Submitted:
21 November 2025
Posted:
24 November 2025
You are already at the latest version
Abstract
Chronic inflammation and the development of cancer are closely linked, with components that comprise the tumour microenvironment—including proinflammatory cytokines—exerting essential tumourigenic effects. These proinflammatory cytokines include IL-1β, which has been reported to be overexpressed in several cancers and shown to activate several signalling pathways. These pathways may involve kinases such as AKT and Src, and have a broad capacity to activate nuclear factors, including NF-κB, which can regulate the transcription of genes encoding proteins such as cIAP1, Bcl-2, and cyclin D, thereby regulating processes like apoptosis and cell cycle inhibition. The objective of this study was to investigate the role of IL-1β in regulating cell death and proliferation in leukaemic lymphoblasts via the PI3K/AKT/NF-κB pathway. We employed flow cytometry to determine the expression levels and phosphorylation status of various proteins, proliferation was assessed using the CCK-8 kit, and apoptosis was evaluated with the Annexin V kit. Our findings indicate that the signalling pathway activated by IL-1β modulates these cellular processes in leukaemic lymphoblasts. We therefore conclude that IL-1β exerts significant effects on cell death and proliferation in leukaemic lymphoblasts through the PI3K/AKT/NF-κB pathway, with the study’s findings indicating that an inflammatory environment may promote such lymphoblasts to acquire neoplastic characteristics. As such, the proteins involved in the effects evaluated in this work could be considered as potential therapeutic targets for the treatment of Acute Lymphoblastic Leukaemia (ALL).
Keywords:
leukaemia
; IL-1β
; inflammation
; proliferation
; apoptosis
; necroptosis
Introduction
Inter- and intracellular events such as inhibition of apoptosis, proliferation, differentiation, and inflammation are involved in the development of neoplastic processes [1]. In the specific case of inflammation, the presence of proinflammatory cytokines such as IL-1β, IL-6, and TNF-alpha has been reported in the tumour environment for various cancers [2]. These cytokines are produced by immune cells that are attracted to the niche in which cancer cells develop and may also be secreted by the same cells that comprise the niche [3,4]. Increased IL-1β expression has been reported in several cancers, including hepatocellular carcinoma, colorectal cancer, and chronic myeloid leukaemia [5,6,7]. Once IL-1β is recognised by its receptor in the cell, it initiates a signalling cascade involving kinases and transcription factors, such as NF-κB, which regulate the expression of various genes involved in apoptosis, proliferation, and inflammation [8].
ALL is a hematopoietic neoplasm that originates in the bone marrow and subsequently spreads to the lymph nodes, spleen, and central nervous system [9].
While the effect of IL-1β on lymphoblasts from ALL has not been reported, it has been found to induce NF-κB and PI3K activation in other types of neoplasia, such as liver cancer, and has also been shown to influence NF-κB activation [10]. Another study reported that Src kinase induces NF-κB activation and IL-6 expression in cells undergoing Src oncogene-dependent transformation [11]. The possible effects of NF-κB activation mentioned above may help neoplastic cells to remain and develop in the niche in which they arose.
This study aimed to investigate whether the inflammatory cytokine IL-1β, through kinases such as PI3K/Akt, Src, and NF-κB, affects cell death and proliferation in leukaemic lymphoblasts. We found that this cytokine inhibits early apoptosis and limits necroptosis in leukaemic lymphoblasts while increasing proliferation, in a manner mediated by the activity and activation of PI3K, AKT, Src, and NF-κB.
Materials and Methods
In this study, the RS4:11 cell line—obtained from the ATCC, a recognised biological resource centre—was employed as the cellular model. RS4:11 cells were originally derived from the bone marrow of a 32-year-old female patient with relapsed acute lymphoblastic leukaemia (ALL). These cells exhibit a biphenotypic profile, co-expressing both B-cell and monocytic markers, indicative of mixed-lineage characteristics typical of this leukaemia subtype. Additionally, RS4:11 cells display features consistent with pre-B lymphoblasts, including expression of CD19, HLA-DR, and terminal deoxynucleotidyl transferase. These cells also demonstrate lineage plasticity upon induction of differentiation, making RS4:11 a robust and valuable model for investigating signalling pathways and evaluating therapeutic strategies in acute lymphoblastic leukaemia [12,13].
Cell Culture
The RS4:11 cell line was cultured in Iscove’s medium supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C and with 5% CO2. Lymphoblasts were harvested and washed with PBS, then seeded overnight in 48-well plates with serum-free culture medium at a cell density of 1.0 × 106 lymphoblasts/mL. The cells were treated with 10 nM of IL-1β (Peprotech) and the following inhibitors were used: for PI3K, Wortmannin at 200 nM [14]; for Src, PP2 at 20 μM [15] (Sigma Co); and for NF-κB, JSH23 (Calbiochem) at 10 µM [16].
Protein Expression via Flow Cytometry
The RS4:11 cells were washed with PBS and subsequently fixed with 2% paraformaldehyde at 37 °C for 10 minutes. They were then permeabilised with 1% SDS for 5 minutes, followed by further washing with PBS. Next, the cells were incubated overnight at 4 °C with the corresponding primary antibodies (mouse monoclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:150 each for anti-p-AKT, anti-p-Src, p65, Bcl-2, cIAP1, and cyclin D1 antibody, at a final concentration of 1.3 µg/µL. After removal of the primary antibodies by washing with PBS, the cells were incubated for 1 hour at room temperature with an FITC-labelled secondary antibody diluted 1:100 (Jackson Immuno Research), followed by additional washing with PBS. Non-specific binding was assessed by incubating the cells with the secondary antibody alone. Detection of FITC-positive leukaemic lymphoblasts was performed using an Aria FACS cytometer (Becton Dickinson) analysing 10,000 events, and the data were analysed using the FlowJo software, version 10.0.
Proliferation and Apoptosis
Leukaemic lymphoblasts were cultured in 96-well plates and incubated for 24, 48, and 72 hours with IL-1β. Proliferation was measured using the CCK-8 Kit (Enzo), and absorbance was detected at 450 nm to evaluate cell proliferation.
For analysis of apoptosis, lymphoblasts were incubated with IL-1β for 48 hours, then washed and stained with Annexin V FITC/IP. Each sample was analysed using a flow cytometer (FACS), and the data were analysed with the FlowJo software to determine the percentages of necrotic cells (Annexin V FITC-/PI+), early apoptotic cells (Annexin (FITC+/PI-), and late apoptotic cells (Annexin V FITC-/PI-).
Statistical Analysis
Three independent experiments were conducted in triplicate for each determination. Data are presented as the mean ± standard deviation. Statistical analyses were conducted using GraphPad Prism version 5.0. One-way analysis of variance (ANOVA) was performed to assess the phosphorylation levels of Akt, Src, and p65 in cells treated with IL-1β alone or in combination with inhibitors. Tukey’s multiple comparison test was applied to evaluate the expression of Bcl-2, cIAP1, and Cyclin D1 under the same treatment conditions, aiming to identify statistically significant differences between groups. Additionally, Student’s t-test was performed to analyse apoptosis and proliferation.
Results
IL-1β Induces AKT, Src, and NF-κB Activation in RS4:11 Leukaemic Lymphoblasts
We tested whether PI3K/AKT and Src were activated by IL-1β at different culture times (24, 48, and 72 h) by determining the phosphorylation of serine 473 of AKT and tyrosine 416 of Src via flow cytometry. When quantifying AKT phosphorylation, we observed that the level was increased 4-fold in comparison with the control group after 24 hours (9,306 vs. 2,296 MFI); meanwhile, at both 48 and 72 hours, the increase was 2.9-fold (4,268 vs. 1,454 MFI and 2,860 vs. 956 MFI, respectively; see Figure 1A and B).
To test whether IL-1β activates PI3K/AKT, and as PI3K activates AKT, we used wortmannin to inhibit PI3K (and, consequently, AKT) in RS4:11 leukaemic lymphoblasts cultured with IL-1β for 24 h. We found that AKT phosphorylation was decreased 2.53-fold in the presence of wortmannin compared with lymphoblasts treated with IL-1β alone (3,945 vs. 9,306 MFI; Figure 1C and D).
Regarding Src activation due to IL-1β in leukaemic lymphoblasts, we observed that Src showed a 2-fold increase at 24 h (3,996 vs. 1,960 MFI) and a 3-fold increase at 48 and 72 h, when compared with the control group (2,403 vs. 798 MFI and 2,810 vs. 621 MFI, respectively; Figure 1E and F). To test whether IL-1β induces the activation of Src, leukaemic lymphoblasts were incubated with IL-1β for 24 hours in the presence of a Src inhibitor. Src phosphorylation was found to be significantly decreased in the presence of PP2, when compared with lymphoblasts treated with IL-1β alone (2,452 vs. 3,996 MFI, 1.62-fold decrease; Figure 1G and H).
To determine NF-κB activation due to IL-1β, leukaemic lymphoblasts were cultured for 24, 48, and 72 h in the presence of IL-1β, and phosphorylation of serine 536 of the p65 subunit of NF-κB was analysed via flow cytometry. We found that after 24 hours of incubation, p65 showed a 3.7-fold increase in phosphorylation (12,384 vs. 3,281 MFI); after 48 hours, it was 1.66-fold higher (5,507 vs. 2,166 MFI); and, after 72 hours, it was 2.9-fold higher (4,953 vs. 1,107 MFI); see Figure 1I and J.
To investigate whether the activation of NF-κB by IL-1β might involve PI3K and Src kinases, lymphoblasts were cultured for 24 h with IL-1β and inhibitors of these proteins. We found that p65 phosphorylation was significantly decreased with both inhibitors compared with IL-1β alone, (8,391 MFI vs. 12,384 MFI, 1.47-fold decrease and 9,544 vs. 12,384 MFI, 1.29-fold decrease, respectively; Figure 1K and L). To prove that IL-1β specifically induces NF-κB activation, lymphoblasts were cultured with the NF-κB inhibitor for 1 h before and with IL-1β for 24 h after treatment. A significant decrease in MFI corresponding to p65 phosphorylation was observed (6,609 vs. 12,384 MFI, 1.87-fold decrease; Figure 1K and L).
IL-1β Influences Expression of Bcl-2 and cIAP1 via PI3K/AKT/NF-κB Pathway in RS4:11 Leukaemic Lymphoblasts
The genes encoding the anti-apoptotic proteins Bcl-2 and cIAP are known to be regulated by NF-κB [17,18]. For this reason, and considering our results above, we investigated whether activation of the PI3K/AKT/NF-κB pathway due to IL-1β had any effect on Bcl-2 and cIAP expression in leukaemic lymphoblasts. For this purpose, we cultured leukaemic lymphoblasts in the presence of IL-1β for 24, 48, and 72 h and assessed Bcl-2 and cIAP expression via flow cytometry.
When quantifying the effect of IL-1β on Bcl-2 expression, we did not observe a significant increase compared with control lymphoblasts at 24 h (2,367 vs. 2,047 MFI), while a 2.2-fold increase was observed at 48 hours (4,754 vs. 2,141 MFI) and a 1.9-fold increase at 72 hours (3,419 vs. 1,775 MFI); see Figure 2A and B. On the other hand, to determine whether IL-1β induces Bcl-2 expression via PI3K/AKT, Src, and NF-κB, we cultured leukaemic lymphoblasts with IL-1β in the presence of inhibitors for PI3K, Src, and NF-κB; as we observed the most significant increase in Bcl-2 expression at 48 h, we cultured the lymphoblasts for the same period. Inhibition of PI3K, Src, and NF-κB decreased Bcl-2 expression 3.9-fold (1,995 vs. 4,754 MFI), 2-fold (2361 vs. 4754 MFI), and 2.9-fold (1,610 vs. 4,754 MFI), respectively; see Figure 2C and D.
Regarding cIAP1 expression, we observed that IL-1β in leukaemic lymphoblasts induced 3.7-fold (13,661 vs. 3,601 MFI), 3.3-fold (7,128 vs. 2,144 MFI), and 3.8-fold (8,717 vs. 2,291 MFI) increases at 24, 48, and 72 hours of culture, respectively; see Figure 2E and F.
To verify whether cIAP1 expression is mediated by PI3K, Src, and NF-κB, we also cultured leukaemic lymphoblasts with IL-1β in the presence of inhibitors of these proteins. As the expression of cIAP1 was similar at all three culture times, we decided to culture the lymphoblasts for 24 hours. Wortmannin, PP2, and JSH23 decreased cIAP1 expression 2.2-fold (5,954 vs. 13,661 MFI), 1.79-fold (7,631 vs. 13,661 MFI), and 3.5-fold (3,824 vs. 13,661 MFI), respectively; see Figure 2G and H.
IL-1β Decreases Apoptosis and Limits Necroptosis via the PI3K/AKT /Src/NF-κB Pathway in RS4:11 Leukaemic Lymphoblasts
Having observed that IL-1β increases the expression of anti-apoptotic proteins in leukaemic lymphoblasts, we investigated whether this interleukin inhibits apoptosis in these cells. To this end, we analysed the presence of phosphatidylserine residues on lymphoblast cell surfaces via labelling with annexin V after 48 hours, as we observed the highest expression of Bcl-2 at this time.
As shown in Figure 3A and Tables S1 and S2 (Supplementary Materials [this section shows the significant differences between the values being compared]) lymphoblasts treated with IL-1β had a 37% greater viable cell rate than control lymphoblasts (89.76% vs. 65.11%). We also observed that these lymphoblasts showed a 5.3-fold decrease in early apoptosis compared with untreated lymphoblasts (0.90% vs. 5.0%). Regarding lymphoblasts in late apoptosis, we observed a comparable percentage in both experimental groups although they exhibit a significant difference, (IL-1β: 0.91%; control group: 0.84%; see Figure 3A, Tables S1 and S2 in Supplementary Materials).
In our analysis of the involvement of the transcription factor NF-κB and the kinases Src and AKT in early apoptosis in leukaemic lymphoblasts, we found that it decreased by 1.7-fold when NF-κB was inhibited (0.90% vs. 0.53%), by 3.9-fold when PI3K was inhibited (0.90% vs. 0.24%), and by 1.5-fold when Src was inhibited (0.90% vs. 0.62%); see Figure 3B and Tables S1 and S3 (Supplementary Materials). Inhibiting these same proteins in the presence of IL-1β had a less pronounced effect on late apoptosis in leukaemic lymphoblasts: wortmannin decreased it by 1.2-fold (from 0.91% to 0.75%), while PP2 decreased it by 1.0-fold (from 0.91% to 0.83%). However, when NF-κB was inhibited in leukaemic lymphoblasts in the presence of IL-1β, we observed a 2-fold increase in late apoptosis compared with lymphoblasts treated only with this interleukin (1.9% vs. 0.91%); see Figure 3B and Tables S1 and S4 (Supplementary Materials).
Regarding necrotic cells, we observed that lymphoblasts treated with IL-1β showed 5.0% necrotic cell death, compared with 13.64% in the control group (Figure 3A, Table 1), indicating that IL-1β limits necrosis by 36.9%. Regarding treatment with inhibitors, we found that when treating lymphoblasts with IL-1β plus JSH3, wortmannin, or PP2, the percentage of lymphoblasts in necrosis was 30.5%, 12.7%, and 10.53%, respectively. These results indicate that inhibiting NF-κB, PI3K, and Src in these lymphoblasts increases necrosis 6-fold, 2.5-fold, and 2.1-fold, respectively; see Tables S1 and S5 (Supplementary Materials).
IL-1β via PI3K/AKT /Src/NF-κB Influences Cyclin D1 Expression and Proliferation of RS4:11 Leukaemic Lymphoblasts
Cyclin D1 overexpression has been reported in various types of cancer [19]; however, it has not yet been established whether IL-1β can affect cyclin D1 expression. For this reason, our study evaluated the potential influence of IL-1β on cyclin D1 expression in leukaemic lymphoblasts via the PI3K/AKT/Src/NF-κB pathway at 24, 48, and 72 hours of culture. When quantifying cyclin D1 protein levels due to IL-1β stimulation in leukaemic lymphoblasts, we found a 1.5-fold increase in cyclin D1 levels at 24 h compared with the control group (6,420 vs. 4,192 MFI), a 1.85-fold increase at 48 h (7,076 vs. 3,813 MFI), and a 2.3-fold increase at 72 h (12,905 vs. 5,3812 MFI); see Figure 4A and B. Subsequently, as we observed the greatest upregulation of cyclin D1 in leukaemic lymphoblasts at 72 hours of culture with IL-1β, we investigated whether the activation of AKT, Src, and NF-κB could influence cyclin D1 expression after this period. Inhibition of PI3K, Src, and NF-κB led to 1.98-fold (6,493 vs. 12,905 MFI), 1.78-fold (7,247 vs. 12,905 MFI), and 2.98-fold (4,328 vs. 12,905 MFI) decreases in cyclin D expression, respectively, as shown in Figure 4C and D.
Since we observed that IL-1β induces overexpression of cyclin D1 in leukaemic lymphoblasts via the PI3K/AKT/Src/NF-κB pathway, we first assessed whether IL-1β induces proliferation of leukaemic lymphoblasts at 24, 48, and 72 h of culture. As shown in Figure 4E, F, and G, we observed 78%, 104%, and 91% increases in IL-1β over the control after 24 h, 48 h, and 72 h, respectively. We then assessed the effects of IL-1β on the proliferation of leukaemic lymphoblasts at these culture times, using the corresponding inhibitors for NF-κB, PI3K, and Src. At all cultivation times, we observed a decrease in proliferation compared with lymphoblasts treated only with IL-1β (at 24 hours: NF-κB, 88%; PI3K, 97%; and Src, 56%. At 48 hours: NF-κB, 93%; PI3K, 98%; and Src, 52%. At 72 hours: NF-κB, 99%; PI3K, 98%; and Src, 55%; see Figure 4 E, F, and G).
Discussion
As they secrete molecules such as proinflammatory cytokines and interferons, promoting a chronic inflammatory state that fosters the formation of a tumour microenvironment, the cells that constitute the immune system play a crucial role in the establishment and development of cancer [20]. The increased expression of IL-1β has been observed in chronic myeloid leukaemia [21], and autocrine secretion of IL-1β has even been reported in cell lines derived from chronic monocytic leukaemia and ALL [22]. Reports have indicated that the presence of IL-1β is related to the activation of PI3K/AKT in different cancers [23]. In addition, in 2007, Lin pointed out the relationship between IL-6 and Src in gastric cancer. Considering this evidence, we tested whether IL-1β induced the activation of AKT and Src in lymphoblasts derived from ALL, discovering that this interleukin effectively induced a significant increase in the activation of both kinases [24]. Therefore, we investigated whether exposure to IL-1β could induce the activation of NF-κB and found that this interleukin induced the highest increase in NF-κB activation in RS4:11 leukaemic lymphoblasts. This finding is consistent with observations in breast cancer, where IL-1β and NF-κB have been reported to be associated with the progression of this type of neoplasm [25].
These results led us to consider a possible signalling pathway in which IL-1β could trigger the activation of NF-κB. Subsequently, we tested whether the activation of these kinases, due to the presence of IL-1β, could be involved in the activation of NF-κB in lymphoblasts. As a result, we found that inhibiting PI3K and Src significantly decreased NF-κB activation. It has been reported separately in breast and cervical cancer cell lines that PI3K–AKT activates NF-κB, and in liver cancer that Src is involved in the activation of this nuclear factor [26,27,28]; therefore, these reports support our findings. However, in this study, we showed in the same ALL cell model that IL-1β triggers a signalling pathway involving PI3K/AKT/Src/NF-κB—an observation that has not yet been reported for any leukaemia.
On the other hand, it has previously been suggested that a general inflammatory environment may be related to some intrinsic characteristics of neoplastic processes, such as the inhibition of apoptosis and proliferation [29]. Therefore, we analysed whether IL-1β induces any changes in the expression of anti-apoptotic proteins, such as Bcl-2 and cIAP1, in leukaemic lymphoblasts. We observed that IL-1β triggers a significant increase in the expression of Bcl-2 and cIAP1 in these lymphoblasts; furthermore, when PI3K, Src, and NF-κB were inhibited, the expression of these anti-apoptotic proteins decreased significantly. Several research groups have reported the presence of these anti-apoptotic proteins in various types of neoplasms [30,31,32], suggesting that they may regulate the process of apoptosis. Concerning the relationship between proinflammatory interleukins and apoptosis-regulating proteins, it has been observed that IL-1β induces phosphorylation of Bcl-2 in acute myeloid leukaemia blasts [21]; on the other hand, in 2002, Spets et al. found that IL-6 induces an increase in the expression of anti-apoptotic proteins such as Bcl-2 and MCL-1 in multiple myeloma cells [33]. These data support our finding that IL-1β is involved in the expression of anti-apoptotic proteins via the PI3K/AKT/NF-κB pathway in leukaemic lymphoblasts.
Considering these results, we tested whether this interleukin has any effect on the death of leukaemic lymphoblasts. Our results indicated that IL-1β significantly decreases the number of lymphoblasts in early apoptosis. These findings are consistent with previous work, as it has been reported that Bcl-2 inhibits early apoptosis by preventing the externalisation of phosphatidylserine in the plasma membrane and can also inhibit the action of caspases by inhibiting the release of cytochrome c. It is well-known that cIAP1 inhibits caspases, which act in the first phase of apoptosis [34,35]. This observation is similar to that reported by Turzanki et al. in 2004, who found that IL-1β produced by blast cells from patients with chronic myeloid leukaemia causes resistance to apoptosis [21].
Regarding late apoptosis, no significant differences were observed between leukaemic lymphoblasts treated with or without IL-1β, nor were any differences found between early and late apoptosis in the presence of IL-1β. Taken together, these findings suggest that IL-1β exerts a general inhibitory effect on apoptosis in leukaemic lymphoblasts.
On the other hand, when NF-κB, PI3K, and Src were inhibited in leukaemic lymphoblasts in the presence of IL-1β, we observed that the percentage of lymphoblasts in early apoptosis decreased in general. These data confirm our earlier observations regarding the effects of these kinases and transcription factor on the expression of Bcl-2 and cIAP1.
Concerning the analysis of late apoptosis in leukaemic lymphoblasts treated with IL-1β in the presence of PI3K and Src inhibitors, we note that there was no significant difference in lymphoblasts at this stage of apoptosis—probably because IL-1β does not promote late apoptosis. On the other hand, we found that leukaemic lymphoblasts treated with IL-1β in the presence of the NF-κB inhibitor showed a modest increase in the percentage of lymphoblasts in late apoptosis; as this factor is known to regulate the transcription of proteins involved in the initiation of apoptosis [8], this increase could be due to the fact that when this transcription factor is inhibited, apoptosis is no longer inhibited and the lymphoblasts can therefore carry it out. Thus, these data also support our observation that IL-1β promotes early apoptosis in leukaemic lymphoblasts via NF-κB.
Likewise, we noted that this interleukin limits necrosis in leukaemic lymphoblasts. As is well-known, the concept of necrosis has evolved into a highly regulated process, now known as necroptosis. Necroptosis involves kinases such as RIPK1, 2, and 3, as well as MLKL [36]. As cIAP1 has a ubiquitin ligase domain, it can induce the degradation of target proteins [37]; one of these is RIPK1, which—when ubiquitinated and degraded by this pathway—can result in a decrease in necroptosis [38]. In this study, we observed that IL-1β induced an increase in the expression of cIAP1 in leukaemic lymphoblasts. On one hand, this protein may prevent early apoptosis; on the other hand, it may limit necrosis by eliminating RIP1K. It is important to note that the expression of RIPK3 and MLKL has been reported in ALL cells and that, when they were genetically suppressed and supplemented with caspase inhibitors, a significant decrease in cell death was observed [39]. Feldmann et al. (2017) have reported that in ALL cells, sorafenib—a multi-targeted tyrosine kinase inhibitor used for the treatment of acute leukaemia—decreased the phosphorylation of MLKL, resulting in a decrease in cell death [40]. These data support our theory and suggest that evaluating the expression of RIPK 1 and 3 in our cell model at a later stage could yield further insights.
When we observed the necroptosis of lymphoblasts previously treated with PI3K, Src, and NF-κB inhibitors followed by IL-1β, we found that necroptosis increased under all these conditions compared with lymphoblasts treated only with IL-1β. However, the most significant increase was noted when NF-κB was directly inhibited. These results may be due to NF-κB having cIAP1 as one of its target genes; therefore, inhibiting this transcription factor also decreases the expression of its target genes, including cIAP1 (this effect was observed in this study), which (as mentioned above) likely affects the elimination of RIPK1, thus causing an increase in necroptosis. This effect was similarly observed when PI3K and Src kinases were inhibited, as these kinases influence the activation of this transcription factor. These results further indicate that the necrosis observed when lymphoblasts were treated with IL-1β was influenced by activation of the PI3K/AKT/NF-κB signalling pathway, which implies regulation of this pathway. This finding allows us to consider the observed necrosis as necroptosis, as the defining feature that differentiates necroptosis from necrosis is the former is a regulated process. In this regard, we emphasise that our work demonstrates that IL-1β prevents cell death by inhibiting early apoptosis and limiting necroptosis in leukaemic lymphoblasts—a phenomenon which has not been previously reported.
In this study, we also investigated whether IL-1β could have any effect on the expression of proteins related to cell cycle regulation, such as cyclin D1. It has not previously been determined that IL-1β induces an increase in cyclin D expression in any disease or pathology; however, Bousserouel et al. (2004) reported that IL-1β slightly induced cyclin D in an in vitro system using rat muscle cells, and this effect was significantly enhanced upon the addition of arachidonic acid [41]. It has also been reported that IL-7 promotes the overexpression of cyclin D1 in lung cancer-derived cell lines [42]. These data align with our finding that a proinflammatory interleukin—namely, IL-1β—induced an increase in cyclin D1 expression in leukaemic lymphoblasts.
On the other hand, the inhibition of PIK3 in myeloma and breast cancer cells resulted in a decrease in cyclin D1 expression [19,43]. These data support our observation that PI3K activation, induced by IL-1β in leukaemic lymphoblasts, contributes significantly to cyclin D1 expression.
Different researchers have found that NF-κB induces the transcription of the cyclin D1 gene [44], supporting our finding that NF-κB inhibition causes a decrease in cyclin D1 expression in IL-1β-treated lymphoblasts. Furthermore, it is known that NF-κB activity may be modulated under the action of PI3K and Src kinases [23], consistent with our finding that lymphoblasts in the presence of IL-1β and the inhibitors of these kinases significantly decreased cyclin D1 expression. We then tested whether this interleukin influenced the proliferation of these lymphoblasts, and observed that this cytokine induces a significant increase in lymphoblast proliferation. Evidence from both in vivo and in vitro models has demonstrated that SPP1+ macrophages secrete IL-1β, which subsequently promotes the proliferation of head and neck squamous cell carcinoma [45]. Similarly, Teixeira et al. (2019) observed that silencing of NF-κB1 gene expression inhibited proliferation through the downregulation of IL-1β in renal cell carcinoma [46]. These data support our finding that IL-1β can induce lymphoblast proliferation; in our model, this effect is most likely related to the observed increase in Cyclin D1 expression.
Finally, we found that the inhibition of PI3K, Src, and NF-κB induced decreases in the proliferation of lymphoblasts treated with IL-1β. These results can also be explained by the fact that these kinases are involved in the activation of NF-κB which, in turn, induces the expression of cyclin D1. Nevertheless, it is important to note that the greatest percentage of inhibition in the expression of this cyclin in leukaemic lymphoblasts treated with IL-1β was observed when NF-κB and PI3K were inhibited. This finding is consistent with reports in other types of cancers regarding the regulation of Cyclin D by PI3K and NF-κB [43,44]. In the case of Src kinase inhibition, it was observed that its inhibition in leukaemic lymphoblasts treated with IL-1β reduced the expression of this cyclin by an average of 50%, indicating that IL-1β does not fully induce the activation of this kinase.
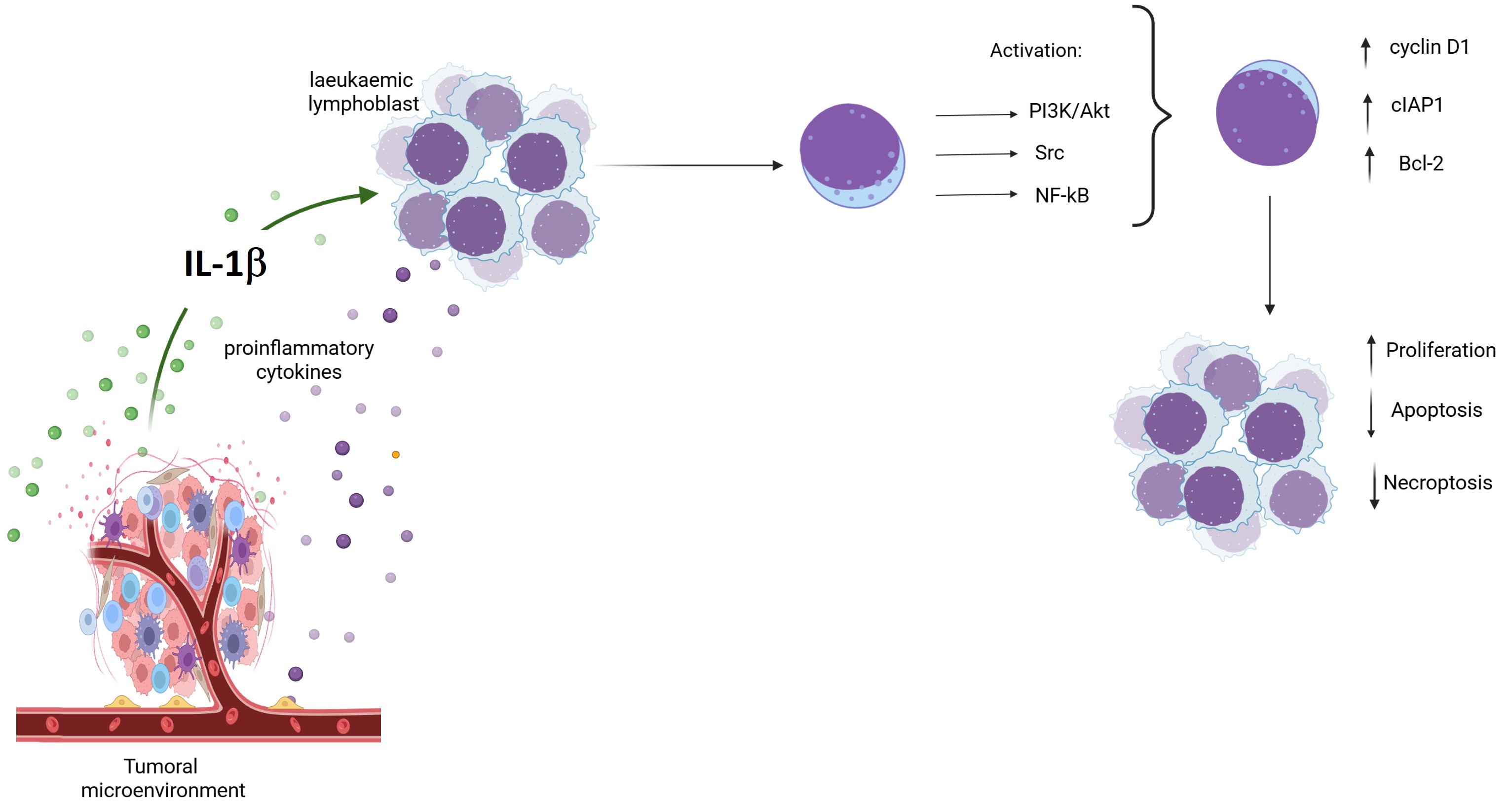
Considering all our results and the data already reported in the literature, we can propose a model of a signalling pathway in leukaemic lymphoblasts which is triggered by IL-1β and involves PI3K/AKT/Src/NF-κB, with effects on the regulation of cell death and lymphoblast proliferation. This pathway can be explained as follows: in the presence of IL-1β, the PI3K/AKT and Src kinases are activated in leukaemic lymphoblasts, which then activate IKKβ. The activation of IKKβ causes p65 translocation to the nucleus [47] and induces the transcription of its target genes, including Cyclin D1, Bcl-2, and cIAP1 [48]. The increase in Cyclin D1 expression could be involved in the observed increase in proliferation. Similarly, the increases in Bcl2 and cIAP1 expression could be related to the decrease in early apoptosis, and cIAP1 may also be involved in limiting necroptosis through the ubiquitination and subsequent degradation of RIPK1 (see Figure 5).
The presence of an inflammatory environment in the cellular niche where leukaemia cells appear or develop may contribute to their neoplastic characteristics. The role of proinflammatory cytokines and the proteins involved in the signalling pathways triggered by these molecules are related to the malignant abilities of these cells; therefore, this field of research points to possible therapeutic targets that may be of great benefit in the treatment of patients with this or other types of neoplasm.
Finally, our work demonstrates how a proinflammatory cytokine can influence the reduction in apoptosis and necroptosis while simultaneously increasing the proliferation of leukaemic lymphoblasts via the PI3K/AKT/NF-κB pathway. However, it is also necessary to determine whether other proinflammatory cytokines, such as IL-6 or TNF-α, have similar effects and through which signalling pathways they may act. Furthermore, understanding the additional effects of proinflammatory cytokines is crucial; for instance, whether they are involved in a possible epithelial–mesenchymal transition, which could enable leukaemic lymphoblasts to migrate to other organs and facilitate metastasis. However, this remains a hypothesis to be validated.
The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Z.V.A. contributed to the conceptualisation of the study and conducted all technical work. E.R.M. critically reviewed the manuscript. M.L.D.L. contributed to the flow cytometry analyses and performed critical revisions of the text. J.V.O. undertook a thorough critical review of the manuscript’s language and style. O.R.A. contributed to the flow cytometry analyses. A.L.R. contributed to implementing the modifications suggested by the reviewers. R.A.L. conceived the study, supervised the overall project, and prepared the initial draft of the manuscript.
Funding
Z.V.A. received a fellowship from CONACYT. Additional support for the authors was provided by scientific projects under SIP-IPN.
Institutional Review Board Statement
This study did not involve human participants, and therefore ethical approval by an Institutional Review Board was not required.
Informed Consent Statement
This study utilised an established cell line and did not involve direct participation of human subjects; therefore, informed consent from individuals was not required.
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
- Braun T., Carvalho G., et al. Targeting NF-kB in hematologic malignancies. Cell Death Differ. 2006. 13, 748-758. [CrossRef]
- Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008.18:19-26. [CrossRef]
- Grivennikov SI, Greten FR., et al. Immunity, inflammation, and cancer. Cell. 2010. 140:883-99. [CrossRef]
- Darnay, B. G., Aggarwal, B. Human cytokines. In Advances in Oncobiology. Pergamon Press.UK. ISBN 0-7623-016-5. 1996. Vol. 1, pp. 179–205.
- Bent R, Moll L., et al. Interleukin-1 Beta—A Friend or Foe in Malignancies. International Journal of Molecular Sciences. 2018. 8: 2155. [CrossRef]
- Krzystek-Korpacka M, Diakowska D., et al. Profiles of circulating inflammatory cytokines in colorectal cancer (CRC), high cancer risk conditions, and health are distinct. Possible implications for CRC screening and surveillance. Cancer Lett. 2013.337:107-14. [CrossRef]
- Matti BF, Saleem MA., et al. Assessment of interleukin 1β serum level in different responder groups and stages of chronic myeloid leukemia patients on imatinb mesylate therapy. Indian J Hematol Blood Transfus. 2014. 30:247-52. [CrossRef]
- Dolcet X, Llobet D, et al. NF-kB in development and progression of human cancer. Virchows Arch. 2005. 446:475-82. [CrossRef]
- Chan KW. Acute lymphoblastic leukemia, Current Problems in Pediatric and Adolescent Health Care. 2002. 32:40-49. [CrossRef]
- Sizemore N, Leung S., et al. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999. 19:4798-805. [CrossRef]
- Iliopoulos D, Hirsch HA., et al. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009. 139:693-706. [CrossRef]
- American Type Culture Collection. (2025). RS4;11 cell line (CRL-1873). Manassas, VA, USA.https://www.atcc.org/products/crl-1873-.
- Cyton. (2025). RS4:11 cells. Available at: https://www.cytion.com/es/RS4-11-Celulas/305360.
- Nakamura I, Takahashi N, et al. Wortmannin, a specific inhibitor of phosphatidylinositol-3 kinase, blocks osteoclastic bone resorption.FEBS Lett. 1995 Mar 13;361(1):79-84. [CrossRef]
- Dai X, Wang LJ, et al. Src kinase inhibitor PP2 regulates the biological characteristics of A549 cells via the PI3K/AKT signaling pathway.Oncol Lett. 2018.16 (4):5059-5065. [CrossRef]
- Chen X., Liu G., et al. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019;10:906. [CrossRef]
- Wang CY, Guttridge DC., et al. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol. 1999. 19:5923-9. [CrossRef]
- Ahmad I, Irfan S., et al. The SMAC mimetic AT-101 exhibits anti-tumor in lung adenocarcinoma cells by the IAPs/ caspase-dependent apoptosis and p65-NFƙB cross-talk. Iran J Basic Med Sci. 2021. 24:969-977. [CrossRef]
- Cai Z, Wang J., et al. Overexpressed Cyclin D1 and CDK4 proteins are responsible for the resistance to CDK4/6 inhibitor in breast cancer that can be reversed by PI3K/mTOR inhibitors. Sci China Life Sci. 2023. 66:94-109. [CrossRef]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002.420:860-7. [CrossRef]
- Turzanski J, Grundy M., et al. Interleukin-1beta maintains an apoptosis-resistant phenotype in the blast cells of acute myeloid leukaemia via multiple pathways. Leukemia. 2004. 18:1662-70. [CrossRef]
- Beaupre DM, Talpaz M., et al. Autocrine interleukin-1beta production in leukemia: evidence for the involvement of mutated RAS. Cancer Res. 1999. 15:2971-80. PMID: 10383163.
- Reddy SA, Huang JH., et al. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J Biol Chem. 1997. 272:29167-73. [CrossRef]
- Lin MT, Lin BR., et al. IL-6 induces AGS gastric cancer cell invasion via activation of the c-Src/RhoA/ROCK signaling pathway. Int J Cancer. 2007. 120: 2600-8. [CrossRef]
- Diep S, Maddukuri M., et al. Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance. Cells. 2022. 11:1673. [CrossRef]
- Zhang W, Ding W., et al. Up-regulation of breast cancer resistance protein plays a role in HER2-mediated chemoresistance through PI3K/AKT and nuclear factor-kappa B signaling pathways in MCF7 breast cancer cells. Acta Biochim Biophys Sin (Shanghai). 2011. 43:647-53. [CrossRef]
- Ghoneum A, Said N. PI3K-AKT -mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers (Basel). 2019. 11:949. [CrossRef]
- Wheeler DL, Iida M., et al. The role of Src in solid tumors. Oncologist. 2009. 14:667-78. [CrossRef]
- Mantovani A, Allavena P., et al. Cancer-related inflammation. Nature. 2008. 454:436-44. [CrossRef]
- Cory S, Huang DC., et al. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003. 22:8590-607. [CrossRef]
- Rahmani M, Aust MM., et al. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013. 73:1340-51. [CrossRef]
- Nachmias B, Ashhab Y., et al. The inhibitor of apoptosis protein family (IAPs): an emerging therapeutic target in cancer. Semin Cancer Biol. 2004. 14:231-43. [CrossRef]
- Spets H, Strömberg T., et al. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: regulation during interleukin-6(IL-6)-induced growth and survival. Eur J Haematol. 2002. 69:76-89. [CrossRef]
- Martin SJ, Reutelingsperger CP., et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995. 82:1545-56. [CrossRef]
- Earnshaw WC, Martins LM., et al. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999. 68:383-424. [CrossRef]
- Kayagaki N, Webster JD., et al. Control of Cell Death in Health and Disease. Annu Rev Pathol. 2024.19:157-180. [CrossRef]
- Kocab AJ, Duckett CS. Inhibitor of apoptosis proteins as intracellular signaling intermediates. FEBS J. 2016. 283:221-31. [CrossRef]
- McComb S, Cheung HH., et al. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 2012.19:1791-801. [CrossRef]
- Gerges S, Rohde K., et al. Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett. 2016. 375:127-132. [CrossRef]
- Feldmann F, Schenk B., et al. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget. 2017. 8:68208-68220. [CrossRef]
- Bousserouel S, Raymondjean M, et al. Modulation of cyclin D1 and early growth response factor-1 gene expression in interleukin-1beta-treated rat smooth muscle cells by n-6 and n-3 polyunsaturated fatty acids. Eur J Biochem.2004. 271(22):4462-73. [CrossRef]
- Ming J, Jiang G., et al. Interleukin-7 up-regulates cyclin D1 via activator protein-1 to promote proliferation of cell in lung cancer. Cancer Immunol Immunother. 2012. 61(1):79-88. [CrossRef]
- Baumann P, Schneider L., et al. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anticancer Drugs. 2012. 23(1):131-8. [CrossRef]
- Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in cancer? Biochem Pharmacol. 2004. 68:1071-80. [CrossRef]
- Liu C, Wu K., et al. SPP1+ macrophages promote head and neck squamous cell carcinoma progression by secreting TNF-α and IL-1β. J Exp Clin Cancer Res. 2024. 43(1):332. [CrossRef]
- Teixeira LFS, Peron JPS., et al. Silencing of nuclear factor kappa b 1 gene expression inhibits colony formation, cell migration and invasion via the downregulation of interleukin 1 beta and matrix metallopeptidase 9 in renal cell carcinoma. Mol Biol Rep. 2020. 47(2):1143-1151. [CrossRef]
- Viatour P, Merville MP., et al. Phosphorylation of NF-kappa B and Ikappa B proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005. 30:43-52. [CrossRef]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006. 25:6680-4. PMID: 17072321. [CrossRef]
Figure 1.
IL-1β induces increased activation of AKT, Src, and NF-κB in RS4:11 leukaemic lymphoblasts. (A, E, I) Representative histograms obtained through flow cytometry showing AKT, Src, and p65 subunit of NF-κB phosphorylation in leukaemic lymphoblasts at 24, 48, and 72 hours of culture with IL-1β. (B, F, J) MFI analysis of phosphorylated AKT, Src, and p65 at 24, 48, and 72 hours of culture with IL-1β. (C, G, K) Representative histograms illustrating phosphorylation of AKT, Src, and p65 after 24 hours of culture with IL-1β in the presence of inhibitors of PI3K (wortmannin), Src (PP2), and NF-κB (JSH23). (D, H, L) MFI analysis of AKT, Src, and phosphorylated p65 after 24 hours of culture with IL-1β in the presence of the inhibitors wortmannin, PP2, and JSH23. All results are the mean from three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
Figure 1.
IL-1β induces increased activation of AKT, Src, and NF-κB in RS4:11 leukaemic lymphoblasts. (A, E, I) Representative histograms obtained through flow cytometry showing AKT, Src, and p65 subunit of NF-κB phosphorylation in leukaemic lymphoblasts at 24, 48, and 72 hours of culture with IL-1β. (B, F, J) MFI analysis of phosphorylated AKT, Src, and p65 at 24, 48, and 72 hours of culture with IL-1β. (C, G, K) Representative histograms illustrating phosphorylation of AKT, Src, and p65 after 24 hours of culture with IL-1β in the presence of inhibitors of PI3K (wortmannin), Src (PP2), and NF-κB (JSH23). (D, H, L) MFI analysis of AKT, Src, and phosphorylated p65 after 24 hours of culture with IL-1β in the presence of the inhibitors wortmannin, PP2, and JSH23. All results are the mean from three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
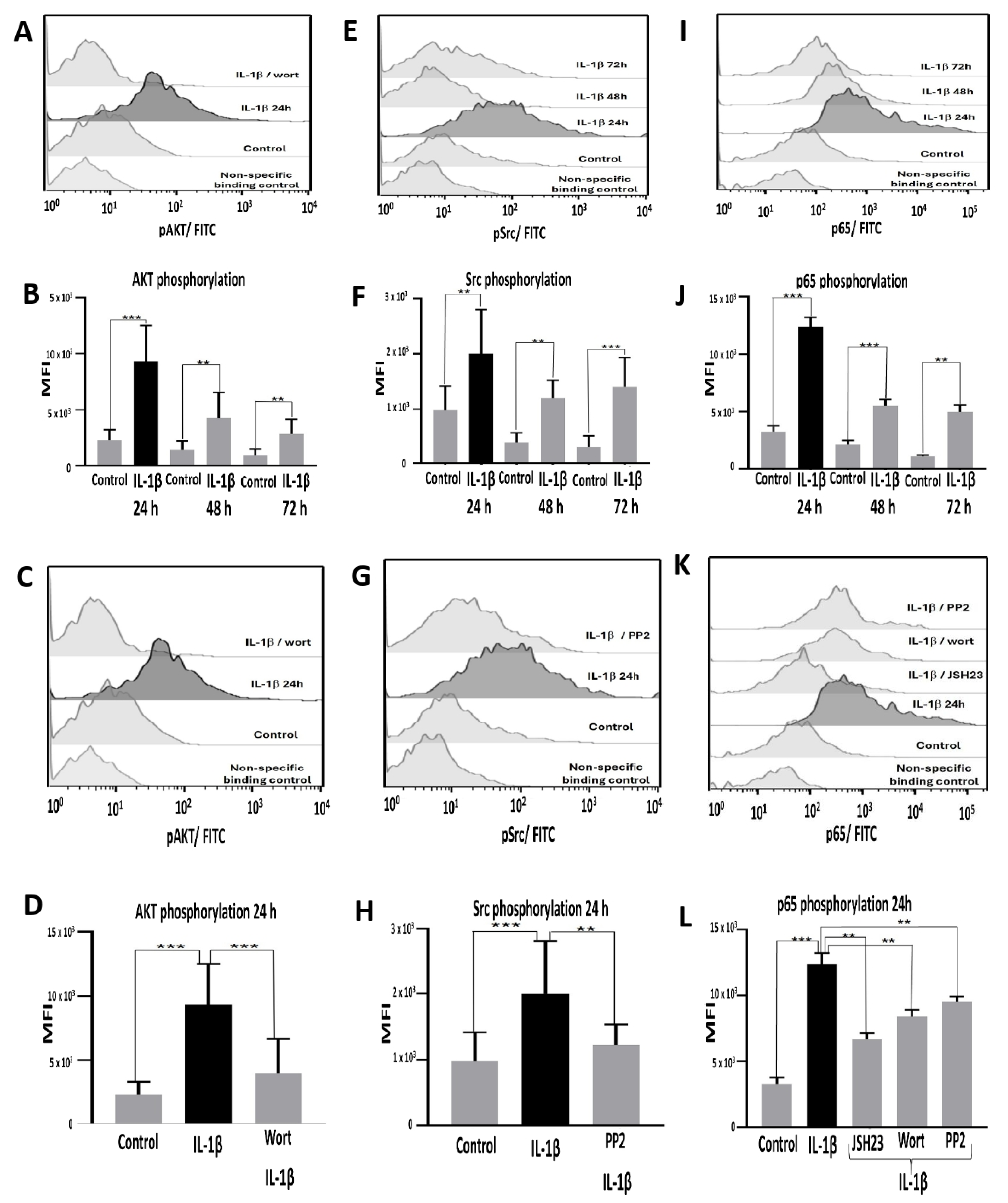
Figure 2.
IL-1β induces Bcl-2 and cIAP1 overexpression in RS4:11 leukaemic lymphoblasts via PI3K/AKT/NF-κB pathway. (A, E) Representative flow cytometry histograms showing Bcl-2 and cIAP1 expression at 24, 48, and 72 hours in leukaemic lymphoblasts treated with IL-1β. (B, F) MFI analysis of Bcl-2 and cIAP1 expression at the same time points with IL-1β. (C, G) Representative histogram of Bcl-2 expression at 48 hours with IL-1β in the presence of the inhibitors wortmannin, PP2, and JSH23. (D, H) MFI analysis of Bcl-2 at 48 hours and cIAP1 at 24 hours under the same conditions. All results are the means of three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
Figure 2.
IL-1β induces Bcl-2 and cIAP1 overexpression in RS4:11 leukaemic lymphoblasts via PI3K/AKT/NF-κB pathway. (A, E) Representative flow cytometry histograms showing Bcl-2 and cIAP1 expression at 24, 48, and 72 hours in leukaemic lymphoblasts treated with IL-1β. (B, F) MFI analysis of Bcl-2 and cIAP1 expression at the same time points with IL-1β. (C, G) Representative histogram of Bcl-2 expression at 48 hours with IL-1β in the presence of the inhibitors wortmannin, PP2, and JSH23. (D, H) MFI analysis of Bcl-2 at 48 hours and cIAP1 at 24 hours under the same conditions. All results are the means of three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
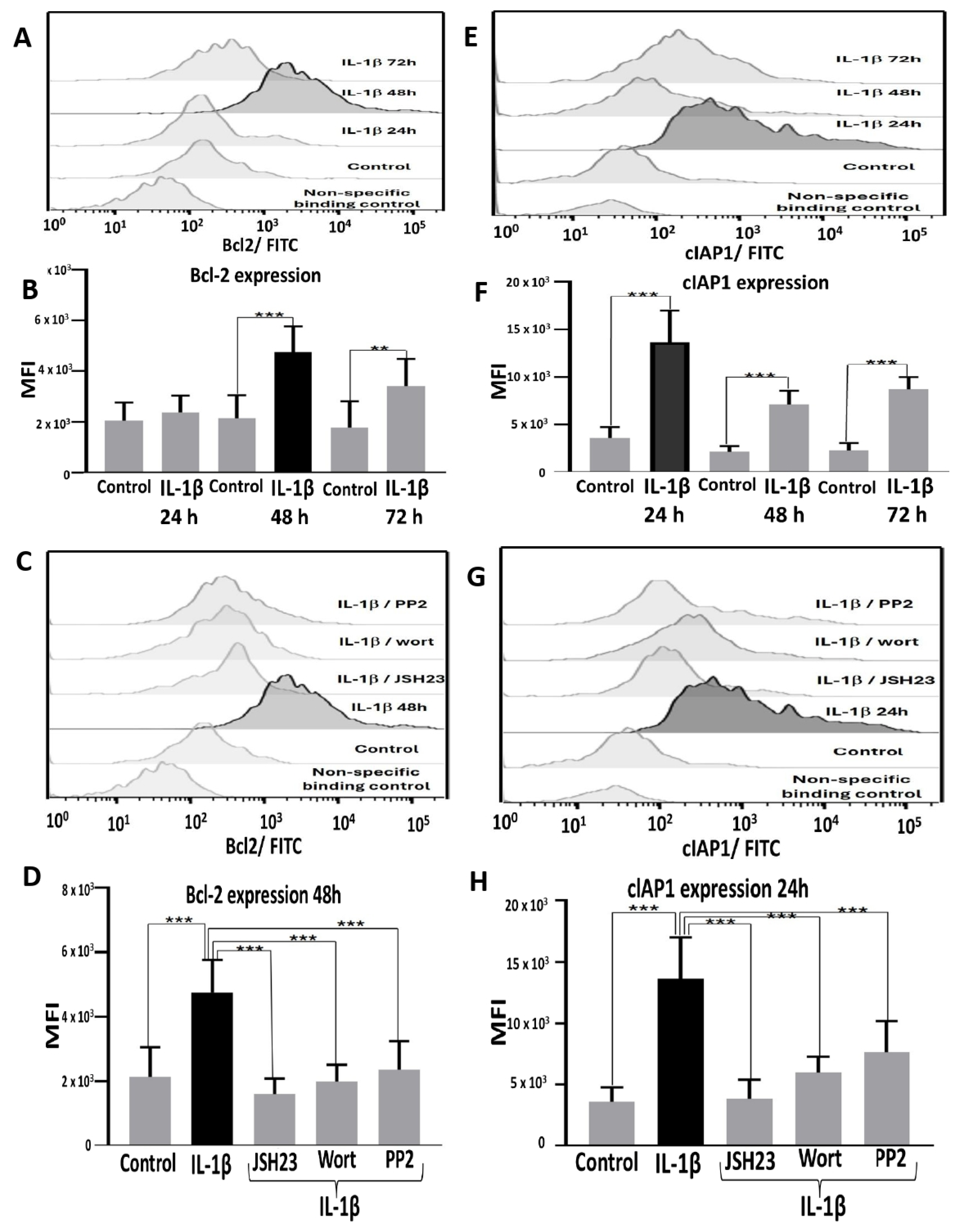
Figure 3.
IL-1β inhibits early apoptosis and limits necroptosis in RS4:11 leukaemic lymphoblasts via PI3K/AKT/Src/ NF-κB. (A) Representative dot plots showing analysis of apoptosis in leukaemic lymphoblasts incubated in the presence and absence (control) of IL-1β at 48 hours. The procedure was performed using flow cytometry with an Annexin V probe and labelling with propidium iodide (PI) and fluorescein isothiocyanate (FITC). (B) Representative dot plots illustrating analysis of apoptosis in leukaemic lymphoblasts incubated for 48 hours with IL-1β in the presence of wortmannin, PP2, and JSH23. All results are the means of three independent experiments, with each experimental condition performed in triplicate.
Figure 3.
IL-1β inhibits early apoptosis and limits necroptosis in RS4:11 leukaemic lymphoblasts via PI3K/AKT/Src/ NF-κB. (A) Representative dot plots showing analysis of apoptosis in leukaemic lymphoblasts incubated in the presence and absence (control) of IL-1β at 48 hours. The procedure was performed using flow cytometry with an Annexin V probe and labelling with propidium iodide (PI) and fluorescein isothiocyanate (FITC). (B) Representative dot plots illustrating analysis of apoptosis in leukaemic lymphoblasts incubated for 48 hours with IL-1β in the presence of wortmannin, PP2, and JSH23. All results are the means of three independent experiments, with each experimental condition performed in triplicate.
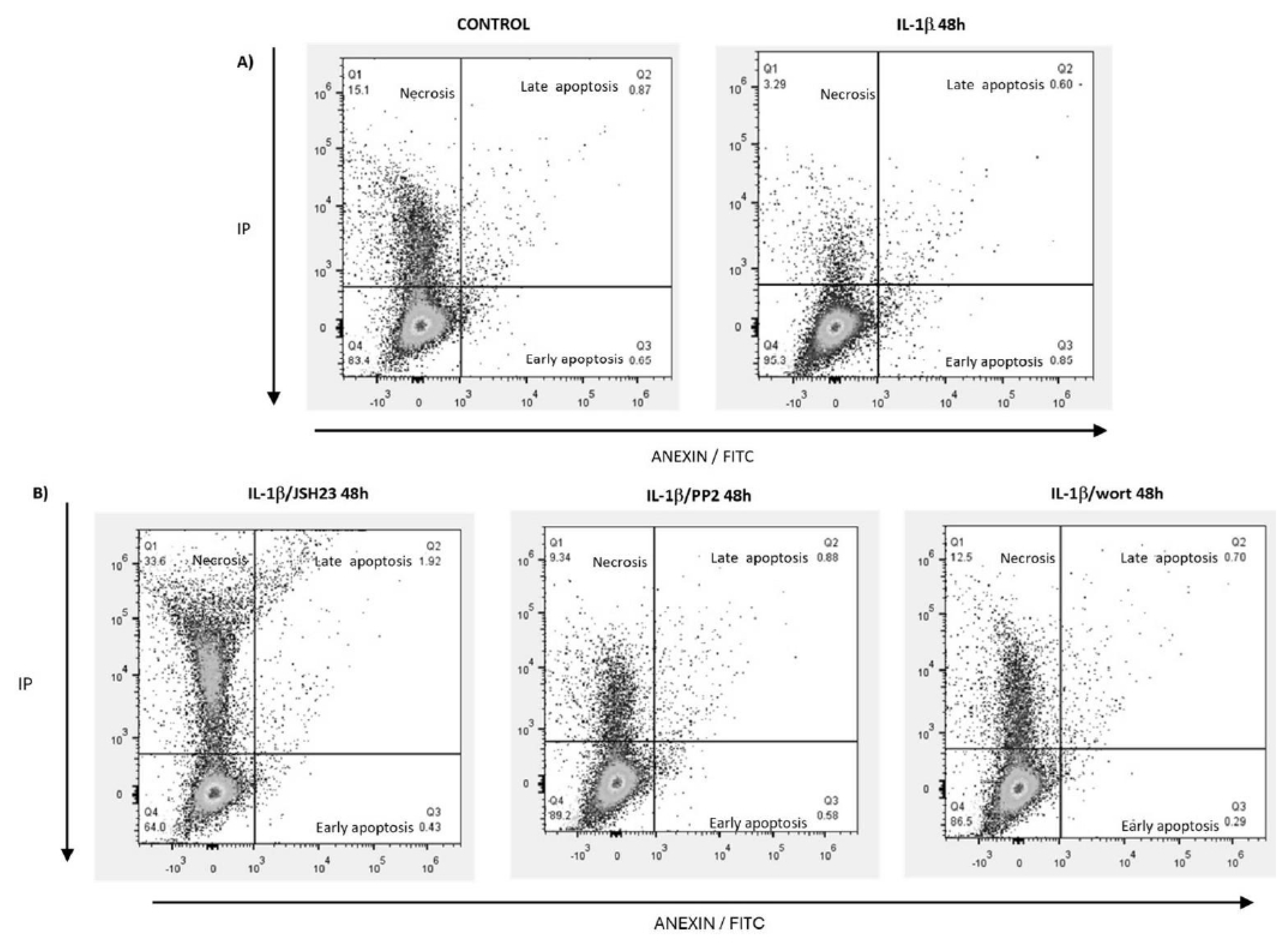
Figure 4.
IL-1β induces cyclin D1 overexpression and increases proliferation in RS4:11 leukaemic lymphoblasts via PI3K/AKT/Src/NF-κB. (A) Representative histogram of cyclin D1 expression in leukaemic lymphoblasts cultured with IL-1β at 24, 48, and 72 hours, measured via flow cytometry. (B) MFI analysis of cyclin D1 expression at 24, 48, and 72 hours with IL-1β. (C) Representative histogram of cyclin D1 expression at 72 hours cultured with IL-1β in the presence of inhibitors wortmannin, PP2, and JSH23. (D) MFI analysis of cyclin D1 expression at 72 hours with IL-1β in the presence of wortmannin, PP2, and JSH23. (E) Percentage analysis of leukaemic lymphoblast proliferation after incubation with IL-1β for 24 hours and IL-1β in the presence of wortmannin, PP2, and JSH23. (F) Percentage analysis of leukaemic lymphoblast proliferation incubated with IL-1β for 48 hours and IL-1β plus the same inhibitors. (G) Percentage analysis of leukaemic lymphoblast proliferation incubated with IL-1β for 72 hours and IL-1β plus the same inhibitors. All results are the means of three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
Figure 4.
IL-1β induces cyclin D1 overexpression and increases proliferation in RS4:11 leukaemic lymphoblasts via PI3K/AKT/Src/NF-κB. (A) Representative histogram of cyclin D1 expression in leukaemic lymphoblasts cultured with IL-1β at 24, 48, and 72 hours, measured via flow cytometry. (B) MFI analysis of cyclin D1 expression at 24, 48, and 72 hours with IL-1β. (C) Representative histogram of cyclin D1 expression at 72 hours cultured with IL-1β in the presence of inhibitors wortmannin, PP2, and JSH23. (D) MFI analysis of cyclin D1 expression at 72 hours with IL-1β in the presence of wortmannin, PP2, and JSH23. (E) Percentage analysis of leukaemic lymphoblast proliferation after incubation with IL-1β for 24 hours and IL-1β in the presence of wortmannin, PP2, and JSH23. (F) Percentage analysis of leukaemic lymphoblast proliferation incubated with IL-1β for 48 hours and IL-1β plus the same inhibitors. (G) Percentage analysis of leukaemic lymphoblast proliferation incubated with IL-1β for 72 hours and IL-1β plus the same inhibitors. All results are the means of three independent experiments, each performed in triplicate. ***p< 0.01 vs. control, **p< 0.02 vs. control.
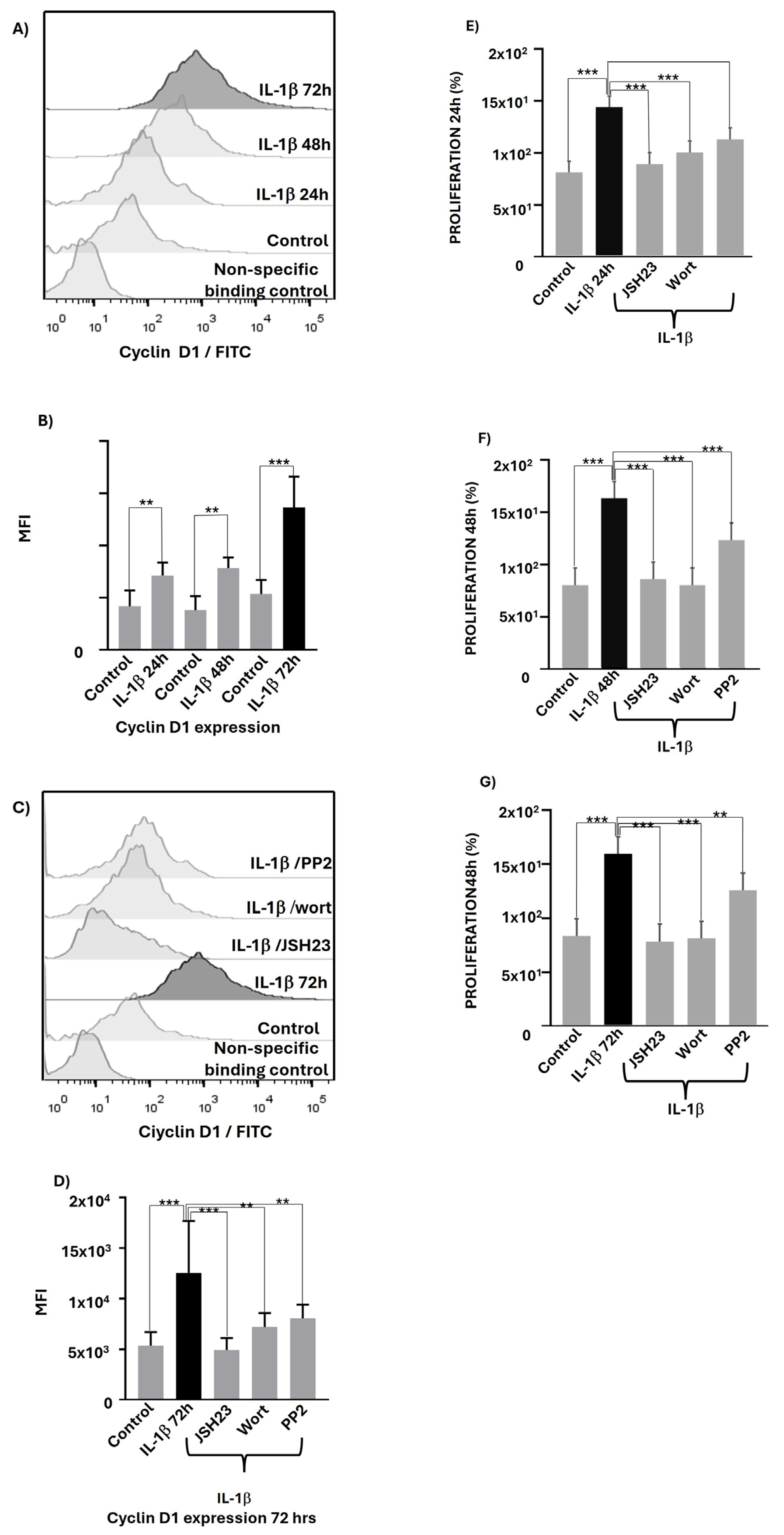
Figure 5.
Signalling pathway triggered by IL-1β affecting proliferation and death in leukaemic lymphoblasts. A model is proposed based on the results obtained in this study and previously reported data regarding targeting of PI3K/AKT on IKKβ, Src on IKKβ, and IKKβ on the p65 subunit of NF-κB, leading to its translocation to the nucleus. IL-1β binds to its receptor on leukaemic lymphoblasts, recruiting and activating PI3K/AKT and Src at this receptor. These kinases then activate IKKβ, which phosphorylates the p65 subunit of NF-κB, causing it to translocate to the nucleus. Once in the nucleus, NF-κB induces the transcription of target genes such as cyclin D1, Bcl-2, and cIAP1. Overexpression of cyclin D1 can increase proliferation. Higher levels of Bcl-2 and cIAP1 may be associated with inhibition of apoptosis, and cIAP1 might also help to limit necroptosis.
Figure 5.
Signalling pathway triggered by IL-1β affecting proliferation and death in leukaemic lymphoblasts. A model is proposed based on the results obtained in this study and previously reported data regarding targeting of PI3K/AKT on IKKβ, Src on IKKβ, and IKKβ on the p65 subunit of NF-κB, leading to its translocation to the nucleus. IL-1β binds to its receptor on leukaemic lymphoblasts, recruiting and activating PI3K/AKT and Src at this receptor. These kinases then activate IKKβ, which phosphorylates the p65 subunit of NF-κB, causing it to translocate to the nucleus. Once in the nucleus, NF-κB induces the transcription of target genes such as cyclin D1, Bcl-2, and cIAP1. Overexpression of cyclin D1 can increase proliferation. Higher levels of Bcl-2 and cIAP1 may be associated with inhibition of apoptosis, and cIAP1 might also help to limit necroptosis.
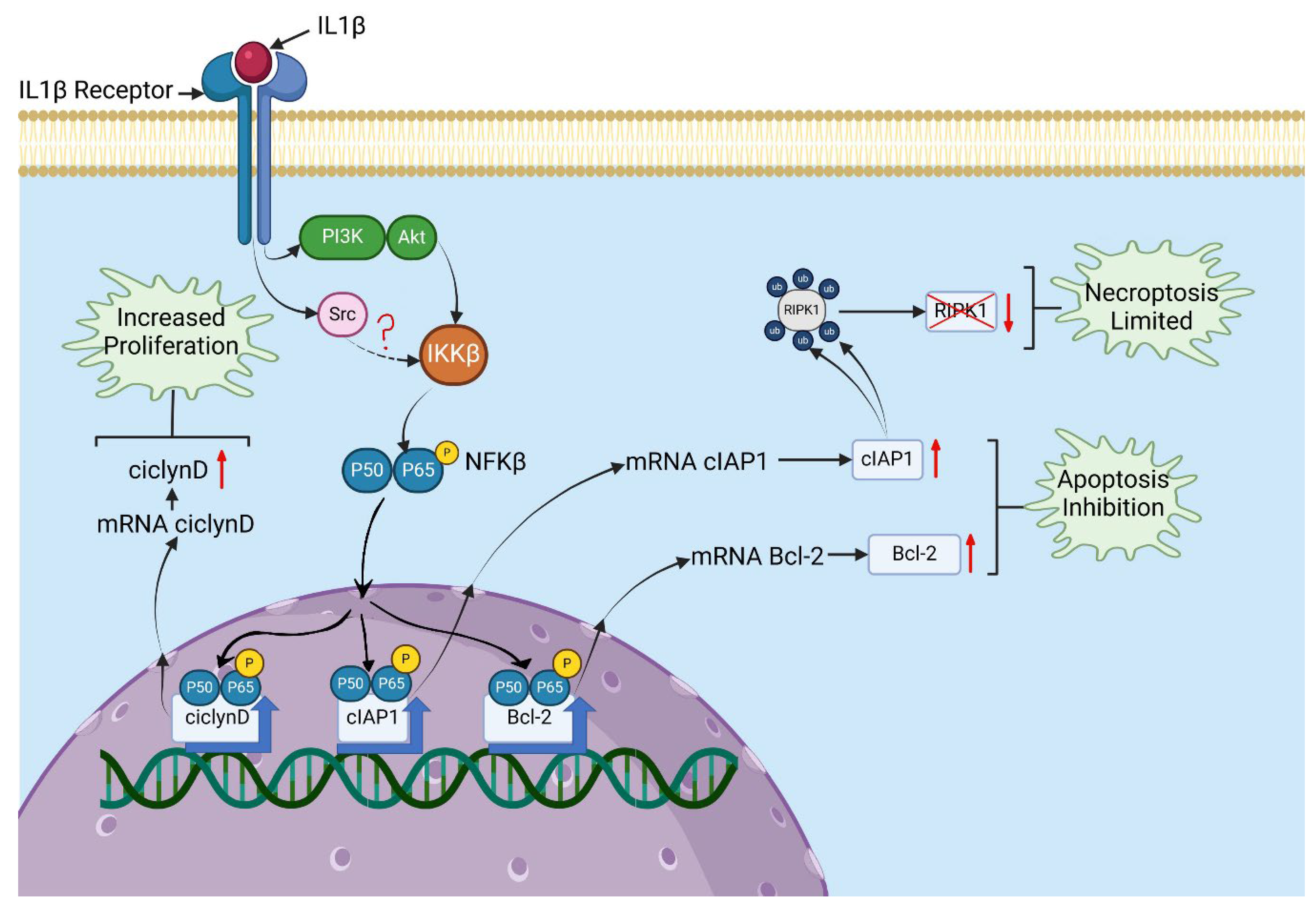

Table 1.
IL-1β inhibits early apoptosis and limits necroptosis through PI3K/AKT/Src/NF-κB.
| Treatment | Viable cells (%) | Early apoptosis (%) | Late apoptosis (%) | Necrosis (%) |
|---|---|---|---|---|
| Control | 66.11+/- 7.0 | 5.0 +/- 0.08 | 0.84 +/- 0.10 | 13.54 +/- 2.7 |
| IL-1β | 89.76 +/- 5.0 | 0.90 +/- 0.06 | 0.91 +/- 0.33 | 5.0 +/- 1.9 |
| IL-1β+JSH23 | 66.47 +/- 7.3 | 0.53 +/- 0.12 | 1.9 +/- 6.16 | 30.5 +/- 3.8 |
| IL-1β + PP2 | 84.90 +/- 6.4 | 0.62 +/- 0.10 | 0.83 +/- 0.09 | 10.53 +/- 2.3 |
| IL-1β + Wort | 85.50 +/- 5.4 | 0.24 +/- 0.05 | 0.75 +/- 0.07 | 12.7 +/- 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



