedures.”[104] To test this hypothesis, separate acetone, MeCN and MeOH solutions of an isothiocyanate sesquiterpene, cadinane (5.28), and the photosensitiser Rose Bengal, were allowed to stand at r.t. for two weeks exposed to sunlight through the laboratory window. The acetone solution yielded 5.29 and 5.30, while MeCN yielded 5.30, and MeOH 5.31 and 5.32. Not only did this study demonstrate the potential for photooxidations, but it also highlighted that artifact pathways can be solvent dependent.
Figure 5.
2.1.
5.3. Photoreactivity
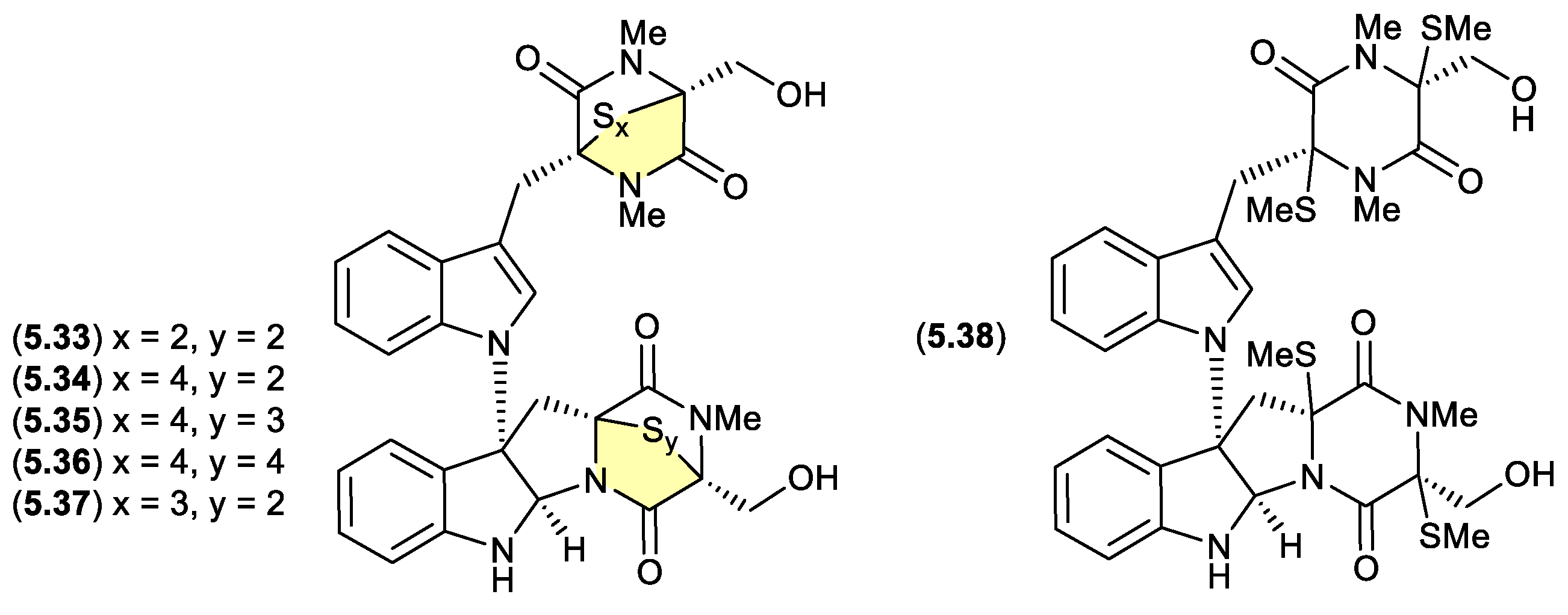
chetomins (Figure 5.3.1)
Chetomin (5.33) was first isolated in 1944 by Wakeman et al from the fungus Chaetomium cochliodes, and exhibits strong cytotoxicity via the transcription factor, hypoxia-inducible factor 1 (HIF-1).[105] Building on this rare structure class, in 2018 Zou et al reported an investigation of a C. cochliodes type strain (CGMCC3.17123), which yielded both 5.33 and the new chetomins A–D (5.34–5.37) and dethio-tetra (methylthio) chetomin (5.38).[106] As 5.33–5.38 proved to be photosensitive, isolation, characterisation and structure elucidation was achieved in darkness. Once isolated, controlled exposure to light indicated a high degree of interconversion between 5.33–5.37, with sulphur heterocycles both enlarging and contracting.
Figure 5.
3.1.
talaromycins/purpactins (Figure 5.3.2)
The South China Sea gorgonian-derived fungus Talaromyces sp. yielded the new diphenyl ether talaromycins A–C (5.39–5.41), along with several known natural products, including tenellic acid A methyl ester (5.42), purpactin C (5.43) and C′ (2.59).[31] Interestingly, 5.43 transformed under daylight to 2.59 and 5.39. Similarly, the extensive use of silica gel (CH2Cl2/MeOH) chromatography likely facilitated transformation (methanolysis) of the lactone ring in 2.59 to the seco methyl ester 5.42, and the benzaldehydes 5.39 and 5.42, to the dimethyl acetals 5.40 and 5.41, respectively (see Section 2.3). Hence, the combination of both sunlight and the use of silica gel suggests that talaromycins A–C (5.39–5.41), tenellic acid A methyl ester (5.42) and purpactin C′ (2.59) are likely artifacts.
Figure 5.
3.2.
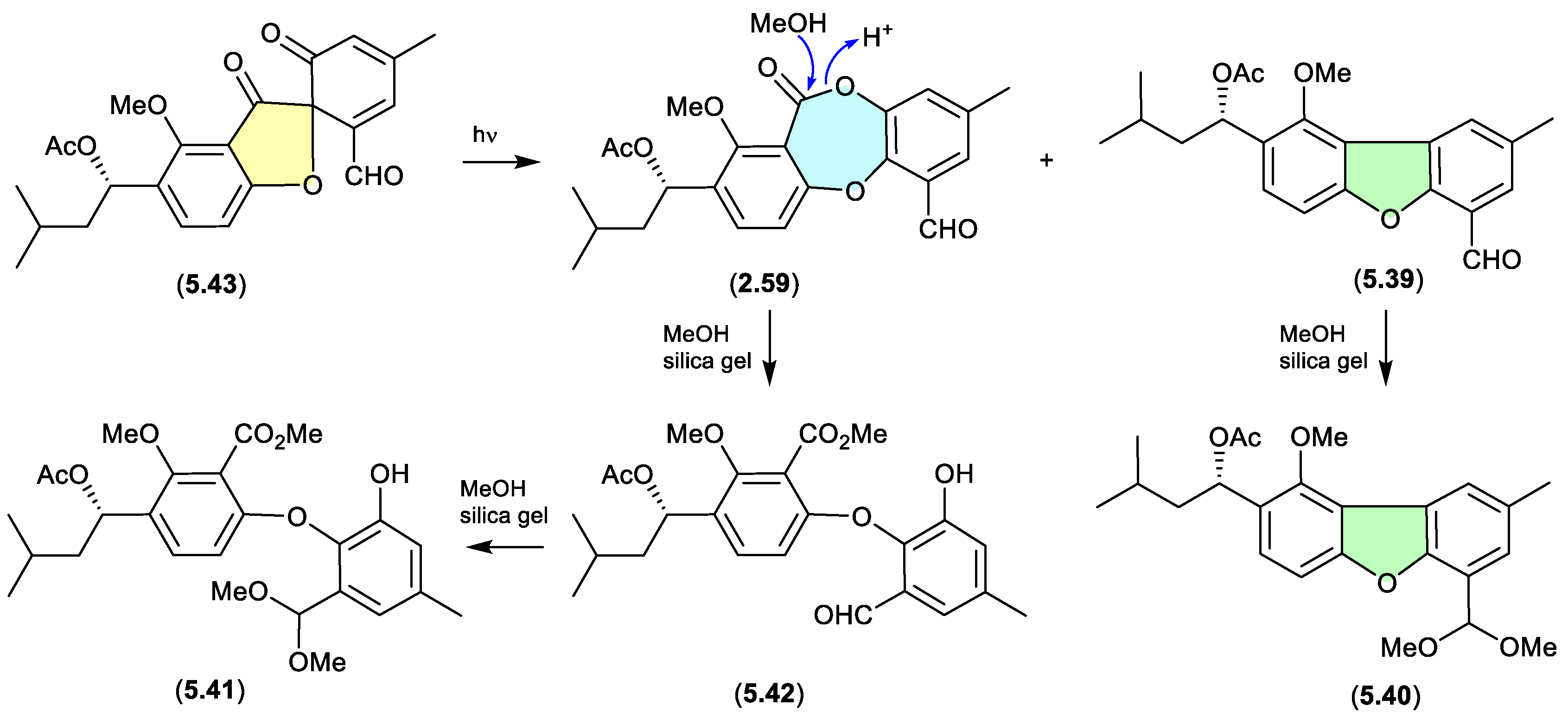
6. Air Oxidation
Many functional groups are prone to air oxidation, including phenols, hydroquinones, alkenes and alcohols. In many cases these oxidation artifacts can be readily predicted, and through cautious laboratory handing, be minimised or even avoided. Air oxidation can also initiate transformations that are less intuitive, potentially leading to artifacts that risk going unnoticed and mischaracterised as natural products.
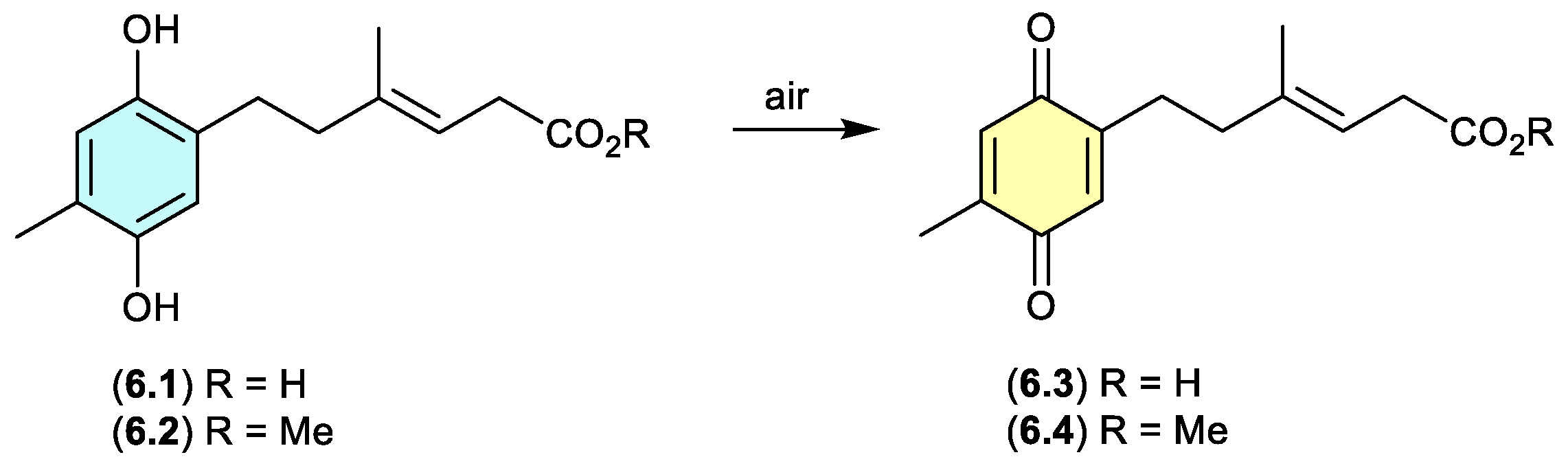
ketidocillinones (Figure 6.1)
On exposure to air, the antibacterial polyketide ketidocillinones A (6.1) and B (6.2), isolated from the Antarctic sponge-derived fungus Penicillium sp. HDN151272, slowly oxidised to the corresponding quinones 6.3 and 6.4, respectively.[107] This is a common transformation among natural products that feature a hydroquinone moiety.
Figure 6.
1.
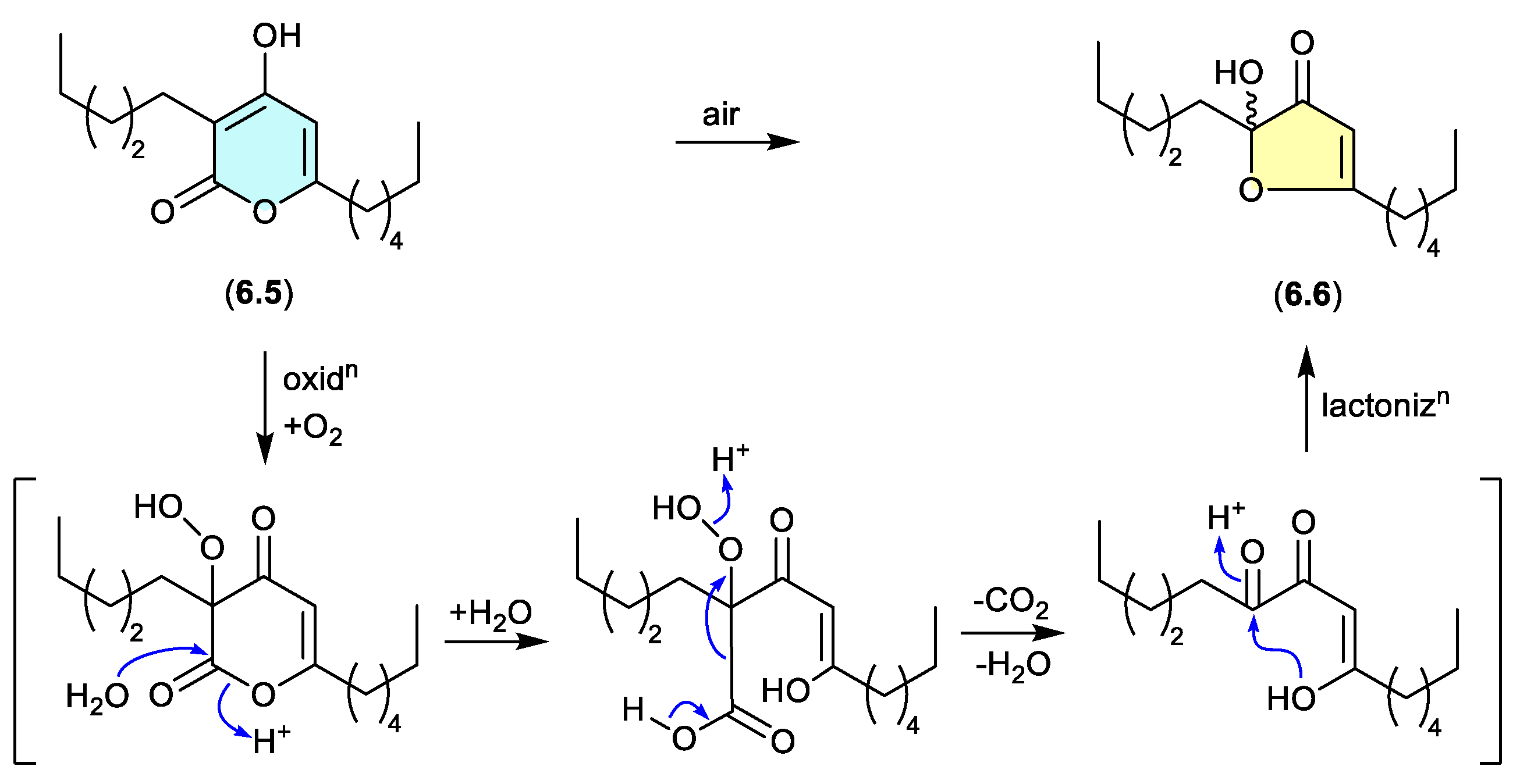
pseudopyronines (Figure 6.2)
The polyketide a-pyrone, pseudopyronine B (6.5), isolated from a Fijian marine sponge-derived Pseudomonas sp. F92S91, undergoes oxidative transformation during handling to the furanone 6.6.[108] A key characteristic of this transformation is the racemic nature of the acetal-furanone moiety, and a plausible mechanism proceeds via autoxidation, hydrolysis, decarboxylation/dehydration and lactonisation (as indicated).
Figure 6.
2.
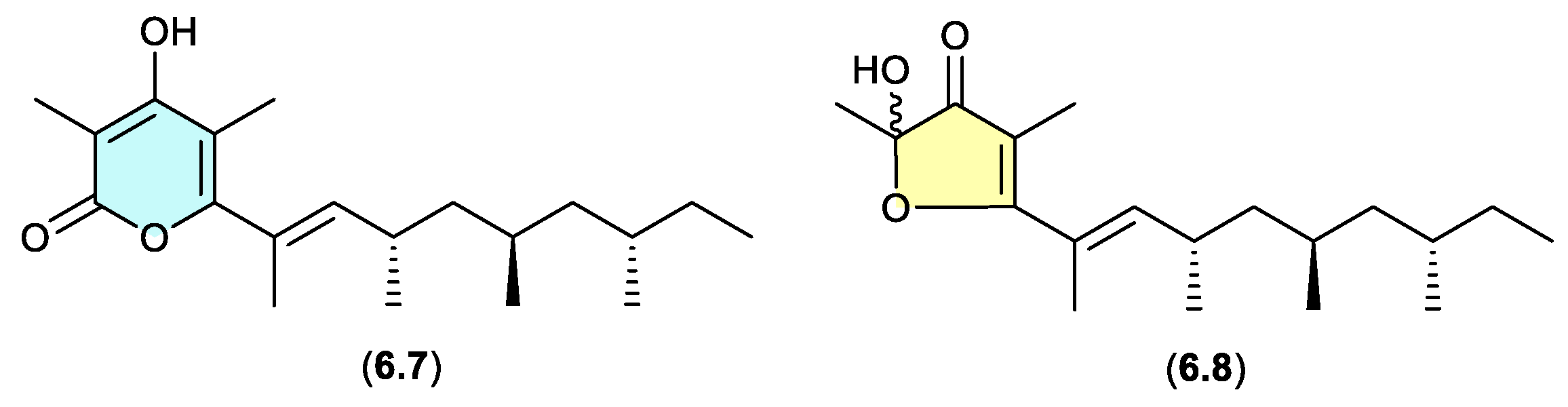
norpectinatone (Figure 6.3)
Natural products featuring the acetal-furanone moiety, as in 6.6, are relatively rare in the scientific literature, and typically co-occur with the corresponding a-pyrones (e.g. 6.5), which is suggestive of an artifact relationship. A clear example is seen with the polyketide a-pyrone norpectinatone (6.7) and ketal-furanone 6.8 recovered from a Chilean collection of the pulmonated mollusc Siphonaria lessonii[109] — the latter potentially an artifact of the former.
Figure 6.
3.
linfuranones/hyafurones/aurafurones (Figure 6.4)
On other occasions, aggressive cultivation and/or isolation conditions may induce the oxidative biotransformation of a prospective (un-isolated) precursor a-pyrones such that only the acetal-furanones are isolated. For example, silica gel fractionation (see Section 4) of extracts prepared from shaken broth (aerated!) cultivations of the actinomycete Sphaermonospora mesophila GMKU 363 yielded linfuranones [e.g. linfuranone A (6.9)],[110,111] while the myxobacterium Hyalangium minutum yielded hyafurones [e.g. hyafurone A1 (6.10)].[112] Biosynthetic studies into the closely related myxobacteria aurafurones [e.g. aurafuron A (6.11)] provided valuable insights, but did not exclude the possibility of post-translational artifact formation as noted above.[113]
Figure 6.
4.
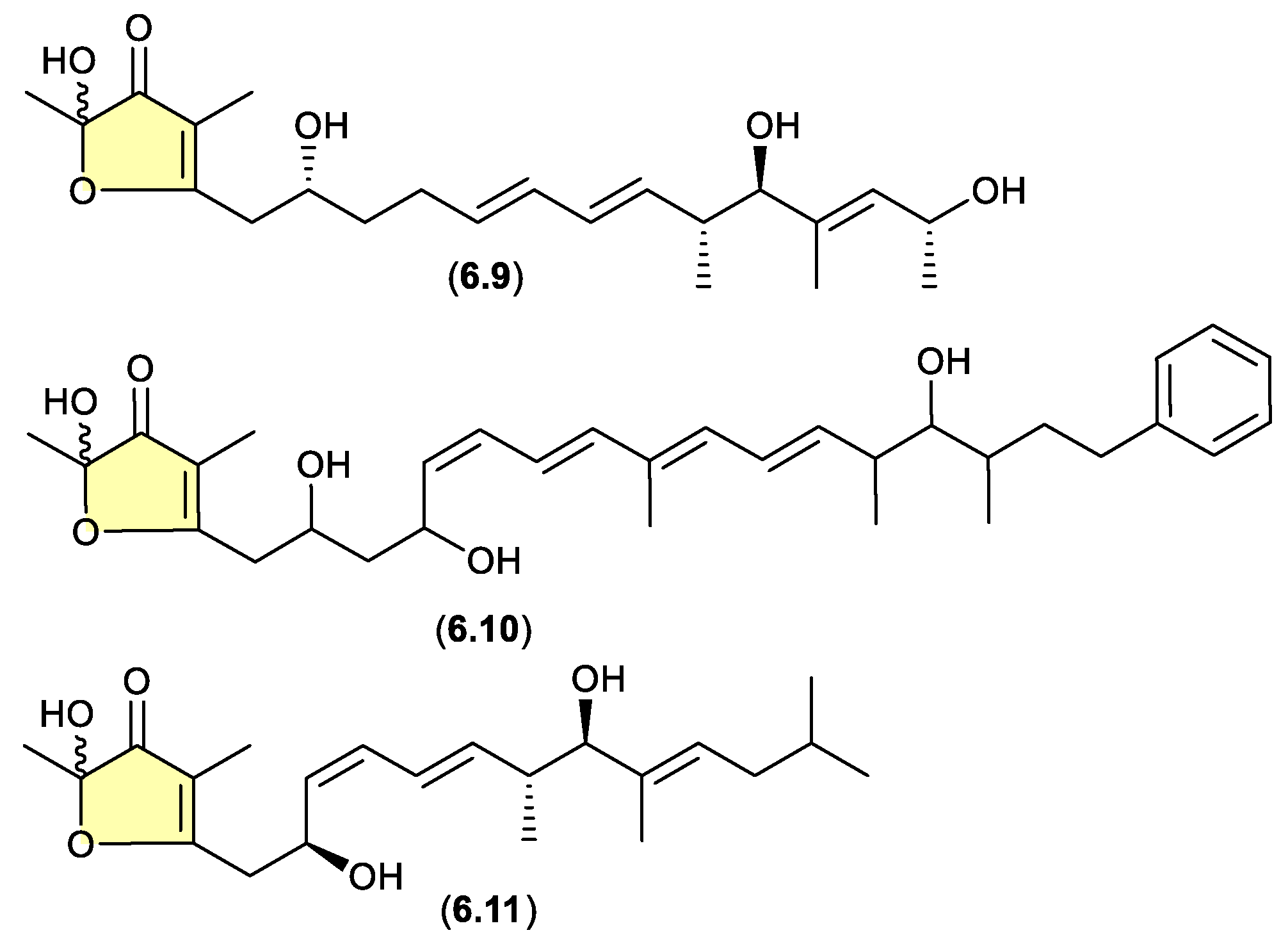
avermectins (Figure 6.5)
Prolonged (multi-year) storage of solid samples of technical abamectin (typically a mixture of avermectin B1a (6.12) and minor avermectins), even under refrigeration and without exposure to light, resulted in oxidative transformation (up to 34%) to the remarkably stable and chiral peroxide 6.13.[114]
Figure 6.
5.
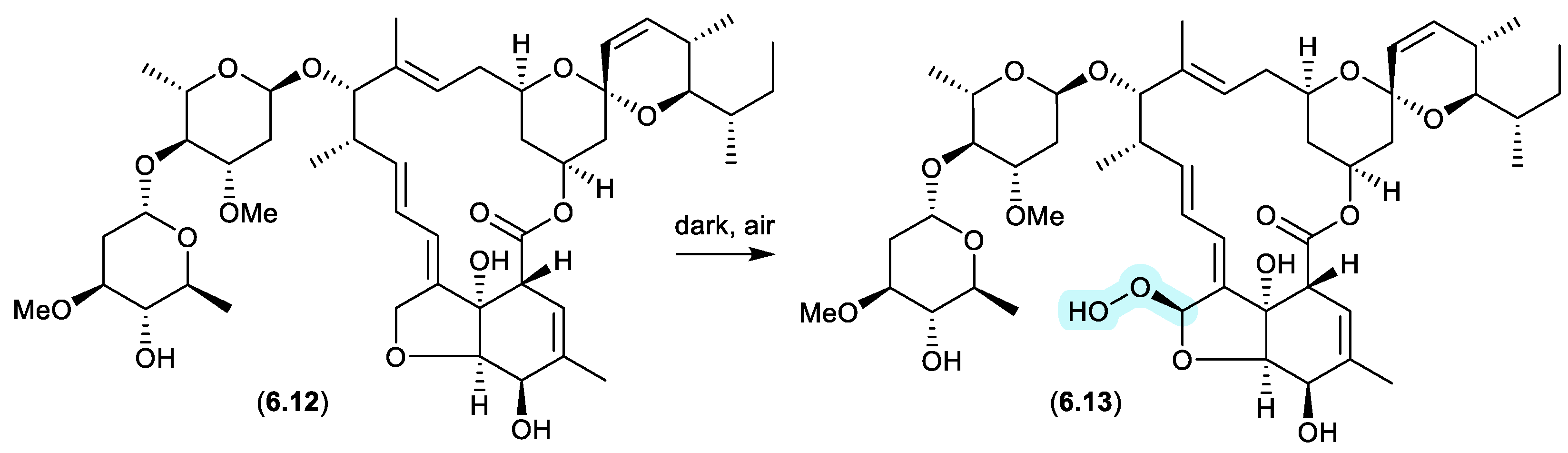
penilumamides (Figure 6.6)
A lumazine-type peptide featuring an l-methionine sulfoxide, penilumamide (6.14), was first reported in 2010 from the marine-derived fungus Penicillium sp. CNL-338,[115] It was later isolated from the mangrove-derived fungus Aspergillus sp. 33241 in 2015,[116] and from the gorgonian-derived fungus Aspergillus sp. XS-20090B15 in 2014.[117] As the latter study also reported the l-methionine sulfone penilumamide C (6.15) as a minor co-metabolite, this prompted speculation that the true natural product may be an as yet undetected cryptic l-methionine analogue 6.16 (see Section 10). This hypothesis was validated when careful examination of an extract of XS-20090B15, avoiding air oxidation, yielded penilumamide B (6.16). As predicted, exposure of 6.16 to air at r.t. resulting in initial transformation to the sulfoxide 6.14, and later to the sulfone 6.15.
Figure 6.
6.
7. Acetal/ketal Equilibration
Not strictly a matter of artifacts, acetal/ketal equilibria — much like equilibration in protic versus non-protic solvents (see Section 2.10) — reveals chemical reactivity that can impact on structure elucidation, as well as consideration of biosynthetic origins, SAR, and biological mechanisms of action. Many examples of acetal and ketal equilibration are relatively trivial (e.g. carbohydrate anomers), but others are less so. To illustrate this, two case studies are outlined below.
okichromanone (Figure 7.1)
Reported in 2024 from the sponge-derived actinomycete Microbispora sp., okichromanone (8.1) exists as an equilibrating mixture of two racemic ketal regioisomers.[118]
Figure 7.
1.
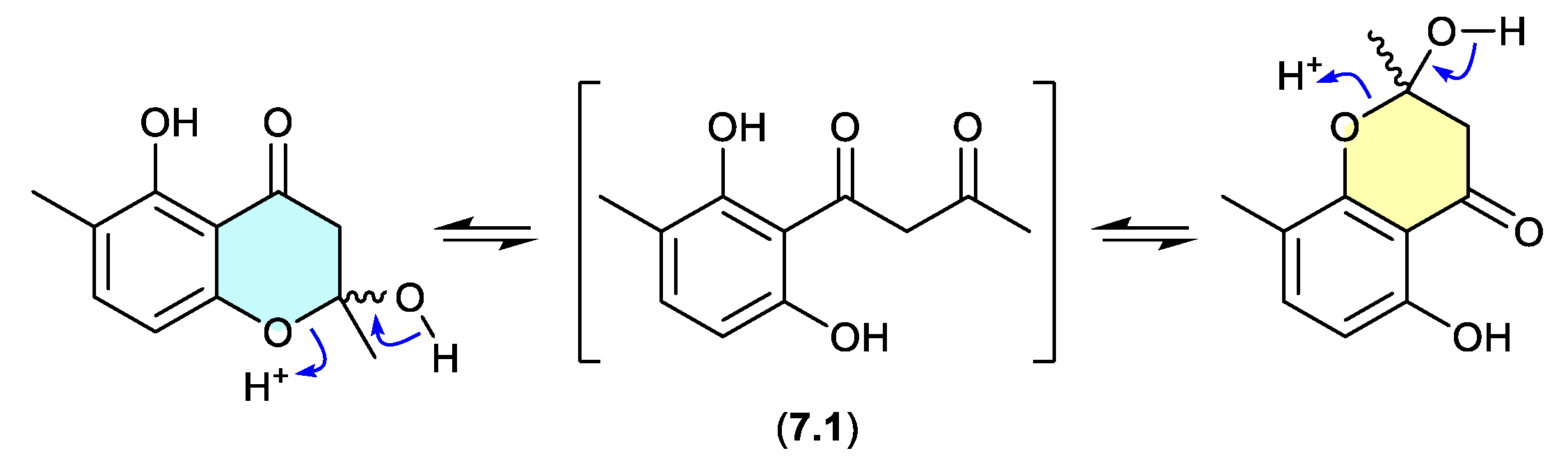
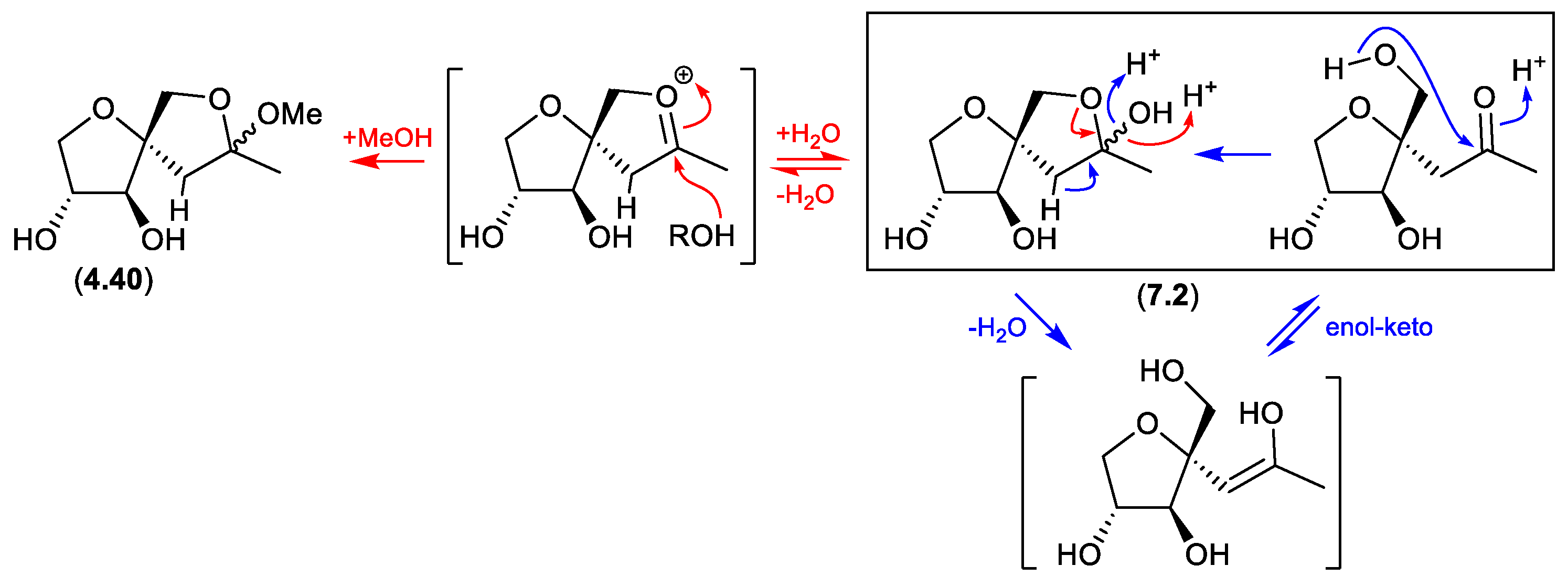
sphydrofuran (Figure 7.2)
The Streptomyces metabolite sphydrofuran (4.40) exists as an equilibrating mixture of ketal epimers and the ring opened ketone (7.2), which readily convert in MeOH to a corresponding epimeric mixture of methyl ketals (4.40).[87]
Figure 7.
2.
8. Trans-Esterification
During extraction, fractionation, handling and/or storage some natural product esters undergo trans-esterification. While trans-esterifications are not especially common, it is nevertheless a potential pathway to artifact formation.
kipukasins (Figure 8.1)
The esterified nucleosides, kipukasins M (8.1) and N (8.2), from the marine-derived fungus Aspergillus versicolor, exhibited a facile trans-esterification to an equilibrating mixture of regioisomers when stored for 4 h in HPLC solvents.[119]
Figure 8.
1.
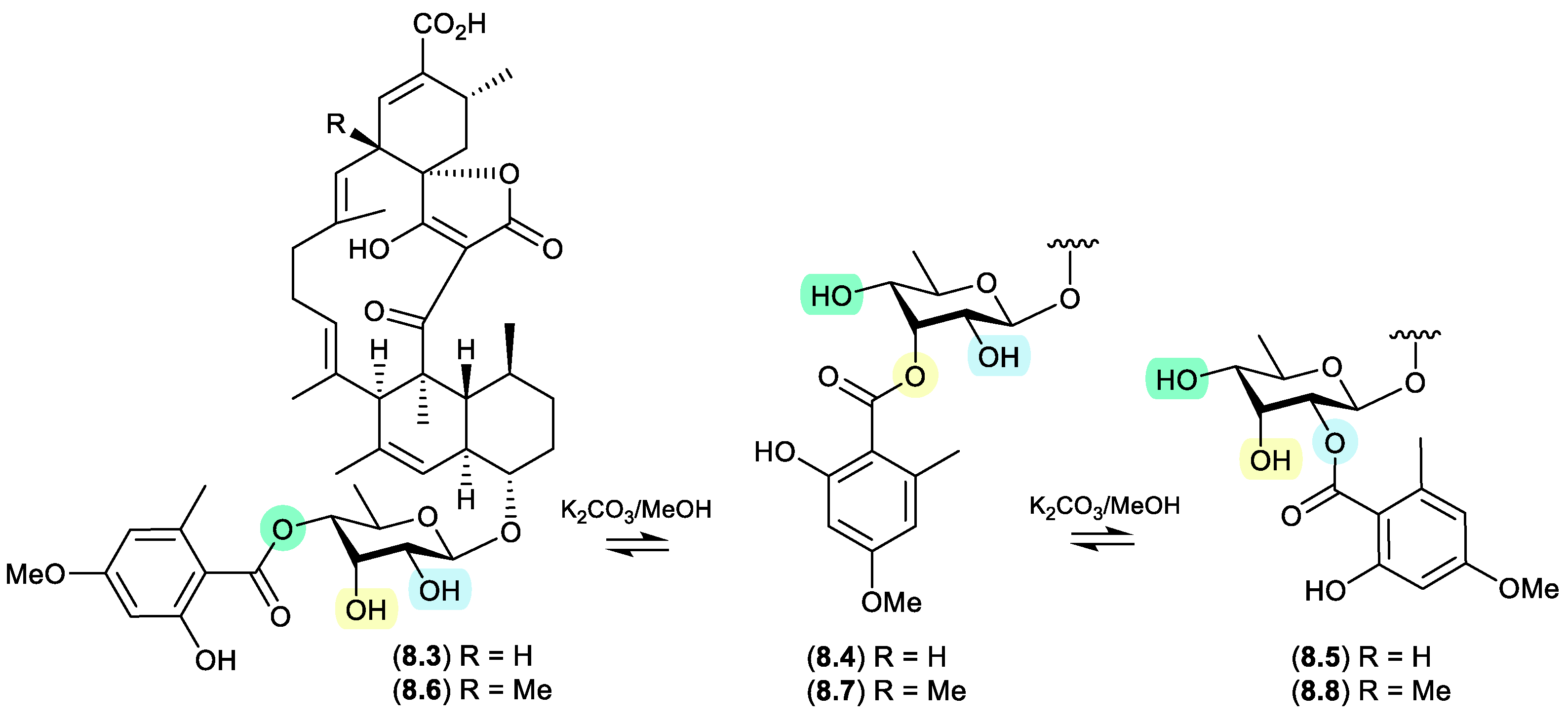
glenthmycins (Figure 8.2)
On exposure to mild base (K2CO3 in MeOH), a trans-esterification was observed between Streptomyces sp. CMB-PB041 derived glenthmycins C (8.3), F (8.4) and H (8.5), and between glenthmycins E (8.6), N (8.7) and M (8.8).[120]
Figure 8.
2.
amaurones (Figures 8.3 and 8.4)
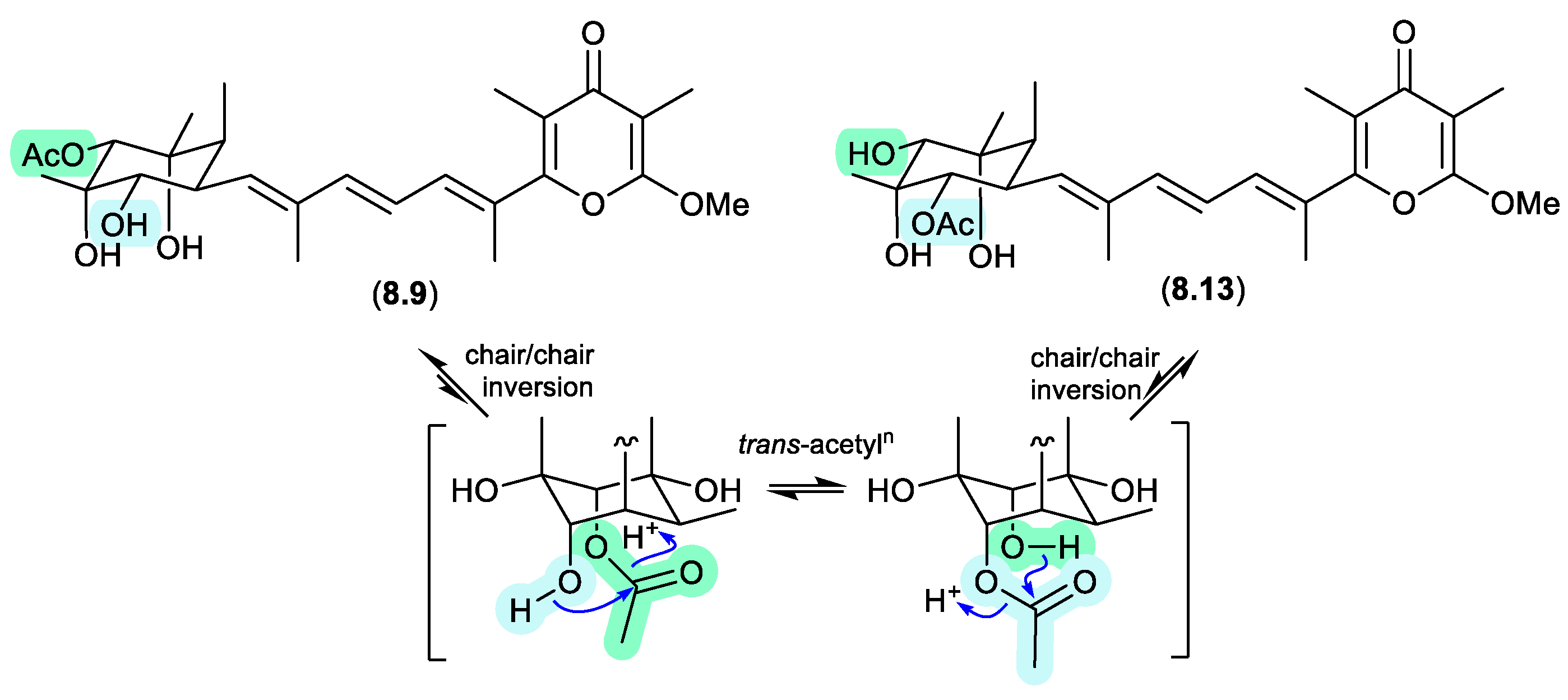
The Australian mullet fish gastrointestinal tract-derived fungus Amauroascus sp. CMB-F713 yielded an array of polyketide pyrones with unprecedented carbon skeletons, including amaurones A–C (8.9–8.11), E (8.12) and J (8.13).[121] Drying a solution of the orthoacetate 8.12 under nitrogen (at 40 °C overnight) resulted in hydrolysis to a 3:2:1 mixture with the monoacetates 8.10 and 8.11, which on further heating (4 d) returned a 1:1 mixture of 8.12 and 8.10, favouring the regioisomer with an equatorially (rather than axially) disposed acetate moiety. A comparable transformation was observed between 8.9 and 8.13, necessitating chair-chair ring inversion to bring the hydroxy and acetate moieties into a suitable 1,3-diaxial arrangement, where on this occasion both regio-isomers featured equatorially disposed acetate groups.
Figure 8.
3.
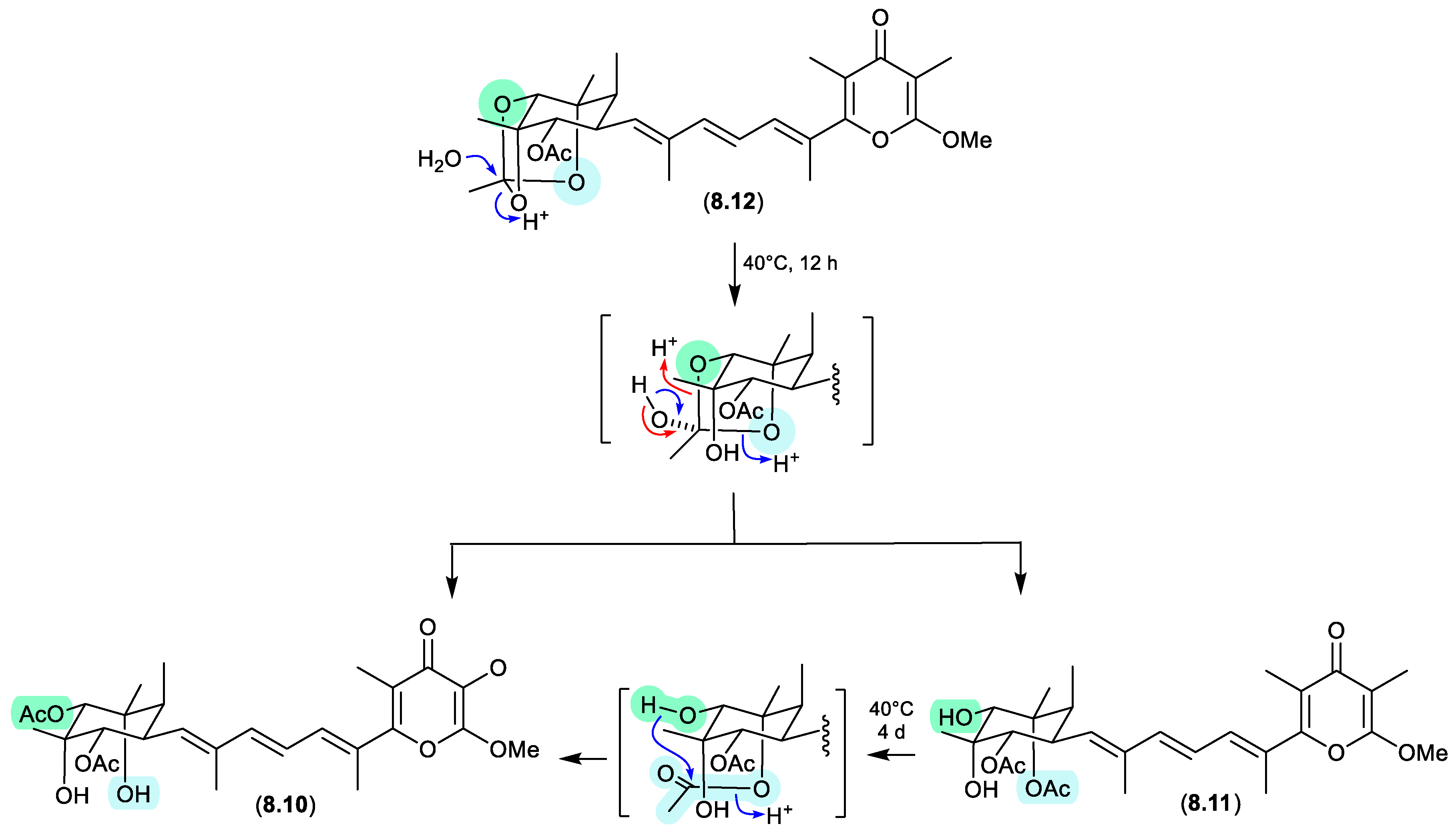
Figure 8.
4.
9. Epimerization
In selected natural products, non-acetal chiral centres can undergo epimerisation, providing insights into the chemical reactivity of unusual functionality and/or scaffolds.
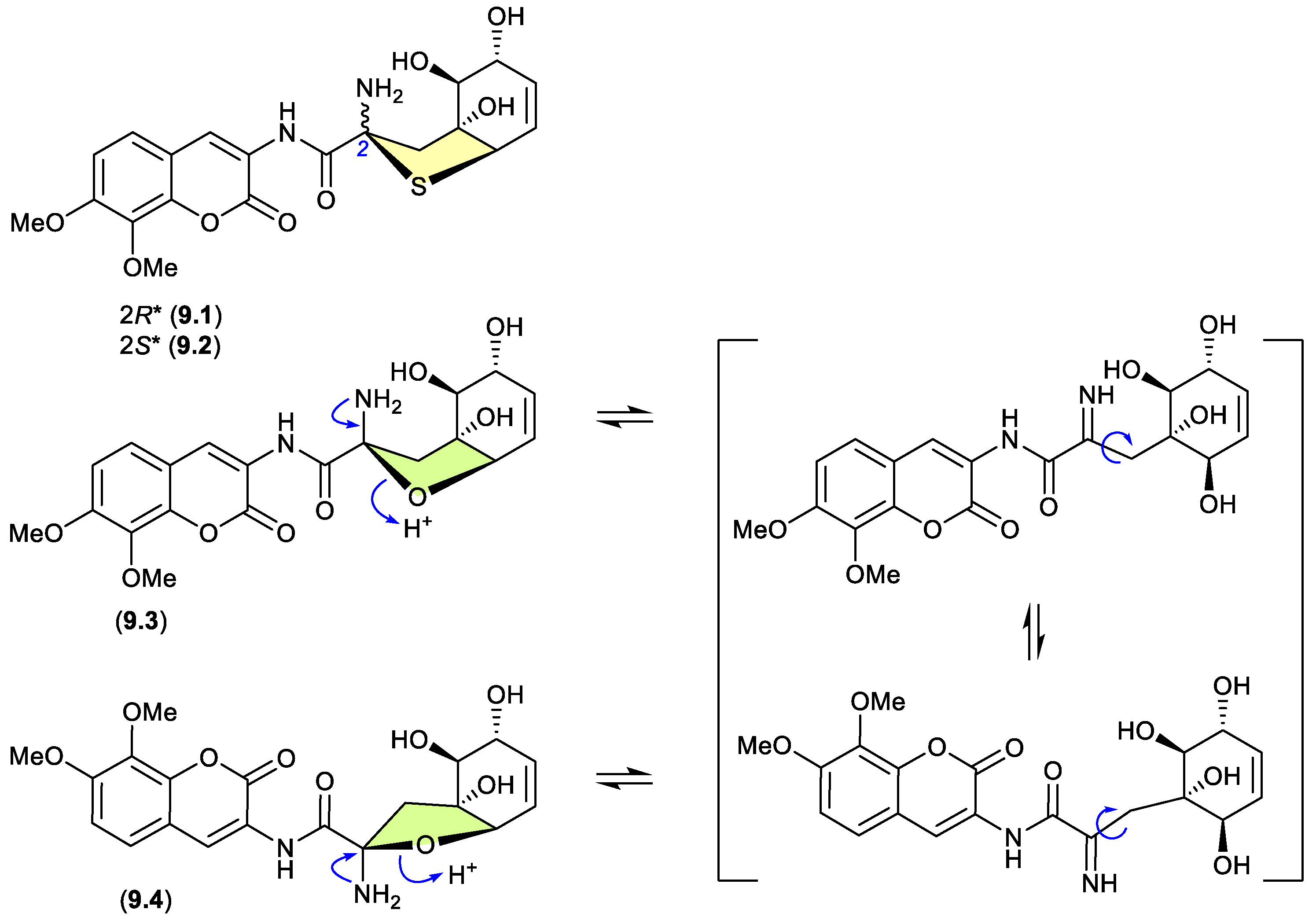
aspergillazines (Figure 9.1)
Indicative of how even slight changes in functionalisation can have a dramatic effect on chemical reactivity, the epimeric and highly modified Aspergillus thiophane dipeptides, aspergillazines B (9.1) and C (9.2), did not undergo equilibration/epimerization on handling, whereas the corresponding tetrahydrofuran analogues, aspergillazines D (9.3) and E (9.4), underwent rapid equilibration/epimerization to a 1:0.85 mixture (most likely via a ring opened imine intermediate).[122]
Figure 9.
1.
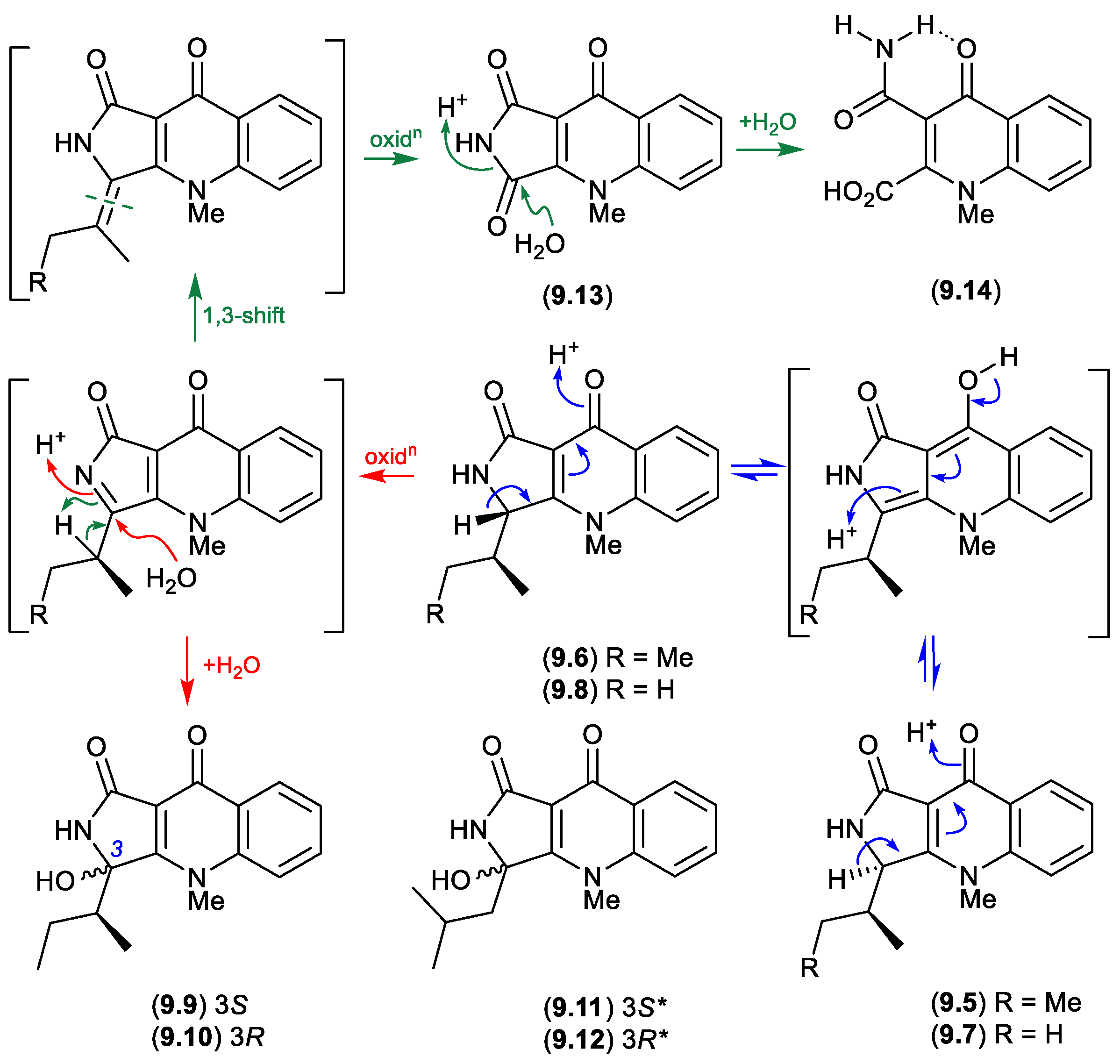
quinolactacins (Figures 9.2 and 9.3)
Planar structures for the tumor necrosis factor (TNF) inhibitory quinolactacins A–C were first reported in 2000, from an undescribed Penicillium sp.[123] A subsequent 2001 account revealed that quinolactacin A from Penicillium citrinum 90684 was a mixture of the epimers A1 (9.5) and A2 (9.6), with the latter being 14-times more potent at inhibiting acetylcholinesterase. [124] Significantly, the stereochemical difference between 9.5 and 9.6 was asserted at that time (incorrectly) to be about the sidechain 2°-methyl, suggestive of biosynthetic incorporation of Ile and allo-Ile. A 2001 biomimetic synthesis of quinolactacin B failed to disclose that it too existed as a mixture of epimers B1 (9.7) and B2 (9.8),[125] while a 2003 total synthesis confirmed the structure for quinolactacin B2 (9.8) but incorrectly affirmed the earlier incorrect stereochemical assignment for A1 (9.5) and A2 (9.6).[126]
A 2006 investigation into the Australian isolate Penicillium citrinum MST-F10130 revealed the true complexity of quinolactacin chemistry and stereochemistry, reporting the known quinolactacins A2 (9.6), B2 (9.8), C2 (9.9) and A1 (9.5), as well as the new quinolactacins B1 (9.7), C1 (9.10), D2 (9.11) and D1 (9.12), along with the novel analogues, quinolonimide (9.13) and quinolonic acid (9.14).[127] Significantly, this study revealed that under mild handling conditions; (i) A2 (9.6) epimerises to a racemic mixture with A1 (9.5), as does B2 (9.8) with (9.7); (ii) A2 (9.6) undergoes a facile oxidation and non-stereospecific H2O addition to yield C2 (9.9) and C1 (9.10), and (iii) undergoes oxidation to yield quinolonimide (9.13); and (iv) 9.13 undergoes rapid and regiospecific hydrolysis to yield quinolonic acid (9.14). These ease with which these epimerisations occurred suggests one of each epimer (i.e. 9.6/9.8 or 9.5/9.7) may be a natural product, and their respective epimer a handling artifact. It also seems likely that D1/D2 (9.11/9.12) are oxidative hydrolysis products of a hitherto undetected quinolactacin bearing a leucyl sidechain (cryptic — see Section 10).
Figure 9.
2.
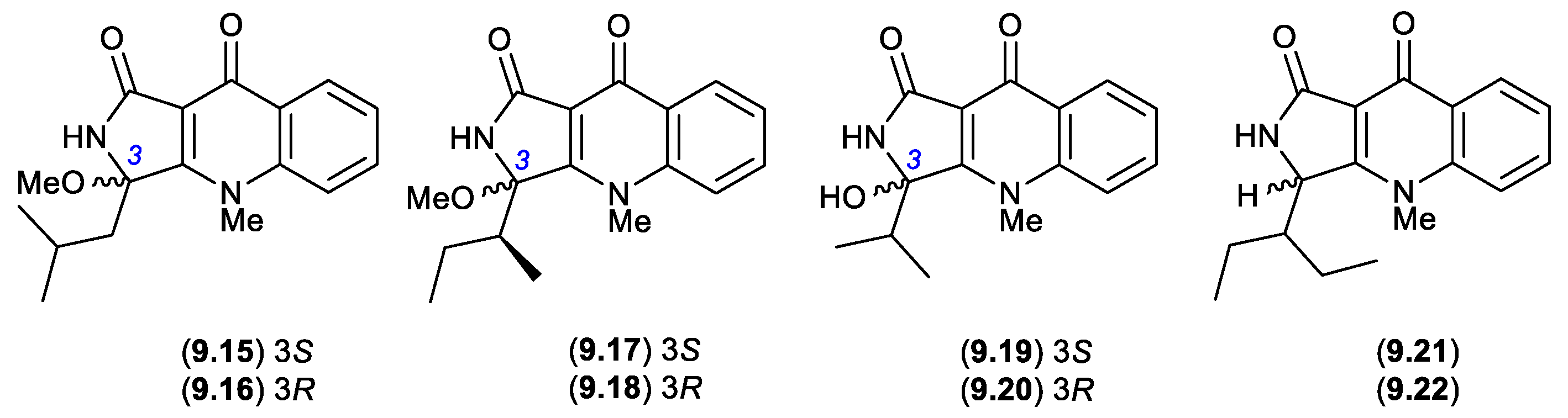
In more recent studies, the known epimeric pairs of quinolactacins A2/A1 (9.6/9.5), B2/B1 (9.8/9.7) and C2/C1 (9.9/9.10), along with the purportedly new epimeric pairs quinolactacins E1/E2 (9.15/9.16), F1/F2 (9.17/9.18) and G1/G2 (9.19/9.20), were reported from a sponge-derived Penicillium sp. SCSIO 41303.[128] Based on the chemical reactivity/artifact studies outlined above, and given the use of silica gel chromatography with a MeOH/CH2Cl2 eluant, it seems highly probable that; (i) E1/E2 (9.15/9.16) are methanolysis artifacts of the hitherto undetected quinolactacin (cryptic — see Section 10) that is the speculated natural product source of D1/D2 (9.11/9.12); (ii) F1/F2 (9.17/9.18) are methanolysis artifacts of the co-metabolites A2/A1 (9.6/9.5); and (iii) G1/G2 (9.19/9.20) are hydrolysis artifacts of the co-metabolites B2/B1 (9.8/9.7). In other words, all the purported new natural products can be rationalised as artifacts.
Similarly, a recent account of an X-ray structure analysis and total synthesis of quinolactacin-H, from a marine-derived Penicillium sp. ENP701, revealed it to be a 1:1 magnesium salt of the epimers (R)-(+)-quinolactacin-H (9.21) and (S)-(–)-quinolactacin-H (9.22).[129] As 9.21 and 9.22 are capable of ready equilibration to a mixture of C-3 epimers, it is interesting to speculate whether one or other, or both are natural products. Most recently, in 2023 quinolactacins C1 (9.10) and C2 (9.9) were reported (incorrectly as new natural products) from the mangrove-derived Penicillium citrinum YX-002.[130]
Figure 9.
3.
10. Cryptic Natural Products
While modern natural products research benefits from highly sensitive and informative analytical technologies that make it possible to detect the complex array of chemistry found in natural extracts, thorough understanding still demands (for the most part) that natural products be extracted, purified, characterised, identified, and their properties evaluated experimentally. This can prove very challenging for that subset of highly chemically reactive natural products that do not survive extraction, and more often than not these unknowns remain unknown. This can be a lost opportunity, as knowledge of these compounds can be remarkably instructive. The following three case studies illustrate the value of pushing the limits to better understand and appreciate cryptic natural products.
N-amino-l-proline methyl ester/prolinimines (Figure 10.1)
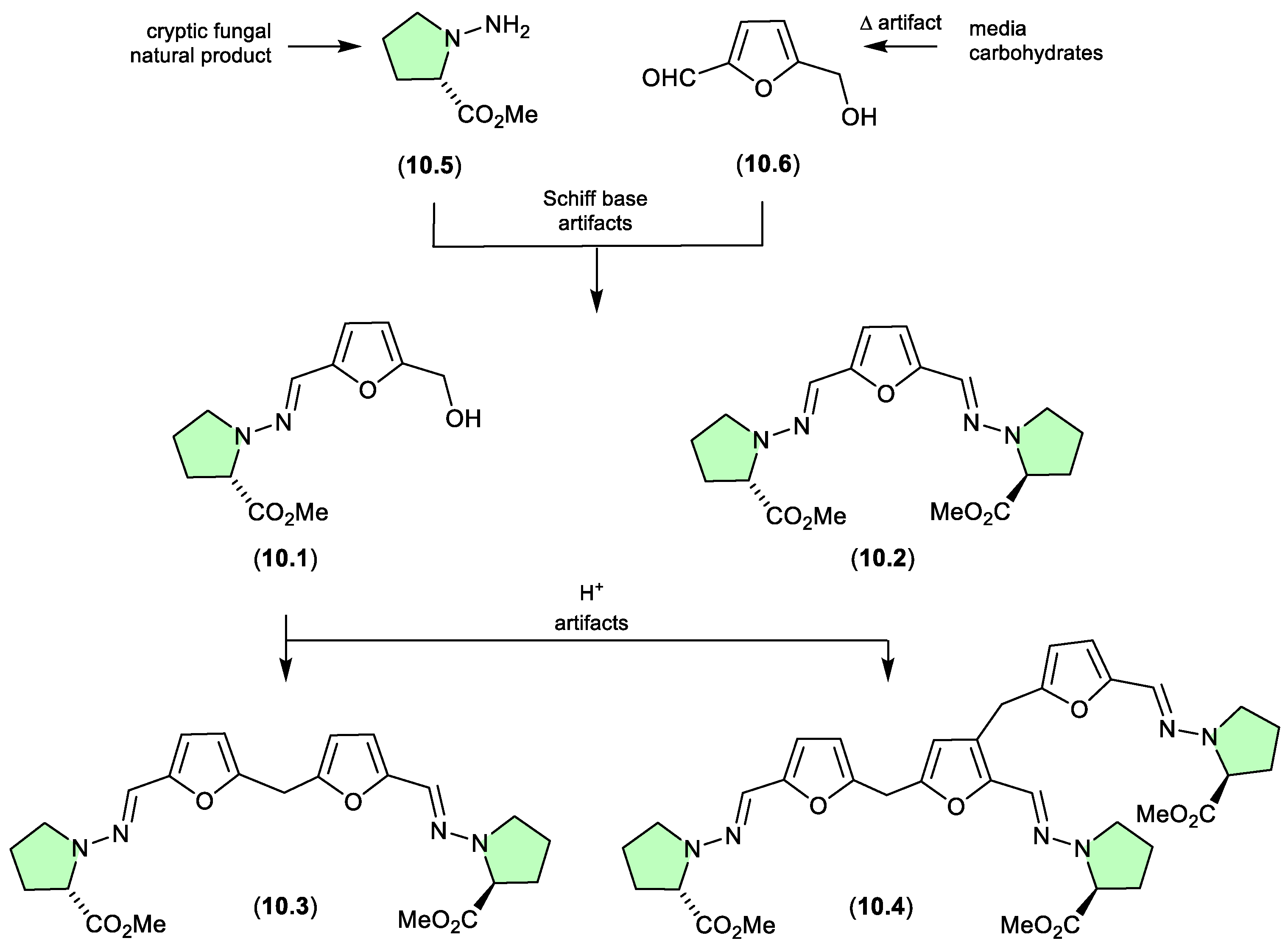
The Australian fish gastrointestinal tract-derived fungus, Trichoderma sp. CMB-F563, yielded a series of unprecedented Schiff bases, prolinimines A–D (10.1–10.4).[131] Of note, although 10.2–10.4 were isolated, characterised and identified from a solvent extract of the fungal culture, 10.1 was not. Nevertheless, it was speculated that 10.1 should be present as a logical biosynthetic precursor of 10.2–10.4 and following chemical analysis of fresh extracts it was detected. Moreover, it was demonstrated that during isolation and handling 10.1 underwent rapid and quantitative acid-mediated transformation to the 10.3 and 10.4. The structures for 10.1–10.4 were confirmed by detailed spectroscopic analysis and a convergent biomimetic total synthesis starting with N-amino-l-proline methyl ester (10.5) and 5-hydroxymethylfurfural (10.6). These observations revealed 10.1 as a cryptic natural product, with 10.3 and 10.4 designated as artifacts. However, in a surprising twist, a follow-up study aimed at better understanding the biosynthetic origins of the prolinimines, revealed that all the prolinimines A–D (10.1–10.4) were artifacts of the “real” cryptic fungal natural product N-amino-l-proline methyl ester (10.5).[132] On solvent extraction of the fungal culture, it was shown that 10.5 was released from the fungal mycelia at which time it came into contact with the media constituent 10.6 — a thermolysis artifact produced during autoclaving of carbohydrate rich media – and underwent rapid and quantitative transformation to the Schiff bases 10.1 and 10.2. As noted above, during isolation and handling 10.1 dimerised and trimerised to 10.3 and 10.4, respectively. The use of culture media depleted in carbohydrates (and hence 10.6) facilitated the detection and isolation of 10.5. It is interesting to speculate whether the cryptic small molecular weight, water soluble 10.5 would have been discovered without the fortuitous choice of a culture media rich in the thermolysis artifact 10.6.
Figure 10.
1.
N-amino-anthranilic acid/penipacids (Figure 10.2–10.3)
Re-consideration of the penipacids A–E, first reported as the acyclic amidines 10.7–10.11 from the South China deep sea sediment-derived fungus Penicillium paneum SD-44,[133] prompted a total synthesis structure revision as the hydrazones 10.12–10.16.[134] This revision proposed that penipacids A (10.12) and B (10.13) were Schiff base artifacts of the cryptic (undetected) natural product N-amino-anthranilic acid (10.17) with diacetone alcohol (10.18) and its corresponding methyl ether 10.19, likely induced by excessive exposure to acetone and methanol under acidic handling conditions. Likewise, penipacids C (10.14) and D (10.15) were viewed Schiff base artifacts of 10.17 and the media constituent pyruvic acid (10.20) and is methyl ester 10.21, while penipacid E (10.16) was viewed as a Schiff base artifact of 10.17 and the media constituent furfural (10.22).
Figure 10.
2.
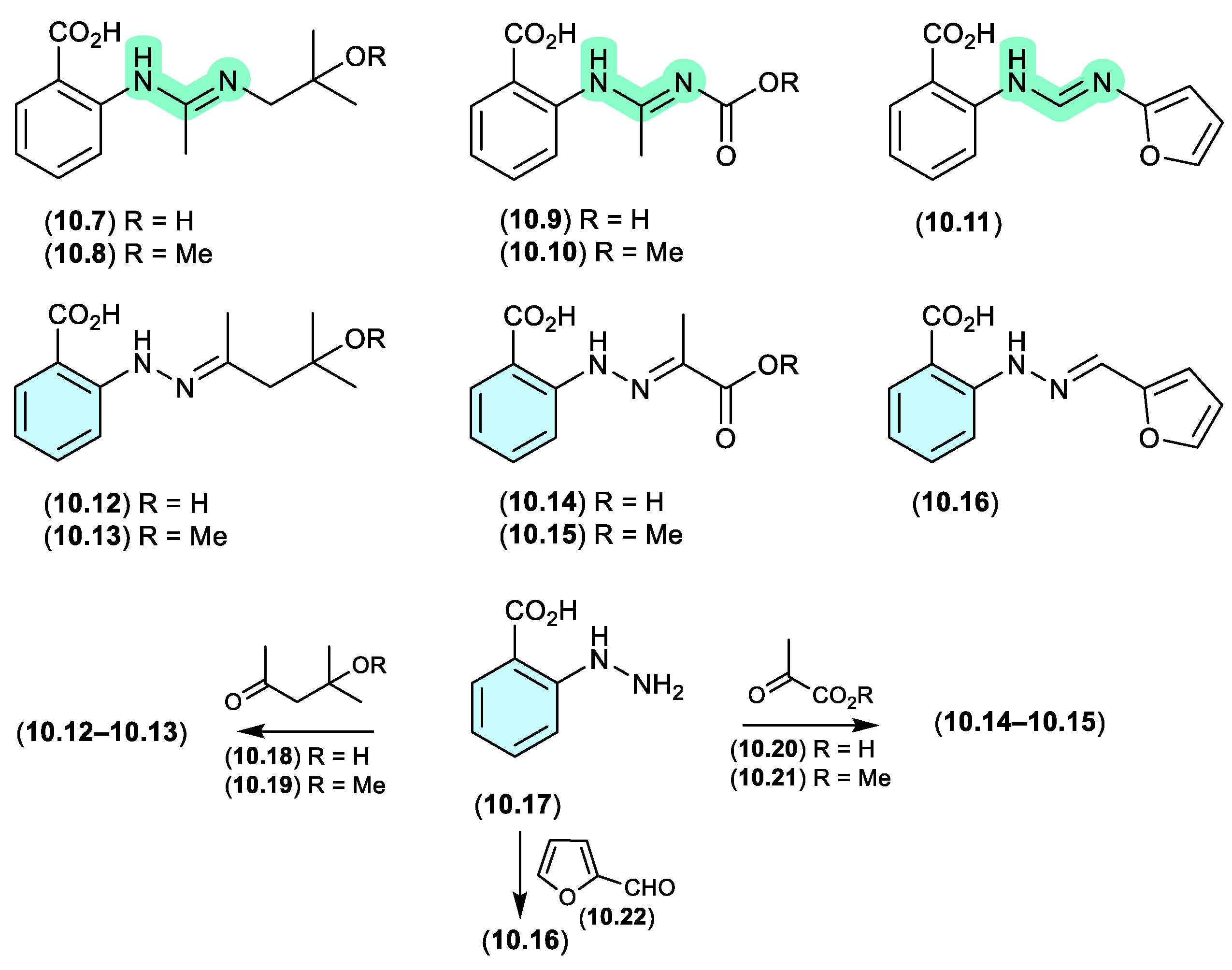
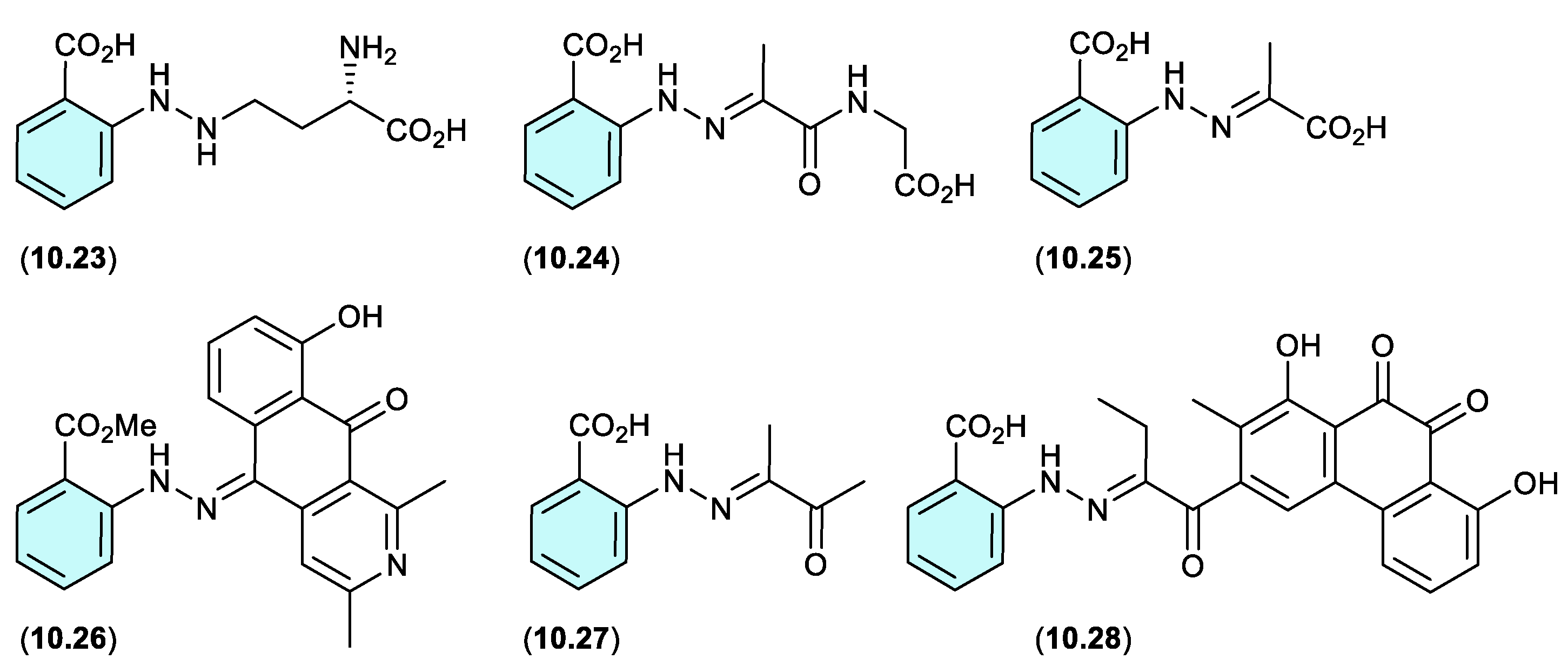
A review of the natural products literature revealed other instances of potential artifacts based on the purported cryptic natural product 10.17. These include; (i) the γ-glutamylphenylhydrazine anthglutin (10.23) as a potential conjugate of 10.17 and L-homoserine, from a Japanese fungus Penicillium oxalicum SANK 10477;[135] (ii) the phenylhydrazone farylhydrazones A (10.24) and B (10.25) from a Tibetan Cordyceps-colonising fungus Isaria farinosa – as potential Schiff base adducts of 10.17 and pyruvylglycine and pyruvic acid, respectively.[136] Note that the revised structure for penipacid C (10.14) is now identical to farylhydrazone B (10.25); (iii) the 2-azoquinone-phenylhydrazine katorazone (10.26) from a Japanese soil-derived Streptomyces sp. IFM 11299 — a potential Schiff base adduct of 10.17 and the known fungal anthraquinone utahmycin A, which was coincidentally reported to be a co-metabolite with 10.26.[137] (iv) farylhydrazone C (10.27), along with farylhydrazone B (10.25), from an Antarctic soil-derived Penicillium sp. HDN14-431 — as potential Schiff base adduct of 10.17 with pyruvic acid and dimethylglyoxal, respectively, where the latter is known to be produced during thermal processing of carbohydrate rich foods, and as such could be a media constituent induced during autoclaving;[138] and (v) the aromatic polyketide murayaquinone C (10.28) from the ant gut-derived Streptomyces sp. NA4286 — a potential Schiff base adduct of 10.17 with a suitably substituted anthraquinone co-metabolite.[139]
Figure 10.
3.
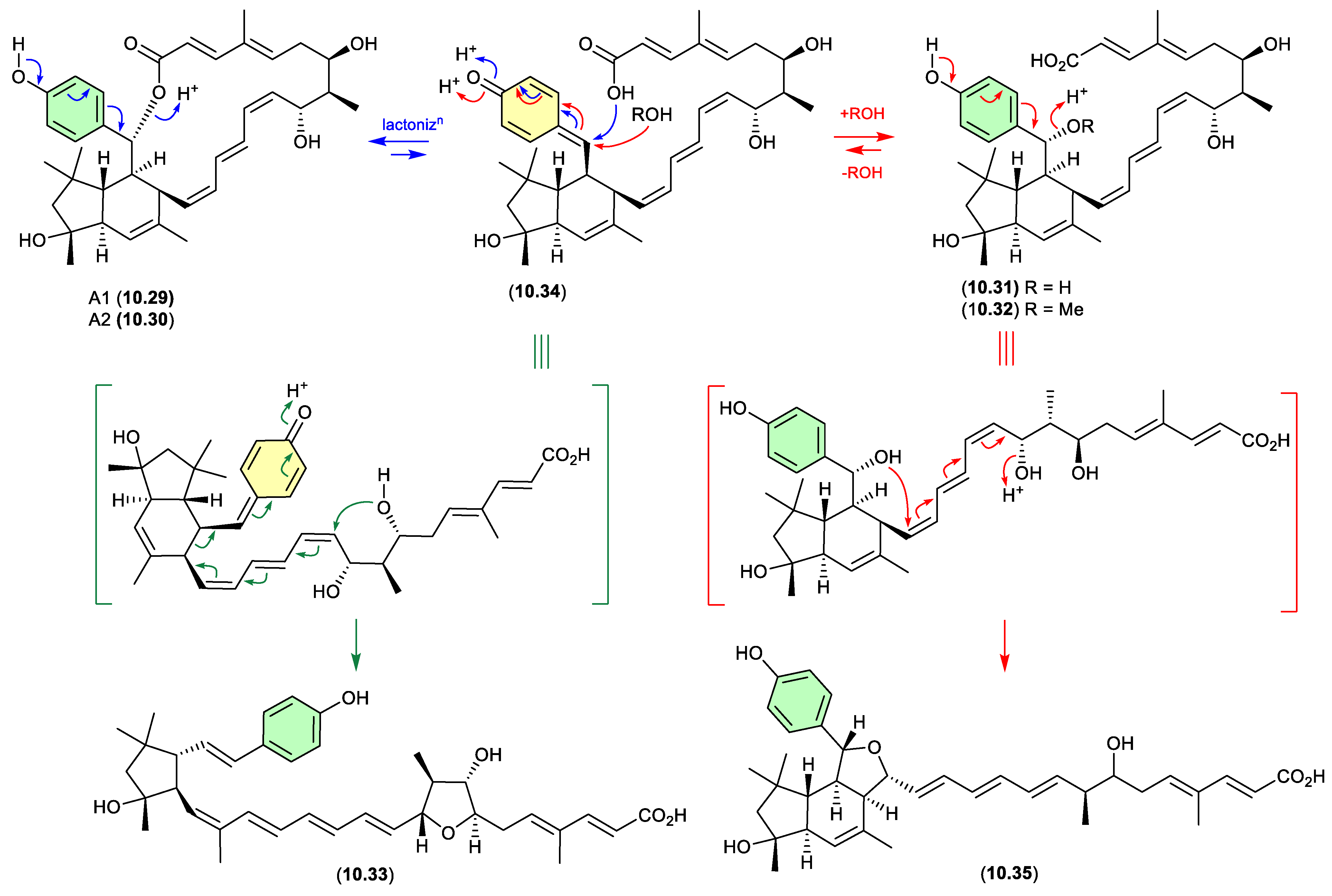
elansolids (Figure 10.4)
In 2011 Müller et al reported on the atropisomeric elansolids A1 (10.29) and A2 (10.30), and the seco analogues, elansolids B1 (10.31) and B2 (10.32), as the first polyketide natural products from the non-myxobacterial gliding bacteria Chitinophaga sancti.[140] Interestingly, where A2 (10.30) exhibited promising antibacterial activity, its atropisomer A1 (10.29) was less active. Supportive of structure assignments, on storage in DMSO-d6 at r.t. A2 (10.30) transformed to A1 (10.29), while on exposure to a 0.1 M NaOH in MeOH/H2O both A2 (10.30) and A1 (10.29) underwent ring opening to B2 (10.32). This latter observation prompted speculation that B2 (10.32) was an artifact of A1 (10.29) and A2 (10.30) brought about by solvolysis during isolation and handling. In a follow-up study, these authors observed that elansolid production was cultivation condition dependent, isolated and identified the new seco analogue elansolid D1 (10.33) and revealed that B1 (10.31) and B2 (10.32) were artifacts induced by the addition of H2O and MeOH, respectively, to a common chemically reactive cryptic metabolite. [141] With exquisite attention to careful isolation and handling, the cryptic metabolite was successfully isolated and identified as the quinone methide elansolid A3 (10.34). Moreover, when exposed to a range of solvents under neutral or mildly acidic or basic conditions, A3 (10.34) could transform into all the other elansolids (A1, A2, B1, B2 and D1). Equally, under mildly basic conditions A1 (10.29) and A2 (10.30) could transform to A3 (10.34). These observations place the chemically reactive quinone methide elansolid A3 (10.34) as the pivotal biosynthetic intermediate, and potentially the sole natural product — with other elansolids being artifacts. In a separate study, Piel et al reported elansolid B1 (10.31) and the new elansolid D2 (10.35) as the sole natural products from another heterotrophic gliding bacterium, C. pinensis DSM 2588.[142] However, Müller et all demonstrated that on exposure to MeCN/H2O (1% TFA) — the HPLC conditions used for the isolation of B1 (10.31) and D2 (10.35) from C. pinensis — A3 (10.34) transformed to both D1 (10.33) and D2 (10.35).
Figure 10.
4.
11. Biotransformation
The biological properties of natural products are often determined by applying different concentrations to live cells (e.g. mammalian, bacterial, fungal) and measuring quantifiable changes in cell morphology/viability and/or reporter outputs. Comparison of bioassay data for related natural products reveals SAR, which in turn define pharmacophores and inform our understanding of mechanisms-of-action. The veracity of any bioassay data relies on the assumption that natural products being tested are stable to assay conditions, and do not convert to other molecules with greater, lesser or even completely different biological properties. While this assumption largely holds true, it is not universally the case. The following case studies illustrate how chemically reactive natural products undergo biotransformation during bioassay.
abyssomicins (Figure 11.1)
Abyssomicin J (11.1), isolated from the South China Sea deep-sea sediment-derived actinomycete, Verrucosispora sp. MS100128, was reported to exhibit activity against bovine tuberculosis bacillus Mycobacterium bovis (BCG).[143] This anti-tubercular activity was unexpected, as earlier SAR studies on the abyssomicin family had pointed to a pharmacophore dependent on an α,β-unsaturated ketone (i.e. a Michael acceptor).[144] To account for the anti-tubercular properties of 11.1 is was speculated to be a pro-drug with in situ biotransformation releasing the true anti-tubercular agent. To test this hypothesis, when exposed to a MeCN/H2O solution of the oxidising reagent Oxone, 11.1 was reported to undergo rapid oxidation to the sulfoxide 11.2, which in turn underwent quantitative oxidation to the sulfone 11.3. Significantly, 11.3 was “primed” for multiple, facile and sequential reverse-Michael additions, and on handling yielded the α,β-unsaturated ketone atropisomers, abyssomicin C (11.4) and atrop-abyssomicin C (11.5), with 11.5 being a particularly potent Michael acceptor. Building on these observations, when exposed to BCG cells for only 1 h, 11.1 was reported to undergo rapid (enzymatic) biotransformation to 11.4 and 11.5. Hence, although not initially apparent, the anti-tubercular activity of 11.1 proved to be dependent on an otherwise cryptic chemical reactivity, and propensity for in situ cellular biotransformation.
Figure 11.
1.
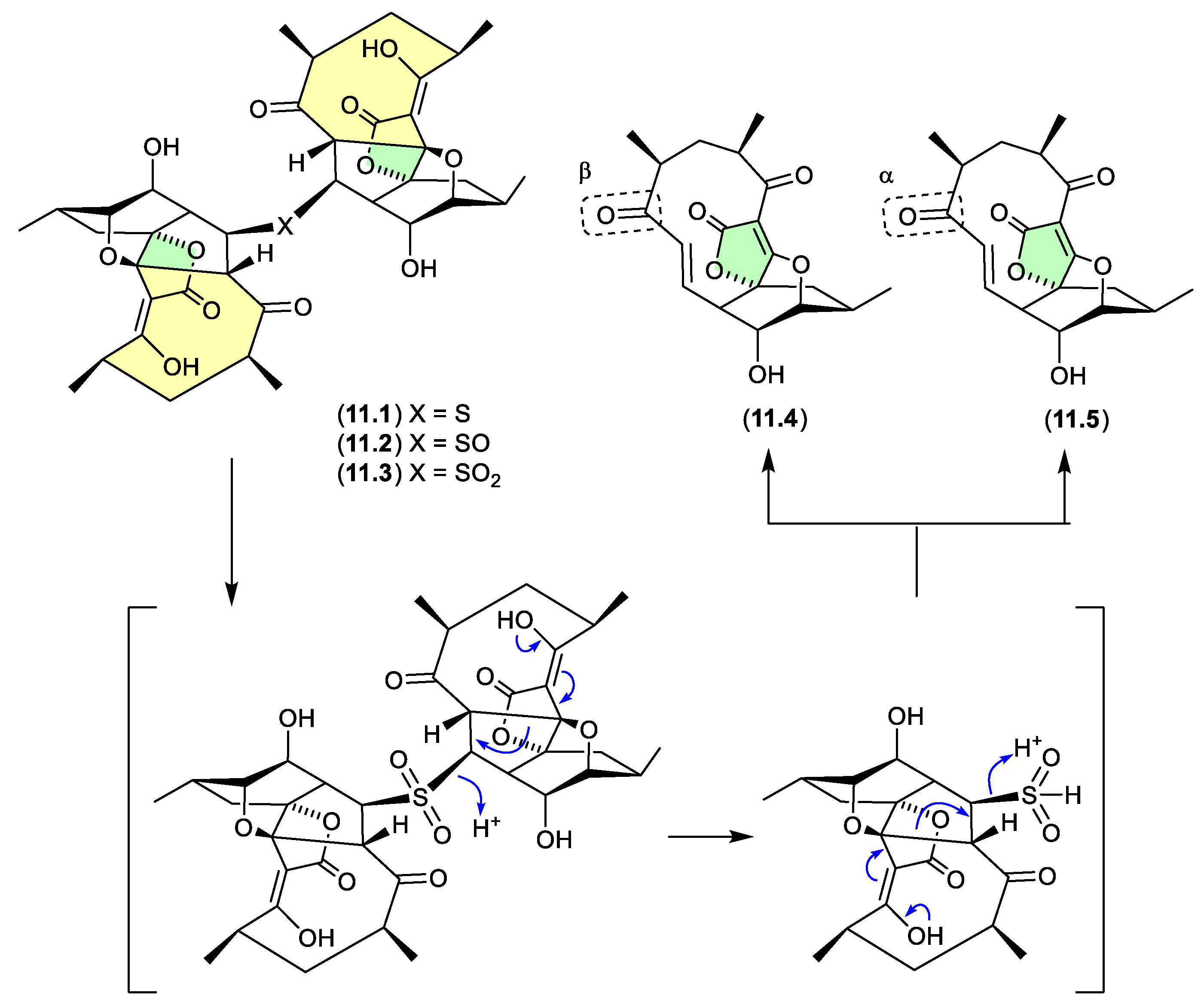
roseopurpurins (Figure 11.2)
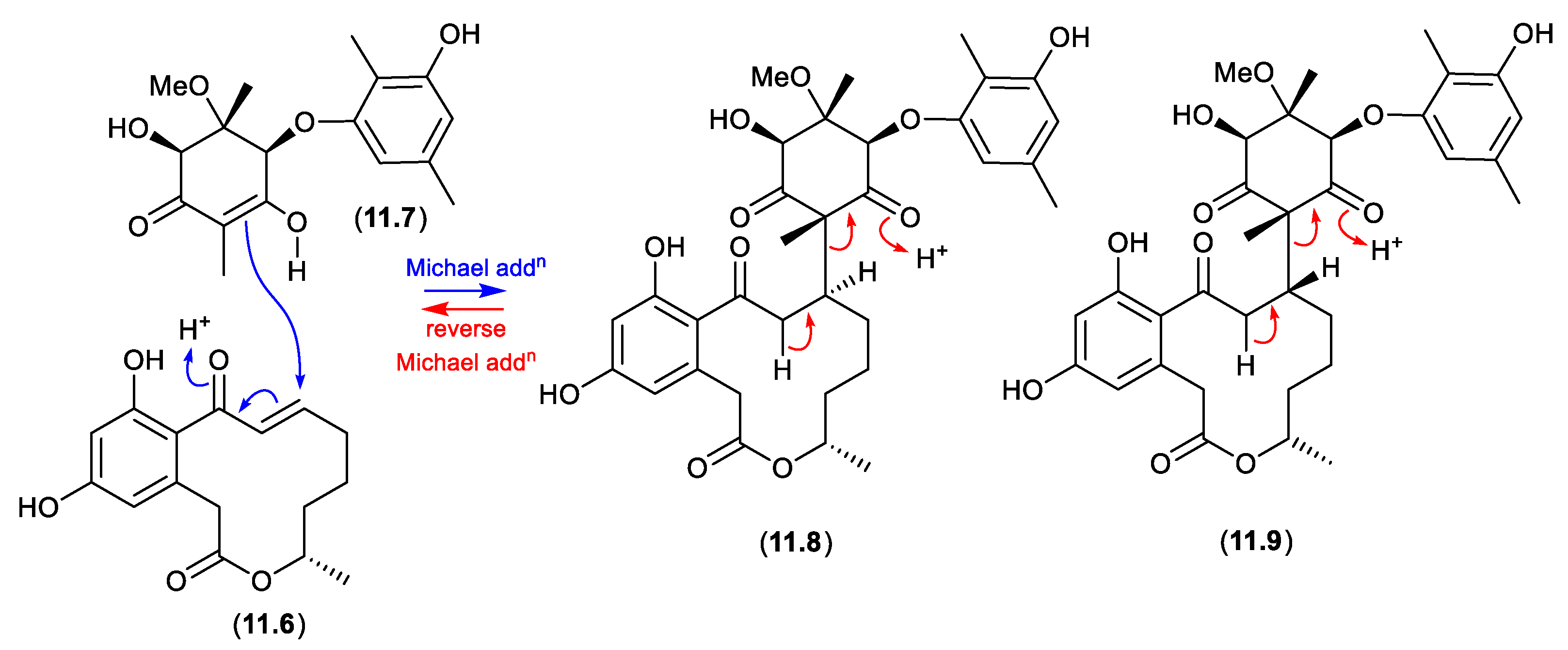
The Australian estuarine marine mud-derived fungus, Penicillium roseopurpureum CMB-MF038, yielded an array of polyketides, including the known 15S-a,b-dehydrocurvularin (11.6) and aculeatusquinone C (11.7), and new roseopurpurins H (11.8) and I (11.9).[145] Although 11.8 and 11.9 are putative Michael adducts of 11.6 and 11.7, as they were detected in the crude unfractionated culture extract it was concluded they were natural products — albeit, ones that could have formed in situ by a non-enzymatic Michael addition. More intriguing, while 11.6 exhibited low mM activity (IC50) against multiple human cancer cell lines, as would be expected of a Michael acceptor, the same was also true of the supposedly inactivated Michael adducts 11.8 and 11.9. This prompted speculation that the latter were pro-drugs, prone to reverse Michael addition leading to 11.6. Indeed, chemical analysis revealed that a 2-6 h exposure to human colon (SW620) and lung (NCI-H460) carcinoma cells, was sufficient for quantitative biotransformation of 11.8 and 11.9, to 11.6. Hence the cytotoxic activity of 11.8 and 11.9 proved to be dependent on an otherwise cryptic chemical reactivity, and propensity for in situ biotransformation.
Figure 11.
2.
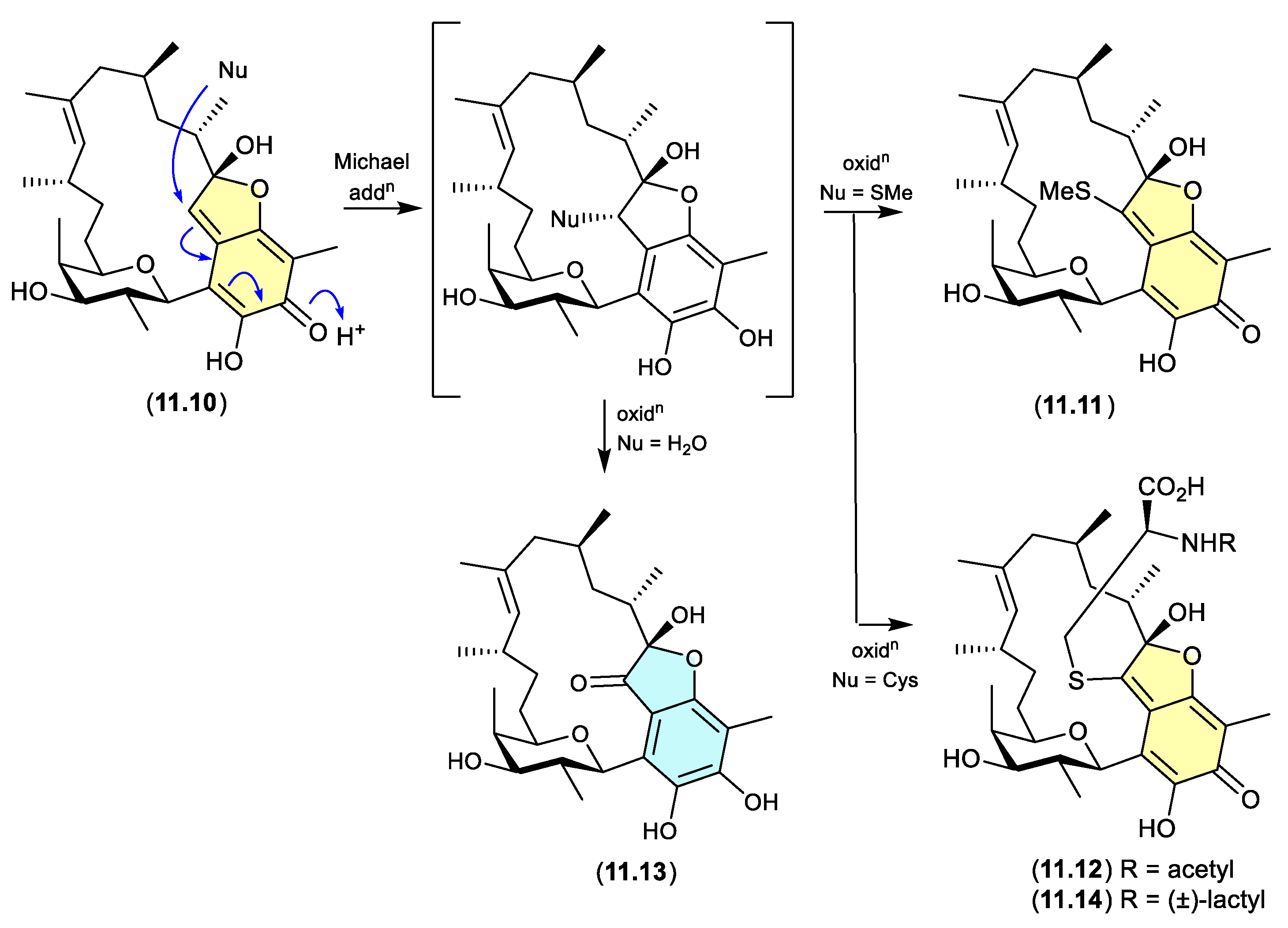
kendomycin/goondomycins (Figure 11.3)
The Australian pasture soil-derived Streptomyces sp. S4S-00052A05 was reported to yield a wide array of polyketides, including the known carbocyclic ansa-polyketide kendomycin (11.10), and new goondomycins A (11.11), B (11.12), E (11.13) and H (11.14) — with all detected in the unfractionated culture extract, confirming their status as natural products. [146] Notwithstanding, overnight exposure of the otherwise chemically stable Michael acceptor 11.10 to Staphylococcus aureus cells, resulted in biotransformation to 11.11–11.14. These biotransformations are likely proceed via Michael additions with subsequent oxidations, and further highlight the challenges associated with acquiring meaningful SAR data on chemically reactive natural products.
Figure 11.
3.
12. Conclusions
As illustrated throughout this review, an awareness of and willingness to both recognise and understand natural product artifacts can open valuable new windows into the innate chemical reactivity of natural product chemical space.
12.1. Distinction Between a Natural Product and an Artifact
To make the case for valuing artifacts, it’s helpful to first agree on a definition of a natural product, versus an artifact. Curiously, although the term “natural product” defines an entire field of science and is in common use across multiple disciplines, it is difficult to find a clear and unambiguous definition of what is a natural product. For the most part of the last century, the chemistry of life was largely viewed as a binary divide between primary metabolites (essential for life), and secondary metabolites or natural products (important to, but not essential for life). Over time, as more knowledge became available, this definition has not fared well. Putting aside the limitations of the primary versus secondary metabolism duopoly, if we accept for the moment that natural products are secondary metabolites, we can move on to the following definitions.
A natural product – is a chemical (secondary metabolite!?) that originates from and can be detected in a fresh unfractionated extract of a source organism – provided the process of extraction and/or detection does not initiate a chemical transformation that is solely responsible for producing the chemical.
The latter “proviso” seeks to account for those cryptic highly reactive natural products that rapidly (sometimes quantitatively) transform to artifacts during extraction (see Section 10), such that the natural product remains cryptic, and the artifact is detected in the extract and erroneously attributed to natural product status.
An artifact – is a natural product that has undergone a chemical transformation during extraction, handling, storage and/or analysis.
As a codicil, natural product and artifact status are not necessarily mutually exclusive. This may seem counter-intuitive but consider for example a compound that meets the definition of a natural product but that can also be obtained by the transformation of a co-metabolite during extraction and handling. This duality reflects the situation where chemical reactivity provides both a path for (i) non-enzyme mediated natural product formation and diversification in situ within the source organism and (ii) artifact formation post extraction.
12.2. How to Recognise the Presence of Artifacts
In the first instance, it’s enough to simply be aware of the various types of artifacts and mechanisms by which they can be formed and be open to the possibility that any isolated compound may (may not) be an artifact. For example, the isolation of acetonides or methyl ethers/esters should immediately raise suspicion, especially if acetone or MeOH was used during handling. Likewise with the presence of an N-chloromethyl from a CH2Cl2 extract, or a dimethyl acetal from a MeOH extract. Depending on the nature of the artifacts and the extraction and isolation conditions, another strategy for recognising the presence of artifacts is to repeat the process using different conditions. For example, if acetone, MeOH or CH2Cl2 extractions are suspected of generating artifacts, repeat the extraction with less reactive solvents (e.g. EtOAc, MeCN).
Another useful strategy is to reflect on the likely biosynthetic origin of any isolated compound, as this may reveal component parts, or structural features, that are not obviously natural and may hint at artifact formation. In addition, the presence of epimers or enantiomers can be indicators of artifacts; however, it’s worth cautioning that such stereo-complexity can also be natural.
At an experimental level it is also good practice to carefully monitor the fractionation process, to detect changes that may inadvertently take place. For example, HPLC analysis of extracts/fractions prior to and after chromatography, especially on silica gel or following the use of acid modified eluants (e.g. TFA), can reveal the appearance of new compounds – artifacts.
Another valuable experimental approach is to set aside a sample of the unfractionated extract as a control. If pure compounds isolated from the fractionated extract are not present in the unfractionated extract, then it’s likely they are artifacts.
Two special cases warrant separate comment. Cryptic natural products (see section 10) by their properties are difficult to detect, as they may not survive extraction under normal laboratory conditions (i.e. r.t., ambient light and air, and exposure to protic solvents). As a result, their transformation artifacts may be isolated and identified, and detected in the unfractionated extract, and be miscategorised as natural products. Revealing the existence of cryptic natural products can be challenging and is usually achieved on a case-by-case basis by those who are receptive to the possibility with the necessary technical skills and initiative.
The biotransformation of natural products during cell-based bioassays (see Section 11) can generate artifacts in situ, that remain undetected and compromise data acquisition and analysis. As routine re-extraction of cell assay cultures to check for biotransformation artifacts is impractical (indeed, almost never done), detection of such occurrences is rare, and limited to those who are alert to and take the initiative to explore this possibility.
Another consideration is that of natural product storage artifacts. Once isolated, characterised and identified, residual samples of natural products are typically stored either dry or in solution (e.g. DMSO) at low temperature in the dark. These samples can then be recovered in the near or distant future and used for further experiments (e.g. bioassays). While tempting, it is risky to assume that a natural product survives storage intact, as they very commonly undergo partial and even complete transformation into one or more artifacts. As a result, it’s prudent to perform an independent analysis (i.e. HPLC-MS) when accessing a stored sample to confirm its integrity. The same also applies to stored artifacts, as prolonged storage can facilitate further transformations.
12.3. How to Control the Formation of Artifacts
The chemical reactivity of some natural products can be such that it is nearly impossible to avoid artifact formation — with cryptic natural products being the extreme example. That said, there are some actions that can be taken to potentially control artifact formation.
Solvent extraction – As noted in Section 2, most extraction solvents bring with them the risk of artifact formation, but some are riskier than others. Particularly problematic solvents include those that are chemically reactive (e.g. acetone, MeOH), and those that have a propensity to become acidic over time (e.g. CH2Cl2, CHCl3). Two solvents that offer the least risk of artifact formation are EtOAc and MeCN. In addition, care needs to be taken to minimise the presence of solvent impurities, some of which can be chemically reactive (e.g. formaldehyde, acetaldehyde, HCl, peroxides).
Chromatography – The classes of chromatography that dominate the modern natural products landscape include silica gel, various forms of reversed-phase MPLC/HPLC (e.g. analytical, semi-preparative and preparative, C8, C18…), and to a lesser degree size exclusion gel (e.g. Sephadex LH-20). That silica gel is inherently acidic is problem enough, but all too often its risk to fragile natural products is compounded by the use of eluants comprised of chlorinated solvents (e.g. CH2Cl2, CHCl3), MeOH and even acetone. Silica gel is overwhelmingly the riskiest chromatography media with respect to artifact formation (see Section 4.3). Also of risk is the use of strong acids such as trifluoracetic acid as an eluant modifier in reversed-phase chromatography. While there are occasions where such a modifier is essential for practical resolution, widespread reporting in the natural products literature suggests (at least for some laboratories) its use is the default position, rather than the exception. We would advocate analytical scale reversed-phase method development with and without a TFA modifier, with follow-up preparative and/or semi-preparative implementation only incorporating TFA when absolutely necessary. Also, where practical, we would advocate MeCN as the organic phase in reversed-phase chromatography, over more reactive solvents such as MeOH, CH2Cl2 and acetone — although clearly solvent choice will be influenced by solubility issues, and the need for compound resolution.
pH, heat, air and light – To a large degree, controlling these variables is attainable through good laboratory practice. The risk posed by acidic/basic pH can be minimised by careful choice of solvents and chromatography conditions (see above). Notwithstanding, it is important to acknowledge that natural products themselves can feature acidic/basic functionality, and when in concentrated/dry form this functionality can auto-activate pH-mediated artifact formation. For particularly pH sensitive molecules auto-activation can be managed by handing/storage in dilute neutral solutions.
It is also common to use rotary evaporators equipped with heating baths to remove solvents and concentrate fractions and pure compounds to dryness. Although water baths can operate to high temperatures near 100 °C, and some take advantage of this, we advocate their operation at reduced temperatures (35–40 °C), relying instead on an effective choice of vacuum and condenser chilling to effect solvent removal. Heat controlled centrifugal evaporators can also be used for solvent removal on a smaller scale.
While nitrogen purging of flasks and sample vials, and reduced lighting can be a useful precaution when dealing with air/light sensitive natural products, understandably, unless needs dictate, most researchers carry out operations under normal ambient air and laboratory lighting. That said, as a general rule, it is prudent to avoid exposing any samples to direct sunlight. Likewise, when drying samples under a stream of dry gas, caution dictates the use of nitrogen rather than air.
NMR data acquisition – The acquisition of NMR data can take many hours, during which time natural products are exposed to deuterated solvents at or near r.t. While all the concerns noted above for solvent use in extraction and chromatography apply, it is noteworthy that virtually all NMR solvents have the potential to facilitate artifact formation. Perhaps the only cautionary advice we can offer is to minimise exposure, store samples in the dark at reduced temperature (~4 °C to avoid freezing and sample precipitation), and avoid exposure to direct sunlight during handling.
12.4. Why and How to Take Advantage of Artifacts
Putting aside notable examples where researchers detect, identify, understand and report valuable knowledge about natural product artifacts (as illustrated in this review), it is clear from the scientific literature that many researchers are not so inclined. In an effort to make the case for this latter cohort, consider the following.
Evolved chemical space — One of the core benefits of studying natural products is to gain insights into the unique regions of chemical space that have co-evolved with life on earth. By mischaracterising artifacts as natural products, we contaminate/dilute our understanding of this chemical space. Similarly, by failing to correctly identify the artifact status of some isolated compounds, we risk overlooking valuable chemical knowledge, and in extreme cases (i.e. cryptic natural products) deny ourselves access to some of the most unique regions of natural product chemical space.
Biosynthesis — Miscategorising artifacts as natural products risks derailing biosynthetic investigations, as ill-informed efforts are made to force a biosynthetic path on compounds that sit outside biosynthetic processes. The same can apply when a single natural product is lost in a sea of artifacts, as biosynthetic focus is diluted and misdirected. On a more positive note, the recognition of artifacts can provide valuable biosynthetic insights.
Chemical and biological properties — Knowledge of the relationship between natural products and artifacts can inform the design of highly efficient and convergent biomimetic syntheses, while simultaneously providing insights into chemical stability, so critical for drug discovery and development. Such knowledge can also enable access to a greater structure diversity, to support bioassays and SAR investigations. The development of any natural product as a drug lead is dependent on understanding its chemical properties, which in turn means, where relevant, understanding its relationship with artifacts.
Author Contributions
The co-authors, M S Bulter and R J Capon, contributed equally.
Funding
This research received no external funding.
Acknowledgments
The authors are appreciative of the support offered by The University of Queensland, and the Institute for Molecular Bioscience.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hanson, J.R. Pseudo-Natural Products, Some Artefacts Formed during the Isolation of Terpenoids. J. Chem. Res. 2017, 41, 497–503. [CrossRef]
- Xu, T.; Chen, W.; Zhou, J.; Dai, J.; Li, Y.; Zhao, Y. Virtual Screening for Reactive Natural Products and Their Probable Artifacts of Solvolysis and Oxidation. Biomolecules 2020, 10, 1486. [CrossRef]
- Tang, Y.; Friesen, J.; Nikolić, D.; Lankin, D.; McAlpine, J.; Chen, S.-N.; Pauli, G. Silica Gel-Mediated Oxidation of Prenyl Motifs Generates Natural Product-Like Artifacts. Planta Med. 2021, 87, 998–1007. [CrossRef]
- Verpoorte, R.; Kim, H.K.; Choi, Y.H. Trivialities in Metabolomics: Artifacts in Extraction and Analysis. Front. Mol Biosci. 2022, 9, 972190. [CrossRef]
- Sauerschnig, C.; Doppler, M.; Bueschl, C.; Schuhmacher, R. Methanol Generates Numerous Artifacts during Sample Extraction and Storage of Extracts in Metabolomics Research. Metabolites 2017, 8, 1. [CrossRef]
- Venditti, A. What Is and What Should Never Be: Artifacts, Improbable Phytochemicals, Contaminants and Natural Products. Nat. Prod. Res. 2020, 34, 1014–1031. [CrossRef]
- Capon, R.J. Extracting Value: Mechanistic Insights into the Formation of Natural Product Artifacts – Case Studies in Marine Natural Products. Nat. Prod. Rep. 2020, 37, 55–79. [CrossRef]
- Woo, E.J.; Starks, C.M.; Carney, J.R.; Arslanian, R.; Cadapan, L.; Zavala, S.; Licari, P. Migrastatin and a New Compound, Isomigrastatin, from Streptomyces platensis. J. Antibiot. 2002, 55, 141–146. [CrossRef]
- Hochlowski, J.E.; Whittern, D.N.; Hill, P.; McAlpine, J.B. Dorrigocins: Novel Antifungal Antibiotics That Change the Morphology of Ras-Transformed NIH/3T3 Cells to That of Normal Cells. II. Isolation and Elucidation of Structures. J. Antibiot. 1994, 47, 870–874. [CrossRef]
- Ju, J.; Lim, S.-K.; Jiang, H.; Shen, B. Migrastatin and Dorrigocins Are Shunt Metabolites of Iso-Migrastatin. J. Am. Chem. Soc. 2005, 127, 1622–1623. [CrossRef]
- Ju, J.; Lim, S.-K.; Jiang, H.; Seo, J.-W.; Shen, B. Iso-Migrastatin Congeners from Streptomyces platensis and Generation of a Glutarimide Polyketide Library Featuring the Dorrigocin, Lactimidomycin, Migrastatin, and NK30424 Scaffolds. J. Am. Chem. Soc. 2005, 127, 11930–11931. [CrossRef]
- Ju, J.; Lim, S.-K.; Jiang, H.; Seo, J.-W.; Her, Y.; Shen, B. Thermolysis of Isomigrastatin and Its Congeners via [3,3]-Sigmatropic Rearrangement: A New Route to the Synthesis of Migrastatin and Its Analogues. Org. Lett. 2006, 8, 5865–5868. [CrossRef]
- Orfanoudaki, M.; Akee, R.K.; Martínez-Fructuoso, L.; Wang, D.; Kelley, J.A.; Smith, E.A.; Henrich, C.J.; Schnermann, M.J.; O’Keefe, B.R.; Grkovic, T. Formation of Trideuteromethylated Artifacts of Pyrrole-Containing Natural Products. J. Nat. Prod. 2024, 87, 415–423. [CrossRef]
- Sala, S.; James, P.J.C.; Nealon, G.L.; Fromont, J.; Gomez, O.; Vuong, D.; Lacey, E.; Flematti, G.R. Dendrillic Acids A and B: Nitrogenous, Rearranged Spongian Nor-Diterpenes from a Dendrilla sp. Marine Sponge. J. Nat. Prod. 2023, 86, 482–489. [CrossRef]
- Yu, G.; Wang, Q.; Liu, S.; Zhang, X.; Che, Q.; Zhang, G.; Zhu, T.; Gu, Q.; Li, D. Methylsulfonylated Polyketides Produced by Neosartorya udagawae HDN13-313 via Exogenous Addition of Small Molecules. J. Nat. Prod. 2019, 82, 998–1001. [CrossRef]
- Shang, Z.; Arishi, A.A.; Wu, C.; Lao, F.; Gilchrist, C.L.M.; Moggach, S.A.; Lacey, E.; Piggott, A.M.; Chooi, Y. Self-Resistance Gene-Guided Discovery of the Molecular Basis for Biosynthesis of the Fatty Acid Synthase Inhibitor Cerulenin. Angew. Chem. 2024, 137, e20241494. [CrossRef]
- Socha, A.M.; Garcia, D.; Sheffer, R.; Rowley, D.C. Antibiotic Bisanthraquinones Produced by a Streptomycete Isolated from a Cyanobacterium Associated with Ecteinascidia turbinata. J. Nat. Prod. 2006, 69, 1070–1073. [CrossRef]
- Samarasekera, K.; Hussein, W.M.; Wu, T.; Salim, A.A.; Capon, R.J. Glyclauxins A-E: Dimeric Oxaphenalenone Aminoglycosides from an Australian Wasp Nest-Derived Fungus Talaromyces sp. CMB-MW102. J. Nat. Prod. 86, 517–525, 2023. [CrossRef]
- Wu, J.; Shui, H.; Zhang, M.; Zeng, Y.; Zheng, M.; Zhu, K.-K.; Wang, S.-B.; Bi, H.; Hong, K.; Cai, Y.-S. Aculeaxanthones A–E, New Xanthones from the Marine-Derived Fungus Aspergillus aculeatinus WHUF0198. Front. Microbiol. 2023, 14, 1138830. [CrossRef]
- Zhen, X.; Gong, T.; Wen, Y.-H.; Yan, D.-J.; Chen, J.-J.; Zhu, P. Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor. Mar. Drugs 2018, 16, 357. [CrossRef]
- Wu, G.; Yu, G.; Kurtán, T.; Mándi, A.; Peng, J.; Mo, X.; Liu, M.; Li, H.; Sun, X.; Li, J.; et al. Versixanthones A–F, Cytotoxic Xanthone–Chromanone Dimers from the Marine-Derived Fungus Aspergillus versicolor HDN1009. J. Nat. Prod. 2015, 78, 2691–2698. [CrossRef]
- Lünne, F.; Köhler, J.; Stroh, C.; Müller, L.; Daniliuc, C.G.; Mück-Lichtenfeld, C.; Würthwein, E.-U.; Esselen, M.; Humpf, H.-U.; Kalinina, S.A. Insights into Ergochromes of the Plant Pathogen Claviceps purpurea. J. Nat. Prod. 2021, 84, 2630–2643. [CrossRef]
- Qin, T.; Iwata, T.; Ransom, T.T.; Beutler, J.A.; Porco, J.A. Syntheses of Dimeric Tetrahydroxanthones with Varied Linkages: Investigation of “Shapeshifting” Properties. J. Am. Chem. Soc. 2015, 137, 15225–15233. [CrossRef]
- Parish, C.A.; Smith, S.K.; Calati, K.; Zink, D.; Wilson, K.; Roemer, T.; Jiang, B.; Xu, D.; Bills, G.; Platas, G.; et al. Isolation and Structure Elucidation of Parnafungins, Antifungal Natural Products That Inhibit mRNA Polyadenylation. J. Am. Chem. Soc. 2008, 130, 7060–7066. [CrossRef]
- Guan, J.; Zhang, P.-P.; Wang, X.-H.; Guo, Y.-T.; Zhang, Z.-J.; Li, P.; Lin, L.-P. Structure-Guided Discovery of Diverse Cytotoxic Dimeric Xanthones/Chromanones from Penicillium chrysogenum C-7-2-1 and Their Interconversion Properties. J. Nat. Prod. 2024, 87, 238–257. [CrossRef]
- Ayers, S.; Graf, T.N.; Adcock, A.F.; Kroll, D.J.; Shen, Q.; Swanson, S.M.; Matthew, S.; Blanco, E.J.C. de; Wani, M.C.; Darveaux, B.A.; et al. Cytotoxic Xanthone–Anthraquinone Heterodimers from an Unidentified Fungus of the Order Hypocreales (MSX 17022). J. Antibiot. 2012, 65, 3–8. [CrossRef]
- Khong, Q.T.; Smith, E.A.; Wendt, K.L.; Dalilian, M.; Goncharova, E.I.; Brownell, I.; Cichewicz, R.H.; Henrich, C.J.; Beutler, J.A.; O’Keefe, B.R.; et al. Chemoreactive 2,5-Diketopiperazines from a Penicillium sp., Structure Revision of Reported Analogues and Proposed Facile Transformation Pathways. J. Nat. Prod. 2024, 87, 1826–1837. [CrossRef]
- Lv, H.; Su, H.; Xue, Y.; Jia, J.; Bi, H.; Wang, S.; Zhang, J.; Zhu, M.; Emam, M.; Wang, H.; et al. Polyketides with Potential Bioactivities from the Mangrove-Derived Fungus Talaromyces sp. WHUF0362. Mar. Life Sci. Technol. 2023, 5, 232–241. [CrossRef]
- Bang, S.; Kwon, H.E.; Baek, J.Y.; Jang, D.S.; Kim, S.; Nam, S.-J.; Lee, D.; Kang, K.S.; Shim, S.H. Colletotrichalactones A-Ca, Unusual 5/6/10-Fused Tricyclic Polyketides Produced by an Endophytic Fungus, Colletotrichum sp. JS-0361. Bioorg. Chem. 2020, 105, 104449. [CrossRef]
- Pang, X.; Lin, X.; Wang, J.; Liang, R.; Tian, Y.; Salendra, L.; Luo, X.; Zhou, X.; Yang, B.; Tu, Z.; et al. Three New Highly Oxygenated Sterols and One New Dihydroisocoumarin from the Marine Sponge-Derived Fungus Cladosporium sp. SCSIO41007. Steroids 2018, 129, 41–46. [CrossRef]
- Chen, M.; Han, L.; Shao, C.-L.; She, Z.-G.; Wang, C.-Y. Bioactive Diphenyl Ether Derivatives from a Gorgonian-Derived Fungus Talaromyces sp. Chem. Biodivers. 2015, 12, 443–450. [CrossRef]
- Li, F.; Sun, C.; Che, Q.; Zhu, T.; Gu, Q.; Guan, H.; Zhang, G.; Li, D. Pyrazinopyrimidine Alkaloids from a Mangrove-Derived Fungus Aspergillus versicolor HDN11-84. Phytochemistry 2021, 188, 112817. [CrossRef]
- Wu, C.-Z.; Li, G.; Zhang, Y.-H.; Yuan, S.-Z.; Dong, K.-M.; Lou, H.-X.; Peng, X.-P. Interconvertible Pyridone Alkaloids from the Marine-Derived Fungus Penicillium oxalicum QDU1. J. Nat. Prod. 2023, 86, 739–750. [CrossRef]
- Davidson, B.S.; Molinski, T.F.; Barrows, L.R.; Ireland, C.M. Varacin: A Novel Benzopentathiepin from Lissoclinum vareau That Is Cytotoxic toward a Human Colon Tumor. J. Am. Chem. Soc. 1991, 113, 4709–4710. [CrossRef]
- Makarieva, T.N.; Stonik, V.A.; Dmitrenok, A.S.; Grebnev, B.B.; Isakov, V.V.; Rebachyk, N.M.; Rashkes, Y.W. Varacin and Three New Marine Antimicrobial Polysulfides from the Far-Eastern Ascidian Polycitor sp. J. Nat. Prod. 1995, 58, 254–258. [CrossRef]
- Jiang, G.; Zhang, P.; Ratnayake, R.; Yang, G.; Zhang, Y.; Zuo, R.; Powell, M.; Huguet-Tapia, J.C.; Abboud, K.A.; Dang, L.H.; et al. Fungal Epithiodiketopiperazines Carrying α,β-Polysulfide Bridges from Penicillium steckii YE, and Their Chemical Interconversion. Chembiochem 2020, 22, 416–422. [CrossRef]
- Lindel, T.; Jensen, P.R.; Fenical, W.; Long, B.H.; Casazza, A.M.; Carboni, J.; Fairchild, C.R. Eleutherobin, a New Cytotoxin That Mimics Paclitaxel (Taxol) by Stabilizing Microtubules. J. Am. Chem. Soc. 1997, 119, 8744–8745. [CrossRef]
- Cinel, B.; Roberge, M.; Behrisch, H.; Ofwegen, L. van; Castro, C.B.; Andersen, R.J. Antimitotic Diterpenes from Erythropodium caribaeorum Test Pharmacophore Models for Microtubule Stabilization. Org. Lett. 2000, 2, 257–260. [CrossRef]
- Britton, R.; Roberge, M.; Berisch, H.; Andersen, R.J. Antimitotic Diterpenoids from Erythropodium caribaeorum : Isolation Artifacts and Putative Biosynthetic Intermediates. Tetrahedron Lett. 2001, 42, 2953–2956. [CrossRef]
- Ortiz-López, F.J.; Oves-Costales, D.; Garzón, J.F.G.; Gren, T.; Sterndorff, E.B.; Jiang, X.; Jo̷rgensen, T.S.; Blin, K.; Fernández-Pastor, I.; Tormo, J.R.; et al. Genome-Led Discovery of the Antibacterial Cyclic Lipopeptide Kutzneridine A and Its Silent Biosynthetic Gene Cluster from Kutzneria Species. J. Nat. Prod. 2024, 87, 2515–2522. [CrossRef]
- Koshino, S.; Koshino, H.; Matsuura, N.; Kobinata, K.; Onose, R.; Isono, K.; Osada, H. A New Cyclic Lipopeptide Antibiotic, Enamidonin. J. Antibiot. 1995, 48, 185–187. [CrossRef]
- Son, S.; Ko, S.-K.; Kim, S.M.; Kim, E.; Kim, G.S.; Lee, B.; Ryoo, I.-J.; Kim, W.-G.; Lee, J.-S.; Hong, Y.-S.; et al. Antibacterial Cyclic Lipopeptide Enamidonins with an Enamide-Linked Acyl Chain from a Streptomyces Species. J. Nat. Prod. 2018, 81, 2462–2469. [CrossRef]
- Namatame, I.; Tomoda, H.; Matsuda, D.; Tabata, N.; Kobayashi, S.; Omura, S. K97-0239A and B, New Inhibitors of Macrophage Foam Cell Formation, Produced by Streptomyces sp. K97-0239. Proc. Jpn. Acad., Ser. B 2002, 78, 45–50. [CrossRef]
- Zhang, H.; Zhang, C.; Li, Q.; Ma, J.; Ju, J. Metabolic Blockade-Based Genome Mining Reveals Lipochain-Linked Dihydro-β-Alanine Synthetases Involved in Autucedine Biosynthesis. Org. Lett. 2022, 24, 5535–5540. [CrossRef]
- Yan, J.-X.; Chevrette, M.G.; Braun, D.R.; Harper, M.K.; Currie, C.R.; Bugni, T.S. Madurastatin D1 and D2, Oxazoline Containing Siderophores Isolated from an Actinomadura sp. Org. Lett. 2019, 21, 6275–6279. [CrossRef]
- Pérez-Bonilla, M.; Sánchez-Hidalgo, M.; González, I.; Oves-Costales, D.; Martín, J.; Murillo-Alba, J.; Tormo, J.R.; Cho, A.; Byun, S.-Y.; No, J.-H.; et al. Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents? Int. J. Mol. Sci. 2023, 25, 301. [CrossRef]
- Oleinikova, G.K.; Zhuravleva, O.I.; Berdyshev, D.V.; Menzorova, N.I.; Popov, R.S.; Denisenko, V.A.; Kirichuk, N.N.; Afiyatullov, S.S. New Dihydrobenzofuranoid from the Marine-Derived Fungus Aspergillus ustus KMM 4664. Nat. Prod. Res. 2019, 35, 3332–3335. [CrossRef]
- Aguilar-Ramírez, E.; Rivera-Chávez, J.; Alvarado-Zacarías, B.D.; Barquera-Lozada, J.E. Exploring the Nonenzymatic Origin of Duclauxin-like Natural Products. J. Nat. Prod. 2024, 87, 2230–2242. [CrossRef]
- Wang, M.; Yang, L.; Feng, L.; Hu, F.; Zhang, F.; Ren, J.; Qiu, Y.; Wang, Z. Verruculosins A–B, New Oligophenalenone Dimers from the Soft Coral-Derived Fungus Talaromyces verruculosus. Mar. Drugs 2019, 17, 516. [CrossRef]
- Lacey, A.E.; Minns, S.A.; Chen, R.; Vuong, D.; Lacey, E.; Kalaitzis, J.A.; Tan, Y.P.; Shivas, R.G.; Butler, M.S.; Piggott, A.M. Talcarpones A and B: Bisnaphthazarin-Derived Metabolites from the Australian Fungus Talaromyces johnpittii sp. Nov. MST-FP2594. J. Antibiot. 2023, 77, 147–155. [CrossRef]
- El-Elimat, T.; Raja, H.A.; Day, C.S.; Chen, W.-L.; Swanson, S.M.; Oberlies, N.H. Greensporones: Resorcylic Acid Lactones from an Aquatic Halenospora sp. J. Nat. Prod. 2014, 77, 2088–2098. [CrossRef]
- Shi, Y.; Zaleta-Pinet, D.A.; Clark, B.R. Isolation, Identification, and Decomposition of Antibacterial Dialkylresorcinols from a Chinese Pseudomonas aurantiaca Strain. J. Nat. Prod. 2020, 83, 194–201. [CrossRef]
- Wibowo, M.; Gotfredsen, C.H.; Sassetti, E.; Melchiorsen, J.; Clausen, M.H.; Gram, L.; Ding, L. Azodyrecins A–C: Azoxides from a Soil-Derived Streptomyces Species. J. Nat. Prod. 2020, 83, 3519–3525. [CrossRef]
- Su, X.; Zhang, L.; Hu, K.; An, Y.; Zhang, Q.; Tang, J.; Yan, B.; Li, X.; Cai, J.; Li, X.; et al. Discovery of Natural Potent HMG-CoA Reductase Degraders for Lowering Cholesterol. Angew. Chem. Int. Ed. 2024, 63, e202313859. [CrossRef]
- Xu, M.; Gessner, G.; Groth, I.; Lange, C.; Christner, A.; Bruhn, T.; Deng, Z.; Li, X.; Heinemann, S.H.; Grabley, S.; et al. Shearinines D–K, New Indole Triterpenoids from an Endophytic Penicillium sp. (Strain HKI0459) with Blocking Activity on Large-Conductance Calcium-Activated Potassium Channels. Tetrahedron 2007, 63, 435–444. [CrossRef]
- Chen, M.; Hao, B.-C.; Zhu, X.-H.; Zhang, L.-K.; Zheng, Y.-Y.; Zhou, X.-J.; Schäberle, T.F.; Shen, L.; Wang, C.-Y.; Liu, Y. Molecular Networking Reveals Indole Diterpenoids from the Marine-Derived Fungus Penicillium sp. N4-3. Mar. Life Sci. Technol. 2025, 7, 302–312. [CrossRef]
- Tuzi, A.; Carbone, M.; Ciavatta, M.L.; Evidente, A. Structure Revision of the Fungal Phytotoxin Cavoxin and of Its Corresponding Chroman-4-One Cavoxone by X-ray Crystallography. J. Nat. Prod. 2024, 87, 1888–1892. [CrossRef]
- Fotso, S.; Wu, S.J.; Qin, S.; Laatsch, H. 5,7-Dihydroxy-5,6,7,8-Tetrahydro-1H-Azocin-2-One from a Marine-Derived Streptomyces sp. Nat. Prod. Commun. 2006, 1, 1934578X0600100102. [CrossRef]
- Miguel-Gordo, M.; Gegunde, S.; Calabro, K.; Jennings, L.K.; Alfonso, A.; Genta-Jouve, G.; Vacelet, J.; Botana, L.M.; Thomas, O.P. Bromotryptamine and Bromotyramine Derivatives from the Tropical Southwestern Pacific Sponge Narrabeena nigra. Mar. Drugs 2019, 17, 319. [CrossRef]
- Shin, A.-Y.; Lee, H.-S.; Lee, Y.-J.; Lee, J.S.; Son, A.; Choi, C.; Lee, J. Oxygenated Theonellastrols: Interpretation of Unusual Chemical Behaviors Using Quantum Mechanical Calculations and Stereochemical Reassignment of 7α-Hydroxytheonellasterol. Mar. Drugs 2020, 18, 607. [CrossRef]
- Bringmann, G.; Lang, G.; Gulder, T.A.M.; Tsuruta, H.; Mühlbacher, J.; Maksimenka, K.; Steffens, S.; Schaumann, K.; Stöhr, R.; Wiese, J.; et al. The First Sorbicillinoid Alkaloids, the Antileukemic Sorbicillactones A and B, from a Sponge-Derived Penicillium chrysogenum Strain. Tetrahedron 2005, 61, 7252–7265. [CrossRef]
- Abe, N.; Sugimoto, O.; Tanji, K.; Hirota, A. Identification of the Quinol Metabolite “Sorbicillinol”, a Key Intermediate Postulated in Bisorbicillinoid Biosynthesis. J. Am. Chem. Soc. 2000, 122, 12606–12607. [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Capon, R.J. Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14. Molecules 2021, 26, 7144. [CrossRef]
- Igarashi, Y.; Zhou, T.; Sato, S.; Matsumoto, T.; Yu, L.; Oku, N. Akaeolide, a Carbocyclic Polyketide from Marine-Derived Streptomyces. Org. Lett. 2013, 15, 5678–5681. [CrossRef]
- Zhang, S.; Yang, Q.; Guo, L.; Zhang, Y.; Feng, L.; Zhou, L.; Yang, S.; Yao, Q.; Pescitelli, G.; Xie, Z. Isolation, Structure Elucidation and Racemization of (+)- and (-)-Pratensilins A-C: Unprecedented Spiro Indolinone-Naphthofuran Alkaloids from a Marine Streptomyces sp. Chem. Comm. 2017, 53, 10066–10069. [CrossRef]
- Zhang, S.; Zhang, L.; Kou, L.-J.; Yang, Q.-L.; Qu, B.; Pescitelli, G.; Xie, Z. Isolation, Stereochemical Study, and Racemization of (±)-Pratenone A, the First Naturally Occurring 3-(1-Naphthyl)-2-Benzofuran-1(3H)-One Polyketide from a Marine-Derived Actinobacterium. Chirality 2020, 32, 299–307. [CrossRef]
- Arabshahi, L.; Schmitz, F.J. Brominated Tyrosine Metabolites from an Unidentified Sponge. J. Org. Chem. 1987, 52, 3584–3586. [CrossRef]
- Calcul, L.; Inman, W.D.; Morris, A.A.; Tenney, K.; Ratnam, J.; McKerrow, J.H.; Valeriote, F.A.; Crews, P. Additional Insights on the Bastadins: Isolation of Analogues from the Sponge Ianthella cf. reticulata and Exploration of the Oxime Configurations. J. Nat. Prod. 2010, 73, 365–372. [CrossRef]
- Birnbacher, J.; Schüffler, A.; Deininger, F.; Opatz, T.; Anke, T. Isolation and Biological Activity of New Norhirsutanes from Creolophus cirrhatus. Z. für Naturforsch. C 2008, 63, 203–206. [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Neobulgarones Revisited: Anti and Syn Bianthrones from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD22. J. Nat. Prod. 2021, 84, 762–770. [CrossRef]
- Cueto, M.; Jensen, P.R.; Kauffman, C.; Fenical, W.; Lobkovsky, E.; Clardy, J. Pestalone, a New Antibiotic Produced by a Marine Fungus in Response to Bacterial Challenge. J. Nat. Prod. 2001, 64, 1444–1446. [CrossRef]
- Li, E.; Jiang, L.; Guo, L.; Zhang, H.; Che, Y. Pestalachlorides A-C, Antifungal Metabolites from the Plant Endophytic Fungus Pestalotiopsis adusta. Bioorg. Med. Chem. 2008, 16, 7894–7899. [CrossRef]
- Slavov, N.; Cvengroš, J.; Neudörfl, J.; Schmalz, H. Total Synthesis of the Marine Antibiotic Pestalone and Its Surprisingly Facile Conversion into Pestalalactone and Pestalachloride A. Angew. Chem. Int. Ed. 2010, 49, 7588–7591. [CrossRef]
- Zheng, L.; Jiang, X.; Zhang, Q.; Zhu, Y.; Zhang, H.; Zhang, W.; Saurav, K.; Liu, J.; Zhang, C. Discovery and Biosynthesis of Neoenterocins Indicate a Skeleton Rearrangement of Enterocin. Org. Lett. 2019, 21, 9066–9070. [CrossRef]
- Xu, L.; Guo, F.-W.; Zhang, X.-Q.; Zhou, T.-Y.; Wang, C.-J.; Wei, M.-Y.; Gu, Y.-C.; Wang, C.-Y.; Shao, C.-L. Discovery, Total Syntheses and Potent Anti-Inflammatory Activity of Pyrrolinone-Fused Benzoazepine Alkaloids Asperazepanones A and B from Aspergillus candidus. Commun. Chem. 2022, 5, 80. [CrossRef]
- Xu, W.-F.; Chao, R.; Hai, Y.; Guo, Y.-Y.; Wei, M.-Y.; Wang, C.-Y.; Shao, C.-L. 17-Hydroxybrevianamide N and Its N1-Methyl Derivative, Quinazolinones from a Soft-Coral-Derived Aspergillus sp. Fungus: 13 S Enantiomers as the True Natural Products. J. Nat. Prod. 2021, 84, 1353–1358. [CrossRef]
- Williams, P.G.; Buchanan, G.O.; Feling, R.H.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. New Cytotoxic Salinosporamides from the Marine Actinomycete Salinispora tropica. J. Org. Chem. 2005, 70, 6196–6203. [CrossRef]
- Kang, H.; Jensen, P.R.; Fenical, W. Isolation of Microbial Antibiotics from a Marine Ascidian of the Genus Didemnum. J. Org. Chem. 1996, 61, 1543–1546. [CrossRef]
- Liu, H.; Chen, Z.; Zhu, G.; Wang, L.; Du, Y.; Wang, Y.; Zhu, W. Phenolic Polyketides from the Marine Alga-Derived Streptomyces sp. OUCMDZ-3434. Tetrahedron 2017, 73, 5451–5455. [CrossRef]
- Xu, D.-B.; Ma, M.; Deng, Z.-X.; Hong, K. Genotype-Driven Isolation of Enterocin with Novel Bioactivities from Mangrove-Derived Streptomyces qinglanensis 172205. Appl. Microbiol. Biotechnol. 2015, 99, 5825–5832. [CrossRef]
- Salim, A.A.; Samarasekera, K.; Khalil, Z.G.; Capon, R.J. Exploring Natural Product Artifacts: The Polyketide Enterocin Warms to a Ballet of Isomers. Org. Lett. 2020, 22, 4828–4832. [CrossRef]
- Schneider, Y.; Jenssen, M.; Isaksson, J.; Hansen, K.Ø.; Andersen, J.H.; Hansen, E.H. Bioactivity of Serratiochelin A, a Siderophore Isolated from a Co-Culture of Serratia sp. and Shewanella sp. Microorganisms 2020, 8, 1042. [CrossRef]
- Zhang, H.; Conte, M.M.; Capon, R.J. Franklinolides A-C from an Australian Marine Sponge Complex: Phosphodiesters Strongly Enhance Polyketide Cytotoxicity. Angew. Chem. Int. Ed. 2010, 49, 9904–9906. [CrossRef]
- Sirirath, S.; Tanaka, J.; Ohtani, I.I.; Ichiba, T.; Rachmat, R.; Ueda, K.; Usui, T.; Osada, H.; Higa, T. Bitungolides A-F, New Polyketides from the Indonesian Sponge Theonella cf. swinhoei. J. Nat. Prod. 2002, 65, 1820–1823. [CrossRef]
- Salim, A.A.; Xiao, X.; Cho, K.-J.; Piggott, A.M.; Lacey, E.; Hancock, J.F.; Capon, R.J. Rare Streptomyces sp. Polyketides as Modulators of K-Ras Localisation. Org. Biomol. Chem. 2014, 12, 4872–4878. [CrossRef]
- Zhong, W.-M.; Wang, J.-F.; Wei, X.-Y.; Zeng, Q.; Chen, X.-Y.; Xiang, Y.; Tian, X.-P.; Zhang, S.; Long, L.J.; Wang, F.-Z. (+)- and (−)-Eurotone A: A Pair of Enantiomeric Polyketide Dimers from a Marine-Derived Fungus Eurotium sp. SCSIO F452. Tetrahedron Lett. 2019, 60, 1600–1603. [CrossRef]
- Bindseil, K.U.; Henkel, T.; Zeeck, A.; Bur, D.; Niederer, D.; Séquin, U. Metabolic Products of Microorganisms. Part 262. The Absolute Configuration of Sphydrofuran, a Widespread Metabolite from Streptomycetes. Helv. Chim. Acta 1991, 74, 1281–1286. [CrossRef]
- Shibata, S.; Ogihara, Y.; Tokutake, N.; Tanaka, O. Duclauxin, a Metabolite of Penicillium duclauxi (Delacroix). Tetrahedron Lett. 1965, 6, 1287–1288. [CrossRef]
- Zheng, M.; Li, Q.; Liao, H.; Li, Y.; Zhou, C.; Zhao, X.; Chen, C.; Sun, W.; Zhang, Y.; Zhu, H. Adpressins A-G: Oligophenalenone Dimers from Talaromyces adpressus. J. Nat. Prod. 2024, 87, 1921–1929. [CrossRef]
- Zang, Y.; Genta-Jouve, G.; Retailleau, P.; Escargueil, A.; Mann, S.; Nay, B.; Prado, S. Talaroketals A and B, Unusual Bis(Oxaphenalenone) Spiro and Fused Ketals from the Soil Fungus Talaromyces stipitatus ATCC 10500. Org. Biomol. Chem. 2016, 14, 2691–2697. [CrossRef]
- Li, Q.; Zhang, M.; Zhang, X.; Li, L.; Zheng, M.; Kang, J.; Liu, F.; Zhou, Q.; Li, X.; Sun, W.; et al. Talaroclauxins A and B: Duclauxin-Ergosterol and Duclauxin-Polyketide Hybrid Metabolites with Complicated Skeletons from Talaromyces stipitatus. Chin. Chem. Lett. 2024, 35, 108193. [CrossRef]
- Chaiyosang, B.; Kanokmedhakul, K.; Sanmanoch, W.; Boonlue, S.; Hadsadee, S.; Jungsuttiwong, S.; Kanokmedhakul, S. Bioactive Oxaphenalenone Dimers from the Fungus Talaromyces macrosporus KKU-1NK8. Fitoterapia 2019, 134, 429–434. [CrossRef]
- Dethoup, T.; Manoch, L.; Kijjoa, A.; Nascimento, M.S.; Puaparoj, P.; Silva, A.M.; Eaton, G.; Herz, W. Bacillisporins D and E, New Oxyphenalenone Dimers from Talaromyces bacillisporus. Planta Med. 2006, 72, 957–960. [CrossRef]
- Chaudhary, N.K.; Crombie, A.; Vuong, D.; Lacey, E.; Piggott, A.M.; Karuso, P. Talauxins: Hybrid Phenalenone Dimers from Talaromyces stipitatus. J. Nat. Prod. 2020, 83, 1051–1060. [CrossRef]
- Dramae, A.; Intaraudom, C.; Bunbamrung, N.; Saortep, W.; Srichomthong, K.; Pittayakhajonwut, P. Heptacyclic Oligophenalenones from the Soil Fungus Talaromyces bacillisporus BCC17645. Tetrahedron 2020, 76, 130980. [CrossRef]
- Cao, P.; Yang, J.; Miao, C.-P.; Yan, Y.; Ma, Y.-T.; Li, X.-N.; Zhao, L.-X.; Huang, S.-X. New Duclauxamide from Penicillium manginii YIM PH30375 and Structure Revision of the Duclauxin Family. Org. Lett. 2015, 17, 1146–1149. [CrossRef]
- Isaka, M.; Sappan, M.; Auncharoen, P.; Srikitikulchai, P. Chromone Derivatives from the Wood-Decay Fungus Rhizina sp. BCC 12292. Phytochem. Lett. 2010, 3, 152–155. [CrossRef]
- Liu, Y.; Stuhldreier, F.; Kurtán, T.; Mándi, A.; Arumugam, S.; Lin, W.; Stork, B.; Wesselborg, S.; Weber, H.; Henrich, B.; et al. Daldinone Derivatives from the Mangrove-Derived Endophytic Fungus Annulohypoxylon sp. RSC Adv. 2017, 7, 5381–5393. [CrossRef]
- Jiang, L.; Huang, P.; Ren, B.; Song, Z.; Zhu, G.; He, W.; Zhang, J.; Oyeleye, A.; Dai, H.; Zhang, L.; et al. Antibacterial Polyene-Polyol Macrolides and Cyclic Peptides from the Marine-Derived Streptomyces sp. MS110128. Appl. Microbiol. Biotechnol. 2021, 105, 4975–4986. [CrossRef]
- Kim, M.C.; Cullum, R.; Machado, H.; Smith, A.J.; Yang, I.; Rodvold, J.J.; Fenical, W. Photopiperazines A-D, Photosensitive Interconverting Diketopiperazines with Significant and Selective Activity against U87 Glioblastoma Cells, from a Rare, Marine-Derived Actinomycete of the Family Streptomycetaceae. J. Nat. Prod. 2019, 82, 2262–2267. [CrossRef]
- Zheng, J.; Xu, Z.; Wang, Y.; Hong, K.; Liu, P.; Zhu, W. Cyclic Tripeptides from the Halotolerant Fungus Aspergillus sclerotiorum PT06-1. J. Nat. Prod. 2010, 73, 1133–1137. [CrossRef]
- Fu, X.; Schmitz, F.J.; Kelly-Borges, M.; McCready, T.L.; Holmes, C.F.B. Clavosines A−C from the Marine Sponge Myriastra clavosa: Potent Cytotoxins and Inhibitors of Protein Phosphatases 1 and 2A. J. Org. Chem. 1998, 63, 7957–7963. [CrossRef]
- Fusetani, N.; Matsunaga, S. Bioactive Sponge Peptides. Chem. Rev. 1993, 93, 1793–1806. [CrossRef]
- Kurnianda, V.; Fujimura, H.; Kanna, Y.; Tanaka, J. Photooxidation Products from a Marine Cadinane Sesquiterpenoid. Chem. Lett. 2021, 50, 220–222. [CrossRef]
- Waksman, S.A.; Bugie, E. Chaetomin, a New Antibiotic Substance Produced by Chaetomium cochliodes. J. Bacteriol. 1944, 48, 527–530. [CrossRef]
- Wang, M.-H.; Hu, Y.-C.; Sun, B.-D.; Yu, M.; Niu, S.-B.; Guo, Z.; Zhang, X.-Y.; Zhang, T.; Ding, G.; Zou, Z.-M. Highly Photosensitive Poly-Sulfur-Bridged Chetomin Analogues from Chaetomium cochliodes. Org. Lett. 2018, 20, 1806–1809. [CrossRef]
- Shah, M.; Sun, C.; Sun, Z.; Zhang, G.; Che, Q.; Gu, Q.; Zhu, T.; Li, D. Antibacterial Polyketides from Antarctica Sponge-Derived Fungus Penicillium sp. HDN151272. Mar. Drugs 2020, 18, 71. [CrossRef]
- Kong, F.; Singh, M.P.; Carter, G.T. Pseudopyronines A and B, α-Pyrones Produced by a Marine Pseudomonas sp. F92S91, and Evidence for the Conversion of 4-Hydroxy-α-Pyrone to 3-Furanone. J. Nat. Prod. 2005, 68, 920–923. [CrossRef]
- Capon, R.J.; Faulkner, D.J. Metabolites of the Pulmonate Siphonaria lessoni. J. Org. Chem. 1984, 49, 2506–2508. [CrossRef]
- Akiyama, H.; Indananda, C.; Thamchaipenet, A.; Motojima, A.; Oikawa, T.; Komaki, H.; Hosoyama, A.; Kimura, A.; Oku, N.; Igarashi, Y. Linfuranones B and C, Furanone-Containing Polyketides from a Plant-Associated Sphaerimonospora mesophila. J. Nat. Prod. 2018, 81, 1561–1569. [CrossRef]
- Indananda, C.; Igarashi, Y.; Ikeda, M.; Oikawa, T.; Thamchaipenet, A. Linfuranone A, a New Polyketide from Plant-Derived Microbispora sp. GMKU 363. J. Antibiot. 2013, 66, 675–677. [CrossRef]
- Okanya, P.W.; Mohr, K.I.; Gerth, K.; Kessler, W.; Jansen, R.; Stadler, M.; Müller, R. Hyafurones, Hyapyrrolines, and Hyapyrones: Polyketides from Hyalangium minutum. J. Nat. Prod. 2014, 77, 1420–1429. [CrossRef]
- Frank, B.; Wenzel, S.C.; Bode, H.B.; Scharfe, M.; Blöcker, H.; Müller, R. From Genetic Diversity to Metabolic Unity: Studies on the Biosynthesis of Aurafurones and Aurafuron-like Structures in Myxobacteria and Streptomycetes. J. Mol. Biol. 2007, 374, 24–38. [CrossRef]
- Gliński, J.A.; Proudfoot, J.; Madura, I.; Zhang, H.; Gleńsk, M.; Day, V.; Dudek, M.K. Spontaneous Stereoselective Oxidation of Crystalline Avermectin B1a to Its C-8a-(S)-Hydroperoxide. J. Nat. Prod. 2019, 82, 3477–3481. [CrossRef]
- Meyer, S.W.; Mordhorst, T.F.; Lee, C.; Jensen, P.R.; Fenical, W.; Köck, M. Penilumamide, a Novel Lumazine Peptide Isolated from the Marine-Derived Fungus, Penicillium sp. CNL-338. Org. Biomol. Chem. 2010, 8, 2158–2163. [CrossRef]
- Zheng, C.; Wu, L.; Li, X.; Song, X.; Niu, Z.; Song, X.; Chen, G.; Wang, C. Structure and Absolute Configuration of Aspergilumamide A, a Novel Lumazine Peptide from the Mangrove-Derived Fungus Aspergillus sp. Helv. Chim. Acta 2015, 98, 368–373. [CrossRef]
- Chen, M.; Shao, C.-L.; Fu, X.-M.; Kong, C.-J.; She, Z.-G.; Wang, C.-Y. Lumazine Peptides Penilumamides B-D and the Cyclic Pentapeptide Asperpeptide A from a Gorgonian-Derived Aspergillus sp. Fungus. J. Nat. Prod. 2014, 77, 1601–1606. [CrossRef]
- Elsbaey, M.; Jomori, T.; Tanaka, J.; Oku, N.; Igarashi, Y. Okichromanone, a New Antiviral Chromanone from a Marine-Derived Microbispora. J. Antibiot. 2024, 77, 389–392. [CrossRef]
- Li, H.; Guo, J.; Zhang, R.; Wang, J.; Hu, Z.; Zhang, Y. Two New Nucleoside Derivatives Isolated from the Marine-Derived Aspergillus versicolor and Their Intramolecular Transesterification. Nat. Prod. Res. 2022, 36, 3346–3352. [CrossRef]
- Wu, T.; Salim, A.A.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Glenthmycins A-M: Macrocyclic Spirotetronate Polyketide Antibacterials from the Australian Pasture Plant-Derived Streptomyces sp. CMB-PB041. J. Nat. Prod. 2022, 85, 1641–1657. [CrossRef]
- Wu, T.; Salim, A.A.; Bernhardt, P.V.; Capon, R.J. Amaurones A–K: Polyketides from the Fish Gut-Derived Fungus Amauroascus sp. CMB-F713. J. Nat. Prod. 2021, 84, 474–482. [CrossRef]
- Capon, R.J.; Ratnayake, R.; Stewart, M.; Lacey, E.; Tennant, S.; Gill, J.H. Aspergillazines A-E: Novel Heterocyclic Dipeptides from an Australian Strain of Aspergillus unilateralis. Org. Biomol. Chem. 2005, 3, 123–129. [CrossRef]
- Takahashi, S.; Kakinuma, N.; Iwai, H.; Yanagisawa, T.; Nagai, K.; Suzuki, K.; Tokunaga, T.; Nakagawa, A. Quinolactacins A, B and C. Novel Quinolone Compounds from Penicillium sp. EPF-6. II. Physico-Chemical Properties and Structure Elucidation. J. Antibiot. 2000, 53, 1252–1256. [CrossRef]
- Kim, W.-G.; Song, N.-K.; Yoo, I.-D. Quinolactacins Al and A2, New Acetylcholinesterase Inhibitors from Penicillium citrinum. J. Antibiot. 2001, 54, 831–835. [CrossRef]
- Tatsuta, K.; Misawa, H.; Chikauchi, K. Biomimetric Total Synthesis of Quinolactacin B, TNF Production Inhibitor, and Its Analogs. J. Antibiot. 2001, 54, 109–112. [CrossRef]
- Zhang, X.; Jiang, W.; Sui, Z. Concise Enantioselective Syntheses of Quinolactacins A and B through Alternative Winterfeldt Oxidation. J. Org. Chem. 2003, 68, 4523–4526. [CrossRef]
- Clark, B.; Capon, R.J.; Lacey, E.; Tennant, S.; Gill, J.H. Quinolactacins Revisited: From Lactams to Imide and Beyond. Org. Biomol. Chem. 2006, 4, 1512–1519. [CrossRef]
- Guo, T.-T.; Song, M.-M.; Han, W.-R.; Zhu, J.-H.; Liu, Q.-C.; Wang, J.-F. New N-Methyl-4-Quinolone Alkaloid and Citrinin Dimer Derivatives from the Sponge-Derived Fungus Penicillium sp. SCSIO 41303. Phytochem. Lett. 2021, 46, 29–35. [CrossRef]
- Zhu, J.X.; Lu, Y.; Chen, J.; Chen, J.; Zhang, H.; Bao, X.; Ye, X.; Wang, H. Total Synthesis of Quinolactacin-H from Marine-Derived: Penicillium sp. ENP701 and Biological Activities. RSC Advances 2020, 10, 24251–24254. [CrossRef]
- Liu, Y.; Xue, X.; Zhou, L.; Yang, W.; She, Z.; Liao, Q.; Feng, Y.; Chen, X.; Zhang, Y. Quinolinones Alkaloids with AChE Inhibitory Activity from Mangrove Endophytic Fungus Penicillium citrinum YX-002. Chem. Biodivers. 2023, 20, e202300735. [CrossRef]
- Mohamed, O.G.; Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. Prolinimines: N-Amino-L-Pro-Methyl Ester (Hydrazine) Schiff Bases from a Fish Gastrointestinal Tract-Derived Fungus, Trichoderma sp. CMB-F563. Org. Lett. 2018, 20, 377–380. [CrossRef]
- Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. N-Amino-L-Proline Methyl Ester from an Australian Fish Gut-Derived Fungus: Challenging the Distinction between Natural Product and Artifact. Mar. Drugs 2021, 19, 151. [CrossRef]
- Li, C.-S.; Li, X.-M.; Gao, S.-S.; Lu, Y.H.; Wang, B.-G. Cytotoxic Anthranilic Acid Derivatives from Deep Sea Sediment-Derived Fungus Penicillium paneum SD-44. Mar. Drugs 2013, 11, 3068–3076. [CrossRef]
- Khalil, Z.G.; Kankanamge, S.; Capon, R.J. Structure Revision of Penipacids A–E Reveals a Putative New Cryptic Natural Product, N-Aminoanthranilic Acid, with Potential as a Transcriptional Regulator of Silent Secondary Metabolism. Mar. Drugs 2022, 20, 339. [CrossRef]
- Minato, S. Isolation of Anthglutin, an Inhibitor of γ-Glutamyl Transpeptidase from Penicillum oxalicum. Arch. Biochem. Biophys. 1979, 192, 235–240. [CrossRef]
- Ma, C.; Li, Y.; Niu, S.; Zhang, H.; Liu, X.; Che, Y. N-Hydroxypyridones, Phenylhydrazones, and a Quinazolinone from Isaria farinosa. J. Nat. Prod. 2011, 74, 32–37. [CrossRef]
- Abdelfattah, M.S.; Toume, K.; Arai, M.A.; Masu, H.; Ishibashi, M. Katorazone, a New Yellow Pigment with a 2-Azaquinone-Phenylhydrazone Structure Produced by Streptomyces sp. IFM 11299. Tetrahedron Lett. 2012, 53, 3346–3348. [CrossRef]
- Zhang, T.; Zhu, M.-L.; Sun, G.-Y.; Li, N.; Gu, Q.-Q.; Li, D.-H.; Che, Q.; Zhu, T.-J. Exopisiod B and Farylhydrazone C, Two New Alkaloids from the Antarctic-Derived Fungus Penicillium sp. HDN14-431. J. Asian Nat. Prod. Res. 2016, 18, 959–965. [CrossRef]
- Cheng, P.; Xu, K.; Chen, Y.C.; Wang, T.T.; Chen, Y.; Yang, C.L.; Ma, S.Y.; Liang, Y.; Ge, H.M.; Jiao, R.H. Cytotoxic Aromatic Polyketides from an Insect Derived Streptomyces sp. NA4286. Tetrahedron Lett. 2019, 60, 1706–1709. [CrossRef]
- Steinmetz, H.; Gerth, K.; Jansen, R.; Schläger, N.; Dehn, R.; Reinecke, S.; Kirschning, A.; Müller, R. Elansolid A, a Unique Macrolide Antibiotic from Chitinophaga sancti Isolated as Two Stable Atropisomers. Angew. Chem. Int. Ed. 2011, 50, 532–536. [CrossRef]
- Jansen, R.; Gerth, K.; Steinmetz, H.; Reinecke, S.; Kessler, W.; Kirschning, A.; Müller, R. Elansolid A3, a Unique p-Quinone Methide Antibiotic from Chitinophaga sancti. Chem. A Eur. J. 2011, 17, 7739–7744. [CrossRef]
- Teta, R.; Gurgui, M.; Helfrich, E.J.N.; Künne, S.; Schneider, A.; Echten-Deckert, G.V.; Mangoni, A.; Piel, J. Genome Mining Reveals trans-AT Polyketide Synthase Directed Antibiotic Biosynthesis in the Bacterial Phylum Bacteroidetes. ChemBioChem 2010, 11, 2506–2512. [CrossRef]
- Wang, Q.; Song, F.; Xiao, X.; Huang, P.; Li, L.; Monte, A.; Abdel-Mageed, W.M.; Wang, J.; Guo, H.; He, W.; et al. Abyssomicins from the South China Sea Deep-Sea Sediment Verrucosispora sp.: Natural Thioether Michael Addition Adducts as Antitubercular Prodrugs. Angew. Chem. Int. Ed. 2013, 52, 1231–1234. [CrossRef]
- Bister, B.; Bischoff, D.; Ströbele, M.; Riedlinger, J.; Reicke, A.; Wolter, F.; Bull, A.T.; Zähner, H.; Fiedler, H.-P.; Süssmuth, R.D. Abyssomicin C - A Polycyclic Antibiotic from a Marine Verrucosispora Strain as an Inhibitor of the p-Aminobenzoic Acid/Tetrahydrofolate Biosynthesis Pathway. Angew. Chem. Int. Ed. 2004, 43, 2574–2576. [CrossRef]
- Shang, Z.; Khalil, Z.; Li, L.; Salim, A.A.; Quezada, M.; Kalansuriya, P.; Capon, R.J. Roseopurpurins: Chemical Diversity Enhanced by Convergent Biosynthesis and Forward and Reverse Michael Additions. Org. Lett. 2016, 18, 4340–4343. [CrossRef]
- Han, J.; Bruhn, D.F.; Roberts, D.C.; Burkman, E.; Moreno, Y.; Salim, A.A.; Capon, R.J. Goondomycins A–H: Carbocyclic Ansa-Polyketides from an Australian Pasture Streptomyces with Selective Activity against Dirofilaria immitis. J. Nat. Prod. 2024, 87, 2810–2821. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



