
Submitted:
23 November 2025
Posted:
25 November 2025
You are already at the latest version
Abstract
Rapid‐acting antidepressants show that mood can lift within hours when glutamatergic circuits are pushed from an "NMDA-dominant" to an "AMPA-dominant" state. Intravenous ketamine achieves this flip but is hampered by dissociative side-effects and clinical logistics, while the oral pairing of dextromethorphan + bupropion (Auvelity®) delivers only the initial NMDA blockade and therefore yields slower, less durable benefit. We propose a fully oral, low-cost, four-component regimen designed to replicate ketamine's entire plasticity cascade: (1) dextromethorphan (DXM) supplies fast NMDA antagonism; (2) a strong CYP2D6 inhibitor (fluoxetine, paroxetine, or high-dose duloxetine) prolongs DXM exposure without relying on bupropion; (3) the AMPA positive allosteric modulator piracetam amplifies the downstream glutamate burst, driving BDNF- and mTOR-dependent synaptogenesis; and (4) micronized L-glutamine restores presynaptic glutamate pools and buffers against excitotoxicity. Preclinical evidence shows that each element—DXM's ketamine-like behavioral effects, piracetam's enhancement of AMPA currents, and glutamine's reversal of stress-induced glutamatergic depletion—synergizes along the same mechanistic axis. This strategy could democratize ketamine-level efficacy using inexpensive, readily available medications.
Keywords:
DXM
; dextromethorphan
; CYP2D6 inhibition
; fluoxetine
; paroxetine
; piracetam
; AMPA positive allosteric modulator
; L-glutamine
; rapid-acting antidepressant
; ketamine-like
; Auvelity
; neuroplasticity
; NMDA antagonism
; AMPA throughput
; BDNF
; mTOR
; synaptogenesis
; glutamate surge
; treatment-resistant depression
; oral regimen
; off-label stack
; polypharmacy
; excitotoxicity risk
; serotonin syndrome risk
Introduction
The past decade has revealed that the fastest antidepressant relief comes not from tweaking serotonin but from jump-starting glutamatergic plasticity. Ketamine's single-dose success proves the concept, yet its dissociation, abuse potential, and infusion logistics keep it out of reach for most patients. Meanwhile, the oral agent Auvelity (dextromethorphan + bupropion) shows that we can deliver an NMDA-blocking "spark" in pill form, but its benefits arrive more slowly and fade more quickly than ketamine's—likely because the combination never fully flips the synapse into the AMPA-dominated state that drives lasting circuit repair. Here we propose a four-component, all-oral regimen designed to recreate ketamine's full plasticity cascade without IV lines or street-drug baggage: (1) dextromethorphan for NMDA antagonism, (2) a potent CYP2D6 inhibitor such as fluoxetine to keep dextromethorphan active, (3) the inexpensive AMPA positive allosteric modulator piracetam to amplify downstream signaling, and (4) dietary L-glutamine to replenish presynaptic glutamate stores and buffer excitotoxic risk. Each element is already marketed, inexpensive, and well characterized, yet together they form a mechanistically coherent strategy to deliver rapid, sustained antidepressant action.
How Ketamine Ignites AMPA-Fueled Neuroplasticity to Lift Depression
Ketamine's antidepressant story begins at the NMDA receptor. At sub-anesthetic doses the drug preferentially blocks NMDA receptors that sit on inhibitory GABA interneurons in the prefrontal cortex and hippocampus. Momentarily silencing these "brakes" disinhibits nearby pyramidal cells and triggers a burst of glutamate release [1]. This glutamate surge is not simply biochemical noise; in patients it coincides with mood lift that can appear within hours and last for days—an effect unmatched by conventional monoaminergic drugs that require weeks [2]. Yet the NMDA story is only half the plot: other NMDA antagonists (e.g., memantine or MK-801) fail to reproduce ketamine's robust clinical benefit, and the less potent NMDA-blocking R-enantiomer of ketamine actually shows the stronger and longer-lasting antidepressant activity [3]. Those clues point beyond NMDA blockade alone.
The glutamate pulse set loose by ketamine lands squarely on postsynaptic AMPA receptors. Rodent work shows that blocking AMPA receptors with NBQX abolishes both the immediate and the sustained behavioral antidepressant effects of ketamine, while positive AMPA modulators ("AMPAkines") mimic or amplify the response [4,5]. Electrophysiology and EEG demonstrate a rapid rise in AMPA-mediated currents and γ-band power, and within an hour the surface expression of GluA1/GluA2 subunits is up-regulated in prefrontal and hippocampal synapses [6]. In short, ketamine appears to flip the glutamatergic balance from "NMDA dominant" to "AMPA dominant," a shift sometimes summarized as increasing the AMPA/NMDA throughput ratio, which is thought to be the proximal signal for its antidepressant action.
Figure 1.
Ketamine initiates its antidepressant effect through The Disinhibition Trigger, by blocking NMDA receptors specifically on GABA interneurons—the brain’s natural "brakes." This disinhibition unleashes a burst of activity in excitatory pyramidal neurons, flooding synapses with glutamate and creating the initial spark. As NMDA receptors are blocked or subdued, the excess glutamate is funneled through AMPA receptors, marking The AMPA Dominance phase, where synaptic signaling shifts toward a "High AMPA / Low NMDA" throughput. Studies confirm that blocking AMPA receptors nullifies Ketamine's antidepressant action, highlighting their central role. Finally, Structural Restoration occurs as AMPA activation leads to calcium influx, which triggers BDNF release and mTOR activation. This cascade rapidly synthesizes new proteins, regrowing dendritic spines lost to stress—and the resulting physical repair of neural circuits closely tracks with the observed clinical improvement in mood.
Figure 1.
Ketamine initiates its antidepressant effect through The Disinhibition Trigger, by blocking NMDA receptors specifically on GABA interneurons—the brain’s natural "brakes." This disinhibition unleashes a burst of activity in excitatory pyramidal neurons, flooding synapses with glutamate and creating the initial spark. As NMDA receptors are blocked or subdued, the excess glutamate is funneled through AMPA receptors, marking The AMPA Dominance phase, where synaptic signaling shifts toward a "High AMPA / Low NMDA" throughput. Studies confirm that blocking AMPA receptors nullifies Ketamine's antidepressant action, highlighting their central role. Finally, Structural Restoration occurs as AMPA activation leads to calcium influx, which triggers BDNF release and mTOR activation. This cascade rapidly synthesizes new proteins, regrowing dendritic spines lost to stress—and the resulting physical repair of neural circuits closely tracks with the observed clinical improvement in mood.
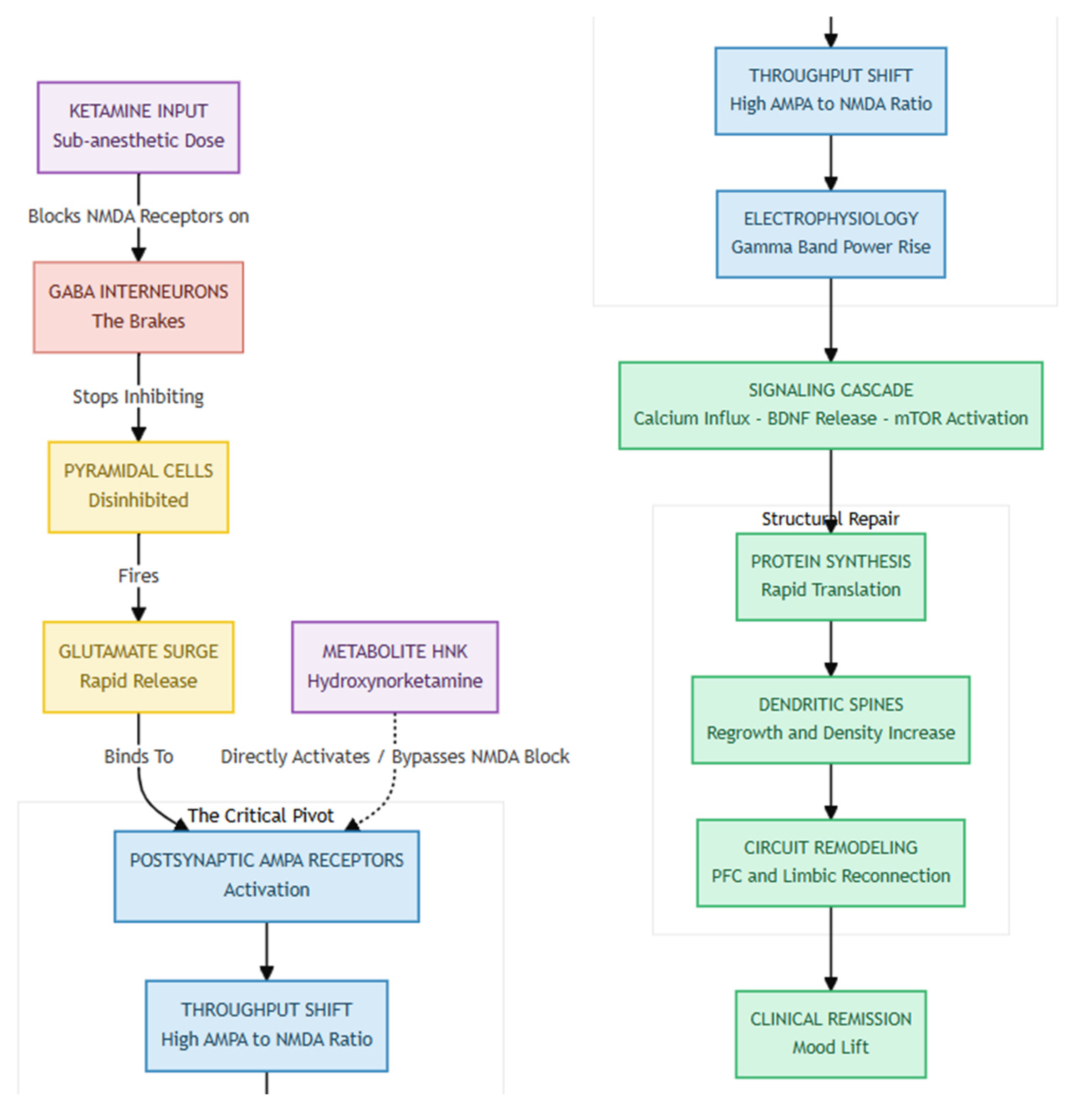
When AMPA receptors fire, the resulting calcium rush prompts the release of brain-derived neurotrophic factor (BDNF) and switches on the mTOR pathway. That, in turn, jump-starts rapid protein synthesis, dendritic spine growth, and circuit remodeling that repairs stress-related synaptic loss [1,6]. Within about 24 h, spine density and the functional connections linking prefrontal and limbic regions rebound, and the extent of this rebound tracks with how long symptom relief lasts. Remarkably, the ketamine metabolite hydroxynorketamine (HNK) can reproduce the same AMPA-, BDNF-, and mTOR-dependent effects without blocking NMDA receptors or causing dissociation, pointing to the possibility of safer next-generation treatments [3]. The evidence indicates that ketamine alleviates depression by temporarily inhibiting the NMDA receptor and activating the AMPA receptor, thereby reactivating the brain's inherent capacity for adaptive neuroplasticity.
Auvelity: Delivering the NMDA Spark but Missing the Full AMPA Flame
Figure 2.
Auvelity achieves its effect through The Kinetic Boost, using a pharmacokinetic strategy where Bupropion not only contributes to mood regulation but also inhibits the CYP2D6 enzyme, preventing the rapid breakdown of Dextromethorphan and allowing it to reach therapeutic concentrations in the brain—levels that would otherwise be unachievable. This sets the stage for The NMDA Spark, as Dextromethorphan, like Ketamine, acts as an NMDA antagonist, reducing NMDA signaling and providing the initial antidepressant "spark," which likely accounts for Auvelity’s faster onset compared to traditional antidepressants. However, The Missing AMPA Flame highlights a key limitation: unlike Ketamine, Dextromethorphan does not trigger a glutamate surge or directly engage AMPA receptors. Without this critical "fuel," the cascade leading to rapid synaptogenesis and structural neural repair may be weaker, potentially explaining Auvelity’s comparatively reduced efficacy and speed.
Figure 2.
Auvelity achieves its effect through The Kinetic Boost, using a pharmacokinetic strategy where Bupropion not only contributes to mood regulation but also inhibits the CYP2D6 enzyme, preventing the rapid breakdown of Dextromethorphan and allowing it to reach therapeutic concentrations in the brain—levels that would otherwise be unachievable. This sets the stage for The NMDA Spark, as Dextromethorphan, like Ketamine, acts as an NMDA antagonist, reducing NMDA signaling and providing the initial antidepressant "spark," which likely accounts for Auvelity’s faster onset compared to traditional antidepressants. However, The Missing AMPA Flame highlights a key limitation: unlike Ketamine, Dextromethorphan does not trigger a glutamate surge or directly engage AMPA receptors. Without this critical "fuel," the cascade leading to rapid synaptogenesis and structural neural repair may be weaker, potentially explaining Auvelity’s comparatively reduced efficacy and speed.
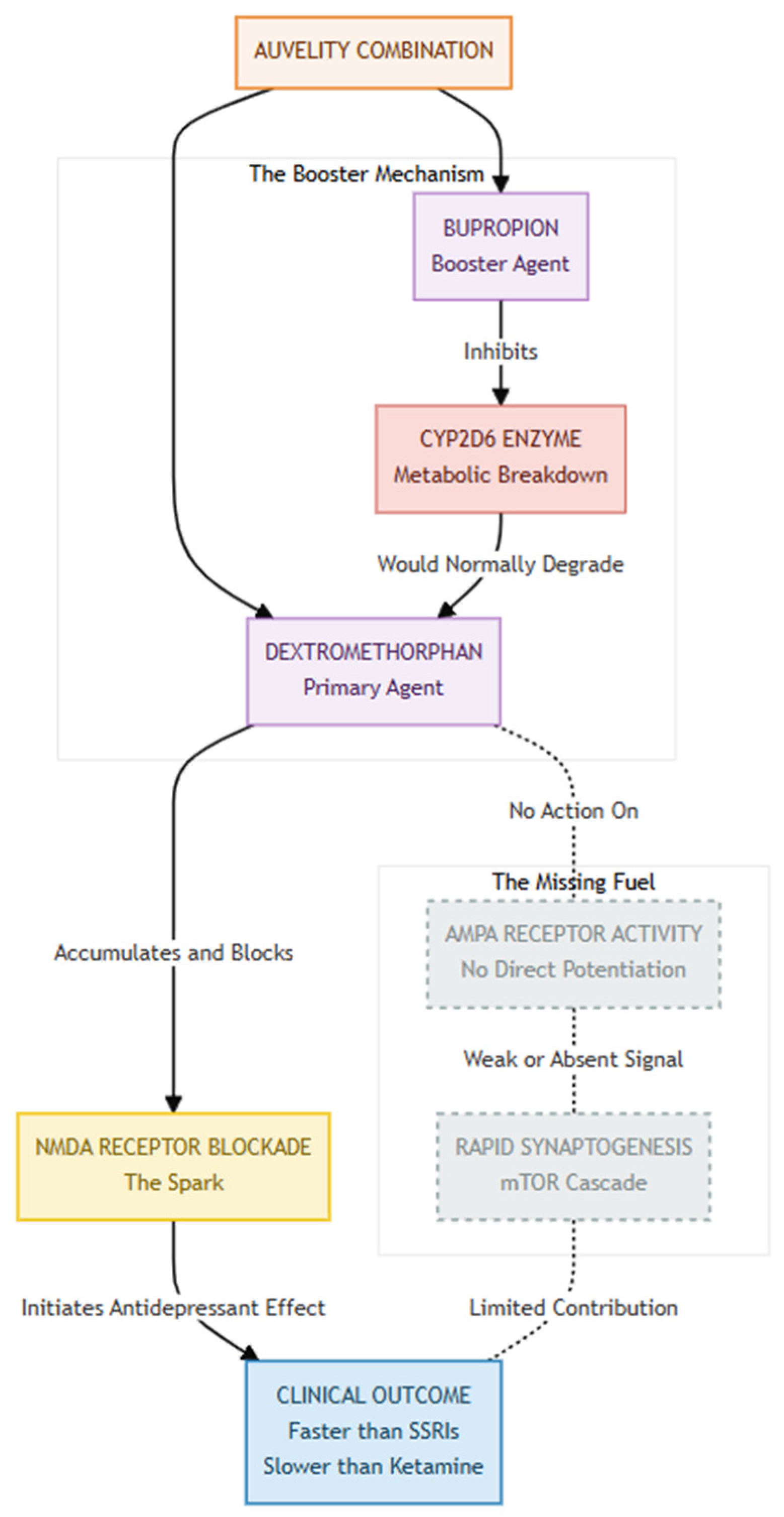
Auvelity®—the fixed-dose combination of dextromethorphan and bupropion—appears to replicate solely the initial phase of ketamine's antidepressant cascade. Dextromethorphan, an uncompetitive NMDA-receptor antagonist like ketamine, reduces NMDA-mediated signalling, while bupropion slows its breakdown by inhibiting CYP2D6, thereby extending exposure [7]. Whether the combination also produces the secondary rise in AMPA-receptor activity believed to drive ketamine-related synaptogenesis and prolonged mood benefits remains an open question. Dextromethorphan has no intrinsic AMPA-potentiating action, and neither ingredient directly augments AMPA currents. In other words, Auvelity may supply the NMDA-blocking "spark" without the AMPA "fuel." This pharmacological gap could help explain why Auvelity acts faster than classic monoaminergic antidepressants yet still lags behind ketamine in both onset and effect size, and why the durability of its benefit is still being studied [7].
Using CYP2D6 Inhibitors to Keep Dextromethorphan Active Without Bupropion
When the bupropion half of Auvelity is unavailable, the simplest way to keep dextromethorphan (DXM) from being chewed up too quickly is to pair it with another psychotropic that blocks the same metabolic "off-ramp," CYP2D6. Fluoxetine and paroxetine stand out because they are high-affinity, mechanism-based inhibitors that can push extensive metabolizers into the poor-metabolizer range within days [8,9]. In practice, adding 20 mg of fluoxetine or 10–20 mg of paroxetine once daily would be expected to raise DXM exposure to a degree comparable with—or greater than—the 105 mg of bupropion found in Auvelity, while simultaneously providing their own SSRI antidepressant activity. Duloxetine offers a middle-ground: it is only a moderate inhibitor, yet at 60 mg daily can still curb CYP2D6 enough to meaningfully boost DXM while delivering dual serotonin–norepinephrine re-uptake benefits [10]. Sertraline could work in a pinch, but only at ≥100 mg, a dose that many patients find activating [11].
Antipsychotics present a more unconventional—but pharmacologically sound—backup plan. Low doses of perphenazine or thioridazine inhibit CYP2D6 with Ki values in the same ballpark as the laboratory reference inhibitor quinidine, suggesting that even 2–4 mg of perphenazine at bedtime could "protect" DXM throughout the day [12]. Risperidone is gentler, acting as a mild inhibitor at routine doses, yet it might suffice in patients who already take a mood-stabilizing atypical and simply need a modest DXM boost [13]. Whichever substitute is chosen, clinicians must remember that blocking CYP2D6 is a double-edged sword: it enhances DXM's rapid-acting antidepressant punch but can also elevate plasma levels of any co-prescribed β-blockers, opioids, or tamoxifen [14]. Close monitoring—especially during the first two weeks—keeps the strategy safe, letting DXM do its NMDA-modulating job while its "pharmacokinetic bodyguard" stands watch.
How Budget- Friendly Piracetam Can Supercharge Auvelity's Antidepressant Spark
Auvelity (dextromethorphan + bupropion) relieves depression by two complementary moves: bupropion boosts cortical catecholamine and glutamate release, while dextromethorphan lowers background "static" at NMDA receptors so that meaningful glutamatergic bursts come through more clearly [15]. What the combo does not directly address is the final hand-off of that glutamate signal to AMPA receptors, the fast synaptic switches that trigger BDNF release and downstream mood-repair cascades. Piracetam—and several other inexpensive AMPA-positive allosteric modulators (e.g., aniracetam, CX-series ampakines)—can fill this gap. In aged animals piracetam increases AMPA receptor density and enhances AMPA-mediated currents, thereby strengthening long-term potentiation and synaptic plasticity [16,17]. Layering a cheap, well-tolerated dose of piracetam (1–3 g/day oral; 18) onto standard Auvelity could therefore push more of the liberated glutamate through a "wide-open" AMPA channel, theoretically translating into a quicker and more robust lift in mood, energy, and cognitive drive.
Figure 3.
The Auvelity "Spark" primes the brain for transformation by combining Bupropion, which boosts glutamate release, with Dextromethorphan, which clears NMDA receptor "static," producing a clean, high-fidelity glutamate signal ready for transmission. However, this signal requires a sensitive target, which is where The Piracetam "Fuel" comes in—acting as an AMPA Positive Allosteric Modulator (PAM), Piracetam increases AMPA receptor density and keeps the channels "wide open," ensuring the glutamate signal is fully captured rather than dissipated. The synergy between the clean signal provided by Auvelity and the sensitized receptors enabled by Piracetam leads to The Supercharged Result: a potent release of BDNF that enhances synaptic plasticity and accelerates mood repair, theoretically delivering a faster lift in energy, cognition, and emotional resilience than Auvelity alone.
Figure 3.
The Auvelity "Spark" primes the brain for transformation by combining Bupropion, which boosts glutamate release, with Dextromethorphan, which clears NMDA receptor "static," producing a clean, high-fidelity glutamate signal ready for transmission. However, this signal requires a sensitive target, which is where The Piracetam "Fuel" comes in—acting as an AMPA Positive Allosteric Modulator (PAM), Piracetam increases AMPA receptor density and keeps the channels "wide open," ensuring the glutamate signal is fully captured rather than dissipated. The synergy between the clean signal provided by Auvelity and the sensitized receptors enabled by Piracetam leads to The Supercharged Result: a potent release of BDNF that enhances synaptic plasticity and accelerates mood repair, theoretically delivering a faster lift in energy, cognition, and emotional resilience than Auvelity alone.
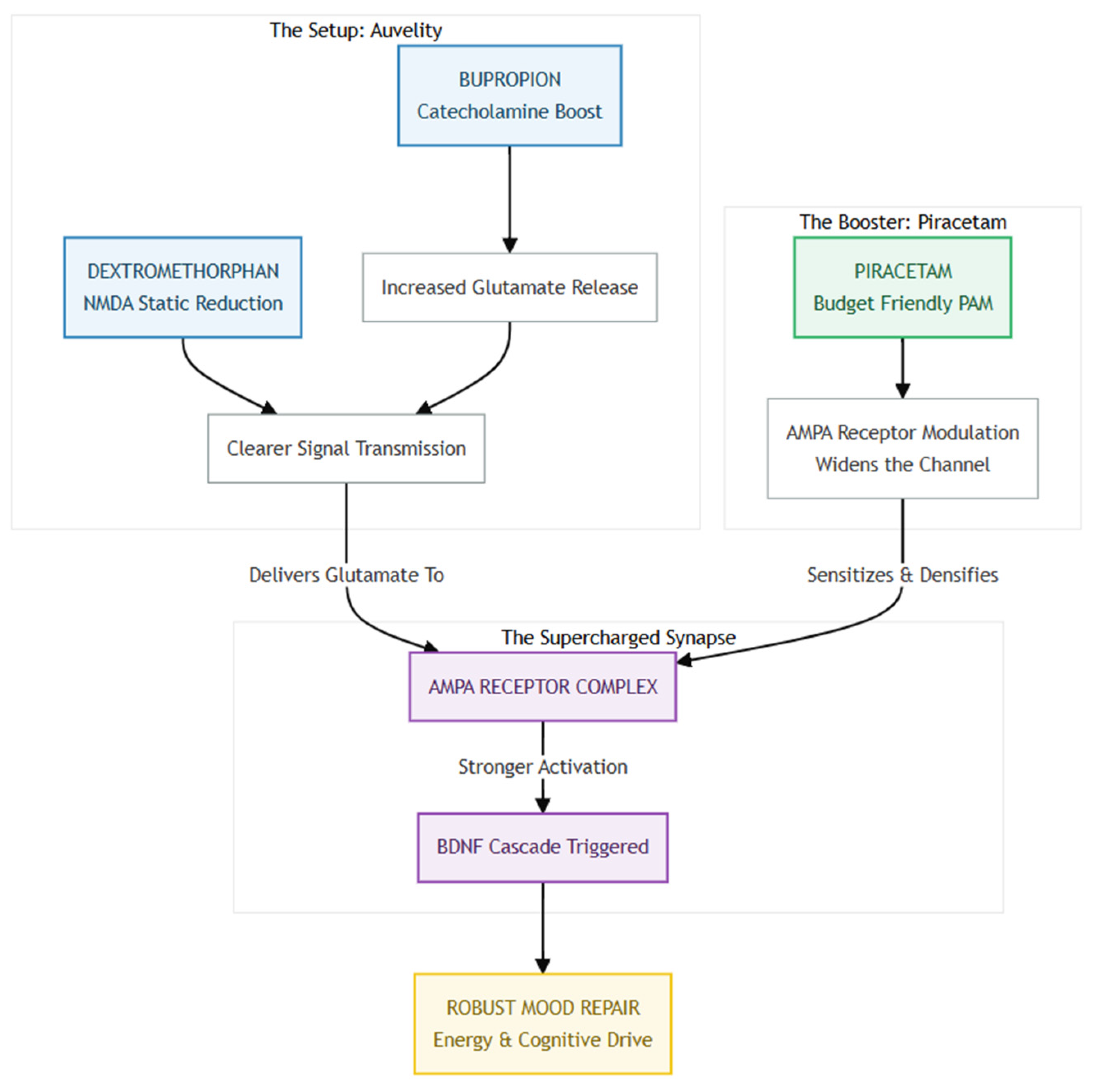
From a practical standpoint the strategy is attractive: piracetam is generic in most countries, costs only a few dollars per week, and carries a side-effect profile that is gentler than many vitamins [18]. Because it lacks significant hepatic metabolism or protein binding, drug–drug interactions with Auvelity are unlikely, and the main safety considerations are mild insomnia or jitteriness—signs that the AMPA gain may simply need dialing back. Until head-to-head trials are run, clinicians could consider a cautious "AMPA-priming" add-on in patients who have a partial response to Auvelity alone, monitoring sleep and anxiety just as they would when titrating bupropion. In short, Auvelity may provide the NMDA "spark," but a low-cost AMPA PAM like piracetam can help the antidepressant flame catch—and stay lit.
How Glutamine Supercharges the New Glutamatergic Stack
Figure 4.
Refilling the Tank is essential because chronic stress depletes the brain’s presynaptic glutamate reserves, and without sufficient Glutamine, there’s simply no fuel to generate healthy glutamatergic bursts. Oral Glutamine replenishes this supply, providing the raw material needed to sustain neurotransmission and potentially replacing the need for Bupropion’s glutamate-releasing effect. As Fueling the Machinery, Glutamine becomes critical in a stack that includes DXM, which clears inhibitory blocks, and Piracetam, which enhances AMPA receptor throughput—both of which increase demand without supplying the signal itself. Glutamine ensures there’s actual neurotransmitter available to be transmitted through the enhanced circuit. Finally, as The Safety Valve, Glutamine adds a protective layer by buffering against excitotoxicity—a known risk when combining NMDA modulation with AMPA amplification. It helps regulate inflammation-driven glutamate spikes and stabilizes cycle dynamics, preventing overshoot and neuronal damage, thereby keeping the stack both effective and sustainable.
Figure 4.
Refilling the Tank is essential because chronic stress depletes the brain’s presynaptic glutamate reserves, and without sufficient Glutamine, there’s simply no fuel to generate healthy glutamatergic bursts. Oral Glutamine replenishes this supply, providing the raw material needed to sustain neurotransmission and potentially replacing the need for Bupropion’s glutamate-releasing effect. As Fueling the Machinery, Glutamine becomes critical in a stack that includes DXM, which clears inhibitory blocks, and Piracetam, which enhances AMPA receptor throughput—both of which increase demand without supplying the signal itself. Glutamine ensures there’s actual neurotransmitter available to be transmitted through the enhanced circuit. Finally, as The Safety Valve, Glutamine adds a protective layer by buffering against excitotoxicity—a known risk when combining NMDA modulation with AMPA amplification. It helps regulate inflammation-driven glutamate spikes and stabilizes cycle dynamics, preventing overshoot and neuronal damage, thereby keeping the stack both effective and sustainable.
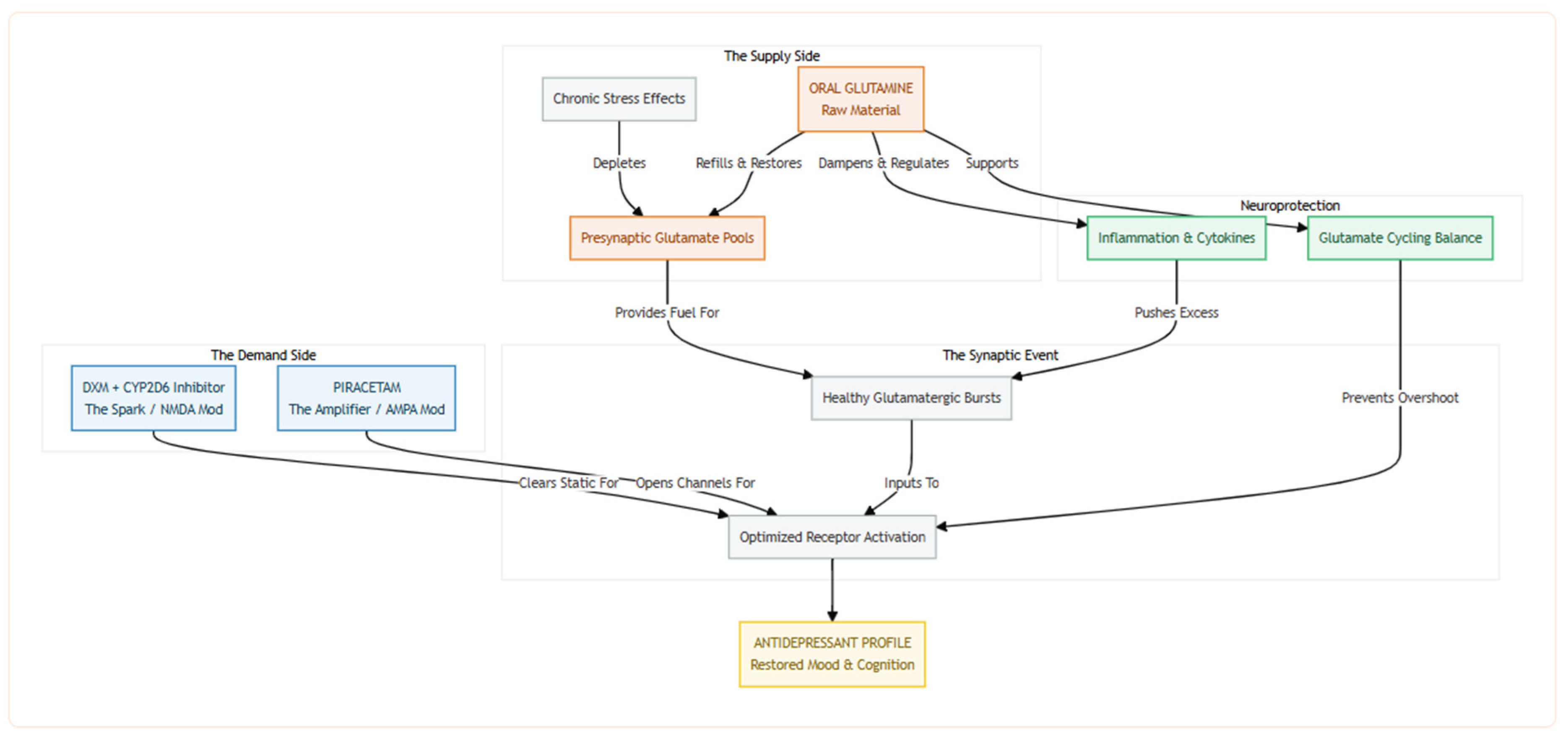
Adding oral glutamine to the new regime (i.e., dextromethorphan prolonged by a CYP2D6 inhibitor plus the AMPA-priming piracetam) can replenish the raw material the brain needs to fire healthy glutamatergic bursts. Chronic stress depletes presynaptic pools of glutamate, blunting excitatory postsynaptic currents in the medial prefrontal cortex and flattening mood; in rodents, just one to two weeks of dietary glutamine reverses these deficits, normalizes glutamate and glutamine levels, and produces a clear-cut antidepressant-like behavioral profile [19,20]. By topping up intracellular glutamine, we ensure that dextromethorphan's NMDA-modulating "spark" and piracetam's AMPA-opening "amplifier" actually have enough neurotransmitter fuel to work with—something especially important now that bupropion (an inhibitor of stimulus-evoked glutamate release) is no longer in the mix [21]. In short, glutamine can restore the supply side of the synapse while the rest of the stack optimizes demand.
Glutamine also seems to protect the brain. When inflammation is brewing, microglial signals like TNF-α and IL-1β push neurons to crank up glutaminase, flooding the space between cells with too much glutamate [22]. Large supplemental doses of glutamine actually dial that problem back: they lower spinal-fluid glutamate in people with mitochondrial encephalopathy [23] and blunt the cognitive hit of cytokines in Alzheimer's mouse models [24]. Keeping glutamate cycling smoothly this way could cut down on the excitotoxic "overshoot" that can happen when NMDA blockers and AMPA boosters are used together.
Summary
By pairing pharmacokinetic protection (CYP2D6 inhibition) with sequential pharmacodynamic "pushes" at NMDA, AMPA, and glutamate-supply checkpoints, the proposed stack aims to compress onset to hours, extend relief to weeks, and minimize adverse effects. Dextromethorphan provides the trigger; fluoxetine (or paroxetine) keeps blood levels therapeutic; piracetam opens the AMPA gate that releases BDNF and engages mTOR-dependent synaptogenesis; and glutamine ensures that chronic stress–drained terminals have the raw material to sustain healthy firing while also dampening cytokine-driven glutamate spikes. Because all four agents are oral, generic, and largely non-overlapping in safety profiles, the regimen is immediately testable in a phase-2, parallel-group study against standard Auvelity, with primary endpoints of 24-hour MADRS change and four-week durability. If successful, this approach could deliver ketamine-like speed and staying power in a prescription bottle, expanding rapid-acting relief from specialized clinics to everyday practice.
References
- Duman RS, Aghajanian GK. Synaptic dysfunction in depression: Potential therapeutic targets. Science. 2012;338(6103):68-72.
- Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351-354.
- Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481-486.
- Maeng S, Zarate CA Jr, Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: Role of AMPA receptors. Biol Psychiatry. 2008;63(4):349-352.
- Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models. Behav Brain Res. 2011;224(1):107-111.
- Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959-964.
- McCarthy B, Bunn H, Santalucia M, et al. Dextromethorphan-bupropion (Auvelity) for the Treatment of Major Depressive Disorder. Clin Psychopharmacol Neurosci. 2023;21(4):609-616.
- Crewe HK, Lennard MS, Tucker GT, et al. The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol. 1992;34(3):262-265.
- Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
- Spina E, Santoro V, D'Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
- Flockhart DA, Thacker D, McDonald C, et al. The Flockhart Cytochrome P450 Drug-Drug Interaction Table. Division of Clinical Pharmacology, Indiana University School of Medicine; 2021.
- Shin JG, Soukhova N, Flockhart DA. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: preferential inhibition of CYP2D6. Drug Metab Dispos. 1999;27(9):1078-1084.
- Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
- Bousman CA, Stevenson JM, Ramsey LB, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6, CYP2C19, CYP2B6, SLC6A4, and HTR2A Genotypes and Serotonin Reuptake Inhibitor Antidepressants. Clin Pharmacol Ther. 2023;114(1):51-68.
- Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of dextromethorphan-bupropion in major depressive disorder: A randomized, double-blind, controlled trial. Am J Psychiatry. 2022;179(7):490-499.
- Cohen SA, Müller WE. Effects of piracetam on N-methyl-D-aspartate receptor properties in the aged mouse brain. Pharmacology. 1993;47(4):217-222.
- Gualtieri F, Manetti D, Romanelli MN, et al. Design and study of piracetam-like nootropics, controversial members of the problematic class of cognition-enhancing drugs. Curr Pharm Des. 2002;8(2):125-138.
- Winblad B. Piracetam: A review of pharmacological properties and clinical uses. CNS Drug Rev. 2005;11(2):169-182.
- Baek JH, Jung S, Son H, et al. Glutamine Supplementation Prevents Chronic Stress-Induced Mild Cognitive Impairment. Nutrients. 2020;12(4):910.
- Son H, Baek JH, Go BS, et al. Glutamine has antidepressive effects through increments of glutamate and glutamine levels and glutamatergic activity in the medial prefrontal cortex. Neuropharmacology. 2018;143:143-152.
- Lin TY, Yang TT, Lu CW, et al. Inhibition of glutamate release by bupropion in rat cerebral cortex nerve terminals. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(2):598-606.
- Ye L, Huang Y, Zhao L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125(6):897-908.
- Guerrero-Molina MP, Morales-Conejo M, Delmiro A, et al. High-dose oral glutamine supplementation reduces elevated glutamate levels in cerebrospinal fluid in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur J Neurol. 2023;30(2):538-547.
- Baek JH, Park H, Kang H, et al. The Role of Glutamine Homeostasis in Emotional and Cognitive Functions. Int J Mol Sci. 2024;25(2):1302.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



