Submitted:
12 November 2025
Posted:
13 November 2025
You are already at the latest version
Abstract
The development of non-noble metal catalysts provides a cost-effective and sustainable route for glucose oxidation to gluconic acid. In this study, a series of catalysts based on inexpensive transition metals (Cr, Cu, Ni, Fe) and/or Au were synthesized using siliceous supports (SiO₂ and MCM-41) and systematically evaluated. The aim was to partially or fully replace noble metals with lower-cost alternatives, while maintaining high catalytic performance. Comprehensive characterization—including ICP-AES for composition, N₂ adsorption–desorption for porosity, XRD for structure, H₂-TPR for reducibility, and NH₃-TPD for acidity—was conducted to establish structure–property relationships. Among the tested catalysts, Ni- and Fe-based systems exhibited superior stability, with NiO/SiO₂ achieving gluconic acid yields comparable to Au. The bimetallic Au–Ni/SiO₂ catalyst displayed enhanced metal–support interactions and minimal leaching (< 2 %), while Au–Fe/SiO₂ improved selectivity, yielding up to 23 % gluconic acid, surpassing 5Fe/SiO₂ (18 %) and 0.3Au/SiO₂ (15 %), albeit with lower stability. These results highlight the potential of low-cost transition-metal and bimetallic catalysts as efficient and eco-nomically viable systems for selective glucose oxidation, providing insights for rational catalyst design in sustainable carbohydrate valorization.
Keywords:
Ni and Fe heterogenous catalysts
; glucose oxidation
; gluconic acid
; catalyst stability
1. Introduction
The oxidation of glucose into value-added carboxylic acids, such as gluconic and glucaric acids, represents a key route toward renewable biomass valorization and sustainable chemical production [1]. These compounds—widely used in food formulation, pharmaceuticals, metal chelation, and as intermediates in fine chemical syntheses—have stimulated continuous research interest in development of efficient heterogeneous catalysts [2,3]. Currently, gluconic acid is predominantly produced via biotechnological cascade oxidation of glucose [4], which, although highly selective, limits large-scale production and increases costs. Consequently, considerable efforts have been directed toward developing chemo-catalytic systems as scalable and economically viable alternatives. Heterogeneous catalysts are particular attractive, as conventional electrochemical or stoichiometric oxidation methods often rely on undesirable reagents such as NaBr, NaOCl, mineral acids, HNO₃, or KMnO₄ [5]. These homogeneous approaches are inherently limited by the use of corrosive and hazardous chemicals, difficulties in catalyst recovery and recycling, and the generation of toxic by-products [6]. Therefore designing catalysts that achieve high glucose conversion, excellent selectivity, and robust catalyst stability under aqueous conditions remains a significant challenge [3]..
Supported noble metal catalysts—particularly gold (Au) nanoparticles—have shown remarkable activity and selectivity for liquid-phase glucose oxidation under relatively mild conditions. For example, Au-based catalysts supported on oxides such as TiO2 [7,8], CeO2 [9] or Al2O3 [10], have demonstrated high glucose conversion and selectivity toward gluconic acid when properly engineered. However, the high cost and limited availability of noble metals, together with scalability concerns, have motivated the search for more economical alternatives.
Recent research has focused on low-cost, environmentally benign, and recyclable heterogeneous catalysts for glucose oxidation. Achieving catalyst stability under oxidative aqueous conditions without compromising activity or selectivity remains a key challenge. Most reported systems still rely on expensive noble metals (Pt, Pd, and Au) on neutral (SiO₂) or acid–base (MgO, Al₂O₃, TiO₂, activated carbon, etc.) supports [3,6,11,12]. Thus, designing catalysts that maintain stable physicochemical properties while minimizing the use of noble metals remains of vital importance.
In this context, low-cost transition metals represent promising alternatives due to their natural abundance, redox versatility, and lower environmental footprint. Although less explored, non-noble metal systems have recently attracted attention for glucose oxidation. For instance, CuO [13] and CuO–CeO₂ [14] can oxidize glucose even in the absence of base, highlighting their intrinsic redox capability. Moreover, bimetallic Au–Cu and Au–Co catalysts supported on TiO₂ have shown that incorporating transition metals can significantly affect activity and selectivity. Amaniampong et al. [15] reported that Au–Cu/TiO₂ achieved gluconic acid yields up to 80 %, outperforming their monometallic counterparts, although catalyst stability decreased upon reuse due to by-product accumulation and surface fouling [15]. Despite these promising advances, systemic studies on low-cost, truly heterogeneous transition-metal catalysts under mild and environmentally benign conditions remain limited, highlighting the need to clarify structure–stability–selectivity relationships.
The choice of support plays a crucial role in heterogeneous catalyst performance. Siliceous materials including amorphous SiO₂ and mesoporous silicas such as MCM-41, offer high surface area, tunable porosity and an ideal platform for dispersing active species while modulating metal–support electronic interactions. The support - metal oxide synergy can influence reaction pathways, product distribution, and catalyst stability [16,17]. High-surface-area mesoporous silicates—including MCM-41, HMS, SBA-15, and MCF—enable improved dispersion of metals, even at higher loadings, compared to non-porous or low-surface-area supports [16,18]. Accordingly, commercial SiO₂ and a synthesized MCM-41-type mesoporous silica, were selected as supports for low-cost transition-metal oxides (Cr₂O₃, CuO, Fe₂O₃ and NiO), to optimize dispersion, enhance stability, and promote selective glucose oxidation.
In this study, we systematically investigate three classes of catalysts: (i) transition metal oxides (CuO, Cr₂O₃, Fe₂O₃, NiO,) supported on SiO₂ and MCM-41; (ii) Au-containing catalysts supported on SiO₂ and MCM-41, and (iii) bimetallic Au–transition metal catalysts supported on the same siliceous supports. All catalysts were evaluated under standard reaction conditions (80 °C, 60 min, pH 8, H₂O₂/glucose ratio 40), for activity, selectivity towards gluconic acid and stability (via leaching analysis). This study provides the first direct comparison of all synthesized low-cost oxidation catalysts, aiming to minimize the use of expensive noble metals while elucidating correlations between catalyst composition, metal-support interaction, stability and product distribution. Overall, the work establishes design guidelines for efficient, stable and economically viable heterogeneous catalysts for selective glucose oxidation to gluconic acid.
2. Results and Discussion
2.1. Textural and Structural Characterization
The chemical composition of the oxidation catalysts is summarized in Table 1. For all supported transition metals, the nominal metal loadings were found to be consistent with the experimentally determined values, indicating that wet impregnation successfully achieved the desired metal contents. Gold deposition on SiO₂ via the PVA-protected method, accompanied by parallel reduction with NaBH₄, also yielded loadings close to the nominal values. Similar trends were observed for the corresponding bimetallic catalysts, demonstrating that Au deposition on SiO₂ was as effective as in the monometallic systems. In contrast, Au deposition on MCM-41 proved less efficient, achieving only 62% of the nominal 1 wt. % Au and dropping to 20 % for 0.3Au/MCM-41. This pronounced metal loss during the PVA-protected method is attributed to the filtration step, where very small metal particles may have passed through the filter.
Previous studies support these observations. Kumar et al. [19] reported Au losses during deposition on MCM-41 via several methods including homogeneous deposition–precipitation, polyol, impregnation, and microemulsion, with production yields of 82, 70, 71, and 73%, respectively. In another work [20], Au deposition on Ti-containing MCM-41 by homogeneous deposition–precipitation using NaOH as precipitating agent showed that higher Ti content in the silica framework increased the experimental Au loading, likely due to an increased zero-point charge facilitating Au nanoparticle deposition. Yields ranged from 28% on pure MCM-41 up to 90 % for MCM-41 containing 10.7 wt.% Ti.
The porosity characteristics of all transition metal oxide and Au catalysts supported on SiO₂ and MCM-41 are presented in Table 1. N₂ adsorption/desorption isotherms and pore size distributions of selected catalysts are shown in Figure 1a and 1b. Overall, deposition of metallic species did not significantly alter the original porous structure of the supports. Isotherms of supported transition metal oxide catalysts were similar to those of the respective supports, suggesting a minor impact of wet impregnation on the siliceous surfaces. The surface areas of the supported catalysts, normalized to the silica content (Table 1, in brackets), were compared with those of the pure SiO₂ and MCM-41 supports. Only minor reductions (0 – 29 %) were observed, indicating that the deposition of metallic or bimetallic species did not appreciably affect the textural properties. Similarly, total pore volumes remained largely unchanged, confirming the preservation of the mesoporous structure. Transition metal oxides supported on MCM-41 retained the high surface areas, large mesopore volumes, and average mesopore sizes (~ 2.5 nm) characteristic of the support, while SiO₂-supported catalysts exhibited the typical porosity of the siliceous substrate with an average pore size of 10 nm (Figure 1a and b) [16,21].
These trends are consistent with prior observations in the literature. Szegedi et al. [22] reported that wet impregnation of Ni acetate on mesoporous MCM-41 reduced the support surface area by up to 27 % for Ni loadings of 2.4 – 5.5 wt. %. Similar surface area reductions were observed for Cr, Cu and Fe catalysts prepared by various deposition methods [23,24,25]. In our case, the most pronounced decreases (28 – 29 %) occurred for Cu and Ni catalysts supported on MCM-41, whereas bimetallic catalysts showed a smaller impact. XRD patterns (low 2θ, not shown for brevity) confirmed the well-ordered mesoporous structure of all MCM-41 supported catalysts. In contrast, surface area reductions were smaller for SiO₂ - supported catalysts (0 – 7 %), consistent with previous reports [25]. For Au-supported catalysts, the PVA-protected deposition with NaBH₄, despite the low Au content, led to a more pronounced decrease in surface area relative to the respective supports. Sequentially prepared bimetallic catalysts, in which Ni or Fe was first deposited followed by Au, exhibited the largest reductions in surface area.
XRD patterns of monometallic oxidation catalysts supported on MCM-41 and SiO₂ are presented in Figure 2a and b, respectively, while patterns of bimetallic and Au-containing catalysts supported on SiO₂ and MCM-41 are shown in Figure 2c and d. Average crystal sizes of detectable phases were calculated using the Scherrer equation and are summarized in Table 1. All supported catalysts exhibited a broad peak at 15 – 35 ° (maximum at 2θ = 22 °), corresponding to amorphous silica [16,26].
Chromium oxide supported catalysts showed diffraction peaks at 2θ = 24.5 °, 33.6 °, 36.2 °, 41.5 °, 50.3 °, 55 °, 63.5 ° and 65.2 °, characteristic of the rhombohedral Cr₂O₃ phase [27]. The average crystal size of Cr₂O₃ was estimated to be ~21 nm on MCM-41 and ~26 nm on SiO₂ (Table 1), comparable to or smaller than values reported for similar systems [25,28]. NiO-supported catalysts presented five distinct diffraction peaks at 2θ = 37 °, 43 °, 63 °, 75 °, and 79 °, corresponding to cubic NiO [29]. The average crystal size of NiO particles was smaller on MCM-41 than on SiO₂, indicating the beneficial effect of the mesoporous structure on nickel dispersion [30]. Diffraction peaks observed for 5Cu/SiO₂ at 2θ = 32.6 ° (111), 35.6 ° (002), 38.8 ° (111), 48.8 ° (202), 53.5 ° (020), 58.3 ° (202), 61.6 ° (113), 66.5 ° (022), and 68.1 ° (220) corresponded to the monoclinic CuO phase [31]. For 5Cu/MCM-41, only the two most intense peaks at 35.6 ° and 38.8 ° were detected, implying a higher dispersion of CuO species [32].
Among the examined oxides, iron oxide exhibited the smallest crystal size and thus the highest dispersion. Weak and broad peaks at 2θ = 32.1 °, 35.5 °, and 54.1 °, corresponding to rhombohedral α-Fe₂O₃ [33], were observed only for Fe₂O₃ on SiO₂, while no diffraction peaks were detected for Fe₂O₃ on MCM-41, suggesting the formation of very small particles (< 4 nm) or the absence of bulk Fe₂O₃ crystallites [16]. Deposition of bimetallic transition metal oxide catalysts on both supports favored metal dispersion, as the coexistence of two metals led to smaller crystal sizes compared to the respective monometallic catalysts (Figure 2c, Table 1).
XRD patterns of Au-containing catalysts exhibited four distinct peaks at 2θ = 38.3 °, 44.3 °, 64.5 °, and 77.7 °, corresponding to metallic Au (111), Au (200), Au (220), and Au (311) reflections, respectively [34,35]. Increasing Au loading led to larger Au crystal sizes (6.4 – 15.3 nm, Table 1). For bimetallic Au–NiO or Au–Fe₂O₃ catalysts, ad-ditional peaks corresponding to NiO or Fe₂O₃ phases were also present, though the overlap between diffraction signals prevented reliable estimation of individual crys-tallite sizes (Figure 2d). Overall, both textural and structural analyses indicate that metal deposition, particularly in bimetallic and Au-containing systems, influences crystal size and surface area, with the mesoporous MCM-41 generally promoting high metal dispersion and structural integrity of the siliceous framework.
2.2. Reducibility of Supported Catalysts
Reducibility of the supported catalysts was evaluated by H₂-TPR experiments (Figure 3). As shown, the extent of reduction strongly depends on both the nature of the active metal and the support, reflecting different metal–support interaction phenomena. The degree of reducibility was estimated from the theoretical H₂ consumption required for complete reduction of CuO, Fe₂O₃, and NiO, and the experimentally measured H₂ uptake (Table 2). Siliceous supports were assumed non-reducible, and thus all H₂ consumption was attributed to metal oxide reduction.
For Cu-based catalysts, three reduction peaks were observed for 5Cu/MCM-41 (Figure 3a). The low-temperature peak at ~ 250 °C corresponds to the reduction of highly dispersed Cu²⁺ species, such as isolated ions or small two- and three-dimensional clusters. The peak at ~ 350 °C is attributed to bulk-like CuO phases, while a broader feature at ~ 550 °C suggests the presence of strongly bound or framework-incorporated Cu²⁺ species due to their very small size [26,27,28,29,30,31,32]. In the case of SiO₂-supported CuO catalysts, the main reduction occurs at 344 °C with a broad shoulder at higher temperatures (Figure 3b). The comparison between 5Cu/MCM-41 and 5Cu/SiO₂ reveals easier reduction and higher copper dispersion on the mesoporous MCM-41, consistent with XRD results showing smaller crystallites on this support.
The 5Fe/MCM-41 catalyst exhibited three well-defined reduction steps (Figure 3a): (i) Fe₂O₃ → Fe₃O₄ at ~ 448 °C, (ii) Fe₃O₄ → FeO at ~ 564 °C, and (iii) FeO → Fe at ~ 774 °C, confirming complete reduction of iron oxide to metallic Fe [37]. The corresponding Fe/SiO₂ catalyst showed similar peaks slightly shifted to higher temperatures (Figure 3b). A small additional feature at ~ 216 °C, detected only for Fe/MCM-41, indicates the presence of highly dispersed Fe species on the mesoporous surface [38].
Ni-based catalysts exhibited distinct reduction behavior. The 5Ni/MCM-41 catalyst presented a broad peak centered at ~ 630 °C, initiating at ~ 300 °C with a shoulder near 450 °C attributed to bulk NiO nanoparticles [39]. The higher-temperature portion of the peak corresponds to NiO species strongly interacting with the silica framework or Ni²⁺ ions partially incorporated into it [40]. The higher experimental H₂ consumption relative to the theoretical value supports this interpretation (Table 2). Conversely, Ni/SiO₂ showed a double reduction feature with maxima at 337 °C and 368 °C (Figure 3b), indicating the presence of NiOx species with intermediate valence states. Although reduction occurs at lower temperatures on SiO₂, the overall reducibility is lower, suggesting incomplete NiO reduction.
Co-impregnation of two transition metals (Cu–Ni, Cu–Fe, and Fe–Ni) significantly enhanced reducibility and shifted reduction peaks to lower temperatures (Table 2), consistent with smaller crystal sizes determined by XRD (Table 1). The formation of mixed oxide species and stronger metal–metal interactions likely facilitated hydrogen activation and oxygen removal.
The H₂-TPR profile of 0.3Au/SiO₂ showed no peaks below 100 °C, indicating that Au was predominantly metallic [36]. A weak, broad signal at ~ 230 °C (H₂ uptake = 277 μmol g⁻¹, Table 2) was assigned to ionic Au species interacting with the SiO₂ surface [41]. A second broad feature centered at ~ 670 °C is related to bulk oxygen reduction and formation of lower silicon oxides, consistent with previous studies demonstrating Au-induced support reduction in Au/CeO₂ and Au/TiO₂ systems [42,43]. Incorporation of Au into 5Ni/SiO₂ and 5Fe/SiO₂ notably improved reducibility (Figure 3c), suggesting that even trace of Au (0.3 wt. %) facilitates hydrogen dissociation and promotes the reduction of NiO and Fe₂O₃ well beyond their theoretical H₂ consumption. The shift of reduction peaks to lower temperatures further supports that metallic Au⁰ sites, confirmed by XRD, act as active centers for H₂ activation and nucleation, enabling easier reduction of transition-metal oxides [44].
2.3. Acidity of Supported Catalysts
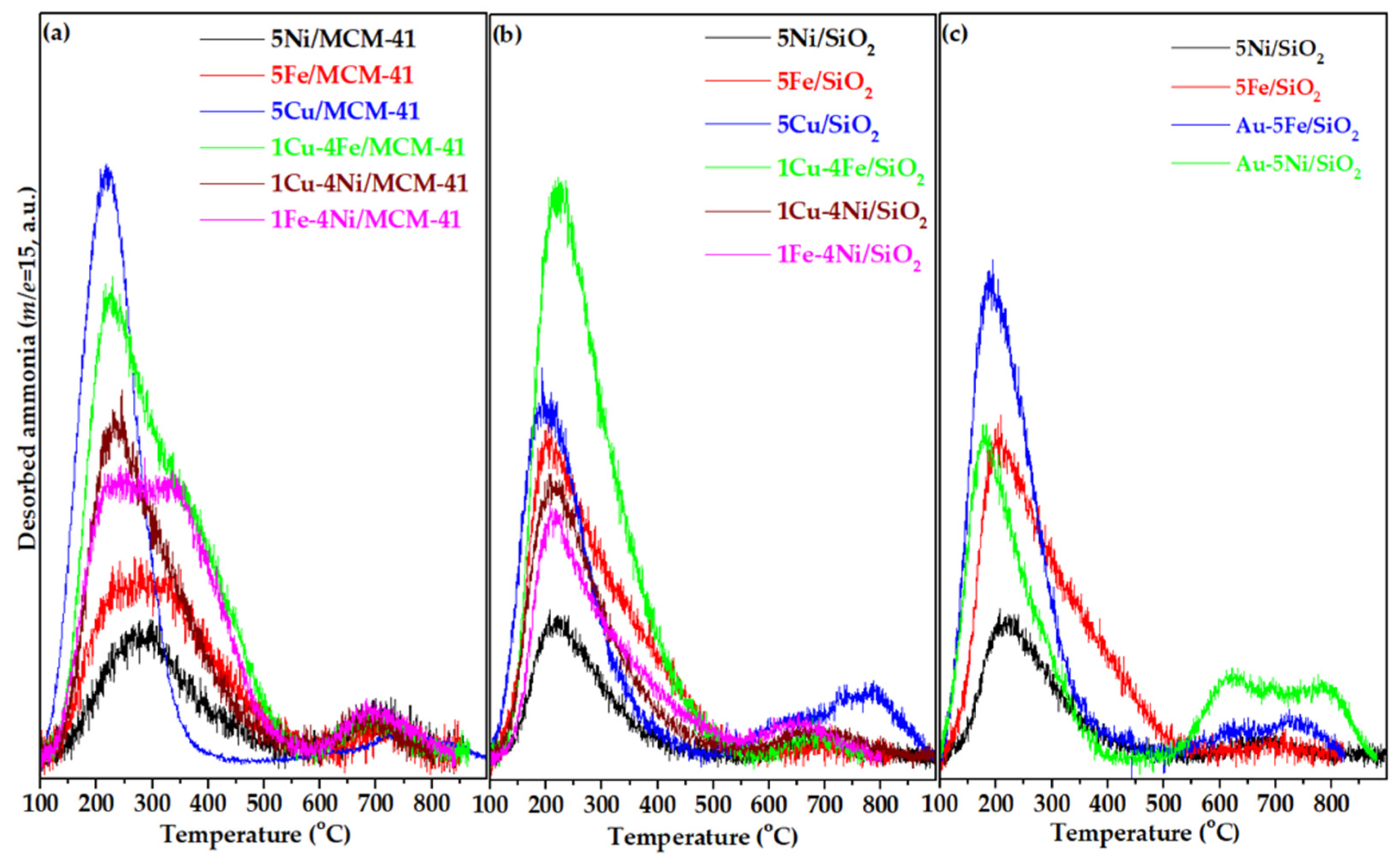
The number and strength of acid sites of the selected catalysts were evaluated by temperature-programmed desorption of ammonia (NH₃ - TPD). Ammonia was used as the probe molecule because of its small size and strong basicity, which allow it to interact with both Brønsted and Lewis acid sites over a wide range of strengths. The amount of ammonia desorbed at characteristic temperatures was taken as a measure of the total number of acid sites, while the temperature range of desorption indicated their strength. According to the literature [45,46], desorption of ammonia in the 100–300 °C range corresponds to weak acid sites, 300–500 °C to medium acid sites, and temperatures above 500 °C to strong acid sites.
The NH₃ - TPD profiles of selected transition metal oxide catalysts supported on MCM-41 and SiO₂, and Au-containing SiO₂-supported catalysts, are presented in Figure 4. The corresponding quantitative data for total acidity and the distribution of weak, medium, and strong acid sites, derived from Gaussian deconvolution, are summarized in Table 2. In all cases, the supported catalysts exhibited similar desorption patterns, characterized by a broad peak extending from 100 to 400 °C—associated with weak and medium acid sites—and a smaller, less intense desorption feature at higher temperatures (700–750 °C), indicative of strong acid sites.
Both pristine supports (MCM-41 and SiO₂) exhibited negligible acidity, with NH₃ uptakes not exceeding ~10 μmol NH₃ g⁻¹ (not shown). These few acid sites likely originate from isolated surface hydroxyl groups or silanol nests on the support surface [47]. Metal oxide deposition markedly increased surface acidity, yielding values between 19 and 70 μmol NH₃ g⁻¹, depending primarily on the nature of the deposited metal (Table 2). Among the monometallic catalysts, CuO deposition generated the highest number of acid sites, while NiO gave rise to a smaller number of sites, mostly of weak and medium strength. CuO/SiO₂ also showed the highest concentration of strong acid sites, as reflected by a broad desorption feature above 550 °C with two distinct maxima at 635 and 789 °C.
Partial substitution of Ni by Cu (1 wt.% Cu – 4 wt.% Ni instead of 5 wt.% Ni) or by Fe (1 wt.% Fe – 4 wt.% Ni) resulted in the formation of additional acid sites compared with the corresponding monometallic catalysts. A similar effect was observed when Fe was partially replaced by Cu, in which case the total acidity reached its maximum value. Moreover, in all bimetallic catalysts, the main desorption peaks shifted slightly to lower temperatures, suggesting an overall increase in the proportion of weaker acid sites. This can be rationalized by the smaller crystallite sizes of bimetallic species (as confirmed by XRD), which generate a higher number of metals – oxygen interface sites with relatively weaker acidity. The enhancement in acidity was most pronounced for the Fe-containing bimetallic catalysts.
2.4. Evaluation of Catalytic Performance Under Standard Reaction Conditions
The catalytic activity of the synthesized catalysts toward the wet oxidation of glucose was evaluated under standard reaction conditions (80 °C, 60 min; Table 3). Before the reaction, pH was adjusted to 8 with NaOH to enhance the reaction rate and prevent catalyst deactivation [48,49] . Hydrogen peroxide (H₂O₂) was used as the oxidant at a molar ratio to glucose equal to 40, offering the advantage of high solubility and minimized mass-transfer limitations [49]. The glucose-to-metal molar ratio was maintained at 64 for transition-metal catalysts and at 1000 for gold-containing systems, reflecting the higher intrinsic reactivity of Au. A blank experiment conducted under the same conditions, resulted in 35 % glucose conversion with 13 % gluconic acid yield, along with minor formation of lactic (2.5 % yield) and formic (3 % yield) acids. Trace amounts of organic acids glucuronic, 5-keto-D-gluconic, oxalic, and tartronic, were also detected, indicating that hydrogen peroxide alone can promote partial oxidative degradation of glucose in aqueous medium. Nevertheless, the overall oxidation extent remains limited in the absence of catalyst, in agreement with previous reports [49].
Under the applied reaction conditions, all tested materials catalyzed glucose oxidation to carboxylic acids to different extents (Table 3). Glucose conversion was above 40 % in all cases, with gluconic acid being the primary product. Additional oxidation and degradation products, including glucaric, formic, lactic, oxalic, 5-keto-D-gluconic, and tartronic acids, were also detected, consistent with the established oxidation pathways [12,50,51] (Table 3). Gluconic acid is formed via oxidation of the aldehyde group (-CHO) on C1 of glucose to a carboxylic acid (-COOH). Further oxidation of the primary alcohol (-OH) at C6 of gluconic acid may lead to the formation of glucuronic acid, which may subsequently oxidize to glucaric acid. Additionally, glucuronic acid can undergo isomerization to form 5-keto gluconic acid (Scheme 1). Side reactions, such as glucose isomerization to fructose under alkaline conditions (not detected in this study) and C–C cleavage of gluconic acid intermediate aldehydes (undetected by our analytical methods) followed by further oxidation to lactic, oxalic, tartronic and formic acids, may also occur (Scheme 1) [12,50,51].
2.4.1. Monometallic Catalysts Supported on SiO₂ and MCM-41
Monometallic catalysts containing transition metal oxides (Cr, Cu, Fe, and Ni) supported on SiO2 and MCM-41 demonstrated notable activity in glucose oxidation, achieving 40 – 100 % conversion in all cases (Table 3). The type of support did not significantly affect the metal’s oxidative performance, as both glucose conversion and gluconic acid selectivity followed similar trends across metals on either support.
CuO-supported catalysts, on both SiO2 and MCM-41, achieved near-complete glucose conversion, attributed to a combination of higher acidity (Section 2.3) and increased Cu leaching (Section 2.5, Table 4), indicating contributions from homogeneous and heterogeneous catalysis. The main products were lactic, oxalic, and formic acids, with respective selectivities of 2.7, 1.3, and 13.3 % for 5Cu/SiO2 and 5.2, 0.5, and 19.5 % for 5Cu/MCM-41. Minor CO2 and CO formation were also detected (data not shown). These results suggest that oxidation extended beyond the C1 aldehyde group, proceeding via deep oxidation pathways facilitated by electrophilic lattice oxygen species (O2−, O22−, O−) on the copper oxide surface, while nucleophilic oxygen (O2−) contributed to both selective and non-selective reactions [52]. Strong adsorption of reaction products on the lattice oxygen further promotes secondary oxidation, reducing gluconic acid selectivity.
For the other transition metals, glucose conversion followed the order Fe > Cr > Ni on both supports, reflecting differences in acid site density, whereas gluconic acid selectivity followed the inverse trend Ni > Cr > Fe, with a maximum gluconic acid yield of 18 % for 5Fe/SiO2. NiO catalysts exhibited increased gluconic acid selectivity due to the presence of weaker nucleophilic oxygen species, facilitating desorption and selective oxidation[53].
Au was employed as a reference noble metal on SiO2 and MCM-41. Au/SiO2 achieved 46.1 % glucose conversion with 36.4 % gluconic acid selectivity, while Au/MCM-41 reached 41.0 % conversion with 40.3 % selectivity at (Table 4). The higher selectivity on MCM-41 is attributed to smaller Au particle sizes, favoring gluconic acid formation [54]. Both Au catalysts produced glucaric acid (5.4 - 6.0 % selectivity) and minor formic acid (< 8 %), and additionally exhibited isomerization to 5-keto-D-gluconic and tartronic acids, transformations that were not observed for the transition-metal catalysts. Gold, as a noble metal, is well recognized for its high selectivity in glucose oxidation to gluconic acid [12,51]. However, due to its high cost, strategies to minimize Au loading, such as partial substitution with transition metals, are desirable to maintain both catalytic efficiency and economic feasibility.
2.4.2. Bimetallic Transition Metal Oxide Catalysts Supported on SiO₂ and MCM-41
Given the high oxidation activity of CuO species, bimetallic transition-metal oxide catalysts were investigated to enhance gluconic acid yield and selectivity. Catalysts containing Cu, Fe, and Ni (overall metal loading ~5 wt.%) were prepared, with Fe and Ni selected for their higher selectivity toward gluconic acid relative to Cu. Experimental results (Table 3) showed that even 1 wt.% Cu in bimetallic catalysts led to complete glucose conversion, predominately forming lactic, oxalic, and formic acids, without detectable gluconic or glucaric acids, accompanied by CO2 and CO formation. This behavior is attributed to the strong electrophilic lattice oxygen species of CuO, which promote deep, non-selective oxidation pathways [52].
In contrast, the Fe–Ni combination improved glucose conversion compared to the corresponding monometallic catalysts, likely due to enhanced dispersion of metal oxides, formation of smaller particles, and an increased number of acid sites via synergistic effects (Table 1 and Table 2). Nonetheless, gluconic acid selectivity and yield remained low due to further oxidation to smaller acids and gases, although minor glucaric acid (<3.5 %) was detected. Among the bimetallic combinations, the 1 wt.% Fe – 4 wt.% Ni catalyst demonstrated the most favorable performance for gluconic acid formation, associated with the generation of weaker, more selective adsorbed oxygen species compared to monometallic CuO catalysts.
Overall, the results highlight the balance between catalyst activity and selectivity: monometallic CuO favors complete glucose oxidation, Ni- and Fe-based catalysts enhance gluconic acid selectivity, and bimetallic combinations provide intermediate behavior, modulated by synergistic oxygen species and dispersion effects.
2.5. Catalyst Stability
Catalyst stability is a key factor influencing performance, affected by hydrothermal conditions and reaction pH. The leaching of transition metals (Cr, Cu, Fe, Ni) and Au from the catalysts was evaluated by ICP-AES (Table 4). All transition-metal catalysts exhibited some leaching, with stability strongly dependent on both metal type and support.
Among the supports tested, transition-metal oxides deposited on SiO2 were more stable than those on MCM-41. Metal-support interactions followed the trend Ni > Fe > Cr > Cu for both SiO2- and MCM-41- supported catalysts (Table 4). In particular, Cu and Cr were the least stable catalysts, with leaching from the support exceeding 26 % and 50 %, respectively. This indicates that, although the metals were successfully deposited on the silica surface, they did not form sufficiently strong bonds and partially migrated into the reaction medium, participating in homogeneous catalysis and contributing to higher glucose conversion. On the contrary, Ni and Fe presented minimal leaching, especially from SiO2. NiO catalysts supported on SiO2 and MCM-41 presented the least leaching of 3.3 % and 7.0 %, respectively.
Bimetallic catalysts followed a similar trend: those containing Cu were less stable than the corresponding Fe–Ni-supported catalysts. While active, bimetallic catalysts showed leaching from the support exceeding 50 % in some cases (Table 4), indicating significant homogeneous catalysis without achieving full glucose conversion. The only supported catalysts showing no detectable leaching were those containing Au, which also demonstrated high activity toward gluconic acid production (Table 3).
In summary, among the various oxidation catalysts tested, 5Ni/SiO2 and 5Fe/SiO2 showed relatively high tolerance to solubility during the reaction. Comparing these two, iron oxide achieved high glucose conversion, while maintaining gluconic acid selectivity around 23 %, yielding the highest amount of product. NiO-supported catalysts proved more selective toward gluconic acid. The Fe-Ni bimetallic system was the only combination showing preference for both gluconic and glucaric acid production, despite very high glucose conversion.
Employing gentler reaction conditions is recommended to further enhance catalyst stability and minimize side reactions. Gold catalysts were the most stable and selective, although deposition on MCM-41 was less efficient, resulting in slightly lower conversion but higher gluconic acid selectivity compared to Au/SiO₂. Future work should focus on improved Au deposition techniques on MCM-41, including heterometal incorporation to enhance support charge [55]. Based on these findings, Au, Fe, and Ni supported on SiO₂ were selected for further investigation.
2.5. Evaluation of Au-Supported Catalysts with or Without Transition Metals on SiO₂
To reduce gold loading while maintaining its remarkable stability and high catalytic activity, a series of Au-containing catalysts supported on SiO₂ were synthesized, with metal loadings ranging from 1 to 0.3 wt.% (Section 4.3). Their performance in glucose oxidation was evaluated under the standard reaction conditions applied for catalyst screening (80 °C, 60 min, pH 8, H₂O₂/Glucose molar ratio 40, Glucose 2 wt.%, Catalyst 0.2 wt.%). As shown in Figure 5, reducing the Au content to 0.5 wt.% had minimal effect, with glucose conversion (46 ± 0.1 %), gluconic acid selectivity (37 ± 1 %) and yield (17 ± 1 %) remaining nearly unchanged. Further decrease the Au loading to 0.3 wt.% slightly decreased glucose conversion to 42 % and gluconic acid yield to 15 %, without affecting selectivity. The main by-products, were glucaric, formic, lactic, oxalic, 5-keto-D-gluconic, and tartronic acids, each with selectivity below 10 %. ICP-AES analysis confirmed that all Au/SiO₂ catalysts were stable, with no detectable leaching. These results indicate that 0.3 wt. %. represents the minimum Au loading that preserves catalytic activity
To improve gluconic acid selectivity in the most promising non-noble catalysts supported on SiO₂ (5Ni/SiO₂ and 5Fe/SiO₂), bimetallic catalysts were prepared by incorporating Au at 0.3 wt.%, denoted as Au-5Ni/SiO₂ and Au-5Fe/SiO₂, respectively (Section 4.3). Under standard reaction conditions (Figure 5), the addition of Au to 5Ni/SiO₂ did not significantly alter catalytic performance, with glucose conversion (43 %), gluconic acid selectivity (32 %), and yield (14 %) remaining comparable to monometallic 5Ni/SiO₂ (Table 3). However, stability improved, as only minor Au leaching (2.0 %, 2.1 ppm) was detected, indicating enhanced metal-support interaction. TPR-H₂ analysis further confirmed the Au–Ni synergy, showing an “over-reduction” effect, where both NiOx and surface oxygen species of the support were more extensively reduced.
In contrast, deposition of Au on 5Fe/SiO₂ positively influenced selectivity, increasing it from 22.5 % to 30.5 %, while glucose conversion slightly decreased from 79.5 % to 76.6 %, leading to a rise in gluconic acid yield from 17.9 % to 23.4 %. This effect is attributed to synergistic oxidation between Au and Fe. Minor formation of by-products (glucaric, formic, lactic, oxalic, 5-keto-D-gluconic, and tartronic acids) was observed, each below 10 % selectivity (Figure 5). However, Au incorporation did not enhance the Fe-support interaction, as Fe leaching increased from 9.1 % (5Fe/SiO₂) to 12.3 % (Au-5Fe/SiO₂, 9.1 ppm detected).
Further study of the effect of reaction parameters — including temperature, time, oxidant type, glucose content, and pH — along with alternative synthetic strategies to enhance metal-support interactions, is required. Additionally, exploring the elimination of Au while retaining the synergistic effect with Ni represents a key objective for future work.
4. Materials and Methods
4.1. Materials and Reagents
D-Glucose was supplied by Sigma-Aldrich and used without further purification. NaOH was also purchased by Sigma-Aldrich and used to control pH of the reaction medium and synthesis of hydrotalcite-like materials. Ammonia solution (NH4OH, 25 % in water, Sigma-Aldrich) was used as pH regulator and TEtraethyl OrthoSilicate (TEOS, Si(OC2H5)4, Sigma-Aldrich) as the silicon source for MCM-41 synthesis. Hydrogen peroxide (H2O2, 30 wt. % in water) and sodium borohydride (NaBH4) used as reducing agents, were purchased from Honeywell. Commercial SiO2 used as support and labeled as SiO2 (Sigma-Aldrich). CetylTrimethylAmmonium Bromide (CTAB, CH3(CH2)15N(Br)(CH3)3, Sigma-Aldrich) and PolyVinyl Alcohol (PVA, [-CH2CHOH-]n, Sigma-Aldrich) were used as templates, while Sodium Carbonate (Na2CO3, Sigma-Aldrich) was used as anion source for the synthesis of hydrotalcite-like materials.
Solids of Copper (II) nitrate trihydrate (Cu(NO3)2 · 3H2O), Chromium (III) nitrate nonahydrate (Cr(NO3)3 · 9H2O), Nickel (II) nitrate hexahydrate (Ni(NO3)2 · 6 H2O), Iron (III) nitrate nonahydrate (Fe(NO3)2·6H2O), and solution of Gold (III) chloride solution (30 wt. % of HAuCl4 in dilute HCl) used as metal precursors were also provided by Sigma-Aldrich.
4.2. Preparation of Mesoporous MCM-41
An alternative procedure was developed for the synthesis of pure mesoporous silica with hexagonal pore structure and narrow pore size distribution known as MCM-41 similar with the typical synthesis method based on self-assembly mechanisms combined with the sol-gel process [56] excluding only the long-lasting ageing step under hydrothermal conditions by replacing it with an ageing step with milder conditions. As a surfactant, aqueous solution of CTAB was used while TEOS represented the silicon source. The simplified method was based on previously reported study [57] with some alterations. In detail, 16.4 g of CTAB were fully dissolved in 820 g of double distilled water. The adjustment of pH was achieved by the appropriate addition of aqueous ammonia in order to retain pH value at 10. Subsequently, the silicon source was added (TEOS) and the suspension was left under stirring for 2 h at room temperature. This mixture had the following molar composition: 1 TEOS:0.147C16TABr:3.04 NH3:160H2O. The synthesis product was recovered by filtration and washed with ~2 L of distilled water. After drying at 100 °C, the samples were calcined in air at 550 °C for 11 h using a heating rate of 2 °C/ min. The obtained white powder was labeled as MCM-41.
4.3. Preparation of Supported Metal Catalysts
Deposition of varying monometallic (Cu, Cr, Ni and Fe) or bimetallic (combination of Ni, Fe or Cu) transition metals via typical wet impregnation with aqueous solutions of metal nitrates at appropriate amounts was performed on the surface of MCM-41 and SiO2 supports. Based on previous studies [58,59], the total metal loading was decided to be of 5 wt. %, while the final catalysts were obtained after calcination at 500 °C for 3 h using a heating rate of 5 °C/min under air.
In addition, Au was deposited on both silicious supports via polyvinyl alcohol (PVA)-protected method with parallel in-situ reduction by using sodium borohydride as a reducing agent [60]. Since gold has higher oxidation activity compared to transition metals, metal composition varied between 0.3-1 wt. %. In a typical procedure, 0.2 g of gold (III) chloride were dissolved in 15 mL of double distilled (DD) H2O. A solution containing 0.0039 g of PVA in 15 mL of DD water was then added to the first solution. Few minutes later, the resulting solution was added to a suspension containing 3.5 g of SiO2 in 60 mL of DD water. The resulting mixture was stirred for 2 h. A freshly prepared solution of sodium borohydride (0.01 g in 5 mL of DD water) was added dropwise to the suspension and stirred for additional 16-20 hours. Filtration and washing were followed. The final powders were obtained after overnight drying at 120 °C and labeled as xAu/SiO2 and xAu/MCM-41 respectively where x represents the nominal wt. % value of Au.
Finally, Au was also combined with Ni and Fe oxidation catalysts supported on SiO2. At first, deposition of Ni or Fe with 5 wt.% via wet impregnation on the surface of SiO2 was executed, followed by calcination of the resulting catalysts (5Ni/SiO2 or 5Fe/SiO2) at 500 oC, while a sequential deposition of 0.3 wt. % Au was performed via (PVA)-protected method with parallel in-situ reduction by NaBH4, as described in the previous paragraph.
4.4. Catalysts Characterization
Inductive Coupled Plasma - Atomic Emission Spectroscopy (ICP-AES) was used for the determination of the metal chemical composition. N2 adsorption-desorption at –196 °C was performed on an Automatic Volumetric Sorption Analyzer (Autosorb-1, Quantachrome) for the determination of surface area (BET method), pore volume and pore size distribution (BJH method) of samples previously outgassed at 250 °C for 16 h under 1.33 x 10-1 Pa vacuum. Powder X-ray diffraction (XRD) experiments were performed on a Siemens D-500 X-ray diffractometer with Cu Kα radiation in the 2θ range of 5-85 °.
Reducibility was studied by temperature-programmed reduction with H2 (TPR-H2). In a typical experiment, 0.1 g of the catalyst sample was loaded in a fixed bed quartz reactor and pre-treated at 500 °C (for supported oxidation catalysts) or 400 °C (for mixed oxides catalysts) for one hour. The catalyst was then cooled down to 35-40 °C and TPR-H2 analysis was carried out from 40 to 850 °C at a heating rate of 10 °C/min in 5 % H2/He flow. The composition of the exit gas was monitored online with a quadrupole mass analyzer (Omnistar, Balzer). The m/z fragments registered were as follows: H2 = 2, H2O = 18 και He = 4. Quantitative analysis of the consumed H2 was based on m/z = 2.
The acidic characteristics of selected catalysts were studied with NH3 - temperature programmed desorption (TPD-NH3). In a typical experiment, 0.1 g of the sample was loaded in a fixed bed quartz reactor and pre-treated at 500 °C (for supported oxidation catalysts) or 400 °C (for mixed oxide catalysts) in He for one hour and then the catalyst was cooled down to 100 °C under He flow. Adsorption of ammonia was then performed with a flow of 5 % NH3/He for 1 h at 100 °C. After flushing with pure helium at 100 °C for 12 hours to remove the physiosorbed ammonia, TPD analysis was carried out from 100 to 850 °C at a heating rate of 10 °C/min in helium. The composition of the exit gas was monitored on line with a quadrupole mass analyser (Omnistar, Balzer). The m/z fragments registered were as follows: NH3 = 17, 16, 15; H2O = 18; N2 = 28; NO = 30; N2O = 44. Quantitative analysis of the desorbed ammonia was based on m/z = 15.
4.5. Catalyst Performance Evaluation and Product Analysis
Τhe catalytic oxidation of glucose took place in a batch, stirred, autoclave reactor (C-276 Parr Inst., USA) using H2O2 as oxidant, after pH adjustment to 8.0 upon addition of NaOH 0.05 mM. In a typical experiment, an aqueous solution of glucose and the corresponding catalyst were charged into the reactor and heated to the desired temperature. Zero time was recorded and the reaction was allowed to proceed for a given time under continuous stirring. The pH was not further controlled during the reaction. At the end of the reaction time the reactor vessel was cooled rapidly and the liquid product obtained after filtration was analyzed by Ion Chromatography (ICS-5000, Dionex, USA).
The quantification was based on external calibration, using standard solutions of sugars (glucose, mannose, xylose, fructose, galactose, arabinose and rhamnose), sugar alcohols (sorbitol and mannitol), hydroxymethylfurfural (HMF) and organic acids (gluconic, lactic, glycolic, propionic, formic, glucuronic, 5-keto-gluconic, glucaric, tartaric, tartronic and oxalic acid). The analysis of sugars was performed using a CarboPac PA1 (10 μm, 4 × 250 mm) column and guard column (10 μm, 4 × 30 mm) connected to a pulsed amperometric detector (PAD). The eluent was 20 mM NaOH at a 0.6 ml/min flow rate and the total analysis time was 75 min. The analysis of the organic acids was performed on an AS-11 (9 μm, 4 × 250 mm) column and pre-column (9 μm, 4 × 30 mm) connected to a conductivity detector (CD). The eluent was 0.5 mM NaOH at a 1 ml/min flow rate and the total analysis time was 65 min.
The conversion of glucose, the yields and the selectivity of the products (mol based) were calculated according to the following Eqs. (1), (2) and (3):
The stability of the synthesized catalysts on terms of metal leaching was evaluated by analyzing the liquid product obtained after the reaction for the presence of Au, Cu, Cr, Fe and/or Ni by means of ICP-AES analysis.
5. Conclusions
Catalytic evaluation of low-cost transition metal oxides (Cr2O3, CuO, Fe2O3, NiO) supported on siliceous materials (SiO2, MCM-41) revealed that glucose oxidation proceeds via multiple pathways, strongly depending on both the type of metal and the support. Metal-support interactions, assessed through metal leaching in the liquid phase, varied from very low (ca. 2 %) to as high as 50 % for Cu catalysts, highlighting the coexistence of heterogeneous and homogeneous catalytic contributions.
NiO and Fe2O3 supported on SiO2, tested for glucose oxidation under aqueous conditions for the first time, exhibited promising stability. Among them, NiO/SiO2 promoted selective gluconic acid formation most effectively. Incorporation of low-load Au loading (0.3 wt.%) into 5Ni/SiO2 (Au-5Ni/SiO2) further enhanced catalyst stability without compromising catalytic activity. Bimetallic catalysts demonstrated distinct synergistic effects: Au addition to Ni-based catalysts reinforced stability, whereas Au incorporation into Fe-based catalysts improved gluconic acid selectivity, attributed to synergistic oxidation activity, though Fe leaching slightly increased.
Overall, these results indicate that minimal Au incorporation can strategically enhance both catalyst stability and selectivity, while careful design of bimetallic transition-metal catalysts allows fine-tuning of catalytic performance. Future work should focus on optimizing reaction parameters—including temperature, time, pH, glucose concentration, and oxidant type — to further enhance activity and minimize metal leaching. The combination of high oxidation activity, tunable selectivity, and relative robustness makes these supported transition metal oxides attractive candidates for glucose oxidation and other oxidative processes, providing a cost-effective and sustainable approach to heterogeneous catalysis.
Author Contributions
Writing—original draft preparation, S.K. and A.M.; Conceptualization, Investigation and Methodology, S.K., A.M. and C.M., writing—review and editing, S.K., A.M. and A.L.; data curation, S.K. and A.M., supervision, S.K. and A.L.; project administration, S.K. and A.L.; funding acquisition and resources, A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under grant agreement No 287-128035/I2.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Mehtiö, T.; Toivari, M.; Wiebe, M.G.; Harlin, A.; Penttilä, M.; Koivula, A. Production and Applications of Carbohydrate-Derived Sugar Acids as Generic Biobased Chemicals. Crit. Rev. Biotechnol. 2016, 36, 904–916. [CrossRef]
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A.; Eliot, D.; Lasure, L.; Jones, S. Top Value Added Chemicals from Biomass Volume I - Results of Screening for Potential Candidates from Sugars and Synthesis Gas. Us Nrel 2004, 76 pages. [CrossRef]
- Shcherbakova-Sandu, M.P.; Meshcheryakov, E.P.; Gulevich, S.A.; Kushwaha, A.K.; Kumar, R.; Sonwane, A.K.; Samal, S.; Kurzina, I.A. Use of Glucose Obtained from Biomass Waste for the Synthesis of Gluconic and Glucaric Acids: Their Production, Application, and Future Prospects. Molecules 2025, 30. [CrossRef]
- Kornecki, J.F.; Carballares, D.; Tardioli, P.W.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Alcántara, A.R.; Fernandez-Lafuente, R. Enzyme Production of D-Gluconic Acid and Glucose Oxidase: Successful Tales of Cascade Reactions. Catal. Sci. Technol. 2020, 10, 5740–5771. [CrossRef]
- Zhang, Z.; Huber, G.W. Catalytic Oxidation of Carbohydrates into Organic Acids and Furan Chemicals. Chem. Soc. Rev. 2018, 47, 1351–1390. [CrossRef]
- Arias, P.L.; Cecilia, J.A.; Gandarias, I.; Iglesias, J.; López Granados, M.; Mariscal, R.; Morales, G.; Moreno-Tost, R.; Maireles-Torres, P. Oxidation of Lignocellulosic Platform Molecules to Value-Added Chemicals Using Heterogeneous Catalytic Technologies. Catal. Sci. Technol. 2020, 10, 2721–2757. [CrossRef]
- Guo, S.; Fang, Q.; Li, Z.; Zhang, J.; Zhang, J.; Li, G. Efficient Base-Free Direct Oxidation of Glucose to Gluconic Acid over TiO 2 -Supported Gold Clusters. Nanoscale 2019, 11, 1326–1334. [CrossRef]
- Cao, Y.; Liu, X.; Iqbal, S.; Miedziak, P.J.; Edwards, J.K.; Armstrong, R.D.; Morgan, D.J.; Wang, J.; Hutchings, G.J. Base-Free Oxidation of Glucose to Gluconic Acid Using Supported Gold Catalysts. Catal. Sci. Technol. 2016, 6, 107–117. [CrossRef]
- Wang, Y.; Van De Vyver, S.; Sharma, K.K.; Román-Leshkov, Y. Insights into the Stability of Gold Nanoparticles Supported on Metal Oxides for the Base-Free Oxidation of Glucose to Gluconic Acid. Green Chem. 2014, 16, 719–726. [CrossRef]
- Delidovich, I. V.; Moroz, B.L.; Taran, O.P.; Gromov, N. V.; Pyrjaev, P.A.; Prosvirin, I.P.; Bukhtiyarov, V.I.; Parmon, V.N. Aerobic Selective Oxidation of Glucose to Gluconate Catalyzed by Au/Al2O3 and Au/C: Impact of the Mass-Transfer Processes on the Overall Kinetics. Chem. Eng. J. 2013, 223, 921–931. [CrossRef]
- Lee, J.; Saha, B.; Vlachos, D.G. Pt Catalysts for Efficient Aerobic Oxidation of Glucose to Glucaric Acid in Water. Green Chem. 2016, 18, 3815–3822. [CrossRef]
- Zhang, Q.; Wan, Z.; Yu, I.K.M.; Tsang, D.C.W. Sustainable Production of High-Value Gluconic Acid and Glucaric Acid through Oxidation of Biomass-Derived Glucose: A Critical Review. J. Clean. Prod. 2021, 312, 127745. [CrossRef]
- Amaniampong, P.N.; Trinh, Q.T.; Wang, B.; Borgna, A.; Yang, Y.; Mushrif, S.H. Biomass Oxidation: Formyl C-H Bond Activation by the Surface Lattice Oxygen of Regenerative CuO Nanoleaves. Angew. Chemie - Int. Ed. 2015, 54, 8928–8933. [CrossRef]
- Jin, X.; Zhao, M.; Shen, J.; Yan, W.; He, L.; Thapa, P.S.; Ren, S.; Subramaniam, B.; Chaudhari, R. V. Exceptional Performance of Bimetallic Pt1Cu3/TiO2 Nanocatalysts for Oxidation of Gluconic Acid and Glucose with O2 to Glucaric Acid. J. Catal. 2015, 330, 323–329. [CrossRef]
- Amaniampong, P.N.; Jia, X.; Wang, B.; Mushrif, S.H.; Borgna, A.; Yang, Y. Catalytic Oxidation of Cellobiose over TiO2 Supported Gold-Based Bimetallic Nanoparticles. Catal. Sci. Technol. 2015, 5, 2393–2405. [CrossRef]
- Karakoulia, S.A.; Triantafyllidis, K.S.; Tsilomelekis, G.; Boghosian, S.; Lemonidou, A.A. Propane Oxidative Dehydrogenation over Vanadia Catalysts Supported on Mesoporous Silicas with Varying Pore Structure and Size. Catal. Today 2009, 141, 245–253. [CrossRef]
- Zhou, X.G.; Yu, X.Q.; Huang, J.S.; Li, S.G.; Li, L.S.; Che, C.M. Asymmetric Epoxidation of Alkenes Catalysed by Chromium Binaphthyl Schiff Base Complex Supported on MCM-41. Chem. Commun. 1999, 41, 1789–1790. [CrossRef]
- Wisniewska, J.; Sobczak, I.; Ziolek, M. Gold Based on SBA-15 Supports – Promising Catalysts in Base-Free Glucose Oxidation. Chem. Eng. J. 2021, 413. [CrossRef]
- Kumar, A.; Kumar, V.P.; Vishwanathan, V.; Chary, K.V.R. Influence of Preparation Methods of Nano Au/MCM-41 Catalysts for Vapor Phase Oxidation of Benzyl Alcohol. J. Nanosci. Nanotechnol. 2015, 15, 9944–9953. [CrossRef]
- Torres, C.C.; Alderete, J.B.; Pecchi, G.; Campos, C.H.; Reyes, P.; Pawelec, B.; Vaschetto, E.G.; Eimer, G.A. Heterogeneous Hydrogenation of Nitroaromatic Compounds on Gold Catalysts: Influence of Titanium Substitution in MCM-41 Mesoporous Supports. Appl. Catal. A Gen. 2016, 517, 110–119. [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [CrossRef]
- Szegedi, Á.; Popova, M.; Mavrodinova, V.; Urbán, M.; Kiricsi, I.; Minchev, C. Synthesis and Characterization of Ni-MCM-41 Materials with Spherical Morphology and Their Catalytic Activity in Toluene Hydrogenation. Microporous Mesoporous Mater. 2007, 99, 149–158. [CrossRef]
- Sánchez-Velandia, J.E.; Villa, A.L. Isomerization of A- and Β- Pinene Epoxides over Fe or Cu Supported MCM-41 and SBA-15 Materials. Appl. Catal. A Gen. 2019, 580, 17–27. [CrossRef]
- Cho, K.S.; Kim, B.J.; Kim, S.; Kim, S.H.; Park, S.J. Reduction Behaviors of Nitric Oxides on Copper-Decorated Mesoporous Molecular Sieves. Bull. Korean Chem. Soc. 2010, 31, 100–103. [CrossRef]
- Li, K.T.; Weng, W.T. Ethylene Polymerization over Cr/MCM-41 and Cr/MCM-48 Catalysts Prepared by Chemical Vapor Deposition. J. Taiwan Inst. Chem. Eng. 2009, 40, 48–54. [CrossRef]
- Lee, B.; Ma, Z.; Zhang, Z.; Park, C.; Dai, S. Influences of Synthesis Conditions and Mesoporous Structures on the Gold Nanoparticles Supported on Mesoporous Silica Hosts. Microporous Mesoporous Mater. 2009, 122, 160–167. [CrossRef]
- Khan, I.A.; Ullah, S.; Nasim, F.; Choucair, M.; Nadeem, M.A.; Iqbal, A.; Badshah, A.; Nadeem, M.A. Cr2O3-Carbon Composite as a New Support Material for Efficient Methanol Electrooxidation. Mater. Res. Bull. 2016, 77, 221–227. [CrossRef]
- Elías, V.; Sabre, E.; Sapag, K.; Casuscelli, S.; Eimer, G. Influence of the Cr Loading in Cr/MCM-41 and TiO 2/Cr/MCM-41 Molecular Sieves for the Photodegradation of Acid Orange 7. Appl. Catal. A Gen. 2012, 413–414, 280–291. [CrossRef]
- Vora, B. V. Development of Catalytic Processes for the Production of Olefins. Trans. Indian Natl. Acad. Eng. 2023, 8, 201–219. [CrossRef]
- Hsu, C.-H.; Wang, Y.-L.; An-Nan Ko Liquid Phase Hydrogenation of t, t, c-1, 5, 9-Cyclododecatriene over Ni/MCM-41 and Ni/SiO2 Catalysts. J. Chinese Chem. Soc. 2009, 56, 908–915.
- Bouazizi, N.; Bargougui, R.; Oueslati, A.; Benslama, R. Effect of Synthesis Time on Structural, Optical and Electrical Properties of CuO Nanoparticles Synthesized by Reflux Condensation Method. Adv. Mater. Lett. 2015, 6, 158–164. [CrossRef]
- Zhang, G.; Long, J.; Wang, X.; Zhang, Z.; Dai, W.; Liu, P.; Li, Z.; Wu, L.; Fu, X. Catalytic Role of Cu Sites of Cu/MCM-41 in Phenol Hydroxylation. Langmuir 2010, 26, 1362–1371. [CrossRef]
- Supattarasakda, K.; Petcharoen, K.; Permpool, T.; Sirivat, A.; Lerdwijitjarud, W. Control of Hematite Nanoparticle Size and Shape by the Chemical Precipitation Method. Powder Technol. 2013, 249, 353–359. [CrossRef]
- Zhu, H.; Lee, B.; Dai, S.; Overbury, S.H. Coassembly Synthesis of Ordered Mesoporous Silica Materials Containing Au Nanoparticles. Langmuir 2003, 19, 3974–3980. [CrossRef]
- Yin, H.; Ma, Z.; Zhu, H.; Chi, M.; Dai, S. Evidence for and Mitigation of the Encapsulation of Gold Nanoparticles within Silica Supports upon High-Temperature Treatment of Au/SiO2 Catalysts: Implication to Catalyst Deactivation. Appl. Catal. A Gen. 2010, 386, 147–156. [CrossRef]
- Dow, W.P.; Wang, Y.P.; Huang, T.J. TPR and XRD Studies of Yttria-Doped Ceria/γ-Alumina-Supported Copper Oxide Catalyst. Appl. Catal. A Gen. 2000, 190, 25–34. [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [CrossRef]
- Mokhonoana, M.P.; Coville, N.J. Highly Loaded Fe-MCM-41 Materials: Synthesis and Reducibility Studies. Materials (Basel). 2009, 2, 2337–2359. [CrossRef]
- Ashik, U.P.M.; Wan Daud, W.M.A. Nanonickel Catalyst Reinforced with Silicate for Methane Decomposition to Produce Hydrogen and Nanocarbon: Synthesis by Co-Precipitation Cum Modified Stöber Method. RSC Adv. 2015, 5, 46735–46748. [CrossRef]
- Son, I.H.; Lee, S.J.; Soon, A.; Roh, H.S.; Lee, H. Steam Treatment on Ni/γ-Al2O3 for Enhanced Carbon Resistance in Combined Steam and Carbon Dioxide Reforming of Methane. Appl. Catal. B Environ. 2013, 134–135, 103–109. [CrossRef]
- Pérez, P.; Soria, M.A.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Application of Au/TiO2 Catalysts in the Low-Temperature Water-Gas Shift Reaction. Int. J. Hydrogen Energy 2016, 41, 4670–4681. [CrossRef]
- Zhang, R.R.; Ren, L.H.; Lu, A.H.; Li, W.C. Influence of Pretreatment Atmospheres on the Activity of Au/CeO2 Catalyst for Low-Temperature CO Oxidation. Catal. Commun. 2011, 13, 18–21. [CrossRef]
- Alencar, C.S.L.; Paiva, A.R.N.; Da Silva, J.C.M.; Vaz, J.M.; Spinace, E.V. One-Step Synthesis of AuCu/TiO2catalysts for CO Preferential Oxidation. Mater. Res. 2020, 23, 2–7. [CrossRef]
- Yuan, G.; Louis, C.; Delannoy, L.; Keane, M.A. Silica- and Titania-Supported Ni-Au: Application in Catalytic Hydrodechlorination. J. Catal. 2007, 247, 256–268. [CrossRef]
- Liu, W.W.; Hu, C.W.; Yang, Y.; Tong, D.M.; Zhu, L.F.; Zhang, R.N.; Zhao, B.H. Study on the Effect of Metal Types in (Me)-Al-MCM-41 on the Mesoporous Structure and Catalytic Behavior during the Vapor-Catalyzed Co-Pyrolysis of Pubescens and LDPE. Appl. Catal. B Environ. 2013, 129, 202–213. [CrossRef]
- Yu, J.; Liu, S.; Cardoso, A.; Han, Y.; Bikane, K.; Sun, L. Catalytic Pyrolysis of Rubbers and Vulcanized Rubbers Using Modified Zeolites and Mesoporous Catalysts with Zn and Cu. Energy 2019, 188, 116117. [CrossRef]
- Rosenholm, J.B.; Rahiala, H.; Puputti, J.; Stathopoulos, V.; Pomonis, P.; Beurroies, I.; Backfolk, K. Characterization of Al- and Ti-Modified MCM-41 Using Adsorption Techniques. Colloids Surfaces A Physicochem. Eng. Asp. 2004, 250, 289–306. [CrossRef]
- Abbadi, A.; Van Bekkum, H. JOURNAL OF MOLECULAR CATALYSIS Effect of PH in the Pt-Catalyzed Oxidation of D-Glucose to D-Gluconic Acid. J. Mol. Catal. A Chem. 1995, 97, 11–18.
- Moreno, T.; Kouzaki, G.; Sasaki, M.; Goto, M.; Cocero, M.J. Uncatalysed Wet Oxidation of D-Glucose with Hydrogen Peroxide and Its Combination with Hydrothermal Electrolysis. Carbohydr. Res. 2012, 349, 33–38. [CrossRef]
- Aggarwal, N.; Aadil Yatoo, M.; Saravanamurugan, S. Glucose Oxidation to Carboxylic Products with Chemocatalysts; Elsevier B.V., 2019; ISBN 9780444643070.
- Jin, X.; Zhao, M.; Vora, M.; Shen, J.; Zeng, C.; Yan, W.; Thapa, P.S.; Subramaniam, B.; Chaudhari, R. V. Synergistic Effects of Bimetallic PtPd/TiO2 Nanocatalysts in Oxidation of Glucose to Glucaric Acid: Structure Dependent Activity and Selectivity. Ind. Eng. Chem. Res. 2016, 55, 2932–2945. [CrossRef]
- Allen, S.E.; Walvoord, R.R.; Padilla-salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. 2013.
- Dry, M.E.; Stone, F.S. Oxidation Reactions Catalyzed by Doped Nickel Oxide. Discuss. Faraday Soc. 1959, 28, 192–200. [CrossRef]
- Ishida, T.; Kinoshita, N.; Okatsu, H.; Akita, T.; Takei, T.; Haruta, M. Influence of the Support and the Size of Gold Clusters on Catalytic Activity for Glucose Oxidation. Angew. Chemie - Int. Ed. 2008, 47, 9265–9268. [CrossRef]
- Walkowiak, A.; Wolska, J.; Wojtaszek-Gurdak, A.; Sobczak, I.; Ziolek, M.; Wolski, L. Modification of Gold Zeolitic Supports for Catalytic Oxidation of Glucose to Gluconic Acid. Materials (Basel). 2021, 14. [CrossRef]
- Catalano, F.; Pompa, P.P.; Barrabino, A.; He, Y.; Li, J.J.; Long, M.; Liang, S.; Xu, H.; Isa, E.D.M.; Ahmad, H.; et al. Novel Pathways for the Preparation of Mesoporous MCM-41 Materials: Control of Porosity and Morphology. Alg. Dagbl. 2017, 457, 47237–47246.
- Ribeiro Carrott, M.M.L.; Conceição, F.L.; Lopes, J.M.; Carrott, P.J.M.; Bernardes, C.; Rocha, J.; Ramôa Ribeiro, F. Comparative Study of Al-MCM Materials Prepared at Room Temperature with Different Aluminium Sources and by Some Hydrothermal Methods. Microporous Mesoporous Mater. 2006, 92, 270–285. [CrossRef]
- Onda, A.; Ochi, T.; Kajiyoshi, K.; Yanagisawa, K. A New Chemical Process for Catalytic Conversion of D-Glucose into Lactic Acid and Gluconic Acid. Appl. Catal. A Gen. 2008, 343, 49–54. [CrossRef]
- Witońska, I.; Frajtak, M.; Karski, S. Selective Oxidation of Glucose to Gluconic Acid over Pd-Te Supported Catalysts. Appl. Catal. A Gen. 2011, 401, 73–82. [CrossRef]
- Solmi, S.; Morreale, C.; Ospitali, F.; Agnoli, S.; Cavani, F. Oxidation of D-Glucose to Glucaric Acid Using Au/C Catalysts. ChemCatChem 2017, 9, 2797–2806. [CrossRef]
Figure 1.
(a) N2 adsorption/desorption isotherms and (b) BJH pore size distribution from adsorption data for selected oxidation catalysts supported on SiO2 and MCM-41.
Figure 1.
(a) N2 adsorption/desorption isotherms and (b) BJH pore size distribution from adsorption data for selected oxidation catalysts supported on SiO2 and MCM-41.
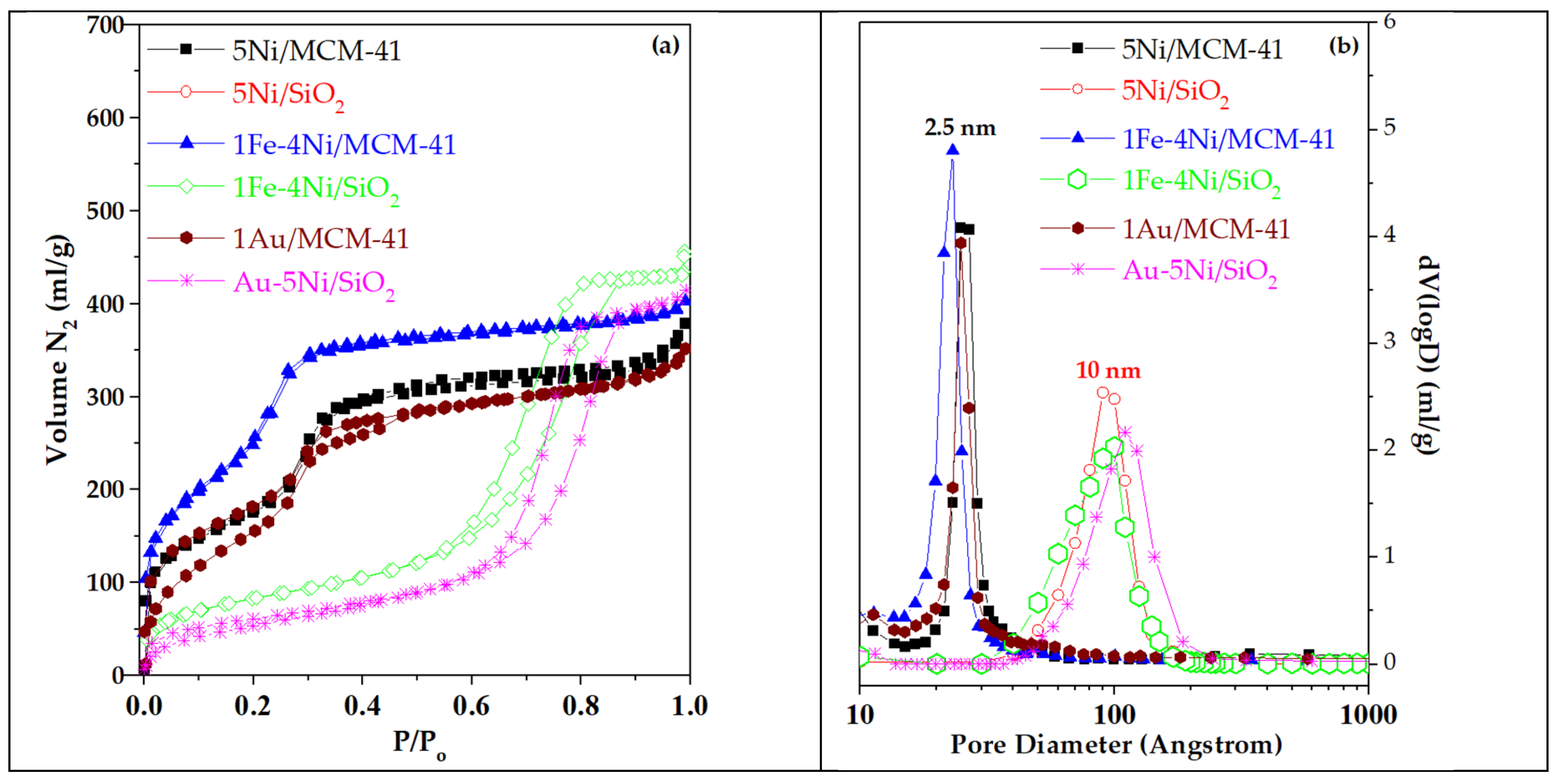
Figure 2.
XRD patterns of (a, b) monometallic, (c) bi-metallic transition metal oxides and (d) gold containing catalysts supported on SiO2 and MCM-41.
Figure 2.
XRD patterns of (a, b) monometallic, (c) bi-metallic transition metal oxides and (d) gold containing catalysts supported on SiO2 and MCM-41.
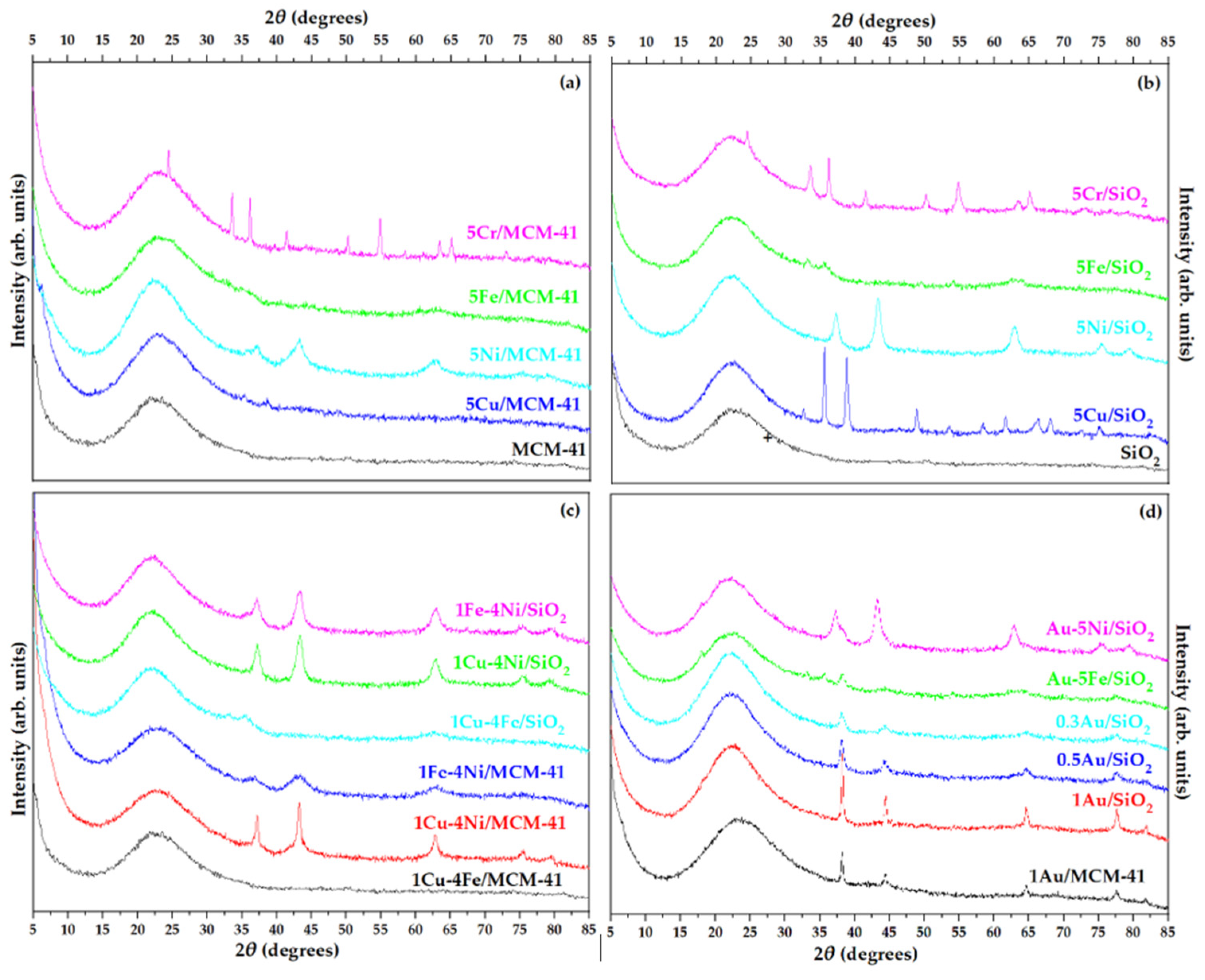
Figure 3.
TPR-H2 diagrams of selected (a) transition metal oxide catalysts supported on MCM-41, (b) transition metal oxide catalysts supported on SiO2 and (c) gold comprising catalysts supported on SiO2.
Figure 3.
TPR-H2 diagrams of selected (a) transition metal oxide catalysts supported on MCM-41, (b) transition metal oxide catalysts supported on SiO2 and (c) gold comprising catalysts supported on SiO2.
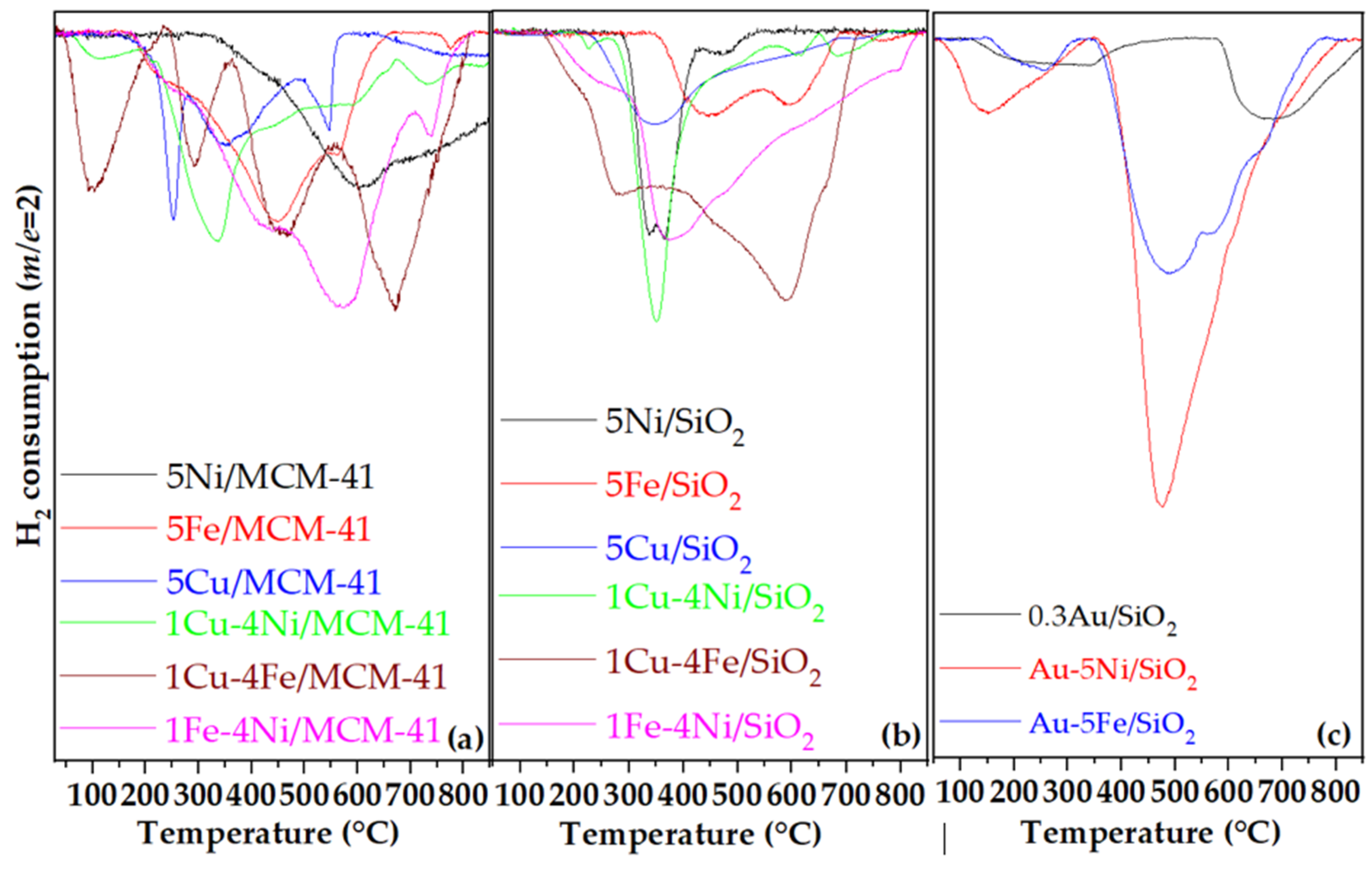
Figure 4.
TPD-NH3 diagrams of selected (a) transition metal oxide catalysts supported on MCM-41, (b) transition metal oxide catalysts supported on SiO2 and (c) gold comprising catalysts supported on SiO2.
Figure 4.
TPD-NH3 diagrams of selected (a) transition metal oxide catalysts supported on MCM-41, (b) transition metal oxide catalysts supported on SiO2 and (c) gold comprising catalysts supported on SiO2.
Scheme 1.
Possible reaction pathway of glucose oxidation (based on the obtained reaction products and the proposed mechanisms in references [12,50,51].
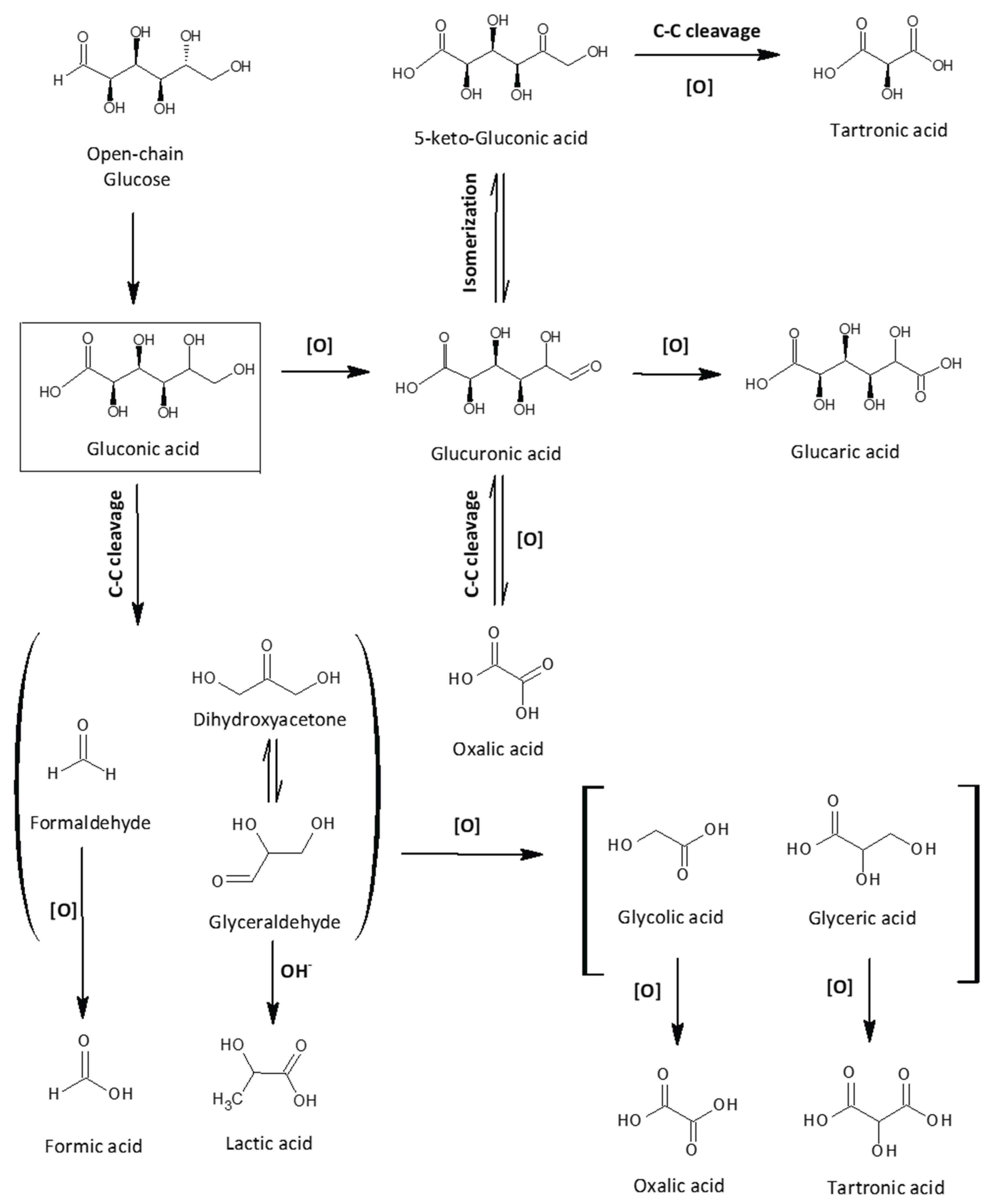
Figure 5.
Effect of gold supported catalysts with or without transition metals on glucose oxidation reaction (Reaction conditions: 80 °C, 60min, pH 8, H2O2/Glucose molar ratio 40, Glucose 2 wt.%, Catalyst 0.2 wt.%).
Figure 5.
Effect of gold supported catalysts with or without transition metals on glucose oxidation reaction (Reaction conditions: 80 °C, 60min, pH 8, H2O2/Glucose molar ratio 40, Glucose 2 wt.%, Catalyst 0.2 wt.%).
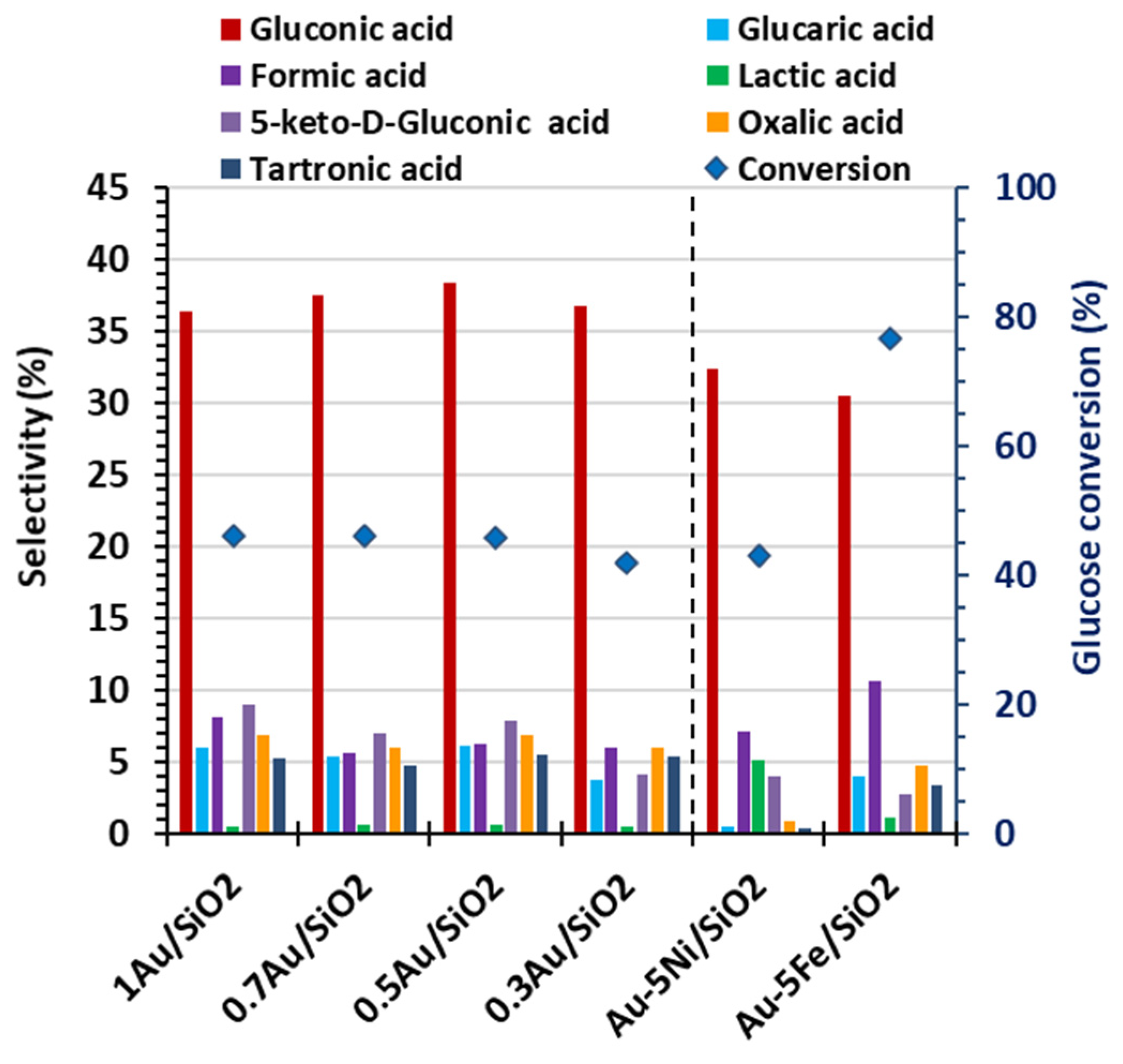

Table 1.
Textural, chemical and structural characteristics of oxidation catalysts.
| Catalysts | ICP-AES | XRD | BET | |||
|---|---|---|---|---|---|---|
| Metal composition (wt.%) (a) | Crystal size (nm) (Reflection angle, degrees) (b) | Total surface area (m2/g) (c) | Total Pore volume (ml/g) (d) | Meso-macro-pore volume (ml/g) | Textural Volume (ml/g) (e) | |
| SiO2 | - | - | 329 | 0.75 | 0.74 | 0.01 |
| 0.3Au/SiO2 | 0.31 | 6.4 (44.4°) | 299 (328) | 0.72 | 0.70 | 0.02 |
| 0.5Au/SiO2 | 0.53 | 9.6 (44.3°) | 318 (327) | 0.77 | 0.74 | 0.03 |
| 0.7Au/SiO2 | 0.64 | 10.5 (44.3°) | 304 (327) | 0.76 | 0.73 | 0.03 |
| 1Au/SiO2 | 1.24 | 15.3 (44.2°) | 311 (325) | 0.75 | 0.72 | 0.02 |
| 5Cr/SiO2 | 4.78 | 25.9 (36.2°) | 290 (306) | 0.64 | 0.62 | 0.01 |
| 5Cu/SiO2 | 4.90 | 37.4 (35.6°) | 298 (309) | 0.92 | 0.68 | 0.24 |
| 5Fe/SiO2 | 4.78 | 8.6 (33.0°) | 308 (308) | 0.69 | 0.68 | 0.01 |
| 5Ni/SiO2 | 5.00 | 12.5 (43.3°) | 287 (308) | 0.78 | 0.68 | 0.10 |
| Au-5Fe/SiO2 | 0.38 - 5.02 | 15.9 (44.3°)(f) | 263 (304) | 0.66 | 0.60 | 0.06 |
| Au-5Ni/SiO2 | 0.32 - 5.68 | n.d. | 279 (304) | 0.68 | 0.66 | 0.02 |
| 1Cu-4Fe/SiO2 | 1.09 - 4.84 | traces | 304 (302) | 0.71 | 0.65 | 0.06 |
| 1Cu-4Ni/SiO2 | 1.25 - 4.48 | 10.1 (43.3°)(g) | 298 (305) | 2.17 | 0.67 | 1.50 |
| 1Fe-4Ni/SiO2 | 1.14 - 4.96 | 10.2 (43.3°)(g) | 303 (303) | 0.71 | 0.66 | 0.05 |
| MCM-41 | - | - | 964 | 0.82 | 0.77 | 0.08 |
| 0.3Au/MCM-41 | 0.06 | n.d. | 705 (963) | 0.72 | 0.69 | 0.05 |
| 1Au/MCM-41 | 0.62 | 13.3 (44.2°) | 651 (958) | 0.75 | 0.72 | 0.03 |
| 5Cr/MCM-41 | 4.68 | 21.2 (36.2°) | 738 (898) | 0.73 | 0.63 | 0.10 |
| 5Cu/MCM-41 | 3.60 | traces | 652 (921) | 0.56 | 0.49 | 0.08 |
| 5Fe/MCM-41 | 4.83 | traces | 743 (897) | 0.65 | 0.57 | 0.09 |
| 5Ni/MCM-41 | 5.30 | 6.5 (43.2°) | 639 (899) | 0.59 | 0.51 | 0.08 |
| 1Cu-4Fe/MCM-41 | 0.93 - 4.15 | traces | 821 (896) | 0.58 | 0.56 | 0.03 |
| 1Cu-4Ni/MCM-41 | 1.07 - 4.03 | 8.3 (43.5°)(g) | 866 (902) | 0.61 | 0.58 | 0.03 |
| 1Fe-4Ni/MCM-41 | 1.02 - 3.97 | 9.7 (43.2°)(g) | 867 (901) | 0.62 | 0.59 | 0.03 |
(a) In the case of bi-metallic catalysts the metal composition series is the same of the metal series of the catalyst name, (b) calculated with the Scherrer equation based on the referenced reflection, (c) from multi-point ΒΕΤ method. The normalized value based on the pure silica content of the catalyst is in the brackets (d) calculated at P/Po=0.99, (e) Textural volume = Total pore volume - volume calculated at P/Po=0.90, (f) for Au crystal and (g) for NiO crystal.
Table 2.
Redox properties of selected oxidation catalysts.
| Catalysts | Acid sites | H2(d) cons. | Reduci-bility(e) | |||
|---|---|---|---|---|---|---|
| Total | Weak(a) | Medium(b) | Strong(c) | |||
| (μmol/g) | (%) | |||||
| 0.3Au/SiO2 | - | - | - | - | 277 | - |
| 5Cu/SiO2 | 53.8 | 33.1 (196) | 8.9 (376) | 11.8(f) | 527 | 68.4 |
| 5Ni/SiO2 | 19.0 | 10.1 (223) | 5.5 (334) | 3.4 (682) | 459 | 55.6 |
| 5Fe/SiO2 | 46.0 | 24.8 (200) | 18.2 (357) | 3.0 (698) | 1050 | 81.8 |
| Au-5Fe/SiO2 | 56.8 | 44.1 (184) | 8.4 (347) | 4.3(g) | 2700 | 200.3 |
| Au-5Ni/SiO2 | 49.4 | 34.2 (180) | 0 | 15.2(h) | 4281 | 442.3 |
| 1Cu-4Fe/SiO2 | 69.9 | 40.5 (215) | 25.1 (339) | 4.3 (677) | 2366 | 160.7 |
| 1Cu-4Ni/SiO2 | 37.3 | 21.4 (212) | 10.7 (354) | 5.2 (691) | 1169 | 121.9 |
| 1Fe-4Ni/SiO2 | 35.6 | 18.7 (209) | 11.6 (393) | 5.3 (653) | 1771 | 153.9 |
| 5Cu/MCM-41 | 61.8 | 58.0 (217) | 0 | 3.8 (756) | 644 | 85.5 |
| 5Fe/MCM-41 | 36.8 | 15.7 (248) | 15.3 (356) | 5.8 (723) | 1239 | 95.5 |
| 5Ni/MCM-41 | 22.6 | 8.3 (268) | 10.0 (382) | 4.3 (708) | 1781 | 197.2 |
| 1Cu-4Fe/MCM-41 | 64.9 | 35.4 (237) | 25.0 (347) | 4.5 (7.2) | 2382 | 188.8 |
| 1Cu-4Ni/MCM-41 | 45.8 | 27.8 (239) | 14.4 (338) | 3.6 (680) | 2033 | 237.3 |
| 1Fe-4Ni/MCM-41 | 51.8 | 24.3 (221) | 20.2 (339) | 7.3 (698) | 2436 | 255.9 |
(a) From 100-300°C (temperature peak value in Celsius in the brackets), (b) from 300-500°C (temperature peak value in Celsius in the brackets), (c) from 500-900°C (temperature peak value in Celsius in the brackets), (d) from TPR-H2 experiments, (e) based on estimation of theoretical H2 consumption required for the complete reduction of each catalyst compared to H2 consumption estimated from TPR experiments, (f) peaks at 631 °C, 729 °C and 789 °C, (g) peaks at 613 °C and 724 °C and (h) peaks at 623 °C, 694 °C and 783 °C.
Table 3.
Catalyst screening for the conversion of glucose in aqueous medium (Reaction conditions: 80 °C, 60min, pH 8, Glucose 2 wt.%, H2O2/Glucose molar ratio 40, Glucose/Metal moral ratio: 64 (1000 for Au)).
Table 3.
Catalyst screening for the conversion of glucose in aqueous medium (Reaction conditions: 80 °C, 60min, pH 8, Glucose 2 wt.%, H2O2/Glucose molar ratio 40, Glucose/Metal moral ratio: 64 (1000 for Au)).
| Catalyst | Glucose conversion (%) | Selectivity (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Gluconic acid | Glucaric acid | Formic acid | Lactic acid | Oxalic acid | 5-keto-D-Gluconic acid | Tartronic acid | ||
| 1Au/SiO2 | 46.1 | 36.4 | 6.0 | 8.1 | 0.5 | 6.8 | 9.0 | 5.3 |
| 5Cr/SiO2 | 65.5 | 25.9 | 1.5 | 14.0 | 9.0 | 0.8 | - | - |
| 5Cu/SiO2 | 100.0 | - | - | 13.3 | 2.7 | 1.3 | - | - |
| 5Fe/SiO2 | 79.5 | 22.5 | 1.7 | 15.1 | 10.3 | 1.5 | - | - |
| 5Ni/SiO2 | 41.1 | 32.2 | 0.5 | 7.7 | 7.1 | - | - | - |
| 1Cu-4Fe/SiO2 | 99.9 | - | - | 7.5 | 2.5 | 9.3 | - | - |
| 1Cu-4Ni/SiO2 | 99.7 | - | - | 16.2 | 3.9 | 1.4 | - | - |
| 1Fe-4Ni/SiO2 | 98.9 | 6.5 | 1.8 | 19.5 | 5.5 | 2.4 | - | - |
| 1Au/MCM41 | 40.3 | 41.0 | 5.4 | 8.2 | 1.2 | 5.9 | 7.1 | 4.9 |
| 5Cr/MCM-41 | 57.5 | 28.8 | 1.8 | 15.3 | 7.8 | 0.9 | - | - |
| 5Cu/MCM-41 | 100.0 | - | - | 19.5 | 5.2 | 0.5 | - | - |
| 5Fe/MCM-41 | 75.5 | 22.2 | 1.4 | 14.1 | 9.2 | 1.6 | - | - |
| 5Ni/MCM-41 | 44.1 | 32.6 | 1.1 | 10.8 | 7.9 | 0.8 | - | - |
| 1Cu-4Fe/MCM-41 | 99.8 | - | - | 13.1 | 3.7 | 6.8 | - | - |
| 1Cu-4Ni/MCM-41 | 99.8 | - | - | 9.2 | 1.1 | 1.7 | - | - |
| 1Fe-4Ni/MCM-41 | 92.7 | 12.0 | 2.9 | 20.3 | 4.2 | 3.4 | - | - |
Table 4.
Metal leaching from supported and mixed oxide catalysts in the reaction medium.
| Catalyst | Leaching (%) * (ICP-AES (ppm)) | ||||
|---|---|---|---|---|---|
| Au | Cu | Cr | Fe | Ni | |
| 1Au/SiO2 | 0 (n.d.) | - | - | - | - |
| 5Cr/SiO2 | - | - | 20.3 (14.7) | - | - |
| 5Cu/SiO2 | - | 24.4 (20.0) | - | - | - |
| 5Fe/SiO2 | - | - | - | 9.1 (6.5) | - |
| 5Ni/SiO2 | - | - | - | - | 3.3 (2.7) |
| 1Cu-4Fe/SiO2 | - | 41.3 (6.9) | - | 16.1(11.9) | - |
| 1Cu-4Ni/SiO2 | - | 63.6 (53.5) | - | 41.7 (126.0) | |
| 1Fe-4Ni/SiO2 | - | - | - | 27.2 (4.0) | 5.3 (3.4) |
| 1Au/MCM41 | 0 (n.d.) | - | - | - | - |
| 5Cr/MCM-41 | - | - | 50.6 (37.0) | - | - |
| 5Cu/MCM-41 | - | 26.0 (22.5) | - | - | - |
| 5Fe/MCM-41 | - | - | - | 20.0 (16.1) | - |
| 5Ni/MCM-41 | - | - | - | 7.0 (5.8) | |
| 1Cu-4Fe/MCM-41 | - | 43.7 (7.3) | - | 16.5 (12.2) | - |
| 1Cu-4Ni/MCM-41 | - | 53.8 (55.0) | - | - | 44.8 (173.0) |
| 1Fe-4Ni/MCM-41 | - | - | - | 39.3 (6.7) | 6.1 (4.0) |
* Calculated on the basis of the metal content in the initial metal supported catalysts prior to reaction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



