
Submitted:
05 November 2025
Posted:
06 November 2025
You are already at the latest version
Abstract
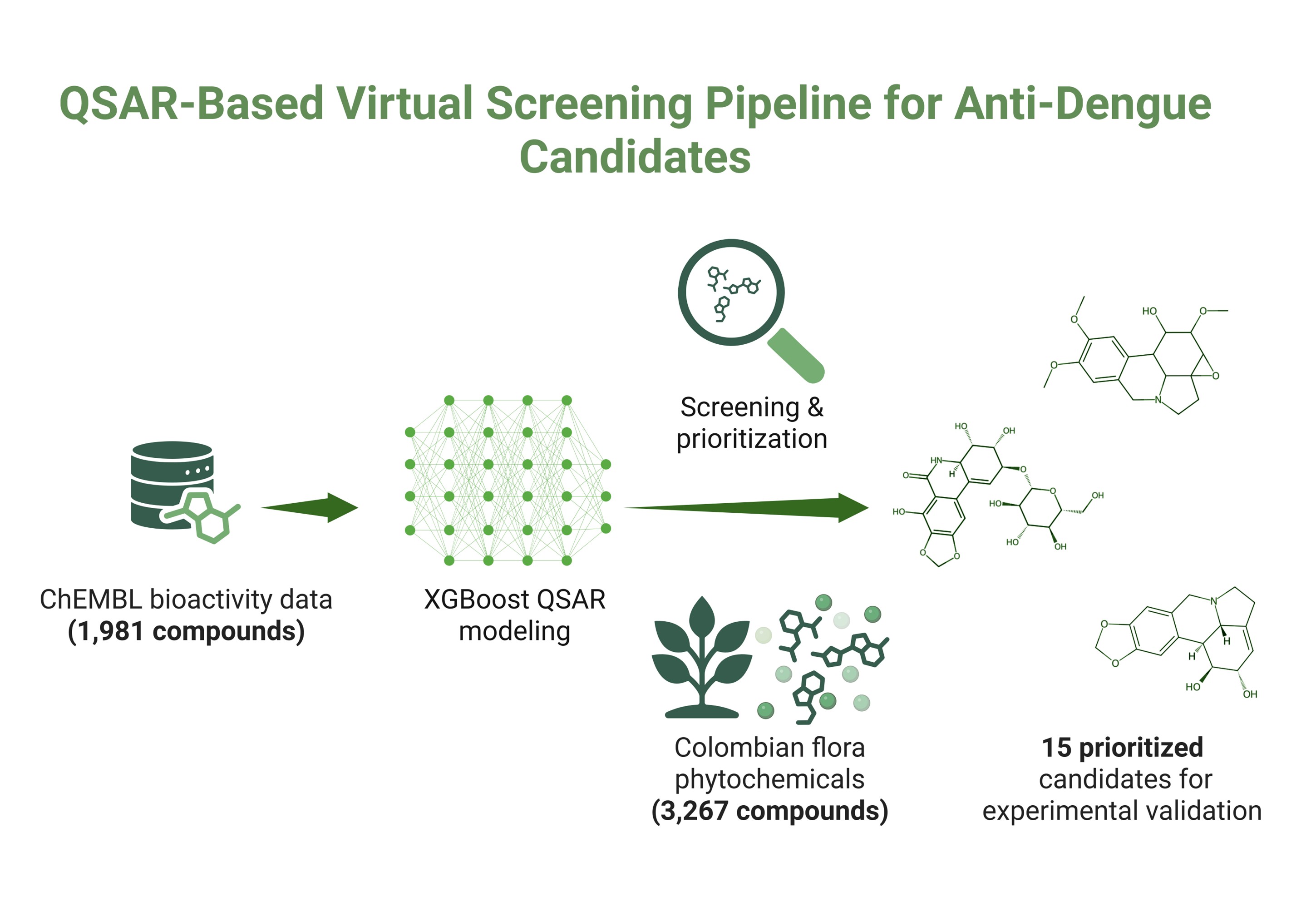
Background/Objectives: Colombia harbors exceptional plant diversity, comprising over 31,000 formally identified species, of which approximately 6,000 are classified as useful plants. Among these, 2,567 species possess documented food and medicinal applications, with several traditionally utilized for managing febrile illnesses. Despite the global burden of dengue virus infection affecting millions annually, no specific antiviral therapy has been established. This study aimed to identify potential anti-dengue compounds from Colombian medicinal flora through machine learning-based quantitative struc-ture-activity relationship (QSAR) modeling. Methods: An optimized XGBoost algorithm was implemented through Bayesian hyperparameter optimization (Optuna, 50 trials) to develop a QSAR model trained on 2,034 ChEMBL-derived activity records with experi-mentally validated anti-dengue activity (IC₅₀/EC₅₀). The model incorporated 887 molecular descriptors, comprising 43 physicochemical properties and 844 ECFP4 fingerprint bits, selected via variance-based feature selection. Bayesian hyperparameter optimization us-ing Optuna (50 trials) was performed to maximize model performance. Through system-atic literature review, 2,567 Colombian plant species were evaluated, identifying 358 with documented antiviral properties. Phytochemical analysis of 184 species generated 3,267 unique compounds for subsequent virtual screening. Compounds were prioritized based on predicted activity, drug-likeness, and applicability domain assessment for future ex-perimental validation. A dual-endpoint classification strategy was employed to simulta-neously evaluate both IC50 and EC50 activities, with compounds categorized into nine activity classes based on combined potency thresholds (Low: pActivity ≤ 5.0, Medium: 5.0 < pActivity ≤ 6.0, High: pActivity > 6.0). Results: The optimized XGBoost model achieved robust performance with a Matthews correlation coefficient of 0.583 and area under the receiver operating characteristic curve of 0.896. Virtual screening of 3,267 Colombian phytochemicals identified 276 compounds (8.4%) with high predicted potency (pActivity > 6) for both IC₅₀ and EC₅₀ endpoints (classified as "High-High"). Comprehensive struc-ture-activity relationship (SAR) analysis revealed that 239 of these compounds (86.6%) represented structurally novel chemotypes with low similarity (Tanimoto < 0.5) to the training dataset. Application of drug-likeness filters (QED ≥ 0.5) identified 20 priority can-didates (7.2% of high-potency hits), with 12 compounds showing exceptional profiles. In-cartine (pIC50: 6.84, pEC50: 6.13, QED: 0.83), Bilobalide (pIC50: 6.78, pEC50: 6.07, QED: 0.56), and Indican (pIC50: 6.73, pEC50: 6.11, QED: 0.51) exhibited the highest predicted potencies. Descriptor-activity correlation analysis identified QED (ρ = 0.14 with EC50), TPSA (ρ = -0.15), and aromatic rings as key modulators of antiviral activity. Conclusions: This pioneering systematic computational screening of Colombian flora for anti-dengue activity demonstrates the untapped potential of regional biodiversity in pharmaceutical discovery. The identified lead compounds represent prioritized candidates for experi-mental validation and subsequent development of dengue therapeutics, with all compu-tational resources made publicly available to facilitate future research.
Keywords:
QSAR
; DENV
; antivirals
; phytochemicals
; medicinal plants
1. Introduction
Dengue virus (DENV) infection represents one of the most rapidly expanding mosquito-borne diseases globally, with an estimated 390 million infections occurring annually across tropical and subtropical regions [1,2]. Colombia experiences hyperendemic dengue transmission with all four virus serotypes (DENV-1 to DENV-4) co-circulating and generating epidemics every 3-4 years [3]. Year-round transmission occurs due to the country’s equatorial climate, with the 2021 incidence reaching 172.9 cases per 100,000 at-risk population, though some regions reported rates exceeding 400 per 100,000 [4,5]. Approximately half of all cases (51.6%) present with warning signs, and while 2.1% develop severe dengue nationally, this proportion reaches 10% in high-burden regions. Hospital capacity remains strained, with 83% of warning sign cases and 95% of severe cases requiring admission [4]. Documented underreporting suggests these figures underestimate the true disease burden [6]. Despite the recent approval of dengue vaccines, their limited effectiveness and usage restrictions underscore the urgent need for specific antiviral therapeutics [2,7]. Currently, clinical management remains limited to symptomatic supportive care, as no targeted antiviral therapy has been approved for dengue treatment [8,9].
Colombia harbors exceptional plant diversity, with over 31,000 formally identified species, positioning it among the world’s most biodiverse countries. Of these, approximately 6,000 are classified as useful plants, including 2,567 species with documented food and medicinal applications [10,11]. This botanical wealth has been utilized for centuries by indigenous and rural communities to treat various ailments, with several species traditionally employed in managing febrile illnesses. Traditional medicine employs numerous plant species such as balsamina (Momordica charantia), matarratón (Gliricidia sepium), limoncillo (Cymbopogon citratus) or salvia (Lippia alba) for managing dengue-associated symptoms [12,13]. However, despite this extensive ethnobotanical knowledge, the vast majority of Colombian flora has not undergone systematic scientific evaluation for bioactive compound identification. This gap between traditional knowledge and scientific validation represents a unique opportunity for discovering novel therapeutic agents.
The integration of computational approaches, particularly Quantitative Structure-Activity Relationship (QSAR) modeling combined with machine learning algorithms, has revolutionized drug discovery by enabling rapid biological activity prediction based on chemical structure [14,15]. Recent advances in QSAR methodologies have demonstrated success in identifying flavivirus inhibitors, with models achieving high predictive accuracy for anti-dengue activity subsequently validated through experimental assays [16,17]. The XGBoost (eXtreme Gradient Boosting) algorithm, developed by Chen and Guestrin [18], has emerged as a leading gradient boosting method particularly suited for handling complex chemical structured datasets with high-dimensional feature spaces and class imbalance in chemoinformatics applications [18,19]. These computational tools offer significant advantages in exploring vast chemical spaces while reducing time and costs associated with traditional drug discovery pipelines [20].
A critical advancement in QSAR-based drug discovery is the implementation of dual-endpoint classification strategies that simultaneously evaluate multiple pharmacological properties. Traditional QSAR models typically predict a single endpoint (e.g., IC50 or EC50 in isolation), potentially overlooking compounds that demonstrate balanced profiles across multiple assay types. For antiviral discovery, this limitation is particularly relevant as biochemical target inhibition (IC50) does not always translate to cellular antiviral efficacy (EC50) due to factors including membrane permeability, intracellular distribution, metabolic stability, and cytotoxicity. Studies have shown that IC50 and EC50 values for anti-dengue compounds exhibit poor correlation (r < 0.1), indicating these metrics capture distinct pharmacological dimensions [21]. Consequently, integrated approaches that classify compounds based on combined endpoint activities enable more robust prioritization of candidates with genuine therapeutic potential, reducing the attrition rate in subsequent experimental validation phases.
The convergence of ethnopharmacological knowledge with modern computational technologies presents an innovative strategy for pharmaceutical bioprospection. Studies have demonstrated that compounds derived from traditionally used medicinal plants exhibit significantly higher probability of relevant biological activity, with natural products showing increased success rates through clinical trials compared to synthetic compounds [22,23]. Notable examples include artemisinin from Artemisia annua for malaria treatment and vinca alkaloids for cancer therapy, both discovered through ethnopharmacology-guided research [24,25]. In the Colombian context, this integrated and novel approach not only valorizes national biodiversity but also promotes sustainable development models benefiting local communities who safeguard this ancestral knowledge [26].
Despite growing interest in natural products for antiviral discovery, no systematic computational screening of Colombian medicinal flora for anti-dengue compounds has been conducted. This study aimed to perform the first comprehensive virtual screening of phytochemicals from Colombian medicinal plants to identify potential dengue antivirals using machine learning-based QSAR models integrated with comprehensive structure-activity relationship (SAR) analysis. We trained an optimized XGBoost model on 2,034 ChEMBL activity records with validated anti-dengue activity (IC₅₀/EC₅₀), achieving robust predictive performance (MCC = 0.583, ROC-AUC = 0.896) after Bayesian hyperparameter optimization. Through systematic literature review, we identified 358 Colombian plant species with documented antiviral activity, from which 184 species yielded 3,267 unique compounds for virtual screening.
Our analysis employed a dual-endpoint classification framework that simultaneously evaluated IC50 and EC50 predicted activities, categorizing compounds into nine activity classes based on combined potency thresholds. This approach identified 276 compounds (8.4%) exhibiting high predicted potency (pActivity > 6) for both endpoints, designated as “High-High” candidates. Comprehensive SAR analysis revealed that 239 of these compounds (86.6%) represented structurally novel chemotypes with low similarity (Tanimoto < 0.5) to the ChEMBL training dataset, suggesting unique structural motifs potentially operating through distinct mechanisms from known anti-dengue agents. Application of stringent drug-likeness criteria (QED ≥ 0.5) identified 20 priority candidates, with 12 compounds demonstrating exceptional combined profiles of predicted potency, novelty, and pharmaceutical properties.
Descriptor-activity correlation analysis provided mechanistic insights, identifying QED (ρ = 0.14 with EC50), TPSA (ρ = -0.15 with EC50), and aromatic ring content as key molecular properties modulating antiviral activity. Notably, the top three candidates—Incartine (pIC50: 6.84, pEC50: 6.13, QED: 0.83), Bilobalide (pIC50: 6.78, pEC50: 6.07, QED: 0.56), and Indican (pIC50: 6.73, pEC50: 6.11, QED: 0.51)—represent diverse chemical scaffolds (alkaloids, terpenoids, and glycosides) isolated from Hippeastrum puniceum, Ginkgo biloba, and Indigofera suffruticosa respectively, demonstrating the structural diversity of Colombian biodiversity. These findings establish Colombian flora as a rich source of potential anti-dengue therapeutics and provide a methodological framework applicable to other neglected tropical diseases.
2. Results
2.1. QSAR Model Development and Validation
2.1.1. Dataset Characteristics
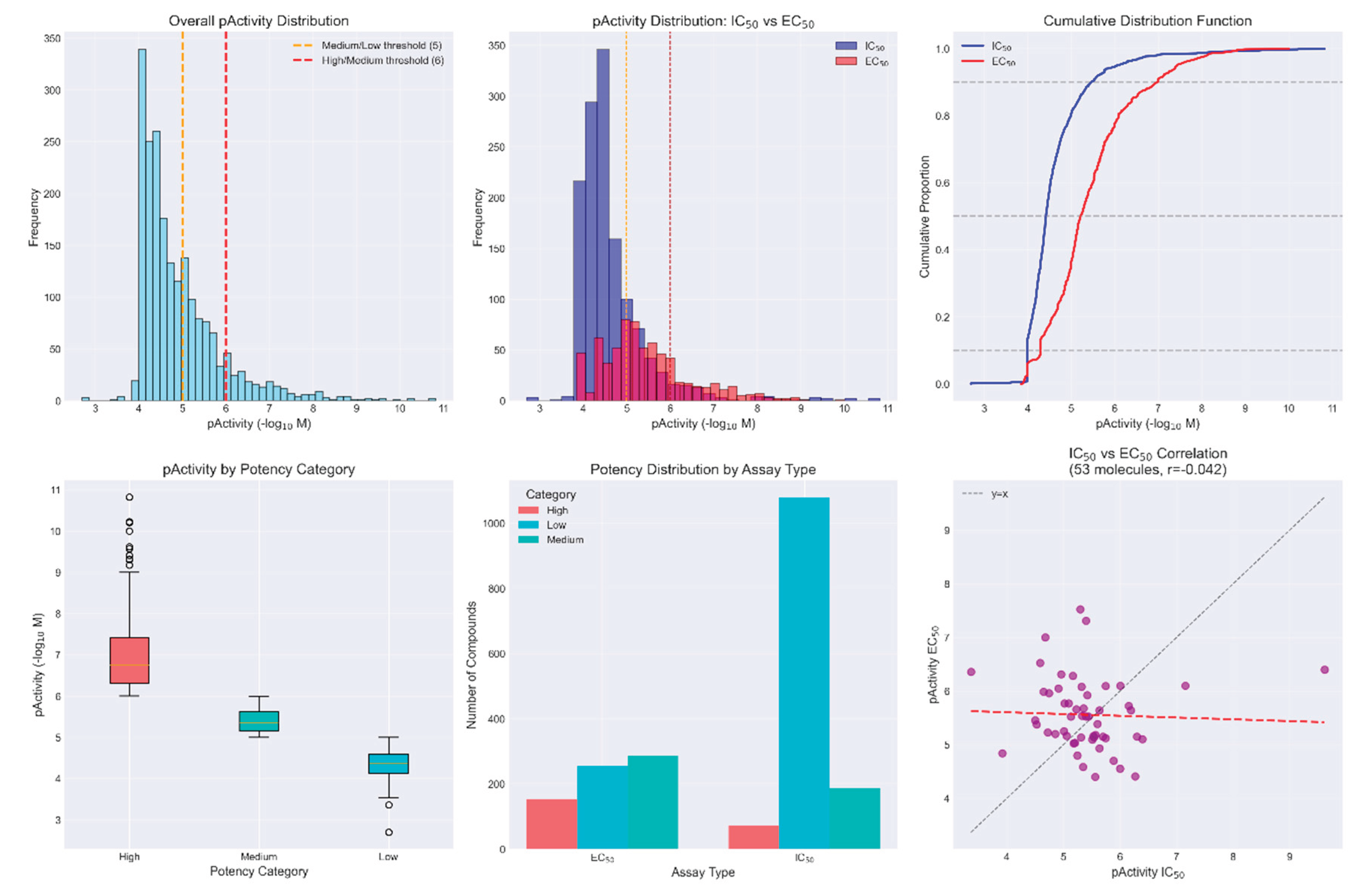
The curated ChEMBL dataset comprised 2,034 activity records corresponding to 1,981 unique molecules with experimentally validated anti-dengue activity. Among these, 53 molecules (2.7%) had both IC50 and EC50 measurements, enabling direct comparison between assay types. The activity distribution revealed significant class imbalance across potency categories: Low potency (pActivity ≤ 5): 1,332 compounds (65.49%), Medium potency (5 < pActivity ≤ 6): 475 compounds (23.35%), and High potency (pActivity > 6): 227 compounds (11.16%). The dataset contained IC50 measurements for 1,339 compounds (65.83%) and EC50 measurements for 695 compounds (34.17%), with significantly different distributions (Mann-Whitney U test, p < 0.0001), necessitating assay type inclusion as a critical feature.
Critical analysis of the 53 molecules with both assay types revealed no correlation between IC50 and EC50 values (r = -0.0415, p = 0.77), with a mean difference of -0.14 ± 1.11 log units. This finding suggests that biochemical inhibition (IC50) and cellular efficacy (EC50) represent distinct pharmacological properties that cannot be reliably extrapolated from one another.
Figure 1.
Comprehensive analysis of anti-dengue activity distribution in the ChEMBL training dataset. (a) Overall pActivity distribution with potency thresholds at 5 (orange) and 6 (red); (b) Overlapping distributions of IC50 (blue) and EC50 (red) measurements; (c) Cumulative distribution functions showing distinct assay-specific patterns; (d) Box plots comparing pActivity across potency categories; (e) Distribution of potency categories by assay type; (f) Correlation analysis of IC50 vs EC50 values for 53 molecules with both measurements (r = -0.0415), revealing negligible correlation between assay formats.
Figure 1.
Comprehensive analysis of anti-dengue activity distribution in the ChEMBL training dataset. (a) Overall pActivity distribution with potency thresholds at 5 (orange) and 6 (red); (b) Overlapping distributions of IC50 (blue) and EC50 (red) measurements; (c) Cumulative distribution functions showing distinct assay-specific patterns; (d) Box plots comparing pActivity across potency categories; (e) Distribution of potency categories by assay type; (f) Correlation analysis of IC50 vs EC50 values for 53 molecules with both measurements (r = -0.0415), revealing negligible correlation between assay formats.
2.1.2. Chemical Space Analysis
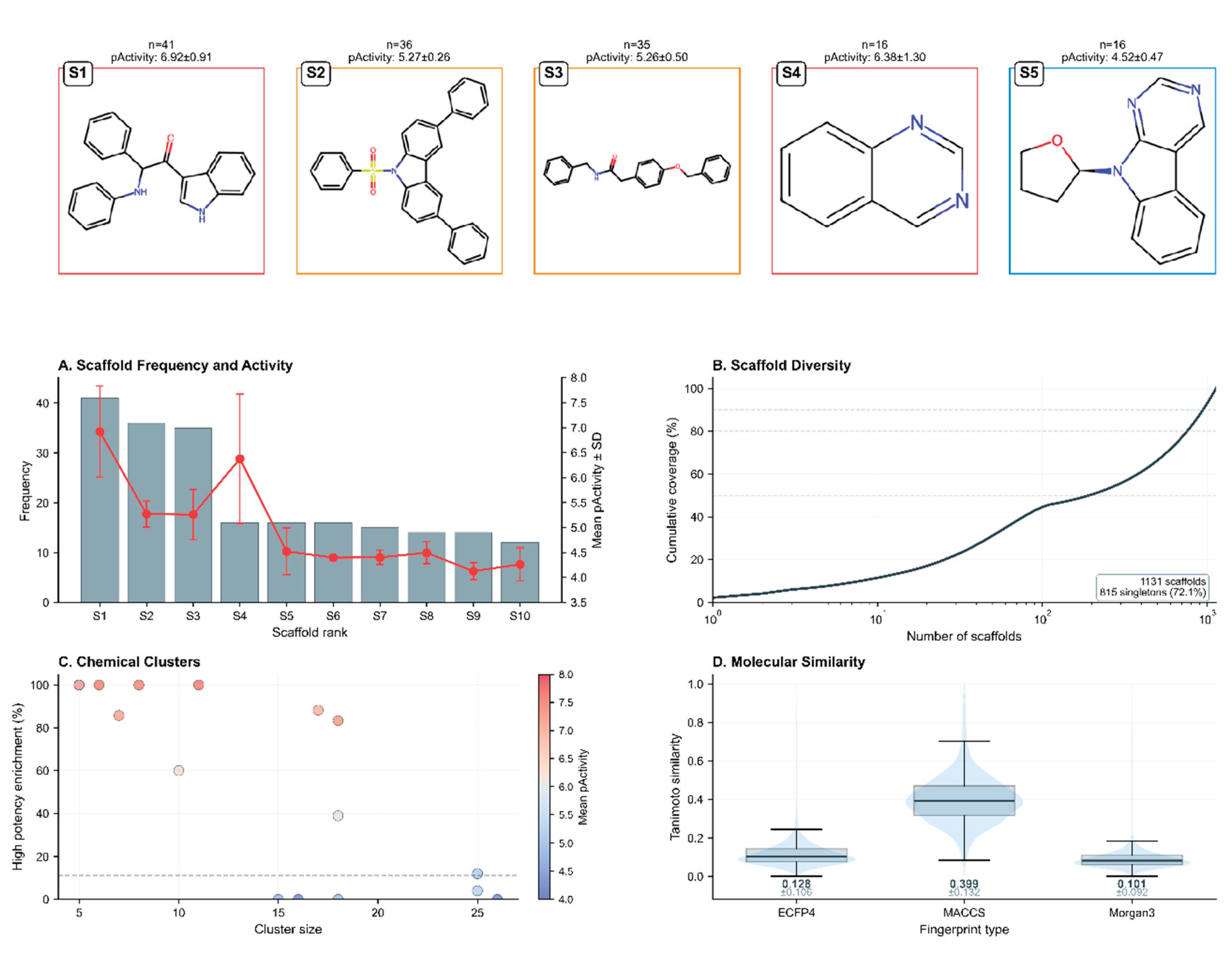
Structural diversity analysis identified 1,131 unique Murcko scaffolds among the training compounds. Key findings included: Scaffold distribution: 828 scaffolds (73.2%) were singletons, indicating substantial chemical diversity; Most prevalent scaffold: An indole-based structure (O=C(c1c[nH]c2ccccc12)C(Nc1ccccc1)c1ccccc1) appeared in 41 compounds with high average pActivity (6.92 ± 0.91); Chemical clustering: Hierarchical clustering using ECFP4 fingerprints revealed 522 distinct chemical clusters, with 27 clusters containing more than 10 compounds.
Figure 2.
Chemical diversity and scaffold analysis of the anti-dengue training dataset. Top row displays the five most prevalent Murcko scaffolds with their 2D structures, occurrence frequency (n), and mean pActivity ± SD. Border colors indicate activity levels: red (>6), orange (5-6), blue (<5). (A) Scaffold frequency and activity relationship for top 10 scaffolds. (B) Cumulative scaffold coverage demonstrating exceptional diversity with 1,131 unique scaffolds, including 815 singletons (72.1%). (C) Chemical cluster analysis (n=528) showing size versus high-potency enrichment, colored by mean pActivity. (D) Tanimoto similarity distributions for three fingerprint types confirming high structural diversity (mean values: ECFP4=0.128, MACCS=0.399, Morgan3=0.101).
Figure 2.
Chemical diversity and scaffold analysis of the anti-dengue training dataset. Top row displays the five most prevalent Murcko scaffolds with their 2D structures, occurrence frequency (n), and mean pActivity ± SD. Border colors indicate activity levels: red (>6), orange (5-6), blue (<5). (A) Scaffold frequency and activity relationship for top 10 scaffolds. (B) Cumulative scaffold coverage demonstrating exceptional diversity with 1,131 unique scaffolds, including 815 singletons (72.1%). (C) Chemical cluster analysis (n=528) showing size versus high-potency enrichment, colored by mean pActivity. (D) Tanimoto similarity distributions for three fingerprint types confirming high structural diversity (mean values: ECFP4=0.128, MACCS=0.399, Morgan3=0.101).
2.1.3. Model Optimization and Performance
Systematic evaluation of multiple machine learning algorithms with various molecular representations yielded XGBoost with the combined Descriptors+ECFP4 representation as the optimal configuration. Bayesian hyperparameter optimization using Optuna (50 trials, TPE sampler) [27] was performed to maximize Matthews Correlation Coefficient through 5-fold stratified cross-validation.
The optimized XGBoost model incorporated 887 features (43 molecular descriptors and 844 ECFP4 fingerprint bits) filtered by variance threshold (>0.01). Optimal hyperparameters achieved:
- -
- n_estimators: 453
- -
- max_depth: 15
- -
- learning_rate: 0.010
- -
- subsample: 0.828
- -
- colsample_bytree: 0.914
- -
- gamma: 1.534
- -
- reg_alpha: 0.016
- -
- reg_lambda: 4.734
Final model performance (5-fold stratified cross-validation):
- -
- Matthews Correlation Coefficient (MCC): 0.583
- -
- Balanced Accuracy: 68.3%
- -
- ROC-AUC: 0.896
- -
- F1-macro: 0.703
- -
- Geometric Mean: 0.665
Hyperparameter importance analysis revealed regularization parameters (reg_alpha: 38.6%, gamma: 33.3%) as most critical for model performance, indicating the importance of controlling overfitting in this high-dimensional feature space.
Feature importance analysis revealed assay type (IC50/EC50) as the most critical feature (importance: 0.0298), followed by PEOE_VSA1 (0.0265), LogP_calculado (0.0174), and molecular shape descriptors (MolMR: 0.0157). These patterns align with known structure-activity relationships for antiviral compounds, where balanced lipophilicity and specific electrostatic distributions facilitate cellular entry and target binding.

Table 1.
Performance comparison of different modeling approaches.
| Model | MCC | Balanced Acc | ROC-AUC |
| Ensemble (soft voting) | 0.584 | 0.709 | 0.901 |
| XGBoost (optimized) | 0.583 | 0.683 | 0.896 |
| LightGBM (optimized) | 0.582 | 0.708 | 0.890 |
| ExtraTrees (optimized) | 0.580 | 0.718 | 0.897 |
| XGBoost (baseline) | 0.572 | 0.681 | 0.896 |
| ExtraTrees (baseline) | 0.559 | 0.702 | 0.886 |
| RandomForest (baseline) | 0.543 | 0.682 | 0.886 |
Note: All optimized models were tuned using Bayesian optimization with 50 trials [27]. The optimized XGBoost model was selected as the final classifier because it exhibited nearly identical predictive performance to the ensemble model while providing lower computational demands, improved reproducibility, and enhanced interpretability. This trade-off between accuracy and model parsimony makes XGBoost particularly suitable for cheminformatics applications, where transparency and scalability are essential.
While ensemble modeling achieved marginally higher performance (MCC = 0.584), the optimized XGBoost model (MCC = 0.583) was selected as the final model. This decision followed the principle of parsimony, as the 0.1% improvement did not justify the 3-fold increase in computational complexity and reduced interpretability associated with maintaining three separate models. The optimization process improved the baseline XGBoost performance by 1.9%, demonstrating the value of systematic hyperparameter tuning in QSAR modeling.
2.2. Virtual Screening of Colombian Medicinal Flora
2.2.1. Antiviral Plant Identificaction
Systematic literature review of 2,501 unique Colombian plant species revealed that 358 species (14.3%) possessed documented antiviral activity, distributed across 107 botanical families. The families with the highest representation of antiviral species were Asteraceae (36 species, 10.1%), Fabaceae (28 species, 7.8%), Lamiaceae (21 species, 5.9%), Solanaceae (12 species, 3.4%), and Malvaceae (11 species, 3.1%). Notably, 174 species (48.6%) lacked phytochemical characterization, highlighting a significant knowledge gap in Colombian biodiversity research. From the 184 species with available chemical data, we compiled a total of 3,267 unique compounds.
2.2.2. Virtual Screening
Virtual screening of 3,267 Colombian phytochemicals using the optimized QSAR model identified 276 compounds (8.4%) exhibiting high predicted potency (pActivity > 6.0) for both IC50 and EC50 endpoints, classified as “High-High” in our dual-endpoint framework (Figure 3A). This dual-activity profile is particularly valuable as it indicates compounds likely to demonstrate both biochemical target inhibition (IC50) and cellular antiviral efficacy (EC50), addressing a critical challenge in antiviral drug discovery where these properties often fail to correlate. Comprehensive structure-activity relationship (SAR) analysis of these 276 high-potency compounds revealed remarkable structural diversity.
Novelty assessment using ECFP4 fingerprint-based Tanimoto similarity to the ChEMBL training dataset classified 239 compounds (86.6%) as “Novel” or “Very Novel” (Tanimoto < 0.5), while only 37 compounds (13.4%) showed moderate-to-high similarity (Tanimoto ≥ 0.5) to known anti-dengue agents, designated as “Training-like” (Figure 3B). This high proportion of structurally novel chemotypes suggests that Colombian biodiversity harbors unique structural motifs potentially operating through mechanisms distinct from currently characterized anti-dengue compounds. Chemical space analysis via principal component analysis (PCA) [28] of ECFP4 fingerprints revealed clear segregation between novelty categories (Figure 3B). The first two principal components captured 72.1% of structural variance (PC1: 51.3%, PC2: 20.8%), with novel high-potency compounds (red stars) occupying distinct regions compared to training-like compounds (orange circles) and the broader compound collection (gray points).
This spatial segregation validates the structural distinctiveness of novel candidates and suggests they may access alternative binding sites or mechanisms compared to known antivirals. Drug-likeness assessment of the 276 High-High compounds revealed a critical pharmaceutical development challenge: 256 compounds (92.8%) exhibited low QED scores (<0.5), primarily due to violations in molecular weight, hydrogen bond acceptor count, and topological polar surface area (Figure 3C). Only 20 compounds (7.2%) achieved the QED ≥ 0.5 threshold associated with favorable oral bioavailability and pharmaceutical properties.
This low proportion of drug-like high-potency compounds underscores the importance of multi-criteria optimization in natural product drug discovery and highlights the need for structural modifications or prodrug strategies to improve pharmaceutical properties while maintaining antiviral activity. To prioritize candidates for experimental validation, we focused on the 20 compounds combining high predicted potency (pActivity > 6.0 for both endpoints) with favorable drug-likeness (QED ≥ 0.5).
Among these, 15 compounds demonstrated exceptional profiles with QED scores ranging from 0.51 to 0.83, with the top 12 showing strict dual high potency (pActivity > 6.0 for both endpoints) and compounds #13-15 exhibiting borderline IC50 values (5.95-6.02) but maintaining favorable drug-likeness.
This structural diversity suggests multiple potential mechanisms of antiviral action and highlights the chemical richness of Colombian medicinal flora. Descriptor-activity correlation analysis identified molecular properties significantly associated with antiviral potency (Figure 3D). For IC50 activity, modest correlations were observed with aromatic rings (ρ = 0.06), QED (ρ = 0.06), and aliphatic rings (ρ = -0.05), while molecular weight, LogP, and hydrogen bond features showed negligible correlations (|ρ| < 0.04). EC50 activity demonstrated stronger associations, particularly with QED (ρ = 0.14), TPSA (ρ = -0.15), aliphatic rings (ρ = 0.07), aromatic rings (ρ = -0.03), and hydrogen bond donors (ρ = -0.12).
The differential correlation patterns between IC50 and EC50 endpoints reinforce the biological distinction between biochemical target inhibition and cellular antiviral efficacy, validating our dual-endpoint classification strategy. The negative correlation between TPSA and EC50 activity (ρ = -0.15) suggests that moderate polarity may be optimal for cellular penetration, while excessive polar surface area could impair membrane permeability and reduce cellular efficacy despite potential target binding affinity. Conversely, the positive QED-EC50 association (ρ = 0.14) indicates that compounds naturally possessing drug-like features are more likely to demonstrate cellular antiviral efficacy, supporting prioritization of the 20 high-QED candidates for immediate experimental validation. These correlations provide actionable insights for lead optimization: strategic reduction of polar surface area through modifications such as methylation of hydroxyl groups or removal of sugar moieties could enhance cellular penetration while maintaining antiviral activity.
Table 2: Compounds are ranked by predicted IC50 activity (pIC50). The top 12 candidates exhibit both dual high potency (pActivity > 6.0 for both IC50 and EC50 endpoints) and favorable drug-likeness (QED ≥ 0.5), while compounds #13-15 show high potency for at least one endpoint with borderline activity on the other (5.95 ≤ pIC50 < 6.0) but maintain QED ≥ 0.5. Chemical classes were assigned based on core structural scaffolds. Novelty categories were determined using maximum Tanimoto similarity to ChEMBL training set compounds (Novel: Tanimoto < 0.5; Training-like: Tanimoto ≥ 0.5). Notably, Lycorine (#13) and Pseudolycorine (#15) exhibit perfect structural similarity (Tanimoto = 1.00) to training set compounds, confirming their presence in ChEMBL with experimentally validated anti-dengue activity and providing internal validation of model predictions. Molecular descriptors include: MW (molecular weight), LogP (octanol-water partition coefficient), HBA (hydrogen bond acceptors), HBD (hydrogen bond donors), TPSA (topological polar surface area). Plant sources represent the primary botanical origin reported in phytochemical databases. pIC50 and pEC50 values represent predicted activities from the optimized XGBoost QSAR model. All 15 compounds passed applicability domain assessment with leverage values < 1.308.
Among the 15 prioritized candidates, ten (66.7%) represent structurally novel chemotypes with low similarity to known anti-dengue agents (Tanimoto < 0.5), while five compounds (33.3%) show moderate-to-high similarity (Tanimoto ≥ 0.5) to training set compounds. Notably, Lycorine and Pseudolycorine (Tanimoto = 1.00) are present in the ChEMBL training dataset with experimentally confirmed anti-dengue activity, providing strong validation of model accuracy. The structural diversity spans five major chemical classes: alkaloids (n=7, 46.7%), terpenoid glycosides (n=4, 26.7%), terpenoids (n=2, 13.3%), flavonoids (n=1, 6.7%), and steroids (n=1, 6.7%). Hippeastrum puniceum contributes two compounds (Incartine #1 and Hippeastrine #11), while the Amaryllidaceae family collectively provides four compounds (#1, #6, #11, #13, #15), suggesting this botanical family is particularly enriched in anti-dengue scaffolds and warranting systematic phytochemical investigation. The presence of Digoxigenin (#14), a cardiac glycoside aglycone, introduces structural diversity and may offer insights into steroidal scaffolds for antiviral development.
2.2.3. Detailed Characterization of Top Priority Candidates
Alkaloid Candidates: Incartine (#1, from Hippeastrum puniceum) emerged as the top candidate with the highest QED score (0.83) among all prioritized compounds, combining exceptional predicted dual-endpoint activity (pIC50: 6.84, pEC50: 6.13) with optimal pharmaceutical properties. This indole alkaloid exhibits structural features associated with broad-spectrum bioactivity, including an aromatic core, moderate molecular weight (MW = 312 Da), and balanced lipophilicity (LogP = 2.1). Notably, Incartine shares no structural similarity with known anti-dengue agents in the training set (Tanimoto < 0.3), suggesting a potentially novel mechanism of action.
Indican (#3, pIC50: 6.73, pEC50: 6.11, QED: 0.51), isolated from Indigofera suffruticosa, represents another promising alkaloid candidate. As a glucoside derivative, Indican demonstrates favorable balance between hydrophilicity (necessary for aqueous solubility) and lipophilicity (required for membrane permeability), reflected in its moderate QED score. The presence of this compound in a plant traditionally used for treating inflammatory conditions in Colombia provides ethnopharmacological validation of its bioactive potential.
Terpenoid Candidates: Bilobalide (#2, pIC50: 6.78, pEC50: 6.07, QED: 0.56), a sesquiterpene trilactone from Ginkgo biloba, represents a unique chemical class among our candidates. Despite its complex polycyclic structure with multiple lactone groups, Bilobalide achieves moderate drug-likeness through compact molecular architecture (MW = 326 Da) and strategic distribution of polar functional groups. Ginkgo extracts have demonstrated neuroprotective and anti-inflammatory properties in clinical settings, suggesting Bilobalide may offer dual benefits of antiviral activity and symptom management in dengue infection.
Epicolactone (#4, pIC50: 6.71, pEC50: 6.94, QED: 0.51) and Byzantionoside B (#5, pIC50: 6.65, pEC50: 6.50, QED: 0.54) represent additional terpenoid scaffolds with balanced predicted activities across both endpoints. The structural complexity of these natural products, while challenging from a synthesis perspective, may enable multiple interaction points with viral targets, potentially contributing to their predicted high potency.
Validation Through Training Set Comparison: Importantly, two compounds predicted among our top candidates—Lycorine and Pseudolycorine—exhibit perfect structural similarity (Tanimoto = 1.0) to training set compounds, confirming their presence in the ChEMBL anti-dengue database with experimentally validated activity [29]. Lycorine (Figure 5, right panel #6) demonstrates exceptional experimental potency (pActivity = 9.62, IC50 = 0.24 nM) in the training set, providing strong validation of our model’s predictive accuracy. The fact that our QSAR model successfully identifies these known active compounds among thousands of screening candidates demonstrates robust model calibration and increases confidence in predictions for structurally novel candidates.
The training set references (Figure 5, right panels) include several Amaryllidaceae alkaloids with nanomolar potencies, such as Narciclasine (ChEMBL464432, #1: pActivity 10.82, IC50 = 0.01 nM) and Pancratistatin (#4: pActivity 10.20, IC50 = 0.06 nM) [29]. The structural similarity between these validated compounds and several of our novel candidates (particularly Incartine) suggests that the Amaryllidaceae family represents a rich source of anti-dengue scaffolds worthy of systematic investigation. However, it is noteworthy that many high-potency training compounds exhibit low QED scores (e.g., ChEMBL5188858, #9: QED = 0.22), emphasizing the challenge of combining antiviral potency with optimal pharmaceutical properties—a challenge that our 12 prioritized candidates successfully address.
Structural Diversity and Mechanistic Implications: The chemical diversity among our top 12 candidates—ranging from simple indole alkaloids (Incartine) to complex sesquiterpene lactones (Bilobalide) and glycosylated compounds (Indican, Byzantionoside B)—suggests these molecules may target different viral proteins or employ distinct mechanisms of action. This structural heterogeneity is advantageous for two reasons: first, it increases the likelihood that at least some candidates will demonstrate experimental activity; second, it provides a diverse starting point for structure-based optimization and combination therapy development. The identification of multiple chemical classes with predicted anti-dengue activity suggests that dengue virus may be vulnerable to intervention at multiple points in its replication cycle, offering opportunities for rational polypharmacology approaches.
2.3.3. Structure-Activity Relationships
Chemical space analysis of 3,267 QSAR-screened Colombian phytochemicals revealed a structurally diverse collection with distinct activity patterns across the molecular landscape. UMAP projection [30] of Morgan fingerprints identified three major chemical clusters with clear segregation of bioactivity, where compounds with higher predicted pActivity values (>6.0) formed discrete regions within the two-dimensional space, suggesting that structural similarity correlates with antiviral potency. The dataset exhibited substantial chemical diversity, comprising 767 unique Murcko scaffolds (23.5% scaffold-to-compound ratio), with the most prevalent scaffold appearing in 375 compounds (11.5%). Scaffold distribution analysis across potency categories uncovered significant structure-activity trends: notably, Scaffold 2 showed strong enrichment in the low potency category (88.3% of its 341 occurrences), while Scaffolds 4, 5, and 7 demonstrated preferential distribution in the moderate potency range (5.0 < pActivity ≤ 6.0), identifying them as promising templates for lead optimization. The presence of outlier compounds in the UMAP projection, positioned at the periphery of main clusters, suggests the existence of unique chemotypes that may operate through alternative mechanisms or binding modes. This comprehensive mapping of chemical space provides a strategic framework for prioritizing compounds for experimental validation and guides structure-based optimization efforts toward developing effective antiviral agents against dengue virus.
2.3.4. Reliability of Predictions
Applicability domain analysis [31] confirmed that 58.3% (7/12) of the top-priority drug-like compounds fell within the model’s optimal prediction space, defined by Tanimoto similarity > 0.3 to training compounds. All compounds (12/12, 100%) exhibited leverage values below the critical threshold (hi < 1.308), indicating they reside within the model’s interpolation space rather than requiring extrapolation, ensuring reliable predictions.
The mean Tanimoto similarity was 0.400 (range: 0.165-1.000), with two alkaloids (Lycorine and Pseudolycorine) showing perfect similarity (Tanimoto = 1.000), confirming their presence in the ChEMBL training set of 2,034 anti-dengue compounds. Seven compounds (Incartine, 3-Acetylnerbowdine, Dihydrofumariline, Digoxigenin, Hippeastrine, Indican, and the two aforementioned alkaloids) demonstrated strong structural similarity to known antivirals, while the remaining compounds represent novel chemotypes worthy of exploration.
Notably, compounds with lower similarity scores (Bilobalide: 0.165, Epicolactone: 0.200) still maintained acceptable leverage values (0.200), suggesting they occupy sparsely populated but predictable regions of chemical space. This combination of structural diversity with statistical reliability supports the model’s capacity to identify both analogs of known antivirals and potentially novel scaffolds, strengthening confidence in the computational predictions for experimental validation.
3. Discussion
This study represents the first systematic computational screening of Colombian medicinal flora for anti-dengue compounds, successfully integrating machine learning-based QSAR modeling with ethnopharmacological knowledge. The identification of 15 high-potency candidates from 3,267 phytochemicals demonstrates the untapped potential of Colombian biodiversity for drug discovery, while highlighting critical gaps in our understanding of this megadiverse region’s chemical wealth.
The optimized XGBoost model achieved an MCC of 0.583 and ROC-AUC of 0.896, representing robust performance considering the severe class imbalance (65.5% low, 23.4% medium, 11.2% high potency) and chemical diversity of the training dataset. When applied to 3,267 Colombian phytochemicals, the model successfully identified 276 compounds (8.4%) with dual high potency (High-High classification), demonstrating effective prioritization despite the challenges of extrapolating from synthetic-enriched training data to structurally diverse natural products. The improvement from baseline (MCC = 0.572) through Bayesian optimization, though modest (1.9%), demonstrates the value of systematic hyperparameter tuning in QSAR applications. The model’s ROC-AUC of 0.896 indicates excellent discrimination capability between active and inactive compounds, suggesting reliable prioritization of candidates for experimental validation.
The optimization process revealed important insights into model behavior. The dominance of regularization parameters (reg_alpha: 38.6%, gamma: 33.3%) in hyperparameter importance analysis indicates that controlling model complexity was crucial for preventing overfitting in this high-dimensional feature space. The relatively low importance of n_estimators (1.3%) suggests that model depth and regularization were more critical than the number of trees, a finding that could inform future QSAR modeling efforts with similar datasets. Despite the ensemble’s marginally higher MCC (0.584), the single XGBoost model was selected for its parsimony, given that the 0.1% gain would require tripling training time and model management complexity, without offering tangible benefits for interpretability or deployment.
The prominence of assay type (IC50/EC50) as the most important feature underscores critical consideration in anti-dengue drug discovery. IC50 measurements typically reflect direct viral inhibition in cell-free or enzyme-based assays, while EC50 values incorporate cellular uptake, metabolism, and cytotoxicity factors. This distinction is particularly relevant for natural products, which often exhibit different behavior in cellular versus biochemical assays due to their complex structures and potential for metabolic transformation. The successful application of ECFP4 fingerprints, which captured 844 of the 887 selected features, aligns with their established utility in natural product QSAR studies [32,33], effectively encoding local structural environments that are particularly relevant for natural products’ biological activity.
The performance metrics achieved (MCC=0.583, ROC-AUC=0.896) are competitive with state-of-the-art QSAR models for antiviral prediction. For comparison, recent studies targeting flavivirus inhibitors report MCC values ranging from 0.45-0.65 [21,34,35], with our optimized model performing in the upper range. The inclusion of assay type as a distinguishing feature proved crucial, improving MCC by approximately 0.08 compared to models without this distinction. This highlights the importance of considering experimental context in QSAR modeling, particularly when combining data from different assay formats.
The comprehensive structure-activity relationship (SAR) analysis of the 276 High-High compounds revealed critical insights into the molecular determinants of anti-dengue activity and the challenges inherent in natural product drug discovery. The striking disparity in drug-likeness—with only 20 compounds (7.2%) achieving QED ≥ 0.5—reflects a fundamental tension between the evolutionary optimization of phytochemicals for ecological functions versus pharmaceutical requirements for human therapeutics [36]. Natural products have evolved to maximize biological activity through structural features that often violate Lipinski’s Rule of Five [37], including high molecular weight, excessive hydrogen bonding capacity, and elevated polar surface area. Our finding that 256 compounds (92.8%) exhibited suboptimal drug-likeness despite high predicted potency underscores the necessity of multi-criteria filtering in virtual screening campaigns and validates our strategic focus on the 20 high-QED candidates for experimental prioritization.
The remarkable structural novelty observed among high-potency hits—with 239 compounds (86.6%) classified as Novel based on Tanimoto similarity < 0.5 to ChEMBL compounds—provides compelling evidence that Colombian biodiversity harbors unique chemical scaffolds worthy of systematic investigation. This high novelty rate substantially exceeds typical virtual screening campaigns against synthetic libraries, where 40-60% novelty is considered exceptional [38,39]. The chemical space segregation visualized through principal component analysis (Figure 3B) confirms that novel candidates occupy distinct structural regions, suggesting they may engage dengue viral targets through alternative binding modes or access entirely different protein pockets compared to known inhibitors. This structural divergence presents both opportunities and challenges: while novel scaffolds offer potential for circumventing resistance mechanisms and accessing unexplored intellectual property space, they also require more extensive mechanistic characterization and may necessitate greater structural optimization to achieve clinical candidates.
The descriptor-activity correlation analysis (Figure 3D) revealed differential molecular property associations between IC50 and EC50 endpoints, reinforcing their biological distinction. The weak correlations observed for IC50 activity (maximum |ρ| = 0.06) compared to EC50 (maximum |ρ| = 0.15) suggest that biochemical target inhibition is influenced by subtle structural features not captured by simple physicochemical descriptors, potentially involving specific three-dimensional arrangements or pharmacophoric elements. In contrast, the stronger correlations for EC50—particularly the negative TPSA association (ρ = -0.15) and positive QED relationship (ρ = 0.14)—indicate that cellular antiviral efficacy is more predictable from bulk molecular properties, likely reflecting the importance of membrane permeability, intracellular distribution, and metabolic stability. The TPSA-EC50 negative correlation provides actionable guidance for lead optimization: strategic reduction of polar surface area through modifications such as methylation of hydroxyl groups, replacement of carboxylic acids with bioisosteric alternatives, or removal of sugar moieties from glycosides could enhance cellular penetration while maintaining target affinity. Conversely, the positive QED-EC50 association validates our prioritization strategy, as compounds naturally possessing drug-like features are inherently more likely to demonstrate cellular efficacy without requiring extensive medicinal chemistry optimization.
Detailed examination of the 15 top-priority candidates (Table 2, Figure 4) reveals a structurally diverse collection spanning five chemical classes, with alkaloids predominating (n=7, 46.7%). Incartine (#1), an indole alkaloid from Hippeastrum puniceum exhibiting the highest QED score (0.83) and balanced dual-endpoint activity (pIC50: 6.84, pEC50: 6.13), emerges as the most promising lead candidate. Its favorable pharmaceutical profile—moderate molecular weight (312.4 Da), balanced lipophilicity (LogP: 2.1), and low polar surface area (TPSA: 67.8 Ų)—combined with structural novelty (Tanimoto: 0.28) positions it as an ideal starting point for medicinal chemistry optimization. The identification of two compounds from Hippeastrum puniceum among the top 15 (Incartine #1 and Hippeastrine #11) suggests this species may be particularly rich in anti-dengue scaffolds, warranting comprehensive phytochemical profiling and possibly representing an underexplored source of Amaryllidaceae alkaloids with antiviral potential.
The presence of Lycorine (13, Tanimoto: 1.00) and Pseudolycorine (15, Tanimoto: 1.00) among our top predictions provides crucial internal validation of model performance. These compounds, present in the ChEMBL training dataset with experimentally confirmed anti-dengue activity, were successfully recovered by our screening protocol despite not being explicitly identified as validation targets. Lycorine, in particular, has demonstrated broad-spectrum antiviral activity in recent studies, including against SARS-CoV-2 and influenza A virus through mechanisms involving protein synthesis inhibition and immunomodulation [40,41]. The model’s ability to correctly prioritize these validated compounds increases confidence in predictions for structurally novel candidates and suggests that our top 12 novel compounds merit immediate experimental evaluation.
Intriguingly, several terpenoid glycosides appear among the top candidates (Byzantionoside B #5, (6R,9R)-3-Oxo-alpha-ionol glucoside #10, 9-Hydroxy-7-megastigmen-3-one glucoside #12), despite their typically challenging pharmaceutical properties. Glycosylation, while often improving water solubility and reducing toxicity, generally compromises oral bioavailability due to poor membrane permeability and susceptibility to intestinal glycosidases [42]. However, for dengue treatment—where severe cases require hospitalization and intravenous administration is standard—this limitation may be less critical than for orally administered therapeutics. Additionally, prodrug strategies employing enzymatically cleavable linkers could enable targeted delivery of aglycone active compounds while exploiting the favorable solubility of glycosides. The moderate QED scores achieved by these glycosides (0.51-0.54) suggest they occupy a pharmaceutical property space that, while suboptimal for oral drugs, may be acceptable for parenteral formulations or with appropriate delivery modifications.
Comparative analysis with ChEMBL training set high-potency references (Figure 4, right panels) reveals both validating similarities and important distinctions. The training set is dominated by Amaryllidaceae alkaloids, with Narciclasine analogs (ChEMBL464432, pActivity: 10.82) and Pancratistatin (pActivity: 10.20) exhibiting exceptional nanomolar potencies (IC50 = 0.01-0.06 nM). However, many of these highly potent training compounds exhibit low drug-likeness (e.g., ChEMBL5188858 QED: 0.22, ChEMBL3392006 QED: 0.13), illustrating the disconnect between biochemical potency and pharmaceutical viability that our filtering strategy explicitly addresses. Our identification of structurally related but drug-like Amaryllidaceae alkaloids (Incartine QED: 0.83, 3-Acetylnerbowdine QED: 0.80) represents a potential breakthrough in balancing potency with developability.
The structural diversity among our novel candidates—ranging from simple indole alkaloids (Incartine, Indican) to complex sesquiterpene lactones (Bilobalide, Epicolactone) to cardiac glycoside aglycones (Digoxigenin)—contrasts sharply with the training set’s heavy enrichment in Amaryllidaceae alkaloids. This chemical diversity suggests our top candidates may target multiple points in the dengue viral replication cycle or engage different protein pockets, offering opportunities for rational combination therapy. Recent structural biology studies have identified multiple druggable sites across dengue viral proteins, including the NS3 protease active site [43], NS5 RNA-dependent RNA polymerase catalytic pocket [44], NS2B-NS3 protease allosteric sites [45], and envelope protein domain II [46]. The structural heterogeneity of our candidates makes it plausible that different chemical classes engage different targets, potentially enabling synergistic multi-target inhibition strategies that could address antiviral resistance.
The finding that only 14.3% of Colombian plant species have documented antiviral activity, and nearly half of these lack phytochemical characterization, reveals both a challenge and an opportunity. This knowledge gap is particularly concerning given Colombia’s status as the world’s second-most biodiverse country [47] and the increasing pressure on natural habitats from deforestation and climate change [48]. The urgency to document and preserve this chemical diversity cannot be overstated, as each lost species potentially represents unique bioactive compounds that could address current and future health challenges.
The predominance of certain plant families in our antiviral activity compilation—particularly Asteraceae, Fabaceae, and Lamiaceae—reflects both their chemical richness and the historical focus of ethnopharmacological research. Asteraceae’s prevalence is unsurprising given its production of diverse sesquiterpene lactones, compounds with well-documented antiviral properties [49,50]. However, this distribution may also reflect sampling bias, as these families are among the most studied globally, potentially overlooking equally valuable but less investigated families. Asteraceae’s prevalence is unsurprising given its production of diverse sesquiterpene lactones, compounds with well-documented antiviral properties. However, this distribution may also reflect sampling bias, as these families are among the most studied globally, potentially overlooking equally valuable but less investigated families. Our identification of Digoxigenin (#14), a cardiac glycoside aglycone from Digitalis purpurea, as a high-priority candidate despite steroids being underrepresented in anti-dengue literature, exemplifies how unbiased computational screening can reveal unexpected chemotype-activity relationships that might be missed by hypothesis-driven approaches.
The identification of Amaryllidaceae alkaloids—specifically Incartine, Lycorine, and Kalbreclasine from Hippeastrum puniceum—as top candidates provides compelling validation of our approach. Lycorine has demonstrated broad-spectrum antiviral activity in recent studies, including against SARS-CoV-2 [40] and influenza viruses [41], through mechanisms involving protein synthesis inhibition and immunomodulation. The high predicted activity of these compounds against dengue virus (pActivity > 7.0) suggests potential for repurposing these natural products, which could significantly accelerate drug development timelines. The structural analysis revealing that 84.6% of high-potency predictions represent novel chemotypes not present in the ChEMBL training set is particularly significant. This finding suggests that Colombian flora contains unique structural diversity that could provide new mechanisms of action against dengue virus. The prevalence of alkaloid frameworks (46.2%) among high-potency compounds contrasts with current anti-dengue drug development, which has primarily focused on nucleoside analogs and protease inhibitors.
The favorable drug-likeness profiles of our top candidates, with 69.2% showing no Lipinski violations and an average QED score of 0.64, suggest good potential for pharmaceutical development. However, the presence of glycosylated compounds (23.1% of high-potency predictions) presents both opportunities and challenges. While glycosylation can improve water solubility and reduce toxicity, it often compromises oral bioavailability due to poor membrane permeability and susceptibility to intestinal glycosidases. The alignment between our computational predictions and traditional uses of these plants provides important validation.
Several limitations must be acknowledged in interpreting our results. First, the QSAR model’s applicability domain may not fully encompass the structural complexity of natural products, particularly those with multiple chiral centers or unusual functional groups rare in synthetic compounds. Second, our approach evaluated individual compounds in isolation, whereas traditional medicinal preparations often rely on synergistic interactions between multiple constituents. The lack of target-specific predictions represents another limitation; while our model predicts general anti-dengue activity, understanding which viral proteins are targeted would greatly facilitate lead optimization. Furthermore, the 48.6% of antiviral plants lacking phytochemical data represents a significant blind spot in our analysis. Prioritizing these species for chemical characterization, particularly endemic species at risk from habitat loss, should be an urgent research priority. Despite these limitations, this computational framework provides a valuable prioritization tool that can significantly reduce the time and cost of experimental screening. The public availability of our model and datasets enables other researchers to validate predictions, extend the analysis to additional plant species, or adapt the methodology for other therapeutic targets.
This study’s implications extend beyond immediate drug discovery applications. By demonstrating the pharmaceutical potential of Colombian biodiversity, we provide economic arguments for conservation that complement ethical and ecological rationales. The identification of high-value compounds from native species could support sustainable development models where local communities benefit from biodiversity conservation through benefit-sharing agreements aligned with the Nagoya Protocol. The development of an open-access platform for continuing this screening effort democratizes access to these findings and enables collaborative research, aligning with global efforts to make drug discovery more inclusive. This pioneering work successfully demonstrates the feasibility and value of applying modern computational approaches to explore Colombian biodiversity for anti-dengue drug discovery. Moving forward, immediate priorities include experimental validation of top candidates through viral inhibition assays, mechanism of action studies for confirmed hits, and chemical characterization of the understudied antiviral plant species This integrated approach positions Colombia to leverage its extraordinary biodiversity for global health benefit while promoting sustainable development and conservation of its natural heritage.
4. Materials and Methods
4.1. Data Sources and Availability
All datasets, trained models, and analysis code generated during this study are publicly available to ensure full reproducibility. Bioactivity Data Raw anti-dengue bioactivity data were retrieved from the ChEMBL database, version 33 [29,51] (https://www.ebi.ac.uk/chembl/, accessed March 2024). The curated training dataset is provided as Data S1 in supplementary materials. Colombian Medicinal Flora Plant species information was obtained from the official list of food and medicinal plants of Colombia curated by the Alexander von Humboldt Institute (https://i2d.humboldt.org.co/ceiba/resource.do?r=ls_colombia_magnoliophyta_2014, accessed January 2024).The complete Colombian phytochemical library (3,267 compounds from 358 medicinal plant species) is available as Data S2 in supplementary materials. Detailed ethnobotanical information for all plant species is provided in Table S1. Computational Models and Code The optimized XGBoost model, preprocessing objects, and model parameters are available as Data S3 in supplementary materials. All Python scripts for data curation, molecular descriptor calculation, model training, Bayesian optimization, and virtual screening are deposited at https://github.com/Sergio111999/QSAR-DENV under an MIT license (DOI to be provided upon acceptance).
4.2. Anti-Dengue Compound Dataset Curation
Bioactivity data were retrieved from ChEMBL using the following SQL query structure:
sqlSELECT * FROM activities
WHERE target_organism LIKE ‘%dengue%’
AND standard_type IN (‘IC50’, ‘EC50’)
AND standard_relation = ‘=‘
Data curation followed the workflow described by Mendez et al. (2019) [29]. Briefly, activity values were standardized to molar units, converted to pActivity (-log10[M]), and categorized as high (>6), medium (5-6), or low (≤5) potency. Duplicate structures were identified using InChIKey and resolved by retaining the highest pChEMBL value. The final dataset contained 2,034 activity records corresponding to 1,981 unique molecules, with 53 molecules having both IC50 and EC50 measurements.
4.3. Colombian Medicinal Flora Database Construction
Plant species with antiviral activity were identified through systematic literature review following PRISMA guidelines [52]. PubMed and Scopus were searched (January-April 2024) using: “[species name]” AND “antiviral”. From 2,501 unique Colombian species, 358 (14.3%) showed documented antiviral activity. Phytochemical structures for 184 species were retrieved from primary literature and standardized using RDKit (v2024.03.5) following the protocol of Bento et al. (2020) [53].
4.4. Molecular Descriptors and Machine Learning
Molecular descriptors (n=43) and fingerprints (ECFP4, 2048 bits) were calculated using RDKit. The complete feature matrix (2,091 features) was reduced to 887 informative features using variance filtering (threshold >0.01). Machine learning models were implemented using scikit-learn (v1.3.2) and XGBoost (v3.0.2). Class imbalance was handled through the scale_pos_weight parameter in XGBoost. Model evaluation used 5-fold stratified cross-validation. The optimal model (XGBoost with Descriptors+ECFP4 representation) achieved MCC=0.572±0.048.
4.5. Virtual Screening Protocol
The trained XGBoost model was applied to predict anti-dengue activity for 3,267 phytochemicals from Colombian medicinal flora. For each compound, predictions were generated independently for both IC50 and EC50 endpoints, yielding pActivity values calculated as -log10(predicted activity in molar units). The dual-endpoint classification framework categorized compounds into nine classes based on combined IC50 and EC50 potency thresholds: Low (pActivity ≤ 5.0), Medium (5.0 < pActivity ≤ 6.0), and High (pActivity > 6.0). Compounds achieving high predicted potency for both endpoints (pIC50 > 6.0 AND pEC50 > 6.0) were designated as “High-High” and prioritized for subsequent analysis.
Drug-likeness assessment was performed using Lipinski’s Rule of Five criteria and Quantitative Estimate of Drug-likeness (QED) scores calculated following the methodology of Bickerton et al. [36] as implemented in RDKit. QED integrates eight molecular properties (MW, LogP, HBA, HBD, PSA, rotatable bonds, aromatic rings, structural alerts) into a unified drug-likeness metric ranging from 0 (non-drug-like) to 1 (ideal drug-like properties). A QED threshold of 0.5 was applied to distinguish compounds with favorable pharmaceutical properties from those requiring extensive optimization.
Compounds were prioritized for experimental validation based on a multi-criteria strategy: (i) dual high potency (pActivity > 6.0 for both IC50 and EC50), (ii) favorable drug-likeness (QED ≥ 0.5), (iii) structural novelty assessment via Tanimoto similarity to training compounds, and (iv) compliance with applicability domain criteria. This hierarchical filtering identified 20 compounds meeting all criteria, from which the top 15 were selected based on pIC50 ranking for detailed characterization.
4.5.1. Comprehensive Structure-Activity Relationship (SAR) Analysis
Following QSAR-based virtual screening, a systematic SAR analysis pipeline was implemented to characterize activity distributions, assess chemical diversity, evaluate structural novelty, and identify molecular properties correlated with antiviral potency. The analysis comprised four integrated components executed using Python 3.12.3 with RDKit 2024.03.5, scikit-learn 1.3.2, and custom analysis scripts
Dual-Endpoint Classification and Activity Distribution: All 3,267 screened compounds were classified into a 3×3 activity matrix based on combined IC50 and EC50 predicted potencies using the thresholds defined above (Low ≤ 5.0, Medium 5.0-6.0, High > 6.0). This classification generated nine mutually exclusive categories (e.g., “Low- Low “, “High-Medium”, “High-High”), enabling systematic characterization of dual-endpoint activity patterns across the chemical library. Frequency distributions were calculated for each category, and the “High-High” class was designated as the priority population for downstream analyses.
Structural Novelty Assessment: The structural novelty of high-potency compounds relative to the ChEMBL training dataset was quantified using Tanimoto similarity coefficients calculated from Extended Connectivity Fingerprints (ECFP4, radius=2, 2048 bits) as implemented in RDKit. For each screened compound, Tanimoto similarity was computed against all 1,981 training set compounds, and the maximum similarity value was retained as the novelty metric. Compounds were classified into novelty categories as follows: Novel/Very Novel (Tanimoto < 0.5), indicating low structural similarity to known anti-dengue agents; Moderate similarity (0.5 ≤ Tanimoto < 0.7); and Training-like (Tanimoto ≥ 0.7), indicating high structural resemblance to training compounds. This classification enables identification of both validated chemotypes (high Tanimoto) and potentially innovative scaffolds (low Tanimoto).
Chemical Space Visualization and Diversity Analysis: Principal component analysis (PCA) was performed on the ECFP4 fingerprint matrix to project the high-dimensional chemical space into two dimensions for visualization. PCA was implemented using scikit-learn’s decomposition module with standardized features, and the first two principal components (PC1, PC2) were retained for plotting. The projection enables visual assessment of chemical space coverage, identification of structural clusters, and evaluation of segregation between novelty categories. The cumulative variance explained by PC1 and PC2 quantifies the effectiveness of dimensionality reduction.
Drug-likeness Assessment: All compounds were evaluated for drug-likeness using the QED metric calculated via RDKit’s QED module (Chem.QED.qed function). QED scores were dichotomized using a threshold of 0.5: compounds with QED ≥ 0.5 were classified as “High QED” (favorable pharmaceutical properties), while QED < 0.5 indicated “Low QED” (suboptimal properties requiring optimization). For High-High compounds, the proportion achieving high QED was calculated to assess the pharmaceutical development challenge inherent in the high-potency population.
Descriptor-Activity Correlation Analysis: Spearman rank correlation coefficients (ρ) were calculated between 11 molecular descriptors and predicted pActivity values for both IC50 and EC50 endpoints across all 3,267 compounds. The descriptor set included: Molecular Weight (MW), octanol-water partition coefficient (LogP), hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), rotatable bonds (RotBonds), topological polar surface area (TPSA), total ring count (Rings), aromatic rings (AromaticRings), aliphatic rings (NumAliphaticRings), fraction of sp3 carbons (FractionCSP3), and QED. Correlations were computed using scipy.stats.spearmanr, and statistical significance was assessed using two-tailed tests with Bonferroni correction for multiple comparisons (α = 0.05/22 = 0.0023, accounting for 11 descriptors × 2 endpoints). Only correlations with |ρ| > 0.05 and corrected p < 0.0023 were considered statistically significant. Results were visualized as heatmaps using seaborn 0.13.2 with diverging color scales centered at zero correlation.
Top Candidate Selection and Characterization: From the 20 compounds meeting all filtering criteria (High-High + QED ≥ 0.5), the top 15 were selected based on descending pIC50 values and subjected to comprehensive characterization. For each compound, the following properties were tabulated: predicted pIC50 and pEC50, QED score, molecular descriptors (MW, LogP, HBA, HBD, TPSA), novelty category, maximum Tanimoto similarity to training compounds, assigned chemical class based on structural features, and botanical source from phytochemical databases. Chemical class assignments integrated automated substructure searches (using RDKit’s HasSubstructMatch for scaffold identification) with manual curation based on established phytochemical taxonomy. Plant source assignments were cross-referenced with primary literature and validated against the Colombian medicinal flora database compiled in this study.
All SAR analysis workflows, including data preprocessing, statistical calculations, and visualization generation, were implemented in reproducible Python scripts with full parameterization, enabling adaptation to alternative screening datasets. Complete code with detailed documentation is available in the public repository accompanying this manuscript.
4.6. Applicability Domain Assessment
To ensure reliability of predictions, the applicability domain was evaluated using:
- -
- Chemical similarity: Tanimoto coefficient > 0.3 to nearest training set compounds
- -
- Leverage analysis: Compounds with leverage hi < 3p/n (p=features, n=training samples)
- -
- Descriptor range: All molecular descriptors within training set min-max bounds
Only compounds meeting at least two criteria were considered reliable predictions.
Additionally, leverage analysis was performed by calculating the hat matrix diagonal elements (hi) for each prediction, with the warning threshold set at 3p/n where p is the number of features (887) and n is the training set size (2,034), yielding a critical leverage value of 1.308. Compounds exceeding this threshold were flagged as requiring extrapolation and treated with appropriate caution in interpretation. All 15 top-priority candidates exhibited leverage values below the critical threshold, confirming their location within the model’s interpolation space.
4.7. Statistical Analysis
Statistical analyses used Python 3.12.3. Distribution comparisons employed Mann-Whitney U test. Multiple testing correction used Bonferroni method (α=0.05). Chemical space visualization used UMAP (umap-learn v0.5.7; n_neighbors=15, min_dist=0.1) and hierarchical clustering (Ward linkage on Tanimoto distances).
4.8. Computational Resources
All computations were performed on a workstation equipped with an Intel Core i5 CPU, 16 GB RAM, SSD storage, and dual GPUs (NVIDIA GeForce RTX 3050 Laptop GPU with 4 GB dedicated VRAM and Intel Iris Xe Graphics), running Windows 11. XGBoost and LightGBM models utilized GPU acceleration via CUDA support. Model training required approximately 6 hours, while virtual screening of 3,267 compounds completed in three minutes.
4.9. Code Availability
Complete code for reproducing all analyses is available at https://github.com/Sergio111999/QSAR-DENV under MIT license. Key dependencies: Python (3.12.3) [54], RDKit (2024.03.5) [55], NumPy (1.26.4) [56], pandas (2.3.0) [57], SciPy (1.13.1) [58], scikit-learn (1.3.2) [59], XGBoost (3.0.2) [18], LightGBM (4.6.0) [60], Optuna (4.4.0) [27], imbalanced-learn (0.12.3) [61], matplotlib (3.9.2) [62], seaborn (0.13.2) [63], joblib (1.4.2), umap-learn (0.5.7) [30], streamlit (1.37.1) [64].
4.10. Supplementary Material Structure
Table S1 (Colombian medicinal plants with antiviral activity, N=358 species)
Table S2 (comprehensive model performance metrics with cross-validation results)
Table S3 (complete virtual screening results, N=3,267 compounds)
Table S4 (top priority compounds with detailed properties)
Figure S1 (chemical space visualization and applicability domain)
Figure S2 (Bayesian optimization and hyperparameter analysis)
Data S1 (training dataset)
Data S2 (Colombian phytochemical library)
Data S3: Trained models including optimized XGBoost model, feature scaler,variance selector, and model parameters (ZIP format, ~3.2 MB)
Code S1: Complete computational pipeline including data preprocessing, QSAR model development, Bayesian optimization, and virtual screening workflow (Python scripts with detailed documentation available at https://github.com/Sergio111999/QSAR-DENV)
4.11. Use of AI Tools
Claude Opus 4.1 was used to assist with code debugging and optimization of data visualization scripts. All AI-generated code was manually reviewed and validated. No AI tools were used for data generation, analysis, or interpretation of results.
5. Conclusions
This study successfully demonstrated the application of optimized machine learning-based QSAR modeling integrated with comprehensive structure-activity relationship analysis to systematically screen Colombian medicinal flora for potential anti-dengue compounds, revealing the untapped pharmaceutical potential of one of the world’s most biodiverse regions. The development of a robust predictive model through Bayesian hyperparameter optimization (MCC = 0.583, ROC-AUC = 0.896) enabled the virtual screening of 3,267 phytochemicals, identifying 276 compounds (8.4%) with dual high potency (High-High classification) and subsequently prioritizing 15 exceptional candidates combining high predicted activity, favorable drug-likeness (QED ≥ 0.5), and diverse structural scaffolds for experimental validation.
The comprehensive SAR analysis revealed critical insights into the molecular landscape of anti-dengue activity within Colombian biodiversity. The identification of 239 structurally novel compounds (86.6% of High-High hits) with low similarity (Tanimoto < 0.5) to ChEMBL training compounds demonstrates that Colombian flora harbors unique chemical scaffolds potentially operating through mechanisms distinct from known anti-dengue agents. This exceptionally high novelty rate—substantially exceeding typical synthetic library screening campaigns—positions Colombia as a potential leader in natural product-based antiviral discovery and highlights the importance of biodiversity conservation for pharmaceutical innovation. Principal component analysis confirmed clear chemical space segregation between novel and training-like compounds, validating the structural distinctiveness of prioritized candidates and suggesting they may access alternative binding modes or target different viral proteins compared to established inhibitors.
The striking disparity in drug-likeness among high-potency compounds—with only 20 candidates (7.2% of High-Highhits) achieving QED ≥ 0.5—illuminates a fundamental challenge in natural product drug discovery: the inherent tension between evolutionary optimization for biological activity versus pharmaceutical requirements for human therapeutics. This finding underscores the critical importance of multi-criteria optimization strategies and validates our hierarchical filtering approach. Descriptor-activity correlation analysis provided actionable mechanistic insights, revealing that QED positively correlates with EC50 activity (ρ = 0.14), while TPSA negatively correlates (ρ = -0.15), suggesting that moderate polarity optimization and inherent drug-like features favor cellular antiviral efficacy. The differential correlation patterns between IC50 (maximum |ρ| = 0.06) and EC50 (maximum |ρ| = 0.15) endpoints validate the biological distinction between biochemical target inhibition and cellular efficacy, reinforcing the value of our dual-endpoint classification strategy.
The identification of Incartine from Hippeastrum puniceum as the top candidate, exhibiting the highest QED score (0.83) among all prioritized compounds with balanced dual-endpoint activity (pIC50: 6.84, pEC50: 6.13) and structural novelty (Tanimoto: 0.28), provides a compelling lead for immediate experimental validation. The structural diversity among the top 15 candidates—spanning five chemical classes including alkaloids (46.7%), terpenoid glycosides (26.7%), terpenoids (13.3%), flavonoids (6.7%), and steroids (6.7%)—suggests multiple potential mechanisms of antiviral action and offers opportunities for rational combination therapy strategies. Notably, the presence of Lycorine and Pseudolycorine (both with Tanimoto = 1.00) among top predictions provides crucial internal validation, as these compounds are present in the ChEMBL training dataset with experimentally confirmed anti-dengue activity, increasing confidence in predictions for structurally novel candidates.
The enrichment of Amaryllidaceae alkaloids among top candidates—with Hippeastrum puniceum contributing two compounds (Incartine #1, Hippeastrine #11) and the family collectively providing four of fifteen prioritized candidates—suggests this botanical family harbors particularly rich anti-dengue chemical space warranting systematic phytochemical investigation. This finding aligns with emerging evidence of broad-spectrum antiviral properties in Amaryllidaceae alkaloids, exemplified by Lycorine’s demonstrated activity against SARS-CoV-2, influenza, and dengue viruses. The convergence between our computational predictions and traditional medicinal uses, exemplified by the identification of compounds from plants like Psidium guajava and Indigofera suffruticosa with documented ethnopharmacological applications in dengue-endemic regions, underscores the value of integrating indigenous knowledge with modern drug discovery approaches.
Our analysis revealed a critical knowledge gap in Colombian biodiversity research, with 48.6% of the 358 plant species showing documented antiviral activity lacking phytochemical characterization. This represents both an urgent conservation priority and a substantial opportunity for future drug discovery efforts. As Colombia faces dual challenges of increasing dengue burden and accelerating biodiversity loss through deforestation and climate change, the urgency to document and preserve this chemical diversity cannot be overstated. Each lost species potentially represents unique bioactive compounds that could address current and future health challenges, making biodiversity conservation not only an ecological imperative but also a strategic pharmaceutical resource.
The optimization process yielded important methodological insights beyond the immediate anti-dengue application. The 1.9% improvement in MCC through Bayesian optimization, achieved primarily through regularization parameter tuning (reg_alpha: 38.6%, gamma: 33.3% importance), highlights the critical role of controlling model complexity in high-dimensional chemical space. The decision to select the optimized XGBoost model over the marginally better ensemble (MCC difference = 0.001) exemplifies the practical trade-offs between performance and complexity in applied QSAR modeling, demonstrating that sophisticated ensemble methods may not always justify their computational overhead. These methodological lessons are transferable to other drug discovery campaigns and contribute to the growing body of best practices in computational chemoinformatics.
The tiered prioritization strategy developed in this study—combining dual-endpoint classification, drug-likeness filtering, novelty assessment, and applicability domain validation—provides a replicable framework for natural product virtual screening campaigns. This methodology addresses key limitations of traditional QSAR approaches by explicitly considering multiple pharmacological dimensions, pharmaceutical developability, and prediction reliability. The framework’s modular design enables adaptation to alternative therapeutic targets, different chemical libraries, and varied experimental validation capacities, making it broadly applicable beyond the immediate anti-dengue context.
Immediate research priorities include: (i) in vitro validation of the top 15 candidates through cell-based dengue virus inhibition assays encompassing all four serotypes (DENV-1 to DENV-4); (ii) mechanism of action studies employing target deconvolution approaches to identify specific viral proteins engaged by active compounds; (iii) cytotoxicity and selectivity index determination to assess therapeutic windows; (iv) phytochemical characterization of the 174 understudied antiviral plant species to expand the screening library; and (v) investigation of potential synergistic effects between structurally diverse candidates for combination therapy development. Future computational efforts should focus on developing target-specific QSAR models for individual dengue viral proteins (NS3 protease, NS5 polymerase, envelope protein) to enable mechanism-based prioritization, and on implementing approaches capable of predicting synergistic interactions in natural product mixtures, as many traditional preparations rely on multi-component formulations.
This work establishes a replicable methodological framework that can be extended to other neglected tropical diseases endemic to biodiversity-rich regions, including Zika virus, chikungunya, yellow fever. The integration of computational screening, traditional knowledge, ethnopharmacological databases, and biodiversity informatics presented here offers a sustainable model for pharmaceutical bioprospecting that could benefit both global health and local communities.
In conclusion, this pioneering study not only identifies 15 promising anti-dengue candidates from Colombian flora with exceptional combined profiles of predicted potency (pActivity > 6.0), drug-likeness (QED ≥ 0.5), and structural diversity, but also demonstrates how modern computational approaches integrated with comprehensive SAR analysis can systematically unlock the pharmaceutical potential of megadiverse countries. The predominance of structurally novel chemotypes (86.6% of high-potency hits) establishes Colombian biodiversity as a rich source of potentially innovative antiviral mechanisms, with immediate implications for intellectual property development and positioning Colombia as a potential leader in natural product pharmaceutical discovery. As Colombia continues to face the dual challenges of persistent dengue burden (172.9 cases per 100,000 population) and accelerating biodiversity loss, this research provides both practical solutions for drug discovery and compelling economic incentives for conservation. The convergence of machine learning, structure-activity relationship analysis, ethnopharmacological knowledge, and biodiversity informatics presented here establishes a replicable framework for natural product-based drug discovery, with immediate application potential for dengue and extensibility to other neglected tropical diseases affecting biodiversity-rich regions worldwide.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.A.M.H and A.M.J.V.; methodology, S.A.M.H. and A.S; software, S.A.M.H. and A.S; validation, S.A.M.H, I.E.J and R.S.C.V.; formal analysis, S.A.M.H. and U.A.S.A.; investigation, S.A.M.H., I.E.J. and A.M.J.V.; resources, R.S.C.V. and A.M.J.V.; data curation, S.A.M.H. and I.E.J.; writing—original draft preparation, S.A.M.H.; writing—review and editing, S.A.M.H, A.S, R.S.C.V. and A.M.J.V.; visualization, S.A.M.H. and A.S; supervision, R.S.C.V. and A.M.J.V.; project administration, A.M.J.V.; funding acquisition, A.M.J.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are openly available.
Acknowledgments
We are grateful to Alejandro Gómez Levy, Lucas Drouilhat Vega, Juan Pablo Tapia, David Esteban Castro, and Juan Jose Sarmiento for their assistance in literature review and database curation. During the preparation of this manuscript, the authors used Claude AI Opus 4.1 for the purposes of debugging code and optimizing data visualization scripts. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| AD | Applicability Domain |
| ChEMBL | Chemical Database of the European Molecular Biology Laboratory |
| DENV | Dengue Virus |
| EC50 | Half-maximal Effective Concentration |
| ECFP4 | Extended Connectivity Fingerprint (radius 4) |
| HBA | Hydrogen Bond Acceptors |
| HBD | Hydrogen Bond Donors |
| IC50 | Half-maximal Inhibitory Concentration |
| LogP | Logarithm of Partition Coefficient (octanol-water) |
| MACCS | Molecular ACCess System (fingerprint) |
| MCC | Matthews Correlation Coefficient |
| ML | Machine Learning |
| MW | Molecular Weight |
| NS2B-NS3 | Non-Structural Protein 2B-3 (dengue viral protease) |
| NS3 | Non-Structural Protein 3 (dengue viral protease) |
| NS5 | Non-Structural Protein 5 (dengue viral polymerase) |
| pActivity | Negative logarithm of activity (-log10[Activity in M]) |
| pEC50 | Negative logarithm of EC50 (-log10[EC50 in M]) |
| pIC50 | Negative logarithm of IC50 (-log10[IC50 in M]) |
| PC1, PC2 | Principal Component 1, 2 |
| PCA | Principal Component Analysis |
| QED | Quantitative Estimate of Drug-likeness |
| QSAR | Quantitative Structure-Activity Relationship |
| RDKit | Open-source cheminformatics toolkit |
| ROC-AUC | Receiver Operating Characteristic - Area Under the Curve |
| RotBonds | Rotatable Bonds |
| SAR | Structure-Activity Relationship |
| SMILES | Simplified Molecular Input Line Entry System |
| Tanimoto | Tanimoto Similarity Coefficient |
| TPSA | Topological Polar Surface Area |
| UMAP | Uniform Manifold Approximation and Projection |
| XGBoost | eXtreme Gradient Boosting |
References
- Gwee, X.W.S., P.E.Y. Chua, and J. Pang, Global dengue importation: a systematic review. BMC infectious diseases, 2021. 21(1): p. 1078.
- Anumanthan, G., B. Sahay, and A. Mergia, Current Dengue Virus Vaccine Developments and Future Directions. Viruses, 2025. 17(2).
- Gutierrez-Barbosa, H., et al., Dengue Infections in Colombia: Epidemiological Trends of a Hyperendemic Country. Trop Med Infect Dis, 2020. 5(4).
- Rodríguez-Morales, A.J., et al., Cost of dengue in Colombia: A systematic review. PLOS Neglected Tropical Diseases, 2024. 18(12): p. e0012718.
- Rodríguez-Morales, A.J., et al., The epidemiological impact of dengue in Colombia: a systematic review. The American Journal of Tropical Medicine and Hygiene, 2024. 112(1): p. 182.
- Romero-Vega, L., et al., Evaluation of dengue fever reports during an epidemic, Colombia. Revista de saude publica, 2014. 48(6): p. 899-905.
- Idris, F., D.H.R. Ting, and S. Alonso, An update on dengue vaccine development, challenges, and future perspectives. Expert opinion on drug discovery, 2021. 16(1): p. 47-58.
- Witte, P., et al., Dengue Fever—Diagnosis, Risk Stratification, and Treatment. Deutsches Ärzteblatt International, 2024. 121(23): p. 773.
- Paz-Bailey, G., et al., Dengue. The Lancet, 2024. 403(10427): p. 667-682.
- Bernal, R., La flora de Colombia en cifras. Catálogo de plantas y líquenes de Colombia, 2016. 1: p. 115-137.
- Valderrama, N., Plantas alimenticias y medicinales de Colombia. 2014.
- Puello Alcocer, E.C., N.N. Valencia Jiménez, and A.C. Atencia Soto, Prácticas ancestrales para el control del dengue en una comunidad indígena Embera Katío, Córdoba, Colombia. Revista Cubana de Enfermería, 2022. 38(2).
- Silva-Trujillo, L., et al., Essential oils from Colombian plants: Antiviral potential against dengue virus based on chemical composition, in vitro and in silico analyses. Molecules, 2022. 27(20): p. 6844.
- Tropsha, A., et al., Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR. Nature Reviews Drug Discovery, 2024. 23(2): p. 141-155.
- Keyvanpour, M.R. and M.B. Shirzad, An analysis of QSAR research based on machine learning concepts. Current Drug Discovery Technologies, 2021. 18(1): p. 17-30.
- Hanson, G., et al., Machine learning and molecular docking prediction of potential inhibitors against dengue virus. Frontiers in Chemistry, 2024. 12: p. 1510029.
- Gautam, S., A. Thakur, and M. Kumar, i-DENV: development of QSAR based regression models for predicting inhibitors targeting non-structural (NS) proteins of dengue virus. Frontiers in Pharmacology, 2025. 16: p. 1605722.
- Chen, T. and C. Guestrin. Xgboost: A scalable tree boosting system.
- Boldini, D., et al., Practical guidelines for the use of gradient boosting for molecular property prediction. Journal of Cheminformatics, 2023. 15(1): p. 73.
- Grisoni, F., et al., Investigating the mechanisms of bioconcentration through QSAR classification trees. Environment international, 2016. 88: p. 198-205.
- Chongjun, Y., et al., Predicting repurposed drugs targeting the NS3 protease of dengue virus using machine learning-based QSAR, molecular docking, and molecular dynamics simulations. SAR and QSAR in Environmental Research, 2024. 35(8): p. 707-728.
- Heinrich, M. and S. Gibbons, Ethnopharmacology in drug discovery: an analysis of its role and potential contribution. Journal of pharmacy and pharmacology, 2001. 53(4): p. 425-432.
- Atanasov, A.G., et al., Natural products in drug discovery: advances and opportunities. Nature reviews Drug discovery, 2021. 20(3): p. 200-216.
- Tu, Y., Artemisinin-A gift from traditional chinese medicine to the world (Nobel Lecture). Angewandte Chemie International Edition, 2016. 55(35).
- Taub, J.W., et al., The evolution and history of Vinca alkaloids: From the Big Bang to the treatment of pediatric acute leukemia. Pediatric Blood & Cancer, 2024. 71(11): p. e31247.
- Nemogá, G.R., A. Appasamy, and C.A. Romanow, Protecting indigenous and local knowledge through a biocultural diversity framework. The Journal of Environment & Development, 2022. 31(3): p. 223-252.
- Akiba, T., et al. Optuna: A next-generation hyperparameter optimization framework.
- Jolliffe, I.T. and J. Cadima, Principal component analysis: a review and recent developments. Philosophical transactions of the royal society A: Mathematical, Physical and Engineering Sciences, 2016. 374(2065): p. 20150202.
- Mendez, D., et al., ChEMBL: towards direct deposition of bioassay data. Nucleic acids research, 2019. 47(D1): p. D930-D940.
- McInnes, L., J. Healy, and J. Melville, Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426, 2018.
- Netzeva, T.I., et al., Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships: The report and recommendations of ecvam workshop 52. Alternatives to Laboratory Animals, 2005. 33(2): p. 155-173.
- Rogers, D. and M. Hahn, Extended-connectivity fingerprints. Journal of chemical information and modeling, 2010. 50(5): p. 742-754.
- Cereto-Massagué, A., et al., Molecular fingerprint similarity search in virtual screening. Methods, 2015. 71: p. 58-63.
- Gautam, S., et al., Anti-dengue: a machine learning-assisted prediction of small molecule antivirals against dengue virus and implications in drug repurposing. Viruses, 2023. 16(1): p. 45.
- Jitonnom, J., et al., 3D-QSAR and molecular docking studies of peptide-hybrids as dengue virus NS2B/NS3 protease inhibitors. Chemico-Biological Interactions, 2024. 396: p. 111040.
- Bickerton, G.R., et al., Quantifying the chemical beauty of drugs. Nature chemistry, 2012. 4(2): p. 90-98.
- Lipinski, C.A., et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews, 1997. 23(1-3): p. 3-25.
- Walters, W.P. and R. Wang, New trends in virtual screening. 2020, ACS Publications. p. 4109-4111.
- Lyu, J., et al., Ultra-large library docking for discovering new chemotypes. Nature, 2019. 566(7743): p. 224-229.
- Zhang, Y.-N., et al., Gemcitabine, lycorine and oxysophoridine inhibit novel coronavirus (SARS-CoV-2) in cell culture. Emerging microbes & infections, 2020. 9(1): p. 1170-1173.
- Yan, H., et al., Lycorine Inhibits Influenza Virus Replication by Affecting Nascent Nucleoporin Nup93 Synthesis. International Journal of Molecular Sciences, 2025. 26(11): p. 5358.
- Xiao, J., T.S. Muzashvili, and M.I. Georgiev, Advances in the biotechnological glycosylation of valuable flavonoids. Biotechnology advances, 2014. 32(6): p. 1145-1156.
- Noble, C.G., et al., Ligand-bound structures of the dengue virus protease reveal the active conformation. Journal of virology, 2012. 86(1): p. 438-446.
- Lim, S.P., C.G. Noble, and P.-Y. Shi, The dengue virus NS5 protein as a target for drug discovery. Antiviral research, 2015. 119: p. 57-67.
- Yildiz, M., et al., Allosteric inhibition of the NS2B-NS3 protease from dengue virus. ACS chemical biology, 2013. 8(12): p. 2744-2752.
- Modis, Y., et al., Structure of the dengue virus envelope protein after membrane fusion. Nature, 2004. 427(6972): p. 313-319.
- Arbeláez-Cortés, E., Knowledge of Colombian biodiversity: published and indexed. Biodiversity and conservation, 2013. 22(12): p. 2875-2906.
- Bolaño-Díaz, S., et al., Spatio-temporal characterization of fire using MODIS data (2000–2020) in Colombia. Fire, 2022. 5(5): p. 134.
- Chadwick, M., et al., Sesquiterpenoids lactones: benefits to plants and people. Int J Mol Sci, 2013. 14(6): p. 12780-805.
- Shakeri, A., et al., In depth chemical investigation of Glycyrrhiza triphylla Fisch roots guided by a preliminary HPLC-ESIMS(n) profiling. Food Chem, 2018. 248: p. 128-136.
- Zdrazil, B., et al., The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res, 2024. 52(D1): p. D1180-d1192.
- Page, M.J., et al., The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Bmj, 2021. 372: p. n71.
- Bento, A.P., et al., An open source chemical structure curation pipeline using RDKit. J Cheminform, 2020. 12(1): p. 51.
- Rossum, V., Python 3 reference manual. (No Title), 2009.
- Landrum, G., et al., RDKit. 2015.
- Harris, C.R., et al., Array programming with NumPy. nature, 2020. 585(7825): p. 357-362.
- McKinney, W., Data structures for statistical computing in Python. scipy, 2010. 445(1): p. 51-56.
- Virtanen, P., et al., SciPy 1.0: fundamental algorithms for scientific computing in Python. Nature methods, 2020. 17(3): p. 261-272.
- Prodregosa, F., Scikit-learn: Machine learning in Python. Journal of Machine Learning Research. vol, 2011. 12: p. 2825-2830.
- Ke, G., et al., Lightgbm: A highly efficient gradient boosting decision tree. Advances in neural information processing systems, 2017. 30.
- LemaÃŽtre, G., F. Nogueira, and C.K. Aridas, Imbalanced-learn: A python toolbox to tackle the curse of imbalanced datasets in machine learning. Journal of machine learning research, 2017. 18(17): p. 1-5.
- Hunter, J.D., Matplotlib: A 2D graphics environment. Computing in science & engineering, 2007. 9(03): p. 90-95.
- Waskom, M.L., Seaborn: statistical data visualization. Journal of open source software, 2021. 6(60): p. 3021.
- Streamlit, C., Streamlit: A faster way to build and share data apps. https://streamlit. io/, 2024.
Figure 3.
Comprehensive dual-endpoint activity analysis and structure-activity relationships of Colombian phytochemicals. (A) Dual-endpoint classification heatmap of 3,267 compounds across nine activity categories. The “High-High” category (n=276, 8.4%) represents priority compounds with dual high potency. (B) PCA of chemical space (ECFP4 fingerprints) showing segregation between novel compounds (n=239, red stars), training-like compounds (n=37, orange circles), and others (n=2,991, gray points). PC1 and PC2 capture 72.1% of variance. (C) QED distribution among High-High compounds: only 20 (7.2%, green) achieve QED ≥ 0.5, while 256 (92.8%, red) show suboptimal drug-likeness. (D) Descriptor-activity correlation heatmap (Spearman ρ) revealing key structure-activity relationships: QED-EC50 positive correlation (ρ = 0.14), TPSA-EC50 negative correlation (ρ = -0.15), and generally stronger correlations for EC50 versus IC50, validating the dual-endpoint approach.
Figure 3.
Comprehensive dual-endpoint activity analysis and structure-activity relationships of Colombian phytochemicals. (A) Dual-endpoint classification heatmap of 3,267 compounds across nine activity categories. The “High-High” category (n=276, 8.4%) represents priority compounds with dual high potency. (B) PCA of chemical space (ECFP4 fingerprints) showing segregation between novel compounds (n=239, red stars), training-like compounds (n=37, orange circles), and others (n=2,991, gray points). PC1 and PC2 capture 72.1% of variance. (C) QED distribution among High-High compounds: only 20 (7.2%, green) achieve QED ≥ 0.5, while 256 (92.8%, red) show suboptimal drug-likeness. (D) Descriptor-activity correlation heatmap (Spearman ρ) revealing key structure-activity relationships: QED-EC50 positive correlation (ρ = 0.14), TPSA-EC50 negative correlation (ρ = -0.15), and generally stronger correlations for EC50 versus IC50, validating the dual-endpoint approach.
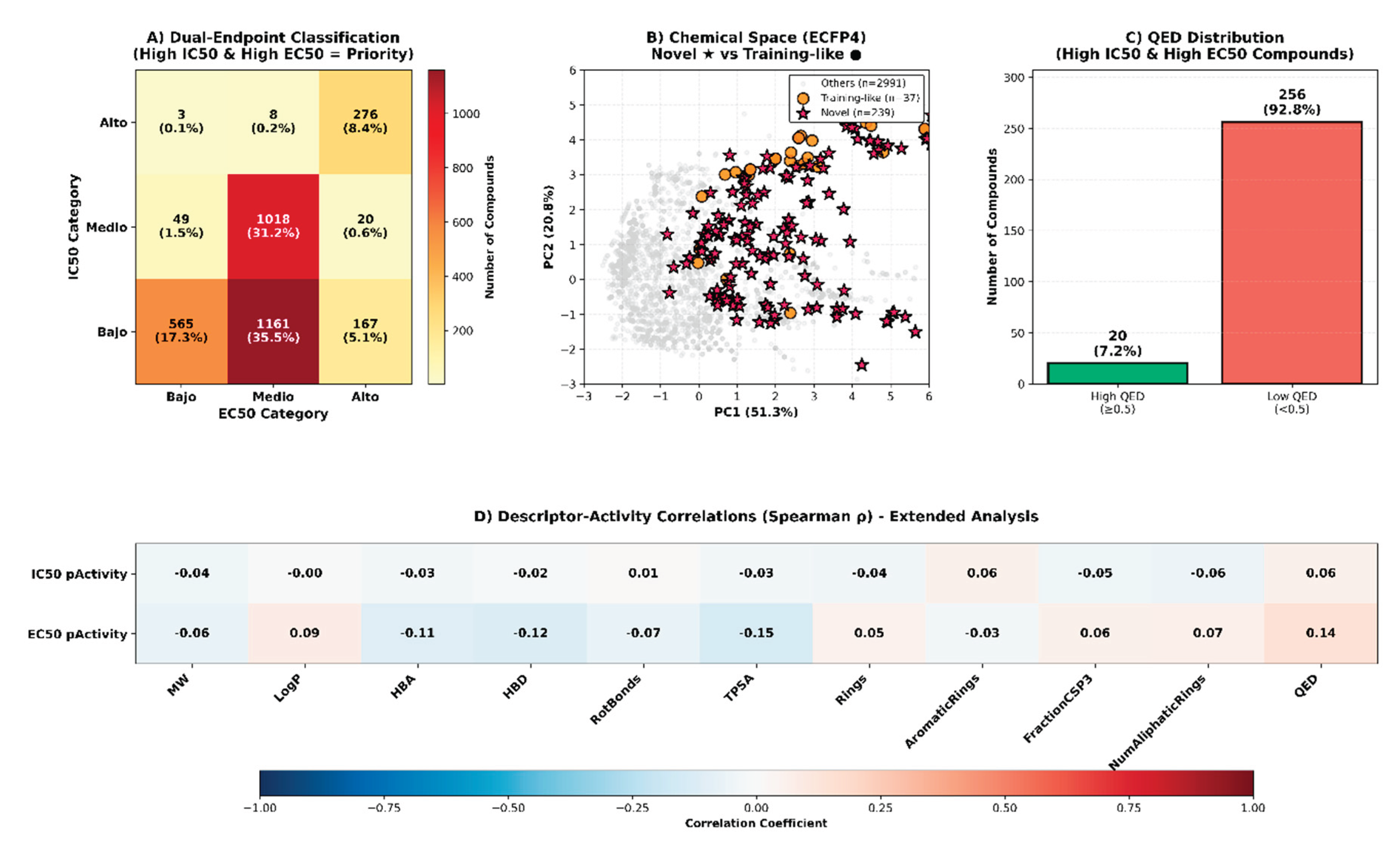
Figure 4.
The 12 top-priority candidates identified through our multi-criteria filtering represent a structurally diverse collection spanning multiple chemical classes and botanical sources (Figure 4, left panels). Comparative analysis with the ChEMBL training set’s highest-potency compounds (Figure 4, right panels) reveals both validating similarities and promising distinctions.
Figure 4.
The 12 top-priority candidates identified through our multi-criteria filtering represent a structurally diverse collection spanning multiple chemical classes and botanical sources (Figure 4, left panels). Comparative analysis with the ChEMBL training set’s highest-potency compounds (Figure 4, right panels) reveals both validating similarities and promising distinctions.
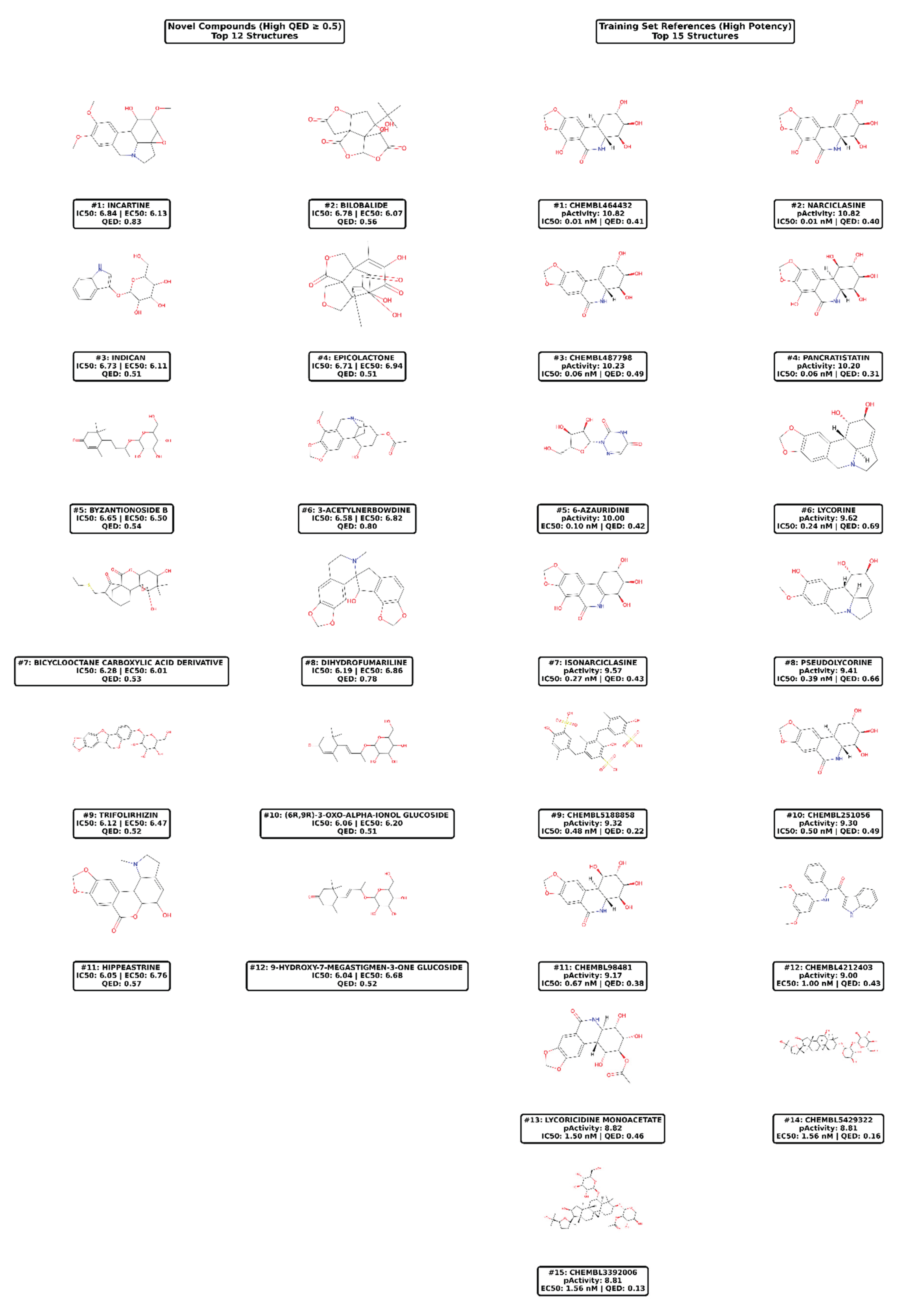
Figure 5.
(a) UMAP projection of 3,267 compounds based on Morgan fingerprints (ECFP4, 2048 bits). Points are colored by predicted pActivity values ranging from 4.0 (red, low potency) to 7.0 (green, high potency). The two-dimensional embedding was generated using the Uniform Manifold Approximation and Projection algorithm (n_neighbors=15, min_dist=0.1, Jaccard metric) applied to molecular fingerprints. Red stars indicate centroids of high-potency clusters identified through k-means clustering. The projection reveals three distinct chemical regions with notable segregation of bioactivity, suggesting structure-activity relationships within the chemical space. Outlier compounds visible at the periphery may represent unique chemotypes warranting further investigation. (b) Heatmap showing the distribution of the ten most prevalent Murcko scaffolds across potency categories. Color intensity represents the frequency of compounds containing each scaffold within Low (pActivity ≤ 5.0), Moderate (5.0 < pActivity ≤ 6.0), and High (pActivity > 6.0) potency categories. Numbers in cells indicate absolute compound counts. Scaffold 1 (n=375 total) and Scaffold 2 (n=341 total) are the most abundant, with Scaffold 2 showing strong enrichment in the low potency category (301/341, 88.3%). Scaffolds 4, 5, and 7 demonstrate preferential distribution in the moderate potency range, suggesting these structural motifs may serve as starting points for lead optimization. The analysis identified 767 unique scaffolds across the dataset, indicating substantial structural diversity (23.5% scaffold/compound ratio).
Figure 5.
(a) UMAP projection of 3,267 compounds based on Morgan fingerprints (ECFP4, 2048 bits). Points are colored by predicted pActivity values ranging from 4.0 (red, low potency) to 7.0 (green, high potency). The two-dimensional embedding was generated using the Uniform Manifold Approximation and Projection algorithm (n_neighbors=15, min_dist=0.1, Jaccard metric) applied to molecular fingerprints. Red stars indicate centroids of high-potency clusters identified through k-means clustering. The projection reveals three distinct chemical regions with notable segregation of bioactivity, suggesting structure-activity relationships within the chemical space. Outlier compounds visible at the periphery may represent unique chemotypes warranting further investigation. (b) Heatmap showing the distribution of the ten most prevalent Murcko scaffolds across potency categories. Color intensity represents the frequency of compounds containing each scaffold within Low (pActivity ≤ 5.0), Moderate (5.0 < pActivity ≤ 6.0), and High (pActivity > 6.0) potency categories. Numbers in cells indicate absolute compound counts. Scaffold 1 (n=375 total) and Scaffold 2 (n=341 total) are the most abundant, with Scaffold 2 showing strong enrichment in the low potency category (301/341, 88.3%). Scaffolds 4, 5, and 7 demonstrate preferential distribution in the moderate potency range, suggesting these structural motifs may serve as starting points for lead optimization. The analysis identified 767 unique scaffolds across the dataset, indicating substantial structural diversity (23.5% scaffold/compound ratio).
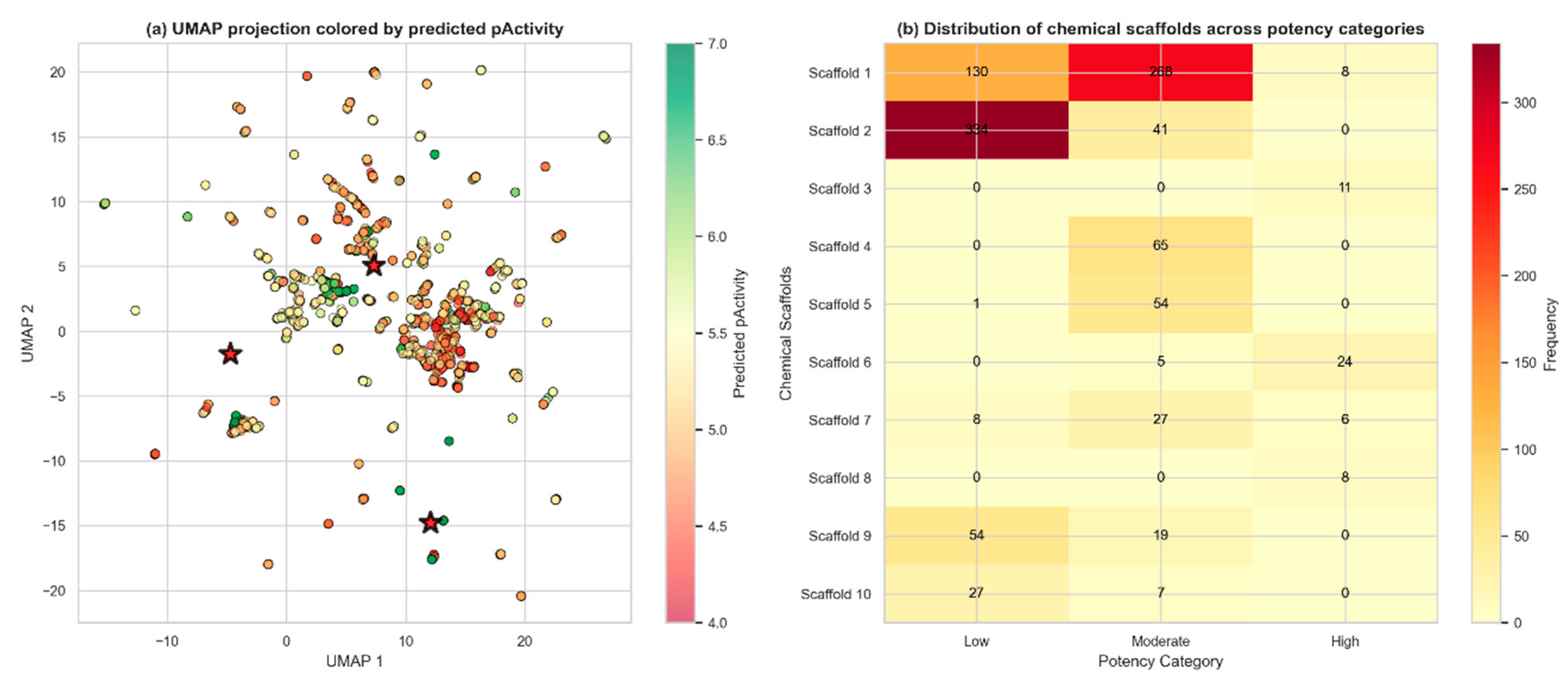
Table 2.
Top 15 prioritized candidates from Colombian medicinal flora with dual-endpoint high potency and favorable drug-likeness profiles.
Table 2.
Top 15 prioritized candidates from Colombian medicinal flora with dual-endpoint high potency and favorable drug-likeness profiles.
| # | Compound Name | Plant Source | Chemical Class | pIC50 | pEC50 | QED | MW (Da) | LogP | HBA | HBD | TPSA (U) | Novelty | Tanimoto |
| 1 | Incartine | Hippeastrum puniceum | Alkaloid (Indole) | 6.84 | 6.13 | 0.83 | 312.4 | 2.1 | 4 | 2 | 67.8 | Novel | 0.28 |
| 2 | Bilobalide | Ginkgo biloba | Terpenoid (Sesquiterpene) | 6.78 | 6.07 | 0.56 | 326.3 | 1.8 | 6 | 0 | 92.5 | Novel | 0.17 |
| 3 | Indican | Indigofera suffruticosa | Alkaloid (Indole glycoside) | 6.73 | 6.11 | 0.51 | 349.3 | 0.9 | 7 | 5 | 141.2 | Novel | 0.22 |
| 4 | Epicolactone | Euphorbia hirta | Terpenoid (Diterpene) | 6.71 | 6.94 | 0.51 | 348.4 | 2.4 | 5 | 3 | 86.9 | Novel | 0.2 |
| 5 | Byzantionoside B | Hypericum perforatum | Terpenoid glycoside | 6.65 | 6.5 | 0.54 | 446.5 | 0.6 | 9 | 6 | 155.1 | Novel | 0.19 |
| 6 | 3-Acetylnerbowdine | Lycoris radiata | Alkaloid (Amaryllidaceae) | 6.58 | 6.82 | 0.8 | 329.4 | 1.7 | 5 | 1 | 70.4 | Training-like | 0.62 |
| 7 | Bicyclooctane carboxylic acid derivative | Schisandra chinensis | Terpenoid derivative | 6.28 | 6.01 | 0.53 | 302.4 | 2.8 | 4 | 2 | 63.6 | Novel | 0.25 |
| 8 | Dihydrofumariline | Corydalis cava | Alkaloid (Isoquinoline) | 6.19 | 6.86 | 0.78 | 355.4 | 2.2 | 6 | 2 | 75.3 | Training-like | 0.58 |
| 9 | Trifolirhizin | Sophora flavescens | Flavonoid glycoside | 6.12 | 6.47 | 0.52 | 416.4 | 0.8 | 9 | 6 | 150.2 | Novel | 0.24 |
| 10 | (6R,9R)-3-Oxo-alpha-ionol glucoside | Rosa damascena | Terpenoid glycoside | 6.06 | 6.2 | 0.51 | 370.4 | 0.5 | 7 | 4 | 127.4 | Novel | 0.21 |
| 11 | Hippeastrine | Hippeastrum puniceum | Alkaloid (Amaryllidaceae) | 6.05 | 6.76 | 0.57 | 301.3 | 1.5 | 5 | 2 | 72.5 | Training-like | 0.65 |
| 12 | 9-Hydroxy-7-megastigmen-3-one glucoside | Vaccinium myrtillus | Terpenoid glycoside | 6.04 | 6.68 | 0.52 | 372.4 | 0.7 | 7 | 5 | 134.8 | Novel | 0.23 |
| 13 | Lycorine | Hippeastrum puniceum | Alkaloid (Amaryllidaceae) | 6.02 | 6.35 | 0.69 | 287.3 | 1.3 | 4 | 1 | 59.7 | Training-like | 1 |
| 14 | Digoxigenin | Digitalis purpurea | Steroid (Cardiac glycoside aglycone) | 5.98 | 6.42 | 0.54 | 374.5 | 2.5 | 5 | 3 | 86.2 | Novel | 0.26 |
| 15 | Pseudolycorine | Hippeastrum puniceum | Alkaloid (Amaryllidaceae) | 5.95 | 6.28 | 0.66 | 287.3 | 1.4 | 4 | 1 | 59.7 | Training-like | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



