1. Introduction
The evolutionary transition from unicellular to multicellular life necessitated the development of sophisticated communication systems capable of integrating metabolism, immunity, and environmental sensing into coherent organismal responses [
1,
2]. Among the most ancient and conserved of these biological information processing systems is the capacity for ketogenesis - the hepatic production of ketone bodies from fatty acid precursors during periods of altered energy availability [
3,
4]. While ketosis has been traditionally conceptualized through the reductionist framework of metabolic compensation for glucose deficiency - a pathological state requiring suppression - recent systems-level investigations have revealed ketone bodies, particularly β-hydroxybutyrate (BHB), as pleiotropic signaling molecules that encode complex physiological information and orchestrate adaptive programs extending far beyond simple energy provision [
5,
6,
7].
The remarkable conservation of ketogenic pathways across vertebrate evolution -from arctic mammals enduring months of food scarcity to migratory birds completing transcontinental flights without feeding - suggests fundamental adaptive significance that transcends the pathological framing dominant in medical and veterinary literature [
1,
8]. This evolutionary persistence implies that ketone bodies function as carriers of survival-relevant information about nutritional status, immune challenges, temporal phase, and environmental conditions - information that must be reliably encoded, transmitted, and decoded to generate appropriate adaptive responses [
5,
7]. In humans, physiological ketosis occurs naturally during multiple life stages and conditions including prolonged fasting, intensive exercise, pregnancy, lactation, and early postnatal development, each representing distinct informational contexts requiring coordinated metabolic-immune adjustments [
3,
9]. Yet conventional medical paradigms have systematically pathologized these natural states, largely based on extrapolation from extreme pathological conditions such as diabetic ketoacidosis - a conceptual error equivalent to condemning fever because hyperthermia is dangerous [
10,
11].
The periparturient dairy cow represents perhaps the most intensive natural model of integrated metabolic-immune information processing, where the physiological demands of late pregnancy, parturition, and early lactation initiate coordinated systemic transitions involving massive energy mobilization, dramatic hormonal restructuring, immune system modulation, and fundamental alterations in microbial ecology [
12,
13]. During this critical transition period, plasma BHB concentrations can increase 5- to 10-fold within days of calving, coinciding with profound alterations in circulating immune cell populations, systemic inflammatory mediator profiles, and calcium homeostasis [
13,
14,
15]. Veterinary medicine has conventionally treated this ketone elevation as primary pathology requiring aggressive suppression, despite limited evidence that such interventions improve long-term outcomes and growing recognition that early ketone elevation may represent adaptive programming [
14,
15]. The temporal sequence and pattern of these metabolic changes - not merely their magnitude - appears to encode diagnostic information that distinguishes adaptive from pathological trajectories, suggesting that biological systems "read" complex metabolic signatures to coordinate appropriate responses [
14,
15].
Recent paradigm-shifting discoveries in molecular immunology and metabolic biochemistry have fundamentally challenged reductionist conceptual boundaries separating metabolism and immune function [
14,
16]. The recognition that immune cells undergo systematic metabolic reprogramming during activation states, that small molecule metabolites function as potent cellular signaling mediators with context-dependent meanings, and that mitochondria serve as critical immune sensing organelles has created entirely new theoretical frameworks for understanding health, disease, and therapeutic intervention [
16,
17]. These discoveries reveal that metabolites are not merely biochemical intermediates in linear pathways but information carriers - molecular symbols in biological languages that enable cellular communication and coordinate distributed decision-making across tissues and organ systems. Yet these insights have been slow to penetrate clinical thinking about ketosis, which remains dominated by threshold-based pathology models focused on metabolite normalization rather than understanding the biological information encoded in metabolic patterns.
This comprehensive review challenges the reductionist pathological paradigm by introducing a systems-level framework for understanding ketone metabolism. We begin by characterizing the
Keto-Inflammatory Network (KIN) - the interconnected system of metabolic, inflammatory, immune, microbial, and circadian pathways through which ketone bodies exert their pleiotropic effects [
5,
6,
7]. This network perspective reveals connections invisible to reductionist approaches: bidirectional metabolic-immune crosstalk, trans-kingdom signaling between host and microbiome, temporal coordination through circadian mechanisms, and multi-scale integration from molecular interactions to whole-organism physiology.
As our analysis deepens, we recognize that network connectivity alone inadequately captures the sophisticated information processing properties exhibited by this system. The evidence compels us toward a more fundamental reconceptualization: the Ketoinflammatory Code - a biological information processing system with true code-like properties including syntax (molecular grammar governing signal patterns), semantics (biological meaning emerging from metabolic signatures), and pragmatics (context-dependent interpretation generating appropriate adaptive responses). This framework positions ketosis not as metabolic failure but as an evolutionarily conserved adaptive program - an encoded biological language through which organisms assess their state, communicate between distributed cellular systems, and coordinate situation-appropriate responses that optimize survival and reproductive success under diverse environmental challenges.
By progressing from network architecture to code comprehension, we provide both deeper mechanistic insights into how ketone signaling operates and more sophisticated therapeutic strategies that support rather than suppress evolved regulatory intelligence. This review synthesizes evidence from molecular biology, immunology, microbiology, chronobiology, and clinical investigation to demonstrate that ketosis represents sophisticated biological adaptation warranting respect and, when necessary, informed modulation - not reflexive pathologization and suppression. The framework we present directly challenges century-old paradigms while providing practical tools for predictive diagnostics and therapeutic innovation in both human and veterinary medicine.
2. The Reductionist Paradigm - Ketosis as Pathology
The dominant medical and veterinary paradigm characterizing ketosis has been shaped by over a century of observations made primarily within pathological contexts, creating a deeply embedded conceptual framework that views any elevation in ketone body concentrations as inherently problematic and requiring therapeutic intervention [
10,
18]. This reductionist perspective emerged from early clinical observations of diabetic ketoacidosis, starvation ketosis in malnourished populations, and periparturient ketosis in high-producing dairy cattle, where extreme ketone elevations were consistently associated with morbidity, reduced performance, and increased mortality risk [
5,
10,
19]. Critically, this pathological framing has systematically ignored the possibility that ketone bodies might function as signaling molecules encoding biological information, instead treating them exclusively as toxic metabolic byproducts requiring elimination.
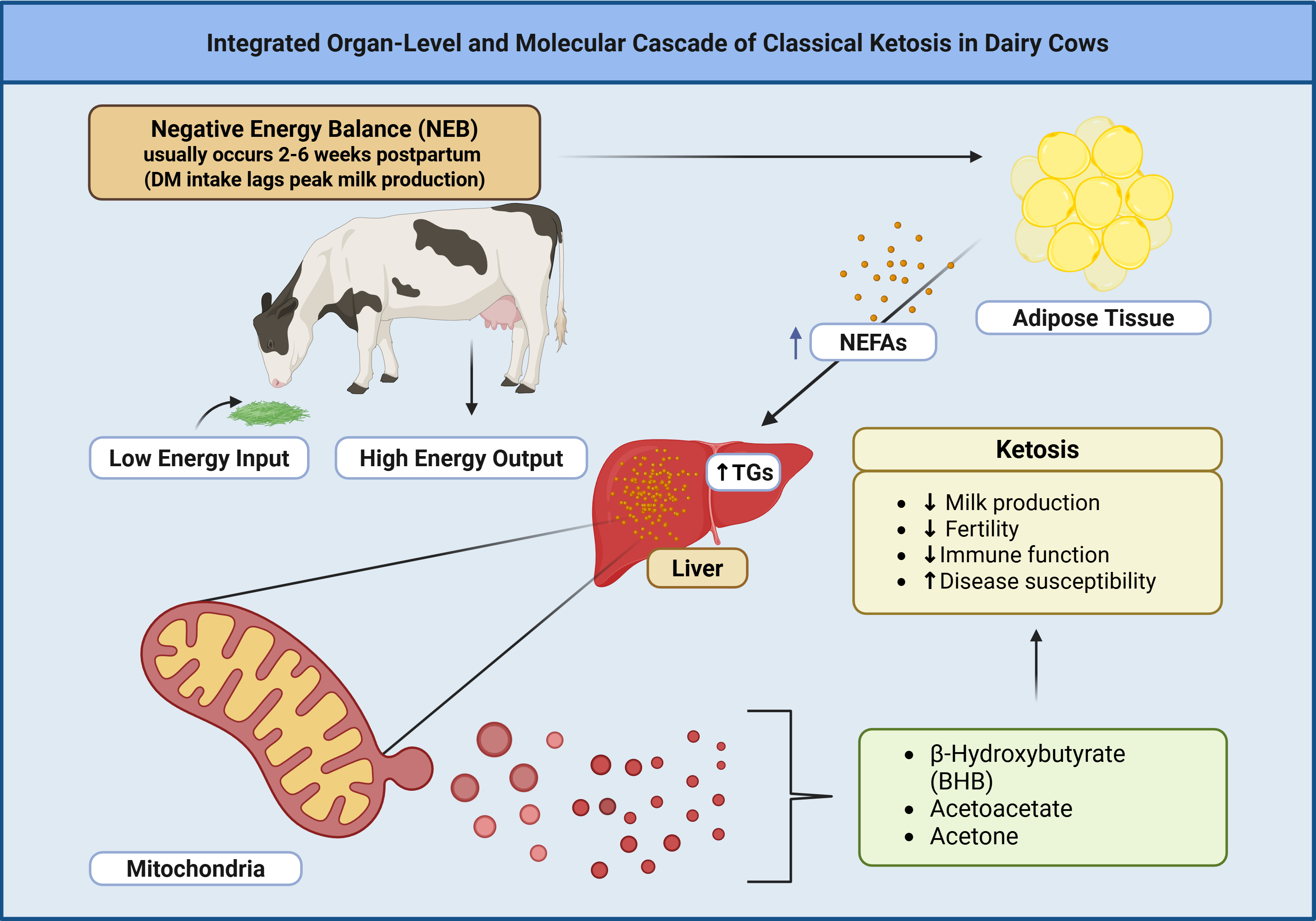
Within the traditional veterinary framework, ketosis in dairy cattle has been conceptualized as a direct consequence of insufficient energy intake relative to the enormous metabolic demands of early lactation milk synthesis (
Figure 1) [
12,
20]. This energy deficit model proposes that when dietary energy intake fails to meet the 40-60% increase in energy requirements associated with peak milk production, stored adipose tissue lipids are mobilized through lipolysis, overwhelming hepatic oxidative capacity and resulting in ketone accumulation [
20,
21]. This framework treats ketone elevation as simple metabolic overflow - analogous to water spilling from an overfilled container - with no consideration that ketone production might be actively regulated to communicate metabolic state to distant tissues. However, recent metabolomics investigations have revealed that the pathophysiology of ketosis is far more complex than simple energy deficiency, with alterations in immune function, inflammatory mediators, and metabolic pathways detectable weeks before clinical disease manifestation [
14]. The predictive power of these early metabolic signatures suggests coordinated biological programming rather than passive metabolic failure - a finding fundamentally incompatible with the overflow model [
14]. The clinical classification system distinguishing between subclinical ketosis (elevated blood BHB concentrations without overt clinical signs) and clinical ketosis has reinforced the perception that any detectable ketone elevation represents pathological departure from normal physiological function [
13,
22].
Epidemiological investigations have consistently documented that 40-60% of periparturient dairy cows experience some degree of ketosis within the first three weeks following calving, with significant economic impacts including direct treatment costs and indirect consequences such as reduced milk yield, impaired reproductive performance, increased susceptibility to infectious diseases, and elevated culling rates [
13,
23]. Importantly, cows with high somatic cell counts prior to dry-off show increased likelihood of developing ketosis, suggesting interconnected disease processes rather than isolated metabolic dysfunction [
24]. These epidemiological associations, while traditionally interpreted as ketosis causing secondary pathologies, are equally consistent with ketosis representing an integrated response to underlying inflammatory or immune dysregulation -a possibility the reductionist paradigm has not adequately explored.
The pathophysiological model underlying traditional ketosis management focuses on the complex endocrine changes accompanying negative energy balance (NEB), including decreased insulin concentrations, elevated glucagon, cortisol, and growth hormone levels that collectively stimulate non-esterified fatty acid (NEFA) mobilization from adipose tissue stores [
25,
26]. These mobilized fatty acids undergo hepatic β-oxidation through the carnitine palmitoyltransferase system, and when the rate of acetyl-CoA production exceeds the oxidative capacity of the tricarboxylic acid cycle, excess acetyl-CoA is diverted into ketogenesis through the mitochondrial enzymes acetyl-CoA acetyltransferase, 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), and HMG-CoA lyase [
4,
27]. While biochemically accurate, this mechanistic description implicitly frames ketogenesis as passive consequence of substrate availability rather than actively regulated biological process - ignoring substantial evidence for transcriptional, post-translational, and allosteric regulation of ketogenic enzymes that would enable ketone production to function as controlled signaling rather than uncontrolled overflow.
Traditional therapeutic approaches have emphasized minimizing energy deficits through strategic supplementation with gluconeogenic precursors including propylene glycol and sodium propionate, energy-dense bypass fats that avoid ruminal fermentation, and feed additives such as monensin that improve feed conversion efficiency and propionate production [
28,
29]. While these approaches have demonstrated efficacy in reducing ketone concentrations and improving short-term clinical outcomes, they fundamentally operate from the assumption that ketosis represents metabolic dysfunction requiring correction rather than adaptive physiology that might serve beneficial biological functions [
5,
30,
31]. More problematically, by focusing exclusively on suppressing ketone production or enhancing clearance, these interventions may inadvertently disrupt biological signaling processes - analogous to treating fever by suppressing thermogenesis without addressing why the organism generated elevated temperature in the first place.
The limitations of this reductionist paradigm have become increasingly apparent as molecular and systems biology reveal sophisticated regulatory networks connecting ketone metabolism with immune function, circadian rhythms, epigenetic regulation, and microbial ecology. The growing recognition that ketone bodies, particularly BHB, function as signaling molecules through G-protein coupled receptors, as histone deacetylase inhibitors influencing gene expression, and as inflammasome regulators modulating immune responses demands fundamental reconceptualization of what ketosis represents biologically [
6,
35]. The remainder of this review presents an alternative framework - grounded in network analysis and information theory - that repositions ketosis from metabolic pathology to sophisticated adaptive program, with profound implications for both mechanistic understanding and therapeutic strategy.
3. The Keto-Inflammatory Network – A Systems Level Framework
The classical reductionist view of ketosis as passive metabolic overflow fundamentally fails to account for the sophisticated regulatory capabilities and evolutionary conservation characterizing ketone body metabolism across diverse species and physiological contexts [
1,
16]. Contemporary systems biology approaches reveal that biological networks operate through complex, multi-layered integration systems capable of generating context-appropriate responses that optimize organismal fitness under varying environmental conditions [
16,
32,
33].
The
Keto-Inflammatory Network (KIN) represents a theoretical framework that reconceptualizes ketosis as a sophisticated, evolutionarily refined biological program in which ketone bodies coordinate metabolic flexibility, immune modulation, stress adaptation, and cellular communication across multiple organ systems (
Figure 2) [
5,
6,
7]. This paradigm shift moves beyond viewing ketones merely as alternative energy substrates or toxic metabolic byproducts to recognizing their roles as regulatory molecules capable of orchestrating complex physiological adaptations that enhance survival and reproductive success under diverse environmental challenges.
3.1. Network Architecture: Molecular Integration and Distributed Processing
The fundamental architecture of the KIN operates through multiple interconnected regulatory nodes that sense, integrate, and respond to diverse physiological signals including energy availability, inflammatory status, microbial ecology, circadian timing, environmental stressors, and developmental stage [
16,
34]. Central to this architecture is BHB, which serves simultaneously as energy substrate, G-protein coupled receptor ligand (GPR109A/HCAR2), histone deacetylase inhibitor, post-translational modifier (β-hydroxybutyrylation), and inflammasome regulator [
6,
7,
35]. This multiplexed functionality creates potential for coordinated responses across multiple biological systems [
35,
36].
The network exhibits architectural features characteristic of evolved biological systems. Redundancy provides fault tolerance through multiple pathways for ketone production (hepatic ketogenesis, astrocytic synthesis) and sensing (receptor-mediated, metabolic, epigenetic). Distributed processing generates emergent behavior from local decisions by hepatocytes, immune cells, neurons, and other cell types, without requiring centralized control. Scale-free topology employs hub nodes such as hepatic ketogenesis and inflammatory mediators to coordinate activity across widely distributed peripheral components [
16,
32]. These properties enable the network to maintain function despite perturbations and to coordinate responses across vastly different biological scales - from molecular interactions within individual cells to whole-organism behavioral and metabolic adjustments.
Critically, the network generates qualitatively different responses depending on the specific combination of input signals, duration of activation, and physiological context [
6,
17]. Identical ketone concentrations produce different biological outcomes based on concurrent inflammatory signals, nutritional status, or circadian phase - a functional plasticity that distinguishes the KIN from simple linear metabolic pathways.
3.2. Predictive Metabolomics: Evidence for Coordinated Programming
The systems veterinary medicine approach has provided compelling experimental evidence that ketosis represents coordinated biological programming rather than acute metabolic failure [
16]. Multi-platform metabolomics investigations reveal that metabolite signatures can distinguish animals that will develop ketosis from those that will remain healthy weeks before clinical manifestation, achieving prediction accuracies exceeding 90% [
93].
The patterns that distinguish pre-ketotic from healthy animals extend far beyond ketone body concentrations themselves, encompassing coordinated alterations in amino acid profiles, lipid species, inflammatory mediators, and microbial metabolites [
93]. This multi-metabolite nature demonstrates that the network operates through coordinated changes across multiple metabolic pathways rather than isolated perturbations in ketone production. The predictive power of these signatures - their ability to forecast outcomes weeks in advance - indicates that the biological system undergoes progressive, orchestrated changes rather than sudden metabolic collapse.
Furthermore, immune system alterations frequently precede rather than follow metabolic dysfunction, challenging assumptions that ketosis represents primary metabolic failure with secondary immune consequences [
14]. This temporal architecture reveals bidirectional metabolic-immune crosstalk, with the network integrating signals from multiple physiological domains to generate adaptive responses. The recognition that immune status modulates metabolic programming fundamentally distinguishes the KIN framework from traditional views of ketosis as purely metabolic pathology.
3.3. Evolutionary Conservation: Fundamental Biological Importance
The evolutionary conservation of KIN components across mammalian species provides compelling evidence for fundamental biological importance extending beyond species-specific metabolic adaptations [
1,
2,
27]. Comparative genomic analyses reveal high conservation of ketogenic enzymes (HMGCS2, BDH1, ACAT1), ketone-sensing receptors (GPR109A), and downstream signaling pathways from rodents to ruminants to primates, suggesting that core network functionality has been maintained through selective pressure over millions of years of vertebrate evolution [
27,
37].
Such deep conservation implies that the network solves fundamental survival problems common to diverse species and ecological niches - coordinating distributed cellular responses to fluctuating nutritional availability while simultaneously modulating immune function, maintaining neural activity, and preserving reproductive capacity. The fact that organisms as metabolically divergent as obligate carnivores and herbivores retain similar ketone signaling machinery suggests that the network's value lies in its capacity to integrate and communicate organismal state across tissues.
The distribution of ketone-responsive elements across functionally diverse tissues -brain, heart, skeletal muscle, immune cells, kidneys, and adipose tissue - indicates that the network evolved as a whole-organism coordination mechanism [
6,
35]. This architectural feature ensures that local cellular decisions align with organism-level metabolic and immunological state, enabling coherent adaptive responses to environmental challenges.
3.4. Implications of the Network Framework
The KIN framework fundamentally challenges reductionist paradigms by demonstrating that ketosis involves coordinated multi-system responses rather than isolated metabolic dysfunction. Network analysis reveals sophisticated regulatory architecture with redundancy, distributed processing, and multi-scale integration capabilities that enable adaptive flexibility under diverse physiological conditions. The predictive power of metabolomics signatures and the evolutionary conservation of network components provide compelling evidence that ketone metabolism represents an adaptive biological program rather than compensatory overflow.
This network perspective provides the foundation for understanding the molecular mechanisms, regulatory interactions, and physiological functions detailed in subsequent sections. The KIN framework enables investigation of specific network components - including calcium integration, circadian coordination, microbial interactions, and epigenetic programming - that collectively generate the sophisticated adaptive responses observed during physiological and pathological ketosis.
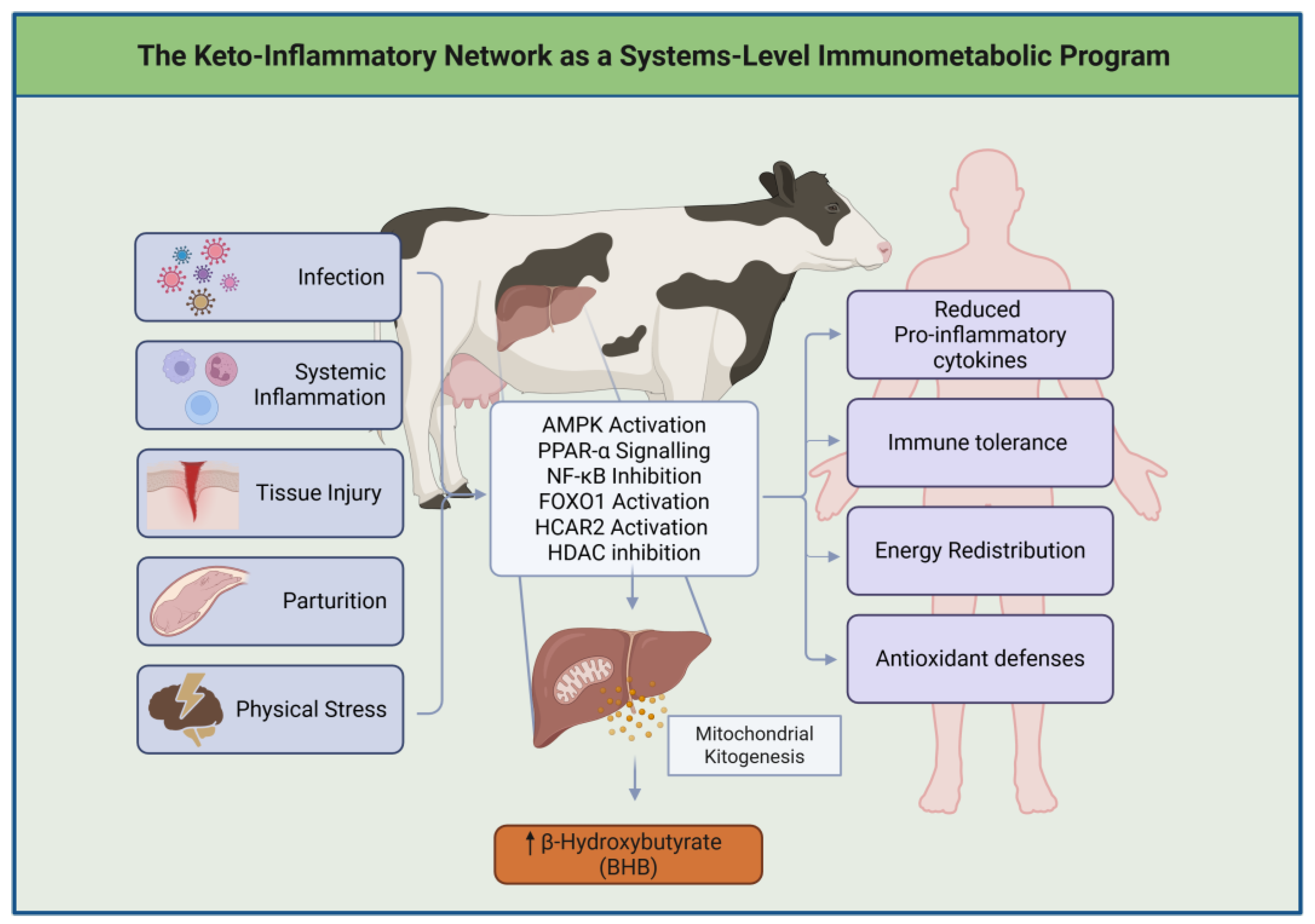
4. Triggers of the Keto-Inflammatory Network
The activation of the KIN extends far beyond carbohydrate scarcity. Rather than functioning solely as a starvation-induced response, the KIN is triggered by a diverse array of physiological and pathological stressors, including systemic inflammation, microbial exposure, tissue injury, intense physical exertion, prolonged fasting, and parturition (
Figure 2) [
38,
39,
40]. These stimuli converge on evolutionarily conserved molecular pathways that coordinate metabolic adaptation with immune regulation.
At the molecular level, these triggers initiate a cascade involving mitochondrial reprogramming, AMP-activated protein kinase (AMPK) activation, peroxisome proliferator-activated receptor alpha (PPAR-α) signaling, and transcriptional networks mediated by NF-κB and FOXO1. These pathways recalibrate energy production, suppress uncontrolled inflammation, and initiate hepatic ketogenesis, placing BHB at the center of an integrated immunometabolic response.
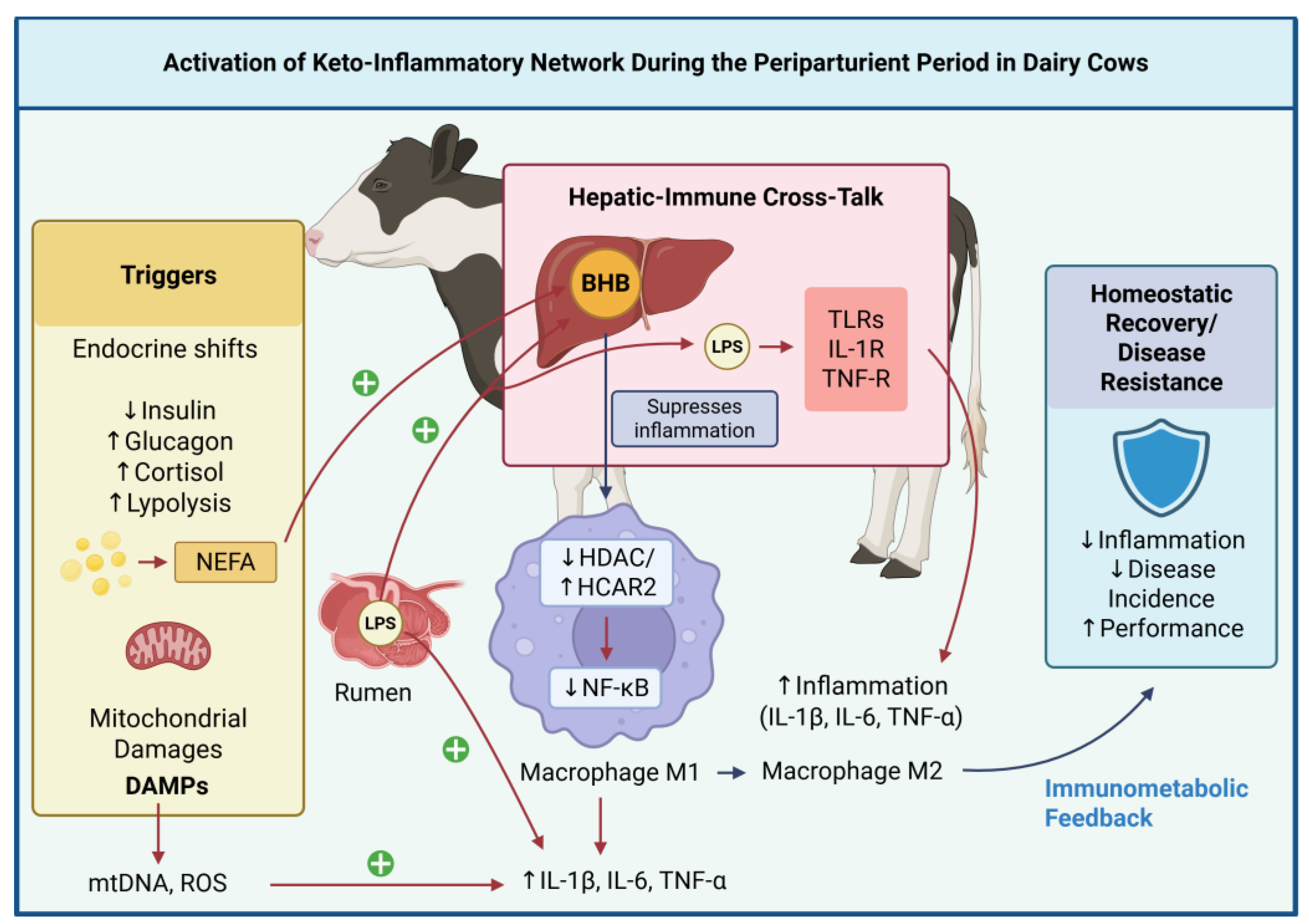
In dairy cows, the periparturient period provides a quintessential model of KIN activation (
Figure 3). During this time, abrupt endocrine shifts, intense lipolysis, microbial components’ translocation (especially lipopolysaccharide, LPS), and elevated proinflammatory cytokines [e.g., Interleukin (IL)-1β, Tumor Necrosis Factor (TNF)-α] converge to stimulate hepatic ketogenesis alongside immune activation [
5,
41,
42]. In humans, similar triggers, such as trauma, infection, caloric restriction, or fasting, activate hepatic oxidative metabolism through PPAR-α and sirtuin pathways, leading to increased BHB synthesis [
7,
40].
Importantly, rising BHB levels often mirror elevations in IL-6 and TNF-α, suggesting a tightly coupled feedback mechanism in which ketogenesis helps suppress excessive immune activation. In this broader framework, ketogenesis is no longer a passive metabolic adaptation to nutrient depletion but a proactive and regulated mechanism of physiological resilience. It coordinates immune tone and metabolic needs in the face of diverse threats, from pathogens to parturition.
The immune system and liver engage in dynamic cross-talk during these events, linking microbial sensing (via pattern recognition receptors such as TLRs, IL-1R, and TNF-R) with hepatic substrate metabolism and mitochondrial ketone biosynthesis [
43,
44]. Within this integrated network, BHB emerges not as a metabolic waste product, but as a central immunometabolic signal that mediates adaptation, preserves tissue integrity, and restores homeostasis. The KIN thus represents a fundamental survival circuit, evolutionarily encoded to maintain balance during metabolic, inflammatory, and environmental perturbations.
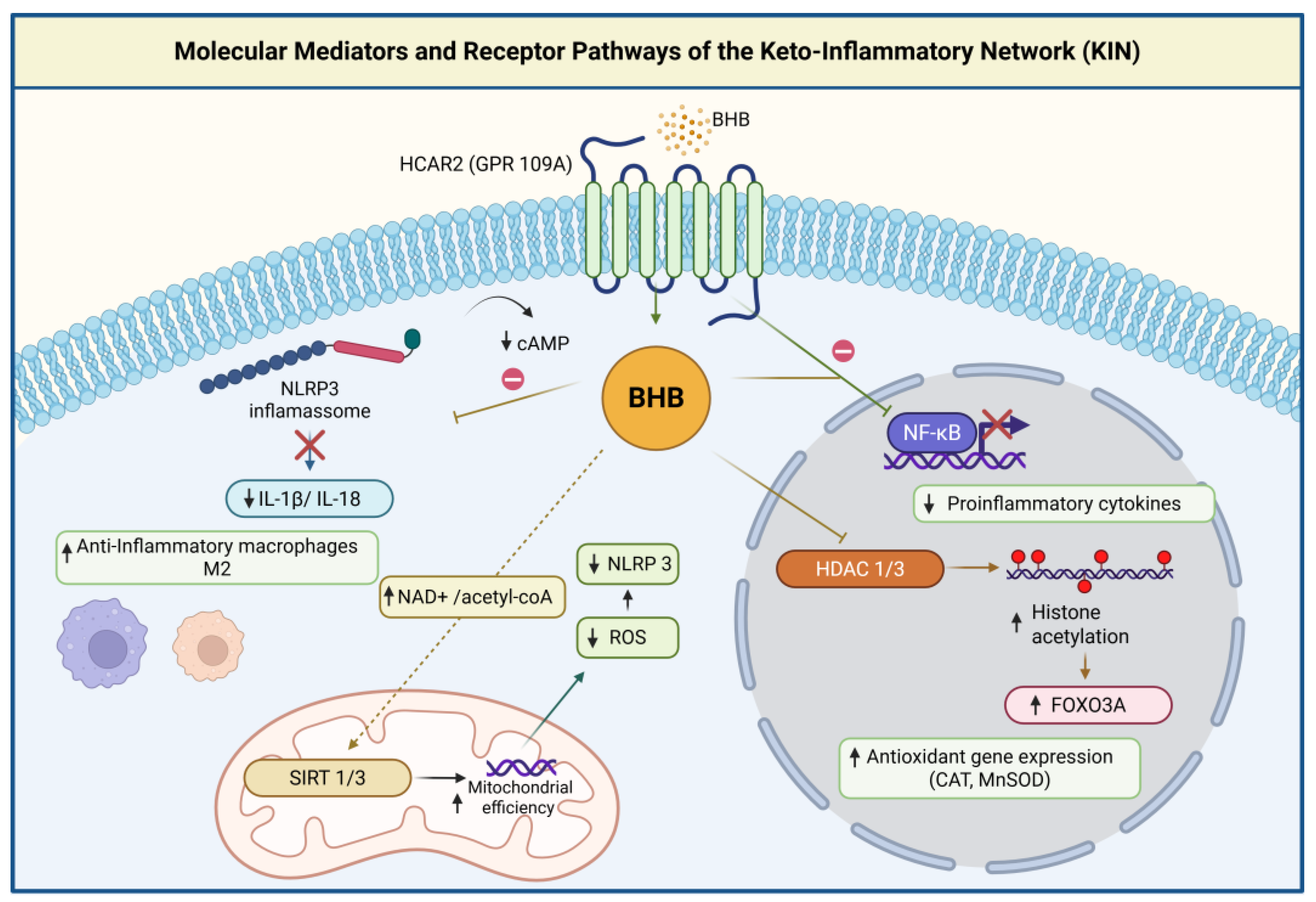
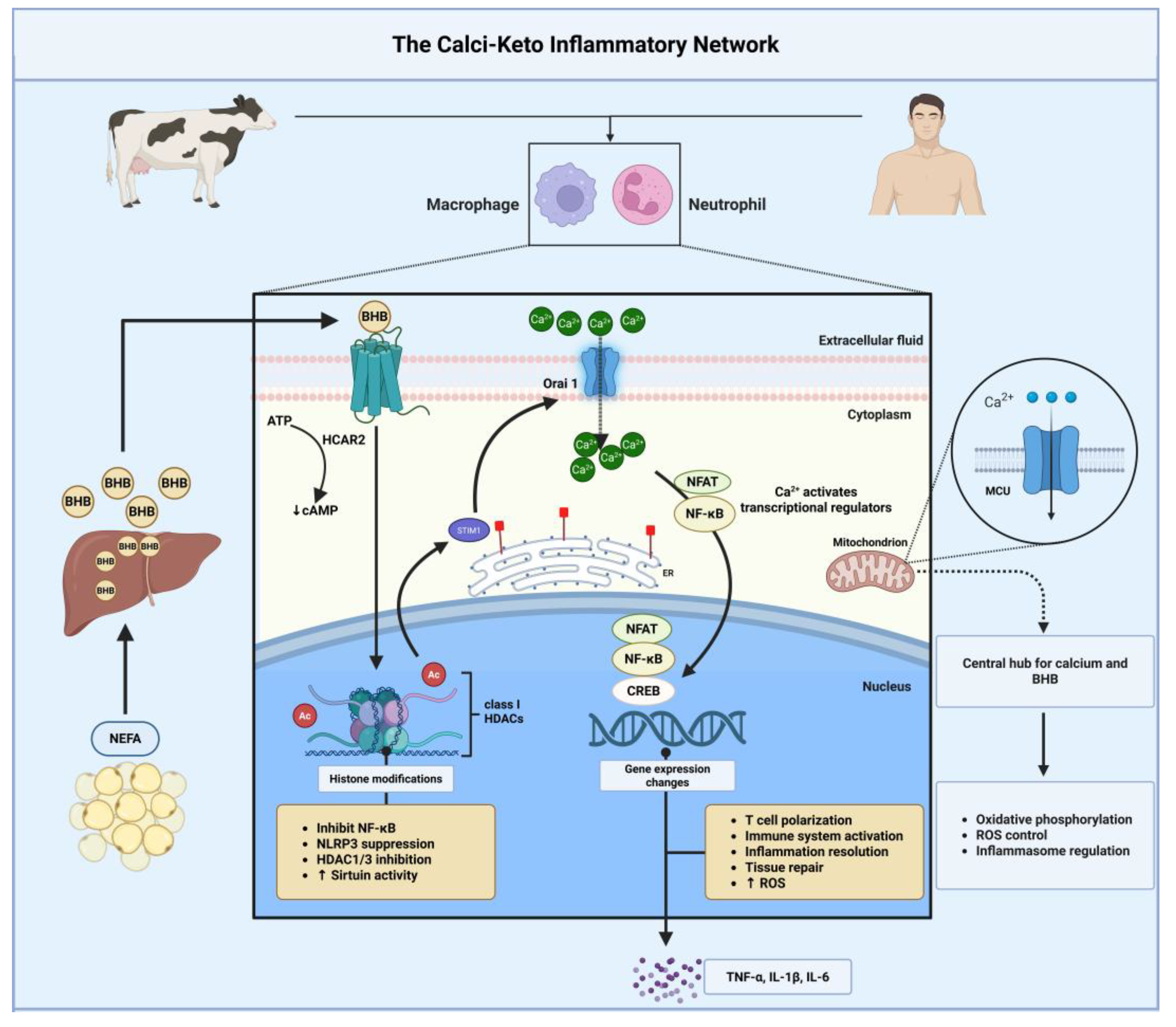
5. Receptors and Molecular Mediators of the Keto-Inflammatory Network
The sophisticated immunomodulatory and metabolic effects of the KIN are mediated through a diverse array of molecular recognition systems and intracellular signaling pathways that enable cells and tissues to detect, interpret, and respond to ketone body availability in highly context-specific ways [
6,
45]. Unlike simple metabolic substrates that primarily influence cellular energetics, ketone bodies, particularly BHB, interact with multiple classes of cellular receptors, enzymes, and regulatory proteins to generate coordinated changes in gene expression, protein function, and cellular behavior (
Figure 4) [
6,
7].
5.1. G-Protein Coupled Receptor Signaling
The hydroxycarboxylic acid receptor 2 (HCAR2, also known as GPR109A) represents the most extensively characterized and clinically relevant receptor system for BHB signaling [
45,
46]. Originally identified as a receptor for nicotinic acid, HCAR2 demonstrates high affinity binding for BHB with physiologically relevant activation occurring at concentrations typically observed during fasting, exercise, or metabolic stress [
45,
47]. HCAR2 expression is particularly abundant in immune cells including macrophages, neutrophils, dendritic cells, and certain T cell subsets, suggesting specialized roles in immune system modulation [
45,
48].
HCAR2 activation triggers Gi/Go-protein coupled signaling cascades that result in decreased intracellular cyclic adenosine monophosphate (cAMP) concentrations, leading to downstream suppression of protein kinase A activity and reduced phosphorylation of cAMP response element-binding protein (CREB) [
33]. This signaling cascade ultimately results in decreased nuclear factor kappa B (NF-κB) activity, reduced transcription of pro-inflammatory cytokine genes including TNF-α, IL-1β, and IL-6, and enhanced expression of anti-inflammatory mediators such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) [
14,
45,
49].
5.2. Epigenetic Regulation Through Histone Deacetylase Inhibition
Among the most significant discoveries in ketone body research has been the identification of BHB as an endogenous inhibitor of class I histone deacetylases (HDACs), particularly HDAC1, HDAC2, and HDAC3 [
35,
50]. This mechanism of action places ketone bodies within the broader category of epigenetic modulators capable of inducing lasting changes in gene expression patterns through alterations in chromatin structure and accessibility [
36,
51].
The selective inhibition of class I HDACs by BHB occurs through competitive binding at the enzyme active site, with inhibition constants in the millimolar range that are readily achieved during physiological ketosis [
35]. HDAC inhibition by BHB leads to increased acetylation of histones H3 and H4 at specific gene loci, resulting in chromatin relaxation and enhanced transcriptional accessibility [
36,
50]. Key target genes include those encoding antioxidant defense enzymes (catalase, superoxide dismutase, glutathione peroxidase), stress response transcription factors (FOXO3a, Nrf2), and anti-inflammatory mediators (IL-10, arginase 1) [
52,
53].
5.3. NAD+-Dependent Sirtuin Activation
The sirtuin family of NAD
+-dependent protein deacetylases represent another critical molecular target through which ketone bodies exert regulatory effects on cellular function [
54,
55]. Sirtuins, particularly SIRT1 and SIRT3, play essential roles in metabolic regulation, stress resistance, and longevity pathways through their ability to deacetylate key regulatory proteins in both nuclear and mitochondrial compartments [
56,
57].
Ketone body metabolism influences sirtuin activity through multiple mechanisms including direct provision of NAD
+ cofactor, enhancement of the NAD
+/NADH ratio, and indirect effects on NAD
+ biosynthetic pathways [
54,
58]. SIRT1 activation by enhanced NAD
+ availability leads to deacetylation of transcription factors including p53, FOXO family members, and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), resulting in enhanced stress resistance, improved mitochondrial biogenesis, and increased oxidative metabolism [
59,
60].
5.4. Inflammasome Modulation and Innate Immune Signaling
The NLRP3 inflammasome represents a critical convergence point where ketone body signaling intersects with innate immune activation pathways [
48,
61]. BHB suppresses NLRP3 inflammasome activation through multiple complementary mechanisms including stabilization of mitochondrial membrane potential, reduction in mitochondrial reactive oxygen species (ROS) production, prevention of potassium efflux, and direct interference with inflammasome complex assembly [
48,
61]. These effects collectively prevent caspase-1 activation and reduce the processing and release of pro-inflammatory cytokines IL-1β and IL-18, which play central roles in sterile inflammation and metabolic dysfunction [
48,
62].
Metabolomics investigations have revealed that cows destined to develop ketosis show elevated concentrations of inflammatory mediators including IL-6 and TNF-α weeks before clinical disease manifestation, suggesting that inflammasome modulation represents a critical early intervention point in ketosis pathophysiology [
14].
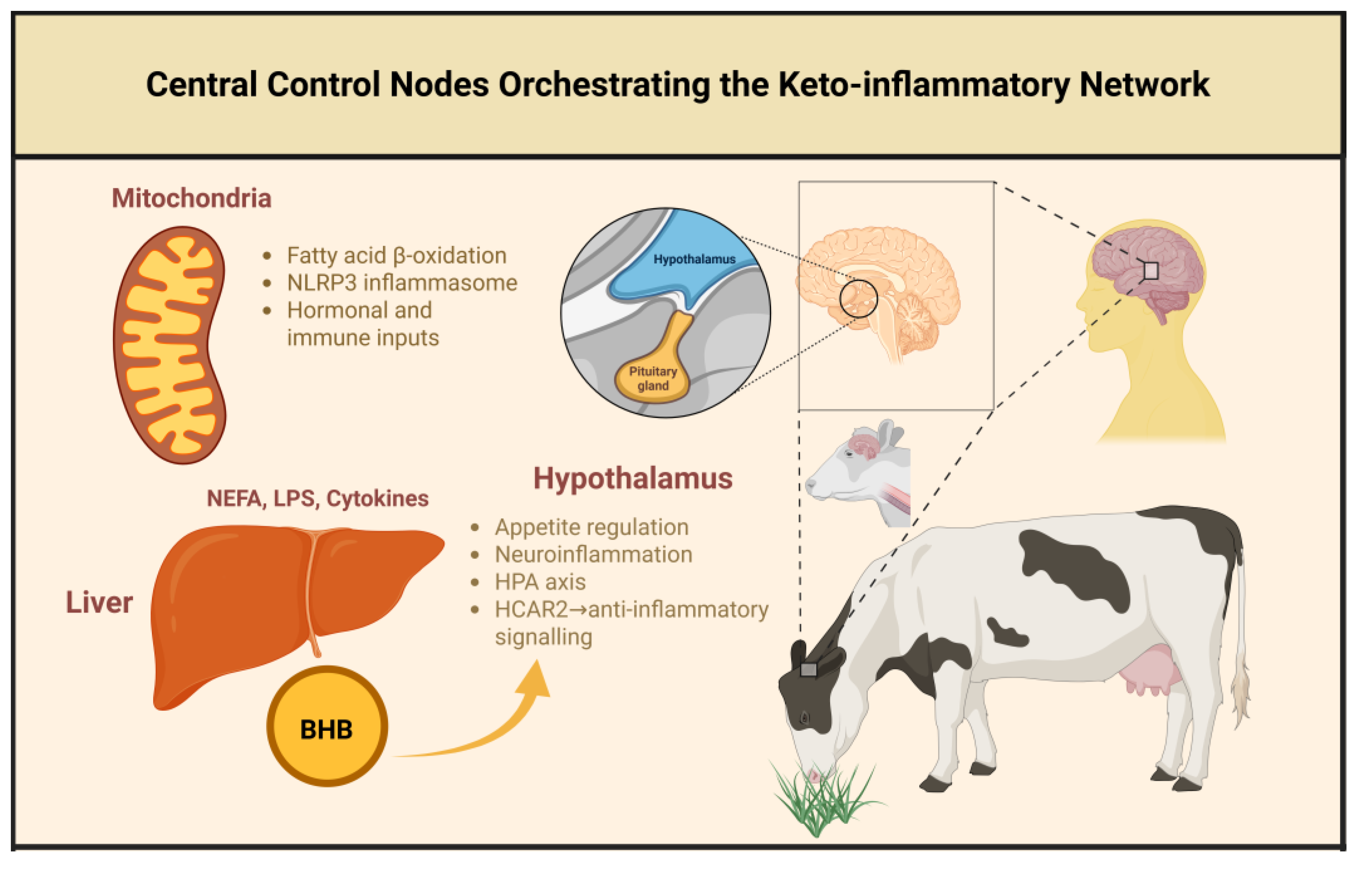
6. Central Control Nodes: Hypothalamic Integration and Circadian Coordination
The sophisticated coordination of KIN responses across multiple organ systems requires centralized integration mechanisms capable of processing diverse physiological signals and generating appropriate systemic responses [
63,
64]. The hypothalamus, functioning as the primary neuroendocrine control center, serves as a critical integration node where metabolic status, immune signals, circadian timing, and environmental information converge to regulate KIN activation and coordinate organism-wide adaptive responses (
Figure 5) [
63,
65].
6.1. Hypothalamic Ketone Sensing and Neuroendocrine Integration
The blood-brain barrier readily permits the passage of ketone bodies, enabling direct access to hypothalamic neurons that express specialized metabolic sensing mechanisms [
63,
66]. Multiple hypothalamic nuclei, including the arcuate nucleus, paraventricular nucleus, and lateral hypothalamus, contain neurons that respond to changes in ketone body concentrations through alterations in membrane potential, neurotransmitter release, and neuropeptide production [
63,
67].
Ketone-sensitive neurons within the arcuate nucleus modulate the activity of key metabolic regulatory circuits including neuropeptide Y (NPY)/agouti-related peptide (AgRP) neurons that stimulate feeding behavior and pro-opiomelanocortin (POMC) neurons that promote satiety and energy expenditure [
63]. During periods of elevated ketosis, the balance between these neuronal populations shifts to favor energy conservation through reduced thermogenesis, decreased locomotor activity, and altered feeding patterns [
63,
68].
6.2. Circadian Clock Integration and Temporal Coordination
The intersection of ketone metabolism with circadian biology represents a fundamental aspect of KIN function that has received increasing scientific attention [
69,
70]. Circadian clocks, present in virtually all cells and tissues, generate approximately 24-hour rhythms in gene expression, protein synthesis, and metabolic activity that optimize physiological function according to predictable daily cycles of feeding, fasting, activity, and rest [
69,
71].
Hepatic ketogenesis exhibits robust circadian rhythmicity, with peak ketone production typically occurring during the fasting phase of the daily cycle [
70,
72]. This temporal pattern is regulated by the coordinated expression of core clock genes including CLOCK, BMAL1, Period (PER), and Cryptochrome (CRY) that directly control the transcription of key ketogenic enzymes including HMGCS2 and enzymes involved in fatty acid oxidation [
69,
70]. The relationship between ketosis and circadian function extends beyond simple temporal regulation to include direct molecular interactions where ketone bodies can influence clock gene expression and circadian rhythm amplitude [
58,
59].
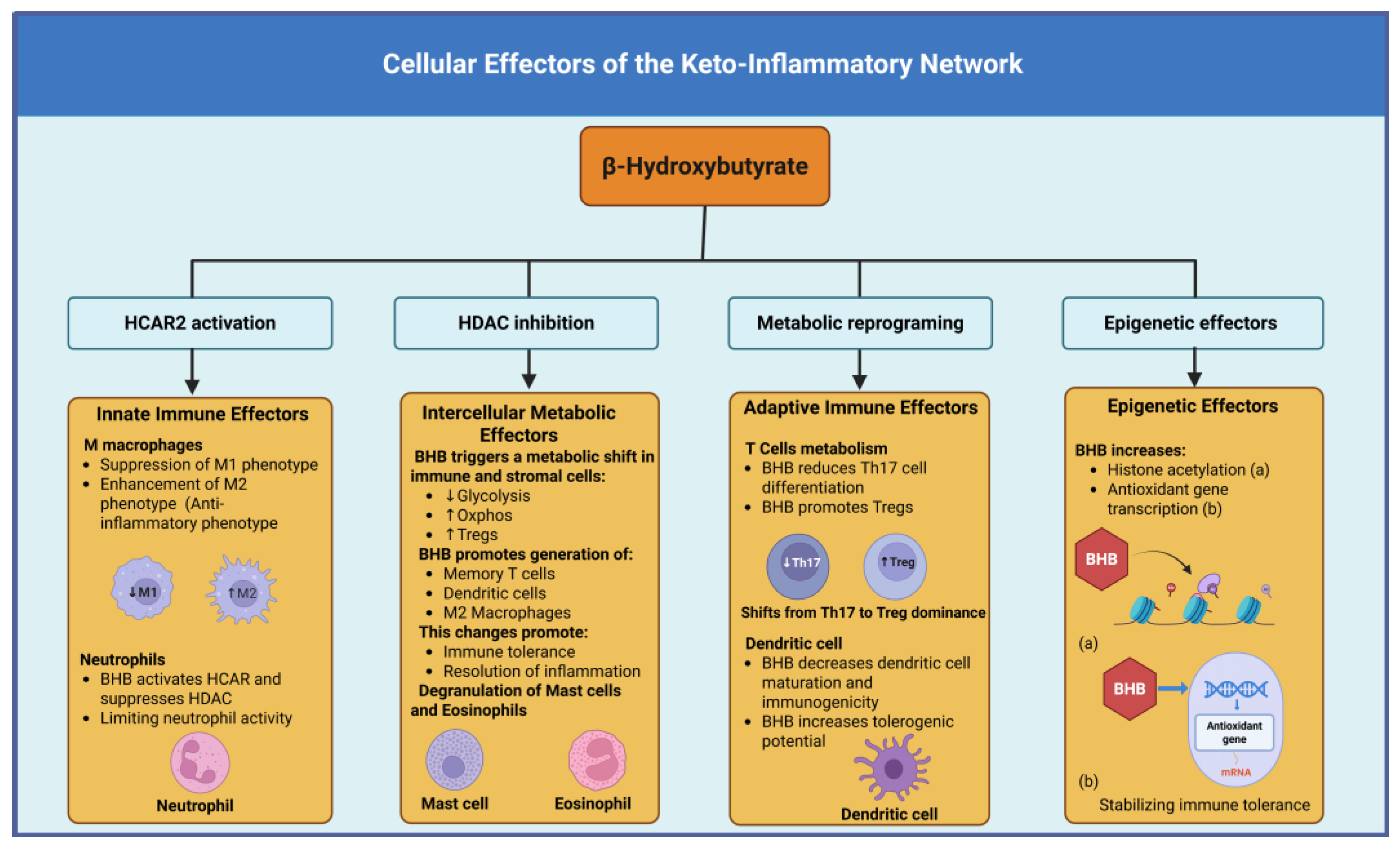
7. Cellular Effectors of the Keto-Inflammatory Network
The functional implementation of KIN signaling occurs through coordinated responses across diverse cellular populations, each contributing specialized capabilities that collectively generate the complex physiological adaptations characteristic of effective ketotic states [
73,
74]. The cellular effectors of the KIN encompass both immune and non-immune cell types, with their responses being shaped by cell-specific expression patterns of ketone-sensing receptors, metabolic enzymes, and downstream signaling components (
Figure 6) [
14,
73].
7.1. Myeloid Cell Populations and Innate Immunity
Macrophages represent primary cellular mediators through which the KIN exerts its anti-inflammatory and tissue-protective effects [
73,
75]. The functional plasticity of macrophages, encompassing a spectrum from classically activated (M1) pro-inflammatory states to alternatively activated (M2) anti-inflammatory and tissue repair phenotypes, provides an ideal cellular system for ketone-mediated immune modulation [
73,
74].
Ketone body exposure shifts macrophage polarization toward M2-like phenotypes characterized by enhanced oxidative metabolism, increased production of anti-inflammatory cytokines including IL-10 and TGF-β, elevated expression of tissue repair mediators such as arginase 1 and insulin-like growth factor 1, and improved phagocytic clearance of apoptotic cells and cellular debris [
45,
76]. These changes occur through multiple molecular mechanisms including HCAR2 receptor activation, metabolic reprogramming toward oxidative phosphorylation, and epigenetic modifications that favor anti-inflammatory gene expression programs [
45,
77].
Remarkably, dairy cows destined to develop ketosis show elevated concentrations of inflammatory mediators including IL-6, TNF-α, and serum amyloid A beginning at -8 and -4 weeks prior to parturition, indicating that immune system activation precedes rather than follows metabolic dysfunction [
14]. This temporal sequence suggests that immune system modulation represents a primary rather than secondary component of ketosis pathophysiology.
7.1. Lymphocyte Populations and Adaptive Immunity
T lymphocyte responses represent critical components of KIN function, with ketone body availability during T cell activation influencing both immediate functional responses and long-term memory formation [
78,
79]. The metabolic requirements of T cell activation, including the need for rapid ATP production, biosynthetic precursor generation, and sustained energy supply for proliferation and effector function, make T cells particularly sensitive to changes in substrate availability and metabolic environment [
78,
80].
CD4
+ T helper cell differentiation represents a key target for ketone-mediated immune modulation, with particular effects on the balance between pro-inflammatory Th17 cells and immunosuppressive regulatory T cells (Tregs) [
81,
82]. Ketone availability during T cell activation favors Treg differentiation through mechanisms including enhanced oxidative metabolism, increased expression of the transcription factor FoxP3, and epigenetic modifications that stabilize the regulatory T cell phenotype [
82,
83].
CD8
+ T cell memory formation represents another important aspect of KIN function with potential implications for vaccine responses, cancer immunotherapy, and long-term immune protection [
79,
84]. Memory CD8
+ T cells rely heavily on oxidative metabolism and fatty acid oxidation for their survival and rapid recall responses, making them well-suited to benefit from ketone body availability [
79,
80]. T cells activated in the presence of elevated ketone concentrations exhibit enhanced memory characteristics including improved survival, increased expression of memory-associated transcription factors, and enhanced functional capacity upon secondary antigen encounter [
84,
85].
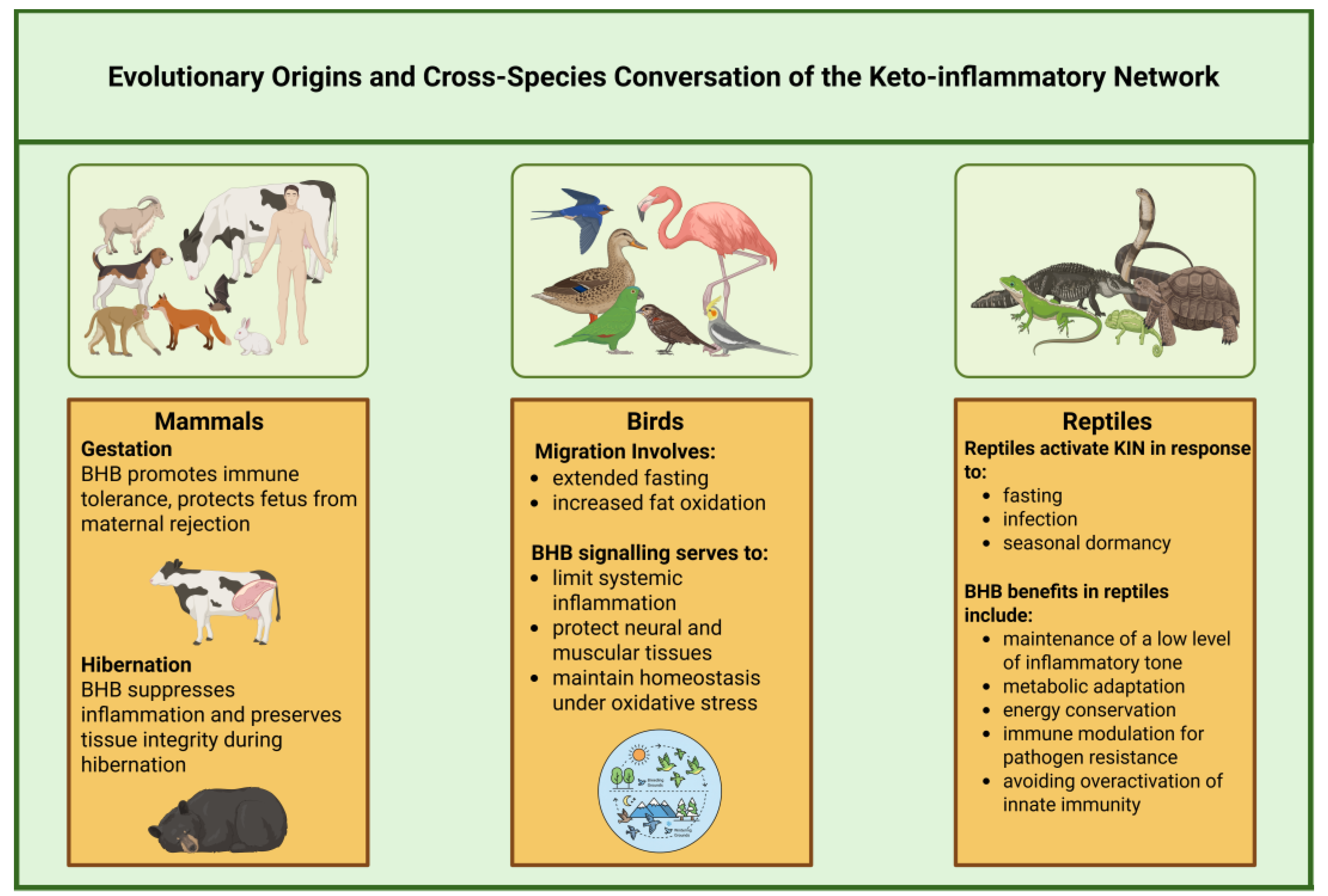
8. Evolutionary Origins and Conservation of the Keto-Inflammatory Network
The remarkable conservation of ketogenic pathways and associated regulatory mechanisms across vertebrate evolution provides compelling evidence for the fundamental adaptive significance of the KIN [
1,
2]. Phylogenetic analyses reveal that core components of ketone metabolism, including the key biosynthetic enzymes, transport proteins, and sensing mechanisms, have been maintained with high fidelity across hundreds of millions of years of evolutionary divergence (
Figure 7) [
27,
37].
8.1. Comparative Physiology Across Taxa
The utilization of ketosis as an adaptive strategy extends across diverse mammalian lineages, each demonstrating species-specific modifications that optimize ketogenic responses for particular ecological niches and life history strategies [
1,
86]. Arctic mammals, including seals, bears, and arctic ground squirrels, rely heavily on ketogenic metabolism during extended periods of food unavailability, with some species maintaining ketotic states for months during hibernation or migration [
1,
87].
Migratory species demonstrate particularly sophisticated ketogenic adaptations that support sustained locomotor performance under conditions of limited food access [
8,
88]. Long-distance migratory birds can maintain ketotic states throughout transcontinental flights lasting days to weeks, with ketone bodies providing a metabolically efficient fuel source that maximizes flight range while minimizing body weight [
8,
89]. The anti-inflammatory effects of ketosis during these extended periods of physical stress help prevent exercise-induced tissue damage and maintain immune function despite the physiological challenges of long-distance migration [
89,
90].
8.2. Domestication and Artificial Selection Pressures
The intensive artificial selection applied to domestic animals, particularly dairy cattle, has created physiological challenges that may exceed the adaptive capacity of natural ketogenic responses [
12,
91]. Modern dairy cows produce 8-10 times more milk than their wild ancestors, creating energetic demands that can overwhelm the regulatory mechanisms that normally coordinate ketosis with immune function and metabolic adaptation [
5,
12]. Understanding the evolutionary context of ketogenic adaptation provides important insights for developing management strategies that work with, rather than against, natural biological processes [
92,
93].
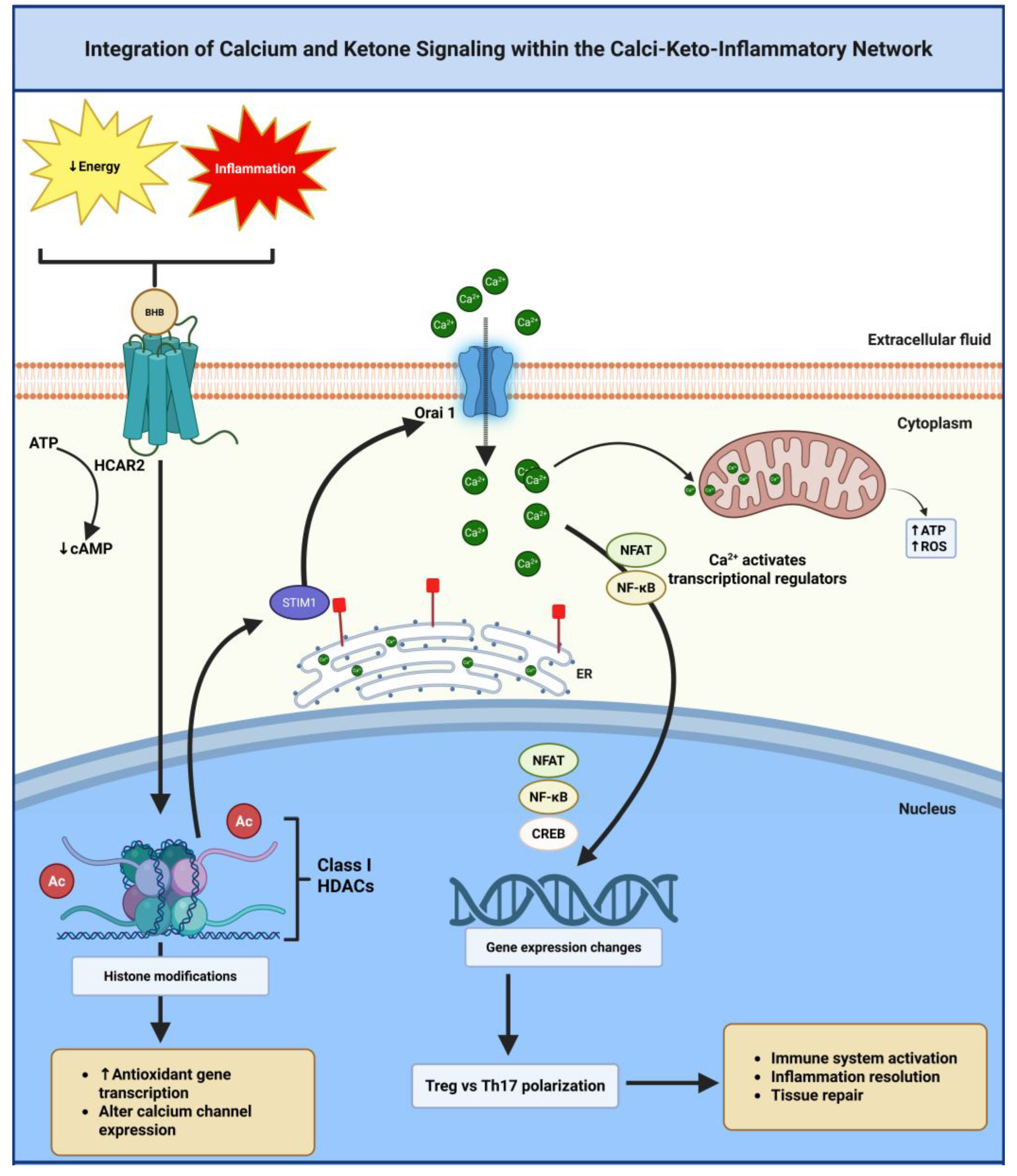
9. The Calcio-Keto-Inflammatory Network: Integrating Calcium and Ketone Signaling
The intersection between calcium homeostasis and ketone metabolism represents a sophisticated regulatory interface that has profound implications for immune function, cellular signaling, and physiological adaptation [
94,
95]. The
Calci-Keto-Inflammatory Network (CKIN) describes the dynamic integration of calcium-dependent signaling pathways with ketone-mediated metabolic and immune responses, creating emergent regulatory properties that exceed the capabilities of either system operating independently (
Figure 8) [
92,
96].
Recent paradigm-shifting research has revealed that the coordinated occurrence of hypocalcemia and hyperketonemia during the periparturient period in dairy cattle may represent adaptive biological programming rather than independent pathological processes [
92]. The calci-inflammatory network (CIN) provides a conceptual framework for understanding how calcium signaling coordinates with inflammatory responses to optimize physiological adaptation during periods of intense metabolic demand [
92].
9.1. Advanced Molecular Mechanisms and Clinical Integration
Calcium (Ca²⁺) serves as a ubiquitous second messenger in nearly all eukaryotic cells, governing gene transcription, cellular excitability, mitochondrial respiration, and immune activation. In immune cells, calcium influx through store-operated calcium entry (SOCE) channels - particularly those formed by STIM1 and Orai1 proteins activates transcription factors such as NFAT, NF-κB, and CREB, which drive cytokine production, cell proliferation, and survival [
97,
98]. Concurrently, BHB modulates immune responses by inhibiting HDACs, suppressing NLRP3 inflammasome activation, enhancing antioxidant defense, and promoting anti-inflammatory macrophage and T cell phenotypes through engagement of receptors such as HCAR2 (
Figure 9) [
38,
39,
99].
The mechanistic convergence between calcium and ketone signaling is particularly evident at the mitochondrial level. Mitochondria serve as central hubs for both Ca²⁺ buffering and oxidative metabolism. BHB maintains mitochondrial membrane potential, enhances respiratory efficiency, and reduces ROS generation - factors that stabilize intracellular calcium flux and protect immune cells from oxidative and inflammatory stress [
44,
100]. Through these effects, BHB modulates the threshold for calcium-induced immune activation and supports bioenergetic integrity.
9.2. Clinical Applications and Therapeutic Implications
The CKIN provides a mechanistic framework for understanding the co-occurrence of hypocalcemia and hyperketonemia in periparturient dairy cows. This convergence reflects coordinated adaptive programming rather than dual pathology, with mitochondria serving as the critical integration point.
Mitochondria function as central hubs integrating calcium and ketone signaling. They simultaneously buffer Ca²⁺ and conduct oxidative metabolism, with BHB playing modulatory roles across both processes. BHB maintains mitochondrial membrane potential, enhances respiratory efficiency, and reduces ROS generation - collectively stabilizing intracellular calcium flux and protecting immune cells from oxidative and inflammatory stress [
44,
100]. Through these mechanisms, BHB modulates the threshold for calcium-induced mitochondrial membrane permeabilization while supporting bioenergetic integration.
9.3. Clinical Applications and Therapeutic Implications
The CKIN provides a mechanistic framework for understanding the co-occurrence of hypocalcemia and hyperketonemia in periparturient dairy cows. This convergence reflects coordinated adaptive programming: a strategy to transiently downregulate immune reactivity, reduce metabolic strain, and allocate energy toward lactation and tissue remodeling [
5,
43].
In humans, disruption of this coordinated axis is increasingly recognized in the pathophysiology of chronic inflammation, metabolic syndrome, autoimmune disease, and neurodegeneration, where calcium mishandling and impaired ketogenesis co-occur [
40,
101]. The enhancement of CD8+ T cell memory and anti-tumor immunity through ketogenic interventions represents a particularly promising therapeutic application [
97], while ketogenic approaches show remarkable efficacy in alleviating colitis through modulation of gut immune cell populations [
99].
9.4. The "Pedal and Brake" System
The CKIN bridges three previously isolated domains - mineral signaling, ketone metabolism, and immune regulation - into an integrated "pedal and brake" system governing immune activation and resolution. This represents a paradigm shift: recognizing that optimal health requires understanding how energy metabolism, calcium dynamics, and immune control function as a co-evolved, adaptive network. CKIN opens new therapeutic avenues for metabolic-inflammatory diseases across species, providing a co-evolved mechanism of physiological resilience during stress [
100,
102].
9.5. Cellular Calcium Dynamics and Ketone Interactions
Calcium serves as a universal intracellular signaling molecule regulating diverse cellular processes including enzyme activation, gene transcription, membrane excitability, vesicle trafficking, and programmed cell death [
95,
97]. Ketone bodies influence cellular calcium dynamics through multiple interconnected mechanisms spanning membrane transport, intracellular buffering, and mitochondrial calcium handling [
94,
103].
BHB enhances the expression and activity of sarcoplasmic/endoplasmic reticulum Ca²⁺-ATPases (SERCA pumps), leading to improved calcium sequestration within intracellular stores and enhanced cellular calcium buffering capacity [
104]. Additionally, HCAR2 receptor activation by BHB modulates Ca²⁺ signaling: it activates phospholipase C and inositol 1,4,5-trisphosphate (IP3) generation, resulting in controlled calcium release from endoplasmic reticulum stores [
45,
105]. These dual mechanisms - enhanced sequestration and modulated release - enable BHB to stabilize calcium homeostasis during metabolic stress.
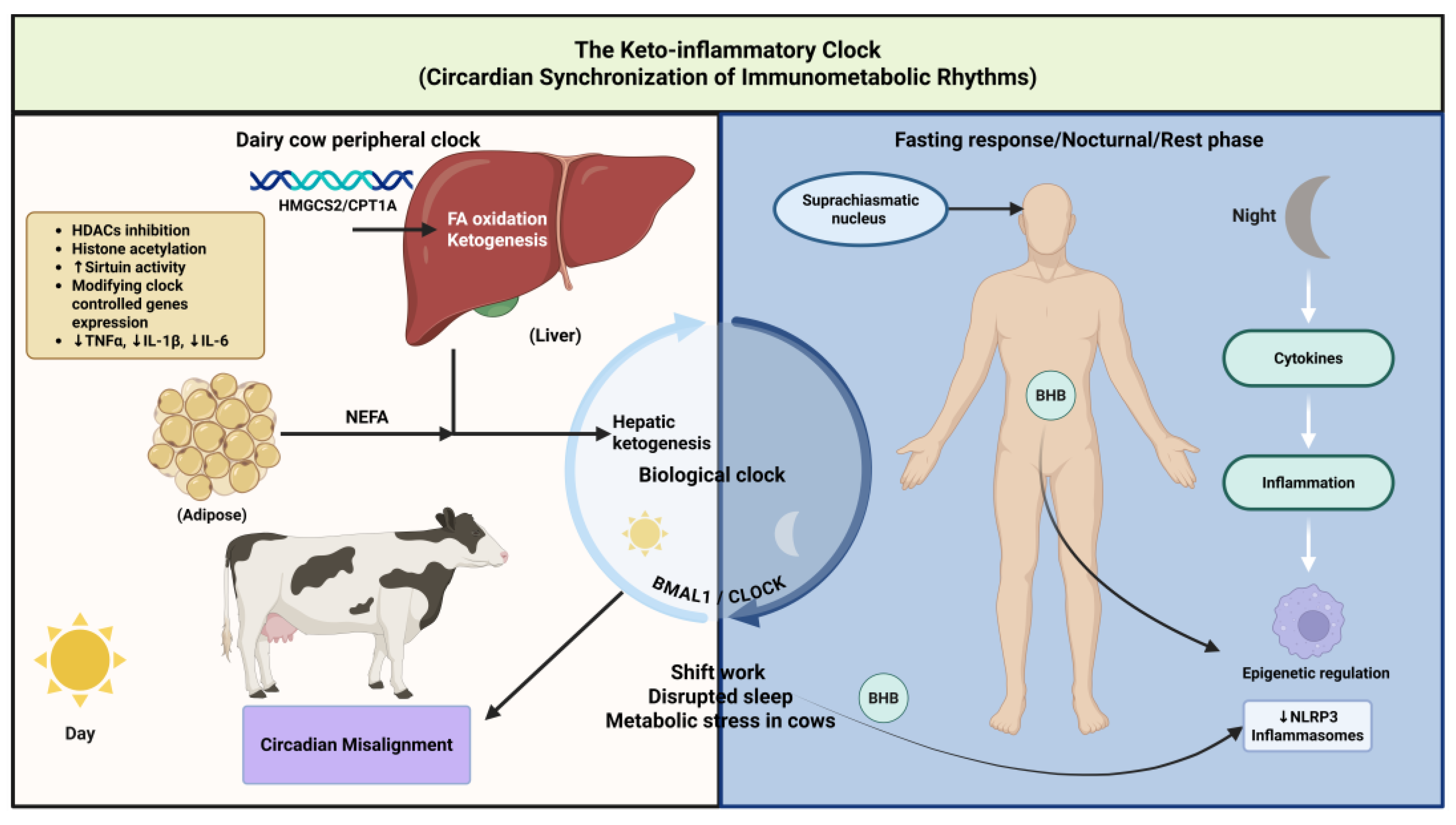
10. The Ketoinflammatory Clock: Circadian Synchronization of Immunometabolic Rhythms
Temporal coordination of metabolic and immune functions represents a fundamental organizing principle in biology, with circadian rhythm disruption contributing to metabolic syndrome, autoimmune disease, and inflammatory disorders [
109,
110]. The intersection of ketone metabolism with circadian biology creates the
Ketoinflammatory Clock - a regulatory system governing daily fluctuations in immune responsiveness, metabolic flexibility, and stress adaptation (
Figure 10) [
16,
69,
70].
10.1. Molecular Circadian Control of Ketogenesis
Hepatic ketone body production exhibits robust circadian rhythmicity coordinated with feeding-fasting cycles [
70,
71]. This temporal regulation emerges from core clock genes including CLOCK, BMAL1, Period (PER), and Cryptochrome (CRY), which generate approximately 24-hour oscillations through interlocking transcriptional-translational feedback loops [
69,
111].
The CLOCK-BMAL1 heterodimer functions as a master transcriptional activator regulating expression of key metabolic genes including those controlling ketogenesis (HMGCS2, BDH1), fatty acid oxidation (CPT1A, ACOX1), and glucose metabolism (G6PC, PCK1) [
69,
111]. This molecular architecture ensures that ketogenic capacity peaks during the typical fasting period, optimizing metabolic efficiency. Additional circadian regulation occurs through rhythmic expression of nuclear receptors including peroxisome proliferator-activated receptor α (PPARα) and REV-ERBα, which serve as molecular links between circadian timing and metabolic regulation [
69,
111].
10.2. Circadian Regulation of Immune Function
Immune cells possess functional circadian clocks that autonomously regulate diverse aspects of immune function including cell trafficking patterns, activation thresholds, cytokine production kinetics, and antimicrobial activity [
110,
113]. The coordination of immune rhythms with metabolic cycles through the Ketoinflammatory Clock provides a mechanism for optimizing immune responses according to predicted pathogen exposure patterns and energy availability [
110,
114]. This temporal alignment ensures that energetically expensive immune surveillance peaks during periods of anticipated pathogen encounter (such as feeding times when microbial exposure increases), while dampening inflammatory responses during fasting periods when metabolic resources are directed toward cellular maintenance and repair. The integration of ketone signaling with circadian immune regulation thus enables strategic allocation of metabolic and immune resources across the 24-hour cycle, balancing pathogen defense against metabolic efficiency.
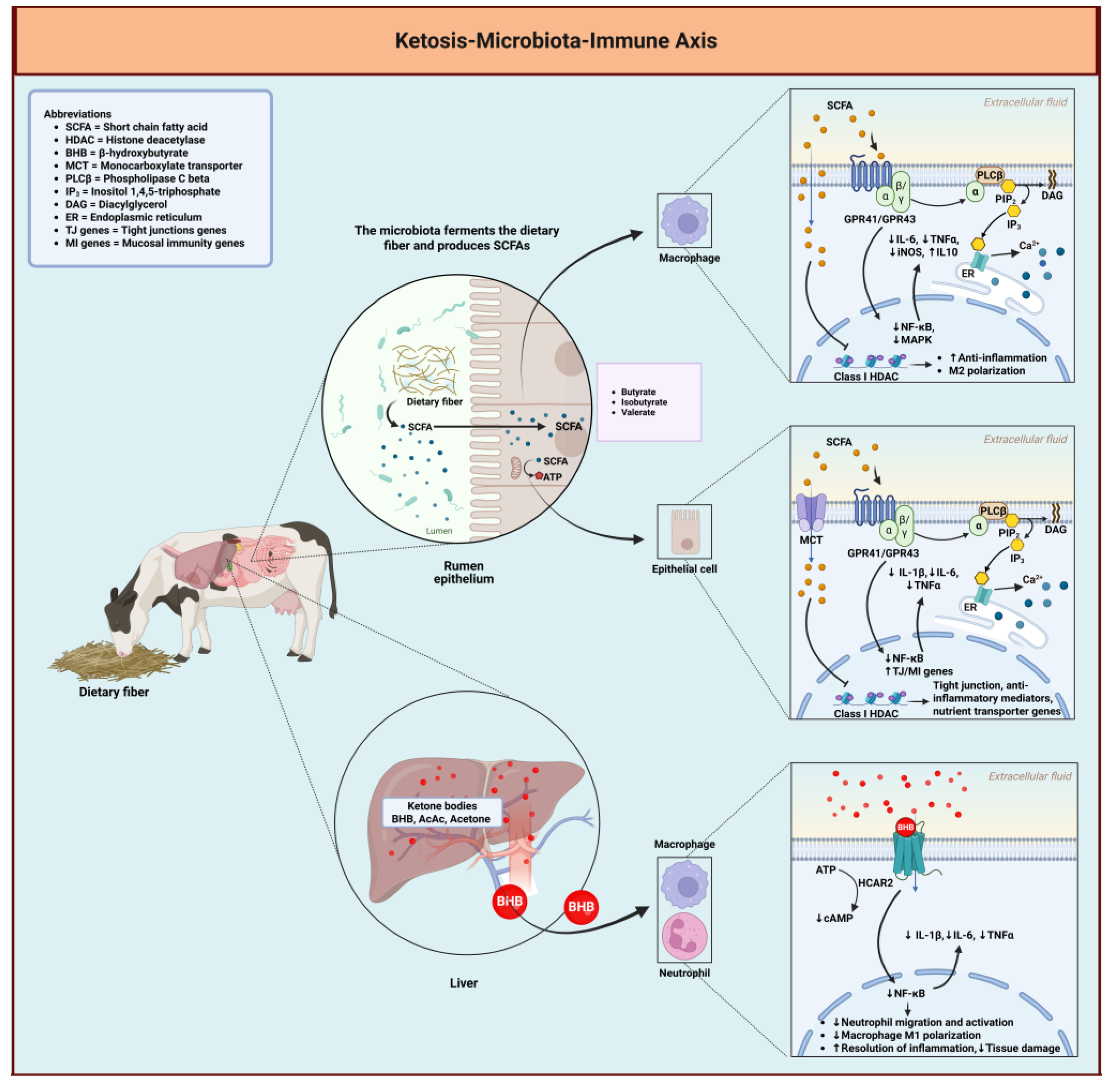
11. The Ketosis-Microbiota-Immune Triad: Trans-Kingdom Signaling
The recognition that host metabolism and microbial ecology function as integrated systems has fundamentally transformed our understanding of health, disease, and metabolic regulation [
34,
115]. The ketosis-microbiota-immune triad exemplifies trans-kingdom signaling, where host-derived ketone bodies and microbe-derived metabolites converge on shared regulatory pathways to coordinate systemic physiological responses (
Figure 11) [
81,
116].
11.1. Evolutionary Origins of Host-Microbe Metabolic Integration
The intimate metabolic relationship between mammals and their microbial communities reflects millions of years of co-evolution that has shaped both host physiology and microbial metabolic capabilities [
34,
116]. The structural similarity between host-derived BHB and microbe-derived butyrate exemplifies this evolutionary convergence [
117,
118]. Both molecules activate HCAR2 receptors despite their distinct biosynthetic origins, suggesting that host signaling systems evolved to recognize and respond to functionally similar metabolites regardless of source [
45,
82].
11.2. Bidirectional Metabolic Communication
Host-derived ketone bodies directly influence microbial growth, with some bacterial species demonstrating enhanced growth while others are inhibited in the presence of ketones [
119,
120]. Ketogenic dietary interventions consistently alter microbiome composition, including reduced abundance of pro-inflammatory taxa such as Enterobacteriaceae and increased representation of beneficial genera including Akkermansia, Bifidobacterium, and Lactobacillus [
114,
120].
Conversely, microbial metabolites significantly influence host ketogenic capacity. Short-chain fatty acids produced by microbial fermentation of dietary fiber enhance hepatic ketogenesis through activation of AMPK and PPARα, key regulators of fatty acid oxidation and ketone production [
121,
122]. The microbiota also influences ketogenesis through effects on systemic inflammation, with balanced microbial communities maintaining intestinal barrier integrity and limiting LPS translocation that can suppress hepatic ketogenic capacity [
49,
116].
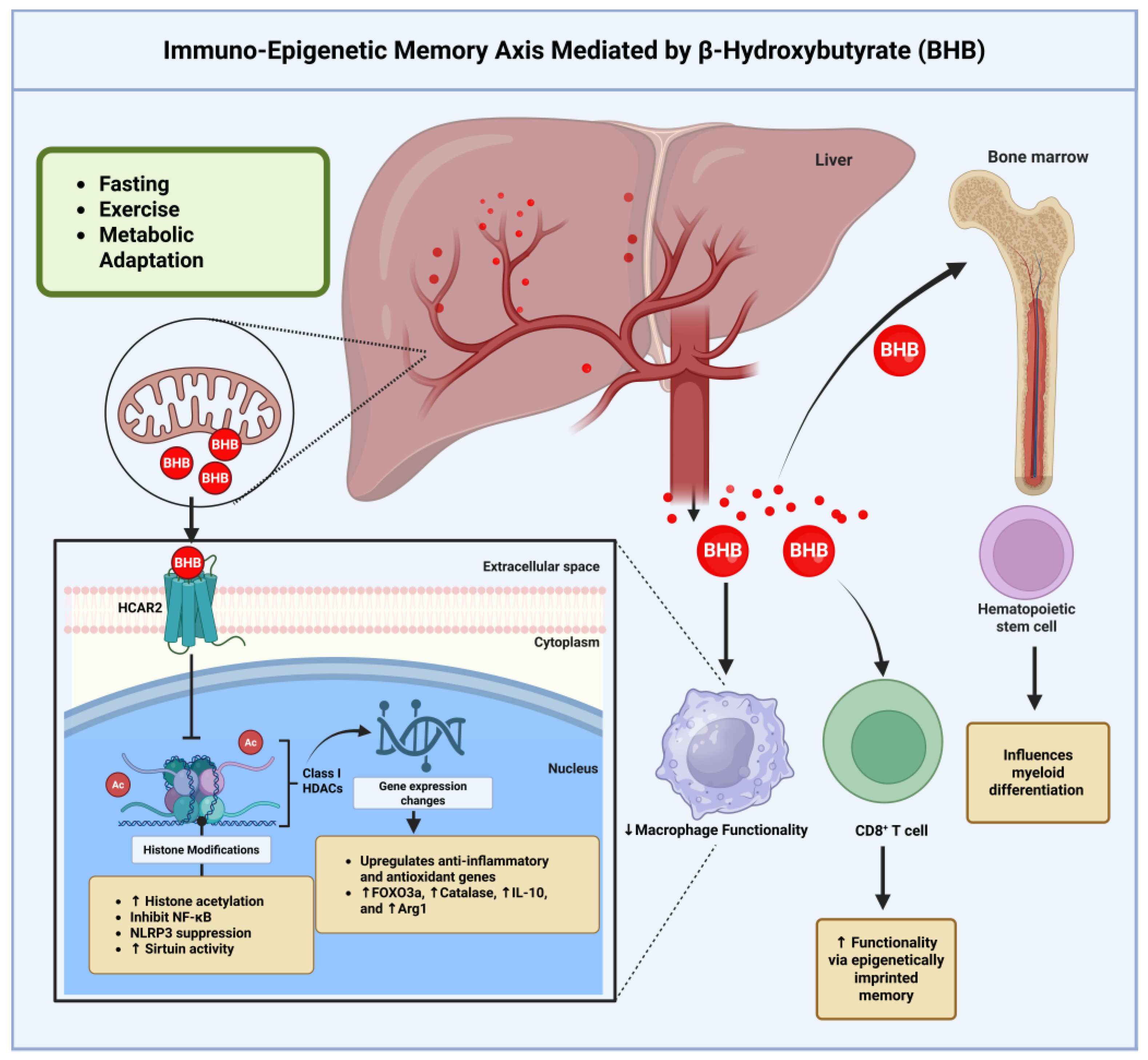
12. Ketosis and Immuno-Epigenetic Memory
The concept of immunological memory has traditionally focused on the adaptive immune system's capacity to retain information about previous antigen encounters through long-lived memory T and B cells [
123]. However, emerging evidence demonstrates that the innate immune system also possesses memory-like properties through epigenetic reprogramming mechanisms that persistently alter immune cell responsiveness [
85]. Ketone body exposure represents a particularly potent stimulus for establishing beneficial epigenetic memory that enhances resistance to inflammatory challenges and improves stress adaptation (
Figure 12) [
35,
36].
12.1. Molecular Mechanisms of Ketone-Induced Epigenetic Memory
The establishment of epigenetic memory by ketone bodies involves coordinated modifications to chromatin structure that persist long after ketone concentrations return to baseline levels [
35,
50]. The primary mechanism involves BHB-mediated inhibition of class I histone deacetylases, leading to increased acetylation of histones H3 and H4 at specific gene loci involved in stress response, antioxidant defense, and anti-inflammatory signaling [
35,
36].
Key target genes for ketone-induced epigenetic modifications include those encoding antioxidant enzymes (catalase, superoxide dismutase, glutathione peroxidase), stress-response transcription factors (FOXO3a, Nrf2), anti-inflammatory cytokines (IL-10), and tissue repair mediators (arginase 1, TGF-β) [
52,
53]. The coordinated upregulation of these protective gene programs creates cellular phenotypes characterized by enhanced resilience to oxidative stress, reduced inflammatory responsiveness, and improved capacity for tissue repair and regeneration [
102,
104].
12.2. Trained Immunity and Metabolic Reprogramming
Unlike classical trained immunity paradigms that typically involve enhanced inflammatory responses, ketone-induced immune training promotes metabolic reprogramming toward oxidative metabolism and anti-inflammatory programming [
75,
85]. Monocytes and macrophages exposed to ketone bodies demonstrate lasting metabolic changes including enhanced oxidative phosphorylation, increased fatty acid oxidation, and improved mitochondrial biogenesis [
75,
76]. The effects of ketone-induced trained immunity extend to hematopoietic stem and progenitor cells within the bone marrow, where epigenetic modifications can influence the differentiation programs that generate mature immune cells [
85].
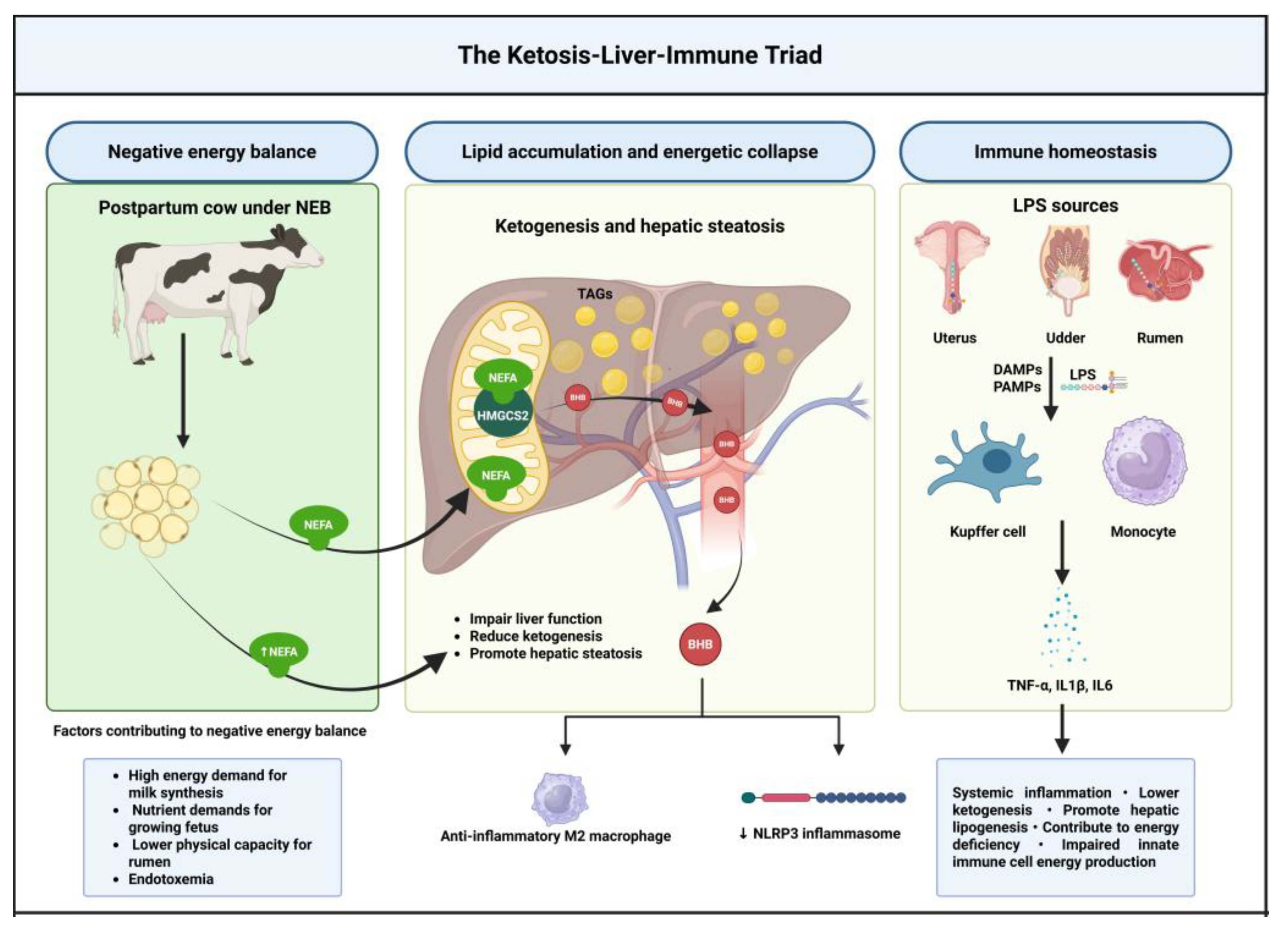
13. The Ketosis-Liver-Immune Triad: Hepatic Surveillance and Metabolic Integration
The liver occupies a unique position in mammalian physiology as the primary site of ketone body synthesis, a major organ for immune surveillance and regulation, and a critical integration hub for metabolic and inflammatory signaling [
124,
125]. The hepatic implementation of KIN responses involves sophisticated coordination between hepatocyte metabolic activity, resident immune cell populations, and circulating immune mediators that collectively determine both ketogenic capacity and systemic immune tone (
Figure 13) [
13,
124].
13.1. Hepatic Architecture and Metabolic-Immune Integration
The structural organization of the hepatic lobule creates distinct metabolic and immunological microenvironments that contribute to the sophisticated regulation of ketogenic responses [
124,
126]. Hepatocytes are arranged in plates extending from portal triads to central veins, creating oxygen and nutrient gradients that influence both metabolic enzyme expression and immune cell distribution [
125,
126].
The liver contains the largest population of tissue-resident macrophages (Kupffer cells) in the body, strategically positioned within hepatic sinusoids to monitor portal blood for potential threats including pathogens, toxins, and inflammatory mediators [
49,
110]. During periods of increased ketogenesis, Kupffer cells demonstrate altered activation patterns characterized by reduced inflammatory responsiveness and enhanced tissue repair functions [
13,
76].
13.2. Pathological Disruption and Therapeutic Restoration
Hepatic dysfunction can significantly compromise KIN function through impaired ketogenic capacity, altered immune regulation, or disrupted integration of metabolic and inflammatory signals [
127,
128]. Fatty liver disease, increasingly prevalent in periparturient dairy cattle and increasingly prevalent in humans, can impair ketogenic enzyme expression and activity while promoting hepatic inflammation [
13,
94].
Metabolomics investigations have revealed that alterations in liver function markers and lipid metabolism pathways are detectable weeks before clinical ketosis manifestation, suggesting that hepatic dysfunction represents an early and potentially reversible component of ketosis pathophysiology [
93]. Therapeutic approaches that support hepatic KIN function focus on optimizing the balance between fatty acid delivery, oxidative capacity, and ketogenic activity [
127,
129].
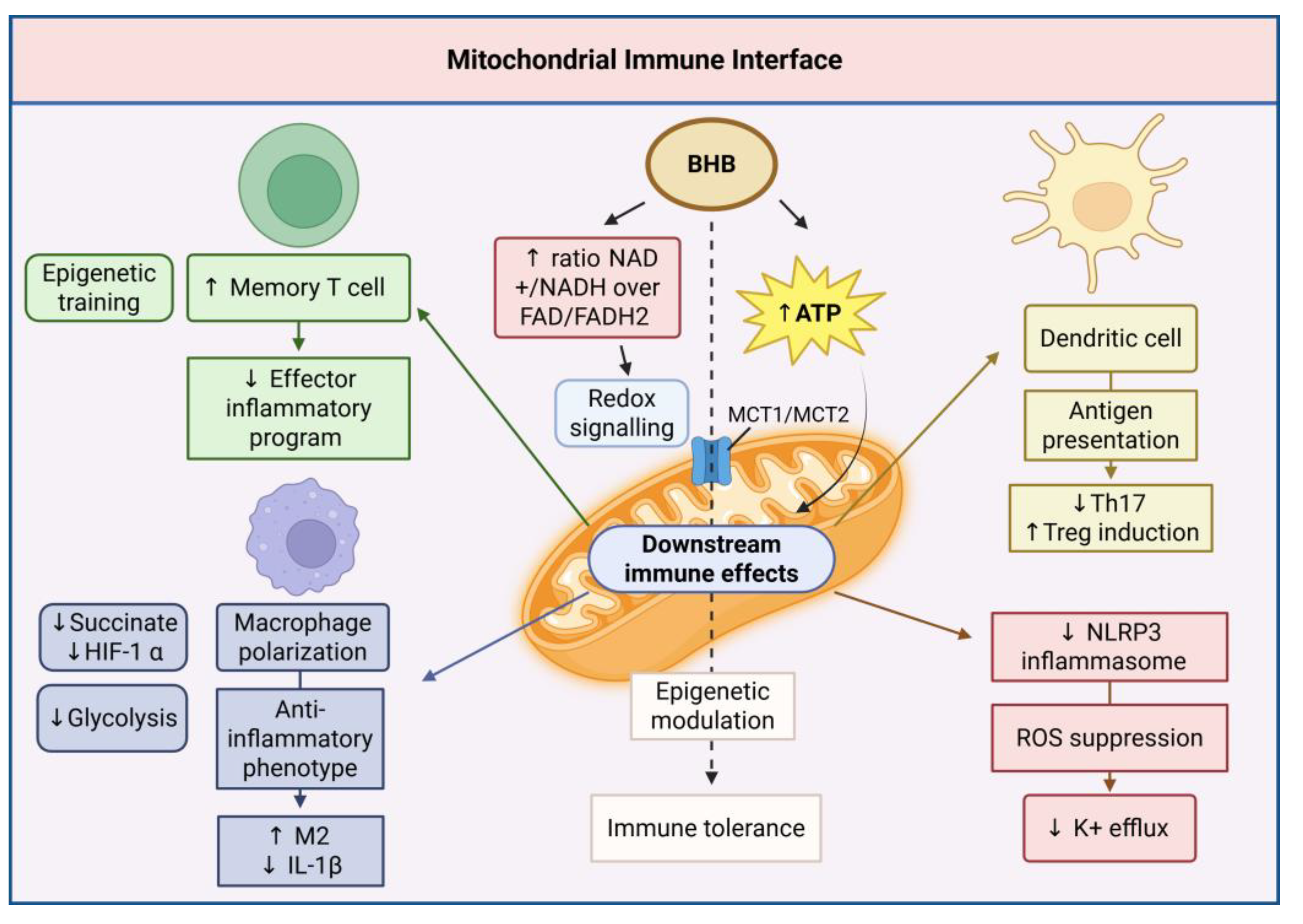
14. The Mitochondrial Interface of Metabolic-Immune Integration
Mitochondria represent the fundamental cellular interface where energy metabolism and immune signaling converge to determine cellular fate decisions, stress responses, and functional adaptations [
17,
62]. Within the context of the KIN, mitochondria serve simultaneously as the primary sites of ketone body production and utilization, critical regulators of immune cell activation and function, and central integrators of metabolic and inflammatory signaling (
Figure 14) [
17,
94].
14.1. Mitochondrial Ketone Metabolism and Bioenergetics
The mitochondrial matrix contains the complete enzymatic machinery for both ketone body synthesis and oxidation, enabling mitochondria to function as both producers and consumers of ketone bodies depending on cellular metabolic status and tissue-specific requirements [
4,
130]. The regulation of mitochondrial ketogenesis involves sophisticated control mechanisms that integrate information about substrate availability, energy demand, and cellular stress status [
27,
130].
In peripheral tissues, mitochondria serve as the primary sites of ketone body oxidation through the action of succinyl-CoA:3-ketoacid CoA transferase (SCOT) and acetyl-CoA acetyltransferase [
102,
131]. The energy yield from ketone oxidation, combined with the metabolic efficiency of ketone utilization, makes ketone bodies particularly valuable fuel sources for metabolically active cells including immune cells, cardiac myocytes, and neurons [
131].
14.2. Immune Cell Metabolic Reprogramming
Different immune cell populations demonstrate distinct metabolic requirements reflecting their specialized functional roles [
78,
79]. Activated effector T cells rely heavily on glycolysis to support rapid proliferation and cytokine production, while memory T cells depend on oxidative phosphorylation and fatty acid oxidation for longevity and rapid recall responses [
78,
80].
Ketone body availability can significantly influence immune cell metabolic programming by providing alternative oxidative substrates that favor anti-inflammatory and memory cell phenotypes [
75,
77]. Macrophages cultured in the presence of ketone bodies demonstrate enhanced oxidative metabolism, reduced glycolytic activity, and increased expression of M2 activation markers including arginase 1, IL-10, and TGF-β [
73,
76]. This metabolic shift is consistent with the observation that cows destined to develop ketosis show early alterations in immune cell function and inflammatory mediator production [
14].
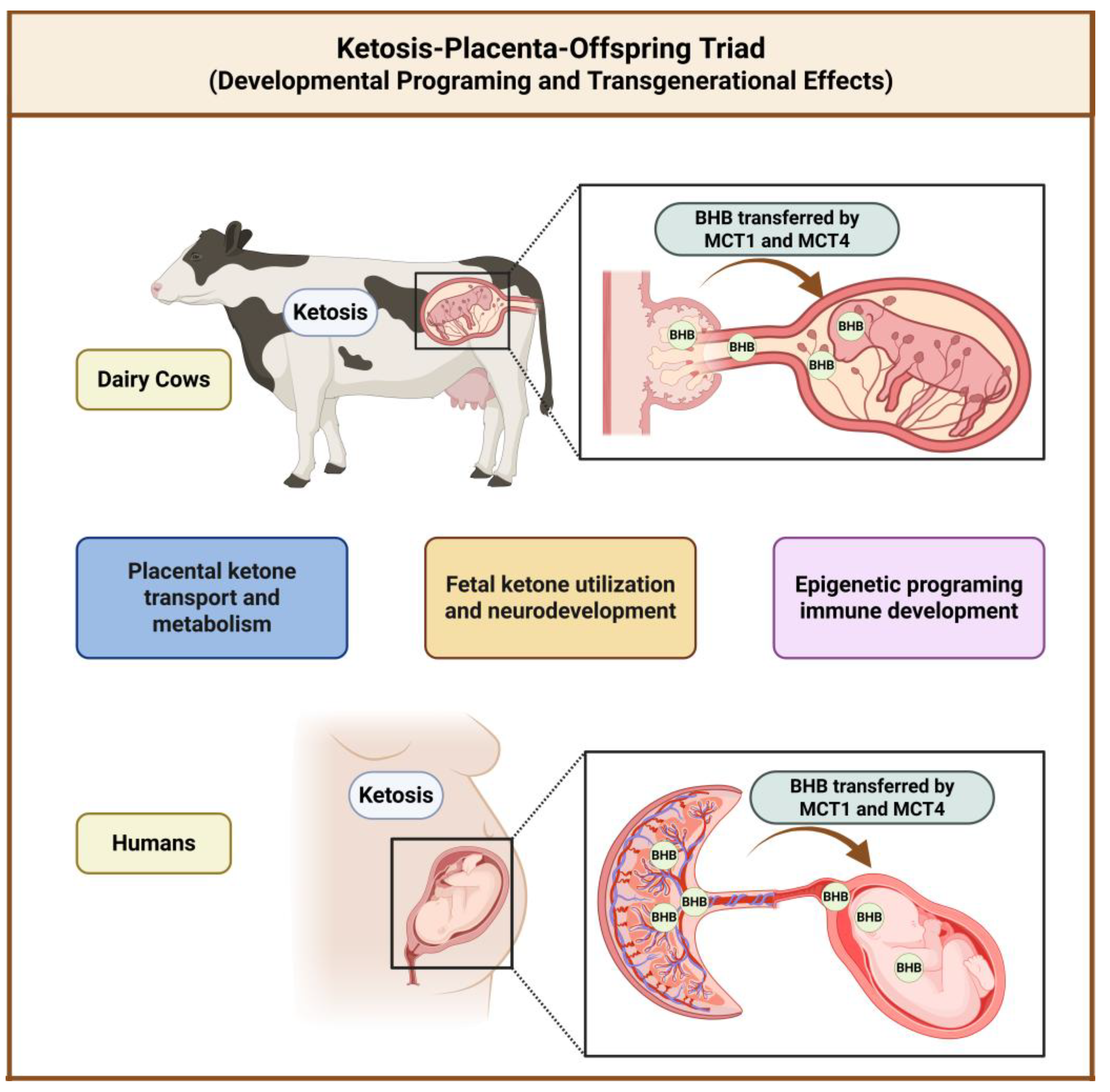
15. The Ketosis-Placenta-Offspring Triad: Developmental Programming and Transgenerational Effects
The maternal-fetal interface represents a critical developmental window where maternal metabolic status profoundly influences fetal growth, organ development, and long-term health outcomes [
5,
132]. Maternal ketosis during pregnancy creates unique opportunities for developmental programming through direct effects of transplacental ketone transfer, epigenetic modifications in developing tissues, and alterations in maternal immune function that influence fetal immune development (
Figure 15) [
133].
15.1. Placental Ketone Transport and Metabolism
The placenta functions as a sophisticated metabolic organ that actively regulates the transfer of nutrients, metabolites, and signaling molecules between maternal and fetal circulation [
132]. Ketone bodies cross the placental barrier through specialized monocarboxylate transporters (MCTs), particularly MCT1 and MCT4, which are expressed on both maternal and fetal sides of the placental interface [
102].
15.2. Fetal Ketone Utilization and Neurodevelopment
During fetal development, ketone bodies serve multiple functions that extend beyond simple energy provision to include roles in brain development, myelination, and neurotransmitter synthesis [
134,
135]. The developing fetal brain demonstrates particularly high capacity for ketone utilization, with ketone oxidation providing up to 25-30% of total brain energy requirements during certain developmental stages [
131,
134].
15.3. Epigenetic Programming and Immune Development
Maternal ketosis can influence fetal development through epigenetic mechanisms that establish lasting changes in gene expression patterns [
36,
133]. The developing epigenome is particularly susceptible to environmental influences during critical developmental windows, and ketone-mediated HDAC inhibition can establish epigenetic modifications that persist throughout life [
35,
133]. The immune system represents one of the most important targets for developmental programming by maternal ketones [
83].
16. Reframing Ketosis: From Pathology to Adaptive Programming
The fundamental reconceptualization of ketosis from a pathological condition requiring correction to an adaptive biological program deserving support represents one of the most significant paradigm shifts in modern metabolic medicine and veterinary science [
5,
6,
11]. This transformation in understanding requires moving beyond reductionist approaches that classify any deviation from baseline metabolism as inherently problematic, instead embracing the sophisticated biological complexity that characterizes ketotic adaptation [
124,
135].
16.1. Historical Context and Conceptual Evolution
The medical and veterinary establishment's historical perspective on ketosis has been profoundly shaped by observations made primarily within extreme pathological contexts where massive ketone elevations were associated with life-threatening conditions including diabetic ketoacidosis and starvation ketosis in humans as well as severe postpartum clinical ketosis in dairy cattle [
10,
18]. This pathocentric perspective was reinforced by the limited analytical capabilities available during the early development of clinical biochemistry [
10,
22].
The evolution toward a more balanced understanding of ketosis has been driven by technological advances that enable precise measurement of ketone concentrations, sophisticated analytical techniques that can characterize the molecular mechanisms of ketone action, and longitudinal studies that reveal the complex relationships between ketosis and health outcomes [
5,
6,
93]. Metabolomics investigations have been particularly valuable in revealing the complex metabolic alterations that precede, accompany, and follow ketosis development, demonstrating that ketosis represents coordinated physiological programming rather than simple metabolic failure [
93].
16.2. Physiological vs. Pathological Ketosis: Critical Distinctions
The recognition of ketosis as an adaptive response does not negate the reality that ketotic states can become pathological under certain circumstances [
11,
19]. The critical distinction lies not in the simple presence or absence of ketone bodies, but rather in the physiological context, magnitude, duration, and associated metabolic conditions that determine whether ketosis serves beneficial or harmful functions [
135,
136].
Predictive metabolomics approaches have demonstrated that it is possible to identify cows at risk for developing pathological ketosis weeks before clinical manifestation, enabling targeted interventions that could support beneficial adaptive ketotic responses while preventing pathological progression [
136]. These findings suggest that the traditional reactive approach to ketosis management could be replaced with proactive strategies that optimize natural adaptive mechanisms.
16.3. Therapeutic Implications of the Adaptive Paradigm
The recognition that ketosis can serve adaptive functions has profound implications for therapeutic approaches across multiple medical and veterinary disciplines [
11]. Rather than focusing exclusively on ketone suppression, optimal therapeutic strategies should aim to support the beneficial aspects of ketotic adaptation while addressing the factors that prevent healthy ketogenic regulation [
30,
127].
This paradigm shift is consistent with the growing recognition that many metabolic "diseases" may represent adaptive responses to environmental challenges that become pathological only when the adaptive capacity is exceeded or when the adaptive response persists beyond its optimal duration [
5]. The challenge for clinicians and researchers is to develop approaches that can distinguish between beneficial and harmful aspects of metabolic adaptation and to design interventions that support optimal physiological function.
17. The Calci-Keto-Inflammatory Code: A Systems Integration Framework
The synthesis of evidence presented throughout this review converges on a fundamental reconceptualization: ketosis represents not merely a metabolic disorder caused by energy deficiency, but rather an integral component of a sophisticated biological information processing system. We propose the Calci-Keto-Inflammatory Code (CKIC) as a unifying framework that integrates calcium homeostasis, ketone metabolism, and immune regulation into a coordinated system capable of encoding, transmitting, and decoding physiological information.
The transition from viewing these processes as independent "pathways" to recognizing them as an integrated "code" is conceptually significant. While network terminology properly describes interconnections between metabolic and inflammatory systems, it inadequately captures the fundamental information-theoretic properties we now recognize as central to biological regulation. A code, by definition, requires vocabulary (molecular signals), grammar (temporal dynamics), semantics (biological meaning), and pragmatics (context-dependent interpretation) [
32,
33]. Preliminary evidence suggests that mammalian metabolic-immune systems exhibit precisely these properties, with calcium functioning as a master regulator that gates cross-talk between subsystems [
137] [manuscript in preparation].
17.1. Calcium as a Master Regulatory Node
Central to this framework is recognition of calcium's dual function as both metabolic signal and immune regulator. Beyond its well-established roles in muscle contraction, nerve transmission, and bone metabolism, calcium serves a previously underappreciated function as a dynamic immune rheostat [
92]. Elevated extracellular calcium concentrations directly activate macrophages and neutrophils through calcium-dependent signaling pathways, stimulating pro-inflammatory cytokine release including TNF-α, IL-1β, and IL-6 [
92]. Conversely, reduced calcium levels dampen immune cell activation, creating a physiological mechanism for controlling inflammatory intensity during metabolic stress [
92].
This regulatory role of calcium, as described in [
92], supports the hypothesis that hypocalcemia during transition periods is an adaptive strategy to prevent excessive immune activation, rather than solely a pathological condition requiring correction. During periparturient period, when tissue remodeling, microbial exposure, and metabolic challenges create inherent inflammatory risk, the organism strategically lowers circulating calcium to prevent inflammatory overactivation - essentially "turning down" the immune system's sensitivity to avoid collateral tissue damage while maintaining sufficient immune function for pathogen defense.
If validated, this concept necessitates fundamental reconceptualization of clinical approaches to hypocalcemia. Rather than reflexively supplementing calcium in all hypocalcemic animals, therapeutic decisions must account for inflammatory status: calcium supplementation in animals with active inflammation may be iatrogenic, activating immune responses at precisely the time when dampening would be protective. Preliminary analyses suggest that integrated assessment of calcium-inflammation interactions provides substantially greater predictive power for disease outcomes than either measurement alone [data not shown], supporting the existence of code-like information processing beyond simple biochemical correlations.
17.2. Clinical and Research Implications
The proposed
CKIC operates through hierarchical information processing layers. At the molecular level, individual metabolites (BHB, NEFA, glucose) and cytokines function as elementary signals - the "vocabulary" of the code. However, their biological meaning emerges not from static concentrations but from temporal patterns: rates of change (first derivatives), acceleration (second derivatives), and trajectory curvature distinguish transient perturbations from sustained threats and predict whether systems will stabilize or progress toward pathology [
39,
40].
Critically, the same biochemical pattern generates different physiological outcomes depending on circumstances - the essence of code-dependent interpretation [
41]. For example, BHB = 1.2 mmol/L coupled with low calcium and controlled inflammation may represent successful adaptive ketosis, whereas identical BHB levels with elevated calcium and rising inflammation signal impending metabolic decompensation. This context-dependency mirrors semantic properties of human language, where identical words convey different meanings in different contexts.
Information-theoretic analyses reveal that metabolic and inflammatory markers exhibit properties characteristic of sophisticated communication systems, including redundancy (multiple signals encoding similar information for error correction), synergistic information (combinations providing greater predictive power than individual measurements), and directed information flow (transfer entropy quantifying causal influences) [
67,
68]. The calcium-inflammation interaction appears to function as a regulatory "control bit" that modulates interpretation of metabolic signals, analogous to header information in digital communication protocols specifying how message content should be decoded [manuscript in preparation].
17.3. Broader Implications and Future Directions
Recognition of code-like properties in metabolic-immune integration has immediate clinical implications. Traditional diagnostic approaches relying on threshold-based classification of individual biomarkers (BHB >1.2 mmol/L for ketosis, Ca²⁺ <2.0 mmol/L for hypocalcemia) obscure the information content embedded in patterns and interactions. Code-based diagnosis requires integrated assessment: not "Is BHB elevated?" but rather "What pattern do BHB, calcium, and inflammatory markers form, and what does this pattern signify about future trajectories?"
Preliminary work developing machine learning algorithms to recognize and interpret these patterns demonstrates prediction of clinical ketosis with >90% accuracy at 14 days before diagnosis [
14]; [manuscript in preparation] - substantially earlier and more accurate than conventional screening approaches. This predictive power derives from the recognition that current biochemical patterns encode information about future trajectories, analogous to how current weather patterns enable meteorological forecasting.
Therapeutically, code-based approaches shift focus from treating abnormal laboratory values to restoring system integrity. If inflammation is driving dysregulation, interventions targeting inflammatory reduction may prove more effective than metabolic support alone. If calcium-dependent immune gating is dysregulated, treatment strategies must account for this regulatory failure rather than simply supplementing deficient metabolites. The framework suggests that successful intervention requires understanding which aspect of code integrity has failed and targeting restoration of regulatory function rather than normalization of individual parameters.
17.4. Clinical Translation: From Code Comprehension to Therapeutic Application
The CKIC framework potentially extends beyond transition dairy cattle to any mammalian system navigating metabolic-immune stress. Similar regulatory challenges occur in human metabolic syndrome, fasting, lactation across species, and critical illness - situations where organisms must coordinate competing demands of energy mobilization and immune defense. The remarkable conservation of calcium signaling, ketone metabolism, and innate immunity across mammalian taxa suggests that this regulatory architecture may represent a fundamental biological organizing principle rather than a species-specific adaptation.
Testing this framework rigorously requires prospective validation studies examining whether code-based diagnostic and therapeutic approaches improve outcomes compared to conventional management. Such studies should assess not only predictive accuracy but also clinical utility: Do earlier predictions enable more effective interventions? Does code-guided therapy reduce disease incidence and severity? What are the economic implications of shifting from reactive disease treatment to predictive code-based management?
From a basic science perspective, critical questions remain regarding mechanisms of calcium-dependent immune regulation during metabolic stress, temporal dynamics of system responses, sources of individual variation in code integrity, and potential for epigenetic memory of prior code disruptions influencing future lactations. Investigation of these questions will require integration of metabolomics, immunology, systems biology, and information theory - truly interdisciplinary approaches matching the integrated nature of the biological system under study.
17.5. Evolutionary Perspective: The Code as Ancient Adaptive Language
The CKIC represents a conceptual evolution from viewing metabolism and immunity as separate systems that occasionally interact, toward recognizing them as components of an integrated information processing architecture. Calcium emerges not merely as a metabolite requiring homeostatic regulation but as a master regulatory signal that gates immune activation and thereby determines whether metabolic adaptations remain physiological or progress to disease. This code-based framework provides both deeper mechanistic understanding and enhanced clinical utility, enabling early prediction of disease trajectories and rational therapeutic decisions guided by underlying regulatory dysfunction rather than superficial laboratory abnormalities.
Perhaps most importantly, this framework exemplifies a broader paradigm shift occurring across biology and medicine: from reductionist approaches studying individual pathways in isolation toward systems-level recognition of integrated networks processing biological information. Health is not the absence of abnormal values but rather the maintenance of code integrity - the capacity to accurately encode, transmit, and decode physiological signals. Disease represents information processing failure - errors that prevent appropriate responses to environmental and internal challenges.
The detailed characterization of the CKIC, including comprehensive metabolomic validation, information-theoretic formalization, prospective clinical trials, and therapeutic protocols, will be presented in forthcoming dedicated publications. The present review establishes the conceptual foundation and synthesizes existing evidence supporting this integrative framework, pointing toward a new era of code-based precision medicine in which treatments are tailored not to population averages or simple laboratory thresholds but to individual animals' unique information processing patterns, physiological contexts, and regulatory capacities.
Funding
This review article received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to express my sincere gratitude to Zohaib Saleem, Jincheol Choi, and Jingyu Choi for their valuable assistance in translating my original figure concepts into high-quality visual illustrations using BioRender. The figures presented in this review article were developed collaboratively, with the scientific design and initial drafts created by myself, and the final versions refined and executed by the students through BioRender. Their diligence, technical skill, and commitment to accuracy greatly enhanced the clarity and visual impact of the article.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Leonard, W.R.; Robertson, M.L.; Snodgrass, J.J.; Kuzawa, C.W. Metabolic correlates of hominid brain evolution. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003, 136, 5–15. [Google Scholar]
- Blackstone, N.W. A units-of-evolution perspective on the endosymbiont theory of the origin of the mitochondrion. Evolution 1995, 49, 785–796. [Google Scholar] [CrossRef]
- Cahill, G.F. Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Zhang, G.; Ametaj, B.N. Ketosis an old story under a new approach. Dairy 2020, 1, 61–90. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Jenni-Eiermann, S.; Jenni, L. Metabolic responses to flight and fasting in night-migrating passerines. J. Comp. Physiol. B 1991, 161, 465–474. [Google Scholar] [CrossRef]
- Hawdon, J.M.; Ward Platt, M.P.; Aynsley-Green, A. Patterns of metabolic adaptation for preterm and term infants in the first neonatal week. Arch. Dis. Child. 1992, 67, 357–365. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Drackley, J.K. Biology of dairy cows during the transition period: The final frontier? J. Dairy Sci. 1999, 82, 2259–2273. [Google Scholar] [CrossRef]
- Bertoni, G.; Trevisi, E.; Han, X.; Bionaz, M. Effects of inflammatory conditions on liver activity in puerperium period and consequences for performance in dairy cows. J. Dairy Sci. 2008, 91, 3300–3310. [Google Scholar] [CrossRef]
- Zhang, G.; Hailemariam, D.; Dervishi, E.; Deng, Q.; Goldansaz, S.; Deng, Q.; Dunn, S.M.; Ametaj, B.N. Dairy cows affected by ketosis show alterations in innate immunity and lipid and carbohydrate metabolism during the dry off period and postpartum. Res. Vet. Sci. 2016, 107, 246–256. [Google Scholar]
- Martinez, N.; Risco, C.A.; Lima, F.S.; Bisinotto, R.S.; Greco, L.F.; Ribeiro, E.S.; Maunsell, F.; Galvão, K.; Santos, J.E. Evaluation of peripartal calcium status, energetic profile, and neutrophil function in dairy cows. J. Dairy Sci. 2012, 95, 7158–7172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ametaj, B.N. Ketosis Under a Systems Veterinary Medicine Perspective. In Periparturient diseases of dairy cows: A systems biology approach; Springer International: Cham, Switzerland, 2017; pp. 201–222. [Google Scholar]
- Mills, E.L.; Kelly, B.; O'Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.D. Primary ketosis in the high-producing dairy cow: Clinical and subclinical disorders, treatment, prevention, and outlook. J. Dairy Sci. 1982, 65, 1–10. [Google Scholar] [CrossRef]
- Mitchell, G.A.; Kassovska-Bratinova, S.; Boukaftane, Y.; Robert, M.F.; Wang, S.P.; Ashmarina, L.; Lambert, M.; Lapierre, P.; Potier, E. Medical aspects of ketone body metabolism. Clin. Invest. Med. 1995, 18, 193–216. [Google Scholar] [PubMed]
- Herdt, T.H. Ruminant adaptation to negative energy balance. Influences on the etiology of ketosis and fatty liver. Vet. Clin. North Am. Food Anim. Pract. 2000, 16, 215–230. [Google Scholar] [CrossRef]
- Grummer, R.R. Impact of changes in organic nutrient metabolism on feeding the transition dairy cow. J. Anim. Sci. 1995, 73, 2820–2833. [Google Scholar] [CrossRef]
- Oetzel, G.R. Monitoring and testing dairy herds for metabolic disease. Vet. Clin. North Am. Food Anim. Pract. 2004, 20, 651–674. [Google Scholar] [CrossRef]
- McArt, J.A.A.; Nydam, D.V.; Oetzel, G.R. Epidemiology of subclinical ketosis in early lactation dairy cattle. J. Dairy Sci. 2012, 95, 5056–5066. [Google Scholar] [CrossRef]
- Egyedy, A.; Rosales, E.B.; Ametaj, B.N. Association of High Somatic Cell Counts Prior to Dry off to the Incidence of Periparturient Diseases in Holstein Dairy Cows. Vet. Sci. 2022, 9, 624. [Google Scholar] [CrossRef]
- Goff, J.P.; Horst, R.L. Physiological changes at parturition and their relationship to metabolic disorders. J. Dairy Sci. 1997, 80, 1260–1268. [Google Scholar] [CrossRef]
- Etherton, T.D.; Bauman, D.E. Biology of somatotropin in growth and lactation of domestic animals. Physiol. Rev. 1998, 78, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef]
- Nielsen, N.I.; Ingvartsen, K.L. Propylene glycol for dairy cows. A review of the metabolism of propylene glycol and its effects on physiological parameters, feed intake, milk production and risk of ketosis. Anim. Feed Sci. Technol. 2004, 115, 191–213. [Google Scholar] [CrossRef]
- Kristensen, N.B.; Raun, B.M. Ruminal and intermediary metabolism of propylene glycol in lactating Holstein cows. J. Dairy Sci. 2007, 90, 4707–4717. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L.; LeBlanc, S.J.; Duffield, T.F. Ketosis treatment in lactating dairy cattle. Vet. Clin. North Am. Food Anim. Pract. 2013, 29, 433–445. [Google Scholar] [CrossRef]
- Walsh, R.B.; Walton, J.S.; Kelton, D.F.; LeBlanc, S.J.; Leslie, K.E.; Duffield, T.F. The effect of subclinical ketosis in early lactation on reproductive performance of postpartum dairy cows. J. Dairy Sci. 2007, 90, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Montminy, M. Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 1997, 66, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Chriett, S.; Dabek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hailemariam, D.; Dervishi, E.; Goldansaz, S.A.; Deng, Q.; Dunn, S.M.; et al. Alterations of innate immunity reactants in transition dairy cows before clinical signs of lameness. Animals 2015, 5, 717–747. [Google Scholar] [CrossRef]
- Peter, T, Ruggiero, M. Ketogenic diet and immunotherapy in cancer and neurological diseases: A new therapeutic paradigm. Am. J. Immunol. 2021, 24, 374–380. [Google Scholar] [CrossRef]
- Thomsen, H.H.; Rittig, N.; Johannsen, M.; Møller, A.B.; Jørgensen, J.O.; Jessen, N.; et al. Effects of 3-hydroxybutyrate and free fatty acids on muscle protein kinetics and signaling during LPS-induced inflammation in humans: Anticatabolic impact of ketone bodies. Am. J. Clin. Nutr. 2018, 108, 857–867. [Google Scholar] [CrossRef]
- Mezzetti, M.; Minuti, A.; Piccioli-Cappelli, F.; Amadori, M.; Bionaz, M.; Trevisi, E. Inflammatory status and metabolic changes at dry-off in high-yield dairy cows. Ital. J. Anim. Sci. 2019, 19(1), 51–65. [Google Scholar] [CrossRef]
- Lei, L.; Gao, W.; Loor, J.J.; Aboragah, A.; Fang, Z.; Du, X.; Zhang, M.; Song, Y.; Liu, G.; Li, X. Reducing hepatic endoplasmic reticulum stress ameliorates the impairment in insulin signaling induced by high levels of β-hydroxybutyrate in bovine hepatocytes. J. Dairy Sci 2021, 104, 12845–12858. [Google Scholar] [CrossRef]
- Mann, S.; Yepes, F.A.; Duplessis, M.; Wakshlag, J.J.; Overton, T.R.; Cummings, B.P.; et al. Dry period plane of energy: Effects on glucose tolerance and insulin sensitivity in transition dairy cows. J. Dairy Sci. 2016, 99, 701–717. [Google Scholar] [CrossRef]
- Nagao, M.; Toh, R.; Irino, Y.; Mori, M.; Nakajima, H.; Hara, T.; et al. β-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 475, 322–328. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Wise, A.; Foord, S.M.; Fraser, N.J.; Barnes, A.A.; Elshourbagy, N.; Eilert, M.; Ignar, D.M.; Murdock, P.R.; Steplewski, K.; Green, A.; et al. Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 2003, 278, 9869–9874. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, S.W.; et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. β-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. Activation and inhibition of sirtuins: From bench to bedside. Med. Res. Rev. 2024, 44, 2496–2533. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Faller, D.V.; Li, T. SIRT3 Activation: A Promise in Drug Development? New Insights into SIRT3 Biology. J. Med. Chem. 2024, 67, 2765–2800. [Google Scholar] [CrossRef]
- Balan, I.; Taube, J.; Zhai, X.; Meador-Woodruff, J.H. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Laeger, T.; Metges, C.C.; Kuhla, B. Role of β-hydroxybutyric acid in the central regulation of energy balance. Appetite 2010, 54, 450–455. [Google Scholar] [CrossRef]
- Dantzer, R.; O'Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.; Pellerin, L. Nutritional Impact on Metabolic Homeostasis and Brain Health. Front. Neurosci. 2021, 15, 767405. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Han, B.; Wang, J.H.; Geng, Y.; Shen, L.; Wang, H.L.; Wang, Y.Y.; Wang, M.W. Chronic stress contributes to cognitive dysfunction and hippocampal metabolic abnormalities in APP/PS1 mice. Cell. Physiol. Biochem. 2017, 41, 1766–1776. [Google Scholar] [CrossRef]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Lo Sasso, G.; Moschetta, A.; Schibler, U. REV-ERBα participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar] [CrossRef] [PubMed]
- Laermans, J.; Depoortere, I. Chronobesity: Role of the circadian system in the obesity epidemic. Obes. Rev. 2016, 17, 108–125. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef]
- Niu, M.; Ying, Y.; Bartell, P.A.; Harvatine, K.J. The effects of feeding time on milk production, total-tract digestibility, and daily rhythms of feeding behavior and plasma metabolites and hormones in dairy cows. J. Dairy Sci. 2014, 97, 7764–7776. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Galván-Peña, S.; O'Neill, L.A. Metabolic reprogramming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [PubMed]
- Contreras, G.A.; Sordillo, L.M. Lipid mobilization and inflammatory responses during the transition period of dairy cows. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 281–289. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O'Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O'Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Song, X.; Greene, M. (2019). FoxP3 in Treg cell biology: a molecular and structural perspective. Clinical and Experimental Immunology, 2019, 199, 255–262. [Google Scholar] [CrossRef]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef]
- Collins, N.; Han, S.J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, E.A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R.; et al. The Bone Marrow Protects and Optimizes Immunological Memory during Dietary Restriction. Cell 2019, 178, 1088–1101. [Google Scholar] [CrossRef]
- Parker, K.L.; Barboza, P.S.; Gillingham, M.P. Nutrition integrates environmental responses of ungulates. Funct. Ecol. 2009, 23, 57–69. [Google Scholar] [CrossRef]
- Nelson, R.A. Protein and fat metabolism in hibernating bears. Fed. Proc. 1980, 39, 2955–2958. [Google Scholar] [PubMed]
- Jeukendrup, A.E.; Aldred, S. Fat supplementation, health, and endurance performance. Nutrition 2004, 20, 678–688. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Dechow, C.D.; Rogers, G.W.; Clay, J.S. Heritabilities and correlations among body condition scores, production traits, and reproductive performance. J. Dairy Sci. 2001, 84, 266–275. [Google Scholar] [CrossRef]
- Ametaj, B.N. The Calci-Inflammatory Network: A Paradigm Shift in Understanding Milk Fever. Dairy 2025, 6, 22. [Google Scholar] [CrossRef]
- Zhang, G.; Mandal, R.; Wishart, D.S.; Ametaj, B.N. A Multi-Platform Metabolomics Approach Identifies Urinary Metabolite Signatures That Differentiate Ketotic From Healthy Dairy Cows. Front. Vet. Sci. 2021, 8, 595983. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Neves, R.C.; Leno, B.M.; Stokol, T.; Overton, T.R.; McArt, J.A. Risk factors associated with postpartum subclinical hypocalcemia in dairy cows. J. Dairy Sci. 2017, 100, 3796–3804. [Google Scholar] [CrossRef] [PubMed]
- Oswald, B.; DeCamp, L.; Longo, J.; Dahabieh, M.; Bunda, N.; Watson, M.; Sheldon, R.; Vincent, M.; Johnson, B.; Ellis, A.; Soper-Hopper, M.; Isaguirre, C.; Shen, H.; Williams, K.; Crawford, P.; Kaech, S.; Jang, H.; Krawczyk, C.; Jones, R. Dietary Restriction Enhances CD8+ T Cell Ketolysis to Limit Exhaustion and Boost Anti-Tumor Immunity. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, J.; Andersen, T.Ø.; Haj-Yasein, N.; et al. A ketogenic diet improves mitochondrial dynamics and function in APP/PS1 mice. Nutrients 2023, 15, 2706. [Google Scholar]
- Kong, C.; Yan, X.; Liu, Y.; Huang, L.; Zhu, Y.; He, J.; Gao, R.; Kalady, M.F.; Goel, A.; Qin, H.; Ma, Y. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct. Target. Ther. 2021, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Myette-Côté, É.; Neudorf, H.; Little, J.P. Ketone monoester supplementation attenuates inflammation and insulin resistance in obese mice. J. Physiol. 2021, 29, 1–16. [Google Scholar] [CrossRef]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Tapia, E.; Pedraza-Chaverri, J. β-Hydroxybutyrate: A signaling metabolite in starvation response? Cell. Signal. 2016, 28, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Kovacs, C.S. Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery. Physiol. Rev. 2016, 96, 449–547. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef]
- Hirota, T.; Okano, T.; Kokame, K.; Shirotani-Ikejima, H.; Miyata, T.; Fukada, Y. Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J. Biol. Chem. 2002, 277, 44244–44251. [Google Scholar] [CrossRef]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407–21412. [Google Scholar] [CrossRef]
- Canaple, L.; Rambaud, J.; Dkhissi-Benyahya, O.; Rayet, B.; Tan, N.S.; Michalik, L.; Delaunay, F.; Wahli, W.; Laudet, V. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor α defines a novel positive feedback loop in the rodent liver circadian clock. Mol. Endocrinol. 2006, 20, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 178, 1102–1114. [Google Scholar] [CrossRef]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gerstner, D.B.; Turnbaugh, J.E.; Wolfe, C.; Triplett, A.V.; Turnbaugh, P.J. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Lima, F.S.; Oikonomou, G.; Lima, S.F.; Bicalho, M.L.; Ganda, E.K.; de Oliveira Filho, J.C.; Lorenzo, G.; Trojacanec, P.; Bicalho, R.C. Prepartum and postpartum rumen fluid microbiomes: Characterization and correlation with production traits in dairy cows. Appl. Environ. Microbiol. 2015, 81, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef]
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism 2016, 7, 37. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef]
- Jungermann, K.; Keitzmann, T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu. Rev. Nutr. 1996, 16, 179–203. [Google Scholar] [CrossRef]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef] [PubMed]
- White, H.M. The role of TCA cycle anaplerosis in ketosis and fatty liver in periparturient dairy cows. Animals 2015, 5, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.M.; Aitken, S.L. Impact of oxidative stress on the health and immune function of dairy cattle. Vet. Immunol. Immunopathol. 2009, 128, 104–109. [Google Scholar] [CrossRef]
- Weber, C.; Hametner, C.; Tuchscherer, A.; Losand, B.; Kanitz, E.; Otten, W.; Singh, S.P.; Bruckmaier, R.M.; Becker, F.; Kanitz, W.; et al. Variation in fat mobilization during early lactation differently affects feed intake, body condition, and lipid and glucose metabolism in high-yielding dairy cows. J. Dairy Sci. 2013, 96, 165–180. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Dahl, G.E.; Tao, S.; Monteiro, A.P. Effects of late-gestation heat stress on immunity and performance of calves. J. Dairy Sci. 2016, 99, 3193 3198. [Google Scholar] [CrossRef]
- Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-Mimicking Diet Promotes Ngn3-Driven β-Cell Regeneration to Reverse Diabetes. Cell 2017, 168, 775–788. [Google Scholar] [CrossRef]
- Crawford, M.A.; Bloom, M.; Broadhurst, C.L.; Schmidt, W.F.; Cunnane, S.C.; Galli, C.; Gehbremeskel, K.; Linseisen, F.; Lloyd-Smith, J.; Parkington, J. Evidence for the unique function of docosahexaenoic acid during the evolution of the modern hominid brain. Lipids 1999, 34, S39–S47. [Google Scholar] [CrossRef]
- Kim, D.Y.; Davis, L.M.; Sullivan, P.G.; Maalouf, M.; Simeone, T.A.; van Brederode, J.; Rho, J.M. Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 2007, 101, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Ametaj, B.N.; Zhang, G.; Dervishi, E.; Dunn, S.M.; Mandal, R.; Wishart, D.S. DI/LC-MS/MS-based metabolomics identifies early predictive serum biomarkers for ketosis in dairy cows. J. Anim. Sci. 2016, 94, 72. [Google Scholar] [CrossRef]
- Dejos, C.; Gkika, D.; Cantelmo, A.R. The Two-Way Relationship Between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol 2020, 8, 573747. [Google Scholar] [CrossRef] [PubMed]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).