Submitted:
20 November 2025
Posted:
21 November 2025
You are already at the latest version
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), is now recognized as the leading cause of chronic liver disease worldwide. MASLD spans a spectrum ranging from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH), and is linked to progressive fibrosis and ultimately hepatocellular carcinoma (HCC). Growing evidence implicates cellular senescence (CS) and lipid droplets (LD) dysregulation as key drivers of disease progression, although their interaction remains poorly characterized. This review synthe-sizes current mechanistic insights into how CS and LD regulation affect the transition from steatosis to MASH. Senescent hepatocytes display altered lipid metabolism, includ-ing upregulation of receptors such as cluster of differentiation (CD) 36, enhancing lipid uptake to meet increased energy demands. Initially, elevated free fatty acid influx can ac-tivate peroxisome-proliferator receptor alpha (PPARα), promoting fatty acid oxidation (FAO) as a compensatory response. Over time, persistent cellular senescence (CS) under steatotic conditions leads to mitochondrial dysfunction and suppression of fatty acid ox-idation (FAO), while the senescence-associated secretory phenotype (SASP), largely driven by nuclear factor – kappa B (NF-κB) signaling, promotes chronic hepatic inflammation. A comprehensive understanding of this dynamic crosstalk between CS and lipid metabo-lism could identify novel therapeutic targets to modulate the MASLD progression.
Keywords:
cellular senescence
; lipid droplets
; metabolic dysfunction-associated steatotic liver disease
; senescence-associated secretory phenotype
; mitochondrial dysfunction
; fatty acid oxidation
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously referred to as non-alcoholic fatty liver disease (NAFLD) is a prevalent chronic liver disorder characterized by abnormal lipid accumulation within lipid droplets (LDs) in over 5% of hepatocytes [1]. The global prevalence of MASLD has increased markedly, affecting more than one third of the adult population worldwide. The burden is particularly high in Western populations, as MASLD is closely associated with type 2 diabetes (T2D) and obesity [2].
In approximately 15% of patients, MASLD progresses to metabolic dysfunction-associated steatohepatitis (MASH), and among these, up to 30% may develop fibrosis, which can further progress to cirrhosis and, ultimately, hepatocellular carcinoma (HCC) [3].
Several studies have highlighted the presence of senescent hepatocytes during the development of MASLD, suggesting that dysregulated cellular senescence (CS) may play a key role in its pathogenesis [4,5]. A chronically diseased liver generates an enormous burden of CS, as up to 80% of hepatocytes have been shown to become senescent in advanced stages [4,6,7]. Changes in the metabolic activity of senescent hepatocytes, particularly in lipid regulation, have been shown to further exacerbate steatotic conditions [8]. Liver function deterioration has also been associated with prolonged CS [6]. However, the causal relationships between CS, lipid dysregulation and MASLD progression are far from being clearly defined. The present review focuses on the complex interplay between CS and LD regulation in the context of MASLD and how this interaction influences disease progression from simple steatosis to MASH.
2. CS
2.1. Stages and Biomarkers
CS, which refers to a stable cell cycle arrest, is a complex, multistep biological process that is often divided into three stages: early, full, and late senescence. CS can be classified into two main types; replicative senescence (RS) is primarily driven by telomere shortening and occurs during normal aging, affecting all somatic cells capable of proliferation. In contrast, telomere-independent senescence is triggered by various internal or external stressors, a process known as stress-induced premature senescence (SIPS), including mitochondrial dysfunction, oxidative stress, DNA damage (mainly induced by chemotherapeutic drugs, also referred to as treatment-induced senescence, TIS), and oncogene activation[1]. Nevertheless, RS and SIPS depend on similar pathways as it is well established that p53-p21 and p16INK4α-Rb signal transduction cascades are induced during the activation of the CS program (early stage), although in some cases, CS can proceed independently [2].
Since no single specific biomarker for CS exists, molecules involved in these pathways, together with the decreased expression of proliferation associated markers such as Ki-67, serve as valuable indicators of CS[3]. Senescence-associated beta-galactosidase (SA- β-gal) is widely regarded as the gold standard for detecting senescent cells. However, recently provided data raised concerns regarding its specificity, as a rapid increase in Sa-b-gal levels is not indicative only of the CS state, but it was also identified in stressed or pre-senescent cells that either recover from stressful stimulus and successfully reenter the cell cycle, or alternatively enter the CS state [3]. Furthermore, it was found to be constitutively expressed in several tissues, including neurons [4]. Lipofuscin, a yellowish-brown pigment referred to as the “age pigment”, is also considered an established biomarker for CS [5]. Lipofuscin is accumulated in senescent cells as a result of lysosomal dysfunction and is composed of incompletely degraded molecules, such as lipids, proteins and sugars [6]. However, lipofuscin is not detected only in senescent cells, since it has also been found in metabolically active postmitotic cells, such as cardiomyocytes and certain neurons, which retain proliferative capacity [7]. SA-β-Gal and lipofuscin were found to co-localize, suggesting that these markers could be used interchangeably to detect CS [8]. Epigenetic changes such as senescence-associated heterochromatin foci (SAHF), regions of heterochromatin that supress proliferation-promoting genes in senescent cells [9], and senescence-associated DNA damage foci (SADF), which contain key proteins for DNA-damage response (DDR) and CS, also serve as potential CS biomarkers [10].
2.2. Senescence-Associated Secretory Phenotype
CS is accompanied by the senescence-associated secretory phenotype (SASP), which includes various proteins, cytokines, chemokines, growth factors, and proteases [11]. SASP is highly complex, dynamic and heterogeneous, varying across different cell types [12], senescence-inducing stimuli [13], and stages of CS [14]. It is considered the most influential feature of senescent cells, enabling their communication with the surrounding microenvironment through paracrine actions [15], while in an autocrine manner, SASP factors reinforce the CS program within the cells themselves.
SASP secretion could be divided into three main phases: an initial rapid phase associated with the DDR pathway, followed by a slower self-amplifying phase, and ultimately the development of a late or “mature” SASP phase [16]. SASP involves modifications to both soluble factors and extracellular vesicles (EVs). Soluble SASP factors include interleukin (IL)-1, IL-6, IL-8, chemokines such as Monocyte Chemoattractant Protein-1 (MCP-1), growth factors such as Hepatocyte Growth Factor (HGF) and proteases such as Matrix Metalloproteinases (MMPs) [17]. Moreover, cellular components, such as proteins and lipids enclosed to EVs serve also as SASP factors by mediating intercellular communication [18]. Senescent cells release a large amount of EVs and induction of CS results in changes in lipid composition of EVs [19].
Early SASP is detectable only a few days following SIPS induction and activation of DDR pathway. During this phase, SASP factors, such as IL-1, can be detected at very low levels in the extracellular milieu [11]. IL-1, has been associated with the paracrine activity of senescent cells, as it can induce normal adjacent cells to enter the CS program [20]. Early senescent cells secrete these factors to meet the increased energy demands associated with the induction of the senescent phenotype and to support wound healing process, highlighting the beneficial effects of the transient presence of senescent cells [21]. The composition of SASP secreted by early senescent cells is highly heterogeneous, while SASP from late senescent cells displays several similarities [21]. This difference is considered to be due to the fact that during early CS cells undergo a transition from their original proliferative state to a senescent phenotype, which depends on the initial cellular conditions and stressors, whereas in later stages of CS, common functional changes emerge, such as metabolic rewiring to support the survival of senescent cells, modifications in the extracellular matrix (ECM), and immune system modulation, thereby resulting in a more universal phenotype, regardless of the initial conditions [21]. The early SASP phase also marks the initiation of slow, self-amplifying autocrine loops that gradually intensify the production and secretion of SASP factors[11].
As cells progress to full senescence, they undergo extensive chromatin remodeling, which is largely attributed to the reduction of lamin B1, a structural protein of the nuclear membrane[22]. This decrease in lamin B1 leads to a “leaky” nucleus promoting shifts in SASP gene expression [23]. During this stage, SASP alterations include the enrichment of mainly pro-inflammatory factors through positive feedback loops, which promote age-related diseases, such as MASLD.[21]. Nuclear factor- kappa B (NF-κΒ) is also implicated in the regulation of key inflammatory SASP components, such as IL-1a, IL-6 and IL-8[24] and these cytokines act within a positive feedback loop to further promote NF-κB expression and enhance SASP signaling [24]. It was recently described that sustained NF-κB activity enhanced inflammatory gene expression through increased NF-κB-DNA binding and slowed the cell cycle [25]. Another regulator of SASP is mTOR, which inhibits SASP- mRNA degrading proteins, thus stabilizing SASP expression and reinforcing CS[26].
Following continuous self-amplification, SASP eventually reaches a mature state, which represents the most prominent feature of late senescent cells [16]. During the late stage of senescence, cells display marked phenotypic heterogeneity, with alterations in SASP composition that may be adjusted to distinct pathophysiological conditions[27]. Further upregulation of p16 is implicated in maintaining the irreversible nature of the senescence state , increasing SA β-gal activity and altering mitochondrial metabolism [28].
Apart from nuclear changes, senescent cells also display prominent cytosolic characteristics. A widely recognized hallmark of CS is the enlarged cellular morphology accompanied by an increased number and mass of cellular organelles, such as lysosomes and mitochondria as well as alteration in plasma membrane composition [29]. Cell enlargement during CS is largely attributed to the activation of the mammalian target of rapamycin (mTOR) pathway [30], facilitating processes, such as nutrient uptake and synthesis of proteins and lipids, both contributing to the enhanced cell size [31]. Additionally, the increased mass of lysosomes has been linked to the accumulation of the lysosomal enzyme SA-β-gal [32,33]. Finally, senescent cells may either undergo immune-mediated clearance or persist in a more profound senescent state.
Various proinflammatory factors, such as the pro-inflammatory cytokines Il-6 and IL-8, ECM remodeling factors and immune evasion mechanisms as well as factors associated with stress response and metabolic regulation, are increased in this late phase [21]. Interestingly, specific microRNAs, such as miR-146a and miR-146b, can regulate SASP secretion to prevent excessive amplification [16]. If this modulation fails to attenuate SASP secretion, a more stable, long-term SASP signature is established [16]. This phase is also described as the stable late SASP phase and typically occurs approximately 30 days or more after the persistence of senescent cells. During this stage, pro-inflammatory cytokines dominate, contributing to the detrimental effects of CS, including chronic inflammation, tumor oncogenesis and aging [21]. However, only a limited number of studies have investigated the dynamic regulation of SASP secretion. Notably, SASP can also exhibit beneficial effects by inducing inflammation and promoting tumor surveillance [11,20,34].
2.3. Lipogenesis in Senescent Cells
Considering the above-mentioned features of CS, such as membrane remodeling, increased cellular mass and SASP secretion, especially through the lipid-enriched EVs, lipids are considered key modulators for the establishment and maintenance of CS [35]. Evidence shows that senescent cells undergo a rewiring of lipid-related pathways, including lipid uptake, biosynthesis and fatty acid oxidation (FAO) [35], thereby displaying a new metabolic phenotype that contributes to LD accumulation within senescent cells.
Lipid intake capacity [36], represents a feature possibly involved in preserving the senescent phenotype, including membrane remodeling and SASP [37]. Senescent cells, mainly in lipid-rich tissues as hepatic and adipose tissue [38], are characterized by increased levels of cluster of differentiation (CD) 36, a membrane receptor involved in fatty acid uptake and lipid metabolism. CD36 is also implicated in the induction of CS of adjacent cells and in the modulation of SASP [39]. Notably, the initial upregulation of CD36, observed rapidly upon CS, is required for the activation of crucial transcription factors, including NF-κB, that drive SASP formation[37]. Importantly, the sustained expression of CD36 promotes the secretion of pro-inflammatory SASP components, such as IL-6 and IL-8, thereby contributing to the establishment of a full senescent state[39].
Lipid biosynthesis is also upregulated during initial stages of CS, as the enlarged cellular mass and accumulation of membranous organelles, such as mitochondria and lysosomes in senescent cells are highly dependent on lipid biosynthesis [29]. This upregulation relies on key rate-liming enzymes, such as fatty-acid synthase (FASN), the levels of which have been found elevated prior to the induction of late-stage senescence [40]. Apart from membranous remodeling, researchers found that downregulation of FASN resulted in inhibition of SASP, highlighting the importance of lipid biosynthesis for the acquisition of secretory features in early senescent cells [40].
LDs are highly dynamic organelles that play a central role in lipid metabolism, storage, and cellular signaling. They are composed of a core of neutral lipids, primarily TGs and cholesteryl esters, surrounded by a phospholipid monolayer embedded with proteins, such as perilipins (PLINs), that regulate lipid metabolism and LDs interactions with other organelles. Thus, LDs are considered protectors of cells from lipid-induced toxicity by sequestering excess fatty acids in the form of triglycerides (TGs) [41]. They are also recognized as significant regulators of cellular homeostasis as they interact with various organelles, including mitochondria, endoplasmic reticulum, and peroxisomes, to regulate lipid flux and energy balance [42]. Beyond their metabolic functions, LDs can also modulate intracellular signaling pathways, immune responses, and membrane trafficking [43] and disruption of their homeostasis can lead not only to dysregulation of lipid metabolism but also to alterations in cellular signaling pathways[44]. Abnormalities in lipid metabolism can further lead to hepatic steatosis, inflammation, and oxidative stress[12].
2.4. Mitochondrial Dysfunction in Senescent Cells
Mitochondria are not only passive responders to metabolic rewiring during CS but also active regulators of its initiation, maintenance, and propagation. During the initial phase of CS, mitochondria undergo rapid and coordinated remodeling events that reflect both structural and functional adaptations. One of the earliest events involves the phosphorylation of specific mitochondrial proteins, altering inner membrane architecture, and increasing the number and density of cristae [13]. This reorganization is not merely structural; it directly contributes to a transient enhancement of FAO, allowing the cell to generate additional pools of acetyl-CoA that are required for metabolic adaptation and epigenetic remodeling. In this context, proteins associated with the DDR, particularly ataxia-telangiectasia mutated (ATM) kinase, play a central role. ATM activation not only propagates canonical DNA repair signals but also transduces stress cues to mitochondria, thereby inducing CS through upregulation of FAO pathways [14]. Furthermore, activation of p53 during the early phase of CS results in upregulation of key enzymes implicated in mitochondrial FAO [45].
As CS progresses toward a more stable and irreversible state, mitochondrial physiology changes dramatically. Mitochondria frequently become enlarged, elongated, and functionally compromised, with impaired oxidative phosphorylation, declining ATP output, and excessive generation of ROS [46]. Interestingly, ROS was shown to promote the activation of proinflammatory components of SASP, mainly via NF-κΒ activation, creating a feed-forward inflammatory loop that reinforces the senescent state [47]. Moreover, prolonged enhanced FAO results in epigenetic alterations such as increased histone acetylation, thereby promoting p16 expression and contributing to epigenetic reinforcement of cell-cycle arrest [14].
3. MASLD Pathogenesis
3.1. Microvescular and Macrovescular Type of Steatosis
Nascent LDs originate from the outer membrane of the ER, where neutral lipids accumulate[17]. Under normal conditions the size of LDs is tightly regulated, and hepatocytes primarily contain smaller LDs which are more metabolically active, allowing for efficient interaction with lipases and cytosolic organelles, such as mitochondria. However, in MASLD, this balance is disrupted. Hepatocytes in an attempt to compensate for the elevated influx of free fatty acids (FFAs) sequester them as TGs within LDs, a process known as steatosis [48]. The type of steatosis observed in MASLD is also influenced by LD structure; earlier stages of MASLD are typically characterized by macrovesicular steatosis, which entails a large droplet of fat within the hepatocyte displacing the nucleus to the periphery of the cytoplasm. The presence of macrovesicular steatosis is mainly associated with a favorable long-term prognosis, as these large LDs serve as long-term lipid reservoirs, thus protecting the liver against lipotoxicity [49]. However, as MASLD progresses to more severe stages, such as MASH, hepatocytes display a coexistence of macrovesicular steatosis along with aggregates (“patches”) of microvesicular steatosis, which are described as enlarged hepatocytes with a foamy, vacuolated cytoplasm, known as balloon cells [49]. Microvesicular steatosis has been associated with more severe grades of macrovesicular steatosis, inflammation, oxidative stress and mitochondrial dysfunction [50]. These LD aggregates may indicate regions of hepatocytes undergoing increased metabolic stress, where defective FAO leads to the formation of amphiphilic compounds, which contribute to the development of microvesicular steatosis [51]. The coexistence of both forms of steatosis suggests a spectrum of hepatocellular injury, where different hepatocytes or liver regions undergo varying degrees of lipid dysregulation.
3.2. The Implication of PLINs
Apart from size, protein composition on LDs surface is also modified during MASLD, playing major role in lipid metabolism and consequently in the propagation of the disease. Notably, PLINs, the most abundant LD-associated proteins, are strongly associated with MASLD progression [52].
The predominance of PLIN2 in the steatotic liver suggests that the balance between lipid storage and mobilization is severely disrupted, favoring lipid retention over lipid utilization[53]. This imbalance not only exacerbates steatosis but also predisposes hepatocytes to additional metabolic stress, contributing to the transition from simple steatosis to more advanced stages of liver disease, including metabolic-associated steatohepatitis (MASH) and fibrosis.PLIN2 was found to be the most upregulated PLIN in the steatotic liver, being responsible for LD formation by promoting the sequestration of TGs within LDs and inhibiting the access of lipolytic mechanisms to LDs [54,55] leading to excessive LDs accumulation and persistence of steatosis [56]. Moreover PLIN2 levels were also related to mitochondrial dysfunction leading to the induction of CS through the upregulation of stress related proteins [57].
PLIN3 was found to be upregulated in human steatotic livers [58] and its silencing was shown to reduce steatosis [17]. PLIN5, which is typically expressed in oxidative tissues including liver and muscle, was also upregulated in MASLD [59]. When cells are overloaded with lipids, PLIN5 inhibits the recruitment of cytosolic lipases to LDs and mitochondrial beta-oxidation, thus protecting the liver against lipotoxicity [60]. Moreover, PLIN5 has been implicated in modulating inflammatory responses, as its deficiency was recognized to increase oxidative stress, which may in turn activate pro-inflammatory pathways, including NF-κB signaling[61].
3.3. PPARa and NF-κΒ
The complex interplay between impaired lipid metabolism and chronic inflammation primarily drives the progression of MASLD from simple steatosis to severe liver damage. Among the key regulatory pathways involved, PPARα, which governs lipid metabolism and NF-κΒ, a key regulator of inflammation, plays a crucial role in the development of the disease.
PPARα is a nuclear receptor that regulates FAO and lipid homeostasis, helping to mitigate lipid accumulation [62]. It is a well-established modulator of MASLD pathogenesis through its complex interaction with mitochondria and inflammatory signaling pathways [62]. Under normal conditions, PPARα activation facilitates mitochondrial biogenesis and function, ensuring efficient FAO and preventing excess lipid accumulation [62]. In early MASLD, PPARα serves as a compensatory mechanism, counteracting lipid overload by promoting FAO[63]. PPARα activation suppresses NF-κB-dependent transcription of cytokines, such as IL-6, IL-1β, and TNF-α, thereby limiting hepatic inflammation [64]. However, as MASLD progresses, PPARα expression declines, leading to impaired mitochondrial function, reduced ATP production, and increased oxidative stress [65]. This mitochondrial dysfunction exacerbates lipid accumulation, contributing to hepatocellular damage and inflammation. Additionally, PPARα downregulation disrupts LD metabolism, resulting in impaired lipolysis and excessive lipid accumulation within hepatocytes[62]. This lipid overload may induce lipotoxicity and promote inflammatory signaling pathways, which further accelerate MASLD progression.
In contrast, NF-κB is a central mediator of inflammatory responses, since its sustained activation drives the secretion of proinflammatory cytokines through SASP, contributing to persistent inflammation, hepatocyte injury, and fibrosis progression [64], representing a key player in the progression of steatosis to MASH. Under normal conditions, NF-κB activity is strictly regulated, ensuring that inflammatory pathways are activated in response to a stressful stimulus, while preventing persistent inflammation once the stimulus has been resolved [66]. This tight regulation prevents chronic inflammation and uncontrolled tissue damage in the liver, maintaining hepatic homeostasis. However, in MASLD, chronic metabolic stress, lipotoxicity, and oxidative damage trigger persistent NF-κB activation, leading to sustained inflammation and hepatocyte injury [67]. Activated NF-κB induces the transcription of proinflammatory cytokines, including IL-6, IL-1β, and TNF-α, which perpetuate hepatic inflammation and immune cell recruitment [68]. Additionally, NF-κB interacts with the SASP, amplifying the release of inflammatory mediators that exacerbate hepatocellular stress and fibrosis [16].
The crosstalk between PPARα and NF-κB determines whether the liver adapts to metabolic stress or succumbs to progressive damage. In the early stages of MASLD, elevated levels of PPARα play a protective role against lipid overload through the activation of FAO[63]. Moreover, PPARα activation has a direct anti-inflammatory effect by suppressing NF-κB-dependent transcription of key cytokines, which are central to the inflammatory response in the liver [64]. Conversely, the downregulation of PPARα in advanced stages of MASLD contributes significantly to the propagation of the disease by promoting excessive activation of NF-κB, resulting in chronic inflammation, hepatocellular senescence, and fibrosis[64]. NF-κB-mediated inflammation enhances the production of SASP factors, which perpetuate a pro-inflammatory microenvironment, thereby accelerating disease progression[24]. Studies have shown that oxidative stress is an important mechanism of NF-κB activation in the liver [69] and it is also associated with DNA damage and genomic instability, thus contributing to hepatocellular dysfunction and disease progression.
3.4. Mitochondrial Dysregulation
Mitochondrial dysfunction, observed by altered mitochondrial morphology and decreased oxidative capacity, represents a well-established driver of hepatοcellular steatosis [70,71] and is considered one of the main hallmarks of MASLD, especially during MASH [72]. Elevated fatty-acid influx towards the liver results in an initial upregulation of mitochondrial fatty-acid oxidation, which acts as a compensatory mechanism to prevent lipid accumulation, a process known as “mitochondrial flexibility” [71]. However, this adaptation in mitochondrial metabolism does not persist in more severe stages like MASH, during which hepatocytes display decreased energy production and impaired mitochondrial respiration [70]. However, prolonged elongated mitochondria ultimately result in higher production of reactive oxygen species (ROS) and decreased mitochondrial respiration activity [46]. Interestingly, ROS was found to promote the activation of proinflammatory components of SASP [47]. Moreover, prolonged FAO results in epigenetic alterations, such as overexpression of p16 [14]. In a recent study overexpression of p16 in primary hepatocytes was shown to reduce mitochondrial activity and FAO and led to accumulation of LDs both in vitro and in vivo[73], pointing out a potential vicious cycle between CS and mitochondrial metabolism.
4. Interplay Between CS and LDs in MASLD
Steatosis is considered as a stressful stimulus for hepatocytes, to which they respond through different pathways. Increased FFA influx towards the liver results in the initial upregulation of PPARα, acting as a compensatory mechanism to enhance β-oxidation and to mitigate LD accumulation [63]. Following increased FFA influx, in a subset of hepatocytes CS develops gradually, competing with cell cycle arrest, cell death and proliferation, resulting in a heterogeneous population of hepatocytes [36]. Stressed hepatocytes exhibit a pre-senescent phenotype, characterized by the expression of both proliferation-associated biomarkers, such as Ki-67, and senescence-associated biomarkers, such as Sa-b-gal [3]. Concomitantly, hepatocytes are marked by the elevated levels of p53/p21[74]. Under these conditions, hepatocytes, which seem not to display any other senescent characteristics apart from temporary cell cycle arrest, try to mitigate the stressful stimulus. In cases where the stressful stimulus of steatosis is transient and hepatocytes can effectively eliminate excess lipids, the resolution of stress may allow them to re-enter the cell cycle[75] (Figure 1a). The transition to the senescent state is marked by increased levels of p16, apart from p53/p21, which subsequently ensures the irreversibility of SIPS [76]. Under steatotic conditions early senescent hepatocytes exhibit distinct metabolic and secretory features, including altered LD metabolism and a mild SASP. Interestingly, scattered senescent hepatocytes display enhanced lipid-associated receptors, such as CD36 [39] and increased lipid biosynthesis[29], as pointed by elevated levels of FASN[40], indicating enhanced lipid uptake and biosynthesis to meet increased energy demands associated with early morphological alterations (e.g., increased cellular size, lysosomes). Therefore, senescent steatotic hepatocytes try also to accumulate lipids to cover elevated energy demands, potentially contributing to further LD accumulation. However, increased FFA influx into the liver may upregulate PPARα, thereby enhancing FAO and acting as an efficient compensatory mechanism against steatosis [63]. Apart from metabolic changes, SASP is a hallmark of CS and a crucial regulator of MASLD pathogenesis [77]. Interestingly, early SASP involves mainly growth factors and cytokines such as IL-1a and b, which are initially secreted at low levels [11,15]. In early MASLD, the upregulation of PPARα helps to attenuate NF-κB activity, limiting the extent of SASP-mediated inflammation[64]. However, the low levels of secreted cytokines as SASP factors can induce immune mediated clearance of the scattered senescent hepatocytes, thereby ensuring the regenerative capacity of the liver and preserving normal hepatic function (Figure 1b).
As steatosis persists, hepatocytes progress to a late senescent state. Of note, increased mitochondrial mass and subsequent increased FA oxidation are associated with significant transcriptional changes, such as epigenetic induction of p16 expression, thus maintaining permanent cell-cycle arrest and promoting CS reprogramming [28]. In contrast, overexpression of p16 results in mitochondrial deregulation and gradual decrease of β- oxidation capacity [73]. This metabolic reprogramming plays a protective role since cells are able to sequester FAs, which are highly reactive species prone to oxidation, and store them away from membranes, thus limiting membrane damage under oxidative conditions [35]. Under these conditions prolonged enhanced influx of FFAs, combined with p16-induced initial decline in mitochondrial function, result in the significant LD accumulation seen during MASLD progression. However, further investigations comparing LD accumulation between senescent and proliferative steatotic hepatocytes are necessary to fully understand the interaction between these two cell populations and determine whether senescent hepatocytes accumulate more LDs than their proliferative counterparts. Apart from LDs, senescent steatotic hepatocytes secrete SASP factors, entering a self-amplifying phase, characterized by significantly increased levels of IL-1a and b [11]. These cytokines act as positive feedback signals, stimulating the production of additional proinflammatory cytokines, such as IL-6 and 8. Moreover, as MASLD progresses. levels of PPARα begin to decrease, contributing further to mitochondrial downregulation and activation of NF-kB, which promotes the secretion of proinflammatory cytokines (Figure 1c).
5. Interaction Between SIPS and RS in MASH
Senescent hepatocytes have been demonstrated to increase in number as chronic liver disease progresses [78]. Persistent liver insults, such as steatosis, drive the continuous production of senescent hepatocytes, leading to their accumulation, a process known as chronic senescence[75]. Notably, senescence biomarkers, such as p21, correlate positively with the MASLD activity score [79], with further elevation in more severe stages, such as MASH[80]. This increase in p21 may be attributed to the progressive induction of senescence in more and more neighboring hepatocytes contributing to the damage of hepatic architecture and the deterioration of liver function.
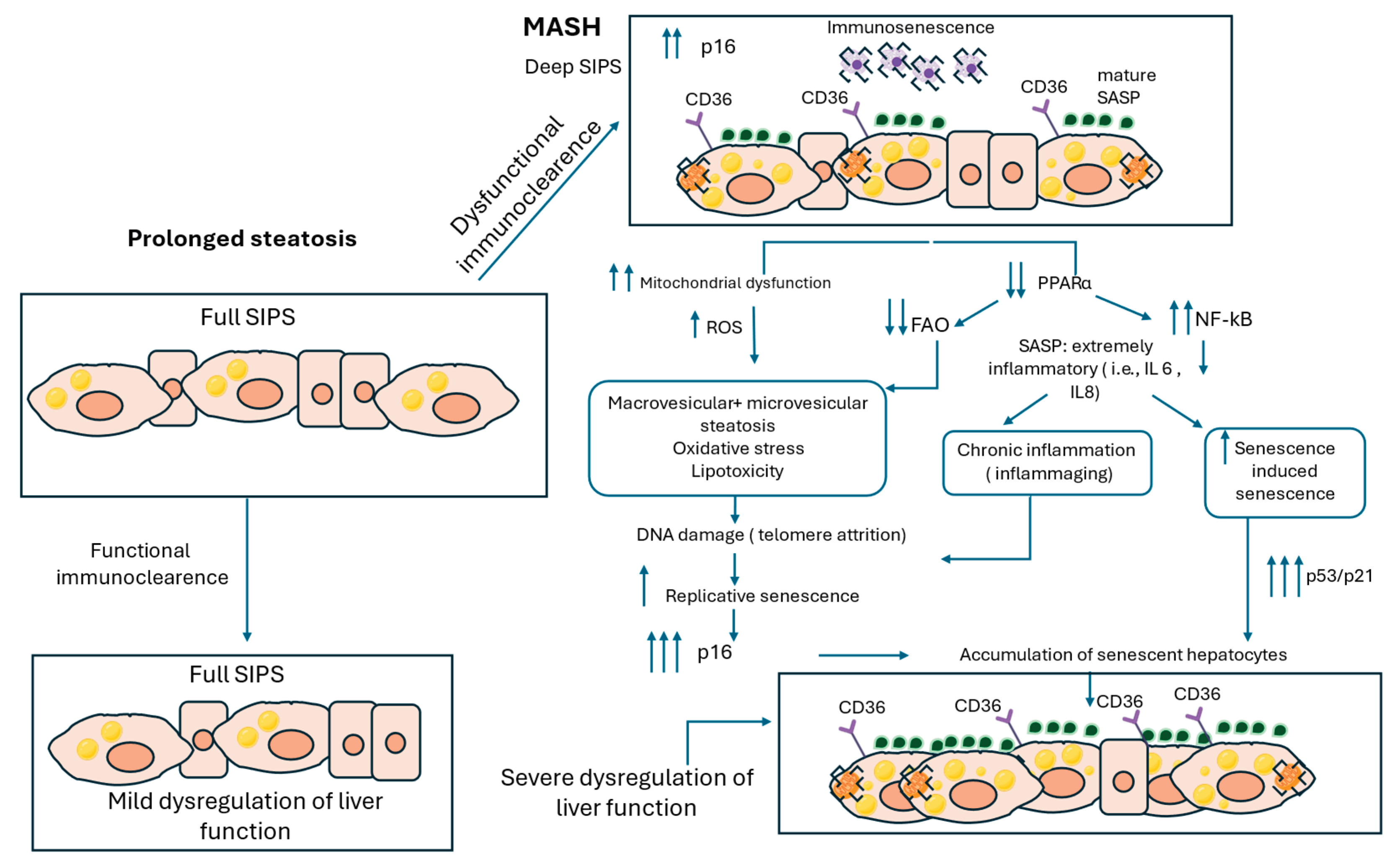
During later stages of SIPS, common functional changes emerge, such as metabolic rewiring to support the survival of senescent cells, modifications in the extracellular matrix (ECM), and immune system modulation, thereby resulting in senescent cells exhibiting a more universal phenotype, regardless of the initial conditions [21]. Concurrently, PPARα levels are further decreased during MASH, thus exacerbating mitochondrial beta-oxidation impairment in senescent hepatocytes [81]. This downregulation of PPARα also elevates NF-κB activity, driving to a sustained production of proinflammatory SASP factors, such as IL-1, IL-6 and TGF-b, which in turn promote CS in adjacent cells[82]. This process referred to as senescence-induced senescence contributes to the clustering of senescent hepatocytes [83]. The prolonged exposure to proinflammatory SASP factors not only sustains the senescent phenotype, but also drives the transition into its 'mature' state following continuous self-amplifying loops, which is the most widely studied feature of senescent cells[16] (Figure 2).
Apart from the well-established role of SASP in the pathogenesis of MASH, metabolic dysregulations such as the impaired mitochondrial function may play also a crucial role in the propagation of the disease [84]. Mitochondrial dysfunction in MASH has been associated with the presence of microvesicular steatosis [85]. The coexistence of regions microvesicular steatosis along with macrovesicular steatosis is a widely known histological feature of MASH livers [50], which can be possibly explained by the impaired mitochondrial function along with lipid metabolic dysregulations. More specifically, it is plausible that the increasing uptake of fatty acids exceeds the capacity of senescent cells to store them in large LDs. The residual non-esterified FAs, which could not be stored in LDs, may form amphiphilic compounds in the cytoplasm shown as fat deposits[51]. Together with the severe mitochondrial dysfunction, this results in the development of patches of microvesicular steatosis, located nearby ballooned hepatocytes [49], which are regions associated with increased inflammation and oxidative stress.
Considering that MASLD is characterized by prolonged oxidative stress and thus increased DNA damage, it is plausible that the accumulation of senescent hepatocytes and the persistent “mature” SASP signaling further contribute to telomere attrition and genomic instability. This, in turn, could amplify RS during MASH. Thus, it is proposed that both SIPS and RS are implicated in the propagation of CS [75], highlighting the multifaceted role of hepatocyte senescence in MASLD progression. In line with this notion, studies have shown that telomere attrition along with DNA damage are more prominent in hepatocytes with more severe stages of steatosis [80], possibly indicating the induction of both RS and SIPS in more chronic steatotic conditions. Both mechanisms share similarities, such as the activation of p53 pathway triggered by both DNA damage and critical shortening of telomeres [86]. The percentage of hepatocytes with DNA damage was found to increase in parallel with steatosis grade, while concomitantly, hepatocytes with DNA damage had also significantly shorter telomeres, as apparently DNA damage disproportionately affects telomere length due to less efficient repair mechanisms[87]. Furthermore, chronic accumulation of fat in the liver can induce both DNA damage and attrition of telomere length in hepatocytes, possibly mediated by persistent oxidative stress [80]. These findings suggest that the initial activation of SIPS by fat accumulation may gradually amplify RS mechanism, thus resulting in a significant accumulation of senescent cells, especially affecting aged patients who already have a substantial number of these cells[88]. premature telomere shortening was detected in hepatocytes in MASLD [80], providing further evidence that persistent liver damage may amplify RS, highlighting the potential interaction between SIPS and RS during MASLD progression.
6. Conclusion
Hepatic steatosis, a hallmark of MASLD, is significantly influenced by the accumulation of senescent hepatocytes. A body of evidence shows an increase in the percentage of senescent hepatocytes, that correlates with inflammatory activity and fibrosis advancement. In addition senescent cells have been shown to undergo significant metabolic changes that affect their ability to process and store lipids efficiently. These metabolic alterations can be further exacerbated under steatotic conditions, leading to excessive lipid accumulation, elevated oxidative stress, and mitochondrial dysfunction, which constitute key contributors to MASLD progression. During the early stages of ΜASLD, hepatocytes seem to respond differently to the stress stimulus of steatosis, resulting in only scattered areas of senescent cells, while overall hepatic function remains preserved. Although the presence of senescent hepatocytes in MASLD is widely accepted, the mechanisms responsible for the development of CS during the different stages of the disease are still under debate. The induction of CS, as indicated by the elevated levels of p21, occurs alongside the accumulation of cytosolic LDs in hepatocytes. Significant changes in lipid metabolism and cellular lipid dynamics have been observed in senescent cells. Thus, lipid dysregulation and inflammatory conditions during MASLD progression seem to contribute to the induction of CS concomitantly. In MASLD these changes are further exacerbated, promoting disease progression and supporting the interaction between SIPS and LDs metabolism. MASH is characterized by an intense accumulation of senescent cells, resulting in a severe dysregulation of normal liver function. Given that the late-stage SASP is associated with chronic inflammation and tissue dysfunction, the persistence of senescent hepatocytes in MASLD may be a key driver to disease severity.
While these findings highlight a crucial link between CS, LD metabolism and MASLD pathogenesis, further research is needed to clarify whether targeting CS or modulating the SASP could serve as viable therapeutic strategies for MASLD management.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Hepatocyte Senescence and Lipid Dysregulation in the Early Stages of Metabolic dysfunction- associated steatotic liver disease (MASLD); Figure S2: Hepatocyte Senescence and Lipid Dysregulation in Metabolic dysfunction-associated steatohepatitis (MASH) Progression.
Author Contributions
Conceptualization, A.V.C.; writing—original draft preparation, E.M.M.; A.V.C., designed the figure, E.M.M.; writing—review and editing, A.V.C., A.C.G., A. K., E.K., G.G, and P.K.; visualization, A.V.C.; supervision, A.V.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ATM | Ataxia-telangiectasia mutated |
| CD36 | Cluster of differentiation 36 |
| CS | Cellular senescence |
| DDR | DNA damage response |
| ECM | Extracellular matrix |
| ER | Endoplasmic reticulum |
| EVs | Extracellular vesicles |
| FA | Fatty acid |
| FAO | Fatty acid oxidation |
| FASN | Fatty acid synthase |
| FFA | Free fatty acid |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| IL | Interleukin |
| LD | Lipid droplet |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| mTOR | mammalian target of rapamycin |
| NAFLD | Non-alcoholic fatty liver disease |
| NF-κB | Nuclear factor kappa B |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| ROS | Reactive oxygen species |
| RS | Replicative senescence |
| SA-β-gal | Senescence-associated β-galactosidase |
| SADF | Senescence-associated DNA damage foci |
| SAHF | Senescence-associated heterochromatin foci |
| SASP | Senescence-associated secretory phenotype |
| SIPS | Stress-induced premature senescence |
| TGF-β | Transforming growth factor beta |
| TG | Triglyceride |
| PLINs | Perilipins |
References
- L. HAYFLICK and P. S. MOORHEAD, "The serial cultivation of human diploid cell strains," (in eng), Exp Cell Res, vol. 25, pp. 1961. [CrossRef]
- A. Prieur, E. A. Prieur, E. Besnard, A. Babled, and J. M. Lemaitre, "p53 and p16(INK4A) independent induction of senescence by chromatin-dependent alteration of S-phase progression," (in eng), Nat Commun, vol. 2, p. 2011; 13. [Google Scholar] [CrossRef]
- N. Alessio, D. N. Alessio, D. Aprile, S. Cappabianca, G. Peluso, G. Di Bernardo, and U. Galderisi, "Different Stages of Quiescence, Senescence, and Cell Stress Identified by Molecular Algorithm Based on the Expression of Ki67, RPS6, and Beta-Galactosidase Activity," (in eng), Int J Mol Sci, vol. 22, no. 2021; 6. [Google Scholar] [CrossRef]
- M. Piechota et al., "Is senescence-associated β-galactosidase a marker of neuronal senescence?," (in eng), Oncotarget, vol. 7, no. 49, pp. 8109. [CrossRef]
- E. A. Georgakopoulou et al., "Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues," (in eng), Aging (Albany NY), vol. 5, no. 1, pp. 2013; -50. [CrossRef]
- A. Terman and U. T. Brunk, "Lipofuscin," (in eng), Int J Biochem Cell Biol, vol. 36, no. 8, pp. 1400; -4. [CrossRef]
- J. R. Sparrow and M. Boulton, "RPE lipofuscin and its role in retinal pathobiology," (in eng), Exp Eye Res, vol. 80, no. 5, pp. 20 May; 05. [CrossRef]
- S. Ferreira-Gonzalez, D. S. Ferreira-Gonzalez, D. Rodrigo-Torres, V. L. Gadd, and S. J. Forbes, "Cellular Senescence in Liver Disease and Regeneration," (in eng), Semin Liver Dis, vol. 41, no. 1, pp. 2021; -66. [Google Scholar] [CrossRef]
- K. M. Aird and R. Zhang, "Detection of senescence-associated heterochromatin foci (SAHF)," (in eng), Methods Mol Biol, vol. 965, pp. 2013; -96. [CrossRef]
- F. d'Adda di Fagagna et al., "A DNA damage checkpoint response in telomere-initiated senescence," (in eng), Nature, vol. 426, no. 6963, pp. 2003; -8. [CrossRef]
- J. C. Acosta et al., "A complex secretory program orchestrated by the inflammasome controls paracrine senescence," (in eng), Nat Cell Biol, vol. 15, no. 8, pp. 2013; -90. [CrossRef]
- M. V. Reid, G. M. V. Reid, G. Fredickson, and D. G. Hepatology, 2024; 30. [Google Scholar] [CrossRef]
- Y. S. Yoon et al., "Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1," (in eng), J Cell Physiol, vol. 209, no. 2, pp. 2006; -80. [CrossRef]
- S. Yamauchi et al., "Mitochondrial fatty acid oxidation drives senescence," (in eng), Sci Adv, vol. 10, no. 43, p. 5887. [CrossRef]
- V. Gorgoulis et al., "Cellular Senescence: Defining a Path Forward," (in eng), Cell, vol. 179, no. 4, pp. 2019; 31. [CrossRef]
- N. Malaquin, A. N. Malaquin, A. Martinez, and F. Rodier, "Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype," (in eng), Exp Gerontol, vol. 82, pp. 2016; -49. [Google Scholar] [CrossRef]
- J. L. Dempsey, G. N. J. L. Dempsey, G. N. Ioannou, and R. M. Carr, "Mechanisms of Lipid Droplet Accumulation in Steatotic Liver Diseases," (in eng), Semin Liver Dis, vol. 43, no. 4, pp. 2023. [Google Scholar] [CrossRef]
- L. Terlecki-Zaniewicz et al., "Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype," (in eng), Aging (Albany NY), vol. 10, no. 5, pp. 19 May 1103. [CrossRef]
- H. Okawa, Y. H. Okawa, Y. Tanaka, and A. Takahashi, "Network of extracellular vesicles surrounding senescent cells," (in eng), Arch Biochem Biophys, vol. 754, p. 1099; 53. [Google Scholar] [CrossRef]
- J. P. Coppé, P. Y. J. P. Coppé, P. Y. Desprez, A. Krtolica, and J. Campisi, "The senescence-associated secretory phenotype: the dark side of tumor suppression," (in eng), Annu Rev Pathol, vol. 5, pp. 2010. [Google Scholar] [CrossRef]
- Y. Oguma et al., "Meta-analysis of senescent cell secretomes to identify common and specific features of the different senescent phenotypes: a tool for developing new senotherapeutics," (in eng), Cell Commun Signal, vol. 21, no. 1, p. 2023; 28. [CrossRef]
- Z. Liao, H. L. Z. Liao, H. L. Yeo, S. W. Wong, and Y. Zhao, "Cellular Senescence: Mechanisms and Therapeutic Potential," (in eng), Biomedicines, vol. 9, no. 2021; 12. [Google Scholar] [CrossRef]
- T. Shimi et al., "The role of nuclear lamin B1 in cell proliferation and senescence," (in eng), Genes Dev, vol. 25, no. 24, pp. 2579; -93. [CrossRef]
- R. Kumari and P. Jat, "Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype," (in eng), Front Cell Dev Biol, vol. 9, p. 64 5593, 2021. [CrossRef]
- S. Tabata et al., "NFκB dynamics-dependent epigenetic changes modulate inflammatory gene expression and induce cellular senescence," (in eng), FEBS J, vol. 291, no. 22, pp. 4951. [CrossRef]
- N. Herranz et al., "mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype," (in eng), Nat Cell Biol, vol. 17, no. 9, pp. 1205; -17. [CrossRef]
- N. Basisty et al., "A proteomic atlas of senescence-associated secretomes for aging biomarker development," (in eng), PLoS Biol, vol. 18, no. 1, p. 3000. [CrossRef]
- C. M. Beauséjour et al., "Reversal of human cellular senescence: roles of the p53 and p16 pathways," (in eng), EMBO J, vol. 22, no. 16, pp. 4212; -22. [CrossRef]
- S. Hamsanathan and A. U. Gurkar, "Lipids as Regulators of Cellular Senescence," (in eng), Front Physiol, vol. 13, p. 79 6850, 2022. [CrossRef]
- E. H. Bent, L. A. E. H. Bent, L. A. Gilbert, and M. T. Hemann, "A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses," (in eng), Genes Dev, vol. 30, no. 16, pp. 1811; -21. [Google Scholar] [CrossRef]
- R. A. Saxton and D. M. Sabatini, "mTOR Signaling in Growth, Metabolism, and Disease," (in eng), Cell, vol. 168, no. 6, pp. 2017; 03. [CrossRef]
- Biran, *!!! REPLACE !!!*; et al. , "Quantitative identification of senescent cells in aging and disease," (in eng), Aging Cell, vol. 16, no. 4, pp. 2017. [Google Scholar] [CrossRef]
- K. Itahana, Y. K. Itahana, Y. Itahana, and G. P. Dimri, "Colorimetric detection of senescence-associated β galactosidase," (in eng), Methods Mol Biol, vol. 965, pp. 2013; -56. [Google Scholar] [CrossRef]
- T.-W. Kang, "Senescence surveillance of pre-malignant hepatocytes limits liver cancer development," vol. 479, Woller Norman Ed., ed. Nature.
- YYY. Millner and G. E. Atilla-Gokcumen, "Lipid Players of Cellular Senescence," (in eng), Metabolites, vol. 10, no. 2020; 9. [CrossRef]
- YYY. C. Flor, D. Wolfgeher, D. Wu, and S. J. Kron, "A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence," (in eng), Cell Death Discov, vol. 3, p. 1 7075, 2017. [CrossRef]
- Q. Zeng, Y. Q. Zeng, Y. Gong, N. Zhu, Y. Shi, C. Zhang, and L. Qin, "Lipids and lipid metabolism in cellular senescence: Emerging targets for age-related diseases," (in eng), Ageing Res Rev, vol. 97, p. 1022; 94. [Google Scholar] [CrossRef]
- R. Quintana-Castro et al., "Cd36 gene expression in adipose and hepatic tissue mediates the lipids accumulation in liver of obese rats with sucrose-induced hepatic steatosis," (in eng), Prostaglandins Other Lipid Mediat, vol. 147, p. 1064; 04. [CrossRef]
- M. Chong et al., "CD36 initiates the secretory phenotype during the establishment of cellular senescence," (in eng), EMBO Rep, vol. 19, no. 2018; 6. [CrossRef]
- J. Fafián-Labora et al., "FASN activity is important for the initial stages of the induction of senescence," (in eng), Cell Death Dis, vol. 10, no. 4, p. 2019; 08. [CrossRef]
- M. Danielli, L. Perne, E. Jarc Jovičić, and T. Petan, "Lipid droplets and polyunsaturated fatty acid trafficking: Balancing life and death," (in eng), Front Cell Dev Biol, vol. 11, p. 110 4725, 2023. [CrossRef]
- H. Fan and Y. Tan, "Lipid Droplet-Mitochondria Contacts in Health and Disease," (in eng), Int J Mol Sci, vol. 25, no. 2024; 13. [CrossRef]
- R. K. Angara, M. F. Sladek, and S. D. Gilk, "ER-LD Membrane Contact Sites: A Budding Area in the Pathogen Survival Strategy," (in eng), Contact (Thousand Oaks), vol. 7, p. 2515256424130 4196, 2024. [CrossRef]
- S. Xu, X. S. Xu, X. Zhang, and P. Liu, "Lipid droplet proteins and metabolic diseases," (in eng), Biochim Biophys Acta Mol Basis Dis, vol. 1864, no. 5 Pt B, pp. 20 May 1968; 18. [Google Scholar] [CrossRef]
- D. Jiang et al., "Analysis of p53 transactivation domain mutants reveals Acad11 as a metabolic target important for p53 pro-survival function," (in eng), Cell Rep, vol. 10, no. 7, pp. 1096. [CrossRef]
- P. V. S. Vasileiou et al., "Mitochondrial Homeostasis and Cellular Senescence," (in eng), Cells, vol. 8, no. 2019; 7. [CrossRef]
- M. G. Vizioli et al., "Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence," (in eng), Genes Dev, vol. 34, no. 5-6, pp. 2020; 01. [CrossRef]
- Y. Li et al., "GSK3 inhibitor ameliorates steatosis through the modulation of mitochondrial dysfunction in hepatocytes of obese patients," (in eng), iScience, vol. 24, no. 3, p. 1021; 49. [CrossRef]
- S. Tandra et al., "Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease," (in eng), J Hepatol, vol. 55, no. 3, pp. 2011. [CrossRef]
- C. W. Germano et al., "Microvesicular Steatosis in Individuals with Obesity: a Histological Marker of Non-alcoholic Fatty Liver Disease Severity," (in eng), Obes Surg, vol. 33, no. 3, pp. 2023. [CrossRef]
- B. Fromenty, A. B. Fromenty, A. Berson, and D. Pessayre, "Microvesicular steatosis and steatohepatitis: role of mitochondrial dysfunction and lipid peroxidation," (in eng), J Hepatol, vol. 26 Suppl 1, pp. 1997; -22. [Google Scholar] [CrossRef]
- P. Chandrasekaran, S. P. Chandrasekaran, S. Weiskirchen, and R. Weiskirchen, "Perilipins: A family of five fat-droplet storing proteins that play a significant role in fat homeostasis," (in eng), J Cell Biochem, vol. 125, no. 6, p. 3057; e9. [Google Scholar] [CrossRef]
- A. E. Libby, E. A. E. Libby, E. Bales, D. J. Orlicky, and J. L. McManaman, "Perilipin-2 Deletion Impairs Hepatic Lipid Accumulation by Interfering with Sterol Regulatory Element-binding Protein (SREBP) Activation and Altering the Hepatic Lipidome," (in eng), J Biol Chem, vol. 291, no. 46, pp. 2423. [Google Scholar] [CrossRef]
- B. K. Straub et al., "Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation," (in eng), Histopathology, vol. 62, no. 4, pp. 2013; -31. [CrossRef]
- J. L. McManaman et al., "Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease," (in eng), J Lipid Res, vol. 54, no. 5, pp. 20 May 1346; -59. [CrossRef]
- R. M. Carr and R. S. Ahima, "Pathophysiology of lipid droplet proteins in liver diseases," (in eng), Exp Cell Res, vol. 340, no. 2, pp. 2016; -92. [CrossRef]
- Chiariello, *!!! REPLACE !!!*; et al. , "Downregulation of PLIN2 in human dermal fibroblasts impairs mitochondrial function in an age-dependent fashion and induces cell senescence via GDF15," (in eng), Aging Cell, vol. 23, no. 5, p. 20 May 1411; e1. [Google Scholar] [CrossRef]
- L. M. Pawella et al., "Perilipin discerns chronic from acute hepatocellular steatosis," (in eng), J Hepatol, vol. 60, no. 3, pp. 2014; -42. [CrossRef]
- S. Y. Ma et al., "Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD," (in eng), Liver Int, vol. 40, no. 10, pp. 2427. [CrossRef]
- C. Wang et al., "Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis," (in eng), Hepatology, vol. 61, no. 3, pp. 2015; -82. [CrossRef]
- M. Cinato et al., "Role of Perilipins in Oxidative Stress-Implications for Cardiovascular Disease," (in eng), Antioxidants (Basel), vol. 13, no. 2024; 2. [CrossRef]
- M. Pawlak, P. M. Pawlak, P. Lefebvre, and B. Staels, "Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease," (in eng), J Hepatol, vol. 62, no. 3, pp. 2015; -33. [Google Scholar] [CrossRef]
- Y. Lin, Y. Wang, and P. F. Li, "PPARα: An emerging target of metabolic syndrome, neurodegenerative and cardiovascular diseases," (in eng), Front Endocrinol (Lausanne), vol. 13, p. 107 4911, 2022. [CrossRef]
- N. Zhang et al., "Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway," (in eng), Oncotarget, vol. 5, no. 18, pp. 8330; -40. [CrossRef]
- S. Todisco et al., "PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH)," (in eng), Biology (Basel), vol. 11, no. 23 May 2022; 5. [CrossRef]
- J. A. Prescott, J. P. J. A. Prescott, J. P. Mitchell, and S. J. Cook, "Inhibitory feedback control of NF-κB signalling in health and disease," (in eng), Biochem J, vol. 478, no. 13, pp. 2619. [Google Scholar] [CrossRef]
- G. Targher, C. D. G. Targher, C. D. Byrne, and H. Tilg, "MASLD: a systemic metabolic disorder with cardiovascular and malignant complications," (in eng), Gut, vol. 73, no. 4, pp. 2024; 07. [Google Scholar] [CrossRef]
- T. Liu, L. T. Liu, L. Zhang, D. Joo, and S. C. Sun, "NF-κB signaling in inflammation," (in eng), Signal Transduct Target Ther, vol. 2, pp. 1702. [Google Scholar] [CrossRef]
- H. P. Glauert et al., "The Role of NF-kappaB in PPARalpha-Mediated Hepatocarcinogenesis," (in eng), PPAR Res, vol. 2008, p. 28 6249, 2008. [CrossRef]
- K. Lee et al., "Hepatic Mitochondrial Defects in a Nonalcoholic Fatty Liver Disease Mouse Model Are Associated with Increased Degradation of Oxidative Phosphorylation Subunits," (in eng), Mol Cell Proteomics, vol. 17, no. 12, pp. 2371. [CrossRef]
- M. Longo, M. M. Longo, M. Meroni, E. Paolini, C. Macchi, and P. Dongiovanni, "Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending?," (in eng), Metabolism, vol. 117, p. 1547; 08. [Google Scholar] [CrossRef]
- B. Fromenty and M. Roden, "Mitochondrial alterations in fatty liver diseases," (in eng), J Hepatol, vol. 78, no. 2, pp. 2023. [CrossRef]
- Y. Deleye et al., "CDKN2A/p16INK4a suppresses hepatic fatty acid oxidation through the AMPKα2-SIRT1-PPARα signaling pathway," (in eng), J Biol Chem, vol. 295, no. 50, pp. 1731. [CrossRef]
- K. Evangelou et al., "Cellular senescence and cardiovascular diseases: moving to the "heart" of the problem," (in eng), Physiol Rev, vol. 103, no. 1, pp. 2023; 01. [CrossRef]
- A. D. Aravinthan and G. J. M. Alexander, "Senescence in chronic liver disease: Is the future in aging?," (in eng), J Hepatol, vol. 65, no. 4, pp. 2016. [CrossRef]
- N. Kudlova, J. B. N. Kudlova, J. B. De Sanctis, and M. Hajduch, "Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs," (in eng), Int J Mol Sci, vol. 23, no. 2022; 8. [Google Scholar] [CrossRef]
- A. S. Meijnikman, H. A. S. Meijnikman, H. Herrema, T. P. M. Scheithauer, J. Kroon, M. Nieuwdorp, and A. K. Groen, "Evaluating causality of cellular senescence in non-alcoholic fatty liver disease," (in eng), JHEP Rep, vol. 3, no. 4, p. 1003; 01. [Google Scholar] [CrossRef]
- A. Aravinthan et al., "Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease," (in eng), Cell Cycle, vol. 13, no. 9, pp. 1489; -94. [CrossRef]
- M. Ogrodnik et al., "Cellular senescence drives age-dependent hepatic steatosis," (in eng), Nat Commun, vol. 8, p. 1569; 1. [CrossRef]
- A. Aravinthan et al., "Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease," (in eng), J Hepatol, vol. 58, no. 3, pp. 2013; -56. [CrossRef]
- S. Francque et al., "PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis," (in eng), J Hepatol, vol. 63, no. 1, pp. 2015; -73. [CrossRef]
- C. Kiourtis et al., "Hepatocellular senescence induces multi-organ senescence and dysfunction via TGFβ," (in eng), Nat Cell Biol, vol. 26, no. 12, pp. 2075. [CrossRef]
- M. Hoare and M. Narita, "Transmitting senescence to the cell neighbourhood," (in eng), Nat Cell Biol, vol. 15, no. 8, pp. 2013; -9. [CrossRef]
- A. Vouilloz et al., "Impaired unsaturated fatty acid elongation alters mitochondrial function and accelerates metabolic dysfunction-associated steatohepatitis progression," (in eng), Metabolism, vol. 162, p. 1560; 51. [CrossRef]
- A. B. Koenig, A. A. B. Koenig, A. Tan, H. Abdelaal, F. Monge, Z. M. Younossi, and Z. D. Goodman, "Review article: Hepatic steatosis and its associations with acute and chronic liver diseases," (in eng), Aliment Pharmacol Ther, vol. 60, no. 2, pp. 2024. [Google Scholar] [CrossRef]
- O. Toussaint et al., "Stress-induced premature senescence or stress-induced senescence-like phenotype: one in vivo reality, two possible definitions?," (in eng), ScientificWorldJournal, vol. 2, pp. 2002; -47. [CrossRef]
- T. von Zglinicki, "Oxidative stress shortens telomeres," (in eng), Trends Biochem Sci, vol. 27, no. 7, pp. 2002; -44. [CrossRef]
- V. Paradis et al., "Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas," (in eng), Hum Pathol, vol. 32, no. 3, pp. 2001; -32. [CrossRef]
Figure 1.
Hepatocyte Senescence and Lipid Dysregulation in the Early Stages of Metabolic dysfunction- associated steatotic liver disease (MASLD). This figure illustrates the interaction between stress-induced premature senescence (SIPS) and lipid droplets (LDs) in early stages of MASLD. (a) Early steatosis is characterized by temporary cell-cycle arrest, with stressed hepatocytes expressing p53/p21, Ki-67, and Senescence-associated beta-galactosidase (Sa-β-gal). These hepatocytes may either undergo apoptosis or recover through fatty acid oxidation (FAO) activation, mediated by increased levels of peroxisome proliferator activated receptor alpha (PPARα). (b) In cases of prolonged steatosis, hepatocytes exhibit SIPS initiation, marked by upregulation of p16, p53/p21, and early senescence-associated secretory phenotype (SASP) cytokines such as IL-1α, which can recruit macrophages for immune-mediated clearance of senescent hepatocytes. However, the increased levels of PPARα are unable to compensate for excess fatty acid influx and along with increased levels of CD36, a lipid associated receptor, lead to lipid accumulation. If steatosis persists and immune clearance of senescent hepatocytes is unsuccessful, these cells enter full SIPS. (c) During this phase, levels of PPARα are downregulated, leading to decreased FAO, while early mitochondrial dysregulation contributes to further lipid accumulation, known as macrovesicular steatosis. This phase is also characterized by self-amplifying SASP, persistent nuclear factor kappa B (NF-κB) activation due to reduced PPARα expression and a positive feedback loop of SASP pro-inflammatory cytokines. Additionally, SASP factors begin to exert paracrine effects, triggering senescence-induced senescence in adjacent normal hepatocytes. Depending on the efficiency of immune clearance, hepatocytes in full SIPS may either be eliminated, allowing partial restoration of liver function, or persist, leading to deep senescence and progressive liver dysfunction.
Figure 1.
Hepatocyte Senescence and Lipid Dysregulation in the Early Stages of Metabolic dysfunction- associated steatotic liver disease (MASLD). This figure illustrates the interaction between stress-induced premature senescence (SIPS) and lipid droplets (LDs) in early stages of MASLD. (a) Early steatosis is characterized by temporary cell-cycle arrest, with stressed hepatocytes expressing p53/p21, Ki-67, and Senescence-associated beta-galactosidase (Sa-β-gal). These hepatocytes may either undergo apoptosis or recover through fatty acid oxidation (FAO) activation, mediated by increased levels of peroxisome proliferator activated receptor alpha (PPARα). (b) In cases of prolonged steatosis, hepatocytes exhibit SIPS initiation, marked by upregulation of p16, p53/p21, and early senescence-associated secretory phenotype (SASP) cytokines such as IL-1α, which can recruit macrophages for immune-mediated clearance of senescent hepatocytes. However, the increased levels of PPARα are unable to compensate for excess fatty acid influx and along with increased levels of CD36, a lipid associated receptor, lead to lipid accumulation. If steatosis persists and immune clearance of senescent hepatocytes is unsuccessful, these cells enter full SIPS. (c) During this phase, levels of PPARα are downregulated, leading to decreased FAO, while early mitochondrial dysregulation contributes to further lipid accumulation, known as macrovesicular steatosis. This phase is also characterized by self-amplifying SASP, persistent nuclear factor kappa B (NF-κB) activation due to reduced PPARα expression and a positive feedback loop of SASP pro-inflammatory cytokines. Additionally, SASP factors begin to exert paracrine effects, triggering senescence-induced senescence in adjacent normal hepatocytes. Depending on the efficiency of immune clearance, hepatocytes in full SIPS may either be eliminated, allowing partial restoration of liver function, or persist, leading to deep senescence and progressive liver dysfunction.
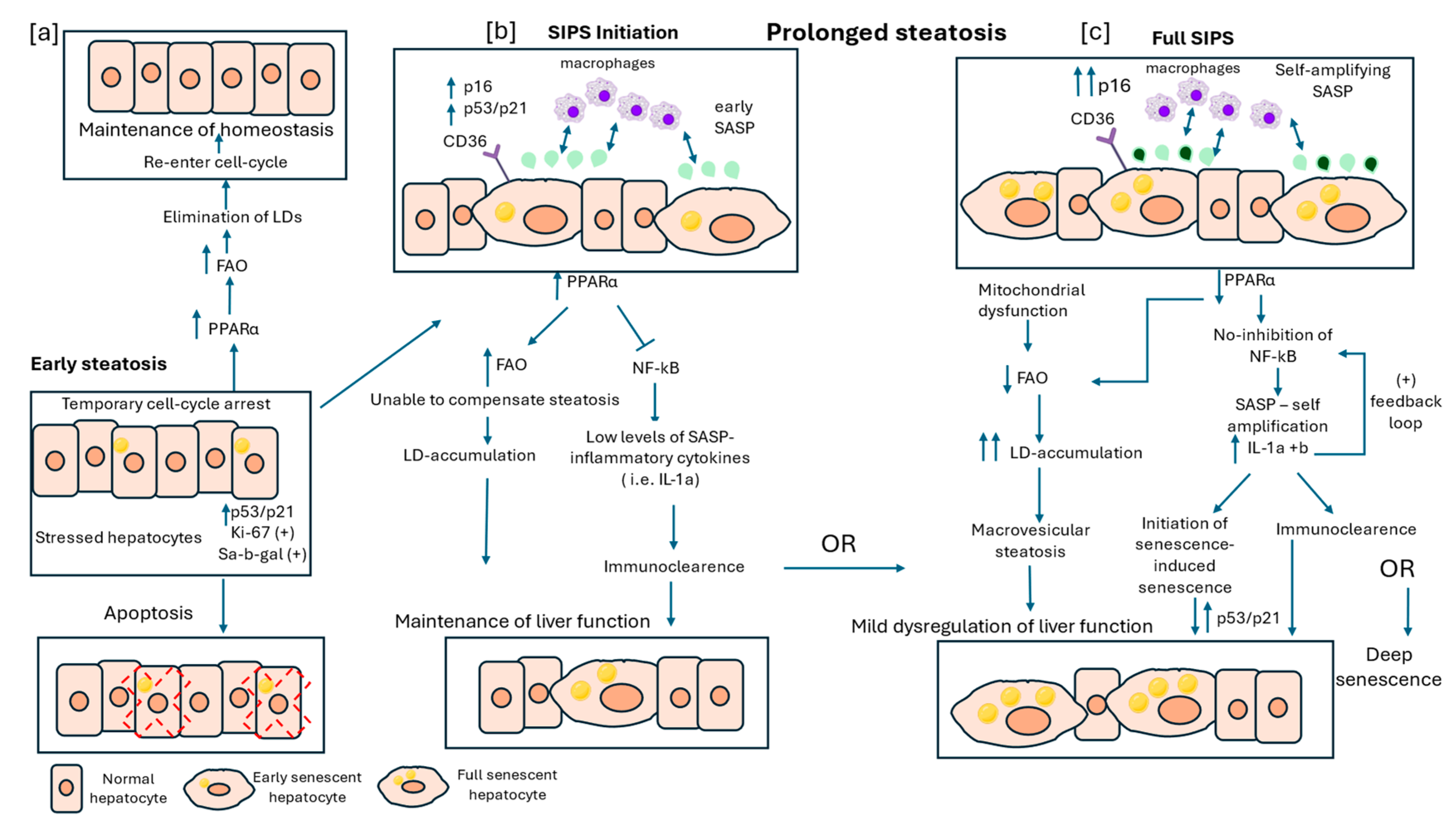
Figure 2.
Hepatocyte Senescence and Lipid Dysregulation in Metabolic dysfunction-associated steatohepatitis (MASH) Progression. This figure illustrates the molecular mechanisms linking interaction of SIPS and LDs to MASH progression. In deep SIPS, hepatocytes exhibit sustained upregulation of p16 and mature SASP factors, promoting a chronic pro-inflammatory environment. Increased CD36 expression further exacerbates lipid accumulation, while immune senescence impairs the clearance of senescent hepatocytes. Mitochondrial dysfunction is another hallmark of deep senescent cells, leading to increased reactive oxygen species (ROS) production. Sustained mitochondrial impairment along with extremely downregulated levels of PPARα and FAO result in both macrovesicular and microvesicular steatosis, oxidative stress, and lipotoxicity, all of which contribute to chronic liver inflammation. Additionally, the amplification of NF-κB signaling enhances SASP-driven secretion of highly inflammatory cytokines, such as IL-6 and IL-8, which further promote senescence-induced senescence of neighboring hepatocytes. Persistent oxidative stress and lipid toxicity drive DNA damage which disproportionately affects telomeres, leading to telomere attrition and triggering replicative senescence marked by p53/p21 upregulation. As senescent hepatocytes accumulate, along with the inability of the immune system to clear them effectively leads to progressive liver dysfunction and exacerbation of MASH pathology.
Figure 2.
Hepatocyte Senescence and Lipid Dysregulation in Metabolic dysfunction-associated steatohepatitis (MASH) Progression. This figure illustrates the molecular mechanisms linking interaction of SIPS and LDs to MASH progression. In deep SIPS, hepatocytes exhibit sustained upregulation of p16 and mature SASP factors, promoting a chronic pro-inflammatory environment. Increased CD36 expression further exacerbates lipid accumulation, while immune senescence impairs the clearance of senescent hepatocytes. Mitochondrial dysfunction is another hallmark of deep senescent cells, leading to increased reactive oxygen species (ROS) production. Sustained mitochondrial impairment along with extremely downregulated levels of PPARα and FAO result in both macrovesicular and microvesicular steatosis, oxidative stress, and lipotoxicity, all of which contribute to chronic liver inflammation. Additionally, the amplification of NF-κB signaling enhances SASP-driven secretion of highly inflammatory cytokines, such as IL-6 and IL-8, which further promote senescence-induced senescence of neighboring hepatocytes. Persistent oxidative stress and lipid toxicity drive DNA damage which disproportionately affects telomeres, leading to telomere attrition and triggering replicative senescence marked by p53/p21 upregulation. As senescent hepatocytes accumulate, along with the inability of the immune system to clear them effectively leads to progressive liver dysfunction and exacerbation of MASH pathology.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



