Submitted:
25 February 2026
Posted:
26 February 2026
Read the latest preprint version here
Abstract
Lipid nanoparticles (LNPs) are central to modern mRNA therapeutics, including COVID‑19 vaccines. Far from passive carriers, their ionizable lipids actively interact with cellular membranes. Evidence from cellular, transcriptomic, and proteomic studies indicates that LNPs, with or without nucleic acid, alter gene and protein expression, thereby initiating inflammatory, detoxification, and stress responses at the membrane. Key pathways affected include lipid metabolism and detoxification, with roles for Peroxisome Proliferator-Activated Receptor γ (PPARγ) and cytochrome P450 enzymes. We hypothesize that the phosphatidylinositol (PI) cycle is the primary site of LNP-induced perturbations, regulating membrane restructuring and organelle trafficking during endocytosis. Disruption of this cycle triggers downstream signaling cascades, including Nuclear Factor κB (NF-κB), Mitogen-Activated Protein Kinases (MAPKs), Janus kinase/signal transducers and activators of transcription (JAK/STAT), and Mechanistic Target of Rapamycin (mTOR). We term this systemic effect lipid-nanoparticle-driven membrane dysfunction (L‑DMD), characterized by dysregulated cellular communication, stress responses, and energy balance. This review provides a mechanistic framework for understanding the persistent biological effects of modified modRNA-LNP exposure and emphasizes a systems-level intracellular perspective.
Keywords:
lipid nanoparticles
; mRNA therapeutics
; oxidative stress
; phosphatidylinositol cycle
; membrane disruption
; signaling cascades
1. Introduction
Lipid nanoparticles (LNPs) used in mRNA vaccine technology are engineered to resemble low-density lipoprotein (LDL) particles, thereby improving cellular penetration and facilitating endosomal escape [1]. Four distinct lipid types comprise LNPs: cationic ionizable lipids, cholesterol, phospholipids, and PEGylated lipids. Cationic ionizable lipids aid endosomal escape, cholesterol stabilizes the membrane and supports fusion, and phospholipids stabilize the LNP. [2] PEGylation, the addition of polyethylene glycol, masks the lipid particle's cationic surface charge and provides a hydrophilic stealth coating. Ionizable lipids interact with negatively charged endosomal phospholipids by forming cone-shaped ion pairs [3]. PEG coatings shield the surface from aggregation, opsonization, and phagocytosis [4].
While LNPs are recognized as biointeractive entities that integrate into cellular membranes and influence membrane structure, leaflet asymmetry, and local electrostatics, current pharmacodynamic and toxicological frameworks treat lipid-mediated effects as secondary or transient to the resulting protein expression. These frameworks focus primarily on nucleic acid payload, generalized innate immune responses, and protein expression, without linking membrane-level perturbations to downstream signaling dysregulation. We propose lipid-nanoparticle-driven membrane dysfunction (L-DMD), a hypothesis centered on phosphatidylinositol (PI) cycle perturbations, as a unifying mechanistic framework connecting structural membrane changes to persistent alterations in intracellular signaling pathways, including Nuclear Factor κB (NF-κB), Mitogen-Activated Protein Kinases (MAPKs), Janus kinase/signal transducers and activators of transcription (JAK/STAT), and Mechanistic Target of Rapamycin (mTOR).
1.1. L-DMD – A Rational Hypothesis
Lipid nanoparticles are often seen as delivery vehicles for nucleic acids, but their biological activity goes beyond payloaded transport. Their physicochemical properties, particularly those of ionizable lipids, render them inherently biointeractive (Figure 1). These lipids are engineered to undergo charge transitions in response to their local environment [5], enabling membrane interaction, bilayer penetration, and endosomal escape [6] [7]. We propose lipid nanoparticles act as active modifiers of membrane structure and function rather than passive carriers; cryogenic transmission electron microscopy (Cryo-TEM) indicates that LNPs lack a hollow aqueous core or stable internal membrane bilayers [2,8]. Instead, the encapsulated ribonucleic acid is intimately associated with ionizable lipids through electrostatic interactions [9]. The negatively charged phosphate backbone of the ribonucleic acid interacts directly with positively charged or protonatable lipid headgroups, forming a compact, disordered lipid-nucleic acid core [10] that reflects a metastable, non-crystalline lipid-nucleic acid assembly capable of structural reorganization, including bleb formation [11].
Lipid nanoparticles are stabilized predominantly by weak, non-covalent interactions, including electrostatic attraction, van der Waals forces, and hydrophobic effects [8,10]. No single interaction maintains structural integrity; stability arises from the collective interactions and flexibility of lipids and nucleic acids. The core is best seen as a dynamic network stabilized by the high configurational freedom of its components rather than from fixed architectural elements [12].
Consequently, the outer lipid shell of a lipid nanoparticle forms through spontaneous self-organization of lipids at the interface, not via an internal scaffold or layered membrane system [2]. This structure is sufficiently stable to permit formulation, storage, and systemic transport [13], yet it remains deliberately metastable [14]. Its metastability enables structural rearrangements in response to environmental cues, such as pH shifts or membrane contact, thereby facilitating cellular uptake and release of nucleic acid payload.
Given these properties, lipid nanoparticles are best described as supramolecular assemblies rather than fixed molecules [15]. Their behaviour reflects their multiparticulate, dynamic, and colloidal nature, with structure arising from collective interactions [16]. This distinction is not merely semantic; it has implications for how lipid nanoparticles are perceived, their interactions with biological systems, their responses to environmental stimuli, and their structural transitions in vivo.
Having outlined key structural and organizational features of LNPs, it is necessary to briefly discuss how these structures function in vivo. Such a discussion is essential to bridge the gap between formulation design concepts derived from physicochemical considerations and the actual biological behavior of LNPs after administration.
In biological environments, lipid nanoparticles are exposed to complex and heterogeneous conditions, including variable pH, ionic strength, protein coronas, and membrane interfaces. Under these conditions, their colloidal organization enables adaptive responses that may not be readily predictable from static structural models alone. As a result, lipid nanoparticles can give rise to emergent nonlinear effects arising from the interplay among particle composition, membrane interactions, and cellular context. Understanding these behaviors requires explicit consideration of both their physicochemical design principles and their dynamic reorganization in vivo. This structural characterization describes LNPs in isolation. To fully evaluate their biological impact, it should be considered how these metastable assemblies may behave when exposed to the complex environment of living systems.
1.2. The Special Properties of Ionizable Lipids
Central to this in vivo reorganization may be the ionizable lipids themselves, whose unique physicochemical properties could drive membrane interaction and integration. At membrane interfaces, the metastable behaviour of LNPs decomposes into the actions of individual lipids, which mediate interactions with cell membranes. Recent studies show that ionizable lipids can directly integrate into phospholipid bilayers [17,18]. Molecular dynamics simulations reveal that ionizable lipid nanodroplets merge spontaneously with model membranes, with insertion of lipid components into the bilayer rather than transient surface adsorption [17]. These simplified models isolate key physicochemical interactions that are otherwise masked in fully biological systems. Free energy profiles indicate several clinically used ionizable lipids favor partitioning into phospholipid membranes, with insertion depth and stability depending on lipid chemistry, bilayer composition, and phase state [18].
These findings are in keeping with the structure-function relationships described by Atmuri et al. [19], who demonstrated experimentally that molecular geometry (i.e., branched vs. linear tails, ionizable head-group, pKa, and linker biodegradability) can influence bilayer disruption and retention. Notably, membranes with higher phase-transition temperatures and greater lipid order (such as liquid-ordered cholesterol-enriched domains) are more susceptible to ionizable lipid penetration, indicating that the membrane physical state critically determines lipid integration. Importantly, such insertion represents the expected behavior of a metastable supramolecular assembly undergoing dynamic interactions with the lipid bilayer.
Ionizable lipids can cause lasting changes in membrane organization beyond insertion, affecting leaflet asymmetry, cholesterol distribution, lipid packing density, and membrane fluidity, often locally, at signaling microdomains [17]. These perturbations may alter the electrostatic and dielectric properties of the inner leaflet [20], crucial for membrane signaling. Since phosphoinositide (PIP)-dependent signaling relies on membrane-protein interactions and exhibits [21], even modest but long-lived perturbations in lipid packing or local charge density can be sufficient to destabilize [22].
Building on this conceptual gap, we present our L-DMD hypothesis, introducing how lipid-induced membrane perturbations may drive system-wide signaling dysregulation.
1.3. Powerful Signaling Effects of Phosphoinositides (PIPs)
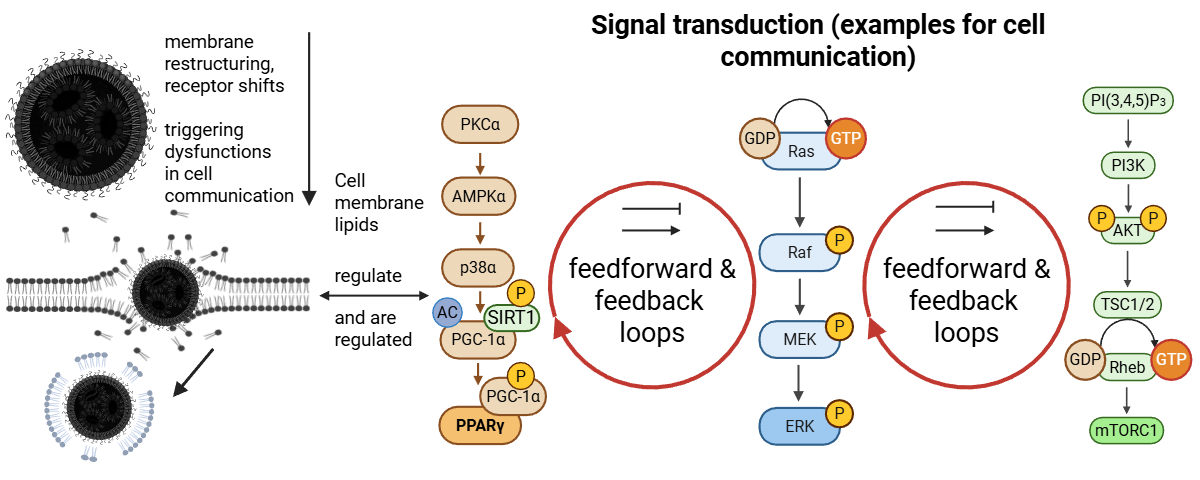
Within this context, phosphoinositides (PIPs; also referred to as PtdInsP) play a central mechanistic role. Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) and Phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) are low-abundance lipids that nonetheless control a disproportionate share of cellular membrane-proximal signaling processes [21]- [24]. They serve as precursors for canonical second messengers, regulate ion channels and vesicular trafficking, organize actin-membrane interactions, and act as obligatory cofactors for enzymes such as phospholipase C and phosphoinositide 3-kinase (PI3K) [21]. Importantly, their signaling function is governed not by bulk concentration but by spatial availability and orientation within the inner plasma membrane.
PI(4,5)P2 organization is dictated by electrostatic interactions between its highly negative headgroup and clusters of basic residues in intrinsically disordered protein domains. Proteins such as Myristoylated Alanine-Rich C Kinase Substrate (MARCKS), Epithelial Sodium Channel (ENaC) and Growth-Associated Protein 43 (GAP43) reversibly bind multiple PI(4,5P)2 molecules through multivalent electrostatic binding, forming separate lipid pools [21,25,26]. This dynamic system depends on input: increased calcium promotes calcium-calmodulin binding, displaces proteins, and releases sequestered lipids, while phosphorylation neutralizes positive charges and releases PI(4,5)P2. Consequently, membrane surface charge and lipid packing regulate signaling complexes formation productively [27].
Perturbations of membrane electrostatics therefore have immediate and predictable consequences for PIP-dependent signalling. Integration of ionizable lipids into the plasma membrane is expected to alter local charge density, modify lipid headgroup spacing, and change the effective dielectric environment experienced by PIPs and their binding partners. Even modest shifts in these parameters can disrupt the balance between sequestration and release of PI(4,5)P2 and PI(3,4,5)P3, thereby reshaping the spatial logic of membrane signaling [28]. Given that ionizable lipids demonstrably integrate into membranes and alter local charge density (as discussed in 1.2), such perturbations represent a mechanistically plausible scenario warranting investigation.
From this electrostatic perspective, signal-regulatory disorder represents a direct physicochemical consequence rather than a secondary biological effect. Signaling pathways depend on precise PIPs positioning, including PI3K and serine/threonine-specific protein kinases (PKB, also known as AKT). Phospholipase C-dependent calcium signaling, small GTPase activation, and actin-regulating cascades are inherently susceptible to disruptions in membrane lipid organization because these pathways are organized at the membrane through lipid–protein co-assembly rather than isolated protein–protein interactions. Therefore, alterations in lipid availability propagate across multiple signaling axes simultaneously [29].
Despite advances in understanding lipid nanoparticle (LNP) physicochemical behaviour and membrane interactions, current pharmacodynamic and toxicological frameworks largely treat LNPs as inert delivery vehicles, attributing adverse effects primarily to the encoded antigen (e.g., the spike protein) or non-specific innate immune activation (e.g., Toll-like receptor (TLR) activation [30]). This view may underestimate the direct bioactive potential of lipid components, both individually and in assemblies, particularly their ability to induce sustained alterations in membrane electrostatics and PIP organization: the integration of exogenous ionizable lipids into cell membranes may lead to permanent changes in local charge density and lipid packing, disrupting spatially limited PI/PI(4,5)P2/PI(3,4,5)P3 pools exceeding thresholds, subsequently dysregulating multiple membrane-proximal signalling pathways (e.g., PI3K/Akt/mTORC1) axis [31], phospholipase C-calcium signaling (PLC-Ca²⁺) [32,33] , small GTPases [34], and actin regulatory cascades [35] in a cell-type- and context-dependent manner.
L-DMD describes physicochemical disruption of membrane-organized signalling rather than nonspecific toxicity or antigen-driven immune activation. Our hypothesis predicts comparable pathway effects from empty and payloaded LNPs, dependence on ionizable lipid partitioning, mitigation via restoration of PI(4,5)P₂/PI(3,4,5)P₃ homeostasis, and cumulative effects after repeated exposure. The following sections review supporting evidence for membrane integration, PIP dysregulation, downstream signalling alterations, and the pharmacokinetic and biodistribution properties underlying LNP in vivo behavior.
2. Lipid Nanoparticles for mRNA Delivery: Biological Properties and Effects on Cellular Systems
Having outlined the conceptual framework of L-DMD, we now examine the physicochemical, pharmacological and biological properties of LNPs that provide the mechanistic basis for this hypothesis.
2.1. Factors Influencing Nanoparticle Bioactivity
Nanoparticle uptake and immune activation arise from their high surface area-to-mass ratio and reflect their integrated physical features, including size, charge, shape, and lipid composition, rather than any individual lipid [36]. Ionizable-lipid chemistry (unsaturation, branching, pKa), formulation conditions and manufacturing variables together influence membrane fusion, biodegradability, persistence, safety, and effectiveness [36,37], producing emergent, process-dependent behaviors that cannot be inferred from individual components.
modRNA-LNP architectures are heterogeneous due to self-assembly, ranging from multilamellar vesicles with blebs to core-shell-like morphologies [8,38]. Across published platforms, estimates suggest approximately 12% [39] to 80% [40] of particles are empty or minimally loaded with modRNA, depending on formulation or assay used (e.g. dye-binding, confocal methods) as well as batch- or manufacturing-related variability [41]. Importantly, these minimally loaded or empty LNPs (eLNPs) exhibit distinct physicochemical and functional properties [3,4,7,11], may be structurally more fluid [3,8], and can exhibit higher per-particle fusogenicity and membrane disruption [8]. Payload content alters ζ potential, corona composition [11,12], and cellular uptake kinetics [40,42], creating a biologically mixed exposure profile [43] that requires human-relevant modeling to define functional response which may differ between eLNPs and loaded particles (see Section 3). Figure 2 schematizes the physicochemical and biological characteristics of a modRNA-loaded lipid nanoparticle (LNP).
2.2. The Biocorona and Biodistribution of the LNPs
Upon entry into biological fluids, LNPs rapidly acquire a dynamic coating, a biocorona, comprised of serum proteins, primarily of lipoproteins, immunoglobulins, albumin, complement, and coagulation factors that confer a new biological identity. It varies by species [45], and is determined by both LNP characteristics (size, charge, PEG density, rigidity) and host or environmental factors [46]. In humans, it remains incompletely defined, with ApoE, vitronectin, C-reactive protein, and alpha-2-macroglobulin consistently present [47], and it can alter LNP structure, biodistribution, and modRNA stability [48].
LNPs remodel in plasma and lymph. Indeed, recent cryo-EM work demonstrates that LNPs do not simply adsorb proteins but rather exchange lipids and apolipoproteins with endogenous lipoproteins, producing hybrid LNP-lipoprotein assemblies resembling physiological LDL [44]. These fusions modify surface potential and fluidity, redirect uptake via the LDL receptor (LDL-R) pathway, apolipoprotein-dependent routes, and influence stability, tissue tropism, and immune recognition [49,50].
Considering these distinctions, it is crucial to evaluate LNP distribution within the body and across cell types, as these factors affect uptake, antigen presentation, and downstream cellular processes. Biodistribution indicates where LNPs or drugs are in the body but does not itself imply cell entry or gene expression. Transfection requires the uptake and endosomal escape of nucleic acids into cells. Gene expression depends on the persistence of intact modRNA translating into protein. These processes are often conflated in studies and regulatory submissions. Recognizing this distinction is crucial for understanding toxicity, durability and efficacy, as biodistribution alone cannot predict which specific cells are transfected or the levels of protein expression.
Among the factors affecting LNP distribution, the route of administration exerts the greatest influence, more so than in small-molecule pharmacology. Factors such as syringe pressure, perfusion rate, proximity to blood and lymphatic vessels, local pH, and temperature can influence particle dispersion during intramuscular (IM) administration [51]. Following intramuscular administration, LNPs remain initially at the injection site before traversing lymphatics to draining nodes, later entering circulation primarily as lipoprotein-hybrids.
After a single intravenous infusion in rats (patisiran-like), 90% of radioactivity was found in the liver within four hours, indicating fenestrated endothelium and ApoE/LDL-R uptake. Conversely, a single intramuscular dose of modRNA-LNP (Moderna-like) remained at the injection site for 24-48 hours and only appeared in the liver after 8-48 hours, showing that the administration route affects timing and distribution patterns, even in controlled conditions. [45].
LNPs primarily accumulate in the liver, spleen, and draining lymph nodes, which are rich in phagocytic cells of the reticuloendothelial system (RES), such as monocytes, Kupffer cells, and macrophages [52]. Such tropism toward the hepatic and lymphoid RES compartments indicates that immune activation and detoxification pathways are primary determinants of systemic LNP fate.
Recruited antigen-presenting cells (APCs) can internalize SARS-CoV-2 LNPs, translate the encoded spike protein, and then subsequently migrate to nearby lymph nodes, where T cell priming occurs [53]. Lesser amounts reach the heart, lungs, adrenal glands, ovaries, eyes, and other tissues via transcytosis or direct penetration [3,7,54]- [58].
In humans, the ionizable lipids and the vaccine modRNA (Moderna®), quantified by mass spectrometry and qPCR, respectively, appeared in plasma, with T max (time to maximum observed concentration) between 4 hours and 2 days, with wide inter-individual variability in timing and magnitude [59]. Buckley et al. used PET-CT in non-human primates to track rapid, stochastic LNP trafficking to lymph nodes after IM injection of a Moderna-like formulation. Quadriceps injections consistently drained to iliac nodes with clear muscle distribution at 4 hours. Deltoid injections varied, draining to axillary, apical, or pectoral nodes, with little spleen signal and no uptake in heart or other tissues (plasma kinetics not measured) [60]. These findings show that intramuscular LNP administration triggers early lymphatic trafficking and distribution prior to immune activation and clearance. Variability in Tmax likely results from administration factors, tissue biology, and particle interactions with cell membranes affecting sequestration and redistribution.
Conventional radiotracing and fluorescence-based labelling approaches blur intact vs degraded components and lack the subcellular resolution within tissues. New peptide-tag imaging confirms the presence of intact hybrid particles in tissue [61] highlighting the extent to which existing approaches may underestimate the complexity and heterogeneity of LNP distribution.
Uptake, or endocytosis, occurs via both receptor-mediated and receptor-independent mechanisms, often simultaneously, with the protein corona modulating these interactions [62,63]. Receptor-independent uptake relies on nonspecific hydrophobic and/or electrostatic forces, whereas receptor-mediated pathways, including clathrin-mediated endocytosis, depend on the local microenvironment and lipid–membrane interactions. Specific lipid components, such as the helper lipid DSPC, can alter membrane conformation and signaling to facilitate G-protein coupled receptor (GPCR) engagement without direct binding, and lipoprotein-enriched coronas further enhance internalization [64]. Overall, the biocorona determines biodistribution, uptake and persistence, linking formulation chemistry to L-DMD risk.
2.3. Endosomal Escape and Membrane Destabilization Due to Ionizable Lipids
Endosomal escape is the rate-limiting step for RNA translation. After endocytosis, progressive endosomal acidification protonates ionizable lipids, thereby altering charge balance and triggering lipid rearrangement and membrane destabilization. Several, not mutually exclusive, mechanisms have been proposed to explain this process, including proton-driven osmotic swelling (i.e., proton-sponge effect) [65,66], promotion of non-bilayer hexagonal phases, and directed lipid mixing or pore formation facilitated by inverted-cone lipid geometries [18]. Endosomal damage, evidenced by galectin recruitment, can occur solely from ionizable lipids without cytosolic RNA delivery [67]. Smaller membrane perturbations induced by LNPs are detected and rapidly sealed by the Endosomal Sorting Complexes Required for Transport-III (ESCRT-III) machinery. Computational and experimental studies suggest that LNPs can transiently tear membranes, disrupt lipid raft organization, and tether to the endosomal membrane, enhancing escape [66,68,69].
Escape is inefficient and remains a central bottleneck for RNA therapeutics [65,70]. Here, ‘endosomal release/escape efficiency’ refers to assay-specific estimates of the fraction of internalized LNP payload that reaches the cytosol in a functionally available form. Values are not directly comparable across assays and formulations [42]. Across representative studies, estimated endosomal release efficiencies are generally low and span roughly ~1-15%, as summarized in Müller et al. [42], based on data drawn from Sabnis et al [71], Maugeri et al [72], and Gilleron et al [73]. These efficiencies depend on the ionizable lipid structure, pKa, particle topology, cell type activation state, and the timing of the transient “burst” fusion window during early endosome acidification (pH<pKa) [42,65,74]- [76]. Escape occurs within minutes to a few hours after uptake, depending on formulation, cargo and assay methodology. If modRNA does not release into the cytoplasm, then maturing endosomes fuse with lysosomes, degrading modRNA and lipids. Accumulation of LNP components in lysosomes can impair function, block receptor recycling, create a cellular “traffic jam”, and prolong lipid retention, which may reduce therapy effectiveness and cause toxicity [65,76]- [79].
Indeed, LNP uptake follows a bell-shaped, nonlinear response influenced by particles and cell-specific factors. Immune cells like monocyte-derived macrophages and THP-1 cells show limited cytotoxicity, likely because their phagocytic pathways better handle LNP uptake and membrane stress. [80]. Uptake and endosomal escape are further influenced by interactions between the LNP surface and surrounding biomolecules [81,82], leading to a stochastic distribution of particle identities and protonation thresholds [76,82]. The relationship between dose and functional delivery is probabilistic, rather than linear, leading to variability in cellular responses and signaling, which are highly dose dependent. [83].
2.4. Spread to Distant Sites via Exosomes
Once the genetic material (RNA/modRNA/DNA) escapes the endosome, it can be packaged into naturally secreted membrane-bound vesicles along with ionizable lipids and intact mRNA. Maugeri [72] showed that, in a murine model, LNPs in recycling endosomes are either expelled intact or partially degraded, thereby affecting transfection efficiency. This process involves the trafficking of a fraction of LNP-delivered mRNA together with LNP-derived lipids into intraluminal vesicles of multivesicular endosomes (MVBs, see also Figure 4), which are subsequently released via extracellular vesicles, thereby enabling intercellular transfer. In such models, these exosomes circulate systemically and have been shown to mediate functional RNA transfer to distant cells and enable protein translation [72] . The evidence for modRNA-LNPs transfer in humans remains preliminary and needs further investigation [84,85].
Exocytosis serves as both a clearance route and a secondary distribution mechanism, extending modRNA or lipid fragments to the surrounding microenvironment in a paracrine manner [86], where functional translation is possible. Importantly, recycling of endosomes, as well as eLNPs or those with blebs, may cause cellular stress, oxidative damage, and chronic inflammation [87]. These factors are not considered in biodistribution studies and may contribute to cumulative toxicity, especially with repeated doses [88].
Spike-carrying exosomes prior to seroconversion have been detected in humans [89], supporting the plausibility of protein relay via vesicular pathways, potentially extending antigen exposure and immune activation beyond initially transfected cells. However, definitive evidence for exosome-mediated transfer of vaccine-modRNA in humans remains limited and warrants further study.
2.5. LNP Metabolism and Oxidative Stress Mechanism
The process by which the LNPs disassemble and are metabolized is typically described as hydrolysis and clearance [90,91]. Fatty acid metabolites derived from these lipids can activate PPARs, initiating lipid-sensing and stress response pathways [78]. The long-term persistence [91] and biodistribution [92] have not been systematically characterized in vivo, particularly with respect to tissue-specific retention and potential for bioaccumulation following repeated dosing.
In silico studies suggest these lipids may localize within the bilayer [18,66], further complicating predictions of LNP stability and in vivo behavior. Intracellularly, LNP components intercalate directly with cell membranes, triggering both repair and oxidative-stress pathways, leading to endosomal damage (detectable by galectins), immune signaling, and the production of reactive oxygen species (ROS) [93]. Oxidative stress can arise from incomplete lipid degradation, membrane perturbation, and lysosomal processing, and it is modulated by lipid structure, intracellular localization, and cellular context [67].
Tertiary amine-based ionizable lipids can generate N-oxides and reactive aldehydes during manufacturing and storage, which may covalently modify modRNA, rendering it untranslatable. Packer et al first identified such adducts in 2021 [94]. They can be mitigated by buffering or novel lipid designs [95]. These adducts may generate aberrant molecules that activate interferon-driven innate immune pathways [96,97] or potentially modify cellular proteins through electrophilic mechanisms. Ionizable lipids commonly incorporate unsaturated acyl chains susceptible to peroxidation, producing electrophilic aldehydes such as 4-hydroxynonenal (4-HNE), a well-established oxidative-stress product known to disrupt protein folding and lysosomal function [78,98]. However, direct evidence that modRNA-LNP administration generates 4-HNE at biologically significant levels in humans is lacking, and any clinical impact remains speculative.
2.6. Activation of the Immune System
LNPs can activate the immune system independently of spike protein expression. Within the adaptive immune system, two mechanisms have been identified. First, IgE-mediated anaphylaxis arises from antibodies induced against PEGylated particles in the biocorona [3,99,100]. Such reactions are rare but potentially life-threatening. Second, exposure to PEGylated LNPs can induce anti-PEG IgM antibodies, leading to the accelerated blood clearance (ABC) phenomenon, in which subsequent doses are rapidly cleared, reducing efficacy and altering exposure profiles [101].
Third, LNPs also activate innate immunity through complement- and receptor-mediated pathways. Complement activation-related pseudo-allergy (CARPA) represents a blood-facing innate immune reaction to nanoparticles, including PEGylated drugs [102,103]. CARPA is mediated by complement activation and anaphylatoxins (Complement (C)3a, C5a), often triggered by anti-PEG IgM antibodies, which can activate mast cells and release histamine, leading to acute infusion reactions [103].
Evaluation of CARPA typically relies on porcine and non-human primate models that recapitulate human cardiopulmonary responses; to date, to our knowledge, only one published study [104] has directly examined modRNA-LNP administration in a porcine CARPA model using intravenous administration, confirming complement activation and CARPA-like hemodynamics. Allergic reactions observed after modRNA COVID-19 vaccination may be mediated by this phenomenon [105] and have prompted calls for alternative PEG excipients to mitigate CARPA [106]. Clinically, patisiran (an siRNA-LNP therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis, Onpattro®) infusions can also trigger CARPA, sometimes severely, through complement activation [107,108].
LNPs can activate the innate immune system by transfecting immune cells, providing an intrinsic adjuvant-like activity [79] that aids in antibody production; LNPs can activate TLR3, 7, 8, and 9 in the endosome [109]. Additionally, LNPs bind to diverse cell membrane receptors like C-reactive protein (CRP), TLR4, and TLR2, and can activate the NLRP3 inflammasome [110]. These immune effects highlight the need to distinguish between biodistribution, transfection, and gene expression, as they are context-dependent and may cause pathogen-like effects beyond just cytotoxicity.
2.7. Delivery Architecture as a Determinant of Membrane Stress and Possible Systemic Risk
The biological effects of nucleic acid therapeutics are determined not solely by RNA modality, but also by delivery architecture and membrane interactions. Comparative analysis of approved platforms reveals a continuum of membrane engagement and persistence not captured by conventional pharmacokinetic or biodistribution frameworks.
This principle is evident when examining clinically approved RNA therapeutics. The siRNA therapeutic inclisiran (Leqvio®) achieves durable LDL-cholesterol lowering through triantennary N-acetylgalactosamine (GalNAc)-mediated receptor-targeted delivery of hepatocytes, rather than lipid nanoparticles, and has been evaluated in large, repeated-dose clinical trials with extended follow-up [111,112]. Its favorable safety profile, supported by precise cellular targeting, low administered doses, and infrequent dosing, mitigates systemic exposure and inflammatory risk, suggesting that the delivery strategy, not the RNA modality itself, may be a key determinant of long-term tolerability. Patisiran (Onpattro®) has a safety profile that includes transient elevations in transaminase levels (often resolved with dose adjustment) and infusion reactions, but little evidence of progressive hepatotoxicity over 18 months of treatment [107,113]. This contrasts with the acute reactogenicity observed with modRNA-LNP vaccines, although direct comparisons are confounded by route (IV vs IM) and dosing frequency.
In contrast, the European Medicines Agency (EMA)’s assessment of the self-amplifying-RNA COVID-19 vaccine zapomeran (Kostaive®), which employs LUNAR (ATX-126) ionizable lipids designed for improved degradability [114], reported very slow clearance of ATX-126 with tissue half-life not determinable for most organs, and an estimated half-life in muscle of 31-64 days [115]. Collectively, these products define a continuum of RNA delivery: from targeted, non-particle delivery (Leqvio®) to hepatotrophic siRNA-LNP delivery (Onpattro®), and to lipid-based vaccine platforms employing ionizable lipids tuned for increased endosomal fusion and cytosolic release (Kostaive®) (see Table 1). This is consistent with experimental structure–activity analyses showing that lipid architecture and ionization state influence bilayer disruption and toxicity [19].
These data indicate that ionizable lipid nanoparticles cannot be regarded as biologically inert carriers whose effects end once RNA is released. They function as supramolecular, membrane-active assemblies that persist within endosomal and lysosomal membranes, engaging repair circuits, oxidative stress pathways, and innate immune signaling. These processes are highly context-dependent, vary across tissues and individual cells [118] and are not captured by conventional biodistribution or transfection metrics. Together, the continuum of observations supports the Lipid-Driven Membrane Dysfunction (L-DMD) hypothesis as a coherent explanatory framework for the persistent, tissue-specific and heterogeneous outcomes associated with modRNA-LNP exposure. These mechanistic insights give rise to the core L-DMD hypothesis: membrane- and vesicle-level perturbations are the missing link that couples delivery architecture to cellular dysregulation.
Building on these considerations, the following section investigates how the biological disposition, pharmacodynamic, and physicochemical properties of modRNA-LNP systems may produce detectable molecular signatures in vivo. If membrane-associated uptake, intracellular trafficking, endosomal escape, or LNP restructuring perturb local signaling environments, such effects are expected to produce coordinated, system-level transcriptomic and proteomic shifts rather than isolated pathway activation. Omics-based profiling therefore provides a suitable framework to evaluate whether modRNA–LNP exposure is associated with broad molecular changes consistent with membrane-centered dysregulation.
3. Omics Data indicating Membrane Dysfunction Secondary to LNP Transfection
Omics approaches, including transcriptomics, metabolomics, lipidomics, and proteomics, are exploratory tools for analyzing cellular and organismal responses in experimental systems. They generate high-dimensional datasets that reveal molecular patterns and pathway alterations, providing reproducible, falsifiable signatures that support hypothesis generation rather than causal proof. In the context of modRNA–LNP COVID-19 vaccines, these datasets enable systematic assessment of intracellular responses to both eLNPs and payloaded formulations following uptake, endosomal processing, and modRNA translation. Although primarily descriptive, omics data provide a foundation for mechanistic interpretation, including evaluation of membrane-associated dysfunction. A key limitation is that transcriptomic changes alone cannot establish functional pathway activation without proteomic validation, while proteomic data alone cannot fully reconstruct upstream regulatory or membrane-level processes.
To date, omics analyses of LNPs remain limited, particularly for longitudinal human comparisons between vaccinated and unvaccinated cohorts. Transcriptomic, lipidomic, and proteomic datasets are therefore of particular interest, as they provide the most direct insights into altered signalling and protein activity states following LNP exposure. Established signalling pathway reconstructions, particularly from cancer research, provide a validated framework for interpreting pathway crosstalk and network dynamics and guide the hypothesis-driven component of this analysis. Accordingly, this section is organized into three parts: (1) omics data from eLNP-treated mouse models, (2) available human omics datasets, and (3) evaluation of whether convergent transcriptomic and proteomic signatures plausibly support the L-DMD hypothesis, while clearly distinguishing exploratory associations from mechanistic inference. Subsequent sections integrate these findings across models and interpret them within the L-DMD framework.
3.1. Mouse Data
In 2021, Ndeupen et al. published a pioneering in vivo study, in which they analyzed how a biological system responds to eLNPs [119]. Their pharmacokinetic investigation, in which wild-type (WT) C57BL/6 (B6) mice were injected intramuscularly with 10 μg of eLNPs, revealed thousands of changes in gene expression. The chosen dosage corresponds to the typical design for pharmacodynamic evaluation [120,121].
Regarding the number of up- or down-regulated genes, the study analysis revealed that “with p < 0.05 and FDR [False Discovery Rate] < 0.05, 9,508 genes and 8,883 genes, respectively, were differentially expressed.” A marked upregulation of genes involved in monocyte and granulocyte development, recruitment, and function (e.g., CCL2, CCL3, CCL4, CCL7) was observed. Gene set enrichment analysis (GSEA) revealed a pronounced induction of inflammatory cytokines, including IL-1β, granulocyte–macrophage colony-stimulating factor (GM-CSF), and IL-6, which are hallmark markers of acute innate and adaptive immune activation [122]- [126].
NF-κB was upregulated by an estimated 2.5–2.6 log₂-fold. In contrast, the TCA cycle and PPAR signaling pathways were markedly downregulated, with an approximate 2.0 log₂-fold reduction in expression. The upregulation of genes associated with TLRs, NOD-like receptors, and RIG I-like receptors suggests a robust system-wide immunological response [127]. Hematopoietic cell lines, in particular, showed a more than 2 log₂-fold increase in gene expression, indicating pronounced activation within stem and progenitor compartments that may be driven by the combined signaling of TLRs [128], NOD receptors [129], and RIG I [130] pathways and/or the internalization pathways of the LNPs. These findings are in line with the single cell-based flow cytometric experiment by Parhiz et al. [131].
Furthermore, the observations of Ndeupen et al. [119] are consistent with the in vitro experiment made by Zelkoski et al. [132], showing that eLNPs trigger TLR4-dependent signaling that bifurcates into a strong Myeloid differentiation factor 88 (MyD88)/NF-κB response and a parallel, though weaker, TRIF-mediated IRF activation. Their Kyoto encyclopedia of genes and genomes (KEGG) gene set enrichment analysis (GSEA) analysis revealed that JUN, a component of the AP-1 transcription factor complex, activated via the c-Jun N-terminal kinase (JNK)-MAPK cascade downstream of TLR4/MyD88 signaling, exhibited a 1.93-fold log₂ expression change, whereas the JAK-STAT axis, typically associated with interferon signaling, showed a more modest 1.13-fold log₂ change [133].
Korzun et al. investigated the reactogenicity of eLNPs compared to Luc mRNA-LNPs in vivo, both as a single dose and for multiple doses in C57BL/6 mice (wild-type) as well as MyD88, TLR4 and TRIF knockout lines [134] (See Table 3). The LNPs were produced microfluidically (Onpattro®-like, Pfizer®-like, Moderna®-like) and injected intraperitoneally. The ionizable lipids were the only variable changed. Reactogenicity was quantified by sickness behaviour (reduced food intake and weight loss). TLR4 dependence was tested by pharmacological inhibition with Resatorvid (TAK-242) (3 mg/kg) given 2 hours prior to each dose in the eLNPs groups. Gene expression was analyzed using the NanoString Counter Mouse Inflammation Panel (liver and hypothalamus tissue) and qRT-PCR, supplemented by ELISA (IL-6, IL-1β, TNF-α, Lipocalin 2 (LCN2) and FACS analysis of peritoneally obtained immune cells.
The authors showed that TLR4 and MyD88 in particular are necessary for the initiation of these reactions and that pharmacological inhibition of TLR4 (e.g. with TAK-242) can attenuate the reactogenicity of eLNPs, and thus also the disease-related behavioural changes. These findings underscore the importance of the TLR4/MyD88 axis not only for proinflammatory gene expression, but also for physiological behavioural responses associated with sickness behaviour. This finding is in line with in vitro experiments performed by Zelkoski et al. [132].
Similarly, Korzun et al. [134] describes an inflammatory signature for ionizable lipids with the induction of the proinflammatory cytokines IL-1β and IL-6 as well as the chemokines CCL2, CCL4, CXCL2 and CXCL10, which is consistent with the observations of Ndeupen et al. [119]
Further examination of the raw transcriptional data provides additional support for the observations of Ndeupen et al. Specifically, analyses of whole-tissue expression reveal substantial regulation of key signaling nodes, including Ras homolog gene family member A (RhoA), phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma (Pik3c2g), Rho-associated coiled-coil containing protein kinase 2 (Rock2), Mapk3 (also known as extracellular signal-regulated kinase 1 (ERK1), hereinafter referred to as ERK), and STAT3. These changes, observed in whole tissue, indicate a systemic activation of RAS/PI3K/ROCK/ERK signaling pathways that likely involves multiple cell types, including immune and epithelial cells. Analyses restricted to isolated immune-cell subsets may not fully capture these effects [133]- [136]. Canonical genes such as KRAS were not significantly altered in immune-cell-enriched subsets, and the downstream and parallel pathways show robust modulation, highlighting the importance of considering global transcriptional responses to eLNPs.
Another study supporting these observations was published by Luo et al. [137]investigating proteomic alterations induced by eLNPs lacking a modRNA payload. Depending on the formulation, the various nanocarriers were administered intranasally, intramuscularly, intravenously, orally, or intradermally, with doses ranging from 0.0005 mg kg⁻¹ to 0.5 mg kg⁻¹ (LNPs, liposomes, polyethylenimine (PEI) complexes) or 50.725 mg kg⁻¹ (DNA origami) and 10¹³ GC ml⁻¹ adeno-associated viruses (AAVs).
The authors compared payload versus no-payload LNPs versus phosphate buffered saline [PBS] controls. They detected 375 differentially expressed proteins (DEPs), of which 240 were upregulated and 135 downregulated in the no-payload LNP group in vivo. These changes were linked to metabolic processes, particularly ribosome function, protein translation, and RNA metabolism, as determined by Reactome pathway analysis. Specific markers, including Ribosomal Protein (Rpl)11, Rpl15, Eukaryotic Initiation Factor (Eif) 4b, Eif2b3, Ribosomal Protein (Rps) 6, and Rps2, were found to be differentially regulated.
These data, derived from in vivo Omics analyses, provide early insights into the broad immunological and signaling effects elicited by lipid nanoparticles.
3.2. Human Data
There are, to the best of our knowledge, only two existing ex vivo human studies providing direct molecular readouts following COVID-19 modRNA-LNP vaccination: Knabl et al. [138] (ex vivo buffy coat transcriptomics) and Hickey et al. [139] (longitudinal serum proteomics). Other studies are cited solely to provide context.
Knabl et al. conducted an ex vivo transcriptomic study to assess the systemic effects of modRNA packaged in LNPs in both comorbid elderly and healthy younger vaccinated individuals [138].
The first study group consisted of nine hospitalized Beta-variant COVID-19 patients, of which four had received the first dose of BNT162b2 8-11 days before symptom onset, while five patients remained unvaccinated; all vaccinated and three of the five unvaccinated patient received dexamethasone. Immune transcriptomes (MSigDB-GSEA) were analyzed at days 7-13, 20-32, and 42-60 after symptom onset (Supplementary Data 3 and 4 in Knabl et al.). In the raw data, thousands of genes in the transcriptomics were statistically significantly changed (up- or downregulated) after the BNT162b2-application.As reported by the authors, COVID-19 symptoms (Beta variant) developed in all patients including those infected shortly after the first BNT162b2 dose [138]. MSigDB-GSEA analysis of the four vaccinated patients revealed acute changes in gene expression across thousands of genes. One elderly comorbid patient died before the end of the study. Signaling pathways affected included JAK-STAT3, Interleukin-6 (Il-6) and Kirsten Rat Sarcoma (KRAS), among others, with many changes reaching statistical significance.
The canonical ERK-MAPK signaling pathway involves several key components: Rat Sarcoma (RAS), Rapidly Accelerated Fibrosarcoma (RAF), Mitogen-Activated Protein Kinase (MEK), and Extracellular Signal-Regulated Kinase (ERK), and it has extensive crosstalk with the mTORC1/2-pathways in feed-forward and feed-back mechanisms [140].
Notably, tumor necrosis factor alpha (TNF-α), via NF-κB signaling, was upregulated in both study groups (see next paragraphs), similar to the activation observed by Zelkoski et al. [132] and Ndeupen et al. [119]. It remains unclear whether this reflects canonical (RELA/p65-IκB-dependent) or non-canonical (NIK-RELB-mediated) NF-κB activation [141].
Moreover, Knabl et al. detected significant mTORC1 signaling across doses 1 to 3, with ten overlapping genes reaching statistical significance, underscoring that both mTORC1 and p53 are co-activated under conditions of physiological and genotoxic stress [138]. KRAS signaling can engage NF-κB through multiple downstream routes (including RAF/MEK/ERK and PI3K/AKT branches) [142], thereby promoting transcriptional programs that support cell survival and stress adaptation. Distinguishing canonical from non-canonical NF-κB activation in these samples requires targeted inspection of pathway components (e.g. RELA/p65 phosphorylation and IκB degradation for the canonical arm; NIK/RelB dynamics for the non-canonical arm) at the transcript and protein level [142]. Moreover, the four elderly patients showed profound upregulation of the JAK-STAT5 pathway with IL-2 and the JAK-STAT3 pathway with IL-6.
The authors later included eight healthy, naive individuals receiving their first vaccine dose and analyzed transcriptional changes 7-10 days post-vaccination. In these naive individuals, selective upregulation of the transcription factor E2F8 and specific interferon-stimulated genes was observed, while E2F transcription factor (E2F)1 and Cyclin A1 (CCNA1) remained largely unchanged. Similarly, only three canonical mTORC1 target genes (Cell Division Cycle 25A (CDC25A), Ribonucleotide Reductase Family Member 2 (RRM2), and BUB1 Mitotic Checkpoint Kinase (BUB1)) were modulated, indicating that global mTORC1 activation was minimal but statistically significant at this early time-point. CDC25A, RRM2, and BUB1 mark the transitions G1/S (CDC25A), DNA synthesis (RRM2), and mitosis (BUB1) [143]. Taken together, these findings indicate that the observed mTORC1-related transcriptional patterns are unlikely to be primarily driven by dexamethasone treatment, particularly given its half-life of 36-54h [144], which is unlikely to account for weeks-long pathway activation. If corticosteroid exposure were the dominant driver, suppression rather than persistence of mTORC1-associated transcriptional programs would be expected.
Hickey et al. conducted one of the first proteomic analyses examining molecular alterations following BNT162b2 and mRNA-1273 vaccination [139]. Serum samples from adults vaccinated with BNT162b2 or mRNA-1273 were analysed longitudinally for proteomic changes. Statistical analyses, including predictive modeling, identified key markers and signaling pathways. One month post-third dose, both modRNA vaccines modulated pathways linked to RAS/MAPK and ubiquitin-mediated protein regulation, with consistent UB2D1/PolyUbiquitin K48 upregulation, indicating shared activation of protein degradation. Phosphoinositide-dependent signaling, including PI3K–PI(3,4,5)P3–AKT, PIP2-dependent calcium mobilization, and PIP-regulated endocytic trafficking, was modulated, reflecting early membrane-proximal post-translational signaling. Downstream effects varied by vaccine platform and gender.
ChaC glutathione specific gamma-glutamylcyclotransferase 1 (CHAC1) and INS upregulation diverged between BNT162b2 and mRNA-1273, suggesting platform-specific LNP/ionizable lipid effects on stress and metabolic signaling. A transient induction of cytochrome P450 Phase I metabolic pathways occurred exclusively in male BNT162b2 recipients, without Constitutive Androstane Receptor (CAR)/Pregnane X Receptor (PXR) enzyme activation, arguing against xenobiotic induction and pointing to sex-specific or context-dependent hepatic modulation. Long-term, six-month assessments show persistent activation of translational and ribosomal pathways, with male mRNA-1273 recipients displaying additional cap-dependent translation initiation and AKT signaling.
3.3. Convergent Findings Across Studies and Platforms
Despite differences in model systems (mouse versus human), omics platforms (transcriptomics versus proteomics), LNP formulations (MC3, ALC-0315, SM-102), and experimental designs, several molecular signatures emerge consistently across studies:
- (1)
- Multiple lines of evidence indicate modulation of phosphoinositide-related pathways. These include downregulation of PIK3C2G (class II PI3K) across independent mouse datasets, phosphoinositide-dependent signaling alterations observed in human serum proteomics, and dysregulation of ESCRT-associated pathways involved in membrane repair and endocytic trafficking.
- (2)
- Inflammatory and stress-associated signaling pathways are reproducibly engaged, including NF-κB activation, upregulation of TNF-α, IL-6, and IL-1β, chemokine induction, and evidence of NLRP3 inflammasome involvement. TLR4/MyD88-dependent signaling is consistently implicated, with indications of pathway bias, depending on cellular and experimental context.
- (3)
- Metabolic and detoxification pathways are affected in a context-dependent manner. These include downregulation of cytochrome P450-associated xenobiotic metabolism, suppression of PPAR and AMPK signaling, and attenuation of TCA cycle activity, consistent with altered lipid and energy homeostasis.
- (4)
- Multiple signaling cascades downstream of membrane-proximal events are activated, including RAS/MAPK, PI3K/AKT/mTOR, and JAK-STAT pathways, as observed in both mouse and human datasets.
Importantly, several of these molecular perturbations are observed following exposure to empty LNPs lacking RNA payload, indicating that lipid nanoparticle components alone are sufficient to induce broad transcriptomic and proteomic responses. The convergence of phosphoinositide-dependent signaling changes, PI3K modulation, membrane repair pathway engagement, and downstream signaling activation provides empirical support for the L-DMD hypothesis, which posits membrane-level dysregulation as a central initiating event.
However, the biggest hurdle is distinguishing between payloaded LNPs and eLNPs due to their altered in vivo behaviour, as outlined in Section 2. In addition, antagonistic and/or synergistic effects are expected during endosomal escape and subsequent payload release and transcription. This was demonstrated by Luo et al. in their proteomics analysis comparing empty versus payloaded LNPs [137].
Taken together, the observed disparities in LNP behavior suggest a shift from empirical description toward a systems-level mechanistic interpretation.
4. Proposed Mechanistic Hypothesis Derived from the Omics Data: L-DMD as a Central Node
In this section, we present several pathways at the level of mechanistic signaling nodes, with direct experimental support. This serves as a reference framework for network dynamics, rather than as a complete causal chain or disease model. The previous omics signatures will be interpreted through three types of evidence: 1) Direct observations (e.g. PIK3C2G downregulation) 2) Mechanistic nodes with cross-study validation (e.g. TLR4/MyD88) and 3) System-level implications requiring prospective study (e.g. cell-cycle dysregulation). Phosphoinositides (PIPs) are emphasized as central organizers of these pathways, providing membrane-proximal coordination of signaling nodes and network integration.
4.1. Disruption of the ESCRT Circuit and Phosphatidylinositol Signaling (Hickey et al.)
The most striking observation from the proteomics study by Hickey et al. [139] concerns a reduction in endocytic activity and a downregulation of the ESCRT-circuit, both of which could indicate a disruption of the PI cycle [145]- [147]. It has been shown that the modRNA-LNPs induce endosomal membrane ruptures [88] which are detected by the ESCRT machinery. Small disruptions are quickly sealed by the ESCRT-circuit [148] in crosstalk with PIPs before disruption accelerates and larger perturbations trigger NLRP3 and galectins repair. Interestingly, Forster III et al. [149] demonstrated that mRNA-carrying lipid nanoparticles can also induce lysosomal rupture, which likewise leads to the activation of NLRP3 inflammasome and reduces mRNA translation efficiency.
This is consistent with the review by Hurley et al. [150], which summarizes ESCRTs as central to cell membrane repair mechanisms and, beyond that, to the organelle transport system. ESCRT complexes are recruited to membrane domains in part through interactions with phosphoinositide pools that define membrane identity, and their function in sorting ubiquitinated payload integrates ubiquitin signaling with membrane remodeling [148]. Distinct ESCRT subunits contain ubiquitin-binding motifs that recognize ubiquitylated membrane proteins and phosphoinositides, allowing coordinated recruitment and assembly of ESCRTs at phosphoinositide-rich membranes, thereby linking ubiquitin-driven payload recognition to membrane deformation and trafficking. In summary this pathway identified by proteomics fully supports our hypothesis.
4.2. Downregulated Xenobiotic Metabolism by Cytochrome P450 Enzymes (Ndeupen et al., Hickey et al.)
A notably downregulated KEGG pathway identified by Ndeupen et al. and sex specific for BNT162b2 in Hickey et al. [139] was the metabolism of xenobiotics by cytochrome P450 (CYP) enzymes [119]. Available data also implicate perturbations in hepatic xenobiotic metabolism, warranting closer examination of CYP enzymes as membrane-embedded pharmacologic sensors. Biochemical and structural studies demonstrate that these enzymes are functionally embedded in biological membranes [151]- [153].
Rises in pro-inflammatory cytokines, including IL-6, TNF-α, IFN-γ, IL-2, IL-1α, and IL-1β further suppresses hepatic CYP1A2, CYP2C9, CYP2C19 and CYP3A4 [154,155] Such reductions usually only occur after marked systemic inflammation (e.g. sepsis, CRP) >100mg/L) [156]. Clinically, case reports and cohort analyses document relevant changes in clozapine pharmacokinetics post vaccination, in some cases leading to neutropenia and hospitalization [157]- [159].
The mechanism is consistent with inflammation-mediated suppression of CYP enzymes, particularly CYP1A2 and CYP3A4, central to clozapine metabolism [160]. Comparable effects have been reported for other CYP-metabolized drugs, including statins, benzodiazepines, antiepileptics, and immunosuppressants, which are predominantly CYP3A4 [161] or CYP2C9 substrates [162], likely reinforced by altered membrane conditions. Together, these findings identify context-dependent modulation of CYP-associated pathways following modRNA-LNP vaccination, prompting mechanistic exploration of membrane-related processes.
CYP enzymes are essential for xenobiotic detoxification and endogenous lipid metabolism. They perform crucial biological functions, including cholesterol and fatty acid metabolism, vitamin D activation, and the synthesis of prostacyclins and thromboxanes from arachidonic acid [163].
ModRNA-LNPs can transiently remodel local cellular and vesicular membranes by altering phospholipid organization and membrane curvature through fusogenic and/or receptor-binding interactions [164]. Ionizable lipids with specific amine headgroups have been reported to directly interact with membrane immune receptors such as TLR4 and Cluster of Differentiation 1d (CD1d), supporting the possibility that LNP components can perturb native lipid microdomains that normally stabilize receptor complexes and membrane-bound enzymes [110].
The exact mechanisms are still under intense investigations. However, such microdomain disruption provides a plausible mechanistic basis for the ‘decoupled’ TLR4 signaling patterns [165] observed in murine studies by Korzun et al. [134] and Ndeupen et al. [119], as well as in luminescence assays by Zelkoski et al. [132], and could additionally contribute to CYP activity at the hepatocellular membrane interface [166]. In addition, independent biophysical studies have further demonstrated rapid pH-responsive structural transformations of ionizable LNPs that induce curvature stress and lipid packing rearrangements consistent with membrane remodeling behavior [74,167].
Furthermore, CYP enzymes are functionally embedded in biological membranes, with their binding, orientation activity depending crucially on the lipid composition: Anionic phospholipids such as phosphatidylserine or phosphatidylglycerol mediate stable docking of positively charged CYP domains via electrostatic interactions [168], while phosphatidylethanolamine, due to its small head group, induces membrane curvature and loosening of the packing, thus enabling partial insertion and correct orientation of the enzyme [49]; this lipid-dependent embedding is dynamic, influences the opening of membrane-side substrate channels and coupling to redox partners [169], and thus directly determines catalytic efficiency, whereby this principle is not limited to ER-localised CYPs, but also applies to mitochondrial steroidogenic CYP enzymes [170]. Therefore, perturbations to membrane composition would be expected to directly alter CYP and catalytic performance.
In summary, CYP downregulation likely reflects both cytokine-mediated transcriptional suppression and direct perturbation of membrane microdomains by ionizable lipids, consistent with the L-DMD prediction that membrane-embedded enzyme function is sensitive to lipid reorganization. Corroborating these molecular events, clinical evidence of post-vaccination hypercholesteremia [171] supports the concept that impaired hepatic detoxification and membrane-associated enzyme modulation translate into measurable serum lipid changes at the population level.
4.3. Are the TLR4 Reactions Biased? What Mouse Data Reveal (Ndeupen et al., Korzun et al.)
In line with the CYP observations, the results by Ndeupen et al. [119] , Korzun et al. [134], and also Zelkoski et al. [132] indicate a biased TLR4 dual-pathway activation pattern, with evidence pointing toward a stronger MyD88-dependent response relative to TRIF-dependent signaling in certain contexts. These observations raise the possibility of divergent or potentially decoupled TLR4 signaling. Such a bias may originate upstream at the cell membrane level and could be linked to early membrane dysfunction, consistent with patterns described by Zelkoski et al. [132] and related literature on ligand-specific TLR4 modulation [172]- [174].
Building on considerations of membrane structure and lipid microdomain organization, it is mechanistically plausible that early plasma membrane alterations could perturb TLR4 receptor localization, orientation, and/or associated phosphoinositide dynamics (e.g., PI(4,5)P₂). This could potentially contribute to biased or divergent dual-pathway activation. As a working hypothesis, this concept may connect our proposed membrane-level dysregulations to the observed TLR4 signaling patterns, given the established interconnection of TLR4 with PI(4,5)P₂ [172]- [174].
ModRNA N1-methylpseudouridine (m1ψ) modifications suppress endosomal TLR recognition [175], but ionizable lipidamine head groups are capable of simultaneously activating membrane-associated TLR4 and CD1d [110], which could lead to a paradoxical state of simultaneous TLR suppression and activation. This should also be considered in omics data. This dual, potentially opposing regulation may contribute to complex or seemingly contradictory immune activation signatures and should therefore be carefully considered when interpreting omics datasets.
Within the context of the L-DMD hypothesis, such a mechanism provides a plausible explanation for divergent TLR4 signaling patterns and supports the concept that supramolecular LNP properties can modulate receptor signaling independently of the RNA payload. While direct experimental evidence linking LNP-induced membrane perturbation to TLR4 pathway bias remains limited, extensive literature demonstrates that TLR4 signaling is highly sensitive to lipid raft integrity and local PI(4,5)P₂ availability, rendering this a testable mechanistic link within the L-DMD framework [176,177].
4.4. Upregulation of Multiple Inflammatory Markers (Ndeupen et al., Korzun et al., Knabl et al.)
IL-6, IL-1β Ndeupen et al. showed a remarkable cytokine and chemokine profile [119]. The simultaneous upregulation of TNFα, and IL-17 is notable, since these cytokines form a pro-inflammatory axis frequently implicated in the pathogenesis of autoimmune and chronic inflammatory conditions [178].
While IL-6 can modulate autophagy in the context of oxidative stress [179], and IL-1β levels may be influenced by autophagic clearance of pro-IL-1β [180], their elevation in this setting more likely reflects a broader integration of NLRP3-inflammasome activation [179], NF-κB signaling, and MAPK pathway dysregulation [181]- [183]. Similarly, β-chemokines such as CCL2, CCL3, CCL4, and CCL7 contribute not only to immune cell recruitment [184,185], but may also intersect with autophagy-related transcriptional regulation (e.g., via FOXK1) [186,187]. Korzun et al. observed comparable pattern [134].
Taken together, the coordinated upregulation of IL-6, IL-1β, TNF-α, IL-17, and β-chemokines (CCL2, CCL3, CCL4, CCL7) indicates activation of converging inflammatory pathways, including NLRP3 inflammasome signaling, NF-κB activation, and MAPK pathway dysregulation.In addition, marked increases were observed in CXC chemokines (CXCL1, CXCL2, CXCL5, CXCL10) and the colony-stimulating factors CSF2 and CSF3, which displayed the highest fold changes. Interestingly, CXCL5 has been shown to orchestrate the recruitment and spatial organization of innate and adaptive leukocytes in the lung during influenza infection, highlighting its role in regulating local inflammatory responses [188].
4.5. Complement Activation (Korzun et al. & Luo et al.)
Notably, Korzun et al. observed in the raw data in all models used C3 activation with a 1.5 log fold change [134]. This suggests that Complement 3 activation was not primarily driven by TLR4. G-Protein Coupled Receptors such as the C3a Receptor are highly dependent on cell membrane integrity [189]- [193]and cholesterol ratios [194].
Interestingly, the study by Luo et al. [137] investigated interactions of different ionizable LNP lipids (MC3, Lung SORT, SM-102, and ALC-0315) with cell surface proteins to identify possible competitors for LNP attachment forming a proteinaceous corona. In this context, vitronectin and ficolin-1 were found to particularly facilitate attachment of LNPs to heart tissue cells. Interactions with vitronectin may promote cell proliferation and are able to enhance tumor growth and metastatic potential [195], while ficolin-1, through binding to transforming growth factor (TGF-β1), may modulate the lectin pathway and the complement component of innate immunity [196]. This suggests that the protein corona itself may already have direct effects on membrane organization.
Notably, analysis of the raw data reported by Korzun et al. [134] revealed that PIK3C2G, which encodes a class II PI3K, was consistently downregulated across all datasets, providing an additional line of evidence in support of our L-DMD hypothesis. The combination of TLR4-independent C3 activation and evidence that protein corona components directly engage membrane-associated receptors supports the L-DMD model, in which LNPs can initiate immune signaling through biophysical membrane reorganization prior to, or in parallel with, classical receptor-mediated pathways.
4.6. Downregulation of PPAR and AMPK Signaling (Ndeupen et al.)
Given that peroxisome proliferator-activated receptor gamma (PPARγ) is a central regulator of lipid metabolism, mitochondrial function, and anti-inflammatory signaling, [119] and is modulated through AMPK-dependent phosphorylation [197,198] , its strong downregulation suggests disruption of homeostatic lipid and energy control. Such suppression, as reported under inflammatory or stress conditions [199]- [201], may exacerbate NF-κB activation (as shown by the data) and contribute to chronic inflammatory and/or unpredictable outcomes. The expression pattern is indicative of MAPK pathway dysregulation that affects PPARγ phosphorylation and signaling [200,201]. Notably, in macrophages, PPARγ regulates inflammatory gene expression [200]. Ballav et al. highlight that partial PPARγ dysregulation can broadly disturb cellular metabolism and signaling, influencing disease fate decisions across multiple tissues [202]. If such PPAR suppression were sustained across tissue documented in Section 2.2. (liver, spleen, lymph nodes), it would be consistent with the systemic, multi-organ distribution pattern of the LNPs and the L-DMD framework. However, direct evidence of persistent PPAR dysregulation in human tissues following modRNA-LNP administration is lacking, and extrapolation to chronic disease outcomes requires longitudinal validation.
4.7. RAS (Rat Sarkoma) Signaling and the MAPK (Hickey et al., Knabl. et al., Korzun et al.)
In summary, the insights of Knabl et al. [138] and Korzun et al [134] support the assumption that the inflammatory properties of LNPs observed in mice by Ndeupen et al. [119] and the findings of Luo et al. [137] are also present following BNT162b2 transfection. Moreover, not every immune cell internalizes LNPs equally, as uptake efficiency is influenced by factors such as the protein corona and the modRNA-payload (Section 2.3).
Such induction is consistent with stress-responsive transcriptional reprogramming. In this context, KRAS activation represents a central signaling hub, as RAS-driven pathways can engage both RAF–MEK–ERK and PI3K–AKT cascades (which was also observed in the proteomics by Hickey et al. [139]), thereby integrating inflammatory, metabolic, and genotoxic stress signals and promoting short-term cellular adaptation. The concurrent upregulation of p53, as also reported by Knabl et al. [138], suggests the activation of compensatory checkpoint mechanisms that counterbalance proliferative or stress-related cues [203,204].
Growing evidence indicates that GTPases, particularly RAS isoforms and RHOA, are spatially localized at the plasma membrane, where they play crucial roles in vesicle trafficking and membrane organization. Additionally, these GTPases can interact with lipids of the inner membrane leaflet, including phosphatidylserine and phosphoinositides, forming lipid–protein interfaces that may influence local signaling nanodomains [34,205]- [210].
The partial heterogeneity of transcriptional changes among different age groups highlights limitations in generalizing the transcriptomic effects observed in Knabl et al. [138] and sets the stage for the subsequent discussion of more controlled and consistent experimental models.
However, these observations position RAS [211,212] and other related GTPases, like RHO [213] as membrane-proximal integrators of phosphoinositide- and voltage-dependent signaling perturbations, providing a direct mechanistic bridge between membrane dysregulation and downstream transcriptional reprogramming, including the translation of altered electrical signaling into sustained cellular state changes [211]. [212]. Interestingly Guillot-Ferriols et al. demonstrated that electric fields have an impact on intracellular signaling pathways [214,215].
4.8. Transcription Factors (E2F1, E2F8) (Knabl et al.) and Mechanistic Target of Rapamycin Complex (mTORC) (Knabl et al., Hickey et al.)
Knabl et al. note: “Interestingly E2F8, but not CCNA1 or E2F1, was modestly upregulated 7 days post-vaccination in the naïve vaccinated individuals (1.5-fold, padj = 0.04).” However, growing evidence suggests that E2F1/2/4 expression correlates with immune cell infiltration and may modulate immune responses in proliferative or stressed cells [216]- [218]. It is known that E2F1 is in tight crosstalk during cell cycle phases with the 7 other members of the E2F transcription factor family [219]- [221]. Furthermore, it was shown that E2F1 plays a crucial role in immune cell differentiation and cloning [222], [223].
These findings support the notion that modRNA-LNP exposure can induce selective, systemic transcriptomic changes even in the absence of active viral infection, with partial activation of cell cycle genes, interferon responses, and partial mTORC1 engagement without coherent downstream cell-cycle coordination, consistent with a fragmented or dysregulated cell cycle program rather than a synchronized proliferative response [224], [225].
Furthermore, the consistent P53 upregulation in both age groups together with a decoupled E2F1 from E2F8 transcription factor transcriptome is indicating that a cell cycle dysfunctionality was observed in the immune cells which was depending on the mTORC1 signaling regulating CDC25A, RRM2 and BUB1 [226], [227]. Another indication that the cell cycle is affected at the transcriptomic level is the coordinated enrichment of gene sets associated with mitosis, checkpoint control, and Rho-GTPase signaling (Supplementary Data 5 in Knable et al.). All analyses were performed using KEGG and showed that the signals were active.
Recent evidence suggests that PI(3,5)P₂ may act as an upstream lipid regulator by forming a triad of lipid-lipid, lipid-protein, and protein-protein interactions, thereby modulating class I PI3K signaling activity—a pathway functionally linked to mTORC1 and mTORC2 regulation [228]- [230].
This decoupled activation pattern indicates a state of cellular stress adaptation rather than controlled expansion, with potential long-term consequences for immune cell homeostasis and functional changes.
4.9. Conceptual Consolidation of Section 3 and Section 4
Of note, Luo et al. also observed the activation of eucaryotic initiation factors (eIF) family [137]. Driven by these data, we suggest that signaling transduction pathways such as mTORC and the MAPKs regulating eIFs may be involved also in the endosomal escape processes [231], [232].
However, in vitro, ex vivo and in vivo data demonstrate that lipid formulated constructs, such as liposomes and LNPs, change cellular transcriptomics and proteomics by altering signal transduction pathways, receptor activation, and phosphorylation cascades [119,132,233]- [235]. This finding is of considerable significance, as the omics data we have discussed here and the shifts in signaling pathways cannot be attributed solely to the presence of modRNA and/or spike proteins.
This is reasonable, given the sequence of events: the LNPs are first taken up via membrane physics and chemistry (time point 0), after which the release of modRNA into the cytosol occurs with a delay. In fact, release rates, estimated to be below 10–15% as suggested by Müller et al. [42], appear far more likely to indicate that most of the modRNA remains within the LNP and is subsequently exocytosed. Importantly, this delayed release and translation may occur at anatomical sites distant from the injection site (Section 2.3).
Our L-DMD hypothesis implicates that LNPs may also drive the immune system’s initial response through membrane-driven effects of transfected cells, thereby impacting systems beyond immune cells alone. This is further supported by the study of Connors et al. [236], who showed that eLNPs induce activation and maturation of monocyte derived dendritic cells (MDDCs) and also upregulated CD40 expression, which led to recruitment of pro T follicular helper (pro-TFH) cell cytokines, IL-6, IL-12, and IL-21. These findings are also consistent with Amor et al. [237].
Furthermore, Qin et al. showed in mice that these effects have transgenerational immunological consequences [233]. Taken together, these findings indicate that LNP-driven immune activation is not restricted to acute innate immune responses or transient antigen-presentation–linked effects, but can also engage epigenetically mediated regulatory programs, consistent with recent evidence demonstrating short-term and persistent epigenetic memory in innate immune cells following BNT162b2 mRNA vaccination [238], [239]. In this context, recent work by Chytla et al., for example, has highlighted that PI(4,5)P₂ signaling is not confined to the plasma membrane but also operates at the nuclear level, where it contributes to transcriptional regulation and chromatin organisation [240], [241].
Table 2 organizes the omics-derived pathway alterations (Section 3 and Section 4) into functional categories reflecting the proposed L-DMD mechanistic cascade: Primary evidence is derived from controlled omics studies, supported by human observations, to demonstrate the functional relevance of molecular findings in human populations. Data are provided as corroborative evidence of pathway engagement rather than as validated clinical outcomes.
Table 3 presents primary omics and functional studies that inform the pathway classifications in Table 2. Each record details the model, material type, route of exposure, data modality, and timeline used to resolve membrane-associated signalling alterations described within the L-DMD framework.
Taken together (Table 2 and Table 3), these findings suggest that the earliest perturbations following LNP exposure likely originate at the plasma membrane, with the PI cycle as a central regulatory hub. Even subtle disturbances can cascade through lipid raft organization, receptor localization, and downstream signaling, producing transcriptomic and proteomic alterations, as shown by Hickey et al. [139] and Luo et al. [137].
Establishing precise in vivo mechanistic patterns remains challenging, especially without large cohorts stratified by age, vaccine type, dose, batch, comorbidities, prior illnesses, genetic predisposition, and LNP formulations. Extrapolation to chronic outcomes (e.g., autoimmunity [243], myocarditis [117]) requires longitudinal validation. The pathway enrichments presented here are hypothesis-generating rather than definitive disease mechanisms.
Omics data from Section 3 and Section 4 demonstrate multi-pathway perturbations following LNP exposure that cannot be attributed to the payload alone. Convergent alterations—including phosphoinositide signaling [139], PIK3C2G downregulation [134], mTOR and MAPK engagement, ESCRT dysregulation [139], [149], and metabolic pathway suppression [119] across studies and platforms support the L-DMD hypothesis.
These observations indicate that early plasma membrane perturbations propagate through lipid rafts, receptor localization, and downstream signalling cascades, resulting in the observed transcriptomic and proteomic changes. Such early membrane-centered dysregulation, though not reflected in conventional lab values, may initiate broader signaling alterations, including paracrine and endocrine effects [244].
5. Breaching the Plasma Membrane: Important Roles for Phosphoinositides
In Section 2, we outlined that LNP/modRNA complexes enter cells via receptor-dependent and receptor-independent endocytosis-like mechanisms, including membrane breaching. LNPs transfect diverse cell types, including epithelial cells, B cells, T cells, macrophages, and dendritic cells, depending on formulation and in vivo transformations, though the contribution of individual cell types to COVID-19 modRNA–LNP transfection remains under investigation. Computational simulations show that various ionizable lipids can integrate into the inner plasma membrane leaflet [17]- [19], making early membrane-level alterations of receptor localization or conformation mechanistically plausible. LNP uptake occurs via endocytosis and/or macropinocytosis, as demonstrated by Panariti et al. [250], Voigt et al. [251], and reviews by Wang et al. [252] and Gerelli [253].
Building on the phosphoinositide-centered signaling framework, we extend the hypothesis to membrane biophysical constraints that may couple LNP exposure to intracellular trafficking dynamics, integrating pathway-level observations with membrane-scale processes. Membrane penetration and direct interactions of ionizable lipids with the bilayer and membrane-bound receptors could drive reorganization events, including changes in lipid raft stability, where GPCRs are embedded.
This aligns with observations by Wang et al. [252] and Aliakbarinodehi et al. [74]. Lavagna et al. [254] suggest that size-dependent aggregation of hydrophobic nanoparticles within lipid membranes induces local curvature, folding, and transient phase transitions, acting as an initial mechanical trigger perturbing phosphoinositide organization and receptor localization, and setting the stage for altered signaling and recycling dynamics.
5.1. Brief Overview of the Phosphatidylinositol(PI)-Cycle
Given that membrane interactions are evident, the membrane structuring and recycling process must be examined by investigating the PI-cycle (Figure 3 and Supplementary Table 1) and the potential effects LNPs may have on it.
The eukaryotic plasma membrane is a phospholipid bilayer forming a stable barrier between aqueous compartments [255]. Phospholipids are amphipathic, with hydrophobic tails oriented inward and hydrophilic heads facing the aqueous environment, thereby enabling selective transport. Membrane fluidity depends on fatty acid composition and cholesterol content. Embedded proteins, such as receptors (e.g., GPCRs), ion channels, and transmembrane enzymes (e.g., ACE2), mediate transport, signaling, and cell-cell recognition via extracellular signals [256]. Fatty acid composition often follows the 18:0/18:1 or 18:1/18:1 rule, where unsaturated chains (e.g., 18:1) reduce packing density and increase lipid mobility [257]. The bilayer should be regarded as a fluid mosaic [257].
Lipid rafts are specialized microdomains enriched in sphingolipids and cholesterol, with specific phospholipid composition, including phosphoinositides, influencing membrane order and permeability [259], [260]. They create local spatial and electrical potential differences, promoting receptor clustering and anchoring of signaling molecules. Phosphoinositides act as first intracellular messengers, initiating downstream cascades at plasma and endosomal membranes after receptor internalization [261]. These cascades regulate the cycle via feedback loops [e.g., via RHO-GTPases (Ras homologous) and cell division cycle 42 (Cdc42 - part of the RHO family)] [262], [263].
Fallahi-Sichani & Linderman showed that lipid rafts as membrane microdomains influence the localization of receptors like GPCRs [264]. With rapid dimerization and monomerization, frequent partner changes lead to the formation of oligomeric receptor clusters. In addition, the effect of ligands varies broadly: “while some GPCRs are stimulated to dimerize by ligands, others are inhibited - indicating a differential, ligand-dependent receptor organization.” [264].
It has been suggested that these membrane microdomains also play a critical role in mediating interactions with exogenous particles. They can facilitate interactions between LNPs and the cellular membrane, thereby enhancing intracellular uptake through endocytosis [265]. Furthermore, the specific organization of these domains improves recognition and internalization processes, coordinates receptor localization and membrane trafficking and influences downstream signaling events, thus highlighting their central role in understanding how LNPs influence cellular homeostasis.
Although not yet demonstrated in vivo, it is conceivable that lipid nanoparticles may interfere with autophagic lysosome reformation (ALR) [266], [267], a process essential for sustaining lysosomal homeostasis and autophagic flux [268,269]. Since ALR depends on phosphoinositide transitions [270] and clathrin-mediated membrane remodeling [271], the insertion of ionizable lipids into these organelles could compromise the delicate balance of lysosomal recycling. Such perturbations may not only disturb autophagic flux but could also contribute to organ-specific toxicities reported in the nanomedicine safety literature [108].
5.2. Signaling Through Phosphorylation States of Phosphatidylinositol
Phosphatidylinositol (PI) are amphiphilic glycerophospholipids that insert into the plasma membrane with their inositol headgroups facing the cytosol. At physiological pH, PI is monoanionic (net charge~ −1), constitutes ~5–10 % of membrane phospholipids [272,273], and serves as a structural membrane component. Phosphorylated derivatives, phosphoinositides (PIPs), are low-abundance signaling lipids (~1–5 %) that remain membrane-bound, define organelle identity, and regulate trafficking, including endocytosis, phagocytosis, and receptor-mediated internalization.
The PI cycle, described by Posor et al. [274] (see Supplementary Table 1 for detailed PIPs-transitions with enzymes and kinases involved), is a dynamic network of phosphorylation transitions, e.g., PI(4,5)P₂ ↔ PI(3,4,5)P3, controlled by kinases (PI3K), phosphatases (INPPs, PTEN), and downstream effectors like Akt. These transitions govern organelle maturation, endosomal-lysosomal trafficking, phospholipid recycling, and membrane integrity [274]- [277], integrating with RAB GTPases and signaling cascades such as AKT/mTOR and ERK. Exposure to LNPs is hypothesized to perturb membrane biophysics, phosphoinositide organization, and endocytic trafficking, with secondary effects on PI-dependent signaling and organelle identity [278,279].
5.3. The LNP Components and Their Effects
5.3.1. Ionizable Lipids
Given that PIPs mark endosomal membranes and tightly regulate organelle maturation and trafficking, disruption of their dynamic phosphorylation-dephosphorylation cycles can compromise membrane integrity. These lipids interact with negatively charged PIPs and the surrounding phospholipid matrix, potentially destabilizing the tightly regulated membrane environment described above. Consequently, this can induce transient endosomal and lysosomal membrane rupture or permeabilization, as demonstrated in recent studies, thereby facilitating the cytosolic release of encapsulated payloads, such as modRNA, while bypassing the canonical degradation pathways normally orchestrated by the PI cycle [88,149]. This is especially notable because only a proportion of payload-containing LNPs (depending on the formulation and cell type) reach the phase of endosomal/lysosomal escape [72], and it is not yet known how many remain in endosomal/lysosomal/autophagosome transition [76] (discussed in Section 2).
Recent spatiotemporal analyses using sensitive LNP labeling platforms further support this notion, showing that continuous endocytosis and endolysosomal trafficking are required to maintain pools of releasable compartments, thereby mechanistically explaining why only a fraction of LNPs achieve cytosolic release while the remainder remain in lysosomes [87]. This partial efficiency highlights the unresolved question of how the remaining LNPs, which do not induce endosomal rupture and are trafficked to lysosomes, are subsequently degraded or recycled within the cell, and whether their presence might disrupt these processes.
LNPs, upon endocytic uptake, introduce high local concentrations of cationic or ionizable lipids into the endosomal membrane, a phenomenon that has been highlighted as a major determinant of both efficacy and toxicity of LNP formulations [90]. Moreover, Jörgensen et al. suggest that even the biodegraded blocks of the protonated tertiary amines (ionizable lipids) like ALC-0315 and SM-102 remain bioactive. The authors note: “However, they are neither synthesized from endogenous or natural structures nor are they degraded into typical biocompatible building blocks” [90].
5.3.2. DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine)
In the context of the recycling processes and membrane organization, another aspect that has received little attention is the phospholipid, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) [280,281], which is used as a helper lipid in LNPs. DSPC (also known as 18:0/18:0) has two saturated stearic acid chains. While structurally similar to the phospholipids naturally found in plasma membranes, DSPCs are more tightly packed, more rigid, and more thermally stable [282] than the typical 18:0/18:1 and 18:1/18:1 phospholipids found in eukaryotic membranes [257]. It has a high gel-liquid transition temperature (Tm approx. 55°C), which confers it with unusual rigidity [283]. It is a natural phospholipid, primarily found in human pulmonary surfactant [284].
This raises the question of how DSPC affects the PI-cycle, given that PCs are recycled into various phospholipids, including PI. Phospholipase D, which converts PCs into phosphatidic acid (PA), plays an essential role in this process [285]- [287]. PA is converted into CDP-diacylglycerol (CDP-DAG) by CDP-DAG synthase, a key step in channeling PA into the biosynthesis of PI and other phospholipids [273,288]. CDP-DAG subsequently serves as the immediate precursor for PI, synthesized by PI synthase at the endoplasmic reticulum, forming the core PI scaffold [273,289].
Data on how DSPC is degraded and whether it can interfere with membrane physiology beyond the transfection process via recycling is sparse. After 24 hours, most DSPC primarily accumulates in the liver, spleen, heart, kidney, and lung, indicating that these organs' plasma membranes take up the lipid [290]. This confirms DSPC's tissue delivery and retention. However, liposomes composed of phospholipids such as DSPC can markedly alter membrane structure, as previously discussed in the review by Lonez et al. [234]. Studies indicate that DSPC-containing liposomes can undergo hydrolytic and oxidative degradation, producing phospholipid-derived products that may alter membrane structure and stability in unpredictable ways [291,292]. However, the exact cellular mechanisms in vivo remain under investigation.
5.3.3. Oxysterol-Binding Proteins
Oxysterol-binding protein (OSBP)-related proteins coordinate PI metabolism with sterol transport [293,294], adding regulatory complexity to the PI cycle. Whether LNP-delivered cholesterol engages this pathway, particularly under inflammatory conditions [294] that alter sterol trafficking and phosphoinositide turnover, warrant systematic investigation to determine if OSBP proteins mediate lipid exchange or signaling events.
5.3.4. A Role for Lipid Impurities
As we suggested in Section 2, LNPs act as bioactive particles with unpredictable biodistribution and transfection pathways. This makes it difficult to determine the membrane conditions under which their proposed mechanisms of action will occur. Furthermore, the process by which lipid impurities or their by-products may influence endosomal-lysosomal transition, autophagosome formation, and the subsequent autophagosome-lysosome fusion that leads to autolysosome formation requires further study.
Covalent RNA-lipid adducts formed during manufacturing [94,95], and possible oxidative degradation products (e.g., 4-HNE), may compound membrane perturbations (see Section 2.5). In particular, 4-HNE, a highly reactive α,β-unsaturated aldehyde generated from peroxidation of omega-6 polyunsaturated fatty acids, readily forms Michael adducts with nucleophilic amino acid residues (cysteine, histidine, lysine) in proteins [296,297] If sufficiently generated in vivo, 4-HNE and related electrophilic lipid species merit systematic investigation as they are established modifiers of MAPK components and other signaling pathways [296,297]. Such modification could disrupt phosphoinositide homeostasis, whether by altering PI(4)P pools at the Golgi, impairing PI(4,5)P₂-mediated membrane dynamics, or modulating PI(3,4,5)P₃ signalling cascades that control vesicle trafficking, autophagy, and immune sensing, or directly modifying phosphoinositide-metabolizing enzymes that may contribute to sustained pathway dysregulation beyond direct membrane perturbations.
5.4. Small Perturbations Can Lead to Major Shifts in PIP Signaling
Recent work by Fung et al. demonstrates that phosphoinositide (PIP) interconversion is governed by feedback and feedforward circuits, excitable and oscillatory regimes, and kinetic thresholds, such that minor perturbations in membrane composition, enzyme activity, or lipid transport can trigger disproportionately large and sometimes paradoxical shifts in PIP balance [22]. This nonlinearity implies that local LNP-associated perturbations (e.g., ionizable lipids, DSPC, or pH changes) may push membranes across critical dynamical thresholds, altering endosomal identity, trafficking, and payload release [298]. Consequently, simple formulation optimization may not ensure predictable delivery across diverse cell types or physiological states.
This perspective highlights that LNP-induced effects are not purely additive but may exceed critical system thresholds that conventional toxicological frameworks, which often inadequately model complex, self-assembling bioactive nanosystems, may fail to capture. Models of lipid nanoparticle activity that neglect intracellular phosphoinositide processing and downstream signalling cascades, including MAPK pathways, therefore remain incomplete. Fung et al.’s [22] findings furthermore align with broader evidence that PIPs function as central hubs integrating signalling and mechanical regulation through feedback interactions with cytoskeletal dynamics and membrane remodeling [299]- [301].
6. Discussion
In this hypothesis-driven review, we proposed Lipid Nanoparticle-driven Membrane Dysfunction (L-DMD) as a mechanistic framework that may explain the persistent biological effects reported after exposure to LNPs. The model focuses on the bioactive properties of the ionizable lipid components and their interactions with cell membranes independent of any nucleic acid payload. In contrast to models which are limited to innate immune activation or modRNA translation kinetics, L-DMD emphasizes how direct physicochemical interactions with cellular membranes can induce sustained dysregulation in cellular signaling, metabolism, and homeostasis.
The core premise of the L-DMD hypothesis is that the metastable, supramolecular nature of LNPs, together with the electrostatic properties of ionizable lipids, can allow them to integrate into cellular membranes, including disruption of endosomal vesicles. This could lead to local changes in charge density, lipid packing, and the dielectric environment. Even minor alterations in these parameters may perturb localized pools of key phosphoinositides (e.g., PI(4,5)P₂, PI(3,4,5)P₃, PI4P, PI(3,4)P₂), which are central to membrane-proximal signaling and organelle identity. Therefore, their dysregulation can propagate to downstream networks including NF-κB, MAPK, JAK/STAT, and mTORC1/2, leading to the broad inflammatory stress-responses and metabolic patterns observed in cellular, transcriptomic, and proteomic analyses.
Experimental lipidomic analyses of nanoparticle–plasma interactions support lipid-mediated membrane remodeling at the nano–bio interface. To the best of our knowledge, only Pepafilippou et al. have examined ex vivo lipidomic profiles of liposome–corona complexes [302]. Using HSPC/cholesterol liposomes incubated with human plasma, they identified selective enrichment of sphingomyelins, ceramides, and acylglycerols in the corona by mass spectrometry. Although not directly transferable to ionizable LNPs due to their pH-dependent biophysics, the study demonstrates selective lipid–lipid interactions at the nano–bio interface and supports the conceptual basis of our hypothesis.
Signal transduction through PI3K-AKT-mTOR and RAS-ERK provide an example of this principle. These cascades are nonlinear, threshold-dependent systems with feedforward and feedback loops, where small shifts in lipid composition or charge distribution can destabilize network homeostasis [303]- [305]. One example, in which LNP-driven alterations at the plasma membrane or endosome could theoretically affect both the amplitude and duration of signaling outputs is shown schematically in Figure 5 (e.g., insulin signalling pathways).
This framework addresses an existing explanatory gap in current pharmacodynamic and toxicological models, which often treat LNPs primarily as delivery vehicles and attribute observed effects mainly to the persistence of the modRNA encoded protein [85,306] or to transient innate immunity [307,308].
6.1. LNP Perturbation and Functional modRNA Persistence
The phenomenon of persistent detection of spike protein and modRNA in certain individuals is consistent with this framework and is of particular interest to us. L-DMD suggests that such persistence may arise from signaling dysregulation rather than intrinsic molecular stability. If LNP-induced perturbations create conditions under which MAPK and RAS signaling platform activation becomes spatially and temporally dysregulated [19], such dysregulation could, in principle, influence RNA turnover, translation shutdown and protein degradation mechanisms, producing functional persistence of modRNA and prolongation of protein half-life, even in the absence of chemical “indestructibility” [309]- [313]. We assume that the decisive factor is not the intrinsic chemical stability of modRNA, but a systems-level failure of cellular homeostatic shutdown mechanisms. As detailed in Section 3, the data point to alterations in phosphoinositide-regulated signaling nodes and do not provide evidence for a single dominant upstream trigger, leaving open the possibility of effects on higher-order signal termination processes.
Experimental human ex vivo data support this interpretation [314]. Sequence-optimized, but chemically unmodified PpLuc mRNA formulated in LNPs demonstrated significantly higher luciferase expression at 24 and 48 hours compared with non-formulated, uridine-unmodified mRNA, suggesting that the supramolecular colloidal structure of the mRNA-LNP complex enhances intracellular persistence.
Furthermore, activation of innate immune sensors such as Toll-like receptor 4 (TLR4) may be associated with modulation of intracellular RNA processing and proteostatic pathways. Experimental studies demonstrate that RNA sensing by endosomal Toll-like receptors requires coordinated RNase-dependent processing, including RNase T2, RNase 2, and OAS-RNase L systems, linking innate immune activation to RNA turnover regulation [315]- [317]. In parallel, TLR4 signaling has been shown to interact with proteolytic and ubiquitin-dependent protein regulatory networks [318]- [320]. Comparable innate immune activation has also been described for cationic lipid transfection systems such as Lipofectamine, supporting the concept that lipid-mediated immune sensing may influence intracellular RNA and protein processing dynamics [321].
Together, these findings may indicate a shift in cellular homeostatic regulation rather than effects solely attributable to nucleoside modification. They support our argument that the functional behavior of modRNA LNP complexes can be influenced by supramolecular and biophysical properties, including entropic aspects of lipid organization, lipid-RNA interactions, phase behavior, and interactions with membrane-bound receptors and with plasma and vesicle membranes, which may in turn affect intracellular processing, RNA stability, and protein persistence.
6.2. Experimental Validation
The L-DMD hypothesis is testable with current tools. Endosomal function and LNP escape can be quantified using galectin-8-GFP or SNAPSwitch reporter assays and confocal microscopy; proteasome and RNase activities can be measured using Proteasome Glo kits, RNase L substrates, or pulse-chase experiments with actinomycin D, each under the control of MAPK inhibitors (e.g., SB203580, U0126) or mTORC blockers (rapamycin). KEGG pathway enrichment analyses of RNA-Seq data (6–48 h post-LNP transfection) can reveal dysregulation of MAPK, mTORC, RNA degradation, and ribosome biogenesis pathways, complemented by phospho-Western blots for key proteins (p-ERK, p-p38, p-S6K).
In in vivo mouse models, bioluminescence imaging combined with tissue qPCR/ELISA enable the correlation of prolonged luciferase or spike persistence with individual signaling variability. By comparing modified vs. unmodified mRNA, empty LNPs, and genetic/pharmacological interventions, it is possible to distinguish effects driven by the supramolecular properties of the LNPs from those linked to the modRNA payload.
6.3. Inter-Individual Variability
Variability in spike/modRNA persistence across individuals may reflect genetic or physiological differences in membrane signaling tolerance, feedback-loop robustness, or stress adaptation thresholds. These factors determine the rate at which a cell restores homeostasis after LNP-induced perturbations. Individuals with reduced ability or tolerance for membrane asymmetry or weaker feedback control could experience prolonged signaling activity and delayed molecular turnover.
6.4. Pharmacovigilance and Monitoring Gaps
Current post-marketing pharmacovigilance frameworks [322] are optimized for detecting and attributing immunologically mediated adverse events with acute temporal profiles, typically within hours to weeks post-vaccination. The L-DMD hypothesis, however, predicts a distinct class of lipid-mediated responses characterized by low-grade activation of signaling pathways, phosphoinositide dysregulation, autophagy impairment, and metabolic imbalance, manifesting as delayed (weeks to months), cumulative, multi-systemic effects of mild-to-moderate severity. These attributes fall outside standard case definitions, temporal windows, and biological plausibility assessments linked to known immune mechanisms and are associated with high confounding in causality assessment in multifactorial pathology [322,323]. Empirical testing of L-DMD requires mechanism-informed biomarker development and prospective cohorts designed to detect non-immunological, delayed manifestations that current surveillance systems are not designed to capture.
6.5. Implications for Nanomedicine Design
Recognizing LNPs as active biointerfaces rather than inert delivery vehicles may enable the rational design of next-generation lipid nanoparticles with improved safety profiles, enhanced controllability, and tunable persistence characteristics. Insights from the L-DMD hypothesis can guide three strategic directions.
- Rational LNP design
Adjusting ionizable lipid composition, charge density, and membrane affinity could allow decoupling of delivery efficiency from prolonged signaling perturbation, enabling safer and more predictable nanomedicine platforms.
- 2.
- Spatiotemporal control of signaling
Determining how LNPs influence membrane signaling domains may facilitate deliberate tuning of MAPK, mTOR, or innate immune pathways for therapeutic benefit, including cancer immunotherapy or transient immune modulation.
- 3.
- Predictive biomarker development
Establishing readouts for phosphoinositide dynamics, MAPK deactivation kinetics, and ubiquitin–protease activity as candidate biomarkers to assess long-term cellular responses to LNP exposure.
6.6. Broader Relevance and Methodological Outlook
L-DMD holds significance beyond modRNA vaccines. Similar lipid-based nanocarriers are increasingly used in gene therapy, oncology, and protein replacement medicine. Understanding how supramolecular lipid organization governs intracellular network behavior will likely have broad implications for nanomedicine, integrating biophysics, physicochemistry, pharmacology, and systems biology. Since open systems like LNPs operate in vivo with open systems like cells, this perspective may be useful in order to develop a consistent model [324]- [327].
Future research should prioritize analytical frameworks that characterize the supramolecular properties of LNPs, which are not fully addressed by current or emerging compendial standards [328,329]. While recent regulatory guidelines establish quality control parameters, including particle size, encapsulation efficiency, and chemical modifications such as adducts, they do not systematically assess biophysical determinants, such as ionizable lipid protonation state [9] lipid domain organization [48], membrane fusion kinetics, or structural heterogeneity [330] that may directly influence the membrane perturbations of the L-DMD hypothesis. Advances in structural and functional assays, including cryo-EM, small-angle X-ray and neutron scattering, and single-particle fluorescence methods, provide nanoscale resolution of lipid packing and particle morphology, informing next-generation quality control strategies [48,331]. In vivo, emerging tracking technologies, such as SPARKLE [61], alongside mass spectrometry imaging, enable spatiotemporal resolution of LNP particle biodistribution that current fluorescent labeling cannot achieve without altering particle biophysics. Standardizing these methodologies will be essential for testing L-DMD predictions, identifying biomarkers of membrane dysregulation, and rationally engineering next-generation LNPs with improved safety and controllability. We see this as one of the largest gaps and one of the greatest opportunities for optimizing future research.
While the L-DMD framework raises important questions about long-term cellular responses to LNP exposure, it remains a hypothesis-generating model awaiting rigorous experimental corroboration. Until such validation is obtained, any extrapolation to clinical risk or public health implications should be approached with caution.
In summary, the evidence compiled here, from structural studies, molecular dynamics simulations, biophysical experiments, and omics datasets, indicates that the lipid components of LNPs possess intrinsic bioactivity that can contribute to longer-term cellular alterations, including effects observed with empty LNPs.
6.7. Limitations
While comprehensive, this hypothesis-driven review remains primarily conceptual. Several limitations must be acknowledged:
1. Evidence hierarchy and model dependence: Most data integrated here derive from in vitro and murine studies employing high LNP doses and simplified cellular systems. Their pharmacodynamic and metabolic contexts differ from human administration, particularly with respect to intramuscular injection, depot kinetics, and systemic clearance. Translation to human physiology, therefore, requires caution.
2. Absence of long-term human datasets: There are currently no controlled longitudinal omics or lipidomic studies comparing vaccinated versus unvaccinated cohorts, nor studies isolating the effects of empty LNPs in humans. As a result, the persistence and mechanistic implications of the proposed L-DMD phenomena remain inferential.
3. Confounding from infection, comorbidities, and drug exposure: The majority of available transcriptomic and proteomic datasets were collected under non-standardized clinical conditions or concurrent illness, complicating attribution of observed molecular patterns specifically to LNP exposure.
4. Methodological gaps in membrane biology and lipid analytics: Quantitative measurement of membrane phosphoinositides, ionizable-lipid partitioning, or phospholipid-domain dynamics in vivo is technically challenging. Present biomarker assays (e.g., lipidomics or phospho-proteomics) lack temporal resolution and standardization across laboratories.
5. Unresolved dose, composition, and batch variability. Manufacturing differences in ionizable lipid identity, particle size distribution, and PEG density can alter biological activity. Without harmonized formulations, cross-study comparison and extrapolation remain uncertain.
6. Speculative extensions to clinical outcomes. Although pathways such as NF-κB, MAPK, JAK/STAT, and mTORC1/2 are well established in stress and immune regulation, linking their modulation by LNPs to specific human disease states or chronic conditions remains speculative. No causal relationships have been established. Similarly, any sex-specific vaccine effects require validation.
7. Omics data require confirmation. Transcriptomics cannot provide information about protein activity, nor can proteomics identify the upstream signals that led to altered protein expression. Furthermore, protein functionality, including correct folding and binding regulation, must also be experimentally determined. Lipidomics should therefore be applied as a complementary tool to trace upstream signaling inputs and the resulting suspected perturbations of the plasma membrane and vesicular transport pathways (see experimental design proposal in the Discussion).
8. ModRNA is based on the concept of suppressing intracellular Pattern Recognition Receptors (PRRs). A crucial aspect of modRNA’s success is that it is chemically modified (e.g., m1ψ) to suppress PRRs such as TLRs, thereby preventing premature mRNA degradation and increasing translation efficiency through nucleoside modifications [332]- [335]. At the same time, however, TLRs are functionally cross-talking with LDL receptors involved in LNP uptake, as well as with the complement system [336]- [339]. In addition, TLRs interact closely with membrane lipids [340,341]. This represents a potential limitation. TLR suppression (via modRNA modifications) combined with simultaneous TLR4 activation (via ionizable lipids) could produce contradictory immunological effects with additive, antagonistic, and/or synergistic consequences. This complicates attribution of observed cellular responses to the LNP structure, the modRNA payload, or their interaction.
Future confirmation of the L-DMD framework will require coordinated systems toxicology studies combining standardized LNP formulations, dose-dependent multi-omic profiling, and mechanistic perturbation experiments to distinguish membrane-driven signaling effects from payload-mediated responses.
7. Conclusions
Collectively, structural, biophysical, and omics data indicate that lipid nanoparticles possess intrinsic bioactivity that can modulate intracellular signaling independent of mRNA payload. The L-DMD framework hypothesis provides a conceptual bridge between colloidal physics and the regulation of cell systems, explaining persistence phenomena following SARS-CoV2-mRNA vaccination, and offering practical routes for design improvement.
By treating lipid nanoparticles as active participants in cellular decision-making rather than passive delivery vehicles, researchers can gain greater control over efficacy, safety, and temporal behavior, paving the way for a new generation of rationally engineered nanomedicines.
Funding
S. Seneff received funding from Quanta Computer, Inc. under contract number 6950759. The other authors received no funding for this work.
Authors’ Contributions
All authors contributed to conceptualization, writing of the original draft, review and editing.
Use of Generative AI
In preparing this work, the authors used ChatGPT and Grammerly to improve the comprehensibility of certain sentences. The authors reviewed and edited the content as necessary and take full responsibility for the published material.
Informed Consent Statement
Not applicable.
Consent for publication: Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank our reviewers for their comments, and Dr. Myra Forster-van Hijfte for reading a draft and offering valuable feedback, which have led to a significantly improved, more focused version of the manuscript. All figures were created by F. Seger using BioRender in accordance with BioRender's terms of use and academic license.
Abbreviations
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
| AAV | adeno-associated viruses |
| AKT | Protein Kinase B |
| AMPK | AMP-Activated Protein Kinase |
| BUB1 | Budding Uninhibited by Benzimidazole 1 |
| CAR | Constitutive Androstane Receptor |
| CARPA | Complement Activation-Related Pseudoallergy |
| CCL(X) | C-C Motif Chemokine Ligand (x) |
| CCNA1 | Cyclin A1 |
| CD1d | Cluster of Differentiation 1d |
| CDC25A | Cell Division Cycle 25A |
| CDP-DAG | CDP-Diacylglycerol |
| CHAC1 | ChaC glutathione specific gamma-glutamylcyclotransferase 1 |
| COVID-19 | Coronavirus Disease 2019 |
| CRP | C-Reactive Protein |
| CSF2 | Colony Stimulating Factor 2 |
| CSF3 | Colony Stimulating Factor 3 |
| CXCL | C-X-C Motif Chemokine Ligand (X) |
| CYP | Cytochrome P450 |
| DEG | Differentially Expressed Gene |
| DEP | Differentially Expressed Protein |
| DSPC | Distearoylphosphatidylcholine |
| E2F | E2F Transcription Factor |
| E2F1 | E2F Transcription Factor 1 |
| E2F8 | E2F Transcription Factor 8 |
| Eif | Eukaryotic Initiation Factor |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| eLNP | Empty Lipid Nanoparticle |
| EMA | European Medicines Agency |
| ENaC | Epithelial Sodium Channel |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-Regulated Kinase |
| ESCRT | Endosomal Sorting Complex Required for Transport |
| FDA | Food and Drug Administration |
| FDR | False Discovery Rate |
| GalNAc | N-Acetylgalactosamine |
| GAP43 | Growth-Associated Protein 43 |
| GO | Gene Ontology |
| GPCR | G-Protein Coupled Receptor |
| GSEA | Gene Set Enrichment Analysis |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| INS | Insulin |
| JAK | Janus Kinase |
| JAK-STAT | Janus Kinase-Signal Transducer and Activator of Transcription |
| JNK | Jun N-terminal Kinase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KRAS | Kirsten Rat Sarcoma |
| L-DMD | Lipid-Driven Membrane Dysfunction |
| LDL | Low-Density Lipoprotein |
| LDLR | Low-Density Lipoprotein Receptor |
| LCN2 | Lipocalin 2 |
| LNP | Lipid Nanoparticle |
| LNPs | Lipid Nanoparticles |
| MAPK | Mitogen-Activated Protein Kinase |
| MARCKS | Myristoylated Alanine-Rich C Kinase Substrate |
| MEK | MAPK/ERK Kinase |
| modRNA | Modified (messenger)RNA |
| mRNA | Messenger RNA |
| mTOR | Mechanistic Target of Rapamycin |
| mTORC1 | mTOR Complex 1 |
| mTORC2 | mTOR Complex 2 |
| MVB | Multivesicular Body |
| NF-κB | Nuclear Factor kappa B |
| NLRP3 | NOD-Like Receptor Protein 3 |
| OSBP | Oxysterol-Binding Protein |
| P38 | p38-mitogenaktivierte Proteinkinasen |
| PA | Phosphatidic Acid |
| PBS | Phosphate Buffered Saline |
| PEG | Polyethylene Glycol |
| PEI | Polyethylenimine |
| PI | Phosphatidylinositol / Phosphoinositide |
| PI(3,4)P₂ | Phosphatidylinositol-3,4-bisphosphate |
| PI(3,4,5)P₃ | Phosphatidylinositol-3,4,5-trisphosphate |
| PI(3,5)P₂ | Phosphatidylinositol-3,5-bisphosphate |
| PI(4,5)P₂ | Phosphatidylinositol-4,5-bisphosphate |
| PI3K | Phosphoinositide 3-Kinase |
| PI4P | Phosphatidylinositol 4-phosphate |
| PIK3C2G | Phosphoinositide 3-Kinase Class 2 Gamma |
| PIP | Phosphoinositide |
| PIP₂ | Phosphoinositide bisphosphate |
| PIP₃ | Phosphoinositide trisphosphate |
| pKa | Negative base-10 logarithm of the acid dissociation constant |
| PKB | Serine/threonine-specific protein kinases |
| PLC | Phospholipase C |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PPARγ | Peroxisome Proliferator-Activated Receptor gamma |
| PRR | Pattern Recognition Receptor |
| PXR | Pregnane X Receptor |
| qPCR | Quantitative Polymerase Chain Reaction |
| RAF | Rapidly Accelerated Fibrosarcoma (kinase) |
| RAS | Rat Sarcoma (GTPase family) |
| RNA-Seq | RNA Sequencing |
| Rps | Ribosomal Protein |
| RRM2 | Ribonucleotide Reductase M2 |
| saRNA | Self-Amplifying RNA |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| siRNA | Small Interfering RNA |
| STAT | Signal Transducer and Activator of Transcription |
| TAK-242 | Resatorvid, a small molecule inhibitor of toll-like receptor 4 signaling |
| TFH | T follicular helper |
| TGA | Therapeutic Goods Administration |
| TLR | Toll-Like Receptor |
| TLR4 | Toll-Like Receptor 4 |
| TLR7 | Toll-Like Receptor 7 |
| TLR8 | Toll-Like Receptor 8 |
| TNF-α | Tumor Necrosis Factor alpha |
| WHO | World Health Organization |
References
- Khurana, A; Allawadhi, P; Khurana, I; Allwadhi, S; Weiskirchen, R; Banothu, AK; et al. Role of nanotechnology behind the success of mRNA vaccines for COVID-19. Nano Today 2021, 38, 101142. [Google Scholar] [CrossRef]
- Hald Albertsen, C; Kulkarni, JA; Witzigmann, D; Lind, M; Petersson, K; Simonsen, JB. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv Drug Deliv Rev 2022, 188, 114416. [Google Scholar] [CrossRef]
- Swetha, K; Kotla, NG; Tunki, L; Jayaraj, A; Bhargava, SK; Hu, H; et al. Recent advances in the lipid nanoparticle-mediated delivery of mRNA vaccines. Vaccines (Basel) 2023, 11(3), 658. [Google Scholar] [CrossRef] [PubMed]
- Suk, JS; Xu, Q; Kim, N; Hanes, J; Ensign, LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016, 99 Pt A, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Kloczewiak, M; Banks, JM; Jin, L; Brader, ML. Perspective on higher-order structure and thermal stability of mRNA vaccines. Mol Pharmaceutics 2022, 19(7), 2022–2031. [Google Scholar] [CrossRef]
- Trollmann, MFW; Böckmann, RA. Decoding pH-driven phase transition of lipid nanoparticles. Small Epub ahead of print. 2026, e11381. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, E; Abolmaali, SS; Dehshahri, A; Mousavi Shaegh, SA; Azarpira, N; Tamaddon, AM. Navigating the intricate in-vivo journey of lipid nanoparticles tailored for the targeted delivery of RNA therapeutics: a quality-by-design approach. J Nanobiotechnology 2024, 22(1), 710. [Google Scholar] [CrossRef]
- Brader, ML; Williams, SJ; Banks, JM; Hui, WH; Zhou, ZH; Jin, L. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys J 2021, 120(14), 2766–2770. [Google Scholar] [CrossRef]
- Eygeris, Y; Gupta, M; Kim, J; Sahay, G. Chemistry of lipid nanoparticles for RNA delivery. Acc Chem Res 2022, 55(1), 2–12. [Google Scholar] [CrossRef]
- Kulkarni, JA; Cullis, PR; van der Meel, R. Lipid nanoparticles enabling gene therapies: From concepts to clinical utility. Nucleic Acid Ther 2018, 28(3), 146–157. [Google Scholar] [CrossRef]
- Simonsen, JB. A perspective on bleb and empty LNP structures. J Control Release 2024, 373, 952–961. [Google Scholar] [CrossRef]
- Trollmann, MFW; Böckmann, RA. mRNA lipid nanoparticle phase transition. Biophys J 2022, 121(20), 3927–3939. [Google Scholar] [CrossRef]
- Hou, X; Zaks, T; Langer, R; Dong, y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, M; Campbell, RA; Arteta, MY; Lawrence, MJ; Sebastiani, F. Review of structural design guiding the development of lipid nanoparticles for nucleic acid delivery. Current Opinion in Colloid & Interface Science 2023, 66, 101705. [Google Scholar] [CrossRef]
- Nele, V; Campani, V; Alia Moosavian, S; De Rosa, G. Lipid nanoparticles for RNA delivery: Self-assembling vs driven-assembling strategies. Adv Drug Deliv Rev 2024, 208, 115291. [Google Scholar] [CrossRef] [PubMed]
- Manning, AM; Tilstra, G; Khan, AB; Couture-Senécal, J; Lau, YMA; Pang, J; et al. Ionizable lipid with supramolecular chemistry features for RNA delivery in vivo. Small 2023, 19(41), e2302917. [Google Scholar] [CrossRef]
- Čechová, P; Paloncýová, M; Šrejber, M; Otyepka, M. Mechanistic insights into interactions between ionizable lipid nanodroplets and biomembranes. J Biomol Struct Dyn 2025, 43(17), 9983–9993. [Google Scholar] [CrossRef]
- Ermilova, I; Swenson, J. Ionizable lipids penetrate phospholipid bilayers with high phase transition temperatures: perspectives from free energy calculations. Chem Phys Lipids 2023, 253, 105294. [Google Scholar] [CrossRef]
- Atmuri, NDP; Saadati, F; Kulkarni, J; Witzigmann, D; Cullis, PR; Ciufolini, MA. Design of cationic ionizable lipids for the delivery of therapeutic nucleic acids. Mol Ther Methods Clin Dev 2025, 33(4), 101585. [Google Scholar] [CrossRef]
- Gurtovenko, AA; Lyulina, AS. Electroporation of asymmetric phospholipid membranes. J Phys Chem B 2014, 118(33), 9909–18. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S; Murray, D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 2005, 438(7068), 605–11. [Google Scholar] [CrossRef] [PubMed]
- Fung, SYS; Xǔ, XJ; Wu, M. Nonlinear dynamics in phosphoinositide metabolism. Curr Opin Cell Biol 2024, 88, 102373. [Google Scholar] [CrossRef]
- Ellenbroek, WG; Wang, YH; Christian, DA; Discher, DE; Janmey, PA; Liu, AJ. Divalent cation-dependent formation of electrostatic PIP2 clusters in lipid monolayers. Biophys J 2011, 101(9), 2178–84. [Google Scholar] [CrossRef]
- Mandala, VS; MacKinnon, R. The membrane electric field regulates the PIP2-binding site to gate the KCNQ1 channel. Proc Natl Acad Sci U S A 2023, 120(21), e2301985120. [Google Scholar] [CrossRef]
- Yue, Q; Al-Khalili, O; Moseley, A; Yoshigi, M; Wynne, BM; Ma, H; Eaton, DC. PIP2 interacts electrostatically with MARCKS-like Protein-1 and ENaC in renal epithelial cells. Biology (Basel) 2022, 11(12), 1694. [Google Scholar] [CrossRef]
- Yamaga, M; Martin, TFJ. PI(4,5)P2 is a master regulator for Ca2+-triggered vesicle exocytosis. Biochim Biophys Acta Mol Cell Biol Lipids 2025, 1870(6), 159651. [Google Scholar] [CrossRef]
- Gada, KD; Logothetis, DE. PKC regulation of ion channels: The involvement of PIP2. J Biol Chem 2022, 298(6), 102035. [Google Scholar] [CrossRef]
- Tariq, K; Luikart, BW. Striking a balance: PIP2 and PIP3 signaling in neuronal health and disease. Explor Neuroprotective Ther 2021, 1, 86–100. [Google Scholar] [CrossRef]
- Clarke, RJ. Electrostatic switch mechanisms of membrane protein trafficking and regulation. Biophys Rev 2023, 15(6), 1967–1985. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T; Legat, A; Adam, E; Steuve, J; Gatot, JS; Vandenbranden, M; et al. DiC14-amidine cationic liposomes stimulate myeloid dendritic cells through Toll-like receptor 4. Eur J Immunol. 2008, 38(5), 1351–7. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A; Foo, ASC; Lam, HY; Yap, KCH; Jacot, W; Jones, RH; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer 2023, 22(1), 138. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A; Atakpa-Adaji, P; Rao, S; Marti-Solano, M; Taylor, CW. Dual regulation of IP3 receptors by IP3 and PIP2 controls the transition from local to global Ca2+ signals. Mol Cell 2024, 84(20), 3997–4015.e7. [Google Scholar] [CrossRef]
- Putney, JW; Tomita, T. Phospholipase C signaling and calcium influx. Adv Biol Reg 2012, 52(1), 152–164. [Google Scholar] [CrossRef]
- Cao, S; Chung, S; Kim, S; Li, Z; Manor, D; Buck, M. K-Ras G-domain binding with signaling lipid phosphatidylinositol (4,5)-phosphate (PIP2): membrane association, protein orientation, and function. J Biol Chem 2019, 294(17), 7068–7084. [Google Scholar] [CrossRef]
- Janmey, PA; Bucki, R; Radhakrishnan, R. Regulation of actin assembly by PI(4,5)P2 and other inositol phospholipids: An update on possible mechanisms. Biochem Biophys Res Commun 2018, 506(2), 307–314. [Google Scholar] [CrossRef]
- Abbasi, R; Shineh, G; Mobaraki, M; Doughty, S; Tayebi, L. Structural parameters of nanoparticles affecting their toxicity for biomedical applications: a review. J Nanopart Res 2023, 25(3), 43. [Google Scholar] [CrossRef]
- Yuan, Z; Yan, R; Fu, Z; Wu, T; Ren, C. Impact of physicochemical properties on biological effects of lipid nanoparticles: Are they completely safe. Sci Total Environ 2024, 927, 172240. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J; Kiss, B; Bozó, T; Turjeman, K; Levi-Kalisman, Y; Barenholz, Y; et al. Insights into the structure of Comirnaty COVID-19 vaccine: A theory on soft, partially bilayer-covered nanoparticles with hydrogen bond-stabilized mRNA-lipid complexes. ACS Nano 2023, 17(14), 13147–13157. [Google Scholar] [CrossRef]
- Münter, R; Larsen, JB; Andresen, TL. The vast majority of nucleic acid-loaded lipid nanoparticles contain cargo. J Colloid Interface Sci 2024, 674, 139–144. [Google Scholar] [CrossRef]
- Li, S; Hu, Y; Li, A; Lin, J; Hsieh, K; Schneiderman, Z; et al. Payload distribution and capacity of mRNA lipid nanoparticles. Nat Commun 2022, 13(1), 5561. [Google Scholar] [CrossRef] [PubMed]
- https. [CrossRef]
- Chen, X; Ye, Y; Li, M; Zuo, T; Xie, Z; Ke, Y; et al. Structural characterization of mRNA lipid nanoparticles (LNPs) in the presence of mRNA-free LNPs. J Control Release 2025, 386, 114082. [Google Scholar] [CrossRef]
- Müller, JA; Schäffler, N; Kellerer, T; Schwake, G; Ligon, TS; Rädler, JO. Kinetics of RNA-LNP delivery and protein expression. Eur J Pharm Biopharm 2024, 197, 114222. [Google Scholar] [CrossRef] [PubMed]
- Liao, S; Wang, S; Wadhwa, A; Birkenshaw, A; Fox, K; Cheng, MHY; et al. Transfection potency of lipid nanoparticles containing mRNA depends on relative loading levels. ACS Appl Mater Interfaces 2025, 17(2), 3097–3105. [Google Scholar] [CrossRef]
- Grumelot, S; Mohammed, N; Colonrosado, J; Sadeghi, SA; Fang, F; Hilsen, K. Lipid nanoparticle protein coronas form via lipoprotein fusion rather than shell-like adsorption. bioRxiv Preprint 2025. [CrossRef]
- Rampado, R; Crotti, S; Caliceti, P; Pucciarelli, S; Agostini, M. Recent advances in understanding the protein corona of nanoparticles and in the formulation of "stealthy" nanomaterials. Front Bioeng Biotechnol 2020, 8, 166. [Google Scholar] [CrossRef]
- Sun, Y; Zhou, Y; Rehman, M; Wang, YF; Guo, S. Protein corona of nanoparticles: Isolation and analysis. Chem Bio Eng. 2024, 1(9), 757–772. [Google Scholar] [CrossRef] [PubMed]
- Voke, E; Arral, ML; Squire, HJ; Lin, TJ; Zheng, L; Coreas, R; et al. Protein corona formed on lipid nanoparticles compromises delivery efficiency of mRNA cargo. Nat Commun 2025, 16(1), 8699. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F; Yanez Arteta, M; Lerche, M; Porcar, L; Lang, C; Bragg, RA; Elmore, CS; et al. Apolipoprotein E binding drives structural and compositional rearrangement of mRNA-containing lipid nanoparticles. ACS Nano 2021, 15(4), 6709–6722. [Google Scholar] [CrossRef]
- Kim, W; Ly, NK; He, Y; Li, Y; Yuan, Z; Yeo, Y. Protein corona: Friend or foe? Co-opting serum proteins for nanoparticle delivery. Adv Drug Deliv Rev 2023, 192, 114635. [Google Scholar] [CrossRef]
- Paunovska, K; Da Silva Sanchez, AJ; Lokugamage, MP; Loughrey, D; Echeverri, ES; Cristian, A; et al. The extent to which lipid nanoparticles require apolipoprotein E and low-density lipoprotein receptor for delivery changes with ionizable lipid structure. Nano Lett 2022, 22(24), 10025–10033. [Google Scholar] [CrossRef]
- Naasani, I. Establishing the pharmacokinetics of genetic vaccines is essential for maximising their safety and efficacy. Clin Pharmacokinet 2022, 61(7), 921–927. [Google Scholar] [CrossRef]
- Hosseini-Kharat, M; Bremmell, KE; Prestidge, CA. Why do lipid nanoparticles target the liver? Understanding of biodistribution and liver-specific tropism. Mol Ther Methods Clin Dev 2025, 33(1), 101436. [Google Scholar] [CrossRef]
- European Medicines Agency. Assessment report: Comirnaty. EMA/707383/2020 Corr.2*1,2. February 19, 2021. 2020. [Google Scholar]
- Available online: https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf.
- Therapeutic Goods Administration. Nonclinical Evaluation Report BNT162b2 [mRNA] COVID-19 vaccine (COMIRNATY); Health, Ed.; Department of Health and Aged Care, 2021; Vol. FOI 2389, Available online: https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf.
- Neves, AR; Queiroz, JF; Costa Lima, SA; Figueiredo, F; Fernandes, R; Reis, S. Cellular uptake and transcytosis of lipid-based nanoparticles across the intestinal barrier: Relevance for oral drug delivery. J Colloid Interface Sci 2016, 463, 258–65. [Google Scholar] [CrossRef] [PubMed]
- Khare, P; Edgecomb, SX; Hamadani, CM; Tanner, EEL; S Manickam, D. Lipid nanoparticle-mediated drug delivery to the brain. Adv Drug Deliv Rev 2023, 197, 114861. [Google Scholar] [CrossRef]
- Chen, J; Xu, Y; Zhou, M; Xu, S; Varley, AJ; Golubovic, A; et al. Combinatorial design of ionizable lipid nanoparticles for muscle-selective mRNA delivery with minimized off-target effects. Proc Natl Acad Sci U S A 2023, 120(50), e2309472120. [Google Scholar] [CrossRef]
- Younis, MA; Sato, Y; Elewa, YHA; Kon, Y; Harashima, H. Self-homing nanocarriers for mRNA delivery to the activated hepatic stellate cells in liver fibrosis. J Control Release 2023, 353, 685–698. [Google Scholar] [CrossRef]
- Kent, SJ; Li, S; Amarasena, TH; Reynaldi, A; Lee, WS; Leeming, MG; et al. Blood distribution of SARS-CoV-2 lipid nanoparticle mRNA vaccine in humans. ACS Nano 2024, 18(39), 27077–27089. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M; Araínga, M; Maiorino, L; Pires, IS; Kim, BJ; Michaels, KK; et al. Visualizing lipid nanoparticle trafficking for mRNA vaccine delivery in non-human primates. Mol Ther 2025, 33(3), 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Balcorta, HV; Mata Corral, MY; Gallegos, A; Chavez, J; Perez, J; Balivada, S; et al. Development of chemical tags for universal lipid nanoparticle visualization and tracking in 2D and 3D imaging. Nano Lett 2025, 25(19), 7682–7689. [Google Scholar] [CrossRef]
- Akhter, MH; Khalilullah, H; Gupta, M; Alfaleh, MA; Alhakamy, NA; Riadi, Y; et al. Impact of protein corona on the biological identity of nanomedicine: Understanding the fate of nanomaterials in the biological milieu. Biomedicines 2021, 9(10), 1496. [Google Scholar] [CrossRef]
- Behzadi, S; Serpooshan, V; Tao, W; Hamaly, MA; Alkawareek, MY; Dreaden, EC; et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev 2017, 46(14), 4218–4244. [Google Scholar] [CrossRef]
- Lavington, S; Watts, A. Lipid nanoparticle technologies for the study of G protein-coupled receptors in lipid environments. Biophys Rev 2020, 12(6), 1287–302. [Google Scholar] [CrossRef]
- Chatterjee, S; Kon, E; Sharma, P; Peer, D. Endosomal escape: A bottleneck for LNP-mediated therapeutics. Proc Natl Acad Sci U S A 2024, 121(11), e2307800120. [Google Scholar] [CrossRef]
- Vermeulen, LMP; Brans, T; Samal, SK; Dubruel, P; Demeester, J; De Smedt, SC; et al. Endosomal size and membrane leakiness influence proton sponge-based rupture of endosomal vesicles. ACS Nano 2018, 12(3), 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Johansson, JM; Du Rietz, H; Hedlund, H; Eriksson, HC; Oude Blenke, E; Pote, A; et al. Cellular and biophysical barriers to lipid nanoparticle mediated delivery of RNA to the cytosol. Nat Commun 2025, 16(1), 5354. [Google Scholar] [CrossRef] [PubMed]
- Er-Rafik, M; Ferji, K; Combet, J; Sandre, O; Lecommandoux, S; Schmutz, M; et al. Tear of lipid membranes by nanoparticles. Soft Matter 2022, 18(17), 3318–3322. [Google Scholar] [CrossRef]
- Pilkington, EH; Suys, EJA; Trevaskis, NL; Wheatley, AK; Zukancic, D; Algarni, A; et al. From influenza to COVID-19: Lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater 2021, 131, 16–40. [Google Scholar] [CrossRef]
- Dowdy, SF. Endosomal escape of RNA therapeutics: How do we solve this rate-limiting problem? RNA 2023, 29(4), 396–401. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, S; Kumarasinghe, ES; Salerno, T; Mihai, C; Ketova, T; Senn, JJ; et al. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther 2018, 26(6), 1509–1519. [Google Scholar] [CrossRef]
- Maugeri, M; Nawaz, M; Papadimitriou, A; Angerfors, A; Camponeschi, A; Na, M; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat Commun 2019, 10, 4333. [Google Scholar] [CrossRef]
- Gilleron, J; Querbes, W; Zeigerer, A; Borodovsky, A; Marsico, G; Schubert, U; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol 2013, 31(7), 638–46. [Google Scholar] [CrossRef]
- Aliakbarinodehi, N; Niederkofler, S; Emilsson, G; Parkkila, P; Olsén, E; Jing, Y; et al. Time-resolved inspection of ionizable lipid-facilitated lipid nanoparticle disintegration and cargo release at an early endosomal membrane mimic. ACS Nano 2024, 18(34), 22989–23000. [Google Scholar] [CrossRef]
- Schlich, M; Palomba, R; Costabile, G; Mizrahy, S; Pannuzzo, M; Peer, D; et al. Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles. Bioeng Transl Med 2021, 6(2), e10213. [Google Scholar] [CrossRef]
- Paramasivam, P; Franke, C; Stöter, M; Höijer, A; Bartesaghi, S; Sabirsh, A; et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J Cell Biol 2022, 221(2), e202110137. [Google Scholar] [CrossRef]
- Sahay, G; Querbes, W; Alabi, C; Eltoukhy, A; Sarkar, S; Zurenko, C; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol 2013, 31(7), 653–8. [Google Scholar] [CrossRef]
- Bitounis, D; Jacquinet, E; Rogers, MA; Amiji, MM. Strategies to reduce the risks of mRNA drug and vaccine toxicity. Nat Rev Drug Discov. 2024, 23(4), 281–300. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, SM; Simberg, D. Pro-inflammatory concerns with lipid nanoparticles. Mol Ther 2022, 30(6), 2109–2110. [Google Scholar] [CrossRef]
- Bates, SM; Munson, MJ; Trovisco, V; Pereira, S; Miller, SR; Sabirsh, A; et al. The kinetics of endosomal disruption reveal differences in lipid nanoparticle induced cellular toxicity. J Control Release 2025, 386, 114047. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L; Bandara, SR; Tan, Z; Leal, C. Lipid nanoparticle topology regulates endosomal escape and delivery of RNA to the cytoplasm. Proc Natl Acad Sci U S A 2023, 120(27), e2301067120. [Google Scholar] [CrossRef] [PubMed]
- Lu, ZR; Sun, D. Mechanism of pH-sensitive amphiphilic endosomal escape of ionizable lipid nanoparticles for cytosolic nucleic acid delivery. Pharm Res 2025l, 42(7), 1065–1077. [Google Scholar] [CrossRef]
- Su, J; Song, Y; Zhu, Z; Huang, X; Fan, J; Qiao, J; et al. Cell-cell communication: new insights and clinical implications. Signal Transduct Target Ther 2024, 9(1), 196. [Google Scholar] [CrossRef]
- Pateev, I; Seregina, K; Ivanov, R; Reshetnikov, V. Biodistribution of RNA vaccines and of their products: Evidence from human and animal studies. Biomedicines 2023, 12(1), 59. [Google Scholar] [CrossRef]
- Fertig, TE; Chitoiu, L; Marta, DS; Ionescu, VS; Cismasiu, VB; Radu, E; et al. Vaccine mRNA can be detected in blood at 15 days post-vaccination. Biomedicines 2022, 10(7), 1538. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U; Karikó, K; Türeci, Ö. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov 2014, 13(10), 759–80. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y; Zhao, E; Yang, X; Luo, C; Zi, G; Wang, R; et al. Entrapment of lipid nanoparticles in peripheral endosomes but not lysosomes impairs intracellular trafficking and endosomal escape. Int J Pharm 2025, 669, 125024. [Google Scholar] [CrossRef]
- Omo-Lamai, S; Wang, Y; Patel, MN; Milosavljevic, A; Zuschlag, D; Poddar, S; et al. Limiting endosomal damage sensing reduces inflammation triggered by lipid nanoparticle endosomal escape. Nat Nanotechnol. 2025, 20(9), 1285–1297. [Google Scholar] [CrossRef]
- Bansal, S; Perincheri, S; Fleming, T; Poulson, C; Tiffany, B; Bremner, RM; et al. Cutting edge: Circulating exosomes with COVID spike protein are induced by BNT162b2 (Pfizer-BioNTech) vaccination prior to development of antibodies: A novel mechanism for immune activation by mRNA vaccines. J Immunol 2021, 207(10), 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Jörgensen, AM; Wibel, R; Bernkop-Schnürch, A. Biodegradable cationic and ionizable cationic lipids: A roadmap for safer pharmaceutical excipients. Small 2023, 19(17), e2206968. [Google Scholar] [CrossRef]
- Knaggs, KLM; Sun, Y; Walz, BA; Pang, J; Khan, OF. The role of excipients in lipid nanoparticle metabolism: implications for enhanced therapeutic effect. Ther Deliv 2025, 16(7), 687–700. [Google Scholar] [CrossRef]
- Wang, W; Deng, S; Lin, J; Ouyang, D. Modeling on in vivo disposition and cellular transportation of RNA lipid nanoparticles via quantum mechanics/physiologically-based pharmacokinetic approaches. Acta Pharm Sin B 2024, 14(10), 4591–4607. [Google Scholar] [CrossRef]
- Yu, Z; Li, Q; Wang, J; Yu, Y; Wang, Y; Zhou, Q; et al. Reactive oxygen species-related nanoparticle toxicity in the biomedical field. Nanoscale Res Lett 2020, 15(1), 115. [Google Scholar] [CrossRef] [PubMed]
- Packer, M; Gyawali, D; Yerabolu, R; Schariter, J; White, P. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat Commun 2021, 12(1), 6777. [Google Scholar] [CrossRef]
- Hashiba, K; Taguchi, M; Sakamoto, S; Otsu, A; Maeda, Y; Ebe, H; et al. Overcoming thermostability challenges in mRNA-lipid nanoparticle systems with piperidine-based ionizable lipids. Commun Biol. 2024, 7(1), 556. [Google Scholar] [CrossRef]
- Cordes, J; Zhao, S; Engel, CM; Stingele, J. Cellular responses to RNA damage. Cell 2025, 188(4), 885–900. [Google Scholar] [CrossRef]
- Maelfait, J; Liverpool, L; Rehwinkel, J. Nucleic acid sensors and programmed cell death. J Mol Biol 2020, 432(2), 552–568. [Google Scholar] [CrossRef]
- Li, Y; Zhao, T; Li, J; Xia, M; Li, Y; Wang, X; et al. Oxidative stress and 4-hydroxy-2-nonenal (4-HNE): Implications in the pathogenesis and treatment of aging-related diseases. J Immunol Res. 2022, 2022, 2233906. [Google Scholar] [CrossRef]
- Zhou, ZH; Stone, CA, Jr.; Jakubovic, B; Phillips, EJ; Sussman, G; Park, J; et al. Anti-PEG IgE in anaphylaxis associated with polyethylene glycol. J Allergy Clin Immunol Pract 2021, 9(4), 1731–1733.e3. [Google Scholar] [CrossRef] [PubMed]
- Kozma, GT; Shimizu, T; Ishida, T; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev 2020, 154-155, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Fu, S; Zhu, X; Huang, F; Chen, X. Anti-PEG antibodies and their biological impact on PEGylated drugs: Challenges and strategies for optimization. Pharmaceutics 2025, 17(8), 1074. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology 2005, 216(2-3), 106–21. [Google Scholar] [CrossRef]
- Szebeni, J; Simberg, D; González-Fernández, Á; Barenholz, Y; Dobrovolskaia, MA. Roadmap and strategy for overcoming infusion reactions to nanomedicines. Nat Nanotechnol 2018, 13(12), 1100–1108. [Google Scholar] [CrossRef]
- Dézsi, L; Mészáros, T; Kozma, G; H-Velkei, M; Oláh, CZ; Szabó, M; et al. A naturally hypersensitive porcine model may help understand the mechanism of COVID-19 mRNA vaccine-induced rare (pseudo) allergic reactions: complement activation as a possible contributing factor. Geroscience 2022, 44(2), 597–618. [Google Scholar] [CrossRef] [PubMed]
- https. [CrossRef]
- Bakos, T; Mészáros, T; Kozma, GT; Berényi, P; Facskó, R; Farkas, H; et al. mRNA-LNP COVID-19 vaccine lipids induce complement activation and production of proinflammatory cytokines: Mechanisms, effects of complement inhibitors, and relevance to adverse reactions. Int J Mol Sci 2024, 25(7), 3595. [Google Scholar] [CrossRef]
- Song, J; Su, D; Wu, H; Guo, J. Implications of anaphylaxis following mRNA-LNP vaccines: It is urgent to eliminate PEG and find alternatives. Pharmaceutics 2025, 17(6), 798. [Google Scholar] [CrossRef]
- European Medicines Agency. Onpattro: European Public Assessment Report (EPAR) EMA/554262/2018; CHMP, Ed.; Amsterdam, NL, 2018. [Google Scholar]
- Wang, J; Ding, Y; Chong, K; Cui, M; Cao, Z; Tang, C; et al. Recent advances in lipid nanoparticles and their safety concerns for mRNA delivery. Vaccines (Basel) 2024, 12(10), 1148. [Google Scholar] [CrossRef]
- Dai, W; Xing, M; Sun, L; Lv, L; Wang, X; Wang, Y; et al. Lipid nanoparticles as adjuvant of norovirus VLP vaccine augment cellular and humoral immune responses in a TLR9- and type I IFN-dependent pathway. J Virology 2024, 98(12), e0169924. [Google Scholar] [CrossRef] [PubMed]
- https. [CrossRef]
- Chaudhary, N; Kasiewicz, LN; Newby, AN; Arral, ML; Yerneni, SS; Melamed, JR; et al. Amine headgroups in ionizable lipids drive immune responses to lipid nanoparticles by binding to the receptors TLR4 and CD1d. Nat Biomed Eng 2024, 8(11), 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Di Fusco, SA; Maggioni, AP; Bernelli, C; Perone, F; De Marzo, V; Conte, E; et al. Inclisiran: A new pharmacological approach for hypercholesterolemia. Rev Cardiovasc Med 2022, 23(11), 375. [Google Scholar] [CrossRef]
- Wright, RS; Koenig, W; Landmesser, U; Leiter, LA; Raal, FJ; Schwartz, GG; et al. Safety and tolerability of Inclisiran for treatment of hypercholesterolemia in 7 clinical trials. J Am Coll Cardiol 2023, 82(24), 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Urits, I; Swanson, D; Swett, MC; Patel, A; Berardino, K; Amgalan, A; et al. A Review of patisiran (ONPATTRO®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020, 9(2), 301–315. [Google Scholar] [CrossRef] [PubMed]
- Thi, TTH; Suys, EJA; Lee, JS; Nguyen, DH; Park, KD; Truong, NP. Lipid-based nanoparticles in the clinic and clinical trials: From cancer nanomedicine to COVID-19 vaccines. Vaccines (Basel) 2021, 9(4), 359. [Google Scholar] [CrossRef]
- European Medicines Agency. Kostaive (zapomeran). Public Assessment report Sections 2.4.3 (Pharmacokinetics) and 2.4.6 (Discussion on non-clinical aspects). 12 December 2024. Available online: https://www.ema.europa.eu/en/documents/assessment-report/kostaive-epar-public-assessment-report_en.pdf.
- Committee for Medicinal Products for Human Use (CHMP). Covid-19 Vaccine Moderna European Public Assessment Report (EPAR). Procedure No. EMEA/H/C/005791/0000. European Medicines Agency. 11 March 2021. Available online: https://www.ema.europa.eu/en/documents/assessment-report/spikevax-previously-covid-19-vaccine-moderna-epar-public-assessment-report_en.pdf.
- Wong, HL; Hu, M; Zhou, CK; Lloyd, PC; Amend, KL; Beachler, DC; et al. Risk of myocarditis and pericarditis after the COVID-19 mRNA vaccination in the USA: a cohort study in claims databases. Lancet 2022, 399(10342), 2191–2199. [Google Scholar] [CrossRef]
- Avraham, R; Haseley, N; Brown, D; Penaranda, C; Jijon, HB; Trombetta, JJ; et al. Pathogen cell-to-cell variability drives heterogeneity in host immune responses. Cell Erratum in: Cell 2015;163(2):523. 2015, 162(6), 1309–21. [Google Scholar] [CrossRef] [PubMed]
- Ndeupen, S; Qin, Z; Jacobsen, S; Bouteau, A; Estanbouli, H; Igyártó, BZ. The mRNA-LNP platform's lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience 2021, 24(12), 103479. [Google Scholar] [CrossRef]
- Finney, DJ. The median lethal dose and its estimation. Arch Toxicol. 1985, 56(4), 215–8. [Google Scholar] [CrossRef] [PubMed]
- Avila, AM; Bebenek, I; Bonzo, JA; Bourcier, T; Davis Bruno, KL; Carlson, DB; et al. An FDA/CDER perspective on nonclinical testing strategies: Classical toxicology approaches and new approach methodologies (NAMs). Regul Toxicol Pharmacol 2020, 114, 104662. [Google Scholar] [CrossRef]
- Luster, AD. The role of chemokines in linking innate and adaptive immunity. Curr Opin Immunol. 2002, 14(1), 129–135. [Google Scholar] [CrossRef]
- Esche, C; Stellato, C; Beck, LA. Chemokines: key players in innate and adaptive immunity. J Invest Dermatol 2005, 125(4), 615–628. [Google Scholar] [CrossRef]
- Tao, T; Jiang, G; Su, Y; He, D; Zhu, L; Jiang, Q; Su, W. The multiple roles of GM-CSF in autoimmune and autoinflammatory uveitis. Biochem Pharmacol. 2025, 240, 117090. [Google Scholar] [CrossRef]
- Rincón, M; Anguita, J; Nakamura, T; Fikrig, E; Flavell, RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exper Med. 1997, 185(3), 461–469. [Google Scholar] [CrossRef]
- de Jong, AJ; Pollastro, S; Kwekkeboom, JC; Andersen, SN; Dorjée, AL; Bakker, AM; et al. Functional and phenotypical analysis of IL-6-secreting CD4+ T cells in human adipose tissue. Eur J Immunol. 2018, 48(3), 471–481. [Google Scholar] [CrossRef]
- Jeannin, P; Jaillon, S; Delneste, Y. Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol 2008, 20(5), 530–537. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y; Garrett, KP; Ohta, S; Bahrun, U; Kouro, T; Akira, S; et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24(6), 801–812. [Google Scholar] [CrossRef] [PubMed]
- Fritz, JH. Nod-like receptors have a grip on stem cells. Cell Host Microbe 2014, 15(6), 659–661. [Google Scholar] [CrossRef]
- Ji, Y; Kumar, R; Gokhale, A; Chao, HP; Rycaj, K; Chen, X; Li, Q; Tang, DG. LRIG1, a regulator of stem cell quiescence and a pleiotropic feedback tumor suppressor. Semin Cancer Biol 2022, 82, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H; Brenner, JS; Patel, PN; Papp, TE; Shahnawaz, H; Li, Q; et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J Control Release 2022, 344, 50–61. [Google Scholar] [CrossRef]
- Zelkoski, AE; Lu, Z; Sukumar, G; Dalgard, C; Said, H; Alameh, MG; et al. Ionizable lipid nanoparticles of mRNA vaccines elicit NF-κB and IRF responses through toll-like receptor 4. NPJ Vaccines 2025, 10(1), 73. [Google Scholar] [CrossRef]
- Raftery, N; Stevenson, NJ. Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway. Cell Mol Life Sci 2017, 74(14), 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Korzun, T; Moses, AS; Jozic, A; Grigoriev, V; Newton, S; Kim, J; et al. Lipid nanoparticles elicit reactogenicity and sickness behavior in mice via toll-like receptor 4 and myeloid differentiation protein 88 axis. ACS Nano 2024, 18(36), 24842–24859. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, MC; Er, EE; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011, 36(6), 320–8. [Google Scholar] [CrossRef]
- Soriano, O; Alcón-Pérez, M; Vicente-Manzanares, M; Castellano, E. The crossroads between RAS and RHO signaling pathways in cellular transformation, motility and contraction. Genes (Basel) 2021, 12(6), 819. [Google Scholar] [CrossRef] [PubMed]
- Luo, J; Molbay, M; Chen, Y; Horvath, I; Kadletz, K; Kick, B; et al. Nanocarrier imaging at single-cell resolution across entire mouse bodies with deep learning. Nat Biotechnol 2025, 43, 2009–2022. [Google Scholar] [CrossRef]
- Knabl, L; Lee, HK; Wieser, M; Mur, A; Zabernigg, A; Knabl, L, Sr.; et al. BNT162b2 vaccination enhances interferon-JAK-STAT-regulated antiviral programs in COVID-19 patients infected with the SARS-CoV-2 Beta variant. Commun Med (Lond) 2022, 2(1), 17. [Google Scholar] [CrossRef]
- Hickey, TE; Mudunuri, U; Hempel, HA; Kemp, TJ; Roche, NV; Talsania, K; et al. Proteomic and serologic assessments of responses to mRNA-1273 and BNT162b2 vaccines in human recipient sera. Front Immunol 2025, 15, 1502458. [Google Scholar] [CrossRef]
- Panwar, V; Singh, A; Bhatt, M; Tonk, RK; Azizov, S; Raza, AS; et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther 2023, 8(1), 375. [Google Scholar] [CrossRef]
- Yu, H; Lin, L; Zhang, Z; Zhang, H; Hu, H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther 2020, 5(1), 209. [Google Scholar] [CrossRef]
- Guo, Q; Jin, Y; Chen, X; Ye, X; Shen, X; Lin, M; et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther 2024, 9(1), 53. [Google Scholar] [CrossRef]
- Bassermann, F; Eichner, R; Pagano, M. The ubiquitin proteasome system - implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta 2014, 1843(1), 150–62. [Google Scholar] [CrossRef]
- Yasir, M; Goyal, A; Sonthalia, S. Corticosteroid Adverse Effects. In StatPearls [Internet]; StatPearls Publishing: Treasure Island (FL), 3 Jul 2023; Available online: https://www.ncbi.nlm.nih.gov/books/NBK531462/.
- Gulluni, F; Martini, M; Hirsch, E. Cytokinetic Abscission: Phosphoinositides and ESCRTs direct the final cut. J Cell Biochem Erratum in: J Cell Biochem 2024;125(8):e30626. https://doi.org/10.1002/jcb.30626. 2017, 118(11), 3561–3568. [Google Scholar] [CrossRef]
- Tan, X; Thapa, N; Choi, S; Anderson, RA. Emerging roles of PtdIns(4,5)P2 -- beyond the plasma membrane. J Cell Sci 2015, 128(22), 4047–56. [Google Scholar] [CrossRef]
- Ajazi, A; Bruhn, C; Shubassi, G; Lucca, C; Ferrari, E; Cattaneo, A; et al. Endosomal trafficking and DNA damage checkpoint kinases dictate survival to replication stress by regulating amino acid uptake and protein synthesis. Dev Cell 2021, 56(18), 2607–2622.e6. [Google Scholar] [CrossRef] [PubMed]
- Giannini, C; Ponzone, L; Barroero, N; Hirsch, E. The interplay between phosphoinositides and ESCRT proteins. Adv Biol Regul 2025, 9, 101126. [Google Scholar] [CrossRef]
- Forster, III J; Nandi, D; Kulkarni, A. mRNA-carrying lipid nanoparticles that induce lysosomal rupture activate NLRP3 inflammasome and reduce mRNA transfection efficiency. Biomater Sci 2022, 10(19), 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- Hurley, JH; Coyne, AN; Miączyńska, M; Stenmark, H. The expanding repertoire of ESCRT functions in cell biology and disease. Nature 2025, 642, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Baylon, JL; Lenov, IL; Sligar, SG; Tajkhorshid, E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013, 135(23), 8542–51. [Google Scholar] [CrossRef]
- Hackett, JC. Membrane-embedded substrate recognition by cytochrome P450 3A4. J Biol Chem 2018, 293(11), 403704046. [Google Scholar] [CrossRef]
- Mukherjee, G; Nandekar, PP; Wade, RC. An electron transfer competent structural ensemble of membrane-bound cytochrome P450 1A1 and cytochrome P450 oxidoreductase. Commun Biol. 2021, 4(1), 55. [Google Scholar] [CrossRef]
- Liu, F; Aulin, LBS; Manson, ML; Krekels, EHJ; van Hasselt, JGC. Unraveling the effects of acute inflammation on pharmacokinetics: A model-based analysis focusing on renal glomerular filtration rate and cytochrome P450 3A4-mediated metabolism. Eur J Drug Metab Pharmacokinet 2023, 48(6), 623–631. [Google Scholar] [CrossRef] [PubMed]
- Stanke-Labesque, F; Gautier-Veyret, E; Chhun, S; Guilhaumou, R; French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol Ther 2020, 215, 107627. [Google Scholar] [CrossRef] [PubMed]
- Koozi, H; Lengquist, M; Frigyesi, A. C-reactive protein as a prognostic factor in intensive care admissions for sepsis: A Swedish multicenter study. J Crit Care 2020, 56, 73–79. [Google Scholar] [CrossRef]
- Bayraktar, İ; Yalçın, N; Demirkan, K. The potential interaction between COVID-19 vaccines and clozapine: A novel approach for clinical trials. Int J Clin Pract 2021, 75(8), e14441. [Google Scholar] [CrossRef]
- Imai, T; Ochiai, S; Ishimaru, T; Daitoku, H; Miyagawa, Y; Fukuhara, R; et al. A case report: Clozapine-induced leukopenia and neutropenia after mRNA COVID-19 vaccination. Neuropsychopharmacol Rep 2022, 42(2), 238–240. [Google Scholar] [CrossRef]
- Thompson, D; Delorme, CM; White, RF; Honer, WG. Elevated clozapine levels and toxic effects after SARS-CoV-2 vaccination. J Psychiatry Neurosci 2021, 46(2), E210–E211. [Google Scholar] [CrossRef]
- Eiermann, B; Engel, G; Johansson, I; Zanger, UM; Bertilsson, L. The involvement of CYP1A2 and CYP3A4 in the metabolism of clozapine. Br J Clin Pharmacol 1997, 44(5), 439–46. [Google Scholar] [CrossRef]
- Villemure, S; Trenaman, SC; Goralski, KB. The impact of COVID-19 infection on cytochrome P450 3A4-mediated drug metabolism and drug interactions. Expert Opin Drug Metab Toxicol 2023, 19(6), 329–332. [Google Scholar] [CrossRef]
- Lim, SYM; Al Bishtawi, B; Lim, W. Role of cytochrome P450 2C9 in COVID-19 treatment: Current status and future directions. Eur J Drug Metab Pharmacokinet 2023, 48(3), 221–240. [Google Scholar] [CrossRef]
- Hossam Abdelmonem, B; Abdelaal, NM; Anwer, EKE; Rashwan, AA; Hussein, MA; Ahmed, YF; et al. Decoding the role of CYP450 enzymes in metabolism and disease: A comprehensive review. Biomedicines 2024, 12(7), 1467. [Google Scholar] [CrossRef] [PubMed]
- Akanchise, T; Luo, F; Angelov, B; Deng, Y; Manna, G; Angelova, A. Rapid structural transformation of ionizable lipid nanoparticles involving Omega-3 polyunsaturated fatty acids enhances antioxidant defense and mitochondrial proteins activity in pH-responsive drug delivery. J Colloid Interface Sci. 2026, 704 Pt 2, 139420. [Google Scholar] [CrossRef]
- Ruysschaert, JM; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim Biophys Acta 2015, 1848(9), 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Chaluvadi, MR; Nyagode, BA; Kinloch, RD; Morgan, ET. TLR4-dependent and -independent regulation of hepatic cytochrome P450 in mice with chemically induced inflammatory bowel disease. Biochem Pharmacol. 2009, 77(3), 464–471. [Google Scholar] [CrossRef]
- Li, Z; Carter, J; Santos, L; Webster, C; van der Walle, CF; Li, P; Rogers, SE; Lu, JR. Acidification-induced structure evolution of lipid nanoparticles correlates with their in vitro gene transfections. ACS Nano 2023, 17(2), 979–90. [Google Scholar] [CrossRef]
- Kim, KH; Ahn, T; Yun, CH. Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4. Biochemistry 2003, 42(51), 15377–15387. [Google Scholar] [CrossRef]
- Mustafa, G; Nandekar, PP; Mukherjee, G; Bruce, NJ; Wade, RC. The Effect of Force-Field Parameters on Cytochrome P450-Membrane Interactions: Structure and Dynamics. Sci Rep. 2020, 10(1), 7284. [Google Scholar] [CrossRef]
- Asady, B; Sampels, V; Romano, JD; et al. Function and regulation of a steroidogenic CYP450 enzyme in the mitochondrion of Toxoplasma gondii. PLoS Pathog 2023, 19(8), e1011566. [Google Scholar] [CrossRef]
- Huang, Z; Xu, X; Zhu, G. Association between coronavirus disease 2019 vaccination and hypercholesterolemia: A cross-sectional study from the 2023 National Health Interview Survey. Medicine (Baltimore) 2025, 104(48), e46221. [Google Scholar] [CrossRef] [PubMed]
- Luo, L; Wall, AA; Yeo, JC; Condon, ND; Norwood, SJ; Schoenwaelder, S; et al. Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun. 2014, 5, 4407. [Google Scholar] [CrossRef]
- Płóciennikowska, A; Hromada-Judycka, A; Dembińska, J; Roszczenko, P; Ciesielska, A; Kwiatkowska, K. Contribution of CD14 and TLR4 to changes of the PI(4,5)P2 level in LPS-stimulated cells. J Leukoc Biol. 2016, 100(6), 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- López-Haber, C; Levin-Konigsberg, R; Zhu, Y; Bi-Karchin, J; Balla, T; Grinstein, S; et al. Phosphatidylinositol-4-kinase IIα licenses phagosomes for TLR4 signaling and MHC-II presentation in dendritic cells. Proc Natl Acad Sci U S A 2020, 117(45), 28251–28262. [Google Scholar] [CrossRef]
- Quan, Y; Yang, H; Li, W; Li, L. mRNA vaccines: immunogenicity and quality characteristics. J Nanobiotechnology 2025, 24(1), 6. [Google Scholar] [CrossRef]
- Shi, Y; Ruan, H; Xu, Y; Zou, C. Cholesterol, eukaryotic lipid domains, and an evolutionary perspective of transmembrane signaling. Cold Spring Harb Perspect Biol 2023, 15(11), a041418. [Google Scholar] [CrossRef]
- Barnett, KC; Kagan, JC. Lipids that directly regulate innate immune signal transduction. Innate Immunity 2019, 26(1), 4–14. [Google Scholar] [CrossRef]
- Zheng, Y; Sun, L; Jiang, T; Zhang, D; He, D; Nie, H. TNFα promotes Th17 cell differentiation through IL-6 and IL-1β produced by monocytes in rheumatoid arthritis. J Immunol Res 2014, 2014, 385352. [Google Scholar] [CrossRef]
- Marasco, MR; Conteh, AM; Reissaus, CA; Cupit, JE, 5th; Appleman, EM; Mirmira, RG; et al. Interleukin-6 reduces β-cell oxidative stress by linking autophagy with the antioxidant response. Diabetes 2018, 67(8), 1576–1588. [Google Scholar] [CrossRef]
- Deretic, V; Saitoh, T; Akira, S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013, 13(10), 722–37. [Google Scholar] [CrossRef]
- Chi, DS; Fitzgerald, SM; Pitts, S; Cantor, K; King, E; Lee, SA; et al. MAPK-dependent regulation of IL-1- and beta-adrenoreceptor-induced inflammatory cytokine production from mast cells: implications for the stress response. BMC Immunol 2004, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- https. [CrossRef]
- Yang, HT; Cohen, P; Rousseau, S. IL-1beta-stimulated activation of ERK1/2 and p38alpha MAPK mediates the transcriptional up-regulation of IL-6, IL-8 and GRO-alpha in HeLa cells. Cell Signal 2008, 20(2), 375–80. [Google Scholar] [CrossRef] [PubMed]
- Tengesdal, IW; Dinarello, A; Powers, NE; Burchill, MA; Joosten, LAB; Marchetti, C; et al. Tumor NLRP3-derived IL-1β drives the IL-6/STAT3 axis resulting in sustained MDSC-mediated immunosuppression. Front Immunol 2021, 12, 661323. [Google Scholar] [CrossRef] [PubMed]
- Li, H; Wu, M; Zhao, X. Role of chemokine systems in cancer and inflammatory diseases. MedComm (2020) 2022, 3(2), e147. [Google Scholar] [CrossRef] [PubMed]
- Unver, N. Macrophage chemoattractants secreted by cancer cells: Sculptors of the tumor microenvironment and another crucial piece of the cancer secretome as a therapeutic target. Cytokine Growth Factor Rev 2019, 50, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Nakatsumi, H; Matsumoto, M; Nakayama, KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1-FOXK1 axis promotes recruitment of tumor-associated macrophages. Cell Rep 2017, 21(9), 2471–2486. [Google Scholar] [CrossRef]
- Huda, N; Khambu, B; Liu, G; Nakatsumi, H; Yan, S; Chen, X; et al. Senescence connects autophagy deficiency to inflammation and tumor progression in the liver. Cell Mol Gastroenterol Hepatol 2022, 14(2), 333–355. [Google Scholar] [CrossRef]
- Guo, L; Li, N; Yang, Z; Li, H; Zheng, H; Yang, J; et al. Role of CXCL5 in regulating chemotaxis of innate and adaptive leukocytes in infected lungs upon pulmonary influenza infection. Front Immunol 2021, 12, 785457. [Google Scholar] [CrossRef]
- Jastrzebska, B; Debinski, A; Filipek, S; Palczewski, K. Role of membrane integrity on G protein-coupled receptors: Rhodopsin stability and function. Prog Lipid Res. 2011, 50(3), 267–77. [Google Scholar] [CrossRef]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem Phys Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef]
- Prasanna, X; Sengupta, D; Chattopadhyay, A. Cholesterol-dependent conformational plasticity in GPCR dimers. Sci Rep 2016, 6, 31858. [Google Scholar] [CrossRef]
- Prasanna, X; Mohole, M; Chattopadhyay, A; Sengupta, D. Role of cholesterol-mediated effects in GPCR heterodimers. Chem Phys Lipids 2020, 227, 104852. [Google Scholar] [CrossRef]
- Kumar, GA; Chattopadhyay, A. Cholesterol-dependent endocytosis of GPCRs: implications in pathophysiology and therapeutics. Biophys Rev 2021, 13(6), 1007–1017. 12. [Google Scholar] [CrossRef]
- Ray, AP; Thakur, N; Pour, NG; Eddy, MT. Dual mechanisms of cholesterol-GPCR interactions that depend on membrane phospholipid composition. Structure 2023, 31(7), 836–847.e6. [Google Scholar] [CrossRef]
- Lin, Y; Bian, L; Zhu, G; Zhang, B. Vitronectin promotes proliferation and metastasis of cervical cancer cells via the epithelial-mesenchymal transition. Front Oncol 2024, 14, 1466264. [Google Scholar] [CrossRef]
- Gao, P; Lu, Y; Tang, K; Wang, W; Wang, T; Zhu, Y; et al. Ficolin-1 ameliorates pulmonary fibrosis via directly binding to TGF-β1. J Transl Med 2024, 22(1), 1051. [Google Scholar] [CrossRef] [PubMed]
- Sozio, MS; Lu, C; Zeng, Y; Liangpunsakul, S; Crabb, DW. Activated AMPK inhibits PPAR-α and PPAR-γ transcriptional activity in hepatoma cells. Am J Physiol Gastrointest Liver Physiol 2011, 301(4), G739–47. [Google Scholar] [CrossRef] [PubMed]
- Sun, H; Zhu, X; Lin, W; Zhou, Y; Cai, W; Qiu, L. Interactions of TLR4 and PPARγ, dependent on AMPK signalling pathway contribute to anti-inflammatory effects of vaccariae hypaphorine in endothelial cells. Cell Physiol Biochem 2017, 42(3), 1227–1239. [Google Scholar] [CrossRef]
- Harada, K; Isse, K; Kamihira, T; Shimoda, S; Nakanuma, Y. Th1 cytokine-induced downregulation of PPARγ in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology 2005, 41(6), 1329–38. [Google Scholar] [CrossRef]
- Yin, R; Dong, YG; Li, HL. PPARγ phosphorylation mediated by JNK MAPK: a potential role in macrophage-derived foam cell formation. Acta Pharmacol Sin 2006, 27(9), 1146–52. [Google Scholar] [CrossRef]
- Su, AC; Zhang, LY; Zhang, JG; Hu, YY; Liu, XY; Li, SC; et al. The regulation of autophagy by p38 MAPK-PPARγ signaling during the brain ischemic tolerance induced by cerebral ischemic preconditioning. DNA Cell Biol 2022, 41(9), 838–849. [Google Scholar] [CrossRef] [PubMed]
- Ballav, S; Biswas, B; Sahu, VK; Ranjan, A; Basu, S. PPARγ partial agonists in disease-fate decision with special reference to cancer. Cells 2022, 11(20), 3215. [Google Scholar] [CrossRef]
- McCubrey, JA; Steelman, LS; Chappell, WH; Abrams, SL; Montalto, G; Cervello, M; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3(9), 954–87. [Google Scholar] [CrossRef]
- Nigam, M; Punia, B; Dimri, DB; Mishra, AP; Radu, AF; Bungau, G. Reactive oxygen species: A double-edged sword in the modulation of cancer signaling pathway dynamics. Cells 2025, 14(15), 1207. [Google Scholar] [CrossRef]
- Worthylake, RA; Burridge, K. RhoA and ROCK promote migration by limiting membrane protrusions. J Biol Chem 2003, 278(15), 13578–13584. [Google Scholar] [CrossRef]
- Yoshizaki, H; Ohba, Y; Parrini, MC; et al. Cell type-specific regulation of RhoA activity during cytokinesis. J Biol Chem 2004, 279(43), 44756–44762. [Google Scholar] [CrossRef]
- Omerovic, J; Prior, IA. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J 2009, 276(7), 1817–1825. [Google Scholar] [CrossRef]
- https. [CrossRef]
- Schmick, M; Vartak, N; Papke, B; Kovacevic, M; Truxius, DC; Rossmannek, L; Bastiaens, PIH. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 10;157(2), 459–471. [Google Scholar] [CrossRef]
- Zhou, Y; Prakash, PS; Liang, H; Gorfe, AA; Hancock, JF. The KRAS and other prenylated polybasic domain membrane anchors recognize phosphatidylserine acyl chain structure. Proc Natl Acad Sci U S A 2021, 118(6), e2014605118. [Google Scholar] [CrossRef] [PubMed]
- Munro, E. Anillin puts RhoA in touch with PIP2. Dev Cell 2019, 49(6), 819–820. [Google Scholar] [CrossRef]
- Hancock, JF; Parton, RG. Ras plasma membrane signalling platforms. Biochem J 2005, 389 Pt 1, 1–11. [Google Scholar] [CrossRef]
- Jang, H; Abraham, SJ; Chavan, TS; Hitchinson, B; Khavrutskii, L; Tarasova, NI; et al. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J Biol Chem 2015, 290(15), 9465–77. [Google Scholar] [CrossRef]
- Mosaddeghzadeh, N; Kazemein Jasemi, NS; Majolée, J; Zhang, SC; Hordijk, PL; Dvorsky, R; et al. Electrostatic forces mediate the specificity of RHO GTPase-GDI interactions. Int J Mol Sci 2021, 22(22), 12493. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Ferriols, M; Lanceros-Méndez, S; Gómez Ribelles, JL; Gallego Ferrer, G. Electrical stimulation: Effective cue to direct osteogenic differentiation of mesenchymal stem cells? Biomater Adv 2022, 138, 212918. [Google Scholar] [CrossRef]
- Chen, Z; Chen, Y; Zhe, M; Jiang, J; Liu, H; Qin, L; et al. Engineered smart piezoelectric materials facilitate bone defect regeneration. Materials & Design 2026, 262, 115501. [Google Scholar] [CrossRef]
- Chen, Y; Gong, W; Dai, W; Jiang, H; Xu, X. E2F1/2/4 mRNA is associated with immune infiltration and are potential biomarkers for the prognosis of human gastric carcinoma. Transl Cancer Res 2021, 10(6), 2801–2811. [Google Scholar] [CrossRef]
- Daigh, LH; Saha, D; Rosenthal, DL; Ferrick, KR; Meyer, T. Uncoupling of mTORC1 from E2F activity maintains DNA damage and senescence. Nat Commun 2024, 15, 9181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y; Hao, X; Han, G; Lu, Y; Chen, Z; Zhang, L; et al. E2F1-mediated GINS2 transcriptional activation promotes tumor progression through PI3K/AKT/mTOR pathway in hepatocellular carcinoma. Am J Cancer Res 2022, 12(4), 1707–1726. [Google Scholar] [PubMed]
- Calzone, L; Gelay, A; Zinovyev, A; Radvanyi, F; Barillot, E. A comprehensive modular map of molecular interactions in RB/E2F pathway. Mol Syst Biol 2008, 4, 173. [Google Scholar] [CrossRef]
- Bertonnier-Brouty, L; Andersson, J; Kaprio, T; Hagström, J; Bsharat, S; Asplund, O; et al. E2F transcription factors promote tumorigenicity in pancreatic ductal adenocarcinoma. Cancer Med 2024, 13(9), e7187. [Google Scholar] [CrossRef]
- Wasserman, D; Nachum, S; Cohen, M; Enrico, TP; Noach-Hirsh, M; Parasol, J; et al. Cell cycle oscillators underlying orderly proteolysis of E2F8. Mol Biol Cell 2020, 31(8), 725–740. [Google Scholar] [CrossRef]
- Timmers, C; Sharma, N; Opavsky, R; Maiti, B; Wu, L; Wu, J; Orringer, D; et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. Erratum in: Mol Cell Biol 2012;32(9):1758. 2007, 27(1), 65–78. [Google Scholar] [CrossRef]
- Spitschak, A; Dhar, P; Singh, KP; Casalegno Garduño, R; Gupta, SK; Vera, J; et al. E2F1-induced autocrine IL-6 inflammatory loop mediates cancer-immune crosstalk that predicts T cell phenotype switching and therapeutic responsiveness. Front Immunol 2024, 15, 1470368. [Google Scholar] [CrossRef] [PubMed]
- Li, J; Ran, C; Li, E; Gordon, F; Comstock, G; Siddiqui, H; et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 2008, 14(1), 62–75. [Google Scholar] [CrossRef]
- Zheng, J; Huang, J; Xia, J; Zhou, W; Dai, L; Lin, S; et al. Transcription factor E2F8 is a therapeutic target in the basal-like subtype of breast cancer. Front Oncol 2023, 13, 1038787. [Google Scholar] [CrossRef] [PubMed]
- Vigo, E; Müller, H; Prosperini, E; Hateboer, G; Cartwright, P; Moroni, MC; et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 1999, 19(9), 6379–95. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M; Washam, CL; Urbaniak, A; Heflin, B; Storey, AJ; Lan, RS; et al. Phosphoproteomics provides novel insights into the response of primary acute lymphoblastic leukemia cells to microtubule depolymerization in G1 phase of the cell cycle. ACS Omega 2021, 6(38), 24949–24959. [Google Scholar] [CrossRef]
- Wang, X; Hills, LB; Huang, YH. Lipid and protein co-regulation of PI3K effectors Akt and Itk in lymphocytes. Front Immunol. 2015, 6, 117. [Google Scholar] [CrossRef]
- Sun, F; Yang, Y; Tu, H; Cai, H. Gradients of PI(4,5)P2 and PI(3,5)P2 jointly participate in shaping the back state of dictyostelium cells. Front Cell Dev Biol 2022, 10, 835185. [Google Scholar] [CrossRef]
- Sun, J; Zalejski, J; Song, S; Sharma, A; Wang, W; Hu, Y; et al. PI(3,5)P 2 controls the signaling activity of class I PI3K. bioRxiv Preprint 2025, 2023.01.25.525550. [Google Scholar] [CrossRef]
- Yang, M; Lu, Y; Piao, W; Jin, H. The Translational Regulation in mTOR Pathway. Biomolecules 2022, 12(6), 802. [Google Scholar] [CrossRef]
- Shveygert, M; Kaiser, C; Bradrick, SS; Gromeier, M. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol Cell Biol 2010, 30(21), 5160–7. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z; Bouteau, A; Herbst, C; Igyártó, BZ. Pre-exposure to mRNA-LNP inhibits adaptive immune responses and alters innate immune fitness in an inheritable fashion. PLoS Pathog 2022, 18(9), e1010830. [Google Scholar] [CrossRef]
- Lonez, C; Vandenbranden, M; Ruysschaert, JM. Cationic liposomal lipids: From gene carriers to cell signaling. Prog Lipid Res 2008, 47(5), 340–7. [Google Scholar] [CrossRef]
- Lonez, C; Vandenbranden, M; Ruysschaert, JM. Cationic lipids activate intracellular signaling pathways. Adv Drug Deliv Rev 2012, 64(15), 1749–58. [Google Scholar] [CrossRef]
- Connors, J; Joyner, D; Mege, NJ; Cusimano, GM; Bell, MR; Marcy, J; et al. Lipid nanoparticles (LNP) induce activation and maturation of antigen presenting cells in young and aged individuals. Commun Biol Erratum in: Commun Biol 2025;8(1):285. https://doi.org/10.1038/s42003-025-07603-0. 2023, 6(1), 188. [Google Scholar] [CrossRef]
- Amor, NP; Guo, K; Zhang, S; Xia, J; Yang, Y; Lin, A. Lipid nanoparticle: Beyond delivery vehicle-unveiling its immunological adjuvant potential. FASEB J 2025, 39(10), e70641. [Google Scholar] [CrossRef]
- Yamaguchi, Y; Kato, Y; Edahiro, R; Søndergaard, JN; Murakami, T; Amiya, S; et al. Consecutive BNT162b2 mRNA vaccination induces short-term epigenetic memory in innate immune cells. JCI Insight 2022, 7(22), e163347. [Google Scholar] [CrossRef]
- Simonis, A; Theobald, SJ; Koch, AE; Mummadavarapu, R; Mudler, JM; Pouikli, A; et al. Persistent epigenetic memory of SARS-CoV-2 mRNA vaccination in monocyte-derived macrophages. Mol Syst Biol 2025, 21(4), 341–360. [Google Scholar] [CrossRef] [PubMed]
- Hifdi, N; Vaucourt, M; Hnia, K; Panasyuk, G; Vandromme, M. Phosphoinositide signaling in the nucleus: Impacts on chromatin and transcription regulation. Biol Cell 2025, 117(1), e2400096. [Google Scholar] [CrossRef]
- Chytła, A; Rattay, S; Akgül, B; Sztacho, M. Plasma membrane and nuclear phosphatidylinositol 4,5-bisphosphate signalling in cancer. Lipids Health Dis 2025, 24(1), 39. [Google Scholar] [CrossRef] [PubMed]
- McColl, ER; Croyle, MA; Zamboni, WC; Honer, WG; Heise, M; Piquette-Miller, M; et al. COVID-19 Vaccines and the Virus: Impact on Drug Metabolism and Pharmacokinetics. Drug Metab Dispos 2023, 51(1), 130–141. [Google Scholar] [CrossRef]
- Jung, SW; Jeon, JJ; Kim, YH; Choe, SJ; Lee, S. Long-term risk of autoimmune diseases after mRNA-based SARS-CoV2 vaccination in a Korean, nationwide, population-based cohort study. Nat Commun 2024, 15, 6181. [Google Scholar] [CrossRef]
- Sellegounder, D; Ferrucci, L; Anbazhagan, R; Basisty, N. Editorial: Molecular crosstalk between endocrine factors, paracrine signals, and the immune system during aging. Front Endocrinol (Lausanne) 2023, 14, 1203755. [Google Scholar] [CrossRef]
- Chen, M; Tan, JX; Sun, Y; Thapa, N; Cryns, VL; Anderson, RA. Agonist- and stress-driven compartmentalized phosphoinositide signaling in cells. Biochim Biophys Acta Mol Cell Biol Lipids 2025, 1870(6), 159662. [Google Scholar] [CrossRef]
- Ebner, M; Sinkovics, B; Szczygieł, M; Ribeiro, DW; Yudushkin, I. Localization of mTORC2 activity inside cells. J Cell Biol 2017, 216(2), 343–353. [Google Scholar] [CrossRef]
- Rädler, PD; Wehde, BL; Wagner, KU. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol Cell Endocrinol 2017, 451, 31–39. [Google Scholar] [CrossRef]
- Flori, E; Cavallo, A; Mosca, S; et al. JAK/STAT inhibition normalizes lipid composition in 3D human epidermal equivalents challenged with Th2 cytokines. Cells 2024, 13(9), 760. [Google Scholar] [CrossRef] [PubMed]
- Koester, AM; Tao, K; Szczepaniak, M; Rames, MJ; Nan, X. Nanoscopic spatial association between Ras and phosphatidylserine on the cell membrane studied with multicolor super resolution microscopy. Biomolecules 2022, 12(8), 1033. [Google Scholar] [CrossRef] [PubMed]
- Panariti, A; Miserocchi, G; Rivolta, I. The effect of nanoparticle uptake on cellular behavior: disrupting or enabling functions? Nanotechnol Sci Appl 2012, 5, 87–100. [Google Scholar] [CrossRef]
- Voigt, J; Christensen, J; Shastri, VP. Differential uptake of nanoparticles by endothelial cells through polyelectrolytes with affinity for caveolae. Proc Natl Acad Sci U S A 2014, 111(8), 2942–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, T; Bai, J; Jiang, X; Nienhaus, GU. Cellular uptake of nanoparticles by membrane penetration: a study combining confocal microscopy with FTIR spectroelectrochemistry. ACS Nano 2012, 6(2), 1251–9. [Google Scholar] [CrossRef]
- Gerelli, Y. Chapter Three: Exploring interactions between lipid membranes and nanoparticles through neutron and X-ray reflectometry techniques. In Advances in Biomembranes and Lipid Self-Assembly; Iglič, A., Rappolt, M., Losada-Pérez, P., Eds.; Academic Press, 2023; Volume 38, pp. 37–61. [Google Scholar] [CrossRef]
- Lavagna, E; Barnoud, J; Rossi, G; Monticelli, L. Size-dependent aggregation of hydrophobic nanoparticles in lipid membranes. Nanoscale 2020, 12, 9452–9461. [Google Scholar] [CrossRef]
- Cooper, GM. The Cell: A Molecular Approach, 2nd edition; Sinauer Associates: Sunderland (MA), 2000. [Google Scholar]
- Hammond, GRV; Burke, JE. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr Opin Cell Biol 2020, 63, 57–67. [Google Scholar] [CrossRef]
- Skotland, T; Kavaliauskiene, S; Sandvig, K. The role of lipid species in membranes and cancer-related changes. Cancer Metastasis Rev 2020, 39(2), 343–360. [Google Scholar] [CrossRef]
- https. [CrossRef]
- Heimburg, T. The excitable fluid mosaic. Biochim Biophys Acta Biomembr 2023, 1865(3), 184104. [Google Scholar] [CrossRef]
- Lupyan, D; Mezei, M; Logothetis, DE; Osman, R. A molecular dynamics investigation of lipid bilayer perturbation by PIP2. Biophys J 2010, 98(2), 240–7. [Google Scholar] [CrossRef] [PubMed]
- Pike, LJ. Lipid rafts: bringing order to chaos. J Lipid Res 2003, 44(4), 655–67. [Google Scholar] [CrossRef]
- Levin, R; Grinstein, S; Schlam, D. Phosphoinositides in phagocytosis and macropinocytosis. Biochim Biophys Acta 2015, 1851(6), 805–23. [Google Scholar] [CrossRef] [PubMed]
- Weiner, OD; Neilsen, PO; Prestwich, GD; Kirschner, MW; Cantley, LC; Bourne, HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 2002, 4(7), 509–13. [Google Scholar] [CrossRef]
- Guan, K; Curtis, ER; Lew, DJ; Elston, TC. Particle-based simulations reveal two positive feedback loops allow relocation and stabilization of the polarity site during yeast mating. PLoS Comput Biol 2023, 19(10), e1011523. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M; Linderman, JJ. Lipid raft-mediated regulation of G-protein coupled receptor signaling by ligands which influence receptor dimerization: a computational study. PLoS One 2009, 4(8), e6604. [Google Scholar] [CrossRef]
- Wang, X; Shi, X; Wang, R. Regulating mRNA endosomal escape through lipid rafts: A review. Int J Pharm 2025, 675, 125571. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S; Carroll, B; Buganim, Y; Maetzel, D; Ng, AH; Cassady, JP; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 2013, 5(5), 1302–15. [Google Scholar] [CrossRef]
- Dall'Armi, C; Devereaux, KA; Di Paolo, G. The role of lipids in the control of autophagy. Curr Biol 2013, 23(1), R33–45. [Google Scholar] [CrossRef]
- Zhang, S; Peng, X; Yang, S; Li, X; Huang, M; Wei, S; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis 2022, 13(2), 132. [Google Scholar] [CrossRef] [PubMed]
- Jarocki, M; Turek, K; Saczko, J; Tarek, M; Kulbacka, J. Lipids associated with autophagy: mechanisms and therapeutic targets. Cell Death Discov 2024, 10(1), 460. [Google Scholar] [CrossRef]
- Karim, M; Mishra, M; Lo, CW; Saul, S; Cagirici, HB; Gourdelier, M; et al. PIP4K2C inhibition reverses autophagic flux impairment induced by SARS-CoV-2. Nat Commun 2025, 16(1), 6397. [Google Scholar] [CrossRef]
- Haucke, V; Kozlov, MM. Membrane remodeling in clathrin-mediated endocytosis. J Cell Sci 2018, 131(17), jcs216812. [Google Scholar] [CrossRef]
- Vaithianathan, T; Bukiya, A; Liu, J; Liu, P; Asuncion-Chin, M; Fan, Z; Dopico, A. Direct regulation of BK channels by phosphatidylinositol 4,5-bisphosphate as a novel signaling pathway. J Gen Physiol 2008, 132(1), 13–28. [Google Scholar] [CrossRef]
- Blunsom, NJ; Cockcroft, S. Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim Biophys Acta Mol Cell Biol Lipids 2020, 1865(1), 158471. [Google Scholar] [CrossRef]
- Posor, Y; Jang, W; Haucke, V. Phosphoinositides as membrane organizers. Nat Rev Mol Cell Biol 2022, 23(12), 797–816. [Google Scholar] [CrossRef]
- Lolicato, F; Nickel, W; Haucke, V; Ebner, M. Phosphoinositide switches in cell physiology - From molecular mechanisms to disease. J Biol Chem 2024, 300(3), 105757. [Google Scholar] [CrossRef]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol Rev 2013, 93(3), 1019–137. [Google Scholar] [CrossRef]
- Eramo, MJ; Mitchell, CA. Regulation of PtdIns(3,4,5)P3/Akt signalling by inositol polyphosphate 5-phosphatases. Biochem Soc Trans 2016, 44(1), 240–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, S; Cao, B; Xuan, G; Xu, S; An, Z; Zhu, C; et al. Function and regulation of Rab GTPases in cancers. Cell Biol Toxicol 2024, 40(1), 28. [Google Scholar] [CrossRef] [PubMed]
- Koike, S; Jahn, R. Rab GTPases and phosphoinositides fine-tune SNAREs dependent targeting specificity of intracellular vesicle traffic. Nat Commun 2024, 15(1), 2508. [Google Scholar] [CrossRef]
- Puranik, A; Lenehan, PJ; Silvert, E; Niesen, MJM; Corchado-Garcia, J; O'Horo, JC; et al. Comparative effectiveness of mRNA-1273 and BNT162b2 against symptomatic SARS-CoV-2 infection. Med 2022, 3(1), 28–41.e8. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, JT; Aquino-Jarquin, G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed Pharmacother 2021, 142, 111953. [Google Scholar] [CrossRef]
- Harvey, RD; Ara, N; Heenan, RK; Barlow, DJ; Quinn, PJ; Lawrence, MJ. Stabilization of distearoylphosphatidylcholine lamellar phases in propylene glycol using cholesterol. Mol Pharm 2013, 10(12), 4408–17. [Google Scholar] [CrossRef]
- Li, J; Wang, X; Zhang, T; Wang, C; Huang, Z; Luo, X; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J Pharm Sci 2015, 10(2), 81–98. [Google Scholar] [CrossRef]
- McMaster, CR. From yeast to humans - roles of the Kennedy pathway for phosphatidylcholine synthesis. FEBS Lett 2018, 592(8), 1256–1272. [Google Scholar] [CrossRef]
- Cummings, R; Parinandi, N; Wang, L; Usatyuk, P; Natarajan, V. Phospholipase D/phosphatidic acid signal transduction: role and physiological significance in lung. Mol Cell Biochem 2002, 234-235(1-2), 99–109. [Google Scholar] [CrossRef]
- Wagner, K; Brezesinski, G. Phospholipase D activity is regulated by product segregation and the structure formation of phosphatidic acid within model membranes. Biophys J 2007, 93(7), 2373–83. [Google Scholar] [CrossRef]
- Bruntz, RC; Lindsley, CW; Brown, HA. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev 2014, 66(4), 1033–79. [Google Scholar] [CrossRef]
- Semenkovich, CF; Goldberg, AC; Goldberg, IJ. Disorders of Lipid Metabolism. Chapter 37 in Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, Eds. Williams Textbook of Endocrinology (Thirteenth Edition). Elsevier; Volume 2016, pp. 1660–1700.
- https. [CrossRef]
- Pearce, B; Jakobson, K; Morrow, C; Murphy, S. Phosphatidic acid promotes phosphoinositide metabolism and DNA synthesis in cultured cortical astrocytes. Neurochem Int 1994, 24(2), 165–71. [Google Scholar] [CrossRef] [PubMed]
- Quick, J; Santos, ND; Cheng, MHY; Chander, N; Brimacombe, CA; Kulkarni, J; et al. Lipid nanoparticles to silence androgen receptor variants for prostate cancer therapy. J Control Release 2022, 349, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Jeschek, D; Lhota, G; Wallner, J; Vorauer-Uhl, K. A versatile, quantitative analytical method for pharmaceutical relevant lipids in drug delivery systems. J Pharm Biomed Anal 2016, 119, 37–44. [Google Scholar] [CrossRef]
- Rezaei, S; Blick, EE; Mineart, KP; Kelley, EG. Chapter Three - Linking chemical degradation and physical instability of lipid vesicles. Advances in Biomembranes and Lipid Self-Assembly 2025, 41, 47–64. [Google Scholar] [CrossRef]
- Nishimura, T; Gecht, M; Covino, R; Hummer, G; Surma, MA; Klose, C; et al. Osh proteins control nanoscale lipid organization necessary for PI(4,5)P2 synthesis. Mol Cell 2019, 75(5), 1043–1057.e8. [Google Scholar] [CrossRef]
- Heckle, LA; Kozminski, KG. Osh-dependent and -independent regulation of PI4P levels during polarized growth of Saccharomyces cerevisiae. Mol Biol Cell 2023, 34(11), ar104. [Google Scholar] [CrossRef]
- Raychaudhuri, S; Prinz, WA. The diverse functions of oxysterol-binding proteins. Annu Rev Cell Dev Biol 2010, 26, 157–77. [Google Scholar] [CrossRef]
- Leonarduzzi, G; Robbesyn, F; Poli, G. Signaling kinases modulated by 4-hydroxynonenal. Free Radic Biol Med. 2004, 37(11), 1694–702. [Google Scholar] [CrossRef]
- Forman, HJ; Fukuto, JM; Miller, T; Zhang, H; Rinna, A; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008, 477(2), 183–95. [Google Scholar] [CrossRef]
- Thiemicke, A; Neuert, G. Rate thresholds in cell signaling have functional and phenotypic consequences in non-linear time-dependent environments. Front Cell Dev Biol 2023, 11, 1124874. [Google Scholar] [CrossRef]
- Hou, X; Chen, Y; Carrillo, ND; Cryns, VL; Anderson, RA; Sun, J; et al. Phosphoinositide signaling at the cytoskeleton in the regulation of cell dynamics. Cell Death Dis 2025, 16(1), 296. [Google Scholar] [CrossRef]
- Adhikari, H; Cullen, PJ. Role of phosphatidylinositol phosphate signaling in the regulation of the filamentous-growth mitogen-activated protein kinase pathway. Eukaryot Cell 2015, 14(4), 427–40. [Google Scholar] [CrossRef]
- Houthaeve, G; De Smedt, SC; Braeckmans, K; De Vos, WH. The cellular response to plasma membrane disruption for nanomaterial delivery. Nano Converg 2022, 9(1), 6. [Google Scholar] [CrossRef]
- Papafilippou, L; Nicolaou, A; Kendall, AC; Camacho-Muñoz, D; Hadjidemetriou, M. The lipidomic profile of the nanoparticle-biomolecule corona reflects the diversity of plasma lipids. Nanoscale 2023, 15, 11038–11051. [Google Scholar] [CrossRef]
- Gual, P; Grémeaux, T; Gonzalez, T; Le Marchand-Brustel, Y; Tanti, JF. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia 2003, 46(11), 1532–1542. [Google Scholar] [CrossRef]
- Minard, AY; Tan, SX; Yang, P; et al. mTORC1 Is a Major Regulatory Node in the FGF21 Signaling Network in Adipocytes. Cell Rep. 2016, 17(1), 29–36. [Google Scholar] [CrossRef]
- Le, TKC; Dao, XD; Nguyen, DV; Luu, DH; Bui, TMH; Le, TH; et al. Insulin signaling and its application. Front Endocrinol (Lausanne) 2023, 14, 1226655. [Google Scholar] [CrossRef]
- Krauson, AJ; Casimero, FVC; Siddiquee, Z; Stone, JR. Duration of SARS-CoV-2 mRNA vaccine persistence and factors associated with cardiac involvement in recently vaccinated patients. NPJ Vaccines 2023, 8(1), 141. [Google Scholar] [CrossRef]
- Rosati, M; Terpos, E; Homan, P; Bergamaschi, C; Karaliota, S; Ntanasis-Stathopoulos, I; et al. Rapid transient and longer-lasting innate cytokine changes associated with adaptive immunity after repeated SARS-CoV-2 BNT162b2 mRNA vaccinations. Front Immunol. 2023, 14, 1292568. [Google Scholar] [CrossRef]
- Lee, Y; Jeong, M; Park, J; Jung, H; Lee, H. Immunogenicity of lipid nanoparticles and its impact on the efficacy of mRNA vaccines and therapeutics. Exp Mol Med. 2023, 55(10), 2085–2096. [Google Scholar] [CrossRef]
- Iordanov, MS; Paranjape, JM; Zhou, A; Wong, J; Williams, BR; Meurs, EF; et al. Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways. Mol Cell Biol. 2000, 20(2), 617–27. [Google Scholar] [CrossRef]
- Jurkiewicz, A; Graczyk, D. MAP kinases are involved in RNA polymerase III regulation upon LPS treatment in macrophages. Gene 2022, 831, 146548. [Google Scholar] [CrossRef]
- Zhang, G; He, LS; Him Wong, Y; Xu, Y; Zhang, Y; Qian, PY. p38 MAPK regulates PKAα and CUB-serine protease in Amphibalanus amphitrite cyprids. Sci Rep. 2015, 5, 14767. [Google Scholar] [CrossRef]
- Tang, T; Zhu, Q; Li, X; Zhu, G; Deng, S; Wang, Y; et al. Protease Nexin I is a feedback regulator of EGF/PKC/MAPK/EGR1 signaling in breast cancer cells metastasis and stemness. Cell Death Dis. Erratum. 2019, 10(9), 649. [Google Scholar] [CrossRef]
- Sala-Gaston, J; Costa-Sastre, L; Pedrazza, L; Martinez-Martinez, A; Ventura, F; Rosa, JL. Regulation of MAPK signaling pathways by the large HERC ubiquitin ligases. Int J Mol Sci. 2023, 24(5), 4906. [Google Scholar] [CrossRef]
- Lutz, J; Lazzaro, S; Habbeddine, M; Schmidt, KE; Baumhof, P; Mui, BL; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 29. [Google Scholar] [CrossRef]
- Pang, SJ; Jiang, TY; Wang, NG; Cui, XW; Wang, H; Pan, YF; et al. RNA polymerase II subunit 5-mediating protein limits TLR4-induced innate immune activation in macrophages by inhibiting IKKβ/NF-κB signaling during sepsis. Cell Commun Signal. 2025, 23(1), 274. [Google Scholar] [CrossRef]
- Ostendorf, T; Zillinger, T; Andryka, K; Schlee-Guimaraes, TM; Schmitz, S; Marx, S; et al. Immune sensing of synthetic, bacterial, and protozoan RNA by toll-like receptor 8 requires coordinated processing by RNase T2 and RNase 2. Immunity 2020, 52(4), 591–605.e6. [Google Scholar] [CrossRef]
- Lee, D; Le Pen, J; Yatim, A; Dong, B; Aquino, Y; Ogishi, M; et al. Inborn errors of OAS-RNase L in SARS-CoV-2-related multisystem inflammatory syndrome in children. Science 2023, 379(6632), eabo3627. [Google Scholar] [CrossRef]
- Bucci, M; Vellecco, V; Harrington, L; Brancaleone, V; Roviezzo, F; Mattace Raso, G; et al. Cross-talk between toll-like receptor 4 (TLR4) and proteinase-activated receptor 2 (PAR(2) ) is involved in vascular function. Br J Pharmacol. 2013, 168(2), 411–20. [Google Scholar] [CrossRef]
- Zhong, B; Liu, X; Wang, X; Liu, X; Li, H; Darnay, BG; et al. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci Signal. 2013, 6(275), ra35. [Google Scholar] [CrossRef]
- Papendorf, JJ; Krüger, E; Ebstein, F. Proteostasis perturbations and their roles in causing sterile inflammation and autoinflammatory diseases. Cells 2022, 11(9), 1422. [Google Scholar] [CrossRef] [PubMed]
- Guo, X; Wang, H; Li, Y; Leng, X; Huang, W; Ma, Y; et al. Transfection reagent Lipofectamine triggers type I interferon signaling activation in macrophages. Immunol Cell Biol. Erratum in: Immunol Cell Biol. 2020;98(1):88. https://doi.org/10.1111/imcb.12300. 2019, 97(1), 92–96. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Causality assessment of an adverse event following immunization (AEFI): user manual for the revised WHO classification, 2nd ed.; Pharmacovigilance 2019 Geneva, Switzerland: Department, 2019; Available online: https://www.who.int/publications/i/item/9789241516990.
- Bellavite, P. Causality assessment of adverse events following immunization: the problem of multifactorial pathology. F1000research 2020, 9, 170. [Google Scholar] [CrossRef]
- Islam, S; Bhattacharya, S. Dynamical systems theory as an organizing principle for single-cell biology. NPJ Syst Biol Appl. 2025, 11(1), 85. [Google Scholar] [CrossRef] [PubMed]
- Philipps, M; Schmid, N; Hasenauer, J. Current state and open problems in universal differential equations for systems biology. NPJ Syst Biol Appl. 2025, 11(1), 101. [Google Scholar] [CrossRef]
- Simons, BD; Karin, O. Cell cycle criticality as a mechanism for robust cell population control. Mol Syst Biol. 2025, 22, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Fischer, DS; Villanueva, MA; Winter, PS; Shalek, AK. Adapting systems biology to address the complexity of human disease in the single-cell era. Nat Rev Genet. 2025, 26, 514–531. [Google Scholar] [CrossRef]
- U. S. Pharmacopeia (USP). Analytical procedures for mRNA Vaccine Quality. 3rd Edition. 2024. Available online: https://go.usp.org/mRNAVaccineQuality (accessed on 13 February 2026).
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the quality aspects of mRNA vaccines: Draft. Euopean Medicines Agency. Amsterdam, 2025. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-aspects-mrna-vaccines_en.pdf.
- Hassett, KJ; Higgins, J; Woods, A; Levy, B; Xia, Y; Hsiao, CJ; et al. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J Control Release 2021, 335, 237–246. [Google Scholar] [CrossRef]
- Arral, ML; Whitehead, KA. Design principles of lipid nanoparticles for RNA delivery. Nat Rev Bioeng Epub ahead of print. 2026. [Google Scholar] [CrossRef]
- Karikó, K; Ni, H; Capodici, J; Lamphier, M; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004, 279(13), 12542–50. [Google Scholar] [CrossRef]
- Karikó, K; Buckstein, M; Ni, H; Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23(2), 165–75. [Google Scholar] [CrossRef]
- Karikó, K; Muramatsu, H; Welsh, FA; Ludwig, J; Kato, H; Akira, S; et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 2008, 16(11), 1833–40. [Google Scholar] [CrossRef]
- Monroe, J; Eyler, DE; Mitchell, L; Deb, I; Bojanowski, A; Srinivas, P; et al. N1-Methylpseudouridine and pseudouridine modifications modulate mRNA decoding during translation. Nat Commun 2024, 15(1), 8119. [Google Scholar] [CrossRef]
- Ghosh, TK; Mickelson, DJ; Solberg, JC; Lipson, KE; Inglefield, JR; Alkan, SS. TLR-TLR cross talk in human PBMC resulting in synergistic and antagonistic regulation of type-1 and 2 interferons, IL-12 and TNF-alpha. Int Immunopharmacol 2007, 7(8), 1111–21. [Google Scholar] [CrossRef]
- Xu, XH; Shah, PK; Faure, E; Equils, O; Thomas, L; Fishbein, MC. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104(25), 3103–8. [Google Scholar] [CrossRef]
- Hovland, A; Jonasson, L; Garred, P; Yndestad, A; Aukrust, P; Lappegård, KT; et al. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241(2), 480–94. [Google Scholar] [CrossRef] [PubMed]
- Luo, L; Wall, AA; Tong, SJ; Hung, Y; Xiao, Z; Tarique, AA; et al. TLR crosstalk activates LRP1 to recruit Rab8a and PI3Kγ for suppression of inflammatory responses. Cell Rep 2018, 24(11), 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Köberlin, MS; Heinz, LX; Superti-Furga, G. Functional crosstalk between membrane lipids and TLR biology. Curr Opin Cell Biol 2016, 39, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kornilov, FD; Shabalkina, AV; Lin, C; Volynsky, PE; Kot, EF; Kayushin, AL; et al. The architecture of transmembrane and cytoplasmic juxtamembrane regions of Toll-like receptors. Nat Commun. 2023, 14(1), 1503. [Google Scholar] [CrossRef]
Figure 1.
Schematic structural representation of an LNP (via Cryo EM). Diverse weak-loaded forces keep the LNP metastable. Created in BioRender. Seger, F. (2026) https://BioRender.com/ysmgilk.
Figure 1.
Schematic structural representation of an LNP (via Cryo EM). Diverse weak-loaded forces keep the LNP metastable. Created in BioRender. Seger, F. (2026) https://BioRender.com/ysmgilk.
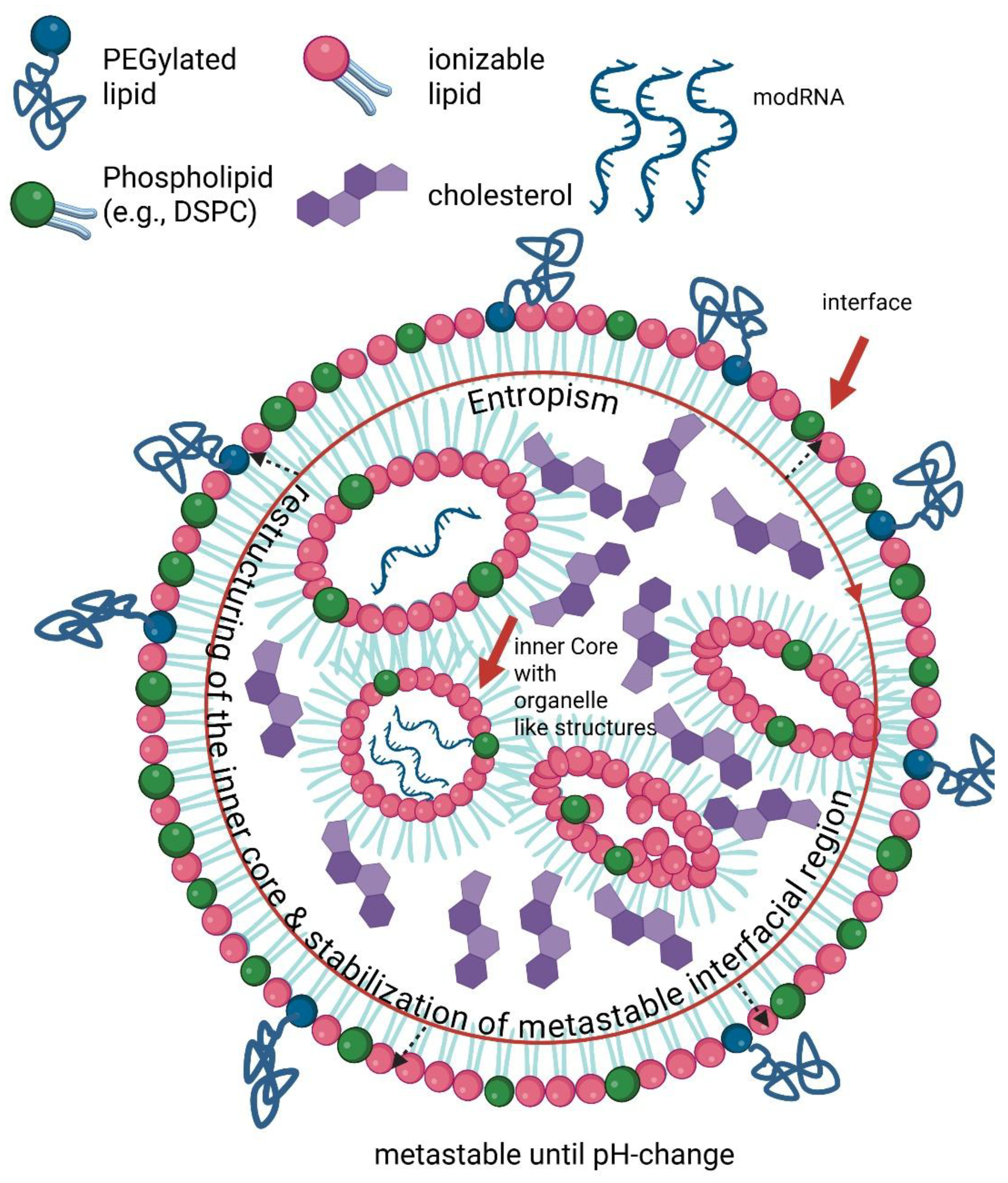
Figure 2.
Schematic representation of the physicochemical and biological characteristics of an mRNA-loaded lipid nanoparticle (LNP). TOP: The lipid components self-assemble into a quasi-bilayer structure, forming ionizable lipid-rich domains that complex with the mRNA payload (black). Under physiological conditions (pH7.4), approximately 10% of the ionizable lipid headgroups are protonated, depending on the lipid’s apparent pKa. BOTTOM: Following exposure to plasma, serum proteins and antibodies rapidly adsorb onto the LNP surface, forming a protein corona (blue/orange/gold/green/yellow) with counterions electrostatically shielding the internal positive potential, producing an apparently neutral surface (near zero- ζ potential). Red plus (+) signs represent the hidden electrostatic potential that fades in density toward the corona, indicating charge attenuation through the lipid shell and adsorbed proteins [44]. This internal-external charge asymmetry influences serum stability, biodistribution, and cellular uptake, and contributes to endosomal escape behavior. Created in BioRender. Seger, F. (2026) https://BioRender.com/mjhdche.
Figure 2.
Schematic representation of the physicochemical and biological characteristics of an mRNA-loaded lipid nanoparticle (LNP). TOP: The lipid components self-assemble into a quasi-bilayer structure, forming ionizable lipid-rich domains that complex with the mRNA payload (black). Under physiological conditions (pH7.4), approximately 10% of the ionizable lipid headgroups are protonated, depending on the lipid’s apparent pKa. BOTTOM: Following exposure to plasma, serum proteins and antibodies rapidly adsorb onto the LNP surface, forming a protein corona (blue/orange/gold/green/yellow) with counterions electrostatically shielding the internal positive potential, producing an apparently neutral surface (near zero- ζ potential). Red plus (+) signs represent the hidden electrostatic potential that fades in density toward the corona, indicating charge attenuation through the lipid shell and adsorbed proteins [44]. This internal-external charge asymmetry influences serum stability, biodistribution, and cellular uptake, and contributes to endosomal escape behavior. Created in BioRender. Seger, F. (2026) https://BioRender.com/mjhdche.
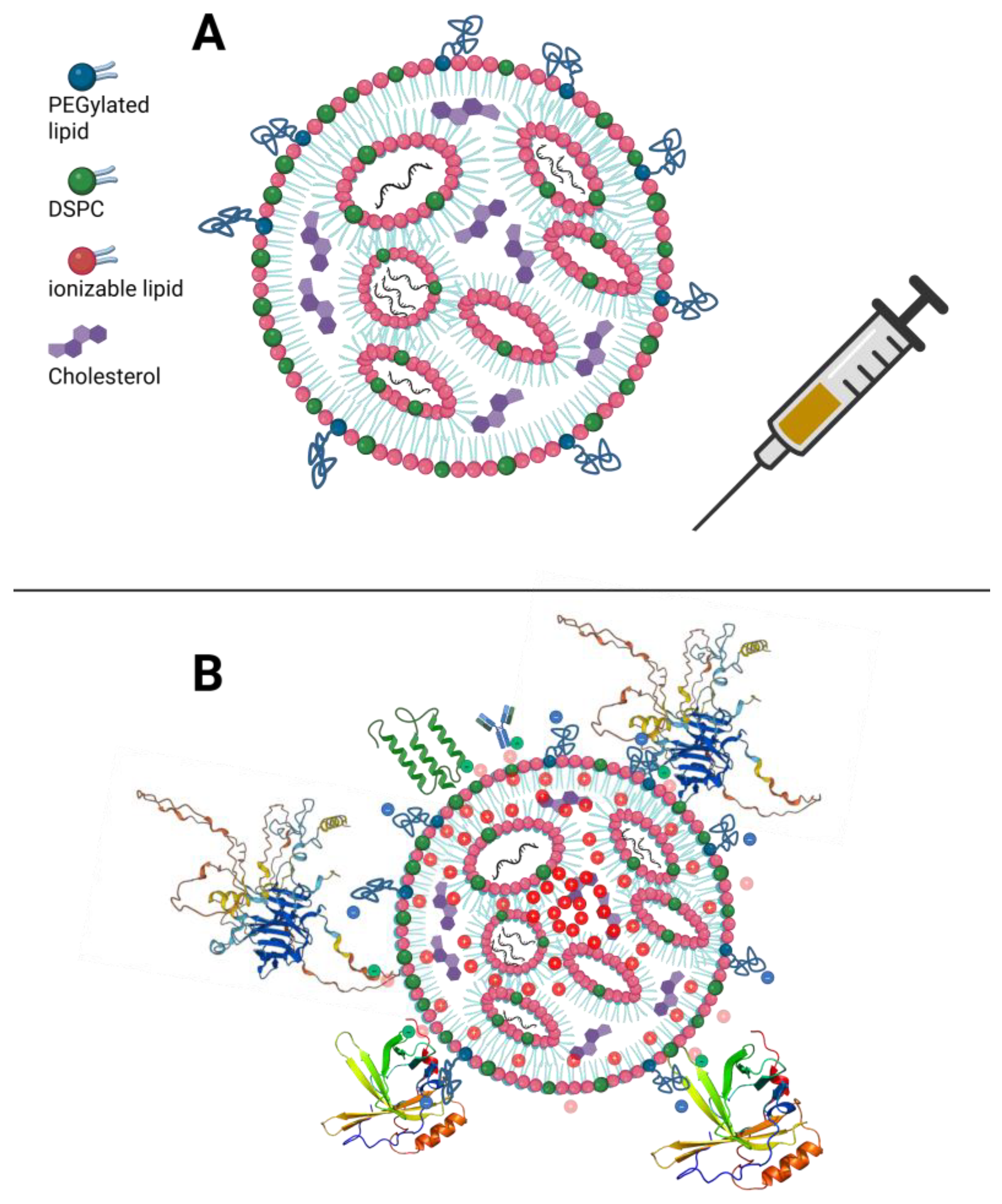
Figure 3.
Cell membrane structure and the Phosphatidylinositol(PI)-cycle. This schematic summarizes established PI-cycle biology and its role in defining membrane identity, vesicle trafficking, and endosomal maturation through spatially restricted PI-species. Distinct phosphorylated PI pools coordinate receptor-proximal signaling and trafficking decisions, thereby linking membrane dynamics to downstream pathways such as PI3K–AKT–mTOR and RAS-ERK/MAPK signaling. (1) Phosphoinositide phosphatases remove phosphate groups, ensuring spatial and temporal signal termination within the PI cycle. (2) Phosphoinositide kinases generate specific PIP-species through site-specific phosphorylation. (3) PIP-species cooperate with Soluble NSF Attachment Protein Receptors (SNARE-proteins) to regulate membrane curvature, fusion, and biophysical properties that underlie vesicular transport and signal compartmentalization. Created in BioRender. Seger, F. (2026) https://BioRender.com/unux4hx.
Figure 3.
Cell membrane structure and the Phosphatidylinositol(PI)-cycle. This schematic summarizes established PI-cycle biology and its role in defining membrane identity, vesicle trafficking, and endosomal maturation through spatially restricted PI-species. Distinct phosphorylated PI pools coordinate receptor-proximal signaling and trafficking decisions, thereby linking membrane dynamics to downstream pathways such as PI3K–AKT–mTOR and RAS-ERK/MAPK signaling. (1) Phosphoinositide phosphatases remove phosphate groups, ensuring spatial and temporal signal termination within the PI cycle. (2) Phosphoinositide kinases generate specific PIP-species through site-specific phosphorylation. (3) PIP-species cooperate with Soluble NSF Attachment Protein Receptors (SNARE-proteins) to regulate membrane curvature, fusion, and biophysical properties that underlie vesicular transport and signal compartmentalization. Created in BioRender. Seger, F. (2026) https://BioRender.com/unux4hx.
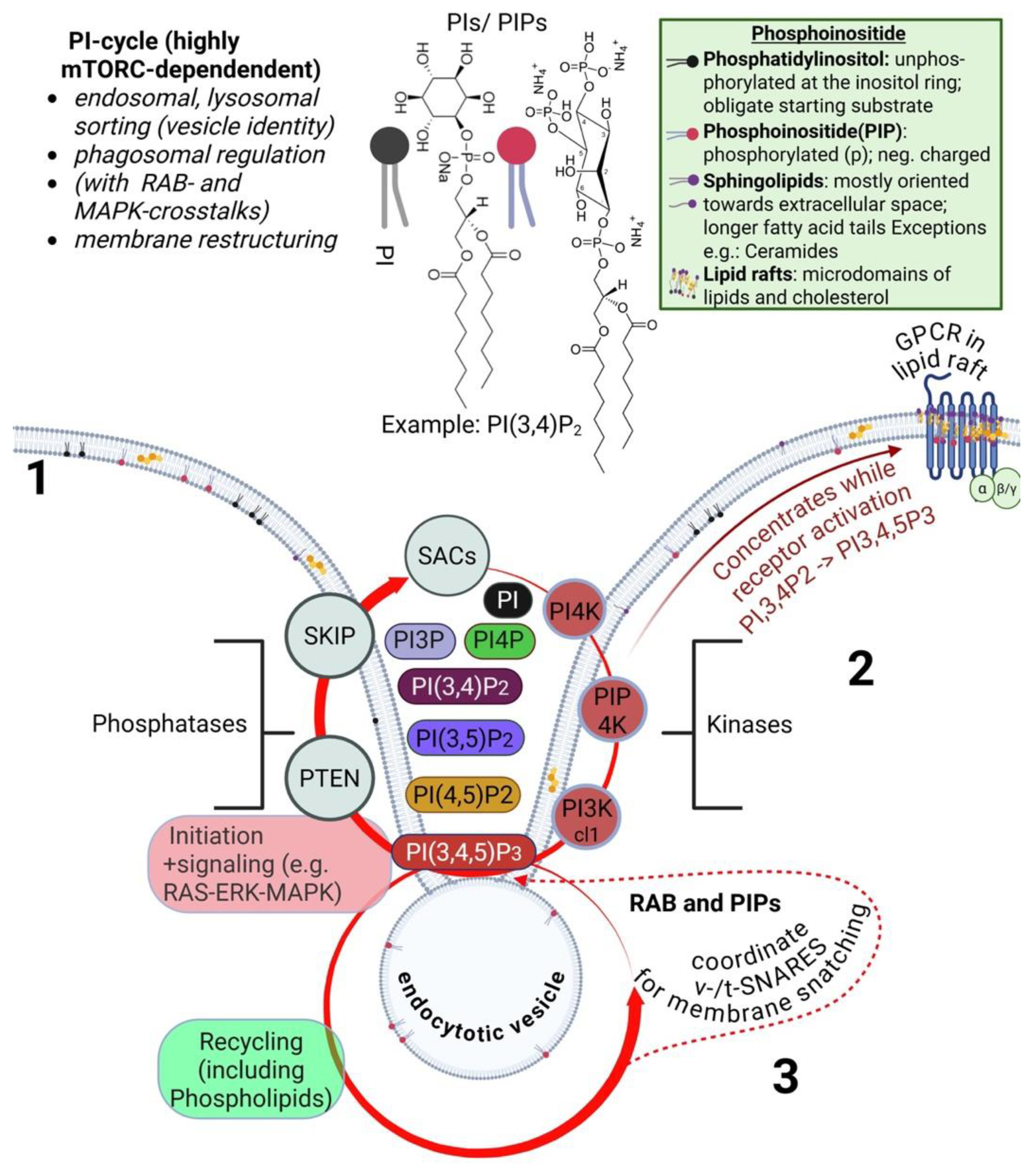
Figure 4.
Organelle trafficking, organelle membrane identity, and lipid nanoparticle influence. (1) Internalization. Lipid nanoparticles are taken up by endocytosis and fuse with early endocytic vesicles. May interfere with the physics of plasma and vesicle membranes during uptake and early endosomal trafficking. (a) Early endosome. Defined predominantly by PI3P, with contributions from phosphatidylinositol-4-phosphate (PI4P). PI3P is generated by class III PI3K, which establishes endosomal identity by recruiting effector proteins, such as ESCRT-0 and sorting nexins (SNX), for the sorting of endosomal cargo. (b) Recycling endosome. Enriched in PI(4,5)P₂, PI4P, and PI3P, supporting membrane recycling, resorting of cargo, and rebalancing of phosphatidylinositol species. (c) Late endosome. Characterized by PI3P with increasing levels of PI(3,5)P₂, generated by phosphoinositide kinase, FYVE-type zinc finger containing (PIKfyve). Regulates payload sorting and MVB formation, and sorting toward exosome release or lysosomal fusion. Incomplete endosomal escape may trap ionizable lipids within these compartments. (d) Endolysosomal transition. Characterized by elevated PI(3,5)P₂ with residual PI3P supporting lysosomal maturation processes, including ion channel activity and vacuolar-type H⁺-ATPase (V-ATPase) function. (e) Lysosome. Is dominated by PI(3,5)P₂ and variable PIPs species depending on functional state. Coordinates degradation of intracellular components and ALR. (f) Autolysosome. Contains PI3P derived from autophagosomes & PI(3,5)P₂ derived from lysosomes. During ALR, PI(4,5)P₂ and PI4P are involved, with PI(4,5)P₂ recruiting clathrin and the adaptor protein-2 (AP-2) complex to support membrane tubulation and lysosome reformation. (2) & (3) Exocytosis pathways. Payload release occurs directly via MVB plasma membrane fusion or via the trans-Golgi network, involving PI4P and PI(4,5)P₂ to regulate vesicle trafficking and exocytosis. (4) Autophagosome formation. Generated at the ER; PI3P produced by class III PI3K recruits WD repeat domain phosphoinositide-interacting protein 2 (WIPI2); promotes phagophore expansion and microtubule-associated protein 1 light chain 3 (LC3) lipidation. Created with BioRender. Seger, F. (2026) https://BioRender.com/s8tc5vs.
Figure 4.
Organelle trafficking, organelle membrane identity, and lipid nanoparticle influence. (1) Internalization. Lipid nanoparticles are taken up by endocytosis and fuse with early endocytic vesicles. May interfere with the physics of plasma and vesicle membranes during uptake and early endosomal trafficking. (a) Early endosome. Defined predominantly by PI3P, with contributions from phosphatidylinositol-4-phosphate (PI4P). PI3P is generated by class III PI3K, which establishes endosomal identity by recruiting effector proteins, such as ESCRT-0 and sorting nexins (SNX), for the sorting of endosomal cargo. (b) Recycling endosome. Enriched in PI(4,5)P₂, PI4P, and PI3P, supporting membrane recycling, resorting of cargo, and rebalancing of phosphatidylinositol species. (c) Late endosome. Characterized by PI3P with increasing levels of PI(3,5)P₂, generated by phosphoinositide kinase, FYVE-type zinc finger containing (PIKfyve). Regulates payload sorting and MVB formation, and sorting toward exosome release or lysosomal fusion. Incomplete endosomal escape may trap ionizable lipids within these compartments. (d) Endolysosomal transition. Characterized by elevated PI(3,5)P₂ with residual PI3P supporting lysosomal maturation processes, including ion channel activity and vacuolar-type H⁺-ATPase (V-ATPase) function. (e) Lysosome. Is dominated by PI(3,5)P₂ and variable PIPs species depending on functional state. Coordinates degradation of intracellular components and ALR. (f) Autolysosome. Contains PI3P derived from autophagosomes & PI(3,5)P₂ derived from lysosomes. During ALR, PI(4,5)P₂ and PI4P are involved, with PI(4,5)P₂ recruiting clathrin and the adaptor protein-2 (AP-2) complex to support membrane tubulation and lysosome reformation. (2) & (3) Exocytosis pathways. Payload release occurs directly via MVB plasma membrane fusion or via the trans-Golgi network, involving PI4P and PI(4,5)P₂ to regulate vesicle trafficking and exocytosis. (4) Autophagosome formation. Generated at the ER; PI3P produced by class III PI3K recruits WD repeat domain phosphoinositide-interacting protein 2 (WIPI2); promotes phagophore expansion and microtubule-associated protein 1 light chain 3 (LC3) lipidation. Created with BioRender. Seger, F. (2026) https://BioRender.com/s8tc5vs.
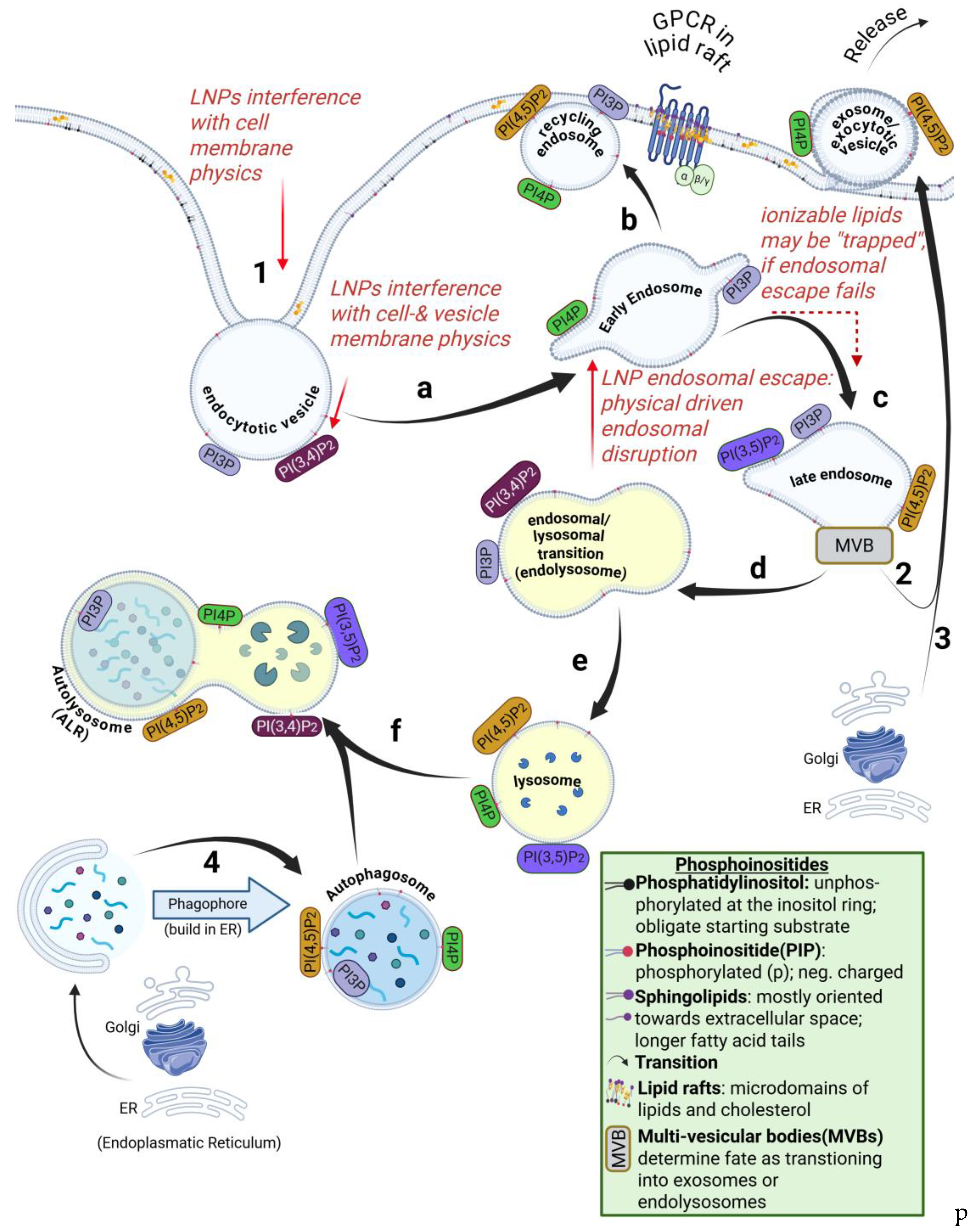
Figure 5.
Nonlinear signaling crosstalk network between the PI3K-AKT-mTOR and RAS-ERK pathways via the phosphatidylinositol (PtdIns) cycle. GPCR activation at the plasma membrane, showing the heterotrimeric G-protein subunits (Gαq, Gβ, and Gγ), triggers PI3K-dependent conversion of PIP₂ to PIP₃, thereby linking receptor signaling to downstream AKT/mTOR and RAS/ERK cascades that regulate cell growth, survival, and metabolic control. Crosstalk to the insulin receptor via RGS proteins is indicated. Created in BioRender. Seger, F. (2025) https://BioRender.com/vl25hmc.
Figure 5.
Nonlinear signaling crosstalk network between the PI3K-AKT-mTOR and RAS-ERK pathways via the phosphatidylinositol (PtdIns) cycle. GPCR activation at the plasma membrane, showing the heterotrimeric G-protein subunits (Gαq, Gβ, and Gγ), triggers PI3K-dependent conversion of PIP₂ to PIP₃, thereby linking receptor signaling to downstream AKT/mTOR and RAS/ERK cascades that regulate cell growth, survival, and metabolic control. Crosstalk to the insulin receptor via RGS proteins is indicated. Created in BioRender. Seger, F. (2025) https://BioRender.com/vl25hmc.
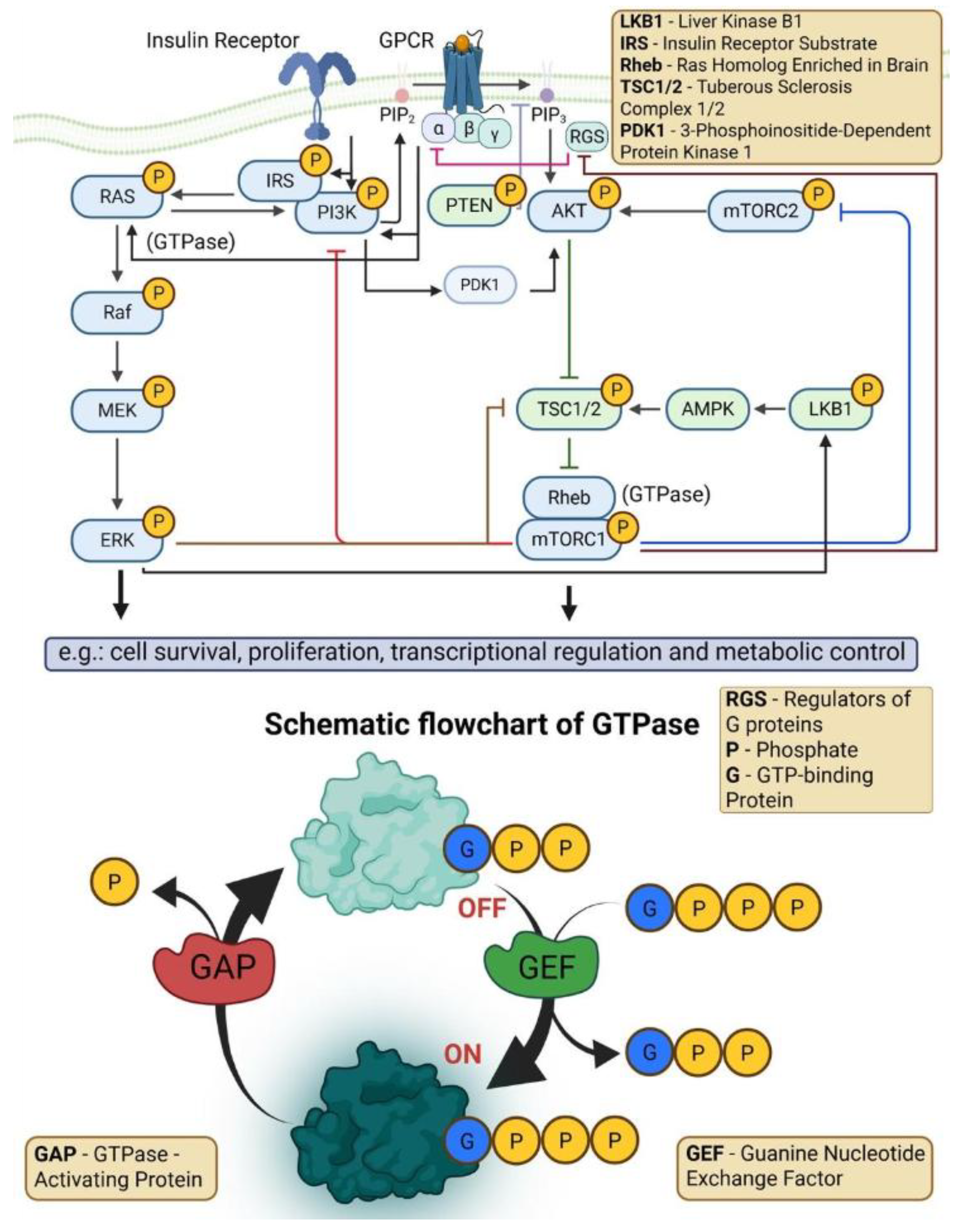

Table 1.
Comparative pharmacology and safety profile of approved RNA-lipid nanoparticle therapeutics.
Table 1.
Comparative pharmacology and safety profile of approved RNA-lipid nanoparticle therapeutics.
| Platform (example) | Route/ Vehicle |
Primary Delivery Mechanism | Approximate Endosomal Escapea | Membrane Activityb | Potential L-DMD Riskc |
Common safety signals e. |
Refs |
|---|---|---|---|---|---|---|---|
| > GalNAc–siRNA (inclisiran) | SC; Ligand–siRNA conjugate; No LNP |
Hepatocyte-targeted receptor uptake (ASGPR) | Sufficient at low doses (no LNP) |
Minimal: negligible disruptive capacity | √ (no ionizable lipids) |
Primarily injection site only | 111, 112 |
| > siRNA–LNP (patisiran) | IV; Ionizable lipid; (DLin-MC3-DMA) |
Hepatic fenestration + ApoE mediated LDL-R | ~1% (model-dependent) | Modest; Transient endosomal lesions repaired by ESCRT |
√√ (hepatotropic, low pKa 6.4) |
↑Transaminases; ↓ vitamin A levels; infusion reactions (CARPA) |
107, 113 |
| >
modRNA–LNP (SARS-CoV-2 vaccines) |
IM; Ionizable lipid; (ALC-0315/ SM-102) |
Immune activation + lymphatic drainage + systemic distribution | ≤~15% (context-dependent) | High; sustained endosomal/ lysosomal engagement |
√√√ (systemic distribution, pKa 6.1-6.7) |
Systemic reactogenicity (fever myalgias); Rare myocarditis; Rare anaphylaxis |
53, 116, 117 |
| >
saRNA–LNP (zapomeran/ SARS-CoV-2 vaccine) |
IM; Ionizable lipid; (ATX-126/LUNAR lipids) |
Prolonged RNA replication + front-loaded lipid-mediated delivery | Variable; amplification design | High; ionizable lipid with saRNA prolonging innate activation |
√√√ (very slow clearance, pKa undefined) |
Systemic reactogenicity ≈modRNA-LNPs; |
19, 115 |
Abbreviations: ApoE, apolipoprotein E; ASGPR, asialoglycoprotein receptor; CARPA, complement activation-related pseudoallergy; EMA, European Medicines Agency; ESCRT, endosomal sorting complexes required for transport; GalNAc, N-acetylgalactosamine; IM, intramuscular; IV, intravenous; LDL-R, low-density lipoprotein receptor; LNP, lipid nanoparticle; LUNAR, Lipid-enabled and Unlocked Nucleomonomer Agent modified RNA; modRNA, nucleotide-modified RNA; pKₐ, acid dissociation constant; SC, subcutaneous; saRNA, self-amplifying RNA; siRNA, small interfering RNA. aEndosomal escape varies by cell type, ionizable lipid pKa and assay method (e.g., fluorescence dequenching, galectin recruitment). Values represent estimates from in vitro and animal models; available human data are sparse. bMembrane activity: Qualitative assessment from EMA/regulatory filings and published mechanistic data; does not imply difference in lipid persistence or systemic half-life. cPotential L-DMD Risk: Based on ionizable lipid pKa, membrane fusogenicity, and biodistribution. √ = minimal, √√√ = highest relative risk category. This represents a proposed mechanistic framework requiring prospective validation, not established clinical risk categories.
Table 2.
Membrane-Associated Signaling Pathways Perturbed by LNP Exposure.
| Section | Pathway | Membrane Connection | Representative Evidence | Reference |
|---|---|---|---|---|
| Membrane Structure and Maintenance | ||||
|
4.1 |
ESCRT/ endocytosis |
Membrane repair following endosomal stress. Recruitment depends on phosphoinositides. |
H-Px: ↓ESCRT proteins and ↓endocytic activity after modRNA-LNP injection. | Hickey et al.139 |
| 4.2 | CYP | ER/mitochondrial embedded membranas. Catalytic geometry depends on local phospholipids and charge. |
↕ M-Tx (eLNP), H-Px, H-Obs: ↓CYP metabolism post IM injection in mice; ↑Phase I enzymes (♂ only, BNT162b2 specific, hepatocellular leakage†); clozapine toxicity post-vaccination (functional corroboration‡). | Ndeupen et al. 119 Hickey et al. 139 Thompson et al. 159 |
| Innate Immune Signaling | ||||
| 4.3 | TLR4 axis (MyD88/ TRIF bias) |
Plasma lipid membrane organization. Intact phosphoinositides needed for balanced signaling. |
M-Tx (eLNP), M-Fx: MyD88-dominant NFκB activation, reversible with TLR-4 blockade (TAK-242) in human THP-1 cells in vitro NFκB.-κB | Ndeupen et al. 119 Korzun et al. 134 *Zelkoski et al.132 |
| 4.4 | Pro-inflammatory cytokines | Membrane-associated PRR signaling (NF-κB/MAPK). Functional organization of opes within inner-leaflet lipid raft. |
↕ (M-Tx, M-Tx (eLNP)), H-Obs: ↑NFκB targets and canonical cytokines in murine eLNP; Concordant upregulation in human datasets; cytokine-mediated drug interactions documented NFκB. -κB |
Ndeupen et al. 119 Korzun et al. 134 McColl et al. 242 |
| 4.4 | Β-Cytokines, CXC- chemokines, IL6, IFNs |
Membrane proximal NF-κB activation. | ↕ (MTx, MFx (eLNP)), H-Tx: Concordant CCL family upregulation in murine eLNP and human buffy-coat transcriptomes (CXCL 9,10).-Tx | Ndeupen et al. 119 Korzun et al. 134 Knabl et al. 138 |
| 4.5 | Complement (C3, lectin) |
Membrane-bound GPCR receptors (C3aR). Protein corona effects. |
M-Tx (eLNP): ↑C3; M-Px: LNP-vitronectin/ficolin-1 binding facilitates C3 activation events |
Korzun et al. 134 Luo et al. 137 |
| Growth and Metabolic Signaling | ||||
| 4.7 | RAS-RAF-MEK-ERK (MAPK) | RAS GTPases lipid anchors at plasma membrane nanodomains. Translate local charge shift to signal amplification. |
↕ M-Tx (eLNP), H-Tx, H-Px: ERK1/KRAS pathway upregulation across species omes. | Korzun et al.134 Knabl et al. 138 Hickey et al. 139 |
| 4.7 | PI3K-AKT-mTOR | Membrane-localized PI3K. Partial mTORC1 engagement indicates upstream dysregulated lipid signaling via RAS. |
↕ M-Tx (eLNP), H-Tx, H-Px: Partial ERK1/KRAS pathway + PI3K modulation post LNP exposure. |
Korzun et al.134 Knabl et al. 138 Hickey et al. 139 |
|
4.4 |
PI3K Class II (PIK3C2G) | Often activated downstream of RTKs + GPCRs. Act independently of regulatory subunits characteristic of PI3Ks. |
M-Tx (eLNP): Consistent ↓PIK3C2G across datasets. | Korzun et al. 134 |
| 4.6 | PPARγ | Integrates lipid-derived signals. Suppression favors NF-κB activation. |
↕ M-Tx (eLNP), H-Obs: ↓PPARγ pathway → loss of anti-inflammatory tone + lipid metabolism imbalance; post-vaccination hypercholesterolemia (OR = 1.54, 95% CI: 1.36–1.74; population level corroboration)§. | Ndeupen et al. 119 Huang et al. 171 |
| 4.6 | AMPK | Senses membrane-linked stress. Suppression favors NF-κB activation. |
M-Tx (eLNP): ↓AMPK transcripts → reduced oxidative metabolism and ↑NFκB. | Ndeupen et al. 119 |
| Cell Cycle and Stress Response | ||||
| 4.8 | p53 + E2F Network | Downstream of RAS-MAPK and PI3K-mTOR signaling. Cell-cycle checkpoint. |
H-Tx: p53 + E2F8; partial MTORC1 activation. | Knabl et al. 138 |
*Not Omics data but included as supportive evidence for the mechanistic inference †Sex- and vaccine-specific enzyme elevation without CAR/PXR indicates leakage, rather than de novo synthesis (Hickey et al) [139]. Section numbers correspond to their respective sections in the main text. ‡Clinical corroboration: Clozapine (CYP1A2/3A4 substrate) toxicity post vaccination provides supports enzyme suppression observed in omics datasets. Observational data included as supportive evidence of pathway engagement. &Cytokine-mediated mechanism reviewed by McColl et al. [242] ; observational reports provide complementary validation. §Population-based association for lipid metabolic imbalance; causality requires further investigation. Abbreviations: H = Human; M = Mouse; Tx = Transcriptomic; Px = Proteomic; Obs = Observational; Fx = Functional.↕ = Crossspecies concordance (Human + Mouse); AKT = Protein kinase B (also PKB); AMPK = AMP-activated protein kinase; C3 = Complement component 3; C3aR = Complement C3a receptor; CAR = Constitutive androstane receptor; CCL = C-C motif chemokine ligand; CI = Confidence interval; CXCL = C-X-C motif chemokine ligand; CYP = Cytochrome P450 enzyme; E2F = E2 transcription factor family; eLNP = Empty lipid nanoparticle (no mRNA payload); ER = Endoplasmic reticulum; ERK = Extracellular signal-regulated kinase; ESCRT = Endosomal sorting complex required for transport; GPCR = G protein-coupled receptor; GTPase = Guanosine triphosphatase; H-Fx, M-Fx = Human / mouse functional (behavioral or physiologic) data; H-Obs = Human clinical or epidemiologic observation; H-Px, M-Px = Human / mouse proteomic data; H-Tx, M-Tx = Human / mouse transcriptomic data; IM = Intramuscular administration; KRAS = Kirsten rat sarcoma viral oncogene homolog; MAPK = Mitogen-activated protein kinase; MEK = Mitogen-activated protein kinase kinase (also MAPK/ERK kinase); modRNA-LNP = Nucleoside-modified mRNA formulation encapsulated in a lipid nanoparticle; mTOR = Mechanistic target of rapamycin; mTORC1 = Mechanistic target of rapamycin complex 1; MyD88 = Myeloid differentiation primary response 88; NF-κB = Nuclear factor kappa B; OR = Odds ratio; p53 = Tumor protein p53; PI3K = Phosphatidylinositol 3-kinase; PIK3C2G = Phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma; PIP = Phosphatidylinositol phosphate; PM = Plasma membrane; PPARγ = Peroxisome proliferator-activated receptor gamma; PRR = Pattern recognition receptor; PXR = Pregnane X receptor; RAF = Rapidly accelerated fibrosarcoma (serine/threonine kinase); RAS = Rat sarcoma (family of small GTPases); RTK = Receptor tyrosine kinase; TLR4 = Toll-like receptor 4; TRIF = TIR-domain-containing adapter-inducing interferon-β; ↕ = Cross-species concordance (Human + Mouse).
Table 3.
Summary of Omic Studies Informing Table 2 (Model, Methods, Route, and Data Type).
Table 3.
Summary of Omic Studies Informing Table 2 (Model, Methods, Route, and Data Type).
| Study and Reference | Model / Species | Material Tested | Route / Dose | Data Type / Readout | Timeline / Endpoints |
|---|---|---|---|---|---|
| Ndeupen et al. [119] | Mouse (C57BL/6, WT). |
eLNP (no mRNA). |
IM 10 µg in PBS |
M-Tx; (Whole tissue RNAseq + GSEA)-seq + GSEA |
~24 h post injection. |
| Korzun et al. [134] | Mouse (WT + KO lines) |
Luc mRNA LNP + eLNP. | Luc mRNA + eLNP groups received 5ug in 100μl PBS; IP single dose (acute); or IP once every 24h for 3 doses (chronic). = 4 groups total eLNP groups received TAK-242 inhibitor 2 hours prior to each dose |
M-Tx, M-FX; (behavior + ELISA). |
6h post last dose |
| Luo et al. [137] | Mouse / Cell models | eLNP (no payload) + comparators. |
Multiple routes (IN, IM, IV, PO, ID); 0.0005–0.5 mg/kg. |
M-Px; (quantitative proteomics). |
≤ 24h post dose-dose |
| Hickey et al. [139] | Human (adults post BNT162b2 / mRNA1273 vaccination)**- | ModRNA-LNP vaccination. | IM; clinical dose. |
H-Px ; (SomaScan v4.1 proteomics). |
1 month and 6 months post-dose dose 3. |
| Knabl et al. [138] | Human (patients and healthy volunteers)** Elderly patients (3 doses; postvaccination or infection, treatment with dexamethasone) Younger healthy group (naive, 1st dose) |
ModRNA-LNP vaccination/ infection cohort. |
IM; clinical dose. |
H-Tx; (whole blood buffy coat transcriptomics) |
Elderly Days 7–60 Younger Days 7–10 |
**Post-vaccination samples; Knabl cohort included patients with concurrent SARS-CoV-2 Beta-variant infection, potentially confounding attribution to LNP-only effects. Hickey cohort represents healthy adults post 3rd dose Abbreviations: H = Human; M = Mouse; Tx = Transcriptomic; Px = Proteomic; Fx = Functional; eLNP, empty lipid nanoparticle; H, human; IM, intramuscular; IP, intraperitoneal; IV, intravenous; Luc, luciferase; SQ, subcutaneous; IN, intranasal; M, mouse; PO, oral; ID, intradermal; Tx, transcriptomic profiling; Px, proteomic profiling; Fx, functional assays; KO, knockout; WT, wild type.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



