Submitted:
29 October 2025
Posted:
29 October 2025
You are already at the latest version
Abstract
Background/Objectives: Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 or PIP2) is a low-abundance plasma membrane phosphoinositide, an essential factor for clathrin-mediated endocytosis. Activation of Gq-protein-coupled receptors (GqPCR) such as protease-activated receptor 2 (PAR2) triggers phospholipase C–mediated hydrolysis of PIP2, creating a paradox in which the lipid is simultaneously consumed for signaling yet required for receptor internalization. I previously showed that β-arrestin promotes PIP2 regeneration by recruiting Phosphatidylinositol-4-phosphate 5-kinase (PIP5K), partially resolving this paradox. However, the modest hyperactivation of PIP5K (~2–3 fold) and the hump-like kinetics of PIP2 recovery following β-arrestin knockdown indicated that additional regulatory mechanisms were involved. Methods: Here, my revised model incorporates contributions from both preexisting and newly forming clathrin-coated pits (CCPs) using Virtual Cell. Results: I find that CCPs provide a spatially confined environment that promotes clustering of receptor–β-arrestin complexes, thereby amplifying cooperatively PIP5K activity through weak interaction between PIP5K and β-arrestins in the receptor–β-arrestin-CCP complexes. This cooperative mechanism accounts for nonlinear PIP2 recovery kinetics, delayed kinase activation under β-arrestin knockdown conditions, and the sufficiency of modest PIP5K activation to restore plasma membrane lipid pools. These findings suggest that transient and spatially restricted protein interactions, often considered weak or biologically insignificant, play decisive roles in lipid metabolism and receptor trafficking. Conclusions: This framework provides a mechanistic resolution to the paradox of simultaneous PIP2 hydrolysis and receptor internalization and highlights the importance of cooperative dynamics in maintaining plasma membrane signaling competence.
Keywords:
cooperative kinase activation
; PIP5K
; β-arrestin
; endocytosis
; GqPCR
1. Introduction
Phosphoinositides are critical regulators of signaling, trafficking, and cytoskeletal dynamics although they are minor constituents of cellular membranes [1]. Among them, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 or PIP2) plays a central role in clathrin-mediated endocytosis by recruiting adaptor proteins, clathrin, and actin regulators [2,3,4,5]. Acute perturbation of PIP2 levels demonstrates its indispensability: optogenetic or enzymatic depletion of PIP2 blocks clathrin-coated pit (CCP) initiation and receptor internalization [6,7,8]. These studies establish PIP2 not only as a structural lipid but also as a dynamic regulator of receptor trafficking.
G-protein–coupled receptors (GPCRs) comprise the largest family of cell-surface receptors and mediate responses to diverse extracellular cues. Upon activation, Gq-coupled GPCRs stimulate phospholipase Cβ (PLCβ), which hydrolyzes PIP2 into diacylglycerol (DAG) and inositol trisphosphate (IP3), thereby reducing plasma membrane PIP2 levels [9,10]. Yet GPCRs such as protease-activated receptor 2 (PAR2) undergo clathrin-dependent endocytosis for desensitization and trafficking, a process that requires PIP2. This apparent contradiction — that Gq signaling consumes PIP2 while GPCR endocytosis depends on it — represents a long-standing paradox in GPCR biology [11,12].
CCPs are increasingly recognized as more than passive vesicle carriers. CCPs serve as multifunctional signaling microdomains that integrate receptor trafficking with downstream signal transduction [13,14]. For example, they provide a spatial platform for MAPK activation downstream of GPCRs [15]. β-arrestins are key to this dual role. They not only terminate G-protein signaling but also scaffold MAPK components and link receptors to clathrin and AP-2 [16]. Live-cell and single-molecule imaging studies have shown that β-arrestin–receptor complexes display dynamic lifetimes and can persist within CCPs, where they regulate both receptor endocytosis and signal propagation [17,18,19].
The previous work provided evidence that β-arrestins contribute directly to the regeneration of PIP2 at the plasma membrane following PAR2 activation [20]. Using lipid biosensors, I found that β-arrestin knockdown impaired PIP2 recovery. Further experimental results suggested that phosphatidylinositol 4-phosphate 5-kinase (PIP5K), the kinase that synthesizes PIP2 from PI(4)P, is recruited to β-arrestin-receptor-clathrin complexes, linking receptor signaling to localized lipid metabolism [21]. These findings offered a potential solution to the PIP2 paradox. However, several observations remain unexplained: the degree of PIP5K hyperactivation was modest (2–3 fold), and β-arrestin knockdown produced a hump-like trajectory in PIP2 recovery inconsistent with linear recruitment models.
In the present study, I tested the hypothesis that PIP5K activity is cooperatively modulated within CCPs using mathematical modeling. I proposed that CCPs can provide spatial confinement and clustering of receptor–β-arrestin complexes that amplify cooperatively the PIP5K activity. Through mathematical modeling, I showed that transient, weak protein–protein interactions at CCPs can be sufficient to reproduce the nonlinear kinetics of PIP2 recovery, the hump phenotype of β-arrestin knockdown, and the modest hyperactivation of PIP5K. These results advance a conceptual framework in which both stable and transient protein interactions at CCPs govern lipid metabolism, thereby ensuring that GPCR signaling and endocytosis remain tightly coupled and reversible.
2. Materials and Methods
2.1. Model Overview
A mathematical model was developed to describe cooperative regulation of PIP5K activity during recovery of PIP2 following activation of Gq-coupled PAR2. The model builds upon our previous framework [20] in which PIP2 hydrolysis and resynthesis kinetics were represented as coupled differential equations describing the balance between phospholipase C (PLC)–mediated degradation and PIP5K-dependent synthesis. The current model introduces additional parameters to account for cooperative enzyme activation and the distinct contributions of newly forming versus preexisting CCPs prior to endocytosis of receptor by dynamin action.
Model Assumptions: 1) Activation of PAR2 by 100 μM activating peptide (AP) induces rapid PIP2 hydrolysis by PLC, leading to transient depletion of the lipid pool at the plasma membrane. 2) PIP5K activity recovers PIP2 levels with two distinct components: one associated with newly forming CCPs and another with preexisting CCPs. 3) β-arrestin–dependent recruitment of PIP5K to CCPs modulates enzyme activity cooperatively, described by Hill-type equations with coefficients n and m, representing newly forming and preexisting CCPs, respectively. 4) The kinetic equations assume well-mixed conditions and constant total enzyme concentration. Spatial heterogeneity and stochastic fluctuations were not explicitly modeled but are implicitly reflected in the cooperative parameters.
2.2. Mathematical Formulation
The cooperative activation term was expressed as:
where, RLPAAPC represents the ligand (L)–bound, phosphorylated (P) GPCR (R) complexed with β-arrestin (A) that nucleates newly forming CCPs through adaptor (AP)–clathrin (C) interactions. RLPA_CCP represents the ligand (L)-bound, phosphorylated (P) GPCR (R) complexed with β-arrestin (A) associated with a preexisting CCP. a and b are weighting constants describing the fractional contributions of newly forming and preexisting CCPs, respectively. Ka and Kb are apparent dissociation constants for each process. n and m are Hill coefficients indicating the degree of cooperativity for each component.
2.3. Parameter Selection
Parameter values were based on previously reported measurements and adjusted to match experimentally observed kinetics of PIP2 depletion and recovery after PAR2 activation [20,21]. The Hill coefficients (n, m) were varied between 1 and 6 to explore the effects of cooperativity. Weighting constants (a, b) were set to evaluate the relative contributions of newly forming and preexisting CCPs. All parameters and equations for reactions were described in Table 1 and Table 2.
Simulation and Analysis: All simulations were performed in Virtual Cell. Plots were generated using the Origin pro (OriginLab Corp. MA, USA) for figures.
β-Arrestin Knockdown Simulations: To simulate β-arrestin knockdown, the amount of cytosolic β-arrestin was reduced by 20%, corresponding to the experimentally observed decrease in PIP2 recovery following β-arrestin depletion. This reduction delayed PIP2 recovery and produced the characteristic “hump” phenotype in the simulated time course.
3. Results
Using a simple diagram, I summarized previous findings [21] as shown in Figure 1A. According to a new simulation result, one modification was localization of PIP5K among the β-arrestin-GPCR complexes at the CCP to describe two major findings: 1) modest hyperactivation of PIP5K after desensitization of PAR2 and 2) hump formation of PIP2 level while β-arrestin was knocked down using siRNA.
To build upon our previous findings, we first examined the dynamics of PIP2 following PAR2 activation. Using a PIP2-sensitive PH probe, we observed that basal PIP2 levels after desensitization of PAR2 exhibited an overshoot [21]. Recovery of PIP2 was markedly impaired when β-arrestin expression was reduced by siRNA, producing a pronounced hump-like trajectory. This data suggested that an additional regulatory mechanism must be incorporated.
Interestingly, PIP5K activity was enhanced only 2–3 fold following PAR2 activation, a degree of hyperactivation that is difficult to reconcile with a simple conformational change mechanism which makes order of magnitude different activity of enzymes. This observation led us to hypothesize that the spatial organization of CCPs could play a critical role in modulating lipid kinase activity. Consistent with this idea, we found that PAR2 associated with both preexisting and newly forming CCPs. However, PAR2 was confined to the periphery of preexisting CCPs, whereas newly forming CCPs dynamically recruited PAR2–β-arrestin complexes together with endocytic machinery. These findings pointed out to de novo CCP formation as a major driver of PIP5K activation.
To capture these features, we developed a revised mathematical model (Figure 1B).
Unlike my previous framework, which treated PIP5K stimulation as a constant, the updated model incorporated dynamic terms reflecting CCP formation. The second and third terms of the equation represent contributions from newly forming and preexisting CCPs, respectively. I further propose that cooperative activation of PIP5K arises within the confined environment of newly forming CCPs rather than through direct conformational changes. Cooperative effects were modeled using Hill equations, with coefficients (n and m) representing newly forming and preexisting CCPs. Parameter tuning allowed the model to reproduce the modest 2–3-fold increase in kinase activity observed experimentally. As the Hill coefficient increased, basal PIP2 levels rose and reached a plateau (Figure 1C, inset).
The model also recapitulated the hump phenotype under conditions of β-arrestin knockdown. At high Hill coefficients (n and m = 6), PIP2 recovery remained incomplete and exhibited a hump-like trajectory (Figure 2A). Correspondingly, PIP5K activation was delayed and sustained at higher levels due to cooperative activation of the kinase (Figure 2B). Knockdown of β-arrestins further prolonged this delay, producing a slow recovery phase followed by accelerated restoration of PIP2. These results suggest that the hump arises from delayed, cooperative clustering of PIP5K mediated by residual β-arrestin–dependent CCP formation.
We next dissected the relative contributions of preexisting and newly forming CCPs by varying constants a and b (Figure 3A). When a = 0, eliminating the contribution of newly forming CCPs, PIP2 recovery was substantially reduced, more so than when b = 0. This indicates that newly forming CCPs play the dominant role in PIP2 homeostasis during desensitization of PAR2. Moreover, the delayed activation pattern of PIP5K disappeared when a = 0 but persisted when b = 0 (Figure 3B), further highlighting the central role of newly forming CCPs in shaping lipid kinase dynamics. As shown in Figure 3C and D, the PIP2 hump and PIP5K activity were reproduced by b = 0 which reflects that only newly forming CCP works for the recovery, suggesting that in the condition, newly forming CCP can be a key locus of the PIP2 recovery. However, it cannot be ruled out the contribution of preexisting CCP due to low degree of PIP5K activation and PIP2 recovery.
Interestingly, constraining m = 1 to reflect the lack of cooperative PAR2–β-arrestin clustering within preexisting CCPs had some effect on either PIP2 recovery or PIP5K activation kinetics (Figure 4). These results indicate that cooperativity associated with newly forming CCPs is the primary contributor to PIP2 regeneration, but not full contributor as previously mentioned, suggesting that the contribution between preexisting and newly forming CCP on the cooperative behavior of the kinase would be balanced by the Hill coefficients (n and m) and weighting constants (a and b) in the individual cases.
4. Discussion
This study identifies a cooperative mechanism by which transient β-arrestin–dependent activation of PIP5K restores PIP2 levels at the plasma membrane following activation of Gq-coupled receptors such as PAR2. Through mathematical modeling, we show that even modest increases in PIP5K activity—amplified by cooperative interactions within CCPs—are sufficient to regenerate PIP2 and maintain receptor signaling competence. This mechanism provides a molecular explanation for the long-standing paradox of how GqPCRs can undergo endocytosis despite hydrolysis of their essential substrate, PIP2.
4.1. Cooperative PIP5K Activation as the Key Regulatory Principle
My model demonstrates that the recovery of PIP2 cannot be explained by linear enzyme activation alone. The inclusion of Hill-type cooperative behavior in PIP5K activity, mediated by β-arrestin–GPCR complexes, reproduces the experimentally observed “hump” pattern of PIP2 recovery following β-arrestin knockdown. The Hill coefficients (n and m) describe the degree of cooperativity within newly forming and preexisting CCPs, respectively. High cooperativity (n & m ≈ 6) in CCPs accounts for both the overshoot in PIP2 recovery and the delayed activation kinetics seen experimentally. Importantly, these Hill coefficients should be viewed as phenomenological parameters that capture emergent cooperative behavior rather than literal molecular stoichiometry. Such cooperativity likely arises from multiple weak and transient interactions among β-arrestin, GPCRs, and endocytic adaptors within spatially confined CCP microdomains. How about effect of contribution constants a (newly forming CCP) and b (preexisting CCP) on the PIP2 recovery? Probably, the relative contribution can be determined by ratio of the newly forming CCP and preexisting CCP depending on cell cycle and classes of GqPCR.
4.2. Functional Difference Between Newly Forming and Preexisting CCPs
By systematically varying contribution parameters (a and b), our simulations revealed that newly forming CCPs are the primary drivers of delayed PIP2 regeneration with nonlinearity, whereas preexisting CCPs play a marginal role for the dynamics. Newly forming CCPs provide an environment where β-arrestin–receptor complexes can nucleate adaptor proteins and clathrin assembly, enabling cooperative clustering of PIP5K which makes delayed kinetics. In contrast, preexisting CCPs are diffusion-limited and recruit the GPCR-β-arrestin complexes at the edge of the CCPs, restricting the formation of cooperative assemblies which make linear saturation kinetics. This distinction explains why β-arrestin–dependent clustering at newly forming CCPs produces robust recovery kinetics, whereas preexisting CCPs cannot fully compensate under knockdown conditions.
4.3. Broader Implications of Transient and Weak Interactions
Beyond this specific system, our findings highlight the broader principle that weak and transient protein–protein interactions can collectively generate robust signaling outcomes. Although individual β-arrestin–PIP5K interactions are short-lived, their repeated and cooperative engagement within confined CCPs amplifies enzymatic activity and ensures lipid homeostasis. This concept aligns with emerging views that weak, dynamic assemblies—rather than stable complexes—underlie efficient information transfer in signaling networks. Similar cooperative mechanisms may regulate other lipid kinases or scaffolded enzymes operating in nanoscale membrane domains.
Transient protein–protein interactions can induce weak or short-lived conformational changes in kinases that enhance their catalytic output without fully stabilizing the active conformation. Such partial structural rearrangements, often involving local loop motions or small domain shifts, can transiently align catalytic residues or promote substrate accessibility, thereby producing a modest but physiologically significant increase in activity [22]. This contrasts with stable conformational changes, such as strong scaffold binding, which lock the enzyme into a fully active state and yield maximal catalytic efficiency [23]. The transiently induced state thus represents a dynamic intermediate between inactive and fully active conformations—sufficient to fine-tune signaling output while maintaining reversibility and rapid responsiveness to fluctuating stimuli. In this framework, transient binding events act as kinetic modulators that bias the conformational ensemble toward catalytically permissive states without requiring full structural rearrangement, allowing cells to modulate kinase activity in a graded, energy-efficient manner.
4.4. Potential Crosstalk of PAR2–PIP2–PIP5K Signaling with Oncogenic KRAS
Our findings describing transient β-arrestin–dependent activation of PIP5K and cooperative recovery of PIP2 at the plasma membrane have potential implications beyond receptor endocytosis. Increasing evidence suggests that PAR2-mediated lipid signaling intersects with KRAS activation and phosphoinositide 3-kinase (PI3K)–Akt pathways, all of which play central roles in cancer progression. Protease activated receptors are aberrantly expressed in many cancers, including breast, pancreatic, and colorectal tumors, where it enhances proliferation, migration, and epithelial–mesenchymal transition [24,25]. KRAS requires stable association with the inner leaflet of the plasma membrane via electrostatic interactions between its C-terminal polybasic domain and negatively charged phosphoinositide, particularly phosphatidylserine (PS) and PIP2 [26]. Thus, transient PIP2 regeneration following PAR2 activation may reinforce KRAS membrane anchoring and preserve its signaling-competent conformation, providing a mechanistic bridge between protease sensing, lipid metabolism, and Ras activation. Once activated, KRAS recruits and activates PI3K, which phosphorylates PIP2 to generate PIP3, leading to Akt recruitment toward the plasma membrane and the initiation of downstream survival signaling [27,28]. Through this cooperative loop, PAR2 signaling could synergize with oncogenic KRAS to sustain PI3K–Akt activity, enhancing tumor cell proliferation, motility, and resistance to stress.
Taken together, the PAR2–PIP2–KRAS–PI3K–Akt axis may represent a molecular hub of cooperative signaling that links receptor desensitization, lipid regeneration, and oncogenic amplification. The cooperative PIP5K activation described in our model provides a plausible kinetic framework for how weak, transient β-arrestin–PIP5K interactions can amplify lipid-dependent signaling in cancer contexts. While each interaction is short-lived, their cumulative effect within spatially confined domains produces a sustained output—analogous to how stochastic, diffusion-limited binding can yield emergent cooperativity. In tumor cells, where PIP5K and PAR2 are often overexpressed, this mechanism could lower the activation threshold for KRAS–PI3K–Akt signaling, enabling persistent survival and invasive behavior even in fluctuating extracellular conditions.
4.5. Limitations and Outlook
While the present model successfully reproduces key kinetic features of PIP2 recovery, it simplifies several aspects of the system. It does not explicitly account for protein diffusion barriers, PIP5K isoform specificity, or dynamic exchange of PIP5Ks within CCPs. Future experimental studies employing single-molecule imaging, optogenetic control of PIP5K, and real-time FRET biosensors could quantitatively test these predictions and reveal the spatiotemporal organization of these transient complexes. Incorporating stochastic spatial or agent-based models will further refine our understanding of how nanoscale cooperativity translates into large-scale lipid regulation.
5. Conclusions
In conclusion, our results demonstrate that transient β-arrestin–dependent activation of PIP5K, though modest in magnitude, is sufficient to restore PIP2 levels through cooperative regulation within CCPs. This cooperative mechanism not only explains the paradox of PIP2-dependent endocytosis during GqPCR activation but also establishes a broader paradigm: cellular homeostasis and signaling fidelity emerge from the collective effects of dynamic, weak interactions that fine-tune enzymatic and trafficking processes.
Author Contributions
S.R.J. designed simulations with Virtual Cell and wrote this manuscript.
Data Availability Statement
mathematical modeling would be open source in the Virtual Cell system.
Acknowledgments
I thank Virtual Cell (NIH grant R24 GM137787) for providing a system to build up mathematical modeling.
Conflicts of Interest
No conflict with JSR Biomedical Science Consulting LLC.
Abbreviations
| PIP5K | Phosphatidylinositol-4-phosphate 5-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) |
References
- Krauss, M.; Haucke, V. Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep. 2007, 8(3), 241–246. [CrossRef]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443 (7112), 651–657. [CrossRef]
- Balla, T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93(3), 1019–1137. [CrossRef]
- He, K.; Marsland III, R.; Upadhyayula, S.; Song, E.; Dand, S.; Capraro, B.R.; Wang, W.; Skillern, W.; Gaudin, R.; Ma, M.; Kirchhausen, T. Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 2017, 552 (7685), 410–414. [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19 (5), 313–326. [CrossRef]
- Zoncu, R.; Perera, R.M.; Sebastian, R.; Nakatsu, F.; Chen, H.; Balla, T.; Ayala, G.; Toomre, D.; De Camilli, P. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. of the Natl. Acad. of Sci. USA 2007, 104 (10), 3793–3798. [CrossRef]
- Stauffer, T.P.; Ahn, S.; Meyer, T. Receptor-induced transient reduction of plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 1998, 8(6), 343–346. [CrossRef]
- Antonescu, C.N.; Aguet, F.; Danuser, G.; Schmid, S.L. Phosphatidylinositol-(4,5)-bisphosphate regulates clathrin-coated pit initiation, stabilization, and size. Mol. Biol. Cell 2011, 22(14), 2588–2600. [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [CrossRef]
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends in Pharmacol. Sci. 2004, 25(8), 413–422. [CrossRef]
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100(6), 603–606. [CrossRef]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 2004, 15(2), 444–455. [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [CrossRef]
- von Zastrow, M.; Sorkin, A. Mechanism for regulating and organizing receptor signaling by endocytosis. Annu. Rev. Biochem. 2021, 90, 709-737. [CrossRef]
- Lauffer, B.E.; Melero, C.; Temkin, P.; Lei, C.; Hong, W.; Kortemme, T.; von Zastrow, M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J. Cell Biol. 2010, 190(4), 565-574. [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32(9), 521–533. [CrossRef]
- Nuber, S.; Zabel, U.; Lorenz, K.; Nuber, A.; Milligan, G.; Tobin, A.B.; Lohse, M.J.; Hoffmann, C. β-arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016, 531(7596), 661–664. [CrossRef]
- Sungkaworn, T.; Jobin, M.-L.; Burnecki, K.; Weron, A.; Lohse, M.J.; Calebiro, D. Single-molecule imaging reveals receptor–G protein interactions at the plasma membrane. Nature 2017, 550(7677), 543–547. [CrossRef]
- Eichel, K.; Jullié, D.; von Zastrow, M. β-arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 2016, 18(3), 303-310. [CrossRef]
- Jung, S.-R.; Seo, J.B.; Deng, Y.; Asbury, C.L.; Hille, B.; Koh, D.S. Contributions of protein kinases and β-arrestin to termination of protease-activated receptor 2 signaling. J. Gen. Physiol. 2016, 147(3), 255-271. [CrossRef]
- Jung, S.-R.; Jiang, Y.; Seo, J.B.; Chiu, T.; Hille, B; Koh, D.-S. β-arrestin-dependent PI(4,5)P2 synthesis boosts GPCR endocytosis. Proc. Natl. Acad. Sci. USA 2021, 118(17), e20111023118. [CrossRef]
- Clark, L.K.; Cullati, S.N.; Activation is only the beginning: mechanisms that tune kinase substrate specificity. Biochem. Soc. Trans. 2025, BST20241420. [CrossRef]
- Hoeflich, K.P.; Ikura, M.; Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 2002, 108(6):739-742. [CrossRef]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 11, 748–754. [CrossRef]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2004, 2(7), 395–402. [CrossRef]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.J.; Gorfe, A.A.; Hancock, J.F. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 2017, 168:239-251.e16. [CrossRef]
- Eser, S.; Reiff, N.; Messer, M.; et al. Selective requirement of PI3K/PDK1 signaling for KRAS oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23(3),193–206. [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 605–635. [CrossRef]
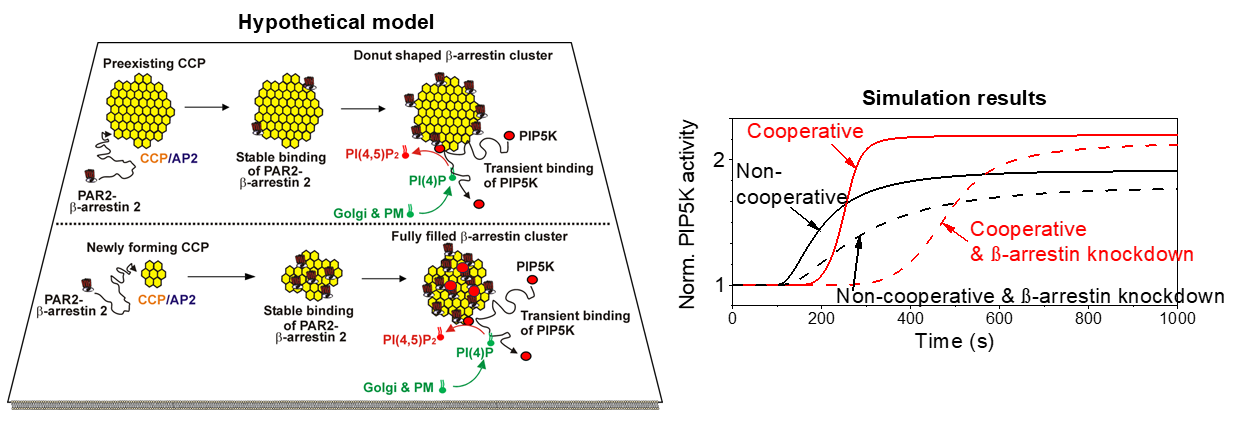
Figure 1.
Cooperative regulation of PIP5K activity by β-arrestin–GPCR–AP2–clathrin-coated pit complexes. (A) Schematic representation of β-arrestin-GPCR complexes binding to clathrin-coated pits (CCPs). This diagram was modified from previous publication by the author [21]. Top: binding to a preexisting CCP. Bottom: recruitment into a newly forming CCP initiated by adaptor protein 2 (AP2)–clathrin assembly. (B) Parameters used to describe cooperative effects of β-arrestin–GPCR complexes within CCPs on PIP5K activity. RLPAAPC denotes the state in which a ligand-bound (RL), phosphorylated (P) GPCR complexed with β-arrestin (A) nucleates a newly forming CCP through AP2–clathrin (APC) assembly. RLPA_CCP represents the state in which the same receptor complex associates with a preexisting CCP. Cooperative constants n and m describe the degree of cooperative activation in each state (RLPAAPC and RLPA_CCP, respectively). The weighting a and b constants indicate relative contribution of preexisting and newly forming CCP, respectively. The Ka and Kb are apparent dissociation constants for cooperative reactions. (C) Simulated hydrolysis of PIP2 in response to PAR2 activation by 100 μM activating peptide (AP), as a function of the cooperative constants (n and m). Each curve represents PIP2 dynamics at values of n and m ranging from 1 (no cooperativity) to 6 (high cooperativity).
Figure 1.
Cooperative regulation of PIP5K activity by β-arrestin–GPCR–AP2–clathrin-coated pit complexes. (A) Schematic representation of β-arrestin-GPCR complexes binding to clathrin-coated pits (CCPs). This diagram was modified from previous publication by the author [21]. Top: binding to a preexisting CCP. Bottom: recruitment into a newly forming CCP initiated by adaptor protein 2 (AP2)–clathrin assembly. (B) Parameters used to describe cooperative effects of β-arrestin–GPCR complexes within CCPs on PIP5K activity. RLPAAPC denotes the state in which a ligand-bound (RL), phosphorylated (P) GPCR complexed with β-arrestin (A) nucleates a newly forming CCP through AP2–clathrin (APC) assembly. RLPA_CCP represents the state in which the same receptor complex associates with a preexisting CCP. Cooperative constants n and m describe the degree of cooperative activation in each state (RLPAAPC and RLPA_CCP, respectively). The weighting a and b constants indicate relative contribution of preexisting and newly forming CCP, respectively. The Ka and Kb are apparent dissociation constants for cooperative reactions. (C) Simulated hydrolysis of PIP2 in response to PAR2 activation by 100 μM activating peptide (AP), as a function of the cooperative constants (n and m). Each curve represents PIP2 dynamics at values of n and m ranging from 1 (no cooperativity) to 6 (high cooperativity).
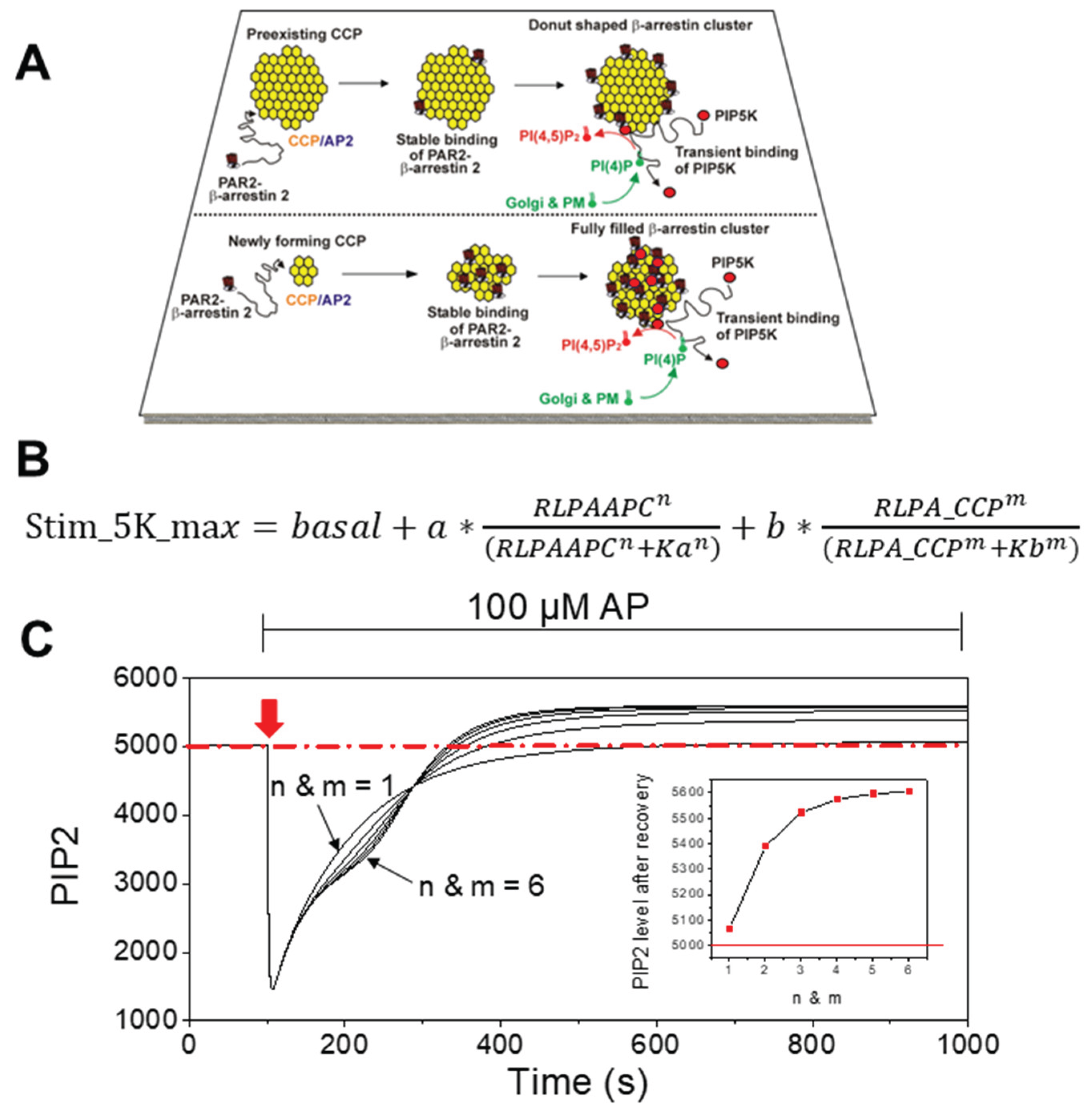
Figure 2.
Cooperative PIP5K activity accounts for hump formation in PIP2 recovery follow ing β-arrestin knockdown. (A) Simulation of PIP2 recovery after PAR2 desensitization at varying cooperative constants. When the cooperative constants (n and m) were set to 6, the hump in PIP2 recovery was most pronounced. (B) Effect of β-arrestin knockdown on PIP5K activity. Stim_5K_max represents maximal PIP5K activity. Following PAR2 activation, β-arrestin knockdown by 20% caused a delayed rise in PIP5K activity and PIP2 recovery. In both control and β-arrestin knockdown conditions, kinase activity was elevated by ~ 30% more compared with the n and m = 1 case, owing to the coherent increase contributed by the two terms representing preexisting and newly forming CCPs.
Figure 2.
Cooperative PIP5K activity accounts for hump formation in PIP2 recovery follow ing β-arrestin knockdown. (A) Simulation of PIP2 recovery after PAR2 desensitization at varying cooperative constants. When the cooperative constants (n and m) were set to 6, the hump in PIP2 recovery was most pronounced. (B) Effect of β-arrestin knockdown on PIP5K activity. Stim_5K_max represents maximal PIP5K activity. Following PAR2 activation, β-arrestin knockdown by 20% caused a delayed rise in PIP5K activity and PIP2 recovery. In both control and β-arrestin knockdown conditions, kinase activity was elevated by ~ 30% more compared with the n and m = 1 case, owing to the coherent increase contributed by the two terms representing preexisting and newly forming CCPs.
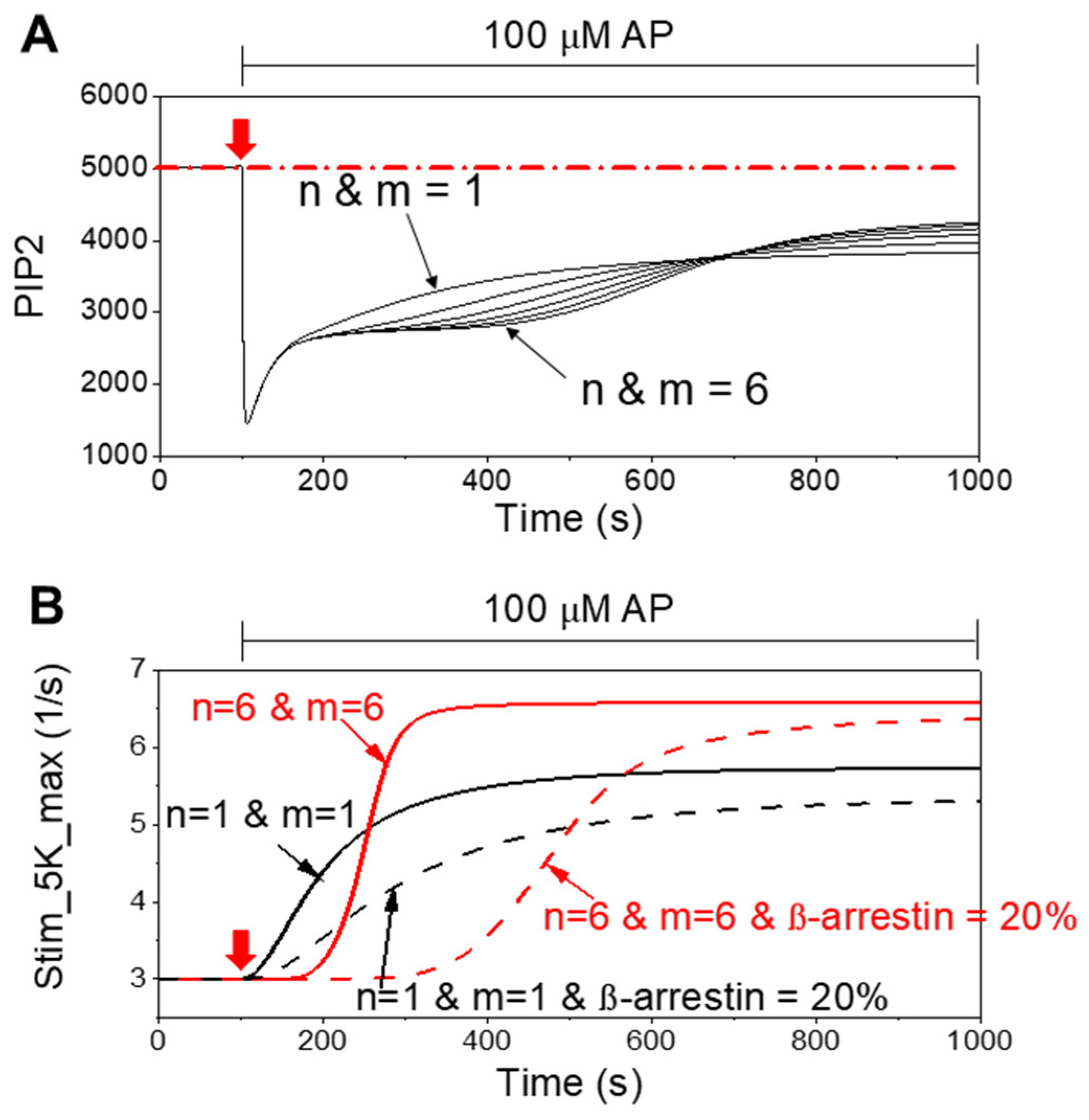
Figure 3.
Newly forming clathrin-coated pits (CCPs) make the dominant contribution to PIP2 recovery. (A) Simulated PIP2 recovery under conditions where contribution constants were set to zero. Setting a = 0 (eliminating the contribution of newly forming CCPs) markedly impaired PIP2 recovery, whereas setting b = 0 (eliminating the contribution of preexisting CCPs) produced a smaller effect. The red broken lines in A & B indicate the level of PIP2 and PIP5K activity in control condition (n & m = 6). (B) PIP5K activity, represented by stim_5K_max, as a function of CCP contribution. Loss of newly forming CCP input (a = 0) strongly reduced and delayed kinase activity, while loss of preexisting CCP input (b = 0) had a relatively marginal effect. These relative constants were optimized to produce marginal increase of PIP5K activity and hump formation during PIP2 recovery induced by β-arrestin knock-down. (C-D) The effect of removal of a or b contribution during β-arrestin knockdown in PIP2 and PIP5K activity. The red broken lines in C & D indicate the level of PIP2 and PIP5K activity during β-arrestin knockdown.
Figure 3.
Newly forming clathrin-coated pits (CCPs) make the dominant contribution to PIP2 recovery. (A) Simulated PIP2 recovery under conditions where contribution constants were set to zero. Setting a = 0 (eliminating the contribution of newly forming CCPs) markedly impaired PIP2 recovery, whereas setting b = 0 (eliminating the contribution of preexisting CCPs) produced a smaller effect. The red broken lines in A & B indicate the level of PIP2 and PIP5K activity in control condition (n & m = 6). (B) PIP5K activity, represented by stim_5K_max, as a function of CCP contribution. Loss of newly forming CCP input (a = 0) strongly reduced and delayed kinase activity, while loss of preexisting CCP input (b = 0) had a relatively marginal effect. These relative constants were optimized to produce marginal increase of PIP5K activity and hump formation during PIP2 recovery induced by β-arrestin knock-down. (C-D) The effect of removal of a or b contribution during β-arrestin knockdown in PIP2 and PIP5K activity. The red broken lines in C & D indicate the level of PIP2 and PIP5K activity during β-arrestin knockdown.
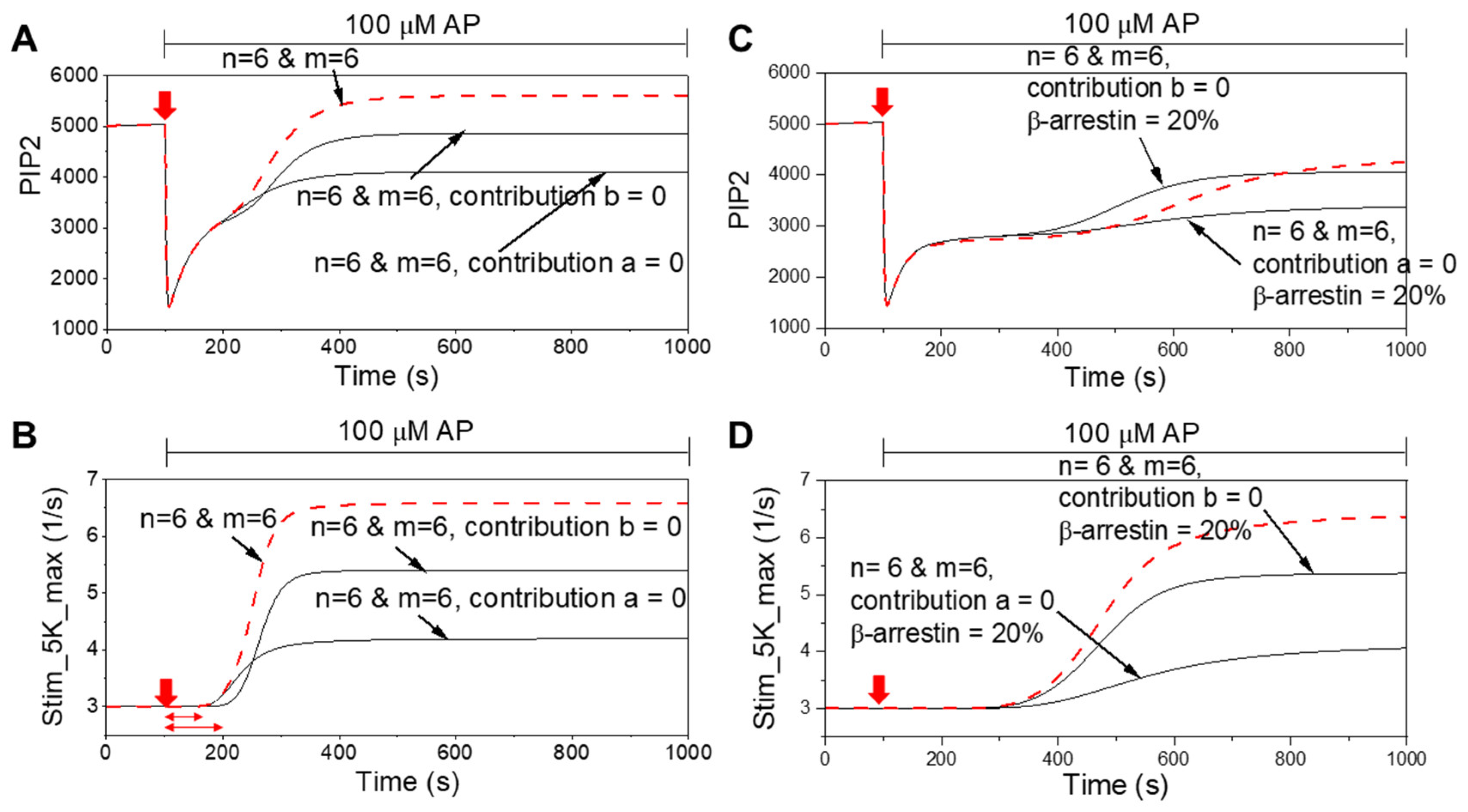
Figure 4.
Both preexisting and newly forming CCPs contribute to PIP2 recovery. (A) Simulated PIP2 hydrolysis and recovery after PAR2 activation (100 μM AP) under varying cooperative constants. The cooperative constant for preexisting CCPs was fixed at m = 1, while the constant for newly forming CCPs (n) was varied from 1 to 6. Increasing n produced progressive enhancement of PIP2 recovery, with n = 6 at m = 1 yielding a recovery profile only ~ 60% compared with the condition where both n and m were set to 6 (red broken line). (B) PIP5K activity, represented by stim_5K_max, as a function of CCP contribution fixed at m = 1. The cooperativity of the kinase activity was not only from newly forming CCP but also from preexisting CCP. (C-D) The cooperativity of newly forming CCP can produce hump of PIP2 recovery by β-arrestin knockdown. The red broken lines in C & D indicate the level of PIP2 and PIP5K activity during β-arrestin knockdown when the n and m = 6 were set.
Figure 4.
Both preexisting and newly forming CCPs contribute to PIP2 recovery. (A) Simulated PIP2 hydrolysis and recovery after PAR2 activation (100 μM AP) under varying cooperative constants. The cooperative constant for preexisting CCPs was fixed at m = 1, while the constant for newly forming CCPs (n) was varied from 1 to 6. Increasing n produced progressive enhancement of PIP2 recovery, with n = 6 at m = 1 yielding a recovery profile only ~ 60% compared with the condition where both n and m were set to 6 (red broken line). (B) PIP5K activity, represented by stim_5K_max, as a function of CCP contribution fixed at m = 1. The cooperativity of the kinase activity was not only from newly forming CCP but also from preexisting CCP. (C-D) The cooperativity of newly forming CCP can produce hump of PIP2 recovery by β-arrestin knockdown. The red broken lines in C & D indicate the level of PIP2 and PIP5K activity during β-arrestin knockdown when the n and m = 6 were set.
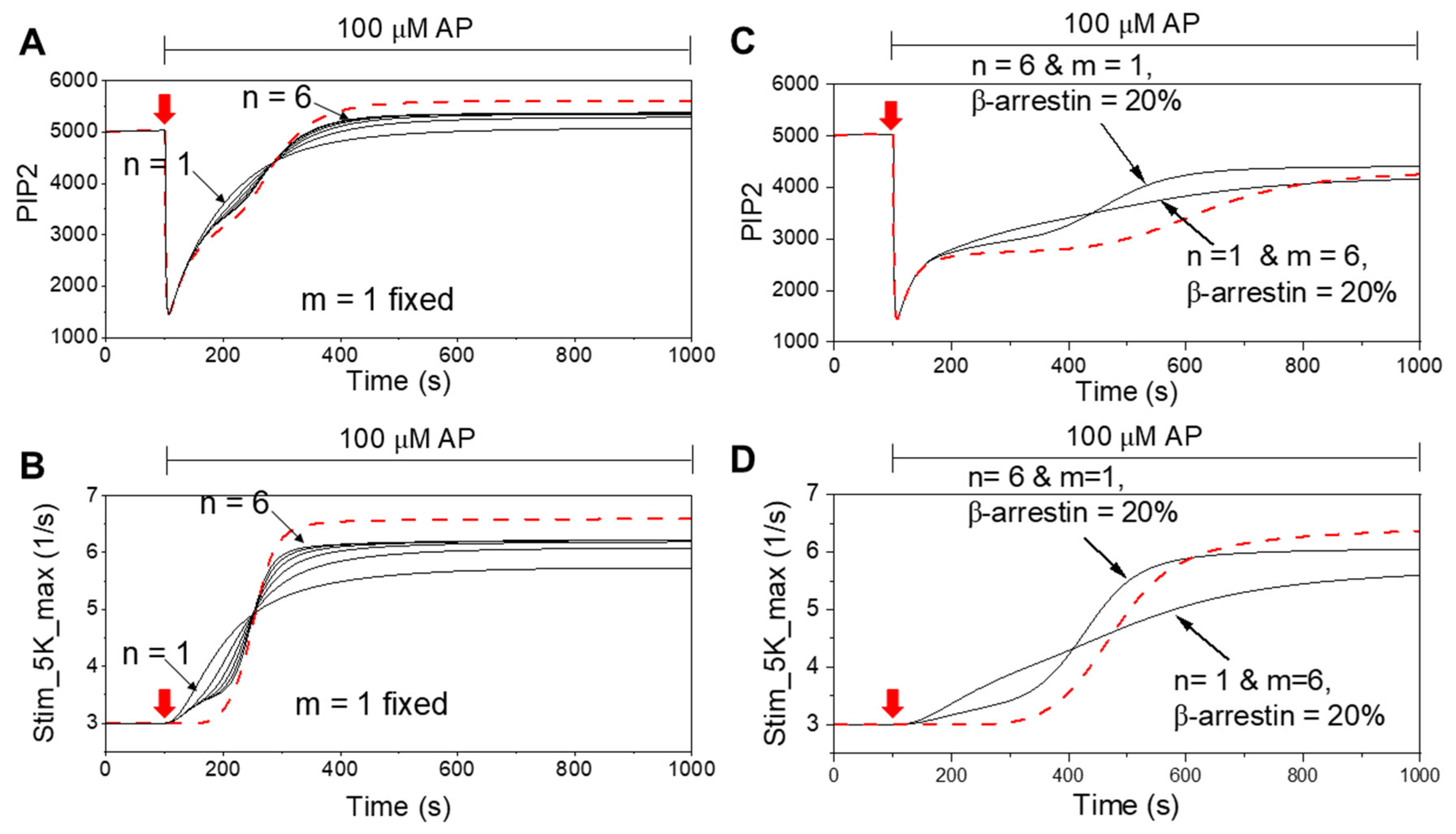

Table 1.
Model initial conditions and parameters.
| Species/constants | Value | Rationale |
| R for PAR2 | 5,000 µm-2 | Receptor density [20]. If the receptors were able to dissolve in cytosol, it would be 5 µM. |
| G (G-protein) | 40 µm-2 | Number of free G-protein at the plasma membrane |
| PLC | 10 µm-2 | Number of free PLC at the plasma membrane |
| PI | 140,000 µm-2 | Number of free PI at the plasma membrane |
| PIP | 3,600 µm-2 | Number free PIP at the plasma membrane |
| PIP2 | 5,000 µm-2 | Number of free PIP2 at the plasma membrane |
| IP3 | 0.015 µM | IP3 at cytosol before receptor activation: Steady state balance from PLC and IP3ase |
| DAG at PM | 23 µm-2 | Number of DAG at the plasma membrane before receptor activation |
| β-arrestin (cytosol) = β-arrestin1+β-arrestin2 |
15 µM | β-arrestin 1 (Arrestin 1) and β-arrestin 2 (Arrestin 2) have 7.5 µM, respectively, to leave proper amount β-arrestins in the cytosol after their binding to the PAR2 at the plasma membrane. |
| PKC_cyto (PKC Cytosol) | 1 µM | The concentration was chosen to make reasonable fitting of DAG bound PKC |
| GRK | 600 | Fixed value to similar to the peak value of PKC_DAG |
| Weighting_factor_PKC_DAG or Weighting_factor_GRK |
0.5 | Active PKC (PKC_DAG) gives the same contribution to phosphorylation of ligand bound receptor as GRK. |
| kf_PKC_DAG | 0.02 µM-1*s-1 | Forward rate constant for binding of PKC and DAG |
| Kr_PKC_DAG | 0.06 s-1 | Reverse rate constant for dissociation of DAG from PKC, giving Kd = 3 µM |
| Ca2+ (cytosol) | 0.13 µM | Typical intracellular Ca2+ level is 0.1-0.2 µM |
| foldPIP2 | 3 | Making the size of the total PIP2 (bound plus free) 3 times the free pool |
| k_4K (basal) | 0.00078 s-1 | FoldPIP2*0.00026 s-1 |
| k_5K (basal) | 0.06 s-1 | FoldPIP2*0.02 s-1 |
| k_4K (stimulated) | 6* k_4K (basal) | To fit the recovery kinetics of PIP2 probe |
| k_5K (stimulated) | 3* k_5K (basal) | To fit the recovery kinetics of PIP2 probe |
| k_4P (basal) | 0.03 s-1 | 0.03 s-1*k4r_basal (k4r_basal = 1) |
| k_5P (basal) | 0.014 s-1 | 0.014 s-1*k5r_basal (k5r_basal = 1) |
| k_4P (stimulated) | 0.19 s-1 | 0.03 s-1*k4r_stim (k4r_stim = 6.25) |
| k_5P (stimulated) | 0.042 s-1 | 0.014 s-1*k5r_stim (k5r_stim = 3) |
| kf_RL | 0.75 µm2*molecules-1*s-1 | Rate constant for phosphorylation of ligand-bound receptor |
| kr_RLP | 0.0125 s-1 | Rate constant for dephosphorylation of ligand-bound phosphorylated receptor |
| kf_R (basal) | 0.0001 s-1 | Basal phosphorylation rate constant of receptor |
| kr_RP (basal) | 2 s-1 | Basal dephosphorylation rate constant of phosphorylated receptor |
| kf_R (stimulated) | 10 s-1 | Stimulated phosphorylation rate constant of receptor |
| kr_RP (stimulated) | 4 s-1 | Stimulated dephosphorylation rate constant of receptor. We assumed that rate of dephosphorylation of RP doubles after ligand treatment |
| kf_L2 | 0.09333 µM-1*s-1 | Binding rate constant of ligand to RP, depending on the dissociation constant (K_L2) and dissociation rate constant (kr_L2) |
| kr_L2 | 5.6 s-1 | Dissociation rate constant of ligand from RP, assuming that it has same dissociation rate constant compared to the native receptor (R) |
| K_L2 | 60 µM | Dissociation constant of ligand bound to RP, which has slightly lower affinity compared to native receptor (R) based on the supplementary data |
| kf_RLP | 0.003 s-1 | Rate constant of β-arrestin 2 binding to phosphorylated ligand-bound receptor to make best fitting compared to Figure 4F and 8F. For β-arrestin 1, we used 0.006 s-1. |
| kr_RLPA1 or kr_RLPA2 | 10-6 s-1 | Dissociation rate constant of β-arrestin from phosphorylated ligand-bound receptor. Same for β-arrestin 1 and 2 |
| Kd_arrestin 1_RLP_PIP2 or Kd_arrestin 2_RLP_PIP2 | 5000 molecules*µm-2 | Dissociation constants of β-arrestin 1 or β-arrestin 2 from RLPA1 or RLPA1 complexes, respectively |
| Kd_PIP2 | 5000 molecules*µm-2 | Dissociation constant of PIP2 dependent clathrin/AP2 binding to the RLPA1 or RLPA2 complexes |
| kf_RLPA1 or kf_RLPA2 | 0.001 µm-2*s-1 | Binding rate constants of RLPA1 or RLPA2 to clathrin/AP2 bound RLPA complexes (RLPAAPC) |
| kr_RLPAAPC | 0.00001 s-1 | Rate constant of clathrin/AP2 dissociation from PLPAAPC complex |
| kf_CCP | 0.01 molecules*µm--2*s-1*µM-1 | Binding rate constant of RLPA1 or RLPA2 to preexisting CCP |
| kr_CCP | 0.00006 s-1 | Dissociation rate constant of RLPA1 or RLPA2 from preexisting CCP |
| kf_ClathrinAP2 | 0.01 molecules*µm--2*s-1*µM-1 | Binding rate constant of clathrin/AP2 to form preexisting CCP |
| kr_ClathrinAP2 | 0.00006 s-1 | Dissociation constant of clathrin/AP2 from preexisting CCP |
| k_endocytosis | 0.001 µM | Binding rate constant of dynamin for receptor endocytosis |
| k_fail | 0.01 molecules*µm-2*s-1 | Dissociation constant of dynamin for failed endocytosis |
| Kd_dynamin | Where, Kd_PIP2 = 2500 molecules/µm2. Dissociation constant of dynamin molecules from endocytic vesicles which have cooperative enhancing mechanisms by PIP2. | |
| The rate constant for the PI4K (k_4K during stimulation) = foldPIP2*(k_4K_rest + (stim_4K – k_4K_rest + k_4K_basal)*scale_4K*(1 – exp(-t/tau_on))). Where, tau_on = 1 s, foldPIP2 = 3, scale_4K = 0.75, k_4K_rest = 0.00026 s-1, k_4K_basal = 0.0002353 s-1, stim_4K = 0.00117 s-1). The rate constant for the PIP5K (k_5K during stimulation) = foldPIP2*(k_5K_rest + (stim_5K – k_5K_rest + k_5K_basal)*scale_5K*(1 – exp(-t/tau_on))), where tau_on = 1 s, foldPIP2 = 3, scale_5K = 1, k_5K_rest = 0.02 s-1, k_5K_basal = 0.0181 s-1, stim_5K = . Where, Stim_5K_max = stim_5K_max_basal+stim_5K_max_constant_a*beta_arrestin_contribution_a* + stim_5K_max_constant_b*beta_arrestin_contribution_b* Where, stim_5K_basal = 3, stim_5K_max_constant_a = 3, beta_arrestin_contribution_a = 0.8, Stim_5K_max_constant_b = 6, beta_arrestin_contribution_b = 0.2. | ||
Table 2.
Model differential equations.
| Reaction | Flux | |
| PI to PI(4)P | k_4K*[PI] – k_4P*[PIP] | |
| PI(4)P to PI(4,5)P2 | k_5K*[PIP] – k_5P*[PIP2] | |
| PLC (basal) | [PIP2]*(PLC_basal + k_ PLC*foldPIP2*Ga_GTP_PLC), where PLC_basal = 0.0001 s-1, k_PLC = 0.2 µm2molecule-1s-1, and foldPIP2 = 3 |
|
| PLC (stimulated) | [PIP2]*(PLC_basal + k_PLC*foldPIP2*Ga_GTP_PLC) + [PIP2]*PLC_stim*Ga_GTP_PLC*[Ca_C/(Ca_C+Kd_PLC_Ca)], where PLC_basal = 0.0001 s-1, k_PLC = 0.2 µm2molecule-1s-1, foldPIP2 = 3, where PLC_stim = 7 s-1, Kd_PLC_Ca = 1 µM | |
| RL to RLP | kf_RL*[RL]*(weighting_factor_PKC_DAG*[PKC_DAG] + weighting_factor_GRK*[GRK]) – kr_RLP*[RLP] | |
| PKC to active PKC | kf_PKC_DAG*[PKC]*[DAG] – kr_PKC_DAG*[PKC_DAG] | |
| R to RP | kf_R*[R]*[PKC_DAG] – kr_RP*[RP] | |
| RP to RLP | kf_L2*AP_Ex*[RP] – kr_L2*[RLP] | |
| RLP to RLPA1 | – kr_RLPA1*[RLPA1] | |
| RLP to RLPA2 | – kr_RLPA2*[RLPA2] | |
| RLPA1 to RLPAAPC | – kr_RLPAAPC*[RLPAAPC] | |
| RLPA2 to RLPAAPC | – kr_RLPAAPC*[RLPAAPC] | |
| RLPA1 & RLPA2 to RLPA_CCP | kf_CCP*[CCP]*[RLPA1]*[RLPA2] – kr_CCP*[RLPA_CCP] | |
| ClathrinAP2 to CCP | kf_ClathrinAP2*[ClathrinAP2] – kr_ClathrinAP2*[CCP] | |
| RLPAAPC to vesicle | – kr_fail*[vesicle] | |
| Many additional reactions which have been already described in our previous paper are not on this table. The parameters are in previous papers [20]. I added arrestin/clathrin/AP2/dynamin dependent endocytosis mechanism in the current model. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



