
Submitted:
05 November 2025
Posted:
07 November 2025
You are already at the latest version
Abstract
Background: Pressure overload-induced heart failure is marked by pathological ventricular remodeling and myocardial fibrosis, contributing to impaired cardiac function and adverse clinical outcomes. Vericiguat, a soluble guanylate cyclase stimulator, has shown therapeutic promise in heart failure with reduced ejection fraction. However, its anti-fibrotic and metabolic effects in pressure overload models remain underexplored. Aim: This study aimed to investigate the anti-fibrotic and metabolic effects of Vericiguat in a murine model of pressure overload-induced cardiac remodeling. Material Method: Male mice (~25 g) underwent transverse aortic constriction (TAC) to induce pressure overload and received oral Vericiguat (10 mg/kg/day) for 14 days. Myocardial fibrosis was evaluated using Masson’s trichrome and Picrosirius red staining. Collagen deposition and wall stress indices were quantified. Proteomic profiling was performed on fibroblast- and myocyte-enriched cardiac tissue to identify differentially expressed proteins (DEPs) across metabolic, structural, mitochondrial, and signaling pathways. Results: Vericiguat treatment significantly reduced myocardial fibrosis and collagen accumulation compared to untreated TAC controls (p< 0.001). Improvements in wall stress indices were observed. Proteomic analysis revealed consistent modulation of DEPs, including reversal of TAC-induced downregulation of mitochondrial and energy-related proteins, indicating enhanced bioenergetic support. Conclusion: Vericiguat mitigates pressure overload-induced cardiac remodeling through anti-fibrotic and metabolic reprogramming mechanisms. These findings support its potential as a therapeutic strategy for heart failure and warrant further clinical investigation.
Keywords:
Vericiguat
; heart failure
; pressure overload
; transverse aortic constriction
; ventricular remodeling
; cardiac fibrosis
; soluble guanylate cyclase
; proteomics
; mitochondrial function
; metabolic reprogramming
1. Introduction
Standard pharmacological therapies for heart failure with reduced ejection fraction (HFrEF)—including angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, beta-blockers, mineralocorticoid receptor antagonists, and sodium-glucose cotransporter 2 inhibitors—have significantly improved patient survival and symptom management. Nonetheless, these agents primarily modulate neurohormonal pathways and often fall short in reversing structural remodeling, particularly myocardial fibrosis and chamber dilation, which are pivotal drivers of disease progression [1]. Even with strict adherence to guideline-directed medical therapy, many patients continue to experience recurrent hospitalizations and worsening cardiac function, underscoring the urgent need for novel interventions that target the underlying pathophysiological mechanisms [2].
Soluble guanylate cyclase (sGC) stimulators constitute a mechanistically distinct class of therapeutics that amplify cyclic guanosine monophosphate (cGMP) signaling independently of nitric oxide. This pathway plays a critical role in regulating vascular tone, mitochondrial function, and anti-fibrotic signaling. Vericiguat, the first sGC stimulator approved for HFrEF, has demonstrated clinical efficacy in lowering cardiovascular mortality and heart failure–related hospitalizations, particularly among patients with recent decompensation [3,4,5]. The VICTORIA trial confirmed its benefit in high-risk populations, while the VICTOR study further established its safety and effectiveness across diverse subgroups, including individuals with renal impairment or diabetes [6]. Beyond symptomatic relief, vericiguat has shown promise in preclinical models for mitigating structural remodeling. In settings of pressure overload and ischemic injury, it has been shown to attenuate myocardial fibrosis, preserve mitochondrial integrity, and suppress inflammatory signaling through modulation of the AMPK/Nrf2/NLRP3 and TGF-β1/Smad2/3 pathways [7,8,9]. These findings suggest that vericiguat may exert direct anti-fibrotic effects by modulating fibroblast activity and extracellular matrix turnover, thereby complementing its functional benefits. Taken together, clinical and experimental evidence supports vericiguat as a promising adjunctive therapy in HFrEF—capable of addressing both symptomatic burden and structural deterioration. Its unique mechanism of action positions it as a valuable addition to the evolving heart failure treatment paradigm.
Although vericiguat has demonstrated clinical efficacy in reducing cardiovascular events among patients with HFrEF, its cellular and histological effects—particularly under pressure-overload conditions—remain insufficiently defined. Most clinical trials, including VICTORIA and VICTOR, have focused on symptomatic relief and event reduction, with limited evaluation of its impact on myocardial architecture and fibrotic remodeling [3,4]. Emerging preclinical studies suggest that vericiguat may exert anti-fibrotic effects via multiple molecular pathways, though these mechanisms are not yet fully elucidated. For instance, modulation of the AMPK/Nrf2/NLRP3 axis has been shown to attenuate pyroptosis and inflammatory signaling in coronary microembolization models [7], while inhibition of TGF-β1/Smad2/3 signaling correlates with reduced collagen deposition in atrial tissue [8]. Whether these pathways are similarly activated in pressure-overload–induced ventricular remodeling remains unclear. Moreover, the potential involvement of CaMKII-related signaling—implicated in arrhythmogenesis and maladaptive remodeling—warrants further exploration. Although some studies propose that vericiguat may stabilize intracellular calcium dynamics and mitigate oxidative stress, direct evidence in pressure-overload settings is currently lacking [1].
To address these knowledge gaps, this study evaluates the therapeutic potential of vericiguat in a transverse aortic constriction (TAC)–induced mouse model that recapitulates key features of hypertensive and valvular heart disease, including elevated wall stress, collagen accumulation, and progressive ventricular dysfunction [5]. Vericiguat was administered postoperatively to assess its capacity to attenuate fibrotic remodeling and preserve myocardial architecture. Histological analyses employed Masson’s trichrome staining to detect general fibrotic tissue and Picrosirius red staining under polarized light to quantify collagen-specific deposition—both established, reproducible methods for assessing extracellular matrix expansion, a hallmark of pathological remodeling. Concurrently, functional parameters, including wall stress indices, were measured to link structural alterations with biomechanical performance. To further elucidate the underlying mechanisms, we conducted proteomic profiling of fibroblast- and myocyte-enriched cardiac tissues. This approach enabled the identification of differentially expressed proteins (DEPs) involved in extracellular matrix turnover, mitochondrial function, and inflammatory signaling. By integrating histological, functional, and proteomic data, this study aims to clarify vericiguat’s role in structural recovery and provide mechanistic insights relevant to its clinical application in heart failure.
2. Material and Method
2.1. Materials
Vericiguat (BAY 1021189) was obtained from Bayer AG (Leverkusen, Germany) and dissolved in sterile saline for in vivo administration. The dosing regimen (10 mg/kg/day) was adapted from previously validated protocols demonstrating efficacy in rodent models of cardiac injury and remodeling [7,10]. To assess myocardial fibrosis, Masson’s trichrome and Picrosirius red staining kits were procured from Sigma-Aldrich and applied according to established procedures for pressure-overload models [8,11]. Proteomic sample preparation employed reagents from Thermo Fisher Scientific (Waltham, MA, USA), including lysis buffers, trypsin digestion kits, and tandem mass tag (TMT) labeling reagents. High-resolution LC-MS/MS analysis was conducted using a Q Exactive Plus Orbitrap system (Thermo Fisher), in alignment with prior cardiac proteomics workflows [9]. All chemicals and reagents were of analytical grade and used in accordance with manufacturer instructions. Experimental procedures conformed to institutional animal care guidelines and were approved by the appropriate ethics committee.
2.2. Animal Model and Experimental Design
Male C57BL/6 mice (8–10 weeks old, ~25 g) were used for all experimental procedures. Animals were housed under standard laboratory conditions, including a 12-hour light/dark cycle, controlled temperature and humidity, and unrestricted access to food and water. All protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Academia Sinica (AS IACUC; protocol number 24-11-2324) and conducted in accordance with ARRIVE guidelines and national regulations governing laboratory animal welfare. To investigate pressure-overload–induced cardiac remodeling, mice were randomly assigned to one of three groups (n = 6–8 per group): (1) sham-operated controls undergoing thoracotomy without aortic constriction; (2) TAC-operated mice subjected to transverse aortic constriction (TAC) to induce left ventricular pressure overload; and (3) TAC + Vericiguat (Veri) mice receiving TAC surgery followed by daily oral administration of vericiguat (10 mg/kg/day) for 14 consecutive days, based on previously validated protocols [10,11]. TAC surgery was performed under isoflurane anesthesia using a 27-gauge needle to standardize constriction between the innominate and left common carotid arteries. Postoperative care included analgesia and routine monitoring to minimize discomfort. Body weight and general health status were assessed regularly throughout the study.
2.3. Induction of Pressure-Overload Heart Failure
Pressure-overload heart failure was induced using the TAC procedure, a well-established model that replicates key features of hypertensive and valvular cardiac stress. Mice were anesthetized with 2% isoflurane in oxygen and positioned supine on a heated surgical platform. Following a midline cervical incision, the transverse aorta was exposed between the innominate and left common carotid arteries. A 27-gauge needle was placed adjacent to the vessel, and a 7-0 silk suture was tied securely around both the needle and aorta. The needle was then gently withdrawn, creating a consistent constriction to elevate left ventricular afterload, as previously described [11]. To verify successful induction of the pressure gradient, Doppler echocardiography was performed 24 hours post-surgery to measure peak flow velocity across the constriction site. Mice with inadequate gradients or signs of surgical complications were excluded from further analysis. Postoperative care included subcutaneous administration of buprenorphine (0.05 mg/kg) every 12 hours for 48 hours, along with daily monitoring for distress, weight loss, or impaired mobility. Animals were housed individually during recovery and returned to group housing once stable. All procedures adhered to institutional animal care guidelines and were approved by the relevant ethics committee.
2.4. Drug Administration
Vericiguat was administered to mice in the TAC + Vericiguat group to assess its therapeutic impact on pressure-overload–induced cardiac remodeling. The compound was freshly prepared in sterile saline and delivered via oral gavage at a dose of 10 mg/kg/day, following dosing protocols validated in previous preclinical studies of cardiac injury and remodeling [11]. Treatment began 24 hours after TAC surgery and continued for 14 consecutive days, a regimen designed to capture early-phase remodeling responses and consistent with prior reports demonstrating vericiguat’s efficacy in reducing fibrosis, oxidative stress, and mitochondrial dysfunction in rodent models [7,9]. To minimize circadian variability, drug administration was performed at the same time each day. Mice were monitored daily for signs of distress, changes in body weight, and treatment tolerance. No adverse effects were observed throughout the treatment period.
2.5. Histological Analysis
Following euthanasia, hearts were excised, rinsed in cold phosphate-buffered saline (PBS), and fixed in 10% neutral-buffered formalin for 24 hours. Tissues were then dehydrated through graded ethanol, cleared in xylene, and embedded in paraffin. Serial transverse sections (5 μm thick) were obtained from the mid-ventricular region using a rotary microtome and mounted on glass slides for histological staining. To assess myocardial fibrosis, sections were stained with Masson’s trichrome, which highlights collagen fibers in blue and muscle tissue in red. For more specific quantification of collagen content, Picrosirius red staining was performed and visualized under polarized light microscopy, enabling enhanced detection of birefringent collagen fibers. These complementary techniques are well validated in pressure-overload models and provide robust assessments of extracellular matrix expansion [11]. Images were acquired using a Nikon Eclipse microscope equipped with a digital camera and analyzed with ImageJ software (NIH, Bethesda, MD, USA). Fibrotic area was quantified as the percentage of stained collagen relative to total myocardial area. At least five fields per section and three sections per heart were evaluated to ensure representative sampling. All image analyses were conducted by investigators blinded to group allocation.
2.6. Functional Assessment
Cardiac function and wall stress parameters were assessed using transthoracic echocardiography and pressure-volume (PV) loop analysis. Echocardiographic imaging was performed with a Vevo 2100 system (VisualSonics, Toronto, Canada) equipped with a high-frequency transducer. Mice were lightly anesthetized with 1–1.5% isoflurane to minimize hemodynamic interference. Standard parasternal long-axis and short-axis views were acquired to evaluate left ventricular (LV) geometry and contractility, including LV end-diastolic diameter (LVEDD), end-systolic diameter (LVESD), fractional shortening (FS), and ejection fraction (EF), as previously described [11]. For PV loop analysis, a 1.2F pressure-volume catheter (Scisense, London, ON, Canada) was inserted into the LV via the right carotid artery under anesthesia. Real-time pressure and volume signals were recorded using LabChart software (ADInstruments, Colorado Springs, CO, USA). Derived indices included end-systolic pressure-volume relationship (ESPVR), preload recruitable stroke work (PRSW), and arterial elastance (Ea), which collectively reflect intrinsic contractility and afterload conditions [9,10]. All functional data were normalized to body weight and heart rate to account for inter-animal variability.
2.7. Proteomic Profiling
Cardiac tissue was harvested 14 days post-surgery and processed for proteomic analysis. Fibroblast- and myocyte-enriched fractions were isolated via enzymatic digestion and differential centrifugation, following protocols adapted from previous studies. Proteins were extracted using RIPA buffer supplemented with protease and phosphatase inhibitors, and quantified by bicinchoninic acid assay. Equal protein amounts from each sample underwent trypsin digestion and tandem mass tag (TMT) labeling for multiplexed quantification. Labeled samples were pooled and analyzed by high-resolution liquid chromatography–tandem mass spectrometry (LC-MS/MS) using a Q Exactive Plus Orbitrap system. Spectral data were processed with Proteome Discoverer software and searched against the UniProt mouse database. DEPs were identified based on fold-change thresholds and adjusted p-values using Benjamini-Hochberg correction. Bioinformatics analysis was conducted using DAVID and Metascape platforms to annotate DEPs and identify enriched biological pathways. Functional clustering emphasized mitochondrial metabolism, cytoskeletal organization, calcium signaling, and fibrosis-related processes. Comparative profiling of fibroblast and myocyte fractions revealed distinct remodeling signatures and cell-type-specific therapeutic targets modulated by vericiguat. These findings align with prior reports of proteomic shifts in cardiac injury models and reinforce the mechanistic relevance of sGC stimulation in pressure-overload heart failure.
2.8. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) or equivalent analytical platforms. Data are presented as mean ± standard deviation (SD), and normality was assessed prior to hypothesis testing. For comparisons involving more than two groups, one-way analysis of variance (ANOVA) was performed, followed by Tukey’s post hoc test to determine intergroup differences. For direct pairwise comparisons, unpaired two-tailed Student’s t-tests were applied. A p-value of less than 0.05 was considered statistically significant.
3. Results
3.1. Ventricular Wall Stress Assessment
Representative M-mode echocardiograms (Figure 1A) revealed distinct wall motion patterns among Sham, TAC, and Vericiguat-treated groups. TAC surgery significantly reduced dynamic wall stress (DWS), indicating compromised ventricular biomechanics and elevated wall tension (***p < 0.001 vs. Sham; Figure 1B). Vericiguat treatment restored DWS toward baseline levels (###p < 0.001 vs. TAC; Figure 1B), reflecting improved contractile function and normalization of stress regulation. These results suggest that pressure overload impairs ventricular mechanics, whereas soluble guanylate cyclase stimulation with Vericiguat effectively reverses this dysfunction and attenuates stress-induced remodeling.
3.2. Left Ventricular Remodeling and Function
TAC induced significant structural remodeling, as evidenced by increased diastolic mass, posterior wall thickness, and septal thickness compared to Sham controls (*p < 0.05 or *p < 0.01; Table 1). Vericiguat treatment attenuated these changes, reducing hypertrophic indices relative to untreated TAC mice (#p < 0.05 or ##p < 0.001). Functionally, TAC elevated end-systolic volume and reduced stroke volume, indicating impaired systolic performance (p < 0.05 or **p < 0.01 vs. Sham). Vericiguat improved both parameters (#p < 0.05 or ##p < 0.001 vs. TAC), with partial normalization of end-diastolic volume and internal diameters, supporting its role in preserving chamber geometry under hemodynamic stress.
3.3. Myocardial Hypertrophy and Fibrosis Assessment
Gross morphological evaluation and heart weight quantification (Figure 2) revealed pronounced cardiac hypertrophy in TAC mice, which was significantly mitigated by Vericiguat. Hearts from the TAC group exhibited increased size and weight compared to Sham (p < 0.001), whereas Vericiguat-treated hearts showed intermediate values (##p < 0.01 vs. TAC), consistent with reduced hypertrophic remodeling. Histological staining with Masson’s trichrome and Picrosirius red further demonstrated extensive collagen deposition in TAC hearts (Figure 3A), with significantly elevated fibrotic area relative to Sham (***p < 0.001; Figure 3B, 3C). Vericiguat markedly reduced fibrosis in both assays (##p < 0.01 vs. TAC), indicating its efficacy in limiting extracellular matrix expansion. Collectively, these results support the dual antifibrotic and antihypertrophic effects of Vericiguat in pressure-overload–induced heart failure.
3.4. Fibroblast Proteome Reprogramming
Hierarchical clustering of differentially expressed proteins (DEPs) in fibroblast-enriched cardiac tissue revealed distinct proteomic profiles across WT, Sham, TAC, and Vericiguat-treated groups (Figure 4). TAC upregulated pro-fibrotic and inflammatory markers while downregulating mitochondrial and reparative proteins. Vericiguat reversed these alterations, restoring expression of bioenergetic and structural components. DEPs were functionally categorized into protein synthesis, folding, degradation, mitochondrial metabolism, and regulatory processes (Table 2). Notably, Vericiguat enhanced mitochondrial enzyme expression and translational machinery, suggesting improved protein homeostasis and extracellular matrix regulation. These findings highlight Vericiguat’s capacity to reprogram fibroblast activity and counteract maladaptive remodeling.
3.5. Myocyte Proteomic Remodeling
Proteomic analysis of myocyte-enriched tissue revealed that TAC disrupted the expression of mitochondrial enzymes, contractile proteins, and calcium-handling components (Figure 5). Vericiguat restored these profiles toward baseline, indicating recovery of cellular integrity. Functional classification of DEPs encompassed metabolic pathways, protein synthesis and modification, cytoskeletal and neuronal elements, mitochondrial regulators, transcriptional control, and vesicular transport (Table 3). Upregulation of mitochondrial and translational components, along with normalization of cytoskeletal and calcium-regulatory proteins, underscores Vericiguat’s role in preserving contractile architecture and intracellular signaling. These data emphasize its multi-pathway engagement in maintaining myocyte function under pathological stress.
4. Discussion
This study demonstrates that Vericiguat exerts a multifaceted therapeutic effect in a pressure-overload model of heart failure, characterized by attenuation of myocardial hypertrophy and fibrosis, restoration of biomechanical integrity, and reversal of proteomic dysregulation at the cellular level. Gross morphological assessment and heart weight analysis revealed significant cardiac enlargement in TAC mice, which was markedly reduced by Vericiguat treatment—indicating suppression of pressure-overload–induced hypertrophic remodeling. Histological evaluation further confirmed Vericiguat’s antifibrotic efficacy, with substantial reductions in left ventricular collagen deposition, as shown by Masson’s trichrome and Picrosirius red staining. These results are consistent with previous reports demonstrating Vericiguat’s capacity to limit extracellular matrix expansion and fibrotic remodeling in various cardiac injury models, including doxorubicin-induced cardiomyopathy and atrial fibrillation [8,10]. The observed reduction in fibrotic burden aligns with Vericiguat’s known modulation of the TGF-β1/Smad2/3 signaling axis, a central pathway in fibroblast activation and collagen synthesis [8]. Functionally, Vericiguat improved dynamic wall stress indices and preserved ventricular geometry, suggesting enhanced myocardial compliance and contractile performance. These biomechanical improvements parallel clinical findings from the VICTORIA and VICTOR trials, where Vericiguat reduced cardiovascular events and supported hemodynamic stability in patients with heart failure and reduced ejection fraction [3,4]. The structural recovery observed in this preclinical model reinforces the hypothesis that Vericiguat’s benefits extend beyond symptomatic relief to include direct modulation of cardiac architecture. Proteomic profiling provided mechanistic insight, revealing that Vericiguat reversed TAC-induced suppression of mitochondrial, metabolic, and cytoskeletal proteins in both fibroblast- and myocyte-enriched cardiac fractions. Notably, Vericiguat restored the expression of bioenergetic regulators and structural components, suggesting improved cellular resilience and energy handling. These findings are supported by prior studies demonstrating Vericiguat’s enhancement of mitochondrial integrity and attenuation of oxidative stress via AMPK/Nrf2/NLRP3 signaling pathways [7,9]. Taken together, the integration of morphological, histological, functional, and proteomic data underscores Vericiguat’s capacity to mitigate pressure-overload–induced cardiac remodeling through coordinated antihypertrophic, antifibrotic, and metabolic reprogramming. This comprehensive therapeutic profile supports its continued development as a structurally restorative agent in the management of heart failure.
The findings of this study align with and extend prior evidence supporting Vericiguat’s role in mitigating cardiac remodeling through antifibrotic and mitochondrial-protective mechanisms. In coronary microembolization models, Vericiguat suppressed pyroptosis and inflammatory signaling via the AMPK/Nrf2/NLRP3 axis, contributing to reduced myocardial injury and enhanced cellular resilience [7]. Similarly, in atrial tissue, Vericiguat attenuated fibrosis by downregulating TGF-β1/Smad2/3 signaling—a pathway also implicated in extracellular matrix turnover in the present pressure-overload model [8]. These mechanistic parallels reinforce Vericiguat’s capacity to modulate key remodeling cascades across diverse cardiac contexts. Importantly, this study provides direct histological and proteomic evidence of Vericiguat’s efficacy in a TAC model, a clinically relevant surrogate for hypertensive and valvular heart disease. While the VICTORIA and VICTOR trials established Vericiguat’s clinical benefit in reducing cardiovascular death and hospitalization among patients with heart failure and reduced ejection fraction [3,4], they did not address its impact on myocardial architecture or cell-type–specific remodeling. Our data fill this gap by demonstrating that Vericiguat not only improves wall stress and ventricular geometry but also reverses TAC-induced proteomic dysregulation in fibroblasts and myocytes. Compared to other injury models, such as doxorubicin-induced cardiomyopathy and ischemia-reperfusion injury, the signaling pathways engaged under pressure-overload conditions converge on mitochondrial integrity, oxidative stress regulation, and cytoskeletal stabilization [9,10]. However, the proteomic signatures observed in this study reveal distinct patterns of DEPs, particularly within structural and energy-related domains, suggesting that Vericiguat’s reparative effects may be context-dependent. For instance, while mitochondrial proteins were consistently restored across models, modulation of calcium-handling and cytoskeletal components was more prominent in the TAC setting, potentially reflecting the biomechanical demands of pressure overload. Together, these comparisons underscore Vericiguat’s versatility in targeting both shared and condition-specific pathways of cardiac remodeling. By integrating histological, functional, and proteomic endpoints, this study advances the mechanistic understanding of Vericiguat and supports its broader application in structurally compromised heart failure phenotypes.
The therapeutic effects of Vericiguat observed in this study are supported by its modulation of multiple signaling pathways implicated in cardiac fibrosis, inflammation, and bioenergetic dysfunction. Notably, Vericiguat attenuated pressure-overload–induced remodeling through suppression of the TGF-β1/Smad2/3 axis—a central driver of fibroblast activation and collagen synthesis. This pathway has been previously associated with atrial fibrosis and arrhythmogenic remodeling, and its downregulation by Vericiguat has been shown to reduce extracellular matrix expansion and improve tissue architecture [8]. In parallel, Vericiguat inhibited components of the AMPK/Nrf2/NLRP3 signaling cascade, which regulates oxidative stress and pyroptotic cell death. In coronary microembolization models, activation of AMPK and Nrf2 by Vericiguat suppressed NLRP3 inflammasome activity, thereby reducing inflammatory injury and preserving myocardial integrity [7]. Our findings extend this mechanism to pressure-overload conditions, suggesting that Vericiguat’s anti-inflammatory effects are broadly applicable across ischemic and hypertensive heart failure phenotypes. Emerging evidence also points to Vericiguat’s potential interaction with CaMKII-related signaling, which governs intracellular calcium dynamics and contributes to maladaptive remodeling and arrhythmogenesis. Although direct modulation of CaMKII was not confirmed in this study, proteomic shifts in calcium-handling proteins and cytoskeletal regulators suggest that Vericiguat may stabilize excitation–contraction coupling and mitigate calcium-driven stress responses [1]. Proteomic profiling revealed distinct patterns of DEPs in fibroblast and myocyte populations, highlighting cell-type–specific responses to Vericiguat. In fibroblasts, Vericiguat reversed TAC-induced upregulation of pro-fibrotic and cytoskeletal proteins while restoring mitochondrial and structural components. In myocytes, treatment enhanced the expression of metabolic enzymes and mitochondrial regulators, consistent with improved bioenergetic support and contractile function. These findings align with prior reports demonstrating Vericiguat’s capacity to preserve mitochondrial quality and reduce oxidative damage in models of doxorubicin-induced cardiomyopathy and mitral regurgitation [7,10]. Together, these mechanistic insights underscore Vericiguat’s multifactorial role in cardiac remodeling. By targeting fibrosis, inflammation, calcium homeostasis, and metabolic resilience, Vericiguat promotes structural recovery and functional improvement in pressure-overload heart failure. The cell-type–specific proteomic shifts further suggest that its therapeutic effects are coordinated across both stromal and contractile compartments, reinforcing its potential as a comprehensive modulator of myocardial remodeling.
The findings of this study reinforce Vericiguat’s emerging role as a dual-action therapeutic agent in HFrEF, offering both symptomatic relief and structural remodeling benefits. While its clinical efficacy in reducing cardiovascular mortality and heart failure–related hospitalizations has been well established through large-scale trials such as VICTORIA and VICTOR [3,4], our data provide complementary mechanistic evidence that Vericiguat also mitigates myocardial fibrosis and restores ventricular architecture. This dual capacity positions Vericiguat as a valuable adjunct to guideline-directed medical therapy, particularly for patients with persistent structural deterioration despite optimal neurohormonal blockade. The pressure-overload model employed in this study closely recapitulates pathophysiological features of hypertensive and valvular heart disease, including elevated wall stress, collagen accumulation, and progressive ventricular dysfunction. These conditions often precede overt heart failure and may benefit from earlier intervention. By demonstrating Vericiguat’s ability to reverse fibrotic remodeling and improve biomechanical indices in this context, our findings suggest that its therapeutic window may extend beyond post-decompensation settings to include patients with subclinical or early-stage structural compromise. This is especially relevant given the limited efficacy of conventional agents in reversing fibrosis and chamber dilation [1,2]. Moreover, proteomic data highlight Vericiguat’s potential utility in patient subgroups characterized by high fibrotic burden or metabolic dysregulation. Restoration of mitochondrial and energy-related proteins in both fibroblast and myocyte populations suggests that Vericiguat may be particularly beneficial in individuals with impaired bioenergetic capacity or systemic comorbidities such as diabetes and renal dysfunction—populations in which Vericiguat has already demonstrated safety and efficacy [5,6]. These molecular insights support a precision medicine approach, where proteomic or biomarker-guided stratification could optimize therapeutic outcomes. In summary, Vericiguat’s ability to address both functional symptoms and structural pathology expands its clinical relevance in HFrEF. Its integration into earlier treatment phases and targeted use in metabolically vulnerable subgroups may enhance long-term outcomes and reduce progression to advanced heart failure.
While this study provides compelling evidence for Vericiguat’s structural and molecular benefits in pressure-overload heart failure, several limitations warrant consideration. First, the treatment duration was restricted to 14 days post-TAC surgery, which may not fully capture the long-term trajectory of myocardial remodeling or the durability of therapeutic effects. Prior studies have demonstrated that chronic Vericiguat administration can influence survival and progressive remodeling in models of volume overload and cardiotoxicity [10,12], suggesting that extended observation periods may uncover additional benefits or compensatory adaptations. Second, although the murine TAC model reproduces key features of hypertensive and valvular heart disease—such as elevated wall stress and fibrotic remodeling—it does not encompass the full complexity of human heart failure. Variables including metabolic comorbidities, neurohormonal activation, and sex-dependent susceptibility are incompletely represented in animal models [13], and caution is advised when extrapolating these findings to clinical populations. Third, while proteomic profiling revealed modulation of several signaling pathways—including TGF-β1/Smad2/3 and AMPK/Nrf2/NLRP3—direct causal validation was not performed. The potential involvement of CaMKII-related signaling, which contributes to calcium dysregulation and arrhythmogenic remodeling, remains speculative. Although previous reports suggest Vericiguat may stabilize intracellular calcium dynamics [1], targeted studies employing pathway-specific inhibitors or genetic knockouts are needed to confirm these mechanistic links. Future investigations should incorporate longer treatment durations, multi-organ assessments, and mechanistic interventions to delineate Vericiguat’s full therapeutic profile. Integration of genetic models and pharmacological modulators will be essential to validate pathway-specific effects and refine patient stratification strategies for clinical application.
Building on the mechanistic and histological insights presented in this study, several avenues for future investigation are warranted to further elucidate and translate the therapeutic potential of Vericiguat in heart failure. First, longitudinal studies are needed to evaluate the durability of Vericiguat’s anti-remodeling effects. While short-term improvements in fibrosis and proteomic profiles were evident, extended treatment protocols may uncover sustained benefits or compensatory adaptations in myocardial structure and function. Prior work in volume-overload and cardiotoxicity models has shown that prolonged stimulation of sGC improves survival and attenuates progressive remodeling [10,12], supporting the rationale for chronic-phase evaluation. Second, validation of differentially expressed proteins (DEPs) and signaling targets identified through proteomic analysis should be pursued using orthogonal methods such as Western blotting and immunohistochemistry. This will confirm the involvement of key pathways—including TGF-β1/Smad2/3, AMPK/Nrf2/NLRP3, and potentially CaMKII—and clarify their spatial and cell-type–specific activation patterns. Mechanistic validation is essential for establishing causal links and identifying therapeutic biomarkers. Third, exploration of Vericiguat’s efficacy in comorbid models is critical for clinical relevance. Heart failure frequently coexists with metabolic and renal dysfunction, which may influence drug responsiveness. Recent studies have demonstrated that Vericiguat maintains cardiovascular benefits in patients with impaired renal function and diabetes [5,6], but preclinical models incorporating these comorbidities could provide deeper insight into molecular adaptations and safety profiles.Finally, clinical translation would benefit from biomarker-guided stratification strategies to identify patients most likely to respond to sGC stimulation. Proteomic signatures, circulating fibrosis markers, and mitochondrial stress indicators may serve as predictive tools for tailoring therapy. This approach aligns with emerging precision medicine frameworks and could enhance the therapeutic index of Vericiguat across diverse heart failure populations [1,14]. In summary, future studies should integrate longitudinal design, mechanistic validation, comorbidity modeling, and translational biomarker development to fully define Vericiguat’s role in heart failure management and optimize its clinical deployment.
In conclusion, this study demonstrates that Vericiguat exerts robust anti-remodeling effects in a pressure-overload model of heart failure, marked by significant attenuation of myocardial hypertrophy and fibrosis, restoration of ventricular geometry, and reversal of proteomic dysregulation in both fibroblast and myocyte populations. Morphological assessment and heart weight quantification confirmed that Vericiguat effectively reduced TAC-induced cardiac hypertrophy, complementing its antifibrotic action observed through Masson’s trichrome and Picrosirius red staining. These structural improvements reflect enhanced myocardial compliance and reduced wall stress, reinforcing Vericiguat’s capacity to preserve biomechanical integrity. Mechanistically, Vericiguat modulated key signaling pathways—including TGF-β1/Smad2/3 and AMPK/Nrf2/NLRP3—thereby suppressing fibrotic and inflammatory responses while supporting mitochondrial and cytoskeletal resilience. Proteomic profiling further revealed cell-type–specific restoration of metabolic and structural proteins, underscoring Vericiguat’s coordinated impact across stromal and contractile compartments. These findings extend prior evidence from ischemic and cardiotoxic models [7,8,10] and address mechanistic gaps left by clinical trials such as VICTORIA and VICTOR, which primarily focused on symptomatic endpoints [3,4]. Importantly, the integration of gross anatomical, histological, and proteomic data supports the repositioning of Vericiguat as a dual-action agent capable of delivering both symptomatic relief and structural remodeling. This therapeutic profile suggests potential utility in patient subgroups with elevated fibrotic burden or metabolic vulnerability. Future studies should investigate extended treatment durations, pathway-specific inhibitors, and comorbid models to further delineate Vericiguat’s clinical scope. Collectively, these findings reinforce the translational relevance of sGC stimulation in heart failure and provide a foundation for precision-guided intervention in structurally compromised cardiac phenotypes.
Author Contributions
Conceptualization, W.-R.H. and T.-H.C.; methodology, W.-R.H.; software, F.-A L; validation, W.-R.H., C.-C.C., and J.-C.L.; formal analysis, W.-R.H.; investigation, W.-R.H.; resources, J.-J.C.; data curation, F.-A L. and H.-Y.C.; writing—original draft preparation, W.-R.H.; writing—review and editing, T.-H.C.; visualization, J.-H.L.; supervision, T.-H.C.; project administration, T.-H.C.; funding acquisition, J.-J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science and Technology Council (NSTC), Tai-609 wan, grant number MOST 111-2314-B-039-014, and by China Medical University, Taiwan, grant number CMU111-MF-102.e
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of Academia Sinica (AS IACUC) (protocol ID 24-11-2324, approved on 29 April 2025).
Informed Consent Statement
Not applicable.
Conflicts of Interest
None
References
- Hao, W.R.; Cheng, C.H.; Chen, H.Y.; Liu, J.C.; Cheng, T.H.; Chen, J.J. Exploring Vericiguat in Heart Failure: Mechanistic Insights, Therapeutic Advantages, and Clinical Validation. Curr Drug Saf 2025. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Germinal, K.; Milfort, A.; Chen, W.H.; Chang, S.H.; Huang, W.; Li, Y.; Lu, Y.; Ahmed, M.M.; Kimmel, S.E.; et al. The most effective combination of pharmacological therapy for heart failure with reduced ejection fraction: a network meta-analysis of randomized controlled trials. BMC Cardiovasc Disord 2024, 24, 666. [Google Scholar] [CrossRef] [PubMed]
- Mentz, R.J.; Butler, J.; McMullan, C.J.; Wojdyla, D.M.; Anstrom, K.J.; Barash, I.; Bonaca, M.P.; Borentain, M.; Corda, S.; Ezekowitz, J.A.; et al. Blood pressure, safety and clinical efficacy of vericiguat in chronic heart failure with reduced ejection fraction: Insights from the VICTOR trial. Eur J Heart Fail 2025. [CrossRef] [PubMed]
- Butler, J.; McMullan, C.J.; Anstrom, K.J.; Barash, I.; Bonaca, M.P.; Borentain, M.; Corda, S.; Ezekowitz, J.A.; Felker, G.M.; Gates, D.; et al. Vericiguat in patients with chronic heart failure and reduced ejection fraction (VICTOR): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet 2025, 406, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, W.; Liu, H.; Zhang, C.; Qi, Y.; Yuan, X.; Yuan, Z.; Sun, L.; She, J.; Lou, B. Initiation of vericiguat and short-term cardiovascular function improvement in heart failure patients with and without worsened renal function. Front Cardiovasc Med 2025, 12, 1628411. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Butler, J.; Young, R.; Lewis, B.S.; Escobedo, J.; Refsgaard, J.; Reyes, E.; Roessig, L.; Blaustein, R.O.; Lam, C.S.P.; et al. Vericiguat and Cardiovascular Outcomes in Heart Failure by Baseline Diabetes Status: Insights From the VICTORIA Trial. JACC Heart Fail 2024, 12, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Khounphinith, E.; Zhou, Y.; Yi, Z.; Li, T.; Li, L. Vericiguat reduces pyroptosis in rats with coronary microembolization by inhibiting the AMPK/Nrf2/NLRP3 signaling pathway. Korean J Physiol Pharmacol 2025. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sheng, Y.; Xu, P.; Peng, Q.; Ruan, Z. Vericiguat reduces atrial fibrillation recurrence by alleviating myocardial fibrosis via the TGF-β1/Smad2/3 pathway. PLoS One 2025, 20, e0328272. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Abuín, X.; Moraña-Fernández, S.; Aragón-Herrera, A.; Sandner, P.; Thomitzek, K.; García-Seara, J.; Bravo, S.B.; Otero-Santiago, M.; de la Fuente-López, P.; Tilves-Bellas, C.; et al. Soluble guanylate cyclase stimulation improves cardiac function and mitochondrial activity in a rat model of early-stage heart failure with preserved ejection fraction. Biomed Pharmacother 2025, 191, 118439. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, Y.; Li, W.; Song, M.; Xu, K.; Wu, M.; Lin, L. Vericiguat improves cardiac remodelling and function in rats with doxorubicin-induced cardiomyopathy. ESC Heart Fail 2025, 12, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.R.; Cheng, C.H.; Chen, H.Y.; Cheng, T.H.; Liu, J.C.; Chen, J.J. Fucoidan Attenuates Cardiac Remodeling by Inhibiting Galectin-3 Secretion, Fibrosis, and Inflammation in a Mouse Model of Pressure Overload. Biomedicines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Gawrys, O.; Husková, Z.; Škaroupková, P.; Honetschlägerová, Z.; Vaňourková, Z.; Kikerlová, S.; Melenovský, V.; Bačová, B.S.; Sykora, M.; Táborský, M.; et al. The treatment with sGC stimulator improves survival of hypertensive rats in response to volume-overload induced by aorto-caval fistula. Naunyn Schmiedebergs Arch Pharmacol 2023, 396, 3757–3773. [Google Scholar] [CrossRef] [PubMed]
- Mira Hernandez, J.; Shen, E.Y.; Ko, C.Y.; Hourani, Z.; Spencer, E.R.; Smoliarchuk, D.; Bossuyt, J.; Granzier, H.; Bers, D.M.; Hegyi, B. Differential sex-dependent susceptibility to diastolic dysfunction and arrhythmia in cardiomyocytes from obese diabetic heart failure with preserved ejection fraction model. Cardiovasc Res 2025, 121, 254–266. [Google Scholar] [CrossRef] [PubMed]
- deFilippi, C.R.; Shah, P.; Shah, S.J.; Alemayehu, W.; Lam, C.S.P.; Butler, J.; Roessig, L.; O'Connor, C.M.; Westerhout, C.M.; Armstrong, P.W. Proteomics Identify Clinical Phenotypes and Predict Functional Outcomes in Heart Failure With Preserved Ejection Fraction: Insights From VITALITY-HFpEF. Circ Heart Fail 2024, 17, e011792. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vericiguat Restores Ventricular Wall Stress in Pressure-Overload Heart Failure. Representative M-mode echocardiographic images (A) and quantitative analysis of dynamic wall stress (DWS; B) are shown for three experimental groups: Sham, transverse aortic constriction (TAC), and TAC treated with Vericiguat (Veri). TAC surgery significantly reduced DWS compared to Sham controls (***p < 0.001), indicating impaired ventricular biomechanics. Vericiguat treatment markedly restored DWS toward baseline levels (###p < 0.001 vs. TAC), reflecting improved myocardial contractility and normalization of wall stress regulation. Data are presented as mean ± SD; n = 5 per group.
Figure 1.
Vericiguat Restores Ventricular Wall Stress in Pressure-Overload Heart Failure. Representative M-mode echocardiographic images (A) and quantitative analysis of dynamic wall stress (DWS; B) are shown for three experimental groups: Sham, transverse aortic constriction (TAC), and TAC treated with Vericiguat (Veri). TAC surgery significantly reduced DWS compared to Sham controls (***p < 0.001), indicating impaired ventricular biomechanics. Vericiguat treatment markedly restored DWS toward baseline levels (###p < 0.001 vs. TAC), reflecting improved myocardial contractility and normalization of wall stress regulation. Data are presented as mean ± SD; n = 5 per group.
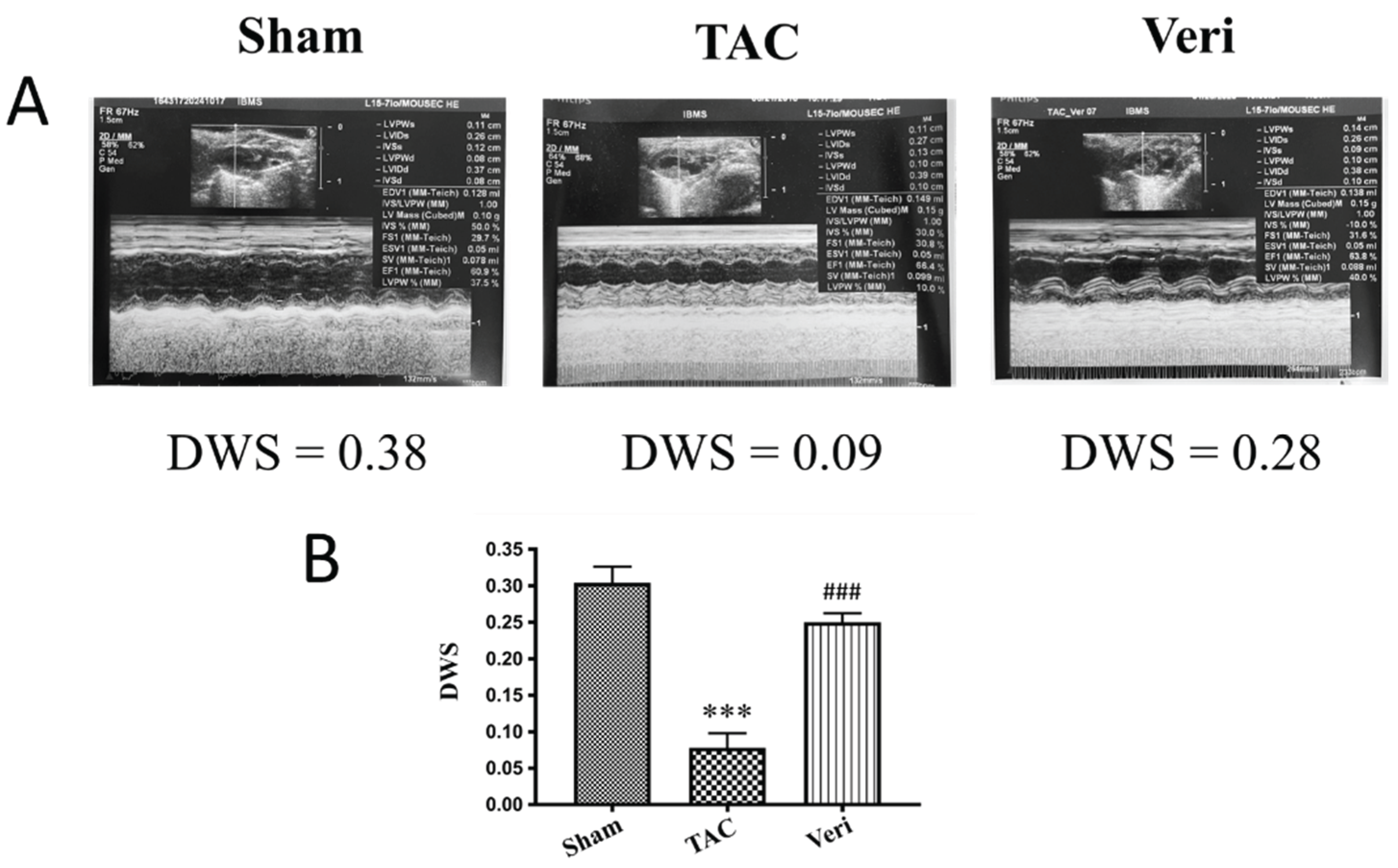
Figure 2.
Vericiguat Attenuates Cardiac Hypertrophy Induced by Pressure Overload. Representative images of excised hearts (A) and corresponding quantification of heart weight (B) from mice subjected to Sham surgery, transverse aortic constriction (TAC), or TAC with Vericiguat treatment (Veri). TAC induced marked cardiac enlargement compared to Sham controls (p < 0.001), while Vericiguat significantly reduced heart size and weight (##p < 0.01 vs. TAC). Bar graph shows mean heart weight ± standard deviation (SD); n = 8 per group.
Figure 2.
Vericiguat Attenuates Cardiac Hypertrophy Induced by Pressure Overload. Representative images of excised hearts (A) and corresponding quantification of heart weight (B) from mice subjected to Sham surgery, transverse aortic constriction (TAC), or TAC with Vericiguat treatment (Veri). TAC induced marked cardiac enlargement compared to Sham controls (p < 0.001), while Vericiguat significantly reduced heart size and weight (##p < 0.01 vs. TAC). Bar graph shows mean heart weight ± standard deviation (SD); n = 8 per group.
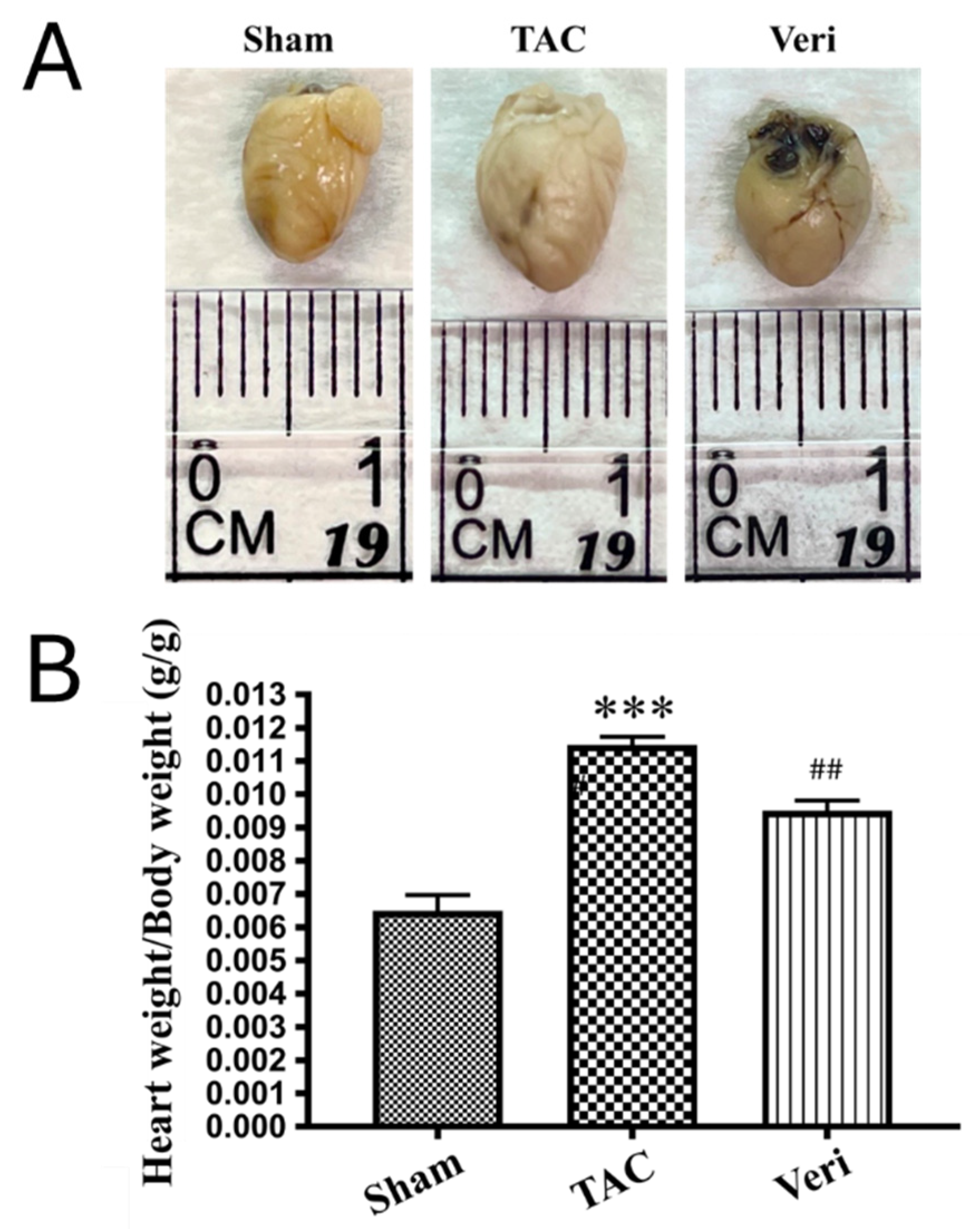
Figure 3.
Vericiguat Reduces Myocardial Fibrosis in Pressure-Overload Heart Failure. (A) Representative histological sections of left ventricular tissue stained with Masson’s trichrome (top row) and Picrosirius red (bottom row) from Sham, transverse aortic constriction (TAC), and TAC + Vericiguat (Veri) groups. Collagen deposition is visualized as blue staining in Masson’s trichrome and red birefringence under polarized light in Picrosirius red. (B) Quantitative analysis of fibrotic area (%) from Masson’s trichrome staining and (C) Picrosirius red staining. TAC significantly increased myocardial fibrosis compared to Sham controls (***p < 0.001), whereas Vericiguat treatment markedly reduced fibrotic burden (##p < 0.01 vs. TAC). Data are presented as mean ± standard deviation (SD); n = 6 per group.
Figure 3.
Vericiguat Reduces Myocardial Fibrosis in Pressure-Overload Heart Failure. (A) Representative histological sections of left ventricular tissue stained with Masson’s trichrome (top row) and Picrosirius red (bottom row) from Sham, transverse aortic constriction (TAC), and TAC + Vericiguat (Veri) groups. Collagen deposition is visualized as blue staining in Masson’s trichrome and red birefringence under polarized light in Picrosirius red. (B) Quantitative analysis of fibrotic area (%) from Masson’s trichrome staining and (C) Picrosirius red staining. TAC significantly increased myocardial fibrosis compared to Sham controls (***p < 0.001), whereas Vericiguat treatment markedly reduced fibrotic burden (##p < 0.01 vs. TAC). Data are presented as mean ± standard deviation (SD); n = 6 per group.
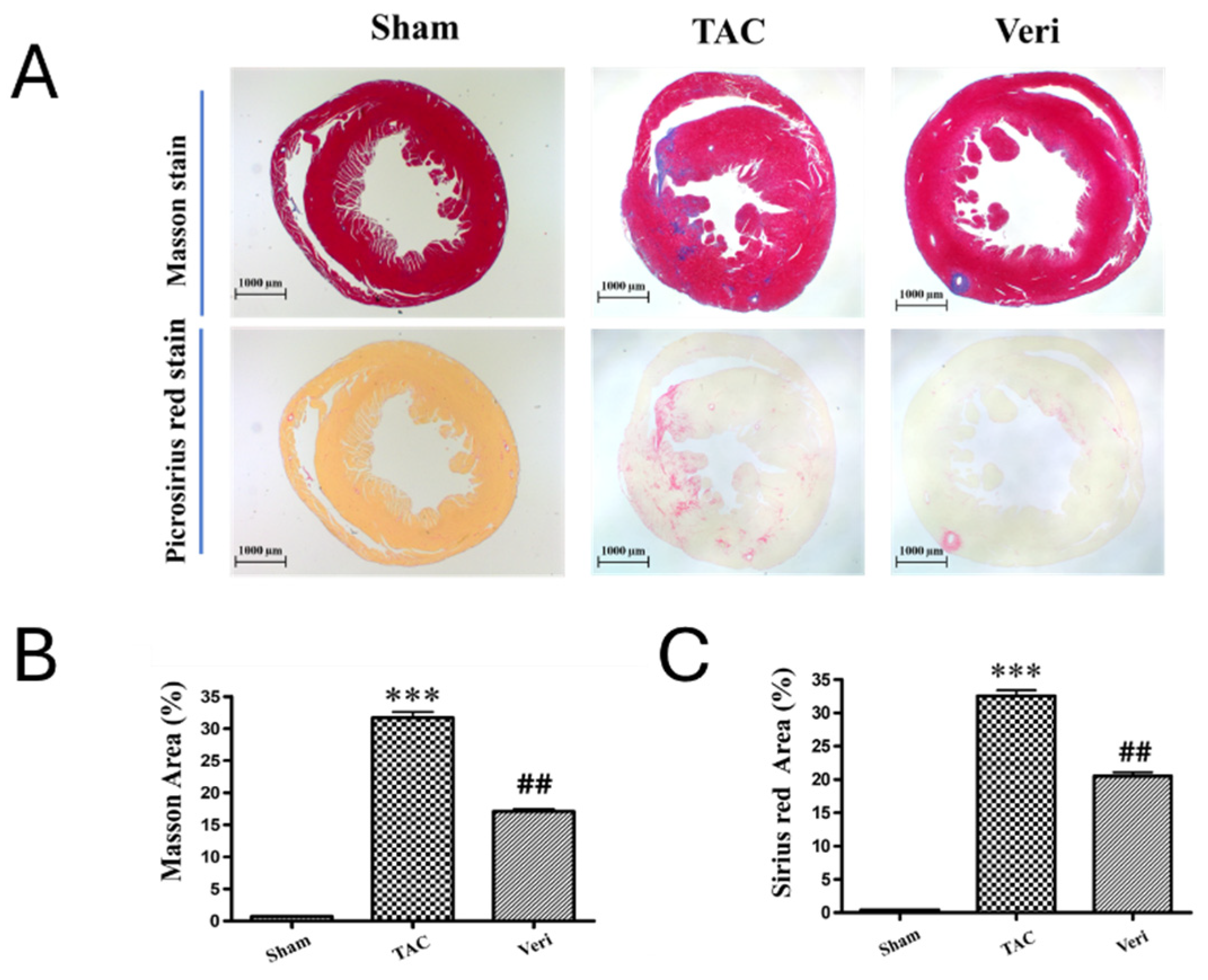
Figure 4.
Vericiguat Reprograms the Fibroblast Proteome in Pressure-Overload Heart Failure. Hierarchical heatmap illustrating differentially expressed proteins (DEPs) in fibroblast-enriched cardiac tissue across four experimental conditions: wild-type (WT), sham-operated (Sham), transverse aortic constriction (TAC), and TAC with Vericiguat treatment (TAC-Ver). Protein abundance is color-coded (green: low; black: intermediate; red: high) and clustered by expression similarity. TAC-induced pressure overload markedly altered fibroblast proteomic profiles, with upregulation of pro-fibrotic and inflammatory markers and suppression of mitochondrial and reparative proteins. Vericiguat treatment reversed these changes, restoring expression of bioenergetic, structural, and reparative components. These findings indicate that Vericiguat modulates fibroblast activity and extracellular matrix remodeling through targeted proteomic reprogramming.
Figure 4.
Vericiguat Reprograms the Fibroblast Proteome in Pressure-Overload Heart Failure. Hierarchical heatmap illustrating differentially expressed proteins (DEPs) in fibroblast-enriched cardiac tissue across four experimental conditions: wild-type (WT), sham-operated (Sham), transverse aortic constriction (TAC), and TAC with Vericiguat treatment (TAC-Ver). Protein abundance is color-coded (green: low; black: intermediate; red: high) and clustered by expression similarity. TAC-induced pressure overload markedly altered fibroblast proteomic profiles, with upregulation of pro-fibrotic and inflammatory markers and suppression of mitochondrial and reparative proteins. Vericiguat treatment reversed these changes, restoring expression of bioenergetic, structural, and reparative components. These findings indicate that Vericiguat modulates fibroblast activity and extracellular matrix remodeling through targeted proteomic reprogramming.
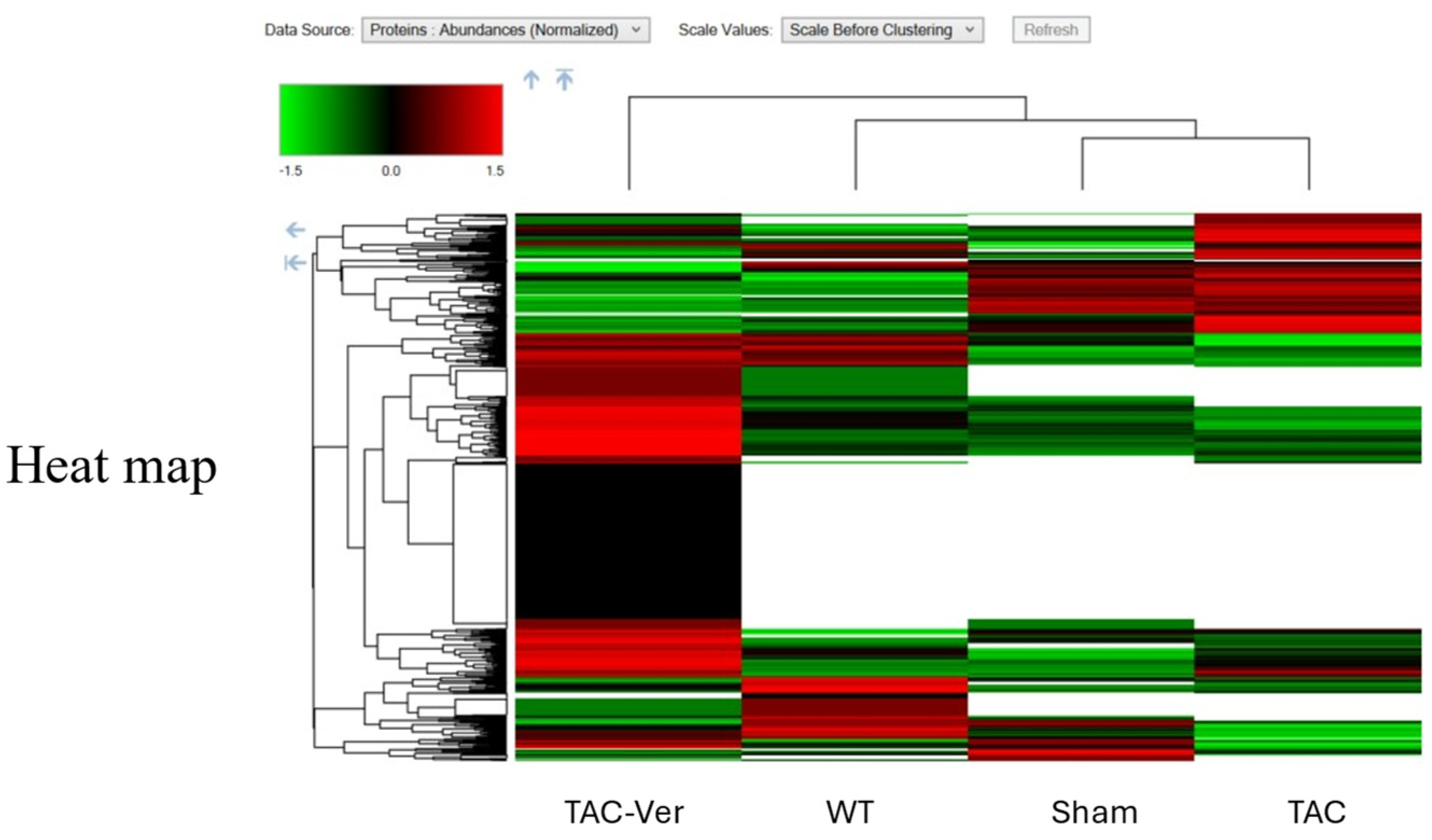
Figure 5.
Vericiguat Modulates Myocyte Proteomic Signatures in Pressure-Overload Heart Failure. Hierarchical heatmap depicting differentially expressed proteins (DEPs) in myocyte-enriched cardiac tissue across four experimental groups: wild-type (WT), sham-operated (Sham), transverse aortic constriction (TAC), and TAC with Vericiguat treatment (TAC-Ver). Protein abundance is color-coded (green: low; black: intermediate; red: high) and clustered by expression similarity. TAC-induced pressure overload disrupted myocyte proteomic profiles, characterized by downregulation of mitochondrial enzymes, contractile proteins, and calcium-handling components. Vericiguat treatment reversed these alterations, restoring expression of bioenergetic and structural proteins. These findings suggest that Vericiguat preserves myocyte integrity and function through targeted proteomic reprogramming under pathological stress.
Figure 5.
Vericiguat Modulates Myocyte Proteomic Signatures in Pressure-Overload Heart Failure. Hierarchical heatmap depicting differentially expressed proteins (DEPs) in myocyte-enriched cardiac tissue across four experimental groups: wild-type (WT), sham-operated (Sham), transverse aortic constriction (TAC), and TAC with Vericiguat treatment (TAC-Ver). Protein abundance is color-coded (green: low; black: intermediate; red: high) and clustered by expression similarity. TAC-induced pressure overload disrupted myocyte proteomic profiles, characterized by downregulation of mitochondrial enzymes, contractile proteins, and calcium-handling components. Vericiguat treatment reversed these alterations, restoring expression of bioenergetic and structural proteins. These findings suggest that Vericiguat preserves myocyte integrity and function through targeted proteomic reprogramming under pathological stress.
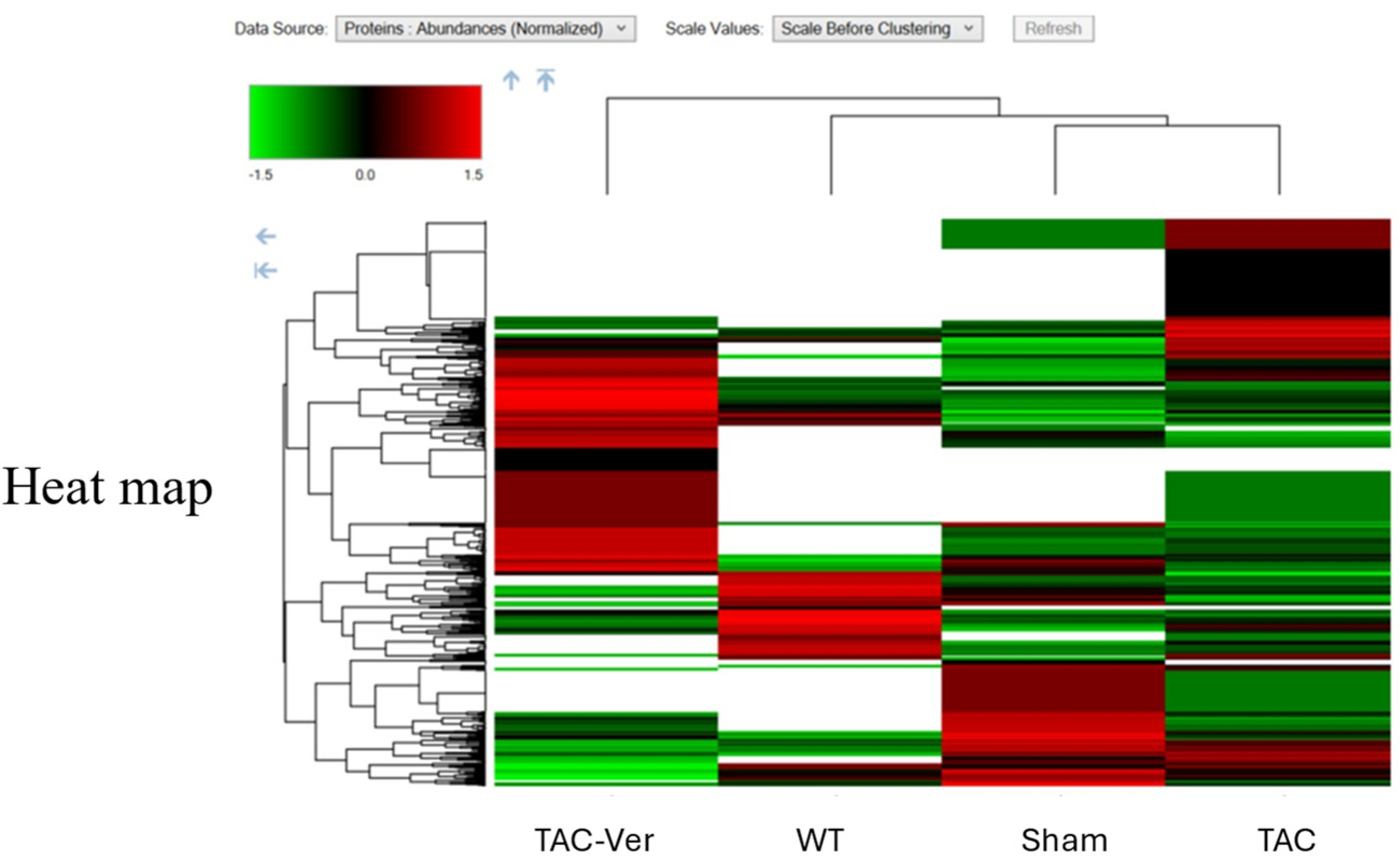

Table 1.
Echocardiographic Parameters Following Vericiguat Treatment in TAC-Induced Cardiac Hypertrophy.
Table 1.
Echocardiographic Parameters Following Vericiguat Treatment in TAC-Induced Cardiac Hypertrophy.
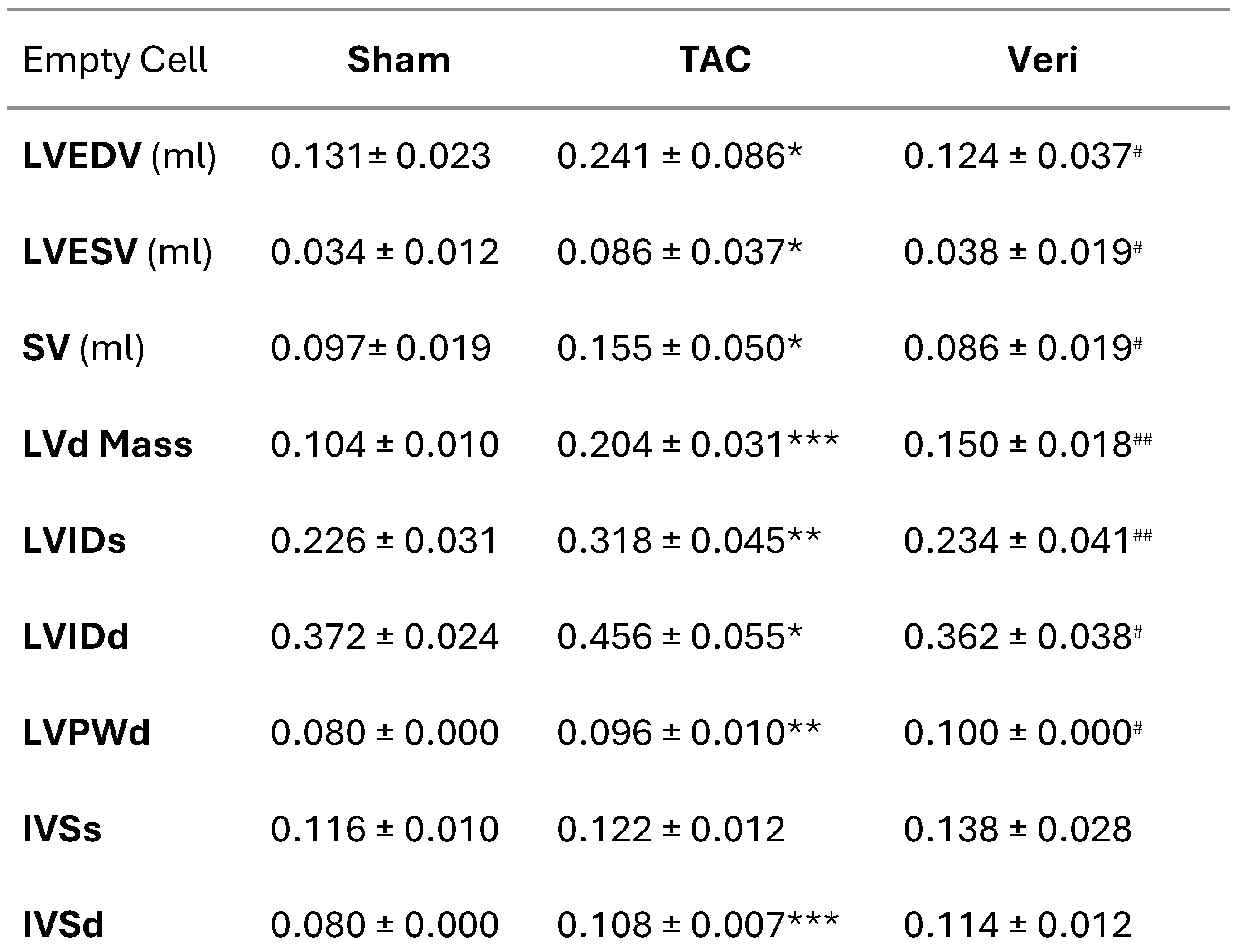 |
Echocardiographic Parameters in Mice Following Transverse Aortic Constriction and Vericiguat Treatment. Summary of left ventricular structural and functional indices measured by echocardiography in four experimental groups: Sham, transverse aortic constriction (TAC), and TAC with Vericiguat treatment (Veri). Parameters include end-diastolic volume (LVEDV), end-systolic volume (LVESV), stroke volume (SV), diastolic mass (LVd Mass), internal diameters (LVIDs, LVIDd), posterior wall thickness (LVPWd), and interventricular septal thickness (IVSd). Data are presented as mean ± SD. \* indicates significant difference vs. Sham; \# indicates significant difference vs. TAC; \\** and \#\# denote p < 0.01 and p < 0.001, respectively.
Table 2.
Functional Classification of Differentially Expressed Proteins in TAC vs. TAC-Vericiguat Comparison (Fibroblast-Enriched Fraction).
Table 2.
Functional Classification of Differentially Expressed Proteins in TAC vs. TAC-Vericiguat Comparison (Fibroblast-Enriched Fraction).
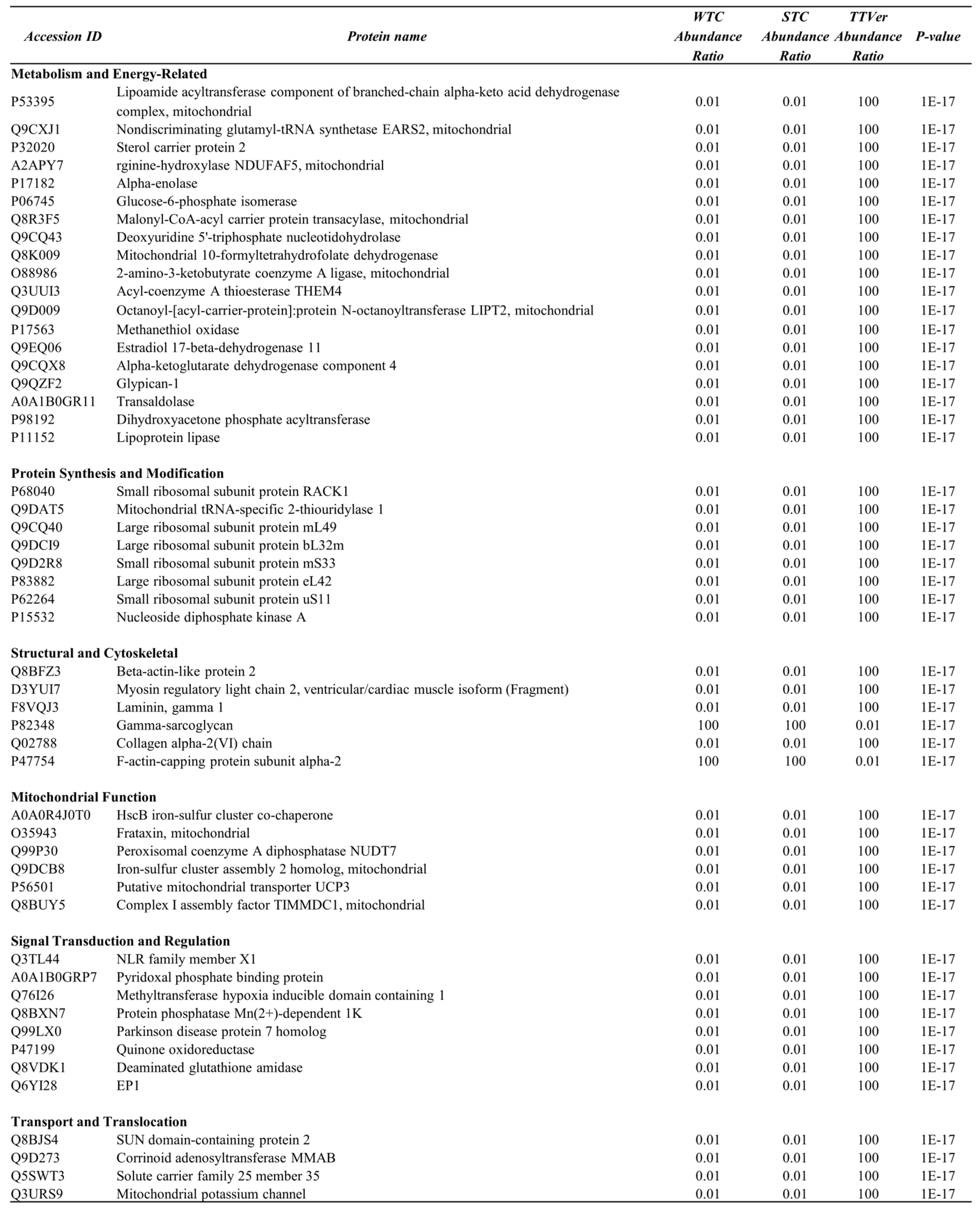 |
This table summarizes the functional categories of differentially expressed proteins (DEPs) identified in fibroblast-enriched left ventricular tissue from mice subjected to transverse aortic constriction (TAC) with or without Vericiguat treatment (TAC-Veri). DEPs were selected based on a log₂ fold change < 0.58 and p < 0.001. Proteins are grouped by biological function, including components of protein synthesis (e.g., ribosomal subunits, translation initiation and elongation factors, aminoacyl-tRNA synthetases), protein folding (molecular chaperones), degradation (proteasome subunits and ubiquitin-related enzymes), mitochondrial metabolism, and other regulatory processes. Vericiguat treatment consistently upregulated mitochondrial enzymes and translational machinery, indicating restoration of bioenergetic and biosynthetic capacity. These proteomic shifts are associated with enhanced protein homeostasis and structural integrity, supporting Vericiguat’s multi-pathway role in reversing pressure overload–induced myocardial remodeling.
Table 3.
Functional Classification of Differentially Expressed Proteins in TAC vs. TAC-Vericiguat Comparison (Myocyte-Enriched Fraction).
Table 3.
Functional Classification of Differentially Expressed Proteins in TAC vs. TAC-Vericiguat Comparison (Myocyte-Enriched Fraction).
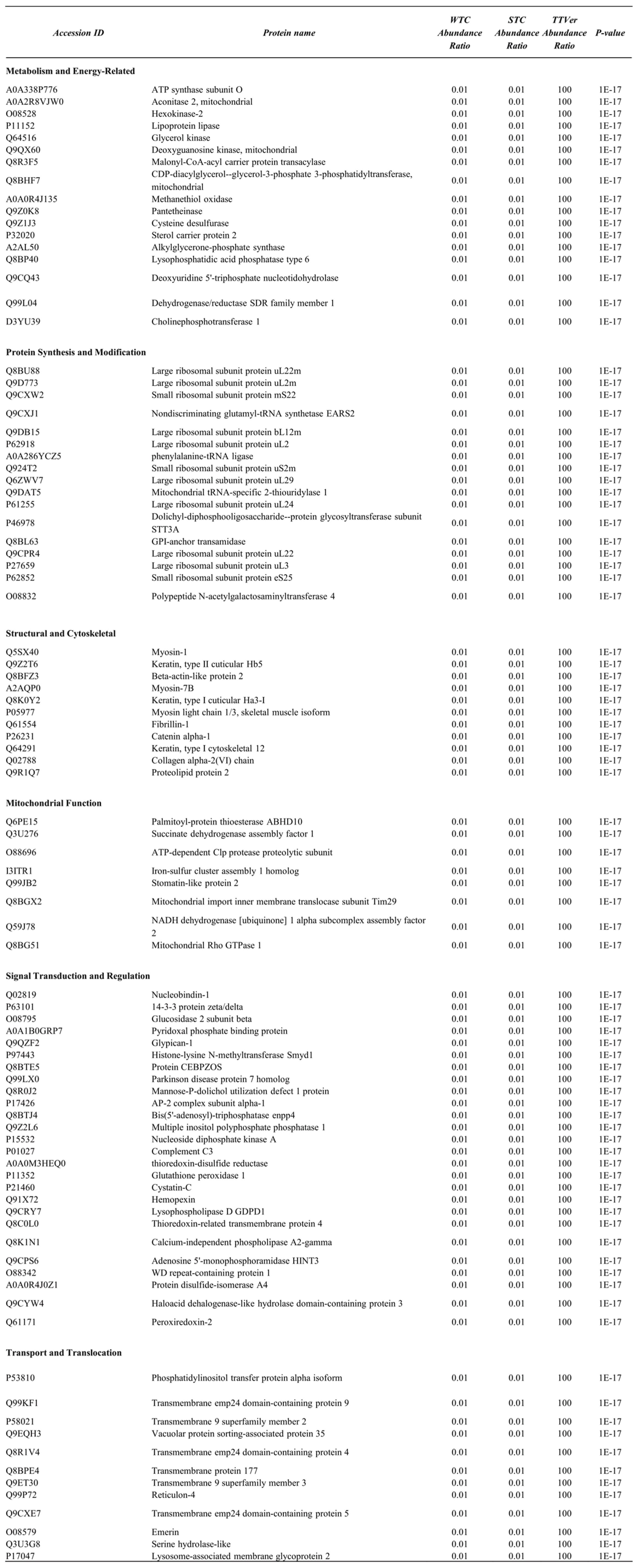 |
This table provides a categorized summary of differentially expressed proteins (DEPs) identified in myocyte-enriched left ventricular tissue from mice subjected to transverse aortic constriction (TAC), with or without Vericiguat treatment (TAC-Veri). DEPs were selected based on a log₂ fold change threshold < 0.58 and statistical significance (p < 0.001). Proteins are grouped by biological function, including metabolism and energy production, protein synthesis and modification, cytoskeletal and neuronal components, mitochondrial regulators, transcriptional control, and vesicular transport. Vericiguat treatment consistently upregulated mitochondrial enzymes and translational machinery, indicating restoration of bioenergetic and biosynthetic capacity. These proteomic shifts support enhanced protein homeostasis and structural integrity, underscoring Vericiguat’s multi-pathway role in counteracting pressure overload–induced myocardial remodeling.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



