
Submitted:
11 November 2025
Posted:
13 November 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since the earliest cases of COVID-19, Long-COVID (LC) has presented as a multi-system disease/disorder with persistent symptoms in individuals after SARS-CoV-2 infection. SARS-CoV-2 is found in the oral cavity and the periodontium during LC; however, its effects have not been fully investigated. This review proposes LC as a novel risk factor for periodontitis. LC possesses systemic mechanisms of immuno-inflammatory dysregulation, which overlap and thus could enhance periodontitis development. Persistent in-creases in neutrophils, elevated pro-inflammatory cytokine production, complement pro-duction from innate immune system activation are involved in both periodontitis and LC, suggesting the potential for interactions between the two. LC leads to dysbiosis of the GI system and lungs, and we consider here the possibility of oral and periodontal dysbiosis. Gingival epithelium and periodontal ligament cells do express the viral receptor, ACE2, which would allow SARS-CoV-2 entry into these cells. Interestingly, ACE2 is increased during active periodontitis. Additionally, LC has been linked to the re-emergence of herpesvirus infections, especially the Epstein-Barr virus (EBV), which has been associated with both autoimmune diseases and periodontitis. In this review, we comprehensively compile the routes by which LC could act as a systemic risk factor for periodontitis. We aim to provide the theoretical foundation for epidemiologic and mechanistic research that could produce the necessary scientific evidence.
Keywords:
COVID-19
; Long-COVID
; SARS-CoV-2
; periodontitis
; immunity
; inflammation
; oral microbiome
; virus
; risk factor
; Epstein-Barr
1. Introduction
As of September 28 2024, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the virus responsible for the COVID-19 pandemic that began in March 2020, has led to over 776 million confirmed cases worldwide, with an increase of more than 84,000/week in recent weeks. In the United States alone, over 103 million infections have been reported[1]. The virus has resulted in over 7.06 million deaths globally, with a significant portion—over 1.2 million—occurring in the United States[1]. Furthermore, a meta-analysis of studies conducted through 2022 indicates that Long Covid-related symptoms affect approximately 43% of COVID-19 subjects worldwide[2]. Based on this meta-analysis and Centers for Disease Control and Prevention data, LC’s prevalence ranges from 20% to 31 % in the United States and is prehaps similarly prevalent worldwide[2,3].
Long COVID (LC; aka, post-acute sequelae of SARS-CoV-2 infection (PASC), post-COVID-19, post-acute COVID-19 syndrome, or long-haul COVID)[4,5,6,7], is a multi-organ condition characterized by persistent symptoms in individuals following infection with SARS-CoV-2. More specifically, it is defined as an ongoing pattern of relapsing and remitting changes in the functionality of multiple systems over time. Some definitions of this condition include any new symptoms or medical conditions lasting 30 days or longer following active SARS-CoV-2 infection[8]. Despite intensive effects, there is still no universally accepted case definition for LC. According to the World Health Organization (WHO) Delphi Consensus case definition, LC is identified in individuals with a history of probable or confirmed SARS-CoV-2 infection who continue to experience a variety of symptoms or impairments, such as cognitive dysfunction, fatigue, shortness of breath or others, for nearly three months or more without an alternative diagnosis to explain these symptoms following the onset of the original acute disease[9].
LC can persist regardless of an individual’s vaccination status and is likely to occur even in cases of reinfection with SARS-CoV-2[10]. The condition may persist for several years, increasing private and public health system costs for those affected[11,12,13,14,15,16]. Individuals with LC also face negative impacts on their health-related quality of life, which can lead to loss of income [17,18] and rising personal healthcare expenses[19,20,21,22].
2. LC Reported Symptoms and Clinical Manifestations
LC encompasses a broad spectrum of physical, psychological, and cognitive symptoms. These include fatigue, malaise, muscle pain, dyspnea, memory loss, hair loss, migraines, along with attention and sleep disorders. LC can lead to adverse clinical manifestations across multiple body systems, including but not limited to the respiratory, neurological, cardiovascular, gastrointestinal, metabolic, renal, and reproductive systems[5,14,16,23,24,25,26,27,28]. The disease is typically more pronounced in individuals with weakened immune systems or those who are immunocompromised[29].
3. LC Clinical Manifestations in the Oral Cavity
Although numerous reports have documented oral complications associated with SARS-CoV-2, few have addressed oral manifestations of LC[30]. The symptoms and clinical manifestations of LC may include taste disorders (e.g., hypogeusia or ageusia), chronic oral dysesthesia, ulceration, discoloration, hemorrhagic changes in the oral mucosa, aphthous-like lesions, atrophic cheilitis, alterations in salivary glands, oral mucormycosis, and actinomycotic osteomyelitis[31,32,33,34,35]. To the best of our knowledge, the potential effects of LC on periodontitis—the most common oral disease after dental caries[36]—have been explored only minimally. However, there is significant potential for a strong relationship between these two conditions due to a shared chronic immune and inflammatory status. One cross-sectional study, which used a limited definition of LC, reported a higher prevalence of "sequelae from COVID-19" in individuals with periodontitis and obesity compared to those with obesity alone[37].
4. Periodontitis: Definition and Systemic Impact
Periodontal diseases affect the tissues that protect and support teeth. Most are induced by dental plaque and can be categorized into gingivitis, a periodontal tissue inflammation without loss of attachment or alveolar bone support, and periodontitis. Gingivitis is a reversible response to supragingival dental plaque and has a universal prevalence[38]. In contrast, periodontitis is a chronic condition that results in the loss of attachment and/or alveolar bone, with a prevalence of 42.2 % in the U.S.[39]. Severe periodontitis often results in tooth loss and impacts approximately 7.8% of the U.S. adult population aged 30 and older[39] and 11.2% globally[40]. It contributes to a poor quality of life and is the most common form of bone pathology in humans[41].
Periodontitis has been linked to various systemic conditions, a subfield termed 'periodontal medicine, ' which explores two primary avenues: (A) the contribution of periodontitis to diseases in other parts of the body, facilitated by bacterial migration and the influence of active periodontitis on systemic inflammation, as evidenced by its association with coronary heart disease[42,43]; and (B) how systemic conditions, such as diabetes, influence the immune system and potentially exacerbate periodontitis. Notably, diabetes is recognized as a significant risk factor for periodontitis[44,45,46,47]. Therefore, examining the mechanisms of LC function and their potential impact on periodontal immunity, leading to subgingival dysbiosis and subsequent periodontal damage, forms a hypothesis modeled on the relationship between diabetes and periodontitis.
While it can be argued that there are differences in the systemic and local immune profiles during infections, it is essential to note that advanced periodontitis results in a destructive immune response at both local (gingival) and systemic levels[48,49], exacerbating any other existing systemic inflammation[42,43]. It might worsen the initial COVID-19 infection or increase the likelihood of leukocyte complications[50]. Conversely, the immuno-inflammatory dysregulation associated with LC could promote the onset, progression, and severity of periodontitis, similar to diabetes[51,52].
5. Epidemiologic Factors Shared Between LC and Periodontitis
LC has been reported to occur in individuals exhibiting specific characteristics, such as smokers, the elderly, women, a lower socio-economic status (SES), ethnic minorities, history of severe symptoms during acute COVID-19, preexistent psychiatric disorders, and preexisting chronic comorbidities such as obesity, diabetes, asthma, cardiovascular disease (CVD), and hypertension[2,6,53,54,55]. Periodontitis shares many of these characteristics, like older age, low SES, and a higher prevalence among ethnic minorities[56,57]. Additionally, periodontitis often coexists with conditions prevalent in the LC group, such as chronic mental health disorders and cardiometabolic diseases (e.g., diabetes, CVD, and obesity)[58,59,60,61,62]. Specific population subgroups also exhibit distinct characteristics related to periodontitis, such as low educational attainment, infrequent dental visits, and poor dietary habits[56,57,63,64].
6. Immuno-Pathophysiology Mechanisms Shared by LC and Periodontitis
6.1. Early Immune Response to the SARS-COV-2
6.1.1. Interferon-Mediated Innate Immune Response to a Viral Infection
Under normal conditions, innate immunity tracks any generic viral infection to its source to combat it. This process is mediated by type I interferon (IFN) expression and related molecules and occurs within a couple of hours[65]. It limits viral replication within the infected cells, slows the virus’s spread, and activates the adaptive immune response, which involves the simultaneous actions of B cells generating neutralizing antibodies alongside T cells, including CD4+ and CD8+, which quickly eliminate infected cells and their viral components[65,66,67].
However, during acute SARS-CoV-2 (or initial) infection, this virus can effectively slow down [68] or evade the activation of the immune system related to type I and type III IFN responses, allowing it to continue replicating[69,70,71]. Additionally, the adaptive immune responses are likewise impaired, as the innate immune system cannot quickly present antigenic information[66]. Along with the impaired or delayed type I and type III IFN innate immune regulation[68,72], low dendritic cell counts, a deficit in IFN-α expression[73,74], and a reduction in the total number of NK cells is observed[75], along with the depletion and exhaustion of the surviving NK cells[76].
Dendritic cells (DCs) are the primary source of type I IFN in the gingival tissue, with higher expression in subjects with periodontitis[77]. However, in the complex etiological model of periodontitis, where bacterial and viral challenges can occur simultaneously, a decrease in type I IFN and IFN-γ is observed due to the co-infection of organisms from these two kingdoms[78]. Studies examining the association between Herpesviruses and periodontitis have also found lower levels of IFN among periodontitis patients who tested positive for these viruses [79] or during active periodontitis[80]. It appears that periodontal infection by Herpesviruses and SARS-CoV-2 may share similar mechanisms for evasion or dampening of IFN expression by dendritic cells.
6.1.2. The Short-Term T-Cell Response to SARS-CoV-2
Among SARS-CoV-2 positive asymptomatic individuals, delays in innate immune responses do not hinder long-term viral clearance. The adaptive immune responses, which include increased CD4+ and CD8+ T cell counts and higher levels of neutralizing antibodies, effectively compensate for the impaired innate response. These antibodies are formed faster than usual to combat the viral infection[81]. The presence of active T cells in infected tissues and antibodies produced by B cells against SARS-CoV-2 signifies disease resolution[66,82,83].
Among patients with acute COVID, there is a functional impairment in adaptive immune responses characterized by a decrease in total B cells, CD4+ T cells, and CD8+ T cells (lymphopenia). It is often accompanied by increased survival but functionally exhausted T cells[66,67,76,83,84]. Immature neutrophils particularly disrupt T-cell polarization, promoting Th17 cell differentiation while suppressing Th1. This causes major disturbances in the immune response against SARS-CoV-2[85,86,87]. Key Th17 effector cytokines, including members of the IL-17 family (primarily IL-17A and IL-22), play crucial roles in the pathogenesis of COVID-19 as well as periodontitis [83,88,89,90,91].
Antigen persistence, antibody production, and T-cell counts are directly linked. The stability of CD4 and CD8 T-cell response is one of the findings related to recovery from acute COVID[92,93,94,95,96], with fewer tissue cells presenting T-cell receptors[97]. However, antibody titers against the SARS-CoV-2 may vary in different screened populations, from reduction [92,93,94,95,96] to somatic effects[98]. Local antibody expression will also be elevated in body parts where persistent antigen release is detected, such as in the small bowel[98].
The role of CD 8+T cells in chronic periodontitis is less noticeable. CD8+ T cells appear to contribute to alveolar bone preservation by suppressing osteoclastogenesis[99,100,101]. Nonetheless, their role can be unnoticed by the higher level of other immuno-inflammatory responses mounted against periodontal microbial infections, dramatically uncoupling bone homeostasis. The overreactive immune response under LC can disguise CD8+ role even further[102].
6.1.3. Neutrophils and the “Cytokine Storm”
The immune system uses various strategies to compensate for the impairments caused by severe SARS-CoV-2 infection, including an increase in the neutrophil count[103] as early as the first week[104,105,106]. Neutrophil activation amplifies inflammation, leads to prothrombotic loops and persistent production of pro-inflammatory cytokines such as Interleukins (IL)-1β, IL-2, IL-6, IL-7, IL-8, IL-10, IL-17, IL-21, IL-23, granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor (TNF), collectively referred to as the 'cytokine storm'[67,75,106,107,108,109,110]. The persistence of high active neutrophil counts with abnormal responses after the acute phase of COVID is considered a marker of severe LC[111,112,113].
In the periodontium, the pathogenic pathways related to plaque-induced periodontal diseases involve many of the cytokines from the storm, which play relevant roles[114,115]. IL-1β, IL-6, IL-17, IL-23, and TNF are well documented in periodontitis, exerting several roles in immune cell recruitment, pro-inflammatory activity, and periodontal tissue destruction. Conversely, IL-2 and IL-10 promote less inflammation and tissue destruction.
Neutrophil-extracellular traps (NETs) are another inflammatory-enhancing and pro-thrombotic mechanism from these cells[116], in addition to NETs’ primary role as traps for pathogens[116,117]. NETs are composed of the extracellular expression of chromatin with histones, proteases, lactoferrin, cathepsins, and myeloperoxidase[118], and are linked to several chronic diseases, most notably Chronic Obstructive Pulmonary Disease (COPD) and asthma[119]. NETs were detected in the blood of severe COVID patients by several studies[120,121,122].
Circulating NETs, identified by cell-free DNA (cfDNA) or myeloperoxidase (MPO)-DNA in blood, are standard markers of acute diseases, including COVID, and are also present in LC[123]. However, cfDNA or MPO-DNA alone is not a precise NET marker, as it also indicates cell necrosis and apoptosis[124]. The best use of circulating cfDNA/MPO-DNA as a NET marker is alongside tissue biopsies. Despite this, it is suggested that the initial SARS-CoV-2 infection may cause epigenetic shifts in parenchymal, vascular, and immune cells, including myeloid progenitor cells in the bone marrow, leading to neutrophil formation and NETs[123]. These hypotheses remain unconfirmed.
NETs are recognized as integral to the immune response against plaque-induced periodontal diseases[125] characterized by their involvement in the acute phases of inflammation. Biopsies indicate a higher expression of NETs in gingivitis compared to periodontitis[126]. Periodontitis and acute COVID possess similarities regarding elevated cfDNA/MPO-DNA levels associated with an inflammatory response with NETs. MPO-DNA levels increased positively with probing depths and clinical attachment loss, with the highest levels among subjects with periodontitis and Rheumatoid Arthritis[127]. This similarity was postulated as one reason why periodontitis could exacerbate the initial COVID infection[118].
6.1.4. Monocytes and Macrophages
Another key component of the immune response to the SARS-CoV-2 that has a relevant role in periodontitis is monocytes and macrophages, which have been reported to be hyperactivated and dysregulated in association with severe COVID[128,129,130]. Monocytes and macrophages are part of virus clearance; however, their dysregulation exacerbates tissue damage[130,131,132]. Like neutrophils, when active, these cells produce cytokine expression and pyroptosis – inflammation-induced cell death[133,134].
Key inflammatory molecules expressed by these two cells when dysregulated are IL-1α, IL-1β, IL-6, IL-7, THF, IFNs I and II, CCL2, CCL3, CXCL10[130], a molecular profile associated with hyperinflammation and severe diseases[131,135]. Particularly, the elevated systemic IL-6 levels led to the use of this cytokine’s inhibitor as therapeutic drugs against COVID, like Tocilizumab and Siltuximab[123].
Hyperactivation of monocytes and macrophages has been reported in LC[136], potentially causing ongoing tissue damage. Active monocytes were detected from eight to fifteen months[137] of LC, regardless of the severity of the initial or acute infection. Macrophages are histological findings in organs impacted by LC, including the brain, liver, lungs, and adipose tissue[138]. In vitro assays with macrophages from subjects recovered from COVID have demonstrated a persistent hyperregulation in the production of inflammatory molecules and a downregulation of pro-resolving factors[139].
Hyperactive monocytes and macrophages also indicate the inflammatory response in periodontitis[140,141].This finding supports the hypothetical pathway linking periodontitis to cardiovascular diseases[142], which suggests that hyperreactive macrophages from active periodontitis sites could enter the bloodstream and initiate endothelial damage or affect other areas of the body. Thus, in patients with periodontitis infected with SARS-CoV-2, a proinflammatory monocyte/macrophage profile is already present. Conversely, the persistence of this proinflammatory profile in patients can also alter the response to dental plaque, leading to a periodontal destructive pattern.
IL-6 is a key cytokine in periodontitis. High levels in the gingival crevicular fluid (GCF) are associated with alveolar bone loss and dysbiotic biofilms[143,144]. However, in an animal model, IL-6 elevation from gingival epithelial cells was observed during common daily episodes, such as chewing[145] in response to mechanical damage. This IL-6-induced mechanism leads to the priming of resident TH17 cells as the key defense mechanism of that specific mucosal barrier, independent of the presence of pathogens.
Elevated IL-6 is a pathway that associates periodontitis with an established systemic risk factor, Diabetes[146], and a path of association with Coronary Heart Disease[147] and COPD[148]. Pre-existing circulating levels of IL-6 before and following the initial COVID infection, owing to its proinflammatory action and regulation of dendritic, B, and T cells[149] would favor LC. Conversely, LC may act as the initial trigger for systemic IL-6 elevation, resulting in damage to various tissues, including the periodontium.
6.2. A Persistent Active Immune Response to the SARS-CoV-2, the Key to LC
6.2.1. The Resolution of the SARS-Cov-2 Infection
Recovery from COVID-19 is common; however, an altered immune phenotype and/or function may persist in convalescent individuals[150,151,152,153]. Neutralizing antibody counts display a continuous decline correlated with the severity of the initial acute COVID-19 infection. Some individuals with a high peak infective viral load have maintained elevated neutralizing antibody titers afterward. In contrast, individuals with lower loads have experienced a gradual decline, returning to baseline levels even more than three months after disease onset[94].
An example of persistent immune response is among elderly individuals who survive severe COVID-19 disease and experience a late increase in circulating CD4+, CD8+, and double-negative B cell populations. Over time, this is characterized by improvements in dysfunctional T and B cells (including alterations in cell memory) and increased frequencies of cell activation and exhaustion, which contrast with the depletion observed during the acute phase[67,74,150,154,155,156].
LC is associated with persistent immunological dysregulation and can affect individuals with initial asymptomatic responses and those with mild, moderate, or severe cases[154,157,158,159,160,161,162,163]. Diminished numbers of DCs, can be detected even seven months post-SARS-CoV-2 infection[73], with continuous neutrophil activity, persistent inflammatory cytokine production, and deficient naive T and B cell numbers[154,155].
Periodontitis is characterized by ongoing elevated neutrophil activity and the production of inflammatory cytokines. In alignment with the continuous activity of neutrophils and persistent inflammatory cytokine production observed in LC, an imbalanced and dysregulated recruitment of neutrophils may occur at periodontal sites due to overwhelming microbial challenges[102,164].
However, the dysregulation observed in periodontitis may lead to an increase in cells that will have a reduced count in LC, such as T cells, B cells, natural killer (NK) cells, macrophages, and DCs, all of which may contribute to the initiation and exacerbation of periodontitis[164,165,166]. The activation and proliferation of these cells will further perpetuate proinflammatory cytokine production.
6.2.2. A Persistent Dysregulation of T-Cells in LC
The behavior of T-cells after the initial COVID infection is likely to be one of the main factors that result in LC. However, the findings regarding these cells are inconsistent. In some cases, a short life cycle of cytotoxic CD8+ was observed[167], and a more significant signal toward cell death from T-cells than the antigenic memory[168]. However, other studies with subjects with areas of persistent SARS-CoV-2 infection, like the lungs, will have larger counts of circulating specific CD8+ T-cells with increased production of IFN Gamma, TNF, IL-6, and C-reactive protein[169]. Similar patients have exhibited dysregulation of tissue-resident CD8+ T-cells from lung biopsies[170].
6.2.3. A Broad and Continuous Adaptive Immune Response Is Part of LC
Although most patients recovering from acute COVID will show a reduced count of antibody-secreting B-cells during this stage, a small percentage will maintain elevated counts of these cells[171]. In line with this finding, LC is characterized by broad adaptive immune activation, with B-cells resuming the production of antibodies against past infections, alongside an increase in CD4+ and CD8+ specific lineages targeting SARS-CoV-2 and cytomegalovirus during convalescence[172]. Other viral infections may (re) emerge during COVID-19, with the Epstein-Barr virus (EBV) relevantly documented in the literature. This co-infection is considered a potential factor for LC and is linked to neurocognitive symptoms such as fatigue[173].
EBV is one of the Herpesviruses associated with periodontitis, similar to Cytomegalovirus. Traditionally, EBV is an etiologic factor for Hairy Leukoplakia, classically regarded as a pathognomonic indicator of HIV infection, but it is not limited to this[174] and is also associated with some oral cancers[175]. EBV is linked to periodontitis in populations across Asia, Europe, and America. It can be detected in subgingival plaque and GCF, showing a positive correlation between the presence of EBV, gingival inflammation, and probing depths[176]. Active periodontitis can act as a reservoir for EBV, which may co-infect with SARS-CoV-2 and is considered a precursor for LC.
Another way the adaptive immune system sustains activity in LC is through COVID-driven autoimmunity. This phenomenon occurs even during the acute phase. It aims to neutralize IFN type I via autoantibodies against it[177,178,179], resulting in a reduced IFN response, failed SARS-CoV-2 clearance, and ongoing local and systemic inflammation[68,70,72,180,181,182,183]. Reports of autoantibodies against other cytokines, chemokines, cell-surface proteins[184], and phospholipids [185] have all been noted.
In LC, autoantibody production is continuous due to the still active adaptive immunity via double-negative B cells, even in subjects with mild initial COVID[171,186]. This pattern resembles several autoimmune diseases, as it involves autoantibodies against components of connective tissue, cytokines, chemokines, and antinuclear antibodies[187]. The co-infection of EBV with SARS-CoV-2 may also contribute to the autoimmunity associated with LC, supported by scientific evidence showing that EBV alone can lead to autoimmunity[188,189,190]. However, there is no consensus on whether COVID triggers autoimmune mechanisms or if these are genetically determined and predate the initial SARS-CoV-2 infection[177]. Furthermore, the ongoing activity of adaptive immunity does not necessarily mean that the autoimmunity process will continue. For instance, autoantibodies against IFN-I are not linked to LC[191].
The role of autoimmunity in LC remains unclear. Studies vary, with some demonstrating a correlation between autoantibodies and LC[192], while others do not[193]. Opinions differ on whether autoimmunity is a risk factor for LC[172], if LC is an autoimmune disease[194], or if autoantibodies could offer protection against LC[195]. There is no consensus on the issue.
6.3. The Complement System
The complement system, an integral part of innate immune regulation that plays a crucial role in immunity and homeostasis by targeting pathogens and damaged cells, has recently been implicated in the activation mechanisms of L[196]. Individuals with LC exhibit an imbalance in terminal complement complex (TCC) formation, which is characterized by elevated levels of soluble C5bC6 complexes, diminished concentrations of C7-containing TCC formations, and associated thrombo-inflammation. Current literature highlights increased markers of tissue injury, red blood cell lysis, platelet activation, and monocyte–platelet aggregates[196]. Active LC is characterized by sustained activation of specific alternative and classical complement pathways.
The complement system might be involved in periodontitis’ pathogenesis as well[197]. Complement proteins are activated in high quantities during active periodontitis, with their components and cleavage products found in the diseased gingival tissues, in contrast to being undetected or present in low levels in healthy gingival tissues. The complement components identified in the affected gingiva or GCF encompass the entire immune cascade, including C1q, factor B, Bb, C3, C3a, C3b, C3c, C3d, C4, C5, C5a, C5b, and C9. A single nucleotide polymorphism in the gene coding for complement C5 and C3 has been linked to periodontitis[198,199]. C3a and C5a, along with mast cells, are involved in osteoclastogenesis and alveolar bone loss[165].
6.4. The Role of IL-17, RANKL, and Matrix Metalloproteinases (MMPs) in Bone Metabolism Related to LC
In the context of LC, the potential direct effects of SARS-CoV-2 are associated with its entry through angiotensin-converting enzyme 2 (ACE-2) receptors on bone cells, including osteoclasts and osteoblasts, which may disrupt the bone remodeling process[200,201]. The heightened production of inflammatory cytokines, recruitment of Th17 cells, and alterations in RANKL and osteoprotegerin (OPG) signaling are among the proposed mechanisms linking COVID-19 to osteoporosis and bone loss observed in affected patients[201,202,203].
In animal models, cytokine storms characterized by elevated serum levels of IL-1β, IL-6, and TNF-α have been shown to lead to trabecular bone loss in long bones and lumbar vertebrae, worsening from the acute to the post-recovery phase in SARS-CoV-2 in hamsters[204]. Th17 cells promote osteoclastogenesis through IL-17-mediated induction of RANKL[205], and As noted, elevated Th17 cell and IL-17 levels have been observed in individuals with LC[201,203].
Consequently, due to the rise in inflammatory markers, increased expression of MMPs, such as MMP-1, 2, 3, 7, 8, and 9—has been noted in various pathophysiological processes, ranging from chronic fatigue symptoms to the rapid progression of pulmonary fibrosis. Specifically, MMP-1, 8, and 9 are significantly elevated levels correlated with disease severity and increased neutrophil degranulation, as well as endothelial function, metabolic dysfunction, and the development of pulmonary fibrosis, which are all integral to the pathophysiology of LC[206,207,208,209,210,211]. MMP-8, primarily secreted by neutrophils, is a major degrading enzyme for interstitial collagens in periodontitis[212,213]. Additionally, MMPs, including MMP-8 and 9, contribute to the severity of COVID-19 infections[214,215,216].
At the periodontal site level, CD4+ T cells (decreased in LC) in gingival tissue are the primary contributors to elevated receptor activator of nuclear factor kappa-B (NFκB) ligand (RANKL) levels in individuals with chronic periodontitis[166]. However, neutrophils can facilitate the progression of periodontitis by inducing the recruitment of Th17 cell-derived CD4+ T cells (elevated in LC), which are activated and differentiated by dendritic cells (DCs) [217,218]in active periodontitis[219], and by promoting the accumulation of B-cells and plasma cells in severe lesions[102].
More specifically, the IL-17 cytokine-derived Th17 cells produced in periodontal tissue [220,221] can stimulate macrophages and other cellular sources, such as endothelial cells, epithelial cells, and fibroblasts, to produce pro-inflammatory mediators (e.g., IL-6, IL-8, TNF-α, IL-1β, PGE2) with partial synergy with LC’s cytokine storm[102,212] and can direct osteoblasts to produce RANKL[222], resulting in bone resorption[212,223,224].
In addition to secreting IL-17, Th17 cells also express RANKL, which activates osteoclasts[225]. Thus, increased IL-17 levels during a COVID-19 infection can potentially enhance osteoclast activity and bone resorption in the development of periodontitis. IL-17 induces the expression of MMP in fibroblasts, leading to the destruction of periodontal connective tissue[91,102].
B cells' production of neutralizing antibodies may directly or indirectly contribute to the destruction of periodontal connective tissue or alveolar bone by expressing pro-inflammatory cytokines, MMPs, and RANKL[102,226,227].
The key immune-inflammatory components shared between LC and periodontitis are summarized in Table 1, highlighting similarities and differences in immune cell activity, cytokine production, and other mediators.
As illustrated in Figure 1, these overlapping mechanisms between initial COVID-19 and periodontitis involve interconnected viral, immune, and inflammatory pathways.
7. The Potential Mechanisms of COVID-19/LC Effects on Periodontitis
Mechanistic understanding of the potential impact of LC on periodontitis development/progression is needed. A recent scoping review suggests that periodontitis and COVID-19 may independently increase serum levels of IL-1β, IL-6, and TNF-α in the same individual[228]. However, it lacks information on the joint or causal effects between the two diseases[228]. As previously mentioned, the association between ‘sequelae’ from COVID-19 and periodontitis has been cross-sectionally assessed[37]. The reverse assessment in a case-control study of COVID-19 indicated an increased salivary IL-6 level measured at two time points in individuals with COVID-19 and periodontitis[229]. The researchers reported that periodontitis was associated with increased salivary levels of RANKL and IL-1β during and after COVID-19[229]. Another study demonstrated that individuals with acute COVID-19 subsequently had an increased serum RANKL/OPG ratio compared to healthy controls. It also showed that in vivo murine coronavirus (MHV-3) can induce an osteoporotic phenotype via TNF and on macrophage/osteoclast infection in a mouse model of SARS-like disease[230].
7.1. The Role of Angiotensin-Converting Enzyme 2 (ACE2) on the LC and Periodontitis Association
Another biological pathway linking LC with periodontitis involves the role of ACE2 cellular receptors, which are abundant in various human tissues[231]. ACE2 is central to the primary SARS-CoV-2 infection pathway for access to and invasion of several organs and tissues, primarily in conjunction with the co-receptor “Transmembrane protease serine 2” (TMPRSS2) in the upper respiratory epithelium[232,233,234,235]. Following initial infection, SARS-CoV-2 induces cells to overexpress ACE2 through 1) direct binding of its spike protein to the ACE2 receptor[236] or (as recently reported) 2) direct or indirect binding and activation of Toll-like receptor 4 (TLR4)[237], followed by fusion of the viral and host cell membranes, promoted by TMPRSS2 and furin molecules[236,238,239], resulting in replication, exacerbation, and persistent inflammation[237].
Both ACE2 and TMPRSS2 are present in the epithelial component of the gingiva, specifically in the sulcular and pocket epitheliums. ACE2 is localized in the nucleus and cytoplasm of the spinous-basal layer, while TMPRSS2 is detected in the gingival epithelium as well as the horny and spinous-basal layers, with its intracellular expression occurring in the cytoplasm and cell membrane[240].
Prior or ongoing infections of mucosal, epithelial, and endothelial cells — associated with inflammation in the respiratory, renal, vascular, gastrointestinal, or cardiac tissues — increase the expression of ACE2, making these tissues more susceptible to SARS-CoV-2 infection[241,242]. Cytokine-mediated tissue stimulation from COVID-19 or other inflammatory processes can upregulate ACE2 expression, potentially creating a positive feedback loop that enhances viral replication[243,244,245,246,247]. SARS-CoV-2 infections of the endothelium introduce the virus to various target organs[248], portraying COVID-19 as a vascular disease[249].
Thus, prolonged or latent chronic SARS-CoV-2 antigen exposure from ACE2-rich tissues could precipitate the immunological dysregulation observed in LC[154,250]. Higher titers of plasma ACE2 activity have been noted in individuals with LC for up to four months after the acute SARS-CoV-2 infection[250]. Similarly, SARS-CoV-2 RNA and/or proteins have been detected long after the acute SARS-CoV-2 infection in gastrointestinal organ tissues, including the stomach, colon, intestine, appendix, gut mucosa, epithelium, and colorectal lamina propria, as well as in other tissues, such as skin, breasts, gallbladder, and olfactory neuroepithelium. Additionally, SARS-CoV-2 viral particles have been identified in the stool and blood of LC patients[251]. The presence of viral RNA in the plasma[172] and the persistence of the virus in “immunologic sanctuaries” like the gastrointestinal tract, olfactory system, and brain[252,253] are regarded as factors that may predict LC. However, when long-term SARS-CoV-2 antigen shedding is the primary subject of cross-sectional epidemiologic studies, associations with LC differ by site. No correlation was found between salivary long shedding and LC[167] or the gastrointestinal tract[252]. However, a connection was noted between SARS-CoV-2 in the olfactory mucosa and local LC symptoms[254].
Evidence of SARS-CoV-2 presence in the oral cavity during LC is still limited. SARS-CoV-2 was found in subsequent tissue biopsies of fungiform papillae from patients with LC sequelae[255]. SARS-CoV-2 has been detected in the supragingival and subgingival biofilms of individuals with acute COVID-19, even those without periodontitis[256], and in the saliva of individuals with asymptomatic or mild COVID-19[257]. Similarly, ACE2 activity in saliva correlates with an individual’s susceptibility to SARS-CoV-2 infection and disease severity[258]. The epithelial cells of the oral mucosa, tongue, and salivary glands exhibit higher ACE2 expression; these cells may serve as initial and late sites for SARS-CoV-2 infection[50,259].
Moreover, similar to other viral infections, e.g., herpesviruses[260,261], periodontal pockets with ulcerated gingival epithelium, exposed connective tissue, and periodontal ligament cells expressing higher levels of ACE2[240] could serve as reservoirs for the SARS-CoV-2 virus[262]. The presence of the virus in these periodontal pockets could exacerbate infections in both periodontal and systemic contexts.
7.2. LC May Affect Oral Dysbiosis, Increasing Susceptibility to Chronic Infections, Including Periodontitis
Persistent immune dysregulation caused by LC creates an optimal environment for an excessive response to other chronic infections. Gut dysbiosis leading to long-term digestive complications has been reported in individuals with acute SARS-CoV-2 infection or L[263,264,265]. More specifically, SARS-CoV-2 RNA was detected in patients’ stool samples ten months after the acute phase of COVID-19, and fecal viral RNA shedding was linked to gastrointestinal symptoms in individuals with L[253]. Similar findings have been noted in recent literature[265,266], suggesting a prolonged and possibly permanent viral presence in fecal material[267].
Moreover, recent findings suggest that the posterior segment of the oropharyngeal microbiome may act as a reservoir for bacteria associated with pneumonia and chronic lung infections in SARS-CoV-2 cases. COVID-19 infection can induce oropharyngeal dysbiosis, which may persist for at least 30 days, regardless of viral clearance or antibiotic treatment[268].
The link between periodontal pathogens and LC remains unclear. Bemquerer et al. (2024) reported no significant changes in subgingival pathogens when examining periodontitis in acute COVID-199[229], whereas Haran et al. (2021), in their longitudinal study, found that higher early levels of Prevotella and Veillonella in acute COVID-19 patients were associated with prolonged symptoms, ultimately leading to LC[269]. Conversely, there have been no assessments regarding the potential effects of chronic LC on subgingival microbial symbiosis that could contribute to periodontitis development.
Figure 2 illustrates the possible bidirectional pathways connecting LC and periodontitis, emphasizing chronic inflammatory feedback loops.
8. Summary
LC, an emerging new disease, can persist after the acute phase of COVID-19. Like diabetes, the immuno-inflammatory dysregulation associated with LC may increase susceptibility to periodontitis, making individuals with this disease a previously untargeted population of interest for preventing and treating periodontitis. Although the epidemiological connection between these two chronic conditions requires further investigation, it is plausible that the onset of periodontitis in previously healthy individuals or the worsening of existing periodontitis in those with a history may represent an oral manifestation of LC.
LC exerts a global pro-inflammatory influence across multiple facets of the immune system, characterized by sustained increases in neutrophils, production of pro-inflammatory cytokines, and activation of immune cells, alongside elevated levels of RANKL, MMPs, and complement factors- features that are common in both periodontitis and LC.
LC can drive gut dysbiosis and may also cause dysbiosis of the oral microbiome, increasing susceptibility to periodontitis through subgingival dysbiosis. While evidence of SARS-CoV-2 presence in the oral cavity related to LC remains limited, the gingiva and periodontal ligament cells in periodontal pockets show increased ACE2 expression, suggesting that areas with active periodontitis may serve as reservoirs for SARS-CoV-2. However, further epidemiological, tissue, cell, and molecular studies confirming a population relationship will be necessary to clarify the mechanisms linking LC and periodontitis.
Further clinical investigations are essential to enhance our understanding of the mechanisms linking LC and periodontitis. By elucidating the connections between chronic comorbid conditions (e.g., diabetes, cardiovascular disease, obesity) and their relationships with COVID-19 or LC, as well as periodontitis, we can focus on preventing and treating periodontitis. This approach may yield dual benefits for both oral health and overall health in individuals with LC. Public educational initiatives about the potential impacts of LC on oral and general health are needed to raise awareness of the possible development of periodontitis and other adverse events associated with LC, such as cardiovascular disease.
Author Contributions
Conceptualization, O.M.A. and M.B.M.; writing—original draft preparation, O.M.A. and M.B.M.; writing—review and editing, O.M.A. and M.B.M.; review and editing, S.W. All authors have read and approved the final version of the review.
Funding
None
Acknowledgments
The authors thank the Center for Oral Health Research (COHR) at the College of Dentistry for supporting this study and the College of Medicine for providing support and the software used to create the figures in this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- (WHO), W.H.O. Coronavirus Disease (COVID-19) Dashboard. 2020; Available from: https://covid19.who.int/.
- Chen, C.; et al. Global Prevalence of Post-Coronavirus Disease 2019 (COVID-19) Condition or Long COVID: A Meta-Analysis and Systematic Review. J Infect Dis 2022, 226, 1593–1607. [Google Scholar] [CrossRef]
- Bull-Otterson, L.B. , S; Saydah, S; Boehmer, TK; Adjei, S; Gray, S; Harris, AM Post–COVID Conditions Among Adult COVID-19 Survivors Aged 18–64 and ≥65 Years — United States, 20–November 2021, M.M.M.W. Rep, Editor. 2022, p. 713–717. 20 March.
- Ladds, E.; et al. Persistent symptoms after Covid-19: qualitative study of 114 "long Covid" patients and draft quality principles for services. BMC Health Serv Res 2020, 20, 1144. [Google Scholar] [CrossRef]
- Nalbandian, A.; et al. Post-acute COVID-19 syndrome. Nat Med 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Subramanian, A.; et al. Symptoms and risk factors for long COVID in non-hospitalized adults. Nat Med 2022, 28, 1706–1714. [Google Scholar] [CrossRef]
- Nath, A. , Long-Haul COVID. Neurology 2020, 95, 559–560. [Google Scholar] [CrossRef]
- Thaweethai, T.; et al. Development of a Definition of Postacute Sequelae of SARS-CoV-2 Infection. JAMA 2023, 329, 1934–1946. [Google Scholar] [CrossRef]
- Soriano, J.B.; et al. A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infect Dis 2022, 22, e102–e107. [Google Scholar] [CrossRef] [PubMed]
- Bowe, B., Y. Xie, and Z. Al-Aly, Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat Med 2022, 28, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Malizos, K.N. , Long COVID-19: A New Challenge to Public Health. J Bone Joint Surg Am 2022, 104, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Danesh, V.; et al. Symptom Clusters Seen in Adult COVID-19 Recovery Clinic Care Seekers. J Gen Intern Med 2023, 38, 442–449. [Google Scholar] [CrossRef]
- Davis, H.E.; et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Huang, C.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.T.; et al. Generalisable long COVID subtypes: findings from the NIH N3C and RECOVER programmes. EBioMedicine 2023, 87, 104413. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.; et al. Multi-organ impairment and long COVID: a 1-year prospective, longitudinal cohort study. J R Soc Med 2023, 116, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Perlis, R.H.; et al. Research Letter: Association between long COVID symptoms and employment status. medRxiv 2022. [CrossRef]
- Admon, A.J.; et al. Assessment of Symptom, Disability, and Financial Trajectories in Patients Hospitalized for COVID-19 at 6 Months. JAMA Netw Open 2023, 6, e2255795. [Google Scholar] [CrossRef]
- Cutler, D.M. , The Costs of Long COVID. JAMA Health Forum 2022, 3, e221809. [Google Scholar] [CrossRef]
- A detailed study of patients with long-haul COVID: an analysis of private healthcare claims. 2021: New York, NY.
- Gottlieb, M.; et al. Severe Fatigue and Persistent Symptoms at 3 Months Following Severe Acute Respiratory Syndrome Coronavirus 2 Infections During the Pre-Delta, Delta, and Omicron Time Periods: A Multicenter Prospective Cohort Study. Clin Infect Dis 2023, 76, 1930–1941. [Google Scholar] [CrossRef]
- Gandjour, A. , Long COVID: Costs for the German economy and health care and pension system. BMC Health Serv Res 2023, 23, 641. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; et al. More than 50 long-term effects of COVID-19: a systematic review and meta-analysis. Sci Rep 2021, 11, 16144. [Google Scholar] [CrossRef]
- Premraj, L.; et al. Mid and long-term neurological and neuropsychiatric manifestations of post-COVID-19 syndrome: A meta-analysis. J Neurol Sci 2022, 434, 120162. [Google Scholar] [CrossRef]
- Ceban, F.; et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav Immun 2022, 101, 93–135. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z., Y. Xie, and B. Bowe, High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [CrossRef]
- Davis, H.E.; et al. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; et al. SARS-CoV-2 and male infertility: from short- to long-term impacts. J Endocrinol Invest 2023, 46, 1491–1507. [Google Scholar] [CrossRef]
- Dagher, H.; et al. Long COVID in cancer patients: preponderance of symptoms in majority of patients over long time period. Elife 2023, 12. [Google Scholar] [CrossRef]
- Zhou, X.; et al. The Oral Complications of COVID-19. Front Mol Biosci 2021, 8, 803785. [Google Scholar] [CrossRef]
- Ahmed, E.; et al. Oral mucormycosis in post-COVID-19 patients: A case series. Oral Dis 2022, 28, 2591–2592. [Google Scholar] [CrossRef]
- Rafalowicz, B., L. Wagner, and J. Rafalowicz, Long COVID Oral Cavity Symptoms Based on Selected Clinical Cases. Eur J Dent.
- Chandwani, N.; et al. Oral Tissue Involvement and Probable Factors in Post-COVID-19 Mucormycosis Patients: A Cross-Sectional Study. Healthcare (Basel).
- Alfaifi, A.; et al. Long-Term Post-COVID-19 Associated Oral Inflammatory Sequelae. Front Cell Infect Microbiol 2022, 12, 831744. [Google Scholar] [CrossRef]
- Arshad, W.; et al. Case of maxillary actinomycotic osteomyelitis, a rare post COVID complication-case report. Ann Med Surg (Lond) 2022, 80, 104242. [Google Scholar] [CrossRef]
- Frencken, J.E.; et al. Global epidemiology of dental caries and severe periodontitis - a comprehensive review. J Clin Periodontol 2017, 44, S94–S105. [Google Scholar] [CrossRef]
- Casarin, M.; et al. Association between sequelae of COVID-19 with periodontal disease and obesity: A cross-sectional study. J Periodontol 2023. [CrossRef]
- Chapple, I.L.C.; et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol 2018, 89, S74–S84. [Google Scholar] [CrossRef]
- Eke, P.I., W. S. Borgnakke, and R.J. Genco, Recent epidemiologic trends in periodontitis in the USA. Periodontol, 82.
- Kassebaum, N.J.; et al. Global burden of severe periodontitis in 1990-2010: a systematic review and meta-regression. J Dent Res 2014, 93, 1045–53. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N. , Epidemiology of periodontal diseases: an update. J Int Acad Periodontol 1999, 1, 110–6. [Google Scholar]
- Friedewald, V.E.; et al. The American Journal of Cardiology and Journal of Periodontology Editors' Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol 2009, 104, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Barbosa De Accioly Mattos, M.; et al. Coronary atherosclerosis and periodontitis have similarities in their clinical presentation. Frontiers in Oral Health 2024, 4. [Google Scholar] [CrossRef] [PubMed]
- American Academy of, P. , American Academy of Periodontology statement on risk assessment. J Periodontol 2008, 79, 202. [Google Scholar]
- Andriankaja, O.M.; et al. Hispanic adults with type 2 diabetes mellitus using lipid-lowering agents have better periodontal health than non-users. Therapeutic Advances in Chronic Disease, 2040. [Google Scholar]
- Andriankaja, O.M.; et al. Periodontal Disease, Local and Systemic Inflammation in Puerto Ricans with Type 2 Diabetes Mellitus. Biomedicines.
- Andriankaja, O.M.; et al. Systemic Inflammation, Endothelial Function, and Risk of Periodontitis in Overweight/Obese Adults. Biomedicines.
- Kinane, D.F.; et al. Humoral immune responses in periodontal disease may have mucosal and systemic immune features. Clin Exp Immunol 1999, 115, 534–41. [Google Scholar] [CrossRef]
- Lappin, D.F., A. M. McGregor, and D.F. Kinane, The systemic immune response is more prominent than the mucosal immune response in the pathogenesis of periodontal disease. J Clin Periodontol.
- Lin, E.C.; et al. Unraveling the Link between Periodontitis and Coronavirus Disease 2019: Exploring Pathogenic Pathways and Clinical Implications. Biomedicines.
- Loe, H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care 1993, 16, 329–34. [Google Scholar]
- Kocher, T.; et al. Periodontal complications of hyperglycemia/diabetes mellitus: Epidemiologic complexity and clinical challenge. Periodontol 2000 2018, 78, 59–97. [Google Scholar] [CrossRef] [PubMed]
- Zimlich, R. What Is Long COVID? 2023.
- Yong, S.J. , Long COVID or post-COVID-19 syndrome: putative pathophysiology, risk factors, and treatments. Infect Dis (Lond) 2021, 53, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Crook, H.; et al. Long covid-mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef] [PubMed]
- Darby, I. , Risk factors for periodontitis & peri-implantitis. Periodontol 2000 2022, 90, 9–12. [Google Scholar]
- Alqahtani, H.M.; et al. Identifying Factors Associated with Periodontal Disease Using Machine Learning. J Int Soc Prev Community Dent 2022, 12, 612–622. [Google Scholar] [CrossRef]
- Sanz, M.; et al. Scientific evidence on the links between periodontal diseases and diabetes: Consensus report and guidelines of the joint workshop on periodontal diseases and diabetes by the International Diabetes Federation and the European Federation of Periodontology. J Clin Periodontol 2018, 45, 138–149. [Google Scholar] [CrossRef]
- Carrizales-Sepulveda, E.F.; et al. Periodontal Disease, Systemic Inflammation and the Risk of Cardiovascular Disease. Heart Lung Circ 2018, 27, 1327–1334. [Google Scholar] [CrossRef]
- Wu, C.Z.; et al. Epidemiologic relationship between periodontitis and type 2 diabetes mellitus. BMC Oral Health.
- Abu-Shawish, G., J. Betsy, and S. Anil, Is Obesity a Risk Factor for Periodontal Disease in Adults? A Systematic Review. Int J Environ Res Public Health.
- Jepsen, S., J. Suvan, and J. Deschner, The association of periodontal diseases with metabolic syndrome and obesity. Periodontol, 83.
- Rivas-Agosto, J.A. , Camacho-Monclova, D.M., Vergara, J.L., Vivaldi-Oliver, J., Andriankaja, O.M., Disparities in periodontal diseases occurrence among Hispanic population with Type 2 Diabetes: the LLIPDS Study. EC. Dental Science.
- Smith, H.; et al. Cross-sectional association among dietary habits, periodontitis, and uncontrolled diabetes in Hispanics: the LLIPDS study. Front Oral Health 2025, 6, 1468995. [Google Scholar] [CrossRef]
- Weaver, C.M. , K, Janeway's immunobiology, in Garland Science. 2016. [Google Scholar]
- Sette, A. and S. Crotty, Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [CrossRef]
- Luo, X.H.; et al. T cell immunobiology and cytokine storm of COVID-19. Scand J Immunol 2021, 93, e12989. [Google Scholar] [CrossRef]
- Galani, I.E.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol 2021, 22, 32–40. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell, 1036. [Google Scholar]
- Laing, A.G.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Hadjadj, J.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Perez-Gomez, A.; et al. Dendritic cell deficiencies persist seven months after SARS-CoV-2 infection. Cell Mol Immunol 2021, 18, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; et al. Distinguishing features of long COVID identified through immune profiling. Nature 2023, 623, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, V.; et al. An Inflammatory Profile Correlates With Decreased Frequency of Cytotoxic Cells in Coronavirus Disease 2019. Clin Infect Dis 2020, 71, 2272–2275. [Google Scholar] [CrossRef]
- Diao, B.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol 2020, 11, 827. [Google Scholar] [CrossRef]
- Sharawi, H.; et al. The Prevalence of Gingival Dendritic Cell Subsets in Periodontal Patients. J Dent Res 2021, 100, 1330–1336. [Google Scholar] [CrossRef]
- Chen, C., P. Feng, and J. Slots, Herpesvirus-bacteria synergistic interaction in periodontitis. Periodontol 2000 2020, 82, 42–64. [Google Scholar] [CrossRef]
- Muzammil, et al., Association of interferon lambda-1 with herpes simplex viruses-1 and -2, Epstein-Barr virus, and human cytomegalovirus in chronic periodontitis. J Investig Clin Dent.
- Kajita, K.; et al. Quantitative messenger RNA expression of Toll-like receptors and interferon-alpha1 in gingivitis and periodontitis. Oral Microbiol Immunol 2007, 22, 398–402. [Google Scholar] [CrossRef]
- Chen, Y.; et al. Immune response pattern across the asymptomatic, symptomatic and convalescent periods of COVID-19. Biochim Biophys Acta Proteins Proteom 2022, 1870, 140736. [Google Scholar] [CrossRef]
- Grifoni, A.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell, 1489. [Google Scholar]
- Kudryavtsev, I.; et al. Dysregulated Immune Responses in SARS-CoV-2-Infected Patients: A Comprehensive Overview. Viruses.
- Silva, M.J.A.; et al. Adaptive immunity to SARS-CoV-2 infection: A systematic review. Front Immunol 2022, 13, 1001198. [Google Scholar] [CrossRef]
- Parackova, Z.; et al. Neutrophils mediate Th17 promotion in COVID-19 patients. J Leukoc Biol 2021, 109, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Martonik, D.; et al. The Role of Th17 Response in COVID-19. Cells.
- De Biasi, S.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun 2020, 11, 3434. [Google Scholar] [CrossRef] [PubMed]
- Shibabaw, T. , Inflammatory Cytokine: IL-17A Signaling Pathway in Patients Present with COVID-19 and Current Treatment Strategy. J Inflamm Res 2020, 13, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; et al. The role of interleukin-22 in lung health and its therapeutic potential for COVID-19. Front Immunol 2022, 13, 951107. [Google Scholar] [CrossRef]
- Alcorn, J.F. , IL-22 Plays a Critical Role in Maintaining Epithelial Integrity During Pulmonary Infection. Front Immunol 2020, 11, 1160. [Google Scholar] [CrossRef]
- Zenobia, C. and G. Hajishengallis, Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol, 69.
- Bilich, T.; et al. T cell and antibody kinetics delineate SARS-CoV-2 peptides mediating long-term immune responses in COVID-19 convalescent individuals. Sci Transl Med.
- Ibarrondo, F.J.; et al. Rapid Decay of Anti-SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N Engl J Med 2020, 383, 1085–1087. [Google Scholar] [CrossRef]
- Seow, J.; et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol 2020, 5, 1598–1607. [Google Scholar] [CrossRef]
- Yamayoshi, S.; et al. Antibody titers against SARS-CoV-2 decline, but do not disappear for several months. EClinicalMedicine 2021, 32, 100734. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.W.; et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Rep Med 2021, 2, 100354. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; et al. Dynamics of TCR repertoire and T cell function in COVID-19 convalescent individuals. Cell Discov 2021, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; et al. Anti-SARS-CoV-2 receptor-binding domain antibody evolution after mRNA vaccination. Nature 2021, 600, 517–522. [Google Scholar] [CrossRef]
- Cardoso, E.M. and F.A. Arosa, CD8(+) T Cells in Chronic Periodontitis: Roles and Rules. Front Immunol 2017, 8, 145. [Google Scholar] [CrossRef]
- John, V.; et al. A role for CD8+ T lymphocytes in osteoclast differentiation in vitro. Endocrinology 1996, 137, 2457–63. [Google Scholar] [CrossRef]
- Choi, Y.; et al. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol 2001, 31, 2179–88. [Google Scholar] [CrossRef]
- Hajishengallis, G. and J.M. Korostoff, Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000 2017, 75, 116–151. [Google Scholar]
- Kuri-Cervantes, L.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol.
- Aschenbrenner, A.C.; et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med 2021, 13, 7. [Google Scholar] [CrossRef]
- Meizlish, M.L.; et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv 2021, 5, 1164–1177. [Google Scholar] [CrossRef]
- Vanderbeke, L.; et al. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat Commun 2021, 12, 4117. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; et al. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front Immunol 2020, 11, 1708. [Google Scholar] [CrossRef]
- Hojyo, S.; et al. How COVID-19 induces cytokine storm with high mortality. Inflamm Regen 2020, 40, 37. [Google Scholar] [CrossRef]
- Tay, M.Z.; et al. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 2020 20, 363–374. [CrossRef]
- Kaiser, R.; et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. JCI Insight.
- Frishberg, A.; et al. Mature neutrophils and a NF-kappaB-to-IFN transition determine the unifying disease recovery dynamics in COVID-19. Cell Rep Med 2022, 3, 100652. [Google Scholar] [CrossRef]
- Wang, X.; et al. Temporal transcriptomic analysis using TrendCatcher identifies early and persistent neutrophil activation in severe COVID-19. JCI Insight.
- Jukema, B.N.; et al. Neutrophil and Eosinophil Responses Remain Abnormal for Several Months in Primary Care Patients With COVID-19 Disease. Front Allergy 2022, 3, 942699. [Google Scholar] [CrossRef] [PubMed]
- Neurath, N. and M. Kesting, Cytokines in gingivitis and periodontitis: from pathogenesis to therapeutic targets. Front Immunol 2024, 15, 1435054. [Google Scholar] [CrossRef]
- Pan, W., Q. Wang, and Q. Chen, The cytokine network involved in the host immune response to periodontitis. Int J Oral Sci 2019, 11, 30. [Google Scholar] [CrossRef]
- Jorch, S.K. and P. Kubes, An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Henriksson, L. and U. Kelter, Ultrasonography and scintigraphy of the liver in focal and diffuse disease. Acta Radiol 1987, 28, 165–8. [Google Scholar] [CrossRef]
- Vitkov, L.; et al. Periodontitis-Derived Dark-NETs in Severe Covid-19. Front Immunol 2022, 13, 872695. [Google Scholar] [CrossRef] [PubMed]
- Twaddell, S.H.; et al. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 2019, 156, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med.
- Zuo, Y.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight.
- Carmona-Rivera, C.; et al. Multicenter analysis of neutrophil extracellular trap dysregulation in adult and pediatric COVID-19. JCI Insight.
- Mohandas, S.; et al. Immune mechanisms underlying COVID-19 pathology and post-acute sequelae of SARS-CoV-2 infection (PASC). Elife 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Grabuschnig, S.; et al. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int J Mol Sci.
- Vitkov, L.; et al. Extracellular neutrophil traps in periodontitis. J Periodontal Res 2009, 44, 664–72. [Google Scholar] [CrossRef]
- Magan-Fernandez, A.; et al. Characterization and comparison of neutrophil extracellular traps in gingival samples of periodontitis and gingivitis: A pilot study. J Periodontal Res 2019, 54, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, C.; et al. Circulating levels of carbamylated protein and neutrophil extracellular traps are associated with periodontitis severity in patients with rheumatoid arthritis: A pilot case-control study. PLoS One 2018, 13, e0192365. [Google Scholar] [CrossRef]
- Silvin, A.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell, 1401. [Google Scholar]
- Zhou, Y.; et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl Sci Rev 2020, 7, 998–1002. [Google Scholar] [CrossRef]
- Merad, M. and J.C. Martin, Author Correction: Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 2020, 20, 448. [Google Scholar] [CrossRef]
- Schulert, G.S. and A.A. Grom, Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med 2015, 66, 145–59. [Google Scholar] [CrossRef]
- Karki, R.; et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. bioRxiv 2020.
- Swanson, K.V., M. Deng, and J.P. Ting, The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019 19, 477–489.
- Junqueira, C.; et al. FcgammaR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K. and M.R. Mehra, COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J Heart Lung Transplant 2020, 39, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front Immunol 2021, 12, 746021. [Google Scholar] [CrossRef]
- Bassler, K.; et al. The Myeloid Cell Compartment-Cell by Cell. Annu Rev Immunol 2019, 37, 269–293. [Google Scholar] [CrossRef]
- Bohnacker, S.; et al. Correction to: Mild COVID-19 imprints a long-term inflammatory eicosanoid- and chemokine memory in monocyte-derived macrophages. Mucosal Immunol 2022, 15, 798. [Google Scholar] [CrossRef]
- Hernichel-Gorbach, E.; et al. Host responses in patients with generalized refractory periodontitis. J Periodontol 1994, 65, 8–16. [Google Scholar] [CrossRef]
- Shapira, L.; et al. The secretion of PGE2, IL-1 beta, IL-6, and TNF alpha by adherent mononuclear cells from early onset periodontitis patients. J Periodontol 1994, 65, 139–46. [Google Scholar] [CrossRef]
- Beck, J.; et al. Periodontal disease and cardiovascular disease. J Periodontol 1996, 67(10 Suppl), 1123–37. [Google Scholar]
- Ptasiewicz, M.; et al. Armed to the Teeth-The Oral Mucosa Immunity System and Microbiota. Int J Mol Sci.
- Kudo, O.; et al. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Dutzan, N.; et al. On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the Oral Barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Bahammam, M.A. and M.S. Attia, Effects of Systemic Simvastatin on the Concentrations of Visfatin, Tumor Necrosis Factor-alpha, and Interleukin-6 in Gingival Crevicular Fluid in Patients with Type 2 Diabetes and Chronic Periodontitis. J Immunol Res 2018, 2018, 8481735. [Google Scholar] [CrossRef]
- Sitompul, S.I.; et al. Analysis of the Effects of IL-6 -572 C/G, CRP -757 A/G, and CRP -717 T/C Gene Polymorphisms; IL-6 Levels; and CRP Levels on Chronic Periodontitis in Coronary Artery Disease in Indonesia. Genes (Basel).
- Hobbins, S.; et al. Is periodontitis a comorbidity of COPD or can associations be explained by shared risk factors/behaviors? Int J Chron Obstruct Pulmon Dis 2017, 12, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Center, D.M. and W. Cruikshank, Modulation of lymphocyte migration by human lymphokines. I. Identification and characterization of chemoattractant activity for lymphocytes from mitogen-stimulated mononuclear cells. J Immunol 1982, 128, 2563–8. 128.
- Loretelli, C.; et al. PD-1 blockade counteracts post-COVID-19 immune abnormalities and stimulates the anti-SARS-CoV-2 immune response. JCI Insight.
- Shuwa, H.A.; et al. Alterations in T and B cell function persist in convalescent COVID-19 patients. Med.
- Szabo, P.A.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity.
- Zhang, J.Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol 2020, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol 2022, 23, 210–216. [Google Scholar] [CrossRef]
- Woodruff, M.C.; et al. Chronic inflammation, neutrophil activity, and autoreactivity splits long COVID. Nat Commun 2023, 14, 4201. [Google Scholar] [CrossRef]
- Files, J.K.; et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J Clin Invest.
- Daugherty, S.E.; et al. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: retrospective cohort study. BMJ 2021, 373, n1098. [Google Scholar] [CrossRef]
- DeVries, A.; et al. One-Year Adverse Outcomes Among US Adults With Post-COVID-19 Condition vs Those Without COVID-19 in a Large Commercial Insurance Database. JAMA Health Forum 2023, 4, e230010. [Google Scholar] [CrossRef]
- Malkova, A.; et al. Post COVID-19 Syndrome in Patients with Asymptomatic/Mild Form. Pathogens.
- Maglietta, G.; et al. Prognostic Factors for Post-COVID-19 Syndrome: A Systematic Review and Meta-Analysis. J Clin Med.
- Sandler, C.X.; et al. Long COVID and Post-infective Fatigue Syndrome: A Review. Open Forum Infect Dis 2021, 8, ofab440. [Google Scholar] [CrossRef]
- Dennis, A. , Wamil, M,Kapur,S, Alberts, J, Badley, A.D., Decker, G.A., Rizza,S.A.,Banerjee, R., Banerjee, A., Multi-organ impairment in low-risk individuals with long COVID. medRxiv.
- Sudre, C.H.; et al. Attributes and predictors of long COVID. Nat Med 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Ryder, M.I. , Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000 2010, 53, 124–37. [Google Scholar] [CrossRef]
- Luntzer, K.; et al. Increased Presence of Complement Factors and Mast Cells in Alveolar Bone and Tooth Resorption. Int J Mol Sci.
- Vernal, R.; et al. High expression levels of receptor activator of nuclear factor-kappa B ligand associated with human chronic periodontitis are mainly secreted by CD4+ T lymphocytes. J Periodontol 2006, 77, 1772–80. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep 2021, 36, 109518. [Google Scholar] [CrossRef] [PubMed]
- Glynne, P.; et al. Long COVID following mild SARS-CoV-2 infection: characteristic T cell alterations and response to antihistamines. J Investig Med 2022, 70, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, K.M.; et al. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PLoS Pathog 2022, 18, e1010359. [Google Scholar] [CrossRef]
- Vijayakumar, B.; et al. Immuno-proteomic profiling reveals aberrant immune cell regulation in the airways of individuals with ongoing post-COVID-19 respiratory disease. Immunity.
- Woodruff, M.C.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol 2020, 21, 1506–1516. [Google Scholar] [CrossRef]
- Su, Y.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell.
- Gold, J.E.; et al. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens.
- Alramadhan, S.A.; et al. Oral Hairy Leukoplakia in Immunocompetent Patients Revisited with Literature Review. Head Neck Pathol 2021, 15, 989–993. [Google Scholar] [CrossRef]
- Ward, B.J.H.; et al. EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses.
- Maulani, C.; et al. Association between Epstein-Barr virus and periodontitis: A meta-analysis. PLoS One 2021, 16, e0258109. [Google Scholar] [CrossRef]
- Bastard, P.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science, 6515. [Google Scholar]
- Combes, A.J.; et al. Publisher Correction: Global absence and targeting of protective immune states in severe COVID-19. Nature 2021, 596, E8. [Google Scholar] [CrossRef]
- Zhang, Q.; et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Kasuga, Y.; et al. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Lowery, S.A., A. Sariol, and S. Perlman, Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. 29.
- Schultze, J.L. and A.C. Aschenbrenner, COVID-19 and the human innate immune system. Cell, 1671. [Google Scholar]
- Merad, M., A. Subramanian, and T.T. Wang, An aberrant inflammatory response in severe COVID-19. Cell Host Microbe 2021, 29, 1043–1047. 29.
- Wang, E.Y.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Knight, J.S.; et al. The intersection of COVID-19 and autoimmunity. J Clin Invest.
- Mishra, P.K.; et al. Vaccination boosts protective responses and counters SARS-CoV-2-induced pathogenic memory B cells. medRxiv 2021.
- Chang, S.E.; et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun 2021, 12, 5417. [Google Scholar] [CrossRef]
- Bjornevik, K.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H. and L. Steinman, Epstein-Barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Peluso, M.J.; et al. Low Prevalence of Interferon alpha Autoantibodies in People Experiencing Symptoms of Post-Coronavirus Disease 2019 (COVID-19) Conditions, or Long COVID. J Infect Dis 2023, 227, 246–250. [Google Scholar] [CrossRef]
- Seessle, J.; et al. Persistent Symptoms in Adult Patients 1 Year After Coronavirus Disease 2019 (COVID-19): A Prospective Cohort Study. Clin Infect Dis 2022, 74, 1191–1198. [Google Scholar] [CrossRef]
- Peluso, M.J.; et al. Lack of Antinuclear Antibodies in Convalescent Coronavirus Disease 2019 Patients With Persistent Symptoms. Clin Infect Dis 2022, 74, 2083–2084. [Google Scholar] [CrossRef]
- Marshall, M. , The four most urgent questions about long COVID. Nature 2021, 594, 168–170. [Google Scholar] [CrossRef]
- Moody, R.; et al. Antibodies against Spike protein correlate with broad autoantigen recognition 8 months post SARS-CoV-2 exposure, and anti-calprotectin autoantibodies associated with better clinical outcomes. Front Immunol 2022, 13, 945021. [Google Scholar] [CrossRef]
- Cervia-Hasler, C.; et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science 2024, 383, eadg7942. [Google Scholar] [CrossRef]
- Hajishengallis, G.; et al. Complement-Dependent Mechanisms and Interventions in Periodontal Disease. Front Immunol 2019, 10, 406. [Google Scholar] [CrossRef]
- Chai, L.; et al. Single nucleotide polymorphisms of complement component 5 and periodontitis. J Periodontal Res 2010, 45, 301–8. [Google Scholar] [CrossRef]
- Zhan, Y.; et al. Prioritization of candidate genes for periodontitis using multiple computational tools. J Periodontol 2014, 85, 1059–69. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; et al. The angiotensin converting enzyme 2/angiotensin-(1-7)/Mas Receptor axis as a key player in alveolar bone remodeling. Bone 2019, 128, 115041. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzawi, I.S. and N.S. Mohammed, The Impact of Angiotensin Converting Enzyme-2 (ACE-2) on Bone Remodeling Marker Osteoprotegerin (OPG) in Post-COVID-19 Iraqi Patients. Cureus 2022, 14, e29926. [Google Scholar]
- Creecy, A.; et al. COVID-19 and Bone Loss: A Review of Risk Factors, Mechanisms, and Future Directions. Curr Osteoporos Rep 2024, 22, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Sapra, L.; et al. Long-term implications of COVID-19 on bone health: pathophysiology and therapeutics. Inflamm Res 2022, 71, 1025–1040. [Google Scholar] [CrossRef]
- Qiao, W.; et al. SARS-CoV-2 infection induces inflammatory bone loss in golden Syrian hamsters. Nat Commun 2022, 13, 2539. [Google Scholar] [CrossRef]
- Sato, K.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006, 203, 2673–82. [Google Scholar] [CrossRef]
- Patel, M.A.; et al. Elevated vascular transformation blood biomarkers in Long-COVID indicate angiogenesis as a key pathophysiological mechanism. Mol Med 2022, 28, 122. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, G.; et al. Reduction in ACE2 expression in peripheral blood mononuclear cells during COVID-19 - implications for post COVID-19 conditions. BMC Infect Dis 2024, 24, 663. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; et al. Post-COVID-19 Pulmonary Fibrosis. Cureus 2022, 14, e22770. [Google Scholar] [CrossRef]
- Tyagi, S.C.; et al. COVID-19 Mimics Pulmonary Dysfunction in Muscular Dystrophy as a Post-Acute Syndrome in Patients. Int J Mol Sci.
- Banerjee, S.; et al. An updated patent review of matrix metalloproteinase (MMP) inhibitors (2021-present). Expert Opin Ther Pat 2023, 33, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Mulet, A.; et al. Biomarkers of Fibrosis in Patients with COVID-19 One Year After Hospital Discharge: A Prospective Cohort Study. Am J Respir Cell Mol Biol 2023, 69, 321–327. [Google Scholar] [CrossRef]
- Balaji, S., P. K. Cholan, and D.J. Victor, An emphasis of T-cell subsets as regulators of periodontal health and disease. J Clin Transl Res.
- Luchian, I.; et al. The Role of Matrix Metalloproteinases (MMP-8, MMP-9, MMP-13) in Periodontal and Peri-Implant Pathological Processes. Int J Mol Sci.
- da Silva-Neto, P.V.; et al. sTREM-1 Predicts Disease Severity and Mortality in COVID-19 Patients: Involvement of Peripheral Blood Leukocytes and MMP-8 Activity. Viruses.
- Gelzo, M.; et al. Matrix metalloproteinases (MMP) 3 and 9 as biomarkers of severity in COVID-19 patients. Sci Rep 2022, 12, 1212. [Google Scholar] [CrossRef]
- Hardy, E. and C. Fernandez-Patron, Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis. Biomolecules.
- Cutler, C.W. and Y.T. Teng, Oral mucosal dendritic cells and periodontitis: many sides of the same coin with new twists. Periodontol 2007, 45, 35–50. [Google Scholar] [CrossRef]
- Wilensky, A.; et al. Dendritic cells and their role in periodontal disease. Oral Dis 2014, 20, 119–26. [Google Scholar] [CrossRef]
- Pelletier, M.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–43. [Google Scholar] [CrossRef]
- Dutzan, N.; et al. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol 2016, 9, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, N.M.; et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med 2014, 6, 229ra40. [Google Scholar] [CrossRef]
- Kotake, S.; et al. Role of osteoclasts and interleukin-17 in the pathogenesis of rheumatoid arthritis: crucial 'human osteoclastology'. J Bone Miner Metab 2012, 30, 125–35. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; et al. The potential role of interleukin-17 in the immunopathology of periodontal disease. J Clin Periodontol 2005, 32, 369–74. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.R.; et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol 2009, 24, 1–6. [Google Scholar] [CrossRef]
- Kikuta, J.; et al. Dynamic visualization of RANKL and Th17-mediated osteoclast function. J Clin Invest 2013, 123, 866–73. [Google Scholar] [CrossRef]
- Berglundh, T., M. Donati, and N. Zitzmann, B cells in periodontitis: friends or enemies? Periodontol 2000 2007, 45, 51–66. [CrossRef]
- Gonzales, J.R. , T- and B-cell subsets in periodontitis. Periodontol 2000 2015, 69, 181–200. [Google Scholar] [CrossRef]
- Mootha, A. , Is There a Similarity in Serum Cytokine Profile between Patients with Periodontitis or 2019-Novel Coronavirus Infection?-A Scoping Review. Biology (Basel).
- Bemquerer, L.M.; et al. Clinical, immunological, and microbiological analysis of the association between periodontitis and COVID-19: a case-control study. Odontology 2024, 112, 208–220. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; et al. Acute coronavirus infection triggers a TNF-dependent osteoporotic phenotype in mice. Life Sci 2023, 324, 121750. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; et al. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; et al. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Torge, D.; et al. Histopathological Features of SARS-CoV-2 in Extrapulmonary Organ Infection: A Systematic Review of Literature. Pathogens.
- Bourgonje, A.R.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Chen, B.; et al. Viral persistence, reactivation, and mechanisms of long COVID. Elife 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; et al. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Aboudounya, M.M. and R.J. Heads, COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflamm 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Hoffmann, M.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell.
- Senapati, S.; et al. Contributions of human ACE2 and TMPRSS2 in determining host-pathogen interaction of COVID-19. J Genet.
- Sakaguchi, W.; et al. Existence of SARS-CoV-2 Entry Molecules in the Oral Cavity. Int J Mol Sci.
- Zou, X.; et al. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Lei, H.Y.; et al. Potential effects of SARS-CoV-2 on the gastrointestinal tract and liver. Biomed Pharmacother 2021, 133, 111064. [Google Scholar] [CrossRef]
- Hou, Y.J.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell.
- Zhuang, M.W.; et al. Increasing host cellular receptor-angiotensin-converting enzyme 2 expression by coronavirus may facilitate 2019-nCoV (or SARS-CoV-2) infection. J Med Virol 2020, 92, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; et al. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev Cell.
- Zhang, J., K. M. Tecson, and P.A. McCullough, Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev Cardiovasc Med.
- Ziegler, C.G.K.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell, 1016. [Google Scholar]
- Iwasaki, M.; et al. Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K., P. Libby, and P.M. Ridker, COVID-19 - A vascular disease. Trends Cardiovasc Med.
- Patel, S.K.; et al. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: implications for COVID-19 pathogenesis and consequences. Eur Respir J.
- Proal, A.D.; et al. SARS-CoV-2 reservoir in post-acute sequelae of COVID-19 (PASC). Nat Immunol 2023, 24, 1616–1627. [Google Scholar] [CrossRef]
- Gaebler, C.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Klein, J.; et al. Distinguishing features of Long COVID identified through immune profiling. medRxiv 2022. [CrossRef]
- de Melo, G.D.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci Transl Med.
- Yao, Q.; et al. Long-Term Dysfunction of Taste Papillae in SARS-CoV-2. NEJM Evid.
- Gomes, S.C.; et al. SARS-CoV-2 RNA in dental biofilms: Supragingival and subgingival findings from inpatients in a COVID-19 intensive care unit. J Periodontol 2022, 93, 1476–1485. [Google Scholar] [CrossRef]
- Marchesan, J.T., B. M. Warner, and K.M. Byrd, The "oral" history of COVID-19: Primary infection, salivary transmission, and post-acute implications. J Periodontol, 1357. [Google Scholar]
- Jimenez, D.; et al. Differences in saliva ACE2 activity among infected and non-infected adult and pediatric population exposed to SARS-CoV-2. J Infect 2022, 85, 86–89. [Google Scholar] [CrossRef]
- Xu, H.; et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 2020, 12, 8. [Google Scholar] [CrossRef]
- Contreras, A., H. Nowzari, and J. Slots, Herpesviruses in periodontal pocket and gingival tissue specimens. Oral Microbiol Immunol.
- Cappuyns, I., P. Gugerli, and A. Mombelli, Viruses in periodontal disease - a review. Oral Dis.
- Badran, Z.; et al. Periodontal pockets: A potential reservoir for SARS-CoV-2? Med Hypotheses 2020, 143, 109907. [Google Scholar] [CrossRef]
- Zhang, D.; et al. Gut Microbiota Dysbiosis Correlates With Long COVID-19 at One-Year After Discharge. J Korean Med Sci 2023, 38, e120. [Google Scholar] [CrossRef]
- Maeda, Y.; et al. Longitudinal alterations of the gut mycobiota and microbiota on COVID-19 severity. BMC Infect Dis 2022, 22, 572. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, V.; et al. Mild SARS-CoV-2 infection results in long-lasting microbiota instability. mBio 2023, 14, e0088923. [Google Scholar] [CrossRef] [PubMed]
- Zollner, A.; et al. Postacute COVID-19 is Characterized by Gut Viral Antigen Persistence in Inflammatory Bowel Diseases. Gastroenterology.
- He, K.Y.; et al. Development and management of gastrointestinal symptoms in long-term COVID-19. Front Microbiol 2023, 14, 1278479. [Google Scholar] [CrossRef] [PubMed]
- Paine, S.K.; et al. Temporal dynamics of oropharyngeal microbiome among SARS-CoV-2 patients reveals continued dysbiosis even after Viral Clearance. NPJ Biofilms Microbiomes 2022, 8, 67. [Google Scholar] [CrossRef]
- Haran, J.P.; et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight.
Figure 1.
Overlapping or concomitant mechanisms between Initial COVID and Periodontitis.
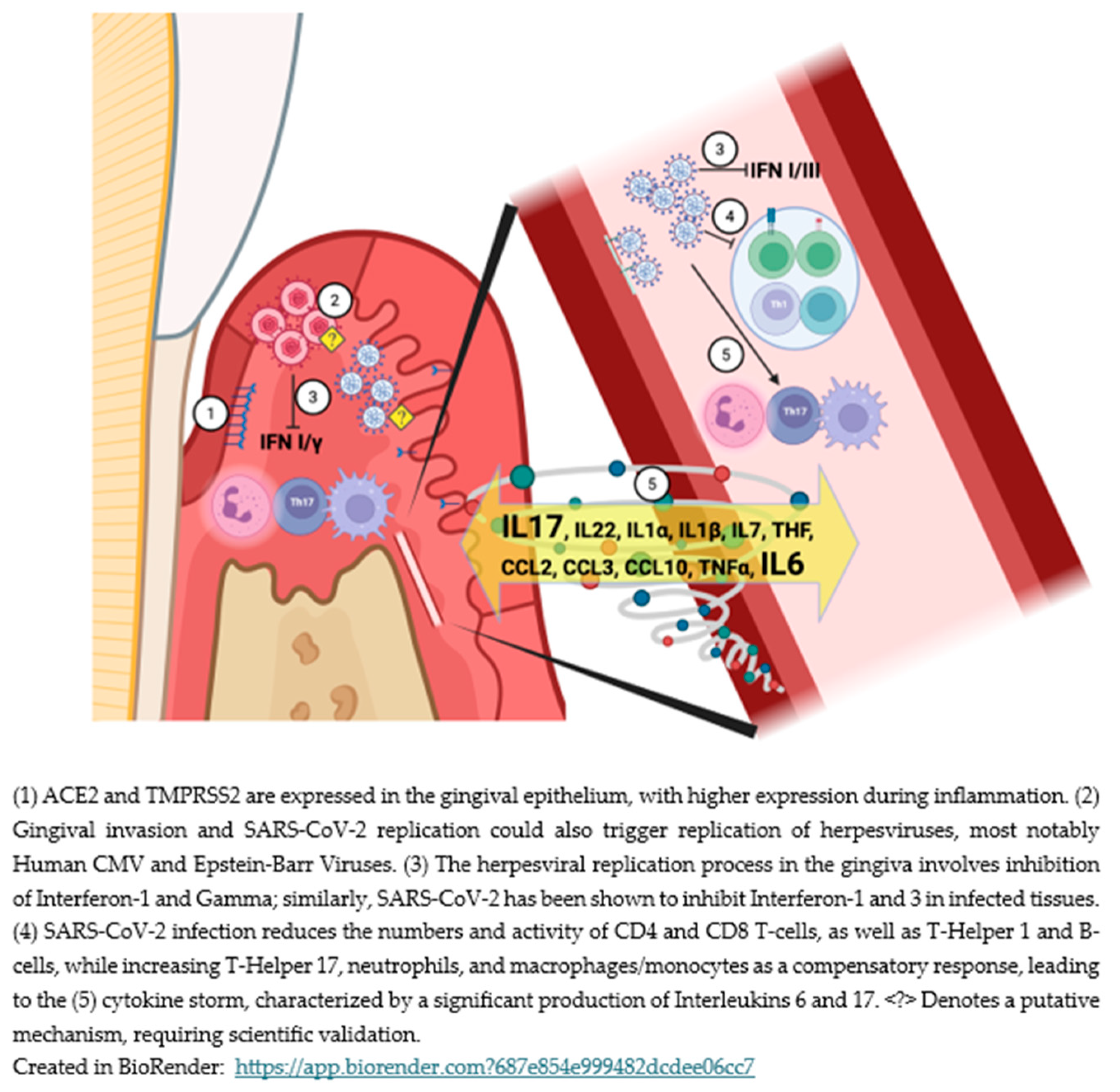
Figure 2.
Pathways through which Long-COVID (LC) can be a systemic risk factor for Periodontitis, and vice versa.
Figure 2.
Pathways through which Long-COVID (LC) can be a systemic risk factor for Periodontitis, and vice versa.
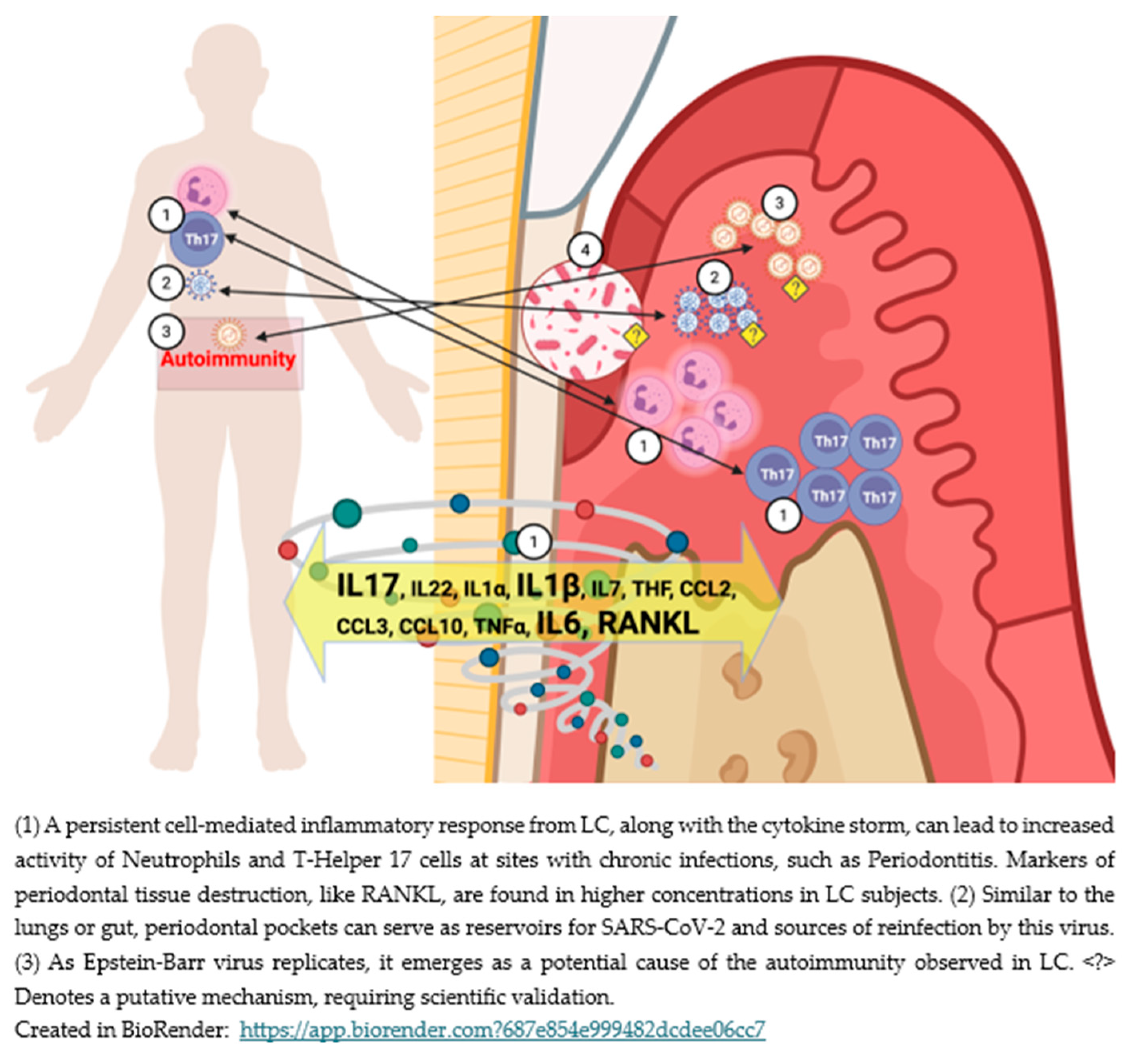

Table 1.
Summary of expression patterns of inflammatory and immune components in LC and periodontitis.
Table 1.
Summary of expression patterns of inflammatory and immune components in LC and periodontitis.
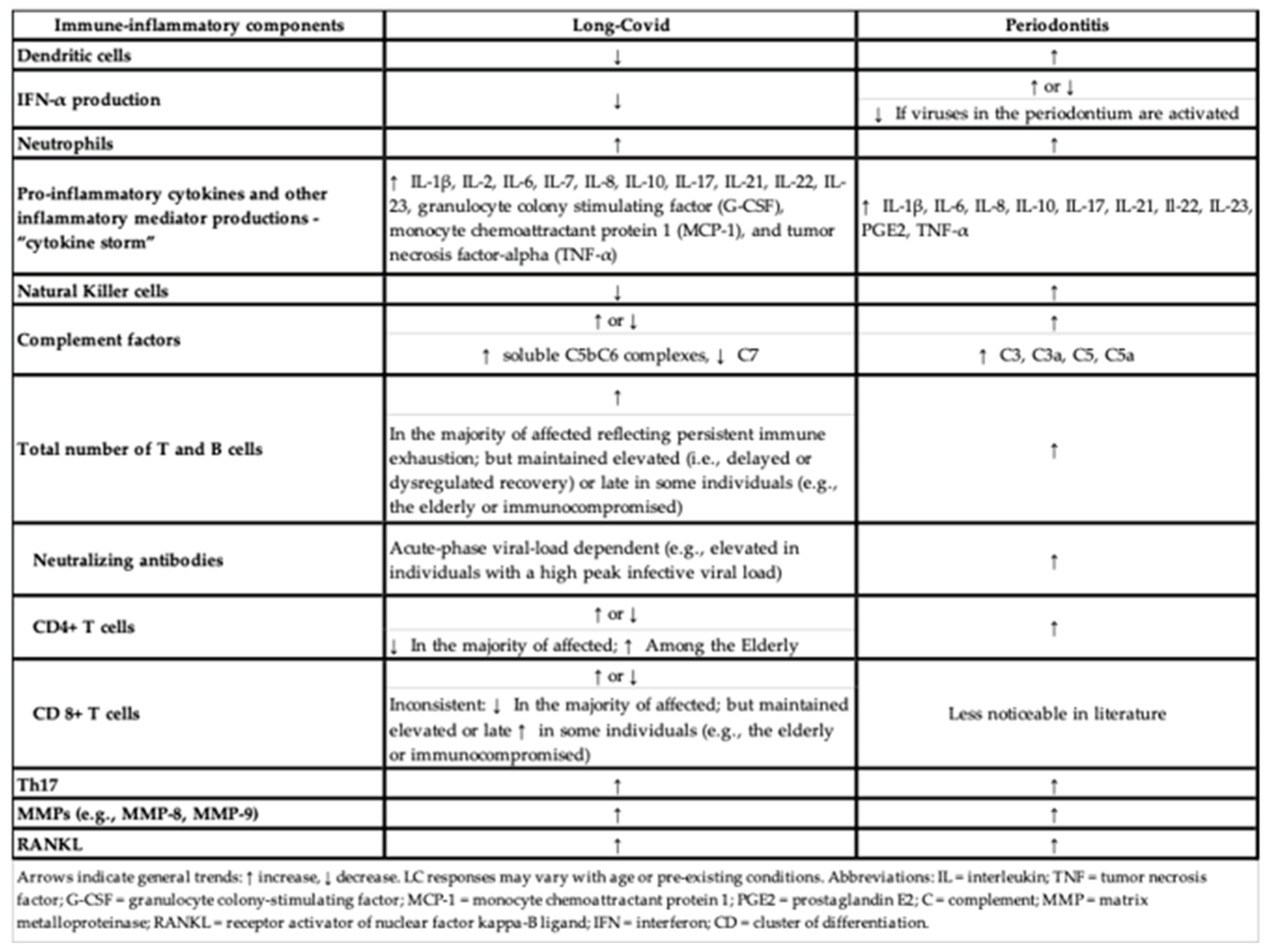 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



