
Submitted:
28 October 2025
Posted:
29 October 2025
You are already at the latest version
Abstract
Post-translational modifications (PTMs) in proteins are essential for cell function. Due to the high cost and time demands of high-throughput sequencing, machine learning and deep learning methods are being rapidly developed for predicting PTM sites. This manuscript presents a comprehensive review of current research on the application of intelligent algorithms for predicting PTM sites. It outlines the key steps for identifying modified sites based on intelligent algorithms, including data preprocessing, feature extraction, dimension reduction and classifier development. The review also discusses potential future research directions in this field, providing valuable insights for advancing the state-of-the-art in PTM site prediction. Collectively, this review provides comprehensive knowledge on PTM identification and contributes to advanced predictors in the future.
Keywords:
post-translational modification
; feature engineering
; machine learning
; deep learning
1. Introduction
Protein synthesis follows the central dogma of genetics and involves three primary processes: replication, transcription, and translation. Protein post-translational modification (PTM) generally occurs during translation. After synthesis, proteins undergo various modifications like phosphorylation [1], acetylation [2], methylation [3], ubiquitination [4], and glycosylation [5]. These modifications expand the functional diversity of proteins and increase their complexity significantly. PTMs play crucial roless in cellular and organismal functions, impacting processes such as cell differentiation, apoptosis, protein degradation, protein-protein interactions, and gene expression and regulation. Furthermore, PTMs are closely linked to human diseases, with current targeted therapies involving regulatory enzymes associated with these modifications. Thus, studying protein PTMs is essential for advancing our understanding of biological processes.
Experimental methods are adept at accurately identifying protein modification sites, with mass spectrometry[6] being the predominant approach, complemented by liquid chromatography[7], and radiochemical methods[8]. However, as sequencing technologies continue to advance, an increasing number of protein sequences have been discovered, rendering traditional experimental methods insufficient for managing the vast scale of data. In this context, computational methods emerge as viable alternatives for analyzing protein sequences and identifying the corresponding modification sites. Machine-learning methods have been successfully used in the field of modification-site identification. However, current prediction methods still need to be improved, such as the simplicity of features and classification methods and the unreliable sequence in the training set[9]. Deep learning methods have been increasingly used in PTM site recognition research to address the limitations of machine learning methods, yielding promising results[10]. The information on modification sites provided by computational prediction is merely speculative, and their biological authenticity must be ultimately confirmed through experimental validation.
Machine learning-based computational identification methods typically consist of six key steps. First, data were obtained from established databases. Second, the acquired data were pre-processing. Third, sequence or structural features are all derived from protein sequences. These features include sequence position information, amino acid physicochemical properties, protein structure information, and so on. Fourth, redundant or insignificant features are eliminated using feature dimension reduction or selection methods. Fifth, a suitable model was chosen for training. Finally, the performance of the model is assessed using a test set. Most machine-learning-based computational identification methods adhere to this fundamental process, as illustrated in Figure 1. This manuscript summarizes the research according to the machine learning process to better understand its application in PTM identification and discusses potential future research directions to enhance the sufficiency of identifying modification sites.
2. Datasets and Data Pre-Processing
2.1. Dataset
With continuous advancements in sequencing technology and proteomics, researchers have developed numerous PTM databases that can be used by other researchers. The following section provides an overview of several popular databases, with additional information in Table 1.
2.1.1. Uniport
UniProt [11,12] is a comprehensive database that offers protein structure, sequence information, functional annotations, Gene Ontology (GO) annotations, subcellular location data, PTM information, similar proteins, and more. UniProt database is an authoritative repository of protein sequence and functional information, systematically integrating annotations of PTMs. These annotations, after manual curation, are stored in Swiss-Prot entries and are centrally displayed in the dedicated “PTM/Processing” module, covering various modification types such as phosphorylation and glycosylation, with specific amino acid residue sites of modification clearly annotated. The data sources include published experimental evidence and reliable computational predictions, often linked to relevant literature. Moreover, PTM information does not exist in isolation but is deeply interconnected with modules such as “Function”, “Disease and Variants”, and “Sequence”, collectively elucidating the biological significance of PTMs in regulating protein activity, localization, interactions, and stability. Simultaneously, UniProt provides cross-references to specialized PTM databases like PhosphoSitePlus and GlyGen, serving as a comprehensive PTM information hub that guides users in further exploration.
2.1.2. dbPTM
dbPTM [13,14] is a comprehensive resource for PTMs of proteins. The database contains 2,235,664 experimental PTM sites and over 70 PTM types integrated into more than 40 databases and includes 30 benchmark datasets. In addition, dbPTMs offer information on the association between modification sites and diseases, which can be valuable for disease research. Researchers can select specific modification sites for data download by clicking on the download bar. Instead of providing the entire protein sequence, the database offers protein fragments with details of the modification site, each fragment being 21 amino acids long.
2.1.3. CPLM 4.0
The Compendium of Protein Lysine Modifications 4.0 (CPLM 4.0) [15] is a comprehensive data resource that builds on previous versions of CPLA [16], CPLM [17] and PLMD [18]. CPLM 4.0 focuses on protein lysine modification. This database includes a significant number of modification events encompassing a wide range of unique sites on various proteins. In total, 105,673 proteins were included in CPLM 4.0, with data pertaining to up to 29 different types of protein lysine modifications across 219 different species [15].
2.2. Data Pre-Processing
Data pre-processing involves three primary steps. First, the protein sequence was segmented to generate fragments. The next step was to collect trustworthy negative data for building the dataset. Finally, the problem of imbalanced datasets was investigated to mitigate the potential adverse effects. The data pre-processing workflow is illustrated in Figure 2.
2.2.1. Sequence Slice
In current studies on PTM, most researchers have chosen the peptide representation method outlined in Equation (1).
signifies the central amino acid in focus for PTM site recognition studies, aiming to determine if these amino acids undergo modifications. For instance, in methylation recognition studies, represents either lysine or arginine. The represents the upstream -th amino acid from the central amino acid , whereas denotes the downstream -th amino acid from the central amino acid . Therefore, the length of the peptide is . It is customary to use ‘-’ or ‘X’ as placeholders when there are insufficient upstream or downstream amino acids.
The lengths of the peptides used in PTM site studies vary from one research project to another. Lai et al. [32] developed an auto-machine learning method to predict lysine lactylation sites in 51 amino acids. Wei et al. [33] used 11 residues to predict the methylation sites. Li et al. [34] used peptides with 31 amino acids. Nie et al. [35] trained on 27-peptide sequence segments and tested 20 sequences. Lyu et al. [36] segmented proteins into 35-residue segments with cysteine at the center. Auliah et al. [37] used a local sliding window of 57 residues to predict pupylation sites. Bao et al. [38] created 27-tuple peptides for K-PTMs. The peptide lengths available in dbPTM and PhosphoSitePlus were 21 and 15 bp, respectively. The choice of peptide length may affect the prediction results, leading researchers to explore various lengths. Khalili et al. [39] investigated window sizes ranging 7–35 and found that a window size of 13 yielded the best performance in their models.
2.2.2. Sequence Redundancy
CD-HIT, a method proposed by Li et al. [40], has been widely used to eliminate homologous protein data. CD-HIT uses a clustering approach to identify and remove similar protein sequences.
The algorithm is as follows [40,41]: The algorithm sorts sequences according to their length. The longest sequence is designated as the representative of the first cluster. Next, the remaining sequences are compared with representatives in the existing class [42]. If the similarity exceeds a predefined threshold, a new sequence is added to the existing class [43]. Otherwise, a new class was created. CD-HIT is widely used in PTM site studies to eliminate the homologous protein sequences and residual fragments. Some studies aimed to remove redundant protein sequences [34,44], whereas others focused on eliminating fragmented residue sequences [45,46].
2.2.3. Selected Reliable Negative Sequences
Although databases commonly provide information on modification sites, they do not provide non-modification site information. Peptides without annotation information (known as unlabeled data) can be categorized into two groups. One is that the site has not been identified as a modification site and the other is that additional research is required to verify whether the site has indeed been modified. Therefore, we obtained reliable positive sequences; however, there may be potential issues with the negative data. The modification site prediction problem can be described as a positive-unlabeled (PU) problem [9,47,48]. Two methods can be used to address the PU problem in modification site prediction.
Segments without modification information are considered negative samples by default [32]. The proposed method is straightforward and manageable. Several studies have used this method to build models; however, it ignores the potential for modifications. Gao et al. [49] established three criteria for identifying non-phosphorylated sites: (1) the fragment must not be labeled as a positive site, (2) the fragment should be within the sequence containing a positive site, and (3) the negative site must be solvent-inaccessible. In [39,50], all residues from a protein with a minimum of three confirmed positive sites were considered negative sites.
Another method aims to build models using limited positive data and large amounts of unlabeled data. Ning et al. [51] used semi-supervised learning and a support vector machine (SVM) to select reliable negative data. Jiang et al. [48] introduced the PUL-PUP algorithm for acquiring negative data. PUL-PUP initially used similarity to identify negative data distant from positive data. Subsequently, PUL-PUP iteratively trains the SVM to expand the reliable negative data.
2.2.4. Balanced Dataset
In PTM studies, the amount of positive data is less than that of negative data, leading to a data imbalance. Imbalanced datasets may have detrimental effects on model training. Various methods have been proposed, such as data-based, algorithm-based, and hybrid-based methods.
(1) Data based methods
Data-based methods can be divided into over-sampling [52], under-sampling [53], and hybrid sampling [54,55,56]. Random over-sampling (ROS) randomly duplicates minority samples to achieve a balanced dataset. The synthetic minority over-sampling technique (SMOTE) [52] generates new samples by considering the k-nearest neighbors of each minority class sample, and is widely used to address imbalanced datasets such as SulSite-GTB [57]. Adaptive synthetic sampling (ADASYN) [53] is an advanced method that can generate samples based on the learning difficulties of individual minority samples. Balanced datasets can also be achieved through undersampling, which involves reducing the number of majority samples. Random under-sampling (RUS) selects sequences from the majority of subsets and is widely used in PTM prediction. NearMiss [58] selects a subset of the majority class samples closest to the minority class samples as representatives. ENN [59] is a KNN-based method that avoids interference samples. Hybrid sampling methods such as SMOTETomek [60,61] and SMOTEENN combine oversampling with under-sampling.
(2) Algorithm-based method
Algorithm-based methods solve imbalanced datasets by shifting the focus toward minority class samples, including cost-sensitive learning [62], ensemble learning [63,64], and one-class classification [64,65]. Cost-sensitive learning uses cost functions to build classifiers by minimizing the misclassification cost, and adjusts the cost of misclassified samples based on a cost matrix [66,67,68]. Commonly used methods include weighted SVM [69], fuzzy SVM [70,71], and cost-sensitive neural networks [72]. Ensemble learning can reduce the bias caused by a single learner and enhance the model efficiency. Bagging, boosting, stacking, and hybrid models are the most representative ensemble-learning models. RUSBOOST [73] trains weak classifiers by constructing multiple balanced datasets by using a combination of RUS and AdaBoost. Jia et al. [74] used an ensemble method to predict the O-GlcNAcylation sites. One-class learning, or novelty detection, is a useful approach for handling significant imbalances between positive and negative samples. This technique builds models for the minority class, for example, a One-class SVM [75].
(3) Hybrid-based methods
Based on the aforementioned methods for handling imbalanced datasets, researchers have proposed hybrid methods that combine different balancing strategies to further enhance model performance. For instance, Islam et al. [76] used undersampling and K-nearest neighbors (KNN) to balance data. In [73] a combination of RUS and AdaBoost was used to balance a dataset. Reference [54] implemented hybrid sampling and a bagging classifier to address imbalanced learning.
2.2.5. Data Splitting
To train a model and objectively assess its generalization capability, the dataset must be divided into training and test sets following specific guidelines. The hold-out method, a widely used and straightforward approach, involves splitting data according to a predefined ratio, such as 70% for training and 30% for testing. Alternatively, a temporal partitioning strategy may be employed, using data collected before 2022 for training and data from 2022 to 2025 for testing. Regardless of the chosen method, it is crucial to ensure that the training and test sets have similar data distributions and that no test data is included in the training set.
3. Feature Engineering
3.1. Feature Extraction
Machine learning cannot recognize sequence data directly. Therefore, researchers must design algorithms to complete feature extraction. Feature extraction methods are categorized into three types: (1) sequence-based methods, (2) physicochemical-based methods, and (3) annotation-based methods. With the development of deep learning, language models have been used to predict PTM sites.
Sequence-based Feature
Sequence-based methods commonly use the composition, position, and other relevant information on amino acids to achieve a numerical representation of protein sequences. In sequence-based method, placeholder (‘-’ or ‘X’) is considered a particular type of amino acid. Therefore, the total number of amino acid residues was 21.
Amino acid composition (AAC) [77,78,79,80] is a common feature extraction method based on the frequency of amino acids in sequence segments. AAC can yield 21 features, including 20 amino acids and one placeholder. The composition of k-spaced amino acid pairs (CKSAAP) uses the frequency of amino acid pairs with the separation of k spaces to represent the protein sequence, where the value of k is available. In particular, CKSAAP has 441 features (from AA, AC, to XX) when k is 0. One-hot encoding [79] commonly used in deep learning, in which each amino acid is converted into a vector of length 21. Finally, a protein fragment of length L was depicted as a two-dimensional (2D) matrix of size L×21. The use of machine-learning methods to extract features is an innovative approach. The K nearest neighbor (KNN) Score [81,82,83] was used to characterize the fragments.
In addition to the methods discussed above, there are several other sequence-based feature-extraction methods. For instance, conjoint triad descriptor (CTriad) [80,84], dipeptide composition (DPC) [80,85], amino acid pair composition (AAPC) [77,79,86], pair potential [49,87], four-body statistical pseudo-potential [49,88], local structural entropy [49,89], information of proximal PTMs [51], position-special amino acid propensity (PSAAP) [51,90,91], enhanced amino acid pair (EAAC), enhanced group amino acid pair (EGAAC) [79,92], and position weight amino acid composition (PWAAC) [57].
3.1.1. Physicochemical Properties
Numerous studies have demonstrated variations in physicochemical properties between sites that undergo PTMs and those that do not. The physicochemical properties of amino acids not only reveal the biological characteristics of PTM sites, but can also aid in predicting the development of identified models. In physicochemical properties, placeholders (‘-’ or ‘X’) are either disregarded or assigned a default value such as 0.5.
The AAindex database, comprising 566 amino acid properties [24,80,93,94,95], can be integrated with computational techniques such as grey models, principal component analysis, and clustering to enhance feature extraction efficiency. Secondary structure (SS) [80,96] is determined using SPIDER2, converting fragments into a 63-dimensional vector that includes probability scores for α-helix, β-helix, and coil for each amino acid (with placeholders). The composition, transition, and distribution (CTD) method [80,97,98] introduced by Dubchak et al. categorized 20 amino acids into three groups based on eight properties, ultimately transforming the fragments into a 188-dimensional vector.
There are some commonly used feature extraction methods, such as backbone torsion angles (BTA) [80,94,99], accessible surface area (ASA) [49,80,100,101,102], physio-chemical properties (PCPs), other binding sites for any chemical groups [94], positively charged amino acid composition (PCAAC), discorded regions by DISOPRED2 [94,103], BioJava [94,104], disorder [49,105], grey pseudo amino acid composition[51], and encoding based on grouped weight (EBGW) [57].
3.1.2. Annotation Information
Protein annotation information typically encompasses basic, structural, and functional details as well as other pertinent information. These data aid in comprehending the structure, function, and significance of proteins within organisms and are frequently used to describe protein fragments.
Numerous annotation-based methods exist, for example, position-specific scoring matrix (PSSM)[76,77,79], evolutionary-based profile bigrams [76,106,107,108], gene ontology (GO) [94,109], InterPro [94,110], KEGG [94,111], Pfam [94,112], STRING [94,113], functional domain [94], active site [94], natural variants [94], BLOSUM62 scoring matrix (B62) [77,114], evolutionary conservation score [49,115,116], and pseudo-position specific scoring matrix (PsePSSM)[57,117].
The PSSM is the most popular method. The PSSM is a matrix, where represents the length of the fragment. Each column corresponds to a residue position in the protein sequence and each row represents one of the 20 possible amino acids. Each element (i, j) in the matrix represents the probability or score of the j-th position in the protein sequence being mutated into the i-th amino acid during evolution. This score typically reflects the degree of conservation and the preference for a specific amino acid at that position.
3.1.3. Network-Based Feature
Deep learning is extensively employed in PTM site recognition research, serving dual purposes: as a classifier for prediction, and as a tool for extracting features from network structures. Convolutional neural networks (CNNs) extract features and reduce dimensionality via convolutional and pooling layers, while recurrent neural networks (RNNs) capture sequence context. Some studies integrate these approaches for hybrid feature extraction.
Natural language processing (NLP) has developed rapidly in recent years. The protein sequences are similar to those of natural languages in several respects. First, both datasets were sequential. Second, both contained contextual information. Several studies have used language models to extract the features of protein fragments. Bidirectional encoder representations from transformers (BERT) are commonly used to predict PTM sites. Alkuhlani et al. [118] used six protein language models, ProtBERT-BFD [119], ProtBERT [119], ProtALBERT [119], ProtXLNet [119], ESM-1b [120], and TAPE [121] to identify PTM sites based on BERT [122], Albert [123], and XLNet [124]. Qiao et al. used BERT to build a novel predictor, BERT-Kcr, for protein Kcr sites prediction [10]. Lyu et al. [36] used word embedding to encode protein fragments, whereas Wang et al. [125] predicted plant ubiquitination using the word2vec feature extraction method.
Post-translational modification is intrinsically linked to the enzyme, with enzyme-substrate relationships deducible from the physical and chemical properties of modification sites through feature engineering. Deep-PLA [126] demonstrates the effective integration of enzyme-specific constraints into deep neural network architectures, offering a pertinent case study for this topic.
3.2. Feature Reduction
We introduce the four types of feature-extraction methods in detail. Several studies have used multiple feature extraction methods to obtain comprehensive feature sets. However, ensemble method often leads to the challenge of an excessive number of feature dimensions. Redundant and nonessential features may reduce the efficiency of the predictor. Important features are retained through feature reduction, whereas those with lower importance are eliminated. Consequently, the final feature vector exhibits a lower dimensionality yet higher relevance. Feature-reduction approaches can be divided into two types: feature selection, which only reduces the number of features, and feature transformation or dimensionality reduction, which focuses on decreasing complexity by transforming existing features. In modification site prediction studies, these two methods are commonly used to optimize feature sets.
Auliah et al. [37] used the chi-squared test to perform feature selection. The chi-square test is a widely used hypothesis testing method that is important in statistics. The chi-square test was used to examine whether the two variables were independent. Maximal-relevance-maximal-distance (MRMD) was used to rank the importance of features in [33]. Li et al. [127] combined analysis of variance (ANOVA) with incremental feature selection (IFS) to find the most vital feature subset. Minimum redundancy maximum relevance (mRMR) [128,129,130,131] was often used to select optimal features from the entire feature set. He et al. [132] proposed a feature selection method called MRMD3.0, which consisted of two steps. The first step contained nine feature rank methods (tree importance, ANOVA, variance threshold, chi-squared, linear model method, mutual information, minimum-redundancy-maximum-relevance, max-relevance-max-distance, and recursive feature elimination) and four-link analysis strategies (PageRank, Trust Rank, Leader Rank, and HITS). The second step uses IFS to select the best feature subset. Ensemble methods are commonly used for feature selection. Yu et al. [133] selected the features via XGboost [134]. Principal component analysis (PCA) is widely used to reduce dimensionality. This method can describe existing high-dimensional feature sets using fewer comprehensive features. Another standard method for feature dimensionality reduction is singular value decomposition (SVD).
In contrast to machine learning, deep learning methods can automatically learn feature information from input data without manual feature extraction. In deep learning, feature-dimensionality reduction is typically not treated as a distinct step. However, in the model structure, some processes are similar to feature selection, such as the pooling layers in convolutional neural networks.
4. Classifiers
In this step, machine-learning methods are trained using the feature set obtained from the previous stage. Currently, popular machine-learning methods in the field of PTM site prediction include support vector machines, naïve Bayes, and decision trees. As research continues to advance, ensemble and deep learning are increasingly being applied in the study of modification site recognition.
4.1. Machine Learning Classifier
Support vector machines (SVM) separate protein fragments by creating an optimal hyperplane for classification, which is particularly effective with small sample data. Xu et al. [135] used SVM to identify protein lysine glycation using sequences. Bao et al. [38] successfully used SVM and multilayer neural networks to predict various PTM sites. Auliah et al. [37] used multiple classifiers to assess the recognition efficiency of PUP-Fuse. Decision Trees use if, then judgment rules for protein fragment classification. K-Nearest Neighbors (KNN) predicts unknown protein fragments based on the nearest samples. Ning et al. [136] used KNN as a classifier to identify formylation sites and explored the impact of different values of K on the experimental results. Artificial Neural Network (ANN) is popular classifiers in bioinformatics that mimic the structures and functions of biological neural networks [137]. Several studies used ANN to predict PTM sites [138,139].
Ensemble methods may enhance the prediction accuracy. Ensemble methods can be categorized into three types: Bagging, Boosting, and Stacking. Bagging resembles voting. Basic classifiers have been used to predict protein fragments, resulting in various outcomes. The category of unknown protein fragments was determined based on the most frequent category. Random Forest (RF) [140,141] is a representative bagging algorithm commonly used in PTM site prediction. Hasan et al. [142] used RF to predict S sulfenylation sites. Cascade Forest [143,144] uses a layered approach comprising multiple forest structures, where the input for each layer is derived from the output feature information of the preceding layer. This methodology facilitated incremental feature extraction and allowed adaptive adjustments to the complexity of the model. Qian et al. [145] proposed a novel predictor, SUMO-forest, based on cascade forests. Boosting refines the base learner by adjusting the data sample weights, and ultimately determines the segment classes through weighted voting. Gradient tree boosting (GTB) [146,147] is a popular boosting method using multiple decision tree (DT) with excellent performance, which has been used in multiple fields. Wang et al. [57] proposed SulSite-GTB to predict the S-sulfenylation sites based on GTB. The stacking method uses multiple-base learners to identify protein fragments and generate classification results. These classification results were then used as features for another learner to learn and produce the final classification results. He et al. [148] used stacking ensemble layers to build a predictor in which the base learners were convolutional neural with different specifications. In addition to the aforementioned ensemble methods, other hybrid methods also exist. Zhang et al. [149] integrated five classifiers–RF, SVM, GBDT, KNN and Logistic Regression–to predict lysine malonylation sites.
4.2. Based on Deep Learning
Deep learning has been extensively used to predict PTM sites. Unlike traditional machine learning techniques that require manual feature design and selection, deep learning models can autonomously learn data feature representations without human intervention.
CNN consists of convolution, pooling, and fully connected layers. The convolution layer extracts features from the input data, and the pooling layer selects these features. The fully connected layer classifies unknown protein fragments. Wang et al. [150] used a CNN to predict multiple PTMs. Zhao et al. [151] used a CNN to predict Kcr. CNN-SuccSite [79] was developed as a CNN model for predicting lysine succinylation sites, comprising an input layer, two convolution layers, two max-pooling layers, two fully connected layers, and an output layer. Wei et al. [152] created a one-dimensional (1D) CNN to predict Kcr sites. However, RNN is advantageous for sequential data due to its ability to handle sequences of varying lengths and capture temporal dependencies. RNN provide rich contextual information and include two common variants: long short-term memory (LSTM) [153] and gated recurrent unit (GRU). Lyu et al. [36] constructed a five-layer LSTM model featuring an input layer, word embedding layer, LSTM layer, dense layer, and output layer to predict cysteine sulfophenylation sites. Li et al. [154] proposed a transfer learning model based on LSTM to predict lysine propionylation, whereas Mul-SNO [155] combined bidirectional long short-term memory (BiLSTM) and bidirectional encoder representations from transformers (BERT) to predict S-nitrosylation sites. Yu et al. [156] used a CNN-LSTM hybrid network for feature extraction and prediction sites. Currently, some studies have integrated deep learning with machine learning to enhance prediction efficiency. Ning et al. [157] combined 4-layer DNN and penalized logistic regression for succinylation site prediction. PROSPECT [158], proposed by Chen et al., integrates two CNNs and an RF to predict phosphorylation sites.
Transformer[159] is a deep learning model that utilizes the self-attention mechanism. Its primary innovation is the ability to capture global dependencies among all elements in a sequence through parallel computation, which enhances the model’s capability to contextualize information within that sequence. The Transformer architecture comprises an encoder and a decoder, with each layer featuring multi-head self-attention mechanisms and feed-forward neural networks. This design enables the model to dynamically assess the significance of each amino acid in the input sequence. Meng et al. [160] proposed TransPTM, a transformer-based neural network model for non-histone acetylation site predication. Liang et al. [161] proposed an effective model named DeepMM-Kcr, which is based on multiple features and multi-head self-attention mechanism.
The core idea of transfer learning is to apply the knowledge, including model parameters and feature representations, acquired from solving one task (the source task) to another related but distinct new task (the target task). This application enhances the learning efficiency and performance of the new task. In some instances, data for certain modification sites may be limited, potentially resulting in insufficient annotated data to train a high-performance model. Utilizing transfer learning methods can effectively address this challenge. Xu et al. [162] developed DTL-NeddSite, a convolutional neural network-based predictor that leverages deep transfer learning and one-hot encoding. The model was first trained on a large dataset of lysine post-translational modification sites, and then fine-tuned using neddylation site data to construct the target model. Soylu et al. [163] developed the DEEPPTM model, integrating a protein embedding approach using ProtBERT with an attention-based Vision Transformer (ViT) to enhance modification prediction accuracy and elucidate the relationships between modification types and protein sequences.
5. Measurement
The prediction of PTM sites is a binary classification problem. The modified sites were divided into positive and unlabeled data. Typically, unlabeled data are considered negative samples. Researchers typically use accuracy (ACC), Matthews correlation coefficient (MCC), F-measure, and area under the receiver operating characteristic curve (AUC) to assess classifier performance. The formulas for these metrics are as follows:
where TP denotes the number of correct classifications in the positive dataset. The TN represents the number of correct classifications in a negative dataset. FN is the number of false-negative results. where FP is the number of false positives.
The receiver operating characteristic (ROC) curve was originally used for radar-signal detection to differentiate between signals and noise. Subsequently, the researchers adopted it for the model evaluation. The horizontal axis of the ROC curve represents the false positive rate (FPR), and the vertical axis represents the true positive rate (TPR). Owing to the curved nature of the ROC, assessing the quality of the model can be challenging. Therefore, in practical applications, the area under the curve (AUC) serves as a measure of the model performance and is particularly beneficial for handling unbalanced data.
6. Summary of Predictors
With advancements in machine and deep learning, there has been a surge in research focusing on predicting PTM sites. Table 2 summarizes various studies, including PTM types, datasets, window sizes, feature extraction methods, prediction models, and web servers.
7. Challenges and Future Directions
Currently, significant progress has been made in the identification of PTM sites using machine learning and deep learning techniques. However, there are still several noteworthy aspects that warrant further attention.
7.1. Data Limitations
The study of PTM sites based on machine learning and deep learning requires a large and accurate dataset for model training. As previously mentioned, there are three main issues concerning the data on modification sites: (1) the absence of completely reliable negative examples; (2) the significant imbalance present in the datasets; and (3) current modification site databases reveal an underrepresentation of certain modification types. For example, the dbPTM database lists only 194 O-palmitoleoylation sites, encompassing both experimentally validated and predicted instances. It is insufficient to support the training requirements of deep learning or machine learning.
Several studies have proposed effective solutions to these issues. However, most of these solutions rely on sampling-based methods for dataset acquisition, often addressing only a single aspect of the problem without thoroughly examining the construction of the dataset. The construction of comprehensive and representative datasets remains a critical challenge in the field of PTM recognition. Negative instance data, which is typically more abundant than positive instance data, is often unreliable and contributes to an imbalanced dataset. To address this issue, further investigation into the application of semi-supervised learning and one-class learning approaches in dataset construction and model building processes is warranted. Transfer learning is a machine learning technique that leverages knowledge gained from one task to improve performance on a related but distinct task. By pre-training a model on a large dataset and then fine-tuning it on a smaller, task-specific dataset, transfer learning can mitigate the challenges posed by limited training data, thereby addressing the issue of underfitting that may arise when working with insufficient data for certain modified sites.
7.2. Interpretability
In recent years, deep learning techniques have shown considerable application value in bioinformatics, particularly in the prediction of protein modification sites. However, these models typically utilize complex nonlinear network architectures, which leads to a lack of interpretability regarding their internal mechanisms. This ‘black box’ nature not only undermines researchers’ trust in the predictive outcomes of these models but also ignites discussions concerning the reliability of algorithmic decisions in biomedical applications.
Establishing interpretable models is crucial in the study of modification sites. Interpretability methods aim to transform the opaque prediction processes of black-box models into specific biological explanations, elucidating the reasons behind the emergence of modified sites. These approaches seek to identify key sequence features that determine modification sites, indicating not only where modifications occur but also explaining why they occur at specific locations. Furthermore, developing interpretable methods that correlate sequence-level importance with three-dimensional protein structures can elucidate the spatial and physicochemical constraints on modifications. This includes explaining why certain sites are more readily recognized and bound by modifying enzymes, often due to their exposure on protein surfaces or within specific domains. Ultimately, the core value of interpretability analysis lies in converting model predictions into experimentally verifiable scientific hypotheses, thereby exploring the relationship between modification sites and biological activity. These models are categorized into post-hoc and ante-hoc interpretability. Post-hoc interpretability encompasses methods such as example-based, attribution-based, latent semantics-based, and rule-based approaches. In contrast, ante-hoc interpretable learning prioritizes interpretability as a fundamental objective during model design and training. This involves adopting a transparent model architecture or imposing interpretability constraints during training. When developing interpretable models, it is essential to design self-transparent neural networks that maintain model performance and efficiency while selecting appropriate interpretability evaluation metrics.
7.3. Interplay Between Modification Sites and Processes
Protein sequences may contain multiple modification sites, which play a crucial role in the functionality and stability of proteins. PTM crosstalk plays a crucial role in organisms and can be categorized into crosstalk within the same protein (intra- protein) and crosstalk between different proteins (inter-protein). Currently, some studies are committed to finding PTM crosstalk pairs[178,179,180]. However, there is limited research on PTM crosstalk mechanisms and crosstalk interaction networks.
Research has shown that changes in certain modification sites may lead to diseases [181]. Therefore, studying the relationship between modification sites and diseases is crucial for a deeper understanding of disease progression, intervention mechanisms, and the development of precision therapeutic drugs. However, there is currently limited research utilizing machine learning or deep learning methods to explore the relationship between modification sites and diseases, making this direction worthy of further exploration.
8. Conclusion
The accurate identification of PTM is crucial for various applications, including drug development, disease diagnosis, and the understanding of molecular processes. Although traditional biological experimental methods are accurate, they are resource intensive. Machine learning can efficiently process large datasets. However, its prediction accuracy may be influenced by various factors. Machine learning methods include traditional methods that require feature extraction and deep learning. This manuscript reviews current PTM site predictors based on machine learning, including datasets, feature extraction, classifiers, and evaluation metrics. This manuscript summarizes the existing methods and explores future research directions. Data play an important role in machine learning. Thus, future research should explore ways to solve imbalanced data and PU problems. The use of few-shot and transfer learning addresses the data scarcity and model complexity. Multilabel learning is conducive to further exploring protein modifications. The association between modification sites and diseases is worth exploring. We hope that our review and analysis will assist research related to PTM.
Author Contributions
Methodology, KY.Q.; writing—original draft preparation, SL.G.; writing—review and editing, KY.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China under Grant, grant number: 62502242; the Henan Provincial Science and Technology Research Project, grant number: 252102220039, the Key Research Project Plan for Higher Education Institutions of Henan Province grant number: 24A520027, and Science and Technology Research Project of Nangyang grant number: 24KJGG059.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PTM | Post-translational modifications |
| DL | Deep Learning |
| ML | Machine learning |
References
- Shrestha, P, Kandel, J, Tayara, H et al. DL-SPhos: Prediction of serine phosphorylation sites using transformer language model. Computers in Biology and Medicine 2024, 169:107925.
- Liang, J-Z, Li, D-H, Xiao, Y-C et al. LAFEM: A Scoring Model to Evaluate Functional Landscape of Lysine Acetylome. Molecular & cellular proteomics : MCP 2024, 23(1):100700.
- Chang, KW, Gao, D, Yan, JD et al. Critical Roles of Protein Arginine Methylation in the Central Nervous System. Molecular Neurobiology 2023, 60(10):6060-6091.
- Dai, XF, Zhang, TX, Hua, D. Ubiquitination and SUMOylation: protein homeostasis control over cancer. Epigenomics 2022, 14(1):43-58.
- Masbuchin, AN, Rohman, MS, Liu, PY. Role of Glycosylation in Vascular Calcification. International Journal of Molecular Sciences 2021, 22(18):9829.
- Wohlschlager, T, Scheffler, K, Forstenlehner, IC et al. Native mass spectrometry combined with enzymatic dissection unravels glycoform heterogeneity of biopharmaceuticals. Nature Communications 2018, 9:1713.
- Park, H, Song, WY, Cha, H et al. Development of an optimized sample preparation method for quantification of free fatty acids in food using liquid chromatography-mass spectrometry. Scientific Reports 2021, 11(1):5947.
- Slade, DJ, Subramanian, V, Fuhrmann, J et al. Chemical and Biological Methods to Detect Post-Translational Modifications of Arginine. Biopolymers 2014, 101(2):133-143.
- Li, FY, Dong, SY, Leier, A et al. Positive-unlabeled learning in bioinformatics and computational biology: a brief review. Briefings in Bioinformatics 2022, 23(1):bbab461.
- Qiao, YH, Zhu, XL, Gong, HP. BERT-Kcr: prediction of lysine crotonylation sites by a transfer learning method with pre-trained BERT models. Bioinformatics 2022, 38(3):648-654.
- Lussi, YC, Magrane, M, Martin, MJ et al. Searching and Navigating UniProt Databases. Current protocols 2023, 3(3):e700.
- Bairoch, A, Bougueleret, L, Altairac, S et al. The Universal Protein Resource (UniProt). Nucleic Acids Research 2008, 36:D190-D195.
- Li, ZY, Li, SF, Luo, MQ et al. dbPTM in 2022: an updated database for exploring regulatory networks and functional associations of protein post-translational modifications. Nucleic Acids Research 2022, 50(D1):D471-D479.
- Lee, TY, Huang, HD, Hung, JH et al. dbPTM: an information repository of protein post-translational modification. Nucleic Acids Research 2006, 34:D622-D627.
- Zhang, WZ, Tan, XD, Lin, SF et al. CPLM 4.0: an updated database with rich annotations for protein lysine modifications. Nucleic Acids Research 2022, 50(D1):D451-D459.
- Liu, ZX, Cao, J, Gao, XJ et al. CPLA 1.0: an integrated database of protein lysine acetylation. Nucleic Acids Research 2011, 39:D1029-D1034.
- Liu, ZX, Wang, YB, Gao, TS et al. CPLM: a database of protein lysine modifications. Nucleic Acids Research 2014, 42(D1):D531-D536.
- Xu, HD, Zhou, JQ, Lin, SF et al. PLMD: An updated data resource of protein lysine modifications. Journal of Genetics and Genomics 2017, 44(5):243-250.
- Hornbeck, PV, Kornhauser, JM, Tkachev, S et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Research 2012, 40(D1):D261-D270.
- Yu, K, Wang, Y, Zheng, YQ et al. qPTM: an updated database for PTM dynamics in human, mouse, rat and yeast. Nucleic Acids Research 2023, 51(D1):D479-D487.
- Tung, CW. PupDB: a database of pupylated proteins. Bmc Bioinformatics 2012, 13:40.
- Duan, GY, Li, X, Köhn, M. The human DEPhOsphorylation database DEPOD: a 2015 update. Nucleic Acids Research 2015, 43(D1):D531-D535.
- Ma, JF, Li, YX, Hou, CY et al. O-GlcNAcAtlas: A database of experimentally identified O-GlcNAc sites and proteins. Glycobiology 2021, 31(7):719-723.
- Dinkel, H, Chica, C, Via, A et al. Phospho.ELM: a database of phosphorylation sites-update 2011. Nucleic Acids Research 2011, 39:D261-D267.
- Rao, RSP, Zhang, N, Xu, D et al. CarbonylDB: a curated data-resource of protein carbonylation sites. Bioinformatics 2018, 34(14):2518-2520.
- Ramasamy, P, Turan, D, Tichshenko, N et al. Scop3P: A Comprehensive Resource of Human Phosphosites within Their Full Context. Journal of Proteome Research 2020, 19(8):3478-3486.
- Hansen, JE, Lund, O, Rapacki, K et al. O-GLYCBASE version 2.0: A revised database of O-glycosylated proteins. Nucleic Acids Research 1997, 25(1):278-282.
- Lee, TY, Chen, YJ, Lu, CT et al. dbSNO: a database of cysteine S-nitrosylation. Bioinformatics 2012, 28(17):2293-2295.
- Li, ZY, Chen, SY, Jhong, JH et al. UbiNet 2.0: a verified, classified, annotated and updated database of E3 ubiquitin ligase-substrate interactions. Database-the Journal of Biological Databases and Curation 2021:baab010.
- Wang, X, Li, Y, He, MQ et al. UbiBrowser 2.0: a comprehensive resource for proteome-wide known and predicted ubiquitin ligase/deubiquitinase-substrate interactions in eukaryotic species. Nucleic Acids Research 2022, 50(D1):D719-D728.
- Durek, P, Schmidt, R, Heazlewood, JL et al. PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Research 2010, 38:D828-D834.
- Lai, FL, Gao, F. Auto-Kla: a novel web server to discriminate lysine lactylation sites using automated machine learning. Briefings in Bioinformatics 2023, 24(2):bbad070.
- Wei, LY, Xing, PW, Shi, GT et al. Fast Prediction of Protein Methylation Sites Using a Sequence-Based Feature Selection Technique. Ieee-Acm Transactions on Computational Biology and Bioinformatics 2019, 16(4):1264-1273.
- Li, ZT, Fang, JY, Wang, SN et al. Adapt-Kcr: a novel deep learning framework for accurate prediction of lysine crotonylation sites based on learning embedding features and attention architecture. Briefings in Bioinformatics 2022, 23(2):bbac037.
- Sua, JN, Lim, SY, Yulius, MH et al. Incorporating convolutional neural networks and sequence graph transform for identifying multilabel protein Lysine PTM sites. Chemometrics and Intelligent Laboratory Systems 2020, 206:104171.
- Lyu, XR, Li, SH, Jiang, CY et al. DeepCSO: A Deep-Learning Network Approach to Predicting Cysteine S-Sulphenylation Sites. Frontiers in Cell and Developmental Biology 2020, 8:594587.
- Auliah, FN, Nilamyani, AN, Shoombuatong, W et al. PUP-Fuse: Prediction of Protein Pupylation Sites by Integrating Multiple Sequence Representations. International Journal of Molecular Sciences 2021, 22(4):2120.
- Bao, WZ, Yuan, CA, Zhang, YH et al. Mutli-Features Prediction of Protein Translational Modification Sites. Ieee-Acm Transactions on Computational Biology and Bioinformatics 2018, 15(5):1453-1460.
- Khalili, E, Ramazi, S, Ghanati, F et al. Predicting protein phosphorylation sites in soybean using interpretable deep tabular learning network. Briefings in Bioinformatics 2022, 23(2):bbac015.
- Li, WZ, Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22(13):1658-1659.
- Li, WZ, Jaroszewski, L, Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17(3):282-283.
- Li, WZ, Jaroszewski, L, Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18(1):77-82.
- Huang, Y, Niu, BF, Gao, Y et al. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010, 26(5):680-682.
- Yu, B, Yu, ZM, Chen, C et al. DNNAce: Prediction of prokaryote lysine acetylation sites through deep neural networks with multi-information fusion. Chemometrics and Intelligent Laboratory Systems 2020, 200:103999.
- Arafat, ME, Ahmad, MW, Shovan, SM et al. Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features. Genes 2020, 11(9):1023.
- Jamal, S, Ali, W, Nagpal, P et al. Predicting phosphorylation sites using machine learning by integrating the sequence, structure, and functional information of proteins. Journal of Translational Medicine 2021, 19(1):218.
- Bekker, J, Davis, J. Learning from positive and unlabeled data: a survey. Machine Learning 2020, 109(4):719-760.
- Jiang, M, Cao, JZ. Positive-Unlabeled Learning for Pupylation Sites Prediction. Biomed Research International 2016, 2016:4525786.
- Gao, Y, Hao, WL, Gu, J et al. PredPhos: an ensemble framework for structure-based prediction of phosphorylation sites. Journal of Biological Research-Thessaloniki 2016, 23:S12.
- Chen, Z, Pang, M, Zhao, ZX et al. Feature selection may improve deep neural networks for the bioinformatics problems. Bioinformatics 2020, 36(5):1542-1552.
- Ning, Q, Ma, ZQ, Zhao, XW et al. SSKM_Succ: A Novel Succinylation Sites Prediction Method Incorporating K-Means Clustering With a New Semi-Supervised Learning Algorithm. Ieee-Acm Transactions on Computational Biology and Bioinformatics 2022, 19(1):643-652.
- Chawla, NV, Bowyer, KW, Hall, LO et al. SMOTE: Synthetic minority over-sampling technique. Journal of Artificial Intelligence Research 2002, 16:321-357.
- He, HB, Bai, Y, Garcia, EA et al: ADASYN: Adaptive Synthetic Sampling Approach for Imbalanced Learning. In: International Joint Conference on Neural Networks: Jun 01-08 2008; Hong Kong, PEOPLES R CHINA. 2008: 1322-1328.
- Lu, Y, Cheung, YM, Tang, YY: Hybrid Sampling with Bagging for Class Imbalance Learning. In: 20th Pacific-Asia Conference on Knowledge Discovery and Data Mining (PAKDD): Apr 19-22 2016; Univ Auckland, Auckland, NEW ZEALAND. 2016: 14-26.
- Seiffert, C, Khoshgoftaar, TM, Van Hulse, J. Hybrid sampling for imbalanced data. Integrated Computer-Aided Engineering 2009, 16(3):193-210.
- Dongdong, L, Ziqiu, C, Bolu, W et al. Entropy-based hybrid sampling ensemble learning for imbalanced data. International Journal of Intelligent Systems 2021, 36(7):3039-3067.
- Wang, MH, Cui, XW, Yu, B et al. SulSite-GTB: identification of protein S-sulfenylation sites by fusing multiple feature information and gradient tree boosting. Neural Computing & Applications 2020, 32(17):13843-13862.
- Wang, MH, Song, LL, Zhang, YQ et al. Malsite-Deep: Prediction of protein malonylation sites through deep learning and multi-information fusion based on NearMiss-2 strategy. Knowledge-Based Systems 2022, 240:108191.
- Wilson, DL. Asymptotic Properties of Nearest Neighbor Rules Using Edited Data. IEEE Transactions on Systems, Man, and Cybernetics 1972, SMC-2(3):408-421.
- Ijaz, MF, Attique, M, Son, Y. Data-Driven Cervical Cancer Prediction Model with Outlier Detection and Over-Sampling Methods. Sensors 2020, 20(10):2809.
- Mbunge, E, Millham, RC, Sibiya, MN et al: Implementation of ensemble machine learning classifiers to predict diarrhoea with SMOTEENN, SMOTE, and SMOTETomek class imbalance approaches. In: Conference on Information-Communications-Technology-and-Society (ICTAS): Mar 08-09 2023; Durban Univ Technol, Durban, SOUTH AFRICA. 2023: 90-95.
- Khan, SH, Hayat, M, Bennamoun, M et al. Cost-Sensitive Learning of Deep Feature Representations From Imbalanced Data. Ieee Transactions on Neural Networks and Learning Systems 2018, 29(8):3573-3587.
- Yuan, ZW, Zhao, P: An Improved Ensemble Learning for Imbalanced Data Classification. In: IEEE 8th Joint International Information Technology and Artificial Intelligence Conference (ITAIC): May 24-26 2019; Chongqing, PEOPLES R CHINA. 2019: 408-411.
- Hu, XS, Zhang, RJ, Ieee: Clustering-based Subset Ensemble Learning Method for Imbalanced Data. In: International Conference on Machine Learning and Cybernetics (ICMLC): Jul 14-17 2013; Tianjin, PEOPLES R CHINA. 2013: 35-39.
- Hayashi, T, Fujita, H. One-class ensemble classifier for data imbalance problems. Applied Intelligence 2022, 52(15):17073-17089.
- Dou, LJ, Yang, FL, Xu, L et al. A comprehensive review of the imbalance classification of protein post-translational modifications. Briefings in Bioinformatics 2021, 22(5):bbab089.
- Branco, P, Torgo, L, Ribeiro, RP. A Survey of Predictive Modeling on Im balanced Domains. Acm Computing Surveys 2016, 49(2):31.
- Kaur, H, Pannu, HS, Malhi, AK. A Systematic Review on Imbalanced Data Challenges in Machine Learning: Applications and Solutions. Acm Computing Surveys 2019, 52(4):79.
- Wang, M, Yang, J, Liu, GP et al. Weighted-support vector machines for predicting membrane protein types based on pseudo-amino acid composition. Protein Engineering Design & Selection 2004, 17(6):509-516.
- Lin, C-F, Wang, S-D. Fuzzy support vector machines. IEEE transactions on neural networks 2002, 13(2):464-471.
- Ju, Z, Wang, SY. Prediction of lysine formylation sites using the composition of k-spaced amino acid pairs via Chou’s 5-steps rule and general pseudo components. Genomics 2020, 112(1):859-866.
- Zhou, ZH, Liu, XY. Training cost-sensitive neural networks with methods addressing the class imbalance problem. Ieee Transactions on Knowledge and Data Engineering 2006, 18(1):63-77.
- Seiffert, C, Khoshgoftaar, TM, Van Hulse, J et al. RUSBoost: A Hybrid Approach to Alleviating Class Imbalance. Ieee Transactions on Systems Man and Cybernetics Part a-Systems and Humans 2010, 40(1):185-197.
- Jia, CZ, Zuo, Y, Zou, Q. O-GlcNAcPRED-II: an integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34(12):2029-2036.
- Wu, XY, Srihari, R, Zheng, ZH: Document representation for one-class SVM. In: Machine Learning: Ecml 2004, Proceedings. Edited by Boulicaut JF, Esposito F, Giannoti F, Pedreschi D, vol. 3201; 2004: 489-500.
- Islam, S, Mugdha, SB, Dipta, SR et al. MethEvo: an accurate evolutionary information-based methylation site predictor. Neural Computing & Applications 2022, 36(1):201-212.
- Huang, KY, Hung, FY, Kao, HJ et al. iDPGK: characterization and identification of lysine phosphoglycerylation sites based on sequence-based features. Bmc Bioinformatics 2020, 21(1):568.
- Sahu, SS, Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Computational Biology and Chemistry 2010, 34(5-6):320-327.
- Huang, KY, Hsu, JBK, Lee, TY. Characterization and Identification of Lysine Succinylation Sites based on Deep Learning Method. Scientific Reports 2019, 9:16175.
- Jiang, PR, Ning, WS, Shi, YS et al. FSL-Kla: A few-shot learning-based multi-feature hybrid system for lactylation site prediction. Computational and Structural Biotechnology Journal 2021, 19:4497-4509.
- Suo, SB, Qiu, JD, Shi, SP et al. Position-Specific Analysis and Prediction for Protein Lysine Acetylation Based on Multiple Features. Plos One 2012, 7(11):e49108.
- Shen, HB, Yang, J, Chou, KC. Fuzzy KNN for predicting membrane protein types from pseudo-amino acid composition. Journal of Theoretical Biology 2006, 240(1):9-13.
- Gao, JJ, Thelen, JJ, Dunker, AK et al. Musite, a Tool for Global Prediction of General and Kinase-specific Phosphorylation Sites. Molecular & Cellular Proteomics 2010, 9(12):2586-2600.
- Shen, JW, Zhang, J, Luo, XM et al. Predicting protein-protein interactions based only on sequences information. Proceedings of the National Academy of Sciences of the United States of America 2007, 104(11):4337-4341.
- Saravanan, V, Gautham, N. Harnessing Computational Biology for Exact Linear B-Cell Epitope Prediction: A Novel Amino Acid Composition-Based Feature Descriptor. Omics-a Journal of Integrative Biology 2015, 19(10):648-658.
- Park, KJ, Kanehisa, M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 2003, 19(13):1656-1663.
- Keskin, O, Bahar, I, Badretdinov, AY et al. Empirical solvent-mediated potentials hold for both intra-molecular and inter-molecular inter-residue interactions. Protein Science 1998, 7(12):2578-2586.
- Liang, SD, Grishin, NV. Effective scoring function for protein sequence design. Proteins-Structure Function and Bioinformatics 2004, 54(2):271-281.
- Chan, CH, Liang, HK, Hsiao, NW et al. Relationship between local structural entropy and protein thermostability. Proteins-Structure Function and Bioinformatics 2004, 57(4):684-691.
- Tang, YR, Chen, YZ, Canchaya, CA et al. GANNPhos: a new phosphorylation site predictor based on a genetic algorithm integrated neural network. Protein Engineering Design & Selection 2007, 20(8):405-412.
- Xu, Y, Wang, XB, Wang, YC et al. Prediction of posttranslational modification sites from amino acid sequences with kernel methods. Journal of Theoretical Biology 2014, 344:78-87.
- Lee, TY, Lin, ZQ, Hsieh, SJ et al. Exploiting maximal dependence decomposition to identify conserved motifs from a group of aligned signal sequences. Bioinformatics 2011, 27(13):1780-1787.
- Kawashima, S, Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Research 2000, 28(1):374-374.
- Li, FY, Li, C, Wang, MJ et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015, 31(9):1411-1419.
- Gong, WM, Zhou, DH, Ren, YL et al. PepCyber:PPEP:: a database of human protein-protein interactions mediated by phosphoprotein-binding domains. Nucleic Acids Research 2008, 36:D679-D683.
- Wagner, M, Adamczak, R, Porollo, A et al. Linear regression models for solvent accessibility prediction in proteins. Journal of Computational Biology 2005, 12(3):355-369.
- Tomii, K, Kanehisa, M. Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein Engineering 1996, 9(1):27-36.
- Dubchak, I, Muchnik, I, Holbrook, SR et al. PREDICTION OF PROTEIN-FOLDING CLASS USING GLOBAL DESCRIPTION OF AMINO-ACID-SEQUENCE. Proceedings of the National Academy of Sciences of the United States of America 1995, 92(19):8700-8704.
- Faraggi, E, Xue, B, Zhou, YQ. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins-Structure Function and Bioinformatics 2009, 74(4):847-856.
- Kabsch, W, Sander, C. DICTIONARY OF PROTEIN SECONDARY STRUCTURE - PATTERN-RECOGNITION OF HYDROGEN-BONDED AND GEOMETRICAL FEATURES. Biopolymers 1983, 22(12):2577-2637.
- López, Y, Dehzangi, A, Lal, SP et al. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Analytical Biochemistry 2017, 527:24-32.
- López, Y, Sharma, A, Dehzangi, A et al. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction. Bmc Genomics 2018, 19:923.
- Ward, JJ, McGuffin, LJ, Bryson, K et al. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20(13):2138-2139.
- Holland, RCG, Down, TA, Pocock, M et al. BioJava:: an open-source framework for bioinformatics. Bioinformatics 2008, 24(18):2096-2097.
- Obradovic, Z, Peng, K, Vucetic, S et al. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins-Structure Function and Bioinformatics 2005, 61:176-182.
- Heffernan, R, Paliwal, K, Lyons, J et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Scientific Reports 2015, 5:11476.
- Islam, MM, Saha, S, Rahman, MM et al. iProtGly-SS: Identifying protein glycation sites using sequence and structure based features. Proteins-Structure Function and Bioinformatics 2018, 86(7):777-789.
- Sharma, A, Lyons, J, Dehzangi, A et al. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition. Journal of Theoretical Biology 2013, 320:41-46.
- Ashburner, M, Ball, CA, Blake, JA et al. Gene Ontology: tool for the unification of biology. Nature Genetics 2000, 25(1):25-29.
- Hunter, S, Jones, P, Mitchell, A et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Research 2012, 40(D1):D306-D312.
- Kanehisa, M, Goto, S, Sato, Y et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research 2012, 40(D1):D109-D114.
- Finn, RD, Tate, J, Mistry, J et al. The Pfam protein families database. Nucleic Acids Research 2008, 36:D281-D288.
- Franceschini, A, Szklarczyk, D, Frankild, S et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research 2013, 41(D1):D808-D815.
- Weng, SL, Huang, KY, Kaunang, FJ et al. Investigation and identification of protein carbonylation sites based on positionspecific amino acid composition and physicochemical features. Bmc Bioinformatics 2017, 18:66.
- Celniker, G, Nimrod, G, Ashkenazy, H et al. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Israel Journal of Chemistry 2013, 53(3-4):199-206.
- Armon, A, Graur, D, Ben-Tal, N. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. Journal of Molecular Biology 2001, 307(1):447-463.
- Shen, HB, Chou, KC. Nuc-PLoc: a new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Engineering Design & Selection 2007, 20(11):561-567.
- Alkuhlani, A, Gad, W, Roushdy, M et al. PTG-PLM: Predicting Post-Translational Glycosylation and Glycation Sites Using Protein Language Models and Deep Learning. Axioms 2022, 11(9):469.
- Ahmed, E, Michael, H, Christian, D et al. ProtTrans: Towards Cracking the Language of Life’s Code Through Self-Supervised Deep Learning and High Performance Computing. 2021; arXiv:2007.06225.
- Rives, A, Meier, J, Sercu, T et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences of the United States of America 2021, 118(15):e2016239118.
- Rao, R, Bhattacharya, N, Thomas, N et al. Evaluating Protein Transfer Learning with TAPE. Advances in neural information processing systems 2019, 32:9689-9701.
- Jacob, D, Ming-Wei, C, Kenton, L et al. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv:1810.04805.
- Lan, ZC, M.; Goodman, S.; Gimpel, K.; Sharma, P.; Soricut, R. Albert: A lite bert for self-supervised learning of language representations. arXiv:1909.11942.
- Yang, Z, Dai, Z, Yang, Y et al. XLNet: Generalized Autoregressive Pretraining for Language Understanding. Arxiv 2020.
- Wang, HF, Wang, Z, Li, ZY et al. Incorporating Deep Learning With Word Embedding to Identify Plant Ubiquitylation Sites. Frontiers in Cell and Developmental Biology 2020, 8:572195.
- Yu, K, Zhang, QF, Liu, ZK et al. Deep learning based prediction of reversible HAT/HDAC-specific lysine acetylation. Briefings in Bioinformatics 2020, 21(5):1798-1805.
- Li, SH, Zhang, J, Zhao, YW et al. iPhoPred: A Predictor for Identifying Phosphorylation Sites in Human Protein. Ieee Access 2019, 7:177517-177528.
- Xu, Y, Ding, YX, Ding, J et al. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Scientific Reports 2016, 6:38318.
- Zhang, N, Zhou, Y, Huang, T et al. Discriminating between Lysine Sumoylation and Lysine Acetylation Using mRMR Feature Selection and Analysis. Plos One 2014, 9(9):e107464.
- Ma, X, Guo, J, Sun, X. Sequence-Based Prediction of RNA-Binding Proteins Using Random Forest with Minimum Redundancy Maximum Relevance Feature Selection. Biomed Research International 2015, 2015:425810.
- Peker, M, Sen, B, Delen, D. Computer-Aided Diagnosis of Parkinson’s Disease Using Complex-Valued Neural Networks and mRMR Feature Selection Algorithm. Journal of Healthcare Engineering 2015, 6(3):281-302.
- He, SD, Ye, XC, Sakurai, T et al. MRMD3.0: A Python Tool and Webserver for Dimensionality Reduction and Data Visualization via an Ensemble Strategy. Journal of Molecular Biology 2023, 435(14):168116.
- Yu, JL, Shi, SP, Zhang, F et al. PredGly: predicting lysine glycation sites for Homo sapiens based on XGboost feature optimization. Bioinformatics 2019, 35(16):2749-2756.
- Chen, TQ, Guestrin, C, Assoc Comp, M: XGBoost: A Scalable Tree Boosting System. In: 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD): Aug 13-17 2016; San Francisco, CA. 2016: 785-794.
- Xu, Y, Li, L, Ding, J et al. Gly-PseAAC: Identifying protein lysine glycation through sequences. Gene 2017, 602:1-7.
- Ning, Q, Ma, ZQ, Zhao, XW. dForml(KNN)-PseAAC: Detecting formylation sites from protein sequences using K-nearest neighbor algorithm via Chou’s 5-step rule and pseudo components. Journal of Theoretical Biology 2019, 470:43-49.
- Dosset, P, Rassam, P, Fernandez, L et al. Automatic detection of diffusion modes within biological membranes using back-propagation neural network. Bmc Bioinformatics 2016, 17:197.
- Butt, AH, Khan, YD. Prediction of S-Sulfenylation Sites Using Statistical Moments Based Features via CHOU’S 5-Step Rule. International Journal of Peptide Research and Therapeutics 2020, 26(3):1291-1301.
- Malebary, SJ, Rehman, MSU, Khan, YD. iCrotoK-PseAAC: Identify lysine crotonylation sites by blending position relative statistical features according to the Chou’s 5-step rule. Plos One 2019, 14(11):e0223993.
- Opitz, D, Maclin, R. Popular ensemble methods: An empirical study. Journal of artificial intelligence research 1999, 11:169-198.
- Rokach, L. Ensemble-based classifiers. Artificial intelligence review 2010, 33:1-39.
- Hasan, MM, Guo, DJ, Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Molecular Biosystems 2017, 13(12):2545-2550.
- Shi, MH, Lin, FX, Qian, Y et al: Research of Imbalanced Classification Based on Cascade Forest. In: IEEE International Conference on Progress in Informatics and Computing (IEEE PIC): Dec 17-19 2021; Electr Network. 2021: 29-33.
- Chu, YY, Kaushik, AC, Wang, XG et al. DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Briefings in Bioinformatics 2021, 22(1):451-462.
- Qian, Y, Ye, SS, Zhang, Y et al. SUMO-Forest: A Cascade Forest based method for the prediction of SUMOylation sites on imbalanced data. Gene 2020, 741:144536.
- Rao, H, Shi, XZ, Rodrigue, AK et al. Feature selection based on artificial bee colony and gradient boosting decision tree. Applied Soft Computing 2019, 74:634-642.
- Friedman, JH. Greedy function approximation: a gradient boosting machine. Annals of statistics 2001:1189-1232.
- He, F, Wang, R, Gao, YX et al: Protein Ubiquitylation and Sumoylation Site Prediction Based on Ensemble and Transfer Learning. In: IEEE International Conference on Bioinformatics and Biomedicine (BIBM): Nov 18-21 2019; San Diego, CA. 2019: 117-123.
- Zhang, YJ, Xie, RP, Wang, JW et al. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Briefings in Bioinformatics 2019, 20(6):2185-2199.
- Wang, DL, Liu, DP, Yuchi, JK et al. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Research 2020, 48(W1):W140-W146.
- Zhao, YM, He, NN, Chen, Z et al. Identification of Protein Lysine Crotonylation Sites by a Deep Learning Framework With Convolutional Neural Networks. Ieee Access 2020, 8:14244-14252.
- Wei, XL, Sha, YT, Zhao, YM et al. DeepKcrot: A Deep-Learning Architecture for General and Species-Specific Lysine Crotonylation Site Prediction. Ieee Access 2021, 9:49504-49513.
- Xiu, QX, Li, DC, Li, HL et al: Prediction Method for Lysine Acetylation Sites Based on LSTM Network. In: 7th IEEE International Conference on Computer Science and Network Technology (ICCSNT): Oct 19-20 2019; Dalian, PEOPLES R CHINA. 2019: 179-182.
- Li, A, Deng, YW, Tan, Y et al. A Transfer Learning-Based Approach for Lysine Propionylation Prediction. Frontiers in Physiology 2021, 12:658633.
- Zhao, Q, Ma, JQ, Wang, Y et al. Mul-SNO: A Novel Prediction Tool for S-Nitrosylation Sites Based on Deep Learning Methods. Ieee Journal of Biomedical and Health Informatics 2022, 26(5):2379-2387.
- Liu, Y, Ye, CF, Lin, C et al. Semi-ssPTM: A Web Server for Species-Specific Lysine Post-Translational Modification Site Prediction by Semi-Supervised Domain Adaptation. Ieee Transactions on Instrumentation and Measurement 2024, 73:2523410.
- Ning, WS, Xu, HD, Jiang, PR et al. HybridSucc: A Hybrid-learning Architecture for General and Species-specific Succinylation Site Prediction. Genomics Proteomics & Bioinformatics 2020, 18(2):194-207.
- Chen, Z, Zhao, P, Li, FY et al. PROSPECT: A web server for predicting protein histidine phosphorylation sites. Journal of Bioinformatics and Computational Biology 2020, 18(4):2050018.
- Vaswani, A, Shazeer, N, Parmar, N et al: Attention Is All You Need. In: 31st Annual Conference on Neural Information Processing Systems (NIPS): Dec 04-09 2017 2017; Long Beach, CA. 2017.
- Meng, LK, Chen, XJ, Cheng, K et al. TransPTM: a transformer-based model for non-histone acetylation site prediction. Briefings in Bioinformatics 2024, 25(3):bbae219.
- Liang, YY, Li, MW. A deep learning model for prediction of lysine crotonylation sites by fusing multi-features based on multi-head self-attention mechanism. Scientific Reports 2025, 15(1):18940.
- Xu, DL, Zhu, YF, Xu, Q et al. DTL-NeddSite: A Deep-Transfer Learning Architecture for Prediction of Lysine Neddylation Sites. Ieee Access 2023, 11:51798-51809.
- Soylu, NN, Sefer, E. DeepPTM: Protein Post-translational Modification Prediction from Protein Sequences by Combining Deep Protein Language Model with Vision Transformers. Current Bioinformatics 2024, 19(9):810-824.
- Lv, H, Dao, FY, Guan, ZX et al. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Briefings in Bioinformatics 2021, 22(4):bbaa255.
- Xu, Y, Ding, J, Wu, LY. iSulf-Cys: Prediction of S-sulfenylation Sites in Proteins with Physicochemical Properties of Amino Acids. Plos One 2016, 11(4):e0154237.
- Liu, S, Xue, C, Fang, Y et al. Global Involvement of Lysine Crotonylation in Protein Modification and Transcription Regulation in Rice. Molecular & Cellular Proteomics 2018, 17(10):1922-1936.
- Sun, HJ, Liu, XW, Li, FF et al. First comprehensive proteome analysis of lysine crotonylation in seedling leaves of Nicotiana tabacum. Scientific Reports 2017, 7:3013.
- Liu, KD, Yuan, CC, Li, HL et al. A qualitative proteome-wide lysine crotonylation profiling of papaya (Carica papaya L.). Scientific Reports 2018, 8:8230.
- Li, SH, Yu, K, Wu, GD et al. pCysMod: Prediction of Multiple Cysteine Modifications Based on Deep Learning Framework. Frontiers in Cell and Developmental Biology 2021, 9:617366.
- Al-barakati, HJ, Saigo, H, Newman, RH et al. RF-GlutarySite: a random forest based predictor for glutarylation sites. Molecular Omics 2019, 15(3):189-204.
- Dou, LJ, Li, XL, Zhang, LC et al. iGlu_AdaBoost: Identification of Lysine Glutarylation Using the AdaBoost Classifier. Journal of Proteome Research 2021, 20(1):191-201.
- Chung, CR, Chang, YP, Hsu, YL et al. Incorporating hybrid models into lysine malonylation sites prediction on mammalian and plant proteins. Scientific Reports 2020, 10(1):10541.
- Liu, Y, Li, A, Zhao, XM et al. DeepTL-Ubi: A novel deep transfer learning method for effectively predicting ubiquitination sites of multiple species. Methods 2021, 192:103-111.
- Long, HX, Sun, Z, Li, MZ et al. Predicting Protein Phosphorylation Sites Based on Deep Learning. Current Bioinformatics 2020, 15(4):300-308.
- Zahiri, Z, Mehrshad, N, Mehrshad, M. DF-Phos: Prediction of Protein Phosphorylation Sites by Deep Forest. Journal of Biochemistry 2023, 175(4):447-456.
- Wang, RL, Wang, Z, Wang, HF et al. Characterization and identification of lysine crotonylation sites based on machine learning method on both plant and mammalian. Scientific Reports 2020, 10(1):20447.
- Li, FY, Zhang, Y, Purcell, AW et al. Positive-unlabelled learning of glycosylation sites in the human proteome. Bmc Bioinformatics 2019, 20:112.
- Dai, YH, Deng, L, Zhu, F. A model for predicting post-translational modification cross-talk based on the Multilayer Network. Expert Systems with Applications 2024, 255:124770.
- Zhu, F, Deng, L, Dai, YH et al. PPICT: an integrated deep neural network for predicting inter-protein PTM cross-talk. Briefings in Bioinformatics 2023, 24(2).
- Deng, L, Zhu, F, He, Y et al. Prediction of post-translational modification cross-talk and mutation within proteins via imbalanced learning. Expert Systems with Applications 2023, 211:118593.
- Simpson, CM, Zhang, B, Hornbeck, P et al. Systematic analysis of the intersection of disease mutations with protein modifications. Bmc Medical Genomics 2019, 12:109.
Figure 1.
Process of machine and deep learning methods. The framework design of a PTM predictor utilizing machine learning and deep learning involves the acquisition of data from existing databases, followed by pre-processing. After pre-processing, the machine learning model necessitates feature extraction and feature selection, which are essential steps before employing a classifier to finalize the model construction. In contrast, deep learning methods do not require manual feature extraction; thus, a deep learning model can be directly utilized to construct the classifier. Ultimately, the model is assessed through various evaluation methods.
Figure 1.
Process of machine and deep learning methods. The framework design of a PTM predictor utilizing machine learning and deep learning involves the acquisition of data from existing databases, followed by pre-processing. After pre-processing, the machine learning model necessitates feature extraction and feature selection, which are essential steps before employing a classifier to finalize the model construction. In contrast, deep learning methods do not require manual feature extraction; thus, a deep learning model can be directly utilized to construct the classifier. Ultimately, the model is assessed through various evaluation methods.
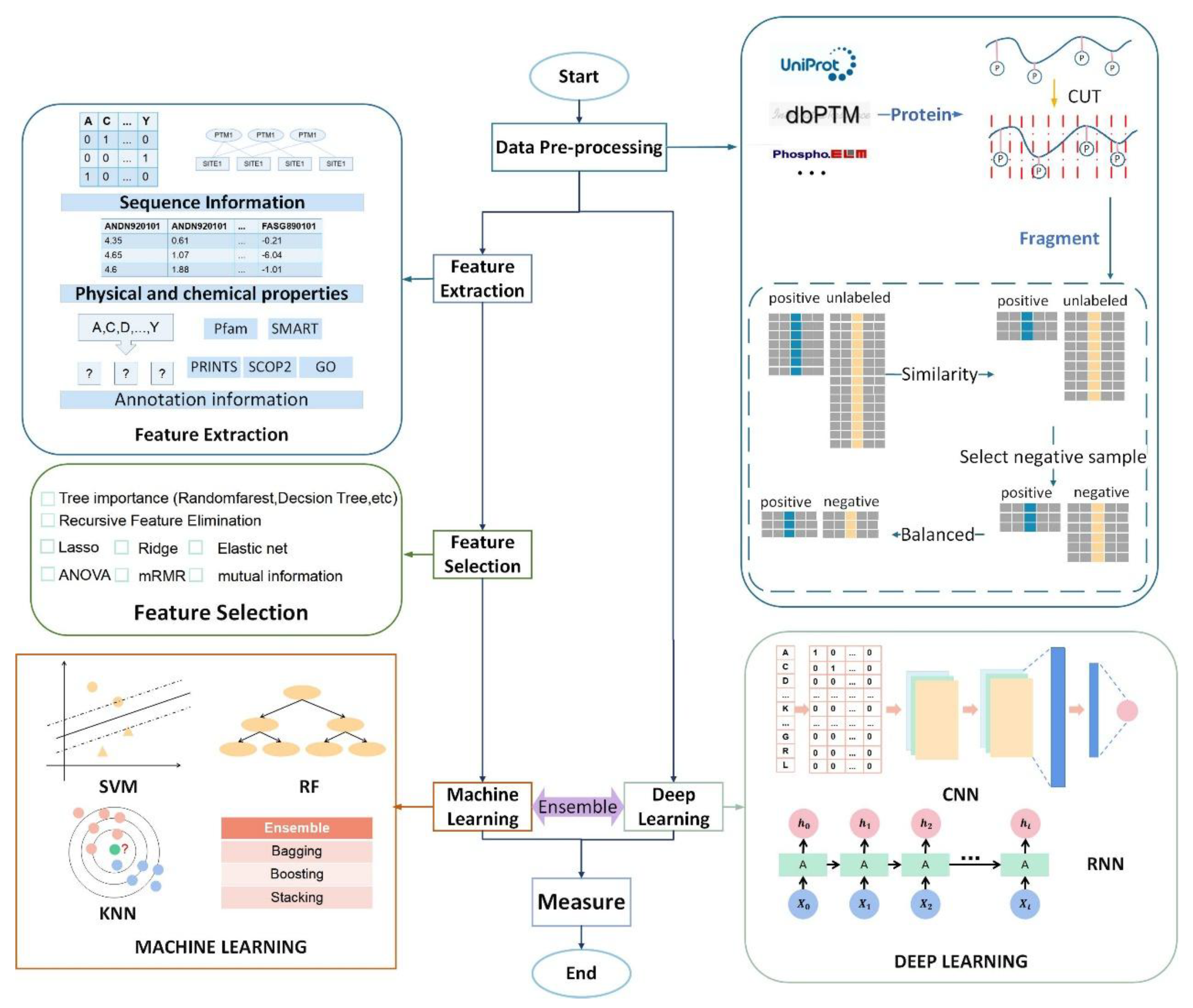
Figure 2.
Schematic diagram of data pre-processing. After segmenting the protein sequences, there are positive examples and unlabeled data. First, reliable negative examples are obtained, and then the dataset is balanced to obtain a benchmark dataset.
Figure 2.
Schematic diagram of data pre-processing. After segmenting the protein sequences, there are positive examples and unlabeled data. First, reliable negative examples are obtained, and then the dataset is balanced to obtain a benchmark dataset.
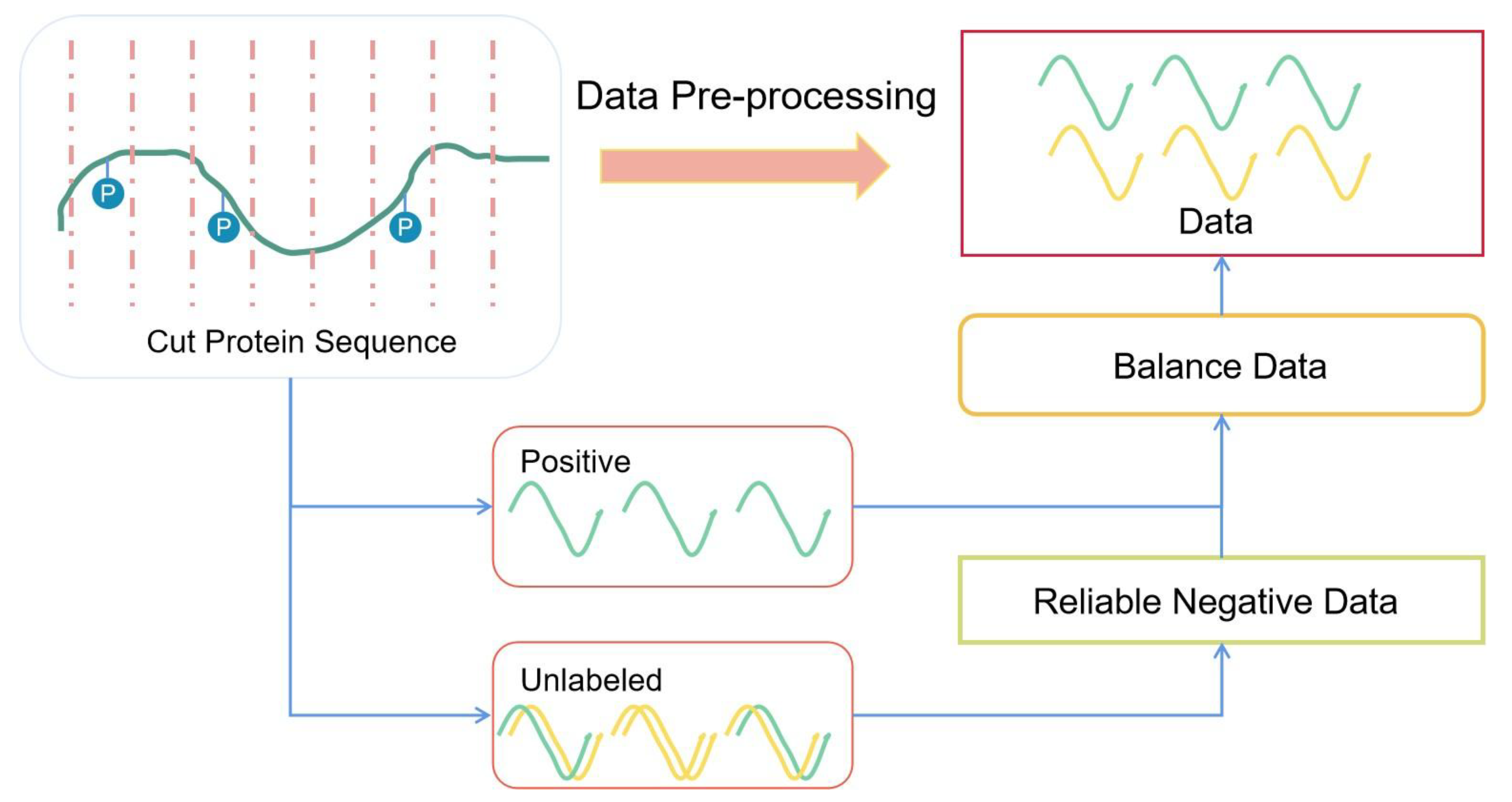

Table 1.
Commonly used datasets for PTM studies.
| Name | Website | PTM Type | Statistics |
|---|---|---|---|
| Uniport[11,12] | https://www.uniprot.org/ | Multiple | 570,420 reviewed proteins, 251,131,639 unreviewed proteins |
| dbPTM[13,14] | https://awi.cuhk.edu.cn/dbPTM/ | Multiple | 2235664 sites, 70+PTM types, 40+ integrated databases, 30+ benchmark datasets |
| PhosphoSitePlus[19] | https://www.phosphosite.org/homeAction | Multiple | 59469 PTM sites, 13 PTM types |
| CPLM 4.0[15] | http://cplm.biocuckoo.cn/ | Multiple | 463,156 unique sites of 105,673 proteins for up to 29 PLM types across 219 species |
| qPTM[20] | http://qptm.omicsbio.info/ | Multiple | 11,482,553 quantification events for 660,030 sites on 40,728 proteins under 2,596 conditions |
| PupDB[21] | https://cwtung.kmu.edu.tw/pupdb/ | Pupylation | 268 pupylation proteins with 311 known pupylation sites and 1123 candidate pupylation proteins |
| DEPOD[22] | https://depod.bioss.uni-freiburg.de/ | Phosphorylation | 194 phosphatases have substrate data |
| O-GlcNAcAtlas[23] | https://oglcnac.org/atlas/ | O-GlcNAcylation | 16877 Unambiguous sites, 10058 ambiguous sites |
| Phospho.elm[24] | http://phospho.elm.eu.org/ | Phosphorylation | 42914 instances, 11224 sequences |
| CarbonylDB[25] | https://carbonyldb.missouri.edu/CarbonylDB/index.php/ | Carbonylation | 1495 proteins, 3781 PTM sites, 21 species |
| Scop3P[26] | https://iomics.ugent.be/scop3p/index | Phosphorylation | 108130 modifications, 20394 proteins |
| O-GlycBase[27] | https://services.healthtech.dtu.dk/datasets/OglycBase/ | O-Glycosylation | 242 proteins |
| dbSNO[28] | http://140.138.144.145/~dbSNO/index.php | S-nitrosylation | 174 experimentally verified S-nitrosylation sites on 94 S-nitrosylated proteins |
| UbiNet 2.0[29] | https://awi.cuhk.edu.cn/~ubinet/index.php | Ubiquitination | 3332 experimentally verified ESIs |
| UbiBrowser 2.0[30] | http://ubibrowser.bio-it.cn/ubibrowser_v3/ | ubiquitination | 1,884,676 predicted high confidence ESIs, 8,341,262 potential E3 recognizing motifs, 4,068 known ESIs from literature |
| PhosPhAt[31] | https://phosphat.uni-hohenheim.de/ | Phosphorylation | 10898 phosphoproteins,64128 serine sites, 13102 threonine sites, 2672 tyrosine sites |
Table 2.
Review of PTM prediction models in recent years.
| PTM | Tools | Dataset | Window Size | Feature Extraction Method | Classifier | Website | Ref |
|---|---|---|---|---|---|---|---|
| lysine crotonylation | BERT-Kcr | used by Lv et al [164] | 31 | BERT | BiLSTM | http://zhulab.org.cn/BERT-Kcr_models/data | [10] |
| Lysine lactylation | Auto-Kla | UniProt | 51 | Token embedding, position embedding, transformer encoder | AutoML, MLP | https://github.com/tubic/Auto-Kla | [32] |
| Cysteine S-sulphenylation | DeepCSO | UniprotKB | 35 | NUM, EAAC, BE, AAindex, CKSAAP, PSSM | LSTM, CNN, RF, SVM | http://www.bioinfogo.org/DeepCSO | [36] |
| phosphorylation | -- | dbPTM | 21 | AAindex, Binary-encoding, ASA, secondary structure (coil, helix and strand), disordered regions, BP, MF, CC, protein functional, domain data from InterPro, KEGG pathway and functional annotation |
RF, SVM | -- | [46] |
| phosphorylation | PredPhos | Phospho.ELM version 9.0, Phospho-POINT and PhosphoSitePlus | -- | PSSM, evolutionary conservation score, disorder, ASA, pair potential, atom and residue contacts, Topographical index, physicochemical features, four-body statistical pseudo-potential, local structural entropy, side-chain energy, Voronoi Contacts, structural conservation score, Two-step feature selectio | Ensemble method, SVM | -- | [49] |
| Succinylation | SSKM_Succ | Training data: PLMD and Uniprot Test data: dbPTM |
21 | Information of Proximal PTMs, Grey Pseudo Amino Acid Composition, K-Space, PSAAP | SVM, RF, NB | https://github.com/yangyq505/SSKM_Succ.git | [51] |
| S-sulfenylation | SulSite-GTB | Carroll Lab, RedoxDB and UniProtKB |
21 | AAC, DPC, EBGW, KNN, PSAAP, PsePSSM, PWAAC | GTB | https://github.com/QUST-AIBBDRC/SulSite-GTB/ | [57] |
| lysine phosphoglycerylation | iDPGK | PLMD | 15 | AAC, PCAAC, AAPC, BLOSUM62, PSSM | DT, RF, SVM | http://mer.hc.mmh.org.tw/iDPGK/. | [77] |
| Succinylation | CNN-SuccSite | PLMD 3.0 | 31 | PspAAC, CKSAAP, PSSM | CNN |
http://csb.cse.yzu.edu.tw/ CNN-SuccSite/ |
[79] |
| Glycosylation and Glycation |
PTG-PLM | UniProt | 31 | ProtBERT-BFD, ProtBERT, ProtALBERT, ProtXLNet, ESM-1b and TAPE | CNN, SVM, LR, RF, and XGBoost | https://github.com/Alhasanalkuhlani/PTG-PLM | [118] |
| Formylation | LFPred | Uniport, PLMD and dbPTM | 41, information entropy | AAC, BPF, AAI | KNN | -- | [136] |
| S-Sulfenylation | S-Sulfenylation | Conducted by Xu et al. [165] and Hasan et al. [142] |
21 | PseAAC, SVV, SM, PRIM, R-PRIM, FV, AAPIV, RAAPIV | BP-NN | https://www.github.com/ahmad-umt/S-Sulfenylation | [138] |
| Sumoylation | SUMO-Forest | UniProt | 21 | PSAAP, PseAAC, SP, BK | Cascade Forest | https://github.com/sandyye666/SUMOForest | [145] |
| Ubiquitylation and sumoylation | DeepUbiSumoPre | Uniprot/Swiss-Prot | 49 | one-hot, PCPs | CNN, DNN, stacking method, transfer learning | https://github.com/ruiwcoding/DeepUbiSumoPre | [148] |
| lysine crotonylation | -- | collected verified Kcr sites on non-histone proteins from papaya |
From 2 to 37 | BE, CKSAAP, AAC, EAAC, EGAAC | CNN | http://www.bioinfogo.org/pkcr | [151] |
| Lysine Crotonylation |
DeepKcrot | Collected from [166,167,168] | 29 | EGAAC, WE | LSTM, CNN, RF | http://www.bioinfogo.org/deepkcrot | [152] |
| Lysine Acetylation Sites | -- | DeepAcet and UniProt | 21 | one-hot encoding, physical and chemical properties including molecular weight, isoelectric point, carboxylic acid dissociation constant and amino acid dissociation constant | LSTM | -- | [153] |
| Lysine propionylation | -- | PLMD and Uniport | 17 | RNN, LSTM | Transfer learning, SVM | http://47.113.117.61/. | [154] |
| Succinylation | HybridSucc | PLMD 3.0, PhosphoSitePlus and dbPTM | -- | PseAAC, CKSAAP, OBC, AAindex, ACF, GPS, PSSM, ASA, SS, and BTA | DNN, PLR | http://hybridsucc.biocuckoo.org/ | [157] |
| -Nitrosylation | Mul-SNO | training set: Li et al. [169], independent test set: DeepNitro | 31 | BiLSTM, BERT | RF, lightgbm, xgboost | http://lab.malab.cn/∼mjq/Mul-SNO/ | [155] |
| phosphorylation | PROSPECT | UniProt | 27 | one-of-K, EGAAC and CKSAAGP | CNNone-of-K, CNNEGAAC and RFCKSAAGP | http://PROSPECT.erc.monash.edu/ | [158] |
| Lysine Glutarylation | iGlu_AdaBoost | Conducted by Al-barakati et al. [170] from PLMD, NCBI, and SWISS-PROT | 23 | 188D, CKSAAP, and EAAC | AdaBoost | -- | [171] |
| Lysine malonylation | Kmalo | PLMD and LEMP | 11~39 | AAC, one hot encoding, Pse-AAC, AAindex, PSSM | hybrid models contain multiple CNNs, random forests and SVM | https://fdblab.csie.ncu.edu.tw/kmalo/home.html | [172] |
| ubiquitination | DeepTL-Ubi | PhosphoSitePlus, mUbiSida and PLMD | 31 | one-hot | transfer deep learning method | https://github.com/USTC-HIlab/DeepTL-Ubi | [173] |
| Phosphorylation | -- | iPhos-PseEn | 13 | BE | CNN, BLSTM | -- | [174] |
| phosphorylation | DF-Phos | dbPAF and Phospho.ELM | 33 | CTD, DDE, EAAC, EGAAC, a series of PseKRAAC, GrpDDE, kGAAC, LocalPoSpKaaF, QSOrder, SAAC, SOCNumber, ExpectedValueGKmerAA, ExpectedValueKmerAA, ExpectedValueGAA, ExpectedValueAA | Deep Forest | https://github.com/zahiriz/DF-Phos | [175] |
| lysine crotonylation | -- | UniProt and pkcr | 31 | AAC, AAPC, BE, CKSAAP, EAAC, EGAAC and PSSM | SVM, RF | -- | [176] |
| glycosylation | GlycoMine_PU | UniProt | 15 | AAC, CTD, AAindex, Pseudo-AAC, Sequence-order, Auto-correlation | RF, SVM, One-SVM | http://glycomine.erc.monash.edu/Lab/GlycoMine_PU/ | [177] |
*NUM: Numerical Representation for Amino Acid; ASA: Solvent accessible area; AAC: amino acid composition; DPC: dipeptide composition; EBGW: encoding based on grouped weight; KNN: k nearest neighbors; PSAAP: position-special amino acid propensity; PsePSSM: Pseudo-position specific scoring matrix; PWAAC: Position weight amino acid composition; PseAAC: pseudo amino acid composition; SP: statistics property; BK: bi-gram and k-skip-bi-gram; PspAAC: position-specifc amino acid composition; BPF: binary profile feature; AAI: amino acid index; PCP: physio-chemical properties; AAPC: amino acid pair composition; PWM: Positional weighted matrix; PSSM: Position specific scoring matrix; B62: BLOSUM62; GPAAC: Grey Pseudo Amino Acid Composition; SVV: site vicinity vector; SM: statistical moments; PRIM: position relative incident matrix; R-PRIM: reverse position relative incident matrix; FV: frequency vector; AAPIV: accumulative absolute position incidence vector; RAAPIV: reverse accumulative absolute position incidence vector; DBPB: di-amino acid BPB; DDE: Dipeptide Deviation from Expected Mean value; EAAC: Enhanced Amino Acid Composition; Enhanced Grouped Amino Acid Composition; PseKRAAC: Pseudo K_tuple Reduced Amino Acid Composition; GrpDDE: Group Dipeptide Deviation from Expected Mean; kGAAC: k Grouped Amino Acid Composition; LocalPoSpKaaF: Local Position Specifi c k Amino Acids Frequency; QSOrder: Quasi Sequence Order; SAAC: Split Amino Acid Composition; SOCNumber: Sequence Order Coupling Number; ExpectedValueKmerAA: Expected Value for K-mer Amino Acid; ExpectedValueGAA: Expected Value for each group Amino Acid; ExpectedValueAA: Expected Value for each Amino Acid; BP: biological process; MF: molecular function; CC: cellular component.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



