Submitted:
15 October 2025
Posted:
16 October 2025
You are already at the latest version
Abstract
Hydroxamic acids are emerging as versatile chiral ligands for metal-catalyzed asymmetric oxidations due to their tunable electronic and steric environments. In this study, we systematically compared the catalytic behavior of C2- and C1-symmetric hydroxamic acid ligands in the vanadium-catalyzed asymmetric epoxidation of allylic alcohols. A series of chiral hydroxamic acids (HA1–HA7) was synthesized and evaluated under varied conditions to elucidate the influence of ligand symmetry on enantioinduction and reactivity. The results demonstrate that C2-symmetric bishydroxamic acids generate a highly organized chiral environment leading to high enantioselectivity but often limited conversion, consistent with the Sabatier principle. Conversely, certain C1-symmetric ligands—particularly HA3—produced notable enantioselectivity (up to 71% ee) and full conversion under optimized conditions with VO(OiPr)₃ in CH₂Cl₂. A quadrant-based stereochemical model is proposed to rationalize the differential performance of these ligands. These findings highlight the critical role of ligand desymmetrization in modulating the chiral environment around vanadium centers, providing valuable design principles for next-generation hydroxamic acid-based catalysts in asymmetric synthesis. The optimized system (VO(OiPr)₃/HA3 in CH₂Cl₂) afforded >99% conversion and 71% ee, providing a basis for extending hydroxamic acid scaffolds to diverse allylic alcohols.
Keywords:
asymmetric epoxidation
; hydroxamic acids
; vanadium catalysis
; ligand symmetry
; enantioselectivity
; C1/C2 ligands
1. Introduction
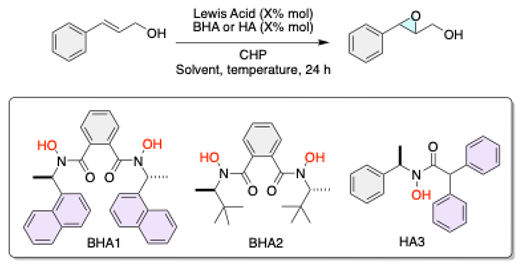
Developing efficient and selective catalysts for asymmetric epoxidation reactions is a significant goal in organic synthesis, as these reactions are pivotal for constructing complex chiral molecules with diverse functional groups. Among the various catalytic systems explored, hydroxamic acids have gained attention due to their ability to form stable metal complexes and facilitate highly enantioselective transformations.1,2 In recent years, hydroxamic acid ligands have been extensively studied in asymmetric catalysis, with a particular focus on their application in metal-catalyzed oxidation reactions.3,4 Hydroxamic acids can coordinate with transition metals, creating catalytic systems capable of achieving high levels of stereoselectivity.5,6 The inherent structural flexibility of hydroxamic acids allows for fine-tuning electronic and steric properties, making them attractive candidates for developing novel asymmetric catalysts.3 The initial phase of our research focused on the synthesis and evaluation of C2-symmetric bishydroxamic acid (BHA) ligands.7 C2-symmetric ligands are known for their ability to create well-defined chiral environments around metal centers, which is crucial for achieving high levels of enantioselectivity. Indeed, preliminary experiments with these C2 BHA ligands, particularly when complexed with vanadium, demonstrated significant enantioselectivity, achieving up to 99% enantiomeric excess under optimal conditions but lower conversions.7
The observation of excellent enantioselectivity but low conversion in the asymmetric epoxidation reactions using chiral C2 hydroxamic acids complexed with vanadium or titanium can be understood through the lens of the Sabatier principle,8 which states that for a catalyst to be effective, the interaction between the catalyst and the substrate should be neither too weak nor too strong. In this case, the specific reaction conditions, particularly the choice of solvent and temperature, appear to create a chiral transition state that strongly favors enantioselectivity but may result in an overly strong interaction between the catalyst and the substrate. This strong interaction could stabilize the intermediate too much, slowing down the overall reaction rate and leading to low conversion. Solvents like acetonitrile may enhance enantioselectivity by stabilizing this chiral environment, yet they can also decrease the reaction kinetics due to the overly stabilized intermediate, thus reducing conversion.9 Similarly, the interaction between the hydroxamic acid ligands and the metal center might create a highly selective catalytic system that binds the substrate too tightly, in line with the Sabatier principle, resulting in low catalytic turnover and, consequently, low conversion. The reaction temperature also plays a crucial role, where lower temperatures can improve selectivity but further slow down the reaction rate by enhancing these strong interactions. Additionally, the inherent properties of the substrate, such as bulkiness or electronic effects, could exacerbate this issue by making the substrate less reactive under the given conditions. Together, these factors suggest that while the system is optimized for enantioselectivity, it may require further adjustments to the catalyst-substrate binding strength and reaction conditions to balance selectivity with reactivity, per the Sabatier principle, to achieve higher overall catalytic efficiency.
Expanding on this idea, Pfaltz described in 2004,10 that while C2-symmetric ligands have proven to be highly effective, there is no inherent reason why they must always outperform their nonsymmetrical counterparts.11 Some nonsymmetrical ligands have been discovered that can achieve even higher enantioselectivities in certain reactions compared to the best C2-symmetric ligands.12 Additionally, there are strong arguments suggesting that nonsymmetrical ligands with electronically and sterically distinct coordinating units, in some cases, may offer more precise enantiocontrol than C2-symmetric ligands.13-15
Building on these observations, the next logical step was to investigate the performance of C1-symmetric HA ligands. Unlike their C2 counterparts, C1-symmetric ligands offer unique geometric and electronic characteristics that could influence catalytic activity and selectivity differently. The exploration of C1-symmetric hydroxamic acid ligands aimed to determine whether these less symmetrical ligands could achieve comparable or even superior enantioselectivity in the epoxidation of allylic alcohols, particularly when complexed with vanadium or titanium.
This investigation into C1-symmetric hydroxamic acid ligands represents a significant extension of our research, aimed at deepening the understanding of how ligand symmetry influences catalytic outcomes in asymmetric epoxidation reactions. By exploring the distinct properties of C1-symmetric ligands, this study seeks to uncover new strategies for optimizing enantioselectivity and reaction efficiency. We anticipate that the insights gained from this research will contribute to developing next-generation chiral ligands, advancing the field of asymmetric synthesis toward more efficient and selective catalytic systems. The study also compares the performance of previously reported C2-symmetric BHA with that of C1-symmetric hydroxamic acid ligands in the epoxidation of allylic alcohols. While C2-symmetric ligands are known for their ability to induce high enantioselectivity through the creation of well-defined chiral environments around the metal center, C1-symmetric ligands present unique geometric and electronic characteristics that could offer distinct advantages in terms of catalytic activity and selectivity.
Herein, we present a detailed analysis of the reactivity and enantioselectivity of bishydroxamic acids (BHA1 and BHA2) and hydroxamic acid ligands (HA1-HA7) in combination with vanadium catalysts for the epoxidation of allylic alcohols. Our study aims to elucidate the role of ligand symmetry in modulating catalytic performance, providing insights into the design of hydroxamic acid-based catalysts for asymmetric synthesis.
2. Experimental
2.1. Material and Methods
- Synthesis of Chiral BHA´s and HAs; General Procedure,
The BHA1 and BHA2 were synthesized following previous literature reports.7 In the case of the HA´s, we use the methodology reported by Yamamoto,16 the chiral hydroxylamines were obtained through oxidation with BPO of commercially available enantioenriched amines, sourced from Sigma–Aldrich, then the hydroxamic acids (HA) were synthesized via a coupling reaction between Acyl chloride and chiral hydroxylamines.
- Synthesis of Synthesis of N-OBz amines (1a to 3a); General Procedure A
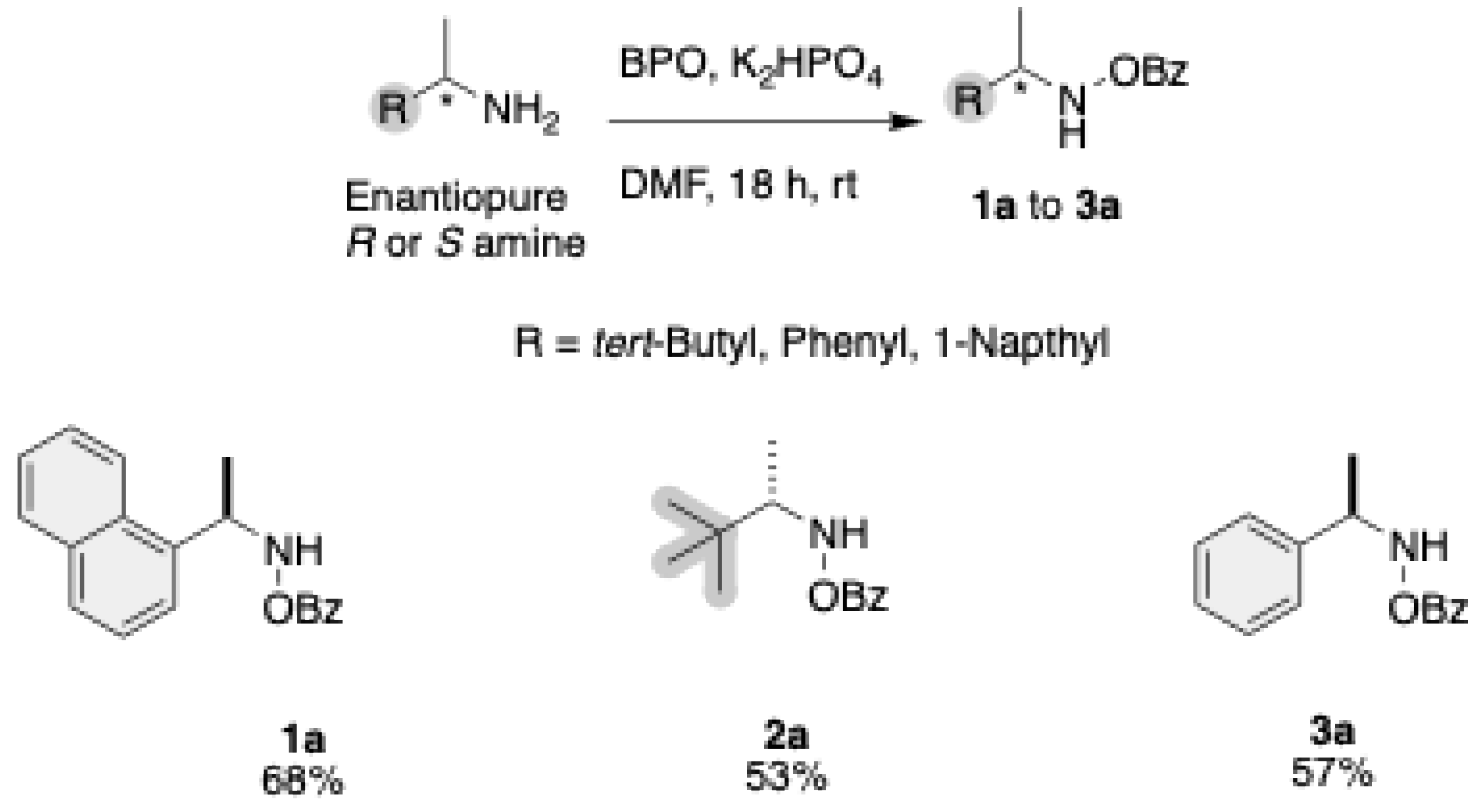
A mixture of BPO (2.0 equiv) and K2HPO3 (3.0 equiv) in DMF was stirred at room temperature for 2 hours. Then, a solution of enantiopure amine (1.0 equiv) in CH2Cl2 was added, and the solution was stirred for 18 hours. Water was added, followed by extraction with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The crude product was purified by silica gel column chromatography using a hexane/EtOAc eluent system. The purified product was obtained after evaporating the solvent.16
Synthesis of ((R)-O-benzoyl-N-(1-(naphthalen-1-yl)ethyl)hydroxylamine) (1a). General procedure A afforded the product 1a as a yellow oil in 68% yield. The product was purified by flash column chromatography (15 % hexane/AcOEt). 1H NMR (500 MHz, CDCl3): δ = 8.20 (d, J = 8.4 Hz, 1H), 8.09 (s, 1H), 7.94 (d, J = 7.9 Hz, 2H), 7.81 (dd, J = 15.5, 7.7 Hz, 2H), 7.53 (dq, J = 14.6, 7.2 Hz, 4H), 7.41 (t, J = 7.6 Hz, 2H), 5.19 (s, 1H), 1.70 (d, J = 6.5 Hz, 3H), 1.58 (s, 1H). 13C NMR (125 MHz, CDCl3) δ = 133.47, 129.52, 129.16, 128.61, 128.38, 126.44, 125.73, 123.86, 122.94, 56.42, 19.74. HRMS (ESI+): m/z [M + Na]+ calcd for C19H17NO2Na+: 314.1157; found: 314.1165.
Synthesis of ((S)-O-benzoyl-N-(3,3-dimethylbutan-2-yl)hydroxylamine) (2a). General procedure A afforded the product 2a as a colorless oil in 53% yield. The product was purified by flash column chromatography (20% hexane/AcOEt). ). 1H NMR (500 MHz, CDCl3) δ = 8.02 (dd, J = 8.2, 1.9 Hz, 3H), 7.58 (tt, J = 7.3, 1.4 Hz, 1H), 7.46 (tt, J = 7.5, 1.6 Hz, 2H), 2.93 (tt, J = 8.1, 4.1 Hz, 1H), 1.17 (dt, J = 6.7, 1.6 Hz, 3H), 1.03 (t, J = 1.6 Hz, 9H). 13C NMR (125 MHz, CDCl3) δ = 167.54, 133.64, 129.67, 128.92, 65.47, 34.13, 27.16, 14.08. HRMS (ESI+): m/z [M + Na]+ calcd for C13H19NO2Na+: 244.1313; found: 244.1316.
Synthesis of ((R)-O-benzoyl-N-(1-phenylethyl)hydroxylamine) (3a). General procedure A afforded the product 3a as a colorless oil in 57% yield. The product was purified by flash column chromatography (15% hexane/AcOEt). ). 1H NMR (500 MHz, CDCl3) δ = 7.93 (m, 3H), 7.54 (m, 1H), 7.41 (m, 4H), 7.35 (m, 2H), 7.29 (m, 1H), 4.31 (m, 1H), 1.53 (d, J= 6.7 Hz, 3H).
13C RMN (125 MHz, CDCl3) δ = 166.8, 141.2, 133.3, 129.3, 128.6, 128.5, 128.3, 127.9, 127.1, 60.9, 19.7. HRMS (ESI+): m/z [M + Na]+ calcd for C15H16NO2: 264.1000; found: 264.1007
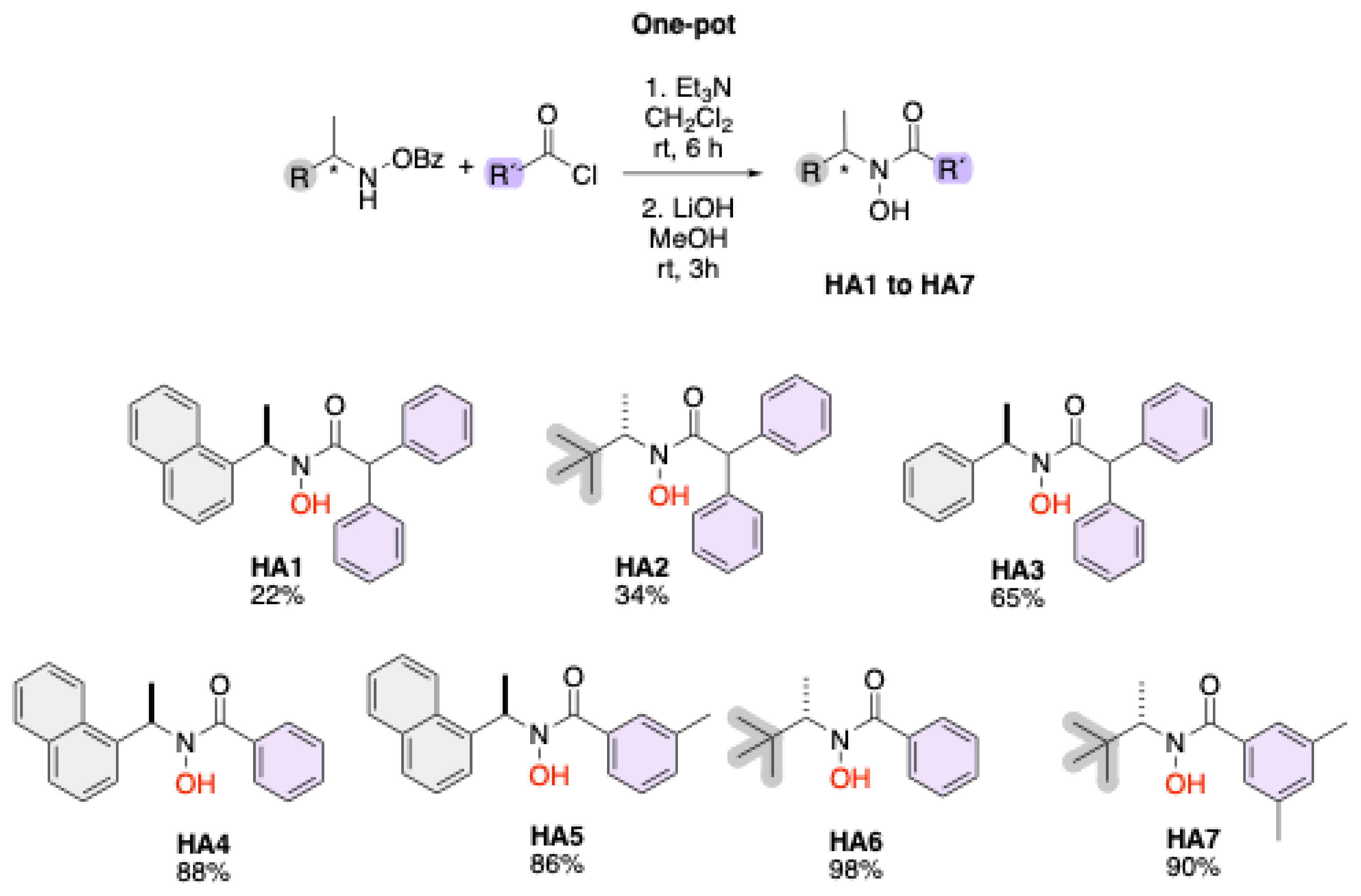
Synthesis of ((R)-N-hydroxy-N-(1-(naphthalen-1-yl)ethyl)-2,2-diphenylacetamide) (HA1). General procedure C afforded the product HA1 as a white solid in 22% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C. 1H NMR (500 MHZ, DMSO-d6): δ = 9.80 (s, 1 H), 8.17-8.08 (m, 1 H), 7.97-7.84 (m, 2 H), 7.50 (ddt, J = 32.4, 18.3, 8.8 Hz, 4 H), 7.37-7.15 (m, 10 H), 6.44 (q, J = 6.8 Hz, 1 H), 5.66 (s, 1 H), 1.58 (d, J = 6.8 Hz, 3 H). 13C NMR (126 MHz, DMSO-d6): δ = 171.36, 140.59, 136.48, 133.80, 131.62, 129.38, 129.33, 129.04, 128.72, 128.52, 128.41, 128.00, 127.94, 127.06, 126.94, 126.65, 126.09, 125.72, 125.44, 123.85, 52.17, 16.79. HRMS (ESI+): m/z [M + Na]+ calcd for C26H23NO2Na+: 404.1626; found: 404.1652
Synthesis of ((S)-N-(3,3-dimethylbutan-2-yl)-N-hydroxy-2,2-diphenylacetamide) (HA2). General procedure C afforded the product HA2 as a white solid in 34% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C. 1H NMR (500 MHZ, DMSO-d6): δ = 7.32-7.18 (m, 10 H), 5.71 (s, 1 H), 4.35 (q, J = 6.9 Hz, 1 H), 1.04 (d, J = 6.9 Hz, 3 H), 0.85 (s, 9 H). 13C NMR (126 MHz, DMSO-d6): δ = 171.17, 140.94, 129.42, 129.28, 128.60, 128.51, 126.89, 57.53, 51.95, 35.15, 27.53, 12.52.
HRMS (ESI+): m/z [M + Na]+ calcd for C20H25NO2Na+: 334.1783; found: 334.1813.
Synthesis of ((R)-N-hydroxy-2,2-diphenyl-N-(1-phenylethyl)acetamide) (HA3). General procedure C afforded the product HA3 as a white solid in 65% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C.1H NMR (500 MHZ, DMSO-d6): δ = 7.37-7.14 (m, 15 H), 5.68 (s, 1 H), 5.67 (q, J = 9.3 Hz,1 H), 1.44 (d, J = 7.1 Hz, 3 H). 13C NMR (126 MHz, 500 MHZ, DMSO-d6): δ = 171.59, 141.53, 140.61, 140.47, 129.34, 129.29, 128.65, 128.55, 128.53, 127.48, 127.01, 126.95, 53.80, 52.22, 17.63. HRMS (ESI+): m/z [M + Na]+ calcd for C22H21NO2Na+: 354.1470; found: 354.1505.
Synthesis of ((R)-N-hydroxy-N-(1-(naphthalen-1-yl)ethyl)benzamide) (HA4). General procedure C afforded the product HA4 as a white solid in 88% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C.1H NMR (500 MHZ, DMSO-d6): δ = δ = 9.64 (s, 1H), 8.23 – 8.11 (m, 1H), 7.97 (dd, J = 7.7, 1.8 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.68 (d, J = 7.1 Hz, 1H), 7.60 – 7.51 (m, 5H), 7.45 – 7.34 (m, 3H), 6.49 (d, J = 7.3 Hz, 1H), 1.69 (d, J = 6.8 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): δ = 168.6, 136.6, 135.5, 133.9, 131.6, 130.4, 129.2, 128.7, 128.5, 128.1, 126.8, 126.1, 125.8, 125.4, 123.7, 50.9, 17.0. HRMS (ESI+): m/z [M + Na]+ calcd for C19H18NO2Na+: 314.1157; found: 314.1180.
Synthesis of ((R)-N-hydroxy-3,5-dimethyl-N-(1-(naphthalen-1-yl)ethyl)benzamide) (HA5). General procedure C afforded the product HA5 as a white solid in 86% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C. 1H NMR (500 MHZ, DMSO-d6): δ = 8.50 (s, 1H), 7.92 (d, J = 8.2 Hz, 1H), 7.87 – 7.80 (m, 1H), 7.54 – 7.43 (m, 3H), 7.36 (s, 1H), 7.25 (dd, J = 5.1, 2.0 Hz, 1H), 6.93 (s, 1H), 6.66 (s, 2H), 5.65 (s, 1H), 2.03 (s, 6H), 1.66 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): δ = 168.9, 154.8, 136.5, 133.8, 130.9, 128.9, 128.8, 126.9, 126.4, 126.4, 125.9, 125.8, 21.2, 16.8. HRMS (ESI+): m/z [M + Na]+ calcd for C21H22NO2Na+: 342.1470; found: 342.1447.
Synthesis of ((S)-N-(3,3-dimethylbutan-2-yl)-N-hydroxybenzamide) (HA6). General procedure C afforded the product HA6 as a white solid in 98% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C. 1H NMR (500 MHZ, DMSO-d6): δ = 9.41 (s, 1H), 7.57 – 7.38 (m, 5H), 4.47 (s, 1H), 1.15 (d, J = 7.0 Hz, 3H), 0.96 (s, 9H). 13C NMR (126 MHz, DMSO-d6): δ = 169.1, 136.3, 130.0, 128.6, 128.1, 57.9, 35.4, 27.6, 12.4. HRMS (ESI+): m/z [M + Na]+ calcd for C13H20NO2Na+: 244.1313; found: 244.1304.
Synthesis of ((S)-N-(3,3-dimethylbutan-2-yl)-N-hydroxy-3,5-dimethylbenzamide) (HA7). General procedure C afforded the product HA7 as a white solid in 90% yield. The product was purified by precipitating the product from a mixture of toluene or benzene and hexane at 0 °C. 1H NMR (500 MHZ, DMSO-d6): δ = 9.28 (s, 1H), 7.14 (s, 2H), 7.04 (s, 1H), 4.45 (s, 1H), 2.28 (s, 6H), 1.14 (d, J = 6.9 Hz, 3H), 0.95 (s, 9H). 13C NMR (126 MHz, DMSO-d6): δ = 169.5, 137.0, 136.5, 131.1, 126.1, 57.7, 35.3, 27.6, 21.3, 12.4. HRMS (ESI+): m/z [M + Na]+ calcd for C15H24NO2Na+: 272.1626; found: 272.1620.
- Asymmetric Epoxidation of Allylic Alcohol; General Procedure D
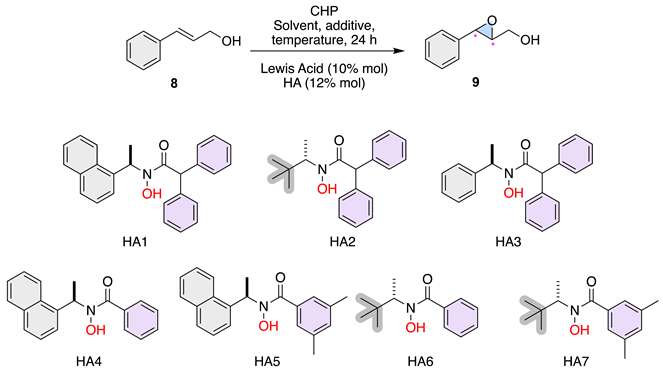
Lewis Acid (10 mol%) in toluene was stirred with HA (11 mol%) for 30 minutes. Allylic alcohol (1.0 equiv) in toluene was added, followed by CHP (CHP = Cumene hydroperoxide, 0.24 mmol, 1.2 equiv). The mixture was stirred for 24 hours at ambient temperature and then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to obtain epoxy alcohol.
- Synthesis of ((2R,3R)-3-Phenyloxiran-2-yl)methanol (9)
To a solution of Lewis acid (0.02 mmol, 10 mol%) and HA (0.022 mmol, 11 mol%) in toluene (2 mL), (E)-3-phenylprop-2-en-1-ol 5a (0.2 mmol, 1 equiv) and CHP (0.24 mmol, 1.2 equiv) were added. General Procedure D afforded product 9 as a yellow oil (8.41 mg, 0.056 mmol, 28% yield). TLC was used to monitor the reaction (hexane/EtOAc 5:1) with visualization by staining. The compound was purified by flash chromatography column (gradient hexane/EtOAc, 9:1 to 5:1). The ee was determined by SFC using a Chiralcel column [OD-H, n-hexane/2-propanol = 90:10]; 1 mL/min, 210 nm, retention time (2S,3S) = 9.9 min, retention time (2R,3R) = 13.0 min, ee >71%. Characterization data are available in previous reports.17
3. Results and Discussion
3.1. Synthesis of N-Obz Amines
The enantiopure amines were transformed to the corresponding N-O Benzoyl amines using potassium phosphate (K₂HPO₄) as a base in dimethylformamide (DMF) as the solvent. This reaction proceeds over 18 hours at room temperature to yield the intermediates 1a, 2a, and 3a, with yields up to 68% (Scheme 1). All intermediates in this pathway share a common benzoyl-protected (OBz) amine group.
Scheme 2 illustrates a one-pot synthetic route for the preparation of hydroxamic acids (HA1–HA7) from benzoyl-protected hydroxamic acid precursors. The reaction proceeds through a two-step process: first, acylation of the hydroxamic acid derivative with an appropriate acyl chloride (R′–COCl) in the presence of triethylamine (Et3N) in dichloromethane (CH₂Cl₂) at room temperature for 6 hours; second, deprotection of the benzoyl group using lithium hydroxide (LiOH) in methanol (MeOH) at room temperature for 3 hours. The resulting compounds (HA1–HA7) exhibit structural diversity in both R and R′ substituents, providing a versatile library of hydroxamic acids, with yields up to 98%.
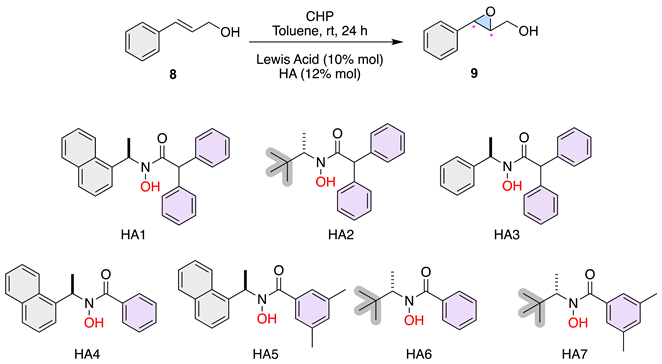
We began our investigation into how ligand symmetry influences enantioinduction by comparing the performance of C2- versus C1-symmetric hydroxamic acid ligands (BHA1, BHA2, and HA3) in the vanadium-catalyzed epoxidation of allylic alcohol 8 (Table 1).
As shown in Table 1, the C2-symmetric ligands BHA1 and BHA2 provided low conversions (14–11%) and poor enantioselectivity (7–9% ee) even at low temperature (–20 °C), indicating that the highly rigid chiral environment imposed by the C2 framework restricts productive substrate binding. This result aligns with the Sabatier principle, suggesting that excessive stabilization of the metal–ligand–substrate complex hinders catalytic turnover. To our delight, the C1-symmetric ligand HA3 and HA7 delivered full conversion (>99%) and markedly higher enantioselectivity (up to 49% ee) at room temperature. Guided by these preliminary results, the subsequent phase of this study focused on optimizing the epoxidation reaction under various conditions.
3.2. Analysis of Epoxidation Reaction Conditions
The epoxidation reactions were carried out using various chiral hydroxamic acids (HAs) and Lewis acids in toluene at room temperature to evaluate their impact on conversion and enantioselectivity. The results are summarized in the table below:
Table 2 presents the results of asymmetric epoxidation reactions conducted using different metal catalysts and hydroxy acids (HAs) in toluene at room temperature. The conversion and enantiomeric excess values are reported for each combination of metal and HA, providing insight into the catalytic efficiency and selectivity of the tested systems. These results reveal a significant disparity in the activity of the two metal catalysts used: Ti(OiPr)4 and VO(OiPr)3. When Ti(OiPr)4 was combined with HA3 or HA2, as shown in entries 1 and 3, the reaction did not proceed, resulting in no conversion. This outcome suggests that Ti(OiPr)4 is not an effective catalyst for this epoxidation reaction under the given conditions. In contrast, VO(OiPr)3 consistently achieved quantitative conversion across all entries where it was tested with various HAs (entries 2, 4, 5-9), indicating its effectiveness as a catalyst for promoting the epoxidation reaction in toluene at room temperature. The enantiomeric excess varied significantly depending on the HA used in conjunction with VO(OiPr)3. HA3, as shown in entry 2, produced an enantioselectivity of 19%, indicating a modest level of enantioselectivity. HA2, in entry 4, resulted in a slightly higher enantiomeric excess of 22%, suggesting that this ligand might provide slightly better chiral induction than HA3 under these conditions. HA1, shown in entry 5, achieved an enantioselectivity of 15%, which is lower than that of HA2 and HA3, indicating less effective enantioselectivity. Notably, HA6, in entry 6, showed a significantly lower enantiomeric excess of 4%, suggesting poor chiral induction. On the other hand, HA7, as presented in entry 7, achieved the highest enantioselectivity of 49%, indicating that HA7 is the most effective ligand for enantioselective epoxidation among those tested. HA4, in entry 8, resulted in an enantiomeric excess of 11%, showing moderate effectiveness in inducing chirality, while HA5, shown in entry 9, produced an enantioselectivity of 30%, which is relatively high compared to most other HAs but still lower than HA7.
Entry 10 presents the results using VO(acac)2 as the metal catalyst with HA2. The conversion remains quantitative, but the enantioselectivity is 19%, which is comparable to the results obtained with VO(OiPr)3 and HA3 (entry 2). This similarity indicates that while VO(acac)2 is effective in achieving conversion, its enantioselectivity is similar to that of VO(OiPr)3 under these conditions. In general, VO(OiPr)3 proves to be a superior catalyst compared to Ti(OiPr)4, achieving full conversion with all HAs tested, while Ti(OiPr)4, under these conditions, fails to catalyze the reaction. The choice of HA significantly influences the enantioselectivity, with HA7 standing out as the most effective ligand, producing the highest enantiomeric excess of 49%. Other HAs show varying degrees of effectiveness, with some like HA6 performing poorly in terms of enantiomeric excess. Despite these findings, the overall enantioselectivity achieved suggests that further optimization is needed. This could involve modifying the ligand structure, adjusting reaction conditions such as temperature or solvent, or exploring other metal catalysts to improve the efficiency and selectivity of the catalytic system.

Table 3.
Analysis of solvent, additive, and temperature effects in the epoxidation reaction.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Lewis Acid (% mol) |
HA (% mol) |
Solvent | Additive | Temperature | Conversion (%) | e.e.b (%) |
| 1 | VO(OiPr)3 (10) | HA2 (12) |
CH2Cl2 | - | r.t. | ≥99 | 23 |
| 2 | VO(OiPr)3 (10) | HA2 (12) |
CH3CN | - | r.t. | ≥99 | 4 |
| 3 | VO(OiPr)3 (10) | HA3 (12) |
CH2Cl2 | - | r.t. | ≥99 | 71 |
| 4 | VO(OiPr)3 (10) | HA3 (12) |
CH3CN | - | r.t. | ≥99 | 63 |
| 5 | VO(OiPr)3 (5) |
HA2 (7) |
Toluene | - | r.t. | ≥99 | 8 |
| 6 | VO(OiPr)3 (5) |
HA2 (7) |
Toluene | - | r.t. | ≥99 | 55 |
| 7 | Ti(OiPr)4 (1.05 eq) |
HA3 (1.05 eq) |
CH2Cl2 | - | r.t. | 0 | n.d. |
| 8 | VO(OiPr)3 (10) | HA2 (12) |
Toluene | MS 4Å | r.t. | ≥99 | 18 |
| 9 | VO(OiPr)3 (10) | HA2 (12) |
Toluene | MgO | r.t. | ≥99 | 22 |
| 10 | VO(OiPr)3 (10) | HA2 (20) |
Toluene | - | r.t. | ≥99 | 25 |
| 11a | VO(OiPr)3 (10) | HA7 (20) |
Toluene | - | r.t. | ≥99 | 7 |
| 12 | VO(OiPr)3 (10) | HA1 (12) |
Toluene | - | 0 °C | ≥99 | 19 |
| 13 | VO(OiPr)3 (10) | HA3 (12) |
Toluene | - | 0 °C | ≥99 | 23 |
| 14 | VO(OiPr)3 (10) | HA2 (12) |
Toluene | - | 0 °C | ≥99 | 21 |
| 15 | VO(OiPr)3 (10) | HA6 (12) |
Toluene | - | 0 °C | ≥99 | 8 |
| 16 | VO(OiPr)3 (10) | HA7 (12) |
Toluene | - | 0 °C | ≥99 | 4 |
| 17 | VO(OiPr)3 (10) | HA4 (12) |
Toluene | - | 0 °C | ≥99 | 14 |
| 18 | VO(OiPr)3 (10) | HA5 (12) |
Toluene | - | 0 °C | ≥99 | 24 |
| 19 | VO(acac)2 (10) | HA2 (12) |
Toluene | - | 0 °C | ≥99 | 17 |
aThe reaction time was 48 hours. bThe enantiomeric excesses were determined by chiral HPLC.
Based on previous results, we decided to analyze the solvent effect. As seen in entries 1 to 4, we explored the impact of different solvents (CH2Cl2 and CH3CN) on the epoxidation reaction using HA2 and HA3 with VO(OiPr)3. In CH2Cl2, HA2 resulted in 99% conversion with a moderate enantioselectivity of 23% (Entry 1). However, the same HA in CH3CN drastically reduced the enantioselectivity to 4% (Entry 2). HA3, on the other hand, exhibited significantly higher enantioselectivity in both solvents, with 71% in CH2Cl2 (Entry 3) and 63% in CH3CN (Entry 4). These results indicate that CH2Cl2 is generally a better solvent for achieving higher enantioselectivity with these HAs. Next, regarding the effect of catalyst loading the entries 5 and 6 examine the impact of reducing the catalyst and ligand loadings using VO(OiPr)3 and HA2 in toluene. Lowering the catalyst and ligand loading to 5% mol and 7% mol, respectively, resulted in a decrease in enantioselectivity to 8% (Entry 5). However, maintaining the same conditions but with an increased loading showed a significant improvement in enantioselectivity to 55% (Entry 6), highlighting the importance of sufficient catalyst and ligand concentrations for optimal performance. Then, entries 8 and 9 investigate the effect of different additives on the reaction outcome with VO(OiPr)3 and HA2 in toluene. Using molecular sieves (MS 4Å) and MgO as additives resulted in slight improvements in enantioselectivity, achieving 18% and 22% respectively (Entries 8 and 9). While these additives showed some positive effects, the improvements were not substantial compared to the reactions without additives. Additionally, entries 12 to 19 explore the influence of temperature on the reaction using various HAs in toluene. Lowering the temperature to 0 °C generally improved enantioselectivity slightly compared to room temperature reactions. For example, HA2 showed a modest improvement from 23% enantiomeric excess at room temperature (Entry 1) to 21% at 0 °C (Entry 14). Similarly, HA5 resulted in 24% enantiomeric excess at 0 °C (Entry 18). Finally, the highest best-performing activity of 71% was observed with HA3 in CH2Cl2 at room temperature (Entry 3). This result suggests that HA3 in combination with VO(OiPr)3 and CH2Cl2 is the optimal condition among those tested for achieving high enantioselectivity in the epoxidation reaction.
Finally, Scheme 4 provides a visual rationale for the enantioselectivity trends observed in our study of hydroxamic acid ligands. Section A illustrates the chiral environment around the vanadium center using a quadrant model, where the ligand's architecture defines equivalently sterically and electronically regions II and III. Section B shows the superior performance often associated with C2-symmetric bishydroxamic acids (BHAs), as their inherent symmetry creates a well-defined and rigid chiral pocket that consistently favors one enantiofacial approach of the allylic alcohol substrate.
In contrast, the C1-symmetric hydroxamic acids (HAs) investigated in this work introduce structural asymmetry, leading to a less uniform and more flexible chiral environment. As depicted in section C, the non-identical substituents in a C1-symmetric ligand result in a less symmetrical arrangement of the quadrants around the metal center. This flexibility can lead to a distribution of substrate-catalyst conformations, which often diminishes enantiocontrol, as reflected in the generally moderate enantiomeric excess values obtained. Nevertheless, the exceptional case of HA3, which achieved 71% ee, suggests that with precise steric and electronic tuning, the unique geometry of a C1-symmetric ligand can create a highly selective chiral pocket that rivals or even surpasses the performance of more symmetrical analogs, underscoring the potential for further optimization in ligand design.
4. Conclusions
This comparative study elucidates how ligand symmetry governs the stereochemical outcome of vanadium-catalyzed asymmetric epoxidations. The results reveal that C2-symmetric bishydroxamic acids (BHAs) establish rigid and well-defined chiral environments that favor high enantioselectivity but can suffer from reduced catalytic turnover. In contrast, C1-symmetric hydroxamic acids (HAs) introduce controlled asymmetry that allows greater substrate flexibility and, in specific configurations (HA3), can achieve competitive enantiocontrol (up to 71% ee). The observed trends are consistent with the Sabatier principle and can be rationalized using a quadrant-based steric model, where desymmetrization modulates the accessibility and stability of the chiral pocket.
These insights emphasize that ligand desymmetrization is not merely a loss of symmetry but a potential tool for tuning both selectivity and activity in asymmetric catalysis. Future work will focus on expanding the substrate scope, exploring solvent–ligand–metal interactions, and integrating computational modeling to validate the proposed transition-state geometries. Collectively, this study contributes to the rational design of hydroxamic acid ligands and reinforces their potential as adaptable scaffolds for high-performance asymmetric oxidations.
Acknowledgements
This work was supported by the Secretaría de Humanidades, Ciencia, Tecnología e Innovación (SECIHTI) through the Frontier and Basic Science 2025 program, under project number CBF-2025-I-246.
References
- Li, Z.; Yamamoto, H. Hydroxamic Acids in Asymmetric Synthesis. Acc. Chem. Res. 2013, 46 (2), 506–518. [CrossRef]
- Pawar, T. J.; Bonilla-Landa, I.; Reyes-Luna, A.; Barrera-Méndez, F.; Enríquez-Medrano, F. J.; Díaz-de-León-Gómez, R. E.; Olivares-Romero, J. L. Chiral Hydroxamic Acid Ligands in Asymmetric Synthesis: The Evolution of Metal-Catalyzed Oxidation Reactions. ChemistrySelect 2023, 8, e202300555. [CrossRef]
- Olivares-Romero, J. L.; Li, Z.; Yamamoto, H. Hf(IV)-Catalyzed Enantioselective Epoxidation of N-Alkenylsulfonamides and N-Tosyl Imines. J. Am. Chem. Soc. 2012, 134 (12), 5440–5443. [CrossRef]
- Li, Z.; Yamamoto, H. Zirconium(IV)- and Hafnium(IV)-Catalyzed Highly Enantioselective Epoxidation of Homoallylic and Bishomoallylic Alcohols. J. Am. Chem. Soc. 2010, 132 (23), 7878–7880. [CrossRef]
- Olivares-Romero, J. L.; Li, Z.; Yamamoto, H. Catalytic Enantioselective Epoxidation of Tertiary Allylic and Homoallylic Alcohols. J. Am. Chem. Soc. 2013, 135 (9), 3411–3413. [CrossRef]
- Wang, C.; Yamamoto, H. Tungsten-Catalyzed Asymmetric Epoxidation of Allylic and Homoallylic Alcohols with Hydrogen Peroxide. J. Am. Chem. Soc. 2014, 136 (4), 1222–1225. [CrossRef]
- Pawar, T. J.; Valtierra-Galván, M. F.; Rodríguez-Hernández, A.; Reyes-Luna, A.; Bonilla-Landa, I.; García-Barradas, O.; Barrera-Méndez, F.; Olivares-Romero, J. L. Synthesis of Novel C₂-Bishydroxamic Acid Ligands and Their Application in Asymmetric Epoxidation Reactions. Synlett 2023, 34, 2496–2502. [CrossRef]
- Medford, A.; Vojvodic, A.; Hummelshøj, J.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J. K. From the Sabatier Principle to a Predictive Theory of Transition-Metal Heterogeneous Catalysis. J. Catal. 2015, 328, 36–42. [CrossRef]
- Suto, Y.; Tsuji, R.; Kanai, M.; Shibasaki, M. Cu(I)-Catalyzed Direct Enantioselective Cross-Aldol-Type Reaction of Acetonitrile. Org. Lett. 2005, 7 (17), 3757–3760. [CrossRef]
- Pfaltz, A.; Drury, W. J. Design of Chiral Ligands for Asymmetric Catalysis: From C₂-Symmetric P,P- and N,N-Ligands to Sterically and Electronically Nonsymmetrical P,N-Ligands. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (16), 5723–5726. [CrossRef]
- Cussó, O.; Cianfanelli, M.; Ribas, X.; Gebbink, R. J. M. K.; Costas, M. Iron-Catalyzed Highly Enantioselective Epoxidation of Cyclic Aliphatic Enones with Aqueous H₂O₂. J. Am. Chem. Soc. 2016, 138 (8), 2732–2738. [CrossRef]
- Trost, B. M.; Donckèle, E. J.; Thaisrivongs, D. A.; Osipov, M.; Masters, J. T. A New Class of Non-C₂-Symmetric Ligands for Oxidative and Redox-Neutral Palladium-Catalyzed Asymmetric Allylic Alkylations of 1,3-Diketones. J. Am. Chem. Soc. 2015, 137 (7), 2776–2784. [CrossRef]
- Trost, B. M.; Hung, C.-I.; Koester, D. C.; Miller, Y. Development of Non-C₂-Symmetric Prophenol Ligands: The Asymmetric Vinylation of N-Boc Imines. Org. Lett. 2015, 17 (15), 3778–3781. [CrossRef]
- Pfaltz, A. Recent Developments in Asymmetric Catalysis. Chimia 2001, 55 (9), 708–714. [CrossRef]
- RajanBabu, T. V.; Casalnuovo, A. L. Role of Electronic Asymmetry in the Design of New Ligands: The Asymmetric Hydrocyanation Reaction. J. Am. Chem. Soc. 1996, 118, 6325–6326. [CrossRef]
- Banerjee, A.; Yamamoto, H. Direct N–O Bond Formation via Oxidation of Amines with Benzoyl Peroxide. Chem. Sci. 2019, 10, 2124–2129.
- Zhang, W.; Basak, A.; Kosugi, Y.; Hoshino, Y.; Yamamoto, H. Enantioselective Epoxidation of Allylic Alcohols by a Chiral Complex of Vanadium: An Effective Controller System and a Rational Mechanistic Model. Angew. Chem. Int. Ed. 2005, 44 (28), 4389–4391. [CrossRef]
Scheme 1.
Synthesis of N-OBz amines.
Scheme 2.
Synthesis of chiral hydroxamic acids.
Scheme 4.
Plausible transition state of the C2-symmetric BHA versus C1-HA and quadrant diagram.
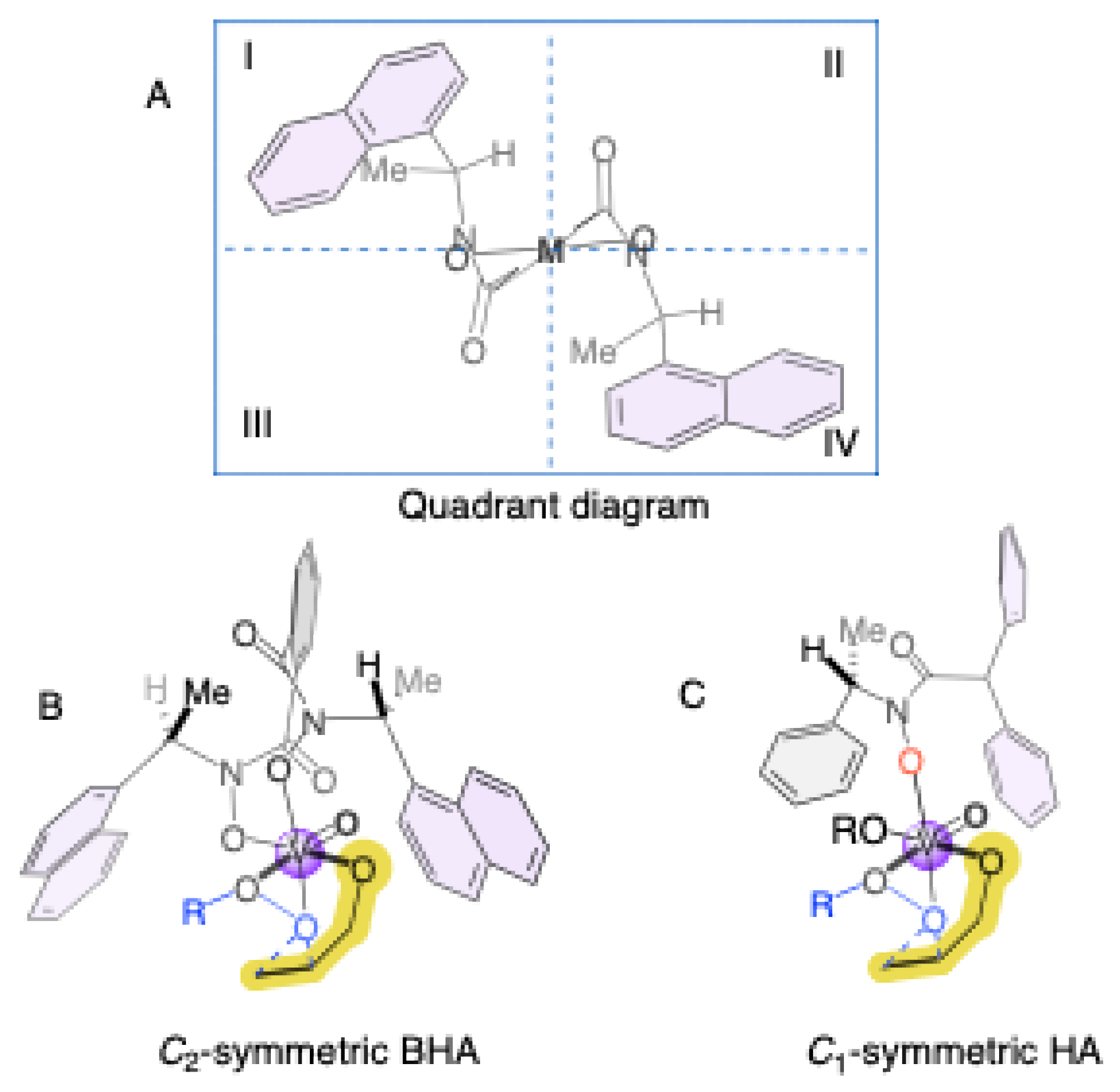
Table 1.
Comparison of enantioselectivities using BHA1 and HA3.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Lewis Acid (% mol) |
HA (% mol) |
Solvent | Temperature (°C) |
Conversion (%) | e.e.a (%) | |
| 1 | VO(OiPr)3 (10) | BHA1 (10) |
CH2Cl2 | -20 | 14 | 9 | |
| 2 | VO(OiPr)3 (10) | BHA2 (10) |
CH2Cl2 | -20 | 11 | 7 | |
| 3 | VO(OiPr)3 (10) | HA3 (12) |
Toluene | r.t. | ≥99 | 19 | |
| 4 | VO(OiPr)3 (10) | HA7 (12) |
Toluene | r.t. | ≥99 | 49 | |
Table 2.
Analysis of chiral hydroxamic acid architecture and Lewis Acids.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Lewis Acid (10% mol) |
HA (12% mol) |
Conversion | e.e.a | |||
| 1 | Ti(OiPr)4 | HA3 | 0 | n.d. | |||
| 2 | VO(OiPr)3 | HA3 | ≥99 | 19 | |||
| 3 | Ti(OiPr)4 | HA2 | 0 | n.d. | |||
| 4 | VO(OiPr)3 | HA2 | ≥99 | 22 | |||
| 5 | VO(OiPr)3 | HA1 | ≥99 | 15 | |||
| 6 | VO(OiPr)3 | HA6 | ≥99 | 4 | |||
| 7 | VO(OiPr)3 | HA7 | ≥99 | 49 | |||
| 8 | VO(OiPr)3 | HA4 | ≥99 | 11 | |||
| 9 | VO(OiPr)3 | HA5 | ≥99 | 30 | |||
| 10 | VO(acac)2 | HA2 | ≥99 | 19 | |||
The enantiomeric excesses were determined by chiral HPLC.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



