Submitted:
13 November 2025
Posted:
18 November 2025
You are already at the latest version
Abstract
Intrinsically disordered regions enable transcription factors (TFs) to undergo structural changes upon ligand binding, facilitating the transduction of environmental signals into gene expression. In this study, we combined molecular modeling methods to explore the hypothesis that unstructured inter-domain and subdomain linkers in bacterial TFs can function as sensors for carbohydrate signaling molecules. We combined molecular dy-namics simulations and carbohydrate docking to analyze six repressors with GntR-type DNA-binding domains, including UxuR, GntR and FarR from Escherichia coli, as well as AraR, NagR and YydK from Bacillus subtilis. Protein models obtained from different time points of the dynamic simulations were subjected to the sequential carbohydrates dock-ing. We found that the inter-domain linker of the UxuR monomer binds D-fructuronate, D-galacturonate, D-glucose, and D-glucuronate with affinities lower that its structured FadR-type effector-binding domain. However, in the monomer, these ligands formed mul-timolecular clusters, a feature absent in the dimer, suggesting that protein dimerization may depend on linker occupancy by cellular carbohydrates. Interacting with linkers con-necting subdomains of the LacI/GalR-type E-domains in GntR and AraR, D-glucose was able to form hydrogen bonds connecting distant structural modules of the proteins, while in NagR, FarR and YydK it bridged the inter-domain linkers and a β-sheet within the HutC-type E-domains. Our results establish flexible linkers as pivotal metabolic sensors that directly integrate nutritional cues to alter gene expression in bacteria.
Keywords:
bacterial transcription factors
; intrinsically disordered regions
; flexible inter-domain linkers
; effector-binding pockets
; metabolic carbohydrates
; sequential molecular docking
1. Introduction
Although bacterial cells employ diverse regulatory mechanisms, the control of transcription is widely regarded as the primary control point for regulating gene expression [1,2]. This process is governed by transcription factors (TFs), which possess evolutionary optimized structures that enable specific response to environmental signals. Nearly 42% of TFs are single-domain proteins [3], built from a limited repertoire of structural modules specialized for DNA sequence recognition at their binding sites [4,5]. Most TFs (50.2%) contain a second domain that interacts with RNA polymerase (RNAP) or other transcriptional machinery components, while the remaining ~8% are organized into three or more domains with distinct or overlapping functions [3]. Beyond these structured domains, TFs often contain flexible inter-domain linkers or unstructured terminal regions. These dynamic segments are critical for allosteric regulation, modulation of DNA/RNA polymerase (RNAP) binding, and signal transduction. For example, in Escherichia coli (E. coli), the AraC protein represses transcription of the araBAD operon in the absence of arabinose. Arabinose binding to its effector domain triggers a conformational shift in the AraC linker from an α helix to a flexible state, enabling the dimeric DNA-binding domains to engage the araBAD promoter and activate transcription [6]. Conversily, maltotriose binding to the effector domain of the TF MalT induces structural ordering of its unstructured N-terminal loop, facilitating MalT oligomerization and subsequent transcription activation [7]. Despite several examples, it is currently unclear to what extent TFs’ regulatory ability depends on the inherent plasticity of their disordered segments.
The functional significance of unstructured protein segments has become widely recognized over the past two decades [8,9]. This led to the formulation of the “disorder–function paradigm” complementing the classical “structure–function paradigm” that equates protein function with well-defined three-dimensional structures [9]. Intrinsically disordered proteins (IDPs) and intrinsically disordered regions of proteins (IDRs) are now considered essential components in a wide variety of cellular processes [10]. Lacking stable secondary and tertiary structure, IDPs/IDRs function as highly plastic molecules that complement the role of globular proteins in cells [11,12]. A central question that has persisted since their discovery is how these proteins recognize their targets and perform specific functions despite their structural heterogeneity [13,14]. Available genomic data indicate a striking difference in the abundance of proteins with unstructured modules across the three domains of life. Whereas only 2.0-4.2% of prokaryotic proteins contain disordered regions longer than 30 residues, this proportion rises to 33% in eukaryotes [15]. However, Molecular Recognition Elements/Features (MoRE/Fs), defined as disordered segments that undergo binding-induced folding [16,17], are present in 29% of prokaryotic IDRs and 21% of eukaryotic IDRs [17]. This comparable ratio suggests a roughly equal propensity for disorder-to-order transitions in pro- and eukaryotes, especially as bacterial transcription factors and other DNA-binding proteins are also significantly enriched among proteins containing MoREs.
IDRs most often exist as localized segments within a protein [11,12], frequently as flexible linkers between globular domains [18], or as unstructured protein termini [19]. When positioned between domains, these linkers not only control domain orientation but also facilitate interactions with small molecules [20]. The structural outcomes of these interactions are diverse, spanning a continuum from rigid, ordered complexes to highly unconstrained states with multiple weak contacts [14,21,22,23]. A key functional consequence is allosteric regulation. It is well-established that IDRs significantly contribute to allosteric modulation in multi-domain proteins, and they can do so without folding into secondary structure elements [12,19,24,25]. By acting solely as flexible linkers that interact with ligands, other linkers or globular domains of the same or other proteins, IDRs can trigger domain reorientation or induce conformational changes within them [26,27,28]. Therefore, flexible polypeptide chains are not simple structural connectors [29,30].
The Dynamic Allostery model posits that flexibility of unstructured modules in proteins provides ensembles of conformations that can be sampled [30]. Transition between these states proceed “along major allosteric propagation pathways” that are pre-encoded within the linker sequences. This process defines a series of sequential transitions between preferred states, where each preceding state determines the subsequent one. Functionally significant linker conformations may represent rare states in the inactive protein that become more populated due to allosteric “maturation”. For IDRs, this directed folding upon ligand binding could stabilize specific conformations [31]. Such stabilization can occur via "conformational selection”, where a pre-existing state is stabilized, or through "induced fit", where ligand binding induces and stabilizes a new conformation [32,33]. Ultimately, binding specificity is thought to arise from distinct frustration patterns encoded within the heterogeneous conformational landscape of the binding region [34].
Allosteric communications between different parts of a protein, provides an efficient way to turn protein function on or off in response to cellular signals. This is particularly important for regulatory proteins, like transcription factors [35,36] and signal transduction proteins [37,38,39]. However, recent studies suggest that the regulation of a protein's function at one site by a change at a distant site is an intrinsic property of virtually all dynamic proteins, moving beyond the classical view that it is exclusive to specialized "allosteric" proteins [40,41]. Decades of research have established fundamental principles for how regulatory signals are perceived and transmitted. It is now widely acknowledged that ligand binding induces local conformational changes promoting a dynamic sampling of the protein’s conformational landscape. This process selects the most stable conformers from a preexisting ensemble [42,43,44,45]. Allosteric signals are then transmitted through transient hydrogen bonds formation [46,47] that propagate along multiple, pre-existing pathways [48], which can be altered by mutations [49]. It is likely that this transmission can occur even without of global alterations in the protein backbone [48,50,51]. Ligand binding thus increases the population of stabilized conformers while preserving the protein’s ability to bind other ligands [52]. This mechanism, known as "conformational selection and population shift" suggests that any ligand can shift the equilibrium of the dynamic conformational ensemble, thereby inducing an allosteric response via evolutionarily tuned dynamic trajectories.
Intrinsically Disordered Proteins (IDPs) are a distinct category of proteins that lack stable tertiary structure entirely. Although less common than proteins containing IDRs, IDPs constitute an average of 1.6% of bacterial proteomes [53]. Structural fluctuations and conformational heterogeneity in IDPs revealed by NMR spectroscopy support the hypothesis that IDPs exist as dynamic conformational assemblies stabilized by transient intramolecular interactions [19,54]. Their primary sequences are grouped into modular elements such as short linear motifs (SLiMs), low-complexity domains (LCD) and MoRFs [55]. Consequently, several IDPs were identified among eukaryotic transcriptional regulators. A key example is the human transcription activating protein HMGN5 (ID P82970), which is fully unstructured [56,57]. HMGN5 counteracts chromatin compaction by binding to nucleosomes [58] and can also interact with cellular RNAs. Its binding site for snoRNA2T2 partially overlaps with its nucleosome-binding site [59]. Furthermore, the transcriptional factor CTCF (CCCTC binding factor) has been identified as an in vivo interaction partner of HMGN5 [59]. These findings illustrate the remarkable functional versatility of a completely unstructured regulator, whose conformational flexibility enables interactions with divers partners. Such binding promiscuity is a hallmark of IDPs. This adaptability is so pronounced that even very short (34–40 residues) unstructured fragments from proteins like PUMA (apoptosis regulator protein) and E1A (the adenovirus hub protein) undergo conformational changes in response to small molecules like glycine, tryptophan, and sarcosine [60]. This suggests that fluctuations in the intracellular solute composition, which occur commonly within cells, can directly modulate the function of proteins with intrinsically disordered regions.
Most TF respond to environmental signals through allosteric regulation, typically triggered by effector molecules such as metabolites from the very pathways they control [61]. Effector binding usually induces conformational changes that modulate either DNA-binding affinity or affect protein-protein interaction interfaces. One of the largest and most widespread TF families in prokaryotes is the GntR-type regulators [62,63,64]. Found across diverse bacterial lineages, these proteins regulate various cellular processes, including carbon metabolism, antibiotic production, and the cell wall stress response. Structurally, GntR family TF consist of primary domains: an N-terminal DNA-binding domain and a C-terminal effector-binding/oligomerization (E-O) domain, connected by flexible linkers [64]. While all proteins from the GntR family share a highly similar structural organization in their N-terminal DNA-binding domains [62], they are classified into four major (FadR, HutC, MocR, YtrA) and two minor subfamilies (AraR and PlmA) based on the structural variations of their C-terminal domains [62,65,66]. Ultimately, effector binding triggers allosteric signals through oligomerization or conformational changes within the E-O domains [49,62,67,68,69].
While unstructured inter-domain linkers are no longer viewed as mere passive connectors, their role in effector binding and intra-domain dynamics remains unclear. Although some ligands can stabilize these flexible linkers into specific conformation, traditional structural biology methods can only capture a limited number of these conformers. Given the vast conformational diversity of intrinsically disordered regions, their potential role in the perceiving the metabolic signals should not be neglected. Therefore, in this study, we evaluate the ability of the intrinsically disordered regions in GntR family of transcription factors to interact with key substrates and intermediates from central bacterial metabolic pathways. We employed molecular dynamic (MD) simulation [70] to sample conformers of the apo-proteins from various time points of the dynamic trajectory. These conformers were then subjected to flexible molecular docking [71] to estimate the binding affinity of carbohydrates for the unstructured protein regions. Performing molecular docking sequentially for ten molecules of each carbohydrate type allowed us to quantify the background level of protein-carbohydrate interactions, sugar preference for unstructured protein chains, and linker occupancy. Finally, we used the Protein-Ligand Interaction Profiler (PLIP) [72] to visualize the hydrogen bonds potentially formed by the carbohydrates that exhibited the highest affinity for the unstructured regions.
2. Results
2.1. Modes of Carbohydrates Interaction with Inter-Domain Linkers of E. coli UxuR TF
The UxuR repressor controls the transcription of genes encoding key enzymes of the Ashwell and Entner–Doudoroff pathways, which integrate hexuronates and hexuronic acids into the glycolytic cycle [73,74]. The protein functions as a homo- or heterodimer with its paralog, ExuR [74,75] and represses the transcription of its own gene as well as several other operons involved in the transport and degradation of sugar acids [61]. Although the crystal structure of UxuR is not yet available, homology modeling and molecular docking have identified a zinc-binding motif within C-terminal domain [76]. In the presence of fructuronate or its isomer glucoronate, the transcription of uxuR is induced and it is hypothesized that this induction is mediated by a conformational transition in UxuR that increases the negative charge on the surface of its N-terminal domains [76]. In addition, UxuR is subjected to catabolic regulation by D-glucose and cyclic AMP [77]. Given this complex regulation, the structural impact of potential carbon effectors has been studied in silico using MD simulation and sequential molecular docking [68,75,78]. In our previous study [68], we identified two predominant sugar-binding sites on the UxuR monomer using eleven small intermediates from the Ashwell and Entner–Doudoroff pathways. The first site, located between N- and C-terminal domains, exhibited nearly similar affinity for all tested sugars. The second site, found within the effector-binding domain (E-domain), had the highest affinity for D-fructuronate and D-glucose [68]. In a subsequent study [78], we employed an additional set of natural carbons to evaluate steric constraints imposed by the conformations of the inter-domain linkers in the UxuR dimer and the size of the protein pocket in the C-terminal domains. This set included small molecules, such as glucose-6-phosphate and fructose-6-phosphate, as well as more complex sugars like lacto-N-fucopentaoses, lacto-N-difucohexaose, and lacto-N-tetraose. As expected, not all complex carbohydrates could enter the CTD pocket. However, the inter-domain linkers interacted with all tested sugars. The main objective of the present study is to evaluate the occupancy of these linkers by the UxuR natural effectors using conformers generated at different time points from molecular dynamic simulations.
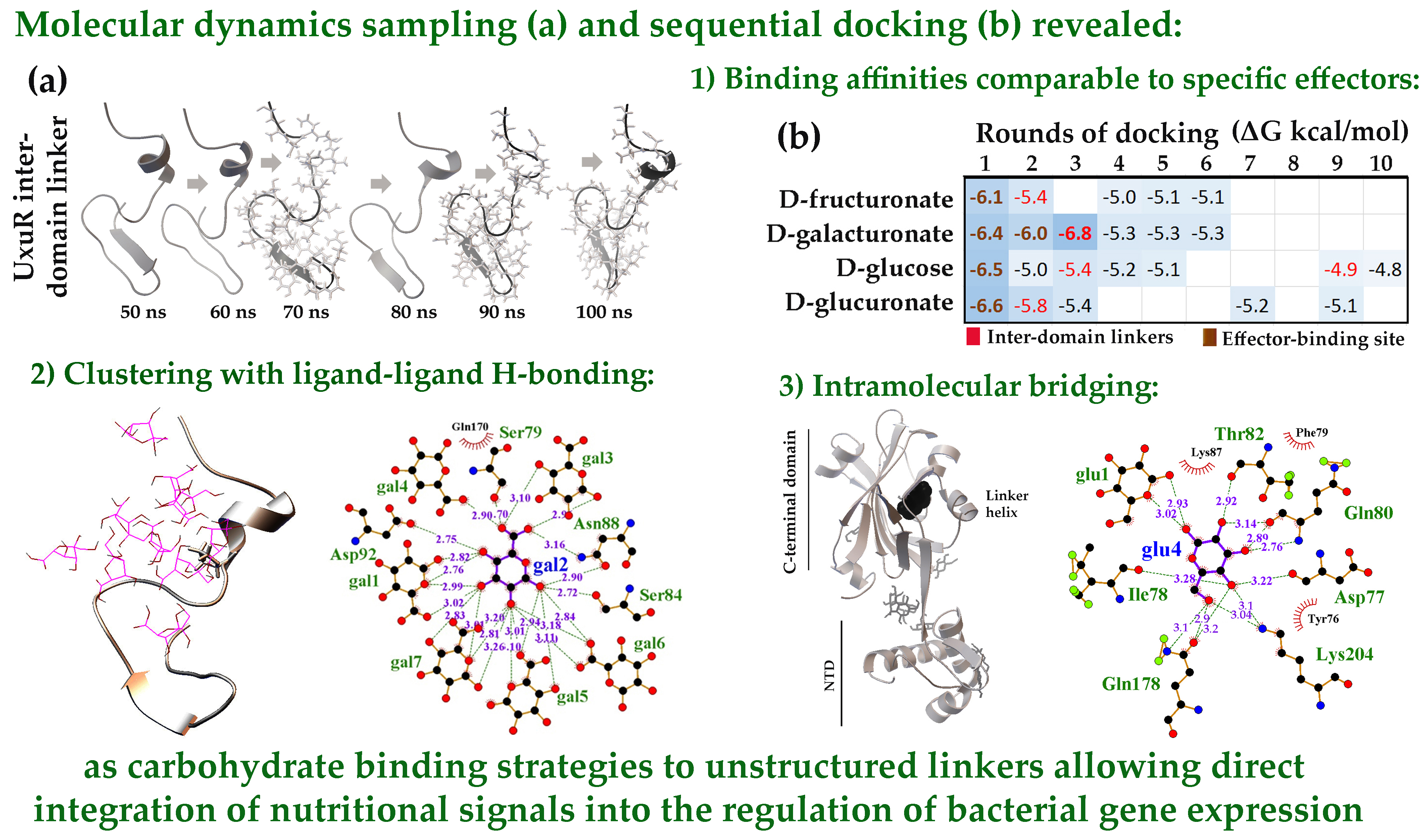
2.1.1. The Flexible Inter-Domain Linker of the UxuR Monomer Exhibited Low Carbohydrate Binding Efficiency but Provided a Platform for their Clustering
Figure 1a shows the structure of the UxuR monomer, predicted by the SWISS-MODEL online server [79,80,81]. It comprises a canonical GntR family N-terminal DNA-binding domain with a winged helix-turn-helix (wHTH) motif and a bulky “all helical” C-terminal FadR-type [65] domain predicted for effector binding. While the inter-domain linker (residues G61 to G94) is unstructured in the UxuR UniProt model P39161 [82] our MD simulations revealed an ability of its amino acid sequence to form short transient alpha-helix and beta-strand during the simulation trajectory (Figure 1b). The results of molecular docking indicated that the inter-domain linker of the UxuR protein interacted with models of every selected carbohydrate types at all time points sampled from the MD trajectory (Figure 1c). Although the binding affinity in most cases did not exceed the average level of random surface binding by more than three standard deviation (StD), at least three molecules of each carbohydrate were consistently positioned in the inter-domain linker region. The highest binding affinity, which was statistically significant against the background, was observed for ligands bound to the linker region of the 50 ns model in the first round of docking. However, the average occupancy of the linker progressively increased in models from later simulation times. Specifically, the linker in the 90 ns UxuR model was associated with 6-9 sugar molecules, while the 100 ns model interacted with 8-9 carbohydrates (Figure 1c). Thus, despite the low binding affinity of most sugars for the linker region, which is typical for non-specific interaction, we observed its clear and predominant occupancy by multiple molecules, which, in contrary, is a hallmark of specific binding.
Figure 2 shows the distribution of D-fructuronate and D-glucose on the surface of the UxuR monomer. Of them, D-fructuronate (Figure 2a) relieves UxuR-mediated repression [76], while D-glucose (Figure 2d) can integrate UxuR into regulatory networks controlling catabolite repression [77]. All ten molecules of each ligand used in molecular docking are shown on the 100 ns molecular model of UxuR. The ligands interacted with the linker region formed clusters (Figure 2a and d), which are potentially stabilized by hydrogen bonds (Figure 2b and c), predicted by Protein-Ligand Interaction Profiler [72].
While the carbohydrate clusters of D-fructuronate and D-glucose were of almost similar size (7 and 8 molecules, respectively, Figure 1c), their topologies within the protein’s inter-domain space differed (Figure 2a,d). In particular, the D-fructuronate molecule with the highest affinity for UxuR (ball model in Figure 2a) was at the hydrogen bond (H-bond) distance from Gln80, Asn81, Thr82, Asp83 and Ser84 (Figure 2b), while the highest-affinity D-glucose molecule, from its predicted location, could form H-bonds only with Ser79, Asp92, and Gln170. Moreover, within these clusters H-bonds can be formed not only between the ligand and the protein but also between different ligand molecules. For instance, the interaction of D-fructuronate with UxuR in the 7th docking round may be stabilized by H-bonds formed with D-fructuronates attached to the protein during the 3rd (fruc3) and 6th (fruc6) rounds (Figure 2b). Similarly, in the glucose binding site, glu3 is stabilized by glu2, which may facilitate the binding of glu7 and glu10 (Figure 2c).
Eight molecules of D-glucuronate (an isomer of D-fructuronate), occupied a nearly identical region in the UxuR inter-domain space (Figure 3a). Among those, the molecules attached in the second (gluc2), third (gluc3) and the sixth (gluc6) rounds of docking (Figure 1d) could form H-bonds with the D-glucuronate molecule that exhibited the highest affinity (gluc4) (Figure 3b). In the case of D-galacturonate (a stereoisomer of D-glucuronate), the network of potential H-bonds was the most diverse (Figure 3c). While Asn88, Asp92 and Gln170 interacted with both stereoisomers, the D-galacturonate molecule from the second docking round can also form H-bonds with Ser79, Ser84 and six other D-galacturonate molecules (Figure 3c,d).
Thus, although the interactions of D-glucose and the three hexuronate isomers with the UxuR inter-domain linker occured at near-background efficiency levels, molecular docking revealed some specific preferences and showed that ligand-ligand interactions can stabilize complex formation. As sequential docking simulates a gradually increasing ligand concentration, the observed clustering suggests that similar structuring in vivo could form structures that restrict the linker’s conformational mobility, thereby stabilizing it in a functionally significant state or, vice versa, limiting intermolecular interactions.
2.1.2. Interacting with the UxuR Dimer, Carbohydrates do not Form Clusters with Inter-Domain Linkers, but Connect them to the α-Helix of the Effector Domain.
In the UxuR dimer, the inter-domain linkers are located in close proximity to each other (Figure 4a–c), creating a spatial hindrance that limits ligand access to these regions. Consequently, a greater number of carbohydrates bind to other surfaces of the dimer (Figure 4d) compared to the monomer (Figure 1d). A primary binding site was the region where the alpha-helices of the C-terminal domain converge, which corresponds to the entrance of the previously identified carbohydrate-binding pocket [68,78]. This site predominantly interacted with ligands in the first rounds of molecular docking and typically exhibited the highest affinities of binding (brawn ΔG values in Figure 4d). Such interaction pattern was observed across protein models derived from various time points of the dynamic trajectory and was most pronounced for the complexes formed with the 100 ns UxuR model (Figure 4d).
Although fewer ligands occupied inter-domain linkers in the UxuR dimer than in its monomer, their average binding affinity for all carbohydrates was slightly higher (Figure 2d and Figure 4d). Moreover, several ligands interacting with the 90 ns and 100 ns models of the UxuR dimer exhibited affinities greater than those bound to the potential effector binding site (indicated by red and brawn ΔG values in Figure 4d, respectively). The highest affinity to this region demonstrated D-galacturonate attached to the 100 ns model of the UxuR dimer at the third round of molecular docking (Figure 4d and Figure 5). Notable, that all ligands bound to this specific site are able to form hydrogen bonds with amino acid residues from both the inter-domain linker and the C-terminal domain of the protein (Figure 5a,b), representing a new mode of carbohydrate interaction with the flexible linkers of UxuR. The observation that such interaction occurred exclusively in protein models from the late stages of the molecular dynamics trajectory suggests that the competent binding site did not appear spontaneously. Instead, its formation likely required a sequential process of conformational selection, which the simulation successfully captured for all ligands.
The limited space at the dimer’s inter-domain linker interface likely explains the absence of active ligand clustering in this region. Nevertheless, an ability of hydrogen bonds formation between ligands attached during different molecular docking rounds was detected. Figure 5b, in particular, shows the arrangement of two D-galacturonate molecules, where first (gal3) bridges the linker and the CTD α-helix, while also facilitating the attachment of a second D-galacturonate molecule (Gal6) to the linker region. Thus, the functional repertoire of inter-domain linkers may extend beyond their ability to adopt specific conformations depending on the bound partner. Multi-molecular clustering of ligands could potentially influence dimerization dynamics, while intramolecular bridging could modulate relative orientation of the domains. Although interactions with linkers generally exhibited low affinity, the spatial proximity of Asn81 to Leu167, Gln170, Ser171 and Gln173 from the UxuR α-helix could enable not only strong but also specific carbohydrate binding for at least D-galacturonate (Figure 5b).
2.2. Similarities and Diversity of D-Glucose Binding Modes with Unstructured Linkers of Other Transcription Factors
In this phase of the study, we investigated whether unstructured linkers from other TFs could participate in potentially functional interactions with carbons. We selected five proteins for modeling, molecular dynamics simulations and subsequent docking as outlined in the Materials and Methods. The selection included two TFs from E. coli (GntR and FarR) and three from Bacillus subtilis (B. subtilis) (AraR, NagR, and YadK). None of the tested carbohydrates is specific effectors for these TFs. However, existing data suggest that the effector-binding sites of many TFs can accommodate small molecules even from different metabolic pathways [83] a feature we anticipated would be more pronounced in IDRs. Because our data on UxuR-ligand complexes showed no statistically significant difference in the interaction with different sugars, we used only D-glucose as a universal probe for linker accessibility. All five selected proteins function as dimers, but we evaluated ligand interactions only with monomeric forms to minimize the likelihood of shielding the linkers at the dimer interface, as it was observed for the UxuR dimer.
2.2.1. The Linker-Separated Subdomains of E. coli GntR and B. subtilis AraR, Formed High-Affinity Binding Pockets for D-Glucose but Differed in its Non-Specific Binding
The GntR repressor of E. coli regulates several operons involved in the uptake of D-gluconate and its catabolism via the Entner-Doudoroff pathway. Similarly, the AraR repressor controls arabinose metabolism in B. subtilis. Both transcription factors contain winged helix-turn-helix DNA-binding domains, which are connected by linkers of 14-15 amino acids to the C-terminal effector-binding domains. In both proteins, E-domains consist of two subdomains separated by short linkers of 3–10 amino acids. The E. coli GntR CTD comprises two subdomains, each with a central parallel β-sheet surrounded by four α-helices (Figure 6a and UniProt model P0ACP5) [82]. Analysis of ligand interactions revealed that, on average, only 2.3 molecules of D-glucose interacted with the inter-domain linker of GntR (black ΔG-values in Figure 6b). Nevertheless, the binding affinity in some models significantly exceeded the level of random attachment to the protein surface (Figure 6b). The UxuR monomer, which has the inter-domain linker more than twice as long, showed a similar occupancy of D-glucose molecules per residue (5.8 molecules per 34 amino acids). However, high-affinity binding with UxuR was observed only for its physiological effectors, D-fructuronate and D-glucuronate (Figure 1d). Thus, the inter-domain linker of GntR possesses a more specific interaction site for D-glucose than the linker of UxuR.
The site with the highest affinity for D-glucose was found in the cavity formed between the two subdomains of the GntR CTD (Figure 6a). The side chains of amino acid residues from the linkers connecting secondary structure elements are exposed within this cavity. Ligand binding to these residues bridges modules that are remote from each other in the protein primary sequence (Figure 6c). Although the cavity itself is relatively small and accommodated a maximum of two ligands (20-ns structure), an average of 3.5 D-glucose molecules were found attached to the inter-subdomain area outside the cavity (red circle in Figure 6a and brown ΔG-values in Figure 6b). The observed affinity for D-glucose in this region may have evolved to capture and retain D-glucose, which if shifted to cavity could potentially act as a structuring factor (Figure 6c).
The effector-binding domain of the AraR protein is organized very similarly to that of GntR (Figure 7a). Although slightly larger (282 versus 267 residues), this domain also comprises two subdomains, each containing a central parallel β-sheet sandwiched by four α-helices. As with GntR, these subdomains form a cavity, where D-glucose bound to AraR with the highest affinity (Figure 7a,b). The amino acid residues forming hydrogen bonds with the ligand molecules within this cavity were located in unstructured regions that connect remote structural modules of the protein (Figure 7c). Furthermore, in six of the seven models, D-glucose was also found at the protein surface outside the inter-subdomain pocket (brown ΔG-values in Figure 7b). However, unlike UxuR and GntR, the low-affinity complexes with D-glucose in AraR formed at sites that do not correspond to its unstructured linkers outside the main pocket (Figure 7b). This indicates that protein chain flexibility alone is insufficient to ensure carbohydrate binding.
It has long been known that the B. subtilis AraR protein has a “mosaic structure” combining the DNA-binding domain of the GntR family TFs with an effector-binding domain homologous to regulators of a GalR/LacI family [62,84]. This fundamental architectural difference could explain the dramatic contrast in how D-glucose interacted with the unstructured linkers of GntR and AraR outside the main pocket (Figure 6b and Figure 7b). However, a pairwise alignment of the E-domains from E. coli GntR and B. subtilis AraR, performed using the Needleman-Wunsch algorithm [85], unexpectedly revealed a higher degree of mutual homology between GntR of E. coli and AraR of B. subtilis (score 243.5) than between the GalR/LacI and AraR (scores 195.5 and 174.0, respectively). Given that the E-domains of both GntR and AraR have low homology scores (ranging from 11.0 to 44.5) to the CTDs of canonical GntR family members such as FarR (E. coli) and NagR (B. subtilis) [65], we propose that the E. coli GntR itself represents another example of a rare modular structure combining functional domains from different evolutionary origins.
2.2.2. The Patterns of the NagR and FarR Interaction with D-glucose Correlate with the Structure of the Polypeptide Chain Forming a High-Affinity Binding Site in the CTD
The NagR (YvoA) protein of B. Subtilis and the FarR (MngR) transcription factor of E. coli are both established members of the GntR family with a HutC-type effector-binding domain. The NagR repressor controls genes responsible for transporting and metabolizing the amino sugar N-acetylglucosamine (GlcNAc) [86]. Its activity is modulated by the effector molecules glucosamine-6-phosphate and/or N-acetylglucosamine-6-phosphate [87]. In contrast, the FarR (MngR) TF regulates only two genes involved in utilizing 2-O-α-mannosyl-D-glycerate (mngA and mngB) [88], however, its potential effector has not yet been identified. Structurally, both proteins possess highly similar 70-residue wHTH DNA-binding domains (alignment score 133). Their E-domains are also highly homologous (alignment score 151). They are built around a central β-sheet of six antiparallel strands, with four short α-helices formed by the linkers connecting the β-strands. At the domain junction, both proteins have a parallel β-sheet formed by the last β-strand from the NTD and a strand from the CTD tail. The length of the polypeptide chains connecting these sheets with the central antiparallel sheets is also similar in both proteins (17 and 18 residues, respectively). Both these linkers contain a short α-helix, which is two residues longer in NagR, spanning Phe89 to Ser95 (Figure 8a and PDB: 2WV0). Given the presence of multiple flexible linkers in the NagR structure, such as loops between secondary structure modules and unstructured N- and/or C-terminal tails, it was not surprising to reveal their frequent interaction with D-glucose (black ΔG values in Figure 8b). Over 10 rounds of molecular docking, we revealed their occupancy by an average of 5.8 ± 2.2 D-glucose molecules.
However, only three ligands, bound to the protein models at 40, 50 and 120 ns, exhibited the binding affinity to linker regions significantly above the background level of -4.46 ± 0.25 kcal/mol. The preferred interaction site was a cavity bordered on one side by the central antiparallel β-sheet of the CTD and by the unstructured polypeptide chain with short α-helix on the other (Figure 8a and red ΔG values in Figure 8b). This cavity was able to accommodate two ligand molecules (30 ns model). Predicted H-bonds were predominantly formed with the residues of β-sheet (Arg135, Gln183, Arg211, Tyr228 and Val236), but positioning of D-glucose allowed simultaneous interaction with Asp92 located on the short α-helix within the linker (Figure 8a,c). Of particular importance is a precise correspondence of this high-affinity D-glucose binding site to the X-ray established effector-binding pocket [87], which accommodates glucosamine-6-phosphate and N-acetylglucosamine-6-phosphate via hydrogen bonds formed with Arg135, Gln183, and Tyr228 along with other residues like Thr87 and Phe89 from the flexible linker.
As in previous cases, models from different points in the MD trajectory varied in their ability to form complexes with D-glucose, both in interaction pattern and affinity. Surprisingly, however, for NagR, molecular docking revealed high-affinity D-glucose binding at the effector site only in models from the early stages of the MD simulation (red ΔG values in Figure 8b). Extending the simulation beyond 100 ns we revealed no trend toward increased D-glucose binding at this site. If the dynamics simulation reflects stabilization into a conformation specific for binding of natural inducers, this may indicate that protein maturation reduces the accessibility of the effector site to a nonspecific ligand.
Although the FarR protein of E. coli has a structural organization highly similar to that of NagR (Figure 9a and P13669 model in UniProt), its interaction pattern with D-glucose was significantly different (Figure 9b). The primary distinction is a markedly higher D-glucose binding at the potential ligand-binding pocket. Over 10 rounds of molecular docking, this pocket sequestered an average 2.60 ± 0.83 D-glucose molecules (Figure 9b). Consequently, fewer D-glucose molecules (2.53 ± 1.55) interacted with unstructured linkers in other protein regions compared to NagR (black ΔG-values in Figure 8b and Figure 9b). Unlike NagR, all structural models of the FarR recruited D-glucose to this pocket (red ΔG-values in Figure 9b) and the amino acid residues within this pocket that formed H-bonds with D-glucose were predominantly from the flexible linker (Asp77, Ile78, Gln80, Thr82) (Figure 9c).
The high-affinity cavity was sufficiently large to accommodate up to four ligand molecules (20 ns model), which often formed H-bonds with each other. Similar to the NagR effector-binding site, the D-glucose molecules attached to the flexible linker of FarR simultaneously interacted with Lys204 and/or Gln178 from two β-strands (Figure 9c). This could mediate conformational transitions affecting relative orientation of N-terminal and C-terminal domains, while the distinct D-glucose interaction patterns observed for FarR and NagR suggest that FarR may be specifically sensitive to the presence of D-glucose.
2.2.3. A putative Transcription Factor YydK with structural Homology to NagR and FarR Interacted with D-Glucose in a NagR-like Manner
The YydK protein from B. subtilis in our set represents an uncharacterized transcription factor with known X-ray structure (PDB: 3bwg). We selected YydK for analysis due to its close structural (Figure 10a) and sequence homology with both NagR and FarR. Similar to these proteins, the winged helix-turn-helix N-terminal domain of YydK is connected by 15 residues linker (Leu77–Asn91) to a HutC-type CTD containing a six-stranded antiparallel β-sheet and four α-helices. This linker is unstructured in the YydK model of UniProt (ID Q45591), in the crystal structure (PDB: 3bwg), and was truncated in our molecular models (Figure 10e).
Over 10 rounds of molecular docking, the flexible linkers outside the potential effector-binding site were occupied by an average of 4.21 ± 1.96 D-glucose molecules (black ΔG values, Figure 10b). This occupancy is intermediate between that of NagR (5.8 ± 2.2) and FarR (2.53 ± 1.55). However, in contrast to FarR but similar to NagR, the CTD pocket of YydK sequestered D-glucose in only three of its structural models (red ΔG values, Figure 10b). The amino acid residues within this pocket that interacted with D-glucose were predominantly located on β-sheet strands (Arg125, Arg127, Glu137, Tyr140, Ile158 and Asp117) (Figure 10c). The interaction pattern, characterized by only a single bridging hydrogen bond with linker residue Leu77, was also observed in NagR (Figure 10c). To check the reproducibility of lower occupancy of the YydK pocket compared to FarR, we repeated the molecular docking using structural models from an independent 70 ns simulation (Figure 10c). As expected, the external linker regions in these new models adopted different, dynamically variable conformations, which altered the surface distribution of D-glucose (Figure 10b,d). However, the pattern of D-glucose interaction with the effector-binding site remained similar to that of NagR (Figure 8a).
Thus, while the inter-domain linker in all FarR models actively participated in the virtual ligand binding primarily in the initial docking rounds, similarly positioned linkers in NagR and YydK exhibited this capability only during the early stages of MD simulation. A key distinction in these interactions is the location of hydrogen bonding: with NagR and YydK, bonds preferentially formed with structured domain regions, primarily β-sheets, whereas with FarR, most bonds were formed with amino acids on the interdomain linker itself.
3. Discussion
The study of proteins with intrinsically disordered regions (IDRs) is an emerging frontier in protein science and molecular biology. Although existing research is predominantly focused on eukaryotes [8,89,90], bioinformatic studies have identified IDRs across various functional classes of prokaryotic proteins [53], including transcription regulators, for which the occurrence of disordered regions was found to be explicitly lower compared to eukaryotic transcription factors [15,90]. This discrepancy is reasonably explained by the greater complexity of transcriptional regulation in higher organisms. Nevertheless, growing evidence demonstrates that IDRs are functional in bacterial regulators [6,7,70] and this study addressed a key aspect of that functionality: the accessibility of IDRs in bacterial transcription factors to regulatory signals from potential effector molecules.
Molecular dynamic simulations [91] were employed to assess the plasticity of unstructured linkers and to generate conformational ensembles for molecular docking. In the UxuR monomer, the inter-domain linker underwent rearrangements characterized by the transient formation and dissolution of short α-helix and β-strand (Figure 1b). A similar variability in β-strand formation was also observed in the UxuR dimer, while the α-helix formed only once in the 100 ns model. However, considering this variance as a virtual structural sampling, we found no correlation between the presence of these transient structured modules and either linker occupancy by carbohydrates or their binding affinity. We did not detect extensive interactions between D-glucose and the AraR inter-subdomain linkers, which remained unstructured in all models. Moreover, among the three structurally similar proteins (FarR, NagR, and YydK), only the inter-domain linker of FarR exhibited preferential and reproducible formation of statistically significant complexes with D-glucose. Therefore, the mere presence of an unstructured region in the polypeptide chain is an insufficient criterion to predict or exclude its capacity for ligand binding.
Nevertheless, for every protein examined, molecular docking revealed conformations of the weakly structured or unstructured regions that were able to form complexes with binding affinities comparable to the specific interaction of carbohydrates with the effector-binding sites. The affinity of D-galacturonate for the UxuR inter-domain linker (-6.8 kcal/mol) was even higher than that of its natural ligands, D-fructuronate and D-glucuronate, for the effector-binding pocket of the UxuR dimer (-6.7 ≤ ΔG ≤ -5.4 kcal/mol). In AraR and GntR, the highest-affinity complexes with D-glucose (-6.4 kcal/mol) were localized in their inter-subdomain spaces, which consist primarily of short 3-10-residue linkers connecting the protein’s structural modules. According to Protein-Ligand Interaction Profiler (PLIP) analysis [72], D-glucose molecules can form seven hydrogen bonds within the AraR CTD pocket, all exclusively with residues from four different linkers. In GntR, only one of the seven H-bonds predicted for the preferred ligand involved an α-helix residue (Lys197), while the rest belonged to the unstructured protein chains. Given that all tested carbohydrates can form extensive hydrogen bond networks with polar/charged residues or the protein backbone, their interactions with the linkers may serve as an important structuring factor that induces or modulates functional conformations.
It is unsurprising that of the 32 hydrogen bonds predicted by the PLIP algorithm in the highest-affinity carbohydrate complexes with unstructured linkers, 25 involved optimal H-bond formers: asparagine, serine, aspartic acid and threonine. In contrast, H-bonds with glutamine and glutamic acid within high-affinity pockets were primarily associated with α-helices and β-strands. In unstructured regions only two H-bonds involved glutamine and one was formed with glutamic acid. This asymmetry between Asn/Asp and Gln/Glu suggests a certain binding “specificity” and corroborates with the underrepresentation of glutamine (and, to a lesser extent, of glutamic acid) in small-molecule metabolite binding to 45 E. coli transcription factors [92]. Given that Gln/Glu possess longer side chains than Asn/Asp, these observations suggest that successful ligands binding within unstructured regions may encounter a size constraint, likely mirroring a geometric selectivity inherent to natural effector binding sites.
Typically, the transient conformations of flexible protein regions do not allow a ligand to form a sufficiently stable complex. Certain conditions, like dynamic conformational adaptation or initial low-affinity ligand interaction, are often required to achieve a binding-competent state. It is therefore surprising that in the UxuR monomer, even background ligand interactions can promote the formation of extended clusters at the dimerization interface. The absence of such clusters in the dimer suggests that a bulky cluster could hinder the UxuR maturation process. As our simulations of multi-ligand binding reflect the natural excess of cellular carbohydrates, these results suggest the hypothesis that UxuR is sensitive to their intracellular concentrations.
On the other hand, when the protein is in a conformation potentially prone to forming a higher-affinity complex, even the accidental formation of such a complex could lock the protein into a functionally significant conformation. The multiple ligand binding observed in the case of the UxuR monomer within the inter-domain linker region, involving the active formation of hydrogen bonds not only with amino acid residues of the protein chain but also with each other, is likely to provide an additional stabilizing effect.
In our docking experiments, where four ligand types were screened against UxuR structures corresponding to different points of the MD trajectory, the different ligands formed their highest-affinity and most populated complexes with different protein conformations. This suggest a biological model where the four carbohydrates compete for the linker conformations that are most favorable for high-affinity binding. Consequently, a regulatory circuit may exist in which protein activity is controlled by the relative concentrations of these carbohydrates. For example, high intracellular concentration of D-glucose, which is not the primary effector for UxuR, could allow it to bind not only to the inter-domain linkers but also to compete with primary effectors, D-fructuronate and D-glucuronate, for the effector site in the C-terminal domain. This would enable glucose to suppress the activation of the Ashwell and Entner-Doudoroff pathways even when primary effectors are abundant. Such a mechanism could represent a significant, so far unrecognized component of the catabolite repression orchestrated by cellular regulatory networks in response to D-glucose.
Extending this regulatory principle to other bacterial proteins with unstructured ligand-binding regions suggests the existence of overlapping networks sensitive to the concentration of key metabolites. The formation of low-affinity complexes with transient interaction sites can maintain a baseline protein activity, while inherent conformational flexibility enables rapid activation upon the arrival of a specific effector. The effectiveness of such networks likely derives from fundamental biophysical properties of the protein chain, minimizing the need for precise evolutionary adaptation to individual ligands.
4. Materials and Methods
4.1. Structural Models of Carbohydrates Used in the Study
4.1. Structural Models of Transcription Factors Used in the Study
The UxuR protein sequence from E. coli K-12 MG1655 (NC_000913) [95] was obtained from the KEGG [97] (entry T00007, K13637). It was selected due to its partially characterized ligand-binding properties [68,75,77,78,83)]. Both monomeric and dimeric three-dimensional structures of UxuR were modeled using the crystal structure of the GntR protein from Streptococcus agalactiae (PDB: 6AZ6) as a template. To select additional transcription factors for analysis, we retrieved the amino acid sequences of 27 candidate proteins from E. coli and B. subtilis using KEGG database [96]. A three-dimensional structure for each candidate was generated using the SWISS-MODEL online server [67,68,69]. Five TFs were subsequently chosen based on two criteria: (1) the presence of unstructured inter-domain linkers, a hallmark of GntR-family regulators, and (2) the quality and completeness of the predicted models, requiring no sequence breaks or major deletions. The selected set included GntR and FarR (MngR) from E. coli K-12 MG1655 (NC_000913) [95], NagR (YvoA) and YydK from B. subtilis subsp. subtilis str. 168 (NC_000964.3) [97] and the AraR protein from B. subtilis strain FX-1 (CP004019) [98].
Among the selected transcription factors, only NagR and YydK have experimentally determined crystal structores of their apo-forms, with resolutions of 2.051 Å (PDB: 2WV0) and 2.09 Å resolution (PDB: 3BWG), respectively. However, the YydK structure contains an engineered expression tag at its N-terminus. NagR has also been co-crystalized with its effector molecules, N-acetylglucosamine-6-phosphate (PDB: 4U0W, 2.001 Å) and glucosamine-6-phosphate (PDB: 4U0V, 2.051 Å). The availability of these ligand-bound structures allowed us to compare our molecular docking results with the natural effector-binding pocket. For GntR, crystal structures are available from three different organisms, though each has limitations. Structures from Thermotoga maritima (PDB: 3FMS, 2.2 Å) [99] and Chromobacterium violacium (PDB: 3H5O, 2.3 Å) contain engineered expression tags, and the 3FMS structure also bears three point substitutions. A structure of a putative GntR from Rhodococcus species RHA1, crystallized from native protein (2.2 Å), possesses only two acetate molecules in its effector-binding site, but features an antiparallel β-sheet within its 16-residue inter-domain linker. For FarR and AraR only AlphaFold-predicted models are available (UniProt entries P13669 and P96711, respectively) [56]. Therefore, to minimize potential artifacts from the heterogeneous experimental conditions and to ensure a consistent structural alignment across all models, we used predicted models for all proteins in this study. The structures of GntR and FarR from E. coli K-12 MG1655 (NC_000913) [95] were modeled using the SWISS model P0ACP6 and PDB template 4U0W, respectively. NagR and YydK from B. subtilis subsp. subtilis str. 168 (NC_000964.3) [98] were modeled using PDB templates 2WV0 and 3BWG, respectively. The AraR protein from B. subtilis strain FX-1 (CP004019) [98] was obtained based on the AlphaFold [56] model D4G1L9 as a template.
4.2. Molecular Dynamics Simulation
The obtained structures were subjected to MD simulations using the Open MMZephyr software package (v. 2.0.3) [100] to assess atomic positional fluctuations and to select conformers for docking. Simulations were performed at 310.15 K using the Amber96 force field and an explicit solvent model (“accurate water”). MD simulations for the UxuR monomer and dimer were carried out independently. A total of 78 structural models, comprising 6 monomers and the UxuR dimer, were collected from subsequent time points along the MD trajectories. For molecular docking, structures were extracted at 10 ns intervals within the pre-optimized time range for each protein (maximal range: 10–150 ns). This sampling strategy enabled the visualization of flexibility in the proteins’ unstructured linkers and allowed comparison of ligand binding patterns across the simulation trajectory.
4.3. Flexible Molecular Docking
Flexible molecular docking was performed using the AutoDock Vina software package [101]. All potentially mobile bonds in the ligands were treated as flexible. For each ligand, preferential binding positions were identified and the corresponding binding affinity was calculated (reported as ΔG in kcal/mol). A sequential docking protocol was employed for all ligands assessed and all 78 protein models. In each subsequent round, the ligand with the highest affinity from the previous round was added to the target protein structure, and the resulting complex was used as the new input for the next docking round. This iterative process was repeated ten times. The specificity of ligand binding was evaluated based on two factors: (1) the calculated binding affinity (ΔG) with unstructured polypeptide chains and (2) the difference in ΔG from ligands binding non-specifically to other protein surfaces. Docking results were visualized using AutoDockTools (v. 1.5.6) [102]. Potential hydrogen bonds in the protein-ligand complexes were predicted and visualized using LigPlot+ (v. 2.2.8) [72].
4.4. Statistics
To assess the statistical significance of carbohydrate interactions with unstructured regions of the studied TF, we calculated the average affinity of nonspecific ligand interaction with protein models outside the linker regions and effector-binding pockets. The binding affinities obtained for protein conformers from different simulation time points were pooled for each ligand. This yielded between 17 and 26 ΔG values for the UxuR monomer and dimer (5 time points). The number of glucose molecules involved in nonspecific interactions with other transcription factors ranged from 36 (GntR, 10 time points) to 74 (YydK, 14 time points). This data provided a baseline interaction level for all protein-carbohydrate pairs, allowing us to identify complexes with binding affinities more than 3 standard deviations above this mean (p-value < 0.05). Pairwise protein sequence alignments were performed using the Needleman-Wunsch algorithm [85] implemented in the European Molecular Biology Open Software Suite (EMBOSS).
Author Contributions
Conceptualization, Y.A.P. methodology, Y.A.P.; software, Y.A.P.; validation, O.N.O.; formal analysis, O.N.O.; investigation, Y.A.P.; resources, O.N.O.; data curation, O.N.O.; writing—original draft preparation, Y.A.P.; writing—review and editing, O.N.O.; visualization, Y.A.P., O.N.O.; supervision, NA; project administration, Y.A.P.; funding acquisition, O.N.O. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation State No. 075-00607-25-02.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Browning, D.F.; Busby, S.J. Local and global regulation of transcription initiation in bacteria. Nat Rev Microbiol 2016, 14, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Busby, S.J.W. Transcription activation in bacteria: ancient and modern. Microbiology (Reading) 2019, 165, 386–395. [Google Scholar] [CrossRef]
- Flores-Bautista, E.; Hernandez-Guerrero, R.; Huerta-Saquero, A.; Tenorio-Salgado, S.; Rivera-Gomez, N.; Romero, A.; Ibarra, J.A.; Perez-Rueda, E. Deciphering the functional diversity of DNA-binding transcription factors in Bacteria and Archaea organisms. PLoS One 2020, 15, e0237135. [Google Scholar] [CrossRef] [PubMed]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor-DNA binding: beyond binding site motifs. Curr Opin Genet Dev 2017, 43, 110–119. [Google Scholar] [CrossRef]
- Schleif, R.F. Modulation of DNA binding by gene-specific transcription factors. Biochemistry 2013, 52, 6755–6765. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Schleif, R. Helical Behavior of the Interdomain Linker of the Escherichia coli AraC Protein. Biochemistry 2019, 58, 2867–2874. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Y.; Richet, E.; Han, Z.; Chai, J. Structural basis for negative regulation of the Escherichia coli maltose system. Nat Commun 2023, 14, 4925. [Google Scholar] [CrossRef]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int J Mol Sci 2022, 23, 14050. [Google Scholar] [CrossRef]
- Davey, N.E. The functional importance of structure in unstructured protein regions. Curr Opin Struct Biol 2019, 56, 155–163. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; Kim, P.M.; Kriwacki, R.W.; Oldfield, C.J.; Pappu, R.V.; Tompa, P.; Uversky, V.N.; Wright, P.E.; Babu, M.M. Classification of intrinsically disordered regions and proteins. Chem Rev 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Sharma, R; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett 2015, 589, 2533–2542. [Google Scholar] [CrossRef]
- Borgia, A.; Borgia, M. B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; Kragelund, B.B.; Best, R.B.; Schuler, B. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555(7694), 61–66. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. ; Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol Biosyst 2016, 12, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.Q.; Otting, G.; Furukubo-Tokunaga, K.; Affolter, M.; Gehring, W.J.; Wüthrich, K. NMR structure determination reveals that the homeodomain is connected through a flexible linker to the main body in the Drosophila antennapedia protein. Proc. Natl. Acad. Sci. USA 1992, 89, 10738–10742. [Google Scholar] [CrossRef]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: emerging interaction specialists. Curr Opin Struct Biol 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Pérez, Y.; Maffei, M.; Igea, A.; Amata, I.; Gairí, M.; Nebreda, A.R.; Bernadó, P.; Pons, M. Lipid binding by the Unique and SH3 domains of c-Src suggests a new regulatory mechanism. Sci Rep 2013, 3, 1295. [Google Scholar] [CrossRef]
- Fuxreiter, M. Classifying the Binding Modes of Disordered Proteins. Int J Mol Sci 2020, 21, 8615. [Google Scholar] [CrossRef]
- Olsen, J.; Teilum, K.; Kragelund, B.B. Behaviour of intrinsically disordered proteins in protein-protein complexes with an emphasis on fuzziness. Cell Mol Life Sci 2017, 74, 3175–3183. [Google Scholar] [CrossRef]
- Miskei, M.; Horvath, A.; Vendruscolo, M.; Fuxreiter, M. Sequence-Based Prediction of Fuzzy Protein Interactions. J Mol Biol 2020, 432, 2289–2303. [Google Scholar] [CrossRef]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev 2016, 116, 6391–6423. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, H.X. Protein allostery and conformationa dynamics. Chem Rev 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, C.; Wang, T.; Quan, J. Structural basis for the inhibition of polo-like kinase 1. Nat Struct Mol Biol 2013, 20, 1047–1053. [Google Scholar] [CrossRef]
- Liu, J.; Nussinov, R. The mechanism of ubiquitination in the cullin-RING E3 ligase machinery: conformational control of substrate orientation. PLoS Comput Biol 2009, 5, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA 2001, 108, 6987–6992. [Google Scholar] [CrossRef]
- Gokhale, R. S.; Khosla, C. Role of linkers in communication between protein modules. Curr Opin Chem Biol 2000, 4, 22–27. [Google Scholar] [CrossRef]
- Ma, B.; Tsai, C-J. ; Haliloğlu, T.; Nussinov, R. Dynamic Allostery: Linkers Are Not Merely Flexible. Structure 2011, 19, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol 2009, 19, 31–38. [Google Scholar] [CrossRef]
- Tsai, C.J.; Ma, B.; Kumar, S.; Son, H. W. L.; Nussinov, R. Protein folding: binding of conformationally fluctuating building blocks via population selection. Crit. Rev. Biochem. Mol. Biol 2001, 36(5), 399–433. [Google Scholar] [CrossRef]
- Hammes, G.G.; Chang, Y.C.; Oas, T.G. Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA. 2009, 106(33), 13737–13741. [Google Scholar] [CrossRef]
- Freiberger, M.I.; Wolynes, P.G.; Ferreiro, D.U.; Fuxreiter, M. Frustration in Fuzzy Protein Complexes Leads to Interaction Versatility. J Phys Chem B 2021, 125, 2513–2520. [Google Scholar] [CrossRef]
- Brüschweiler, S.; Schanda, P.; Kloiber, K.; Brutscher, B.; Kontaxis, G.; Konrat, R.; Tollinger. M. Direct observation of the dynamic process underlying allosteric signal transmission. J. Am. Chem. Soc 2009, 131, 3063–3068. [Google Scholar] [CrossRef]
- Lei, M.; Velos, J.; Gardino, A.; Kivenson, A.; Karplus, M.; Kern, D. Segmented transition pathway of the signaling protein nitrogen regulatory protein C. J Mol Biol 2009, 392, 823–836. [Google Scholar] [CrossRef]
- Friedmann, D.R.; Wilson, J.J.; Kovall, R. A RAM-induced allostery facilitates assembly of a notch pathway active transcription complex. J. Biol. Chem 2008, 283, 14781–14791. [Google Scholar] [CrossRef] [PubMed]
- Kar, G.; Keskin, O.; Gursoy, A.; Nussinov, R. Allostery and population shift in drug discovery. Curr. Opin. Pharmacol 2010, 10, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Miller, L.J. Seven transmembrane receptors as shape-shifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev 2010, 62, 265–304. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins 2004, 57, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nussinov, R. Allostery: An Overview of Its History, Concepts, Methods, and Applications. PLoS Comput Biol 2016, 12, e1004966. [Google Scholar] [CrossRef]
- Tobi, D.; Bahar, I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA 2005, 102, 18908–18913. [Google Scholar] [CrossRef]
- Nussinov, R.; Liu, Y.; Zhang, W.; Jang, H. Protein conformational ensembles in function: roles and mechanisms. RSC Chem Biol 2023, 4, 850–864. [Google Scholar] [CrossRef]
- Clark, J.J.; Benson, M.; Smith, R.D.; Carlson, H.A. Inherent versus induced protein flexibility: Comparisons within and between apo and holo structures. PLoS Comput Biol 2019, 15, e1006705. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput Biol 2016, 12, e1005067. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D. During transitions proteins make fleeting bonds. Cell 2009, 139, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Gardino, A.K.; Villali, J.; Kivenson, A.; Lei, M.; Liu, C.F.; Steindel, P.; Eisenmesser, E.Z.; Labeikovsky, W.; Wolf-Watz, M.; Clarkson, M.W.; Kern, D. Transient non-native hydrogen bonds promote activation of a signaling protein. Cell 2009, 139, 1109–1118. [Google Scholar] [CrossRef]
- del Sol, A.; Tsai, C.J.; Ma, B.; Nussinov, R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure 2009, 17, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Arya, G.; Mishra, R.; Singla, S.; Pratap, A.; Upadhayay, K.; Sharma, M.; Chaba, R. ; Molecular mechanisms underlying allosteric behavior of Escherichia coli DgoR, a GntR/FadR family transcriptional regulator. Nucleic Acids Res 2025, 53, gkae1299. [Google Scholar] [CrossRef]
- Zhang, J.; Sapienza, P.J.; Ke, H.; Chang, A.; Hengel, S.R.; Wang, H.; Phillips, G.N.; Lee, A.L. Crystallographic and nuclear magnetic resonance evaluation of the impact of peptide binding to the second PDZ domain of protein tyrosine phosphatase 1E. Biochemistry 2010, 49, 9280–9291. [Google Scholar] [CrossRef]
- Tsai, C.J.; del Sol, A.; Nussinov, R. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol 2008, 378, 1–11. [Google Scholar] [CrossRef]
- Kenakin, T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J. Recept. Signal Transduct. Res 2010, 30, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Pavlović-Lažetić, G.M.; Mitić, N.S.; Kovačević, J.J.; Obradović, Z.; Malkov, S.N.; Beljanski, M.V. Bioinformatics analysis of disordered proteins in prokaryotes. BMC Bioinformatics 2011, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Piovesan, D.; Heller, G. T.; Bonomi, M.; Kukic, P.; Camilloni, C.; Fuxreiter, M.; Dosztanyi, Z.; Pappu, R.V; Babu, M.M.; Longhi, S.; Tompa, P.; Dunker, A.K.; Uversky, V.N; Tosatto, S.C.E; Vendruscolo, M. Simultaneous quantification of protein order and disorder. Nat. Chem. Biol 2017, 13, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Morris, O.M.; Torpey, J.H.; Isaacson, R.L. Intrinsically disordered proteins: modes of binding with emphasis on disordered domains. Open Biol 2021, 11, 210222. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; Žídek, A.; Green, T.; Tunyasuvunakool, K.; Petersen, S.; Jumper, J.; Clancy, E.; Green, R.; Vora, A.; Lutfi, M.; Figurnov, M.; Cowie, A.; Hobbs, N.; Kohli, P.; Kleywegt, G.; Birney, E.; Hassabis, D.; Velankar, S. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 2022, 50(D1), D439–D444. [Google Scholar] [CrossRef]
- Ahmad, S.; da Costa Gonzales, L.J.; Bowler-Barnett, E.H.; Rice, D.L.; Kim, M.; Wijerathne, S.; Luciani, A.; Kandasaamy, S.; Luo, J.; Watkins, X.; Turner, E.; Martin, M.J. UniProt Consortium The UniProt website API: facilitating programmatic access to protein knowledge. Nucleic Acids Res 2025, 53(W1), W547–W553. [Google Scholar] [CrossRef]
- Nizovtseva, E.; Polikanov, Y.; Kulaeva, O.; Clauvelin, N.; Postnikov, Y. V.; Olson, W. K.; Studitsky, V.M. Opposite effects of histone H1 and HMGN5 protein on distant interactions in chromatin. Mol. Biol 2019, 53, 1038–1048. [Google Scholar] [CrossRef]
- Araya, I.; Nardocci, G.; Schwartz, U.; Babl, S.; Barros, M.; Carrasco-Wong, I.; Imhof, A.; Montecino, M.; Längst, G. HMGN5, an RNA or Nucleosome binding protein-potentially switching between the substrates to regulate gene expression. bioRxiv 2022, 2022–07. [Google Scholar]
- Moses, D.; Yu, F.; Ginell, G.M.; Shamoon, N.M.; Koenig, P.S.; Holehouse, A.S.; Sukenik, S. Revealing the Hidden Sensitivity of Intrinsically Disordered Proteins to their Chemical Environment. J Phys Chem Lett 2020, 11, 10131–10136. [Google Scholar] [CrossRef]
- Salgado, H.; Gama-Castro, S.; Lara, P.; Mejia-Almonte, C.; Alarcón-Carranza, G.; López-Almazo, A.G.; Betancourt-Figueroa, F.; Peña-Loredo, P.; Alquicira-Hernández, S.; Ledezma-Tejeida, D.; Arizmendi-Zagal, L.; Méndez-Hernández, F.; Díaz-Gómez, A.K.; Ochoa-Praxedis, E.; Muñiz-Rascado, L.J.; García-Sotelo, J.S.; Flores-Gallegos, F.A.; Gómez, L.; Bonavides-Martínez, C.; del Moral-Chávez, V.M.; Hernández-Álvarez, A.J.; Santos-Zavaleta, A.; Capella-Gutiérrez, S.; Gelpí, J.L.; Collado-Vides, J. RegulonDB v12.0: a comprehensive resource of transcriptional regulation in E. coli K-12. Nucleic Acids Res 2024, 52(D1), D255–D264. [Google Scholar] [CrossRef]
- Suvorova, I.A.; Korostelev, Y.D.; Gelfand, M.S. GntR Family of Bacterial Transcription Factors and Their DNA Binding Motifs: Structure, Positioning and Co-Evolution. PLoS One 2015, 10, e0132618. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Hernandez-Guerrero, R.; Mendez-Monroy, P.E.; Martinez-Nuñez, M.A.; Ibarra, J.A.; Pérez-Rueda, E. Evaluation of the Abundance of DNA-Binding Transcription Factors in Prokaryotes. Genes (Basel) 2020, 11, 52. [Google Scholar] [CrossRef]
- Pérez-Rueda, E.; Hernandez-Guerrero, R.; Martínez-Núñez, M.A.; Armenta-Medina, D.; Sanchez, I.; Ibarra, J.A. Abundance, diversity and domain architecture variability in prokaryotic DNA-binding transcription factors. PLoS One 2018, 13, e0195332. [Google Scholar] [CrossRef]
- Rigali, S.; Derouaux, A.; Giannotta, F.; Dusart, J. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J Biol Chem 2002, 277, 12507–12515. [Google Scholar] [CrossRef]
- Rigali, S.; Schlicht, M.; Hoskisson, P.; Nothaft, H.; Merzbacher, M.; Joris, B.; Titgemeyer, F. Extending the classification of bacterial transcription factors beyond the helix-turn-helix motif as an alternative approach to discover new cis/trans relationships. Nucleic Acids Res 2004, 32, 3418–3426. [Google Scholar] [CrossRef]
- Jain, D. Allosteric control of transcription in GntR family of transcription regulators: A structural overview. IUBMB Life 2015, 67, 556–563. [Google Scholar] [CrossRef]
- Tutukina, M.N.; Potapova, A.V.; Vlasov, P.K.; Purtov, Y.A.; Ozoline, O.N. Structural modeling of the ExuR and UxuR transcription factors of E. coli: search for the ligands affecting their regulatory properties. J Biomol Struct Dyn 2016, 34, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Arya, G.; Pal, M.; Sharma, M.; Singh, B.; Singh, S.; Agrawal, V.; Chaba, R. Molecular insights into effector binding by DgoR, a GntR/FadR family transcriptional repressor of D-galactonate metabolism in Escherichia coli. Molecular Microbiology. 2021, 115, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.; Schroeder, M. PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res 2021, 49(W1), W530–W534. [Google Scholar] [CrossRef]
- Ritzenthaler, P.; Mata-Gilsinger, M. , Use of in vitro gene fusions to study the uxuR regulatory gene in Escherichia coli K-12: direction of transcription and regulation of its expression. J Bacteriol 1982, 150, 1040–1047. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Mironov, A.A.; Rakhmaninova, A.B.; Gelfand, M.S. Transcriptional regulation of transport and utilization systems for hexuronides, hexuronates and hexonates in gamma purple bacteria. Mol Microbiol 2000, 38, 673–683. [Google Scholar] [CrossRef]
- Purtov, Y.A.; Tishchenko, S.V.; Nikulin, A.D. Modeling the Interaction of the UxuR-ExuR Heterodimer with the Components of the Metabolic Pathway of Escherichia coli for Hexuronate Utilization. Biophysics 2021, 66, 753–763. [Google Scholar] [CrossRef]
- Almeida, B.C.; Kaczmarek, J.A.; Figueiredo, P.R.; Prather, K.L.J.; Carvalho, A.T.P. Transcription factor allosteric regulation through substrate coordination to zinc. NAR Genom Bioinform 2021, 3, lqab033. [Google Scholar] [CrossRef] [PubMed]
- Bessonova, T.A.; Fando, M.S.; Kostareva, O.S.; Tutukina, M.N.; Ozoline, O.N.; Gelfand, M.S.; Nikulin, A.D.; Tishchenko, S.V. Differential Impact of Hexuronate Regulators ExuR and UxuR on the Escherichia coli Proteome. Int J Mol Sci. 2022, 23, 8379. [Google Scholar] [CrossRef]
- Purtov, Y.A.; Tutukina, M.N.; Nikulin, A.D.; Ozoline, O.N. The topology of the contacts of potential ligands for the UxuR transcription factor of Escherichia coli as revealed by flexible molecular docking. Biophysics 2019, 64, 49–56. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; Lepore, R.; Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018, 46(W1), W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository - new features and functionality. Nucleic Acids Res 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Research, 2025; 53(D1), D609–D617.
- Purtov, Y.A.; Ozoline, O.N. Neuromodulators as Interdomain Signaling Molecules Capable of Occupying Effector Binding Sites in Bacterial Transcription Factors. Int J Mol Sci 2023, 24, 15863. [Google Scholar] [CrossRef]
- Franco, I.S.; Mota, L.J.; Soares, C.M.; de Sá-Nogueira, I. Functional domains of the Bacillus subtilis transcription factor AraR and identification of amino acids important for nucleoprotein complex assembly and effector binding. J Bacteriol 2006, 188, 3024–3036. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. (1970). A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins. Journal of Molecular Biology 1970, 48, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Bertram, R.; Rigali, S.; Wood, N.; Lulko, A.T.; Kuipers, O.P.; Titgemeyer, F. Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J Bacteriol 2011, 193, 3525–3536. [Google Scholar] [CrossRef] [PubMed]
- Fillenberg, S.B.; Grau, F.C.; Seidel, G.; Muller, Y.A. Structural insight into operator dre-sites recognition and effector binding in the GntR/HutC transcription regulator NagR. Nucleic Acids Res 2015, 43, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, M.M.; Chevance, F.; Dippel, R.; Eppler, T.; Schlegel, A.; Boos, W.; Lu, Y.J.; Rock, C.O. Phosphotransferase-mediated transport of the osmolyte 2-O-alpha-mannosyl-D-glycerate in Escherichia coli occurs by the product of the mngA (hrsA) gene and is regulated by the mngR (farR) gene product acting as repressor. J Biol Chem 2004, 279, 5537–5548. [Google Scholar] [CrossRef]
- Jiapei, M.; Chong, S. Roles of intrinsically disordered protein regions in transcriptional regulation and genome organization. Current Opinion in Genetics & Development 2025, 90, 102285. [Google Scholar]
- Liu, J.; Perumal, N.B.; Oldfield. CJ.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef]
- Hadži, S.; Živič, Z.; Kovačič, M.; Zavrtanik, U.; Haesaerts, S.; Charlier, D.; Plavec, J.; Volkov, A.N.; Lah, J.; Loris, R. Fuzzy recognition by the prokaryotic transcription factor HigA2 from Vibrio cholerae. Nat Commun 2024, 15, 3105. [Google Scholar] [CrossRef]
- Peng, H.; Kotelnikov. S.; Egbert, M.E.; Ofaim, S.; Stevens, G.C.; Phanse, S.; Saccon, T.; Ignatov, M.; Dutta, S.; Istace, Z.; Moutaoufik, M.T.; Aoki, H.; Kewalramani, N.; Sun, J.; Gong, Y.; Padhorny, D.; Poda, G.; Alekseenko, A.; Porter, K.A.; Jones, G.; Rodionova, I.; Guo, H.; Pogoutse, O.; Datta, S.; Saier, M.; Crovella, M.; Vajda, S.; Moreno-Hagelsieb, G.; Parkinson, J.; Segre, D.; Babu, M,; Kozakov, D.; Emili, A. Ligand interaction landscape of transcription factors and essential enzymes in E. coli. Cell 2025, 188, 1441–1455.e15. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res 2023, 51(D1), D1373–D1380. [Google Scholar] [CrossRef]
- Hanwell, M. D.; Curtis, D. E.; Lonie, D. C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. R. Avogadro: anadvanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G. 3rd.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; Gregor, J.; Davis, N.W.; Kirkpatrick, H.A.; Goeden, M.A.; Rose, D.J.; Mau, B.; Shao, Y. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res 2025, 53, D672–D677. [Google Scholar] [CrossRef]
- Borriss, R.; Danchin, A.; Harwood, C.R.; Médigue, C.; Rocha, E.P.C.; Sekowska, A.; Vallenet, D. Bacillus subtilis, the model Gram-positive bacterium: 20 years of annotation refinement. Microb Biotechnol 2018, 11, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Li, X.; He, P.; Ho, H.; Wu, Y.; He, Y. Whole-genome sequencing of Bacillus subtilis XF-1 reveals mechanisms for biological control and multiple beneficial properties in plants. J Ind Microbiol Biotechnol 2015, 42, 925–937. [Google Scholar] [CrossRef]
- Zheng, M.; Cooper, D.R.; Grossoehme, N.E.; Yu, M.; Hung, L.W.; Cieslik, M.; Derewenda, U.; Lesley, S.A.; Wilson, I.A.; Giedroc, D.P.; Derewenda, Z.S. Structure of Thermotoga maritima TM0439: implications for the mechanism of bacterial GntR transcription regulators with Zn2+-binding FCD domains. Acta Crystallogr D Biol Crystallogr 2009, 65, 356–365. [Google Scholar] [CrossRef]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; LeGrand, S. Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande V.S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comp Chem 2009, 30, 864–872. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. Journal of Computational Chemistry 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R.K.; Goodsell, D. S.; Olson, A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MD simulation revealed conformational flexibility of the E. coli UxuR inter-domain linker, which affected carbon binding affinity. (a) Monomer structure of the UxuR predicted by the SWISS-MODEL. (b) Sequential rearrangements of the UxuR inter-domain linker at 50–100 nanoseconds of the MD trajectory. The linker from the 100 ns model (a) and linkers lacking α-helix are shown with amino acid side groups. (c) Heat maps representing affinity of carbohydrates interaction with UxuR monomer’s inter-domain linker (ΔG kcal/mol). White cells correspond to random, non-specific complexes on other protein surfaces, which had average ΔG values of -4.52 ± 0.26 (D-fructuronate), -4.85 ± 0.20 (D-galacturonate), -4.45 ± 0.23 (D-glucose) and -4.82 ± 0.18 (D-glucuronate) kcal/mol. ΔG-values greater than 3 StDs above the mean non-specific binding are shown in bold.
Figure 1.
MD simulation revealed conformational flexibility of the E. coli UxuR inter-domain linker, which affected carbon binding affinity. (a) Monomer structure of the UxuR predicted by the SWISS-MODEL. (b) Sequential rearrangements of the UxuR inter-domain linker at 50–100 nanoseconds of the MD trajectory. The linker from the 100 ns model (a) and linkers lacking α-helix are shown with amino acid side groups. (c) Heat maps representing affinity of carbohydrates interaction with UxuR monomer’s inter-domain linker (ΔG kcal/mol). White cells correspond to random, non-specific complexes on other protein surfaces, which had average ΔG values of -4.52 ± 0.26 (D-fructuronate), -4.85 ± 0.20 (D-galacturonate), -4.45 ± 0.23 (D-glucose) and -4.82 ± 0.18 (D-glucuronate) kcal/mol. ΔG-values greater than 3 StDs above the mean non-specific binding are shown in bold.
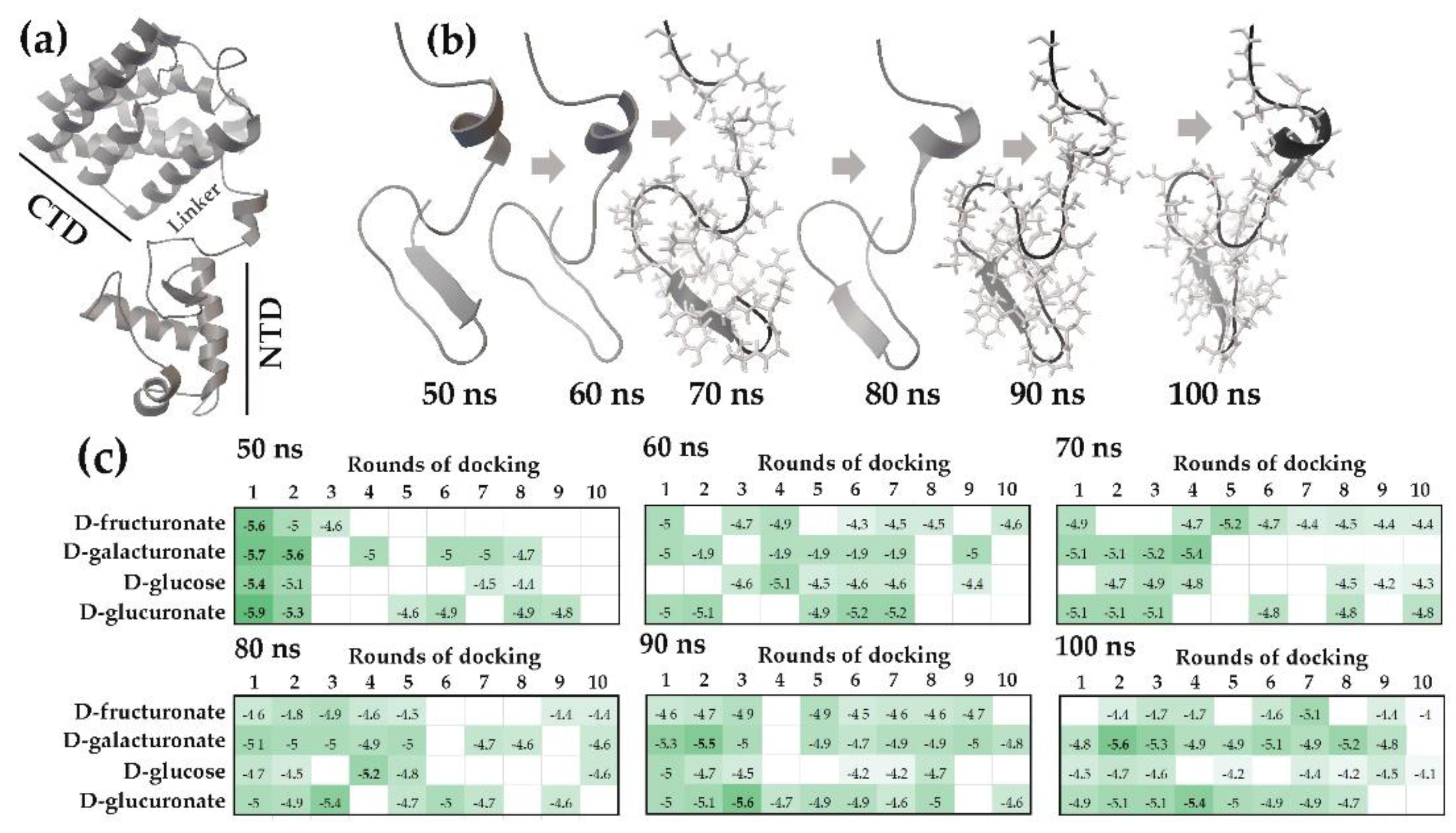
Figure 2.
Preferential binding and cluster formation of D-fructuronate and D-glucose in the UxuR monomer linker region is potentially stabilized by hydrogen bonds formed between ligand molecules. (a,d): The UxuR model (ribbon) from a 100 ns of MD trajectory with bound D-fructuronate and D-glucose molecules (gray rods). The ligand with the highest binding affinity is shown in black ball model. (b,c): LigPlot+ diagrams (PLIP v.2.2.8) predicting the hydrogen-bond interactions for D-fructuronate from the 7th round of molecular docking and for D-glucose from the 3rd round of docking. Hydrogen bond distances are given here and below in Ångstroms.
Figure 2.
Preferential binding and cluster formation of D-fructuronate and D-glucose in the UxuR monomer linker region is potentially stabilized by hydrogen bonds formed between ligand molecules. (a,d): The UxuR model (ribbon) from a 100 ns of MD trajectory with bound D-fructuronate and D-glucose molecules (gray rods). The ligand with the highest binding affinity is shown in black ball model. (b,c): LigPlot+ diagrams (PLIP v.2.2.8) predicting the hydrogen-bond interactions for D-fructuronate from the 7th round of molecular docking and for D-glucose from the 3rd round of docking. Hydrogen bond distances are given here and below in Ångstroms.
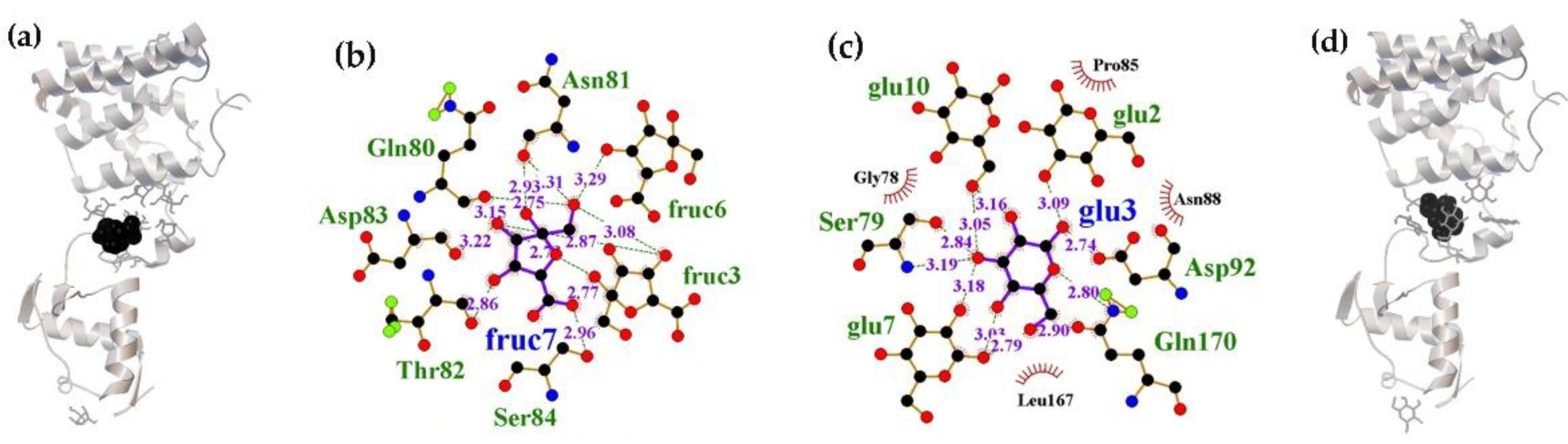
Figure 3.
Preferential binding and cluster formation of D-glucuronate and D-galacturunate in the UxuR monomer linker region is potentially stabilized by hydrogen bonds formed between ligand molecules. (a,d) The UxuR model from a 100 ns of MD trajectory with bound D-glucuronate and D-galacturunate molecules (gray rods). The ligand with the highest binding affinity is shown in black ball model. (b,c): LigPlot+ diagrams (PLIP v.2.2.8) predicting the H-bond interactions for D- glucuronate from the 4th round of molecular docking (b) and for D- galacturunate from the 2nd round of docking.
Figure 3.
Preferential binding and cluster formation of D-glucuronate and D-galacturunate in the UxuR monomer linker region is potentially stabilized by hydrogen bonds formed between ligand molecules. (a,d) The UxuR model from a 100 ns of MD trajectory with bound D-glucuronate and D-galacturunate molecules (gray rods). The ligand with the highest binding affinity is shown in black ball model. (b,c): LigPlot+ diagrams (PLIP v.2.2.8) predicting the H-bond interactions for D- glucuronate from the 4th round of molecular docking (b) and for D- galacturunate from the 2nd round of docking.
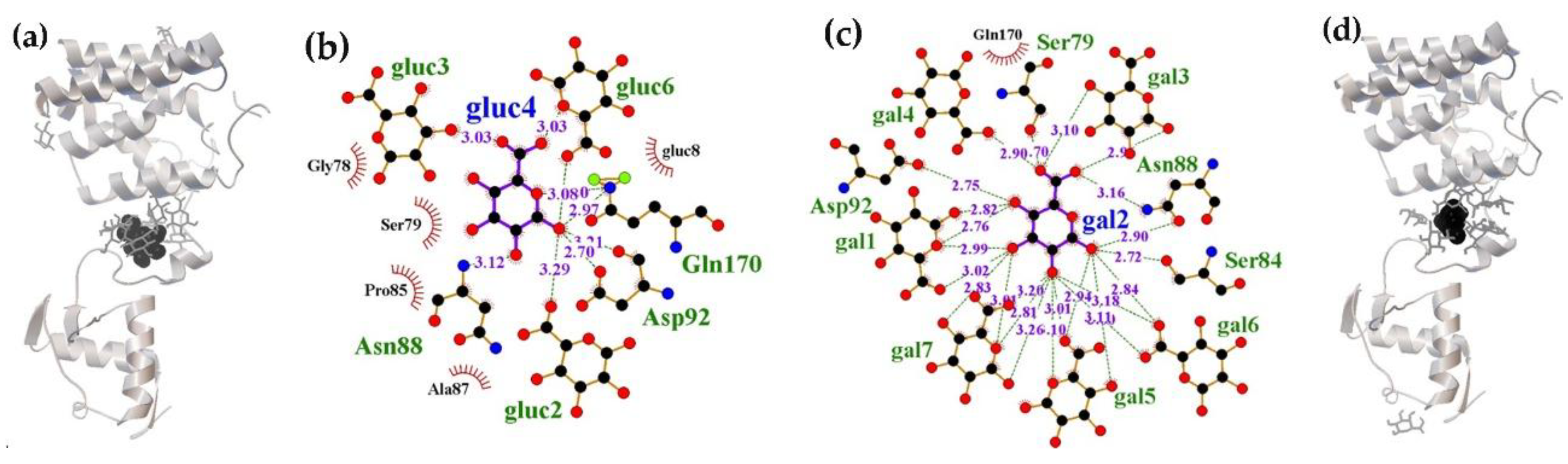
Figure 4.
Confirming the previously observed interaction of carbohydrates with the UxuR effector-binding domain [68,78], molecular docking revealed a novel binding mode, where ligands connect linkers with CTD. (a–c) Dimer structures of UxuR (ribbon) from a 100 ns MD trajectory with bound D-fructruronate (a), D-glucose (b) and D-glucuronate (c) molecules (gray rods). The ligands with the highest binding affinity are shown as black ball models. Red circles indicate carbons that form contacts with the linker and the CTD α-helix. (d) Heat maps representing the binding affinity (ΔG kcal/mol) of hexuronates for the UxuR dimer over successive molecular docking rounds. ΔG-values are categorized by binding site: CTD pocket (brown), inter-domain linkers only (black), and linkers connected by sugar to the CTD (red). White cells correspond to non-specific complexes formed on other protein surfaces, which had average ΔG values of -4.88 ± 0.24 (D-fructuronate), -5.26 ± 0.21 (D-galacturonate), -4.74 ± 0.26 (D-glucose) and -5.22 ± 0.22 (D-glucuronate) kcal/mol. ΔG values corresponding to statistically significant binding are bolded.
Figure 4.
Confirming the previously observed interaction of carbohydrates with the UxuR effector-binding domain [68,78], molecular docking revealed a novel binding mode, where ligands connect linkers with CTD. (a–c) Dimer structures of UxuR (ribbon) from a 100 ns MD trajectory with bound D-fructruronate (a), D-glucose (b) and D-glucuronate (c) molecules (gray rods). The ligands with the highest binding affinity are shown as black ball models. Red circles indicate carbons that form contacts with the linker and the CTD α-helix. (d) Heat maps representing the binding affinity (ΔG kcal/mol) of hexuronates for the UxuR dimer over successive molecular docking rounds. ΔG-values are categorized by binding site: CTD pocket (brown), inter-domain linkers only (black), and linkers connected by sugar to the CTD (red). White cells correspond to non-specific complexes formed on other protein surfaces, which had average ΔG values of -4.88 ± 0.24 (D-fructuronate), -5.26 ± 0.21 (D-galacturonate), -4.74 ± 0.26 (D-glucose) and -5.22 ± 0.22 (D-glucuronate) kcal/mol. ΔG values corresponding to statistically significant binding are bolded.
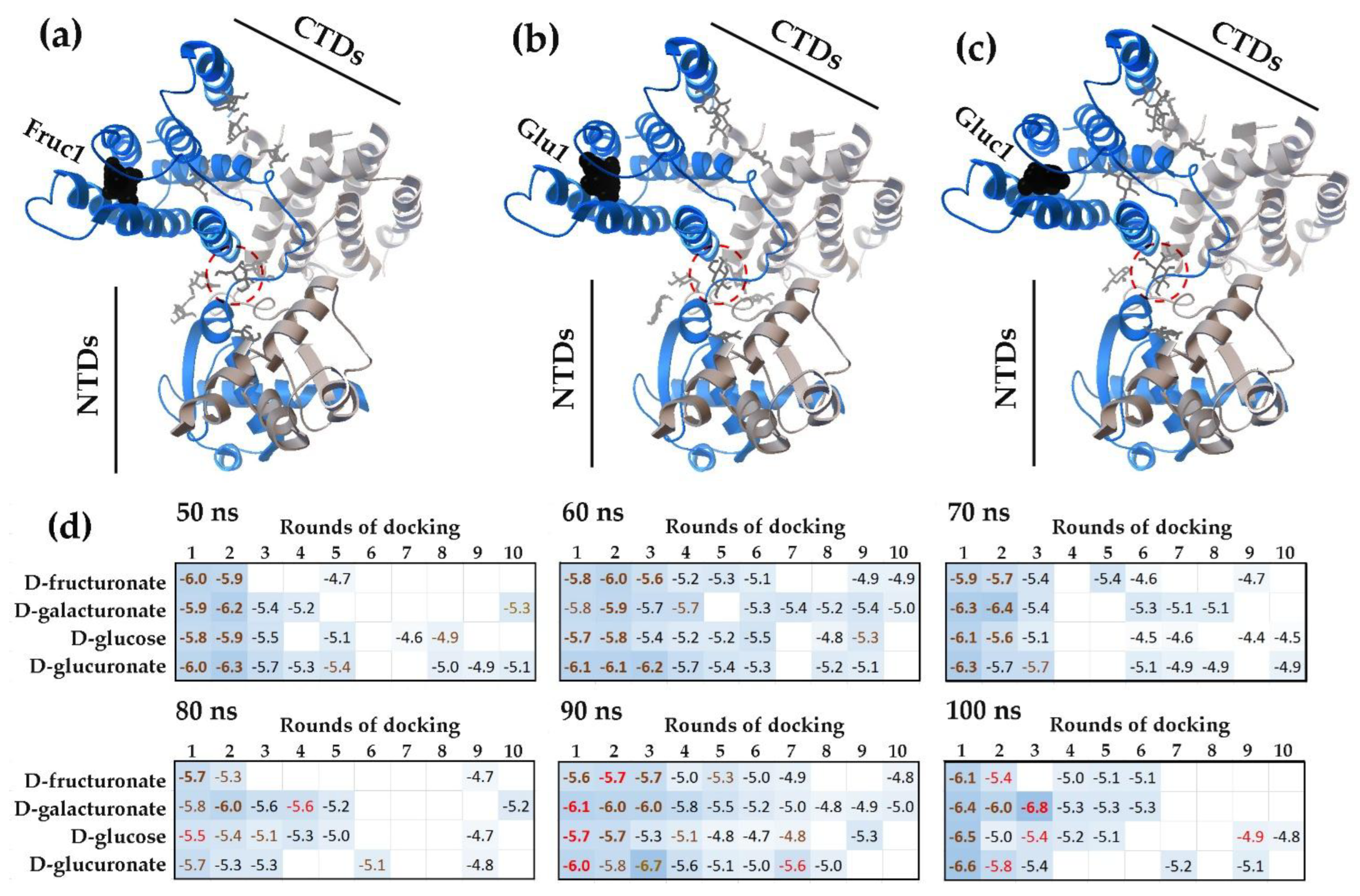
Figure 5.
Carbohydrates can form intramolecular bridges connecting linkers to the UxuR CTD. a) Structures of UxuR dimer from a 100 ns MD trajectory, showing bound D-galacturonate molecules. The ligand connecting Asn81 from the linker to residues Leu167, Gln170, Ser171 and Gln173 on the CTD α-helix is shown as black ball model. (b) Correspondng LigPlot+ diagram visualising predicted H-bond interctions with bond distances provided in Ångströms.
Figure 5.
Carbohydrates can form intramolecular bridges connecting linkers to the UxuR CTD. a) Structures of UxuR dimer from a 100 ns MD trajectory, showing bound D-galacturonate molecules. The ligand connecting Asn81 from the linker to residues Leu167, Gln170, Ser171 and Gln173 on the CTD α-helix is shown as black ball model. (b) Correspondng LigPlot+ diagram visualising predicted H-bond interctions with bond distances provided in Ångströms.
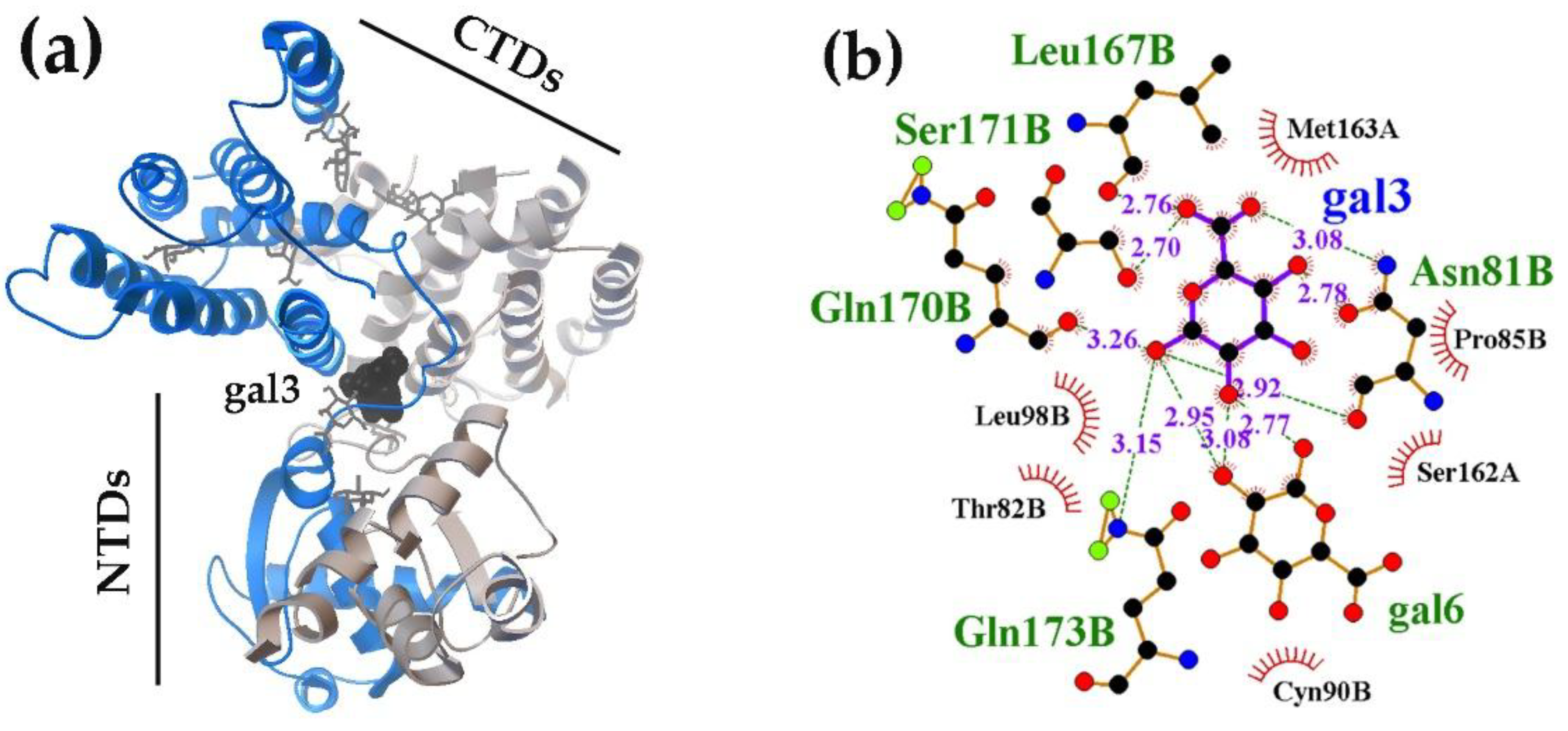
Figure 6.
Accommodating D-glucose on the short inter-domain linker, GntR provides inter-subdomain cavity of CTD for high-affinity interaction with ligand. (a) GntR monomer structures from a 100 ns MD trajectory with bound D-glucose molecules. The red circle outlines the carbons that accumulate on the outer surface of the subdomain cavity. a) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-100 ns molecular models over successive molecular docking rounds (color code indicated). White cells correspond to random, non-specific complexes on other protein surfaces, which had average ΔG value of -4.49 ± 0.20 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. (d) LigPlot+ diagram (PLIP v.2.2.8) predicting the H-bond interactions for D-glucose from the first round of molecular docking.
Figure 6.
Accommodating D-glucose on the short inter-domain linker, GntR provides inter-subdomain cavity of CTD for high-affinity interaction with ligand. (a) GntR monomer structures from a 100 ns MD trajectory with bound D-glucose molecules. The red circle outlines the carbons that accumulate on the outer surface of the subdomain cavity. a) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-100 ns molecular models over successive molecular docking rounds (color code indicated). White cells correspond to random, non-specific complexes on other protein surfaces, which had average ΔG value of -4.49 ± 0.20 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. (d) LigPlot+ diagram (PLIP v.2.2.8) predicting the H-bond interactions for D-glucose from the first round of molecular docking.
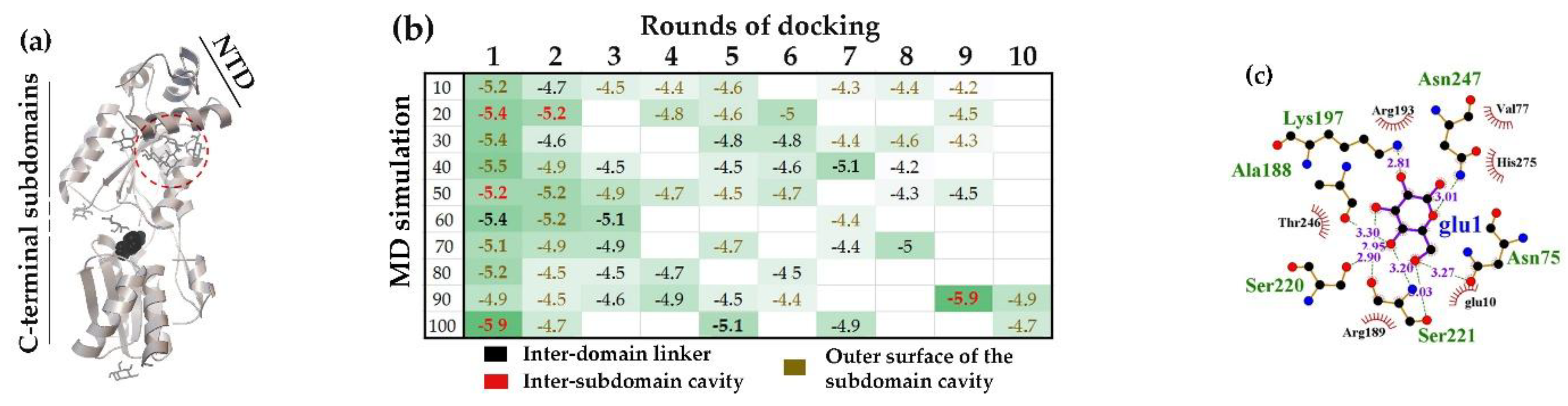
Figure 7.
The inter-subdomain pocket of AraR CTD exhibited the highest affinity for interaction with D-glucose. (a) The AraR monomer structures from a 10 ns MD trajectory with bound D-glucose molecules. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-70 ns molecular models over successive molecular docking rounds (color code indicated). White cells correspond to non-specific complexes on other protein surfaces, which had average ΔG value of -4.70 ± 0.31 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. (c) LigPlot+ diagram (PLIP v.2.2.8) predicting the H-bond interactions for D-glucose from the second round of molecular docking.
Figure 7.
The inter-subdomain pocket of AraR CTD exhibited the highest affinity for interaction with D-glucose. (a) The AraR monomer structures from a 10 ns MD trajectory with bound D-glucose molecules. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-70 ns molecular models over successive molecular docking rounds (color code indicated). White cells correspond to non-specific complexes on other protein surfaces, which had average ΔG value of -4.70 ± 0.31 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. (c) LigPlot+ diagram (PLIP v.2.2.8) predicting the H-bond interactions for D-glucose from the second round of molecular docking.
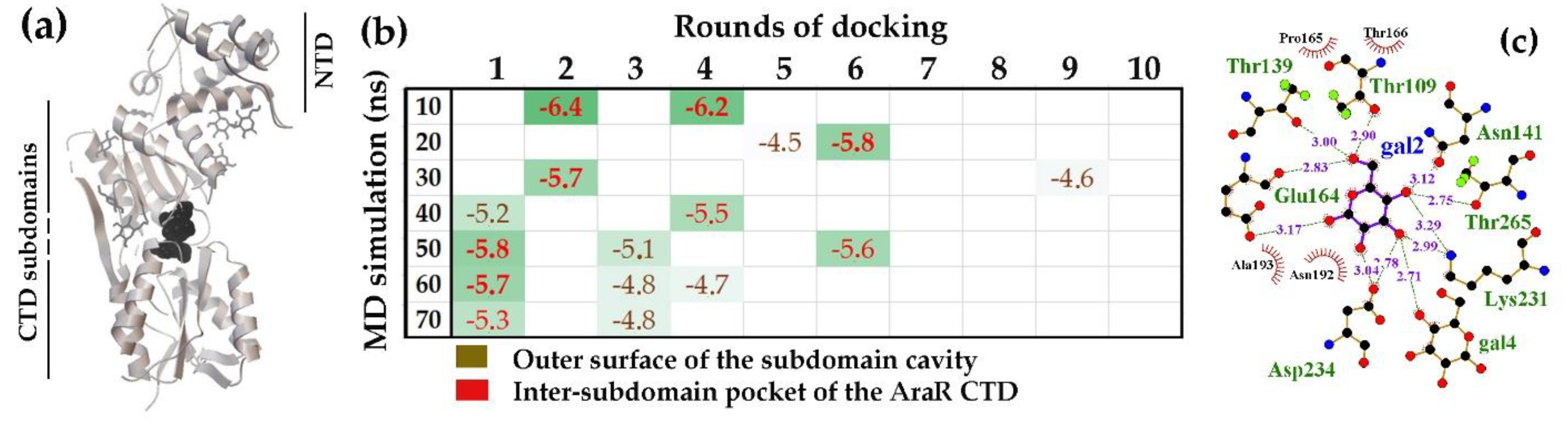
Figure 8.
The pocket within NagR CTD exhibited the highest affinity for interaction with D-glucose. (a) The NagR monomer structure from a 30 ns MD trajectory with bound D-glucose molecules. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. (b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-150 ns molecular models over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are displayed in black, while those, which entered the CTD pocket, are highlighted in red. White cells correspond to non-specific complexes on other protein surfaces, which had and average ΔG value of -4.46 ± 0.25 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. (c) LigPlot+ diagrams (PLIP v.2.2.8) predicting the H-bonds formation for D-glucose from the tenth round of molecular docking.
Figure 8.
The pocket within NagR CTD exhibited the highest affinity for interaction with D-glucose. (a) The NagR monomer structure from a 30 ns MD trajectory with bound D-glucose molecules. The ligand with the highest binding affinity is shown as black ball models. Other 9 molecules are displayed as gray rods. (b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-150 ns molecular models over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are displayed in black, while those, which entered the CTD pocket, are highlighted in red. White cells correspond to non-specific complexes on other protein surfaces, which had and average ΔG value of -4.46 ± 0.25 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the mean non-specific binding are shown in bold. (c) LigPlot+ diagrams (PLIP v.2.2.8) predicting the H-bonds formation for D-glucose from the tenth round of molecular docking.
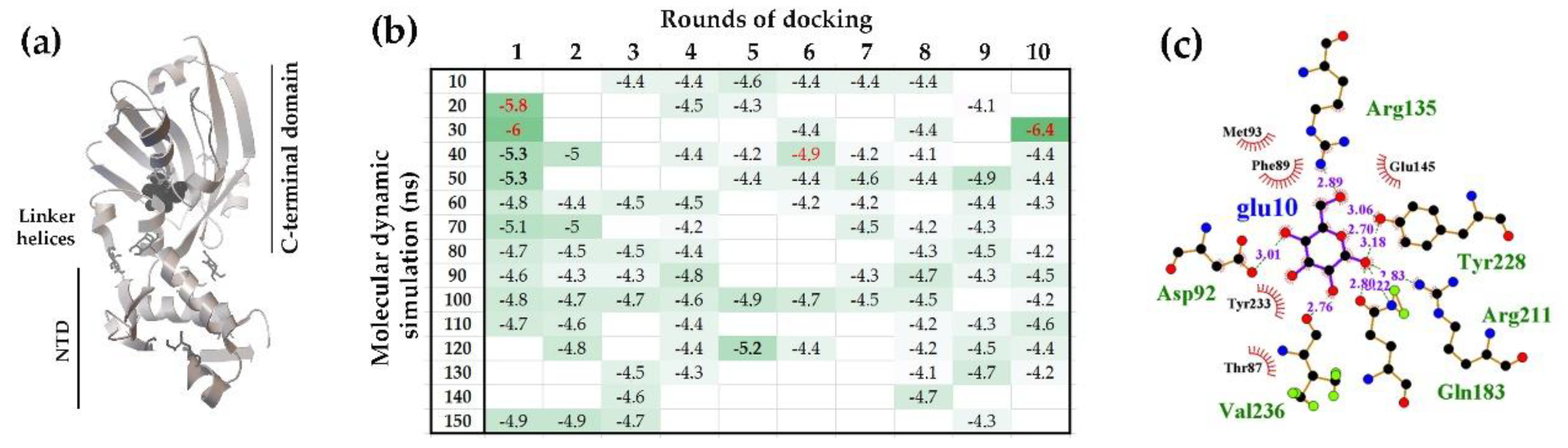
Figure 9.
The pocket within FarR CTD bordered with inter-domain linker exhibit the highest affinity for D-glucose. (a) The FarR monomer structure from a 10 ns MD trajectory with bound D-glucose molecules. Two ligands bound to the high-affinity site are shown as black ball models. (b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-150 ns molecular models over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are shown in black. Those, which entered the high-affinity pocket, are displayed in red. Non-specific complexes on other protein surfaces had and average ΔG value of -4.53 ± 0.26 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the background are shown in bold. (c) LigPlot+ diagrams (PLIP v.2.2.8 [72]) showing the H-bonds potentially formed by D-glucose from the 4th round of molecular docking.
Figure 9.
The pocket within FarR CTD bordered with inter-domain linker exhibit the highest affinity for D-glucose. (a) The FarR monomer structure from a 10 ns MD trajectory with bound D-glucose molecules. Two ligands bound to the high-affinity site are shown as black ball models. (b) Heat map representing the binding affinity (ΔG kcal/mol) of D-glucose for the 10-150 ns molecular models over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are shown in black. Those, which entered the high-affinity pocket, are displayed in red. Non-specific complexes on other protein surfaces had and average ΔG value of -4.53 ± 0.26 kcal/mol. Ligands with binding efficiency greater than 3 StDs above the background are shown in bold. (c) LigPlot+ diagrams (PLIP v.2.2.8 [72]) showing the H-bonds potentially formed by D-glucose from the 4th round of molecular docking.
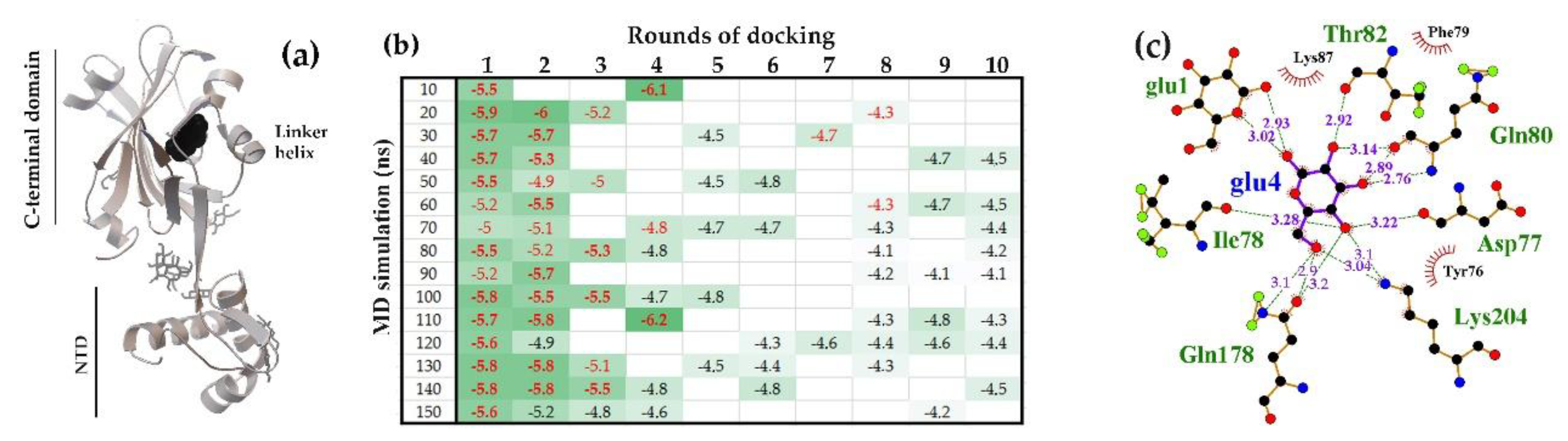
Figure 10.
A putative transcription factor YydK with structural and sequence homology to NagR and FarR interacts with D-glucose in a NagR-like manner. (a) The YydK monomer structure from a 40 ns MD trajectory (panel d) with bound D-glucose molecules. The ligand bound to the highest-affinity site is shown as black ball model. (b,d) Heat maps representing the binding affinity (ΔG kcal/mol) of D-glucose for the molecular models from different time points of dynamic simulation trajectory over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are shown in black. Those, which entered the high-affinity pocket, are displayed in red. Non-specific complexes on other protein surfaces had and average ΔG value of -4.45 ± 0.22 (b) and -4.53 ± 0.29 kcal/mol (d). Ligands with binding efficiency greater than 3 StDs above the background are shown in bold. (c) LigPlot+ diagram (PLIP v.2.2.8 [72]) predicting the H-bonds formation for D-glucose from the first round of molecular docking (40 ns MD trajectory, panel d).
Figure 10.
A putative transcription factor YydK with structural and sequence homology to NagR and FarR interacts with D-glucose in a NagR-like manner. (a) The YydK monomer structure from a 40 ns MD trajectory (panel d) with bound D-glucose molecules. The ligand bound to the highest-affinity site is shown as black ball model. (b,d) Heat maps representing the binding affinity (ΔG kcal/mol) of D-glucose for the molecular models from different time points of dynamic simulation trajectory over successive molecular docking rounds. ΔG-values of ligands attached to the flexible linkers are shown in black. Those, which entered the high-affinity pocket, are displayed in red. Non-specific complexes on other protein surfaces had and average ΔG value of -4.45 ± 0.22 (b) and -4.53 ± 0.29 kcal/mol (d). Ligands with binding efficiency greater than 3 StDs above the background are shown in bold. (c) LigPlot+ diagram (PLIP v.2.2.8 [72]) predicting the H-bonds formation for D-glucose from the first round of molecular docking (40 ns MD trajectory, panel d).
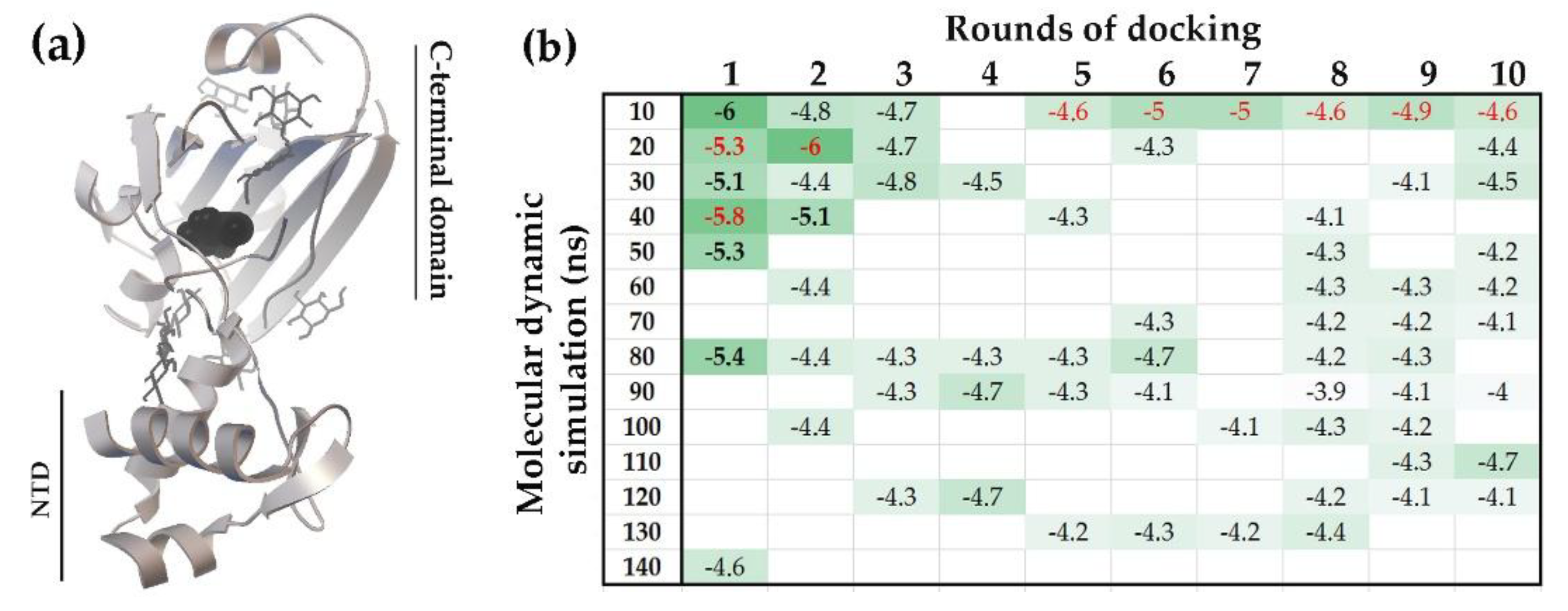
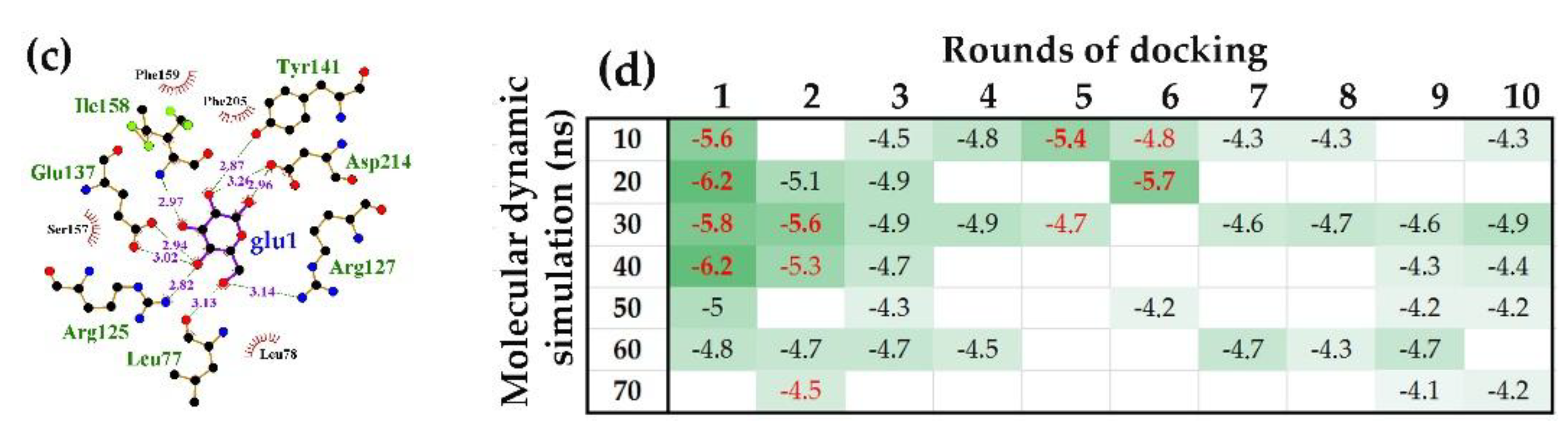
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



