
Submitted:
17 October 2025
Posted:
22 October 2025
You are already at the latest version
Abstract
The aim of this study was to identify differentially expressed microRNAs (miRNAs) in serum and synovial fluid (SF) samples of control horses and those with osteoarthritis (OA), to identify potential candidates for biomarkers of disease. Total RNA was extracted from serum and SF samples of control (n=4) and OA (n=9) horses, and sequenced. Differential expression analysis, pathway analysis and miRNA target prediction were performed. A group of six miRNAs (eca-miR-199a-3p, eca-miR-148a, eca-miR-99b, eca-miR-146a, eca-miR-423-5p and eca-miR-23b) were selected for validation in an independent cohort (serum, n=46; SF, n=88). The effect of clinical variables on miRNA expression was also assessed. Sequencing analyses found 43 and 23 differentially expressed miRNAs in serum and SF samples, respectively. Pathway analysis showed miRNA were involved in inflammatory disease/response and associated with OA pathways. miRNA expression in serum was strongly associated with the horses’ workload, while age had a pronounced influence on miRNA expression in SF. Distinct patterns of miRNA differential expression were observed in serum and SF samples from horses with OA compared to controls. miR-199a-3p and miR-148a warrant further investigation as potential biomarkers of equine OA. Further characterization of these molecular changes could provide novel insights into the mechanisms of early OA.
Keywords:
biomarkers
; equine
; microRNA
; osteoarthritis
; serum
; small RNA sequencing
; synovial fluid
1. Introduction
Genetic research has long put RNA at the center of the field of molecular biology, yet the importance of the regulatory functions of non-coding RNAs for organismal development has only been brought to light in the past two decades [1]. The transcriptional landscape is comprised of several RNA classes with varying functions and is broadly separated into coding RNAs, long non-coding RNAs (lncRNAs) and small non-coding RNAs (sncRNAs). The sncRNA group includes transfer RNAs (tRNAs), which are involved in protein translation; small nuclear RNAs (snRNAs), which are involved in splicing events; small nucleolar RNAs (snoRNAs), which are mainly involved in the modification of other RNAs; and short regulatory non-coding RNAs, such as piwi-associated RNAs (piRNAs), small interfering RNAs (siRNAs) and microRNAs (miRNAs), which regulate gene expression [2]. Among these, the miRNA class is the best characterized. These evolutionarily conserved molecules are on average 22 nucleotides in length and participate in the post-transcriptional regulation of genes [3]. miRNAs target messenger RNAs (mRNAs) and generally repress their translation or promote mRNA decay [3]. On rare occasions, miRNAs can also increase the expression of specific mRNAs [4]. miRNAs are important contributors to many biological functions including cell differentiation, embryogenesis, metabolism and organogenesis. Of particular relevance is their contribution to intercellular communication [3] – miRNAs influence multiple physiological and pathophysiological processes, and their expression can be altered as a result of cellular damage and tissue injury [5]. Therefore, miRNA “signatures” can be detected both in damaged tissues and in the circulation in biofluids, including blood or synovial fluid (SF) [5]. This, along with the fact that they are remarkably stable in the circulation, makes miRNAs prime candidates for use as non-invasive biomarkers in molecular diagnostics of disease [5].
The inability to detect pre-clinical changes in osteoarthritis (OA) is one of the main barriers to the development of effective therapies against this disease. OA is one of the most common causes of lameness in horses, and a significant welfare concern due to pain and disability [6,7]. Currently, when a clinical diagnosis of OA is reached, the changes that have occurred in the joints are generally advanced and irreversible, often leading to early retirement, and can lead to euthanasia of these animals. Therefore, developing a technique that allows for early diagnosis and intervention is a major goal of OA research [8]. miRNAs are promising candidates in the search for diagnostic biomarkers of OA and have been extensively investigated in human studies [8]. Previous work has demonstrated that miRNAs are involved in the regulation of cell apoptosis, inflammation and chondrocyte homeostasis and metabolism, displaying protective or destructive roles, and sometimes both [9]. Furthermore, researchers have investigated the levels of circulating miRNAs in OA patients and found differentially expressed molecules with potential clinical use as biomarkers of disease, and as predictors of OA severity [10,11]. For example, miR-140-3p, miR-33b-3p and miR-671-3p, which are downregulated in the articular cartilage of human OA patients compared with healthy patients, were also found to be downregulated in the serum of these OA patients, showing potential as diagnostic tools [12]. Another example is miR-378a-5p, which is commonly detectable in SF in late-stage OA and mostly undetectable early-stage OA, showing a potential for disease stratification [13]. Additionally, our group previously found a pattern of differentially expressed sncRNAs in equine OA SF linked to OA pathogenesis, which included miR-23b, let-7a-2 and miR-223 [14].
The objective of the present study was to identify differentially expressed miRNAs in equine serum and SF which have potential as biomarkers of OA. For this, we used a small RNA sequencing approach to investigate the miRNA profile of matched (i.e., from the same horse) equine serum and SF in OA. Additionally, we used bioinformatic tools to explore associated pathways and potential mRNA targets. Finally, we validated our findings in a larger, independent cohort and assessed the influence of different clinical variables on the expression of a selected group of miRNAs.
2. Results
This study was carried out in two stages – an exploration stage and a validation stage, which are summarized in Figure 1.
2.1. Exploration Stage
2.1.1. Sample and Group Characterization
A total of 13 matched samples of serum and SF (OA, n=9; Control, n=4) were collected for sequencing (Table 1). The horses’ age and proportion of neutered males/females was similar among groups (p=0.605 and p=0.500, respectively), however the absolute number of neutered males included in the entire sequencing cohort was double of that of females (n=8 and n=4, respectively). Most SF samples were collected from carpal joints. For most horses with available data in the OA group (60%), OA was classified as mild; details on sample classification are described in the methods.
2.1.2. Data Overview
The total number of reads varied between 37 to 45 million following spike-in exclusion (Figure 2). The number of reads were similar for both serum and SF, with serum samples averaging 48 million reads/sample and SF samples averaging 47 million reads/sample. Several different types of RNA molecules were identified including yRNA, snoRNA, snRNA, messenger RNA (mRNA), lncRNA, ribosomal RNA (rRNA), tRNA and miRNA (Figure 2). The relative abundance of miRNAs varied between 0.04–57.03% in serum and 0.03–33.64% in SF (Figure S1). The tRNA class had the highest mean percentage of relative reads in both serum (34.54%) and SF (49.31%).
A total of 308 miRNAs were identified in all samples, with 298 identified in serum and 203 in SF. Figure S2 shows the distribution of the number of miRNA molecules per group and per sample type. Briefly, 105 miRNAs were unique to serum, of which 73 were exclusive to OA samples. A total of 10 miRNAs were uniquely identified in SF, all of which were exclusive to the OA group. An additional group of 28 miRNAs were exclusively present in OA samples and detected in both SF and serum.
A heatmap was built on reads per million (RPM) normalized and scaled reads using the unit variance method for visualization in heatmaps (Figure 3A). Principal component analyses (PCA) was performed using RPM normalized miRNA reads, which were scaled using unit variance. Principal components (PCs) were calculated using singular value decomposition with imputation (Figure 3B).
2.1.3. Differential Expression Analysis
For serum, a total of 300 miRNAs were analyzed and 43 were found to be significantly different between the OA and Control groups (p<0.05; Table 2). Of these, 15 were also differentially expressed at a false discovery rate (FDR)-adjusted p<0.05. For SF, a total of 203 miRNAs were analyzed and 23 of these were significantly different between the OA and Control groups (p<0.05; Table 2). Of these, seven were also differentially expressed at an FDR-adjusted p<0.05.
Of the differentially expressed miRNAs, nine were common for serum and SF: eca-miR-1291a, eca-miR-148a, eca-miR-1892, eca-miR-199a-3p, eca-miR-199b-3p, eca-miR-206, eca-miR-23b, eca-miR-27b and eca-miR-423-5p. Fold changes (FC) values for each of the molecules highlight the similar directionality of change for most miRNAs in both sample type, except for eca-miR-206 and eca-miR-27b (Table 2).
2.1.4. Target Prediction and Pathway Analysis
Target prediction revealed 104 experimentally validated targets for 22 of the 66 differentially expressed miRNAs found in serum and SF. IPA Core Analysis of the combined list (interactome) of differentially expressed miRNAs and miRNA targets revealed involvement of inflammatory disease (p<4.79×10-2) and inflammatory response (p<4.49×10-2). The top two canonical pathways associated with these molecules were “Ribonucleotide Reductase Signaling Pathway” (p=2.96×10-2, with 1.2% overlap [2/170 molecules]) and “Role of chondrocytes in Rheumatoid Arthritis Signaling Pathway” (p=2.20×10-1, with 0.7% overlap [1/141 molecules], Figure S3). Figure 4 shows some of the diseases and functions predicted to be associated with the differentially expressed miRNAs, including osteoarthritis (p=2.93×10-3), rheumatic disease (p=8.11×10-5), atrophy of skeletal muscle (p=9.45×10-3), maturation of chondrocyte cell lines (p=6.14×10-8), apoptosis (p=2.23×10-6) and fibrosis (p=5.25×10-25), as well as the specific miRNAs associated with them.
2.1.5. Selection of miRNAs for Validation
Six miRNAs were selected for further validation using the criteria described in the Methods. These were eca-miR-199a-3p, eca-miR-148a, eca-miR-99b and eca-miR-146a, which were increased in the OA group compared with the Control group; and eca-miR-423-5p and eca-miR-23b, which were decreased in the OA group compared with the Control group (Table 2; Figure 5).
2.2. Validation Stage
2.2.1. Sample and Group Characterization
To estimate the required sample size for miRNA validation, a sample size calculation was performed in R using the package pwr v1.3-0, with results indicating a total of 46 serum samples (Control, n=23; OA, n=23) and 88 SF samples (Control, n=44; OA, n=44). These samples were collected, and the horses’ demographics and clinical characteristics for the validation cohort, by group and by sample type, are in Table 3.
For serum samples, there were no significant differences in mean age or sex between the Control and OA groups (p=0.6582 and p=0.2603, respectively). Most horses had a body condition score (BCS) of 5 in both groups; however, BCS was only assessed in nine OA horses. BCS, breed and occupation were reported descriptively but not statistically compared because the sample numbers for some of the categories were too low. Of note, there were more racing horses in the OA group than horses of any other occupation (9 out of 18 with available information); in contrast, there were no racing horses in the Control group. The horses’ level of work was significantly different between the Control and OA groups (p=0.0214). In the Control group, 52.2% of horses were described as a work level of 1 (light work) compared with 22.2% in the OA group. In contrast, 56.6% of horses in the OA group were described as a work level of 3 (intense work) compared with 8.7% of horses in the Control group.
For SF samples, mean age (SD) was significantly higher in the OA vs Control group (15.7 [6.7] vs 10.2 [6.8] years, respectively; p=0.0003). The proportion of females and neutered males was significantly different among groups (p=0.0227), with the OA group presenting a higher proportion of neutered males vs females (69.2% vs 30.8%, respectively) and the Control group having a lower proportion of neutered males vs females (32.0% vs 68.0%, respectively). As with the serum samples, breed distribution was described descriptively; however, the sample sizes for certain categories were insufficient for meaningful statistical analysis. Of note, there were more Thoroughbred horses in each group than horses of any other breed (Control, n=7; OA, n=9). Mean joint macroscopic score was higher for the OA vs Control group (3.4 ± 1.2 vs 0.5 ± 0.5, p<0.0001); similarly, mean articular cartilage microscopic score was higher for the OA vs Control group (5.0 ± 2.0 vs 3.2 ± 1.7, p=0.015).
2.2.2. Relative Gene Expression
A select group of candidate miRNA molecules (described in the Methods) were tested as reference genes, and the most appropriate normalization strategy for RT-qPCR was calculated using NormFinder. Normalization using the geometric mean of all tested reference genes (eca-miR-107b, eca-miR-423-3p, eca-miR-4865p, cel-mir-39, UniSp4 for serum; eca-miR-486-5p, eca-miR-423-3p, eca-miR-215, eca-miR-107b UniSp4 for SF) was deemed adequate to compare RT-qPCR data in both sequencing and validation cohorts.
Relative expression of the previously selected miRNAs (eca-miR-199a-3p, eca-miR-148a, eca-miR-99b, eca-miR-146a, eca-miR-423-5p and eca-miR-23b) were calculated for the sequencing and the validation cohorts, for both serum and SF samples.
For serum samples, RT-qPCR data in the sequencing cohort followed the same trend as the sequencing data, with a numerical increase in the expression of eca-miR-199a-3p, eca-miR-148a, eca-miR-99b and eca-miR-146a in the OA vs Control, and a decreased expression of eca-miR-423-5p in the OA group (Figure 6A). As with the sequencing data, eca-miR-99b presented low expression in most samples. This was particularly relevant in the Control group, where this miRNA was not expressed in any of the samples. Expression of eca-miR-423-5p was the highest among all miRNAs, which was similar to sequencing data. In contrast to the sequencing findings, eca-miR-23b expression was numerically increased in the OA group compared with the Control group. However, none of the differences were statistically significant.
The relative expression of the selected miRNAs was also investigated in the serum validation cohort (Figure 6B). No significant differences in miRNA expression were found between the Control and OA groups in serum samples. Similar to the sequencing cohort, expression of eca-miR-199a-3p, eca-miR-99b and eca-miR-23b were numerically higher in the OA group compared with the Control group. In contrast to the sequencing cohort, expression of eca-miR-148a and eca-miR-423-5p were generally similar among groups, and eca-miR-146a was numerically decreased in the OA group. Of note, when comparing the overall levels of expression, miRNAs that displayed numerically lower expression in the sequencing cohort (eca-miR-199a-3p and eca-miR-99b) also displayed a numerically lower expression in the validation cohort; miRNAs with average expression in the sequencing cohort (eca-miR-148a and eca-miR-146a) also displayed average expression in the validation cohort; and miRNAs with numerically higher expression in the sequencing cohort (eca-miR-423-5p and eca-miR-23b) also displayed a numerically higher expression in the validation cohort.
For SF samples, RT-qPCR data in the sequencing cohort showed low miRNA expression in the Control group, with one miRNA (eca-miR-99b) not being detected in any of the control samples (Figure 6C). While no statistically significant differences were found between the Control and OA groups, the expression of eca-miR-199a-3p, eca-miR-148a, eca-miR-99b and eca-miR-146a were increased in OA group, which was in line with the sequencing results. In contrast, eca-miR-423-5p and eca-miR-23b expression were also increased in the OA group, which was contrary to sequencing results. Furthermore, eca-miR-23b expression in the OA group displayed the most variability amongst all tested miRNAs.
The relative expression of the selected miRNAs was also investigated in the SF validation cohort (Figure 6D). Overall, miRNA expression was variable among samples in each group. In agreement with the sequencing results, eca-miR-199a-3p expression was numerically increased in the OA group compared with Control, however these differences were not statistically significant. The remaining miRNAs were generally similar in the Control and OA groups. As observed in the SF sequencing cohort, and in line with the results from the serum samples, expression of eca-miR-99b was the lowest in all tested miRNAs.
2.2.3. Influence of Clinical Variables
Figure S4 shows the association between the PCs of miRNA expression, group category (OA vs Control), and some of the clinical variables in the serum and SF validation cohorts. For serum, PC1 was significantly associated with level of work (FDR p-value =0.01) and PC6 was significantly associated with both level of work and group category (FDR p-value=0.01 and 0.02, respectively). For SF, PC1 was significantly associated with age (FDR p-value=0.01), PC4 was significantly associated with group category (FDR p-value=0.01) and joint gross score (FDR p-value=0.01), and PC6 was significantly associated with joint gross score (FDR p-value=0.02).
The associations between miRNA expression and level of work in serum and age in SF were explored further using PCA plots (Figure S5). For serum, samples from horses with a lower level of work (0–2) appeared to cluster together and were separate from those with higher levels of work (3; Figure S5A). For SF samples, there were two distinct clusters separated by PC1. While this separation could not be solely explained by age, samples from younger horses were more abundant in the left cluster (Figure S5B).
The influence of age on miRNA expression was also analyzed (Figure S6A–F). To help with visualization, horses were separated into four age classes: <6 years (n=27), 6–12 years (n=8), 13–20 years (n=37) and >20 years (n=9). Horses of unknown age were excluded from further analyses. Most samples in the <6 age class (19/27) were in the Control group, and most samples in the 13–20 age class (23/37) were in the OA group. Overall, miRNA expression was numerically higher for horses <6 years old in all studied molecules. For eca-miR-199a-3p (Figure S6A), miRNA expression was significantly higher for horses <6 years old compared with 13–20 years old (FDR p=0.0131). For eca-miR-148a (Figure S6B), no significant differences were found between age classes; however, control samples in the 13–20 years age class appeared to have lower expression than OA samples in that same class. For eca-miR-99b (Figure S6C), miRNA expression was significantly higher in the <6 age class compared with 6–12 (FDR p=0.0021) and 13–20 (FDR p=0.0008). For eca-miR-146a-5p (Figure S6D), miRNA expression was significantly higher in the <6 class compared with 6-12 (FDR p=0.0259), 13-20 (FDR p=0.0116) and >20 (FDR p=0.0255). For eca-miR-423-5p (Figure S6E), no significant differences were found among classes; however, in the 13-20 class the expression in OA samples was generally lower than in Control samples. For eca-miR-23b (Figure S6F), expression was significantly higher for <6 age class compared with 6-12 (FDR p=0.0263), 13-20 (FDR p=0.0014) and >20 (FDR p=0.0354).
3. Discussion
With advances in molecular biomarker research offering the potential for new diagnostic tools, OA research has been shifting its focus from disease management to detection and prevention of early disease [16]. This study investigated the miRNA profile of equine serum and SF in OA and Control groups and identified a panel of 43 miRNAs that were differentially expressed in serum, as well as 23 miRNAs that were differentially expressed in SF samples. Pathway analysis revealed that altered miRNAs were possibly involved in inflammatory responses, cell apoptosis, maturation of chondrocytes and atrophy of skeletal muscle, with a significant association with OA. Additionally, the level of work and age were identified as the main clinical variables influencing miRNA expression in serum and SF, respectively, in horses.
SF is the biofluid which more closely reflects the local processes in the joint, whereas serum provides an overview of systemic events. To compare the molecular patterns between SF and serum, matched samples were collected for each horse in the sequencing cohort. Results showed a wide variety of small RNA molecules in serum and SF samples, which is in line with previous findings [14,17]. The relative abundance of miRNAs was highly variable, but generally higher in serum samples compared with SF samples. This difference may be due to the systemic origin of the miRNAs present in the serum, as opposed to the more local, likely articular origin of the miRNAs contained in SF.
Unsupervised analysis of miRNA revealed some overlap among OA and Control samples. Still, there were 111 miRNAs exclusively present in OA samples, of which 73 were exclusive to serum, 10 exclusive to SF and 28 present in both serum and SF. Differential expression analyses of sequencing data revealed a total of 43 significantly altered miRNAs in serum and 23 in SF, between the OA and Control groups. Of these, all but 18 miRNAs (miR-1291a, miR-1892, miR-2483, miR-30e, miR-342-5p, miR-423-3p, miR-490-3p, miR-598, miR-615-5p, miR-7177b, miR-744, miR-8951, miR-8954, miR-8977, miR-8986b, miR-8992, miR-9048, miR-92b) had been previously linked to OA in some capacity [14,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32].
Bioinformatic analysis of the differentially expressed miRNAs revealed a significant association with inflammatory disease and response. Additionally, target prediction using IPA found over a hundred experimentally validated targets for the tested miRNAs and revealed gene associations of interest to osteochondral biology.
In this study, miRNAs were selected for validation based on differential expression in OA compared with Control, both in serum and in SF. Analysis of miR-199a-3p expression showed a numerical increase in the OA group compared with the Control group (not statistically significant), both in the sequencing cohort and the validation cohort, for serum and for SF. This is in line with the results from an experimental model of equine OA, that reported miR-199b-3p (which has the same mature sequence as eca-miR-199a-3p) to be increased in the SF of OA vs Control joints [33]. miR-199a-3p appears to be implicated in chondrogenic processes, being upregulated during chondrogenesis of human adipose-derived stem cells [30] and downregulated in IL-1β-treated chondrocytes [34]. Additionally, chondrocyte transfection with miR-199a-3p mimics decreased expression of COL2, aggrecan and SOX9 [35]. While miR-199a-3p is a promising candidate as a biomarker of disease, the potential effect of ageing on miR-199a-3p expression should be considered. In this study, miR-199a-3p expression was upregulated in young (<6 years) horses when compared with middle-aged (13–20 years) horses. When looking specifically at the 13–20 age class, miR-199a-3p expression was upregulated in OA compared with Control samples. These findings indicate that age is a cofounding factor for miR-199a-3p expression and should be taken into consideration in future studies.
Expression of eca-miR-148a was increased in OA serum and SF when compared with Control, and RT-qPCR analysis in the sequencing cohorts confirmed these results (albeit not statistically significant, and despite differences not being as marked in the validation cohorts). eca-miR-148a is homologous to hsa-miR-148a-3p, which is thought to have a role in cartilage regeneration and is decreased in OA cartilage compared with healthy cartilage [36]. Overexpression of miR-148a in human chondrocytes resulted in increased ECM deposition and promoted a downregulation of metalloproteinase (MMP) 13 and A disintegrin and metalloproteinase with thrombospondin type 1 motif 5 (ADAMTS5) [36]. Due to these chondroprotective functions, it is possible that the increased expression of miR-148a in SF and serum detected in the present study occurred in response to injury, in an attempt to prevent further tissue alterations. Interestingly, age also appeared to affect miR-148a expression, which was particularly evident for the horses in the SF validation cohort between 13 and 20 years old.
Previous studies found miR-99b-5p to be upregulated in OA and demonstrated that overexpression of this miRNA in chondrocytes increased MMP13 and senescence-related factors, and decreased collagen 2 [37]. While the precise role of miR-99b-5p is not yet known, this molecule is part of an evolutionarily conserved cluster (miR-99b/let-7e/miR-125a) that is highly upregulated during the early stages of osteoclastogenesis and has a direct relationship with Nuclear Factor kappa B [38]. However, eca-miR-99b expression was consistently low in the validation stage of this study, both in serum and SF; these results suggest that eca-miR-99b might not be an appropriate biomarker for equine OA.
In the present study, eca-miR-146a-5p was increased in serum in the OA group when compared with Control in the sequencing cohort; however, these results were not consistent with the validation cohorts. In previous reports, miR-146a was reported to play a protective role in OA [39], evidenced by a reduction in reduced IL-1β and induced tumor necrosis factor α production in human chondrocytes overexpressing miR-146-5p [40]. Remarkably, in the present study, expression of miR-146a-5p was statistically upregulated in the SF of young (<6 years) horses when compared with all other age classes, revealing a strong association between age and miRNA expression. Furthermore, analysis of study groups 13–20 years age class showed a distinction in miRNA expression between OA and Control in this class.
miR-423-5p has been previously identified as a negative regulator of osteoblastogenesis [41,42], and women with bone pathologies display higher serum expression of miR-423-5p than controls [22]. However, a study looking at miRNA expression in cartilage of OA patients used miR-423-5p, a reference gene due to its stability across both OA and Control groups [43]. While this miRNA might contribute to OA pathogenesis, previous findings are contradictory, and further research is needed to elucidate its precise roles. The validation results in this study revealed that miR-423-5p expression in serum was increased in the Control group compared with OA (despite not reaching statistical significance), but was similar among groups in SF. Strikingly, this was one of the few miRNAs whose expression was not increased in younger (<6 years) horses compared with older horses.
Finally, analysis of sequencing data revealed eca-miR-23b expression as decreased in OA compared with Control. This was an unexpected finding, as previous studies have found this miRNA to be upregulated in OA tissues [44,45]. However, validation results for eca-miR-23b were inconsistent with sequencing results, and this miRNA was generally upregulated in OA compared with Control in both cohorts and both sample types. While sequencing is a powerful tool, it is subject to bias, particularly during library amplification [46]; it is possible that this was the case for eca-miR-23b.
Clinical characteristics and lifestyle factors are known to influence different epigenetic mechanisms, including miRNA expression [47]. This study investigated the influence of different clinical variables in the expression of a selected group of miRNAs and found that the level of exercise placed upon horses is significantly associated with variations in miRNA expression in their serum. In accordance with these findings, previous studies have shown that exercise is associated with alterations in circulating miRNAs in horses [48,49,50]. A study investigating the miRNA population of plasma extracellular particles in horses reported increased levels of eca-miR-486-5p during and after an endurance race and decreasing levels of eca-miR-9083 [50]. Interestingly, eca-miR-486-5p was identified in the present study as one of the most stable miRNAs in serum and SF; further studies may help elucidate the precise role of this miRNA.
Age was also found to be significantly associated with miRNA levels in SF in this study. Numerous studies have previously described variations in miRNA patterns with age [51], including in joint tissues [52,53]. This is particularly relevant for OA studies because age is a critical factor in OA development [54]. In the present study, most molecules were upregulated in younger horses (<6 years) when compared with older horses. These findings highlight the importance of accounting for variations in age in OA studies, especially if attempting to validate potential OA biomarkers.
Achieving robustness and reproducibility of molecular biomarkers across different populations, settings, and laboratories is a major challenge. miRNA profiling data is only partially reproducible between different platforms, and even within the same platform variation is common [55]. In this study, it was not possible to validate the sequencing findings at an FDR-adjusted p<0.05, which might have been related with the distinct set of samples use in sequencing and in validation. Horses in the OA group of the sequencing cohort were mostly young and from a racing background, suggesting that OA alterations could have arisen after repetitive microtrauma. Meanwhile, horses in the validation cohorts were generally older and originated from more diverse backgrounds. This suggests that, at least for some of these horses, OA might have developed from mechanisms not associated with repetitive microtrauma. Given that the level and type of exercise may be associated with alterations in miRNA expression in serum, it is possible that the type of work and the associated injuries indirectly influence miRNA regulatory mechanisms and can partially explain the disparity in the results of this study. The fact that all the samples in the Control group of the sequencing cohort were collected from abattoir specimens while almost half of the samples in the OA group were collected from live animals may have also influenced the results. Furthermore, the SF samples in this study were collected from different joints, and it is currently unclear whether different joints inherently exhibit different miRNA profiles, which could increase miRNA variability and affect the validation results.
An additional limitation of this study was the small set of molecules selected for validation, which was restricted by finances. Restricting the selection to miRNAs that were differentially expressed in both serum and SF may have limited the findings, as some molecules present exclusively in one sample type could still possess valuable biomarker properties. Future studies investigating the miRNA and the overall sncRNA profile of equine osteoarthritis would benefit from analyzing larger cohorts of patients with similar clinical characteristics, particularly age and level of work. Furthermore, investigating information from a panel of multiple biomarkers as opposed to a single biomarker is more likely to achieve better performance [56].
4. Methods
4.1. Exploration Stage
4.1.1. Sample Collection (Sequencing Cohort)
Serum and SF samples for the sequencing cohort consisted of either post-mortem collections or excess clinical samples. Post-mortem samples were collected from racehorses slaughtered at an abattoir and processed by a veterinary surgeon, as previously described [15]. Briefly, serum samples and SF samples (from the left middle carpal joint) were collected from the same horses. Samples were centrifuged at 1800 g for 20 min to remove cellular debris, snap frozen in liquid nitrogen and stored at -70°C until further processing. Post-mortem collection of abattoir samples was performed at a Swedish institution in accordance with local legislation.
Clinical samples consisted of excess serum and SF obtained by veterinary surgeons from horses during lameness examinations in the UK. Serum samples were obtained from blood samples collected in plain tubes that were allowed to clot and then centrifuged at 2000 g for 20 min at 4°C. The supernatant was frozen and stored at -80°C until further processing. SF was sterilely obtained from different joints, depending on the joint being assessed by the clinician. SF samples were processed as previously described [14]. Collection of excess clinical samples was approved by the University of Liverpool’s Veterinary Research Ethics Committee (VREC660a).
4.1.2. Group Categorization (Sequencing Cohort)
Samples were assigned to two groups (Control and OA), as described in the next paragraphs.
For samples that were collected at post-mortem, a previously described approach was used to determine the presence of OA [15]. In brief, the left middle carpal joint was opened and dissected, and the articular cartilage of the proximal surface of the carpal bones in the proximal row was inspected macroscopically and photographed. The area of all lesions (including superficial fraying, erosions or cleft formation) was measured using a digitized image system (Kontron System 100, Kontron, Germany). Horses with no evidence of macroscopic lesions were included in the Control group. Horses with macroscopic lesions were included in the OA group, and the severity of disease was classified according to the size of lesions: if a cartilage area smaller than 40 mm2 was affected, OA was classified as mild; if lesions extended over an area between 40 mm2 and 230 mm2, OA was considered moderate; and for horses exhibiting lesions on an area exciding 400 mm2, OA was considered severe [15].
For excess clinical samples, group characterization was determined based on the clinical diagnosis made by the attending veterinary surgeon. Horses diagnosed with OA were included in the OA group, and horses that were not diagnosed with OA and that also did not present any signs of joint infection were included in the Control group. Diagnosis of OA was made through a combination of medical history, presence of clinical signs such as lameness, joint effusion or pain on joint flexion, positive response to intra-articular anesthesia, presence of osteoarthritic changes on imaging exams such as radiography, and the exclusion of differential diagnoses. The severity of OA was not classified.
4.1.3. Sample Pre-Processing and RNA Isolation
Briefly, SF samples were thawed, treated with hyaluronidase (H3884, Sigma-Aldrich, Gillingham, UK) and filtered through a Costar® Spin-X® polypropylene microcentrifuge tube filter with 0.22 μm pore cellulose acetate membrane (Corning, Flintshire, UK). Serum was thawed and centrifuged to remove cryoprecipitates. Total RNA was extracted from a 200 μl sample using the miRNeasy Serum/Plasma Advanced Kit (Qiagen, Manchester, UK), with the addition of glycogen (ThermoFisher Scientific, Paisley, UK). No RNA spike-ins were added at this stage. RNA was eluted in 18 μl of RNase-free water.
4.1.4. Library Preparation and Sequencing
Small RNA library preparation was undertaken with the CleanTag® Small RNA Library Preparation kit (TriLink Biotechnologies, California, USA) according to manufacturer’s instructions, using 2 μl of total RNA as input. Adapter-ligated libraries from SF were amplified with 26 PCR cycles and libraries from serum with 23 PCR cycles, using barcoded Illumina reverse primers in combination with the Illumina forward primer (Illumina, California, USA). Library quality control was performed using DNA1000 Chip (Agilent, California, USA). An equimolar pool consisting of all sequencing libraries was prepared and sequenced on an Illumina Novaseq with 75 base pairs (bp) paired end reads.
4.1.5. Data Analysis
Demographics were analyzed descriptively and compared between groups (OA vs Control). Data was tested for normality of distribution and statistical tests were selected for the specific data types, as appropriate; all tests were performed using GraphPad Prism version 8.0 for Windows, with significance set at a p-value of 0.05.
4.1.5.1. Small RNA Sequencing Data Processing and Analysis
Sequencing data were processed and analyzed by TAmiRNA (TAmiRNA GmbH, Vienna, Austria) following the miND pipeline, adapted to equine samples [57]. Briefly, after demultiplexing, the overall quality of the sequencing data were evaluated automatically and manually with fastQC v0.11.8 and multiQC v1.7 [58]. For miRNA analysis, reads from all quality samples were adapter trimmed, and quality filtered using cutadapt v2.3 [59] and filtered for a minimum length of 17 nucleotides. Mapping steps were performed with bowtie v1.2.2 [60] and miRDeep2 v2.0.1.2 [61]. Reads were mapped first against the genomic reference EquCab.3.0 provided by Ensembl [62] allowing for two mismatches and subsequently miRBase v22.1 [63], filtered for equine (eca) miRNAs only, allowing for one mismatch. For a general RNA composition overview, non-miRNA mapped reads were mapped against RNAcentral [64] and then assigned to various RNA species of interest. Statistical analyses of pre-processed sequencing data were done with R v3.6 and the packages pheatmap v1.0.12, pcaMethods v1.78 and genefilter v1.68, to generate heatmaps and principal component analysis (PCA) plots. Differential expression analysis with edgeR v3.28 [65] used the quasi-likelihood negative binomial generalized log-linear model functions provided by the package. The independent filtering method of DESeq2 [66] was adapted for use with edgeR to remove low abundant miRNAs and thus optimize FDR correction.
4.1.6. Pathway Analysis
Prediction of functional targets for the differentially expressed miRNAs was carried out using IPA software (Qiagen, Manchester, UK). For this, equine miRNAs that were differentially expressed in serum and in SF were matched to their human equivalents, and the corresponding miRbase IDs were input together into IPA as identifiers. Data were analyzed using the “Target Prediction” function and results were robustly filtered for experimentally observed targets in chondrocytes and osteoblasts. The list of predicted mRNA targets was combined with the list of differentially expressed miRNAs along with their expression values (logFC) and significance levels, and input back into IPA. Data were analyzed using the “Core Analysis” function, which calculates the p-value of overlap between the molecules in the dataset with the disease and functions contained in the Ingenuity Knowledge Base using a right-tailed Fisher’s exact test. Results were filtered for experimentally observed associations only and used to algorithmically generate a network of canonical pathways, biological functions, diseases and network-eligible molecules based on their connectivity.
4.1.6. Selection of Differentially Expressed miRNAs for Validation
All differentially expressed miRNAs were reviewed, and a limited group was selected for validation through RT-qPCR. miRNAs were selected if they were differentially expressed (FDR<0.05 or p<0.05; Table 2) in both SF and serum with the same directionality in change, with no cut-off for logFC. Of these, four miRNAs that had been previously identified in OA-related studies were selected for validation. Two additional miRNAs (eca-miR-99b and eca-miR-146a) were also selected for further analyses. While these two molecules did not meet the previously mentioned criteria, a preliminary analysis in which data were mapped against the human genome (results not published) found hsa-miR-99b-5p and hsa-miR-146a-5p to be differentially expressed in serum and SF, and so these molecules were selected for further validation.
4.2. Validation Stage
Validation was carried out in two separate cohorts: the sequencing cohort (same samples used in the exploration stage) and an independent cohort (validation cohort). Collection of samples and group categorization for the sequencing cohort are described in the previous sections. Collection of samples and group categorization for the validation cohort are described in the following sections.
4.2.1. Sample Collection (Validation Cohort)
For the validation cohort, serum samples consisted of excess clinical samples obtained by veterinary surgeons in one equine clinic in the UK prior to routine dental examination, and two other centers either during lameness examination or at post-mortem.
Serum samples were obtained from blood samples collected in plain tubes that were allowed to clot and then centrifuged at 2000g for 20 min at 4°C. The supernatant was frozen within 24 hours of collection and stored at -80°C until further processing.
SF was collected at post-mortem from the metacarpophalangeal joints of horses from an abattoir in the UK as a by-product of the agricultural industry, or from horses donated to research. Briefly, the joints were aseptically dissected to allow visual inspection of the metacarpus, the proximal phalanx, and the sesamoids. All joints were photographed, and SF was aseptically collected from the open joints, centrifuged to remove cells and debris, and stored at -80°C.
Ethical collection of excess clinical samples was approved by the University of Liverpool’s Veterinary Research Ethics Committee (VREC660a). Hong Kong Jockey Club samples were collected under the regulations of the club with owner consent. For abattoir samples, ethical review and approval were waived due to samples being obtained as a by-product of the agricultural industry – the Animal (Scientific procedures) Act 1986, Schedule 2, does not define collection from these sources as scientific procedures and ethical approval was therefore not required.
4.2.2. Group Characterization (Validation Cohort)
Samples were assigned to one of two groups: Control and OA. For serum excess clinical samples, group characterization was based on owner reported clinical history: if owners reported their horse to have a diagnosis of OA made by a veterinary surgeon, the horses were included in the OA group; if owners reported no history of OA, horses were included in the Control group; cases in which owners were unsure about an OA diagnosis but reported a history of joint corticosteroid injections or chronic intermittent lameness of unknown origin were also included in the OA group. Samples obtained from horses presenting for lameness examination and that were diagnosed with OA by the attending veterinary surgeon were included in the OA group; horses that were not diagnosed with OA and had no previous history of OA were included in the Control group.
Additional information collected for each horse included age, sex, BCS, breed, current occupation, current level of work, presence of shoes, and affected joints. Level of work was assessed on a 0–3 scale based on owner reports (0, out in the field, no work; 1, light work; 2, mild work; and 3, intense work). BCS was determined by a trained veterinary surgeon based on a defined 9-point scale [67].
For SF samples, joints were photographed and scored at post-mortem by two independent researchers using the Osteoarthritis Research Society International (OARSI) equine macroscopic grading system [68], and the scores were averaged for each horse. Horses were assigned to groups based on their macroscopic scoring: horses with gross scores of 0–1 were included in the Control group, and gross scores ≥3 were included in the OA group. Additional information collected for each horse included age, sex, year of sample collection and place of sample collection (abattoir or hospital/clinic). Histologic/microscopic scoring was carried out on wedge sections of articular cartilage/subchondral bone from the palmar aspect of one of the metacarpal condyles, that were processed and stained with hematoxylin and eosin or Safranin O and graded using OARSI microscopic grading system [69].
4.2.3. Sample Size Calculation
To estimate the required sample size for miRNA validation, a sample size calculation was performed in R using the package pwr v1.3-0. miRNA sequencing data were input as reference for data variation and expected size effect, assuming a similar behavior of qPCR data. Significance level was set at 0.05 and statistical power at 95%. One sample t-tests were performed individually for each miRNA in the selected panel, for both SF and serum. The final sample size corresponded to the test that required the highest n number (i.e., if one miRNA required 20 samples for validation and the following miRNA required 30 samples, the final sample size corresponded to 30 samples).
4.2.4. RNA Extraction, cDNA Synthesis and RT-qPCR
Briefly, serum was centrifuged, and SF was treated hyaluronidase, filtered, and diluted 1:1 in RNase-free water. Total RNA was extracted from 200 μl of diluted SF or 200 μl of serum using miRNeasy Serum/Plasma Advanced Kit, with the addition of RNA spike-ins (RNA Spike-in Kit, for RT, Qiagen, Manchester, UK) and glycogen. cDNA synthesis was performed using miRCURY LNA RT Kit (Qiagen, Manchester, UK) using 4 μl of total RNA as input. qPCR was performed using miRCURY® LNA® miRNA SYBR® Green PCR (Qiagen, Manchester, UK) using 3 μl of diluted cDNA (1:30) as input on a LightCycler® 96 (Roche Life Science, Penzberg, Germany). Bench-validated, equine compatible miRNA assays were obtained from Qiagen (Table S1).
4.2.5. RT-qPCR Data Analysis
Selection of candidate reference genes
Five miRNAs were selected to be tested as reference genes: two were manually selected, two were selected using NormFinder [70,71], and one was a synthetic RNA spike-in (exogenous control). Manual selection was based on miRNA stability across all study samples, considering only molecules that were not differentially expressed (p>0.05) with a logFC close to 0. For selection using NormFinder, miRNAs were first filtered based on a minimum of 15 RPM per sample, and the software was used for SF and serum samples separately. The two most stable genes selected by NormFinder for each sample type were tested as reference genes.
Normalization strategies for RT-qPCR and comparison of performance
Four different normalization methods were compared to find the most robust strategy for analysis of miRNA data. These were normalizing data to a single most stable reference gene that was manually selected; normalizing data to a single most stable reference gene or combination of genes as ranked by NormFinder algorithm; normalizing data to the geometric mean of all tested reference genes; and normalizing data to an exogenous oligonucleotide (spike-in). To assess the performance of these normalization strategies, data for all tested miRNAs (including candidate reference miRNAs and differentially expressed miRNAs) were normalized using the 2-ΔCq method [72] and compared with the log transformed raw (not normalized) Cq data (2-rawCq). The coefficient of variation (CV) was calculated for each individual miRNA after normalization, and CVs for all miRNAs were averaged for each of the normalization techniques. Average CV was used as a normalization performance measure, with lower CVs representing a better removal of experimentally induced noise. The cumulative distribution of the individual CV values was plotted for both raw and normalized data. Samples for which Cq>39 were removed from further analyses.
Relative gene expression
Following the determination of the best normalization strategy, the 2-ΔCq method was used for analysis of relative gene expression [72]. Statistical analyses were undertaken with GraphPad Prism version 8.0.1 for Windows. Data was tested for normality of distribution, and the corresponding parametric or non-parametric tests were performed. Statistical significance was set at an FDR-adjusted p-value of 0.05.
Analysis of clinical variables
The influence of clinical variables on miRNA expression in serum and SF was investigated through analysis of PCs using linear models or one-way ANOVAs, depending on data type. Briefly, miRNA data were normalized and scaled, and PCs were calculate using the prcomp function in R. For SF miRNA data, singular value decomposition was applied prior to PC calculation. Ordinal variables such as level of work, BCS, gross score and microscopic score were assessed as numerical values. Age, level of work, BCS, gross score and microscopic score were assessed against each PC using linear models; sex, group and presence of shoes were numerically encoded and assessed against each PC using One-way ANOVA tests. All p-values were adjusted for multiple comparisons using FDR (Benjamini-Hochberg) correction. Results were presented as a heatmap, generated with the pheatmap function in R.
Where needed, additional PCA plots and/or boxplots were generated to further explore the relationship between the clinical variables and gene expression. For SF, one-way ANOVAs followed by Tukey’s multiple comparison tests were used to compare miRNA expression between age classes.
5. Conclusions
OA is a heterogenous disease where a complex interplay of internal and external variables contributes to altered sncRNA expression in a range of sample types. In this study, we identified a panel of miRNAs that were differentially expressed in serum and SF samples, two of which (miR-199a-3p and miR-148a) warrant further investigation. Further characterization of these molecular changes and their potential targets will contribute to a better understanding of the biological processes they regulate and could provide novel insights into the mechanisms of early OA. While clinical application is still limited, this study is one of the first steps towards the use of sncRNAs as biomarkers of equine OA.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, C.I.G.D.C., S.T., P.D.C. and M.J.P.; methodology, C.I.G.D.C., S.T., E.S., L.M.R-M., P.M., M.H., and M.J.P.; formal analysis, C.I.G.D.C. and M.H. All authors were involved in the writing, reviewing, and editing, and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Horse Trust, grant number G5018. This work was supported by the Medical Research Council (MRC) and Versus Arthritis as part of the Medical Research Council Versus Arthritis Centre for Integrated Research into Musculoskeletal Ageing (CIMA) (MR/P020941/1). The MRC Versus Arthritis Centre for Integrated Research into Musculoskeletal Ageing is a collaboration between the Universities of Liverpool, Sheffield, and Newcastle. Mandy Peffers was funded by Wellcome Trust Clinical Intermediate Fellowship 107471/Z/15/Z.
Institutional Review Board Statement
For abattoir samples, ethical review and approval were waived due to samples being obtained as a by-product of the agricultural industry—the Animal (Scientific procedures) Act 1986, Schedule 2, does not define collection from these sources as scientific procedures and ethical approval was therefore not required. Collection of excess clinical samples was approved by the University of Liverpool’s Veterinary Research Ethics Committee (VREC660ab) approved 19.08.2020. Hong Kong Jockey Club samples were collected under the regulations of the Hong Kong club with owner consent. Clinical samples from Sweden are covered by C206/10, Uppsala djurförsöksetiska nämnd approved 2010.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data has been submitted to NCBI´s gene expression omnibus (GEO).
Acknowledgments
The authors would like to thank James Anderson, Andrew Peffers, and the staff at the abattoir for assistance in sample collection and processing. The authors would also like to thank Daniel Green for his assistance with data analysis.
Conflicts of Interest
Matthias Hackl is an employee of TAmiRNA, GmBH. The remaining authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| ADAMTS5 | A disintegrin and metalloproteinase with thrombospondin type 1 motif 5 |
| BCS | Body condition score |
| cDNA | Complementary DNA |
| CPM | Counts per million |
| Cq | Quantification cycle |
| CS | Clinical sample |
| CV | Coefficient of variation |
| ECM | Extracellular matrix |
| FC | Fold change |
| FDR | False discovery rate |
| IL | interleukin |
| IPA | Ingenuity Pathway Analysis |
| lncRNA | Long non-coding RNA |
| Max | Maximum |
| Min | Minimum |
| MMP | Metalloproteinase |
| mRNA | Messenger RNA |
| miRNA | microRNA |
| OA | Osteoarthritis |
| OARSI | Osteoarthritis Research Society International |
| PC | Principal component |
| PCA | Principal component analysis |
| piRNA | Piwi-interfering RNA |
| PM | Post-mortem |
| QC | Quality control |
| qPCR | Quantitative polymerase chain reaction |
| RPM | Reads per million |
| rRNA | Ribosomal RNA |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| scRNA | Small conditional RNA |
| SD | Standard deviation |
| SF | Synovial fluid |
| siRNA | Small interfering RNA |
| snRNA | Small nuclear RNA |
| snoRNA | Small nucleolar RNA |
| tRNA | Transfer RNA |
References
- Morris, K. V; Mattick, J.S. The Rise of Regulatory RNA. Nat Rev Genet 2014, 15, 423–437. [Google Scholar] [CrossRef]
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. In Non-coding RNAs in Colorectal Cancer; Slaby, O., Calin, G.A., Eds.; Springer International Publishing: Cham, 2016; ISBN 978-3-319-42059-2. [Google Scholar]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An Overview of MicroRNAs: Biology, Functions, Therapeutics, and Analysis Methods. J Cell Physiol 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of MiRNA-Mediated Gene Regulation from Common Downregulation to MRNA-Specific Upregulation. Int J Genomics 2014, 2014, 970607. [Google Scholar] [CrossRef]
- Blondal, T.; Jensby Nielsen, S.; Baker, A.; Andreasen, D.; Mouritzen, P.; Wrang Teilum, M.; Dahlsveen, I.K. Assessing Sample and MiRNA Profile Quality in Serum and Plasma or Other Biofluids. Methods 2013, 59, S1–S6. [Google Scholar] [CrossRef]
- Ireland, J.L.; Clegg, P.D.; McGowan, C.M.; McKane S A; Chandler, K. J.; Pinchbeck, G.L. Disease Prevalence in Geriatric Horses in the United Kingdom: Veterinary Clinical Assessment of 200 Cases. Equine Vet J 2012, 44, 101–106. [Google Scholar] [CrossRef]
- van Weeren, P.R.; de Grauw, J.C. Pain in Osteoarthritis. Veterinary Clinics of North America: Equine Practice 2010, 26, 619–642. [Google Scholar] [CrossRef]
- Rocha, F.A.C.; Ali, S.A. Soluble Biomarkers in Osteoarthritis in 2022: Year in Review. Osteoarthritis Cartilage 2023, 31, 167–176. [Google Scholar] [CrossRef]
- Stanciugelu, S.I.; Homorogan, C.; Selaru, C.; Patrascu, J.M.; Patrascu, J.M.; Stoica, R.; Nitusca, D.; Marian, C. Osteoarthritis and MicroRNAs: Do They Provide Novel Insights into the Pathophysiology of This Degenerative Disorder? Life 2022, 12. [Google Scholar] [CrossRef]
- Beyer, C.; Zampetaki, A.; Lin, N.-Y.; Kleyer, A.; Perricone, C.; Iagnocco, A.; Distler, A.; Langley, S.R.; Gelse, K.; Sesselmann, S.; et al. Signature of Circulating MicroRNAs in Osteoarthritis. Ann Rheum Dis 2015, 74, e18. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Bapat, S.; Hubbard, D.; Hunter, M.; Kolhe, R.; Fulzele, S. Advances in Molecular Biomarker for Early Diagnosis of Osteoarthritis. 2019; 10, 111–119. [Google Scholar] [CrossRef]
- Ntoumou, E.; Tzetis, M.; Braoudaki, M.; Lambrou, G.; Poulou, M.; Malizos, K.; Stefanou, N.; Anastasopoulou, L.; Tsezou, A. Serum MicroRNA Array Analysis Identifies MiR-140-3p, MiR-33b-3p and MiR-671-3p as Potential Osteoarthritis Biomarkers Involved in Metabolic Processes. Clin Epigenetics 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Tavallaee, G.; Tokar, T.; Nakamura, A.; Sundararajan, K.; Weston, A.; Sharma, A.; Mahomed, N.N.; Gandhi, R.; Jurisica, I.; et al. Identification of Synovial Fluid MicroRNA Signature in Knee Osteoarthritis: Differentiating Early- and Late-Stage Knee Osteoarthritis. Osteoarthritis Cartilage 2016, 24, 1577–1586. [Google Scholar] [CrossRef]
- Castanheira, C.; Balaskas, P.; Falls, C.; Ashraf-Kharaz, Y.; Clegg, P.; Burke, K.; Fang, Y.; Dyer, P.; Welting, T.J.M.; Peffers, M.J. Equine Synovial Fluid Small Non-Coding RNA Signatures in Early Osteoarthritis. BMC Vet Res 2021, 17, 26. [Google Scholar] [CrossRef]
- Skiöldebrand, E.; Lorenzo, P.; Zunino, L.; Rucklidge, G.J.; Sandgren, B.; Carlsten, J.; Ekman, S. Concentration of Collagen, Aggrecan and Cartilage Oligomeric Matrix Protein (COMP) in Synovial Fluid from Equine Middle Carpal Joints. Equine Vet J 2001, 33, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. The Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Baker, M.E.; Lee, S.; Clinton, M.; Hackl, M.; Castanheira, C.; Peffers, M.J.; Taylor, S.E. Investigation of MicroRNA Biomarkers in Equine Distal Interphalangeal Joint Osteoarthritis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Cao, Z.; Liu, W.; Qu, X.; Bi, H.; Sun, X.; Yu, Q.; Cheng, G. MiR-296-5p Inhibits IL-1β-Induced Apoptosis and Cartilage Degradation in Human Chondrocytes by Directly Targeting TGF-Β1/CTGF/P38MAPK Pathway. Cell Cycle 2020, 19, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, C.; James, V.; Taylor, S.; Skiöldebrand, E.; Clegg, P.D.; Peffers, M.J. Synovial Fluid and Serum Small Non-Coding RNA Signatures in Equine Osteoarthritis. Osteoarthritis Cartilage 2021, 29, S162. [Google Scholar] [CrossRef]
- Lao, T.D.; Le, T.A.H. Data Integration Reveals the Potential Biomarkers of Circulating MicroRNAs in Osteoarthritis. Diagnostics 2021, 11. [Google Scholar] [CrossRef]
- Le, L.T.T.; Swingler, T.E.; Clark, I.M. Review: The Role of MicroRNAs in Osteoarthritis and Chondrogenesis. Arthritis Rheum 2013, 65, 1963–1974. [Google Scholar] [CrossRef]
- Pertusa, C.; Tarín, J.J.; Cano, A.; García-Pérez, M.Á.; Mifsut, D. Serum MicroRNAs in Osteoporotic Fracture and Osteoarthritis: A Genetic and Functional Study. Sci Rep 2021, 11, 19372. [Google Scholar] [CrossRef] [PubMed]
- Sondag, G.R.; Haqqi, T.M. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr Rheumatol Rep 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Wu, Y.; Li, X.; Jia, Y. MiR-142-5p Protects against Osteoarthritis through Competing with LncRNA XIST. J Gene Med 2020, 22, e3158. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kang, S.; Pei, S.; Sang, C.; Huang, Y. MiR93-5p Inhibits Chondrocyte Apoptosis in Osteoarthritis by Targeting LncRNA CASC2. BMC Musculoskelet Disord 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Tavallaee, G.; Rockel, J.S.; Lively, S.; Kapoor, M. MicroRNAs in Synovial Pathology Associated With Osteoarthritis. Front Med (Lausanne), 2020; Volume 7-2020. [Google Scholar]
- Tu, J.; Han, D.; Fang, Y.; Jiang, H.; Tan, X.; Xu, Z.; Wang, X.; Hong, W.; Li, T.; Wei, W. MicroRNA-10b Promotes Arthritis Development by Disrupting CD4+T Cell Subtypes. Mol Ther Nucleic Acids 2022, 27, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.; Barter, M.J.; Elliott, H.R.; McGillivray, C.M.; Birch, M.A.; Clark, I.M.; Young, D.A. MiR-324-5p Is up Regulated in End-Stage Osteoarthritis and Regulates Indian Hedgehog Signalling by Differing Mechanisms in Human and Mouse. Matrix Biology 2019, 77, 87–100. [Google Scholar] [CrossRef]
- Yu, X.-M.; Meng, H.-Y.; Yuan, X.-L.; Wang, Y.; Guo, Q.-Y.; Peng, J.; Wang, A.-Y.; Lu, S.-B. MicroRNAs’ Involvement in Osteoarthritis and the Prospects for Treatments. Evidence-Based Complementary and Alternative Medicine 2015, 2015, 236179. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, P.; Hu, W.; Yin, W.; Guo, F.; Chen, A.; Huang, H. Downregulated MicroRNA-340-5p Promotes Proliferation and Inhibits Apoptosis of Chondrocytes in Osteoarthritis Mice through Inhibiting the Extracellular Signal-Regulated Kinase Signaling Pathway by Negatively Targeting the FMOD Gene. J Cell Physiol 2019, 234, 927–939. [Google Scholar] [CrossRef]
- Zhou, M.; Zhai, C.; Shen, K.; Liu, G.; Liu, L.; He, J.; Chen, J.; Xu, Y. MiR-1 Inhibits the Ferroptosis of Chondrocyte by Targeting CX43 and Alleviates Osteoarthritis Progression. J Immunol Res 2023, 2023, 2061071. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, Y.; Wang, C.; Zhang, X.; He, D. CircGCN1L1 Promotes Synoviocyte Proliferation and Chondrocyte Apoptosis by Targeting MiR-330-3p and TNF-α in TMJ Osteoarthritis. Cell Death Dis 2020, 11, 284. [Google Scholar] [CrossRef]
- Walters, M.; Skovgaard, K.; Heegaard, P.M.H.; Fang, Y.; Kharaz, Y.A.; Bundgaard, L.; Skovgaard, L.T.; Jensen, H.E.; Andersen, P.H.; Peffers, M.J.; et al. Identification and Characterisation of Temporal Abundance of MicroRNAs in Synovial Fluid from an Experimental Equine Model of Osteoarthritis. Equine Vet J 2025, 57, 1138–1150. [Google Scholar] [CrossRef]
- Rasheed, Z.; Rasheed, N.; Al-Shobaili, H.A. Epigallocatechin-3-O-Gallate up-Regulates MicroRNA-199a-3p Expression by down-Regulating the Expression of Cyclooxygenase-2 in Stimulated Human Osteoarthritis Chondrocytes. J Cell Mol Med 2016, 20, 2241–2248. [Google Scholar] [CrossRef]
- Ukai, T.; Sato, M.; Akutsu, H.; Umezawa, A.; Mochida, J. MicroRNA-199a-3p, MicroRNA-193b, and MicroRNA-320c Are Correlated to Aging and Regulate Human Cartilage Metabolism. Journal of Orthopaedic Research 2012, 30, 1915–1922. [Google Scholar] [CrossRef]
- Vonk, L.A.; Kragten, A.H.M.; Dhert, W.J.A.; Saris, D.B.F.; Creemers, L.B. Overexpression of Hsa-MiR-148a Promotes Cartilage Production and Inhibits Cartilage Degradation by Osteoarthritic Chondrocytes. Osteoarthritis Cartilage 2014, 22, 145–153. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, L.; Pan, J.; Luo, B.; Zeng, H.; Shao, Y.; Zhang, H.; Guan, H.; Guo, D.; Zeng, C.; et al. MFG-E8 Regulated by MiR-99b-5p Protects against Osteoarthritis by Targeting Chondrocyte Senescence and Macrophage Reprogramming via the NF-ΚB Pathway. Cell Death Dis 2021, 12, 533. [Google Scholar] [CrossRef] [PubMed]
- de la Rica, L.; García-Gómez, A.; Comet, N.R.; Rodríguez-Ubreva, J.; Ciudad, L.; Vento-Tormo, R.; Company, C.; Álvarez-Errico, D.; García, M.; Gómez-Vaquero, C.; et al. NF-ΚB-Direct Activation of MicroRNAs with Repressive Effects on Monocyte-Specific Genes Is Critical for Osteoclast Differentiation. Genome Biol 2015, 16, 2. [Google Scholar] [CrossRef]
- Liu, J.-N.; Lu, S.; Fu, C.-M. MiR-146a Expression Profiles in Osteoarthritis in Different Tissue Sources: A Meta-Analysis of Observational Studies. J Orthop Surg Res 2022, 17, 148. [Google Scholar] [CrossRef]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M. V; Needham, M.R.C.; Read, S.J.; Newham, P. The Identification of Differentially Expressed MicroRNA in Osteoarthritic Tissue That Modulate the Production of TNF-α and MMP13. Osteoarthritis Cartilage 2009, 17, 464–472. [Google Scholar] [CrossRef]
- Chang, C.-C.; Venø, M.T.; Chen, L.; Ditzel, N.; Le, D.Q.S.; Dillschneider, P.; Kassem, M.; Kjems, J. Global MicroRNA Profiling in Human Bone Marrow–Skeletal Stromal or Mesenchymal–Stem Cells Identified Candidates for Bone Regeneration. Molecular Therapy 2018, 26, 593–605. [Google Scholar] [CrossRef]
- Xu, J.-F.; Zhang, S.-J.; Zhao, C.; Qiu, B.-S.; Gu, H.-F.; Hong, J.-F.; Cao, L.; Chen, Y.; Xia, B.; Bi, Q.; et al. Altered MicroRNA Expression Profile in Synovial Fluid from Patients with Knee Osteoarthritis with Treatment of Hyaluronic Acid. Mol Diagn Ther 2015, 19, 299–308. [Google Scholar] [CrossRef]
- Kopańska, M.; Szala, D.; Czech, J.; Gabło, N.; Gargasz, K.; Trzeciak, M.; Zawlik, I.; Snela, S. MiRNA Expression in the Cartilage of Patients with Osteoarthritis. J Orthop Surg Res 2017, 12, 51. [Google Scholar] [CrossRef]
- Guo, Y.; Min, Z.; Jiang, C.; Wang, W.; Yan, J.; Xu, P.; Xu, K.; Xu, J.; Sun, M.; Zhao, Y.; et al. Downregulation of HS6ST2 by MiR-23b-3p Enhances Matrix Degradation through P38 MAPK Pathway in Osteoarthritis. Cell Death Dis 2018, 9, 699. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, Y.; Cai, P.; Fu, W.; Wang, J.; Wei, Q.; Li, X. Downregulation of MicroRNA-23b-3p Alleviates IL-1β-Induced Injury in Chondrogenic CHON-001 Cells. Drug Des Devel Ther 2019, Volume 13, 2503–2512. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and Minimizing PCR Amplification Bias in Illumina Sequencing Libraries. Genome Biol 2011, 12, R18. [Google Scholar] [CrossRef]
- Becker, N.; Lockwood, C.M. Pre-Analytical Variables in MiRNA Analysis. Clin Biochem 2013, 46, 861–868. [Google Scholar] [CrossRef]
- Cappelli, K.; Mecocci, S.; Capomaccio, S.; Beccati, F.; Palumbo, A.R.; Tognoloni, A.; Pepe, M.; Chiaradia, E. Circulating Transcriptional Profile Modulation in Response to Metabolic Unbalance Due to Long-Term Exercise in Equine Athletes: A Pilot Study. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Mandal, S.; Denham, M.M.; Spencer, S.J.; Denham, J. Exercise Regulates Shelterin Genes and MicroRNAs Implicated in Ageing in Thoroughbred Horses. Pflugers Arch 2022, 474, 1159–1169. [Google Scholar] [CrossRef]
- de Oliveira Jr, G.P.; Porto, W.F.; Palu, C.C.; Pereira, L.M.; Reis, A.M.M.; Marçola, T.G.; Teixeira-Neto, A.R.; Franco, O.L.; Pereira, R.W. Effects of Endurance Racing on Horse Plasma Extracellular Particle MiRNA. Equine Vet J 2021, 53, 618–627. [Google Scholar] [CrossRef]
- Hooten, N.N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. MicroRNA Expression Patterns Reveal Differential Expression of Target Genes with Age. PLoS One 2010, 5, 1–13. [Google Scholar] [CrossRef]
- Balaskas, P.; Green, J.A.; Haqqi, T.M.; Dyer, P.; Kharaz, Y.A.; Fang, Y.; Liu, X.; Welting, T.J.M.; Peffers, M.J. Small Non-Coding RNAome of Ageing Chondrocytes. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, C.I.G.D.; Anderson, J.R.; Fang, Y.; Milner, P.I.; Goljanek-Whysall, K.; House, L.; Clegg, P.D.; Peffers, M.J. Mouse MicroRNA Signatures in Joint Ageing and Post-Traumatic Osteoarthritis. Osteoarthr Cartil Open 2021, 3, 100186. [Google Scholar] [CrossRef]
- Loeser, R.F. The Role of Aging in the Development of Osteoarthritis. Trans Am Clin Climatol Assoc 2017, 128, 44–54. [Google Scholar]
- Chugh, P.; Dittmer, D.P. Potential Pitfalls in MicroRNA Profiling. WIREs RNA 2012, 3, 601–616. [Google Scholar] [CrossRef]
- Ou, F.-S.; Michiels, S.; Shyr, Y.; Adjei, A.A.; Oberg, A.L. Biomarker Discovery and Validation: Statistical Considerations. Journal of Thoracic Oncology 2021, 16, 537–545. [Google Scholar] [CrossRef]
- Diendorfer, A.; Khamina, K.; Pultar, M.; Hackl, M. MiND (MiRNA NGS Discovery Pipeline): A Small RNA-Seq Analysis Pipeline and Report Generator for MicroRNA Biomarker Discovery Studies. F1000Res 2022, 11, 233. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. MiRDeep2 Accurately Identifies Known and Hundreds of Novel MicroRNA Genes in Seven Animal Clades. Nucleic Acids Res 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. The MicroRNA Registry. Nucleic Acids Res 2004, 32, D109–D111. [Google Scholar] [CrossRef]
- The RNAcentral Consortium RNAcentral: A Hub of Information for Non-Coding RNA Sequences. Nucleic Acids Res 2019, 47, D221–D229. [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Henneke, D.R.; Potter, G.D.; Kreider, J.L.; Yeates, B.F. Relationship between Condition Score, Physical Measurements and Body Fat Percentage in Mares. Equine Vet J 1983, 15, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Kawcak, C.E.; Frisbie, D.D.; Werpy, N.M.; Park, R.D.; McIlwraith, C.W. Effects of Exercise vs Experimental Osteoarthritis on Imaging Outcomes. Osteoarthritis Cartilage 2008, 16, 1519–1525. [Google Scholar] [CrossRef]
- McIlwraith, C.W.; Frisbie, D.D.; Kawcak, C.E.; Fuller, C.J.; Hurtig, M.; Cruz, A. The OARSI Histopathology Initiative – Recommendations for Histological Assessments of Osteoarthritis in the Horse. Osteoarthritis Cartilage 2010, 18, S93–S105. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Tonge, D.P.; Gant, T.W. Evidence Based Housekeeping Gene Selection for MicroRNA-Sequencing (MiRNA-Seq) Studies. Toxicol Res (Camb) 2013, 2, 328–334. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
Figure 1.
Overview of methodology.
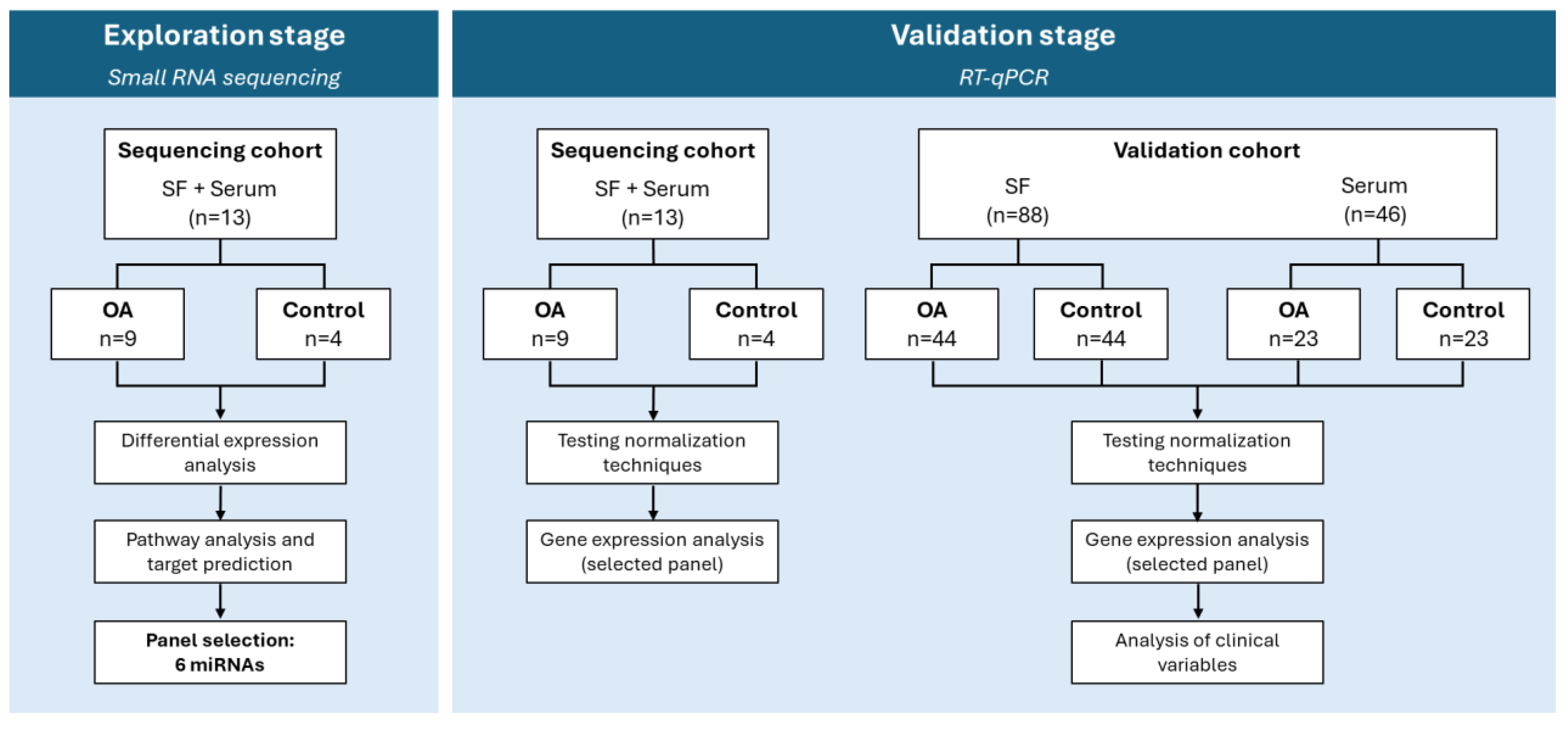
Figure 2.
Absolute reads composition of samples in the sequencing cohort. Bar colors represent different RNA types, and labels with lower opacity correspond to RNA types that were not identified in this dataset. Each bar represents a different sample. QC, quality control; scRNA, small conditional RNA.
Figure 2.
Absolute reads composition of samples in the sequencing cohort. Bar colors represent different RNA types, and labels with lower opacity correspond to RNA types that were not identified in this dataset. Each bar represents a different sample. QC, quality control; scRNA, small conditional RNA.
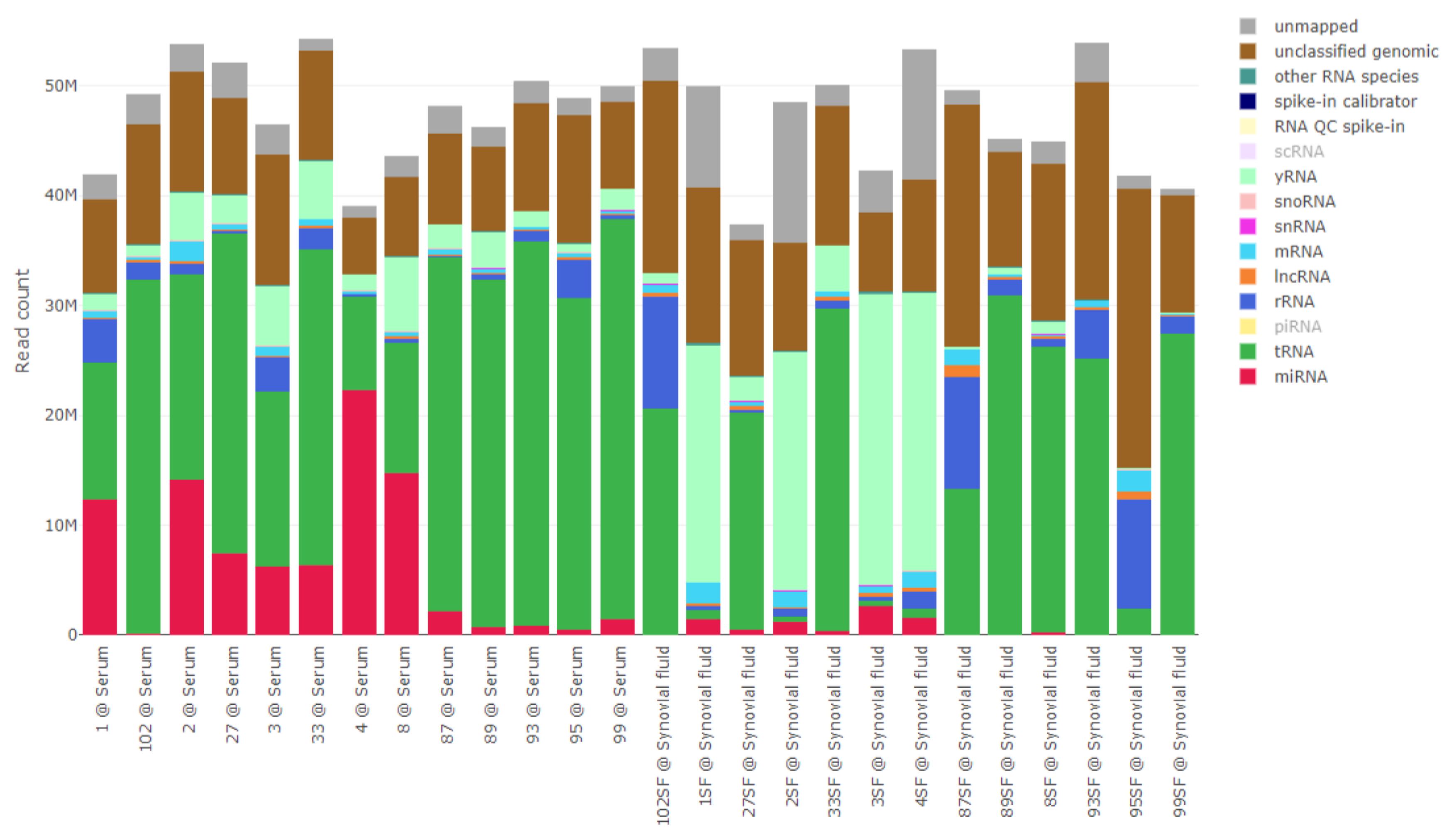
Figure 3.
Heatmap (A) and PCA plots (B) of normalized and scaled miRNA reads. Ellipses in the PCA plots represent a confidence level of 0.95. Images were created with R v3.6 and the packages pheatmap v1.0.12, pcaMethods v1.78 and genefilter v1.68. CS, clinical samples; PM, post-mortem.
Figure 3.
Heatmap (A) and PCA plots (B) of normalized and scaled miRNA reads. Ellipses in the PCA plots represent a confidence level of 0.95. Images were created with R v3.6 and the packages pheatmap v1.0.12, pcaMethods v1.78 and genefilter v1.68. CS, clinical samples; PM, post-mortem.
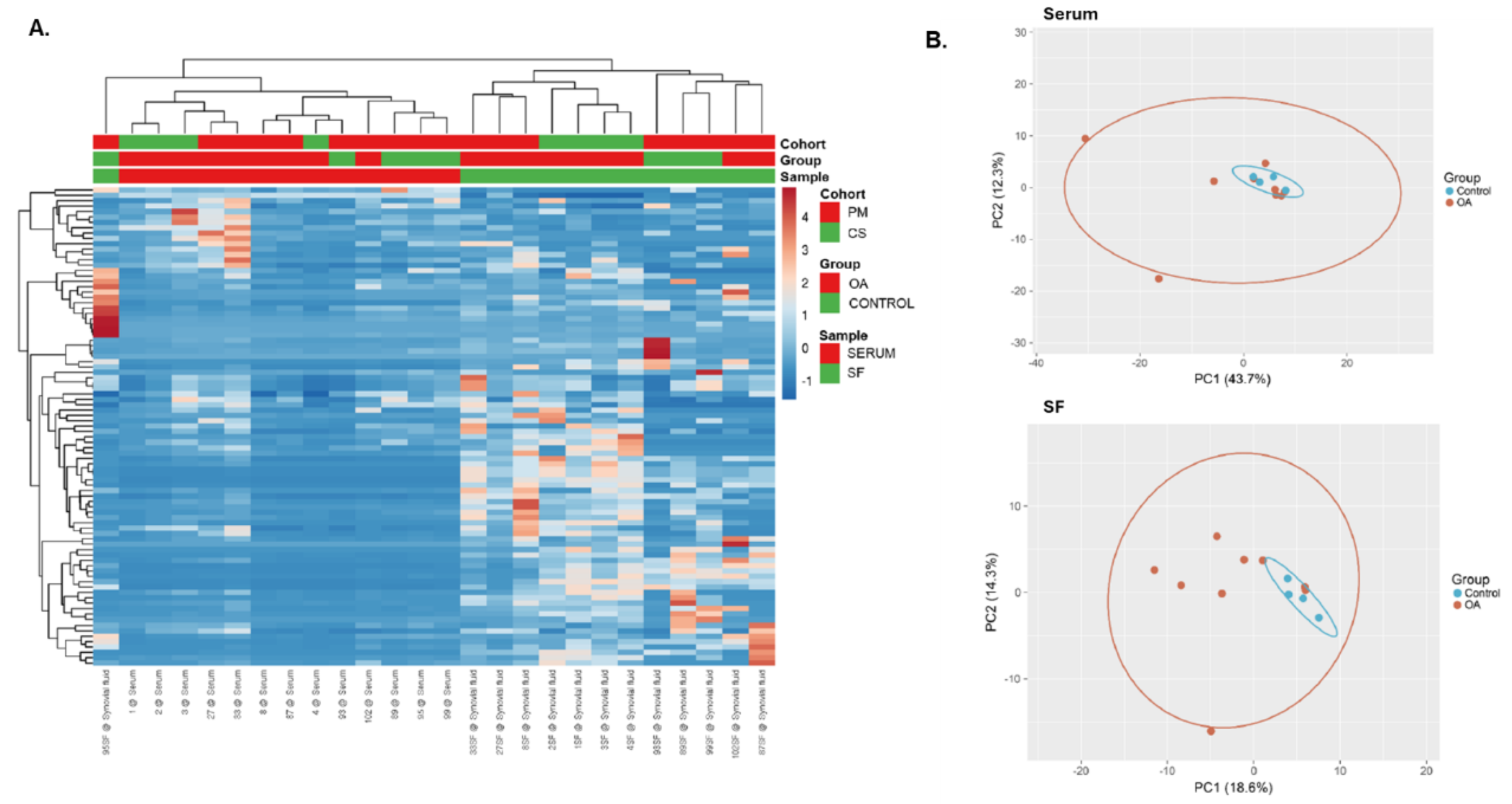
Figure 4.
Selected list of diseases and functions predicted to be associated to the differentially expressed miRNAs in either serum or SF.miRNAs are represented in blue and predicted cellular functions and diseases are represented in grey. Dashed lines represent the relationships between the molecules and predicted functions. Image made in IPA.
Figure 4.
Selected list of diseases and functions predicted to be associated to the differentially expressed miRNAs in either serum or SF.miRNAs are represented in blue and predicted cellular functions and diseases are represented in grey. Dashed lines represent the relationships between the molecules and predicted functions. Image made in IPA.
Figure 5.
Expression (CPM) of selected miRNAs in the (A) serum and (B) SF sequencing cohorts. Box plots show the interquartile data range as box boundaries, with error bars showing the minimum and maximum data range, and the horizontal line showing the median. Images were made using GraphPad Prism version 8.0 for Windows. CPM, counts per million.
Figure 5.
Expression (CPM) of selected miRNAs in the (A) serum and (B) SF sequencing cohorts. Box plots show the interquartile data range as box boundaries, with error bars showing the minimum and maximum data range, and the horizontal line showing the median. Images were made using GraphPad Prism version 8.0 for Windows. CPM, counts per million.
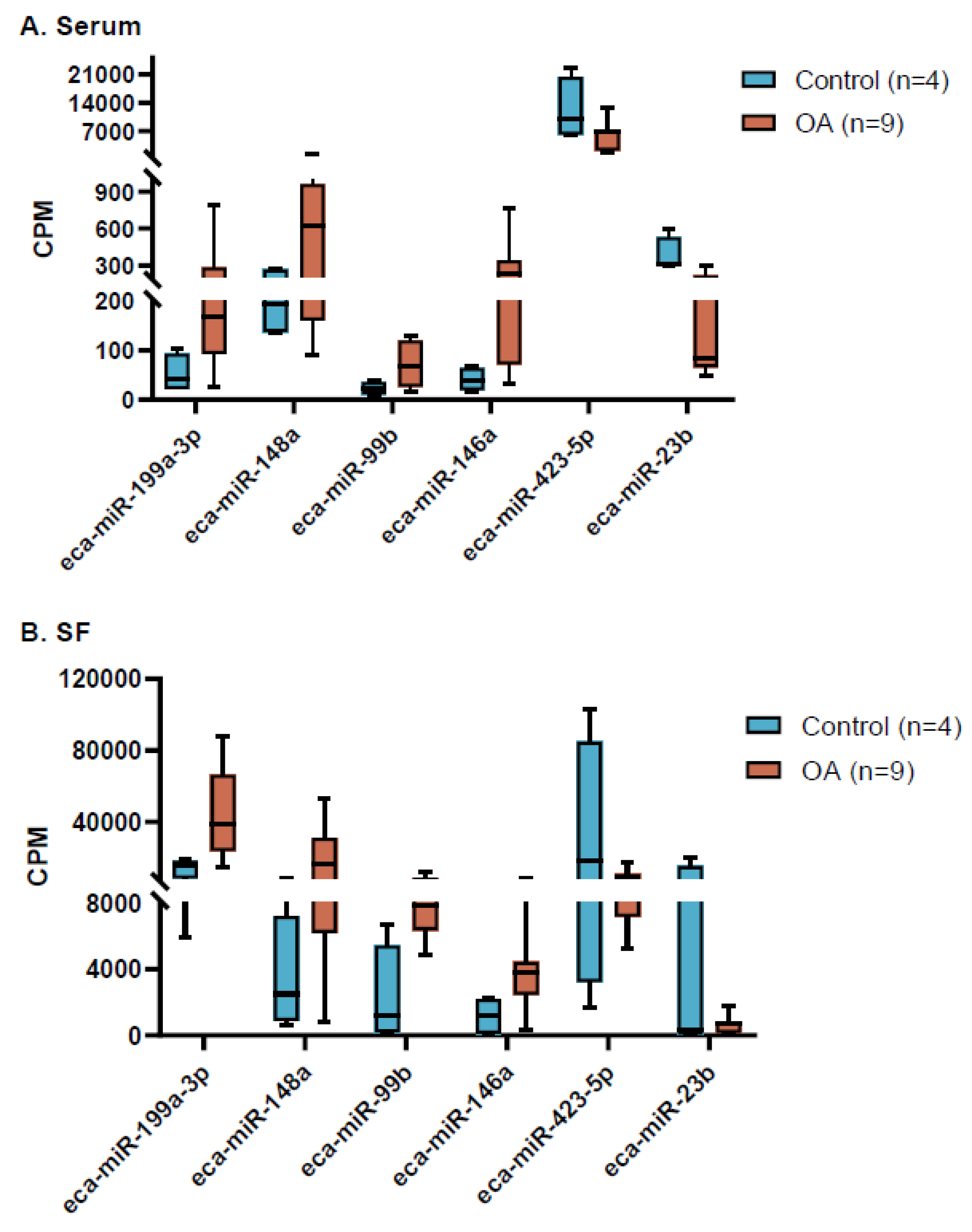
Figure 6.
Relative expression of selected miRNAs in the (A) serum sequencing cohort, (B) serum validation cohort, (C) SF sequencing cohort and (D) SF validation cohort, using RT-qPCR. 1 miRNA expression not detected in 3 samples in the Control group. 2 miRNA expression not detected in any of the samples in the Control group. 3 miRNA expression not detected in 2 samples in the Control group. 4 miRNA expression not detected in 1 sample in the Control group and 6 samples in the OA group. 5 miRNA expression not detected in 1 sample in the OA group. 6 miRNA expression not detected in 1 sample in the Control group. Box plots show the interquartile data range as box boundaries, with error bars showing the minimum and maximum data range, and the horizontal line showing the median. Images were made using GraphPad Prism version 8.0 for Windows. Cq, quantification cycle.
Figure 6.
Relative expression of selected miRNAs in the (A) serum sequencing cohort, (B) serum validation cohort, (C) SF sequencing cohort and (D) SF validation cohort, using RT-qPCR. 1 miRNA expression not detected in 3 samples in the Control group. 2 miRNA expression not detected in any of the samples in the Control group. 3 miRNA expression not detected in 2 samples in the Control group. 4 miRNA expression not detected in 1 sample in the Control group and 6 samples in the OA group. 5 miRNA expression not detected in 1 sample in the OA group. 6 miRNA expression not detected in 1 sample in the Control group. Box plots show the interquartile data range as box boundaries, with error bars showing the minimum and maximum data range, and the horizontal line showing the median. Images were made using GraphPad Prism version 8.0 for Windows. Cq, quantification cycle.
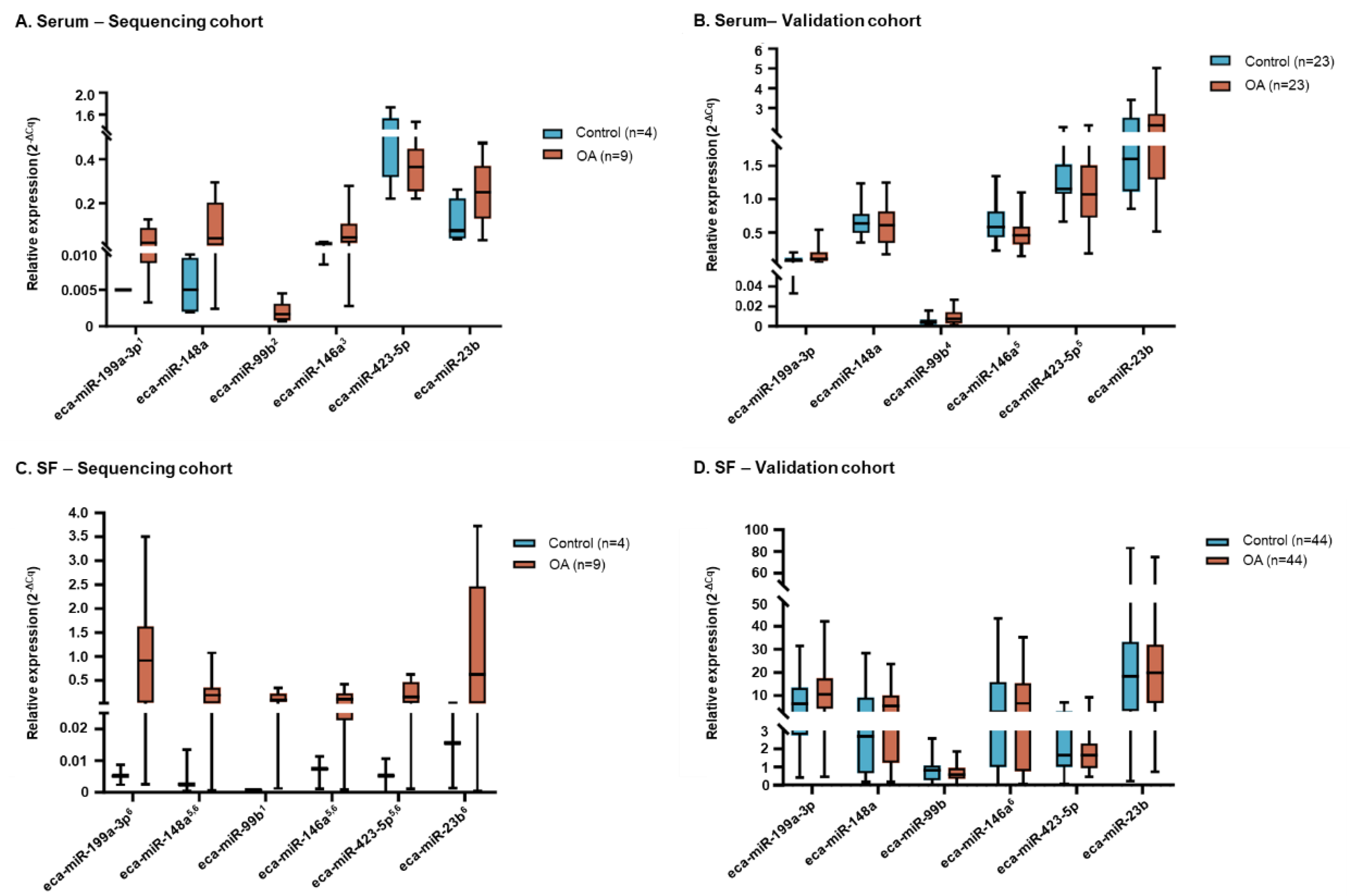

Table 1.
Demographics and clinical characteristics of the sequencing cohort, stratified by group.
|
Control (N=4) |
OA (N=9) |
|
| Collection site, n (%) | ||
| Abattoir1 | 4 (100) | 5 (55.5) |
| Hospital2 | 0 | 4 (44.4) |
| Age, years | ||
| n | 3 | 9 |
| Mean (SD) | 6.3 (7.5) | 6.6 (3.5) |
| Min; Max | 2; 15 | 2; 14 |
| Missing | 1 | 0 |
| Sex, n (%) | ||
| n | 3 | 9 |
| Female | 2 (66.7) | 2 (22.2) |
| Neutered male | 1 (33.3) | 7 (77.8) |
| Missing | 1 | 0 |
| Breed, n (%) | ||
| n | 3 | 8 |
| Arab | 1 (33.3) | 0 |
| Friesian | 0 | 1 (12.5) |
| Standardbred | 1 (33.3) | 1 (12.5) |
| Swedish Warmblood | 4 (50.0) | |
| Thoroughbred | 0 | 2 (25.0) |
| Missing | 1 | 1 |
| Occupation, n (%) | ||
| n | 3 | 8 |
| Racing | 3 (100) | 8 (100.0) |
| Missing | 1 | 1 |
| OA severity, n (%)3 | ||
| n | 4 | 5 |
| Control | 4 (100.0) | 0 |
| Mild | 0 | 3 (60.0) |
| Moderate | 0 | 1 (20.0) |
| Severe | 0 | 1 (20.0) |
| Missing | 0 | 4 |
| SF collection site, n (%) | ||
| n | 4 | 9 |
| Carpal | 4 (100.0) | 6 (66.7) |
| Metacarpophalangeal | 0 | 3 (33.3) |
Percentages were calculated based on the number of samples with available data for the corresponding characteristic. 1 Samples collected post-mortem. 2 Samples collected during lameness examination. 3 Classification of OA severity was performed as previously described by Skiöldebrand et al [15]. SD, standard deviation.
Table 2.
Differentially expressed miRNAs between Control and OA samples, in serum and SF.
| miRNA | logFC1 | p-value | FDR | Significance |
| Serum | ||||
| eca-miR-9048 | -8.74 | <0.0001 | 0.0164 | Decreased in OA |
| eca-miR-143 | 4.09 | 0.0001 | 0.0164 | Increased in OA |
| eca-miR-25 | 1.99 | 0.0002 | 0.0164 | Increased in OA |
| eca-miR-146a | 2.67 | 0.0004 | 0.0242 | Increased in OA |
| eca-miR-1291a | -7.35 | 0.0007 | 0.0242 | Decreased in OA |
| eca-miR-8986b | -7.35 | 0.0007 | 0.0242 | Decreased in OA |
| eca-miR-1892 | -7.26 | 0.0008 | 0.0242 | Decreased in OA |
| eca-miR-8954 | -7.26 | 0.0008 | 0.0242 | Decreased in OA |
| eca-miR-330 | -6.93 | 0.0008 | 0.0242 | Decreased in OA |
| eca-miR-490-3p | -7.47 | 0.0008 | 0.0242 | Decreased in OA |
| eca-miR-191a | 1.76 | 0.0010 | 0.0255 | Increased in OA |
| eca-miR-345-5p | -6.91 | 0.0010 | 0.0255 | Decreased in OA |
| eca-miR-16 | 2.24 | 0.0014 | 0.0296 | Increased in OA |
| eca-miR-133a | 3.80 | 0.0014 | 0.0296 | Increased in OA |
| eca-miR-223 | 4.32 | 0.0022 | 0.0446 | Increased in OA |
| eca-miR-129b-3p | -5.60 | 0.0033 | 0.0580 | Decreased in OA |
| eca-miR-8951 | -5.60 | 0.0033 | 0.0580 | Decreased in OA |
| eca-miR-199a-3p | 2.15 | 0.0038 | 0.0597 | Increased in OA |
| eca-miR-199b-3p | 2.15 | 0.0038 | 0.0597 | Increased in OA |
| eca-miR-483 | -5.01 | 0.0049 | 0.0729 | Decreased in OA |
| eca-miR-142-5p | 1.69 | 0.0065 | 0.0935 | Increased in OA |
| eca-miR-15a | 3.61 | 0.0076 | 0.1032 | Increased in OA |
| eca-miR-148a | 1.63 | 0.0087 | 0.1094 | Increased in OA |
| eca-miR-423-5p | -1.06 | 0.0088 | 0.1094 | Decreased in OA |
| eca-miR-23b | -1.56 | 0.0106 | 0.1271 | Decreased in OA |
| eca-miR-93 | 1.77 | 0.0112 | 0.1287 | Increased in OA |
| eca-miR-744 | 2.64 | 0.0122 | 0.1351 | Increased in OA |
| eca-miR-130a | 3.42 | 0.0132 | 0.1410 | Increased in OA |
| eca-miR-8992 | -3.88 | 0.0143 | 0.1444 | Decreased in OA |
| eca-miR-8977 | -5.19 | 0.0144 | 0.1444 | Decreased in OA |
| eca-miR-423-3p | -1.12 | 0.0217 | 0.2064 | Decreased in OA |
| eca-miR-206 | 3.59 | 0.0221 | 0.2064 | Increased in OA |
| eca-miR-194 | 2.59 | 0.0227 | 0.2064 | Increased in OA |
| eca-miR-1 | 4.53 | 0.0235 | 0.2077 | Increased in OA |
| eca-let-7f | -1.31 | 0.0278 | 0.2358 | Decreased in OA |
| eca-miR-30e | 1.84 | 0.0283 | 0.2358 | Increased in OA |
| eca-miR-98 | -1.88 | 0.0292 | 0.2371 | Decreased in OA |
| eca-miR-340-5p | 2.30 | 0.0312 | 0.2464 | Increased in OA |
| eca-miR-140-3p | 4.07 | 0.0323 | 0.2482 | Increased in OA |
| eca-miR-23a | -1.24 | 0.0340 | 0.2547 | Decreased in OA |
| eca-miR-27b | 1.55 | 0.0470 | 0.3437 | Increased in OA |
| eca-miR-2483 | 3.24 | 0.0492 | 0.3465 | Increased in OA |
| eca-miR-7177b | 8.21 | 0.0497 | 0.3465 | Increased in OA |
| Synovial Fluid | ||||
| eca-miR-324-5p | -6.66 | <0.0001 | <0.0001 | Decreased in OA |
| eca-miR-296 | -5.98 | <0.0001 | 0.0002 | Decreased in OA |
| eca-miR-615-5p | -9.25 | 0.0001 | 0.0072 | Decreased in OA |
| eca-miR-671-3p | -4.69 | 0.0004 | 0.0187 | Decreased in OA |
| eca-miR-27a | -4.42 | 0.0005 | 0.0187 | Decreased in OA |
| eca-miR-184 | -4.47 | 0.0006 | 0.0187 | Decreased in OA |
| eca-miR-1291a | -9.55 | 0.0006 | 0.0187 | Decreased in OA |
| eca-miR-148a | 2.45 | 0.0026 | 0.0646 | Increased in OA |
| eca-miR-423-5p | -1.90 | 0.0032 | 0.0646 | Decreased in OA |
| eca-miR-23b | -3.04 | 0.0032 | 0.0646 | Decreased in OA |
| eca-miR-598 | -7.76 | 0.0059 | 0.1090 | Decreased in OA |
| eca-miR-206 | -7.24 | 0.0075 | 0.1270 | Decreased in OA |
| eca-miR-199a-3p | 1.63 | 0.0122 | 0.1770 | Increased in OA |
| eca-miR-199b-3p | 1.63 | 0.0122 | 0.1770 | Increased in OA |
| eca-miR-31 | -6.76 | 0.0149 | 0.1950 | Decreased in OA |
| eca-miR-92b | -2.66 | 0.0153 | 0.1950 | Decreased in OA |
| eca-miR-99a | 4.81 | 0.0169 | 0.2020 | Increased in OA |
| eca-miR-1892 | -6.60 | 0.0360 | 0.3930 | Decreased in OA |
| eca-miR-10b | 0.89 | 0.0368 | 0.3930 | Increased in OA |
| eca-miR-27b | -2.15 | 0.0437 | 0.4350 | Decreased in OA |
| eca-miR-151-5p | 10.72 | 0.0465 | 0.4350 | Increased in OA |
| eca-miR-342-5p | 10.82 | 0.0480 | 0.4350 | Increased in OA |
| eca-miR-211 | 10.67 | 0.0495 | 0.4350 | Increased in OA |
1A negative logFC corresponds to decreased miRNA expression in the OA group compared with the Control group. logFC, log fold change.
Table 3.
Demographics and clinical characteristics of the validation cohort, stratified by sample type and group.
Table 3.
Demographics and clinical characteristics of the validation cohort, stratified by sample type and group.
| Serum | SF | |||
|
Control (N=23) |
OA (N=23) |
Control (N=44) |
OA (N=44) |
|
| Collection site, n (%) | ||||
| Abattoir1 | 0 | 0 | 39 (88.6) | 40 (90.9) |
| Hospital/Clinic2 | 23 (100) | 23 (100) | 5 (11.4) | 4 (9.1) |
| A | 23 (100) | 9 (39.1) | 0 | 0 |
| B | 0 | 5 (21.7) | 5 (100) | 4 (100) |
| C | 0 | 9 (39.1) | 0 | 0 |
| Age, years | ||||
| n | 23 | 9 | 41 | 41 |
| Mean (SD) | 9.8 (4.2) | 11.1 (5.4) | 10.2 (6.8) | 15.7 (6.7) |
| Min; Max | 3; 18 | 4; 23 | 2; 20 | 3; 25 |
| Missing | 0 | 14 | 3 | 3 |
| p-value | 0.65821 | 0.00031 | ||
| Sex, n (%) | ||||
| n | 23 | 14 | 25 | 26 |
| Female | 8 (34.8) | 2 (14.3) | 17 (68.0) | 8 (30.8) |
| Neutered male | 15 (65.2) | 12 (85.7) | 8 (32.0) | 181 (69.2) |
| p-value | 0.26032 | 0.02272 | ||
| Missing | 0 | 9 | 19 | 18 |
| Body Condition Score, n (%) | ||||
| n | 23 | 9 | 0 | 0 |
| 1–3 | 0 | 0 | – | – |
| 4 | 0 | 1 (11.1) | – | – |
| 5 | 19 (82.6) | 6 (66.6) | – | – |
| 6 | 3 (13.0) | 2 (22.2) | – | – |
| 7 | 1 (4.3) | 0 | – | – |
| 8–9 | 0 | 0 | – | – |
| Missing | 0 | 14 | 44 | 44 |
| Breed, n (%) | ||||
| n | 233 | 233 | 194 | 164 |
| Appaloosa | 0 | 1 (4.3) | 0 | 0 |
| Cob5 | 1 (4.3) | 0 | 2 (10.5) | 1 (6.3) |
| Connemara5 | 4 (17.4) | 1 (4.3) | 0 | 0 |
| Dales5 | 0 | 1 (4.3) | 0 | 0 |
| Dutch Warmblood | 0 | 1 (4.3) | 0 | 0 |
| Hanoverian | 0 | 2 (8.7) | 0 | 0 |
| Holsteiner | 0 | 1 (4.3) | 0 | 0 |
| Irish cob | 0 | 1 (4.3) | 0 | 0 |
| Irish Draught | 2 (8.7) | 0 | 0 | 0 |
| Irish Sport Horse5 | 9 (39.1) | 4 (17.4) | 2 (10.5) | 3 (18.8) |
| Lusitano | 1 (4.3) | 0 | 0 | 1 (6.3) |
| Pony | 1 (4.3) | 1 (4.3) | 4 (21.4) | 0 |
| Thoroughbred5 | 2 (8.7) | 9 (39.1) | 7 (36.8) | 9 (56.3) |
| Warmblood5 | 1 (4.3) | 0 | 0 | 2 (12.5) |
| Welsh Pony/Cob5 | 2 (8.7) | 1 (4.3) | 4 (21.4) | 0 |
| Missing | 0 | 0 | 25 | 25 |
| Occupation, n (%)3 | ||||
| n | 23 | 18 | 0 | 0 |
| Not in work (out in the field’) | 2 (8.7) | 2 (11.1) | – | – |
| All-rounder | 5 (21.7) | 1 (5.6) | – | – |
| Dressage | 1 (4.3) | 0 | – | – |
| Eventing | 2 (8.7) | 0 | – | – |
| Hacking | 5 (21.7) | 32 (16.5) | – | – |
| Hunting | 3 (13.0) | 1 (5.6) | – | – |
| Leisure | 1 (4.3) | 0 | – | – |
| Racing | 0 | 9 (50.0) | – | – |
| Schooling | 4 (17.4) | 1 (5.6) | – | – |
| Showjumping | 0 | 1 (5.6) | – | – |
| Missing | 0 | 5 | 44 | 44 |
|
Current level of work (0–3), n (%)5 |
||||
| n | 23 | 18 | 0 | 0 |
| 0 (‘out in the field’) | 2 (8.7) | 2 (11.1) | – | – |
| 1 (‘light work’) | 12 (52.2) | 4 (22.2) | – | – |
| 2 (‘medium work’) | 7 (30.4) | 2 (11.1) | – | – |
| 3 (‘intense work’) | 2 (8.7) | 10 (55.6) | – | – |
| Missing | 0 | 10 | 44 | 44 |
| p-value | 0.02146 | – | ||
| Shoes, n (%) | ||||
| n | 18 | 8 | 0 | 0 |
| All four feet | 13 (72.2) | 3 (37.5) | n/a | n/a |
| Front feet | 2 (11.1) | 1 (12.5) | n/a | n/a |
| Unshod | 3 (16.7) | 4 (50.0) | n/a | n/a |
| Missing | 5 | 15 | 44 | 44 |
| Joints affected, n (%)7,8 | ||||
| Distal interphalangeal | – | 2 (9.1)3 | 0 | 0 |
| Intervertebral | – | 2 (9.1) | 0 | 0 |
| Metacarpophalangeal | – | 7 (31.8)3 | 44 (100)8 | 44 (100)8 |
| Metatarsophalangeal | – | 4 (18.1)3 | 0 | 0 |
| Sacroiliac | – | 2 (9.1)3 | 0 | 0 |
| Scapulohumeral | – | 1 (4.5)3 | 0 | 0 |
| Tarsometatarsal | – | 2 (9.1)3 | 0 | 0 |
| Front limb9 | – | 2 (9.1)3 | 0 | 0 |
| Joint gross score, n (%)10 | ||||
| n | 0 | 0 | 44 | 44 |
| 0 | – | – | 21 (47.7) | 0 |
| 1 | – | – | 23 (52.3) | 0 |
| 2 | – | – | 0 | 10 (22.7) |
| 3 | – | – | 0 | 16 (36.4) |
| 4 | – | – | 0 | 11 (25.0) |
| 5 | – | – | 0 | 4 (9.1) |
| 6 | – | – | 0 | 2 (4.5) |
| 7 | – | – | 0 | 1 (2.3) |
| 8–9 | – | – | 0 | 0 |
| Mean (SD) | – | – | 0.5 (0.5) | 3.4 (1.2) |
| Min; Max | – | – | 0; 1 | 2; 7 |
| p-value | – | <0.00016 | ||
| Missing | 23 | 23 | 0 | 0 |
|
Articular cartilage microscopic score, n (%)11 |
||||
| n | 0 | 0 | 34 | 30 |
| 0 | – | – | 0 | 0 |
| 1 | – | – | 5 (14.7) | 0 |
| 2 | – | – | 8 (23.5) | 5 (16.7) |
| 3 | – | – | 7 (20.6) | 4 (13.3) |
| 4 | – | – | 9 (26.5) | 5 (16.7) |
| 5 | – | – | 2 (5.9) | 5 (16.7) |
| 6 | – | – | 1 (2.9) | 5 (16.7) |
| 7 | – | – | 1 (2.9) | 4 (13.3) |
| 8 | – | – | 1 (2.9) | 1 (3.3) |
| 9–15 | – | – | 0 | 0 |
| 16 | – | – | 0 | 1 (3.3) |
| 17–20 | – | – | 0 | 0 |
| Mean (SD) | – | – | 3.2 (1.7) | 5.0 (2.9) |
| Min; Max | – | – | 1; 8 | 2; 16 |
| p-value | – | 0.00156 | ||
| Missing | 23 | 23 | 10 | 14 |
1 Calculated using Mann-Whitney U test. 2 Calculated suing Fisher’s exact test. 3 As reported by owner. 4 As reported in the passport. 5 Including breed crosses. 6 Calculated using Chi-square test for trend. 7 Horses could be included in more than one category. 8 For SF, samples were only collected from the metacarpophalangeal joint; information regarding other joints was not available. 9 Owner could not recall affected joint but reported the affected limb. 10 Assessed at post-mortem. –, not applicable/available; BCS, body condition score.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



