Submitted:
13 October 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
To those who did not follow the invention and development of enantioselective catalysis, this mini-review introduces some pertinent historical aspects of the field and presents the scientific concepts of asymmetric bio- and organocatalysis. They are powerful technologies applied in organic laboratories and industry. They realize chiral amplification by converting inexpensive achiral substrates and reagents into enantiomerically enriched products using readily recoverable solvents, if any. Racemic substrates can also be deracemized catalytically. More sustainable fabrications are now available that require neither toxic metallic species, nor costly reaction conditions in terms of energy, atmosphere control, product purification and safety. Nature has been the source of the first asymmetric catalysts (microorganisms, enzymes, alkaloids, amino acids, peptides, terpenoids, sugars and their derivatives). They act as temporarily chiral auxiliaries and reduce reaction activation free enthalpies by changing their mechanisms. Reductions, oxidations, carbon-carbon and carbon-heteroatom bond forming reactions are part of the process panoply. Asymmetric catalyzed multicomponent and domino reactions are becoming common. Typical modes of activation are proton transfers, hydrogen bonded complex formation, charged or uncharged acid/base pairing (e.g., -hole catalysts), formation of equilibria between achiral aldehydes and ketones with their chiral iminium salt or/and enamine intermediates, Umpolung of aldehydes and ketones by reaction with N-heterocyclic carbenes (NHCs), phase transfer catalysis (PTC), etc. Often, best enantioselectivities are observed with polyfunctional catalysts derived from natural compounds, or not. They may combine in a chiral structure nitrogen, phosphorus, sulfur, selenium, iodine functional moieties. Today, man made enantiomerically enriched, if not enantiomerically pure, catalysts are available in both their enantiomeric forms. Being robust, they are recovered and reused readily.
Keywords:
chiral amplification
; enzymes
; man made polyfunctional catalysts
; natural products
; non-metallic catalyzed enantioselective reactions
; reaction mechanisms
1. Introduction
The invention and development of catalytic enantioselective reactions [1] are among the most studied topics of stereochemistry and chemical synthesis. Enantioselective catalysis has become routine in the pharmaceutical industry [2,3,4] and other industries (crop protection, fragrances) [5,6,7,8]. Before 2000, asymmetric catalysis relied mostly upon organometallic complexes (not discussed here) and biocatalysis (Chap. 2) [9]. During the last twenty-five years, organocatalysis has dominated the field of asymmetric synthesis (Chap. 3-11) [10,11,12]. A large number of enantioselective reactions are available. This mini-review presents a selection of them. It has some educational value for chemists with only basic training in stereochemistry.
Early contributions to asymmetric synthesis [13,14] were reported by Emil Fischer [15], Markwald [16], and McKenzie [17] in the early 1900s. Most natural products (terpenes, steroids, alkaloids, lactic acid, tartaric acid, etc.) are chiral and enantiomerically enriched, if not enantiomerically pure. Life's building blocks (amino acids, nucleic acids, carbohydrates, etc.) are chiral and enantiomerically pure (enantiopure). The biopolymers resulting from their combination (e.g., peptides, proteins, oligosaccharides, polysaccharides, glycoproteins, DNA, RNA) are also chiral and enantiopure. Most biomolecules and their biopolymers are homochiral, i.e., they contain only one of two possible enantiomers. Homochirality is a signature of life [18,19,20,21]. It has been a dream for chemists to imitate Nature that, through photosynthesis, converts water and carbon dioxide (two achiral reagents) into homochiral compounds such as D-glucose.
(+)-(R)-Limonene is responsible for the orange flavor, while its enantiomer, (-)-(S)-limonene, is a constituent of the lemon flavor. (+)-(S)-Carvone is found in the smell of cumin sheaths, (-)-(R)-carvone is a component of spearmint smell. D-Asparagine has a sweet taste, L-asparagine is bitter [22,23]. When a chiral drug interacts with its receptor (e.g., an enzyme), it is no surprise that its two enantiomers interact differently and may lead to different biological effects [24,25,26,27,28]. Your right foot fits better in a right shoe than in a left shoe. In the 1960s, racemic Thalidomide (1:1 mixture of two possible enantiomers) had been administered to pregnant women who gave birth to babies with physical deformations. This disaster is a convincing example of the dependence of pharmacological response toward both enantiomers of a chiral drug. The (R)-enantiomer of Thalidomide exhibits desirable analgesic properties; however, the (S)-enantiomer does not and is responsible for the terrible teratogenic effects [29]. Metabolite racemates are also present in Nature [30].
1.1. Discovery of Biocatalysis and Chirality
In 1858, Louis Pasteur reacted the racemic mixture of ammonium tartrate with a microorganism called penicillum glaucum [31]. An enzyme of this microorganism breaks down the (2R,3R)-enantiomer of ammonium tartrate (salt derived from L-(+)-tartaric acid) completely and leaves the (2S,3S)-enantiomer of ammonium tartrate almost unchanged (salt of D-(-)-tartaric acid). This observation has led to a biochemical method of discrimination between two enantiomers widely used today in industry [32,33,34,35,36,37,38].
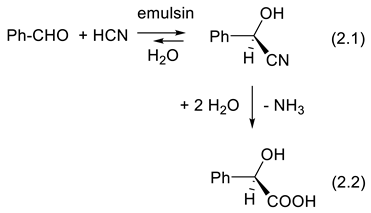
In 1894, Emil Fischer proposed his “lock and key” model for enzyme-catalyzed reactions [39]. A right hand enters a right glove more easily than a left hand. Enzymes display remarkable catalytic activity. This makes them very much used today as catalysts in the industry when mild reaction conditions (temperature, pH) are required [9,40]. After Pasteur's discoveries, the production of enantiomerically enriched compounds from racemic mixtures or achiral substrates was attempted. In 1908, Rosenthaler reported the first example: the addition of HCN to benzaldehyde (PhCHO), which yields mandelonitrile (see below). The chiral catalyst was emulsin, an enzyme isolated from the almond. Today, catalysts made by chemists that are not enzymes or microorganisms are becoming as effective, and often they are easier to use [10]. The enantioselectivities are very high, as are their TON (turnover number: moles of product formed per mole of catalyst) and their TOF (turnover number per unit of time). They can work under conditions (temperature, solvent, nature of reactants) that can be varied much more than with enzymes or microorganisms. Often, an enzyme allows the preparation of only one of the two possible enantiomers. Chemists produce enantiomerically enriched catalysts; they enable us to obtain either one or the other enantiomer of a chiral product. Nevertheless, enzymes and microorganisms remain interesting as catalysts for ecological reasons (no heavy metals, water as solvent, green chemistry).
According to the IUPAC, “asymmetry” in chemistry is a property of three-dimensional objects, such as molecules and crystals, devoid of any symmetry element. These objects belong to the symmetry point group C1 (e.g., (R)- and (S)-lactic acid: MeCH(OH)COOH). Like your left hand and right hand, the mirror image of a chiral object is not superimposable with itself. Molecules with a two-fold axis of rotation, such as (2R,3R)- and (2S,3S)-tartaric acid: HOOC-CH(OH)-CH(OH)-COOH), are also chiral (symmetry point group C2). Objects sharing a plane of symmetry are achiral. “Asymmetric induction” describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other due to the influence of a chiral feature (chiral promoter) present in the substrate, reagent, catalyst, or environment. “Asymmetric syntheses” are those reactions, or sequences of reactions, which produce chiral non-racemic substances from achiral compounds with the intermediate use of chiral non-racemic materials, but excluding a separation operation [41]. With a chiral promoter, the achiral substrate (single reactant reaction, or multiple reactant reaction, one of the reactant called substrate, the other(s) reactant(s)) enters equilibria with complexes and/or aggregates that are diastereomeric (two diastereomers have different physical and chemical properties, Figure 1.1). This is the case for catalysts that are ion pairs (highly polar species), Brønsted–Lowry acids (protic acids), Lewis acids, bases, enzymes, neutral molecules, or metallic species (less polar systems). The chiral promoter and the complexes it forms also interact with solvent molecules. The chiral promoter can be a soluble substance or a solid interacting with its surface [42]. In 1902, Henri proposed a general theory for the sucrase-catalyzed hydrolysis of sucrose (saccharose) into fructose and glucose (reaction called inversion because the angle of specific rotation of the plane-polarized light changes from a positive to a negative value) in which he recognized that the enzyme equilibrates with a complex containing the substrate (sucrose) [43]. In 1913, Michaelis and Menten proposed their classical mechanism for the enzyme (E) catalyzed reaction (eq. (1.1)) which converts substrate S into product P, via an intermediate X [44,45].
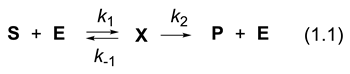
The intermediates complexes or/and aggregates invoked above (correspond to two different intermediates X and X’) can have different stability or/and rates for their transformation into chiral products that are enantiomers. They evolve to at least two transition states that can be associated with different free enthalpy of activation (Δ‡GT). If the chiral promoter is enantiomerically enriched two enantiomeric products can be obtained in proportion (er = enantiomeric ratio) different from 50/50. One defines enantiomeric excess ee = [fraction of the major enantiomer - fraction of the minor enantiomer]/[sum of the fractions of major and minor enantiomers]. An enantiomerically enriched product is called scalemic mixture (er > 50/50 and ee > 0%). An enantiomerically pure compound (ee = 100%) does not exist because no analytical method can measure er = 100/0. The detection limit for the minor enantiomer cannot be zero, but it can be relatively small. For instance, if the detection limit for a minor enantiomer is 1/1000, er > 99.9/0.1, or ee > 99.8%. The process becomes catalytic and enantioselective if the enantiomerically enriched promoter is used in sub-stoichiometric quantities. Asymmetric catalysis might be accompanied by chiral amplification (ee of the product is higher than that of the catalysts, non-linear effect), chiral loss (ee of the product is lower than that of the catalysts), or not [46,47,48,49,50,51]. Under kinetic control, a racemic chiral promoter leads to a racemic mixture of the product. Its enantiomeric ratio er =50/50 and its enantiomeric excess ee = 0%.
1.2. Kinetic Enantioselectivity
A large number of catalysts may be soluble in the reaction medium (homogeneous catalysis). Others, such as crystalline quartz, zeolites, or metal surfaces impregnated with chiral molecules, are not (heterogeneous catalysis). Since two enantiomers have the same stability in an achiral environment, the rate of their equilibration (racemization) might compete with the rates of their formation, and thus lead to loss of enantioselectivity. The catalyst catalyzes the forward reaction as well as the backward (opposite) reaction (microscopic reversibility principle). The highest enantioselectivities are obtained under non-equilibrating conditions (exergonic equilibria, KT > 1), i.e., under kinetic control. The transition states of these reactions are composed of diastereomeric transition structures. For the addition of achiral HCN (linear structure) to achiral PhCHO (shares a plane of symmetry) to give mandelonitrile (PhCH(OH)CN), one of the two achiral starting materials must be prochiral to lead to chiral products. HCN is not prochiral, but PhCHO is for this reaction. It is also the case for the hydrogenation of PhCHO with deuterium (D2) into chiral PhCH(D)OD, but not for the hydrogenation with H2, giving achiral PhCH2OH. The mandelonitrile contains an asymmetric carbon center and has no symmetry elements. The drawings in Figure 1.1 illustrate this point with two different bars welded together (no symmetry element). Note that dimerization may generate chiral products that have a C2 axis of rotation. An example is the oxidative dimerization of 2-naphthols into 1,1’-binaphthols [52]. Another example of C2-symmetrical chiral compound is (-)-(2R,3R)-2,3-butanediol (CH3-CH(OH)-CH(OH)-CH3, see reaction (2.6)). If the two bars of the drawings in Figure 1.1 were identical, and if their points or surfaces of binding were in the same location for the two bars, this characteristic would be illustrated.
1.3. Chiral Catalysts Are Temporary Chiral Auxiliaries
A catalyzed reaction is faster than the same uncatalyzed reaction. The catalyst interacts with the reactants and substrates; it is released at the end of the reaction. It changes the reaction mechanism in several possible ways. We consider the case of a prochiral substrate A that combines with achiral reactant B to generate products (R)-A-B and (S)-A-B. The chiral catalyst C* interacts first with A and equilibrates with intermediate A-C*. Several options are considered. In one of them, this intermediate combines with B and equilibrates with a complex in which A and B approach together and are “forced” to react. This situation corresponds to Diels-Alder reactions ((4+2)-cycloadditions) of alkyl-substituted 1,3-dienes adding to α,β-unsaturated carbonyl compounds (dienophiles) to generate cyclohexene or cyclohexa-1,4-diene derivatives. In the absence of a catalyst, the reaction is a one-step process in which the two new σ-bonds form in the transition state in concert, not necessarily to the same extent. In 1942, Wassermann observed that protic acids accelerate these reactions [53]. In 1960, Yates reported that AlCl3 accelerates the Diels-Alder reactions of maleic anhydride, dimethyl fumarate and benzoquinone. The protic acid protonates the carbonyl group of the dienophiles, AlCl3 forms Lewis carbonyl/AlCl3 complexes that activate the dienophiles. According to the PMO (perturbation of molecular orbitals) theory the rate of the cycloaddition is dominated by the LUMO(dienophile)/HOMO(diene) interaction (normal electronic demand). The smaller is the energy gap between these frontier orbitals, the faster is the reaction. Upon protonation of the carbonyl group of the dienophile, or formation of a Lewis carbonyl/AlCl3 complex, the energy of the LUMO(dienophile) is reduced, thus accelerating the cycloaddition. This occurs without significant change in the nature (nearly the same chemical functions) and geometry of the dienes and dienophiles compared with the non-catalyzed reaction [54].
In another option, intermediate A-C* transforms A chemically, generating a second intermediate A’-C*, which then combines with B. A’-C* reacts quickly with B, much quicker than A + B or A-C* + B. The catalyst not only brings A and B together, but also activates A chemically. One speaks here of catalysis by chemical activation. If B is prochiral, intermediate A’-C* reacts, forming two possible enantiomers (R)-A-B and (S)-A-B with two different rates, thus leading to enantioselectivity. Figure 1.2 illustrates an attempt to explain this concept with macroscopic objects. This mini-review describes several examples of this type of catalysis. To illustrate this concept, the reader may consider a chiral catalyst with an amine function that reacts with an aldehyde to equilibrate with a chiral iminium salt, a better electrophile than the starting aldehyde (see e.g., MacMillan’s enantioselective Diels-Alder reaction catalyzed by a secondary chiral amine, Section 3.1.5, Figure 3.7). Furthermore, the amine catalyst might react with an aldehyde or a ketone equilibrating with a chiral enamine, which is more nucleophilic than the corresponding enol (see e.g., Section 3.5). Another example is the Umpolung of aldehydes with heterocyclic nucleophilic carbenes into acyl anion equivalents (Breslow intermediates), which then can add to electrophiles such as aldehydes (homo-benzoin condensation, Section 8.1), other carbonyl compounds (cross-benzoin condensation, Section 8.1), aldimines (aza-benzoin condensation, Section 8.2), or Michael acceptors (Stetter reaction, Section 7.3).
2. Biocatalysis
When the catalyst is an enzyme or a microorganism, one speaks of enzymatic catalysis or biocatalysis. Depending on substrates, reagents, and conditions, these reactions might be chemoselective, regioselective, diastereoselective, and enantioselective [55]. They are applied more and more frequently in organic chemistry [56,57,58,59,60] and in industry [9,61,62,63]. Biocatalysis is a sustainable approach, a green tool (no heavy metals, water as solvent) for the obtainment of enantiomerically enriched substances of high value, such as drugs [64].
Enzymes involved in primary metabolism may accelerate chemical reactions by up to 26 orders of magnitude [65]. In 1975, the Chemistry Nobel Prize was granted to John W. Cornforth for his work on the stereochemistry of enzyme-catalyzed reactions [66], and to Vladimir Prelog for his research into the stereochemistry of organic molecules and reactions [67]. Apart from possible chemical reactions between the catalyst, the reagent, and/or the substrate, the enzyme interacts with them and the solvent through an array of weak and/or strong electrostatic interactions [68]. Their relative importance increases from induced-dipole/induced dipole (London dispersion forces), dipole-induced dipole, dipole-dipole (e.g., hydrogen bonds [69]), ion-dipole ligation, to ion pairing [42]. Enzymes have structural scaffolds with active sites that provide a complementary structural and electrostatic environment to substrates, reactants, and transition structures of their reaction. The catalytic effect results from the fact that enzymes bind and stabilize transition structures better than starting materials, intermediates, and final products [70,71]. Jean-Marie Lehn, [72,73] Charles Pedersen, [74] and Donald Cram [75,76] were awarded the 1987 Chemistry Nobel Prize for their development and use of molecules with structure-specific interactions of high selectivity[77]. Their findings helped chemists to understand biocatalysis and to design new non-enzymatic catalysts [78]. Recently, protein engineering created non-natural enzymes with excellent activity and stereoselectivity [79,80,81]. Due to their solubility and instability, enzymes are not recovered easily after use. To overcome these problems, they are immobilized by combining them with various possible supports. These biocatalysts can be readily recovered by simple filtration, centrifugation, or other extractive techniques [82,83].
The vigorous development of asymmetric organocatalysis (Chap. 3-11) during the last 25 years was inspired by enzymatic catalysis [84]. Many asymmetric reactions applied enzymes first. Then, much smaller molecules were found to catalyze the same enantioselective reactions. An exception to this rule is the Stetter reaction, which condenses conjugated enones or ene-esters with aldehydes, forming 1,4-dicarbonyl compounds (nucleophilic acylation of Michael acceptors). In 1996, Enders and co-workers reported the first asymmetric version of an intramolecular Stetter reaction. In 2008, they realized enantioselective intermolecular Stetter reactions applying triazolium-derived N-heterocyclic carbenes as chiral catalysts. The Rovis group also contributed to the development of this important C-C bond-forming reaction (see Section 8.3). In 2009, Müller and co-workers reported the first examples of thiamine diphosphate (ThDP)-dependent enzyme-catalyzed asymmetric Stetter reactions [85,86].
2.1. Hydroxynitrile Lyases
More than 2000 plants possess cyanogenic glycosides that liberate hydrogen cyanide (HCN) upon hydrolysis or under the action of an enzyme (e.g., β-glucosidase). This is the case with bitter almonds. Eating 50 bitter almonds may kill an adult. Fortunately, this is not the case with sweet almonds [87]. In 1837, using an open flask, Wöhler and Liebig applied a crude hydroxynitrile lyase on mandelonitrile (PhCH(OH)CN) to liberate HCN and generate benzaldehyde (PhCHO) (backward reaction of equilibrium (2.1)) [88]. Rosenthaler realized the opposite reaction in 1908 (forward reaction of equilibrium (2.1)). Using a crude enzyme extracted from almonds (emulsin), a relatively high concentration of PhCHO and HCN in water, in a closed flask at 20-30 oC he obtained an optically active cyanohydrin in good yield. Under acidic conditions (HCl/H2O), (+)-(R)-mandelonitrile extracted from the reaction mixture with CHCl3 was hydrolyzed into (-)-(R)-mandelic acid (reaction (2.2)). These were the first enantioselective syntheses ever from achiral substrates [89].
Today, hydroxynitrile lyases of various sources are available. They allow the enantioselective synthesis of a large number of important industrial intermediates in both enantiomeric forms [90,91,92,93,94]. Furthermore, new enzymes are engineered through directed evolution [95,96,97,98,99], and some of them catalyze different reactions that follow similar mechanisms, or not. For instance, hydroxynitrile lyases catalyze the enantioselective condensation of nitroalkanes to aldehydes to form β-nitroalcohols (equilibrium (2.3)) [100,101,102,103].
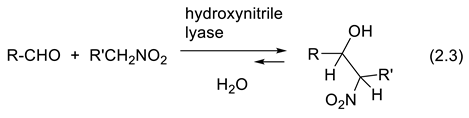
Arabidopsis thaliana hydroxynitrile lyase (AtHNL) engineering has uncovered variants with up to 12-fold improved catalytic efficiency compared to the wild type towards asymmetric Henry reaction with up to 99% ee [104]. Enzyme promiscuity is the property of enzymes that can catalyze unexpected reactions, and/or transform unexpected substrates, and work under unexpected reaction conditions [105,106,107,108].
2.2. Esterases
In 1975, Huang and coworkers reported that dimethyl glutarate (meso-compound) was hydrolyzed with PLE (pig liver esterase) in high enantioselectivity into the corresponding monoester (reaction (2.4)) [109]. Several reports on the enzyme-catalyzed desymmetrization of meso-diesters followed [110].
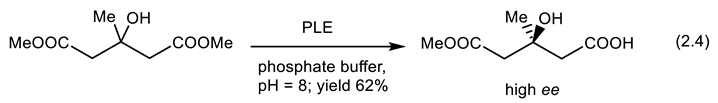
Desymmetrization of malonic esters is predominated by the catalytic hydrolysis using crude pig liver esterase (PLE, EC 3.1.1, enzyme that hydrolyzes esters) to give α-quaternary carboxylic acids [111,112]. Very important reactions such as aldol condensation, Knoevenagel condensation, Hantzsch reaction, Michael addition, Canizzaro reaction, Mannich reaction, Morita-Baylis-Hillman (MBH) reaction, Ugi reaction, and oxidation have been catalyzed by lipases (esterases that hydrolyze fat) [113].
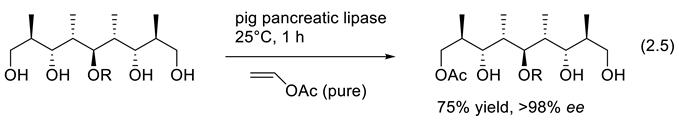
The opposite reaction to esterase hydrolysis is acylation. It requires enol esters as reagents. An example reported by Chênevert is reaction (2.5) that does the desymmetrization of an advanced intermediate in the total synthesis of natural polypropionates [114,115], including Rifamycin S [116].
Enzymatic ester hydrolyses generally proceed by acylation of the enzyme, followed by hydrolysis of the acylated species. With enol esters, this hydrolysis occurs with concomitant acyl group cleavage and C-protonation of the liberated enols. In 1990, Ohta and coworkers reported that the yeast Pichia farinosa IAM4682 catalyzes the enantioselective hydrolysis of prochiral enol esters [117]. Since then, carboxylases, esterases, and lipases were shown to catalyze the enantioselective enolate protonation and enol isomerization [118].
2.3. Baker’s Yeast
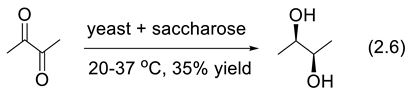
The asymmetric reduction of ketones by baker’s yeast (Saccharomyces cerevisiae) is one of the most used reactions induced by a microorganism. Sometimes it is catalytic; it uses at least one equivalent of a reducing agent such as glucose, sucrose (saccharose), glycerol, or malic acid [119,120,121,122,123,124,125]. The asymmetric reduction of diacetyl (2,3-butanedione) into levorotatory 2,3-butanediol ((-)-(2R,3R)-2,3-butanediol, 35% yield) was reported in 1919 by Neuberg and Nord (reaction (2.6)) [126].
Most yeast-induced reductions of prochiral monoketones (R1-CO-R2, R1 different from R2) are highly enantioselective, leading to the corresponding (S)-alcohols. Formally, the reaction adds a molecule of H2 onto the carbonyl (C=O) moiety, giving the corresponding secondary alcohol (R1-CH(OH)-R2). The re face of dialkylketones is preferred, giving (S)-alcohols (Figure 2.1). Often, formation of ethanol competes (up to 2000 moles per mole of secondary alcohol, which requires up to 1000 moles of glucose) [127,128].
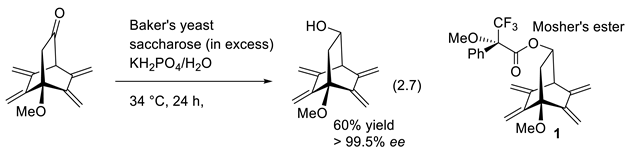
Baker’s yeast reduction of complicated ketones can be highly enantioselective. One example is reaction (2.7) [130]. Mosher’s esters (e.g., 1) permit accurate measurement of the optical purity of alcohols (er), through HPLC, 1H-NMR, 13C-NMR or 19F-NMR (satellite signals 13C-X of major ester compared with 1H, 19F signals of the minor enantiomer) [131]. Alternatively, HPLC with a chiral stationary phase permits the determination of er (chiral column chromatography) [132].
Reductions of C=C double bonds of (E)- and (Z)-methyl 2-chloro-2-alkenoate by baker’s yeast give (R)- and (S)-2-chloroalkanoic acid in 25-92% ee and >98% ee, respectively [133]. These reactions show the reaction promiscuity of this microorganism.
2.4. Fermentative Oxidation
In 1968, Gibson and coworkers showed that Pseudomonas putida, grown with toluene as the sole source of carbon, oxidized chloro, bromo-, iodo-, and fluorobenzenes to their respective 3-halogenated catechol derivatives. They also found that oxidation of 4-chlorotoluene generates (+)-cis-4-chloro-2,3-dihydroxy-1-methylcyclohexa-4,6-diene next to 4-chloro-2,3-dihydroxytoluene [134]. Since then, high-yielding whole-cell fermentations of arenes into substituted cis-cyclohexa-3,5-dienes have been obtained with high ee’s [135,136,137]. These compounds have been tansformed into all kinds of products of biological interest (see e.g., Figure 2.2) [138,139,140,141,142,143,144].
2.5. Aldolases
In 1934, Meyerhof and Lohmann discovered that aldolases are ubiquitous enzymes in plants and animals [145,146]. They catalyze the backward reaction of aldol equilibrium (2.8) in vivo (Figure 2.3). Thus, at high concentrations D-glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) are condensed to generate fructose 1,6-diphosphate FDP (forward reaction of equilibrium (2.8)). Warburg and Christian first recognized the difference in yeast and muscle aldolase properties [147,148,149]. Type I aldolases (from higher plants and animals) such as muscle aldolases are not inhibited by metal-chelating agents and do not contain divalent metals, and type II aldolases (e.g., yeast aldolase from microorganisms [150]) are inhibited by divalent metal-chelating agents and contain Zn++ [151]. For type I, aldol condensation occurs via a Schiff base resulting from the reaction of the aldehyde and the ε-amino group of a lysine unit of the enzyme (Mech. 1, Figure 2.3). In the case of type II aldolases, the Zn++ cation activates the aldehyde via co-coordination with a histidine unit (Mech. 2, Figure 2.3).
An early case of rabbit muscle aldolase (RAMA, E.C.4.1.2.13) catalyzed aldol reaction was reported in 1960 by Jones and Sephton with the synthesis of octulose derivatives through condensation of 1,3-dihydroxy-2-propanone phosphate with pentoses [152]. Then, this biocatalyst was used to catalyze asymmetric aldol condensations with achiral aldehydes. Wide explorations have been carried out by the groups of Whitesides [153,154], Wong, [155,156,157,158] and of others [159,160,161]. The RAMA accepts all aldehydes as electrophilic, except those derivatives congested in the α position, the α,β-unsaturated aldehydes, or those that easily lead to them by water elimination. The products have the D-threo (3S,4R) stereochemistry. As a rule, the enzyme accepts DHAP as the privileged nucleophile. This permitted the asymmetric total synthesis of carbohydrates and analogs of biological interest [162,163].
In Nature, deoxy-D-ribose-5-phosphate aldolase (DERA, EC 4.1.2.4) is a part of the nucleoside salvage pathway; it catalyzes the retro-aldol reaction, forming glyceraldehyde-3-phosphate and acetaldehyde from 2-deoxy-D-ribose-5-phosphate (sometimes assigned to Type III aldolases) [164]. DERA possesses the unique function to catalyze the aldol reaction between two aldehydes instead of an aldehyde and a ketone [165,166]. It accepts a large variety of aldehydes as electrophiles. In addition, DERA can catalyze sequential addition of acetaldehyde, resulting in the formation of more than one stereogenic center (e.g., equilibrium (2.9), Figure 2.4). The entropically disfavored double condensation is overcome through rapid cyclization, generating stable cyclic hemiacetals (pyranose). The sequential aldol reaction has been applied successfully in industry, for example, in the synthesis of statins (Figure 2.4), cholesterol-lowering agents [167,168]. It also permitted the asymmetric synthesis of key building blocks in the synthesis of epothilone [169]. Enantioselective side-chain intermediates in the statin synthesis rely upon whole-cell systems [170]. For other applications of biocatalysts in the industry, see: [171].
L-Threonine aldolase (LTA) is a 5′-phosphate (PLP)-dependent aldolase that catalyzes the formation of β-hydroxy-α-amino acids from glycine (Gly) and aromatic or aliphatic aldehydes [172,173]. β-Hydroxy-α-amino acids are chiral building blocks for pharmaceuticals (antibiotics and proteasome inhibitors) [174]. One recognizes four types of LTAs [175,176,177].
An aldolase called hydratase-aldolase (NahE) catalyzes the stereoselective Michael addition of pyruvate to β-nitrostyrenes. It is a type I aldolase. After oxidative decarboxylation, β-aryl-γ-nitrobutyric acids can be isolated. They give access to precursors of γ-aminobutyric acid (GABA) analogues that possess high pharmacological activity [178].
Other efficient aldolases from natural sources, or engineered through directed evolution or/and design are now available [179,180,181,182,183,184,185]. Furthermore, enamine catalytic aldolase antibodies developed in the laboratories of Lerner catalyze asymmetric aldol reactions [186,187,188]. Enzymes have been engineered to catalyze asymmetric Michael additions [189]. As we shall see below, simple chiral amines such as amino acids and small peptides and analogs can be excellent catalysts for the asymmetric intermolecular aldol and Michael reactions.
2.6. Other Asymmetric Biocatalyzed C-C Bond-Forming Reactions.
The introduction of a methyl substituent into medicinal agents can dramatically enhance their bioactivity due to the “magic methyl effect” [190]. Methods allowing for chemoselective, regioselective and stereoselective methylation of carbonyl compounds are precious [191]. More generally, catalyzed α-alkylation, α-allylation and α-propargylation of carbonyl compounds are very much desired reactions. SgvMVAV was engineered as a general biocatalyst for these reactions (2.10). The catalytic system includes a Lewis acidic Zn site that triggers substrate enolization and an adjacent SAM (S-adenosylmethionine) that permits stereoselective methyl transfer (Figure 2.5b) [192].
3. Asymmetric Amino-Catalysis: The Queen of Organocatalysis
When the catalyst is a chiral organic compound that is not an enzyme, one speaks of asymmetric organocatalysis [193,194,195,196,197,198,199,200]. Asymmetric organocatalysis is the most innovative field in fine chemistry [10]. Most organocatalysts are robust, non-toxic, and affordable. They imply easy reaction manipulations, simple purification procedures, and do not always require an inert atmosphere. Asymmetric organocatalysis realizes greener chemistry [201] (no toxic metals, less solvent) with atomic economy (fewer co-products and side-products to take care of) [202]. Organocatalysts can be utilized without solvent, or with non-toxic ones such as water, ethanol, ethyl acetate, tetrahydrofuran, methyl t-butyl ether, and toluene. They require less energy and produce less waste than organometallic and metal catalysts. Nature provides us with inexpensive enantiomerically pure compounds such as alkaloids, amino acids, terpenes, and carbohydrates. Asymmetric synthesis is also an endless source of new chiral compounds used in the construction of chiral organocatalysts. In 2021, Benjamin List and David W. C. MacMillan received the Nobel Prize in Chemistry for the development of asymmetric organocatalysis [203,204]. When the catalyst is a chiral amine, one refers to asymmetric amino-catalysis [203,205,206,207,208,209].
In 1896, Knoevenagel found that primary and secondary amines, as well as their salts, catalyze the aldol condensation of β-ketoesters or malonic diesters with aldehydes and ketones [210,211]. As we shall see, chiral amines can abstract selectively one or the other proton from enantiotopic C-H or X-H bonds (Section 3.1-1). Protonated chiral amines (ammonium salts) protonate planar enolates and other related π-systems with enantioselectivity (Section 3.1-2). The catalytic effect can also result through other modes of activation [196]. For instance, primary and secondary amines activate aldehydes and ketones through the formation of iminium ion intermediates (C=N(+)R2, electrophiles, Section 3.1-4, 3.5), through the formation of enamines (C=C-NR2, nucleophiles, Section 3.1-5, 3.6)), or through the formation of radical ammonium radical-cation intermediates (R3N(.+), Section 3.7). Moreover, chiral quaternary ammonium salts (e.g.: R3N(+)R’-X(-)) are enantioselective catalysts in phase transfer reactions (Section 3.2). Enhanced electrophilicity of α,β unsaturated acyl chloride is possible through the formation of acylammonium chloride intermediates (Section 3.8). Furthermore, the amine catalyst may work in concert with co-catalysts, or additives that contribute as H-bond donors or acceptors (Chap. 9) [212,213], as halogen (Section 11.6), chalcogen-bonding, staking, or ion-pairing [199,214,215,216]. Amino-catalysis can be combined with other types of catalysis (synergistic catalysis, see e.g., [217]) such as metal catalysis [218], or with photo-redox processes [219,220] (see Section 3.5-6 for an example). Light brings another dimension to enantioselective organocatalysis [221].
3.1. Cinchona Alkaloids and Derivatives as Catalysts
Asymmetric amino-catalysis dates back to 1912, when Bredig and Fiske added HCN to benzaldehyde (PhCHO) in the presence of cinchona alkaloids (0.3 M). For instance, with (-)-quinine (2), the corresponding cyanohydrin (PhC(CN)(OH)H was obtained in 1-2% yield and 2% ee. With (+)-quinidine (4) as catalyst, ee increased to ca. 10% (er < 55/45, forward reaction of equilibrium (2.1)) [222]. This was a groundbreaking report: asymmetric catalysis does not need a microorganism or an enzyme taken from the living world. In 1960, Pracejus added methanol (0.11 M) at - 111 oC to methyl phenylketene (Ph(Me)C=C=O, 0.1 M) in toluene. In the presence of O-acetyl quinine (3, 0.001 M), he obtained 2-phenylpropanoic methyl ester (PhCH(Me)-COOMe) in 60-70% yield and 73% ee [223]. Following this pioneer work, cinchona alkaloids became the most privileged asymmetry inducers in the area of enantioselective catalysis [224]. As illustrated in Figure 3.1 with derivative 3 (functionalization of the secondary alcohol moiety and exchange of the methoxy substituent for another group), the cinchona alkaloids can be modified easily and generate a large variety of chiral catalysts. They are chiral Lewis base catalysts (Section 3.1-1), hydrogen-bond (HB) donors (Section 3.1-2), chiral protic acids for their conjugate acids (Section 3.1-3), nucleophilic activators (Section 3.1-4), iminium ion activators (Section 3.1-4), and enamine activators (Section 3.1-6). Bifunctional catalysts with halogen-bond (XB) donors (Section 3.1-7) lead to ligand-accelerated catalysis. Quaternized ammonium salts serve as phase-transfer catalysts (Section 3.2) and as activators through hydrogen bridging (Section 3.3). Other derivatives are electron-donating agents (Section 3.4) [225,226,227,228,229].
3.1.1. Cinchona Alkaloids as Chiral Brønsted Bases
In 1980, Whitesell and Felman showed that stoichiometric amounts of a strong base, such as chiral lithium amide, induce the enantioselective conversion of cyclohexene oxide into lithium cyclohex-2-enolate (e.g., with [(S)-PhCH(Me)]2NLi, 65% yield, 31% ee) [230]. This work demonstrated that a chiral strong base can distinguish between two enantiotopic C-H bonds. The same year, Vogel and Hagenbuch reported the first examples of enantioselective catalyzed isomerization of endoperoxides into the corresponding γ-hydroxy-α,β unsaturated aldehydes (Kornblum-DeLaMare rearrangement [231]) applying cinchona-alkaloids 2-5 (Figure 3.1). For instance, in the presence of 0.0045 molar (+)-quinidine (4) a 10% solution of endoperoxide 6 in CH3CN was isomerized at 20 oC (> 95% yield) into 7 with 46% ee (reaction (3.1)). A relatively weak base such as (+)-quinidine (4) can distinguish between enantiotopic protons that are three carbon centers away from the chiral centers (C(1), C(4)) of the 7-oxanorbornane skeleton [232].
In 2006, Toste and co-workers reported the cinchona-alkaloid-catalyzed asymmetric Kornblum DeLaMare rearrangement of endoperoxides resulting from the photooxidation of cyclic dienes and obtained corresponding γ-hydroxycycloenones in good yield and up to 99% ee. For instance, the endoperoxides of cycloocta-1,3-diene were isomerized into 4-hydroxycyclooct-2-en-1-one in 99% yield and 99% ee in the presence of the quinidine derivative 5 [233].
In 2010, Barbas and coworkers reported that dimeric forms of cinchona alkaloids are better base catalysts than their monomeric forms (Figure 3.2a). For instance, in the presence of catalyst 8, aminooxydation of 2-oxindoles furnished the corresponding 3-hydroxyoxindole precursors (reaction (3.2)), in good yield and ee. This reaction implies the formation of an enolate intermediate (by deprotonation), which forms a chiral ion-pair with the cinchona analogue [234]. In the presence of 3 equivalents of D-diethyl tartrate, asymmetric (4+2)-annulations of isatins (C=O electrophile) with but-3-yn-2-one used a dimeric cinchona-alkaloid organocatalyst (DHQD)2-phthalazine 9 (reaction (3.3)) [235]. These reactions imply the formation of chiral ion-pairs composed of the buty-3-yn-2-one enolate and protonated catalyst 9. In the same time, the isatins are hydrogen-bridged to D-diethyl tartrate. The first C-C bond forms by the addition of the enolate to the keto group of isatins.
(DHQD)2PHAL (9, 1,4-bis(9-O-dihydroquinidinyl)phthalazine; other name: dihydroquinidine 1,4-phtalazine diether) and other cinchona alkaloid-based catalysts were developed by Sharpless and co-workers for their enantioselective and catalytic osmylation of alkenes to produce vicinal diols (Sharpless asymmetric dihydroxylation) [236,237]. In 1980, Hentges and Sharpless reported that (-)-quinine (2) and (+)-quinidine O-acetylated with their vinyl group hydrogenated are ligands (1 equiv.) for OsO4 (1 equiv.) in the osmylation of alkenes (1 equiv.). They induce enantioselectivity in the formation of vicinal diols [238]. Later, a method using catalytic amounts of toxic OsO4 was developed [239]. For his work on chirally catalyzed oxidation reactions, Barry Sharpless received his first chemistry Nobel award in 2001, which he shared (1/2) with Ryoji Noyori (1/4) and William Standish Knowles (1/4) for their work on asymmetric catalyzed hydrogenation reactions. In 2022, Sharpless received his second Nobel award (1/3) that he shared with Carolyn R. Bertozzi (1/3) and Morten Meldal (1/3) for the development of click chemistry and bio-orthogonal chemistry. (DHQD)2PHAL has been used as a chiral ligand in Pd-catalyzed Suzuki-Miyaura coupling of aryl/heteroaryl halides with aryl boronic acids in aqueous medium and in the absence of phosphine/organic solvent [240], and in Cu(I)-catalyzed azide-alkyne cycloaddition reaction to synthesize 1,2,3-triazoles in water [241]. We shall not discuss these reactions further in this mini-review as they engage metallic species in their catalysts.
Catalytic asymmetric oxidations, most notably dihydroxylation, epoxidation and amino-hydroxylation, are powerful methods for the installation of chiral functionality onto achiral alkene substrates. Halolactonization of unsaturated carboxylic acid is another useful way to install two functionalities on the alkene moiety of unsaturated carboxylic acids. In 2004, Gao and co-workers reported a first catalytic protocol for the iodolactonization of alkenoic acids, whereby trans-5-aryl-4-pentenoic acids were cyclized in the presence of iodine and 30 mol% of a cinchonidine-derived quaternary ammonium salt under phase transfer catalysis (see Section 3.2) [242]. In 2010, Borhan and co-workers reported a first efficient asymmetric chlorolactonization catalyzed (DHQD)2PHAL mixed with benzoic acid [243]. Similarly, enantioselective bromolactonization [244], intermolecular alkene bromoesterification, bromoetherification, bromoamination, and bromohydroxylation [245] have applied the same catalyst. A large number of modern enantioselective organocatalysts contain a Lewis base alone, or a base together with other functions such as hydrogen-bond (HB) or halogen-bond (XB) donors. Examples are presented below.
3.1.2. Cinchona Alkaloids and Derivatives Are Capable of Providing Hydrogen Bonding
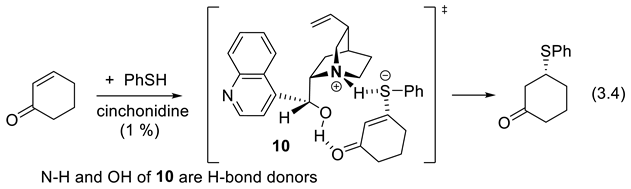
In 1981, Hiemstra and Wynberg reported that the cinchona alkaloid which bear free OH groups in proximity to the basic quinuclidine nitrogen, catalyze enantioselective conjugate addition reactions to conjugated enones (e.g.: reaction (3.4)). The results were interpreted in terms of a transition structute 10 implying of double hydrogen-bridging [246].
The Henry condensation of aldehydes and nitromethane (e.g., reaction (3.5), Figure 3.3) was little enantioselective in the presence of phenolic catalyst 11. However, replacing the 6’-phenolic functional group of 11 by a thiourea moiety (see Section 8.1) led to a substantially improved catalyst 12 [247]. Compounds 11-13 are examples of multifunctional catalysts; they interact with substrates and reactants through the basic and nucleophilic amine function and H-bond donors (electrophilic activation, see Chap. 9) [248,249].
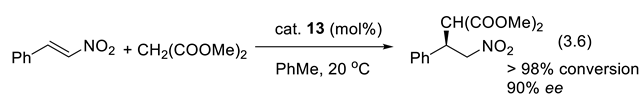
With the quinine analog 13 (Figure 3.3), nucleophilic 1,4-additions of dialkyl malonates to aryl, heteroaryl, and alkyl substituted nitroalkenes are highly enantioselective (e.g., reaction (3.6)) [250].
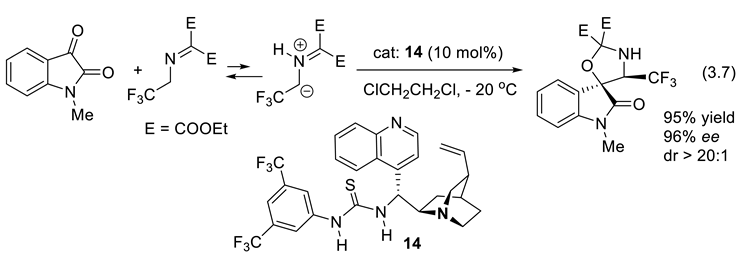
In 2020, Lu and co-workers reported the catalytic asymmetric oxa-1,3-dipolar cycloaddition (3.7) of N-methylisatin with azomethine ylide equilibrating with diethyl 2-(2,2,2-trifluoroethylimino)malonate. It applies cinchona-derived bifunctional thiourea 14 as catalyst [251].
Analogues of 13 and 14 in which the thiourea moieties are exchanged for other H-bond donors such as urea, guanidine, squaramide, and sulfonamide groups are also good asymmetric catalysts (Chap. 9) [252]. Reaction (3.6) is analogous to Michael addition (nucleophilic 1,4-addition). Possible electrophiles are α,β-unsaturated aldehydes, ketones, carboxylic esters, carbonitriles, nitroalkanes, and their yne analogues. Possible nucleophiles are enamines, enols of carbonyl compounds, carbonitriles and nitroalkanes, or their carbanions. These reactions can be enantioselective in the presence of chiral amine catalysts [249,253].
3.1.3. Conjugate Acids of Cinchona Alkaloids and Simpler Chiral Amines as Enantioselective Protonation Catalysts
In 1976, Lucette Duhamel introduced the concept of deracemization [254]. A racemic chiral R1(R2)(R3)C-H compound is deprotonated into the corresponding planar achiral carbanion ion pair R1(R2)(R3)C(-)M(+). The latter is then protonated enantioselectively by an enantiomerically enriched chiral acid, either under thermodynamic or kinetic control [255].
A successful example of asymmetric catalyzed protonation was reported in 1994 by Fehr and Galindo with the enantioselective protonation of the lithium enolate of 1-(2,6,6-trimethylcyclohex-2-enyl)-pentan-1-one in the presence of 20 mol% of (-)-N-isopropylephedrine to generate its (S) enantiomer in 94% yield and 94% ee (compare with Figure 3.4 and Figure 3.5) [256,257]. Fehr also showed that enols are isomerized selectively to one or the other enantiomeric carbonyl compound [258]. In 1991, Kumar, Dike, and coworkers described a synthesis of (S)-Naproxen (used to treat pain and inflammation) based on the catalytic enantioselective protonation of an enolate anion intermediate resulting from the Michael addition of PhSH to an acrylic ester catalyzed by quinine (2). The counter-ion of the enolate anion is the quininium ion (protonated quinine) which protonates the carbanion enantioselectively (reaction (3.8), Figure 3.4). After desulfurization of the Michael adduct (S)-Naproxen was isolated in 85% yield and with 46% ee. A single recrystallization gave (S)-Naproxen with 85% ee [259].
In 2001, Tomioka and co-workers reported a similar asymmetric 1,4-addition, the enantioselectivity (up to 92% ee) being induced by a chiral tertiary amine catalyst ((1R,2S)-(Me2N)-CH(Ph)-CH(Ph)-O-C6H4-2-OMe, 1-2 mol%) [260,261]. Intermolecular oxa-Michael additions are less exothermic than their thia-analog. They are reversible at room temperature [262]. They are difficult to realize in a catalytic enantioselective version. Because cyclization entropy cost is lower than condensation entropy cost, asymmetric intramolecular catalytic alcohol addition to an alkene moiety is possible. An example is the enantioselective cyclization of 4-(2-hydroxyphenyl)-2-butenoates into benzo-2,3-dihydrofuran-2-yl acetates [263]. Related to the conjugate addition of thiols, the asymmetric conjugate addition (CH3)3SiN3 + (E)-R-CH=CHNO2 → RCH(N3)-CH2NO2 has been catalyzed by cinchona-alkaloids [264].
In 1993, Yasukata and Koga [265] found that one equivalent of a 1:1 mixture of acetic acid and chiral triamine 15 converts 2-methyl-1-tetralone lithium enolate into (S)-2-methyl-1-tetralone in 88% yield and 91% ee. When the chiral base was used in a catalytic amount (10 mol%), ee dropped to 28% (reaction (3.9), Figure 3.5a) [266]. In 2021, Łowicki and co-workers reported that the more elaborated chiral diamine-diphenol 16 is a better catalyst (Figure 3.5b) [267]. Amino acids also catalyze the enantioselective protonation of lithium enolates [268].
Silyl enolates are protonated enantioselectively by HF + cinchona alkaloids as catalysts [269] or betaines derived from cinchona alkaloids [270].
Formally, decarboxylation of malonic acid hemiesters generates planar carbanion intermediates. In the presence of a chiral ammonium ion, their protonation can be enantioselective. For instance, heating (70 oC, THF) 2-N-acetylamino-2-ethoxycarbonyl-3-phenylpropionic acid in the presence of 10 mol% N-(9-deoxyepicinchonine-9-yl)-4-methoxybenzamide as the chiral base generates ethyl N-acetyl-L-phenylalaninate in 70% ee [271].
3.1.4. Cinchona Alkaloid Derivatives as Nucleophilic Activators
The construction of the medium-sized ring systems, such as eight-membered ones, is difficult compared to five or six-membered rings. One finds eight-membered lactam moiety in natural products and pharmaceutically important substances [272]. Applying a chiral phosphine for the catalysis of a (4+4) annulation reaction, Lu reported, in 2017, a first enantioselective azocane synthesis (Section 5.1, Figure 5.4). Later, Romo and coworkers reported an asymmetric (5+3) annulation via a Michael addition/lactamization cascade catalyzed by tertiary amines [273]. In 2019, the Coquerel group developed an asymmetric Michael addition/four-atom ring-expansion cascade reaction of activated cyclobutanones and 2-amino nitrostyrenes for the synthesis of benzazocinones in a (4+4) annulation manner [274]. In 2020, Chen and coworkers reported an asymmetric (4+4) annulation reaction of 1-azadienes and cyclobutenones under the catalysis of modified cinchona alkaloids. Among several chiral amines assayed cinchona-derived catalyst 9 gave the best yields and enantioselectivities (reactions (3.10), Figure 3.6) [275]. This approach exploits the ability of amine catalysts to add reversibly to unsaturated systems (here the carbonyl group of cyclobutenone 17), generating zwitterionic intermediates that undergo γ-addition to the electron-poor azadiene 18 permitting the formation of the medium ring final products.
3.1.5. Iminium Ion Mode of Activation by Cinchona Alkaloids
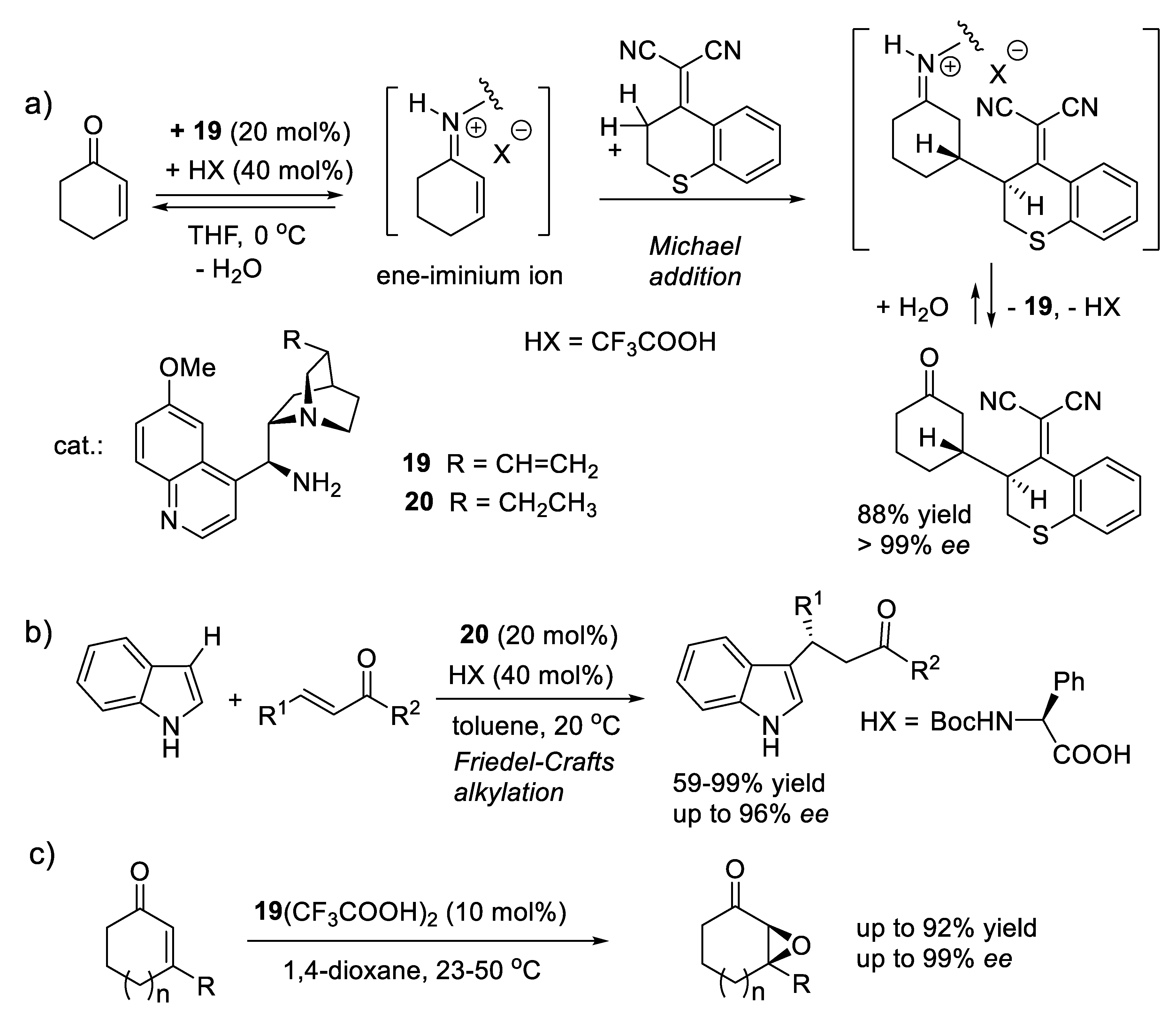
In 2007, the groups of Chen [276] (Figure 3.7a) and Melchiorre [277] (Figure 3.7b) independently introduced 9-amino-9-deoxy-epi-cinchona alkaloids (e.g., 19, 20) as effective catalysts of the stereoselective functionalization of hindered carbonyl compounds through iminium ion mode of activation. The primary amine of the catalyst reacts with conjugated enones and forms corresponding chiral ene-iminium ion intermediates that are more electrophilic in 1,4-nucleophilic additions than the achiral enones themselves (see also Section 3.5) [278]. In 2008, the List group reported that a variety of cyclic enones are epoxidized with H2O2 using catalysts such 19.(CF3COOH)2 (Figure 3.7c). The tertiary amine acts as a general base to promote the conjugate addition of H2O2 to the ene-iminium ion intermediate [279].
Figure 3.7.
Examples of iminium mode of activation of α,β-unsaturated ketones. The substrates form ene-iminium ion intermediates with the primary amine of the chiral catalyst. a) Asymmetric Michael addition (Chen, 2007); b) Asymmetric Friedel-Crafts alkylations (Melchiorre, 2007); c) Asymmetric epoxidation of cyclic enones (List, 2008).
Figure 3.7.
Examples of iminium mode of activation of α,β-unsaturated ketones. The substrates form ene-iminium ion intermediates with the primary amine of the chiral catalyst. a) Asymmetric Michael addition (Chen, 2007); b) Asymmetric Friedel-Crafts alkylations (Melchiorre, 2007); c) Asymmetric epoxidation of cyclic enones (List, 2008).
3.1.6. Enamine Mode of Activation by Cinchona Alkaloids
With primary amine 20, ketones and aldehydes equilibrate with their corresponding enamines, which are more nucleophilic than their corresponding enols (Figure 3.8). Asymmetric reactions (3.11) reported in 2007 by Connon and McCooey illustrate this mode of activation (see also Section 3.6) [280].
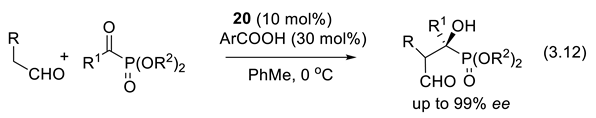
In 2011, Perera and coworkers reported the first asymmetric cross-aldol reactions (3.12) of enolizable aldehydes and α-ketophosphonates catalyzed by 20 [281].
In 2011, MacMillan and coworkers reported the first highly enantioselective α-fluorination of ketones. Best yields, diastereoselectivities, regioselectivities, and enantioselectivities (up to 99% ee) were obtained using cinchona alkaloid derived primary amine 20 as a catalyst, and NFSI ((PhSO2)2NF).as an electrophilic fluorinating agent (see also Section 3.6-4). [282].
3.1.7. Cinchona Alkaloid Derivatives with Halogen Bond (XB) Donors
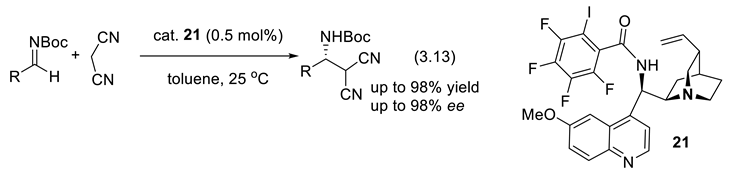
Halogen bonding (XB) has become increasingly relevant in catalytic processes. The halogen bond occurs when there is a net attractive interaction between an electrophilic region (Lewis acid) associated with a halogen atom in a molecular entity, and a nucleophilic region (Lewis base) in another, or the same, molecular entity [283,284,285]. In 2008, Bolm and co-workers demonstrated the catalytic ability of haloperfluoroalkanes in the reduction of 2-substituted quinolines (activation of type C=(R)N:…I-(CF2)nCF3) [286]. In 2018, Arai and co-workers found that quinidine-derived iodoarene 21 catalyzes the asymmetric Mannich reaction (3.13) of malononitrile and various N-Boc imines with high ee. The bifunctional catalyst (amine, C-I) interacts with both the substrate and the reagent. This renders the catalyst highly active, limiting its amount to 0.5 mol% [287,288].
Noncovalent interactions such as halogen (F, Cl, Br, I), chalcogen (O, S, Se, Te), and pnictogen (N, P, As, Sb) bonding have been known for a very long time. Organic compounds containing these elements have a region with positive electrostatic potential on the heteroatoms (so-called σ-hole; anisotropic distribution of the electron density around these atoms). They mimic metal-containing Lewis acids in electrophilic catalysis [289]. Halogen-bond donors like ICF3 have only one σ-hole, while the chalcogen-bond donor Te(CF3)2 has two, and the pnictogen-donor Sb(CF3)3 has three σ-holes. Theoretical investigations show that, besides these electrostatic contributions, charge transfer and dispersion are also important for a proper description of these noncovalent interactions [290]. These σ-hole donors (SHD) exhibit notable catalytic activity accompanied by high tolerance to water and oxygen [285,291].
3.2. Cinchona Alkaloid-Derived Ammonium Salts: Enantioselective Phase Transfer Catalysts (PTCs)
A reagent soluble in water only (e.g., salt M(+)Nu(-), M(+) counter-ion of an anionic nucleophile Nu(-)) does not react readily with an electrophile (e.g., R-X) soluble only in an organic solvent that is non-miscible with water. Classically, alkylation of indene requires the formation of indenyl carbanion and its quenching with an alkyl, or an allyl halide under anhydrous conditions. In 1966, Makosza found that indenyl sodium salt obtained from indene with aqueous NaOH is alkylated with alkyl halides if benzyltriethylammonium chloride (Bn(Et)3N(+)Cl(-)) is added to the reaction mixture [292]. In 1971, Starks also found that the addition of a small amount of a tetraalkylammonium salt (R4N(+)/Y(-)) accelerates nucleophilic displacements dramatically [293]. The quaternary ammonium salt is a phase transfer catalyst (PTC) that exchanges its counter-ion Y(-)with the nucleophilic anion Nu(-) and transports it from the aqueous phase to the organic phase, permitting a fast displacement reaction R4N(+)/Nu(-) + R-X → R4N(+)/X(-) + R-Nu. Ammonium salts have dominated in the field of ion-pair catalysis.
3.2.1. Early Enantioselective Phase Transfer Catalyzed Applications
In 1976, Wynberg and co-workers [294] utilized a cinchona alkaloid-derived quaternary ammonium salt to catalyze the epoxidation of α,β-unsaturated ketones with up to 45% ee [295]. Further studies and applications of cinchona-derived catalysts for alkene epoxidation followed this pioneer work [296]. Asymmetric alkene epoxidation is a powerful entry to highly valuable polyfunctional compounds [297]. More methods are represented below. In 1984, a practical asymmetric alkylation of an α-phenylindanone substrate under the influence of N-(p-trifluoromethylbenzyl)cinchoninium bromide (PTC: 22, reaction (3.14)) was reported by Dolling and co-workers (Figure 3.9a) [298]. In 1989, O’Donnell and coworkers at Merck presented their synthesis of α-amino acids using various cinchona alkaloid-derived PTCs such 23 (reactions (3.15), Figure 3.9b) [299]. In 1996, Prabhakar and co-workers reported that up to 61% ee was obtained for aziridination of methyl acrylate with 24 as catalyst and N-aryl hydroxamic acids as nitrogen sources (reaction (3.16), Figure 3.9c). In 2005, Murugan and Siva reported that up to 95% ee is realized with the aziridination of t-butyl acrylate with PTC 24 [300].
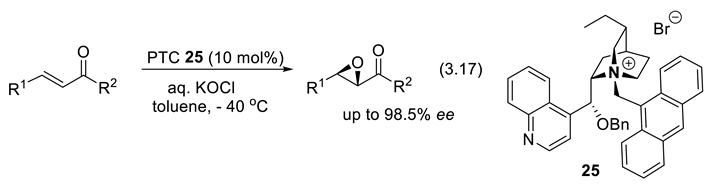
In 1987, and independently of each other, the groups of Lygo [301] and Corey [302] contributed significantly the asymmetric PTC field. When the benzyl group of the quinuclidinium salts is exchanged for an anthracen-9-ylmethyl moiety, higher ee’s are observed. In 1999, Corey and co-workers demonstrated high enantioselectivity for epoxidation of enones with catalyst 25 using KOCl as oxidant (reactions (3.17)) [303]. Many more PTCs have then proposed for the same epoxidations [296].
Lyke an enzyme, N-9-anthracenylmethyl cinchonidinium chloride (2 mol%) catalyzed the asymmetric hydrolysis of enol esters [304].
3.2.2. Industrial Applications of Chiral PTCs
During the last 35 years, asymmetric synthesis using PTCs has become very successful; a large number of chiral ammonium salts have been prepared and used in academia and industry [305,306,307]. Because the cinchona-derived PTCs are easily prepared, they remain attractive catalysts [308,309,310]. Their immobilization on polymeric supports has significantly influenced their use in catalysis. Polymer-anchored cinchona alkaloids permit their easy recovery, recyclability, and reusability, and reduce their environmental impact. They have found applications in enantioselective Michael addition, aldol condensation, Henry reaction, dimerization reaction, dihydroxylation, and benzylation. Anchoring cinchona alkaloids onto polymers includes their covalent attachment in the polymer side chain or main chain, and their ionic attachment via quaternization in the main chain or side chain of the polymer [311]. A synthesis of Pregabalin (central nervous system inhibitor, to treat epilepsy and anxiety), which applies the cinchona-derived PTC 26 for an enantioselective Michael addition of nitromethane (reaction (3.18), Figure 3.10) has been developed. The counter-ion of the quaternary ammonium ion is solid polysulfonated polystyrene (beads), which permits the recovery of the catalyst by simple filtration (99% yield of recovery of the catalyst) [312,313].
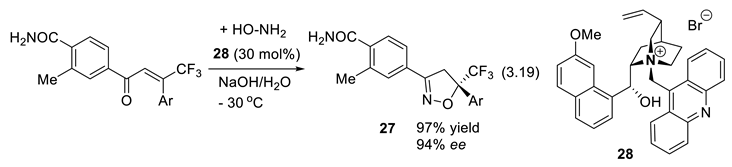
In 2013, Toyama and coworkers at Nissan Chemical Industries Ltd. disclosed their asymmetric synthesis of 2-isoxazoline derivative 27 (a pest-control agent), applying the PTC 28 (reaction (3.19); 1,4-addition of hydroxylamine to the enone, formation of imine) [12].
In 2024, Deng and coworkers presented an asymmetric α-alkylation of N-arylidene-protected alkyl amines (reaction (3.20)). Because of the electron-withdrawing cyano and 4-nitrophenyl groups, substrates A-H (Figure 3.11) are acidic enough to be deprotonated by a phenolate, the counter-anion of the PTC 29. This generates a chiral ion-pair A(-)/R*4N(+) that adds to conjugated enones (Michael addition) with good trans-stereoselectivity and ee [314].
3.2.3. Polyfunctional Cinchona-Derived Polyfunctional PTCs
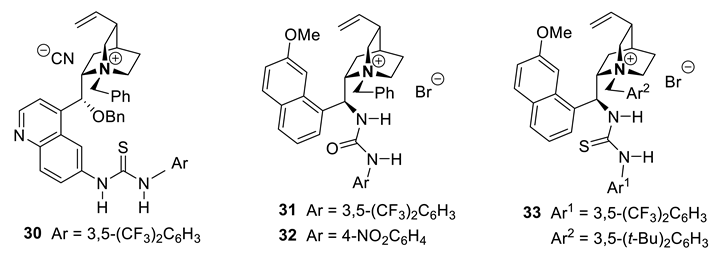
As we shall see below for other type of catalysts, a recent trend has been to add functionalities to the chiral ammonium (or other onium PTCs, see Section 5.3 and 6.4), generating bi- or polyfunctional PTCs [307,310,315]. Cinchona-derived ammonium salts with a free hydroxy group are bifunctional catalysts as they possess, next to the ammonium ion (ion-pairing catalysis), a hydrogen bond (H-bond) donor that can intervene in the activation process and the chiral recognition (for a better enantioselectivity). Obviously, the invention of cinchona derivatives with more H-bond donors (see Chap. 9) as permitted the development of better yielded and more enantioselective organocatalyzed reactions. The thiourea moiety containing catalyst 30 (10 mol%) reported in 2010 by the group of Fernández and Lasseletta has been applied to the cyanosilylation of nitroalkenes [316]. Catalyst 31 (5 mol%) developed by Dixon and coworkers in 2012 has been applied to the enantio- and diastereoselective nitro-Mannich reaction of amidosulfones [317]. The enantioselective synthesis of quaternary-substituted indolenines via a 5-endo-dig cyclization of an α-cyanocarbanion onto an isonitrile applying catalysts 32 (5 mol%) has been reported in 2013 by the group of Smith [318]. In 2018, Duan and co-workers presented an enantioselective aza-Henry reaction of aryl α-ketoester-derived ketimines (ArC(=NTs)COOR + CH3NO2 → ArC(-NHTs)(COOR)-CH2NO2, 99% yield, 99% ee) has been catalyzed by 33 (10 mol%) [319].
3.2.4. Non-Cinchona-Derived Chiral Ammonium Phase Transfer Catalysts
Chiral quaternary ammonium and diammonium salts catalyze numerous reactions, including enantioselective nucleophilic addition reactions [320]. Early examples derived from tartaric acids (e.g., Figure 3.12a) [321,322] or BINOLs (e.g., 34, 35, Figure 3.12b; see Section 9.1 for BINOLs). Other PTCs or ion-pairing catalysts are guanidinium salts such as 36, 37, (N-H bond donors) [323,324] pentanidiniums 38 (e.g.: Figure 3.12c) [325] and 1,2,3-triazolium salts (e.g., Figure 3.12d) [326]. PTCs that are chiral phosphonium, sulfonium salts, and crown ethers are introduced in Section 5.3, Section 6.4, and Section 10.3, respectively [310,315].
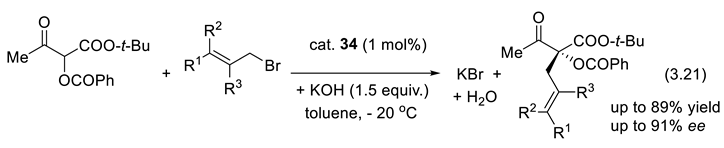
An illustration is the asymmetric allylic allylation (AAA) reported by Maruoka and coworkers in 2003 which applies the Maruoka type of catalyst 34, a monofunctional PTC (reactions (3.21)) [327]. The method was extended to benzyl and propargyl electrophiles [328,329,330].
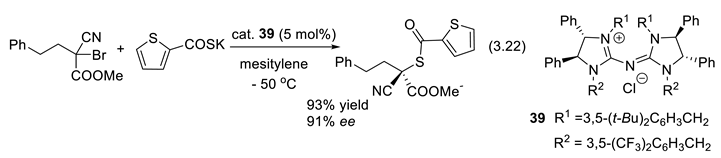
In a typical SN2 displacement reaction (R3C-X + Nu(-) + M(+) → R3CC-Nu + X(-)/M(+)) the nucleophile attacks the carbon face opposite the C–X bond, X being the nucleofugal group (leaving group). When SN1 or SRN1 processes are not competitive, a less common variant, involves initial nucleophilic attack of the X group from the front in a mechanism noted SN2X. It occurs when steric hindrance retards the backside attack by the nucleophile. Thus, if X = halogen it interacts with the nucleophile (Nu(-)/M(+)) to generate a carbanion and a new electrophilic intermediate (Nu–X). The carbanion then displaces X from the Nu–X species to generate the desired substitution product R3C-Nu. In 2019, Lee and co-workers reported an enantioconvergent substitution reaction of activated tertiary bromides by thiocarboxylates or azides. They applied 39 and analogues as PTCs. An example is reaction (3.22). The carbanion intermediate R3C(-)/M(+) reacts with the pentadinidium salt catalyst Q(+)/Cl(-) and equilibrates with M(+)/Cl(-)+ chiral tight-ion pair R3C(-)/Q(+). One of its faces attacks the Nu-X (2-thiophenyl-COS-Br) intermediate leading to enantioselective SN2X reaction [331].
As for cinchona-derived catalysts, the trend in the recent years has been to develop multifunctional PTCs bearing hydrogen bond donors (HB-PTCs) [310,315,332,333]. Figure 3.13 shows selected examples.
A new trend is to add halogens in the ammonium salts to make them halogen bond (XB) donors. An example will be presented with the bis(imidazolium) derivative (228) catalyzed Mukaiyama aldol reaction (11.12) (Section 11.6) [334].
3.3. Asymmetric Hydrogen Atom Abstraction with Cinchona Alkaloid Derivatives
In 2015, MacMillan and co-workers demonstrated that quinuclidinium radical-cations can abstract selectively hydrogen atoms from the C-H bonds of primary (R1CH(OH)-H) and secondary alcohols (R1R2C(OH)-H). The hydroxyalkyl radical intermediate so-obtained can abstract a hydrogen atom from a hydrogen atom donor, or add to unsaturated compounds making possible the direct C-substitution of alcoholic carbon centers [335]. Phosphate anion as additive enhances the selectivity and yield of the reaction because it forms a strong H-bond with the OH group of the alcohols [336]. Using cinchona alkaloid analogs as catalysts, Phipps and co-workers realized an enantioselective epimerization of meso-1,2-diols (reactions (3.23), Figure 3.14) and meso-1,3-diols. They applied a triple catalyst system including 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (photocatalyst, Act) a cinchona alkaloid derivative as chiral H atom abstractor, and 1-dodecanethiol as an H atom donor catalyst. Electron abstraction from the quinuclidine base to the photocatalyst (Act) is induced under blue light irradiation and tetrabutylammonium dihydrogen phosphate as additive (10 mol%). The enantiomerically enriched radical intermediates 40 could be quenched by Michael acceptors permitting selective alkylation (e.g.: Giese reactions (3.24)) of the C-H moieties of alcohols [229]. For reviews on radical-mediated asymmetric organocatalysis, see: [337,338]. For other processes involving a combination of organo-catalysis and photo-redox catalysis, see: [219,220].
3.4. Iminium Ion Mode of Activation by Simple Chiral Amines
In 2000, MacMillan demonstrated that chiral secondary amines (e.g., 41) can catalyze enantioselective Diels-Alder reactions of dienophiles which are conjugated enals (e.g.: reaction (3.25), Figure 19a) [204]. The enals equilibrate with the secondary amine generating α,β-unsaturated iminium ion intermediates (e.g., 42) that are more electrophilic dienophiles than the starting enals. They add to 1,3-dienes (e.g., Diels-Alder reaction (3.25) producing adduct 43). As with other electrophilic dienophiles, the kinetic reactivity of 42 (irreversible reactions) depends mostly on the energy (or electron affinity) and shape of their LUMO (Lowest Unoccupied Molecular Orbital). The water formed during the iminium ion generation hydrolyses the iminium ion in the adduct (e.g.: 43). One speaks here of an “iminium-ion” mode of activation.
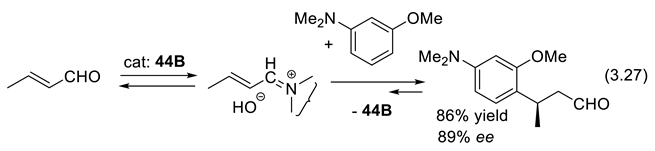
Iminium-ion catalysis is a powerful methodology to activate α,β-unsaturated ketones as dienophiles in a similar asymmetric Diels–Alder reactions (3.26) (Figure 3.15b) as shown by Northrup and MacMillan using imidazolidinone 44 as catalyst [339]. The iminium ion intermediates react with nucleophiles generating β-substituted carbonyl compounds (e.g., asymmetric Michael additions of conjugated enals and enones). The iminium-ion mode of activation facilitates Knoevenagel-type condensations, cycloadditions, nucleophilic additions [340,341,342]. In 2002, Paras and MacMillan reported the enantioselective conjugate addition (3.27) of an electron-rich arene to conjugated (E)-but-2-enal, the latter rendered more electrophilic through the formation of a chiral iminium ion intermediate [343].
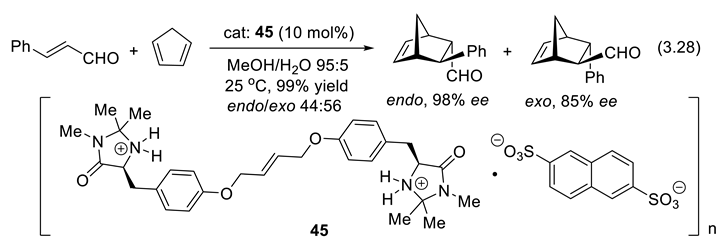
Iminium activation is also reported for 1,6-additions to conjugated dienones (see Section 3.6). Chiral imidazolidin-4-one derivatives such as 41 and 44 catalyze also 1,3-dipolar cycloadditions [344]. Friedel–Crafts alkylation, indole alkylation, α-chlorination of aldehydes, α-fluorination of aldehydes, direct aldol reactions, intramolecular Michael reactions, epoxidation, α-allylation of aldehydes, α-allylation of ketones, and α-alkylation of aldehydes.. To facilitate the separation, recovery, and reuse of the catalyst, chiral imidazolidinones are grafted to polymeric or inorganic supports. Alternatively a main-chain ionic polymer of a tyrosine-derived chiral imidazolidinone with 1,5-naphthalenedisulfonate with 2,6-naphthalenedisulfonate (45) exhibited excellent enantioselectivity for the Diels-Alder reaction (3.28) of (E)-cinnamaldehyde and 1,3-cyclopentadiene (98% ee for the endo isomer). The polymeric chiral organocatalyst was recovered and reused several times, maintaining its high enantioselectivity [345].
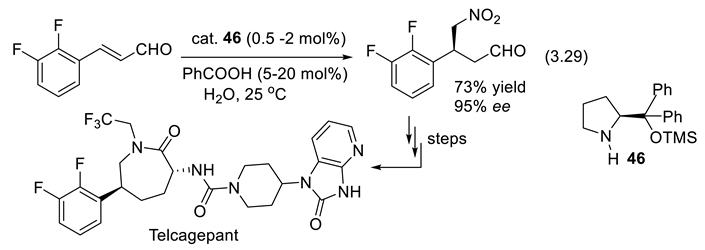
A synthesis of a synthetic intermediate in the preparation of Telcagepant, a CGRP receptor antagonist for the treatment of migraine, applies the diphenylprolinol trimetylsilyl ether (46; a Hiyashi-Jørgensen catalyst) to catalyze the 1,4-addition of nitromethane to (E)-3-(2,3-difluorophenyl)prop-2-enal (reaction (3.29)). The work developed at Merck represents an early (2010) industrial application of iminium organocatalysis. Benzoic acid is a co-catalyst [346].
Melchiorre and coworkers have developed the asymmetric 1,4-additions (3.30) in which electron-rich radical intermediates add to the conjugated enones (Figure 3.16). The catalyst is the primary amine 47 that equilibrates with the iminium ion intermediate 48. The latter absorbs visible light to generate an electronically excited state 49 that implies an intramolecular electron transfer from the carbazole to the iminium ion moieties. In an initiation step, a silane (R-SiMe2Ph) undergoes intermolecular electron transfer to 49 and generates intermediate 50 (a relatively stable 1-amino-3-alkylcyclohex-2-en-1-yl radical) next to the corresponding radical-cation [R-SiMe2Ph] +. The latter reacts with the medium (benzoic acid or salicylic acid in water) and breaks into radical R and PhMe2SiOH/PhMe2SiOOCAr’). The electron-rich radical intermediate adds to iminium ion 48 generating radical 51. The propagating cycle starts now. 51 is converted into radical-cation intermediate 52 by tautomerization and intramolecular electron transfer. Then 52 absorbs an electron from the silane generating imine 53. This imine is hydrolyzed into the final product with recovery of the catalyst (47). Radical-cation [RSiMe2Ph](+ ) formed in this reaction furnishes radical R. which starts the addition process again [221,347].
3.5. Enamine Mode of Activation by Simple Chiral Amines
Enamine catalysis involves catalytically generated enamine intermediates resulting from deprotonation of iminium ions. The enamine intermediates are nucleophiles that react with various electrophiles to generate α-substituted carbonyl compounds. The enols that add to the carbonyl moieties and other electrophiles in uncatalyzed reactions are exchanged for more nucleophilic, faster-reacting enamines. One speaks here of the “enamine” mode of activation [348,349]. As for other nucleophiles, their kinetic reactivity (irreversible reactions) depends very much on the energy (or ionization energy) and shape of their HOMO (Highest Occupied Molecular Orbital). Several valuable and broadly applicable transformations are asymmetric via enamine catalysis. They include cross-aldol [350,351], Michael reactions (conjugate 1,4-additions of nucleophiles to alkenes activated by an electron-withdrawing substituent), [352,353,354], Mannich condensations (three-component reactions combining a primary amine with an enolizable carbonyl compound and an aldehyde, or ketone, producing β-amino carbonyl compounds) [355,356]. Diels–Alder reactions ((2+4)-cycloadditions) [357,358,359], and α-carbon–carbon and α-carbon–heteroatom (C-O, N, S, F, Cl, Br, I) bond formation of aldehydes [360,361,362,363,364].
3.5.1. Asymmetric Michael Additions
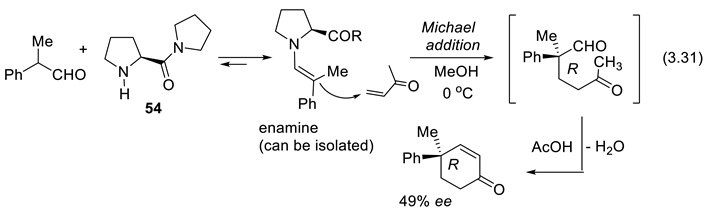
In 1969, Yamada and Otani reported a first example of enantioselective Michael addition catalyzed by L-proline derivatives (e.g., 54, reaction (3.31)) [365].
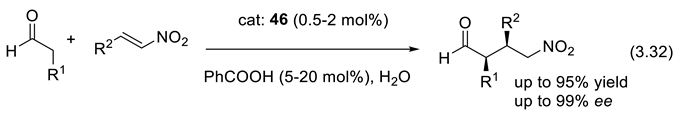
Various effective catalytic systems for asymmetric Michael additions were developed [366]. Reactions (3.32) are examples that apply a Hayashi-Jørgensen catalyst (diarylprolinol trimethyl silyl ether, e.g., 46) [367,368].
3.5.2. Asymmetric Aldol Reactions
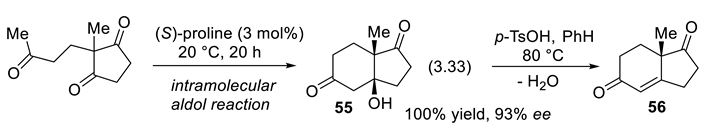
In 1971, Hajos and Parrish at Hoffmann-La Roche,[369,370,371] on one side, and Eder, Sauer, and Wiechert at Schering [372], on the other side, found that a catalytic amount of readily available L-(-) proline (S-Pro) catalyzes asymmetric intramolecular aldol reactions (reaction (3.33)) producing bicyclic compound 55. Crotonization (water elimination from 55) provides enone 56.
We had to wait nearly 30 years for the same catalyst to promote enantioselective intermolecular aldol reactions. The intramolecular aldol reactions have an entropy variation of ca. -20 eu (entropy units = calK-1mol-1), which is less negative than the entropy variation of intermolecular aldol reactions for which typically ΔSrT = -35 eu. Because of that, the intermolecular aldol reactions must be run at a temperature low enough to avoid reversibility (ΔGrT = ΔHrT–TΔSrT). At a given temperature T, concurrent epimerization may happen in competition with the aldol formation. Indeed, when ΔGrT approaches zero, or becomes positive, equilibration of both enantiomeric aldols occurs. The term –TΔSrT is positive, and more so for the intermolecular than for the intramolecular reaction. Catalysts for the aldol reaction also catalyze the opposite reaction, the retro-aldol reaction. In order to be successful with the intermolecular amine-catalyzed cross-aldol reaction, conditions must be found that avoid concurrent elimination of water to generate the corresponding α,β-unsaturated ketones, and homoaldol reactions. In 2000, List, Lerner, and Barbas reported the first examples of direct, asymmetric intermolecular aldol reactions of acetone with various aldehydes. For instance, acetone and 4-nitrobenzaldehyde were condensed into (S)-4-NO2-C6H4-CH(OH)-CH2-CH3 in the presence of 30 mol% of L-proline, in DMSO and at 25 oC (68% yield, 76% ee) [203]. The same year, Notz and List reported the catalyzed synthesis of anti-1,2-diols. In the presence of 30 mol% of L-proline (DMSO; 25 oC) cyclohexanecarboxaldehyde and hydroxyacetone were condensed into anti-4-cyclohexyl-3,4-dihydroxybutan-2-one with high regioselectivity (> 20;1), diastereoselectivity (> 20:1) and ee > 98% [373]. Since this pioneering work, proline and its derivatives have become the most applied catalysts in asymmetric organo-catalysis [374,375,376,377]. In 2024, the group of García-Álvarez reported on the combination of an aerobic and chemoselective Cu-catalyzed oxidation of primary alcohols with a concomitant and compatible enantio- and diastereoselective cross aldol reaction catalyzed by a binary guanidium salt/(S)-proline system. Under air/moisture and in the absence of any organic solvent, stereodivergent and direct conversion of primary alcohols into asymmetric aldols is possible through an economical one-pot/two-steps process [378]. Other chiral α amino acids, small peptides, chiral amines and diamines, amides, sulfonamides and their combinations, as well as chiral metal complexes and Lewis acids can also catalyze the direct enantioselective aldol reactions [379,380]. Examples reported in 2006 by Córdova and coworkers are presented with reactions (3.34) that applies various catalysts including 57 [381].
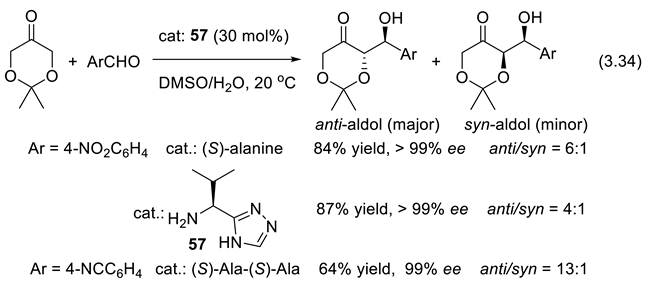
The recovery of the soluble organo-catalysts might be a problem. Immobilized organo-catalysts for the heterogeneous asymmetric direct aldol reaction have been proposed [382,383,384].
The Agami-Houk-List mechanism [385,386,387] is now widely accepted (Figure 3.17) [388,389]; it has been confirmed by recent high-level quantum calculations [390]. L-proline is a difunctional catalyst as it activates aldol condensations through its amine moiety that forms with aldehydes and ketones (the substrates) ene-amine intermediates that are better nucleophiles than the corresponding enols, and through its carboxylic group, which is a H-bond donor (see Chap. 9) that activates aldehydes (the reactants) by rendering them more electrophilic. Debate exists about which of the transition structures 58 (ketone-derived iminium ion formation), 59 (enamine formation, intervention of a second molecule of L-proline (Agami mechanism [391]), and 60 (enamine addition to the aldehyde, C-C bond formation) shown in Figure 3.17 is associated with the rate-determining step and whether other intermediates form or not [392,393]. This depends on the nature of substrates and reactants and on the reaction conditions (temperature, solvent, concentration). Despite intense interest in amine-catalyzed stereoselective reactions, catalyst loadings > 10 mol% are sometimes necessary as undesired aldol reactions can compete [394].
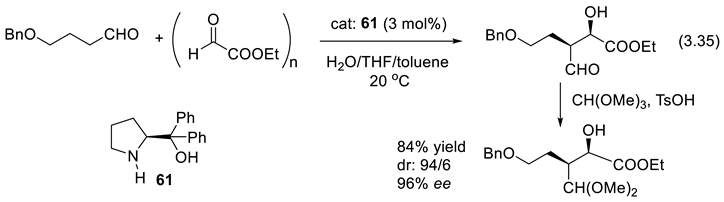
Reaction (3.35) is an asymmetric organocatalyzed direct cross-aldol reaction that produces a key building block of an HIV protease inhibitor. Its preparation is amenable to industrialization [395]. The catalyst is diphenylprolinol (61). Like L-Pro, 61 reacts with 5-benzyloxybutanal to generate an enamine intermediate. The hydroxy group of 61 offers an H-bond to the aldehyde moiety of ethyl glyoxylate. It brings it close to the enamine for a diastereoselective and face-selective addition [396].
3.5.3. Asymmetric Mannich Condensations
The direct catalytic asymmetric addition of unmodified carbonyl compounds to preformed or in situ-generated imines has emerged as a promising new route to optically enriched α- and β-amino acid derivatives, β-lactams, and 1,2- and γ-amino alcohols [397,398]. In 2000, List reported the first example of direct organocatalytic asymmetric three-component Mannich reaction [399,400]. The Mannich reaction of aldehydes, pyruvic esters and 4-methoxyaniline (Figure 3.18) can be either syn (e.g., reactions (3.36)) or anti selective (e.g., reactions (3.37)) depending on the nature of the amino acid catalyst [208].
3.5.4. Asymmetric Diels-Alder Reactions with Inverse-Electron-Demand
Enamine intermediates formed in situ between an amine catalyst and enolizable aliphatic aldehydes can act as electron-rich dienophiles in inverse-electron-demand Diels–Alder reactions (reactivity dominated by their LUMO(diene)/HOMO(dienophile) interactions). Examples are reactions (3.38) that apply the Hayashi-Jørgensen catalyst 46 [358].
3.5.5. Asymmetric α Halogenations
Amino-catalysis has been successful for asymmetric α-fluorination, α-chlorination, α-bromination, and α-iodination of carbonyl compounds [360]. The α-fluorination (Figure 3.19a) reported in 2005 by Enders and Hüttl is an early example [401]. On their side and the same year, the groups of Jørgensen [402], Barbas [403], and MacMillan [404] presented their enantioselective α-fluorination of aldehydes (see also the asymmetric α-fluorination of ketones, Section 3.1.6) [282]. In 2005, Jørgensen and coworkers reported their asymmetric α bromination of aldehydes and ketones (Figure 3.19b) [405]. Reaction conditions that limit the formation of products of α-disubstitution were found.
3.5.6. Asymmetric α-Benzylation of Aldehydes with Alcohols
An application of the combination of organocatalysis with a photo-redox catalysis is given with the α-benzylation of aldehyde (3.39) developed by the group of MacMillan in 2018 (Figure 3.20). Alcohols are cheaper, but much less electrophilic than corresponding chlorides, bromides, iodides, and sulfonic esters. If converted into radical intermediates, the latter adds to activated alkenes such as Michael acceptors (Giese reaction) and electron-rich alkenes like enamines. Iridium(III) complex 62 absorbs a photon, forming the corresponding excited state. The latter takes an electron from the 4-(hydroxymethyl)pyridinium cation, generating an iridium(IV) species and a heterocyclohexadienyl radical that undergoes C-O cleavage of its benzylic alcohol moiety, simultaneously with the formation of water and of a benzylic radical. The latter adds to the imine intermediate resulting from the reaction of the organo-catalyst 41 (Figure 3.15) and the starting aldehyde. A relatively stable amino-substituted radical forms, which is then oxidized by the iridium(IV) complex, restoring the iridium(III) catalyst in its initial state [406]. In 2010, the same group showed that the same double catalytic cycle can be applied to the α-benzylation of aldehydes with benzyl bromides [407].
3.6. Dienamine and Trienamine Mode of Activation
In 2006, Jørgensen and co-workers presented the concept of asymmetric dienamine catalysis in which a secondary amine converts conjugated enals into γ-substituted enals (e.g., reaction (3.40)) via 1-aminoalk-1,3-diene intermediates (Figure 3.21). With diethyl azodicarboxylate as the electrophile, a possible mechanism is the Diels-Alder reaction of the latter (dienophile) with the electron-rich diene resulting from the condensation of the enal with the secondary amine catalyst. Hydrolysis of the adduct liberates the final products [408].
In 2011, Jørgensen and co-workers reported that optically active secondary amines and dienenals generate reactive trienamine intermediates, which readily participate in Diels−Alder reactions with different classes of dienophiles (Figure 3.22). For example, the Diels−Alder reactions with 3-olefinic oxindoles (reactions (3.41)) produce spirocyclic oxindoles in high yields, and up to 98% ee [409]. A cinchona-based primary amine catalyst enables the extension of this activation mode to a highly selective asymmetric Diels–Alder reaction of dienones with electron-deficient dienophiles [410].
In the presence of chiral amines, 2,4-dienones are activated toward the attack of a nucleophile at the δ position, a mode of activation called vinylogous iminium ion catalysis. Asymmetric and stereoselective 1,6-additions of alkanethiols to β-substituted cyclic dienones are catalyzed by a cinchona-based primary amine [411].
3.7. Asymmetric Amine-Catalyzed Domino Reactions
Domino reactions [412] are two-reactant processes in which two or more successive reactions occur in the same pot. Typically A + B (starting materials) → C (first intermediate, which is not isolated) → D (second intermediate, which is not isolated) → P (final product). The successive reactions might be catalyzed, or not, by a single catalyst or might require more than one catalyst introduced at the beginning of the process or in sequence. Domino reactions (also called cascade reactions) that form multiple carbon–carbon bonds and stereocenters are powerful and economical routes to construct complex organic compounds; they require no purification of synthetic intermediates, no time and product consuming protection/deprotection steps, and less solvent to recover [413,414,415,416]. For reviews see: [417,418,419,420,425]. In 2000, Barbas and Bui reported an early example in which L-proline catalyzes the sequence of Michael addition of 2-methylcyclohexa-1,3-dione to methyl vinyl ketone, giving adduct 63. Follows asymmetric intramolecular aldol reaction generating diketone 64. Then water elimination provides S-(+)-65 in 49% yield and 76% ee (reaction (3.42)) [421]. Racemic 65 is Wieland-Miescher ketone. This sequence of three reactions is the Robinson annulation.
A similar asymmetric intermolecular Michael addition, followed by an intramolecular aldol reaction producing tetrasubstituted cyclohexanones have been catalyzed by (S)-1-methyl-4-benzylimidazolidine-2-carboxylic acid [422]. In 2007, the group of Hayashi reported the one-pot enantioselective synthesis of polysubstituted cyclohexanecarbaldehydes (e.g., 68, reaction sequence (3.43)) with control of the configuration of four carbon stereocenters (Figure 3.23). The catalyst is the Hayashi-Jorgensen catalyst (46) that generates enamine intermediate 66 by condensation with pentanedial (enamine activation). Asymmetric Michael addition of 66 to (E)-2-nitrostyrene furnishes adduct 67. The latter undergoes an intramolecular Henry reaction activated by the formation of an iminium ion intermediate (iminium ion activation). Next to 68, three other diastereomers form as minor products [423].
3.8. Asymmetric Amine Catalyzed Multicomponent Synthesis
Since the pioneer work of List and coworkers (see Section 3.5-3) [399], several groups have developed asymmetric three-component Mannich reactions [400,424]. The highly chemoselective reactions between two different unmodified aldehydes and one aromatic amine are efficient routes to β-amino aldehydes with diastereoselectivities syn/anti >19:1 and up to 99% ee. In several examples, the two new carbon centers form with absolute stereocontrol [355,425,426,427,428,429]. In 2001, List and Castello reported the asymmetric three-component reaction of ketones, aldehydes and Meldrum’s acid (reaction (3.44)) [430].
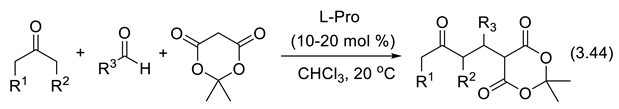
Imitating enzyme-catalyzed aldol reactions of three aldehydes (see Section 2.5, Figure 2.5) Barbas and co-workers reported in 2002 the L-proline catalyzed asymmetric assembly of three acetaldehyde molecules to provide in a single step, (+)-(5S)-hydroxy-(2E)-hexenal with ee up to 90% [431]. They also reported the asymmetric catalyzed asymmetric synthesis of carbohydrates (hexoses) and polypropionates (reactions (3.45)) [432].
In 2007, Enders and coworkers reported the asymmetric construction of cyclohexene-carbaldehydes 69 with perfect control of the configuration of four carbon stereocenters. The process engages three components (an aldehyde, a conjugated enone, a nitroalkene) and catalyst 46 in the same pot (Figure 3.24). In a first step, enamine activation of the aldehyde permits its Michael addition to the nitroalkene. Then the nitroalkane so-obtained undergoes a Michael addition to the enone activated by formation of an iminium ion intermediate by condensation with catalyst 46. A dialdehyde is formed which undergoes intramolecular crotonalization providing 70 with a diastereomeric ratio > 100:1 and 99% ee. Without purification crude 69 with substituent R = (E,E)- hept-3,5-dienyl unit undergoes a stereoselective intramolecular Diels-Alder reaction in the presence of Me2AlCl catalyst in CH2Cl2. This produces the tricyclic compound 70 that contains eight carbon stereocenters [433].
3.9. Ene-Aminium Radical-Cation Mode of Activation
MacMillan and coworkers have developed the concept of singly occupied molecular orbital (SOMO) catalysis. An electron-rich intermediate such as an ene-amine (nucleophile, the reactivity of which is dominated by their HOMO (highest occupied molecular orbitals) can be oxidized into the corresponding ene-aminium radical cation (electrophilic radical, the reactivity of which is dominated by its SOMO). The latter can react with electron-rich compounds such as enol silyl ethers. By comparison, the LUMO (lowest unoccupied molecular orbitals) controls the reactivity of iminium cation intermediates electrophiles [434]. Using catalysts 44B and two equivalents of CAN (cerium ammonium hexanitrate) as oxidant they realized the first asymmetric aldehyde α-enolation (reaction (3.46), Figure 3.25a). The process combines simple aldehydes and enoxysilanes into enantioenriched γ-ketoaldehydes. Figure 3.25b shows a simplified, possible mechanism of the process. SOMO activation can also realize enantioselective allylations and arylations of aldehydes and ketones [337,435,436].
3.10. Asymmetric Morita-Baylis-Hillman Reactions Catalyzed by Amines
The Morita-Baylis-Hillman (MBH) reaction is an atom-economical coupling reaction of activated alkenes and aldehydes in the presence of a nucleophilic catalyst (Figure 3.26a). It corresponds to the hydrocarbation of the aldehyde with the α-C-H bond of the activated alkene. The reaction involves, formally, a sequence of Michael addition of the nucleophilic catalyst, aldol reaction, and a β-elimination with recovery of the catalyst. The reaction was first reported by Morita and coworkers in 1968 [437], and in 1972, by Baylis and Hillman [438]. Phosphines were applied first as nucleophilic activators (Section 5.1). Activated alkenes include acrylic esters, acrylonitriles, vinyl ketones, phenyl vinyl sulfones, phenyl vinyl sulfonates, vinyl phosphates, and acrolein. β-Substituted alkenes require more forcing conditions due to the slow addition of the nucleophilic catalyst. A variety of electrophiles can be used, such as aldehydes, α-ketoesters, 1,2-diketones, aldimines, methyl α-bromoesters, and arenes [439]. Much effort has been devoted to the development of catalytic enantioselective MBH reactions. The catalyst can be a chiral tertiary phosphine (Section 5.1), a chiral Lewis acid, a chiral Brønsted acid, or a chiral amine [440,441,442]. In an early example, N-methylprolinol was applied as a catalyst [443]. More recently, MBH reaction (3.47) was catalyzed by L-proline-derived carboxamide 71 (Figure 3.26b) [444].
3.11. Chiral Lewis Bases That Are Not Amines as Organo-Catalysts
Free guanidines have found widespread use as strong base catalysts in asymmetric synthesis (Section 3.11.1). Guanidinium and 1,2,3-triazolium salts are employed as weak Brønsted acids, hydrogen-bond donor catalysts, or chiral counterions (PTCs, ion pairing catalysis, Section 3.2-3). Lewis bases, such as chiral phosphines, play a central role in asymmetric catalysis as ligands for transition metal catalysts (topic not treated here). They are also organic catalysts (P-nucleophiles) as outlined in Sections 5.1-2). Catalysts that contain one or several phosphine oxide moieties (R3P=O, Section 5.6) or one or several phosphoramide moieties (R3NP=O, Section 5.4) are O-nucleophiles that will be presented later. Chiral catalysts containing thioether (R2S, Sections 6.1-2) or thiol (RSH, Sections 6.3-4) functions are S-nucleophiles that will also be presented later together with examples of asymmetric reactions catalyzed by chiral sulfoxides (R2S=O, Section 6.6), aminothiocarbonates (Section 8.7), sulfinamides (R’(R2N)S=O, Section 6.8) and thiophosphoramides (Section 6.7). Lewis bases that contain selenium, such as diselenides (Section 7.1), selenophosphoramides (Section 7.2), and selenides (Section 7.3), will be presented later. Here we sketch a few examples of reactions catalyzed by chiral Lewis bases such as guadinines, isothioureas, or carbamates.
3.11.1. Chiral Guadinines
Since the first report of chiral guanidine-catalyzed Henry (nitroaldol) reaction by the group of Nájera in 1994 [445], various asymmetric reactions applying this type of organo-catalysts have been proposed [324,446,447,448]. In particular, the Strecker-type reaction developed by Corey represented a significant conceptual advancement in the design of guanidine-catalyzed reactions. Reaction (3.48) utilized a chiral bicyclic guanidine 72 in which two N-H groups interact with cyanide as a nucleophile and with imine as an electrophile to construct the asymmetric environment, as shown for intermediate 73 [449]. Subsequently, Tan and coworkers developed a number of useful and practical reactions catalyzed by chiral bicyclic-type guanidines [450].
Figure 3.27.
Example of enantioselective Strecker-type reaction (Corey, 1999).
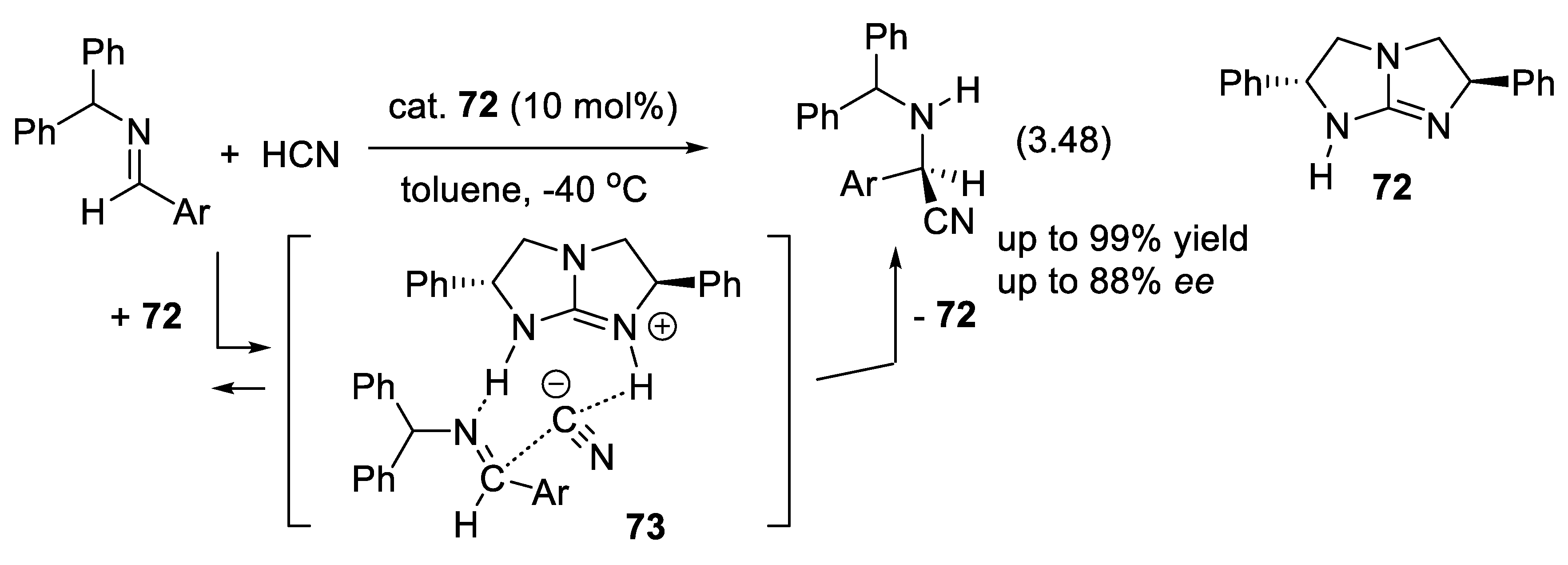
3.11.2. Benzotetramisoles
In 2006, Birman and Li found that the pharmaceutical Tetramisole (74, 10 mol%) and its benzo analogue (75, 4 mol%) catalyze the enantioselective acylation of secondary alcohols (reaction (3.49)). The imine moiety of the isothiourea backbone serves as the active site, and the chiral center adjacent to this nitrogen atom provides the stereo-control (see 76) [451].
Figure 3.28.
Isothiourea-catalyzed enantioselective acylation of secondary alcohols (Birman, 2006).
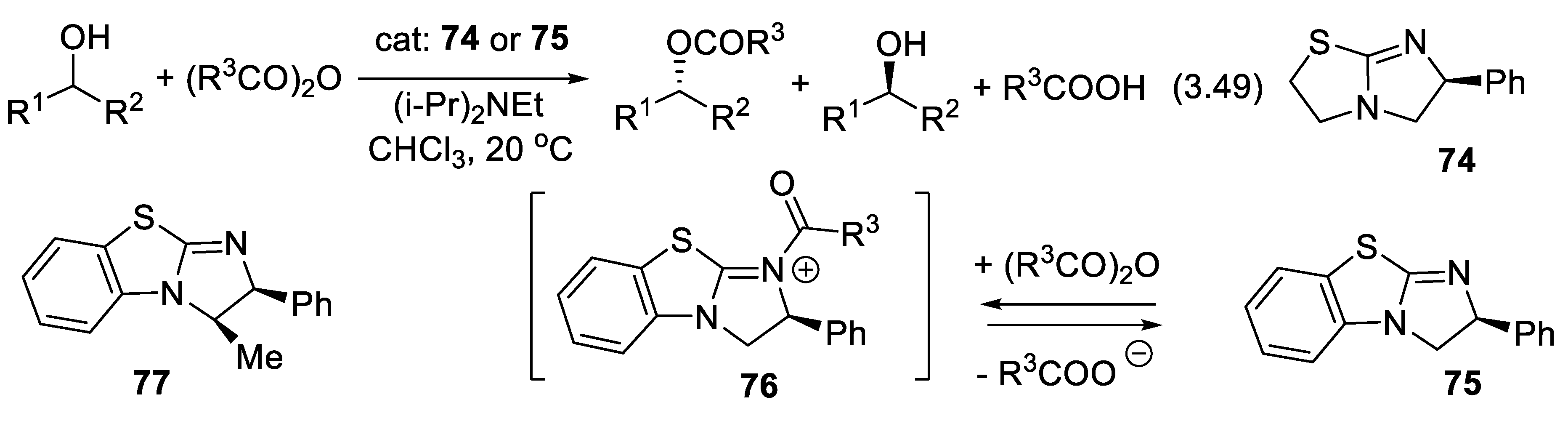
With 77 as a catalyst, Birman and Straub realized the enantioselective (2+2)-cycloaddition of fluoroketene (engendered by elimination of TsOH from the mixed anhydride resulting from the mixing of fluoroacetic acid and p-toluenesulfonyl chloride (TsCl)) to imines. The β-lactams so-obtained were alcoholyzed into corresponding α-fluoro-β-amino acid derivatives in good yield and high diastereo- and enantioselectivity [452]. In 2024, Zi and coworkers developed a new class of benzotetramisole Lewis base catalysts (e.g., 78) that have both central and axial chirality. They proved to be better than simpler benzotetramisoles to catalyze (2+2)-cycloadditions of ketenes to imines (reactions (3.50)) [453].
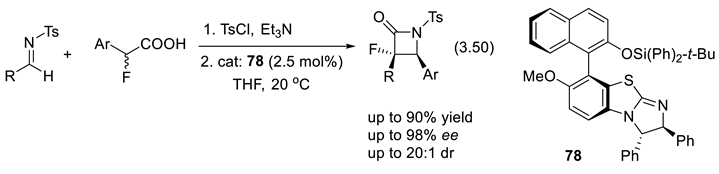
3.11.3. Acylammonium Mode of Activation
Several asymmetric catalyzed Diels Alder reactions ((4+2)-cycloadditions) are known and permit the construction of very important polysubstituted carbocyclic and heterocyclic compounds. A first example is MacMillan’s iminium salt-activated dienophiles (Figure 3.15, Section 3.4). Here one describes the work of Romo and coworkers disclosed in 2014 (Figure 3.29). They activated α,β-unsaturated acyl chloride dienophiles as acylium ammonium salts. Reaction (3.51) illustrates the concept. Low ee’s were observed applying cinchona alkaloid-derived tertiary amines as catalysts. With an isothiourea catalyst such as (-)-benzotetramisole (75) good yield and high ee’s were obtained [454].
3.11.4. Carbamates
In 2015, the Yeung group reported that the enantioselective reaction (3.52) can be catalyzed by a carbamate [455]. Bromolactamization, using a chiral carbamate as a catalyst, resulted in the formation of lactams with up to two stereogenic centers. EtOH additive enhances the reaction rate and enantioselectivity. Reaction (3.52), catalyzed by 79 (Figure 3.30a,b), is a typical example. A switch to the pseudoenantiomeric form of the catalyst led to the formation of the lactam enantiomer. Thiocarbamates catalyze bromo-amino-cyclization of O-cinnamyl tosylcarbamates [456].
4. Asymmetric Organo-Catalysis with Peptides
The high rate enhancements and substrate selectivity exhibited by enzymes have stimulated chemists to generate simpler molecules than enzymes with similar chemical properties. With the discovery that natural amino acids like L-proline and analogs can be powerful asymmetric catalysts, a summit was conquered. Often, these catalysts are required in relatively large amounts, and their recovery may be low-yielded as they can form all kinds of secondary products. Peptides combine through peptidic bonds [-C(=O)-N(-H)-] two or more amino acids. They provide a huge diversity in structures and functionality, including arrays of H-bond donors (Chap. 9) and acceptors. With 20 proteogenic amino acids, one can prepare 202 = 400 dipeptides, 203 = 8000 tripeptides, 204, = 160’000 tetrapeptides, etc. The diversity is augmented by the exchange of natural amino acids by non-natural analogs. Moreover, their side chains can be modified chemically to improve their catalytic properties. Combinatorial chemistry has played a key role in establishing effective asymmetric peptidic catalysts [457,458,459]. They are asymmetric catalysts for enantioselective protonation of enolates (see Sections 2.2; 3.1.2), acylation reactions (Section 2.2), oxidations (Section 2.4), reductions, hydrolytic reactions (Section 2.2), and C–C bond forming reactions such as hydrocyanation (Sections 2.1; 3.1), Michael additions (Sections 3.1.3; 3.5; 3.6), aldol reactions (Sections 2.5; 3.6), the Morita-Baylis-Hillman reactions (Section 3.11), and acyl anion equivalent chemistry (Chap. 8). Many peptides and analogues catalyze the same reactions as simpler chiral amines. Peptides with a functional side chain (e. g.: free NH2 group of lysine, imidazole group of histidine, indole group of tryptophan, guanidinium group of arginine, CONH2 group of asparagine and glutamine, OH group of serine, threonine. and tyrosine, COOH group of aspartic and glutamic acids, SH group of cysteine) can activate substrates and reagents by forming more reactive intermediates. Peptides possess secondary functions and arrays of H-bond donors and acceptors, which provide more specific steric and electrostatic interactions with the substrates and reagents than simpler amines and analogs. Thus, they can lead to higher enantioselectivities using smaller amounts of catalyst and applying milder reaction conditions [460,461,462,463,464]. Simple proteogenic amino acids and peptides are less toxic than catalysts containing transition metals. Since the synthesis of peptide catalysts is costly (production of waste, use of toxic solvents), their recovery and reuse are considered. One method is to immobilize them by cross-linking with hydrophobic polystyrene, hydrophilic TentaGel, polyethylene glycol-polyacrylamide, mesoporous silica, chitosan, or by mixing them with hydroxyapatite or ceria nanocrystals. The recovery of the catalyst requires a simple filtration or centrifugation. Alternatively, the catalysts are grafted onto magnetic nanoparticles. Their recovery applies magnetic separation. Another option is to equip the catalysts with a perfluorinated tag; their recovery relies upon fluorous extraction [465,466].
With the enantioselective conjugate addition of thiols to enones, Inoue and coworkers reported the first example of asymmetric poly-amino acid-catalyzed reaction in 1975. [467,468,469]. Polyaminoacids have played an important role in the early days of organocatalysis. They probably have been catalysts of prebiotic enantioselective reactions [470,471,472,473,474].
Self-assembly of molecules often results in new properties. Even very short peptides can self-assemble into structures with a variety of physical and structural characteristics. Remarkably, many peptide assemblies show high catalytic activity in model reactions, reaching efficiencies comparable to those obtained with natural enzymes by weight [475]. Foldamers are oligomers with a strong tendency to adopt a specific conformation. They approach principles of enzyme functions. For instance, helical structures provide foldamer families that offer a basis for asymmetric catalysis [476,477]. Despite the great advantages of peptidomimetic scaffolds as potential catalysts, only a few examples of their industrial application have been described [478].
4.1. Asymmetric Hydrocyanations
In 1979, Inoue and coworkers found that diketopiperazine 80 (cyclo(L-phenylalanine-L-histidine) catalyzed the asymmetric addition of HCN to arenecarbaldehydes. For instance, with benzaldehyde (reaction (2.1)), the corresponding cyanohydrin was obtained in 97% ee for 87% conversion [479,480,481].
Figure 4.1.
Inoue’s (1979) asymmetric hydrocyanation.
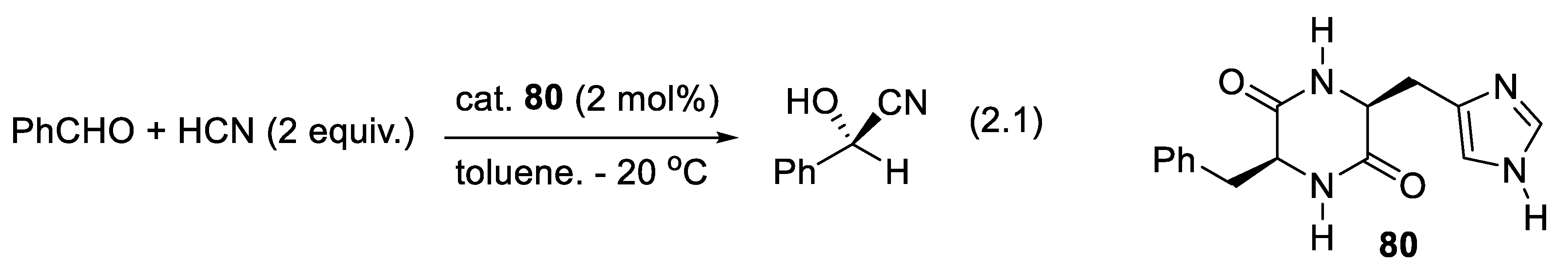
In 1996, Shvo and coworkers found a second-order kinetic dependence on 80 for reaction (2.1), consistent with a mechanism in which two molecules of the catalyst intervene in the transition state, and for which both HCN and the aldehyde are activated by the histidine moiety of 80. Furthermore, better enantioselectivities are observed when the catalyst forms polymers through hydrogen bonding. This also depends on the solvent, concentration, temperature, and mode of preparation of 80 [482]. In 1996, Lipton and coworkers reported a catalytic enantioselective version of the Strecker amino acid synthesis (reactions (4.1)). Cyclic dipeptide cyclo[(S)-His-(S)-NorArg] (2 mol%) in methanol catalyzed the addition of hydrogen cyanide to N-alkylimines to afford α-amino nitriles in high yield and high ee. Acid hydrolysis of N-benzhydryl-α-amino nitriles afforded the corresponding α-amino acids [483]. In 1998, Jacobsen and coworkers found that 81 is an excellent catalyst for the asymmetric Strecker reaction (4.1) of aromatic and aliphatic aldimines [484,485,486]. Applying the readily available catalyst 82 (0.5 mol%), Jacobsen described in 2009 scalable asymmetric Strecker reactions that used KCN in AcOH/H2O/toluene (at 0 oC) instead of dangerous HCN for the addition to aliphatic benzyl aldimines. For R = t-Bu, 1-methylcyclohexyl, and 3-methylpent-3-yl, yields of the final N-Boc protected amino acids varied between 48-65% with 98-99% ee [487].
Figure 4.2.
Examples of asymmetric Strecker reaction catalyzed by short peptide analogues. a) With catalyst 81 (Jacobsen, 1998); b) With catalyst 82 (Jacobsen, 2000).
Figure 4.2.
Examples of asymmetric Strecker reaction catalyzed by short peptide analogues. a) With catalyst 81 (Jacobsen, 1998); b) With catalyst 82 (Jacobsen, 2000).
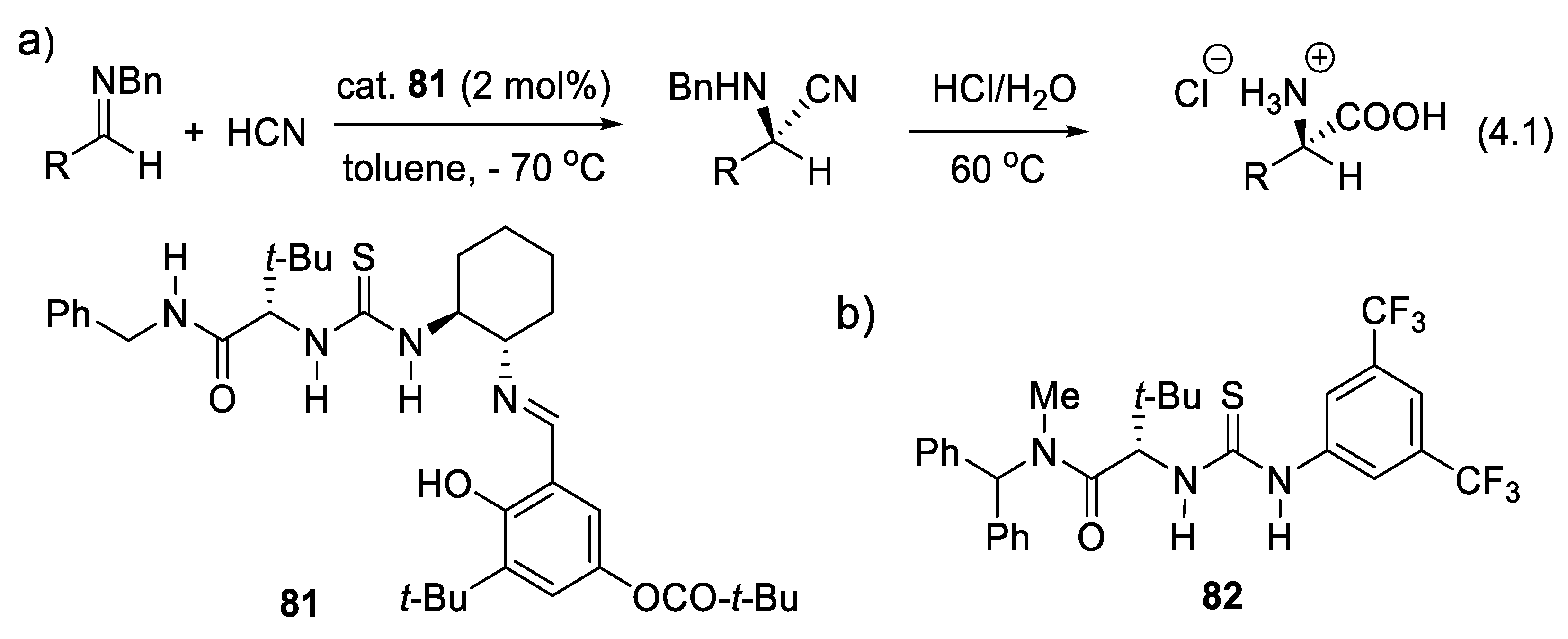
Other chiral organocatalysts, such as bicyclic guanidine, ureas and thioureas, carbohydrates, N-oxides, bisformamides, ammonium salts, Brønsted acids, amino acids, alkaloids, and phase-transfer catalysts and oxazolines (section 11.5) have been applied to the Strecker reaction [488].
4.2. Asymmetric Oxidations
Asymmetric alkene epoxidation is an extremely powerful entry to highly valuable polyfunctional compounds [297]. In 1980, the Spanish Juliá group reported the enantioselective epoxidation of chalcones using poly-L-alanine as catalyst [489]. In 1983, poly-L-isoleucine (83) was found to be a better catalyst as shown with the now called Juliá-Colonna epoxidation of electron-deficient alkenes [490]. It is a triphasic system for which alternative diphasic and monophasic systems have been proposed [491,492,493]. The latter nucleophilic epoxidation is complementary to other asymmetric organocatalyzed epoxidations [296] and to electrophilic epoxidations, such as the Katzuki-Sharpless [494,495,496] and Jacobsen-Katzuki epoxidation [497,498,499].
The stereo-induction of the Juliá–Colonna epoxidation (4.2) depends on the a-helical secondary structure of the poly-leucine catalyst [500]. A 10-mer leucine polypeptide is of sufficient length to provide significant enantioselectivity (Figure 4.3) [501].
In 1990, Itsuno and coworkers reported the enantioselective epoxidation of chalcones using poly-l-alanine and poly-l-leucine immobilized on cross-linked aminomethyl polystyrene (CLAMPS) resin. Without substantial loss of catalytic activity, the polymer-supported catalyst was recovered and reused several times [502]. Miller and coworkers generated peracids (RCO3H) in situ from the carboxylic group of Asp-COOH side moiety of short peptides. They epoxidize allylic carbamates that are not Michael acceptors, as shown with reactions (4.3) [461,503]. Similar Asp-containing peptides permitted enantioselective catalyzed Bayer-Villiger oxidations [504,505].
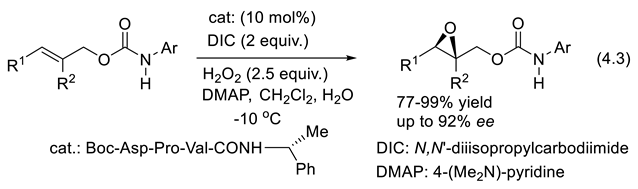
4.3. Asymmetric Intermolecular Aldol Reactions
In 2003, the groups of Reymond [506], Gong [507], and List reported that L-proline amides and dipeptides catalyze asymmetric direct aldol reactions. With L-proline or aldolases applied as catalysts, the direct aldol reactions of hydroxyacetone with aldehydes provided mostly 1,2-diols. In contrast, isomeric 1,4-diols are the major products when using L-proline derived tetra- (reactions (4.4)) or pentapeptides as catalysts [509]. Peptide dendrimers also catalyze direct asymmetric aldol reactions [510].
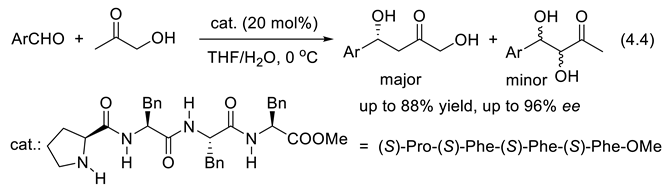
4.4. Peptide-Catalyzed Conjugate Additions
In 2000, Miller and coworkers have reported the enantioselective conjugate addition of azide to unsaturated carbonyl compounds. An example is reaction (4.5). The azidocarboxamides so-obtained are converted into β-amino acids and triazoles with conservation of enantiomeric purity [511].
Examples of dipeptide-catalyzed Michael additions reported in 2006 (Figure 4.4) are reactions (4.6) [512] and (4.7) [513]. Tripeptides such as H-D-Pro-Pro-Asp-NH2 are efficient catalysts for conjugate addition reactions between aldehydes and nitroolefins (reaction (4.8)) [514].
4.5. Peptide-Catalyzed Morita-Baylis-Hillman Reaction
In 2003, Miller and coworkers reported the catalyzed asymmetric Morita-Baylis reaction of 2-nitrobenzaldehyde and methyl vinyl ketone (reaction (4.9)). As found earlier by other authors, better yields and enantioselectivities were observed when the peptide catalyst was used with L-proline as co-catalyst. The larger the peptide, the better is the enantioselectivity [515].
In 2009, Miller also reported enantioselective aza-MBH reactions of N-acyl imines with allenic esters. Reactions (4.10) were catalyzed by peptide 84 containing a Lewis basic pyridylalanine residue [516,517].
4.6. Catalytic Enantioselective Enolate Protonation by Peptides
Nearly three decades after Inoue’s work (Section 4.1) on peptide-catalyzed enantioselective protonation, Yanagisawa and coworkers reported in 2006 a complementary strategy for asymmetric peptide-catalyzed protonation of lithium enolates derived from tetralone derivatives. In the presence of stoichiometric amount of achiral acid (2,6-di-t-butyl cresol, BHT) and a catalytic amount of dipeptide N-β-L-aspartyl-L-phenylalanine methyl ester lithium enolates were protonated furnishing the corresponding ketones with ee up to 88%. Under the same conditions, the lithium enolate of 2-benzylcyclohexanone produced the (R)-enantiomer in 65% yield and 69% ee (reaction (4.11)) [518].
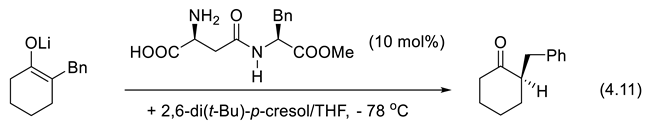
5. Phosphorous-Based Catalysis
Phosphines (R3P), phosphine oxides (R3P=O), phosphoramides ((RN)3P=O) and phosphonium salts (R4P(+)X(-)) are effective organo-catalysts because of their thermal stability, recyclability, and unique catalytic properties [519,520]. Chiral Brønsted acids like diolphosphates and phosphoramides are discussed in Chapter 9.
5.1. Nucleophilic Activation by Chiral Phosphines
Nucleophilic phosphine catalysis implies nucleophilic addition of a phosphine to an electrophilic alkene, allene or alkyne producing a reactive zwitterionic intermediate, generally under mild conditions [519]. Triethylphosphine (Et3P) is less basic, but more nucleophilic than triethylamine (Et3N). In water, pKa(Et3PH(+)) = 8.7, pKa(Et3NH(+)) = 10.7; quaternization of Et3P with MeI is 100 times as fast as Et3N + MeI → Et3(Me)N(+)I(-). Trialkylphosphines burn in the air; they are pyrophoric. Trialkylamines are not. On exchanging their alkyl substituents by aryls the oxidability of phosphines is lower. However, this renders the phosphines much less nucleophilic. For instance, Et3P reacts with MeI 106 times as fast as Ph3P [521]. One simple solution to that problem is to use the corresponding phosphonium salts that are air stable together with an appropriate base, for instance: a 1:1 mixture of (n-Bu)3PH(+)/BF4(-) and (i-Pr)2Net [522].
In 1962, Takashina and Price obtained a hexamer of acrylonitrile on treating acrylonitrile with triphenylphosphine (Figure 5.1a) [523]. In 1963, Rauhut and Currier disclosed the trialkylphosphine-catalyzed dimerization of acrylic esters (Figure 5.1b) [524]. The reactions imply the phosphine additions to the activated alkenes, generating zwitterion intermediates that then add (Michael reactions) to another molecule of the alkene. In 1968, Morita reported that the above zwitterion intermediates engendered by the addition of tri(cyclohexyl)phosphine to acrylic esters can be quenched by aldehydes. This was the first example of the now called Morita-Baylis-Hillman or MBH reaction presented in Section 3.10 (see Figure 3.26a for a mechanism) [437].
Chiral phosphines are the most important ligands in asymmetric metal catalysis. [525,526]. Many chiral phosphine catalysts used as ligands in metal catalysis have been explored first in asymmetric organocatalysis. Others were modified known phosphine ligands (for a review, see [527]). In 1995, Lu’s group reported phosphine-catalyzed (3+2)-cycloadditions of electron-poor allenes to alkenes that produced cyclopentenes [528]. In 1997, Zhang and coworkers reported the enantioselective phosphine-catalyzed version of these reactions. Reactions (5.1) catalyzed by chiral phosphine 85 (Figure 5.2) are an example [529].
Applying 7-phosphanorbonane catalysts 85 or 86, Zhang and coworkers reported in 1998 the first asymmetric γ-addition of dicarbonyl compounds to alkynoates and allenoates (e.g., reactions (5.2), Figure 5.3) [530].
Following Zhang’s reports, several other asymmetric phosphine-catalyzed reactions have been developed, such as (3+2)-, (4+1)-, (4+2)-, and (4+3)-annulations of electron-poor allene and acetylene derivatives, (2+2)-annulation of ketenes, MBH and Rauhut-Currier reactions, and their combinations with other reactions [531]. In 2017, the group of Lu reported an unprecedented example of (4+4)-annulation of allene ketones and 1-azadienes affording azocane derivatives with high enantioselectivity (reaction (5.3), Figure 5.4) [532,533].
Figure 5.4.
a) Asymmetric (4+4)-annulations catalyzed by chiral phosphines (Lu, 2017); b) Possible mechanism.
Figure 5.4.
a) Asymmetric (4+4)-annulations catalyzed by chiral phosphines (Lu, 2017); b) Possible mechanism.
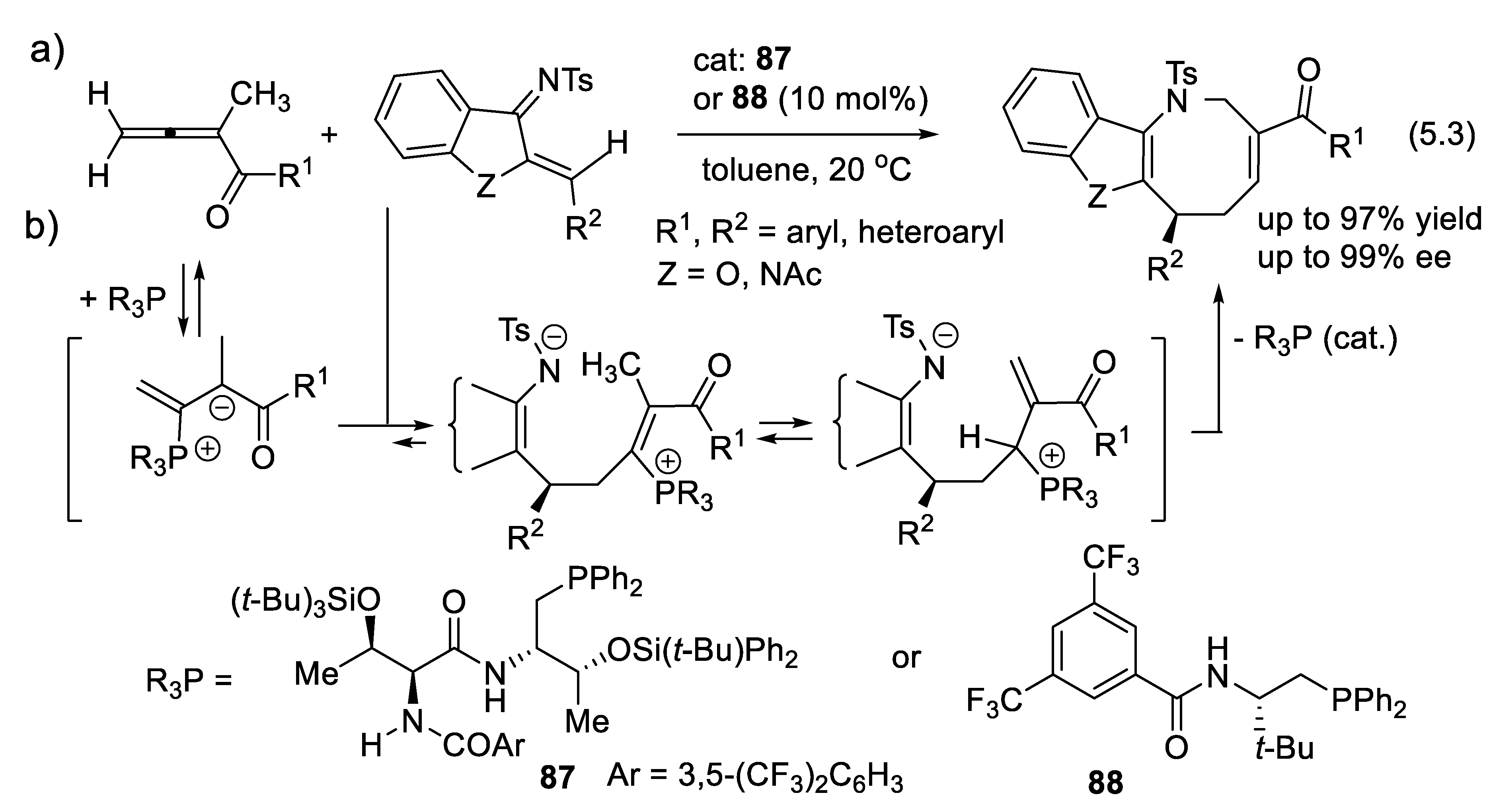
Patureau and coworkers presented a phosphine-catalyzed (3+2)-annulation of benzoxazoles with 1,2-diphenylcyclopropenone (up to 48% ee) [534]. In 2025, You and coworkers described similar asymmetric reactions with benzimidazoles and 1,2-diarylcyclopropenone (reaction (5.4), Figure 5.5) catalyzed by diphosphine 89 [535].
5.2. Enantioselective Wittig and Staudinger-Aza-Wittig Reactions
Tertiary phosphines are powerful reagents for transforming the C=O bonds of ketones and aldehydes into C=C through Wittig reactions. They also convert carbonyl compounds into imines through Staudinger-aza-Wittig reactions. The development of catalytic PIII/PV=O redox processes has transformed the Wittig and Staudinger-aza-Wittig reactions, traditionally plagued by stoichiometric phosphine oxide by-products, into an efficient and more benign method for synthesizing alkenes [536]. In 2014, Werner and coworkers reported the low-yielding, first catalytic asymmetric Wittig reaction through meso-desymmetrization of cyclopentane-1,3-dione with Me-DuPhos as the catalyst and phenylsilane as the terminal reductant (converts the resulting phosphine oxide back into phosphine catalyst, see Figure 5.6) [537]. The Christmann group reinvestigated Werner’s asymmetric transformations (5.5) and applied them in the total synthesis of propellanes [538,539].
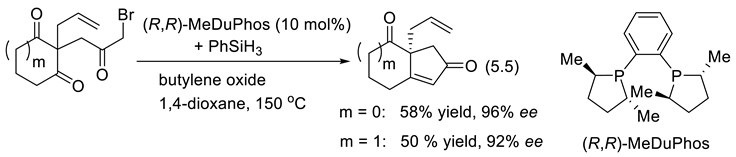
In 2019, Voituriez and coworkers reported the enantioselective phosphine-catalyzed reactions (5.6) via a chemoselective in situ phosphine oxide reduction by PhSiH3. Starting with 4,4,4-trifluoroalkane-1,3-diones and dialkyl acetylenedicarboxylates, highly functionalized fluorinated cyclobutenes were obtained applying the HypPhos catalyst 90. Using the same methodology, CF3-spirocyclobutene derivatives were also synthesized (up to 95% ee) [540].
In 1990, Wilson and Pasternak applied their C2-symmetrical (2R,5R)-1-phenylphospholane to the enantioselective reduction of racemic benzyl azides into primary amines (R1-N3 + R3P → N2 + R1-N=R3; R1-N=PR3 + H2O → R1-NH2 + R3P=O) [541]. In 2006, Marsden and co-workers reported the first enantioselective Staudinger−aza-Wittig reaction applying stoichiometric amounts of chiral oxazaphospholane or diazaphospholane derivatives [542]. In 2019, Kwon and co-workers demonstrated that a catalytic, stereoselective Staudinger-aza-Wittig reactions (5.7) could be achieved under mild conditions using a chiral l-hydroxyproline-derived HypPhos catalyst 91 (endo-phenyl-HypPhos) [543]. Additives like nitrobenzoic acid promote the PhSiH3 reduction of the phosphine oxide intermediate, permitting to run the reaction at room temperature. In 2022, the Kwon group further demonstrated the synthesis of oxindoles featuring a quaternary carbon center, starting from azidoaryl malonates a catalytic stereoselective Staudinger–aza-Wittig reaction. A bulkier HypPhos oxide catalyst (analogue of 91 oxy-substituted at C(7) and with larger aryl group than Ph), in conjunction with [Ir(cod)Cl]2 as an additive, was employed to achieve high enantioselectivity [544]. Using a C2-symmetric chiral bisphosphine, the Tang group realized recently stereoselective Staudinger–aza-Wittig reactions [545].
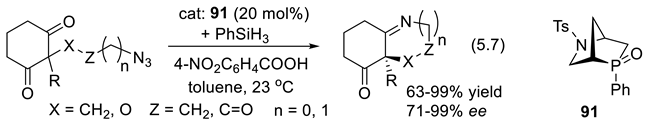
5.3. Chiral Phosphonium Salts as Phase Transfer Catalysts
In 1998, in a pioneer work, Manabe synthesized chiral phosphonium salts having two NH and two OH groups as hydrogen-bond donors. They were PTCs for asymmetric alkylation of β-keto esters to give the corresponding products with up to 50% ee [546]. Since then, many chiral quaternary phosphonium salts have been developped as organocatalysts [547]. In 2007, the group of Ooi developed quaternary tetraaminophosphonium salts [548]. In 2009, Maruoka and co-workers proposed binaphthyl-derived phosphonium salts for asymmetric Michael and Mannich reactions [549]. These phosphonium salts exhibit high stability under strong alkaline conditions and resist attacks by nucleophilic anions. During the last 15 years, the exploration focused on the development and application of amino acid-derived bifunctional phosphonium catalysts [547,550,551]. In 2019, Wang and Xu presented the enantioselective synthesis of benzofuran derivatives (reactions (5.8)). The catalyst 92 was prepared in seven steps from L-valine [552].
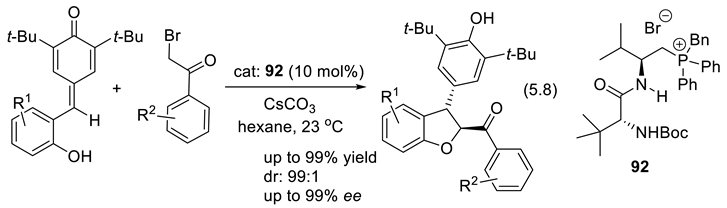
5.4. Chiral Phosphoramides as Lewis Base Catalysts
The classical catalytic enantioselective allyl addition of aldehydes and ketones that produce allylic alcohols applies a chiral Lewis acid that binds to the aldehyde or ketone (electrophile) and activates it toward nucleophilic attack by an allylmetal reagent [553]. In 1994, Denmark and coworkers found that sub-stoichiometric amounts of chiral phosphoramide 93 induces enantioselective allylation of benzaldehyde with allyltrichlorosilane (reaction (5.9)). In this case, the high oxyplilicity of silicon nucleophile permits its coordination with the P=O bond of the Lewis base (LB) and, in the same time, with the oxygen atom of the aldehyde. A second molecule of LB diplaces a chloride forming a hexacoordinated silicon species 95 that adopts a chair like transition state for the allylation [554]. This mechanism explain the high diastereoselectivity and enantioselectivity observed in many aldehyde allylations with allyltrichlorosilanes (Figure 5.7). Reaction (5.10) that applies the double Lewis base 94 is an example. With its two phosphoramide moieties it reduces the entropy cost of the formation of the hexacoordinated silicon intermediates analogues of 95 [555,556].
Denmark and Beutner recognized that the combination of Lewis bases with Lewis acids is a general mode of activation of reactants of type Y3MX (Figure 5.8). These combinations can realize efficient catalysts, including asymmetric ones [557].
In 1999, the group of Denmark reported a new approach to the asymmetric aldol reaction. An example is shown with reaction (5.11) that applies phosphoramide 96 as Lewis base to catalyze the condensation of an aldehyde with a trichlorosilyl enol ether [558].
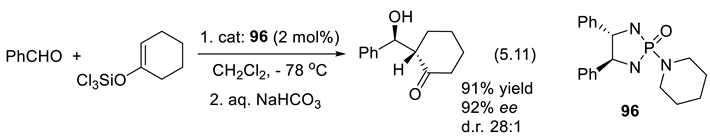
Silicon tetrachloride (SiCl4) is a weak Lewis acid. It can be activated by binding of a strongly Lewis base such a chiral phosphoramide (LB), leading to in situ formation of a chiral Lewis acid of type [(LB)2SiCl3](+)Cl(-) (Figure 5.8). This species catalyzes asymmetric aldol additions of acetate-, propanoate-, and isobutyrate-derived silyl ketene acetals to conjugated and nonconjugated aldehydes. Aldol reactions of silyl dienol ethers are also enantioselective in the presence of this species [559,560]. Ring opening of meso-oxiranes with SiCl4 are enantioselective in the presence of chiral phosphoramides and other chiral LB [561]. In 2003, Denmark and Fan described the first, catalytic, enantioselective α-additions of isocyanides to aldehydes (reactions (5.12)) that applies Lewis base catalyst 97 [562].
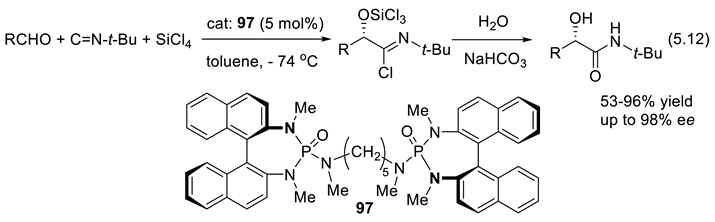
5.5. Chiral Phosphine Oxides as Organocatalysts
In 2004, Kobayashi and co-workers reported that stoichiometric amounts of bis-(diphenylphosphanyl)-binaphthyl dioxide (BINAPO, a chiral Lewis base) induces diastereoselective and enantioselective allylation of a-hydrazono esters using allyltrichlorosilanes [563]. With substoichiometric amounts of BINAPO (20 mol%), yield (11%) and ee (56%) were low. In 2005, Nakajima and coworkers reported the enantioselective allylation of aldehydes (reactions (5.13)) catalyzed by (S)-BINAPO, wherein the combination of diisopropylethylamine and tetrabutylammonium iodide as additives was crucial for accelerating the catalytic cycle [564].
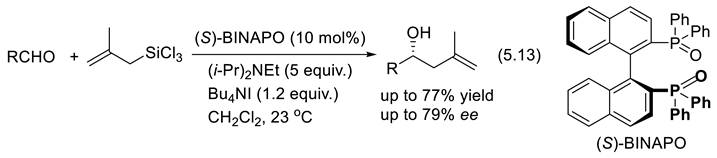
In 2009, the same group showed that (S)-BINAPO catalyzes the direct asymmetric aldol reaction of cyclohexanones + ArCHO + SnCl4 [565]. Since then, further phosphine oxides have been developed and applied as chiral catalysts in Lewis base-catalyzed reactions such as aldehyde allylations, aldol reactions, epoxide openings, and halocyclizations [566,567,568,569]. An example reported by Dogan and coworkers in 2015 is given with the aldehyde allylations (5.14) catalyzed by 98 [570].
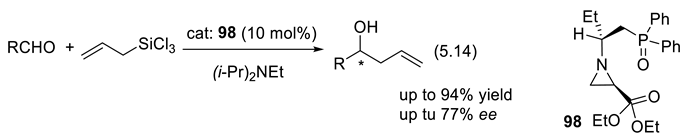
6. Asymmetric Organocatalysis by Sulfur Compounds
In this Chapter, we provide a few examples of asymmetric organic reactions catalyzed by S-nucleophiles such as thioethers (RSR’) and thiols (RSH), by O-nucleophiles like chiral sulfoxides (R2S=O) and sulfinamides (R’(R2N)S=O). Chiral sulfonium salts (R3S(+)X(-)) have been applied as asymmetric phase transfer catalysts.
6.1. Asymmetric Aldehyde Epoxidations Catalyzed by Chiral Dialkylsulfides
In 1958, Johnson and LaCount reported the first example of a sulfur ylide-mediated epoxidation reaction [571]. Sulfides are nucleophiles that add to aldehydes. Reaction of 9-dimethylsulfonium fluorenylide with 4-nitrobenzaldehyde did not afford an alkene (Wittig-like olefination), but an epoxide instead. In 1965, Corey and Chaykovsky found that simpler dimethyl sulfonium ylide and dimethyloxosulfonium ylide can be used in the epoxidation of aldehydes [572]. In 1989, Furukawa and coworkers reported the first example of a catalytic, asymmetric epoxidation reaction. It features reaction (6.1) and catalyst 99 (Figure 6.1) [573].
Then a large number of alternative catalysts have been proposed (Figure 6.2, reaction (6.2)) [574,575,576].
6.2. Asymmetric Halo- and Chalcolactonization Catalyzed by Sulfides
A wide range of chiral organocatalysts has been developed and applied to enantioselective halolactonization [577]. They include Lewis bases, Lewis acids, Brønsted acids, and phase-transfer catalysts containing basic N-, O-, S-, Se-, and P-heteroatoms. They activate electrophilic halogenating reagents such as NIS, NBS, and NCS (N-I, N-Br, and N-Cl-succinimide). These catalysts interact with the epihalonium ion (haliranium ion) intermediate resulting from the reaction of the alkene with the halonium ion. Good enantioselectivities are observed when this process is irreversible, i.e., when the nucleophilic quenching of the three-membered cyclic intermediate is rapid and irreversible [578]. A recent illustration (Figure 6.3) is bromolactonization (6.3) reported by Shirikawa and coworkers. The sulfide catalyst 102 is polyfunctional. Its phenolic group is essential for good ee’s and the diphenylmethanol moiety improves them [579].
In 2016, Zhao and coworkers reported the first enantioselective trifluoromethylthiolating lactonization of alkenes (6.4) by using an indane-based bifunctional chiral sulfide catalyst 103 [580].
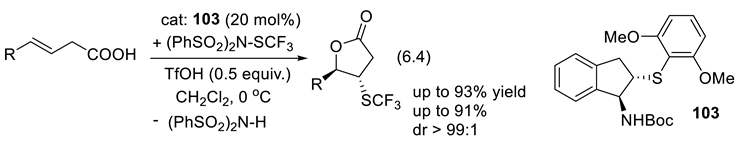
As for other halogen and chalcogen compounds, thioethers are nucleophiles and electrophiles. Their electrophilic property results from their σ-holes (Section 3.1.7). If halide catalysts are halogen bond (XB) donors, thioether catalysts are chalcogen bond (ChB) donors [581,582,583].
6.3. Enantioselective Hydrogen-Atom Transfer Catalyzed by Chiral Thiols
In 1998, Roberts and coworkers reported asymmetric radical-chain additions of triphenylsilane and of tris(trimethylsilyl)silane to prochiral alkenes catalyzed by chiral thiols (e.g., reaction (6.5)) [584]. The initiation step of this radical process engages di-tert-butyl hyponitrite as an initiator, which abstracts an H atom from the thiol catalyst 104 derived from D-mannose. Then the thiyl radical abstracts an H atom from the silane, forming a silyl radical that adds to the least sterically hindered carbon center of the alkene. The resulting carbon-centered prochiral radical abstracts an H-atom from the chiral thiol with face selectivity, forming an enantiomerically enriched product (Figure 6.4) [585].
6.4. Asymmetric Rauhut-Currier Reaction Catalyzed by a Chiral Thiols
Aroyan and Miller have found that N-acetylcysteine methyl ester catalyzed intramolecular enantioselective Rauhut-Currier reactions, such as reactions (6.6) (Figure 6.5) [586].
6.5. Sulfonium Salts as Chiral PTCs
The formation of sulfonium ylides from sulfonium salts does not occur under base-free, neutral conditions. Under such conditions, chiral sulfonium salt catalysts can efficiently promote asymmetric neutral phase-transfer reactions with a high level of enantioselectivity as demonstrated in 2017 by the group of Shirakawa. An example is given with the enantioselective Michael addition (6.7) catalyzed by 105 [587].
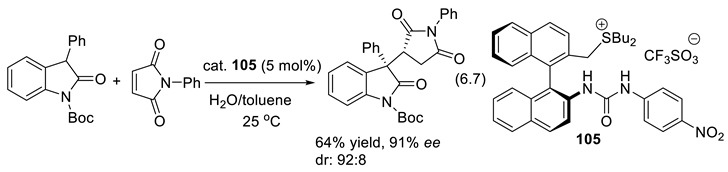
6.6. Chiral Sulfoxides
Chiral, enantiomerically pure sulfoxides are easily accessible. A large number of them have been prepared. They are powerful chiral auxiliaries in asymmetric synthesis and as chiral ligands in organometallic complexes [588]. Those possessing various additional functional groups proved to be efficient organocatalysts in asymmetric synthesis [589,590]. The group of Rachwalski developed chiral organocatalysts 106 that possess three functions: the hydroxy group, the stereogenic enantiomerically pure sulfinyl group, and enantiomeric amine moieties. Sulfoxides such as 106 catalyze the enantioselective Henry reaction, aza-Henry reaction, direct aldol condensation, Mannich reaction, and the asymmetric aziridination. They also catalyze the reduction of aromatic ketimines into the corresponding amines, as shown with reaction (6.8) (see also reaction (6.10)) [591].
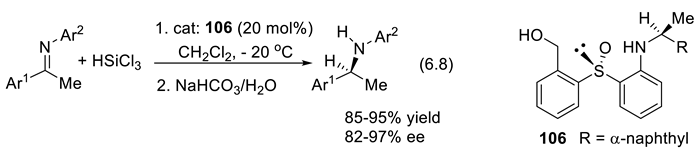
6.7. Chiral Amino-Thiocarbamates
In 2010, Yeung and coworkers realized enantioselective bromo-lactonizations (6.9) of 4-substituted pent-4-enoic acids applying the cinchonidine-derived amino-thiocarbamate 107 as a catalyst. The halophilic character of the C=S moiety is expected to promote the N-Br dissociation of NBS. At the same time, the N-H moiety stabilizes the succinimide anion by H-bond donation, forming a chiral brominating reagent as shown with 108 (see also Section 3.11.4) [592]. The enantioselective bromocyclization of olefinic amides into 2-substituted 3-bromopiperidines apply the same catalyst 107 [593].
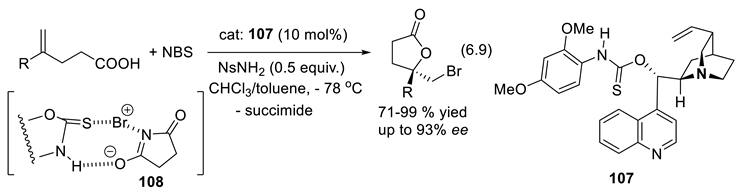
6.8. Chiral Sulfinamides
Chiral sulfinamides have emerged as versatile organocatalysts showing both Lewis base and Lewis acid activation modes [594]. In 2006, Sun and coworkers reported the first example of an organocatalytic reaction. They used a tert-butanesulfinamide-based organocatalyst 109 (readily obtained from the Ellman’s chiral auxiliary t-BuS(=O)NH2 [595] ), in which prochiral aromatic ketimines were reduced to amines using trichlorosilane as the hydrogen source (reaction (6.10)). Catalyst 109 must be seen as bifunctional: its S=O moiety is the Lewis base that interacts with the strongly oxyphilic Si center of the reagent HSiCl3, and the phenolic OH group provides a H-bond to the substrate imine moiety of the substrate [596]. In fact, a dimer of 109 was suspected to be the actual catalysts. This led Sun and coworkers to develop S-chiral bissulfinamides as highly enantioselective organocatalysts for the reduction of ketimines [597].
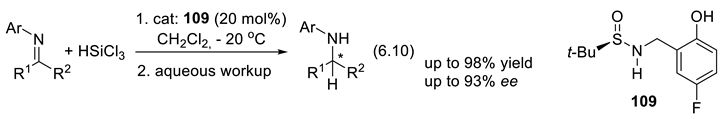
Kureshy and coworkers reported the ring opening reaction of meso-epoxides with anilines catalyzed by an easily available enantiomerically pure sulfinamide 110 (reaction (6.11)) [598].
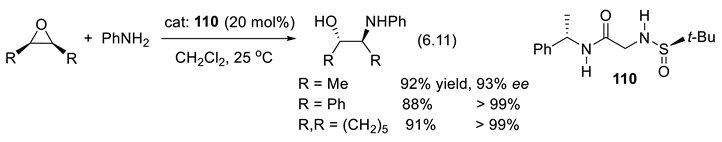
6.9. Chiral Thiophosphoramides
In 2010, the Denmark group realized the first enantioselective selenofunctionalization of unactivated olefins (reaction (6.12)) applying thiophosphoramide 111 as a catalyst. A chiral seleniranium ion-pair intermediate is formed, which is coordinated to the P=S base of the catalyst. It undergoes an intramolecular displacement reaction with its alcoholic moiety (transition structure 112) [599].
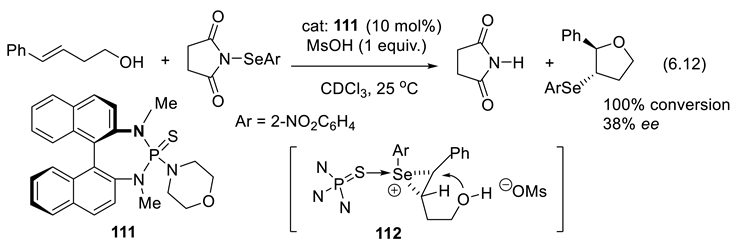
7. Asymmetric Catalysis with Organo-Selenium Compounds
Chiral organo-selenium catalysts have been prepared, and several of them have permitted to develop highly enantioselective asymmetric reactions [600,601,602].
7.1. Aminodiselenides as Chiral Pre-Catalysts
In 1998, Wirth and coworkers catalyzed the sequence (7.1) of methoxyselenylation and oxidative β-hydride elimination of trans-β-methylstyrene with chiral base 113 resulting from the oxidation of an amino-diselenide with potassium peroxodisulfate [603].
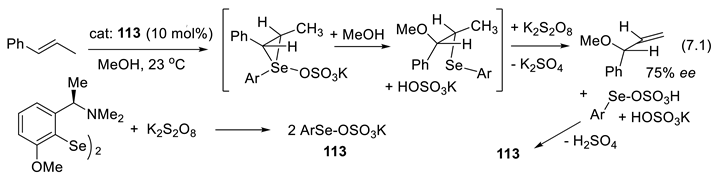
The Se-Se dimer of the natural amino acid l-selenocysteine is responsible for the catalytic activity of glutathione peroxidase. It is a pre-catalyst for the one-pot dihydroxylation of olefins employing H2O2 as a stoichiometric oxidant and water as a reaction medium. In some cases, the reaction affords enantiomerically enriched 1,2-diols [604]. In 2003, Braga and coworkers prepared chiral aliphatic amino-diselenides. They are pre-catalysts for the enantioselective addition of diethylzinc to aldehydes [605]. Examples of chiral selenium-catalysts developed since then are listed in Figure 7.1. They induce enantioselective hydrosilylation of ketones, addition of diorganozinc to aldehydes, conjugate addition to enones, Pd-catalyzed allylic alkylations, and halocyclization of alkenylamides, alcohols, and carboxylic acids [606,607]. Applying a chiral diselenide catalyst, the Denmark group realized an enantioselective syn-dichlorination of alkenes [608].
7.2. Selenophosphoramides as Chiral Catalyts
In 2011, the Denmark group reported the first catalytic, asymmetric sulfenylation of simple alkenes using chiral thio- and selenophosphoramide catalysts. Both inter- and intramolecular thiofunctionalizations are possible for a variety of olefins. An example is shown with reaction (7.2) catalyzed by selenophosphoramide114. The proposed mechanism (Figure 7.2) involves activation of the sulfur electrophile (N-phenylsulfenyl-phthalimide) by the Lewis base catalyst 114 with formation of the chiral and enantiopure selenosulfonium ion intermediate that adds to the prochiral alkene with face selectivity forming a enantioenriched chiral episulfonium ion intermediate (of formal C2-symmetry). Attack of the nucleophile (MeOH) on either one or the other carbon center of the latter give the same final product of anti-addition. In 2014, Denmark disclosed an enantioselective intramolecular sulfenamination of the tosylamide of alk-4-enamines applying catalyst 115 (Figure 7.2) [609,610,611].
7.3. Selenides as Chiral Catalysts
In 2013, Yeung and coworkers catalyzed the asymmetric bromocyclization (7.3) of trisubstituted olefinic amides with the C2-symmetric D-mannitol-derived tetrahydroselenophene 116 [612]. The mechanism of the reaction implies, probably, coordination of the selenide Lewis base with N-bromophthalimide (NBP). An activated and chiral electrophilic brominating agent forms as an intermediate that adds to the prochiral alkene with face selectivity. A cyclic epibromonium cation intermediate is generated, which is quenched intramolecularly by the amino moiety.
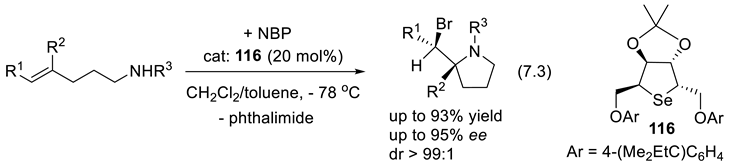
In 2017, Zhao and coworkers reported the enantioselective CF3S-aminocyclization of internal alkenes to afford saturated azaheterocycles (reactions (7.4)) catalyzed by the bifunctional selenide catalyst 117 [613]. It also catalyzed the enantioselective cyclizations (7.5) [614].
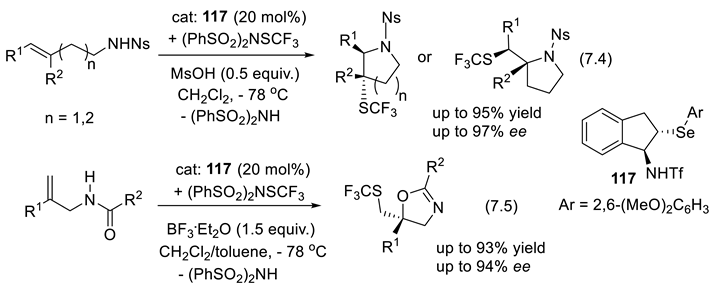
In 2019, the same group reported an efficient protocol for the intermolecular oxysulfenylation (7.6) applying chiral bifunctional selenide 118 as catalyst. Transition state 119 was proposed for these reactions [615].
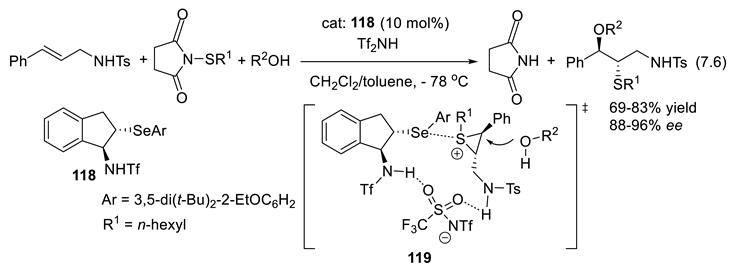
7.4. Chiral Trivalent Selenonium Salts as PTCs
As for sulfonium salts, chiral trivalent selenonium salts are expected to be asymmetric phase transfer catalysts. The Michael addition (7.7) catalyzed by 120 is an example [607].
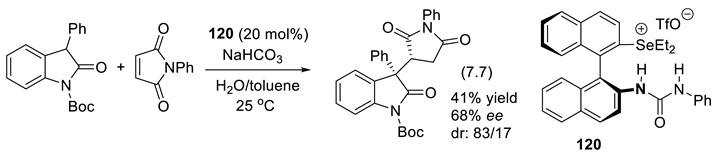
Trisubstituted selenonium salts are organic Lewis acid catalysts in electrophilic halogenation and aldol-type reactions. Chiral analogues of such catalysts will appear soon [616].
8. Asymmetric Organocatalysis with Electron-Rich Carbene Intermediates
In 1943, Ukai reported the first thiazolium salt-catalyzed reaction with the condensation of benzaldehyde into benzoin (2 PhCHO → racemic PhC(OH)H-COPh) [617]. In 1958, Breslow published his mechanism that implies the formation of an electron-rich heterocyclic carbene intermediate. The latter adds to the aldehyde forming an acyl anion equivalent (Breslow intermediate, Figure 8.1b) [618]. Following these seminal works, Stetter [619,620,621] and several other groups have reported other N-heterocyclic carbene (NHC) precursors including chiral ones (see e.g., Figure 8.1). The latter catalyze enantioselective reactions such as homo- and cross-benzoin, and -acyloin condensations (Section 8.1), cross-aza-benzoin condensations (Section 8.2), Stetter reactions (Section 8.3), aza-Stetter reactions (Section 8.4), cycloadditions (Section 8.5), (3,3)-sigmatropic rearrangements (Section 8.6), Michael acceptor Umpolung (Section 8.7), radical reactions (Section 8.8), and synthesis of atropisomers (Section 8.9).[622,623,624,625,626]. Figure 8.1presents typical NHC precursors (pre-catalysts) used today. The triazolium salts employed first by Enders are dominating the scene. As for other types of organo-catalysts, the trend now is to create polyfunctional NHCs [627].
NHCs have singlet ground states with a sp2-divalent carbon center. Their stability arises from electronic and steric factors. The lone pair of electrons is stabilized by inductive electron withdrawal by the proximal electronegative atoms (sp2(C:)/σ*(C-Z) interactions) and by the mesomeric donation of electron density from the filled p orbitals of the heteroatoms (Z = N, S) to the vacant 2p-orbital at the carbene center (n(Z:)/2p(carbene) interactions). This renders NHCs electron-rich: they are nucleophiles. Figure 8.2 displays typical intermediates generated by reactions of NHCs resulting from triazolium salts. Except for acyl azolium salts, these intermediates are nucleophiles that undergo several possible reactions with electrophiles, as illustrated in the following sections. If NHCs are chiral, these intermediates are also chiral (temporary chiral auxiliary).
8.1. NHC-Catalyzed Asymmetric Benzoin Condensations
In 1966, Sheehan (he had developed the first practical synthesis of penicillin V) and Hunneman reported the first enantioselective asymmetric benzoin condensation (8.1) using zwitterion/carbene intermediates as catalysts generated from chiral thiazolium salts (e.g., 138, Figure 8.3a, up to 51% ee) [628]. Based on the catalytic cycle proposed by Breslow (Figure 8.3b [629]), Sheehan and Hara later studied the stereochemical effect of various enantiopure thiazolium salts on the benzoin condensation. Although the authors did not improve the yield, they observed some ee increases [630]. An electrophilic carbonyl moiety becomes a nucleophilic acyl anion equivalent (Umpolung). In analogy to bio-catalysis that uses thiamine (vitamin B1) as a co-factor, under basic conditions, the thiazolium salt 138 (the pre-catalyst) undergoes 1,1-elimination of HBr, generating thiazolylidene carbene 139 (the catalyst) that adds to an aldehyde equilibrating with a zwitterionic intermediate which, after tautomerization, furnishes the crucial Breslow intermediate 140 [631]. The latter is a nucleophile (an enamine). It adds to a second aldehyde molecule and furnishes the benzoin product with regeneration of the active catalyst 139 (Figure 8.3b). Note: cyanide anion also catalyzes the condensation of benzaldehyde into racemic benzoin through a similar mechanism.
Several reports followed this pioneer work, but with deceiving yields and ee’s. In 1996, Enders and coworkers found that NHC 122 (Figure 8.1a) generated on treating triazolium salt 121 (1.25 mol%) with K2CO3 (THF, 25 oC) is a good catalyst (22-72% yield, 20-86% ee) for the asymmetric homo-benzoin condensation of aromatic aldehydes (X-C6H4CHO, X = H, 3-Me, 4-Me, 3-MeO, 4-MeO,4-F3C. 4-Cl, 4-Br) [632]. With the bicyclic triazolium salt 123 (10 mol%) and t-BuOK as a base, up to 95% ee was observed for the same reactions [633]. In 2006, the Enders group realized asymmetric intramolecular cross-benzoin reactions of aldehydes and ketones in the presence of tetracyclic triazolium salt 130 as pre-catalyst (10 mol%, THF or toluene, 25 oC). For instance, 2-(3-oxohept-1-yl)benzaldehyde was converted into (S)-2-butyl-2-hydroxy-1-tetralone in 85% yield and 98% ee [634]. Nearly at the same time, Bode’s group reported similar enantioselective intramolecular cross-benzoin reactions [635] applying Rovis pre-catalysts 127 and 128 (Figure 8.1a) [636].
8.2. NHC-Catalyzed Asymmetric Aza-Benzoin Condensations
In 1988, López-Calahorra reported the condensation of iminium salts generated in the reaction of paraformaldehyde, morpholine, or piperidine, and aromatic aldehydes. α-Aminoketones were obtained in moderate yields [637]. In 2001, Murry, Frantz, and coworkers reported the first example of an aldehyde-imine cross-benzoin reaction catalyzed by thiazolylidene carbenes, using arylsulfonylamides as imine precursors [638]. In 2005, Miller and coworkers disclosed the enantioselective (up to 87% ee) cross-aza-benzoin reaction between aldehydes and N-acylimines. Their pre-catalyst was the peptide-derived thiazolium salt 134 (Figure 8.1b) [639]. In 2012, Rovis and Dirocco reported the direct asymmetric coupling of aliphatic aldehydes and N-Boc-protected imines (reaction (8.2)).The cross-aza-benzoin reaction used triazolium salts 141 as pre-catalysts [640]. Asymmetric aza-benzoin condensation with ketimines as substrates is also possible [641].
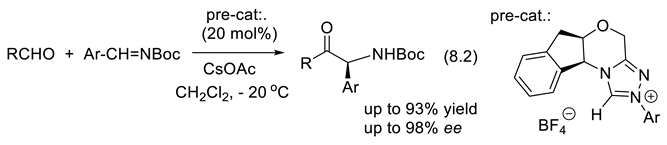
In 2017, Biju and coworkers reported a NHC-catalyzed enantioselective synthesis of dihydroquinoxalines applying Bode’s pre-catalyst (reaction (8.3)). This reaction proceeds via Umpolung at one of the imine moieties (electrophilic carbon center C=N) in the substrate, generating a nucleophilic 1,2-diaminoalkene intermediate (aza-Breslow intermediate). The latter reacts with the other imine moiety [642]. The process is a catalytic asymmetric intramolecular aza-aza-benzoin condensation [643]. The first isolation of an aza-Breslow intermediate was made by the group of Douthwaite [644] and later, by Rovis and coworkers [645]. The Biju group has also reported enantioselective intramolecular additions of aza-Breslow intermediates to carbonyl compounds [646].
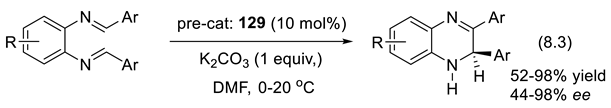
8.3. NHC-Catalyzed Asymmetric Stetter Reactions
In 1974, Stetter and Kuhlmann found that a thiazolium salt in the presence of a base catalyzes the reaction of α,β-unsaturated ketones, esters, and nitriles with aliphatic, aromatic, and heterocyclic aldehydes (Figure 8.4a). The now-named “Stetter reaction” implies Umpolung of the aldehyde into the corresponding Breslow intermediate that undergoes 1,4-addition (instead of a 1,2-addition as for the benzoin condensation) to Michael acceptors [647]. In 1996, Enders and coworkers reported the first asymmetric intramolecular Stetter reaction. With their pre-catalyst 121, they obtained modest yields (22-73%) and ee’s (41-74%) [648,649]. In 2002, Rovis and coworkers reported a more efficient version (reaction (8.4), Figure 8.4b) [650]. Several alternative pre-catalysts have been proposed for this reaction, as shown with reaction (8.5) (Figure 8.4c) [651].
Before 1999, Enders and coworkers found two examples of catalytic asymmetric intermolecular Stetter reaction utilizing a chiral thiazolium pre-catalyst. Their best overall yields (4%) and ee (39%) were deceiving. [652,653]. In 2005, by modifying thiazolylalanine, Miller and coworkers obtained pre-catalysts for the asymmetric intramolecular Stetter reaction (up to 81% ee) [654]. In 2008, the Enders group reported better efficiencies for asymmetric intermolecular Stetter reaction between aryl aldehydes and chalcones applying a triazolium pre-catalyst [649,655]. The same year, Rovis and coworkers reported on the combination of glyoxamides with alkylidene malonates to afford Stetter products in high yields and high ee, also by applying triazolium pre-catalysts [656]. Further NHC pre-catalysts that permit the efficient asymmetric intermolecular Stetter reaction of nitroalkenes and aldehydes were prepared [657]. In 2011, the same group reported catalytic asymmetric intermolecular Stetter reaction between enals and nitroalkenes [658,659].
8.4. NHC-Catalyzed Asymmetric Aza-Stetter Reactions
In 2019, Lupton and coworkers reported a catalytic asymmetric intermolecular imino-Stetter or aza-Stetter reaction (8.5) that condenses aldimines and 3-methylenechroman-2-ones to afford enantioenriched γ-iminolactones (Figure 8.5) [660]. The process involves the formation of an aza-Beslow intermediate with the protected imine that adds then to Michael acceptors [661].
8.5. NHC-Catalyzed Asymmetric Cycloadditions
In 2006, Bode and coworkers reported the first NCH-catalyzed asymmetric aza- and oxo-Diels-Alder reactions (8.7). They applied their pre-catalyst 129. In the case of azadiene-Diels–Alder reaction (Figure 8.6a), the NHC catalyst adds to β-substituted α,β-unsaturated aldehydes and forms the corresponding alkenyl Breslow intermediates. The latter undergo tautomerization into enolates, the dienophiles that react with alkenyl imines (the aza-dienes) in an endo mode of addition. After elimination of NHC (recovery of the catalyst), the cycloadducts are isolated (Figure 8.6b) [662].
The oxa-diene-Diels–Alder reaction reported the same year by the same group occurred in a similar way. In this case, α-chloro-aldehydes and β-substituted α,β-unsaturated ketones or unsaturated α-ketoesters were used as substrates [663]. In 2011, Chi and coworkers reported similar NHC-catalyzed hetero-Diels-Alder reactions between chalcones (oxa-dienes) and enals (converted into enolate intermediates as dienophiles) [664]. Highly enantioselective formal hetero-Diels-Alder reactions between chalcones and formylcyclopropanes have also been reported by the same group [665].
The first NHC-catalyzed Staudinger cycloaddition (8.8) was presented in 2008 by Ye and coworkers (pre-cat: 141, Figure 8.7a) [666]. Ketenes react with NHCs giving enolates, nucleophiles that can add to imines forming zwitterions. The latter cyclize into β-lactams. This is a NHC-catalyzed Staudinger reaction, a formal (2+2)-cycloaddition (Figure 8.7b) [667].
The same group, the same year, reported a NHC-catalyzed asymmetric (2+2) cycloaddition of alkyl (aryl) ketenes with 2-oxoaldehydes to afford β-lactones (up to 99% ee) [668]. In 2009, this group reported a diastereo- and enantioselective synthesis of β-trifluoromethyl-β-lactones bearing two contiguous stereocenters. Chiral NHC-catalyzed formal (2+2)-cycloaddition of alkyl(aryl)ketenes and trifluoromethyl ketones was realized [669].
In 2012, the group of Bode presented highly enantio- and diastereoselective annulations of α- and β,β′-substituted enals with cyclic sulfonylimines as nucleophiles catalyzed by 129. Reaction (8.9) is an example. The NHC derived from the HCl elimination of 129. It reacts with the enal forming the corresponding Breslow intermediate that is then oxidized into an acyl azolium chloride. The latter electrophile reacts with the enamine tautomer of the sulfonylimine in a intermolecular N-acylation reaction. Follows a fast oxy-accelerated aza-Cope rearrangement and a final intramolecular N-acylation (Figure 8.8) [670].
8.6. NHC Catalyzed Asymmetric (3,3)-Sigmatropic Rearrangements
In 2007, Bode and coworkers realized a NHC-catalyzed enantioselective cyclopentene synthesis (reaction (8.10), Figure 8.9a) by condensing cinnamaldehydes and analogues ((E)-β-substituted enals) with (E)-3-oxo-alk-2-ene-carboxylic esters (chalcones and analogues). As already mentioned, the NHC form first homoenolate equivalents with α,β-unsaturated aldehydes (alkenyl-Breslow intermediates). The latter add to the ketone moiety of 3-oxo-alk-2-ene-carboxylic esters (crossed benzoin condensations) forming adducts that are anionic 3,4-dihydroxyhexa-1,5-diene entities ready for oxy-Cope rearrangements furnishing ketones adjacent enolate moieties [671]. Intramolecular aldol reactions, and decarboxylation, provide the final cyclopentenes (Figure 8.9b).
Claisen rearrangements (R1CH=CH(A)-CH(B)-O-C(X)=CHR2 → (B)CH=C(A)-CH(R1)-CH(R2)-C(=O)-X) and their variations (nature of substituents A, B, X ) are powerful tools for the stereoselective construction of C-C bonds. Asymmetric versions have been described that use chiral Lewis acids [672,673,674] and Jacobsen’s ureas [675]. In 2010, Bode and coworkers disclosed the enantioselective Coates-Claisen rearrangement (8.11) catalyzed by NHCs [676]. With conjugated ynals, NHCs equilibrate with α,β-unsaturated acyl azolium intermediates that react with enols (Figure 8.10), 2-oxopropanoic esters, phenols to produce dihydropyranones [677]. In 2011, the same group reported similar enantioselective synthesis of dihydropyridinones via NHC-catalyzed aza-Claisen reactions [678]. Further high yielding and highly enantioselective NHC-catalyzed Coates-Claisen reactions were then reported by the groups of Bode [679] and You [680].
8.7. Umpolung of Michael Acceptors by NHCs: Conversion of Enones into Homoenolates
In 2006, Scheidt and coworkers reported the enantioselective of α,α-disubstituted cyclopentenes by NHC-catalysed desymmetrization of 2-(4-oxobut-2-en-1-yl)-1,3-diketones. In this work, 1,2-addition of an NHC to the enal moiety generates an alkenyl-Breslow intermediate, which, after protonation, undergoes an intramolecular aldol reaction. Then an intramolecular acylation produces a β-lactone that is decarboxylated into cyclopentenes (up to 94% ee) [681]. The Umpolung of Michael by 1,4-addition of NHC acceptors [682] is an unusual activation mode, compared with the more commonly observed 1,2-addition chemistry (formation of alkenyl-Breslow intermediates) [683]. The first example of Umpolung of Michael acceptors by 1,4-addition of an NHC was reported by Fu and co-workers in 2006 for the intramolecular alkylation of α,β-unsaturated esters to afford cyclopentenes [684].
In 2016, Lupton and coworkers disclosed enantioselective reactions (8.12) based on Michael acceptor Umpolung by NHCs. In the example of Figure 8.11a, a bis-Michael acceptor substrate is cyclo-isomerised enantioselectively to (S)-2-aryl propionate esters. The nucleophilic carbene undergoes a 1,4-addition with the conjugated enone, equilibrating with a zwitterion that is composed of an enolate moiety. The latter isomerises into the corresponding homoenolate that is stabilised by the vicinal triazolium cation. The homoenolate adds intramolecularly to the α,β-unsaturated ester group forming another enolate intermediate that is protonated with face selectivity, the crucial step responsible for asymmetry (Figure 8.11b) [685].
Applying the NHC-catalyzed Umpolung to 6-methylidene-(E)-hept-2-enedioic esters and 4,4-disubstituted derivatives, Lupton and coworkers developed an enantioselective cyclopentene synthesis [686] and (5+1) annulation reactions [687]. In 2019, Tobisu and co-workers demonstrated the polarity inversion of aryl acrylamides with NHCs [688]. Later, the same group reported a Truce–Smiles rearrangement reaction using the same strategy. [689]. A cross-coupling reaction involving a deoxy-Breslow intermediate was reported in 2022 by the Biju group [690].
8.8. Radical Reactions Promoted by NHC Catalysts
Breslow intermediates are oxidized into acylazolium salts that react with all kinds of nucleophiles, including alcohols, to generate the corresponding carboxylic esters (NHC-catalyzed oxidation of aldehydes) [691]. As seen in the preceding sections, NHCs undergo preferentially 1,2-additions with α,β-unsaturated aldehydes, equilibrating with alkenyl-Breslow intermediates that can undergo all kinds of intramolecular or intermolecular reactions. Two-electron oxidation of alkenyl-Breslow intermediates generates α,β-unsaturated acylazolium salts that are reactive Michael acceptors useful for many reactions [692,693,694,695], including enantioselective ones when applying chiral NHCs [696,697].
Upon deprotonation, Beslow intermediates (enols) equilibrate with Breslow enolates that are oxidised easily (electron-rich enolates). In the presence of an oxidant capable to abstract one electron (single electron transfer: SET) relatively stable radical-cations form that are persistent [698,699]; they can be quenched by other radicals in bimolecular processes [626]. This is also the case for alkenyl-Breslow intermediates. In 2014, Rovis and coworkers used that property to develop an asymmetric oxidation of enals into chiral β-hydroxy carboxylic esters applying pre-catalyst 142 (reaction (8.13), Figure 8.12) [700].
In 2022, Hong and coworkers described an NHC-catalyzed radical reaction that converts enals to chiral β-pyridyl esters with excellent pyridyl C(4)-regioselectivity and ee (reaction (8.14), Figure 8.13). A pyridinium salt forms a complex with the Breslow enolate resulting from the addition of the NHC-catalyst to the conjugated enal. Under blue light irradiation, a single electron transfer (SET) generates two radicals that combine to form a C-C bond between C(4) of the pyridine moiety and C(β) of the enal [701].
Catalytic enantioselective synthesis of α-aryl ketones includes α-arylation of ketones/enones catalyzed by transition metals, benzylic acylation, and hydroacylation of alkenes via either organocatalysis or reductive copper catalysis [702]. Recently, the group of Ye presented (Figure 8.14) a photocatalytic process that does the direct enantioselective oxidative acylation of benzylic C-H’s (e.g. reaction (8.15)) applying pre-catalyst 143. The photocatalyst is 9,10-dicyanoanghracene (DCA). Under light irradiation, the electronically excited state DCA* is formed. It abstracts an electron from the benzylic substrate (Ar1CH2CH3) forming an radical-cation (Ar1Et(·+)) and radical-anion DCA(·−). The benzylic radical cation reacts with face selectivity with the chiral radical-anion generated by electron transfer from DCA(·−) to the acyl azonium salt resulting from the condensation of the chiral NHC catalyst and the acyl fluoride reagent (Ar2COF), and reduction by radical-anion DCA(·−) [702].
8.9. NHC-Catalyzed Enantioselective Synthesis of Atropisomers
Axially chiral molecules are useful ligands in transition metal-catalyzed reactions and powerful organo-catalysts [703,704]. They are present also in Nature [705] and are often bioactive [706]. In 2018, Wang and coworkers reported the first NHC-catalyzed atroposelective (3+3) annulation (8.16) between ynals and cyclic 1,3-diketones that applies 144 as pre-catalyst to provide chiral α-pyrone-aryls (Figure 8.15) [707]. Further asymmetric syntheses of atropisomers catalyzed by NHCs have been developped (for a review, see [626]).
In 2019, Zhu and coworkers reported a formal (4+2)-cyclization of conjugated dienals and α-arylketones catalyzed by chiral NHCs. The enantiomerically enriched products obtained undergo oxidation into atropoisomeric bis-aryls with 90-98% ee [708]. In 2022, Chi and coworkers reported the enantioselective preparation (up to 98% ee) of axially chiral styrenes bearing a chiral axis between a sterically non-congested acyclic alkene and an aryl ring. The reaction involves ynals, sulfinic acids, and phenols as the substrates with a chiral NHC as the catalyst [709]. Recently, the Biju group have reported NHC-catalyzed amidification leading to N-N axially chiral 3-amino-quinazolinoes (up to 99% yield, up to 98% ee) [710].
9. Enantioselective Organocatalysis by Hydrogen Bonding
The mechanism of action of various enzymes relies upon hydrogen bonding (H-bond) for the binding of substrates, reactants, and for the binding of transition structures (electrophile activation) [70]. In 1942, Wassermann showed that weak protic acids (carboxylic acids, phenols) accelerate the cycloaddition of cyclopentadiene to benzoquinone (Diels Alder reaction with normal electron-demand, electrophilic dienophiles, Chap. 1) [53]. In 1979, Inoue and coworkers found that a compound smaller than an enzyme like cyclo(L-phenylalanine-L-histidine catalyzes the asymmetric addition of HCN to arenecarbaldehydes (Section 4.1). The cyclic dipeptides possess N-H bonds (H-bond donors). In 1981, Wynberg found that cinchona alkaloids that bear a free OH group in proximity to the basic quinuclidine nitrogen moiety, catalyze enantioselective addition reactions to conjugated enones (Section 3.1.2, reaction (3.4)). In 1984, N-alkyl cinchona alkaloid derivatives bearing a free OH group showed to be highly efficient enantioselective PTCs for the alkylation and Michael addition of indanone (Section 3.2.1, Figure 3.9, cat. 22, 23). L-proline catalyzed aldol reaction involves the intervention of H-bond from the COOH group (Section 3.5.2). In 1985, Hine and coworkers demonstrated that 1,8-biphenylenediol (a biphenol) is a better catalyst than monophenols of similar Brønsted acidity for the reaction of diethylamine with phenyl glycidyl ether that produces PhOCH2C(OH)H-CH2-NEt2. This was explained in terms of double electrophilic activation of the oxirane moiety of glycidyl ether by the two O-H bonds provided by 1,8-biphenylenediol [711]. In 1999, Kita and coworkers reported the asymmetric hypervalent-iodine-mediated oxidation of sulfides to sulfoxides (ArSMe + 0.5 PhIO2 → ArSOMe + 0.5 KI, up to 72% ee) catalyzed by O,O’-bis(2-methoxybenzoyl) (2R,3R)-tartaric acid [712]. In 2010, Terada and coworkers found that O,O’-diacyl tartaric acid derivatives catalyze the enantioselective Friedel–Crafts reaction of indoles with an a-imino ester (up to 88% ee [713]). (+)-Tartaric acid was found to catalyze enantioselective (4+2)-cycloaddition of isochromeneacetals and vinylboronates furnishing dihydronaphthalenes and dihydrobenzofluorene products with up to 97% ee employing 10 mol% of (+)-tartaric acid as the catalyst, in combination with 5 mol% of Ho(OTf)3 [714]. Chiral diols such as TADDOLs (C2-symmetrical tetraaryl-1,3- dioxolane-4,5-dimethanols derived from enantiomerically pure L-(+)-(2R,2R)-tartaric acids (natural) and its enantiomer) and BINOLs (C2-symmetrical 1,1’-bi-2-naphthol and substituted derivatives, Figure 9.1) [715,716] were available in the 1980’s already. They are H-bond donors. Optically pure 1,1′-bi-2-naphthol (BINOL) (introduced by Noyori in 1980 [717]) and its derivatives (BINOLs) are the most important sources of chirality for organic synthesis and material sciences (Figure 9.1) [718]. In the beginning, they served as diolates of metallic reagents, or as ligands in organometallic catalysts. Their direct use as diols in asymmetric organocatalysis started after 2000. Because of their greater stability and larger variability of their substitution, they have taken a higher importance relative to simpler tartaric acid derivatives. In general, BINOLs and vaulted analogues VANOLs and TADOLs (Figure 9.1) have served as the most efficient catalysts [719].
In 1995, Curran and coworkers showed that ureas accelerate Claisen rearrangements. These additives modified the stereoselectiviy of the reactions [720]. In 1998, the group of Jacobsen reported asymmetric Strecker reactions catalyzed by short peptide analogues containing a urea moiety or a thiourea unit (see Section 4.1). This opened a new era to asymmetric organocatalysis with chiral ureas, thioureas and, then with squaramides [69,212,721]. These catalysts are double H-bond donors that activate electrophilic substrates toward nucleophilic reactions (e.g., Figure 9.2b). Alternatively, the catalyst binds strongly to anions and form diastereomeric ion-pair intermediates as suggested by Schreiner and coworkers in 2006 (e.g., Figure 9.2c) [722]. This is asymmetric ion-pairing catalysis [723].
As already mentioned (Chap. 2) in their ground state and in solution, uncharged (neutral) substrates, reactants, catalysts and solvent molecules are associated through electrostatic forces that include dipole/dipole and dipole/induced dipole interactions. All compounds with A-H bonds possess local dipole moments associated with each A-H moiety. When A is more electronegative than H, the dipole places a partial negative charge on A and a partial positive charge on H. Therefore, a molecule with one, two, three, etc. A-H moieties tends to stick to the negatively charged extremities of another molecule B with its H-atom(s); it forms a complex of type A-H….B. This makes the structural and dynamic properties of living (e.g., enzymes, proteins, glycoproteins, DNA, etc.) and non-living matter (e.g., water, ice, methane/water clathrates, cellulose, etc.). If the dipole/dipole interaction between A-H and B is strong the H-bond between H and B is shorten and partial covalent bonding occurs. At the limit, bonding in complex A-H….B is a covalent three-center four-electron bond [724]. With a weakly acidic catalyst, Y-H, that possesses a single A-H moiety, and a substrate R-X: with a partial negative charge on X, an equilibrium of type R-X: + Y-H ⇄ R-X…H-Y is realized for which H-bridging is established between X and H without proton transfer. The relative amount of the complex at equilibrium is low if the X…H bond is weak. For an equilibrium constant close to unity (ΔGT = 0) the heat of reaction (ΔHT) must be about -10 kcal mol-1 to compensate for the cost of entropy (-TΔST) of this equilibrium (condensation) at 25 oC. This is the case only if the solvent does not form competitive H-bonds with R-X: and Y-H. If Y-H is a strong Brønsted acid, it may protonate R-X: according to equilibrium R-X: + Y-H ⇄ R-X(+)-H + Y(-) in which a cation and an anion are formed. They form a tight ion-pair implying H-bridging between cation and anion (R-X(+)-H….Y(-)). If H-Y is chiral, quenching of this ion-pair by a nucleophilic reagent can be enantioselective. One speaks of asymmetric ion-pair catalysis [723] and anion recognition [725,726,727]. A tight ion-pair may equilibrate with a solvent separated ion-pair or free ion pair for which hydrogen bridging is not possible between cation and anion, but is possible for both of them with the solvent. The relative concentrations of these species depend on solvent and temperature. There is a spectrum of intermediate species between the two above limiting situations. For instance, in their ground state, substrates and reactants make H-bridged complexes with the catalyst Y-H without proton transfer, and only in the transition state, a proton transfers fully. The catalytic effect of Y-H results from the fact that it stabilizes transition structures more than substrates and reactants. A good catalyst must not interact strongly with the final product, otherwise the latter inhibits the catalytic process. What we discussed above for R-X: substrates also apply to double-bonded compounds bearing carbonyl (C=O:) or imine (C=(R)N:) functions. In these cases, the catalytic effect (rate increase and enantioselectivity) arises from extra stabilization of the tetrahedral intermediates resulting from the nucleophilic addition to the carbonyl or imine moiety (Figure 9.2b): The strength of the hydrogen bonding offered by the catalyst increases from substrate + reactant to the transition structures of their reaction. Indeed, the dipole moment in the latter is generally increased. H-Bond donors form relatively stable complexes with anions (e.g., X(-) of Figure 9.2c). Thioureas are often stronger H-bond donors than ureas and diols. Because of the greater electronegativity of the O atom compared with the S atom, one would expect the opposite. The greater size and polarizability of the S atom compared with the O atom explain the observations [728,729]. Squaramides form stronger complexes with halide anions than thioureas. Guanidinium [324] and amidinium ions are stronger H-bond donors because of their higher acidity. Being positively charged, they transfer a proton to a substrate and transition structures more readily.
9.1. Applications of Chiral Diols as Organocatalysts
(S)-BINOL forms readily by asymmetric oxidative coupling of 2-naphthol with CuCl2 in the presence of (S)-(+)-amphetamine as ligand [730]. When combined with metallic species, BINOLs form diolates that are extremely potent chiral Lewis acid catalysts. One application reported in 2002 by Ding and coworkers is the enantioselective hetero-Diels-Alder reaction of Danishefsky’s diene with aldehydes at room temperature, without solvent, in the presence of only 0.1-0.005 mol% catalyst made by mixing Ti(O-i-Pr)4 with one or two different BINOLs. The reaction affords dihydropyrones derivatives in high yield (up to 99%) and ee (up to 99.8%) [731].
In 2003, McDougal and Schaus reported the first example of a highly enantioselective asymmetric Morita-Baylis-Hillman (MBH) reaction of cyclohexenone with aldehydes using a chiral Brønsted acid 145 (a H8-BINOL) as the catalyst, and triethylphosphine as the nucleophilic promoter (reactions (9.1)) [732].
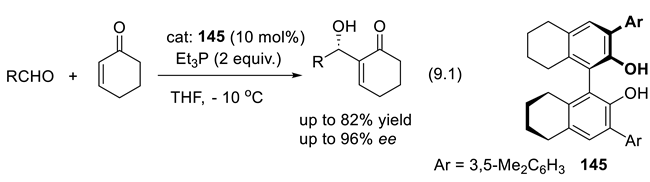
In 2006, Schaus and coworkers reported the highly diastereo- and enantioselective allylboration of ketones (reactions (9.2)) catalyzed by (S)-3,3’-dibromo-BINOL (146) [733].
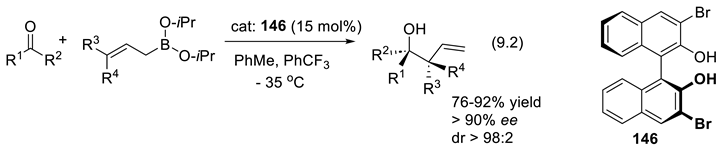
In 2009, the same group found BINOL derivatives that catalyze the enantioselective additions of boronates to acylimines [734]. In 2015, Schaus and coworkers realized enantioselective multicomponent condensation reactions of phenols, aldehydes, and boronates also catalyzed by chiral biphenols. Initial Friedel-Crafts alkylation of the aldehyde and phenol yields an ortho-quinone methide that undergoes an enantioselective boronate addition [735]. Aldehydes and amines are combined with allylboronates in a one-pot two-step procedure to provide chiral homoallylic amines (Petasis-borono-Mannich allylation). In 2017, Jiang and Schaus reported an asymmetric version of this reaction catalyzed by (R)-3,3’-diphenyl-BINOL [736]. For more cases, see [719]. In 1993, the Wulff group introduced vaulted biaryl ligands VANOL and VAPOL (Figure 9.1) as chiral ligands for a metal-catalyzed asymmetric Diels–Alder reaction [737]. They are inexpensive diols that can be prepared on a large scale [738]. The chiral metallic diolates catalyze the enantioselective amidation of imines, iminoallylation of aldehydes, aza-Darzens reaction, cis-aziridination of imines, trans-aziridination of imines, Baeyer–Villiger reaction, benzoyloxylation of aryloxindoles, desymmetrization of aziridines, Diels–Alder reactions, hetero-Diels–Alder reactions, hydroarylation of alkenes, hydrogenation of alkenes, imidation of imines, Mannich reactions, multicomponent aziridination of aldehydes, Petasis reaction, propargylation of ketones, reduction of imines, chlorination and Michael reactions of oxindoles, hydroacylation of alkenes, pinacol rearrangement, reduction of aminals and Michael addition of alkynes. Many modified VANOL and VAPOL derivatives are crucial chiral ligands for all kinds of catalysts, including Brønsted acids (Chapter 10) [739].
Ferrocene-based diols (Figure 9.1) catalyze asymmetric hetero-Diels-Alder reactions CH2=C(OTMS)-CH=CH-NMe2, + RCHO achieving up to 92% ee when R =1-naphthyl. [740,741]. In 2004, Ding and coworkers reported the enantioselective hetero-Diels-Alder reaction of Brassard's diene (Me3SiO(MeO)C=CH-C(OMe)=CH2) with aldehydes catalyzed by 147 and other TADDOL derivatives. They realized a one-step synthesis of (S)-(+)-dihydrokawain [742]. In 2005, the Rawal group reported that TADDOL derivatives such as 147 are better asymmetric organocatalysts than BINOLs for the enantioselective Mukaiyama aldol reaction of silyl dienol ethers with a range of aldehydes (e.g., reaction (9.3)) [743].
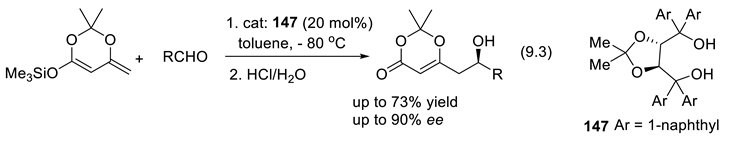
In 2006, the same group reported the Mukaiyama aldol reactions of O-silyl-N,O-ketene acetals + RCHO mediated by 147 (up to 96% ee for the major syn product) or by cyclohexylidene-(1-naphthyl)-TADDOL derivative (up to 98% ee) as an organocatalyst [744]. They also found that 147 catalyzes the diastereoselective and enantioselective Mukaiyama reaction (9.4) with acetyl dimethyl phosphonate.
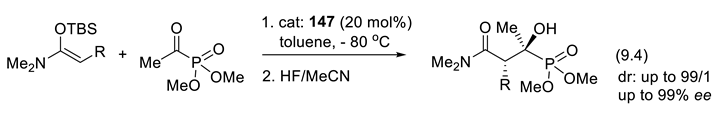
In 2005, Yamamoto and Momiyama reported that carboxylic acids catalyze the reactions of enamines with nitrosobenzene give O-nitroso-aldols. In contrast, with 147 as catalyst, N-nitroso-aldol were produced (reaction (9.5)) [745].
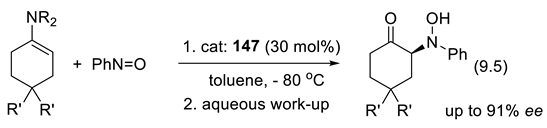
9.2. Silane-1,1-Diols as Chiral Catalysts
In 2006, the group of Kondo reported that silane-1,1-diols recognize anionic species [746]. In 2011, Mattson and coworkers reported on the nitro-group rendered more electrophilic through H-bonding with silane-1,1-diol catalysts [747]. On their side, Franz and coworkers showed that these catalysts are capable to activate electrophiles such as carbonyl compounds [748]. In 2013, the Mattson group exploited silane-1,1-diols such as 148 ((R)-3,5-dihydro-4H-dinaphtho[2,1-c:1’,2’-e]silepine-4,4-diol, or BINOL-silanediol) for their ability to bind small anions in asymmetric reactions (9.6) (Figure 9.3) [749]. In 2016, Mattson and Kondo reported similar asymmetric silane-1,1-diol-catalyzed addition of silyl ketene acetals to benzopyrylium triflate intermediates. With the chiral catalyst that binds the triflate anion, diastereomeric ion-pair intermediates form that explain the enantioselectivity [750,751].
9.3. Applications of Chiral Urea and Thiourea Catalysts
In 1998, Sigman and Jacobsen reported that urea and thiourea derivatives (e.g., 149) catalyze enantioselective hydrocyanation reactions of imines derived from both aromatic and aliphatic aldehydes (reactions (9.7)) [484].
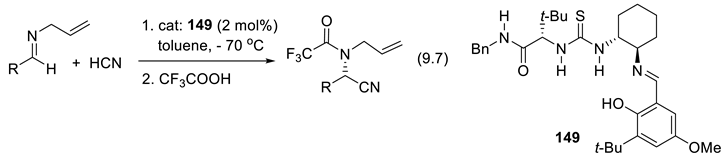
In 2003, Takemoto and coworkers reported a first examples of highly enantioselective 1,4-conjugate addition of malonic esters to nitroalkenes (reactions (9.8)) catalyzed by amino-thiourea derivative 150 [752]. With its amine and thiourea moieties, 150 is an example of bifunctional Brønsted base/H-bonding catalyst (BB catalyst). It catalyzes the enantioselective addition of sulfinates salts to enones [753]. Other reactions apply cinchona-derived thioureas [754,755].
In 2006, Berkessel and coworkers derived the bis(thio)urea catalyst 151 from readiy available isophoronediamine [3-(aminomethyl)-3,5,5-trimethyl-cyclohexylamine, IPDA] produced industrially in a multiton scale. It is also a good catalyst for asymmetric Morita-Baylis-Hillman (MBH) reactions (9.9) together with the base (N,N,N‘,N‘-tetramethylisophoronediamine (TMIPDA) [756].
In 2008, Liu and Shi prepared bis(thio)urea organocatalysts from axially chiral (R)-(−)-5,5‘,6,6‘,7,7‘,8,8‘-octahydro-1,1‘-binaphthyl-2,2‘-diamine (H8-BINAM). For instance derivative 152 (electrophilic activator) catalyzes the enantioselective MBH reaction of 2-cyclohexen-1-one or 2-cyclopenten-1-one with a wide range of aromatic aldehydes in combination with DABCO (nucleophilic activator). Reaction (9.10) led to the best yields and enantioselectivities [757].
The same year, Wang and coworkers applied a chiral bifunctional multiple hydrogen-bonding-donor amine-thiourea to catalyze a highly anti-selective and enantioselective nitro-Mannich reaction (R1CH=NBoc + R2-CH2NO2 → R1-CH(NHBoc)-CH(NO2)-R2 [758]. In 2012 and 2015, Groselj and coworkers prepared several camphor-derived thioureas [759,760]. In 2013, the Dixon group developed bifunctional organocatalysts 153 containing iminophosphorane units (as super bases) and a thiourea moiety (dual H-bond donor). This is an example of bifunctional base (BB) catalyst. They are much better catalysts than analogues possessing only a cinchonine moiety as a base. They catalyze asymmetric additions (9.11) [761].
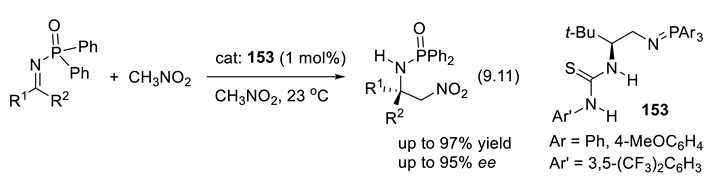
In 2020, Yeung and coworkers reported the highly enantio- and diastereoselective halocyclization and spiroketalization of olefinic keto-acids (e.g., reaction (9.12)) applying BB-catalyst 154. The resulting spiro compounds are privileged cores of many drugs and natural products [762].
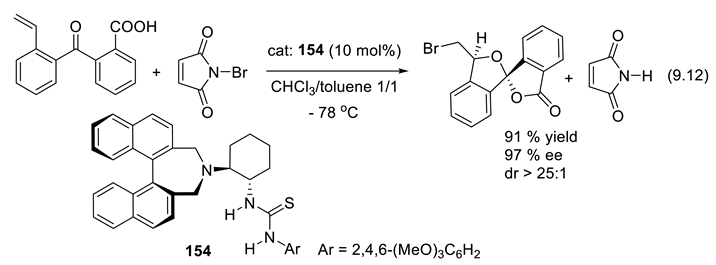
Phase transfer catalysts (PTCs) that transport anions from a solid or aqueous phase to an organic phase see their efficiency improved by attaching them urea or thiourea units. Figure 9.4 presents early examples of such hydrogen bonding phase transfer catalysts (HB PTC). In 1998, Manabe catalyzed the benzylation of β-ketoesters with phoshonium bromide 155 [763]. In 2005, Nagasawa and coworkers catalyzed the asymmetric Henry (nitro-aldol) reaction with the guanidium-thiourea chloride 156 [764]. In 2014, the Duan group reported a highly enantioselective nitro-Mannich reaction that applies cinchonium-urea 157 as catalyst [765].
Fluorine has become a widespread and important element in drugs [766,767] and agrochemicals [768]. With the number of fluorinated compounds on the market increasing, there is a growing demand for enantioselective fluorination methods, with safe and inexpensive sources of fluorine [769]. Typical mehods apply costly reagents (sources of F(+)) which represent a problem for large-scale production (Sections 3.5.5 & 10.3)[361,770]. Alternatively, alkali metal fluorides are safe and inexpensive source of fluoride anion that are available in large amounts [771]. In 2018, Gouverneur and coworkers developed such catalysts for the asymmetric nucleophilic fluorination using safe and readily accessible fluoride salts (Figure 9.5) [772]. Binaphthyl derivative 158 that offers three urea N-H bonds is a good catalyst of reaction (9.13). A racemic product of thiobromination of cis-stilbene is ionised into a meso-episulfonium ion intermediate. Its bromide counter-ion is “solvated” by the urea catalyst forming a large anion soluble in the organic phase and capable to exchange its bromide anion with a fluoride anion taken from CsF. This brings the fluoride anion closed to the episulfonium ion forming two possible diastereomeric tight ion-pairs in proportion different from 1/1. They react forming either one or the other enantiomer of the product of fluorination. This metal-free organocatalyzed asymmetric fluorination applied successfully to the synthesis of a trans-2-fluorocyclohexan-1-amine motif found in several bioactive pharmaceutical molecules [773,774].
Catalytic synthesis of α-fluorinated carbonyl compounds rely primarily on the transition-metal-catalyzed or organocatalytic reaction of a nucleophile (e.g., enolates or enamines) with diverse highly reactive electrophilic fluorinating reagents. In 2025, Sun and coworkers presented the enantioselective nucleophilic displacement reaction (9.14) catalyzed by the thiourea 159 [775].
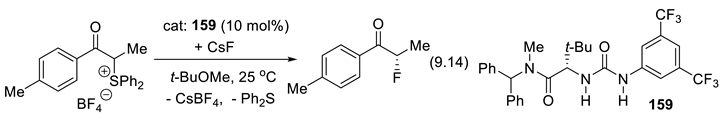
In 2014, Shibata and coworkers presented an iodoarene-catalyzed nucleophilic fluorination of β-dicarbonyl compounds and intramolecular aminofluorination of ω-amino-alkenes using the same reaction conditions. The key for these reactions is the in situ generation of a hypervalent iodine compound ArIF2 by hydrogen fluoride, mCPBA and a catalytic amount of iodoarene [776]. In 2025, Shi and coworkers realized the first example of enantioselective α-flurorination of ketone (47% yield, 46% ee) using cyclopropyl malonoyl peroxide as oxidant, pyr.9HF (Olah’s reagent) as fluoride anion source, and 2,2’-diiodo-1,1’-binaphthyl as chiral catalyst [777].
9.4. Applications of Chiral Squaramide-Containing Catalysts
In 2008, Rawal and coworkers presented chiral squaramides as new H-bond donor catalysts. The (−)-cinchonine modified squaramide 160 is easily prepared through a two-step process from methyl squarate. It is an example of a bifunctional base (BB) catalyst. With loadings as low as 0.1 mol% it catalyzes the asymmetric conjugate additions of 1,3-dicarbonyl compounds to β-nitrostyrenes (e.g., reaction (9.15)) [779]. Since then, a large number of thioureas or squaramide BB catalysts have been developed [721,780].
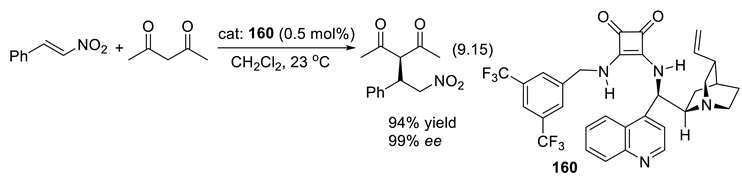
Similar to reaction (9.15), reaction (9.16) catalyzed by 161 starts the synthesis of central nervous system inhibitor Pregabalin (see Figure 3.10, Section 3.2.2 for an alternative synthesis) [781].
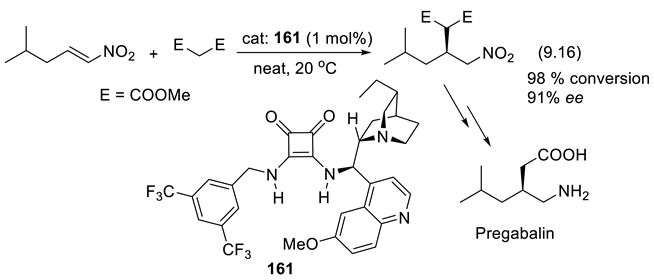
Chiral squaramides catalyze enantioselective conjugate additions of diphenyl phosphite to assorted nitroalkenes [782]. They catalyze also enantioselective Friedel-Crafts reactions of indoles with imines (e.g., reactions (9.17) with catalyst 162) [783].
The trifunctional catalyst 163 catalyzes asymmetric Henry reactions (9.18) [784].
In 2018, Jacobsen and coworkers reported the enantioselective nucleophilic displacement reaction (9.19) catalyzed by 164. Formation of a tight ion-pair intermediate 165 in which the tertiary carbenium ion resulting from the TNSOTf-promoted SN1 ionization is associated with the counter-ion, which is hydrogen-bridged by the chiral catalyst. Carbocation of intermediate 165 is attacked face-selectively by the nucleophile, explaining the enantioselectivity observed [785].
Figure 9.6.
a) An enantioselective SN1 displacement reaction (Jacobsen, 2018); b) Possible mechanism; formation of a tight ion-pair with a hydrogen bridged chiral anion.
Figure 9.6.
a) An enantioselective SN1 displacement reaction (Jacobsen, 2018); b) Possible mechanism; formation of a tight ion-pair with a hydrogen bridged chiral anion.
9.5. Amidinium Ions as Chiral Organo-Catalysts
In 2000, Göbel and coworkers showed that a stoichiometric amount of amidinium salt 166 induced an enantioselective Diels-Alder reaction [786]. In 2010, they reported that 166 can be applied in a sub-stoichiometric amount to catalyze reaction (9.20) that produces an advanced synthetic intermediate of estrone [787].
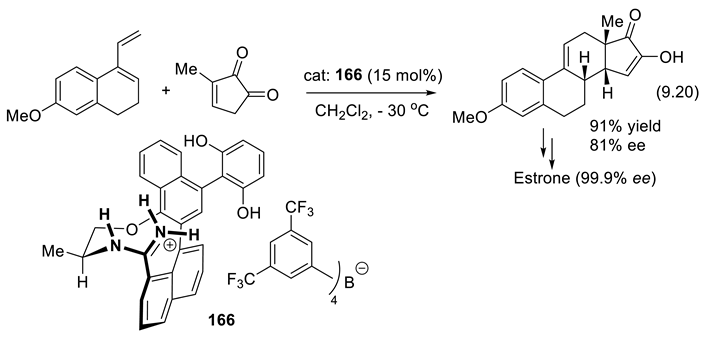
Analogous to amidinium salts H-bond donors are the 2-aminopyridinium salts. The asymmetric Diels-alder reactions (9.21) catalyzed by the pyridinium phosphoramide triflate 167 is an example example in 2016 by Nashikawa and coworkers [788].
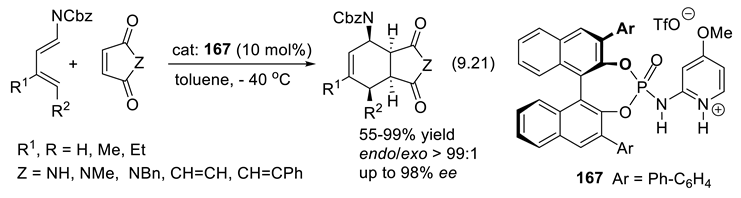
9.6. Other H-Bond Donor Chiral Catalysts
In 2015, Zhou and coworkers developed the chiral phosphoramide catalyst 168 and applied it to the enantioselective Mukaiyama–Michael addition (9.22) of difluoroenol silyl ethers to tetrasubstituted olefins. The secondary amine moiety of 168 is expected to activate the desilylation of the silyl enol ether through the formation of a hypervalent silicate intermediate. In contrast, the phorphoramide N-H moiety activates a carbonitrile group of the Michael acceptor [789].
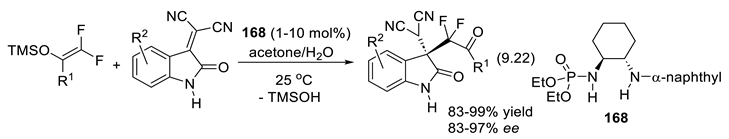
10. Acid Catalysis by Chiral Brønsted Acids
Akiyama [790] and Terada [791] initiated Brønsted acid organocatalysis in 2004 (Section 10.1.1). Brønsted acids, such as diethyl phosphate, (EtO)2P(=O)OH, pKa = 1.3), are general catalysts to activate electrophiles like carbonyl and imine compounds. Chiral phosphoric acid (CPA) catalysts have permitted many important enantioselective reactions [792]. Other chiral Brønsted acids are also developed, such as borates and boroxinates (pKa(HBF4) = - 0.44) [793] and disulfonimides (pKa(benzene-1,2-disulfoimide) = -4.1).
10.1. Chiral Diol Phosphate Catalysts
Chiral phosphoric acids are ideal catalysts for the enantioselective activation of basic substrates, including imines and ketimines, through hydrogen bonding or ion pairing in a bifunctional fashion, and for the strongest acids, for the activation of aldehydes and ketones [794,795]. A large number of structurally different chiral phosphoric (CPAs) [792,796] and bisphosphoric acids [797] have been developed (Figure 10.1).
Diol phosphates are bifunctional catalysts that activate electrophiles by a partial or complete proton transfer, forming positively charged intermediates. The latter can be stabilized by the vicinal free phosphoryl moiety (P=O) which is a Lewis base (P=O ↔ P(+)-O(-)) (Figure 10.2).. CPAs transfer chiral information from the catalyst to the products through either double H-bonds or synergistic hydrogen bonding (Figure 10.2a) and ion-pair interactions (Figure 10.2b) [798,799].
Structural variation of the chiral backbone of the CPA catalyst provides an efficient chiral pocket for almost any achiral electrophilic substrates and their asymmetric reactions. The latter react with face selectivity with a large variety of nucleophilic partners. Today, we dispose of highly enantioselective reactions (see Figure 10.3 for examples) such as enamine additions to imines and carbonyl compounds, electron-rich alkene additions to carbonyl compounds and imines, Friedel–Crafts-type reactions, Strecker reaction, aza-ene, aza-Henry, aza-Diels-Alder reactions, 1,3-dipolar reactions, Pictet-Spengler reactions, transfer hydrogenation and reductive amination, Biginelli reactions, tandem/cascade reactions, multicomponent reactions, conjugate additions, dearomatization reactions. Furthermore, CPA catalysts have also received attention in dynamic kinetic resolution processes. They serve as chiral ligands for metal-catalyzed reactions and have promising prospects as chiral inducers in photocatalytic reactions [800,801]. Enantioselective nucleophilic additions to indole imine 5-methides have been realized recently [802].
10.1.1. BINOL Phosphates
In 2004, Uraguchi and Terada reported that phosphoric acid derivatives like 169 (a BINOL phosphate) catalyze the addition of acetyl acetone to N-Boc-protected arylimines (reaction (10.1) [791]. The same year, and independently, Akiyama and co-workers showed that the BINOL-phosphate 170 catalyzes the enantioselective Mannich reaction (10.2) [790]. In 2005, Antilla and coworkers described an asymmetric acylimine amidation catalyzed by (R)- or (S)-VAPOLphosphate [803]. As already mentioned earlier, the hydrocyanation of imines (Strecker reaction) is a practical and direct method to α-amino acids. In 2006, Rueping and coworkers reported on reaction (10.3) catalyzed by the BINOL phosphate 171 [804]. The same group found that TADDOL-catalyzed Strecker reactions are less enantioselective than those catalyzed by BINOL-phosphate [805]. In 2006, Terada and coworkers found that a small amount of 171 accelerates the asymmetric aza-ene-type reaction of N-benzoylimines with enecarbamates, providing β-aminoimines (reactions (10.4)) [806]. They also reported on the asymmetric Friedel-Crafts reactions with imines (e.g., reaction (10.5) catalyzed by 172 and/or by 173) with carbonyl compounds and electron-rich alkenes [796]. In 2016, Nagorny and coworkers reported the asymmetric spiroketalization (10.6). With deuterated enol ethers, chiral phosphoric acid 174 induced a highly diastereoselective syn-selective protonation/nucleophile addition, thus ruling out long-lived oxycarbenium intermediates. Calculations supported an asynchronous concerted mechanism with a polar transition state [807].
In 2011, Shi and coworkers catalyzed the enantioselective bromocyclization with N-bromosuccimide (NBS) of olefins applying chiral phosphoric acid ent-174 as catalyst (10 mol%, CH2Cl2, 0 oC). Various cis-, trans-, or trisubstituted γ-hydroxy-alkenes and γ-amino-alkenes are cyclized into 2-substituted tetrahydrofurans and tetrahydropyrroles in up to 91% ee [808]. Nearly at the same time, Denmark and Burk demonstrated that the enantioselective bromocycloetherifications of 5-arylpentenols are catalyzed by a combination of chiral Brønsted acid and achiral Lewis base. An example is shown with reaction (10.7) catalyzed by 174 and Ph3P=S. The Lewis base (LB = Ph3P=S:) activates the electrophilic Br of NBS. In the same time the chiral protic acid (HY = 174) protonates a carbonyl group of NBS and promotes the formation of succimide and ion pair [LB-Br](+)Y(-). The latter equilibrates with LB and a chiral hypobromite that adds to the alkene with face selectivity forming a chiral epi-bromonium ion intermediate. The latter undergoes then an intramolecular addition of the alcoholic moiety forming the final bromo-ether and liberating the catalyst 174 (Figure 10.4) [809,810].
In 2009, Wang, Zhu, and coworkers presented an organocatalyzed asymmetric version of a three-component reaction combining aldehydes, anilines, and α-isocyanoacetamides. In the presence of CPAs (e.g., 175), the 5-(1-aminoalkyl)-5-aminooxazole were obtained in excellent yields and moderate to good ee (Figure 10.5, reaction (10.8)) [811].
In 2015, Tan and coworkers presented the first examples of organocatalyzed asymmetric Passerini reactions (10.9). In this three-component reaction, an isonitrile adds to an aldehyde forming a nitrilium ion that is quenched by the carboxylic acid. Follows an acyl group migration producing the correspond ester of a α-hydroxycarboxamide. In the presence of a CPA such as 176, chiral transition structure 178 forms. It leads to intermediate 179. Subsequent acyl group migration is probably also assisted by the CPA (Figure 10.6) [812].
Another multi-component reaction is the Biginelli reaction, which permits the quick access to biologically active DHPM (3,4-dihydropyrimidin-2(1H)-one or -thione) derivatives. Several catalytic enantioselective versions of the reaction were developed. In 2018, Neto and coworkers combined asymmetric counter-anion-directed catalysis (ACDC) with ionic liquid effect and applied it to the enantioselective Biginelli reactions (10.10) that require no reagent in excess, no solvent, no additive, and permit the easy recovery of the catalyst (Figure 10.7) [813]. All proposed mechanisms of the enantioselective version of the Biginelli reaction described so far imply the iminium mechanism, as reexamined by Kappe [814].
In 2018, List and coworkers realized the first direct and enantioselective α-aryloxylation of cyclic ketones with quinones (reaction (10.11)) catalyzed by 177. Preliminary mechanistic experiments suggest that this reaction proceeds via a proton-coupled electron transfer (PCET) followed by radical coupling (Figure 10.8). This explains why the expected 1,4-addition of the enol to benzoquinone is not observed [815].
In 2024, Shi and coworkers reported an asymmetric (2+4) annulation of achiral furan-indoles with 2,3-indolyldimethanols with uncommon regioselectivity under the catalysis of CPA, thereby generating furan-indole compounds bearing both axial and central chirality in high yields with excellent regio-, diastereo-, and enantioselectivities [816]. In the presence of a Brønsted acid, 2-indolylmethanols and 3-alkyl- (aryl)-2-indolylmethanols are ionized onto carbocationic species via dehydration. These intermediates are three-atom building blocks able to enter into (3+n)-cycloadditions with other reaction partners. These reactions can be enantioselective by applying chiral Brønsted acid catalysts such 180. An example of application reported in 2024 by the Shi group is given in Figure 10.9 with the (3+3)-annulation (10.12) [817].
10.1.2. Other Diol Phosphates
The four-component Ugi reaction is one of the most important multicomponent reactions (MCRs). It produces in one-pot operations α-amido amides (dipeptide analogues) carrying four points of diversity as they arise from the combination of four components: an aldehyde, an amine, a carboxylic acid, and an isocyanide. The reaction can be enantioselective when catalyzed by CPAs such as 181 or 182. In 2018, Tan, Houk and coworkers showed that CPAs catalyze reaction (10.13) with good ee. Quantum calculations suggest that a hydrogen-bonded complex 183 involving the phosphoric acid and carboxylic acid sets the stereochemistry for isocyanide attack on an imine intermediate [818].
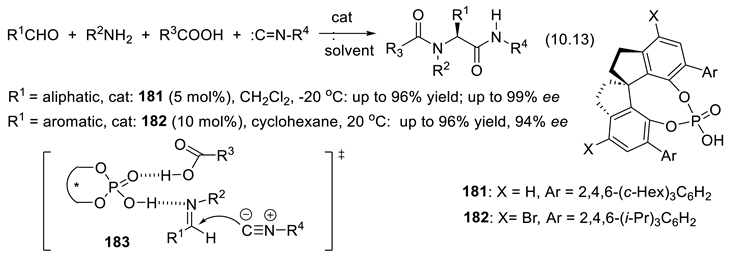
Alkenylation of arenes (aromatic electrophilic substitution) implies that the direct protonation of an alkyne produces alkenyl-arene derivatives. Arenes and alkynes containing large substituents form separable mixtures of atropisomers. Reactions (10.14) between sterically crowded alkynes and electron-rich naphthalene derivatives catalyzed by the SPINO(spirobiindane)-derived phosphoric acids 184 are enantioselective. They produce EBINOLs (1,1’-(ethylene-1,1-diyl)binaphthyl) and derivatives. Chiral ion pair intermediates of the type 1-naphthylalkylidenyl cation/SPINOLphosphate form [819].
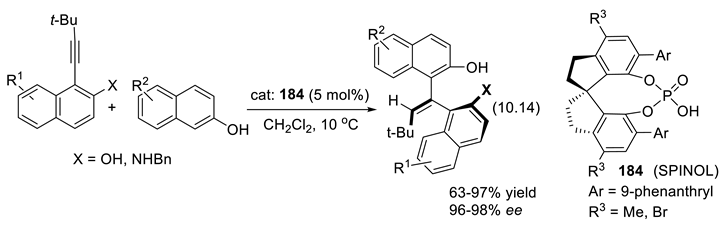
10.2. Stronger Acids Than Diolphosphates
Chiral phosphoric acid catalysts activate basic substrates such as imines or carbonyl compounds. For the functionalization of non-polarized C=C bonds, such as olefins or arenes, stronger acids are necessary [820]. Diaryl phosphates are not acidic enough for a strong activation of carbonyl compounds. The second and third pKa values of N-triflyl phosphoramide ((HO)2P(=O)-NHTf) are about 4 and 5 pKa units lower than those of H3PO4 (7.21 and 12.31, respectively) [821]. This led Yamamoto and Nakashima to develop in 2006 chiral N-triflyl phosphoramides. An example is BINOL-derived 185 that catalyzes enantioselective Diels-Alder reactions with normal electronic demand (10.15) [822].
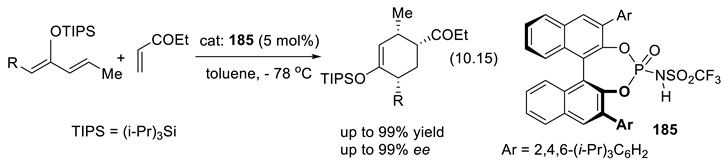
In 2012, List and Coric developed Brønsted acid-promoted asymmetric spiroacetalizations applying various BINOL-derived phosphoric acids and imidophosphoric acids on cyclic enol ethers bearing and alcoholic side-chain. They found that C2-symmetric imidodiphosphoric acid 186 was the best catalyst that permitted the enantioselective synthesis of 6,6- (e.g.: reaction (10.16)), 6,5-, 5,5-, 6,7- and 5,7-spiroacetals [823]. List and coworkers also found that enantioselective oxidation with H2O2 of sulfides into sulfoxides [824] and acetalization can be catalyzed by imidophosphoric acids such as 186 [825]. In the same years, Zhang and coworkers reported the asymmetric Mannich reactions (10.17) catalyzed by the enantiomer of 187 [826]. They also showed this type of Brønsted acid to catalyze Friedel-Crafts reactions of indoles with imines [827]. In 2014, they reported the enantioselective Biginelli condensations (10.18) [828].
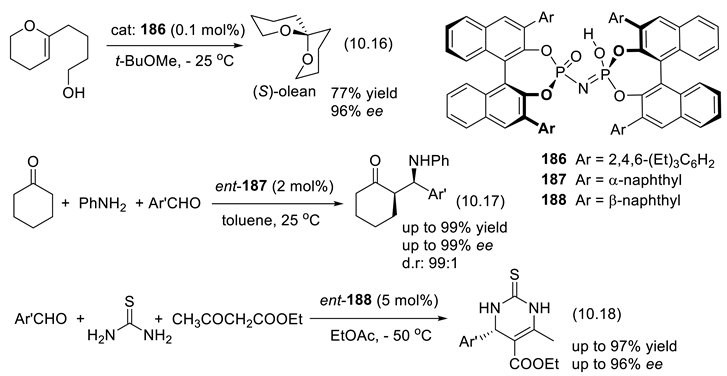
In 2016, the List group reported asymmetric Prins reactions catalyzed by iminoimidodiphosphate Brønsted acids 189. Reaction (10.19).is an example. It is a sequence of a Prins condensation between the aldehyde and the alkene. Follows a facile cyclization that implies an intramolecular displacement of a homoallylic secondary alcohol by a primary alcohol. At least one of these two reactions must be enantioselective [829].
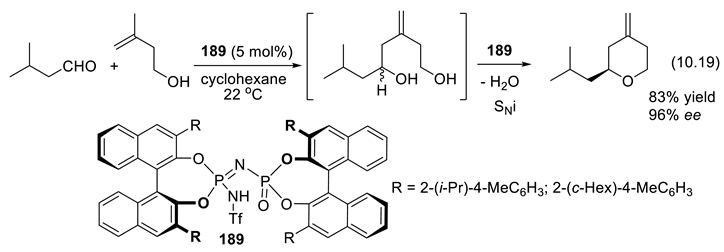
In 2018, the same group disclosed the enantioselective intramolecular hydroalkoxylation reaction (10.20) which applies Brønsted acid 190 as catalyst (Figure 10.10). The latter protonates the alkene forming an enantioenriched tight ion-pair 191. Whether this ion-pair equilibrates or not with the covalent species 192, its intramolecular reaction with the alcoholic moiety is enantioselective [830].
Enantioselective cyclopropanation of alkenes with diazoalkanes are catalyzed both with chiral metal complexes and with engineered metalloenzymes [831]. In 2025, List and coworkers presented a catalytic cycle (Figure 10.11a) in which alkenes are activated as radical cations upon single electron transfer to a photoexcited cationic organocatalyst of type [A(-)X(+)]* associated with an enantiopure counter-anion A(-). The radical-cation (e.g., 195) forms an ion-pair with the chiral counter-anion and react enantioselectively and stereoselectivily with the diazoalkanes, to furnish cyclopropane radical cations (e.g., 196) and loss of N2. A final single electron transfer from the catalyst radical to the cyclopropane radical-cations regenerate the cationic photocatalyst and deliver the desired products (Figure 10.11b). An example is the cyclopropanation of trans-anethole with ethyl diazoacetate (10.21) [831]. The photocatalysts A(-)X(+) is generated by reaction of acid 193 with the thioxanthylium triflate 194 and NaHCO3 (193 + 194 + NaHCO3 → A(-)X(+) + TfONa + H2O + CO2).
In a recent report Wulff, Borhan and coworkers developed VANOL-imidodiphosphorimidate catalyst 197 and applied it to the asymmetric bromonium ion induced spiroketalyzation (10.22). A large chiral ion-pair is formed between the catalyst and the reagent 1,3-dibromo-5,5-dimethylhydantoin which brominates the olefin with face selectivity producing a β-bromocarbenium ion intermediate that forms a tight ion-pair with the counter-ion of the acid catalyst [832].
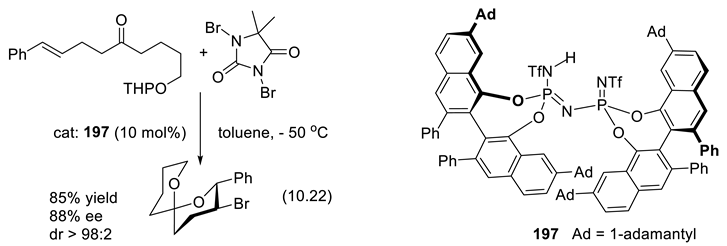
In 2008, Gong and coworkers reported that the bisphosphate 198 is a better catalyst than monophosphoric derivatives such as 170 in the one-pot, three-component dipolar cycloadditions (10.23). This is due to the higher acidity of the bisphosphoric acid compared to monophosphoric acid catalysts (Figure 10.12a). Assisted by the Brønsted acid, the aldehydes and primary amines equilibrate with the corresponding imines, precursors of the azomethine ylid intermediates that are H-bonded with 198 and undergo stereoselective 1,3-dipolar cycloadditions (Figure 10.12b) [833]. In the meantime, bisarylphosphates have been shown to catalyze asymmetric Diels-Alder reactions, aza-Friedel-Crafts reactions, and transfer hydrogenation reactions [797].
On introducing 2,4,6-trimethyl-3,5-dinitrophenyl substituents at C(2) and C(2’) of BINOLphosphate, Zhou and Yamamoto obtained a powerful catalyst for the enantioselective Mukayiama-Mannich reaction [834].
10.3. Diolphosphates as Chiral Anionic Phase Transfer Catalysts for Electrophilic Fluorination
In 2011, Toste and coworkers developed chiral anionic phase transfer catalysts (PTC) and applied them to the electrophilic fluorination of alkenes. An example is reaction (10.24) that uses Selectfluor as electrophilic fluorinating reagent. The latter is insoluble in fluorobenzene, the solvent chosen for this reaction. In the presence of chiral lypophilic diolphosphate 199 and a base (e.g., proton sponge) at least one tetrafluoroborate counter-ion of Selectfluor is exchanged by a phosphate anion generating a species that is soluble and reacts with the alkenes with face selectivity (Figure 10.13) [835].
Chiral anion PTC catlayze the enantioselective fluorinative dearomatization of phenols [836]. In 2024, Wang and coworkers presented new chiral bis-phosphate macrocycles that possess cavities with two cooperative phosphate sites. With Selectfluor, they form lipophilic inclusion complexes that are also good catalysts for electrophilic fluorinations [837].
10.4. Chiral Borate and Boroxinate Brønsted Acids
Wulff and coworkers showed that diols such as VAPOL and VANOL (Figure 9.1) formed polyborate anionic catalysts when combined with borates. For instance, the reaction of VAPOL with triphenylborate generates a Brønsted acid of the type boroxinate which, after addition of an imine (or an amine), forms the catalyst VAPOL-BOROX 200 (reaction (10.25)). Similarly, catalyst VANOL-BOROX 201 derives from VANOL [838,839,840].
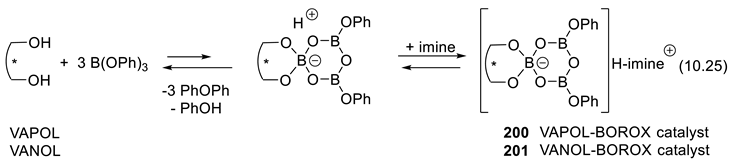
An early application is the asymmetric aziridination of imines (reaction 10.26)) reported in 1999 by Antilla and Wulff. In this case, the catalyst resulted from the reaction of (S)-VAPOL with BH3.THF [841]. Later, catalysts of type 200 and 201 have been applied [842,843,844].
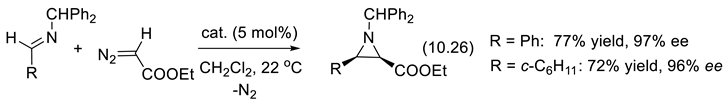
In 2011, Ren and Wulff reported the direct asymmetric aminoallylations of aldehydes (10.27) that implies imine formation and subsequent aza-Cope rearrangement. For good enantioselectivities the benzoic acid additive was necessary. Its role, probably, is to combine with the boroxinate intermediates [845].
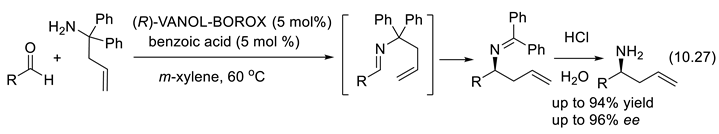
The combination of VANOL or VAPOL with B(OPh)3 catalyzes several other asymmetric reactions. They include: the Diels–Alder, hetero-Diels-Alder, Mannich, Baeyer–Villiger, Michael, aza-Darzens, Petasis and Ugi reactions, the amidation and imidation of imines, the reduction of imines and of aminals, the desymmetrization of aziridines and of diesters, the hydroarylation of alkenes, the cis- and trans-aziridination of imines and alkenes, the benzoyloxylation of aryloxindoles, the pinacol rearrangement, the hydrogenation of alkenes, the propargylation of ketones, the hydroacylation of alkenes, the α-imino rearrangement, the carbonylative spirolactonization of enallenols, and the controlled switching in a foldamer [793].
In 2020, the group of Ooi presented the robust hydrogen borate 202 as a chiral catalyst for the Prins type of cyclization (10.28) which implies face-selective protonation of the enol ether with formation of a oxycarbenium ion intermediate that is quenched intramolecularly by the arylalkene moiety. The resulting benzylic cation intermediate eliminates then a proton [846].
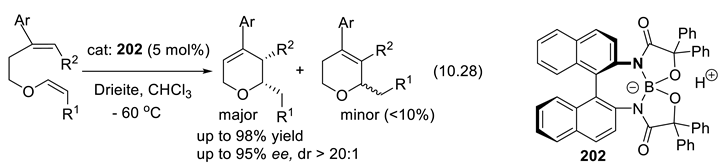
10.5. Chiral Disulfonic Acids and Their Ammonium Salts
In 2008, Ishihara and coworkers prepared chiral 1,1′-binaphthyl-2,2′-disulfonic acid (BINSA, 203) from BINOL. Mixed with a 2,6-diarylpyridine it forms salts that are catalysts of the direct asymmetric Mannich reaction of 1,3-diketones and of 1,3-ketoester equivalent with arylaldimines. Reaction (10.29) is an example [847,848].
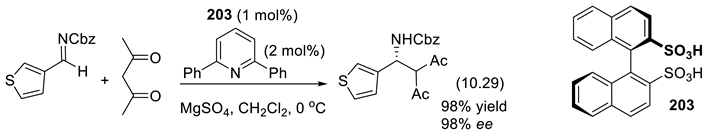
In 2011, Ishihara and coworkers reported a catalytic enantioselective Friedel-Crafts aminoalkylation between aromatic aldimines and N-benzylpyrrole applying (R)-BINSA-N,N-dimethylbutylamine, as a dynamic Brønsted acid-Brønsted base catalyst [848,849]. In 2018, the same group described the synthesis of chiral 1,1′-spirobiindane-7,7′-disulfonic acid (SPISA) and 1,1′-spirobiindane-7,7′-disulfonimide from 1,1′-spirobiindane-7,7′-diol (SPINOL, Figure 9.1) in 4 steps. They catalyze the asymmetric aminalization of aromatic aldimines with primary carboxamides [850].
10.6. Chiral Disulfonimides
In 2009, Giernoth and coworkers reported the first synthesis of (R)-1,1′-binaphthyl-2,2′-bis(sulfon)amide, 204 (BINBAM, more acidic than CPA’s [851]). It was obtained in 4 steps from BINOL [852]. The same year, the List group prepared the 3,3’-dirarylated analogue 205 and found it to be a catalyst of the asymmetric Mukaiyama aldol reaction (10.30) as efficient as BINOL-derived phosphate (Section 10.1.1), N-triflylphosphoramide (Section 10.2), or 1,1’-binaphtlyl-2,2’-disulfonic acid (Section 10.6) [853]. Although TfOH (pKa(H2O) = -5.9) is a stronger Brønsted acid than Tf2NH (pKa(H2O) = 1.7), TMSNTf2 is a stronger Lewis acid than TMSOTf [854]. The actual catalyst of the Mukayiama aldol reaction is not the Brønsted acid 205, but the Lewis acid 206 ⇄ 206’ resulting from the reaction of 205 (pre-catalyst) with the silyl enol ether (Figure 10.14) [855].
By pairing a silylium ion equivalent with a chiral enantiopure counter-anion (e.g., 206 ⇄ 206’), one realizes asymmetric counter-anion-directed catalysis [856,857]. Applications of this concept involve silicon-containing nucleophiles such as silyl ketene acetals (e.g., reaction (10.30),, Mukaiyama-Mannich reaction, Danishefsky-type dienes (hetero-Diels-Alder reaction [858]), allylsilanes (Hosomi-Sakurai reaction), silyl phosphites (Abramov reaction), and silyl cyanide. In the latter case, a small amount of catalysts (0.050 – 0.005 mol%) is sufficient. An example is reaction (10.32), which engages 156 g of naphthalene-2-carbaldehyde [859,860].
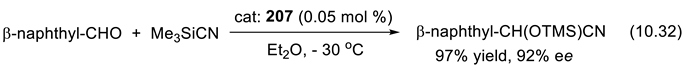
In 2017, Chen and coworkers reported the first example of asymmetric Mukaiyama–Michael reaction (10.33) catalyzed by 205 [861]. In 2018, the List group reported the enantioselective reactions of silyl ketene acetals with Michael acceptors [862].
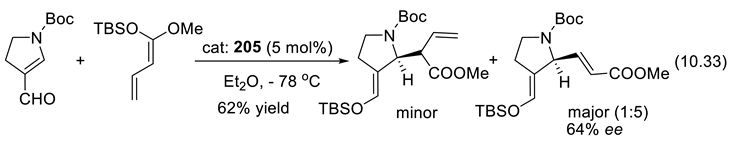
In 2011, Lee and coworkers demonstrated that disulfonimide 205 is an efficient chiral Brønsted acid catalyst (a DSI catalyst, not a pre-catalyst) as illustrated with the enantioselective Friedel–Crafts reaction (10.34) between a wide range of aldimines and indoles [863].
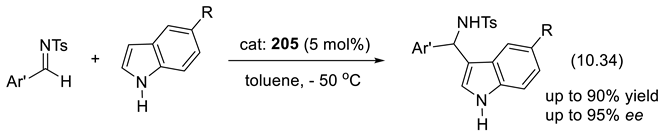
11. Other Asymmetric Organo-Catalysts
Several other functional organic compounds than those presented in the preceeding chapters have been envisonned as asymmetric organocatalysts. We cannot be exhaustive here with them, but we shall concentrate on some of the most useful asymmetric reactions applied that include chiral ketones, imines, amine oxides, oxazolines, and crown ethers. Newly developed halogen bond (XB) donors, such as neutral polyhalogenated compounds and halonium salts, will end this minireview.
11.1. Asymmetric Alkene Epoxidation Catalyzed by Chiral Ketones
Asymmetric alkene epoxidation is a powerful entry to highly valuable polyfunctional compounds [297]. Methods based on organocatalysis are described above (Sections 3.2.1, 4.2) [296]. Those applying ketones as catalysts contribute to green chemistry [864,865]. In 1984, Curci reported the first catalytic dioxirane-promoted alkene epoxidation, but with deceiving enantioselectivities (up to 12.5% ee) for (E)-β-methylstyrene [866]. Their chiral dioxirane (up to 50 mol%) resulted from the reaction of oxone (potassium hydrogen persulfate, KHSO5.KHSO4.K2SO4) as the oxidant and a chiral ketone (Figure 11.1) [867]. Numerous efforts, such as those of Yang [868,869], Denmark [870], and Armstrong [871,872] have led to better catalysts. In 1996, the Shi group realized highly enantioselective epoxidation of (E)-alkenes (reaction (11.1)) with oxone, applying easy-to-prepare D-fructose derivative 209 as the organocatalyst [873]. Catalyst ent-209 is also readily available [874,875]. In 1999, the Shi found that H2O2 can be utilized as a primary oxidant when pure acetonitrile is used as a solvent [876]. The asymmetric epoxidation of di- and trisubstituted olefins is now possible with ketone 209 as a catalyst. It tolerates different types of substituents, and it can be applied to the epoxidation of conjugated dienes and enynes, of enol silyl ethers or enol esters, of hydroxyalkenes, of vinylsilanes, and of fluoroalkenes. Several other chiral ketones have been proposed as catalysts since then. Shi’s ketones 209 and ent-209 apply successfully in the asymmetric synthesis of many drugs, natural products, and analogues of biological interest [296,865] (see also: [877]).
11.2. Asymmetric Alkene Epoxidation Catalyzed by Chiral Iminium Salts
Oxaziridinium salts, first reported by Lusinchi in 1976, [878,879] are also reagents for oxygen transfer to nucleophilic substrates such as sulfides [880] and alkenes, and may be generated catalytically by use of iminium salts in the presence of a stoichiometric oxidant such as oxone. In 1983, Davis and coworkers introduced chiral 2-sulfonyloxaziridines for the stoichiometric asymmetric epoxidation of unfunctionalized alkenes. These reagents transfer an oxygen atom to the alkene in a similar way as dioxiranes [881]. In 2004, Bulman Page and coworkers succeeded in developing a highly enantioselective alkene epoxidation (11.2) with oxone applying their BINOL-derived azepinium salt 210 as catalyst [882,883]. H2O2 [884] or NaOCl [885] can replace oxone as oxidant in this reaction. In the meantime, more chiral iminium salts have been proposed as catalysts for the enantioselective epoxidation of alkenes [886].
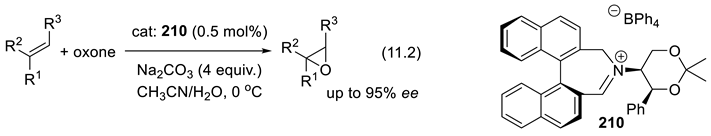
Hydroxylative dearomatization reactions of phenols offer an efficient way to assemble complex, biologically relevant compounds. In 2022, Buckley and coworkers disclosed an enantioselective method applying iminium salt 210 as catalyst for the oxidation of 2,6-dialkylphenol, 2,4,5- and 2,4,6-trialkylphenols. In situ oxidation of amine pre-catalysts 211 by H2O2 generates the catalyst 210. The reactions (e.g., (11.3)) engender cyclohexa-2,4-dienone intermediates that are (4+2)-cyclodimerized [887].
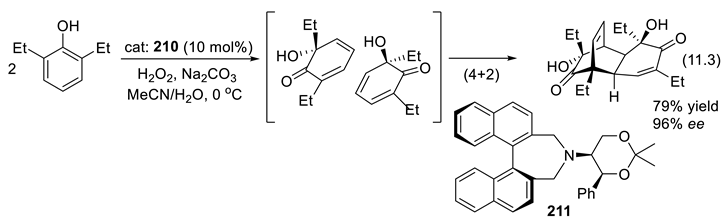
11.3. Crown Ethers
Since their discovery in 1967 by Pedersen [888], macrocyclic crown ethers have been applied in asymmetric transformations in parallel with cyclodextrins and calixarenes [889]. One speaks of asymmetric supramolecular catalysis, which often represents green chemistry. Crown ethers and their derivatives equilibrate with cationic complexes in the presence of alkali cations, ammoniums, and pyridiniums ions. This increases the nucleophilicity of their counter-anions (more “naked” anions, more separated from their cationic partners) [890]. In1981, Cram and Sogah reported the first examples of chiral crown ether-catalyzed Michael additions. Catalyst 212 + t-BuOK deprotonates the β-keto-ester forming a chiral enolate anion/cation pair that adds with one of its two faces to methyl vinyl ketone (reaction (11.4)) [891]. Such pioneering work created a new area of asymmetric supramolecular catalysis. Unfunctional crown ethers like 212 and 213 (see below) are chemically stable catalysts that can be engaged in reactions with aggressive reagents and high temperature.
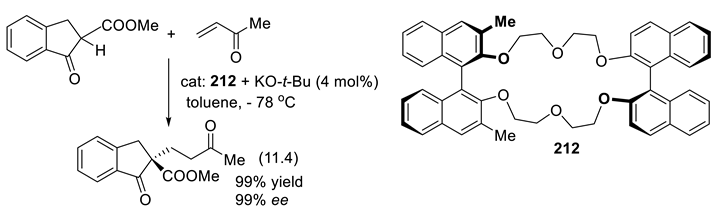
In 2000, Nagayama and Kobayashi reported that complexes of crown ethers and uncommonly used metal ions also serve as asymmetric supramolecular catalysts. They showed that the mixture of Pb(OTf)2 and 213 was an effective chiral catalyst for asymmetric Mukayama aldol reactions of (Z)-silyl enol ether of propiophenone with isovaleraldehyde in aqueous solvent (reaction (11.5)) [892].
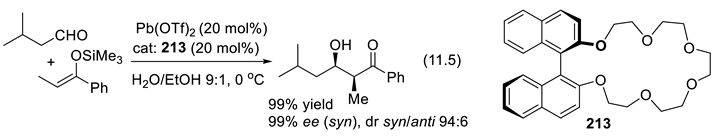
In 1986, the group of Penadés prepared lactose-derived crown ethers such as 214. The 1:1 mixture of 214 and t-BuOK is a PTC that induced the asymmetric Michael addition (11.6) [893]. In the meantime, many more sugar-derived crown ethers have been prepared. Combined with a strong base such t-BuOK they catalyze the asymmetric Michael reactions of enones, acrylic esters, acrylonitriles, and α,β-unsaturated nitro compounds as well as alkene epoxidation, cyclopropanation and Darzens condensations [890].
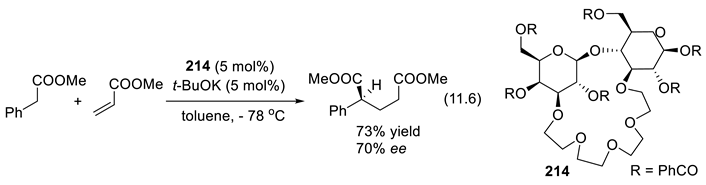
In 2015, the Kobayashi group catalyzed the direct asymmetric Michael additions of simple amides with α,β-unsaturated carbonyl compounds with a catalyst consisting of potassium bis(trimethylsilyl)amide (KHMDS) and a chiral crown ether [894]. In 2006, epoxidation (11.7) of 1,3-diaryl enones with H2O2 and azacrown ether-type catalyst 215 was examined by Hori and coworkers [895]. The alkyl groups on the nitrogen were shown to have a significant effect on the epoxidation yield and ee.
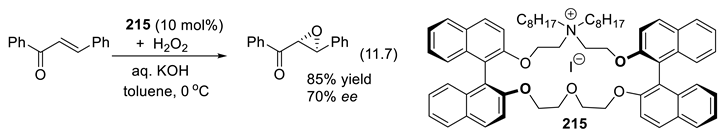
11.4. Chiral N-Amine Oxides
Following Denmark findings (Section 5.4) that a Lewis base such as phosphoramide 93 catalyzes the enantioselective allylation of benzaldehyde with allyltrichlorosilane (reaction (5.9)), the group of Nakajima in 1998 found that this reaction is catalyzed by N-amine oxides. With (S)-3,3-dimethyl-2,2’-biquinoline N,N’-dioxide (216) and in the presence of diisopropylethylamine good yield and enantioselectivity were observed for allylation (11.8) [896]. In 2002, Hayashi and coworkers found that only 0.01-0.1 mol% of 2,2’-bipyridine-N,N’-dioxide (MeCN, - 45 oC) was sufficient for a 96% yield and 94% ee of reaction (11.8) [897]. Since then, several other chiral N-amine oxides such a 217-220 (Figure 11.2) have been prepared. They catalyze the asymmetric allylation of aldehydes [898,899,900].
(S,S)-N,N-Bis (α-methylbenzyl) formamide catalyzes the enantioselective addition of allyl-and crotyltrichlorosilanes to aliphatic aldehydes. [901.]
11.5. Chiral Oxazolines and Bisoxazolines
Chiral oxazolines [902] and bisoxazolines are ligands of metallic species. They play an important role in asymmetric catalysis [903]. In 2013, Ganguly, Khan and coworkers found chiral oxazoline 221 to be a good catalyst of the asymmetric Strecker reaction (11.9) (see also Section 4.1). The catalyst is bifunctional, with an acidic sulfonamide group and a weakly basic N atom of the oxazoline moiety. Activation implies interaction of H-CN (reagent) with the oxazoline (hydrogen bond acceptor) and hydrogen bridging between the imine (substrate) and N-H group of the sulfonamide [488].
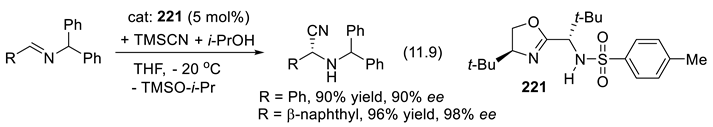
In 2018, the group of Ma reported the asymmetric aldol reaction (11.10) catalyzed by the C2-symmetrical bisoxazoline 222. Activation implies hydrogen bridging between the N atom of 222 and the OH groups of substrate and reactant [904].
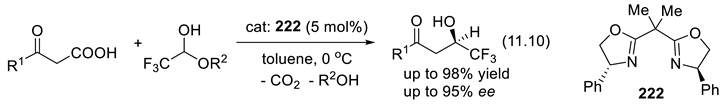
The Hofmann-Löffler-Freytag reaction uses N-halogenated amines to construct pyrrolidine rings and other nitrogen-containing heterocycles. It involves a radical-mediated cyclization, where a nitrogen-centered aminyl radical abstracts a hydrogen atom from a carbon atom generating a carbon-centered radical and an amine [905]. Under well-chosen conditions, the carbon-centered radical is converted into the corresponding carbenium ion intermediate by an oxidative radical-polar crossover (ORPC) process. In the presence of bisoxazoline catalysts such as 223, Lu and coworkers showed in 2025 that the intramolecular quenching of the carbenium ion intermediate by the amine moiety can be enantioselective (Figure 11.3). In their reactions such as (11.11), alkyl benzoyloxycarbamates are converted into oxazolidinones. Blue light irradiation of neutral red (a dye used in biological research and histology, the photocatalyst) generates an electronically excited state that donates an electron to the RNH-OCOAr moiety. The latter splits into benzoyl anion and aminyl radical 224. This radical is hydrogen-bridged with the catalyst 223. Follows a (1,5)-hydrogen shift with formation of the carbon-centered radical intermediate 225. The latter is oxidized by the radical-cation of the dye forming the corresponding carbenium ion intermediate. In the same time, the photocatalyst is recovered. The carbenium ion reacts with the tertiary amine (R3N) giving the corresponding ammonium ion 226. Intramolecular displacement of the ammonium ion by the bisoxazoline engenders chiral intermediate 227, which undergoes face-selective intramolecular quenching by the primary amine [906].
11.6. Chiral Iodine(I) Lewis Acids (XB Donor, σ-Hole Catalysts)
Reissert-type dearomatization of quinoline with a silylketene acetal as nucleophile is catalyzed by the helical tetrakis-iodotriazole 228. The XB donor 228 forms a complex anion 229 with the chloride anion [907]. It equilibrates with a chiral ion-pair containing the iminium ion (electrophile). The latter is attacked face selectively by the nucleophile (silylketene acetal). This is an example of halogen bonding catalysis reported by the group of García-Mancheño in 2023 (Figure 11.4) [908].
Catalyst 228 is an iodine(I) XB donor. Other examples are the bidentate iodine(I) donors 230 prepared by Huber and co-workers. Derivative 230 with Y = B(C6F5)4, R = NHCOCF3 catalyzes the asymmetric Muyaiyama aldol reaction (11.12) [909]. In this case, the two C-I groups are σ-hole Lewis acids activating the carbonyl group of the aldehyde [910].
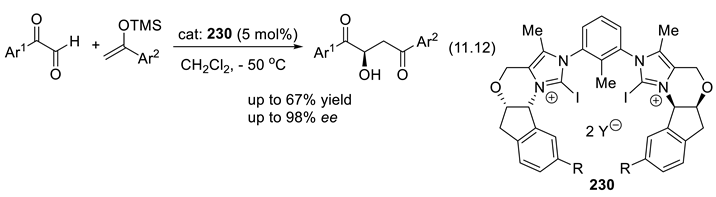
11.7. Chiral Halonium Salts as σ-Hole Catalysts
Bromonium and iodonium salts are hypervalent halogen compounds. They are Br(III)- and I(III)-XB donors (σ-hole Lewis acids). In 2023, Yoshida and coworkers disclosed their synthesis of chiral bromonium salts with N-nitrosamine functionalities as Lewis bases. These catalysts (e.g., 231) are Lewis acid and Lewis bases in the same time. The enantioselective Mannich reaction (11.13) of malonic esters with ketimines represents an application [911]. In 2022, Yoshida and coworkers had reported the enantioselective formation of N,S-acetals by condensation of thiols to imines catalyzed by a binaphthol-derived iodonium salts similar to 231 [912].
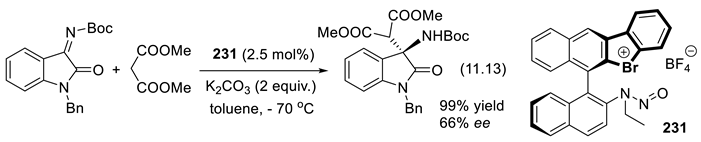
In 2025, Nachstheim and coworkers prepared chiral triazole-substituted iodoniums salts (e.g.: 232) as XB donors. As for reaction (11.13) they catalyze the enantioselective vinylogous Mannich reaction of cyanomethyl coumarin with an isatin-derived ketimine. Nucleophilic additions to imines (e.g., reaction (11.14)) are activated through N….I(III) interactions [913].
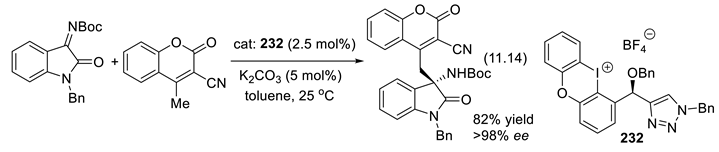
In 2025, the groups of List and Huber found that achiral bidentate iodine(III) based catalyst combined with chiral disulfonimides as counter-anions (e.g., 233) catalyzes the enantioselective Diels-Alder reaction (11.15) of cyclopentadiene and nitrostyrene. This is an example of halogen bonding (N=O…I(+)) with asymmetric counter-anion-directed catalysis [914].
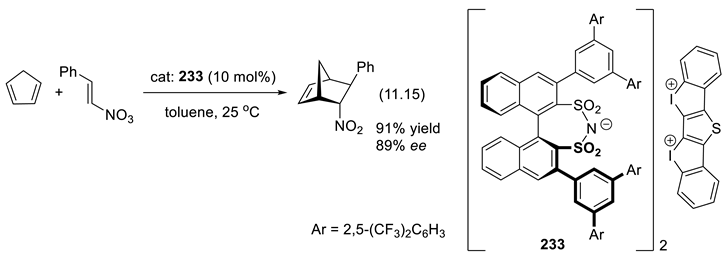
12. Epilogue
Asymmetric organocatalysis has kept busy a great number of scientists for the last forty five years. Extremely useful methods of asymmetric synthesis not relying on toxic transition metals, nor on practically demanding biocatalysis, are now available. Both enantiomers of a large number of economically important compounds (drugs, crop protection agents, fragrances, flavours) can be prepared with the same ease and this with high yield and enantioselectivity. The studies have contributed a lot in our understanding of reaction mechanisms. Chemical space (the number of stable compounds that can be constructed with a given number of light atoms (H, B, C, N, O, F, Si, P, S, Cl, Br, I) is huge and probably will never be explored fully for the search of new asymmetric catalysts [915]. Chiral and non-chiral additive to chiral catalysts might have benificial effets on enantioselectivities and yields. This adds another dimension to the structural space to be explored.
The ideal chiral catalyst must be inexpensive (easy to make enantiomerically pure, low molecular weight), nontoxic, and easy to recover. As seen, recovery and recycling of a catalyst can be achieved by immobilization on a high surface solid polymer, such as polystyrene, nylon, chitosan, polymethylhydrosiloxane, glass beads, or magnetic nanoparticles [916]. An example from the List group combines a cinchona alkaloid-derived catalyst with a nylon-6,6 support through UV irradiation. The textile-immobilized organo-catalyst 234 was recycled more than 250 times for the enantioselective mono-addition of MeOH to meso-anhydrides (e.g., reaction (11.1)) with no loss in efficiency [917].
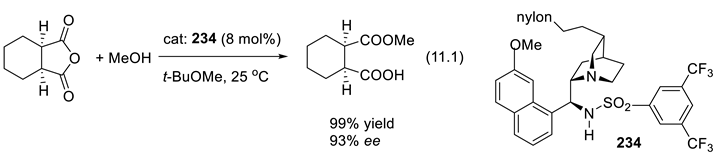
Costly catalysts should work in less than 1% (weight) compared with the substrate. Very often the organo-catalyst is nontoxic and thus can be left (e.g., < 0.1% by weight) in the final product to be sold. The reaction should be run in harmless solvent, if any, and under conditions economical in energy (no ccoling below 0 oC, no heating above 60 oC). When a solvent is necessary it must be nontoxic, easy to recover and should be inert (e.g., water, t-butyl methyl ether, tetrahydrofuran, pentane, ethanol, ethyl acetate, toluene), i.e., it does not generate side-products by action of the substrate, reagents and catalyst. When the catalytic process furnishes a final product that is crystalline with less than 99.9% ee, as required for a drug, its crystallization or co-crystallization with an inert inorganic chemical might improve its enantiopurity [918]. The method might be too costly to develop when a relatively small amount of the compound is required (e.g., for exploratory pharmaceutical or/and medical studies). Thus, chiral chromatographic methods are used instead. If the compound does not crystallize, one makes a solid derivative of it, recrystallize the latter, and do the necessary chemistry to recover the desired compound. Often, racemates crytallize better than enantiopure enantiomers. Alternatively, separation methods that rely upon the formation of crystalline diastereomeric derivatives (salts, complexes, readily hydrolyzable derivatives) with a readily available enantiomerically pure recyclable chiral auxilliary might then be applied.
From what precedes, it is obvious that there is still a lot of opportunities for inventing new catalyts and developing greener and more economical asymmetric processes. The search of new catalysts has contributed a lot in improving our understanding of chemistry in general. Nucleophilic carbenes and σ-hole catalysts are pertinent examples.
References
- Mila, A.; Ooi, T.; Bach, T. Modern enantioselective catalysis in organic chemistry. J. Org. Chem. 2023, 88, 7615–7618. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J, Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem Soc Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Mondal, D. Asymmetric Synthesis in Medicinal Chemistry, Encyclopedia Phys. Org. Chem. First edition; Wang, Z. Ed.; John Wiley & Sons, Inc. 2017, pp. 1-150; ISBN 978—1-118-46858-6.
- Yang, H.; Yu, H-x; Stolarzewicz, I. A.; Tang, W-j. Enantioselective transformations in the synthesis of therapeutic agents. Chem. Rev. 2023, 123, 9397–9446. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A. M.; Zanotti-Gerosa, A. Homogenous asymmetric hydrogenation: Recent trends and industrial applications. Curr. Opin. Drug. Discov. Develop. 2010, 13, 698–716. [Google Scholar]
- Bulger, P. G. Industrial applications of organocatalysis. 9, 2012; 9. [Google Scholar] [CrossRef]
- Gröger, H. Shibasaki catalysts and their use for asymmetric synthetic applications by the chemical industry. Eur. J. Org. Chem. 2016, 24, 4116–4123. [Google Scholar] [CrossRef]
- Dylong, D.; Hansoul, P. J. C.; Palkovits, R.; Eisenacker, M. Synthesis of (-)-menthol: industrial synthesis routes and recent development. Flavour Fragrance J. 2022, 37, 195–209. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J. C.; Baldenius, K.; Bornscheuer, U. T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Aukland, Miles, H. ; List, B. Organocatalysis emerging as a technology. Pure Appl. Chem. 2021, 93, 1371–1381. [Google Scholar] [CrossRef]
- Hughes, D. L. Highlights of the recent patent literature: Focus on asymmetric organocatalysis. Org. Process Res. Dev. 2022, 26, 2224–2239. [Google Scholar] [CrossRef]
- Sharma, S. K. The importance of organocatalysis (asymmetric and non-asymmetric) in agrochemicals. ChemistrySelect 2023, 8, e202300204, pp. 1-10. [Google Scholar] [CrossRef]
- Helmchen, G. Definition of the term asymmetric synthesis—History and revision. Chirality 2024, 35, 465–476. [Google Scholar] [CrossRef]
- Talapatra, S. K. , Talapatra, B. Asymmetric Synthesis. In Basic Concepts in Organic Stereochemistry. Springer Nature, Cham, CH, 2022, pp. [CrossRef]
- Fischer, E.; Slimmer, M. Versuche über asymmetrische Synthese. Ber. dtsch. Chem. Ges. 1903, 36, 2575–2587. [Google Scholar] [CrossRef]
- Markwald, W. Ueber asymmetrische Synthese. Ber. dtsch. Chem. Ges. 1904, 37, 1368–1370. [Google Scholar] [CrossRef]
- McKenzie, A. Studies in asymmetric synthesis. I/II J Chem Soc. 1904, 85, 1249–1262. [Google Scholar] [CrossRef]
- Bada, J. L. Origins of homochirality. Nature 1995, 374, 594–595. [Google Scholar] [CrossRef]
- Meinert, C.; Filippi, J-J. ; Nahon, L.; Hoffmann, S. V.; d’Hendecourt, L.; de Marcellus, P.; Bredehöft, J. H.; Thiemann, W. H-P.; Meierhenrich, U. J. Photochirogenesis: photochemical models on the origin of biomolecular homochirality. Symmetry 2010, 2, 1055–1080. [Google Scholar] [CrossRef]
- Blackmond, D. G. The origin of biological homochirality. Cold Spring Harb. Perspect. Biol. 2019, 11, article nb. 032540, pp. 1-12; [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M. On the emergence of homochirality and life itself. Biochem. 2021, 43, 4–12. [Google Scholar] [CrossRef]
- Gal, J. The discovery of stereoselectivity at biological receptors: Arnaldo Piutti and the taste of the asparagine enantiomers-History and analysis on the 125th anniversary. Chirality 2012, 24, 959–976. [Google Scholar] [CrossRef]
- Kandula, J. S.; Rayala, V. V. S. P. K.; Pullapanthula, R. Chirality: An inescapable concept for the pharmaceutical, bio-pharmaceutical, food, and cosmetic industries. Sep. Sci. plus 2023, 6, 1–18. [Google Scholar] [CrossRef]
- Brooks, W. H.; Guida, W. C.; Daniel, K. G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Alkadi, H.; Jbeily, R. Role of chirality in drugs: An overview. Infect. Disord. Drug Targets 2018, 18, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Ceramella, J.; Iacopetta, D.; Franchini, A.; De Luca, M.; Saturnino, C.; Andreu, I.; Sinicropi, M. S.; Catalano, A. A look at the importance of chirality in drug activity: Some significative examples. Appl. Sci. 2022, 12, article–nb.10909. [Google Scholar] [CrossRef]
- Peng, S-j. ; Zhu, Y-y.; Luo, C-y.; Zhang, P.; Wang, F-y.; Li, R-x.; Lin, G-q.; Zhang, J-g. Chiral drugs: Sources, absolute configuration identification, pharmacological applications, and future research trends. LabMed Discovery 2024, 1, article nb. 100008, pp. 1-9; [Google Scholar] [CrossRef]
- McVicker, R. U.; O’Boyle, N. M. Chirality of new drug approvals (2013–2022): Trends and perspectives. J. Med. Chem. 2024, 67(4), 2305–2320. [Google Scholar] [CrossRef]
- Teo, S. K.; Colburn, W. A.; Tracewell, W. G.; Kook, K. A.; Stirling, D. I.; Jaworsky, M. S.; Scheffler, M. A.; Thomas, S. D.; Laskin, O. L. Clinical pharmacokinetics of thalidomide. Clin. Pharmacokinetics 2004, 43, 311–327. [Google Scholar] [CrossRef]
- Bitchagno, G. T. M.; Nchiozem-Ngnitedem, V. A.; Melchert, D; Fobotou, S. A. Demystifying racemic natural products in the homochiral world. Nat. Rev. Chem. 2022, 6, 806–822. [Google Scholar] [CrossRef]
- Pasteur, L. Mémoire sur la fermentation de l'acide tartrique. Compt. rend. hebd. séa. Acad. Sci. 1858, 46(13), 615-618; see also: Gal, J. The discovery of biological enantioselectivity: Louis Pasteur and the fermentation of tartaric acid, 1857--A review and analysis 150 yr later. Chirality 2008, 20(1), 5-19; [CrossRef]
- Contesini, F. J.; Lopes, D. B.; Macedo, G. A.; Nascimento, M. d. G.; Carvalho, P. d. O. Aspergillus sp lipase: Potential biocatalyst for industrial use. J. Mol. Catal. B-Enzymatic. [CrossRef]
- Patel, R. N. Biocatalysis: Synthesis of key intermediates for development of pharmaceuticals. ACS Catal. 2011, 1, 1056–1074. [Google Scholar] [CrossRef]
- Kuriata-Adamusiak, R.; Strub, D.; Lochynski, S. Application of microorganisms towards synthesis of chiral terpenoid derivatives. Appl. Microbiol. Biotechn. 2012, 95, 1427–1436. [Google Scholar] [CrossRef]
- Ghislieri, D.; Green, A. P.; Pontini, M.; Willies, S. C.; Rowles, I.; Frank, A.; Grogan, G.; Turner, N. J. Engineering an enantioselective amine oxidase for the synthesis of pharmaceutical building blocks and alkaloid natural products. J. Am. Chem. Soc. 2013, 135, 10863–10869. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M. T. Biocatalysis in organic chemistry and biotechnology: past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Farnberger, J. E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 32, 6965–6982. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.; Hayashi, T.; Buller, R. M. Directed evolution of carbon-hydrogen bond activating enzymes. Curr. Opin. Biotechnol. 2019, 60, 29–38. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber. dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- a) Chapman, J.; Ismail, A. E.; Zoica Dinu, C. Industrial applications of enzymes: Recent advances, techniques, and outlooks. Catalysts 2018, 8(6), article nb. 238, pp. 1-26; https://doi.org/10.3390/catal8060238; b) Wu, S.; Snajdrova, R.; Moore, J. C.; Baldenius, K.; Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2020, 60(1), 88-119. [CrossRef]
- IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. [CrossRef]
- Micha, D. A. Molecular interactions: concepts and methods. John Wiley & Sons, Inc. New York, USA, 2020, pp. 9: 1-385; Print ISBN: 9780470290743; Online ISBN, 9780. [Google Scholar] [CrossRef]
- Henri, V. Théorie générale de l'action de quelques diastases, Compt. rend. hebd. séa. Acad. Sci. 1902, 135, 916–919. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M. L. Die Kinetik der Invertinwirkung, Biochemische Zeitschrift 1913, 49, 333-369. 49.
- Michaelis, L.; Menten, M. L.; Johnson, K. A.; Goody, R. S. The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef]
- Puchot, C.; Samuel, O.; Dunach, E.; Zhao, S.; Agami, C.; Kagan, H. B. Nonlinear effects in asymmetric synthesis. Examples in asymmetric oxidations and aldolization reactions. J. Am. Chem. Soc. 1986, 108, 2353–2357. [Google Scholar] [CrossRef]
- Guillaneux, D. , Zhao, S.-H., Samuel, O., Rainford, D.; Kagan, H. B. Nonlinear effects in asymmetric catalysis. J. Am. Chem. Soc. 1994, 116, 9430–9439. [Google Scholar] [CrossRef]
- Soai, K. , Shibata, T., Morioka, H.; Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. [Google Scholar] [CrossRef]
- Blackmond, D. G. ; Kinetic aspects of nonlinear effects in asymmetric catalysis. Acc. Chem. Res. 2000, 33, 402–411. [Google Scholar] [CrossRef]
- Athavale, S.; Simon, A.; Houk, K. N.; Denmark, S. E. Demystifying the asymmetry-amplifying, autocatalytic behaviour of the Soai reaction through structural, mechanistic and computational studies. Nat. Chem. 2020, 12, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Thierry, T.; Geiger, Y.; Bellemin-Laponnaz, S. Observation of hyperpositive non-linear effect in asymmetric organozinc alkylation in presence of N-pyrrolidinyl norephedrine. Molecules 2022, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Suh, S-E. ; Kang, H. Advances in exploring mechanisms of oxidative phenolic coupling reactions. Adv. Synth. Catal. 2024, 366, 4347–4384. [Google Scholar] [CrossRef]
- Wassermann, A. Homogeneous catalysis of diene syntheses. A new type of third-order reaction. J. Chem. Soc, 1942; 618–621. [Google Scholar] [CrossRef]
- Vogel, P.; Houk, K. N. Organic Chemistry, Theory, Reactivity and Mechanisms in Modern Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, Chap. 5.3.9-11, pp. 387-396, Chap. 7.6.6-7, pp. 849-853; ISBN 978-3-527-34532-8.
- Gröger, H.; Asano, Y. Introduction – Principles and historical landmarks of enzyme catalysis in organic synthesis. In “Enzyme Catalysis in Organic Synthesis”. Drauz K.; Gröger, H.; May, O. Eds., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, Print: ISBN: 9783527325474; Online ISBN: 978352763 9861, 2012, Chap. [Google Scholar] [CrossRef]
- Koeller, K. , Wong, C-H. Enzymes for chemical synthesis. Nature 2001, 409, 232–240. [Google Scholar] [CrossRef]
- Itoh, T.; Hanfeld, H. Enzyme catalysis in organic synthesis. Green Chem. 2017, 19, 331–332. [Google Scholar] [CrossRef]
- Sugai, T.; Higashibayashi, S.; Hanaya, K. Recent examples of the use of biocatalysts with high accessibility and availability in natural product synthesis. Tetrahedron 2018, 74, 3469–3487. [Google Scholar] [CrossRef]
- Hanfeld, U.; Hollmann, F.; Paul, C. E. Biocatalysis making waves in organic chemistry. Chem. Soc. Rev. 2022, 51, 594–627. [Google Scholar] [CrossRef]
- Zhao, H. Recent advances in enzymatic carbon-carbon bond formation. RSC Adv. 2024, 14, 25932–25974. [Google Scholar] [CrossRef] [PubMed]
- a) O’Connell, A.; Barry, A.; Burke, A. J.; Hutton, A: E.; Bell, E. L.; Green, A: P.; O’Reilly, E. Biocatalysis: landmark discoveries and applications in chemical synthesis. Chem. Soc. Rev. 2014, 53(6), 2828-2850; https://doi.org/10.1039/d3cs00689a; b)Wu, S-k.; Snajdrova, R.; Moore, J. C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2021, 60(1),88-119; [CrossRef]
- Gilio, A. K.; Thorpe, T. W.; Turner, N.; Grogan, G. Reductive aminations by imine reductases: from milligrams to tons. Chem. Science 2022, 13, 4697–4713. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Mateos, A. I.; Roura, P. D.; Francesca, P. Multistep enzyme cascades as a route towards green and sustainable pharmaceutical syntheses. Nat. Chem. 2022, 14, 489–499. [Google Scholar] [CrossRef]
- Fernandes, G. F. S.; Seong-Heun Kim, S.-H.; Castagnolo, D. Harnessing biocatalysis as a green tool in antibiotic synthesis and discovery. RSC Adv. 2024, 14, 30396–30410. [Google Scholar] [CrossRef]
- Edwards, D. R.; Lohman, D. C.; Wolfenden, R. Catalytic proficiency: The extreme case of S–O cleaving sulfatases. J. Am. Chem. Soc. 2012, 134, 525–531. [Google Scholar] [CrossRef]
- Hanson, J. John Cornforth (1917–2013). Nature 2014, 506, 35. [Google Scholar] [CrossRef]
- Dunitz, J. Vladimir Prelog (1906-98). Nature 1998, 391, 542. [Google Scholar] [CrossRef]
- Maji, R.; Mallojjala, S: C. ; Wheeler, S: E. Electrostatic interactions in asymmetric organocatalysis. Acc. Chem. Res. 2023, 56, 1990–2000. [Google Scholar] [CrossRef]
- Taylor, M. S.; Jacobsen, E. N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Agarwal P., K. A biophysical perspective on enzyme catalysis. Biochemistry 2019, 58, 438–449. [Google Scholar] [CrossRef]
- Copeland, R. A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. 3rd. Ed., John Wiley & Sons, Inc. New Jersey, USA, 2023; ISBN: 9781119793250.
- Goodman, C. Jean-Marie Lehn. Nat. Chem. Biol. 2007, 3, 685. [Google Scholar] [CrossRef]
- Ball, P. The shape of things. Nat. Mater. 2024, 23, 578. [Google Scholar] [CrossRef]
- Desiraju, G. Chemistry beyond the molecule. Nature 2001, 412, 397–400. [Google Scholar] [CrossRef]
- Hawthorne, M. Donald J. Cram (1919–2001). Nature 2001, 412, 696. [Google Scholar] [CrossRef]
- Liu, F.; Helgeson, R. C.; Houk, K. N. Building on Cram’s legacy: stimulated gating in hemicarcerand. Acc. Chem. Res. 2014, 47, 2168–2176. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, D. A universal receptor. Nature Nanotech. 2013, 8, 896–897. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C. R.; Ling, S.; Jacobsen, E. N. The cation-pi interaction in small-molecule catalysis. Angew. Chem. Int. Ed. 2016, 55, 12596–12624. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U. T.; Huisman, G-W. ; Kazlauskas, R.; Lutz, S.; Moore, J. C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485(7397), 185–194. [Google Scholar] [CrossRef]
- Packer, M. S.; Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef]
- Sandoval, B. A.; Hyster, T. K. Emerging strategies for expanding the toolbox of enzymes in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. [Google Scholar] [CrossRef]
- Thompson, M. P.; Peñafel, I.; Cosgrove, S. C.; Turner, N. J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process Res. Dev. 2019, 23, 9–18. [Google Scholar] [CrossRef]
- Mirsalami, S. M.; Mirsalami, M.; Ghodousian, A. Techniques for immobilizing enzymes to create durable and effective biocatalysts. Results Chem. 2024, 7, 1–10. [Google Scholar] [CrossRef]
- Murray, J.; Hodgson, D. R. W.; O'Donoghue, A. C. Going full circle with organocatalysis and biocatalysis: The latent potential of cofactor mimics in asymmetric synthesis. J. Org. Chem. 2023, 88, 7619–7629. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Gocke, D.; Pohl, M. Exploitation of ThDPdependent enzymes for asymmetric chemoenzymatic synthesis. FEBS J. 2009, 276, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Kasparyan, E.; Richter, M.; Dresen, C.; Walter, L. S.; Fuchs, G.; Leeper, F. J.; Wacker, T.; Andrade, S. L.; Kolter, G.; Pohl, M.; Müller, M. Asymmetric Stetter reactions catalyzed by thiamine diphosphate-dependent enzymes. Appl. Microbiol. Biotechnol. 2014, 98, 9681–9690. [Google Scholar] [CrossRef]
- Chaouali, N.; Gana, I.; Dorra, A.; Khelifi, F.; Nouioui, A.; Masri, W.; Belwaer, I.; Hayet Ghorbel, H.; Hedhili, A. Potential toxic levels of cyanide in almonds (Prunus amygdalus), apricot kernels (Prunus armeniaca), and almond syrup. Hindawi Publishing Corporation ISRN Toxicology (Hindawi Publ. Corp.) 2013, article nb. 610648, pp. [CrossRef]
- Wöhler, F.; Liebig, J. Ueber die Bildung des Bittermandelöls. Ann. Pharm. 1837, 2, 1–24. [Google Scholar] [CrossRef]
- Rosenthaler, L. Durch Enzyme bewirkte asymmetrische Synthesen. Biochem. Z., 1908, 14, 238–253. [Google Scholar]
- Effenberger, F. Synthesis and reactions of optically active cyanohydrins. Angew. Chem. Int. Ed. 1994, 33, 1555–1564. [Google Scholar] [CrossRef]
- Effenberger, F.; Förster, S.; Wajant, H. Hydroxynitrile lyases in stereoselective synthesis. Curr. Opin. Biotechnol. 2000, 11, 532–539. [Google Scholar] [CrossRef]
- Purkarthofer, T.; Skranc, W. , Schuster, C.; Griengl, H. Potential and capabilities of hydroxynitrile lyases as biocatalysts in the chemical industry. Appl. Microbiol. Biotechnol. 2007, 76, 309–320. [Google Scholar] [CrossRef]
- Dadashipour, M.; Asano, Y. Hydroxynitrile lyases insights into biochemistry, discovery, and engineering. ACS Catal. 2011, 1, 1121–1149. [Google Scholar] [CrossRef]
- Prixa, B. V.; Padhi, S K. Hydroxynitrile lyase discovery, engineering, and promiscuity towards asymmetric synthesis: Recent progress. Eur. J. Org. Chem. 2023, 26, 1–16. [Google Scholar] [CrossRef]
- Reetz, M. Directed evolution of enantioselective enzymes: An unconventional approach to asymmetric catalysis in organic chemistry. J. Org. Chem. 2009, 74, 5767–5778. [Google Scholar] [CrossRef]
- 96. Arnold, F. H. Innovation by evolution: Bringing new chemistry to life (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14420–14426. [CrossRef]
- Bornscheuer, U. T.; Hauer, B.; Jaeger, K. E.; Schwaneberg, U. Directed evolution empowered redesign of natural proteins for the sustainable production of chemicals and pharmaceuticals. Angew. Chem. Int. Ed. 2019, 58, 36–40. [Google Scholar] [CrossRef]
- Zheng, J. K.; Li, F-L. ; Lin, Z.; Lin, G-Q.; Hong, R.; Yu, H-L.; Xu, J-H. Structure-guided tuning of a hydroxynitrile lyase to accept rigid pharmaco aldehydes. ACS Catal. 2020, 10, 5757–5763. [Google Scholar] [CrossRef]
- Reetz, M. T. ; García-Borràs. The unexplored importance of fleeting chiral intermediates in enzyme-catalyzed reactions. J. Am. Chem. Soc. 2021, 143, 14939–14950. [Google Scholar] [CrossRef] [PubMed]
- Purkarthofer, T.; Gruber, K.; Gruber-Khadjawi, M.; Waich, K.; Skranc, W.; Mink, D.; Griengl, H. A biocatalytic Henry reaction--the hydroxynitrile lyase from Hevea brasiliensis also catalyzes nitroaldol reactions. Angew. Chem. Int. Ed. 2006, 45, 3454–3456. [Google Scholar] [CrossRef]
- Fuhshuku, K.; Asano, Y. Synthesis of (R)-β-nitro alcohols catalyzed by R-selective hydroxynitrile lyase from Arabidopsis thaliana in the aqueous-organic biphasic system. J. Biotechnol. 2011, 153(3-4),153-159. [CrossRef]
- von Langermann, J.; Nedrud, D. M.; Kazlauskas, R. J. Increasing the reaction rate of hydroxynitrile lyase from Hevea brasiliensis toward mandelonitrile by copying active site residues from an esterase that accepts aromatic esters. Chembiochem. 2014, 15, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Bekerle-Bogner,M. ; Gruber-Khadjawi, M.; Wiltsche, H.; Wiedner, R.; H. Schwab, H.; Steiner, K. (R)-Selective nitroaldol reaction catalyzed by metal-dependent bacterial hydroxynitrile lyases ChemCatChem, 2016, 8, 2214–2216. [CrossRef]
- Priya, B. V.; Rao, D. H. S.; Chatterjee, A.; Padhi, S. K. Hydroxynitrile lyase engineering for promiscuous asymmetric Henry reaction with enhanced conversion, enantioselectivity and catalytic efficiency. Chem. Commun. 2023, 12274–12277. [Google Scholar] [CrossRef] [PubMed]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Hydrolases: catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, (11), 4504–4523. [Google Scholar] [CrossRef]
- López-Iglesias, M,.; Gotor-Fernández, V. Recent advances in biocatalytic promiscuity: hydrolase-catalyzed reactions for nonconventional transformations. Chem. Rec, 2015; 743–759. [CrossRef]
- Poddar, H.; Rahimi, M.; Geertsema, E. M.; Thunnissen, A. M.; Poelarends, G. J. Evidence for the formation of an enamine species during aldol and Michael-type addition reactions promiscuously catalyzed by 4-oxalocrotonate tautomerase. Chembiochem. 2015, 16, 738–41. [Google Scholar] [CrossRef]
- Copley, S.D. Shining light on enzyme promiscuity. Curr. Opin. Struct. Biol. 2017, 47, 167–175. [Google Scholar] [CrossRef]
- Huang, F.-C.; Lee, L. F. H.; Mittal, R. S. D.; Ravikumar, P. R.; Chan, J. A.; Sih, C. J.; Capsi, E.; Eck, C. R. Preparation of (R)- and (S)-mevalonic acids. J. Am. Chem. Soc. 1975, 97, 4144–4145. [Google Scholar] [CrossRef]
- Mohr, P.; Waespe-Sarevi, N.; Tamm, C.; Gawronska, K.; Gawronski, J. A study of stereoselective hydrolysis of symmetrical diesters with pig liver esterase. Helv. Chim. Acta 1983, 66, 2501–2511. [Google Scholar] [CrossRef]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective enzymatic desymmetrization in organic synthesis. Chem. Rev. 2005, 105(1), 313–354. [Google Scholar] [CrossRef]
- Zutter, U.; Iding, H.; Spurr, P.; Wirz, B. New, efficient synthesis of Oseltamivir phosphate (Tamiflu) via enzymatic desymmetrization of a meso-1,3-cyclohexanedicarboxylic acid diester. J. Org. Chem. 2008, 73, 4895–4902. [Google Scholar] [CrossRef]
- Dwivedee, B. R.; Soni, S.; Sharma, M.; Bhaumik, J.; Laha, J. K.; Banerjee, U. C Promiscuity of lipase-catalyzed reactions for organic synthesis: A recent update. ChemistrySelect 2018, 3, 2441–2466. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Zhang, W. Natural polypropionates in 1999–2020: An overview of chemical and biological diversity. Mar. Drugs 2020, 18, 569. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Sordo Gonzalo, J. A. Expeditious asymmetric synthesis of polypropionates relying on sulfur dioxide-induced C–C bond forming reactions. Catalysts 2021, 11, article nb.1267, pp. 1-20. [Google Scholar] [CrossRef]
- Chênevert, R.; Rose, Y. S. Enzymatic desymmetrization of a meso polyol corresponding to the C(19)-C(27) segment of Rifamycin S. J. Org. Chem. 2000, 65, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Tsutsumi, S.; Ihori, T.; Ohta, H. Enzyme-mediated enantioface-differentiating hydrolysis of alpha-substituted cycloalkanone enol esters. J. Am. Chem. Soc. 1990, 112, 9614–9619. [Google Scholar] [CrossRef]
- Mohr, J. T.; Hong, A. Y.; Stoltz, B. M. Enantioselective protonation. Nat. Chem, 2009; 359–369. [Google Scholar]
- Neuberg, C. Biochemical reductions at the expense of sugars. Adv. Carbohyd. Chem. 1949, 4, 75–117. [Google Scholar] [CrossRef]
- Sih, C. J.; Chen, C-S. Microbial asymmetric catalysis—Enantioselective reduction of ketones [New Synthetic Methods]. Angew. Chem. Int. Ed. 1984, 23, 570–578. [Google Scholar] [CrossRef]
- Seebach, D.; Roggo, S.; Maetzke, T.; Braunschweiger, H.; Cercus, C.; Krieger, M. Diasterio- und enantioselektive Reduktion von β-Ketoestern mit Cyclopentanon-, Cyclohexanon-, Piperidon- und Tetralon-Struktur durch nicht fermentierende Bäcker-Hefe. Helv. Chim. Acta 1987, 70, 1605–1615. [Google Scholar] [CrossRef]
- Inoue, T.; Hosomi, K.; Araki, M.; Nishida, K.; Node, M. Enantioselective reduction of σ-symmetric bicyclo[3.3.0]octane-2,8-diones with baker's yeast. Tetrahedron: Asymmetry 1995, 6, 31–34. [Google Scholar] [CrossRef]
- Pereira, R. de S. The use of Baker’s yeast in the generation of asymmetric centers to produce chiral drugs and other compounds. Crit. Rev. Biotechn. [CrossRef]
- Wolfson, A.; Dlugy, C.; Tavor, D.; Blumefeld, J. Baker’s yeast catalyzed asymmetric reduction in glycerol. Tetrahedon Asymmetry 2006, 17, 2043–2045. [Google Scholar] [CrossRef]
- Moore, J. C.; Pollard, D. J.; Kosjek, B.; Devine, P. N. Advances in the enzymatic reduction of ketones. Acc. Chem. Res. 2007, 40, 1412–1419. [Google Scholar] [CrossRef]
- Neuberg, C.; Nord, F. F. Die phytochemische Reduktion der Ketone. Biochemische Darstellung optisch-aktiver sekundärer Alkohole. Ber. deutch. chem. Ges. 1919, 52, 2237–2248. [Google Scholar] [CrossRef]
- MacLeod, R.; Prosser, H.; Fickentscher, L.; Lanyi, J.; Mosher, H. S. Asymmetric Reductions. XII. Stereoselective Ketone Reductions by Fermenting Yeast. Biochemistry 1964, 3, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Prelog, V. Specification of the stereospecificity of some oxido-reductases by diamond lattice sections. Pure Appl. Chem. 1964, 9, 119–130. [Google Scholar] [CrossRef]
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "prochirality". [CrossRef]
- Burnier, G. , Vogel, P. Synthesis and circular dichroism of optically pure (-)-( 1S,2S,5S)-5-methoxy-3,4,6,7-tetramethylidenebicyclo[3.2.l]oct-2-yl derivatives. Baker's yeast reduction of 4-methoxy-5,6,7,8-tetramethylidenebicyclo[2.2.2]octan-2-one. Helv. Chim. Acta 1990, 73, 985–1001. [Google Scholar] [CrossRef]
- Dale, J. A.; Dull, D. L.; Mosher, H. S. J. Org. Chem. 1969, 34(9), 2543-2549. [CrossRef]
- Liu, H.; Wu, T.; Chen, J.; Wang, J.; Qiu, H. Recent advances in chiral liquid chromatography stationary phases for pharmaceutical analysis. J. Chromat. A. 2023, 1708, 1–16. [Google Scholar] [CrossRef]
- Utaka, M.; Konishi, S.; Mizuoka, A.; Ohkubo, T.; Sakai, T.; Tauboi, S. ; Takeda, A: Asymmetric reduction of the prochiral carbon-carbon double bond of methyl 2-chloro-2-alkenoates by use of fermenting bakers' yeast J. Org. Chem. 1989, 54, 4989–4992. [Google Scholar] [CrossRef]
- Gibson, D. T.; Koch, J. R.; Schuld, C. L.; Kallio, R. E. Oxidative degradation of aromatic hydrocarbons by microorganisms. II. Metabolism of halogenated aromatic hydrocarbons. Biochemistry 1968(11), 3795–3802. [CrossRef]
- Zylstra, G. J.; Gibson, D. T. Toluene degradation by Pseudomonas putida F1. Nucleotide sequence of the todC1C2BADE genes and their expression in Escherichia coli. J. Biol. Chem. 1989, 264, 14940–14946. [Google Scholar] [CrossRef]
- Endoma, M. A.; Bui, V. P.; Hansen, J.; Hudlicky, T. Medium-scale preparation of useful metabolites of aromatic compounds via whole-cell fermentation with recombinant organisms. Org. Process Res. Dev. 2002, 6, 525–532. [Google Scholar] [CrossRef]
- Johnson, J. A. Microbial arene oxidations. Org. React, 2004. [Google Scholar] [CrossRef]
- Carless, H. A. J. The use of cyclohexa-3,5-diene-1,2-diols in enantiospecific synthesis. Tetrahedron: Asymmetry 1992, 31, 795–826. [Google Scholar] [CrossRef]
- Hudlicky, T.; Gonzalez, D.; Gibson, D. T. Enzymatic dihydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichim. Acta 1999, 32, 35–62. [Google Scholar] [CrossRef]
- Banwell, M. G.; Edwards, A: J. ; Harfoot, G. J.; Jolliffe, K. A.; McLeod, M. D.; McRae, K. J.; Stewart, S. G.; Vögtle, M. Chemoenzymatic methods for the enantioselective preparation of sesquiterpenoid natural products from aromatic precursors. Pure Appl. Chem. 2003, 75, 223–229. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J. W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 8, 3117–3132. [Google Scholar] [CrossRef]
- Taher, E. S.; Banwell, M. G.; Buckler, J. N.; Yan, Q.; Lan, P. The exploitation of enzymatically-derived cis-1,2-dihydrocatechols and related compounds in the synthesis of biologically active natural products. Chem. Rec. 2018, 18, 239–264. [Google Scholar] [CrossRef]
- Hudlicky, T. Benefits of unconventional methods in the total synthesis of natural products. ACS Omega, 2018, 3, 17326–17340. [Google Scholar] [CrossRef]
- Goulart Stollmaier, J.; Hudlicky, T. Sequential enzymatic and electrochemical functionalization of bromocyclohexadienediols: Application to the synthesis of (−)-conduritol C. Tetrahedron 2020, 76, 1–7. [Google Scholar] [CrossRef]
- Meyerhof, O. , Lohmann, K. Über den Nachweis von Triosephosphorsäure als Zwischenprodukt bei der enzymatischen Kohlehydratspaltung. Naturwissenschaften 1934, 22, 134–135. [Google Scholar] [CrossRef]
- Kresge, N.; Simoni, R. D.; Hill, R. L. Otto Fritz Meyerhof and the elucidation of the glycolytic pathway. J. Biol. Chem. 2005, 280, e1–e3. [Google Scholar] [CrossRef]
- Warburg, O. , Christian, W. Isolierung und Kristallisation des Gärungsferments Enolase. Naturwissenschaften 1941, 29, 589–590. [Google Scholar] [CrossRef]
- Warburg, O.; Christian, W. Isolierung und Kristallisation des Gärungsferments Zymohexase. Naturwissenschaften 1942, 30, 731–731. [Google Scholar] [CrossRef]
- Warburg, O.; Christian, W. Isolierung und Kristallisation des Gärungsferments Zymohexase Biochem. Z. 1943, 314, 149–176. [Google Scholar]
- Richards, O. C.; Rutter, W. J. Preparation and properties of yeast aldolase, J. Biol. Chem. 1961, 236, 3177–3184. [Google Scholar] [CrossRef]
- Warburg, O.; Gawehn, K. Z.; Lange, G. Ueber das Verhalten von Ascites-Kbresbzellen zu Zymhexase. Naturforsch. 1954, 9b, 109–114. [Google Scholar] [CrossRef]
- Jones, J. K.; Sephton, H. H. Synthesis of D-glycero-D-altro-, L-glycero-L-galacto-, D-glycero-L-gluco-, and D-glycero-L-galato-octulose. Can J. Chem. 1960, 38, 753–760. [Google Scholar] [CrossRef]
- Bednarski, M. D.; Simon, E. S.; Bischofberger, N.; Fessner, W.-D.; Kim, M.-J.; Lees, W.; Saito, T.; Waldmann, H.; Whitesides, G. M. J. Am. J. Am. Chem. Soc. 1989, 111, 627–635. [CrossRef]
- Toone, E. J.; Simon, E. S.; Bednarski, M. D.; Whitesides, G. M. Enzyme-catalyzed synthesis of carbohydrates. Tetrahedron 1989, 45, 5365–5422. [Google Scholar] [CrossRef]
- Wong, C.-H.; Halcomb, R. L.; Ichikawa, Y.; Kajimoto, T. Enzymes in organic synthesis: Application to the problems of carbohydrate recognition (Part 2). Angew. Chem. Int. Ed. 1995, 34, 521–546. [Google Scholar] [CrossRef]
- Wong, C-H. ; Halcomb, R. L.; Ochikawa, Y.; Kajimoto. Enzymes in organic synthesis: Application to the problems of carbohydrate recognition (Part 1). Angew. Chem. Int. Ed. 1995, 34, 412–432. [Google Scholar] [CrossRef]
- Machajewski, T. D.; Wong, C.-H. The catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed. 2000, 39, 1352–1374. [Google Scholar] [CrossRef]
- Dean, S. M.; Greenberg, W. A.; Wong, C-H. Recent advances in aldolase-catalyzed asymmetric synthesis. Adv. Synth. Catal. 2007, 349, 1308–1320. [Google Scholar] [CrossRef]
- Clapés, P.; Garrabou, X. Current trends in asymmetric synthesis with aldolases. Adv. Synth. Catal. 2011, 353, 2263–2283. [Google Scholar] [CrossRef]
- Hélaine, V; Gastaldi, C; Lemaire, M.; Clapès, P.; Guérard-Hélaine, C Recent advances in the substrate selectivity of aldolases. ACS Catal 2022, 12, 733–761.
- Lee, S-H. ; Yeom, S-J.; Kim. S-E.; Oh, D-K. Development of aldolase-based catalysts for the synthesis of organic chemicals. Trends Biotechnol. 2022, 40, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Schmid, W.; Whitesides, G. M. A New approach to cyclitols based on rabbit muscle aldolase (RAMA). J. Am. Chem. Soc. 1990, 112, 9670–9671. [Google Scholar] [CrossRef]
- Sugai, T., Kajimoto, T. Synthesis of biologically relevant monosaccharides. In: Fraser-Reid, B. O.; Tatsuta, K.; Thiem, J. (eds). Glycoscience: Chemistry and Chemical Biology I–III. Springer, Berlin, Heidelberg, 2001, Chapt. 4.3, 907-1021. [CrossRef]
- Tozzi, M. G. , Camici, M., Mascia, L., Sgarrella, F., Ipata, P. L. Pentose phosphates in nucleoside interconversion and catabolism. FASEB J. 2006, 273, 1089–1101. [Google Scholar] [CrossRef]
- Gijsen, H. J. M. , Wong, C.-H., Sequential one-pot aldol reactions catalyzed by 2-deoxyribose-5-phosphate aldolase and fructose-1,6-diphosphate aldolase. J. Am. Chem. Soc. 1995, 117, 2947–2948. [Google Scholar] [CrossRef]
- Gijsen, H. J. M. , Wong, C.-H., Sequential three- and four-substrate aldol reactions catalyzed by aldolases. J. Am. Chem. Soc. 1995, 117, 7585–7591. [Google Scholar] [CrossRef]
- Greenberg, W. A.; Varvak, A.; Hanson, S. R.; Wong, K.; Huang, H.; Chen, P.; Burk, M. J. , Development of an efficient, scalable, aldolase-catalyzed process for enantioselective synthesis of statin intermediates. Proc. Natl. Acad. Sci. USA 2004, 101, 5788–5793. [Google Scholar] [CrossRef]
- Müller, M. Chemoenzymatic synthesis of building blocks for statin side chains. Angew. Chem. Int. Ed. 2005, 44, 362–365. [Google Scholar] [CrossRef]
- Liu, J.; Wong, C. H. Aldolase-catalyzed asymmetric synthesis of novel pyranose synthons as a new entry to heterocycles and epothilones. Angew. Chem. Int. Ed. 2002, 41, 1404–1407. [Google Scholar] [CrossRef]
- Ošlaj, M.; Cluzeau, J.; Orkić, D.; Kopitar, G.; Mrak, P.; Časar,Z. A highly productive, whole-cell DERA chemoenzymatic process for the production of key lactonized side-chain intermediates in statin synthesis. PLoS ONE 2013, 8, e62250. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Liu, J.; Chen, Z. Some recent examples in developing biocatalytic pharmaceutical processes, in Asymmetric Catalysis, Eds.: Blaser, H-U.; Federsel, H-J. Wiley Online Books, 2010. Chap. 1. pp. 1-12. [CrossRef]
- Dückers, N.; Baer, K.; Simon, S.; Gröger, H.; Hummel, W. Threonine aldolases-screening, properties and applications in the synthesis of non-proteinogenic beta-hydroxy-alpha-amino acids. Appl. Microbiol. Biotechnol. 2010, 88, 409−424; [Google Scholar] [CrossRef] [PubMed]
- Franz, S. E.; Stewart, J. D. Threonine aldolases. Adv. Appl. Microbiol. 2014, 88, 57–101. [Google Scholar] [CrossRef] [PubMed]
- Ligibel, M.; Moore, C.; Bruccoleri, R.; Snajdrova, R. Identification and application of threonine aldolase for synthesis of valuable α-amino, β-hydroxy-building blocks. Biochim Biophys Acta Proteins Proteom, 2020; 1868, 1–8. [Google Scholar] [CrossRef]
- Fesko, K.; Reisinger, C.; Steinreiber, J.; Weber, H.; Schürmann,M. ; Griengl, H. Four types of threonine aldolases: Similarities and differences in kinetics/thermodynamics. J. Mol. Catal. B: Enzym. 2008, 52, 19–26. [Google Scholar] [CrossRef]
- Scott, T. A.; Heine, D.; Qin, Z.; Wilkinson, B. An l-threoninetransaldolase is required for l-threo-β-hydroxy-α-amino acid assembly during Obafluorin biosynthesis. Nat. Commun. 2017, 8, article. [Google Scholar] [CrossRef]
- Franz, S. E.; Stewart, J. D. Threonine aldolases. Adv. Appl. Microbiol. 2014, 88, 57–101. [Google Scholar] [CrossRef]
- Fansher, D. J.; Palmer, D. R. J. A. Type 1 aldolase, NahE, catalyzes a stereoselective nitro-Michael reaction: Synthesis of β-aryl-γ-nitrobutyric acids. Angew. Chem. Int. Ed. 2023, 62, e292214539. [Google Scholar] [CrossRef]
- Williams, G. J.; Domann, S.; Nelson, A.; Berry, A. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 3143–3148. [Google Scholar] [CrossRef]
- Junker, S.; Kullartz, I.; Pietruszka, J. Cloning and characterisation of a new 2-deoxy-D-ribose-5-phosphate aldolase from Rhodococcus erythropolis. J. Biotechn. 2012, 161, 174–180. [Google Scholar] [CrossRef]
- Junker, S.; Roldan, R.; Joosten, H. J.; Clapés, P.; Fessner, W. D. Complete switch of reaction specificity of an aldolase by directed evolution in vitro: Synthesis of generic aliphatic aldol products. Angew. Chem. Int. Ed. 2018, 57, 57(32),10153–10157. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Feng, J.; Wu, Q.; Zhu, D.; Ma, Y. Improving and inverting cβ-stereoselectivity of threonine aldolase via substrate-binding-guided mutagenesis and a stepwise visual screening. ACS Catal, 2019; 9, 4462–4469. [Google Scholar] [CrossRef]
- Widersten, M. Engineering aldolases for asymmetric synthesis. Methods Enzymol. 2020, 644, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yu, H.; Fang, S.; Chen, K.; Wang, Z.; Chen, X.; Xu, G.; Yang, L.; Wu, J. Directed evolution of L-threonine aldolase for the diastereoselective synthesis of β-hydroxy-α-amino acids. ACS Catal. 2021, 11, 3198–3205. [Google Scholar] [CrossRef]
- Li, L.; Zhang, R.; Xu, Y.; Zhang, W. Comprehensive screening strategy coupled with structure-guided engineering of l-threonine aldolase from Pseudomonas putida for enhanced catalytic efficiency towards l-threo-4-methylsulfonylphenylserine. Front. Bioeng. Biotechnol. 2023, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Lerner, R. A.; Barbas, C. F. III. Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995, 270, 1797–1800. [Google Scholar] [CrossRef]
- List, B.; Shabat, D.; Barbas, C. F. III.; Lerner, R. A. Enantioselective total synthesis of some Brevicomins using aldolase antibody 38C2. Chem. Eur. J. 1998, 4, 881–885. [Google Scholar] [CrossRef]
- List, B.; Shabat, D.; Zhong, G.; Turner, J. M.; Li, T.; Bui, T.; Anderson, J.; Lerner, R. A.; Barbas, C. F. III. A catalytic enantioselective route to hydroxy-substituted quaternary carbon centers: resolution of tertiary aldols with a catalytic antibody. J. Am. Chem. Soc. 1999, 121, 7283–7291. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, Q.; Fu, Y.; Tong, Y.; Zhou, Z. Design and evolution of an enzyme for the asymmetric Michael addition of cyclic ketones to nitroolefins by enamine catalysis. Angew. Chem. Int. Ed. 2024, 63, 1–6. [Google Scholar] [CrossRef]
- Schönherr, H.; Cernak, T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
- Zetzsche, L. E.; Narayan A. R., H. Broadening the scope of biocatalytic C-C bond formation. Nat. Rev. Chem, h: 334-346, 1038. [Google Scholar]
- Ju, S. , Kuzelka, K. P., Guo, R.; Krohn-Hansen, B.; Wi, J.; Nair, S. K.; Yang, Y. A biocatalytic platform for asymmetric alkylation of α-keto acids by mining and engineering of methyltransferases. Nat. Commun. 2023, 14, article. [Google Scholar] [CrossRef]
- List, B. Introduction: organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- MacMillan, D. W. C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Guidotti, M. Organocatalysts for enantioselective synthesis of fine chemicals: definitions, trends and developments. ScienceOpen Res. 2015, 0, 1–14. [Google Scholar] [CrossRef]
- Xiang, S. H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, article. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, J. M. Spotting trends in organocatalysis for the next decade. Nat. Commun, 3787; 11. [Google Scholar] [CrossRef]
- Han, B.; He, X-H. ; Liu, Y-Q.; He, G.; Peng, C.; Li, J-L. Asymmetric organocatalysis: an enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 122–1586. [Google Scholar] [CrossRef]
- Garcia Mancheño, O.; Waser, M. Recent developments and trends in asymmetric organocatalysis. Eur. J. Org. Chem. 2023, 26, 1–8. [Google Scholar] [CrossRef]
- Reyes, E.; Prieto, L.; Milelli, A. Asymmetric organocatalysis: A survival guide to medicinal chemists. Molecules 2023, 28, 1–24. [Google Scholar] [CrossRef]
- Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice, Oxford, 2000; online edn, Oxford Academic, 31 Oct. 2023. [Google Scholar] [CrossRef]
- Trost, B. M. The atom economy-a search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef]
- List, B.; Lerner, R. A.; Barbas, C. F. III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122. [Google Scholar] [CrossRef]
- Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels−Alder reaction, J. Am. Chem. Soc. 2000, 122(17), 4243–4244. [Google Scholar] [CrossRef]
- List, B. Asymmetric aminocatalysis, Synlett 2001(11), 1675-1686; https//doi.org/ 10.1055/s-2001-18074.
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric aminocatalysis - Gold rush in organic chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jorgensen, K. A. Mechanisms in aminocatalysis. Chem. Commun. 2011, 47, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F. Amines as catalysts: Dynamic features and kinetic control of catalytic asymmetric chemical transformations to form C−C bonds and complex molecules. Chem. Rec. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Retamosa, M. de G.; Blanco-López, E.; Poyraz, S.; Döndas, H. A.; Sansano, J. M.; Cossio. F. R. Chap.3: Enantioselective C–C, C–halogen and C–H bond forming reactions promoted by organocatalysts based on non-proteinogenic α-amino acid derivatives. Adv. Catal. 2023, 73, 143–210. [Google Scholar] [CrossRef]
- Knoevenagel, E. Ber .dtsch. chem. Ges. 1896, 29, 172–174. [CrossRef]
- List, B. Emil Knoevenhagel and the roots of aminocatalysis. Angew. Chem. Int. Ed. 2010, 49, 1730–1734. [Google Scholar] [CrossRef]
- Doyle, A. G.; Jacobsen, E. N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- Lin, S.; Deiana, L.; Zhao, G. L.; Sun, J.; Córdova, A. Dynamic one-pot three-component catalytic asymmetric transformation by combination of hydrogen-bond-donating and amine catalysts. Angew. Chem. Int. Ed. 2011, 50, 7624–7630. [Google Scholar] [CrossRef]
- Knowles, R. R.; Jacobsen, E. N. Attractive noncovalent interactions in asymmetric catalysis: links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA. 2010, 107, 20678–20685. [Google Scholar] [CrossRef]
- Del Vecchio, A.; Sinibaldi, A.; Nori, V.; Giorgianni, G.; Di Carmine, G.; Pesciaioli, F. Synergistic strategies in aminocatalysis. Chem. Eur. J. 2022, 28, 1–27. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Q-Q. ; Du, W.; Chen, Y-C. Asymmetric organocatalysis involving double activation. Tetrahedron Chem. 2022, 2, article. [Google Scholar] [CrossRef]
- Cai, M.; Zhang, L.; Zhang, W.; Lin, Q.; Luo, S. Enantioselective transformations by “1 + x” synergistic catalysis with chiral primary amines. Acc. Chem. Res. 2024, 57, 1523–1537. [Google Scholar] [CrossRef]
- Samson Afewerki, S.; Córdova. A. Combinations of aminocatalysts and metal catalysts: A powerful cooperative approach in selective organic synthesis. Chem. Rev. 2016, 116, 13512–13570. [Google Scholar] [CrossRef]
- Nicewicz, D. A.; MacMillan, D. W. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Welin, E. R.; Warkentin, A. A; Conrad, J. C.; MacMillan, D. W. C. Enantioselective α-alkylation of aldehydes by photoredox organocatalysis: Rapid access to pharmacophore fragments from β-cyanoaldehydes. Angew. Chem. Int. Ed. 2015, 54, 968–9672. [Google Scholar] [CrossRef] [PubMed]
- Silvi, M.; Melchiorre, P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018, 554, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Bredig, G.; Fiske, P. S. Biochem. Z. 1912, 46, 7–23; Fiske, P. S. Durch katalysatoren bewirkte asymmetrische synthese (Doctoral dissertation, ETH Zurich) 1911; https://www.researchcollection. ethz.ch/handle/20.500.11850/136453.
- Pracejus, H. Organische Katalysatoren. LXI. Asymmetrische Synthesen mit Ketenen. I. Alkaloid-katalysierte asymmetrische Synthesen von α phenyl-proponsaüreestern. Liebigs Ann. Chem, 1960; 634, 9–22. [Google Scholar] [CrossRef]
- Singh, G. S. , Yeboah, E. M. Recent applications of Cinchona alkaloid-based catalysts in asymmetric addition reactions. Rep. Org.Chem. 2016, 6, 47–75. [Google Scholar] [CrossRef]
- Yeboah, E. M.O.; Yeboah, S. O.; Singh, G. S. Recent applications of Cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Saito, H.; Miyairi, S. Recent advances in cinchona alkaloid catalysis for enantioselective carbon-nitrogen bond formation reactions. Curr. Top. Med. Chem. 2014, 14, 224–228. [Google Scholar] [CrossRef]
- Portolani, C.; Centonze, G.; Righi, P.; Bencivenni, G. Role of cinchona alkaloids in the enantio- and diastereoselective synthesis of axially chiral compounds. Acc. Chem. Res. 2022, 55, 3551–3571. [Google Scholar] [CrossRef] [PubMed]
- Carreiro, E. P.; Burke, A: J. ; Hermann, G. J.; Federsel, H-J. Continuous flow enantioselective processes catalyzed by cinchona alkaloid derivatives. Tetrahedron Green Chem. 2023, 1, article. [Google Scholar] [CrossRef]
- Lahdenpera, A. S. K.; Dhankhar, J.; Davies, D. J.; Lam, N. Y. S.; Bucos, P. D.; De la Vega-Hernández, K.; Philipps, R. J. Chiral hydrogen atom abstraction catalyst for the enantioselective epimerization of meso-diols. Science 2024, 386, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, J. K.; Felman, S. W. Asymmetric induction. 3. Enantioselective deprotonation by chiral lithium amide bases. J. Org. Chem. 1980, 45, 755–756. [Google Scholar] [CrossRef]
- Kornblum, N.; DeLaMare, H. E. The base-catalyzed decomposition of dialkyl peroxide. J. Am. Chem. Soc. 1951, 73, 880–881. [Google Scholar] [CrossRef]
- Hagenbuch, J.-P.; Vogel, P. Asymmetric induction in the rearrangement of monocyclic endoperoxides into γ-hydroxy-α,β-unsaturated aldehydes. Chem. Commun. 1980, (22), 1062–1063. [Google Scholar] [CrossRef]
- Staben, S. T.; Linghu. ; Toste, F. D. Enantioselective synthesis of gamma-hydroxyenones by chiral base-catalyzed Kornblum DeLaMare rearrangement. J. Am. Chem. Soc. 2006, 128, 12658–12659. [Google Scholar] [CrossRef]
- Bui, T.; Candeias, N. R.; Barbas III, C. F. Dimeric quinidine-catalyzed enantioselective aminooxygenation of oxindoles: An organocatalytic approach to 3-hydroxyoxindole derivatives. J. Am. Chem. Soc. 2010, 132, 5574–5575. [Google Scholar] [CrossRef]
- Wang, Q.; Lian, Z.; Xu, Q, Shi, M. Asymmetric [4+2] annulations of isatins with but-3-yn-2-one. Adv. Synth. Catal. 2013, 355, 3344–3350. [Google Scholar] [CrossRef]
- Kolb, H. C.; Van Nieuwenhze, M. S.; Sharpless, K. B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- DelMonte, A. J.; Haller, J.; Houk, K. N.; Sharpless, K. B.; Singleton, D. A.; Strassner, T.; Thomas, A. A. Experimental and theoretical kinetic isotope effects for asymmetric dihydroxylation. Evidence supporting a rate-limiting "(3 + 2)" cycloaddition. J. Am. Chem. Soc. 1997, 119, 9907–9908. [Google Scholar] [CrossRef]
- Hentges, S. G.; Sharpless, K. B. Asymmetric induction in the reaction of osmium tetroxide with olefins. J. Am. Chem. Soc. 1980, 102, 4263–4265. [Google Scholar] [CrossRef]
- Jacobsen, E. N.; Markó, I.; Mungall, W. S.; Schröder, G.; Sharpless, K. B. Asymmetric dihydroxylation via ligand-accelerated catalysis. J. Am. Chem. Soc. 1988, 116, 1968–1970. [Google Scholar] [CrossRef]
- Saikia, B.; Ali, A. A.; Boruah, P. R.; Sarma, D.; Barua, N. C. Pd(OAc)2 and (DHQD)2PHAL as a simple, efficient and recyclable/reusable catalyst system for Suzuki-Miyaura cross-coupling reactions in H2O at room temperature. New J. Chem. 2015, 39, 2440–2443. [Google Scholar] [CrossRef]
- Ali, A. A.; Chetia, M.; Saikia, P. J.; Sarma, D. (DHQD)2PHAL ligand-accelerated Cu-catalyzed azide–alkyne cycloaddition reactions in water at room temperature. RSC Adv. 2014, 4, 64388–64392. [Google Scholar] [CrossRef]
- Wang, M.; Gao, L. X.; Mai, W. P.; Xia, A. X.; Wang, F.; Zhang, S. B. Enantioselective iodolactonization catalyzed by chiral quaternary ammonium salts derived from cinchonidine, J. Org. Chem. 2004, 69, 2874–2876. [Google Scholar] [CrossRef]
- Whitehead, D. C.; Yousefi, R.; Jaganathan, A.; Borhan, B. An organocatalytic asymmetric chlorolactonization. J. Am. Chem. Soc. 2010, 132, 3298–300. [Google Scholar] [CrossRef]
- Armstrong, A.; Braddock, D. C.; Jones, A. X.; Clark, S. Catalytic asymmetric bromolactonization reactions using (DHQD)2PHAL-benzoic acid combinations. Tetrahedron Lett. 2013, 54, 7004–7008. [Google Scholar] [CrossRef]
- Braddock, D. C.; Lancaster, B. M. J.; Tighe, C. J.; White, A. J. P. Surmounting byproduct inhibition in an intermolecular catalytic asymmetric alkene bromoesterification reaction as revealed by kinetic profiling. J. Org. Chem. 2023, 88, 8904–8914. [Google Scholar] [CrossRef]
- Hiemstra, H.; Wynberg, H. Addition of aromatic thiols to conjugated cycloalkenones, catalyzed by chiral beta-hydroxy amines. A mechanistic study of homogeneous catalytic asymmetric synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Marcelli, T.; van der Haas, R. N. S.; van Maarseveen, J. H.; Hiemstra, H. Asymmetric organocatalytic Henry reaction. Angew. Chem. Int. Ed. 2006, 45, 929–931. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, L. Organocatalytic applications of cinchona alkaloids as a potent hydrogen-bond donor and/or acceptor to asymmetric transformations: A review. Mini-Rev. Org. Chem. 2016, 13, 184–197. [Google Scholar] [CrossRef]
- Joshi, H.; Singh, V. K. Cinchona derivatives as bifunctional H-bonding organocatalysts in asymmetric ninylogous conjugate addition reactions. Asian J. Org. Chem. 2022, 11, e202100053. [Google Scholar] [CrossRef]
- McCooey, S. H.; Connon, S. J. Urea- and thiourea-substituted cinchona alkaloid derivatives as highly efficient bifunctional organocatalysts for the asymmetric addition of malonate to nitroalkenes: inversion of configuration at C9 dramatically improves catalyst performance. Angew. Chem. Int. Ed. 2005, 44, 6367–6370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W-R. ; Su, Q.; Lin, N.; Chen, Q.; Zhang, Z-W.; Weng, J.; Lu, G. Organocatalytic synthesis of chiral CF3-containing oxazolidines and 1,2-amino alcohols: asymmetric oxa-1,3-dipolar cycloaddition of trifluoroethylamine-derived azomethine ylides: Org. Chem. Front. 2020, 7, 3452–3458. [CrossRef]
- Zhu, Y-R. ; Xu, J.; Jiang, H-F., Fang, R-J., Zhang, Y-J., Chen, L., Sun, C., Eds.; Xiong, F. Bifunctional sulfonamide as hydrogen bonding catalyst in catalytic asymmetric reactions: Concept and application in the past decade. Eur. J. Org. Chem. 2022, article nb. e202201081. [Google Scholar] [CrossRef]
- Mondal, A.; Bhowmick, S.; Ghosh, A.; Chanda, T.; Bhowmick, K. C. Advances on asymmetric organocatalytic 1,4-conjugate addition reactions in aqueous and semi-aqueous media. Tetrahedron: Asymmetry 2017, 28, 849–875. [Google Scholar] [CrossRef]
- a) Duhamel, L. C. R. C. R. Acad. Sci. 1976, 282C, 125-127 ; b) Duhamel, L.; Plaquevent, J.-C. Déracemisation par protonation enantioselective. Tetrahedron Lett. [CrossRef]
- Duhamel, L.; Duhamel, P.; Plaquevent, J.-C. Enantioselective protonations: fundamental insights and new concepts. Tetrahedron Asymmetry 2004, 15, 3653–3691. [Google Scholar] [CrossRef]
- Fehr, C.; Galindo, J. Catalytic enantioselective protonation of enolates. Angew. Chem. Int. Ed. 1994, 33, 1888–1889. [Google Scholar] [CrossRef]
- Fehr, C. Enantioselective protonation of enolates and enols. Angew. Chem. Int. Ed. 1996, 35, 2566–2587. [Google Scholar] [CrossRef]
- Fehr, C. Catalytic enantioselective tauromerization of isolated enols. Angew Chem, Int Ed. 2007, 46, 7119–7121. [Google Scholar] [CrossRef]
- Kumar, A. S.; Salunkhe, R. V.; Rane, R. A.; Dike, S. Y. Novel catalytic enantioselective protonation (proton transfer) in Michael addition of benzenethiol to α-acrylacrylates: synthesis of (S)-naproxen and α-arylpropionic acids or esters. Chem. Commun. 1991, (7), 485–486. [Google Scholar] [CrossRef]
- Nishimura, K.; Ono, M.; Nagaoka, Y.; Tomioka, K. Catalytic enantioselective protonation of lithium ester enolates generated by conjugate addition of arylthiolate to enoates. Angew. Chem. Int. Ed. 2001, 40, 440–442. [Google Scholar] [CrossRef]
- Phelan, J. P.; Ellman, J. A. Conjugate addition-enantioselective protonation reactions. Beilstein J. Org. Chem. 2016, 12, 1203–1228. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Houk, K. N. Organic Chemistry: Theory, Reactivity, and Mechanisms in Modern Synthesis. Wiley VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, Chap. 1.6.1, pp. 10-11; ISBN: 978-3-527-34532-8.
- Hintermann, L.; Ackerstaff, J.; Boeck, F. Inner workings of a cinchona alkaloid catalyzed oxa-Michael cyclization: Evidence for a concerted hydrogen-bond-network mechanism. Chem. Eur. J. 2013, 19, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Zhuang, W.; Jørgensen, K. A. Asymmetric conjugate addition of azide to α,β-unsaturated compounds catalyzed by cinchona alkaloids. Tetrahedron 2007, 63, 5849–5853. [Google Scholar] [CrossRef]
- Yasukata, T.; Koga, K. Enantioselective protonation of achiral lithium enolates using a chiral amine in the presence of lithium bromide. Tetrahedron Asymmetry, 1993, 4, 35–38. [Google Scholar] [CrossRef]
- Riviere, P.; Koga, K. An approach to catalytic enantioselective protonation of prochiral lithium enolates. Tetrahedron Lett. 1997, 38, 7589–7592. [Google Scholar] [CrossRef]
- Łowicki, D.; Watral, J.; Jelecki, M.; Bohusz, W.; Kwit, M. Stereoselective protonation of 2-methyl-1-tetralone lithium enolate catalyzed by salan-type diamines. Tetrahedron 2021, 86, 1–11. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Ito, R.; Arai, T.; Yanagisawa, A. Catalytic asymmetric protonation of lithium enolates using amino acid derivatives as chiral proton sources. Org. Lett. 2006, 8, 1721–1724. [Google Scholar] [CrossRef]
- Poisson, T.; Dalla, V.; Marsais, F.; Dupas, G.; Oudeyer, S.; Levacher, V. Organocatalytic enantioselective protonation of silyl enolates mediated by cinchona alkaloids and a latent source of HF. Angew. Chem. Int. Ed. 2007, 46, 7090–7093. [Google Scholar] [CrossRef]
- Claraz, A.; Landelle, G.; Oudeyer, S.; Levacher, V. Asymmetric organocatalytic protonation of silyl enolates catalyzed by simple and original betaines derived from cinchona alkaloids. Eur. J. Org. Chem. 2013, 7693–7696. [Google Scholar] [CrossRef]
- Brunner, H,; Baur, M. A. α-Amino acid derivatives by enantioselective decarboxylation. Eur. J. Org. Chem, 2854. [CrossRef]
- George, K. M.; Frantz, M.; Bravo-Altamirano, K.; LaValle, C. R.; Tandon, M.; Leimgruber, S.; Sharlow, E.R.; Lazo, J. S.; Wang, Q. J.; Wipf, P. Pharmaceutics 2011, 3(2), 186-22. [CrossRef]
- Kang, G.; Yamagami, M.; Vellalath, S.; Romo, D. Angew. Chem. Int. Ed. 2018, 57, 6527–6531. [CrossRef]
- Zhou, Y.; Wei, Y-L. ; Rodriguez, J.; Coquerel, Y. Angew. Chem. Int. Ed. 2019, 58, 456–460. [Google Scholar] [CrossRef]
- Jiang, B.; Wei Du, W. ; Chen, Y-C. Modified cinchona alkaloid-catalysed enantioselective [4+4] annulations of cyclobutenones and 1-azadienes. Chem. Commun, 7257. [Google Scholar] [CrossRef]
- Xie, J. W.; Chen, W.; Li, R.; Zeng, M.; Du, W.; Yue, L.; Chen, Y. C.; Wu, Y.; Zhu, J.; Deng, J. G. Highly asymmetric Michael addition to alpha,beta-unsaturated ketones catalyzed by 9-amino-9-deoxyepiquinine. Angew. Chem. Int. Ed. 2007, 46, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Carlone, A.; Pesciaioli, F.; Sambri, L.; Melchiorre, P. Organocatalytic asymmetric Friedel-Crafts alkylation of indoles with simple alpha,beta-unsaturated ketones. Org. Lett. 2007, 9, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J. L.; Badia, D.; Carrillo, L. Organocatalytic enantioselective Michael and hetero-Michael reactions. Synthesis 2007, (14), 2065–2092. [Google Scholar] [CrossRef]
- Wang, X.; Reisinger, C. M.; List, B. Catalytic asymmetric epoxidation of cyclic enones. J. Am. Chem. Soc. 2008, 130, 6070–6071. [Google Scholar] [CrossRef]
- Cooey, S. H.; Connon, S. J. Readily accessible 9-epi-amino cinchona alkaloid derivatives promote efficient, highly enantioselective additions of aldehydes and ketones to nitroolefins. Org Lett. 2007, 9, 599–602. [Google Scholar] [CrossRef]
- Perera, S.; Naganaboina, V. K.; Wang, L.; Zhang, B.; Guo, Q.; Rout, L.; Zhao, C-G. Organocatalytic highly enantioselective synthesis of β-formyl-α-hydroxyphosphonates. Adv. Synth. Catal. 2011, 353, 1729–1734. [Google Scholar] [CrossRef]
- Kwiatkowski, P.; Beeson, T. D.; Conrad, J. C.; MacMillan, D. W. C. Enantioselective organocatalytic α-fluorination of cyclic ketones. J. Am. Chem. Soc. 2011, 133, 1738–1741. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. [CrossRef]
- Beer, P. D.; Brown, A. Halogen bonding anion recognition. Chem. Commun. 2016, 52, 8645–8658. [Google Scholar] [CrossRef] [PubMed]
- Sutar, R. L.; Huber, S. M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Bruckmann, A.; Pena, M. A.; Bolm, C. Organocatalysis through halogen-bond activation. Synlett 2008, (6), 900–902. [Google Scholar] [CrossRef]
- Kuwano, S.; Suzuki, T.; Hosaka, Y.; Arai, T. A chiral organic base catalyst with halogen-bonding-donor functionality: asymmetric Mannich reactions of malononitrile with N-Boc aldimines and ketimines. Chem. Commun. 2018, 54, 3847–3850. [Google Scholar] [CrossRef]
- Kuwano, S.; Nishida, Y.; Suzuki, T.; Arai, T. Catalytic asymmetric Mannich-type reaction of malononitrile with N-Boc α-ketiminoesters using chiral organic base catalyst with halogen bond donor functionality. Adv. Synth. Catal. 2020, 362, 1674–1678. [Google Scholar] [CrossRef]
- McGarrigle, E.; Myers, E. L.; Illa, O.; Shaw, M. A, Riches S. L, Aggarwal, V. K. Chalcogenides as organocatalysts. Chem Rev. 2007, 107, 5841–5883. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J. J. σ-Hole interactions in catalysis. Eur. J. Org. Chem. 2020, 34, 5473–5487. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, M.; Xu, C.; Chen. F.-E. Harnessing noncovalent interaction of chalcogen bond in organocatalysis: From the catalyst point of view. Green Synth. Catal. 2021, 2, 329–336. [Google Scholar] [CrossRef]
- Makosza, M. Reactions of organic anions II. Catalytic alkylation of indene. Tetrahedron Lett. 1966, 38, 4621–4624. [Google Scholar] [CrossRef]
- Starks, C. M. Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts. J. Am. Chem. Soc. 1971, 93, 195–199. [Google Scholar] [CrossRef]
- Helder, R.; Hummelen, J. C.; Laane, R. W. P. M.; Wiering, J. S.; Wynberg, H. Catalytic asymmetric induction in oxidation reactions. The synthesis of optically active epoxides. Tetrahedron Lett. 1976, 17, 1831–1834. [Google Scholar] [CrossRef]
- Pluim, H.; Wynberg, H. Catalytic asymmetric induction in oxidation reactions. Synthesis of optically active epoxynaphthoquinones. J. Org. Chem. 1980, 45, 2498–2502. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Q.; Cornwall, R. G.; Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef] [PubMed]
- Moschona, F.; Savvopoulou, I.; Tsitopoulou, M.; Tataraki, D.; Rassias, G. Epoxide syntheses and ring-opening reactions in drug development. Catalysts 2020, 10, 1–65. [Google Scholar] [CrossRef]
- Dolling, U. H.; Davis, P.; Grabowski, E. J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. [Google Scholar] [CrossRef]
- O’Donnell, M. J.; Bennett, W. D.; Wu, S. The stereoselective synthesis of α-amino acids by phase-transfer catalysis. J. Am. Chem. Soc. 1989, 111, 2353–2355. [Google Scholar] [CrossRef]
- Murugan, E.; Siva, A. Synthesis of asymmetric N-arylaziridine derivatives using a new chiral phase-transfer catalyst. Synthesis, 2005; 2022–2028. [Google Scholar]
- Lygo, B.; Wainwright, P. G. A new class of asymmetric phase-transfer catalysts derived from Cinchona alkaloids–application in the enantioselective synthesis of α-amino acids. Tetrahedron Lett. 1997, 38, 8595–8598. [Google Scholar] [CrossRef]
- Corey, E. J.; Xu, F.; Noe, M. C. A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar] [CrossRef]
- Corey, E. J.; Zhang, F.-Y. Mechanism and conditions for highly enantioselective epoxidation of alpha,beta-enones using charge-accelerated catalysis by a rigid quaternary ammonium salt. Org. Lett. 1999, 1, 1287–1290. [Google Scholar] [CrossRef]
- Yamamoto, E.; Nagai, A.; Hamasaki, A.; Tokunaga, M. Catalytic asymmetric hydrolysis: Asymmetric hydrolytic protonation of enol esters catalyzed by phase-transfer catalysts. Chem. Eur. J. 2011, 17, 7178–7182. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Tan, J.; Yasuda, N. Transfer catalysts: Large-scale industrial applications. Org. Proc. Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- Albanese, D. C. M.; Penso, M. New trends in asymmetric phase transfer catalysis. Eur. J. Org. Chem. 2023, 26, 1–17. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yasuda, N. Contemporary asymmetric phase transfer catalysis: Large-scale industrial applications. Org. Process Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- Lee, H. J.; Maruoka, K. Asymmetric phase-transfer catalysis. Nat. Rev. Chem. 2024, 8, 851–869. [Google Scholar] [CrossRef]
- Haripriya, P. , Rai, R. Vijayakrishna, K. Polymer anchored cinchona alkaloids: Synthesis and their applications in organo-catalysis. Top. Curr. Chem. (Z) 2025, 383, 1–70. [Google Scholar] [CrossRef]
- Moccia, M.; Cortigiani, M.; Monasterolo, C.; Torri, F.; del Fiandra, C.; Fuller, G.; Kelly, B.; Adamo, M. F. A. Development and scale-up of an organocatalytic enantioselective process to manufacture (S)-Pregabalin. Org. Process Res. Dev. 2015, 19, 1274–1281. [Google Scholar] [CrossRef]
- Giorgianni, G.; Bernardi, L.; Fini, F.; Pesciaioli, F.; Secci, F.; Carlone, A. Asymmetric organocatalysis—A powerful technology platform for academia and industry: Pregabalin as a case study. Catalysts 2022, 12, 912. [Google Scholar] [CrossRef]
- Deng, T.; Han, X-L. ; Yu, Y.; Cheng, C.; Liu, X.; Gao, Y.; Wu, K.; Li. Z.; Luo, J.; Deng, L. Organocatalytic asymmetric α-C–H functionalization of alkyl amines. Nat. Catal. 2024, 7, 1076–1085. [Google Scholar] [CrossRef]
- Wang, H. Y. Chiral phase-transfer catalysts with hydrogen bond: A powerful tool in the asymmetric synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef]
- Bernal, P.; Fernández, R.; Lassaletta, J. M. Organocatalytic asymmetric cyanosilylation of nitroalkenes. Chemistry 2010, 16, 7714–7718. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K. M.; Rattley, M. S.; Sladojevich, F.; Barber, D. M.; Nuñez, M. G.; Goldys, A. M.; Dixon, D. J. A new family of cinchona-derived bifunctional asymmetric phase-transfer catalysts: application to the enantio- and diastereoselective nitro-Mannich reaction of amidosulfones. Org. Lett. 2012, 14, 2492–2495. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Woods, P. A.; Smith, M. D. Cation-directed enantioselective synthesis of quaternary-substituted indolenines. Chem. Science 2013, 4, 2907–2911. [Google Scholar] [CrossRef]
- Lu, N.; Fang, Y.; Gao. ; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Bifunctional thiourea-ammonium salt catalysts derived from cinchona alkaloids: Cooperative phase-transfer catalysts in the enantioselective aza-Henry reaction of ketimines. J. Org. Chem. 2018, 83, 1486–1492. [Google Scholar] [CrossRef]
- Yang, X. Q.; Deng, Y. L.; Ling, D.; Li, T. T.; Chen, L. Z.; Jin, Z. C. ; Recent progress in chiral quaternary ammonium salt-promoted asymmetric nucleophilic additions. ACS Catal. 2025, 15, 1973–2001. [Google Scholar] [CrossRef]
- Ohshima, T.; Shibuguchi, T.; Fukuta, Y.; Shibasaki, M. Catalytic asymmetric phase-transfer reactions using tartrate-derived asymmetric two-center organocatalysts. Tetrahedron 2004, 60, 7743–7754. [Google Scholar] [CrossRef]
- Waser, M.; Gratzer, K.; Herchl, R.; Müller, N. Design, synthesis, and application of tartaric acid derived N-spiro quaternary ammonium salts as chiral phase-transfer catalysts. Org. Biomol. Chem. 2012, 10(2), 251–254. [Google Scholar] [CrossRef]
- Zong, L.; Tan, C.-H. Phase-transfer and ion-pairing catalysis of pentanidiums and bisguanidiniums. Acc. Chem. Res. 2017, 50, 842–856. [Google Scholar] [CrossRef]
- Dong, S.; Feng, X.; Liu, X. Chiral guanidines and their derivatives in asymmetric synthesis. Chem. Soc. Rev. 2018, 47, 8525–8540. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, Z, Ye, X. ; Tan, C.-H, Ion-pair catalysts-pentanidinium. Chem. Record 2023, 23, 1–14. [Google Scholar] [CrossRef]
- Yang, J. S.; Lu, K.; Li, C. X.; Zhao, Z, H. ; Zhang, X. M.; Zhang, F. M.; Tu, Y. Q. Chiral 1,2,3-triazolium salt catalyzed asymmetric mono- and dialkylation of 2,5-diketopiperazines with the construction of tetrasubstituted carbon centers. Angew. Chem. Int. Ed. 2022, 61, 1–7. [Google Scholar] [CrossRef]
- Ooi, T.; Kameda, M.; Maruoka, K. ; Design of N-spiro C2-symmetric chiral quaternary ammonium bromides as novel chiral phase-transfer catalysts: synthesis and application to practical asymmetric synthesis of alpha-amino acids. J. Am. Chem. Soc. 2003, 125, 5139–5151. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Sasaki, K.; Fukumoto, K.; Murase, Y.; Abe, N.; Ooi, T.; Maruoka, K. Phase-transfer-catalyzed asymmetric alkylation of α-benzoyloxy-β-keto esters: Stereoselective construction of congested 2,3-dihydroxycarboxylic acid esters. Chem. Asian. J. 2010, 5, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Ha, H. W.; Choi, S.; Kim, S.; Lee, J. Y.; Lee, J. K.; Lee, J.; Hong, S.; Park, H.-g. Phase-transfer catalyzed enantioselective α-alkylation of α-acyloxymalonates: construction of chiral α-hydroxy quaternary stereogenic centers. RSC Adv. 2016, 6, 77427–77430. [Google Scholar] [CrossRef]
- Griffiths, C. M.; Franckevicius, V. The catalytic asymmetric allylic alkylation of acyclic enolates for the construction of quaternary and tetrasubstituted stereogenic centres. Chem. Eur. J. 2024, 20, e202304289. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, J.; Tan, S. M.; Tan, D.; Lee, R.; Tan, C. H. An enantioconvergent halogenophilic nucleophilic substitution (SN2X) reaction. Science 2019, 363, 400–404. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Syntheses and applications of (thio)urea-containing chiral quaternary ammonium salt catalysts. Eur. J. Org. Chem. 2014, 2014(4), 802–809. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Waser, M.; Grošelj, U. Synthesis and catalytic activity of bifunctional phase-transfer organocatalysts based on camphor. Molecules 2023, 28, article. [Google Scholar] [CrossRef]
- Sutar, R. L.; Engelage, E.; Stoll, R.; Huber, S. M. Bidentate chiral bis(imidazolium)-based halogen-bond donors: Synthesis and applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef]
- Jeffrey, J- L. ; Terrett, J. A.; MacMillan, D. W. C. O-H hydrogen bonding promotes H-atom transfer from α-C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [Google Scholar] [CrossRef] [PubMed]
- Turner, J. A.; Adrianov, T.; Taylor, M. S. Understanding the origins of site selectivity in hydrogen atom transfer reactions from carbohydrates to the quinuclidinium radical cation: A computational study. J. Org. Chem. 2023, 88, 5713–5730. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, T.; Lin, Y-M. ; Gong, L. Advancements in organocatalysis for radical-mediated asymmetric synthesis: A recent perspective. Chem Catal. 2024, 4, 1–53. [Google Scholar] [CrossRef]
- Nagib, D. A. Asymmetric catalysis in radical chemistry. Chem. Rev. 2022, 122, 15989–15992. [Google Scholar] [CrossRef]
- Northrup, A. B.; MacMillan, D. W. C. The first general enantioselective catalytic Diels-Alder reaction with simple α,β-unsaturated ketones. J. Am. Chem. Soc. 2002, 124, 2458–2460. [Google Scholar] [CrossRef]
- Lelais, G.; MacMillan, D. W. C. Modern strategies in organic catalysis: The advent and development of iminium activation. Aldriquimica Acta 2006, 39, 79–87. [Google Scholar]
- Erkkilä, A.; Majander, I.; Pihko, P. M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Bartoli, G.; Melchiorre, P. A novel organocatalytic tool for the iminium activation of α,β-unsaturated ketones. Synlett 2008, 2008(12), 1759–1772. [Google Scholar] [CrossRef]
- Paras, N. A.; MacMillan, D. W. C. The enantioselective organocatalytic 1,4-Addition of electron-rich benzenes to α,β-unsaturated aldehydes. J. Am. Chem. Soc. 2002, 124, 7894–7895. [Google Scholar] [CrossRef]
- Pronina, Y.A.; Teglyai, L. A.; Ponyaev, A. I.; Petrov, M. L.; Boitsov, V. M.; Stepakow, A. V. Organocatalysis in 1,3-dipolar cycloaddition reactions (A review). Russ. J. Gen. Chem. 2024, 94, 1–44. [Google Scholar] [CrossRef]
- Haraguchi, N.; Takenaka, N.; Najwa, A.; Takahara, Y.; Mun, M. K.; Itsuno, S. Synthesis of main-chain ionic polymers of chiral imidazolidinone organocatalysts and their application to asymmetric Diels–Alder reactions. Adv. Synth. Catal. 2018, 2018(1), 112–123. [Google Scholar] [CrossRef]
- Xu, F.; Zacuto, M.; Yoshikawa, N.; Desmond, R.; Hoerrner, S.; Itoh, T.; Journet, M.; Humphrey, G. R.; Cowden, C.; Strotman, N.; Devine, P. Asymmetric synthesis of telcagepant, a CGRP receptor antagonist for the treatment of migraine. J. Org. Chem. 2010, 75(22), 7829–7841. [Google Scholar] [CrossRef]
- Cao, Z. Y.; Ghosh, T.; Melchiorre, P. Enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion-based EDA complex. Nat. Commun. 2018, 9, article. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Burés, J.; Armstrong, A.; Blackmond, D. G. Explaining anomalies in enamine catalysis: “downstream species” as a new paradigm for stereocontrol. Acc. Chem. Res. 2016, 49, 214–222. [Google Scholar] [CrossRef]
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F. III. Amino acid catalyzed direct asymmetric aldol reactions: a bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267. [Google Scholar] [CrossRef]
- Ghosh, S. K. Efficient catalytic methods for asymmetric cross-aldol reaction of aldehydes. ChemistrySelect 2024, 9, 1–46. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinolsilyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef]
- Kamal, S.; Zahoor, A. F.; Ahmad, S.; Akhtar, R.; Qurban, W.; Mehreen, A. Recent trends in the development of novel catalysts for asymmetric Michael reaction. Curr. Org. Chem. 2020, 24, 1397–1458. [Google Scholar] [CrossRef]
- Pasuparthy, S. D.; Maiti, B. Enantioselective organocatalytic Michael addition reactions catalyzed by proline/prolinol/supported proline based organocatalysts: An overview. ChemistrySelect 2022, 7, 1–36. [Google Scholar] [CrossRef]
- Córdova, A. The direct catalytic asymmetric Mannich reaction. Acc. Chem. Res. 2004, 37, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.; Schaus, S. E. Organocatalytic asymmetric Mannich reactions: New methodology, catalyst design, and synthetic applications. Eur. J. Org. Chem. 2007, (35), 5797–5815. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas, C. F. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels-Alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef]
- Li, J-L. ; Liu, T-Y.; Chen, Y-C. Aminocatalytic asymmetric Diels–Alder reactions via HOMO activation. Acc. Chem. Res. 2012, 45, 1491–1500. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, R. Recent developments in catalytic asymmetric inverse-electron-demand Diels-Alder reaction. Chem Rev. 2013, 113, 5515–5546. [Google Scholar] [CrossRef]
- Ueda, M.; Kano, T.; Maruoka, K. Organocatalyzed direct asymmetric α-halogenation of carbonyl compounds. Org. Biomol. Chem. 2009, 7, 2005–2012. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef]
- Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Enantioselective organocatalyzed α-sulfenylation of aldehydes. Angew. Chem. Int. Ed. 2005, 44, 794–797. [Google Scholar] [CrossRef]
- Duthaler, R. O. Proline-catalyzed asymmetric α-amination of aldehydes and ketones-An astonishing simple access to optically active α-hydrazino carbonyl compounds. Angew. Chem. Int. Ed. 2003, 41, 975–978. [Google Scholar] [CrossRef]
- Vera, S.; Landa, A.; Mielgo, A.; Ganboa, I.; Oiarbide, M.; Soloshonok, V. Catalytic asymmetric α-functionalization of α-branched aldehydes. Molecules 2023, 28, 1–48. [Google Scholar] [CrossRef]
- Yamada, S.; Otani, G. Asymmetric synthesis with amino acid. 2. Asymmetric synthesis of optically active 4,4-disubstituted -2-cyclohexenone. Tetrahedron Lett. 1969, 10, 4237–42401. [Google Scholar] [CrossRef]
- Liu, F.; Wang, S.; Wang, N.; Peng, Y. Prolinol tert-butyldiphenylsilyl ether as an organocatalyst for the asymmetric Michael addition of cyclohexanone to nitroolefins. Synlett 2007, (15), 2415–2419. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.; Marigo, M.; Fielenbach, D.; Wabnitz, T. C.; Kjærsgaard, A.; Jørgensen, K. A. A general organocatalyst for direct α-functionalization of aldehydes: Stereoselective C−C, C−N, C−F, C−Br, and C−S bond-forming reactions. Scope and mechanistic insights. J. Am. Chem. Soc. 2005, 127, 18296–18304. [Google Scholar] [CrossRef] [PubMed]
- Hajos, Z. G.; Parrish, D. R. Asymmetric synthesis of polycyclic organic compounds. German Patent DE 2102623 ( 1971.
- Hajos, Z. G.; Parrish, D. R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Hajos, Z. G.; Parrish, D. R. Synthesis and conversion of 2-methyl-2-(3-oxobutyl)-1,3-cyclopentanedione to isomeric racemic ketols of [3.2.1]bicyclooctane of perhydroindan series. J. Org. Chem. 1974, 39, 1612–1615. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Weichert, R. Total synthesis of optically active steroids. 6. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Notz, W.; List, B. Catalytic asymmetric synthesis of anti-1,2-diols. J. Am. Chem. Soc. 2000, 122, 7386–7387. [Google Scholar] [CrossRef]
- Jensen, K. L.; Dickmeiss, G.; Jiang, H.; Albrecht, L.; Jørgensen, K. L. The diarylprolinol silyl ether system: A general organocatalyst. Acc. Chem. Res. 2012, 45, 248–264. [Google Scholar] [CrossRef]
- Meninno, S.; Lattanzi, A. Asymmetric organocatalysis mediated by α,α-L-diaryl prolinols: Recent advances. Chem. Commun. 2013, 49, 3821–3832. [Google Scholar] [CrossRef]
- Donslund, B. S.; Johansen, T. K.; Poulsen, P. H.; Halskov, K. S.; Jørgensen, K, A. The diarylprolinol silyl ethers: Ten years after. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef]
- Valapil, G. G.; Kadagathur, M.; Shankaraiah, N. Stereoselective aldol and conjugate addition reactions mediated by proline-based catalysts and its analogues: A concise review. Eur. Org. Chem. 2021, (37), 5288–5311. [Google Scholar] [CrossRef]
- López-Aguilar, M.; Ramos-Martín, M.; Ríos-Lombardía, N.; Cicco, L.; García-Álvarez, J.; Concellón, C.; del Amo, V. Synergistic one-pot tandem combination of Cu and proline catalysis: Stereodivergent synthesis of chiral aldols from primary alcohols. ChemCatChem 2024, 16, 1–7. [Google Scholar] [CrossRef]
- Kamanna, K. Amino acids and peptides organocatalysts: A brief overview on its evolution and applications in organic asymmetric synthesis. Curr. Organocatal. 2021, 8, 126–146. [Google Scholar] [CrossRef]
- Kamanna, K. Organocatalysts based on natural and modified amino acids for asymmetric reactions. Phys. Sci. Rev. 2022, 7, 429–467. [Google Scholar] [CrossRef]
- Córdova, A.; Zou, W.; Dziedzic, P.; Ibrahem, I.; Reyes, E.; Xu, Y. Direct asymmetric intermolecular aldol reactions catalyzed by amino acids and small peptides. Chem. Eur. J. 2006, 12, 5383–5397. [Google Scholar] [CrossRef]
- Bartók, M. Advances in immobilized organocatalysts for the heterogenous asymmetric direct aldol reactions. Catal. Rev. 2015, 57, 192–255. [Google Scholar] [CrossRef]
- Shajahan, R.; Sarang, R.; Saithalavi, A. Polymer supported proline-based organocatalyts in asymmetric aldol reactions. A review. Curr. Organocatal. 2022, 9, 124–146. [Google Scholar]
- Álvarez-Bermúdez, O.; Landfester, K.; Zhang, K. A. I.; Muñoz-Espi, R. Proline-functionalized magnetic nanoparticles as highly performing asymmetric catalysts. Macromol. Rap. Commun. 2024, 45. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Houk, K. N.; Martin, H. J.; List, B. Quantum mechanical predictions of the stereoselectivities of proline-catalyzed asymmetric intermolecular aldol reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef] [PubMed]
- Clemente, F. R.; Houk, K. N. Computational evidence for the enamine mechanism of intramolecular aldol reactions catalyzed by proline. Angew. Chem. Int. Ed. 2004, 116, 5890–5892. [Google Scholar] [CrossRef]
- Zhu, H.; Clemente, F. R.; Houk, K. N.; Meyer, M. P. Rate limiting step precedes C–C bond formation in the archetypical proline-catalyzed intramolecular aldol reaction. J. Am. Chem. Soc. 2009, 131, 1632–1633. [Google Scholar] [CrossRef]
- Schmid, M. B.; Zeitler, K.; Gschwind, R. M. The elusive enamine intermediate in proline-catalyzed aldol reactions: NMR detection, formation pathway, and stabilization trends. Angew. Chem. Int. Ed. 2010, 49, 4997–5003. [Google Scholar] [CrossRef]
- Yu, L-J, Coote, M. L. Electrostatic switching of stereoselectivity in aldol reactions. J. Org. Chem. 2021, 86, 9076–9083. [Google Scholar] [CrossRef]
- Yu, L-J. ; Blyth, M. T.; Coote, M. L. Re examination of proline catalyzed intermolecular aldol reactions: An ab Initio kinetic modelling study. Top. Catal. 2022, 65, 354–365. [Google Scholar] [CrossRef]
- Agami, C.; Meynier, F.; Richot, C. New insights into the mechanism of the proline-catalyzed asymmetric Robinson cyclization; structure of two intermedaites. Asymmetric dehydration. Tetrahedron 1984, 40, 1031–1038. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A. K.; Badine, D. M.; Limbach, M.; Eschenmoser, A.; Treasurywala, A. M.; Hobi, R.; Prikosvovich, W.; Linder, B. Are oxazolidinones really unproductive, parasitic species in proline catalysis? Thoughs and experiments pointing to an alternative view. Helv. Chim. Acta 2007, 90, 425–471. [Google Scholar] [CrossRef]
- Sharma, A. K,.; Sunoj, R. B. Enamine versus oxazolidinone: what controls stereoselectivity in proline-catalyzed asymmetric aldol reactions? Angew. Chem. Int. Ed. 2010, 49, 6373–6377. [Google Scholar] [CrossRef]
- Schnitzer, T.; Wennemers, H. Deactivation of secondary amine catalysts via aldol reaction-amine catalysis under solvent-free conditions. J. Org. Chem. 2020, 85, 7633–7640. [Google Scholar] [CrossRef]
- Hayashi, Y.; Aikawa, T.; Shimasaki, Y.; Okamoto, H.; Tomioka, Y.; Miki, T.; Takeda, M.; Ikemoto, T. Research and development of an efficient synthesis of a key building block for anti-AIDS drugs by diphenylprolinol-catalyzed enantio- and diastereoselective direct cross aldol reaction. Org. Process Res. Dev. 2016, 20, 1615–1620. [Google Scholar] [CrossRef]
- Urushima, T.; Yasui, Y.; Ishikawa, H.; Hayashi, Y. Polymeric ethyl glyoxylate in an asymmetric aldol reaction catalyzed by diarylprolinol. Org. Lett. 2010, 12, 2966–2969. [Google Scholar] [CrossRef] [PubMed]
- Córdova, A. The direct catalytic asymmetric Mannich reaction. Acc. Chem. Res. 2004, 37, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Verkade, J.; van Hemert, L. J.; Quaedflieg, P, J. ; Rutjes, F. P. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41. [Google Scholar] [CrossRef]
- List, B. The direct catalytic asymmetric three-component Mannich reaction. J. Am. Chem. Soc. 2000, 122, 9336–9337. [Google Scholar] [CrossRef]
- List, B.; Porjalev, P.; Biller, W. T.; Martin, H. J. The proline-catalyzed direct asymmetric three-component Mannich reaction: Scope, optimization, and application to the highly enantioselective synthesis of 1,2-amino alcohols. J. Am. Chem. Soc. 2002, 124, 827–833. [Google Scholar] [CrossRef]
- Enders, D.; Hüttl, M. R. H. Direct organocatalytic α-fluorination of aldehydes and ketones. Synlett 2005, 2005(6), 991–993. [Google Scholar] [CrossRef]
- Marigo, M.; Fielenbach, D.; Braunton, A.; Kjaersgaard, A.; Jørgensen, K. A. Enantioselective formation of stereogenic carbon-fluorine centers by a simple catalytic method. Angew. Chem. Int. Ed. 2005, 44, 3703–3706. [Google Scholar] [CrossRef]
- Steiner, D. D.; Mase, N.; Barbas, C. F. III. Direct asymmetric alpha-fluorination of aldehydes. Angew. Chem. Int.Ed. 2005, 44, 3706–3710. [Google Scholar] [CrossRef]
- Beeson, T. D.; Macmillan, D. W. Enantioselective organocatalytic alpha-fluorination of aldehydes. J. Am. Chem. Soc. 2005, 127, 8826–8828. [Google Scholar] [CrossRef]
- Bertelsen, S.; Halland, N.; Bachmann, S.; Marigo, M.; Braunton, A.; Jørgensen, K. A. Organocatalytic asymmetric α-bromination of aldehydes and ketones. Chem. Commun, 4821. [Google Scholar]
- Nacsa, E. D.; MacMillan, D. W. C. Spin-center shift-enabled direct enantioselective α-benzylation of aldehydes with alcohols. J. Am. Chem. Soc. 2018, 140, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Shih, H. W.; Van der Wal, M. N.; Grange, R. L.; MacMillan, D. W. Enantioselective α-benzylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 2010, 132, 13600–13603. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, S.; Marigo, M.; Brandes, S.; Dinér, P.; Jørgensen, K. A. Dienamine catalysis: organocatalytic asymmetric gamma-amination of alpha,beta-unsaturated aldehydes. J. Am. Chem. Soc. 2006, 128, 12973–12980. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z-J. ; Jiang, H.; Li, J-L.; Gschwend, B.; Li, Q-Z.; Yin, X.; Grouleff, J.; Chen, Y-C.; Jørgensen, K. A. Trienamines in asymmetric organocatalysis: Diels Alder and tandem reactions. J. Am. Chem. Soc. 2011, 133, 5053–5061. [Google Scholar] [CrossRef]
- Arceo, E.; Melchiorre, P. Extending the aminocatalytic HOMO-raising activation strategy: where is the limit? Angew. Chem. Int. Ed. 2012, 51, 5290–5292. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Y.; Melchiorre, P. Aminocatalytic enantioselective 1,6-additions of alkyl thiols to cyclic dienones: vinylogous iminium ion activation. Angew. Chem. Int. Ed. 2012, 51, 6439–6442. [Google Scholar] [CrossRef]
- Tietze, L. F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Chapman, C. J.; Frost, C. G. Tandem and domino catalytic strategies for enantioselective synthesis. Synthesis, 2007; 1–21. [Google Scholar] [CrossRef]
- Zhao, G-L. ; Ibrahem, I.; Dziedzic, P.; Sun, J.; Bonneau, C.; Córdova, A. One-pot catalytic enantioselective domino nitro-Michael/Michael Synthesis of cyclopentanes with four stereocenters. Chem. Eur. J. 2008, 14, 10007–10011. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Enders, D. Achieving molecular complexity via stereoselective multiple domino reactions promoted by a secondary amine organocatalyst. Acc Chem Res. 2017, 50, 2809–2821. [Google Scholar] [CrossRef]
- Chando, T.; Gui Zhao, J. C. Recent progress in organocatalytic asymmetric domino transformations. Adv. Synth. Catal. 2017, 360, 2–79. [Google Scholar] [CrossRef]
- Enders, D.; Grondal, C.; Hüttl, M. R. Asymmetric organocatalytic domino reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in asymmetric organocatalytic domino reactions. Adv. Synth. Catal. 2012, 354, 237–294. [Google Scholar] [CrossRef]
- Volla, C. M.; Atodiresei, I.; Rueping, M. Catalytic C-C bond-forming multi-component cascade or domino reactions: pushing the boundaries of complexity in asymmetric organocatalysis. Chem. Rev. 2014, 114, 2390–2431. [Google Scholar] [CrossRef]
- Faghtmann, J.; Andresen, P. S. S.; Østergaard, R. A, Jørgensen, A. K. Enantioselective cascade reactions of aminocatalytic dienamines and trienamines initiated by a cycloaddition reaction. Chem. Eur.J. 2024; e202403656,. [Google Scholar] [CrossRef]
- Bui, T.; Barbas, C. F. III. A proline-catalyzed asymmetric Robinson annulation reaction. Tetrahedron Lett. 2000, 41(36), 6951–6954. [Google Scholar] [CrossRef]
- Halland, N.; Aburel, P. S.; Jørgensen, K. A. Highly Enantio- and diastereoselective organocatalytic asymmetric domino Michael–aldol reaction of β-ketoesters and α,β-unsaturated ketones. Angew. Chem. Int. Ed. 2004, 43, 1272–1277. [Google Scholar] [CrossRef]
- Hayashi, Y.; Okano, T.; Aratake, S.; Hazelard, D. Diphenylprolinol silyl ether as a catalyst in an enantioselective, catalytic, tandem Michael/Henry reaction for the control of four stereocenters. Angew. Chem. Int. Ed. 2007, 46, 4922–4925. [Google Scholar] [CrossRef]
- Barbas, W.; Notz, K.; Sakthivel, T.; Bui, G.; Zhong, G.; Barbas, C. F. III Amine-catalyzed direct asymmetric Mannich-type reactions. Tetrahedron Lett. 2001, 42, 199–201. [Google Scholar] [CrossRef]
- Córdoba, A. The direct catalytic asymmetric cross-Mannich reaction: A highly enantioselective route to 3-amino alcohols and α-amino acid derivatives. Chem. Eur. J. 2004, 10, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Urushima, T.; Shoji, M.; Uchimaru, T.; Shiina, I. The direct, enantioselective, one-pot, three-component, cross-Mannich reaction of aldehydes: The reason for the higher reactivity of aldimine versus aldehyde in proline-mediated Mannich and aldol reactions. Adv. Synth. Catal. 2005, 347, 1595–1604. [Google Scholar] [CrossRef]
- Dziedzic, P.; Ibrahem, I.; Córdova, A. Direct catalytic asymmetric three-component Mannich reactions with dihydroxyacetone: enantioselective synthesis of amino sugar derivatives. Tetrahedron Lett. 2008, 49, 803–807. [Google Scholar] [CrossRef]
- Yang, J.; Chandler, C.; Stadler, M.; Kampen, D.; List, B. Proline-catalysed Mannich reactions of acetaldehyde. Nature 2008, 452, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Teo, Y-C. ; Lau, J-J.; Wu, M-C. Direct asymmetric three-component Mannich reactions catalyzed by a siloxy serine organocatalyst in water. Tetrahedron Asymmetry 2008, 19, 186–190. [Google Scholar] [CrossRef]
- List, B.; Castello, C. A novel proline-catalyzed three-component reaction of ketones, aldehydes, and Meldrum’s acid. Synlett 2001, 11, 1687–1689. [Google Scholar] [CrossRef]
- Córdova, A.; Notz, W.; Barbas, C. F. III. J. Org. Chem. 2002, 67, 301–303. [Google Scholar] [CrossRef]
- Chowdari, N. S.; Ramachary, D. B.; Córdova, A.; Barbas, C. F. III. Proline-catalyzed asymmetric assembly reactions: enzyme-like assembly of carbohydrates and polyketides from three aldehyde substrates. Tetrahedron Lett. 2002, 43, 9591–9595. [Google Scholar] [CrossRef]
- Enders, D.; Hüttl, M. R.; Runsink, J.; Raabe, G.; Wendt, B. Organocatalytic one-pot asymmetric synthesis of functionalized tricyclic carbon frameworks from a triple-cascade/Diels-Alder sequence. Angew. Chem. Int. Ed. 2007, 46, 467–469. [Google Scholar] [CrossRef]
- Jang, H-Y. ; Hong, J-B.; MacMillan, D. W. C. Enantioselective organocatalytic singly occupied molecular orbital activation: The enantioselective α-enolation of aldehydes. J. Am. Chem. Soc. 2007, 129, 7004–7005. [Google Scholar] [CrossRef]
- Beeson, T. D.; Mastracchio, A.; Hong, J-B. ; Ashton, K.; MacMillan, D. W. C. Enantioselective organocatalysis using SOMO activation. Science 2007, 316, 582–585. [Google Scholar] [CrossRef]
- Mečiarová, M.; Pavol Tisovský, P.; Sebesta, R. Enantioselective organocatalysis using SOMO activation. New J. Chem. 2016, 40, 4855–4864. [Google Scholar] [CrossRef]
- Morita, K.; Suzuki, Z.; Hirose, H. A tertiary phosphine-catalyzed reaction of acrylic compounds with aldehydes. Bull. Chem. Soc. Jpn. 1968, 41, 2815–2815. [Google Scholar] [CrossRef]
- 4Baylis, A. B.; Hillman, M. E. D. Acrylic compounds. Chem. Abstr. 1972, 77, 34174q. [Google Scholar]
- Maneesha, M.; Haritha, S. H.; Aneeja, T.; Anilkumar, G. Recent progress and prospects in the organocatalytic Morita–Baylis–Hillman reaction. RSC Adv. 2024, 14, 14949–14963. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, M. Recent advances in organocatalytic asymmetric Morita–Baylis–Hillman/aza-Morita–Baylis–Hillman reactions. Chem. Rev. 2013, 113, 6659–6690. [Google Scholar] [CrossRef]
- Vogel, P.; Houk, K. N. Organic Chemistry: Theory, Reactivity and Mechanisms in Modern Synthesis. Wiley VCH GmbH & Co. KGaA, Weinheim, Germany, 2019, Chap. 7.5.56,pp. 9: 826-828; ISBN, 3453. [Google Scholar]
- Pélissier, H. Asymmetric organocatalytic Morita−Baylis−Hillman reaction and asymmetric organocatalytic transformations of Morita−Baylis−Hillman adducts. An update. Tetrahedron 2025, 172, article. [Google Scholar] [CrossRef]
- Krishna, P. R.; Kannan, V.; Reddy, P. V. N. N-methylprolinol catalysed asymmetric Baylis-Hillman reaction. Adv. Synth. Catal. 2004, 346, 603–606. [Google Scholar] [CrossRef]
- Menkudle, M. S.; Pendalwar, S. S.; Goswami, S. V.; Jadhav, W. N.; Bhusare, S. R. SN Appl. Sci. 2020, 2, 672–677. [CrossRef]
- Chinchilla, R.; Nájera, C.; Sánchez-Agulló, P. Enantiomerically pure guanidine-catalysed asymmetric nitroaldol reaction. Tetrahedron Asymmetry 1994, 5, 1393–1402. [Google Scholar] [CrossRef]
- Selig, P. Guanidine organocatalysis. Synthesis 2013, 45, 703–718. [Google Scholar] [CrossRef]
- Chou, H. C.; Leow, D.; Tan, C. H. Recent advances in chiral guanidine-catalyzed enantioselective reactions. Chem. Asian J. 2019, 14, 3803–3822. [Google Scholar] [CrossRef] [PubMed]
- Wei, S. Q.; Bao, X. Z.; Nawaz, S.; Qu, J. ; Wang, B, Identification of a tartrate-based modular guanidine towards highly asymmetric Michael addition of 3-aminooxindoles to nitroolefins. Tetrahedron Lett. 2021, 64, 1–4. [Google Scholar] [CrossRef]
- Corey, E. J.; Grogan, M. J. Enantioselective synthesis of alpha-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Leow, D.; Tan, C. H. Chiral guanidine catalyzed enantioselective reactions. Chem Asian J. 2009, 4, 488–507. [Google Scholar] [CrossRef]
- Birman, V. B.; Li, X. Benzotetramisole: a remarkably enantioselective acyl transfer catalyst. Org Lett. 2006, 8, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Straub, M. R.; Birman, V. B. Organocatalytic enantioselective synthesis of α-fluoro-β-amino acid derivatives. Org Lett. 2018, 20, 7550–7553. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yu, Y.; Liu, R.; Zi, W. Design, preparation, and implementation of axially chiral benzotetramisoles as Lewis base catalysts for asymmetric cycloadditions. Angew. Chem. Int. Ed. 2024, 63, 1–8. [Google Scholar] [CrossRef]
- Abbasov, M. E.; Hudson, B. M.; Tantillo, D. J.; Romo, D. Acylammonium salts as dienophiles in Diels−Alder/lactonization organocascades. J. Am. Chem. Soc. 2014, 136, 4492–4495. [Google Scholar] [CrossRef]
- Cheng, Y. A.; Yu, W. Z.; Yeung, Y. Y. Carbamate-catalyzed enantioselective bromolactamization. Angew. Chem. Int. Ed. 2015, 54, 12102–12106. [Google Scholar] [CrossRef]
- Moreira Ribeiro, F. W.; Matsushima, J. E.; Obeid, G.; Vinhato, E.; Rodrigues, A.; Correra, T. C. Mechanistic Investigation of an organocatalytic bromocyclization of O-allyl carbamates. ChemCatChem 2025, 17, 1–10. [Google Scholar] [CrossRef]
- Berkessel, A. The discovery of catalytically active peptides through combinatorial chemistry. Curr. Opin. Chem. Biol, 1016. [Google Scholar]
- Revell, J. D.; Wennemers, H. Peptidic catalyst developed by combinatorial screening methods. Curr. Opin. Chem. Biol. 2007, 11, 269–278. [Google Scholar] [CrossRef]
- Shu, K.; Kodadek, T. Solid-phase synthesis of β-hydroxy ketones via DNA-compatible organocatalytic aldol reactions. ACS Comb. Sci. 2018, 20, 277–281. [Google Scholar] [CrossRef]
- Miller, S. J. In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004, 37, 601–610. [Google Scholar] [CrossRef]
- Davie, E. A.; Mennen, S. M.; Xu, Y.; Miller, S. J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Commun. 2011, 47, 12036–12041. [Google Scholar] [CrossRef]
- Metrano, A. J.; Chinn, A. J.; Shugrue, C. R.; Stone, E. A.; Kim, B.; Miller, S. J. Asymmetric catalysis mediated by synthetic peptides, version 2.0: Expansion of scope and mechanisms. Chem. Rev. 2020, 120, 11479–11615. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K. ; Asymmetric peptide catalysis. In Catalytic Asymmetric Synthesis, 4th edition, John Wiley & Sons., Inc. 2022, Chap. 5, pp. [CrossRef]
- He, T.; Valagussa, B.; Boanoni, E.; Gentilucci, L. Expedient recycling of peptide organocatalysts using nanocrystalline hydroxyapatite catching systems. Sust. Chem. Pharm. 2024, 37, 157–198. [Google Scholar] [CrossRef]
- Halder, M.; Chawla, V.; Singh, Y. Ceria nanoparticles immobilized with self-assembling peptide for biocatalytic applications. Nanoscale 2024, 16, 16887–16899. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Ohashi, S.; Inoue, S. Asymmetric synthesis catalyzed by poly(5-benzyl L-glutamate). Makromol. Chem. 1975, 176, 2751–2753. [Google Scholar] [CrossRef]
- Fukushima, H.; Inoue, S. Asymmetric synthesis catalyzed by poly(4-benzyl L-aspartate). Makromol. Chem. 1975, 176, 3609–3611. [Google Scholar] [CrossRef]
- Ueyanagi, K.; Inoue, S. Asymmetric synthesis catalyzed by poly-L-alanine. Makromol. Chem. 1976, 177, 2807–2817. [Google Scholar] [CrossRef]
- Carrea, G.; Colonna, S.; Kelly, D. R.; Lazcano, A:, Ottolina, G-; Roberts, S. M. Polyamino acids as synthetic enzymes: mechanism, applications and relevance to prebiotic catalysis. Trends Biotechnol. 2005, 23, 507–513. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Samanta, M.; Ashkenasy, C.; Leman, L. J. Prebiotic peptides: Molecular hubs in the origin of life. Chem. Rev. 2020, 120, 4707–4765. [Google Scholar] [CrossRef]
- Wei, W.; Chu, F.; Chen, G.; Zhou, S.; Sun, C.; Feng, H.; Pan, Y. Prebiotic formation of peptides through bubbling and arc plasma. Chem. Eur. J. 2024, 30, 1–8. [Google Scholar] [CrossRef]
- Zhang, L.; Ying, J. Amino acid analogues provide multiple plausible pathways to prebiotic peptides. J. R. Soc. Interface, 2024. [Google Scholar] [CrossRef]
- Hlouchová, K. Peptides en route from prebiotic to biotic catalysis. Acc. Chem. Res. 2024, 57, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Zozulia, O.; Dolan, M. A.; Korendovych, I. V. Catalytic peptide assemblies. Chem. Soc. Rev. 2018, 47, 3621–3639. [Google Scholar] [CrossRef] [PubMed]
- Girvin, Z. C.; Gellman, S. H. Foldamer catalysis. J. Am. Chem. Soc. 2020, 142, 17211–17223. [Google Scholar] [CrossRef] [PubMed]
- Brown, R. O.; Demapan, D.; Cui, Q. Complete computational reaction mechanism for foldamer-catalyzed aldol condensation. ACS Catal. 2024, 14, 7624–7638. [Google Scholar] [CrossRef]
- Khettar, I.; Araszczuk, A.M.; Schettini, R. Peptidomimetic-based asymmetric catalysts. Catalysts 2023, 13, article. [Google Scholar] [CrossRef]
- Oku, J.-i.; Ito, N.; Inoue, S. Asymmetic cyanohydrin synthesis catalyzed by synthetic dipeptides, 1. Makromol. Chem. 1979, 180, 1089–1091. [Google Scholar] [CrossRef]
- Inoue, W.; Oku, J.-i. Asymmetric cyanohydrin synthesis catalysed by a synthetic cyclic dipeptide. Chem. Commun. 1981, 1981(5), 229–230. [Google Scholar] [CrossRef]
- Ranaky, K.; Mori, A.; Inoue, S. The cyclic dipeptide cyclo[(S)-phenylalanyl-(S)-histidyl] as a catalyst for asymmetric addition of hydrogen cyanide to aldehydes. J. Org. Chem. 1990, 55, 181–185. [Google Scholar] [CrossRef]
- Shvo, Y.; Gal, M.; Becker, Y.; Elgavi, A. Asymmetric hydrocyanation of aldehydes with cyclo-dipeptides: A new mechanistic approach. Tetrahedron Asymmetry 1996, 7, 911–924. [Google Scholar] [CrossRef]
- Iyer, M.; Gigstad, K. M.; Namdev, N. D.; Lipton, M. Asymmetric catalysis of the Strecker amino acid synthesis by a cyclic dipeptide. Amino Acids, 1996; 259–268. [Google Scholar] [CrossRef]
- Sigman, M. S.; Jacobsen, E. N. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Sigman, M. S.; Vachal, P.; Jacobsen, E. N. Angew. Chem. Int. Ed. 2000, 39, 1279–1281. [CrossRef]
- Vachal, P.; Jacobsen, E. N. Structure-based analysis and optimization of a highly enantioselective catalyst for the Strecker reaction. J. Am. Chem. Soc. 2002, 124, 10012–10014. [Google Scholar] [CrossRef]
- Zuend, S. J.; Coughlin, M. P.; Lalonde, M. P.; Jacobsen, E. N. Nature, 2009; 461, 968–971. [CrossRef]
- Sadhukhan, A.; Sahu, D.; Ganguly, B.; Khan, N. U.; Kureshy, R. I.; Abdi, S. H.; Suresh, E.; Bajaj, H. C. Oxazoline-based organocatalyst for enantioselective Strecker reactions: a protocol for the synthesis of levamisole. Chemi. Eur. J. 2013, 19, 14224–14232. [Google Scholar] [CrossRef]
- Juliá, S.; Masana, J.; Vega, J. "Synthetic Enzymes". Highly stereoselective epoxidation of chalcone in a triphasic toluene/water-poly[(S)-alanine system. Angew. Chem. Int. Ed. 1980, 19, 929–931. [Google Scholar] [CrossRef]
- Colonna, S.; Molinari, H.; Banfi, S.; Juliá, S.; Masana, J.; Alvarez, A. Synthetic enzymes—4: Highly enantioselective epoxidation by means of polyaminoacids in a triphase system: Influence of structural variations within the catalysts. Tetrahedron 1983, 39, 1635–1641. [Google Scholar] [CrossRef]
- Allen, J. V.; Bergeron, S.; Griffiths, M. J.; Mukherjee, S.; Roberts, S. M.; Williamson, N. M.; Wu, L. E. Juliá–Colonna asymmetric epoxidation reactions under non-aqueous conditions: rapid, highly regio- and stereo-selective transformations using a cheap, recyclable catalyst. J. Chem. Soc., Perkin Trans. 1, 1998; 3173–3180. [Google Scholar] [CrossRef]
- Flood, R. W.; Geller, T. P.; Petty, S. A.; Roberts, S. M.; Skidmore, J.; Volk, M. Efficient asymmetric epoxidation of α,β-unsaturated ketones using a soluble triblock polyethylene glycol−polyamino acid catalyst. Org. Lett. 2001, 3, 683–686. [Google Scholar] [CrossRef]
- Geller, T.; Gerlach, A.; Krüger, C. M.; Militzer, H.-C. The Juia-Colonna epoxidation: Access to chiral, non-racemic epoxides. J. Mol. Catal. A; Chem. 2006, 252, 71–77. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K. B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Guo, H.-C.; Shi, X.-Y.; Wang, X.; Liu, S.-Z.; Wang, M. Liquid-phase synthesis of chiral tartrate ligand library for enantioselective Sharpless epoxidation of allylic alcohols. J. Org. Chem. 2004, 69, 2042–2047. [Google Scholar] [CrossRef]
- Bach, R. D.; Schlegel, H. B. Mechanism of the Sharpless epoxidation reaction: A DFT study. J. Phys. Chem. A 2024, 128, 2072–2091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Loebach, J. L.; Wilson, S. R.; Jacobsen, E. N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–280. [Google Scholar] [CrossRef]
- Jacobsen, E. N.; Zhang, W.; Muci, A. R.; Ecker, J. R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins using chiral (salen)manganese(III) complexes. Tetrahedron: Asymmetry 1991, 2, 481–494. [Google Scholar] [CrossRef]
- Berkessel, A.; Gasch, N.; Glaubitz, K.; Koch, C. Highly enantioselective enone epoxidation catalyzed by short solid phase-bound peptides: Dominant role of peptide helicity. Org. Lett. 2001, 3, 3839–3842. [Google Scholar] [CrossRef]
- Bentley, P. A.; Cappi, M. W.; Flood, R. W.; Roberts, S. M.; Smith, J. A. Towards a mechanistic insight into the Julia-Colonna asymmetric epoxidation of α,β-unsaturated ketones using discrete lengths of poly-leucine. Tetrahedron Lett. 1998, 39, 9297–9300. [Google Scholar] [CrossRef]
- Itsuno, S.; Sakakura, M.; Ito, K. Polymer-supported poly(amino acids) as new asymmetric epoxidation catalyst of alpha,beta.-unsaturated ketones. J. Org. Chem. 1990, 55, 6047–6049. [Google Scholar] [CrossRef]
- Peris, G.; Jakobsche, C. E.; Miller, S. J. Aspartate-catalyzed asymmetric epoxidation reactions. J. Am. Chem. Soc. 2007, 129, 8710–8711. [Google Scholar] [CrossRef]
- Peris, G.; Miller, S. J. A nonenzymatic acid/peracid catalytic cycle for the Baeyer−Villiger oxidation. Org. Lett. 2008, 10, 3049–3052. [Google Scholar] [CrossRef]
- Huth, S. E.; Stone, E.; Miller, S. J. Selective oxidation of disparate functional groups mediated by a common aspartic acid-based peptide catalyst platform. Acc. Chem. Res. 2025. [Google Scholar] [CrossRef]
- Kofoed, J.; Nielsen, J.; Reymond, J.-L. Discovery of new peptide-based catalysts for the direct asymmetric aldol reaction. Bioorg. Med. Chem. Lett. 2003, 12, 2445–2447. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Jiang, F.; Yu, L.-T; Cui, X.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z.; Wu, Y.-D. Novel small organic molecules for a highly enantioselective direct aldol reaction. J. Am. Chem.Soc. 2003, 125, 5262–5263. [Google Scholar] [CrossRef] [PubMed]
- Martin, H. J.; List, B. Mining sequence space for asymmetric aminocatalysis: N-Terminal prolyl-peptides efficiently catalyze enantioselective aldol and Michael reactions. Synlett 2003, 1901–1902. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, Z.-H.; Cui, L.-F.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z. Small peptides catalyze highly enantioselective direct aldol reactions of aldehydes with hydroxyacetone: Unprecedented regiocontrol in aqueous media. Org. Lett. 2004, 6, 2285–2287. [Google Scholar] [CrossRef]
- Kofoed, J.; Darbre, T.; Reymond, J.-L. Artificial aldolases from peptide dendrimer combinatorial libraries. Org. Biomol. Chem. 2006, 4, 3268–328. [Google Scholar] [CrossRef]
- Horstmann, T. E.; Guerin, D. J.; Miller, S. J. Asymmetric conjugate addition of azide to alpha,beta-unsaturated carbonyl compounds catalyzed by simple peptides. Angew. Chem. Int. Ed. 2000, 39, 3635–3638. [Google Scholar] [CrossRef]
- Xu, Y.; Zou, W.; Sundén, H.; Ibrahem, I.; Córdova, A. Small peptide-catalyzed enantioselective addition of ketones to nitroolefins. Adv. Synth. Catal. 2006, 348, 418–424. [Google Scholar] [CrossRef]
- Tsogoeva, S. B.; Jagtap, S. B.; Ardemasova, Z. A. 4-trans-Amino-proline based di- and tetrapeptides as organic catalysts for asymmetric C–C bond formation reactions. Tetrahedron Asymmetry 2006, 17, 989–992. [Google Scholar] [CrossRef]
- Wiesner, M.; Revell, J. D.; Wennemers, H. Tripeptides as efficient asymmetric catalysts for 1,4-addition. Reactions of aldehydes to nitroolefins–A rational approach. Angew. Chem. Int. Ed. 2008, 47, 1871–1874. [Google Scholar] [CrossRef]
- Imbriglio, J. E.; Vasbinder, M. M.; Miller, S. J. Dual catalyst control in the amino acid-peptide-catalyzed enantioselective Baylis-Hillman reaction. Org Lett. 2003, 5, 3741–3743. [Google Scholar] [CrossRef] [PubMed]
- Cowen, B. J.; Saunders, L. B.; Miller, S. J. Pyridylalanine (pal)-peptide catalyzed enantioselective allenoate additions to N-acyl imines. J. Am. Chem. Soc. 2009, 131, 6105–6107. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L. B.; Cowen, B. J.; Miller, S. J. Pyridylalanine (Pal)-peptide catalyzed enantioselective allenoate additions to N-acyl imines proceed via an atypical “aza-Morita-Baylis-Hillman” mechanism. Org. Lett. 2010, 12, 4800–4803. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, K.; Ito, R.; Arai, T.; Yanagisawa, A. Catalytic asymmetric protonation of enolates using amino acid derivatives as chiral proton sources. Org. Lett. 2006, 8, 1721–1724. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Y. C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef]
- Xie, C.; Smaligo, A. J.; Song, X. R.; Kwon, O. Phosphorus-based catalysis. ACS Cent. Sci. 2021, 7(4), 536–558. [Google Scholar] [CrossRef]
- Methot, J. L.; Roush, W. R. Nucleophilic phosphine organocatalysis. Adv. Synth. Catal. 2004, 346, 1035–1050. [Google Scholar] [CrossRef]
- Netherton, M. R.; Fu, G. C. Air-stable trialkylphosphonium salts: Simple, practical, and versatile replacements for air-sensitive trialkylphosphines. Applications in stoichiometric and catalytic processes. Org. Lett. 2001, 3, 4295–4298. [Google Scholar] [CrossRef]
- Takashina, N.; Price, C. C. A Crystalline hexamer from acrylonitrile. J. Am. Chem. Soc. 1962, 84, 489–491. [Google Scholar] [CrossRef]
- Rauhut, M. M.; Currier, H. Dialkyl 2-methyleneglutarates. US. Patent 3 074 999, 1958. Chem. Abstr. 1963, 58, 66109. [Google Scholar]
- Tang, W.; Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 2003, 103, 3029–3070. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M. M.; Calvete, M. J. F.; Carrilho, R. M. B.; Abreu, A. R. Synthesis of binaphthyl based phosphine and phosphite ligands. Chem. Soc. Rev. 2013, 42, 6990–7027. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Chen, W-L. ; Lu, Y. Phosphine-catalyzed asymmetric organic reactions. Chem. Rev. 2018, 118, 9344–9411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, X. Phosphine-catalyzed cycloaddition of 2,3-butadienoates or 2-butynoates with electron-deficient olefins. A novel [3 + 2] annulation approach to cyclopentenes. J. Org. Chem. 1995, 60, 2906–2908. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, Z.; Jiang, Q.; Xiao, D.; Cao, P.; Zhang, X. Asymmetric [3 + 2] cycloaddition of 2,3-butadienoates with electron-deficient olefins catalyzed by novel chiral 2,5-dialkyl-7-phenyl-7-phosphabicyclo[2.2.1]heptanes. J. Am. Chem. Soc. 1997, 119, 3836–3837. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, G.; Jiang, Q.; Xiao, D.; Cao, P.; Zhang, X. Asymmetric formation of quaternary carbon centers catalyzed novel chiral 2,5-dialkyl-7-phenyl-7-phosphabicyclo[2.2.1]heptanes. J. Org. Chem. 1998, 63. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, M. Applications of chiral phosphine-based catalysts in catalytic asymmetric synthesis. Chem. Asian J. 2014, 9, 2720–2734. [Google Scholar] [CrossRef]
- Ni, H.; Tang, X.; Zheng, W.; Yao, W.; Ullah, N.; Lu, Y. Enantioselective phosphine-catalyzed formal [4 + 4] annulation of α,β-unsaturated imines and allene ketones: Construction of eight-membered rings. Angew. Chem. Int. Ed. 2017, 56, 14222–14226. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, Z.; Zhou, L.; Lv, X. Recent advances in [4 + 4] annulation of conjugated heterodienes with 1,4-dipolar species for the synthesis of eight-membered heterocycles. Org. Biomol. Chem. 2024, 22, 252–268. [Google Scholar] [CrossRef]
- Wang, X.; Yu, C.; Atodiresei, I. L.; Patureau, F. W. Phosphine-catalyzed dearomative [3 + 2] cycloaddition of benzoxazoles with a cyclopropenone. Org Lett. 2022, 24, 1127–1131. [Google Scholar] [CrossRef]
- Zhang, S-S. ; Wang, R-X.; Gu, Q.; You, S-L. Phosphine-catalyzed asymmetric dearomative [3+2] annulation reaction of benzimidazoles with cyclopropenones CCS Chem. 2025, 7, 194–204. [CrossRef]
- Jana, K.; Sparr, C. Enantioselective Wittig reactions controlled by PIII/PV=O redox catalysis. Synlett 2025, 1130–1134. [Google Scholar] [CrossRef]
- Werner, T.; Hoffmann, M.; Deshmukh, S. First enantioselective catalytic Wittig reaction. Eur. J. Org. Chem. 2014, 6630–6633. [Google Scholar] [CrossRef]
- Schneider, L. M.; Schmiedel, V. M.; Pecchioli, T.; Lentz, D.; Merten, C.; Christmann, M. Asymmetric synthesis of carbocyclic propellanes. Org. Lett. 2017, 19, 2310–2313. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, V. M.; Hong, Y. J.; Lentz, D.; Tantillo, D. J.; Christmann, M. Synthesis and structure revision of dichrocephones A and B. Angew. Chem. Int. Ed. 2018, 57, 2419–2422. [Google Scholar] [CrossRef]
- Lorton C; Castanheiro T; Voituriez A Catalytic and asymmetric process via PIII/PV=O redox cycling: Access to (trifluoromethyl)cyclobutenes via a Michael addition/Wittig olefination reaction. J. Am. Chem. Soc. 2019, 141, 10142–10147. [CrossRef]
- Wilson, S. R.; Pasternak, A. Preparation of a new class of C2-symmetric chiral phosphines: The first asymmetric Staudinger reaction. Synlett 1990, 199–200. [Google Scholar] [CrossRef]
- Lertpibulpanya, D.; Marsden, S. P.; Rodriguez-García, I.; Kilner, C. A. Asymmetric aza-Wittig reaction: Enantioselective synthesis of β-quaternary azacycles. Angew. Chem. Int. Ed. 2006, 45, 5000–5002. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, K.; Chen, S.; Lepage, R. J.; Houk, K. N.; Krenske, E.; Kwon, O. Catalytic asymmetric Staudinger-aza-Wittig reaction for the synthesis of heterocyclic amines. J. Am. Chem. Soc. 2019, 141, 9537–9542. [Google Scholar] [CrossRef]
- Xie, C.; Kim, J.; Mai, B, K. ; Cao, S.; Ye, R.; Wang, X. Y.; Liu, P.; Kwon, O. Enantioselective synthesis of quaternary oxindoles: Desymmetrizing Staudinger-aza-Wittig reaction enabled by a bespoke HypPhos oxide catalyst. J. Am. Chem. Soc. 2022, 144, 21318–21327. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.; Zhang, S.; Xue, Z.; Hu, S.; Chen, Y.; Tang, Y. Chiral bisphosphine-catalyzed asymmetric Staudinger/aza-Wittig reaction: An enantioselective desymmetrizing approach to Crinine-type Amaryllidaceae alkaloids. J. Am. Chem. Soc. 2024, 146, 14136–14148. [Google Scholar] [CrossRef]
- Manabe, K. Synthesis of novel chiral quaternary phosphonium salts with a multiple hydrogen-bonding site, and their application to asymmetric phase-transfer alkylation. Tetrahedron 1998, 54, 14465–14476. [Google Scholar] [CrossRef]
- Chen, L.; Deng, Y.; Li. T.; Hu, D.; Ren, X.; Wang, T. Asymmetric nucleophilic additions promoted by quaternary phosphonium ion-pair catalysts. CCS Chem. 2024, 6, 2110–2130. [Google Scholar] [CrossRef]
- Uraguchi, D.; Sakaki, S.; Ooi, T. Chiral tetraaminophosphonium salt-mediated asymmetric direct Henry reaction. J. Am. Chem. Soc. 2007, 129, 12392–12393. [Google Scholar] [CrossRef] [PubMed]
- He, R. J.; Ding, C. H.; Maruoka, K. Phosphonium salts as chiral phase-transfer catalysts: Asymmetric Michael and Mannich reactions of 3-aryloxindoles. Angew. Chem. Int. Ed. 2009, 48, 4559–4561. [Google Scholar] [CrossRef]
- Cao, D. D.; Chai, Z.; Zhang, J. X.; Ye, Z. Q.; Xiao, H.; Wang, H. Y.; Chen, J. H.; Wu, X. Y.; Zhao, G. Thiourea-phosphonium salts from amino acids: Cooperative phase-transfer catalysts in the enantioselective aza-Henry reaction. Chem. Commun. 2013, 49, 5972–5794. [Google Scholar] [CrossRef]
- Liu, S.; Kumatabara, Y.; Shirakawa, S. Chiral quaternary phosphonium salts as phase-transfer catalysts for environmentally benign asymmetric transformations. Green Chem. 2016, 18, 331–341. [Google Scholar] [CrossRef]
- Wang, T.; Xu, Z. Chiral quaternary phosphonium salt phase transfer catalyst, preparation method and application thereof. CN109718851A, May 7, 2019.
- Denmark, S. E.; Fu, J. Catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 2003, 103, 2763–2794. [Google Scholar] [CrossRef]
- Denmark, S. E.; Coe, D. M.; Pratt, N. E.; Griedel, B. D. Asymmetric allylation of aldehydes with chiral Lewis bases. J. Org. Chem. 1994, 59, 6161–6163. [Google Scholar] [CrossRef]
- Denmark, S, E. ; Fu, J. Catalytic, enantioselective addition of substituted allylic trichlorosilanes using a rationally-designed 2,2'-bispyrrolidine-based bisphosphoramide. J. Am. Chem. Soc. 2001, 123, 9488–9489. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S. E.; Fu, J.; Lawler, M. J. Chiral phosphoramide-catalyzed enantioselective addition of allylic trichlorosilanes to aldehydes. Preparative studies with bidentate phosphorus-based amides. J. Org. Chem. 2006, 71, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S. E.; Beutner, G. L. Lewis base catalysis in organic chemistry. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef]
- Denmark, S. E.; Stavenger, R. A.; Wong, K-T. ; Su, X. Chiral phosphoramide-catalyzed aldol additions of ketone enolates. Preparative aspects. J. Am. Chem. Soc. 1999, 121, 4982–4991. [Google Scholar] [CrossRef]
- Denmark, S. E.; Beutner, G. L.; Wynn, T.; Eastgate, M. D. Lewis base activation of Lewis acids: catalytic, enantioselective addition of silyl ketene acetals to aldehydes. J. Am. Chem. Soc. 2005, 127, 3774–3789. [Google Scholar] [CrossRef]
- Denmark, S. E.; Heemstra, J. R. Jr.; Beutner, G. L. Catalytic, enantioselective, vinylogous aldol reactions. Angew. Chem. Int. Ed. 2005, 44, 4682–4698. [Google Scholar] [CrossRef]
- Denmark, S. E.; Barsanti, P. A.; Beutner, G. L.; Wilson, T. W. Enantioselective ring opening of epoxides with silicon tetrachloride in the presence of a chiral Lewis base: Mechanism studies. Adv. Synth. Catal. 2007, 349, 567–582. [Google Scholar] [CrossRef]
- Denmark, S. E.; Fan, Y. The first catalytic, asymmetric alpha-additions of isocyanides. Lewis-base-catalyzed, enantioselective Passerini-type reactions. J. Am. Chem. Soc. 2003, 125, 7825–7827. [Google Scholar] [CrossRef]
- Ogawa, C.; Sugiura, M.; Kobayashi, S. Stereospecific, enantioselective allylation of alpha-hydrazono esters by using allyltrichlorosilanes with BINAP dioxides as neutral-coordinate organocatalysts. Angew. Chem. Int. Ed. 2004, 43, 6491–6493. [Google Scholar] [CrossRef]
- Nakajima, M.; Kotani, S.; Ishizuka, T.; Hashimoto, S. Chiral phosphine oxide BINAPO as a catalyst for enantioselective allylation of aldehydes with allyltrichlorosilanes. Tetrahedron Lett. 2005, 46, 157–159. [Google Scholar] [CrossRef]
- Kotani, S.; Shimoda, Y.; Sugiura, M. ; Novel enantioselective direct aldol-type reaction promoted by a chiral phosphine oxide as an organocatalyst. Tetrahedron Lett. 2009, 50, 4602–4605. [Google Scholar] [CrossRef]
- Benaglia, M.; Rossi, S. Chiral phosphine oxides in present-day organocatalysis. Org. Biomol. Chem. 2010, 8, 3824–3830. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Nakajima, M. Recent advances in asymmetric phosphine oxide catalysis. Tetrahedron Lett. 2020, 61, 1–8. [Google Scholar] [CrossRef]
- Rossi, S.; Benincori, T.; Raimondi, L.; Benaglia, M. 3,3′-Bithiophene-based chiral bisphosphine oxides as organocatalysts in silicon-derived Lewis acid mediated reactions. Synlett 2020, 31, 535–546. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, J.; Zhang, J.; Ma, X. Direct covalent immobilizaion of chiral phosphine oxide: Hollow mesoporous polystyrene nanospheres as a platform for efficient heterogeneous enantioselective reductive aldol reaction. Mol. Catal. 2023, 547, 1–7. [Google Scholar] [CrossRef]
- Dogan, O.; Bulut, A.; Recimer, M. Chiral phosphine oxide aziridinyl phosphonate as a Lewis base catalyst for enantioselective allylsilane addition to aldehydes. Tetrahedron Asymmetry 2015, 26, 966–969. [Google Scholar] [CrossRef]
- a) Johnson, A. W.; LaCount, R. B. 9-Dimethylsulfonium fluorenylide. Chem. Ind. (London) 1958, 1440−1441; b) Johnson, A. W.; LaCount, R. B. The chemistry of ylids. VI. Dimethylsulfonium fliorenylide-A synthesis of epoxides. J. Am. Chem. Soc, 1961; 83, 417–423. [Google Scholar] [CrossRef]
- Corey, E. J.; Chaykovsky, M. J. Dimethyloxosulfonium methylide ((CH3)2SOCH2) and dimethylsulfonium methylide ((CH3)2SCH2). Formation and application to organic synthesis. J. Am. Chem. Soc. 1965, 87. [Google Scholar] [CrossRef]
- Furukawa, N.; Sugihara, Y.; Fujihara, H. Camphoryl sulfide as a chiral auxiliary and a mediator for one-step synthesis of optically active 1,2-diaryloxiranes. J. Org. Chem. 1989, 54, 4222–4224. [Google Scholar] [CrossRef]
- Julienne, K.; Metzner, P.; Henyron, V. Asymmetric synthesis of epoxides from aldehydes mediated by (+)-(2R,5R)-2,5-dimethylthiolane. J. Chem. Soc., Perkin Trans. 1999; 731–735. [Google Scholar] [CrossRef]
- Winn, C. L.; Bellanie, B.; Goodman, J. M. A highly enantioselective one-pot sulfur ylide epoxidation reaction. Tetrahedron Lett. 2002, 43, 5427–5430. [Google Scholar] [CrossRef]
- Aggarwal, V. K.; Winn, C. L. Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: Scope, selectivity, and applications in synthesis. Acc. Chem. Res. 2004, 37, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S. E.; Kuester, W. E.; Burk, M. T. Catalytic, asymmetric halofunctionalization of alkenes: A critical perspective. Angew. Chem. Int. Ed. 2012, 51, 10938–10953. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. ; Zhang, B-Q.; Xiao, W-Y.; Li, Y-L.; Deng, J. Recent advances in catalytic asymmetric syntheses of functionalized heterocycles via halogenation/chalcogenation of carbon-carbon unsaturated bonds. Adv. Synth. Catal, 2022; 364, 3974–4005. [Google Scholar] [CrossRef]
- Furuya, Y.; Nishiyori, R.; Shirakawa, S. Synthesis of a BINOL-derived sulfide catalyst bearing a diphenylmethanol unit and its application in asymmetric bromolactonizations. Asian J. Org. Chem. 2025, 14, 1–5. [Google Scholar] [CrossRef]
- Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X. Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane-based chiral sulfide. Angew. Chem. Int. Ed. 2016, 55, 5846–5850. [Google Scholar] [CrossRef] [PubMed]
- Benz, S.; López-Andarias. J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds. Angew. Chem. Int. Ed. 2017, 56, 812–815. [Google Scholar] [CrossRef]
- Sekar, G.; Nair, V. V.; Zhu, J. Chalcogen bonding catalysis. Chem. Soc. Rev. 2024, 53, 586–605. [Google Scholar] [CrossRef]
- Gao, G.; Xie, D.; Zhou, P-P. Chalcogen bonding catalysis in irganic synthesis. Asian J. Org. Chem. 2025, 14, 1–14. [Google Scholar] [CrossRef]
- Haque, B.; Roberts, B. P.; Tocher, D. A. Enantioselective radical-chain hydrosilylation of alkenes using homochiral thiols as polarity-reversal catalysts. J. Chem. Soc., Perkin Trans. 1, 1998; 2881–2890. [Google Scholar]
- Cai, Y.; Roberts, B. P.; Tocher, D. A. Carbohydrate-derived thiols as protic polarity-reversal catalysts for enantioselective radical-chain reactions. J. Chem. Soc., Perkin Trans. 1, 2002; 1376–1386. [Google Scholar] [CrossRef]
- Aroyan, C. E.; Miller, S. J. Enantioselective Rauhut-Currier reactions promoted by protected cysteine. J. Am. Chem. Soc. 2007, 129, 256–257. [Google Scholar] [CrossRef]
- Liu, S.; Maruoka, K.; Shirakawa, S. Chiral tertiary sulfonium salts as effective catalysts for asymmetric base-free neutral phase-transfer reactions. Angew. Chem. Int. Ed. 2017, 56, 4819–4823. [Google Scholar] [CrossRef]
- Trost, B. M.; Rao, M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem. Int. Ed. 2015, 54, 5026–5043. [Google Scholar] [CrossRef]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral organosulfur ligands/catalysts with a stereogenic sulfur atom: Applications in asymmetric synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef]
- Wang, C-R. ; Sun, J-N.; Li. Y.; Li, J-H. Recent advances in catalytic asymmetric synthesis of chiral sulfinyl compounds. Eur. J. Org. Chem. 2025, 28, 1–13. [Google Scholar] [CrossRef]
- Wujkowska, Z. , Leśniak, S., Kiełbasiński, P., Rachwalski, M. Highly enantioselective asymmetric reduction of aromatic ketimines promoted by chiral enantiomerically pure sulfoxides as organocatalysts. J. Sulfur Chem. 2018, 39, 380–387. [Google Scholar] [CrossRef]
- Zhou, L.; Tan, C, K. ; Jiang, X.; Chen, F.; Yeung, Y. Y. Asymmetric bromolactonization using amino-thiocarbamate catalyst. J. Am. Chem. Soc. 2010, 132, 15474–15476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Tay, D. W.; Chen, J.; Leung, G. Y. C.; Yeung, Y.-Y. Enantioselective synthesis of 2-substituted and 3-substituted piperidines through a bromoaminocyclization process. Chem. Commun. 2013, 49, 4412–4414. [Google Scholar] [CrossRef]
- Dinér, P.; Sadhukhan, A.; Blomkvist, B. Chiral sulfinamides as highly enantioselective organocatalysts. ChemCatChem 2014, 6, 3063–3066. [Google Scholar] [CrossRef]
- Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. [CrossRef]
- Pei, D.; Wang, Z.; Wei, S.; Zhang, Y.; Sun, J. S-chiral sulfinamides as highly enantioselective organocatalysts. Org. Lett. 2006, 8, 5913–5915. [Google Scholar] [CrossRef]
- Pei, D.; Zhang, Y.; Wei, S.; Wang, M.; Sun, J. Rationally-designed S-chiral bissulfinamides as highly enantioselective organocatalysts for reduction of ketimines. Adv. Synth. Catal. 2008, 350, 619–623. [Google Scholar] [CrossRef]
- Kumar, M.; Kureshy, R. I.; Saravanan, S.; Verma, S.; Jakhar, A.; Khan, N. H.; Abdi, S. H. R.; Bajaj, H. C. Org. Lett. 2014, 16. [CrossRef]
- Denmark, S. E.; Kalyani, D.; Collins, W. R. Preparative and mechanistic studies toward the rational development of catalytic, enantioselective selenoetherification reactions. J. Am. Chem. Soc. 2010, 132, 15752–15765. [Google Scholar] [CrossRef] [PubMed]
- Nishiyori, R.; Maynard, J. R. J.; Shirakawa, S. ; Chiral bifunctional selenide catalysts for asymmetric bromolactonization. Asian J. Org. Chem. 2020, 9, 192–196. [Google Scholar] [CrossRef]
- Stadel, J. T.; Back, T. G. Asymmetric synthesis with organoselenium compounds – The past twelve years. Chem. Eur. J. 2024, 30, 2202304074. [Google Scholar] [CrossRef] [PubMed]
- Jian, Y.; Singh, T.; Andersson, P. G.; Zhou, T. Asymmetric synthesis and applications of chiral organoselenium compounds: A review. Molecules 2024, 29, 1–37. [Google Scholar] [CrossRef]
- Wirth, T.; Häupli, S.; Leuenberger, M. Catalytic asymmetric oxyselenenylation–elimination reactions using chiral selenium compounds. Tetrahedron Asymmetry 1998, 9, 547–558. [Google Scholar] [CrossRef]
- Santi, C.; Di Lorenzo, R.; Tidei, C.; Bagnoli, L.; Wirth, T. Stereoselective selenium catalyzed dihydroxylation and hydroxymethoxylation of alkenes. Tetrahedron 2012, 68, 10530–10535. [Google Scholar] [CrossRef]
- Braga, A. L.; Paixão, M. W.; Lüdtke, D. S.; Silveira, C. C.; Rodrigues, O. E. D. Synthesis of new chiral aliphatic amino diselenides and their application as catalysts for the enantioselective addition of diethylzinc to aldehydes. Org. Lett. 2003, 5, 2635–2638. [Google Scholar] [CrossRef]
- Braga, A. L.; Lüdtkr, D. S.; Vargas, F.; Braga, R. C. Catalytic applications of chiral organoselenium compounds in asymmetric synthesis. Synlett, 1453. [Google Scholar]
- Okuno, K.; Nakamura, T.; Shirakawa, S. ; Asymmetric catalysis of chiral bifunctional selenides and selenonium salts bearing a urea group. Asian J. Org. Chem. 2021, 10, 655–659. [Google Scholar] [CrossRef]
- Gilbert, B. B.; Eey, S. T.-C.; Ryabchuk, P.; Garry, O.; Denmark, S. E. catalyzed enantioselective syn-dichlorination of unbiased alkenes. Tetrahedron, 2019; 75, 4089–4098. [Google Scholar] [CrossRef]
- 609. Denmark, S. E.; Kornfilt, D. J. P.; Vogler, T. Catalytic asymmetric thiofunctionalization of unactivated alkenes. J. Am. Chem. Soc. 2011, 135, 15308–15311. [Google Scholar] [CrossRef]
- Matviitsuk, A.; Panger, J. L.; Denmark, S. E. Catalytic, enantioselective sulfenofunctionalization of alkenes: Development and recent advances. Angew. Chem. Int.. Ed. 2020, 59, 19796–19819. [Google Scholar] [CrossRef]
- Matviitsuk, A.; Lee Panger, J.; Denmark, S. E. Enantioselective inter- and intramolecular sulfenofunctionalization of unactivated cyclic and (Z)-alkenes. ACS Catal. 2022, 12, 7377–7385. [Google Scholar] [CrossRef]
- Chen, F.; Tan, C. K.; Yeung, Y. Y. C2-symmetric cyclic selenium-catalyzed enantioselective bromoaminocyclization. J. Am. Chem. Soc. 2013, 135, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, Y.; Zhao, X. Chiral selenide-catalyzed enantioselective construction of saturated trifluoromethylthiolated azaheterocycles. Org. Lett. 2017, 19, 3434–3437. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Jiang, Q.; Ji, J.; Luo, J.; Zhao, X. Chiral selenide-catalyzed enantioselective construction of saturated trifluoromethylthiolated azaheterocycles. Org. Biomol. Chem. 2019, 17, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, X. Enantioselective construction of chiral sulfides via catalytic electrophilic azidothiolation and oxythiolation of N-allyl sulfonamides. ACS Catal. 2019, 9, 6896–6902. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Tse, Y. S.; Ke, Z.; Yeung, Y. Y. Applications of selenonium cations as Lewis acids in organocatalytic reactions. Angew. Chem. Int. Ed. 2018, 57, 12869–12873. [Google Scholar] [CrossRef]
- Ukai, T.; Tanaka, R.; Dokawa, T. A. A new catalyst for acyloin condensation. J. Pharm. Soc. Jpn. 1943, 63, 296–300. [Google Scholar]
- Breslow, R. On the mechanism of thiamine action. IV.1 Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Stetter, H. ; Rämsch, R, Y.; Kuhlmann, H. Über die präparative Nutzung der Thiazoliumsalz-katalysierten Acyloin- und Benzoin-Bildung, I. Herstellung von einfachen Acyloinen und Benzoinen. Synthesis, 1976; 733–735. [Google Scholar] [CrossRef]
- Stetter, H.; Dämbkes, G. Über die präparative Nutzung der Thiazoliumsalz-katalysierten Acyloin- und Benzoin-Bildung; II 1. Herstellung unsymmetrischer Acyloine und α-Diketone. Synthesis, 1977; 403–404. [Google Scholar] [CrossRef]
- Stetter, H.; Dämbkes, G. Über die präparative Nutzung der 1,3-Thiazoliumsalz-katalysierten Acyloin- und Benzoin-Bildung; III 1. Eine neue Methode zur Herstellung von substituierten Enol-trimethylsilyl-ethern des 1,2-Cyclopentandions. Synthesis, 1980; 309–310. [Google Scholar]
- Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef]
- Dzieszkowski, K.; Barańska, I.; Mroczyńska, K.; Słotwiński, M.; Rafiński, Z. Organocatalytic name reactions enabled by NHCs. Materials (Basel) 2020, 13, 1–30. [Google Scholar] [CrossRef]
- Barik, S.; Biju, A. T. N-Heterocyclic carbene (NHC) organocatalysis using aliphatic aldehydes. Chem. Commun. 2020, 56, 15484–15495. [Google Scholar] [CrossRef]
- Bellotti, P.; Koy, M.; Hopkinson, M.; Glorius, F. Recent advances in the chemistry and applications of N-heterocyclic carbenes. Nat. Rev. Chem. 2021, 5, 771–725. [Google Scholar] [CrossRef]
- Chakraborty, S.; Barik, S.; Biju, A. T. N-Heterocyclic carbene (NHC) organocatalysis: from fundamentals to frontiers. Chem. Soc. Rev. 2025, 54, 1102–1124. [Google Scholar] [CrossRef]
- Chen, X-Y. ; Cao, Z-H.; Ye, S. Bifunctional N-heterocyclic carbenes derived from L-pyroglutamic acid and their applications in enantioselective organocatalysis. Acc. Chem. Res. 2020, 53, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J. C.; Hunneman, D. H. J. Am. Chem. Soc. 1966, 88, 3666–3667. [CrossRef]
- Breslow, R. The mechanism of thiamine action: predictions from model experiments. Ann. N. Y. Acad. Sci. 1962, 98, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J. C.; Hara, T. Asymmetric thiazolium salt catalysis of the benzoin condensation. J. Org. Chem. 1974, 39, 1196–1199. [Google Scholar] [CrossRef]
- Berkessel, A.; Yatham, V. R.; Elfert, S.; Neudörfl, J. M. Characterization of the key intermediates of carbene-catalyzed umpolung by NMR spectroscopy and X-ray diffraction: breslow intermediates, homoenolates, and azolium enolates. Angew. Chem. Int. Ed. 2013, 52, 11158–11162. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Teles, J.H. A novel asymmetric benzoin reaction catalyzed by a chiral triazolium salt: Preliminary communication. Helv. Chim. Acta 1996, 79, 1217–1221. [Google Scholar] [CrossRef]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Balensiefer, T. Asymmetric intramolecular crossed-benzoin reactions by N-heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 2006, 45, 1463–1467. [Google Scholar] [CrossRef]
- Takikawa, H.; Hachisu, Y.; Bode, J. W.; Suzuki, K. Catalytic enantioselective crossed aldehyde-ketone benzoin cyclization. Angew. Chem. Int. Ed. 2006, 45, 3492–3494. [Google Scholar] [CrossRef]
- Kerr, M. S.; Rovis, T. Enantioselective synthesis of quaternary stereocenters via a catalytic asymmetric Stetter reaction. J. Am. Chem. Soc. 2004, 126(29), 8876–8877. [Google Scholar] [CrossRef]
- Castells, J.; López-Calahorra, F.; Bassedas, M.; Urrios, P. A new thiazolium salt-catalyzed synthesis of α-aminoketones from aldehydes and iminium salts. Synthesis. [CrossRef]
- Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J.; Reider P., J. Synthesis of alpha-amido ketones via organic catalysis: thiazolium-catalyzed cross-coupling of aldehydes with acylimines. J. Am. Chem. Soc. 2001, 123, 9696–9697. [Google Scholar] [CrossRef]
- Mennen, S. M.; Gipson, J. D.; Kim, Y. R.; Miller, S. J. Thiazolylalanine-derived catalysts for enantioselective intermolecular aldehyde-imine cross-couplings. J. Am. Chem. Soc. 2005, 127, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D. A.; Rovis, T. Catalytic asymmetric cross-aza-benzoin reactions of aliphatic aldehydes with N-Boc-protected imines. Angew. Chem. Int. Ed. 2012, 51, 5904–5906. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Mou, C.; Zhu, T.; Song, B. A.; Chi, Y. R. N-heterocyclic carbene-catalyzed chemoselective cross-aza-benzoin reaction of enals with isatin-derived ketimines: Access to chiral quaternary aminooxindoles. Org. Lett. 2014, 16, 3272–3275. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Mukherjee, S.; Das, T. K.; Jain, S.; Gonnade, R. G.; Biju, A. T. N-Heterocyclic-carbene-catalyzed Umpolung of imines. Angew. Chem. Int. Ed. 2017, 56, 2730–2734. [Google Scholar] [CrossRef]
- Das, T. K.; Ghosh, A.; Balanna, K.; Behera, P.; Gonnade, R. G.; Marelli, U. K.; Das, A. K.; Biju, A. T. ACS Catal. 9, 4065. [Google Scholar] [CrossRef]
- Simonovic, S.; Frison, J-C. ; Koyuncu, H.; Whitwood, A. C.; Douthwaite, R. E. Addition of N-heterocyclic carbenes to imines: phenoxide assisted deprotonation of an imidazolium moiety and generation of breslow intermediates derived from imines. Org Lett. 2009, 11, 245–247. [Google Scholar] [CrossRef]
- DiRocco, D. A.; Oberg, K. M.; Rovis, T. Isolable analogues of the Breslow intermediate derived from chiral triazolylidene carbenes. J. Am. Chem. Soc. 2012, 134, 6143–6145. [Google Scholar] [CrossRef]
- Das, R. C.; Barik, S.; Kunhiraman, A. A.; Goswani, A.; Mondal, A.; De, M.; Biju, A: T. N-Heterocyclic carbene-catalyzed imine Umpolung/semipinacol rearrangement cascade for the synthesis of indoxyls. ACS Catal. 2024, 14, 4202–4210. [Google Scholar] [CrossRef]
- Stetter, H.; Kuhlmann, H. Addition of aliphatic aldehydes to activated double bonds. Angew. Chem. Int. Ed. 1974, 13, 539. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Runsink, J,; Teles, J. H. The first asymmetric intramolecular Stetter reaction. Preliminary communication. Helv. Chim. Acta 1996, 79, 1899–1902. [Google Scholar] [CrossRef]
- Enders, D.; Han, J.; Henseler, A. Asymmetric intermolecular Stetter reactions catalyzed by a novel triazolium derived N-heterocyclic carbene. Chem. Commun. 2008, 3989–3991. [Google Scholar] [CrossRef]
- Kerr, M. S.; Read de Alaniz, J.; Rovis, T. A highly enantioselective catalytic intramolecular Stetter reaction. J. Am. Chem. Soc. 2002, 124, 10298–10299. [Google Scholar] [CrossRef] [PubMed]
- de Alaniz, J. R.; Rovis, T. The catalytic asymmetric intramolecular Stetter reaction. Synlett 2009, 1189–1207. [Google Scholar] [CrossRef]
- Enders, D, Breuer, K. Addition of acyl carbanion equivalents to carbonyl groups and enones. In: Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H, editors. Comprehensive Asymmetric Catalysis I–III. Vol. 3. Springer-Verlag; Berlin, Germany: 1999, p. 1093. [CrossRef]
- Enders, D.; Balensiefer, T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res. 2004, 37, 534. [Google Scholar] [CrossRef]
- Mennen, S. M.; Blank, J. T.; Tran-Dube, M. B.; Imbriglio, J. E.; Miller, S. J. A Peptide-catalyzed asymmetric Stetter reaction. Chem. Commun. 2005, 195–197. [Google Scholar] [CrossRef]
- Enders, D.; Han, J. Asymmetric intermolecular Stetter reactions of aromatic heterocyclic aldehydes with arylidenemalonates. Synthesis 2008, (23), 3864–3868. [Google Scholar] [CrossRef]
- Liu, Q.; Perreault, S.; Rovis, T. Catalytic asymmetric intermolecular Stetter reaction of glyoxamides with alkylidenemalonates. J. Am. Chem. Soc. 2008, 130, 14066–14067. [Google Scholar] [CrossRef]
- DiRocco, D. A.; Oberg. K. M.; Dalton, D. M.; Rovis, T. Catalytic asymmetric intermolecular Stetter reaction of heterocyclic aldehydes with nitroalkenes: Backbone fluorination improves selectivity. J. Am. Chem. Soc. 2009, 131(31), 10872–10874; [CrossRef]
- DiRocco, D. A.; Rovis, T. Catalytic asymmetric intermolecular Stetter reaction of enals with nitroalkenes: Enhancement of catalytic efficiency through bifunctional additives. J. Am. Chem. Soc. 2011, 133, 10402–10405. [Google Scholar] [CrossRef]
- DiRocco, D: A. ; Noey, E. L.; Houk, K. N.; Rovis, T. Catalytic Asymmetric intermolecular Stetter reactions of enolizable aldehydes with nitrostyrenes: Computational study provides insight into the success of the catalyst. Angew. Chem. Int. Ed. 2012, 51, 2391–2394. [Google Scholar] [CrossRef]
- Fernando, J. E. M.; Nakano, Y.; Zhang, C.; Lupton, D. W. Enantioselective N-heterocyclic carbene catalysis that exploits imine Umpolung. Angew. Chem. Int. Ed. 2019, 58, 4007–4011. [Google Scholar] [CrossRef]
- Das, T. K.; Biju, A. T. Imines as acceptors and donors in N-heterocyclic carbene (NHC) organocatalysis. Chem. Commun. 2020, 56, 8537–8552. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Struble, J. R.; Bode, J. W. Highly enantioselective azadiene Diels-Alder reactions catalyzed by chiral N-heterocyclic carbenes. J. Am. Chem. Soc. 2006, 128, 8418–8420. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Uc, G. J.; Bode, J. W. Chiral N-heterocyclic carbene catalyzed, enantioselective oxodiene Diels-Alder reactions with low catalyst loadings. J. Am. Chem. Soc. 2006, 128, 15088–15089. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Chen, X.; Chi, Y. R. Enantioselective Diels-Alder reactions of enals and alkylidene diketones catalyzed by N-heterocyclic carbenes. Org. Lett. 2011, 13, 4708–4711. [Google Scholar] [CrossRef]
- Lv, H.; Mo, J.; Fang, X.; Chi, Y. R. Formal Diels-Alder reactions of chalcones and formylcyclopropanes catalyzed by chiral N-heterocyclic carbenes. Org. Lett. 2011, 13, 5366–5369. [Google Scholar] [CrossRef]
- Zhang, Y. R.; He, L.; Wu, X.; Shao, P. L.; Ye, S. Chiral N-heterocyclic carbene catalyzed Staudinger reaction of ketenes with imines: Highly enantioselective synthesis of N-boc β-lactams. Org. Lett. 2008, 10, 277–280. [Google Scholar] [CrossRef]
- Hans, M.; Wouters, J.; Demonceau, A.; Delaude, L. Mechanistic insight into the Staudinger reaction catalyzed by N-heterocyclic carbenes. Chem. Eur. J. 2013, 19, 9668–9676. [Google Scholar] [CrossRef]
- He, L.; Lv, H.; Zhang, Y. R.; Ye, S. Formal cycloaddition of disubstituted ketenes with 2-oxoaldehydes catalyzed by chiral N-heterocyclic carbenes. J. Org. Chem. 2008, 73, 8101–8103. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shao, P.; Lv, H.; Ye, S. Enantioselective synthesis of β-trifluoromethyl-β-lactones via NHC-catalyzed ketene−ketone cycloaddition reactions. Org. Lett. 2009, 11, 4029–4031. [Google Scholar] [CrossRef] [PubMed]
- Kravina, A. G.; Mahatthananchai, J.; Bode, J. W. Enantioselective, NHC-catalyzed annulations of trisubstituted enals and cyclic N-sulfonylimines via α,β-unsaturated acyl azoliums. Angew. Chem. Int. Ed. 2012, 51, 9433–9436. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Houk, N. Organic Chemistry. Theory, Reactivity an Mechanisms in Modern Synthesis. Wiley-VCH Verlag GmbH & Co., KGaA, Weinheim, Germany, 2019, Chap. 5.5.9, pp. 476-495; ISBN 978-3-527-34532-8. [CrossRef]
- Maruoka, K.; Banno, H.; Yamamoto, H. Asymmetric Claisen rearrangement catalyzed by chiral organoaluminum reagent. J. Am. Chem. Soc. 1990, 112, 7791–7793. [Google Scholar] [CrossRef]
- Yoon, T. P.; MacMillan, D. W. C. Enantioselective Claisen rearrangements: Development of a first generation asymmetric acyl-Claisen reaction. J. Am. Chem. Soc. 2001, 123, 2911–2912. [Google Scholar] [CrossRef]
- Abraham, L.; Czerwonka, R.; Hiersemann, M. The Catalytic enantioselective Claisen rearrangement of an allyl vinyl ether. Angew. Chem. Int. Ed. 2001, 40, 4700–4703. [Google Scholar] [CrossRef]
- Uyeda, C.; Jacobsen, E. N. Enantioselective Claisen rearrangements with a hydrogen-bond donor catalyst. J. Am. Chem. Soc. 2008, 130, 9228–9229. [Google Scholar] [CrossRef]
- Coates, R. M.; Shah, S. K.; Mason, R. W. Stereoselective total synthesis of (±)-Gymnomitrol via reduction-alkylation of a-cyano ketones. J. Am. Chem. Soc. 1982, 104, 2198–2208. [Google Scholar] [CrossRef]
- Kaeobamrung, J.; Mahatthananchai, J.; Zheng, P.; Bode, J. W. An enantioselective Claisen rearrangement catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc. 2010, 132, 8810–8812. [Google Scholar] [CrossRef]
- Wanner, B.; Mahatthananchai, J.; Bode, J. W. Enantioselective synthesis of dihydropyridinones via NHC-catalyzed aza-Claisen reaction. Org. Lett. 2011, 13, 5378–5381. [Google Scholar] [CrossRef]
- Mahatthananchai, J.; Kaeobamrung, J.; Bode, J. W. Chiral N-heterocyclic carbene-catalyzed annulations of enals and ynals with stable enols: A highly enantioselective Coates-Claisen rearrangement. ACS Catal. 2012, 2, 494–503. [Google Scholar] [CrossRef]
- Li, G. T.; Gu, Q.; You, S. L. Enantioselective annulation of enals with 2-naphthols by triazolium salts derived from L-phenylalanine. Chem. Sci. 2015, 6, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Wadamoto, M.; Phillips, E. M.; Reynolds, T. E.; Scheidt, K. A. Enantioselective synthesis of α,α-disubstituted cyclopentenes by an N-heterocyclic carbene-catalyzed desymmetrization of 1,3-diketones. J. Am. Chem. Soc. 2007, 129, 10098–10099. [Google Scholar] [CrossRef]
- Nguyen, X. B.; Nakano, Y.; Lupton, D. W. Polarity inversion catalysis by the 1,4-addition of N-heterocyclic carbenes. Aust. J. Chem. 2020, 73, 1–8. [Google Scholar] [CrossRef]
- Biju, A. T.; Padmanaban, M.; Wurz, N. E.; Glorius, F. N-Heterocyclic carbene catalyzed umpolung of Michael acceptors for intermolecular reactions. Angew. Chem. Int. Ed. 2011, 50, 8412–8415. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Smith, S. W.; Powell, D. A.; Fu, G. C. Umpolung of Michael acceptors catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc. 2006, 128, 1472–1473. [Google Scholar] [CrossRef]
- Nakano, Y.; Lupton, D. W. Enantioselective N-heterocyclic carbene catalysis by the Umpolung of α,β-unsaturated ketones. Angew. Chem. Int. Ed. 2016, 55, 3135–3139. [Google Scholar] [CrossRef]
- Scott, L.; Nakano, Y.; Zhang, C.; Lupton, D. W. Enantioselective N-heterocyclic carbene catalyzed cyclopentene synthesis via the β-azolium ylide. Angew. Chem. Int. Ed. 2018, 57, 10299–10303. [Google Scholar] [CrossRef]
- Breugst, M.; Lupton, D. W. N-Heterocyclic carbene catalyzed (5+1) annulations exploiting a vinyl dianion synthon strategy. Angew. Chem. Int. Ed. 2019, 58, 11483–11490. [Google Scholar] [CrossRef]
- Yasui, K.; Kamitani, M.; Tobisu, M. N-Heterocyclic carbene catalyzed concerted nucleophilic aromatic substitution of aryl fluorides bearing α,β-unsaturated amides. Angew. Chem. Int. Ed. 2019, 58, 14157–14161. [Google Scholar] [CrossRef]
- Yasui, K.; Kamitani, M.; Fujimoto, H.; Tobisu, M. N-Heterocyclic carbene-catalyzed Truce–Smiles rearrangement of N-arylacrylamides via the cleavage of unactivated C(aryl)–N bonds. Org. Lett. 2021, 23, 1572–1576. [Google Scholar] [CrossRef]
- Barik, S.; Shee, S.; Biju, A. T. N-Heterocyclic carbene-catalyzed Umpolung of cyclopent-4-ene-1, 3-diones for activated olefin–isatin cross-coupling. Org. Lett. 2022, 24, 6066–6071. [Google Scholar] [CrossRef]
- Maki, B. E.; Scheidt, K. A. N-Heterocyclic carbene-catalyzed oxidation of unactivated aldehydes into esters. Org. Lett. 2008, 10, 4331–4334. [Google Scholar] [CrossRef]
- Samanta, R. C.; Maji, B.; De Sarkar, S.; Bergander, K.; Fröhlich, R.; Mück-Lichtenfeld, C.; Mayr, H.; Studer, A. Nucleophilic addition of enols and enamines to α,β-unsaturated acyl azoliums: mechanistic studies. Angew. Chem. Int. Ed. 2012, 51, 5234–5238. [Google Scholar] [CrossRef] [PubMed]
- Yetra, S. R.; Kaicharla, T.; Kunte, S. S.; Gonnade, R.; Biju, A. T. Asymmetric N-heterocyclic carbene (NHC)-catalyzed annulation of modified enals with enolizable aldehydes. Org. Lett. 2013, 15, 5202–5205. [Google Scholar] [CrossRef] [PubMed]
- Dzieszkowski, K.; Rafiński, Z. N-Heterocyclic carbene catalysis under oxidizing conditions. Catalysts 2018, 8, 1–16. [Google Scholar] [CrossRef]
- He, Y.; Chen, J.; Jiang, Y.; Fang, X.; Liu, J.; Yan, J-L. N-Heterocyclic carbene catalyzed reactions involving acetylenic Breslow and/or acylazolium as key intermediates. Chem. Rec. 2024, 24, 1–20. [Google Scholar] [CrossRef]
- Mukherjee, S.; Shee, S.; Poisson, T.; Besset, T.; Biju, A.T. Enantioselective N-heterocyclic carbene-catalyzed cascade reaction for the synthesis of pyrroloquinolines via N–H functionalization of indoles. Org. Lett. 2018, 20, 6998–7002. [Google Scholar] [CrossRef]
- Mondal, S.; Yetra, S.; Mukherjee, S.; Biju, A. T. NHC-Catalyzed generation of α,β-unsaturated acylazoliums for the enantioselective synthesis of heterocycles and carbocycles. Acc. Chem. Res. 2019, 52, 425–436. [Google Scholar] [CrossRef]
- De Sarkas, S.; Studer, A. Oxidative amidation and azidation of aldehydes by NHC catalysis. Org. Lett. 2010, 12, 1992–1995. [Google Scholar] [CrossRef]
- Maki, B. E.; Scheidt, K. A. N-Heterocyclic carbene-catalyzed oxidation of unactivated aldehydes to esters. Org. Lett. 2008, 10, 4331–4334. [Google Scholar] [CrossRef]
- White, N. A.; Rovis, T. Enantioselective N-heterocyclic carbene-catalyzed β-hydroxylation of enals using nitroarenes: an atom transfer reaction that proceeds via single electron transfer. J. Am. Chem. Soc. 2014, 136, 14674–14677. [Google Scholar] [CrossRef]
- Choi, H.; Mathi, G. R.; Hong, S.; Hong, S. Enantioselective functionalization at the C4 position of pyridinium salts through NHC catalysis. Nat. Commun. 2022, 13, 1–8. [Google Scholar] [CrossRef]
- Li, C. B.; Li, X. N.; Li, Z. C.; Li, J.; Wang, Z. X.; Gao, Z. H.; Ye, S. Enantioselective acylation of benzylic C(sp3)-H bond enabled by a cooperative photoredox and N-heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 2025, 64, e202421151. [Google Scholar]
- Takizawa, S. ; Salem, M, Editors; Atropisomerism in Asymmetric Organic Synthesis: Challenges and Applications, Wiley-VCH GmbH, Weinheim, Germany, 2024; pp. 1–336. ISBN 9783527352838. [Google Scholar] [CrossRef]
- Schmidt, T. A.; Hutskalova, V.; Sparr, C. Atroposelective catalysis. Nat. Rev. Chem. 2024, 8, 497–517. [Google Scholar] [CrossRef]
- Smyth, J. E.; Butler, N. M.; Keller, P. A. A twist of nature--the significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef]
- Liu, S-J. ; Zhao, Q.; Liu, X-C.; Gamble, A. B.; Huang, W.; Yang, Q-Q.; Han, B. Bioactive atropisomers: Unraveling design strategies and synthetic routes for drug discovery. Med. Res. Rev. 2024, 44, 1971–2014. [Google Scholar] [CrossRef]
- Zhao, C.; Guo, D.; Munkerup, K.; Huang, K-W. ; Li, F.; Wang, J. Enantioselective [3+3] atroposelective annulation catalyzed by N-heterocyclic carbenes. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Xu, K.; Li, W.; Zhu, S.; Zhu, T. Atroposelective arene formation by carbene-catalyzed formal [4+2] cycloaddition. Angew. Chem. Int. Ed. 2019, 58, 17625–17630. [Google Scholar]
- Yan, J. L.; Maiti, R.; Ren, S. C.; Tian, W.; Li, T.; Xu, J.; Mondal, B.; Jin, Z.; Chi, Y. R. Carbene-catalyzed atroposelective synthesis of axially chiral styrenes. Nat Commun. 2022, 13, 1–8. [Google Scholar] [CrossRef]
- Balanna, K.; Barik, S.; Barik, S.; Shee, S.; Manoj, N.; Gonnade, R. G.; Biju, A. T. N-Heterocyclic carbene-catalyzed atroposelective synthesis of N–N axially chiral 3-amino quinazolinones. ACS Catal. 2023, 13, 8752–8759. [Google Scholar] [CrossRef]
- Hine, J.; Linden, S. M.; Kanagasabapathy, V. M. Double-hydrogen-bonding catalysis of the reaction of phenyl glycidyl ether with diethylamine by 1,8-biphenylenediol. J. Org. Chem. 1985, 50, 5096–5099. [Google Scholar] [CrossRef]
- Tohma, H.; Takizawa, S.; Watanabe, H.; Fukuoka, Y.; Maegawa, T.; Kita, K. Hypervalent iodine(V)-induced asymmetric oxidation of sulfides to sulfoxides mediated by reversed micelles: Novel nonmetallic catalytic system. J. Org. Chem. 1999, 64, 3519–3523. [Google Scholar] [CrossRef]
- Ube, H.; Fukuchi, S.; Terada, M. Enantioselective aza-Friedel–Crafts reaction catalyzed by a water inclusion complex of O,O’-diacyl tartaric acid. Tetrahedron Asymmetry 2010, 21, 1203–1205. [Google Scholar] [CrossRef]
- Luan, Y.; Barbato, K. S.; Moquist, P. N.; Kodama, T.; Schaus, S. E. Enantioselective synthesis of 1,2-dihydronaphthalene-1-carbaldehydes by addition of boronates to isochromene acetals catalyzed by tartaric acid. J. Am. Chem. Soc. 2015, 137, 3233–3236. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.-M. BINOL: A versatile chiral reagent. Chem. Rev. 2005, 105, 857–898. [Google Scholar] [CrossRef] [PubMed]
- Pu, L. Regioselective substitution of BINOL. Chem. Rev. 2024, 124, 6643–6689. [Google Scholar] [CrossRef]
- Miyashita, A.; Yasuda, A.; Takaya, H.; Toriumi, K.; Ito, T.; Souchi, T.; Noyori, R. Synthesis of 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (BINAP), an atropisomeric chiral bis(triaryl)phosphine, and its use in the rhodium(I)-catalyzed asymmetric hydrogenation of α-acylamino)acrylic acids. J. Am. Chem. Soc. 1980, 102, 7932–7934. [Google Scholar] [CrossRef]
- Pu, L. 1,1‘-Binaphthyl dimers, oligomers, and polymers: Molecular recognition, asymmetric catalysis, and new materials. Chem. Rev. 1998, 98, 2405–2494. [Google Scholar] [CrossRef]
- Nguyen, T. N.; Chen, P.-A.; Setthakarn, K.; May, J. A. Chiral diol-based organocatalysts in enantioselective reactions. Molecules 2018, 23, 2317. [Google Scholar] [CrossRef]
- Curran, D. P.; Kuo, L. H. Acceleration of a dipolar Claisen rearrangement by hydrogen bonding to a soluble diaryl urea. Tetrahedron Lett. 1995, 36, 6647–6650. [Google Scholar] [CrossRef]
- Vera, S.; García-Urricelqui, A.; Mielgo, A.; Oiarbide, M. Progress in (thio)urea and squaramide-based Brønsted base catalysts with multiple H-bond donors. Eur. J. Org. Chem. 2023, 26, 1–31. [Google Scholar] [CrossRef]
- Kotke, M.; Schreiner, P. R. Acid-free, organocatalytic acetalization. Tetrahedron 2006, 62, 434–439. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E. N. Asymmetric ion-pairing catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef]
- Dereka, B.; Yu, Q.; Lewis, N. H.; Carpenter, W. B.; Bowman, J. M.; Tokmakoff, A. Crossover from hydrogen to chemical bonding. Science 2021, 371, 160–164. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P. R. (Thio)urea organocatalysis – What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef]
- Evans, N. H.; Beer, P. D. Advances in anion supramolecular chemistry: From recognition to chemical applications. Angew. Chem. Int. Ed. 2014, 53, 11716–11754. [Google Scholar] [CrossRef]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P. A. Applications of supramolecular anion recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K. M.; Schreiner, P. R. (Thio)urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef]
- Nieuwland, C.; Fonseca Guerra, C. How the chalcogen atom size dictates the hydrogen-bond donor capability of carboxamides, thioamides, and selenoamides. Chem. Eur. J. 2022, 28, 1–10. [Google Scholar] [CrossRef]
- Brussee, J.; Jansen, A. C. A. A highly stereoselective synthesis of S-(−)-[1,1′-binaphthalene]-2,2′-diol. Tetrahedron Lett. 1983, 24, 3261–3262. [Google Scholar] [CrossRef]
- Long, J.; Hu, J.; Shen, X.; Ji, B.; Ding, K. Discovery of exceptionally efficient catalysts for solvent-free enantioselective hetero-Diels-Alder reaction. J. Am. Chem. Soc. 2002, 124(1), 10-1. [Google Scholar] [CrossRef] [PubMed]
- McDougal, N. T.; Schaus, S. E. Asymmetric Morita-Baylis-Hillman reactions catalyzed by chiral Brønsted acids. J. Am. Chem. Soc. 2003, 125, 12094–12095. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Moquist, P. N.; Schaus, S. E. Asymmetric allylboration of ketones catalyzed by chiral diols. J. Am. Chem. Soc. 2006, 128, 12660–12661. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J. A.; Lou, S.; Schaus, S. E. Enantioselective addition of boronates to acyl imines catalyzed by chiral biphenols. Angew. Chem. Int.. Ed. 2009, 48, 4337–4340. [Google Scholar] [CrossRef]
- Barbato, K. S.; Luan, Y.; Ramella, D.; Panek, J. S.; Schaus, S. E. Enantioselective multicomponent condensation reactions of phenols, aldehydes, and boronates catalyzed by chiral biphenols. Org. Lett. 2015, 17, 5812–5815. [Google Scholar] [CrossRef]
- Jiang, Y.; Schaus, S. E. Asymmetric Petasis borono-Mannich allylation reactions catalyzed by chiral biphenols. Angew. Chem. Int. Ed. 2017, 56, 1544–1548. [Google Scholar] [CrossRef]
- Bao, J.; Wulff, W. D. Vaulted biaryls as chiral ligands for asymmetric catalytic Diels-Alder reactions. J. Am. Chem. Soc. 1993, 115, 3814–3815. [Google Scholar] [CrossRef]
- Ding, Z.; Osminski, W. E. G.; Ren, H.; Wullf, W. D. Scalable syntheses of the vaulted biaryl ligands VAPOL and VANOL via the cycloaddition/electrocyclization cascade. Org. Process Res. Dev. 2011, 15, 1089–1107. [Google Scholar] [CrossRef]
- Guan, Y.; Ding, Z.; Wulff, W. D. Vaulted biaryls in catalysis: A structure-activity relationship guided tour of the immanent domain of the VANOL ligand. Chem. Eur. J. 2013, 19, 15565–15571. [Google Scholar] [CrossRef]
- Nottingham, C.; Müller-Bunz, H.; Guiry, P. J. A family of chiral ferrocenyl diols: Modular synthesis, solid-state characterization, and application in asymmetric organocatalysis. Angew. Chem. Int. Ed. 2016, 55, 11115–11119. [Google Scholar] [CrossRef]
- Cunningham, L.; Guiry, P. J. Design and synthesis of a ferrocene-based diol library and application in the hetero-Diels-Alder reaction. Chem. Eur. J, 2022; 29, 1–9. [Google Scholar] [CrossRef]
- Du, H.; Zhao, D.; Ding, K. Enantioselective catalysis of the hetero-Diels–Alder reaction between Brassard's diene and aldehydes by hydrogen-bonding activation: A one-step synthesis of (S)-(+)-Dihydrokawain. Chem. Eur. J. 2004, 10, 5964–5970. [Google Scholar] [CrossRef] [PubMed]
- Bhasker Gondi, V.; Gravel, M.; Rawal, V. H. Hydrogen bond catalyzed enantioselective vinylogous Mukaiyama aldol reaction. Org. Lett. 2005, 7, 5657–5660. [Google Scholar] [CrossRef] [PubMed]
- McGilvra, J. D.; Unni, A. K.; Modi, K.; Rawal, V. H. Angew. Chem. Int. Ed. 2006, 45, 6130–6133. [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Brønsted acid catalysis of achiral enamine for regio- and enantioselective nitroso aldol synthesis. J. Am. Chem. Soc. 2005, 127, 1080–1081. [Google Scholar] [CrossRef]
- Kondo, S.-i.; Harada, T.; Tanaka, R.; Unno, M. Anion recognition by a silanediol-based receptor. Org. Lett. 2006, 8, 4621–4624. [Google Scholar] [CrossRef]
- Schafer, A. G.; Wieting, J. M.; Mattson, A. E. Silanediols: a new class of hydrogen bond donor catalysts. Org. Lett. 2011, 13, 5228–5231. [Google Scholar] [CrossRef]
- Tran, N. T.; Min, T.; Franz, A. K. Silanediol hydrogen bonding activation of carbonyl compounds. Chem. Eur.J. 2011, 17, 9897–9900. [Google Scholar] [CrossRef]
- Mattson, A. E.; Andrew, A. G.; Wieting, J. M.; Fisher, T. J. Chiral silanediols in anion-binding catalysts. Angew. Chem. Int. Ed. 2013, 52, 11321–11324. [Google Scholar] [CrossRef]
- Hardman-Baldwin, A. M.; Visco, M. D.; Wieting, J. M.; Stern, C.; Kondo, S.; Mattson, A. E. Silanediol-catalyzed chromenone functionalization. Org. Lett. 2016, 18, 3766–3769. [Google Scholar] [CrossRef]
- Visco, M. D.; Attard, J.; Guan, Y.; Mattson, A. E. Anion-binding catalyst designs for enantioselective synthesis. Tetrahedron Lett. 2017, 58, 2623–2628. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Wang, S-f. ; Yan, M.; Shi, J-y.; Li, G-x.; Zhao, J-z. Enantioselective sulfonation of enones with sulfinates by thiourea/tertiary-amine catalysis. Synlett 2024, 35, 1441–1445. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. Organocatalytic enantioselective conjugate additions to enones. J. Am. Chem. Soc. 2006, 128, 12652–12653. [Google Scholar] [CrossRef]
- Guong, J.; Zhao, C.-G. Organocatalytic enantioselective tandem aldol-cyclization reaction of α-isothiocyanato imides and activated carbonyl compounds. Tetrahedron Asymmetry 2011, 22, 1205–1211. [Google Scholar] [CrossRef]
- Berkessel, A.; Roland, K.; Neudörfl, J. M. Asymmetric Morita-Baylis-Hillman reaction catalyzed by isophoronediamine-derived bis(thio)urea organocatalysts. Org. Lett. 2006, 8, 4195–4198. [Google Scholar] [CrossRef]
- Shi, M.; Liu, X-G. Asymmetric Morita-Baylis-Hillman reaction of arylaldehydes with 2-cyclohexen-1-one catalyzed by chiral bis(thio)urea and DABCO. Org. Lett. 2008, 10, 1043–1046. [Google Scholar] [CrossRef]
- Wang, C. J.; Dong, X. Q.; Zhang, Z. H.; Xue, Z. Y.; Teng, H. L. Highly anti-selective asymmetric nitro-Mannich reactions catalyzed by bifunctional amine-thiourea-bearing multiple hydrogen-bonding donors. J. Am. Chem. Soc. 2008, 130, 8606–8607. [Google Scholar] [CrossRef]
- Grošelj, U.; Golobič, A.; Stare, K.; Svete, J.; Stanovnik, B. Synthesis and structural elucidation of novel camphor-derived thioureas. Chirality 2012, 24, 307–317. [Google Scholar] [CrossRef]
- Ricko, S.; Golobič, A.; Svete, J.; Stanovnik, K. B.; Grošelj, U. Synthesis of novel camphor-derived bifunctional thiourea organocatalysts. Chirality 2015, 27, 39–52. [Google Scholar] [CrossRef]
- Núñez, M. G.; Farley, A. J.; Dixon, D. J. Bifunctional iminophosphorane organocatalysts for enantioselective synthesis: application to the ketimine nitro-Mannich reaction. J. Am. Chem. Soc. 2013, 135, 16348–16351. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, X.; Ng, W-H. ; Tse, Y-L. S.; Yeung, Y-Y. Catalytic enantio- and diastereoselective domino halocyclization and spiroketalization. Nature Catal. 2020, 3, 993–1001. [Google Scholar] [CrossRef]
- 763. Manabe, K. 763. Manabe, K. Asymmetric phase-transfer alkylation catalyzed by a chiral quaternary phosphonium salt with a multiple hydrogen-bonding site.Tetrahedron Lett. 1998; 135, 5807–5810. [Google Scholar] [CrossRef]
- Sohtome, Y.; Hashimoto, Y. ; Nagasawa, K Guanidine-thiourea bifunctional organocatalyst for the asymmetric Henry (nitroaldol) reaction. Adv. Synth. Catal. 2005, 347, 1643–1648. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Sun, C.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric phase-transfer catalysts bearing multiple hydrogen-bonding donors: highly efficient catalysts for enantio- and diastereoselective nitro-Mannich reaction of amidosulfones. Org. Lett. 2014, 16, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contributions of organofluorine compounds to pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. Science 2020, 23, 101467. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Modern approaches for asymmetric construction of carbon–fluorine quaternary stereogenic centers: Synthetic challenges and pharmaceutical needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Rozatian, N.; Hodgson, D. R. W. Reactivities of electrophilic N–F fluorinating reagents. Chem. Commun. 2021, 57, 683–712. [Google Scholar] [CrossRef]
- Britton, R.; Gouverneur, V.; Lin, J.-H.; Meanwell, M.; Ni, C.; Pupo, G.; Xiao, J.-C.; Hu, J. Contemporary synthetic strategies in organofluorine chemistry. Nat. Rev. Meth. Primers 2021, 1, 47. [Google Scholar] [CrossRef]
- Pupo, G.; Ibba, F.; Ascough, D. M. H.; Vicini, A. C.; Ricci, P.; Christensen, K. E.; Pfeifer, L.; Morphy, J. R.; Brown, J. M.; Paton, R. S.; Gouverneur, V. Asymmetric nucleophilic fluorination under hydrogen bonding phase-transfer catalysis. Science 2018, 360, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Vicini, A. C.; Alozie, D. M.; Courtes, P.; Roagna, G.; Aubert, C.; Certal, V.; El-Ahmad, Y.; Roy, S.; Gouveneur, V. Scalable synthesis of (R,R)-N,N-dibenzyl-2-fluorocyclohexan-1-amine with CsF under hydrogen bonding phase-transfer catalysis. Org. Process Res. Dev. 2021, 25, 2730–2737. [Google Scholar] [CrossRef]
- Pupo, G.; Gouverneur, V. Hydrogen bonding phase-transfer catalysis with alkali metal fluorides and beyond. J. Am. Chem. Soc. 2022, 144, 5200–5213. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fan, S.; Zhang, C.; Sun, J. Catalytic enantioselective α-fluorination of ketones with CsF. J. Am. Chem. Soc. 2025, 147, 10059–10065. [Google Scholar] [CrossRef]
- Suzuki, S.; Kamo, T.; Fukushi, K.; Hiramatsu, T.; Tokunaga, E.; Dohi, T.; Kita, Y.; Shibata, N. Iodoarene-catalyzed fluorination and aminofluorination by an Ar-I/HF·pyridine/mCPBA system. Chem. Sci. 2014, 5, 2754–2760. [Google Scholar] [CrossRef]
- Zhong, S.; Yu, Z.; Zhu, Y.; Shi, L. Catalytic fluorination of α-branched ketones with nucleophilic fluorine. Org. Lett. 2025, 27, 3452–3458. [Google Scholar] [CrossRef]
- Parvin, T.; Yadav, R.; Choudhury, L. H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef]
- Malerich, J. P.; Hagihara, K.; Rawal, V. H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Squaramides: bridging from molecular recognition to bifunctional organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef]
- 781. Carlone, A.; Bernardi, L.; McCormack, P.; Warr, T.; Oruganti, S.; Cobley, C. J. Asymmetric organocatalysis and continuous chemistry for an efficient and cost-competitive process to Pregabalin. Org. Process Res. Dev. 2021, 25, 2795–2805. [Google Scholar] [CrossRef]
- Zhu, Y.; Malerich, J. P.; Rawal, V. H. Squaramide-catalyzed enantioselective Michael addition of diphenyl phosphite to nitroalkenes. Angew. Chem. Int. Ed. 2010, 49, 153–156. [Google Scholar] [CrossRef]
- Qian, Y.; Ma, G.; Lv, A.; Zhu, H. L.; Zhao, J.; Rawal, V. H. Squaramide-catalyzed enantioselective Friedel-Crafts reaction of indoles with imines. Chem. Commun. 2010, 46, 3004–3006. [Google Scholar] [CrossRef]
- Alegre-Requena, J. V.; Marqués-López, E.; Herrera, R. P. Trifunctional squaramide catalyst for efficient enantioselective Henry reaction activation. Adv. Synth. Catal. 2016, 358, 1801–1809. [Google Scholar] [CrossRef]
- Wendlandt, A.; Vangal, P.; Jacobsen, E.N. Quaternary stereocentres via an enantioconvergent catalytic SN1 reaction. Nature 2018, 556, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Schuster, T.; Bauch, M.; Durner, G.; Göbel, M, W. Axially chiral amidinium ions as inducers of enantioselectivity in Diels-Alder reactions. Org. Lett. 2000, 2, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Weimar, M.; Dürner, G.; Bats, J. W.; Göbel, M. M. Enantioselective synthesis of (+)-estrone exploiting a hydrogen bond-promoted Diels−Alder reaction. J. Org. Chem. 2010, 75, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Nakano, S.; Tahira, Y.; Terazawa, K.; Yamazaki, K.; Kitamura, C.; Hara, O. Chiral pyridinium phosphoramide as a dual Brønsted acid catalyst for enantioselective Diels-Alder reaction. Org Lett. 2016, 18, 2004–2007. [Google Scholar] [CrossRef]
- Yu, J-S. ; Liao, F-M.; Gao, W-M.; Liao, K.; Zuo, R-L.; Zhou, J. Michael addition catalyzed by chiral secondary amine phosphoramide using fluorinated silyl enol ethers: Formation of quaternary carbon stereocenters. Angew. Chem. Int. Ed. 2015, 54, 7381–7385. [Google Scholar] [CrossRef]
- Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed. 2004, 43, 1566–1568. [Google Scholar] [CrossRef]
- Uraguchi, D.; Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. [Google Scholar] [CrossRef]
- Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chiral phosphoric acid catalysis: from numbers to insights. Chem. Soc. Rev. 2018, 47, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gupta, A. K.; Huang, L.; Zhao, W.; Yin, X.; Osminski, W. E. G.; Huang, R. H.; Wulff, W. D.; Izzo, J. A.; Vetticatt, M. J. Pyro-borates, spiro-borates, and boroxinates of BINOL-assembly, structures, and reactivity. J. Am. Chem. Soc. 2017, 139, 10267–10285. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- a) Zhu, C.; Saito, K.; Yamanaka, M.; Akiyama, T. Benzothiazoline: Versatile hydrogen donor for organocatalytic transfer hydrogenation. Acc. Chem. Res. 2015, 48, 388–398; https://doi.org/10.1021/ar500414x; b) Jiménez, E. I. An update on chiral phosphoric acid organocatalyzed stereoselective reactions. Org. Biomol. Chem. 2023, 21, 3477-3502; https://doi.org/10.1039/D3OB00212H.
- Terada, M. Binaphthol-derived phosphoric acid as a versatile catalyst for enantioselective carbon–carbon bond forming reactions. Chem. Commun. 2008, 4097–4112. [Google Scholar] [CrossRef]
- Han, Z. Y.; Gong, L. Z. Chiral bisphosphoric acids and their applications in asymmetric catalysis. Chem. Rec, 2023. [Google Scholar] [CrossRef]
- Rahman, A.; Lin, X. Development and application of chiral spirocyclic phosphoric acids in asymmetric catalysis. Org. Biomol. Chem. 2018, 16, 4753–4777. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, J.; Lin, X. Atroposelective phosphoric acid catalyzed three-component cascade reaction: Enantioselective synthesis of axially chiral N-arylindoles. Angew. Chem. Int. Ed. 2019, 58, 15824–15828. [Google Scholar] [CrossRef]
- Kampen, D.; Reisinger, C. M.; List, B. Chiral Brønsted acids for asymmetric organocatalysis. Top. Curr. Chem. 2010, (291), 395–456. [Google Scholar] [CrossRef]
- Woldegiorgis, A. G.; Han, Z.; Lin, X. Recent advances in chiral phosphoric acid catalyzed asymmetric organic reactions: An overview. J. Mol. Struct. 2024, 1297, 1–69. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Han, Z.; Huang, H.; Hong, B.; Sun, J. Organocatalytic enantioselective nucleophilic addition of indole imine 5-methides. Org. Lett. 2024, 26, 396–400. [Google Scholar] [CrossRef]
- Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. Brønsted acid-catalyzed imine amidation. J. Am. Chem. Soc. 2005, 127, 15696–15697. [Google Scholar] [CrossRef]
- Rueping, M.; Sugiono, E.; Azap, C. A highly enantioselective Brønsted acid catalyst for the Strecker reaction. Angew. Chem. Int. Ed. 2006, 45, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Sugiono, E.; Moreth, S. A. Adv. Synth. Catal. 2007, 349, 759–764. [CrossRef]
- Terada, M.; Machioka, K.; Sorimachi, K. High substrate/catalyst organocatalysis by a chiral Brønsted acid for an enantioselective aza-ene-type reaction. Angew. Chem. Int. Ed. 2006, 45, 2254–2257. [Google Scholar] [CrossRef]
- Khomutnyk, Y, Y. ; Argüelles, A. J.; Winschel, G. A.; Sun, Z.; Zimmerman, P. M.; Nagorny, P. Studies of the mechanism and origins of enantioselectivity for the chiral phosphoric acid-catalyzed stereoselective spiroketalization reactions. J. Am. Chem. Soc. 2016, 138, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Enantioselective bromocyclization of olefins catalyzed by chiral phosphoric acid. Org Lett. 2011, 13, 6350–6353. [Google Scholar] [CrossRef]
- Denmark, S.; Burk, M. T. Enantioselective bromocycloetherification by Lewis base/chiral Brønsted acid cooperative catalysis. Org Lett. 2012, 14, 256–259. [Google Scholar] [CrossRef]
- Denmark, S. E.; Burk, M. T. Development and mechanism of an enantioselective bromocycloetherification reaction via Lewis base/chiral Brønsted acid cooperative catalysis. Chirality 2013, 26, 344–355. [Google Scholar] [CrossRef]
- Yue, T.; Wang, M-X.; Wang, D-X.; Masson, G.; Zhu, J. Brønsted acid catalyzed enantioselective three-component reaction involving the alpha addition of isocyanides to imines. Angew. Chem. Int. Ed. 2009; 48, 6717–6721. [CrossRef]
- Zhang, J.; Lin, S-X. ; Cheng, D-J.; Liu, X-Y.; Tan, B. Phosphoric acid-catalyzed asymmetric classic Passerini reaction. J. Am. Chem. Soc. 2015, 137, 14039–14042. [Google Scholar] [CrossRef]
- Alvim, H. G. O.; Pinheiro, D. L. J.; Carvalho-Silva, V. H.; Fioramonte, M.; Gozzo, F. C.; da Silva, W. A.; Amarante, G. W.; Neto, B. A. D. Combined role of the asymmetric counteranion-directed catalysis (ACDC) and ionic liquid effect for the enantioselective Biginelli multicomponent reaction. J. Org. Chem. 2018, 83(19), 12143–12153. [Google Scholar] [CrossRef]
- Kappe, C. O. A reexamination of the mechanism of the Biginelli dihydropyrimidine synthesis. Support for an N-acyliminium ion intermediate (1). J. Org. Chem. 1997, 62, 7201–7204. [Google Scholar] [CrossRef]
- Shevchenko, G. A.; Oppelaar, B.; List, B. An unexpected α-oxidation of cyclic ketones with 1,4-benzoquinone by enol catalysis. Angew. Chem. Int. Ed. 2018, 57, 10756–10759. [Google Scholar] [CrossRef]
- Wang, J. Y.; Gao, C. H.; Ma, C.; Wu, X. Y.; Ni, S. F.; Tan, W.; Shi, F. Design and catalytic asymmetric synthesis of furan-indole compounds bearing both axial and central chirality. Angew. Chem. Int. Ed. 2024, 63, 1–9. [Google Scholar] [CrossRef]
- Li, T.; Liu, S.; Wu, S.; Cheng, Q.; Chen, Q.; Jiao, Y.; Zhang, Y.; Shi, F. Catalytic asymmetric (3+3) cycloaddition between different 2-indolylmethanols. Sci. China Chem. 2024, 67, 2629–2636. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, P.; Li, S-Y. ; Sun, H.; Xiang, S-H.; Wang, J.; Houk, K. N.; Tan, B. Asymmetric phosphoric acid–catalyzed four-component Ugi reaction. Science 2018, 361, pp. 1-1. [Google Scholar] [CrossRef]
- Wang, J.-B.; Yu, P.; Zhou, Z.-P.; Zhang, J.; Wang, J.; Luo, S.-H.; Q. -S. Gu, Q.-S.; Houk, K. N.; Tan, B. Rational design, enantioselective synthesis and catalytic applications of axially chiral EBINOLs. Nat. Catal. 2019, 2, 504–513. [Google Scholar] [CrossRef]
- Michelet, B.; Martin-Mingot, A.; Rodriguez, J.; Thibaudeau, S.; Bonne, D. Enantioselective organocatalysis and superacid activation: Challenges and opportunities. Chem. Eur. J. 2023, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burlingham, B. T.; Widlanski, T. S. Synthesis and biological activity of N-sulfonylphosphoramidates: probing the electrostatic preferences of alkaline phosphatase. J. Org. Chem. 2001, 66, 7561–7567. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, D.; Yamamoto, H. Design of chiral N-triflyl phosphoramide as a strong chiral Brønsted acid and its application to asymmetric Diels-Alder reaction. J. Am. Chem. Soc. 2006, 128, 9626–9627. [Google Scholar] [CrossRef]
- Čorić, I.; List, B. Asymmetric spiroacetalization catalysed by confined Brønsted acids. Nature 2012, 483, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Čorić, I.; Wang, Q.; List, B. Activation of H2O2 by chiral confined Brønsted acids: a highly enantioselective catalytic sulfoxidation. J. Am. Chem. Soc. 2012, 134, 10765–10768. [Google Scholar] [CrossRef]
- Kim, J. H.; Čorić, I.; Vellalath, S.; List, B. The catalytic asymmetric acetalization. Angew. Chem. Int Ed. 2013, 52, 4474–4477. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y-Y. ; Jiang, Y-J.; Fan, Y-S.; Sha, D.; Wang, Q.; Zhang, G.; Zheng, L.; Zhang, S-Q. Double axially chiral bisphosphorylimides as novel Brønsted acids in asymmetric three-component Mannich reaction. Tetrahedron Asymmetry 2012, 23, 904–909. [Google Scholar] [CrossRef]
- Wu, K. ; Jiang, Y-J.; Fan, Y-S.; Sha, D.; Zhang, S-Q. Double axially chiral bisphosphorylimides catalyzed highly enantioselective and efficient Friedel–Crafts reaction of indoles with imines. Chem. Eur. J, 2013; 19, 474–478. [Google Scholar] [CrossRef]
- An, D.; Fan, Y-S. ; Gao, Y.; Zhu, Z-Q.; Zheng, L-Y.; Zhang, S-Q. Highly enantioselective Biginelli reaction catalyzed by double axially chiral bisphosphorylimides. Eur. J. Org. Chem. 2014, 302–306. [Google Scholar] [CrossRef]
- Liu, L.; Kaib, P. S.; Tap, A.; List, B. A general catalytic asymmetric Prins cyclization. J. Am. Chem. Soc. 2016, 138, 10822–10825. [Google Scholar] [CrossRef]
- Tsuji, N.; Kennemur, J. L.; Buyck, T.; Lee, S.; Prévost, S.; Kaib, P. S. J.; Bykov, D.; Farès, C.; List, B. Activation of olefins via asymmetric Brønsted acid catalysis. Science 2018, 359, 1501–1505. [Google Scholar] [CrossRef]
- Zhu, C.; Das, S.; Guin, A.; De, C. K.; List, B. Organocatalytic regio- and stereoselective cyclopropanation of olefins. Nat. Catal. 2025, 8, 487–494. [Google Scholar] [CrossRef]
- Mohammadlou, A.; Chakraborty, A.; Maday, M.; Yin, X.; Zheng, L.; Gholami, H.; Ashtekar, K.; Staples, R.; Wulff, W. D.; Borhan, B. Structure guided design of VANOL-imidodiphosphorimidate catalysts for the catalytic enantioselective bromospiroketalization reaction. ACS Catal. 2024, 14, 426–436. [Google Scholar] [CrossRef]
- Chen, X.-H.; Zhang, W.-Q.; Gong, L.-Z. Asymmetric organocatalytic three-component 1,3-dipolar cycloaddition: control of stereochemistry via a chiral Brønsted acid activated dipole. J. Am. Chem. Soc. 2008, 130, 5652–5653. [Google Scholar] [CrossRef]
- Zhou, F.; Yamamoto, H. A powerful chiral phosphoric acid catalyst for enantioselective Mukaiyama-Mannich reactions. Angew. Chem. Int. Ed. 2016, 55, 8970–8974. [Google Scholar] [CrossRef]
- Rauniyar, V.; Lackner, A. D.; Hamilton, G. L.; Toste, F. D. Asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst. Science 2011, 334, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R. J.; Toste, F. D. Chiral anion phase-transfer catalysis applied to the direct enantioselective fluorinative dearomatization of phenols. J. Am. Chem. Soc. 2013, 135, 1268–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. W.; Wang, X. D.; Ao, Y. F.; Wang, D. X.; Wang, Q. Q. Chiral bis-phosphate macrocycles for catalytic, efficient, and enantioselective electrophilic fluorination. Chem. Eur. J. 2024, 30, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Huang, L.; Huang, R. H.; Wulff, W. D. Evidence for a boroxinate based Brønsted acid derivative of VAPOL as the active catalyst in the catalytic asymmetric aziridination reaction. J. Am. Chem. Soc. 2009, 131, 15615–15617. [Google Scholar] [CrossRef]
- Desai, A. A.; Wulff, W. D. Controlled diastereo- and enantioselection in a catalytic asymmetric aziridination. J. Am. Chem. Soc. 2010, 132, 13100–13103. [Google Scholar] [CrossRef]
- Osminski, W. E. G.; Lu, Z.; Zhao, W.; Mohammadlou, A.; Yin, X.; Matthews, E. C.; Canestraight, V. M.; Staples, R. J.; Allen, C. J.; Hirschi, J, S. ; Wulff, W. D. Probing catalyst function - Electronic modulation of chiral polyborate anionic catalysts. J. Org. Chem. 2021, 86, 17762–17773. [Google Scholar] [CrossRef]
- Antilla, J. C.; Wulff, W. D. Catalytic asymmetric aziridination with a chiral VAPOL-boron Lewis acid. J. Am. Chem. Soc. 1999, 121, 5099–5100. [Google Scholar] [CrossRef]
- Antilla, J. C.; Wulff, W. D. Catalytic asymmetric aziridination with arylborate catalysts derived from VAPOL and VANOL ligands. Angew. Chem. Int. Ed. 2000, 39, 4518–4521. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Y.; Wulff, W. D. Direct access to N-H-aziridines from asymmetric catalytic aziridination with borate catalysts derived from vaulted binaphthol and vaulted biphenanthrol ligands. J. Am. Chem. Soc. 2007, 129, 7185–7194. [Google Scholar] [CrossRef]
- Zhang, Y.; Desai, A.; Lu, Z.; Hu, G.; Ding, Z.; Wulff, W.D. Catalytic asymmetric aziridination with borate catalysts derived from VANOL and VAPOL ligands: Scope and mechanistic studies. Chem. Eur. J. 2008, 14, 3785–3803. [Google Scholar] [CrossRef]
- Ren, H.; Wulff, W. D. Direct catalytic asymmetric aminoallylation of aldehydes: synergism of chiral and nonchiral Brønsted acids. J. Am. Chem. Soc. 2011, 133, 5656–5659. [Google Scholar] [CrossRef]
- Uraguchi, D.; Ueoka, F.; Tanaka, N.; Kizu, T.; Takahashi, W.; Ooi, T. A structurally robust chiral borate ion: Molecular design, synthesis, and asymmetric catalysis. Angew. Chem. Int. Ed. 2020, 59, 11456–11461. [Google Scholar] [CrossRef]
- Hatano, M.; Maki, T.; Moriyama, K.; Arinobe, M.; Ishihara, K. Pyridinium 1,1'-binaphthyl-2,2'-disulfonates as highly effective chiral Brønsted acid-base combined salt catalysts for enantioselective Mannich-type reaction. J. Am. Chem. Soc. 2008, 130, 16858–16860. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Ishihara, K. Chiral 1,1′-binaphthyl-2,2′-disulfonic acid (BINSA) and its derivatives for asymmetric catalysis. Asian. J. Org. Chem. 2014, 3, 352–365. [Google Scholar] [CrossRef]
- Hatano, M.; Sugiura, Y.; Akakura, M.; Ishihara, K. Enantioselective Friedel-Crafts aminoalkylation catalyzed by chiral ammonium 1,1′-binaphthyl-2,2′-disulfonates. Synlett 2011, (9), 1247–1250. [Google Scholar] [CrossRef]
- Kurihara, T.; Satake, S.; Hatano, M.; Ishihara, K.; Yoshino, T.; Matsunaga, S. Synthesis of 1,1'-spirobiindane-7,7'-disulfonic acid and disulfonimide: Application for catalytic asymmetric aminalization. Chem. Asian J. 2018, 13, 2378–2381. [Google Scholar] [CrossRef]
- Rothermel, K.; Zabka, M.; Hioe, J.; Gschwind, R. M. Disulfonimides versus phosphoric acids in Brønsted acid catalysis: The effect of weak hydrogen bonds and multiple acceptors on complex structures and reactivity. J. Org. Chem. 2019, 84, 13221–13231. [Google Scholar] [CrossRef]
- Treskow, M.; Neudörfl, J.; Giernoth, R. BINBAM – A new motif for strong and chiral Brønsted acids. Eur. J. Org. Chem. 2009, (22), 3693–3697. [Google Scholar] [CrossRef]
- Garcίa-Garcίa, P.; Lay, F.; Rabalakos, C.; List, B. A powerful chiral counteranion motif for asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 4363–4366. [Google Scholar] [CrossRef]
- Mathieu, B.; Ghosez, L. N-Trimethylsilyl-bis(trifluoromethanesulfonyl)imide: A better carbonyl activator than trimethylsilyl triflate. Tetrahedon Lett. 1997, 38, 5407–5000. [Google Scholar] [CrossRef]
- Ratjen, L.; García-García, P.; Lay, F.; Beck, M. E.; List, B. Disulfonimide-catalyzed asymmetric vinylogous and bisvinylogous Mukaiyama aldol reactions. Angew. Chem. Int. Ed. 2011, 50, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Mahlau, M.; List, B. Asymmetric counteranion-directed catalysis: concept, definition, and applications. Angew. Chem. Int. Ed. 2013, 52, 518–533. [Google Scholar] [CrossRef] [PubMed]
- James, T.; van Gemmeren, M.; List, B. Development and applications of disulfonimides in enantioselective organocatalysis Chem. Rev. 2015, 115, 9388–9409. [Google Scholar]
- Guin, J.; Rabalakos, C.; List, B. Highly enantioselective hetero-Diels-Alder reaction of 1,3-bis(silyloxy)-1,3-dienes with aldehydes catalyzed by chiral disulfonimide. Angew. Chem.. Int. Ed. 2012, 51, 8859–8863. [Google Scholar] [CrossRef]
- Zhang, Z.; Bae, H. Y.; Guin, J.; Rabalakos, C.; van Gemmeren, M.; Leutzsch, M.; Klussmann, M.; List, B. Asymmetric counteranion-directed Lewis acid organocatalysis for the scalable cyanosilylation of aldehydes. Nat. Commun, 1247; 8, 1–8. [Google Scholar] [CrossRef]
- Benda, M. C.; France, S. Chiral disulfonimides: a versatile template for asymmetric catalysis. Org. Biomol. Chem. 2020, 18, 7485–7513. [Google Scholar] [CrossRef]
- Lee, G, S. ; Namkoong, G.; Park, J.; Chen, D. Y. Total synthesis of strychnine. Chem. Eur. J. 2017, 23, 16189–16193. [Google Scholar] [CrossRef]
- Gatzenmeier, T.; Kaib, P. S. J.; Lingnau, J. B.; Goddard, R.; List, B. The catalytic asymmetric Mukaiyama-Michael reaction of silyl ketene acetals with α,β-unsaturated methyl esters. Angew. Chem. Int. Ed. 2018, 57, 2464–2468. [Google Scholar] [CrossRef]
- Chen, L. Y.; He, H.; Chan, W. H.; Lee, A. W. Chiral sulfonimide as a Brønsted acid organocatalyst for asymmetric Friedel-Crafts alkylation of indoles with imines. J. Org. Chem. 2011, 76, 7141–7147. [Google Scholar] [CrossRef]
- Triandafillidi, I.; Tzaras, D. I.; Kokotos, C. G. Green organocatalytic oxidative methods using activated ketones. ChemCatChem 2018, 10, 2521–2535. [Google Scholar] [CrossRef]
- Feng, X.; Du, H. Shi epoxidation: A great shortcut to complex compounds. Chin. J. Chem. 2021, 39, 2016–2026. [Google Scholar] [CrossRef]
- Curci, R.; Fiorentino, M.; Serio, M. R. Asymmetric epoxidation of unfunctionalized alkenes by dioxirane intermediates generated from potassium peroxomonosulfate and chiral ketones. Chem. Commun. 1984, 155–156. [Google Scholar] [CrossRef]
- Denmark, S. E; Wu, Z. Dioxiranes are the active agents in ketone-catalyzed epoxidations with oxone. J. Org. Chem. 1997, 62, 8964–8965. [Google Scholar] [CrossRef]
- Yang, D.; Yip, Y-C. ; Tang, M-W.; Wong, M-K.; Zheng, J-H.; Cheung, K-K. A C2 Symmetric chiral ketone for catalytic asymmetric epoxidation of unfunctionalized olefins. J. Am. Chem. Soc. 1996, 118, 491–492. [Google Scholar] [CrossRef]
- Yang, D. ; Wong, M-K. ; Yip, Y-C.; Wang, X-C.; Tang, M-W.; J.-H. Zheng, J-H.; Cheung, K-K. Design and synthesis of chiral ketones for catalytic asymmetric epoxidation of unfunctionalized olefins. J. Am. Chem. Soc. 1998, 120, 5943–5952. [Google Scholar] [CrossRef]
- Denmark, S. E.; Wu, Z. 6-Oxo-1,1,4,4-tetramethyl-1,4-diazepinium salts. A new class of catalysts for efficient epoxidation of olefins with oxone. J. Org. Chem. 1998, 63, 2810–2811. [Google Scholar] [CrossRef]
- Armstrong, A. Catalytic enantioselective epoxidation of alkenes with a tropinone-derived chiral ketone. Chem. Commun. 1998, 621–622. [Google Scholar] [CrossRef]
- Armstrong, A.; Ahmed, G.; Dominguez-Fernandez, B.; Hayter, B. R.; Wailes, J. S. Enantioselective epoxidation of alkenes catalyzed by 2-fluoro-N-carbethoxytropinone and related tropinone derivatives. J. Org. Chem. 2002, 67, 8610–8617. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.-X.; Shi, Y. An efficient asymmetric epoxidation method for trans-olefins mediated by a fructose-derived ketone. J. Am. Chem. Soc. 1996, 118, 9806–9807. [Google Scholar] [CrossRef]
- Zhao, M.-X.; Shi, Y. Practical synthesis of an L-fructose-derived ketone catalyst for asymmetric epoxidation of olefins. J. Org. Chem. 2006, 71, 5377–5379. [Google Scholar] [CrossRef]
- Li, J.; Song, X.; Wu, T.; Zhao, L.; Qin, Q.; Cheng, M.; Liu, Y.; Zhao, D. Scalable, efficient and rapid chemical synthesis of L-fructose with high purity. Carbohydr. Res. 2019, 480, 67–72. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. Asymmetric epoxidation using hydrogen peroxide (H2O2) as primary oxidant. Tetrahedron Lett. 1999, 40, 8721–8724. [Google Scholar] [CrossRef]
- Ager, D. J.; Anderson, K.; Oblinger, E.; Shi, Y.; VanderRoest, J. An epoxidation approach to a chiral lactone: Application of the Shi epoxidation. Org. Process Res. Dev. 2007, 11, 44–51. [Google Scholar] [CrossRef]
- Picot, A.; Milliet, P.; Lusinchi, X. Action de l'acide p-nitroperbenzoïque et de l'eau oxygénée sur un sel d'immonium hétérocyclique stéroïdique et sur l'énamine correspondante. Tetrahedron Lett. 1976, 17, 1577–1780. [Google Scholar] [CrossRef]
- Bohé, L.; Lusinchi, M.; Lusinchi, X. Oxygen atom transfer from a chiral oxaziridinium salt. Asymmetric epoxidation of unfunctionalized olefins. Tetrahedron 1999, 55, 141–154. [Google Scholar] [CrossRef]
- 880. del Río, R. E.; Wang, B.; Achab, S.; Bohé, L. Highly enantioselective oxidation of sulfides to sulfoxides by a new oxaziridinium salt. Highly enantioselective oxidation of sulfides to sulfoxides by a new oxaziridinium salt. Org Lett. 2007, 9(12), 2265–2268. [Google Scholar] [CrossRef]
- Davis, F. A.; Harakal, M. E.; Awad, S. B. Chemistry of oxaziridines. 4. Asymmetric epoxidation of unfunctionalized alkenes using chiral 2-sulfonyloxaziridines: evidence for a planar transition state geometry. J. Am. Chem. Soc. 1983, 105, 3123–3126. [Google Scholar] [CrossRef]
- Bulman Page, P. C.; Buckley, B. R.; Blacker, A. J. Iminium salt catalysts for asymmetric epoxidation: the first high enantioselectivities. Org. Lett, 1543. [Google Scholar] [CrossRef]
- Bulman Page, P. C.; Buckley, B. R.; Heaney, H.; Blacker, A. J. Asymmetric epoxidation of cis-alkenes mediated by iminium salts: highly enantioselective synthesis of levcromakalim. Org. Lett. 2005, 7, 375–357. [Google Scholar] [CrossRef] [PubMed]
- Bulman Page, P. C.; Parker, P.; Rassias, G. A.; Buckley, B. R.; Bethell, D. Iminium salt-catalysed asymmetric epoxidation using hydrogen peroxide as stoichiometric oxidant. Adv. Synth. Catal. 2008, 350, 1867–1874. [Google Scholar] [CrossRef]
- Bulman Page, P. C.; Parker, P.; Buckley, B.; Rassias, G. A.; Bethell, D. Organocatalysis of asymmetric epoxidation by iminium salts using sodium hypochlorite as the stoichiometric oxidant. Tetrahedron 2009, 65, 15–11. [Google Scholar] [CrossRef]
- Bulman Page, P. C.; Bartlett, C. J.; Chan, Y.; Allin, S. M.; McKenzie, M. J.; Lacour, J.; Jone, G. A. New biphenyl iminium salt catalysts for highly enantioselective asymmetric epoxidation: role of additional substitution and dihedral angle. Org. Biomol. Chem. 2016, 14, 4220–4232. [Google Scholar] [CrossRef]
- D'Arcy, T. D.; Elsegood, M. R. J.; Buckley, B. R. Organocatalytic enantioselective synthesis of bicyclo[2.2.2]octenones via oxaziridinium catalysed ortho-hydroxylative phenol dearomatization. Angew. Chem. Int. Ed. 2022, 61(30), article nb. e202205278, pp. 1-6; [CrossRef]
- Pedersen, C. J. J. Am. Chem. Soc. 1967; 89(26), 7017-7036. [CrossRef]
- Cunningham, L. Supramolecular chemistry: An enabling tool for asymmetric catalysis. Synlett 2025, 36, 1189–1200. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, Y.; Tang, J.; Jiang, J.; Wang, L.; Li, S. Supramolecular asymmetric catalysis mediated by crown ethers and related recognition systems. Green Synth. Catal. 2021, 2, 156–164. [Google Scholar] [CrossRef]
- Cram, D. G.; Sogah, G. D. Y. Chiral crown complexes catalyse Michael addition reactions to give adducts in high optical yields. Chem. Commun. 1981, (13), 625–628. [Google Scholar] [CrossRef]
- Nagayama, S.; Kobayashi, S. A novel chiral lead(II) catalyst for enantioselective aldol reactions in aqueous media. J. Am. Chem. Soc. 2000, 122, 11531–11532. [Google Scholar] [CrossRef]
- Alonso-López, M.; Martín-Lomas, M.; Penadés, S. Asymmetric Michael reaction using macrocyclic lactose-derivatives as chiral catalysts. Tetrahedron Lett. 1986, (27), 3551–3554. [Google Scholar] [CrossRef]
- Suzuki, H.; Sato, I.; Yamashita, Y.; Kobayashi, S. Catalytic asymmetric direct-type 1,4-addition reactions of simple amides. J. Am. Chem. Soc. 2015, 137, 4336–4339. [Google Scholar] [CrossRef]
- Hori, K.; Tamura, M.; Tani, K.; Nishiwaki, N.; Ariga, M.; Tohda, Y. Asymmetric epoxidation catalyzed by novel azacrown ether-type chiral quaternary ammonium salts under phase-transfer catalytic conditions. Tetrahedron Lett. 2006, (47), 3115–3118. [Google Scholar] [CrossRef]
- Nakajima, M.; Saito, M.; Motoo, S.; Hashimoto, S-i. (S)-3,3‘-Dimethyl-2,2‘-biquinoline N,N‘-dioxide as an efficient catalyst for enantioselective addition of allyltrichlorosilanes to aldehydes. J. Am. Chem. Soc. 1998, 120, 6419–6420. [Google Scholar] [CrossRef]
- Shimada, T.; Kina, A.; Ikeda, S.; Hayashi, T. A novel axially chiral 2,2'-bipyridine N,N'-dioxide. Its preparation and use for asymmetric allylation of aldehydes with allyl(trichloro)silane as a highly efficient catalyst. Org. Lett. 2002, 4, 2799–2801. [Google Scholar] [CrossRef]
- Malkov, A. V.; Orsini, M.; Pernazza, D.; Muir, K. W.; Langer, V.; Meghani, P.; Kocovský, P. Chiral 2,2'-bipyridine-type N-monoxides as organocatalysts in the enantioselective allylation of aldehydes with allyltrichlorosilane. Org Lett. 2002, 4, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A. V.; Dufková, L.; Farrugia, L.; Kocovský, P. ; Quinox, a quinoline-type N-oxide, as organocatalyst in the asymmetric allylation of aromatic aldehydes with allyltrichlorosilanes: the role of arene-arene interactions. Angew. Chem. Int. Ed. 2003, 42, 3674–3677. [Google Scholar] [CrossRef] [PubMed]
- Romero-Arenas, A.; Ramírez-López, P.; Iglesias-Sigüenza, J.; Fernández, R.; Ros, A.; Lassaletta, J. M. Axially chiral N-oxide catalysts for the allylation and crotylation of aromatic aldehydes: Exploiting nonlinear effects. ChemCatChem 2024, 16, 1–6. [Google Scholar] [CrossRef]
- Iseki, K.; Mizuno, S.; Kuroki, Y.; Kobayashi, Y. Asymmetric allylation with chiral formamide catalysts. Tetrahedron 1999, 55, 977–988. [Google Scholar] [CrossRef]
- Meyers, A. I. Chiral oxazolines and their legacy in asymmetric carbon-carbon bond-forming reactions. J. Org. Chem. 2005, 70, 6137–6151. [Google Scholar] [CrossRef]
- Connon, R.; Roche, B.; Rokade, B. V.; Guiry, P. J. Further developments and applications of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev. 2021, 121, 6373–6521. [Google Scholar] [CrossRef]
- Yang, Z. Y.; Zeng, J. L.; Ren, N.; Meng, W.; Nie, J.; Ma, J. A. C2-Symmetric chiral bisoxazolines as hydrogen-bond-acceptor catalysts in enantioselective aldol reaction of β-carbonyl acids with trifluoroacetaldehyde hemiacetals. Org. Lett. 2016, 18, 6364–6367. [Google Scholar] [CrossRef]
- Wolff, M. E. Cyclization of N-halogenated amines (the Hofmann–Löffler reaction). Chem. Rev. 1963, 63, 55–64. [Google Scholar] [CrossRef]
- Guo, Q.; Mao, Y.; Liu, J.; Zhu, L.; Hong, X.; Lu, Z. Asymmetric Hofmann–Löffler–Freytag-type reaction via a transient carbenium ion complex merging organocatalysis and photocatalysis. Nat. Catal. 2025, 8, 448–456. [Google Scholar] [CrossRef]
- Ostler, F.; Piekarski, D. G.; Danelzik, T.; Taylor, M. S.; García Mancheño, O. Neutral chiral tetrakis-iodo-triazole halogen-bond donor for chiral recognition and enantioselective catalysis. Chem. Eur. J. 2021, 27, 2315–2320. [Google Scholar] [CrossRef]
- Keuper, A. C.; Fengler, K.; Ostler, F.; Danelzik, T.; Piekarski, D. G.; García Mancheño, O. Fine-tuning substrate-catalyst halogen-halogen interactions for boosting enantioselectivity in halogen-bonding catalysis. Angew. Chem. Int. Ed, 2023; 62, 1–7. [Google Scholar] [CrossRef]
- Wolf, J.; Mohanan, M. P.; Robidas, R.; Sutar, R.; Engelage, E.; Legault, C. Y.; Huber, S. M. Highly enantioselective organocatalysis with bidentate halogen bond donors. Angew. Chem. Int. Ed. 2025, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dreger, A.; Wonner, P.; Engelage, E.; Walter, S. M.; Stoll, R.; Huber, S. M. A halogen-bonding-catalysed Nazarov cyclisation reaction: Chem. Commun. 2019, 55, 8262–8265. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ao, T.; Mino, T.; Sakamoto, M. Chiral bromonium salt (hypervalent bromine(III)) with N-nitrosamine as a halogen-bonding bifunctional catalyst. Molecules 2023, 28, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Fujimura, T.; Mino, T.; Sakamoto, M. Chiral binaphthyl-based iodonium salt (hypervalent iodine(III)) as hydrogen- and halogen-bonding bifunctional catalyst: Insight into abnormal counteranion effect and asymmetric synthesis of N,S-acetals. Adv. Synth. Catal. 2022, 364, 1091–1098. [Google Scholar] [CrossRef]
- Damrath, M.; Scheele, T.; Duvinage, D.; Neudecker, T.; Nachtsheim, B. J. Chiral triazole-substituted iodonium salts in enantioselective halogen bond catalysis. ACS Catal. 2025, 15, 422–431. [Google Scholar] [CrossRef]
- Reinhard, D. L.; Iniutina, A.; Reese, S.; Shaw, T.; Merten, C.; List, B.; Huber, S. M. Asymmetric counteranion-directed halogen bonding catalysis. J. Am. Chem. Soc. 2025, 147(10), 8107–8112. [Google Scholar] [CrossRef]
- Reymond, J. L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef]
- Fehér, Z.; Richter, D.; Dargó, G.; Kupai, J. Factors influencing the performance of organocatalysts immobilized on solid supports: A review. Beilstein J. Org. Chem. 2024, 20, 2129–2142. [Google Scholar] [CrossRef]
- Lee, J.-W.; Mayer-Gall, T.; Opwis, K.; Song, C. E.; Gutmann, J. S.; List, B. Science, 2013; 341, 1225–1229. [CrossRef]
- Putman, J. I.; Armstrong, D. W. Recent advances in the field of chiral crystallisation. Chirality 2022, 34, 1338–1354. [Google Scholar] [CrossRef]
Figure 1.1.
Representation of enantioselective catalysis for the combination of a substrate A (represented by the black bar) with reagent B (represented by the red bar) in the presence of a chiral catalyst C* (represented by a right hand). The starting materials A + B are converted into chiral products (R)-A-B or (S)-A-B. They have the same stability in a non-chiral environment. All the species that combine A, B, and C*, such as short-lived intermediates or transition structures, are diastereomeric and have different stabilities and/or reactivities (reaction rates). Their transition states are associated with different free activation enthalpies (ΔG‡(R), ΔG‡(S)). The reader may use Lego® bricks (rectangular parallelepipeds) to construct similar objects and illustrate chirality.
Figure 1.1.
Representation of enantioselective catalysis for the combination of a substrate A (represented by the black bar) with reagent B (represented by the red bar) in the presence of a chiral catalyst C* (represented by a right hand). The starting materials A + B are converted into chiral products (R)-A-B or (S)-A-B. They have the same stability in a non-chiral environment. All the species that combine A, B, and C*, such as short-lived intermediates or transition structures, are diastereomeric and have different stabilities and/or reactivities (reaction rates). Their transition states are associated with different free activation enthalpies (ΔG‡(R), ΔG‡(S)). The reader may use Lego® bricks (rectangular parallelepipeds) to construct similar objects and illustrate chirality.
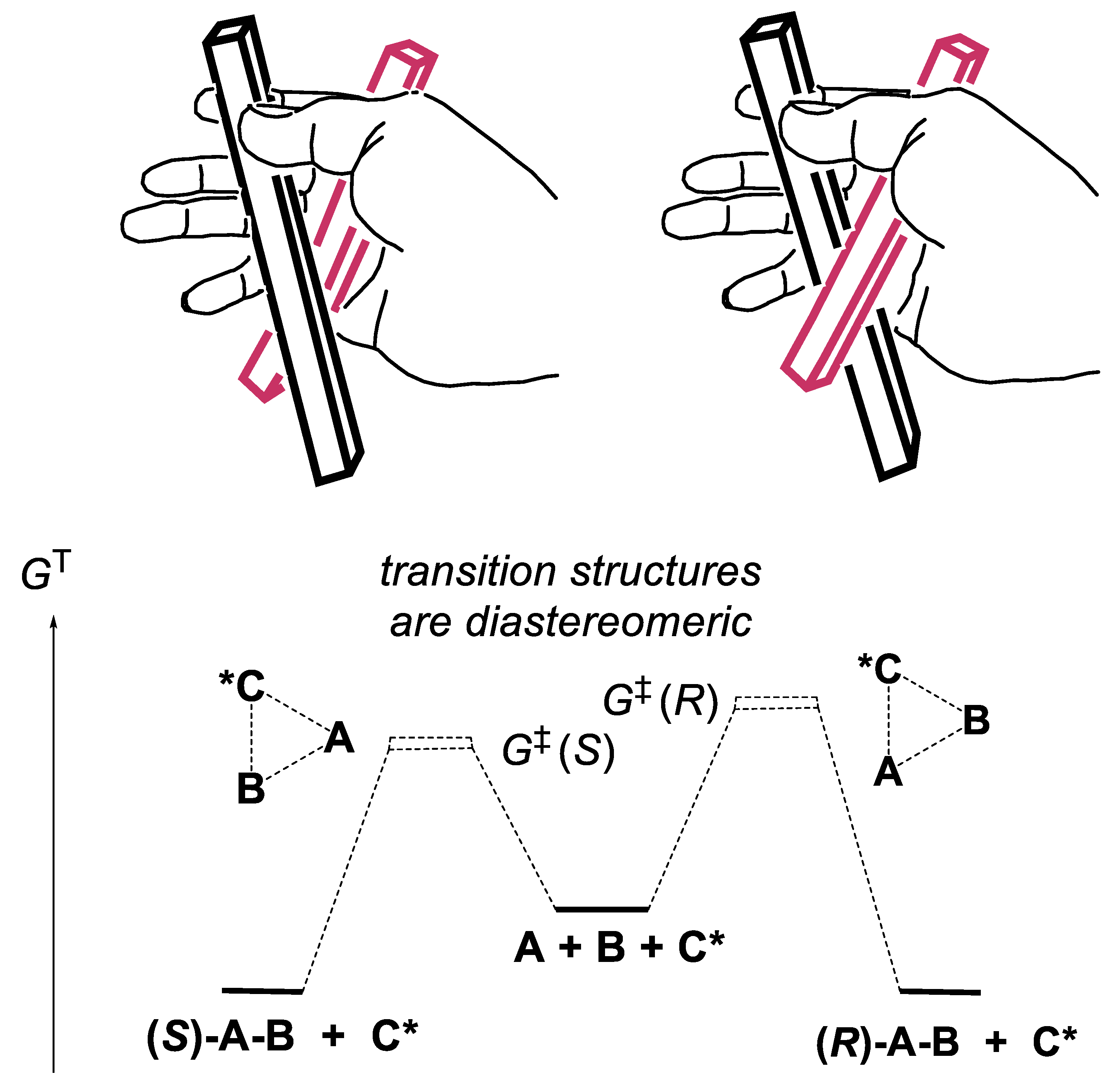
Figure 1.2.
Representation of a catalyzed asymmetric reaction implying activation of an achiral reactant A through a chemical transformation with the chiral catalyst C*. A chiral intermediate A-C* forms first which equilibrates then with the more reactive intermediate A’-C*. The latter reacts preferentially with one face of the prochiral substrate B (represented by an irregular triangle), leading preferentially to enantiomer (R)-A-B of the product. This implies that intermediate A’-C* combines with prochiral B forming two possible diastereomeric complexes or intermediates (R)-A-B-C* and (S)-A-B-C*; they do not have the same stability and/or react with different rates. In the case represented, (R)-A-B-C* and (S)-A-B-C* equilibrate with the starting materials, but the concentration of (R)-A-B-C*, which is higher than that of (S)-A-B-C*. The energy barrier for the transformation of (R)-A-B-C* to product (R)-A-B is nearly the same as that of the conversion (S)-A-B-C* into (S)-A-B. The catalyst is represented by a right hand which grabs first A and then activates it into A’. The hand does this with its dumb that presses down the piston (change of chemical function), liberating the pointer that help to find the place where A has to be attached to B (around the hole). Only one of the two faces of B can react. Because of its shape (irregular triangle), it can fit into the cavity realized by the bottom of the hand for only one of the two possible orientations. When A’ of A’-C* and B have been glued together the hand suppresses the pressure on the piston and let product (R)-A-B to swim away.
Figure 1.2.
Representation of a catalyzed asymmetric reaction implying activation of an achiral reactant A through a chemical transformation with the chiral catalyst C*. A chiral intermediate A-C* forms first which equilibrates then with the more reactive intermediate A’-C*. The latter reacts preferentially with one face of the prochiral substrate B (represented by an irregular triangle), leading preferentially to enantiomer (R)-A-B of the product. This implies that intermediate A’-C* combines with prochiral B forming two possible diastereomeric complexes or intermediates (R)-A-B-C* and (S)-A-B-C*; they do not have the same stability and/or react with different rates. In the case represented, (R)-A-B-C* and (S)-A-B-C* equilibrate with the starting materials, but the concentration of (R)-A-B-C*, which is higher than that of (S)-A-B-C*. The energy barrier for the transformation of (R)-A-B-C* to product (R)-A-B is nearly the same as that of the conversion (S)-A-B-C* into (S)-A-B. The catalyst is represented by a right hand which grabs first A and then activates it into A’. The hand does this with its dumb that presses down the piston (change of chemical function), liberating the pointer that help to find the place where A has to be attached to B (around the hole). Only one of the two faces of B can react. Because of its shape (irregular triangle), it can fit into the cavity realized by the bottom of the hand for only one of the two possible orientations. When A’ of A’-C* and B have been glued together the hand suppresses the pressure on the piston and let product (R)-A-B to swim away.
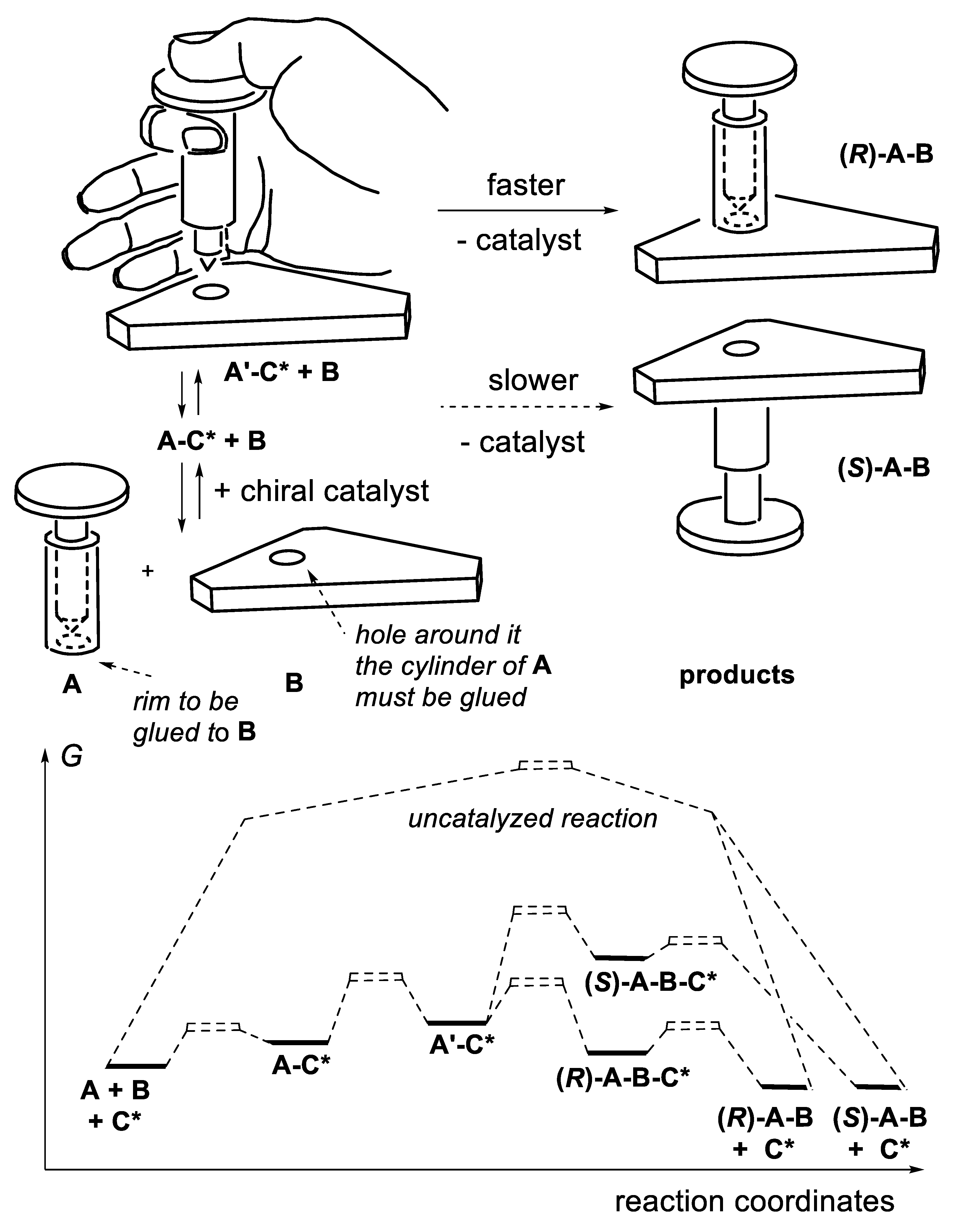
Figure 2.1.
The prochiral carbonyl compound (2-butanone) has a re face, and a si face. H2 addition onto the si face produces the (R)-2-butanol, and the H2 addition to the re face gives (S)-2-butanol [129].
Figure 2.1.
The prochiral carbonyl compound (2-butanone) has a re face, and a si face. H2 addition onto the si face produces the (R)-2-butanol, and the H2 addition to the re face gives (S)-2-butanol [129].
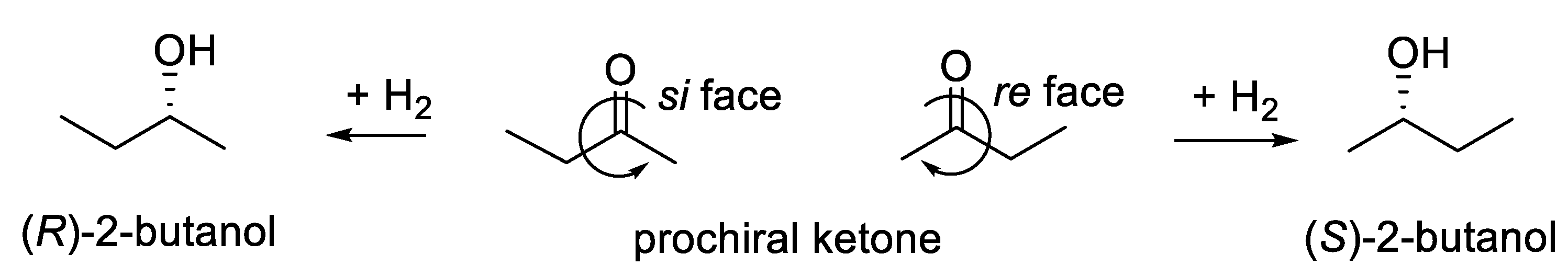
Figure 2.2.
Example of fermentative oxidation of bromobenzene into a chiral diol, synthetic intermediate in the stereoselective synthesis of a 6-amino-5-fluorocyclohexane-1,2,3,4-tetrol derivative.
Figure 2.2.
Example of fermentative oxidation of bromobenzene into a chiral diol, synthetic intermediate in the stereoselective synthesis of a 6-amino-5-fluorocyclohexane-1,2,3,4-tetrol derivative.
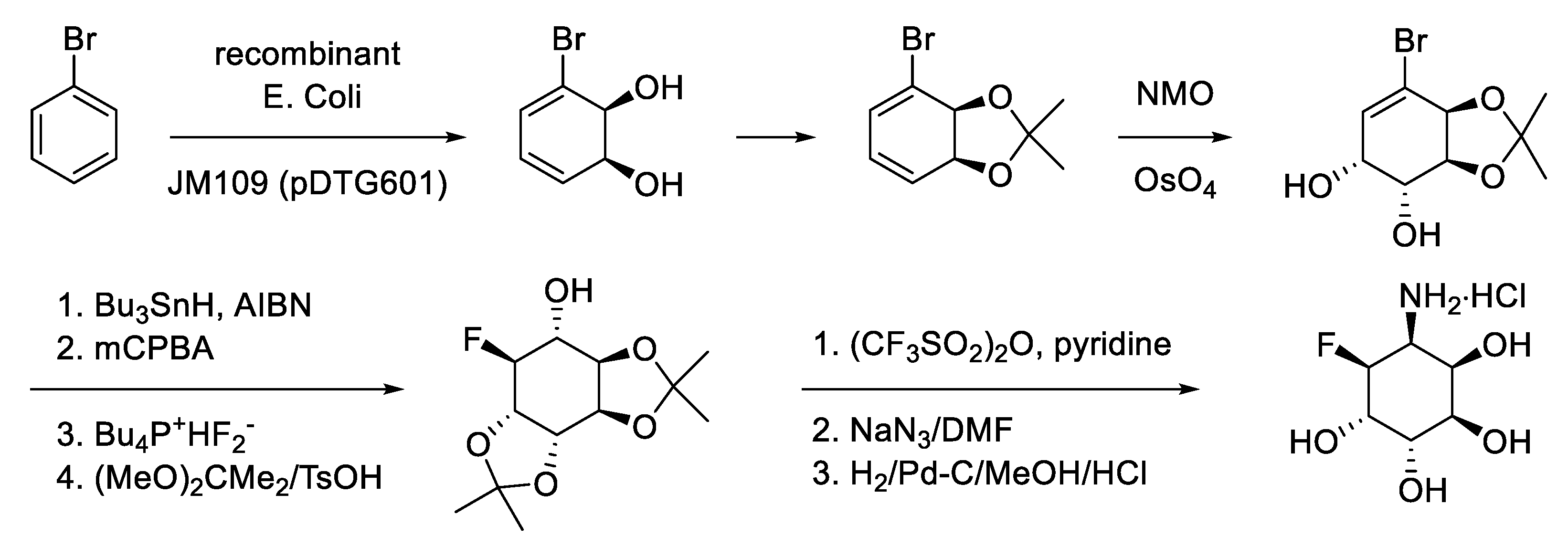
Figure 2.3.
Mechanism of Type I or Type II aldolase-catalyzed aldol and retro-aldol reactions (2.8). Type I involves the addition of an intermediate enamine onto the carbonyl group. Type II implies the enolate addition onto the carbonyl group activated with an imidazolyl complexed Lewis acid.
Figure 2.3.
Mechanism of Type I or Type II aldolase-catalyzed aldol and retro-aldol reactions (2.8). Type I involves the addition of an intermediate enamine onto the carbonyl group. Type II implies the enolate addition onto the carbonyl group activated with an imidazolyl complexed Lewis acid.
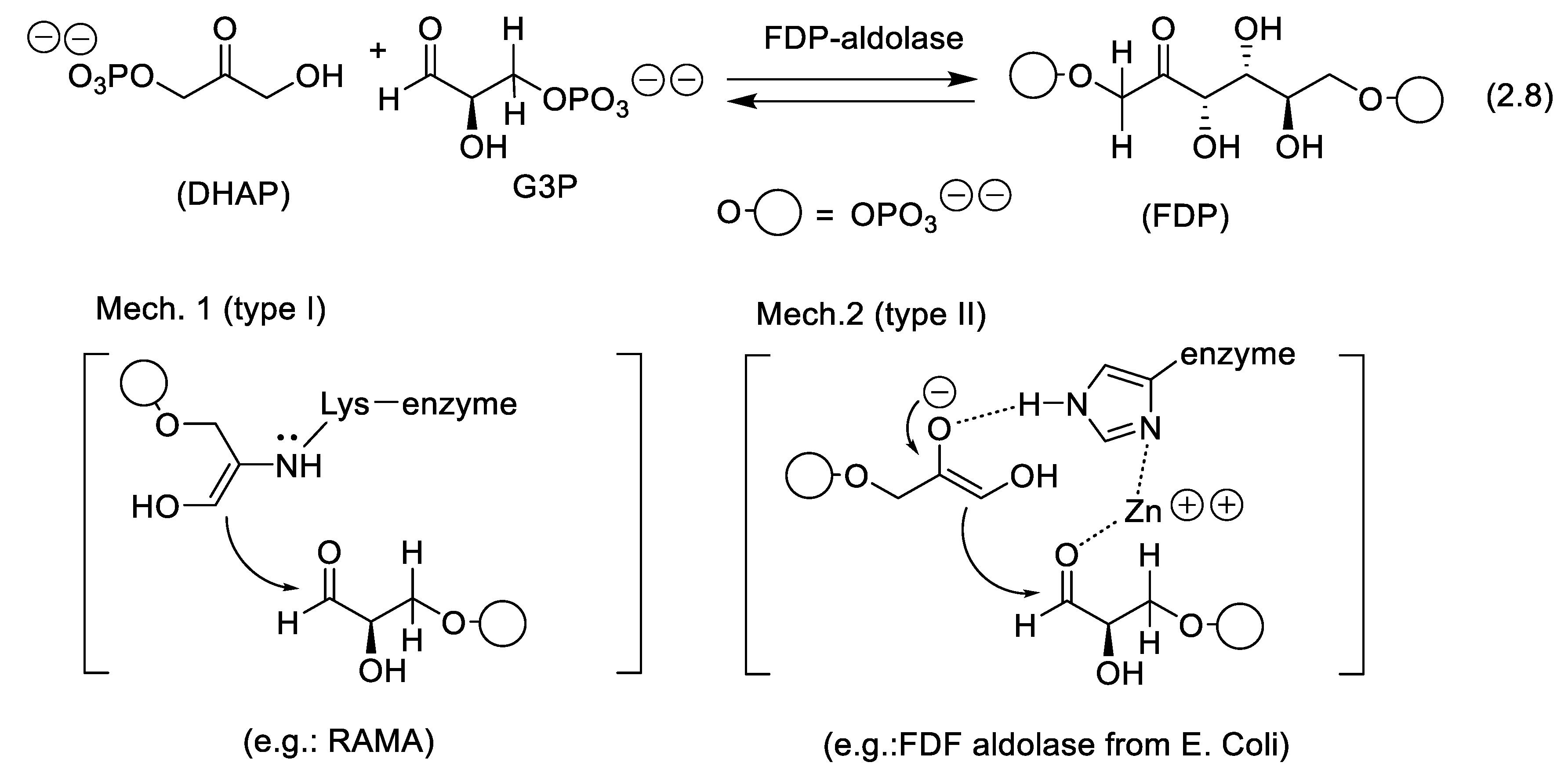
Figure 2.4.
DERA-induced asymmetric synthesis of the side-chain of Atorvastatin, the drug that has brought the highest financial benefits to the pharma industry.
Figure 2.4.
DERA-induced asymmetric synthesis of the side-chain of Atorvastatin, the drug that has brought the highest financial benefits to the pharma industry.
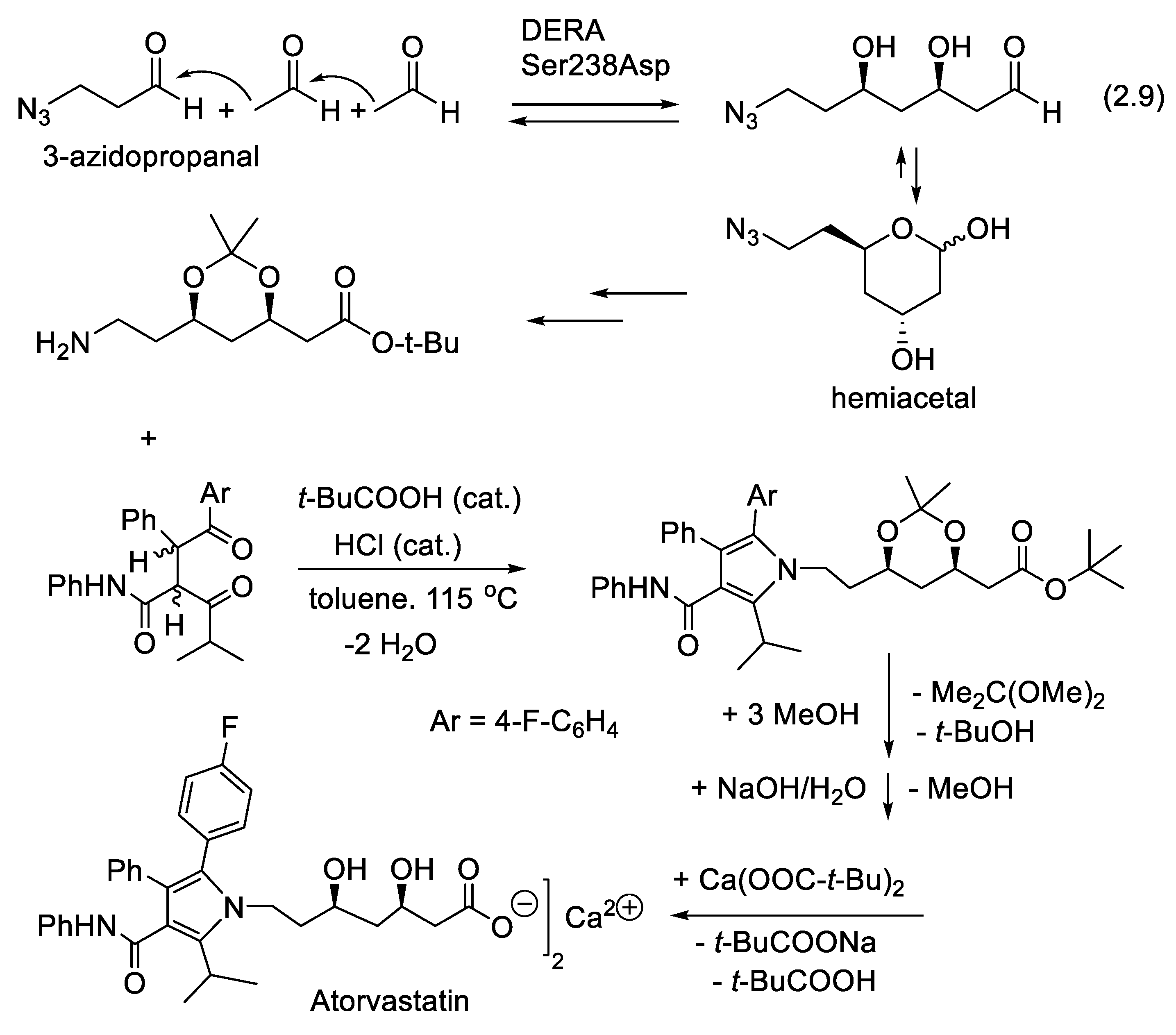
Figure 2.5.
a) An example of asymmetric α-alkylation of α-ketocarboxylic acids catalyzed by a mutant alkyltransferase; b) Proposed transition state for the enzyme-catalyzed α-methylation of pyruvates by S-adenosinsylmethionine (SAM).
Figure 2.5.
a) An example of asymmetric α-alkylation of α-ketocarboxylic acids catalyzed by a mutant alkyltransferase; b) Proposed transition state for the enzyme-catalyzed α-methylation of pyruvates by S-adenosinsylmethionine (SAM).
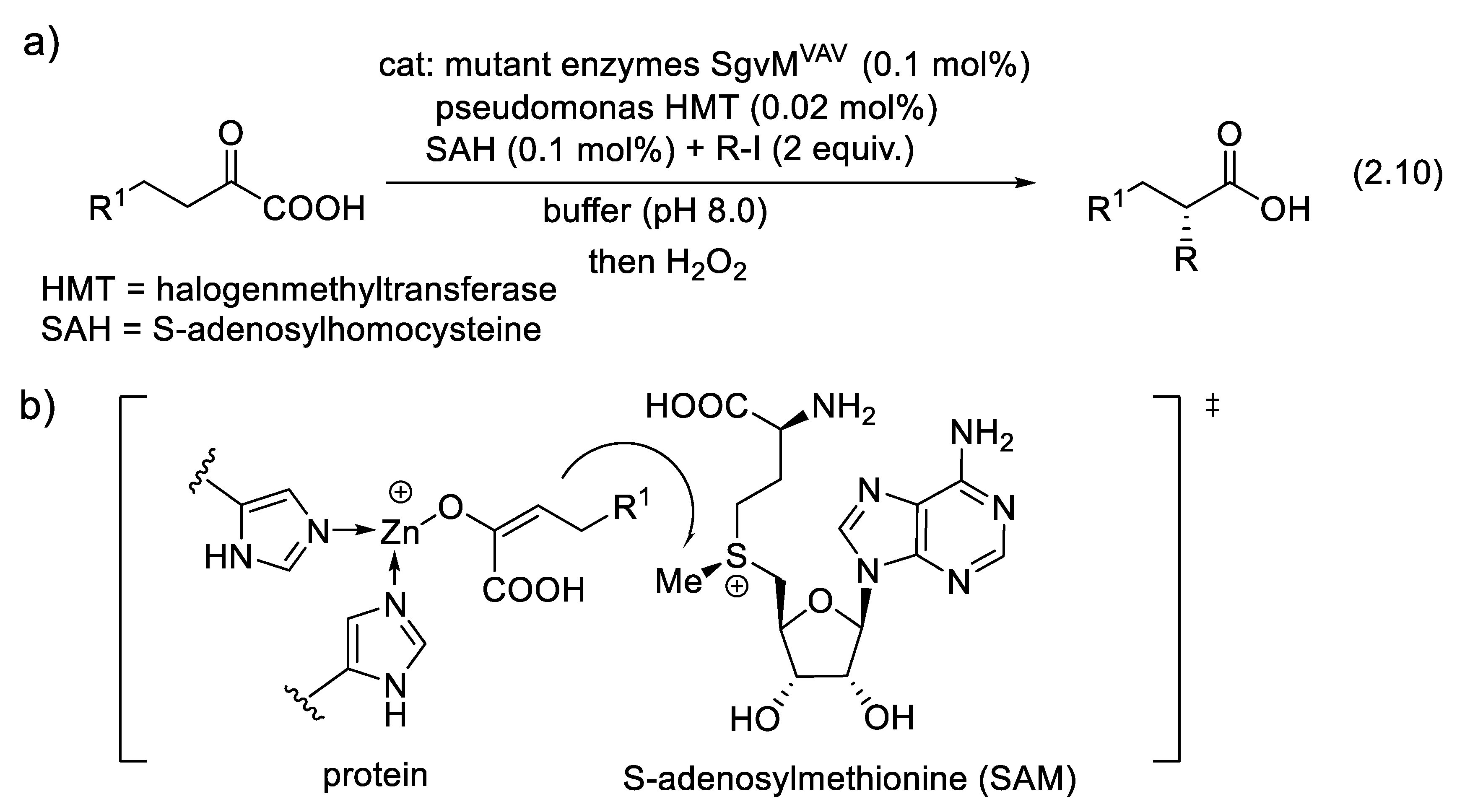
Figure 3.1.
a) Structures of natural (-)-quinine, (+)-quinidine, and derivatives used as chiral base catalysts; b) First example of an enantioselective amine-catalyzed Kornblum-DeLaMare rearrangement (Vogel, 1980).
Figure 3.1.
a) Structures of natural (-)-quinine, (+)-quinidine, and derivatives used as chiral base catalysts; b) First example of an enantioselective amine-catalyzed Kornblum-DeLaMare rearrangement (Vogel, 1980).
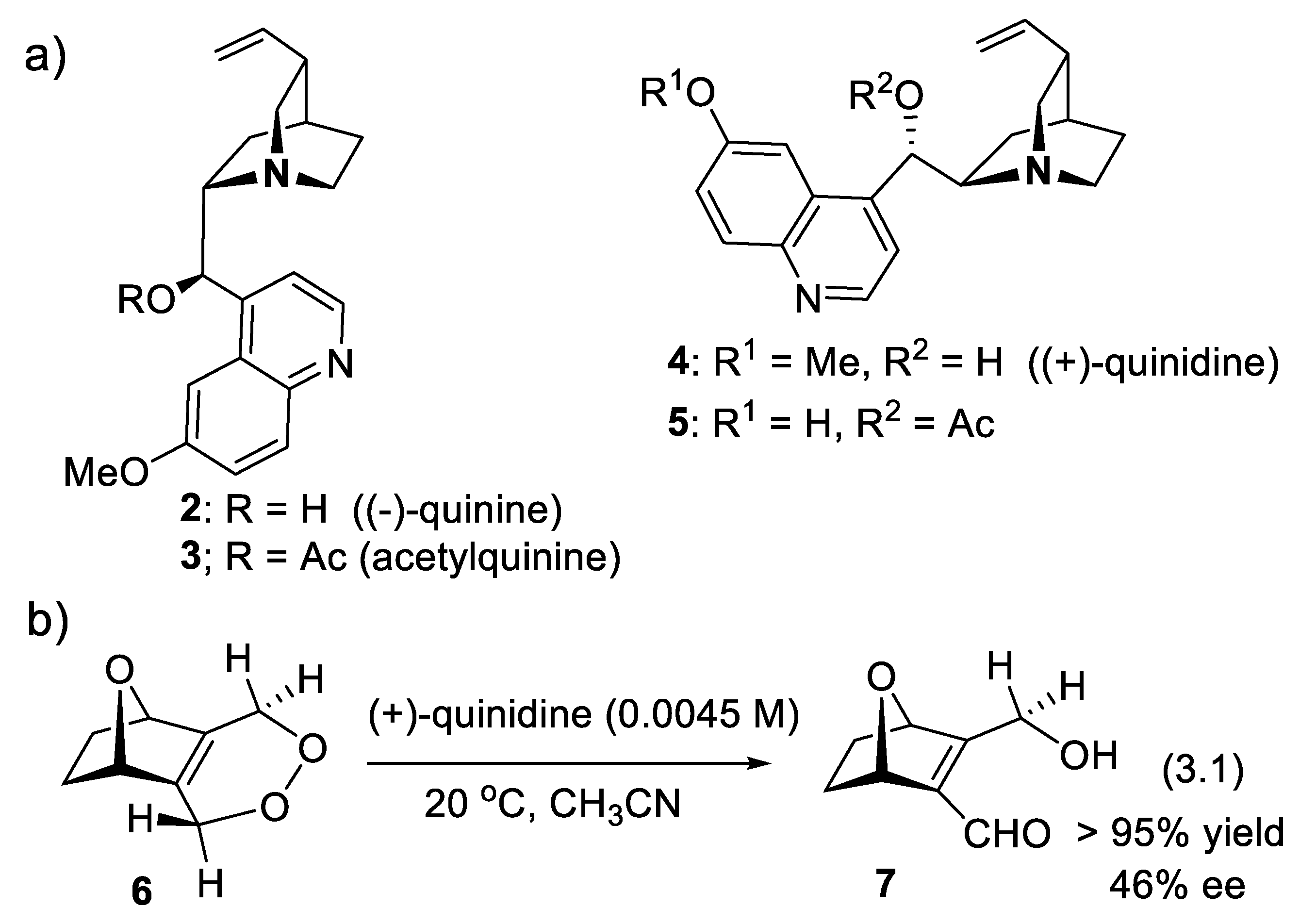
Figure 3.2.
Typical cinchona alkaloid derivatives used as chiral bases: examples of applications. a) α-Amino-oxidation of a ketone (Barbas, 2010); b) (4+2)-annulation of the enol of buty-3-yn-2-one to a highly electrophilic ketone (Xu & Shi, 2013).
Figure 3.2.
Typical cinchona alkaloid derivatives used as chiral bases: examples of applications. a) α-Amino-oxidation of a ketone (Barbas, 2010); b) (4+2)-annulation of the enol of buty-3-yn-2-one to a highly electrophilic ketone (Xu & Shi, 2013).
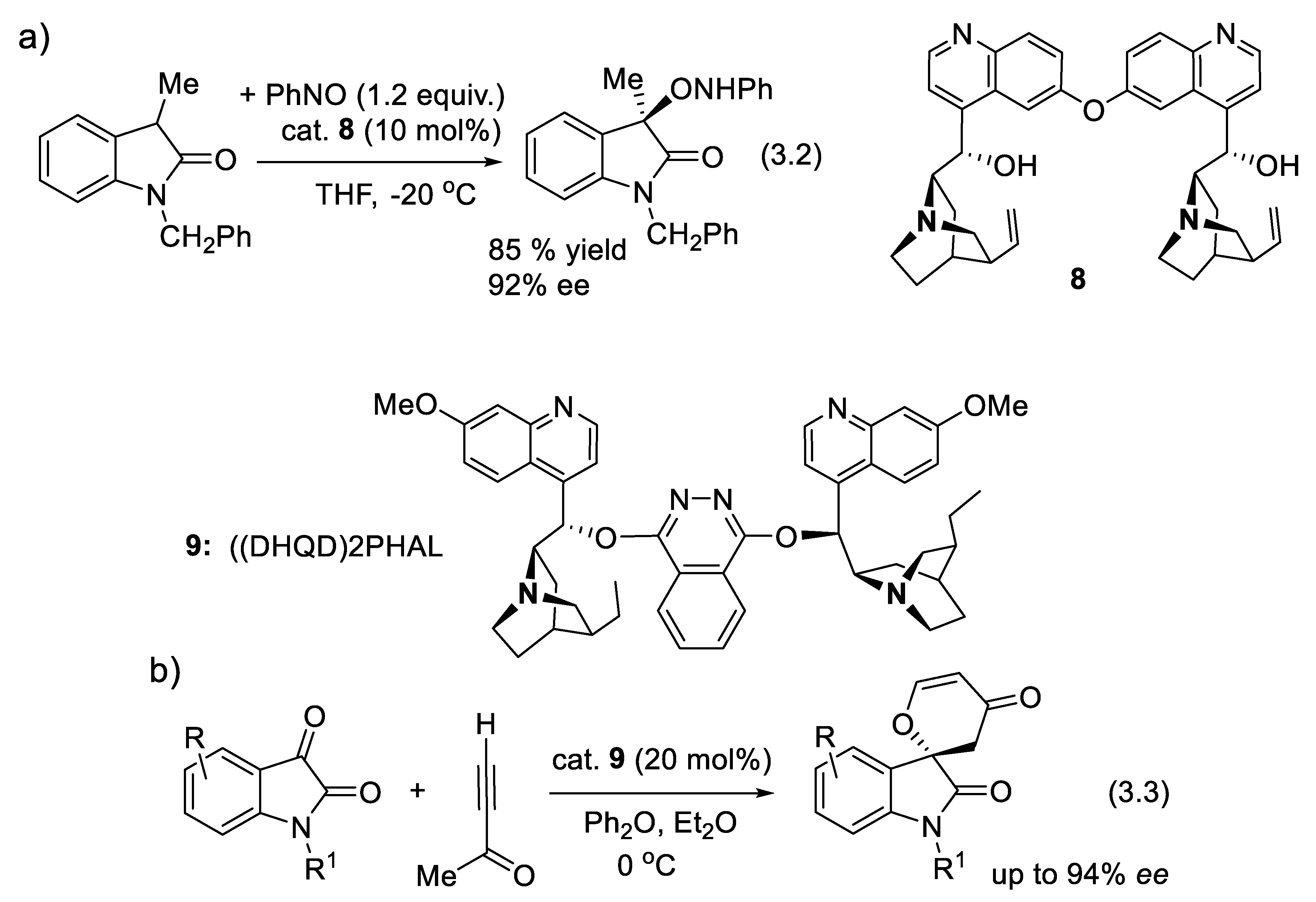
Figure 3.3.
Examples of cinchona alkaloid-derived catalysts providing H-bonding activation (see Chap. 9). They are examples of multifunctional catalysts (Brønsted base and H-bond donors).
Figure 3.3.
Examples of cinchona alkaloid-derived catalysts providing H-bonding activation (see Chap. 9). They are examples of multifunctional catalysts (Brønsted base and H-bond donors).
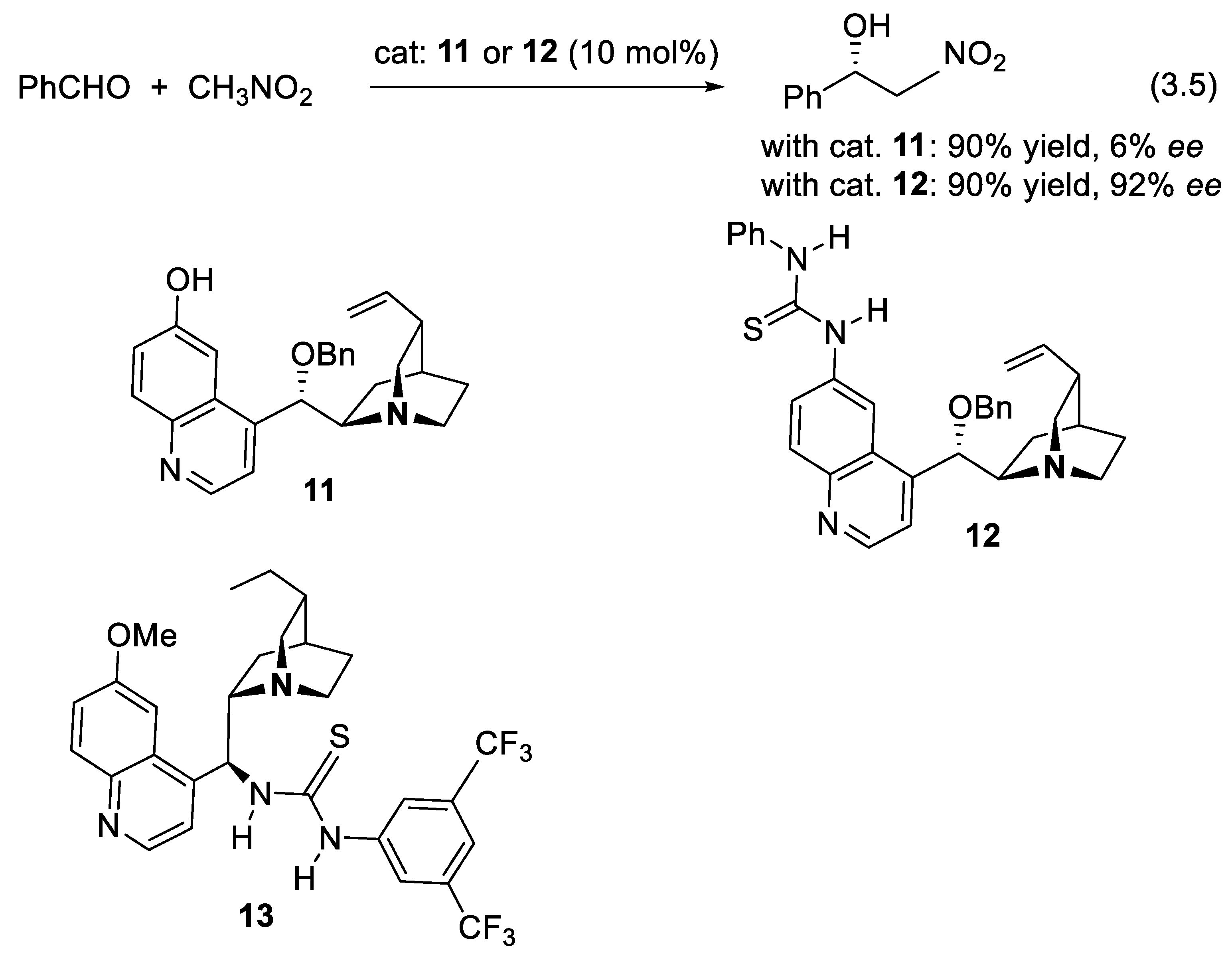
Figure 3.4.
An early example of asymmetric protonation of a carbanion intermediate with (-)-quinine (2) as catalyst (Kumar & Dyke, 1991).
Figure 3.4.
An early example of asymmetric protonation of a carbanion intermediate with (-)-quinine (2) as catalyst (Kumar & Dyke, 1991).
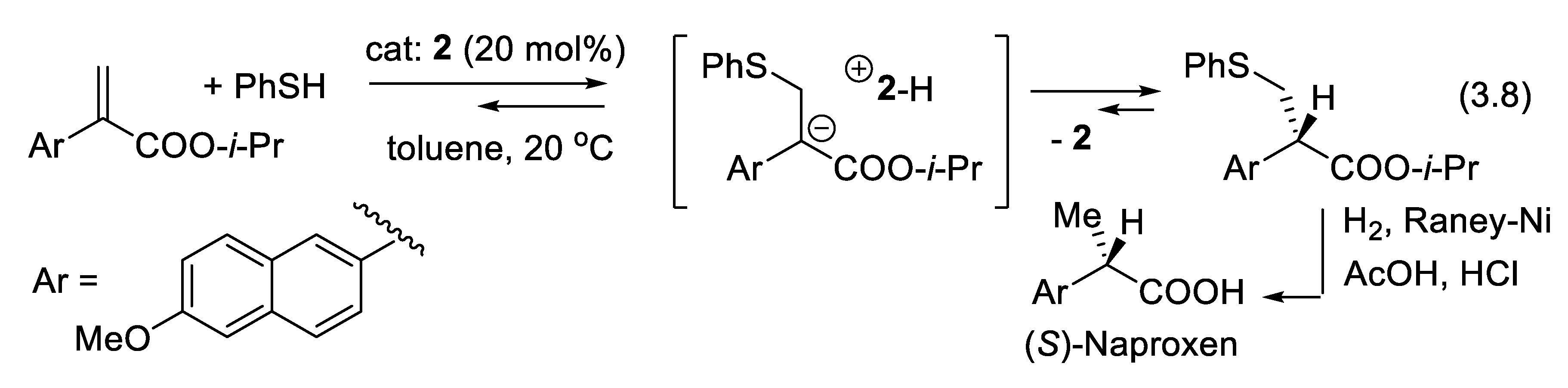
Figure 3.5.
Example of asymmetric lithium enolate protonation with synthetic polyamines: a) (Koga, 1993); b) (Łowicki , 2021).
Figure 3.5.
Example of asymmetric lithium enolate protonation with synthetic polyamines: a) (Koga, 1993); b) (Łowicki , 2021).
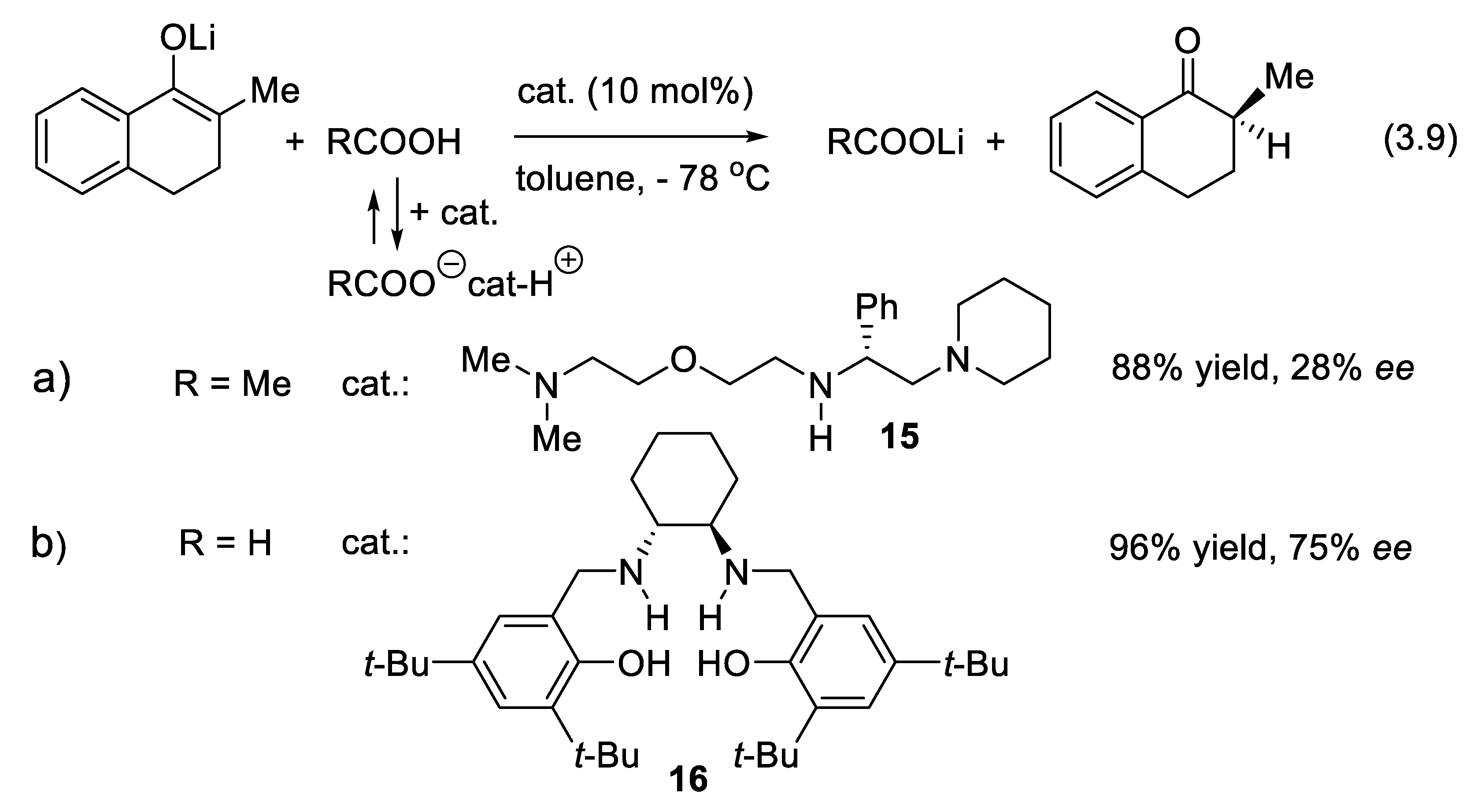
Figure 3.6.
Asymmetric synthesis of benzazocinones (Chen, 2020).
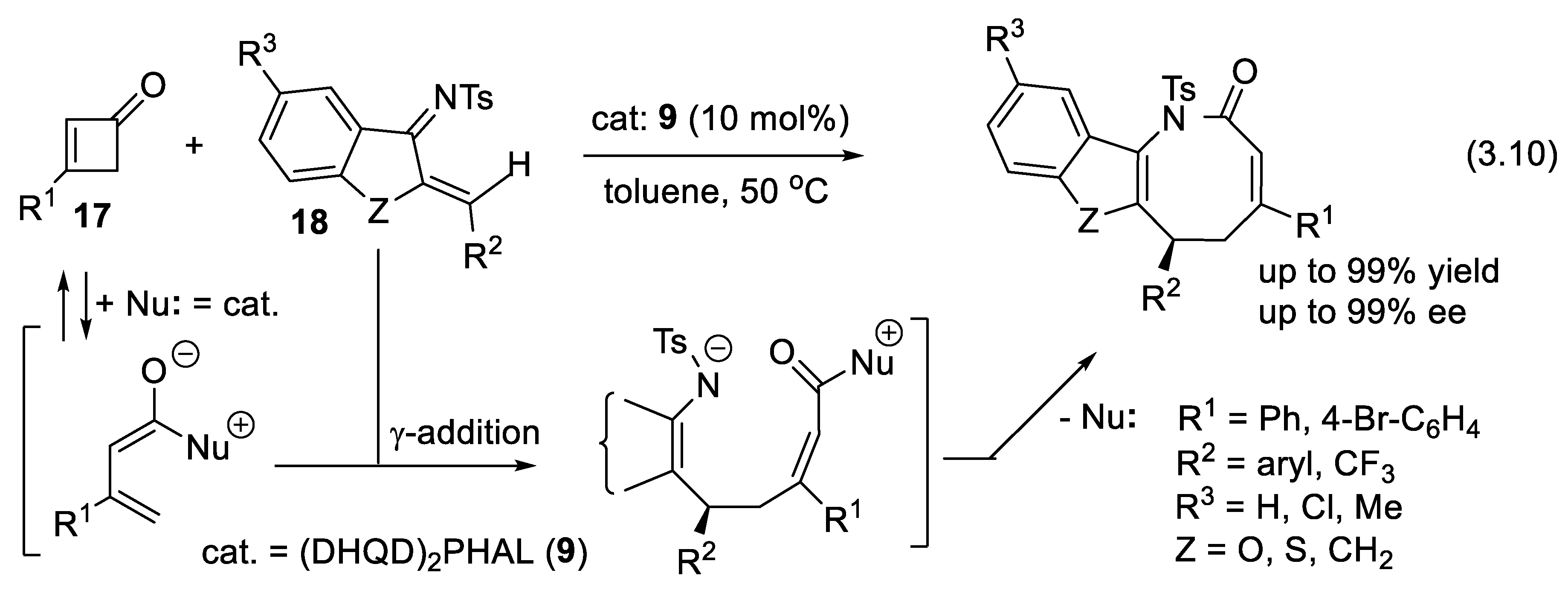
Figure 3.8.
Asymmetric Michael additions of enolizable carbonyl compounds activated through formation of enamine intermediates with the primary amine of the chiral catalyst (Connon, 2007).
Figure 3.8.
Asymmetric Michael additions of enolizable carbonyl compounds activated through formation of enamine intermediates with the primary amine of the chiral catalyst (Connon, 2007).
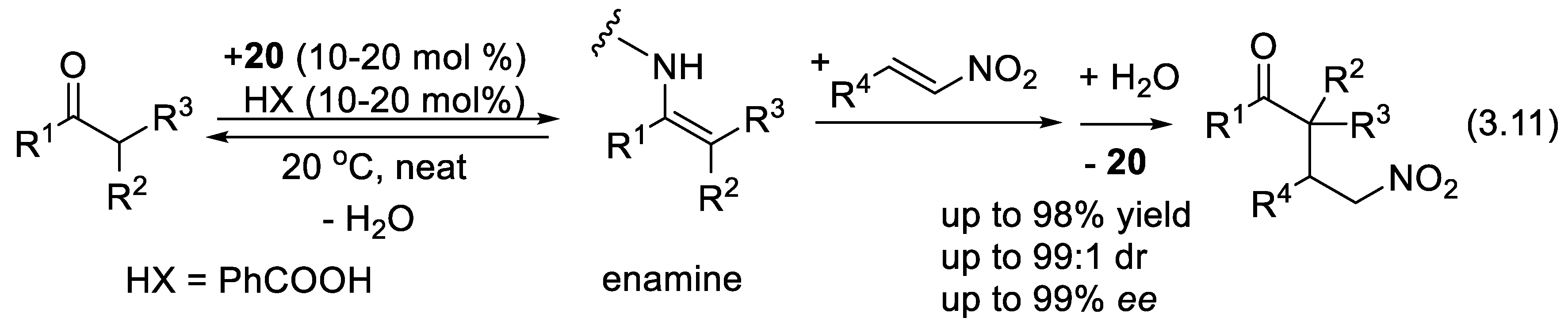
Figure 3.9.
Examples of asymmetric nucleophilic substitutions catalyzed by phase transfer catalysts. a) Asymmetric α-methylation of a ketone (Dolling, 1984); b) Asymmetric synthesis of α-amino acids (O’Donnel, 1989); formation of chiral carbanion/ammonium ion pairs as reactive intermediates; c) Asymmetric aziridination (Prabhakar, 1996); formation of chiral nucleophilic nitrogen intermediates.
Figure 3.9.
Examples of asymmetric nucleophilic substitutions catalyzed by phase transfer catalysts. a) Asymmetric α-methylation of a ketone (Dolling, 1984); b) Asymmetric synthesis of α-amino acids (O’Donnel, 1989); formation of chiral carbanion/ammonium ion pairs as reactive intermediates; c) Asymmetric aziridination (Prabhakar, 1996); formation of chiral nucleophilic nitrogen intermediates.
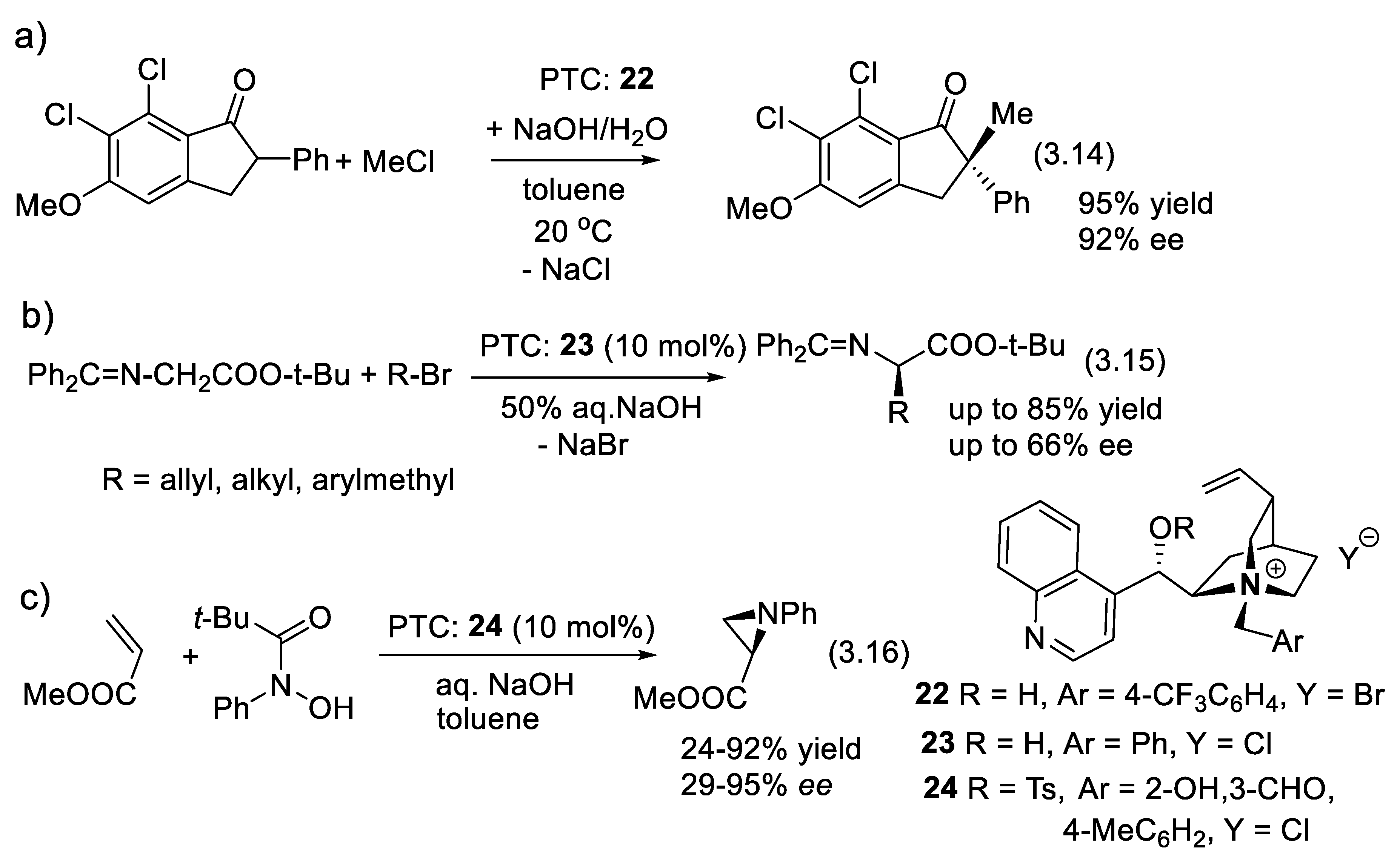
Figure 3.10.
Asymmetric synthesis of Pregabalin.
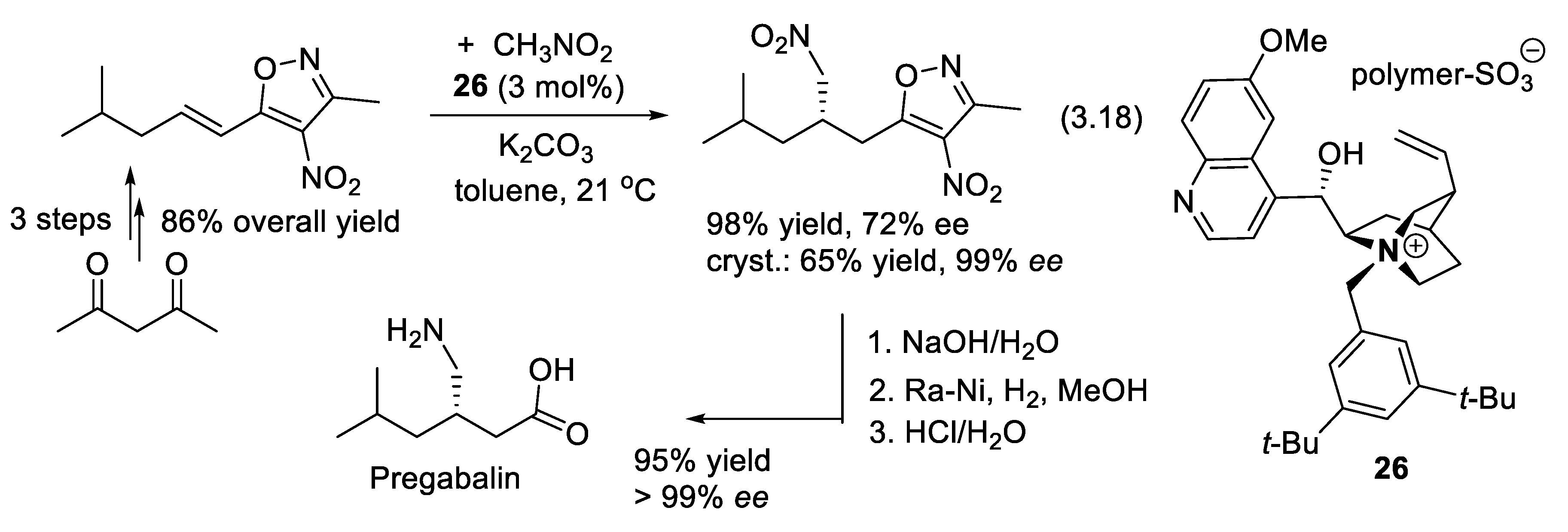
Figure 3.11.
a) PTC-catalyzed asymmetric α-alkylation of protected amines (Zheng, 2024); b) Possible mechanism.
Figure 3.11.
a) PTC-catalyzed asymmetric α-alkylation of protected amines (Zheng, 2024); b) Possible mechanism.
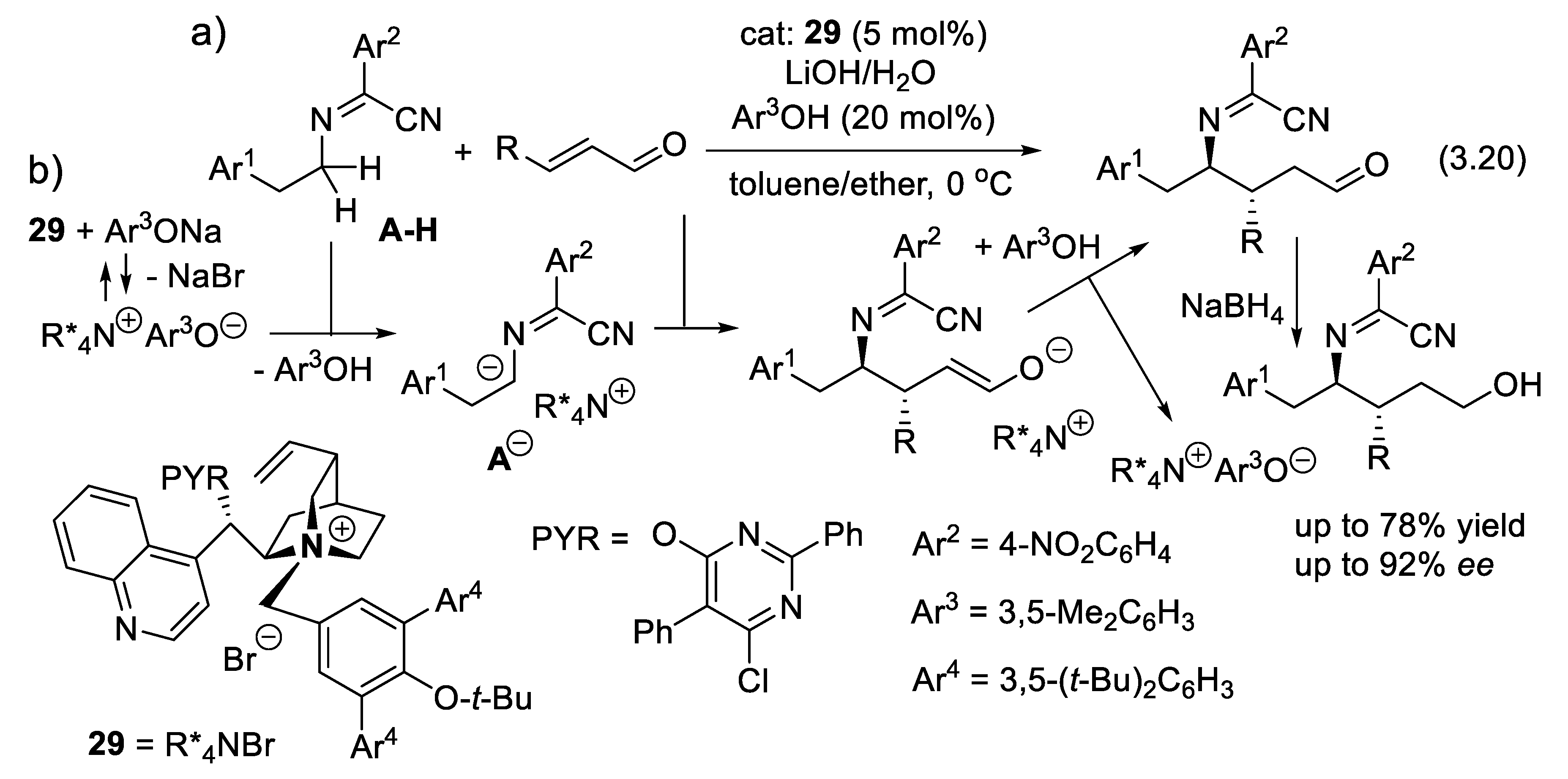
Figure 3.12.
Examples of non-cinchona-derived PTCs; a) ammonium and bisammonium salts derived from tartaric acids; b) ammonium salts derived from BINOLs; c) guadinidium salts; d) 1,2,3-triazolium salts with one amido N-H bond.
Figure 3.12.
Examples of non-cinchona-derived PTCs; a) ammonium and bisammonium salts derived from tartaric acids; b) ammonium salts derived from BINOLs; c) guadinidium salts; d) 1,2,3-triazolium salts with one amido N-H bond.
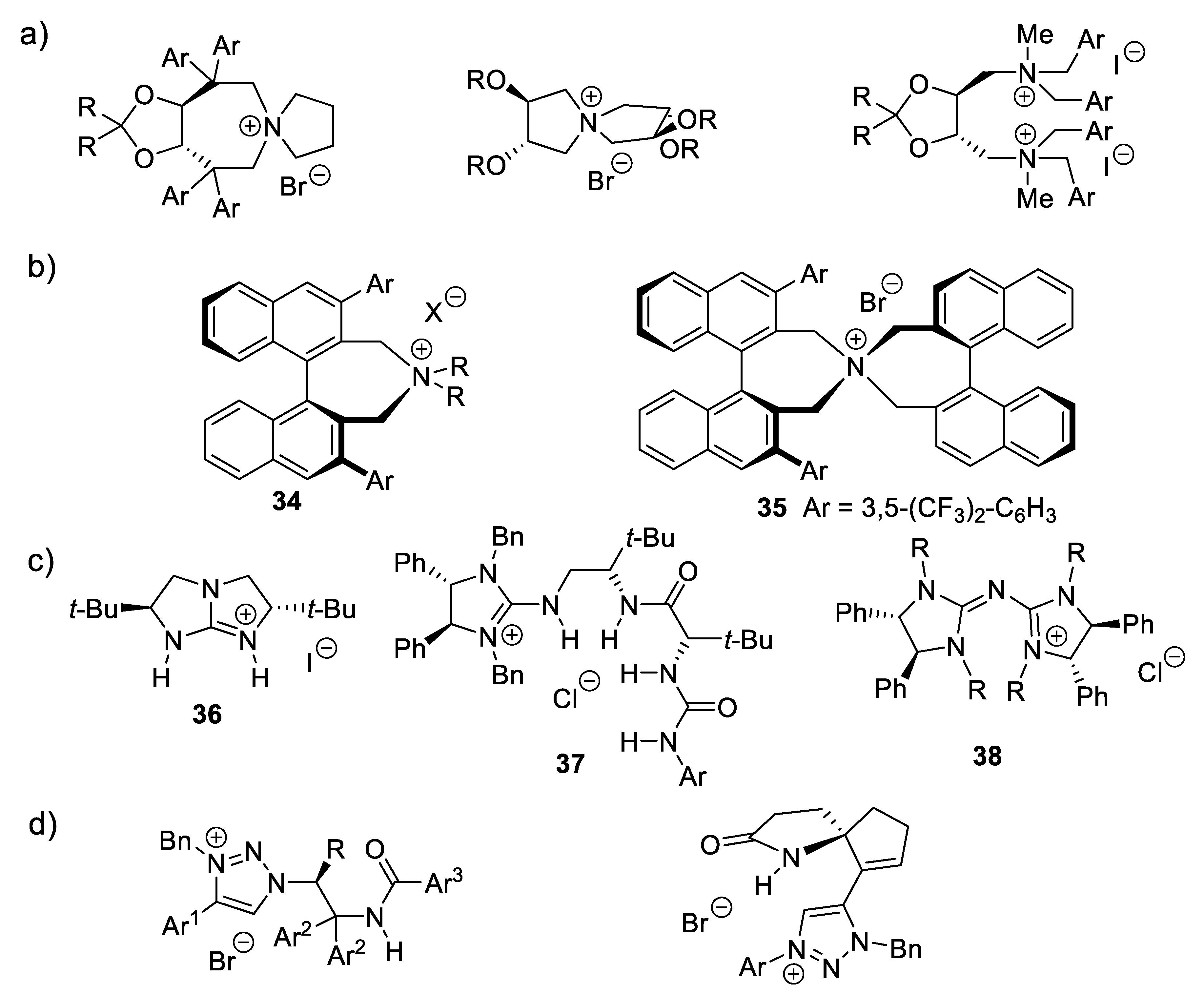
Figure 3.13.
Examples of non-cinchona-derived ammonium phase-transfer catalysts that are H-bond donors (HB-PTCs).
Figure 3.13.
Examples of non-cinchona-derived ammonium phase-transfer catalysts that are H-bond donors (HB-PTCs).
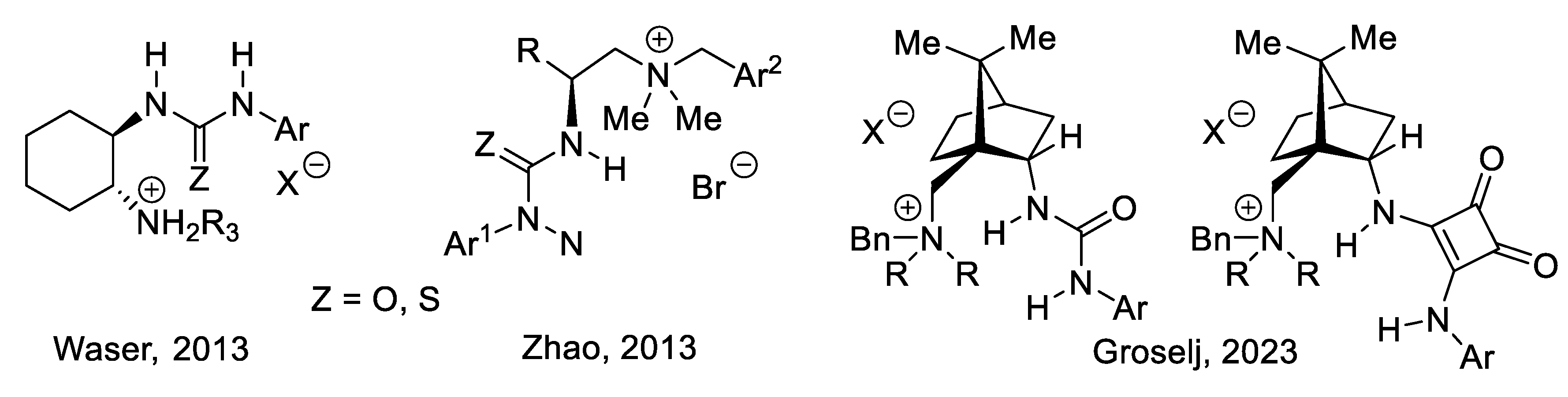
Figure 3.14.
Phipps’ (2024) enantioselective epimerization of meso-1,2-diols; application of MacMillan’s (2015) enantiotopic H-atom abstraction through activation by ammonium radical-cation intermediates.
Figure 3.14.
Phipps’ (2024) enantioselective epimerization of meso-1,2-diols; application of MacMillan’s (2015) enantiotopic H-atom abstraction through activation by ammonium radical-cation intermediates.
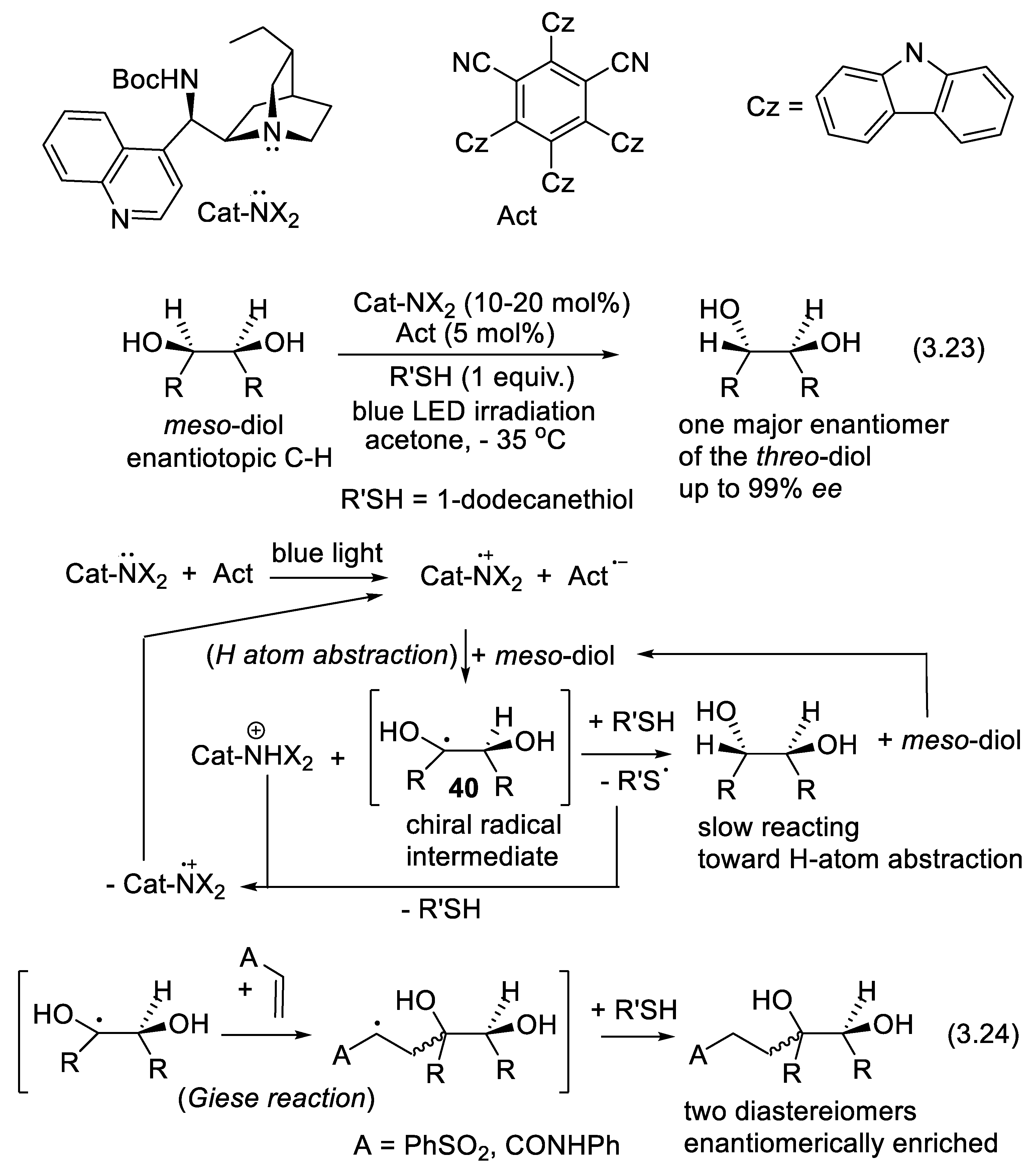
Figure 3.15.
a) Enantioselective Diels-Alder reaction of cyclohexa-1,3-diene and acrolein catalyzed by chiral secondary amine 41 (MacMillan, 2000); b) Enantioselective Diels-Alder reactions of acyclic 1,3-dienes and conjugated enones, catalyzed by a chiral secondary amine 44. These catalyzed reactions are iminium ion LUMO driven (MacMillan, 2002).
Figure 3.15.
a) Enantioselective Diels-Alder reaction of cyclohexa-1,3-diene and acrolein catalyzed by chiral secondary amine 41 (MacMillan, 2000); b) Enantioselective Diels-Alder reactions of acyclic 1,3-dienes and conjugated enones, catalyzed by a chiral secondary amine 44. These catalyzed reactions are iminium ion LUMO driven (MacMillan, 2002).
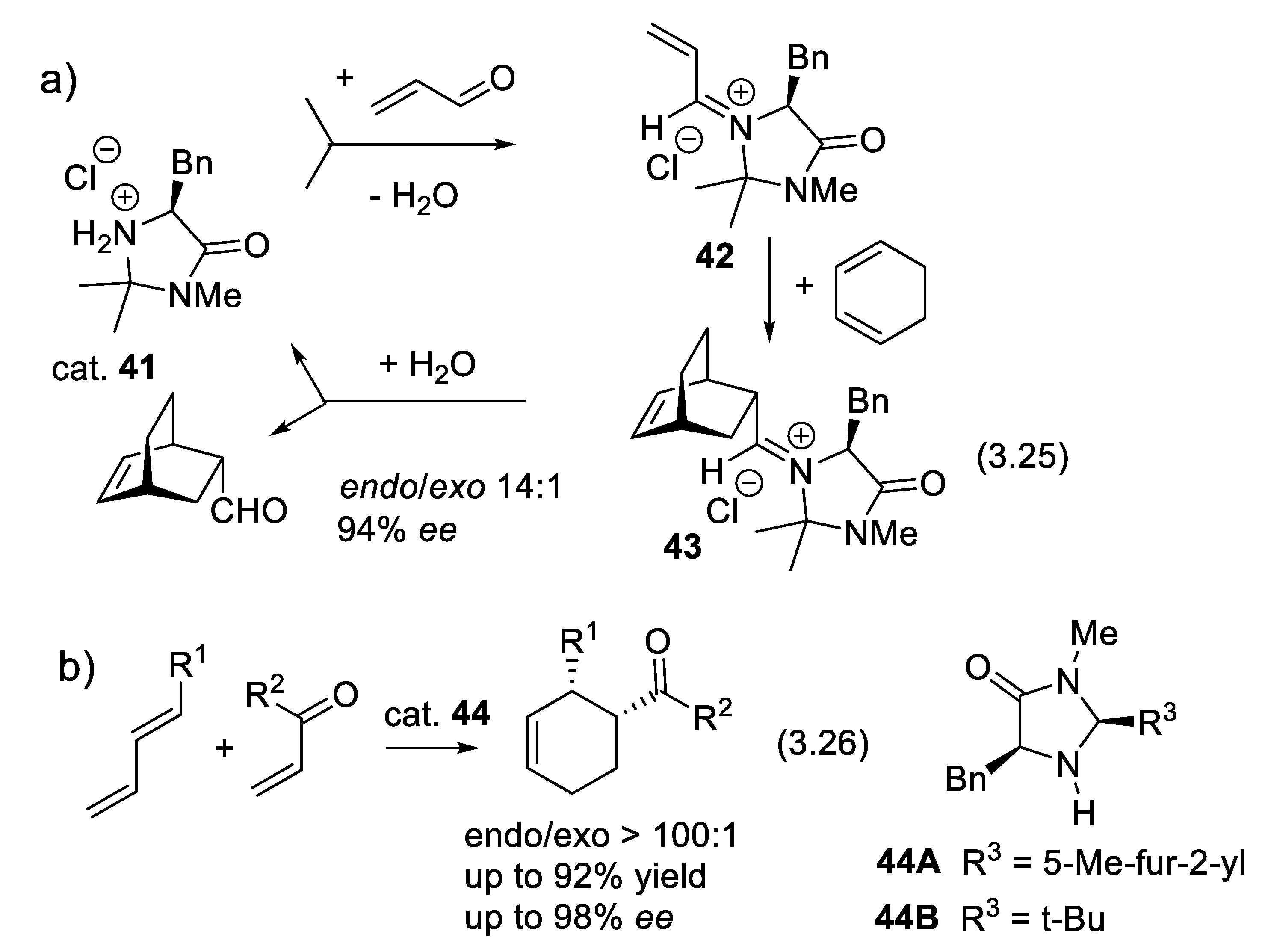
Figure 3.16.
a) Enantioselective 1,4-alkylation of enones by alkylsilanes induced by visible light (Melchiorre, 2018); b) Possible mechanism implying enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion/carbazole complex 48.
Figure 3.16.
a) Enantioselective 1,4-alkylation of enones by alkylsilanes induced by visible light (Melchiorre, 2018); b) Possible mechanism implying enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion/carbazole complex 48.
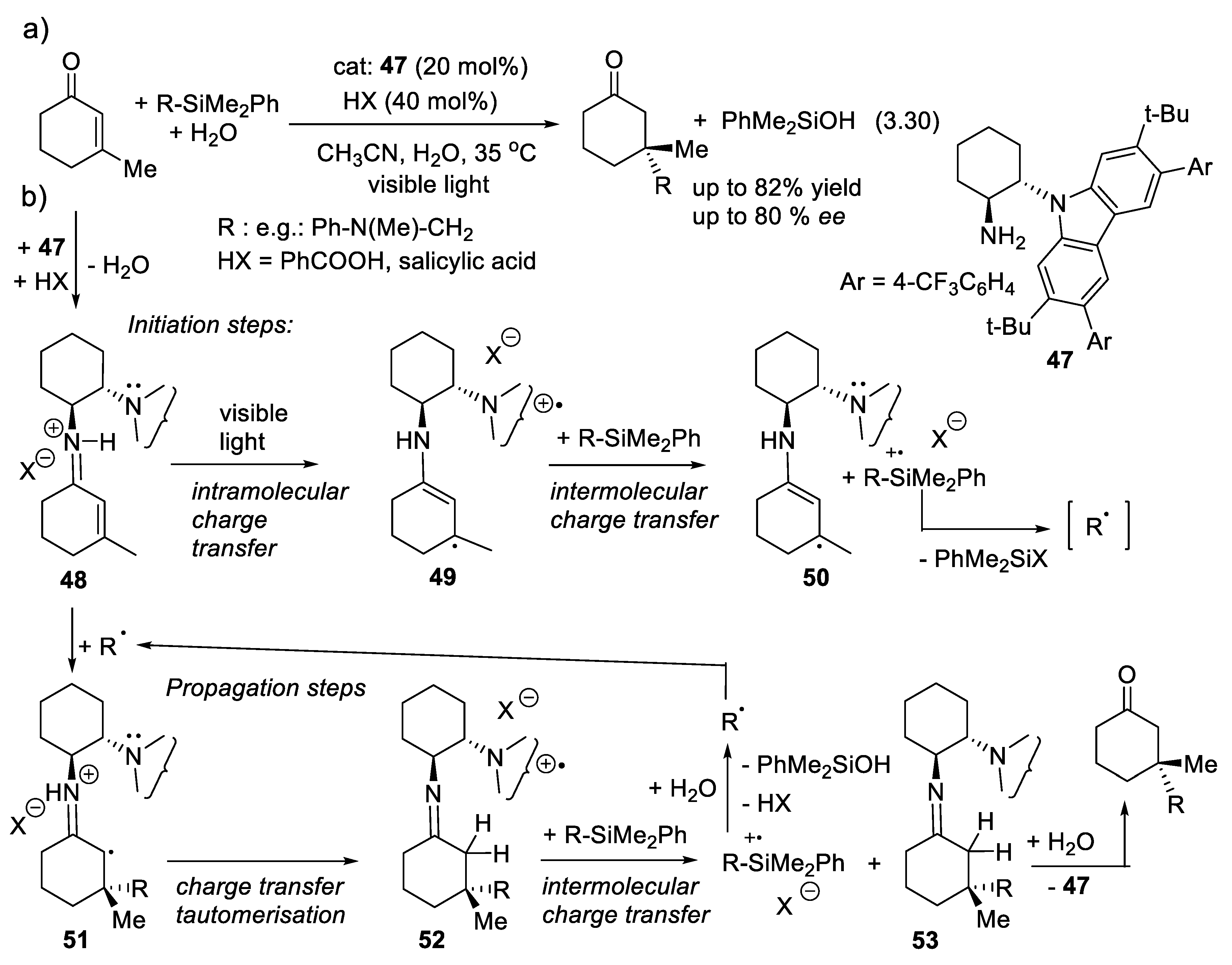
Figure 3.17.
Simplified Agami-Houk-List mechanism of the proline-catalyzed intermolecular aldol reactions (enamine HOMO driven catalysis).
Figure 3.17.
Simplified Agami-Houk-List mechanism of the proline-catalyzed intermolecular aldol reactions (enamine HOMO driven catalysis).
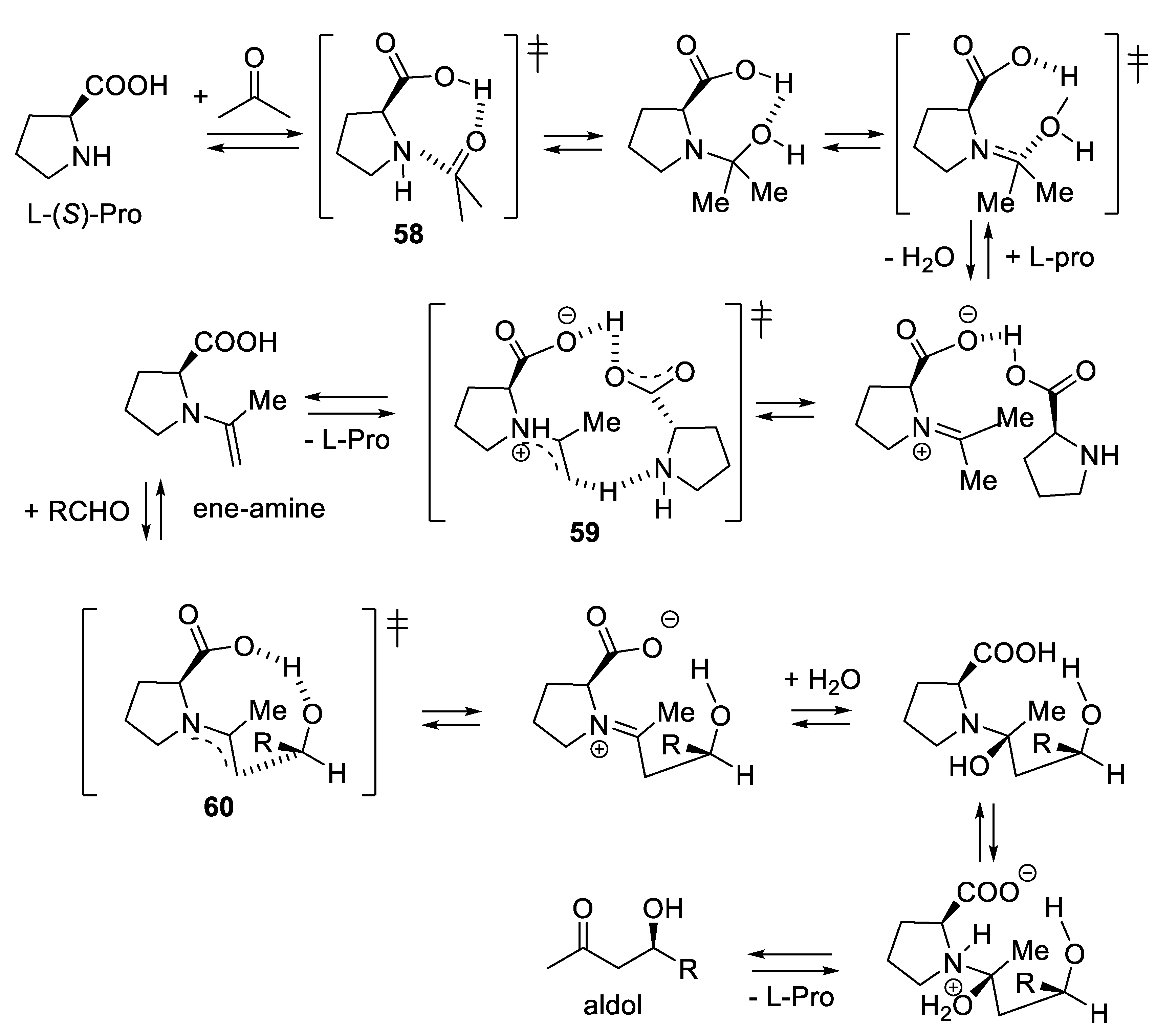
Figure 3.18.
Examples of asymmetric Mannich condensations catalyzed by amino acids (List, 2000).
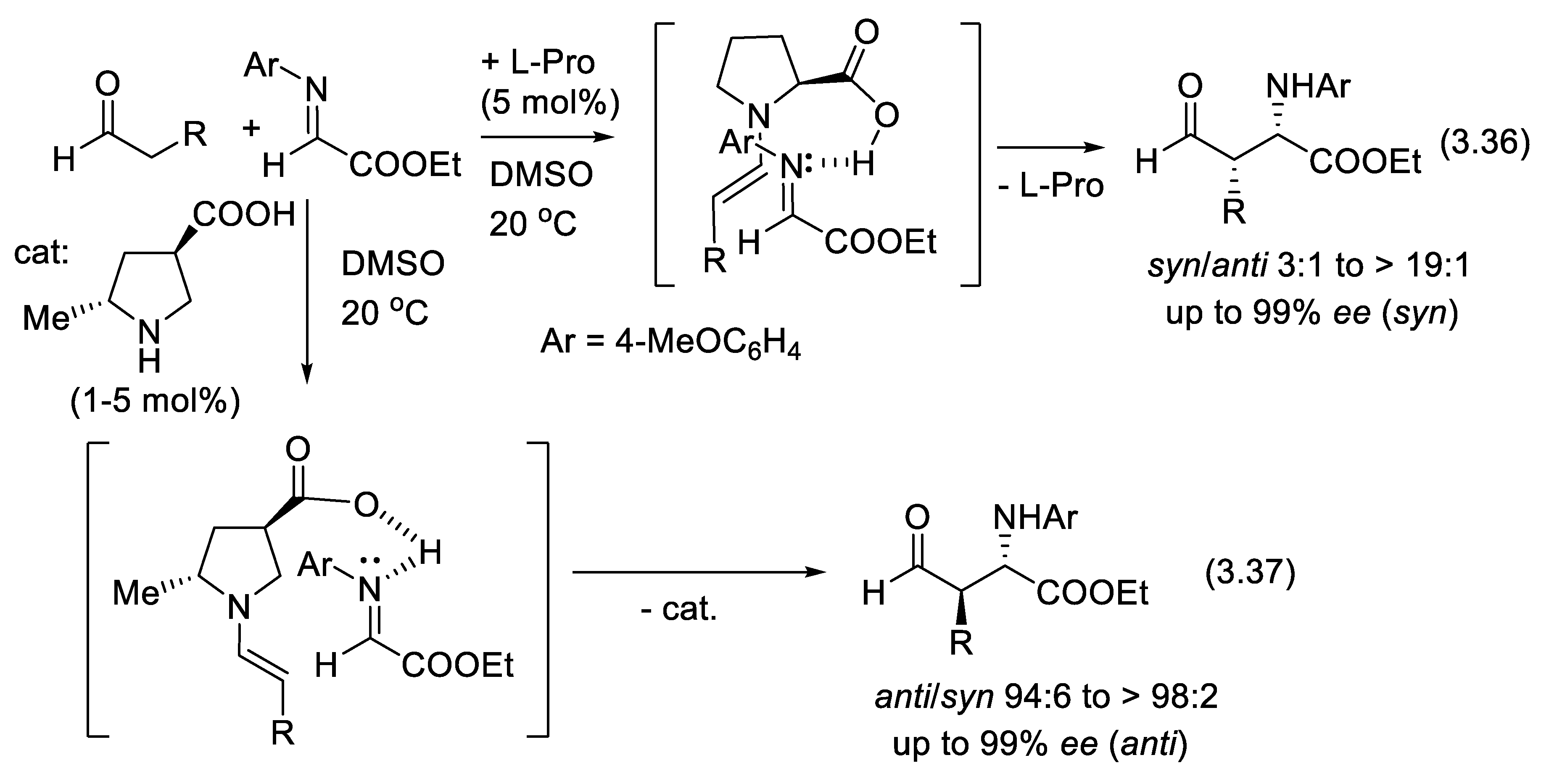
Figure 3.19.
a) Asymmetric α-fluorination of ketones (Enders, 2005); b) Enantioselective α-bromination of aldehydes (Jørgensen, 2005).
Figure 3.19.
a) Asymmetric α-fluorination of ketones (Enders, 2005); b) Enantioselective α-bromination of aldehydes (Jørgensen, 2005).
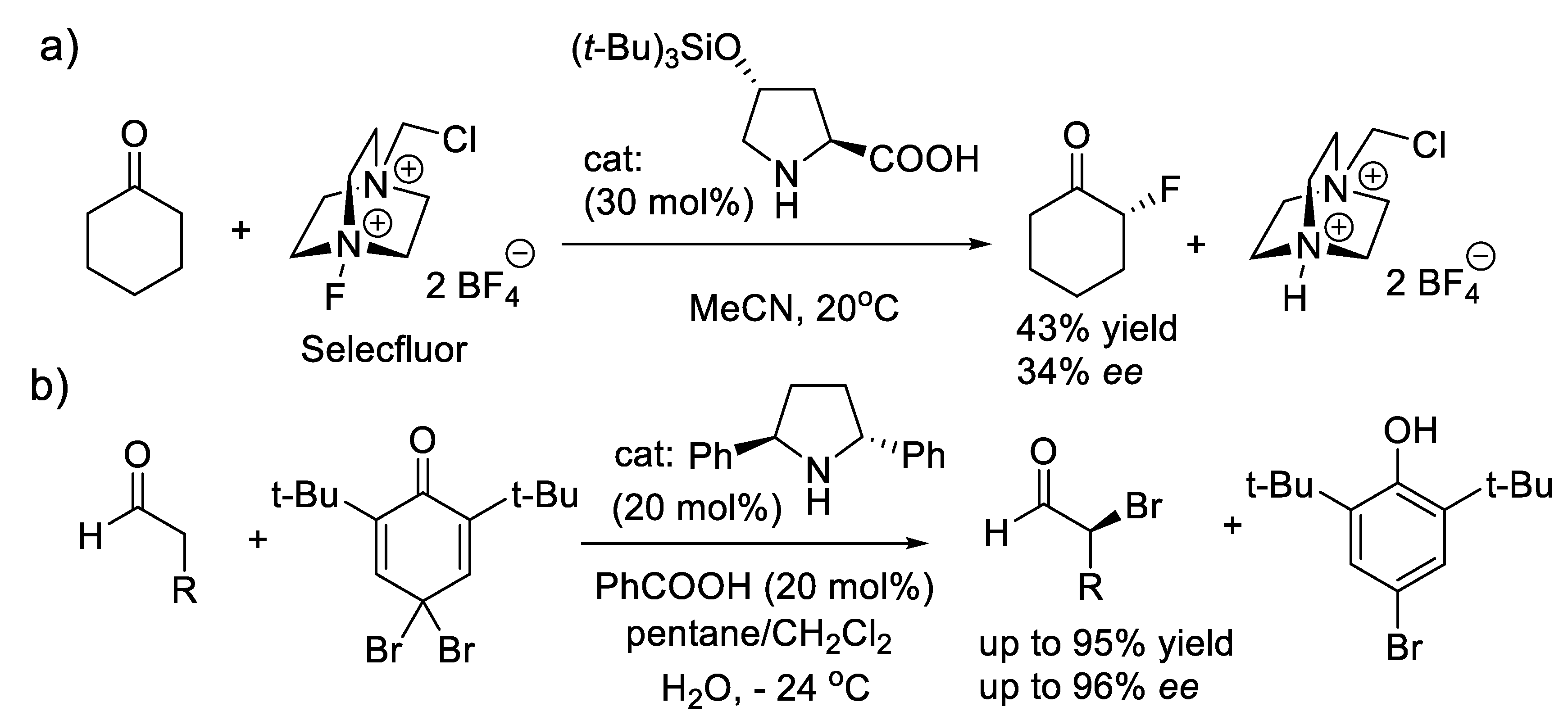
Figure 3.20.
a) Example of asymmetric benzylation of aldehydes with 4-(hydroxymethyl)pyridine applying the combination of amino-catalysis and photo-redox catalysis (MacMillan, 2018); b) Possible mechanism.
Figure 3.20.
a) Example of asymmetric benzylation of aldehydes with 4-(hydroxymethyl)pyridine applying the combination of amino-catalysis and photo-redox catalysis (MacMillan, 2018); b) Possible mechanism.
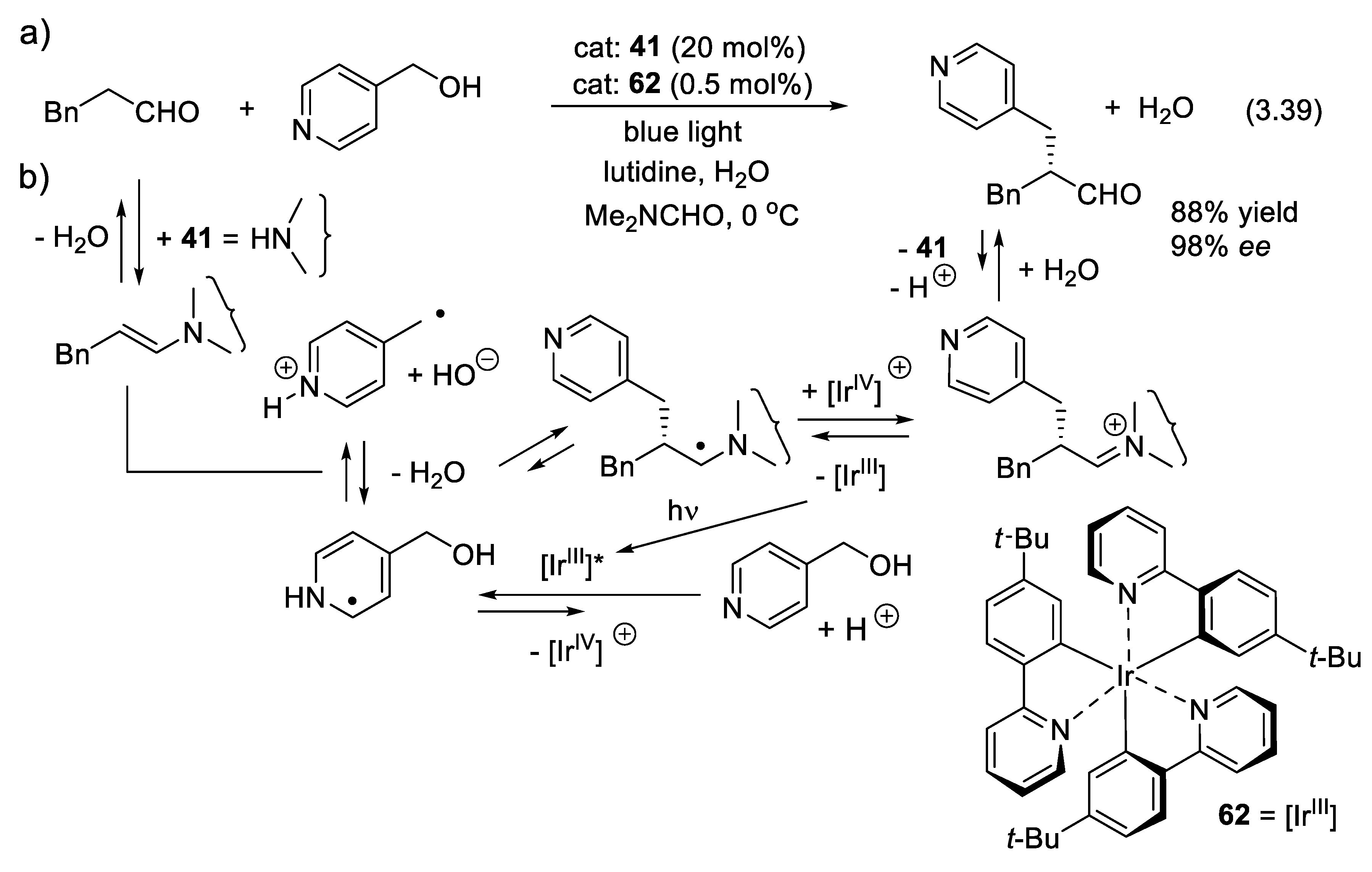
Figure 3.21.
Example of dienamine mode of activation (Jørgensen, 2006).
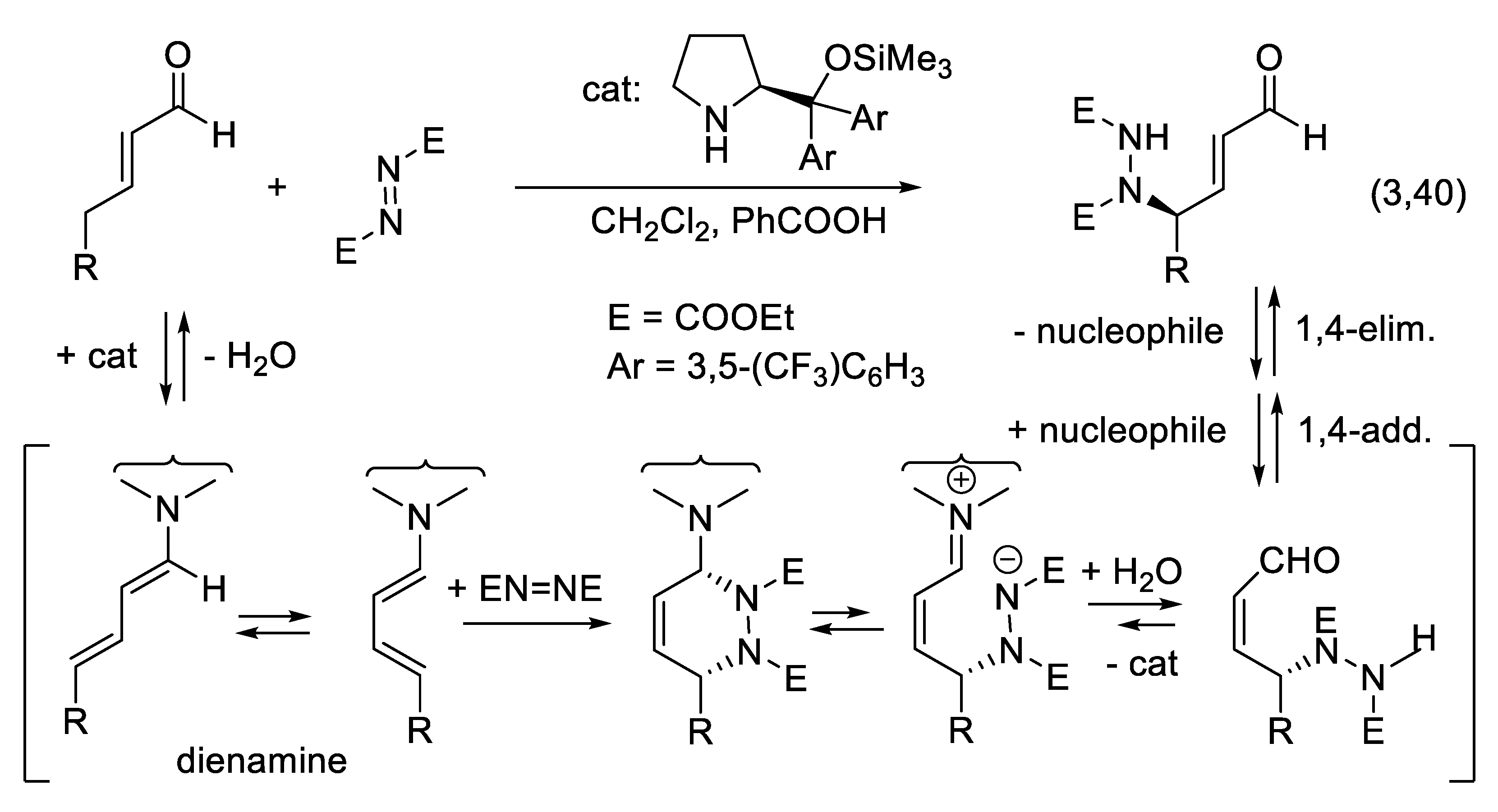
Figure 3.22.
Trienamine mode of activation (Jørgensen, 2011).
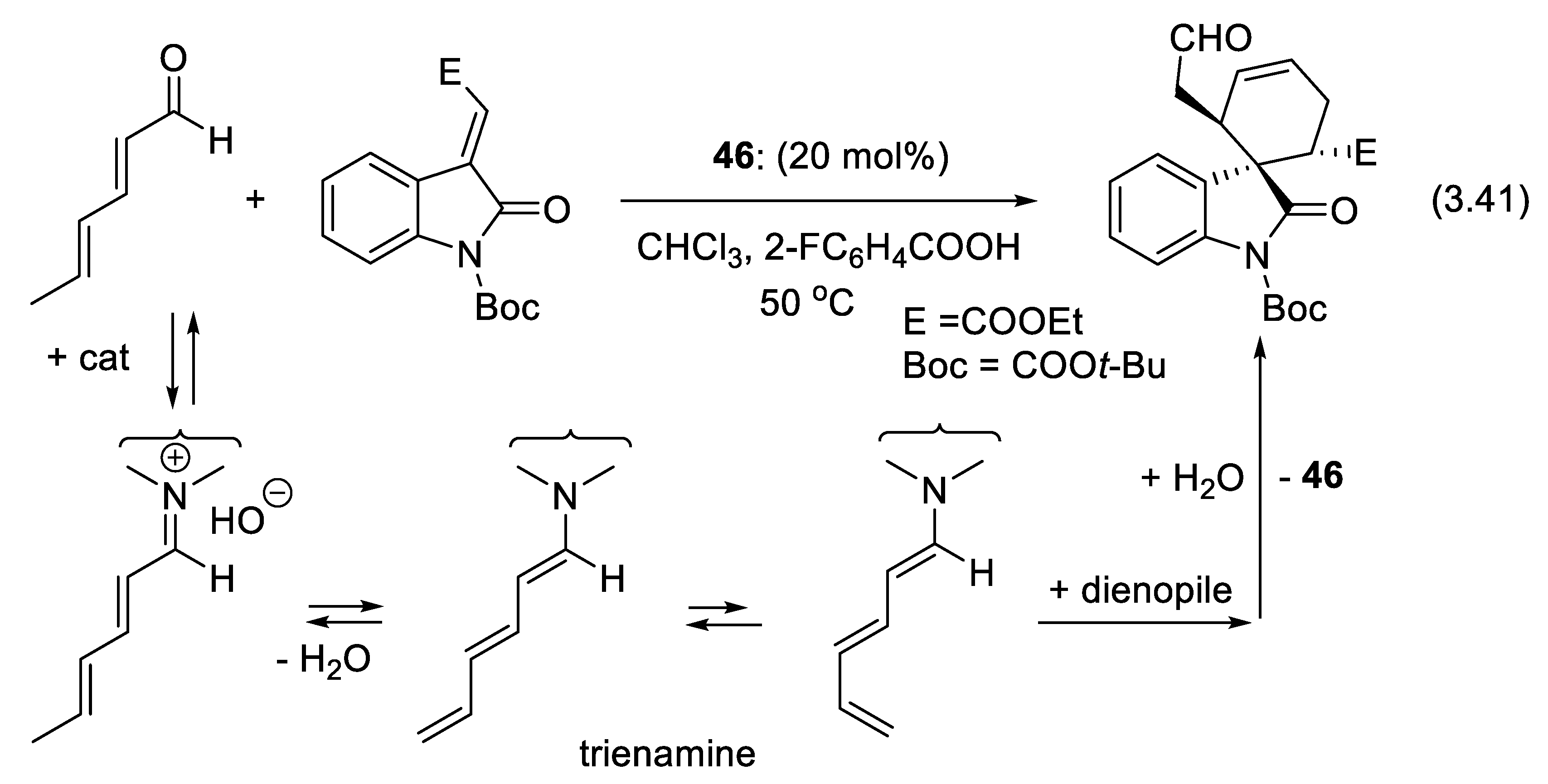
Figure 3.23.
One-pot asymmetric tandem Michael/Henry reaction through enamine/iminium activation (Hayashi, 2007).
Figure 3.23.
One-pot asymmetric tandem Michael/Henry reaction through enamine/iminium activation (Hayashi, 2007).
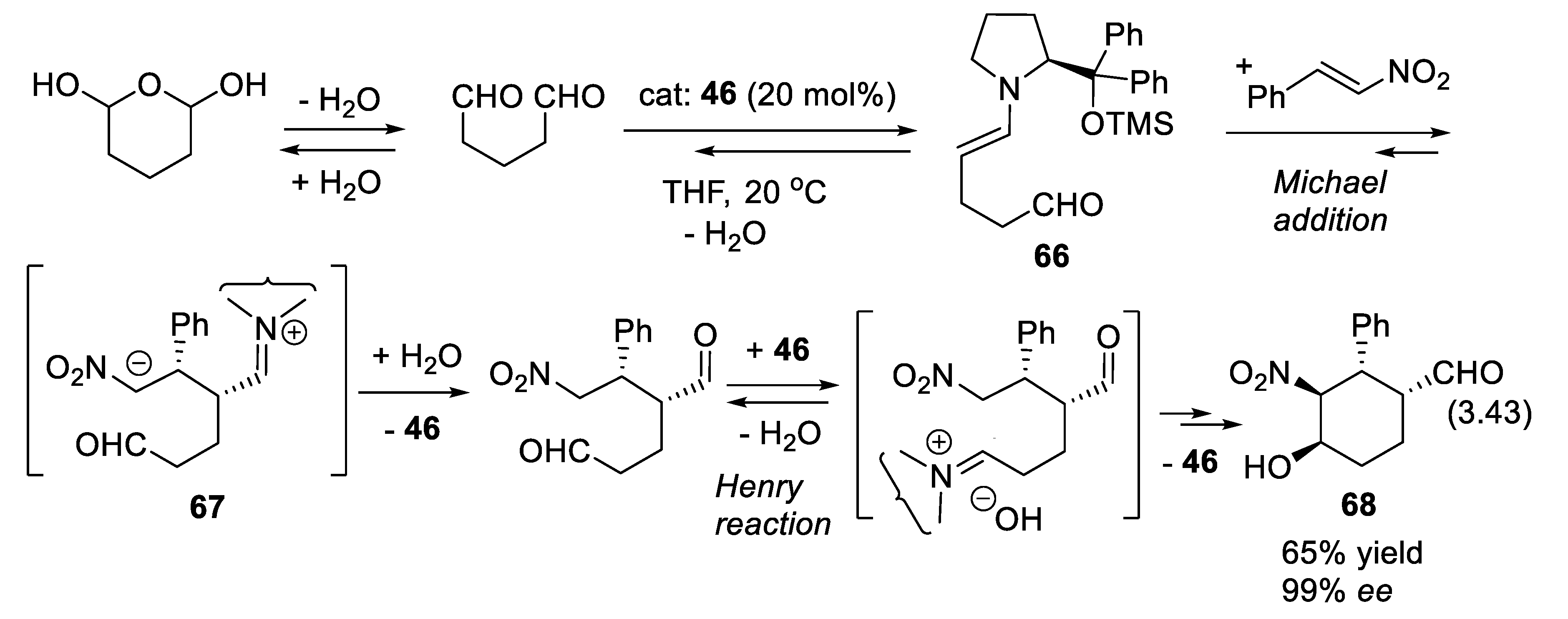
Figure 3.24.
Asymmetric synthesis of tricyclic compound with eight stereocenters obtained in one-pot from three simple achiral starting materials (Enders, 2007).
Figure 3.24.
Asymmetric synthesis of tricyclic compound with eight stereocenters obtained in one-pot from three simple achiral starting materials (Enders, 2007).
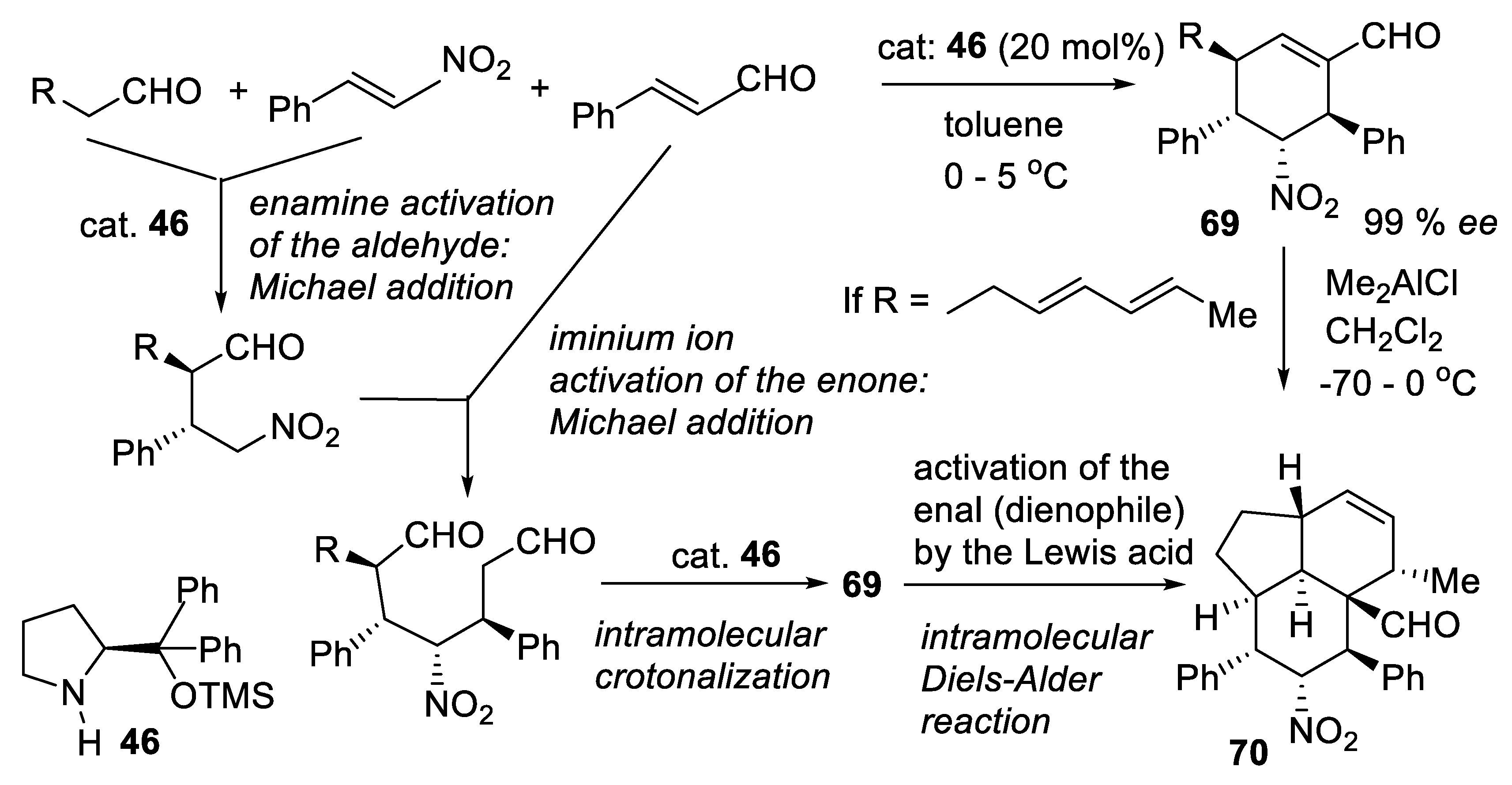
Figure 3.25.
a) Asymmetric α-enolation of aldehydes (ene-aminium radical cation SOMO driven catalysis, MacMillan, 2007); b) Possible simplified mechanism.
Figure 3.25.
a) Asymmetric α-enolation of aldehydes (ene-aminium radical cation SOMO driven catalysis, MacMillan, 2007); b) Possible simplified mechanism.
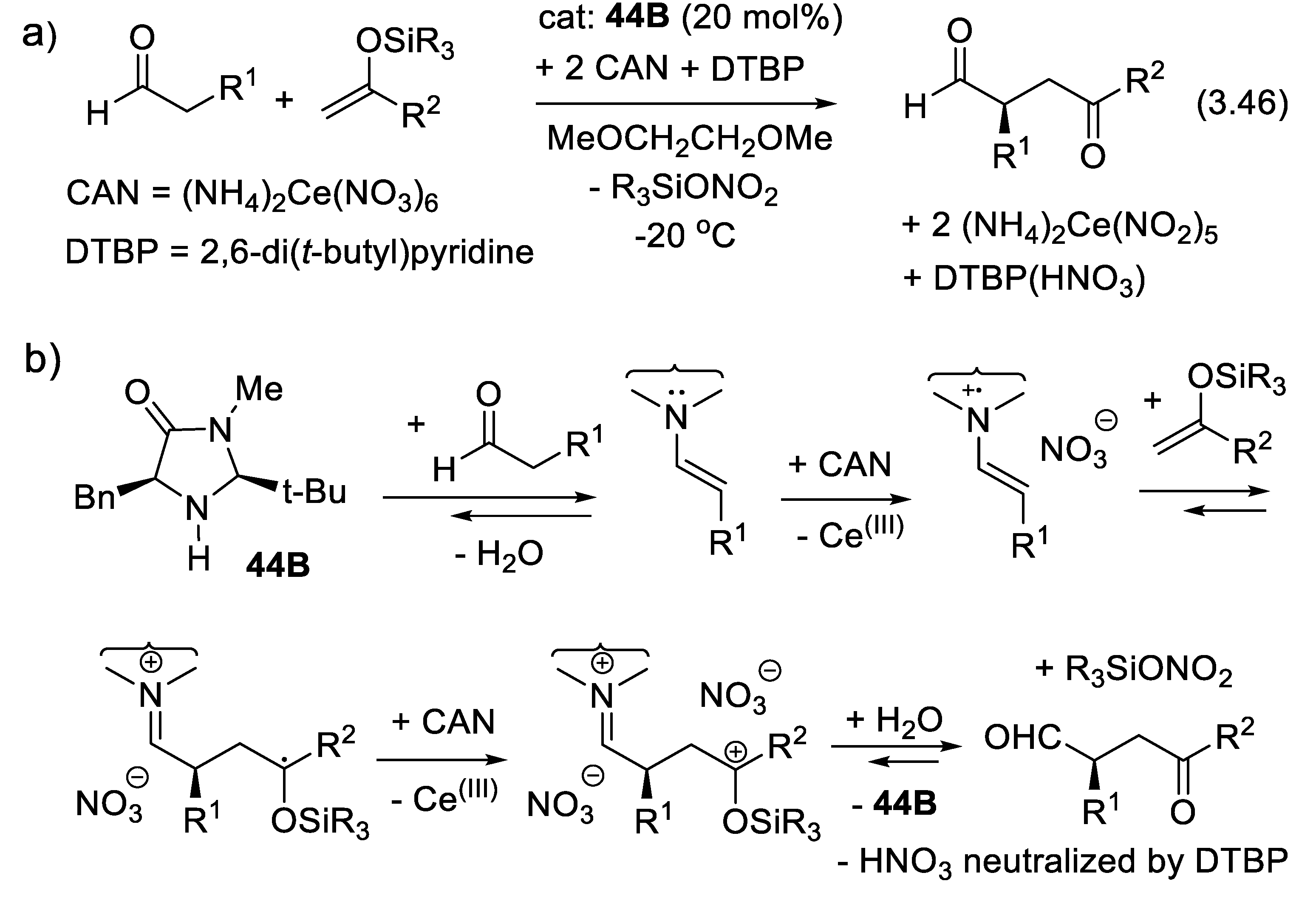
Figure 3.26.
a) General scheme for MBH and aza-MBH reaction catalyzed by nucleophile; b) Example of asymmetric MBH reaction catalyzed by a L-proline carboxamide (Bhuzare, 2020).
Figure 3.26.
a) General scheme for MBH and aza-MBH reaction catalyzed by nucleophile; b) Example of asymmetric MBH reaction catalyzed by a L-proline carboxamide (Bhuzare, 2020).
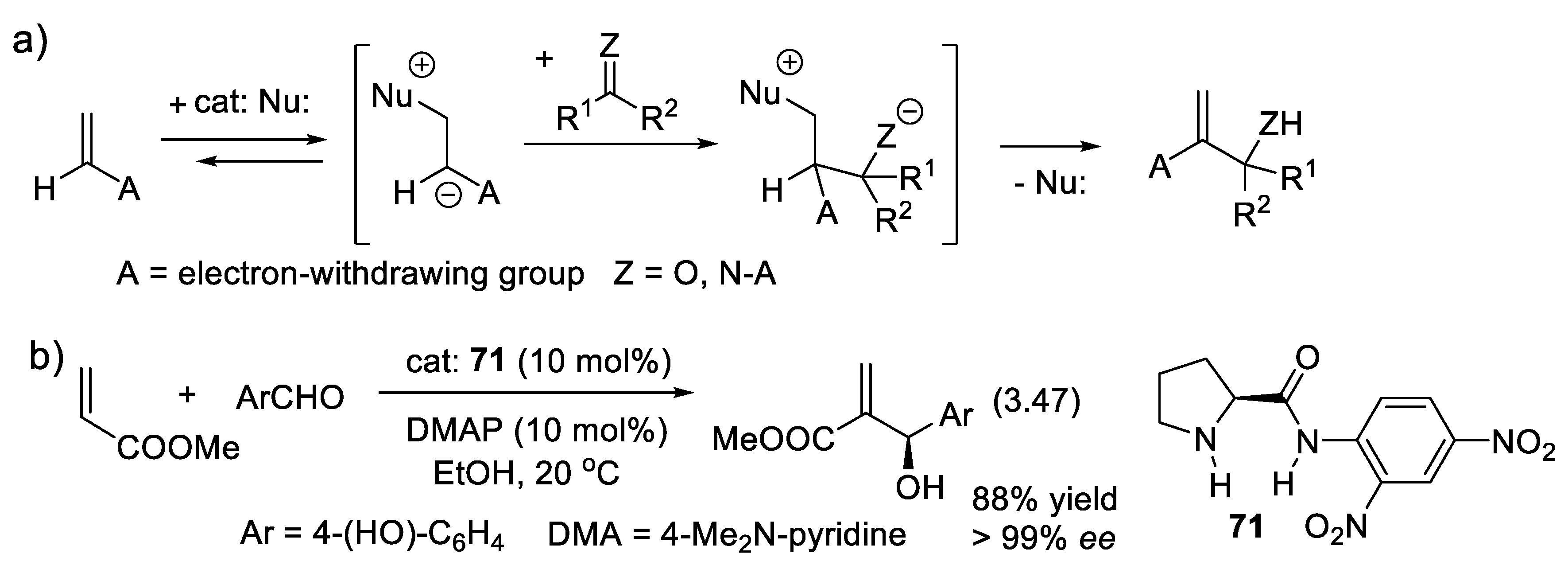
Figure 3.29.
Acylammonium salt mode of activation of asymmetric catalyzed Diels-Alder reaction (Romo, 2014).
Figure 3.29.
Acylammonium salt mode of activation of asymmetric catalyzed Diels-Alder reaction (Romo, 2014).
Figure 3.30.
a) Example of carbamate catalyzed asymmetric bromolactamization (Yeung, 2015); b) Proposed mechanism of reaction; formation of a chiral tight bromonium ion-pair reacting regioselectively with the nucleophile.
Figure 3.30.
a) Example of carbamate catalyzed asymmetric bromolactamization (Yeung, 2015); b) Proposed mechanism of reaction; formation of a chiral tight bromonium ion-pair reacting regioselectively with the nucleophile.
Figure 4.3.
Example of Juliá-Colonna (1983) asymmetric epoxidation.
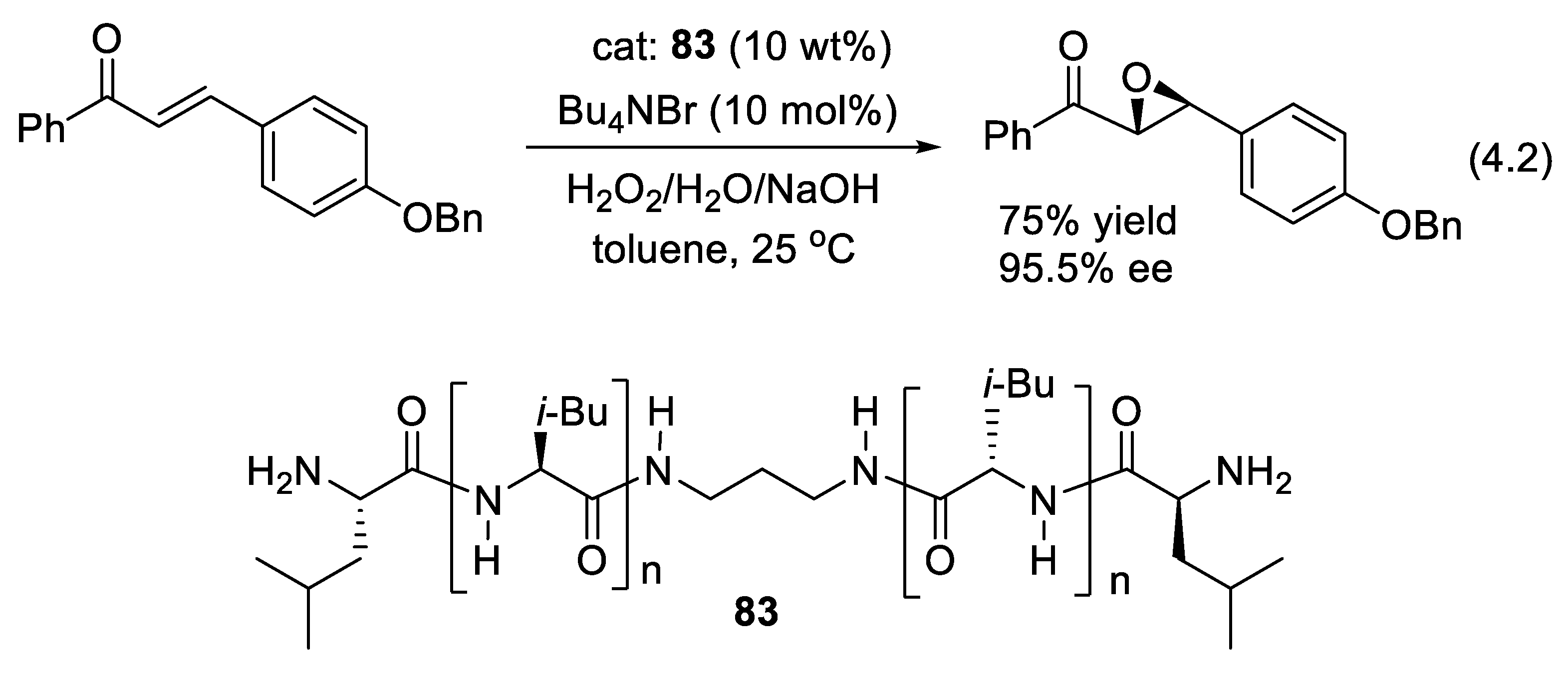
Figure 4.4.
Examples of asymmetric conjugate 1,4-additions catalyzed by short peptides. a) Córdoba, 2006; b) Tsogoeva, 2006; c) Wennemers, 2007.
Figure 4.4.
Examples of asymmetric conjugate 1,4-additions catalyzed by short peptides. a) Córdoba, 2006; b) Tsogoeva, 2006; c) Wennemers, 2007.
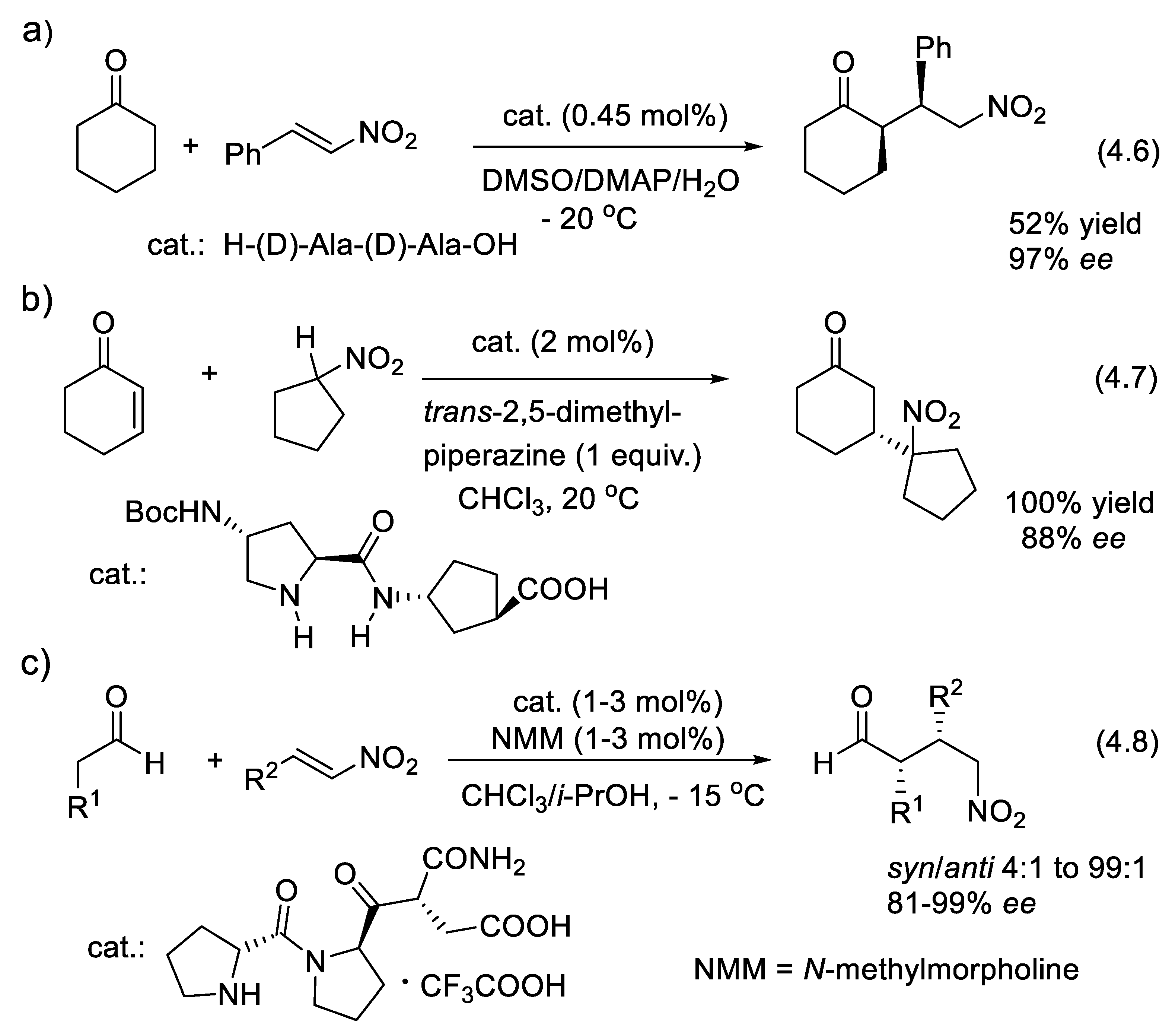
Figure 5.1.
Phosphine-induced oligomerizations of electro-poor alkenes: a) formation of a hexamer (Price, 1962); b) Formation of a dimer (Rauhut-Currier, 1963).
Figure 5.1.
Phosphine-induced oligomerizations of electro-poor alkenes: a) formation of a hexamer (Price, 1962); b) Formation of a dimer (Rauhut-Currier, 1963).
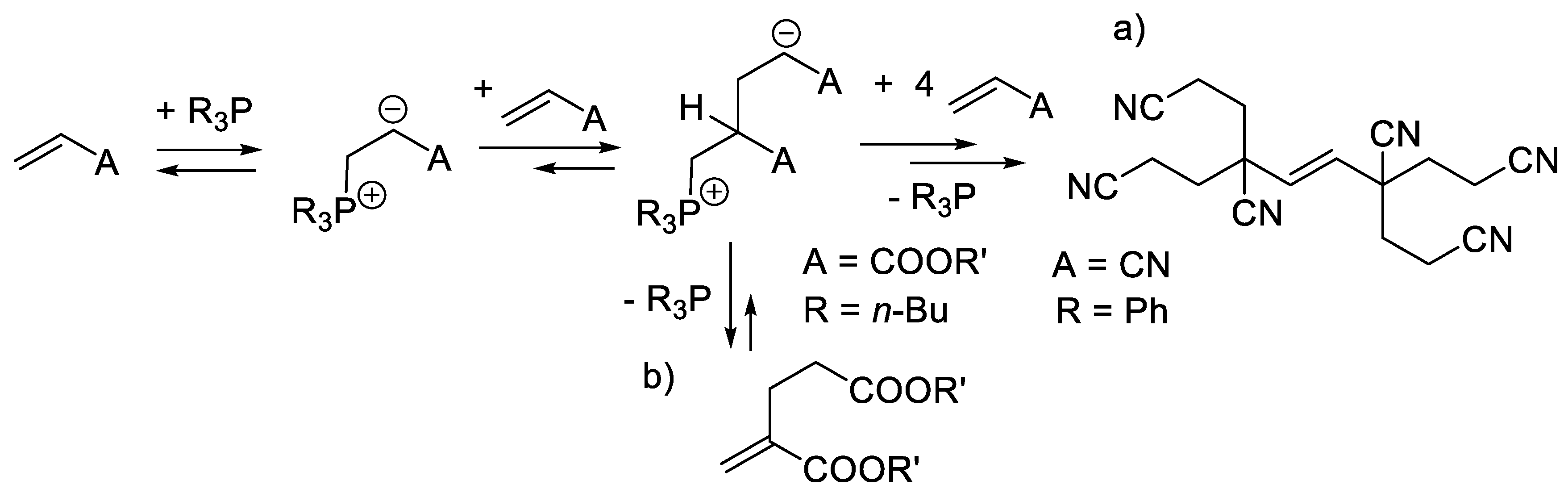
Figure 5.2.
a) Asymmetric (3+2)-cycloaddition of a 2,3-butadienoate to an acrylic ester catalyzed by a chiral phosphine (Zhang, 1997); b) Possible mechanism.
Figure 5.2.
a) Asymmetric (3+2)-cycloaddition of a 2,3-butadienoate to an acrylic ester catalyzed by a chiral phosphine (Zhang, 1997); b) Possible mechanism.
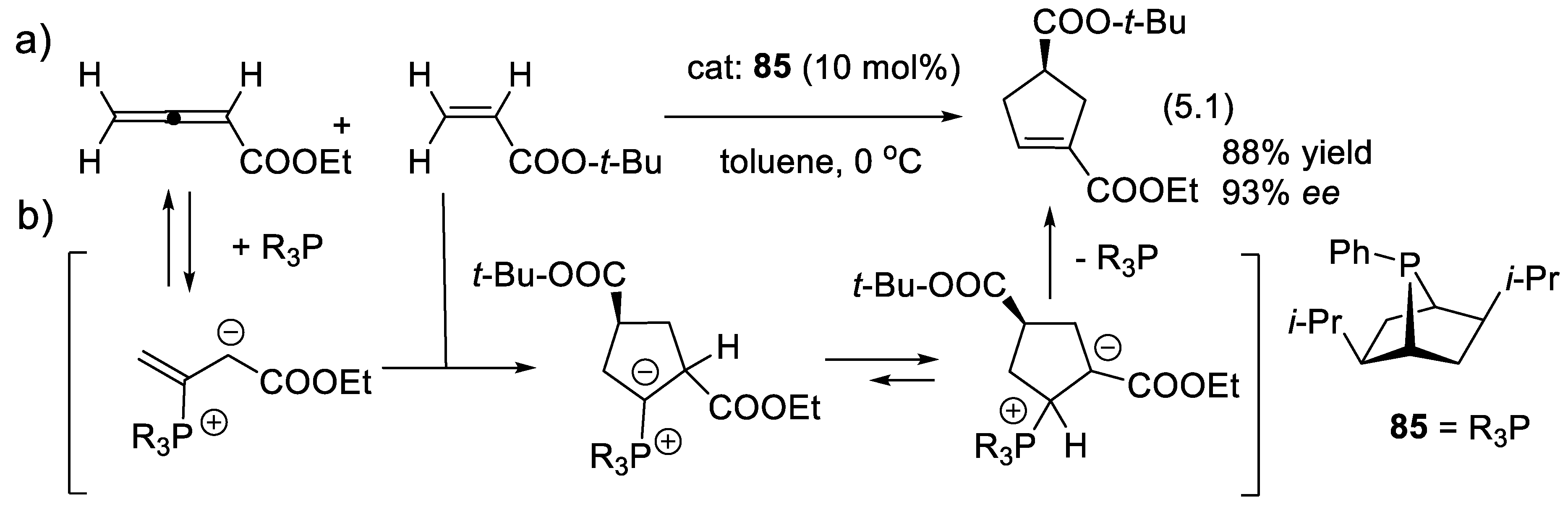
Figure 5.3.
a) Asymmetric γ-allylation of acidic carbonyl compounds by alkynoate and allenoate esters (Zhang, 1998); b) Possible mechanism.
Figure 5.3.
a) Asymmetric γ-allylation of acidic carbonyl compounds by alkynoate and allenoate esters (Zhang, 1998); b) Possible mechanism.
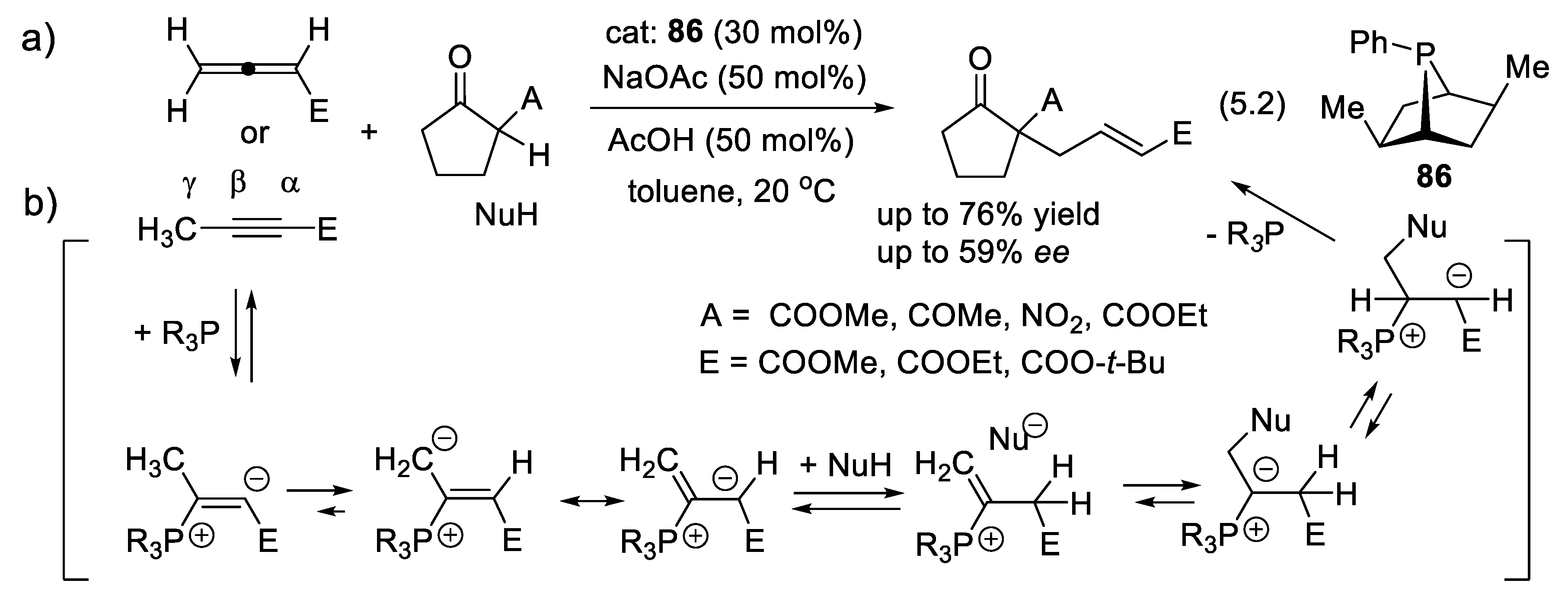
Figure 5.5.
a) Phosphine-catalyzed asymmetric (3+2) annulation of benzimidazoles with diarylcyclopropenones (You, 2025); b) Possible mechanism.
Figure 5.5.
a) Phosphine-catalyzed asymmetric (3+2) annulation of benzimidazoles with diarylcyclopropenones (You, 2025); b) Possible mechanism.
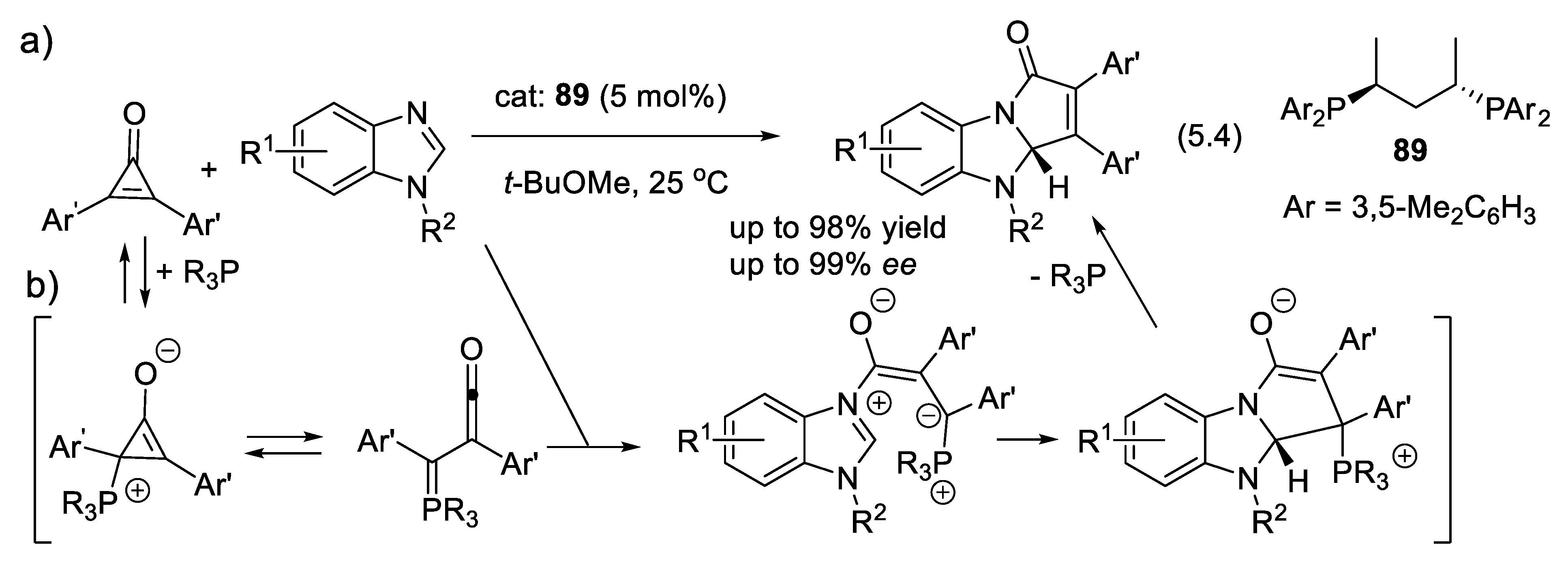
Figure 5.6.
a) Asymmetric synthesis of fluorinated cyclobutenes (Voituriez, 2019); b) Possible mechanism.
Figure 5.6.
a) Asymmetric synthesis of fluorinated cyclobutenes (Voituriez, 2019); b) Possible mechanism.
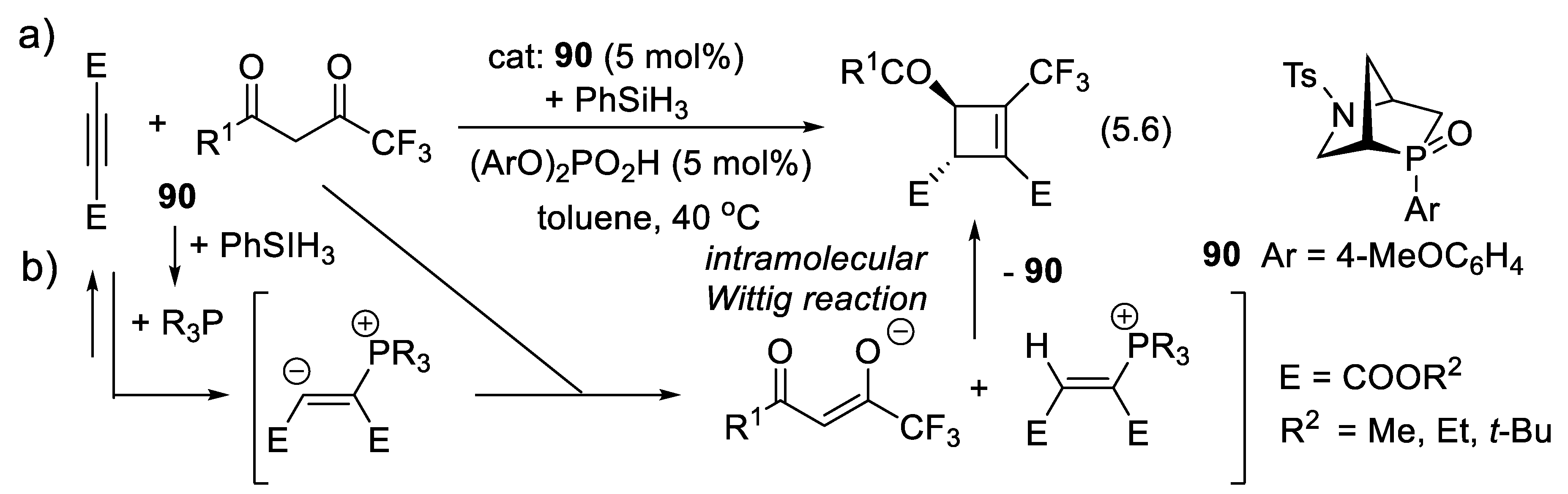
Figure 5.7.
Early examples of phosphoramide induced enantioselective allylation of benzaldehyde with allyltrichlorosilanes (Denmark, 1994, 2001).
Figure 5.7.
Early examples of phosphoramide induced enantioselective allylation of benzaldehyde with allyltrichlorosilanes (Denmark, 1994, 2001).
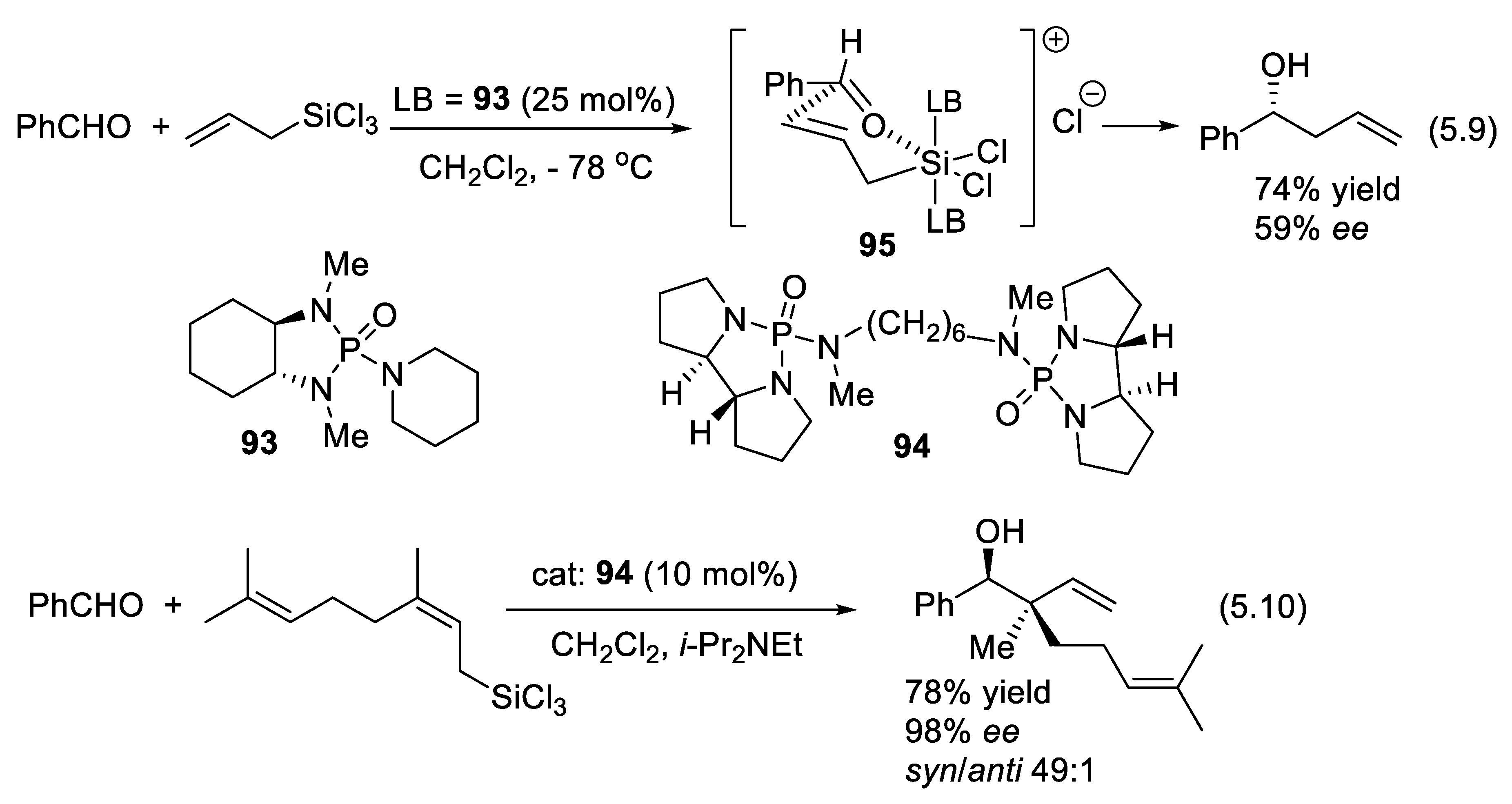
Figure 5.8.
Electron-pair donors (Lewis bases) can influence chemical reactions by enhancing either (or both) electrophilic or nucleophilic character of reagent Y3MX (Denmark, 2008).
Figure 5.8.
Electron-pair donors (Lewis bases) can influence chemical reactions by enhancing either (or both) electrophilic or nucleophilic character of reagent Y3MX (Denmark, 2008).
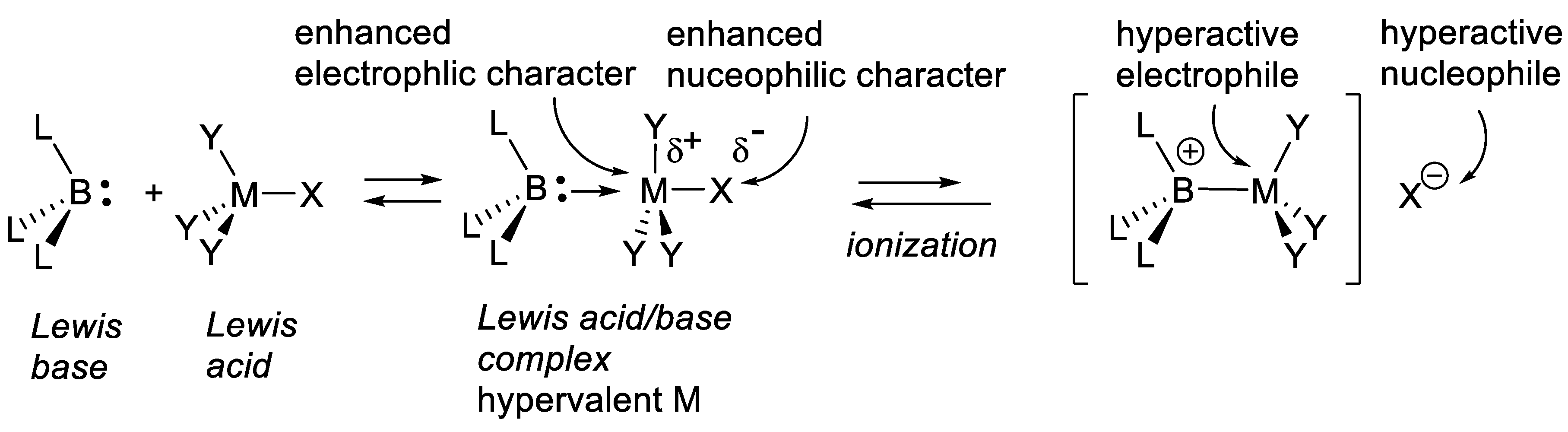
Figure 6.1.
a) Early examples of sulfur-ylide-mediated asymmetric epoxidation of aldehydes (Furukawa, 1989); b) Possible mechanism.
Figure 6.1.
a) Early examples of sulfur-ylide-mediated asymmetric epoxidation of aldehydes (Furukawa, 1989); b) Possible mechanism.
Figure 6.2.
Asymmetric epoxidation of aldehydes; selected examples of sulfide catalysts for the asymmetric synthesis of trans-stilbene epoxide.
Figure 6.2.
Asymmetric epoxidation of aldehydes; selected examples of sulfide catalysts for the asymmetric synthesis of trans-stilbene epoxide.
Figure 6.3.
Example of sulfide-catalyzed enantioselective bromolactonization.
Figure 6.4.
a) Example of enantioselective radical-chain hydrosilylation of alkenes using chiral thiols as catalysts (Roberts, 1998); b) Possible mechanism.
Figure 6.4.
a) Example of enantioselective radical-chain hydrosilylation of alkenes using chiral thiols as catalysts (Roberts, 1998); b) Possible mechanism.
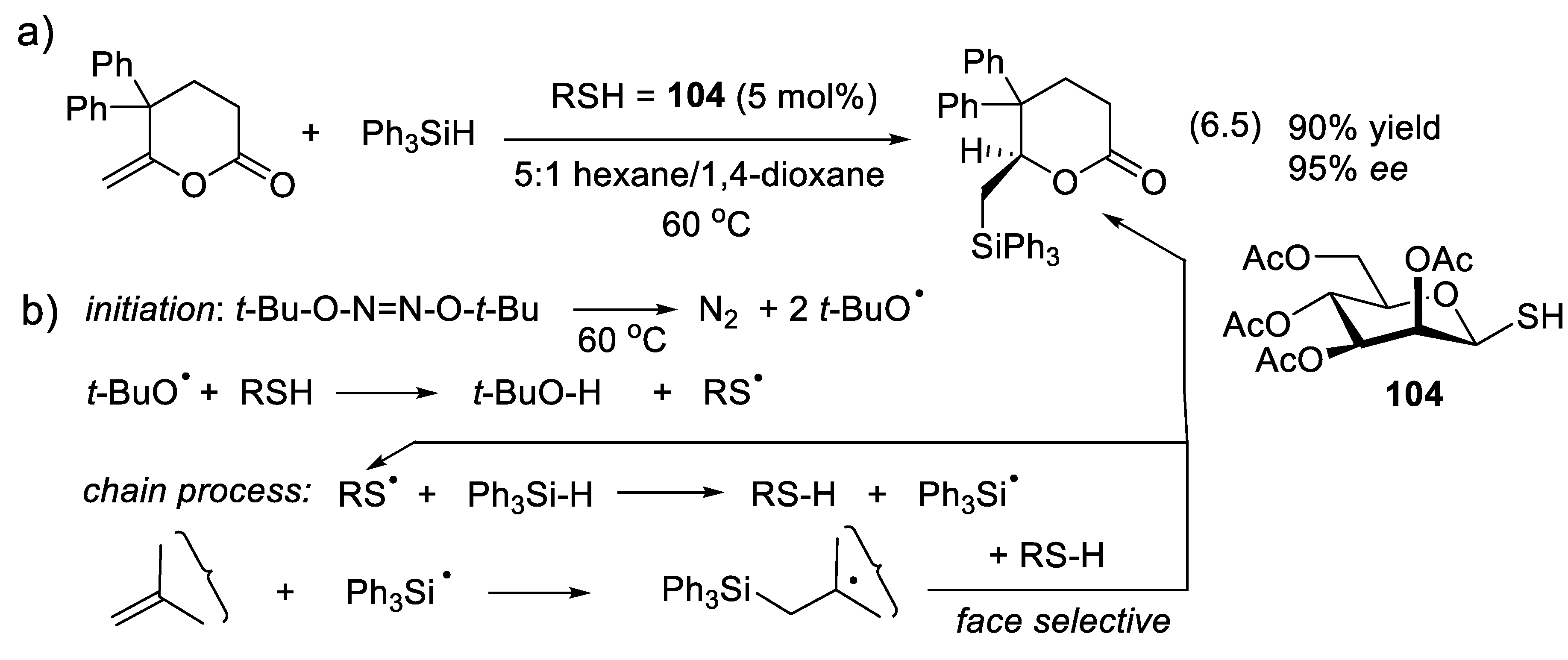
Figure 6.5.
a) Asymmetric Rauhut-Currier reaction catalyzed by a thiol (Miller, 2007); b) Possible mechanism.
Figure 6.5.
a) Asymmetric Rauhut-Currier reaction catalyzed by a thiol (Miller, 2007); b) Possible mechanism.
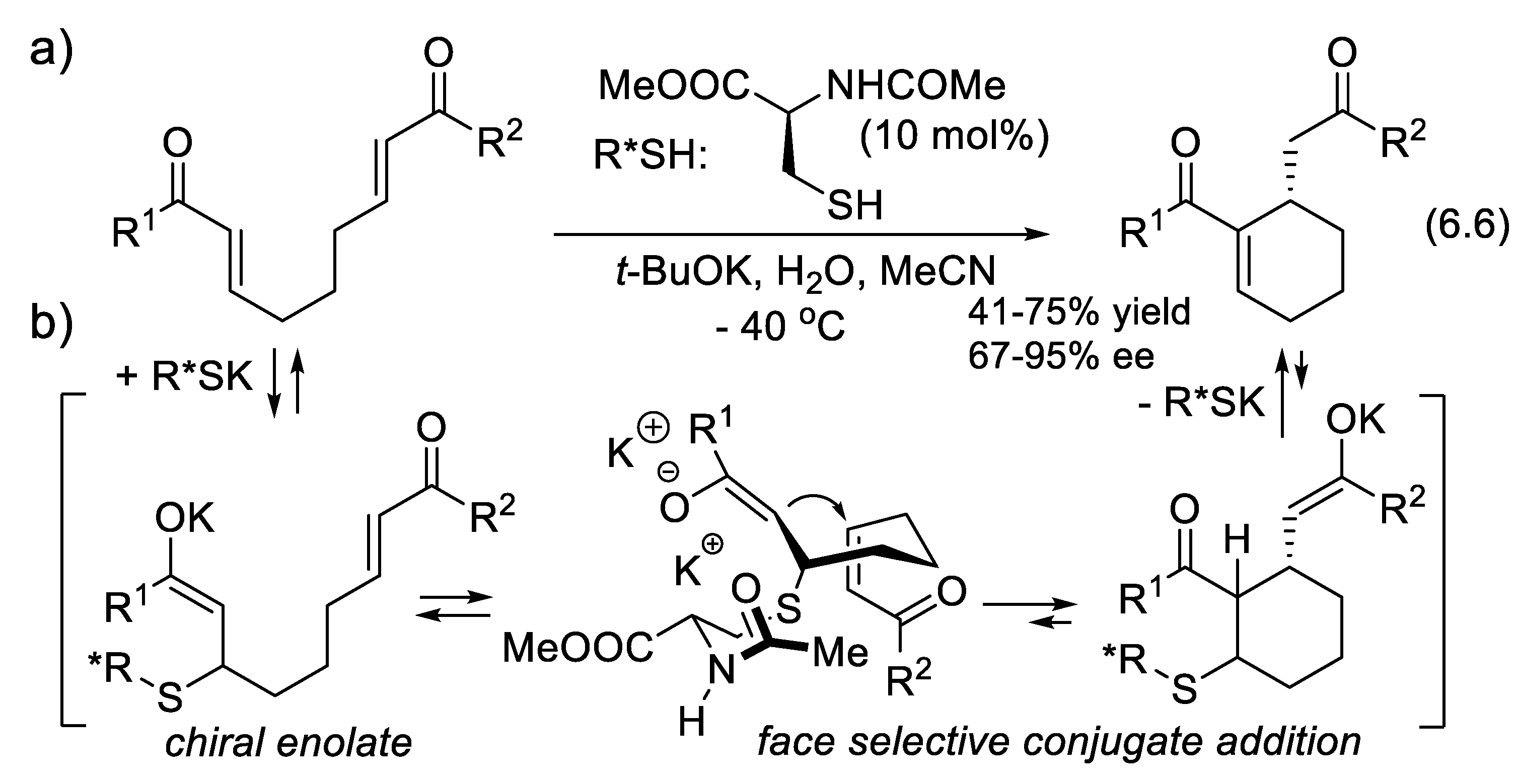
Figure 7.1.
Examples of diselenides and selenides (Lewis basis) used chiral catalysts.
Figure 7.2.
a) Example of asymmetric thiofunctionalization of unactivated alkene (Denmark, 2011); b) Proposed reaction mechanism.
Figure 7.2.
a) Example of asymmetric thiofunctionalization of unactivated alkene (Denmark, 2011); b) Proposed reaction mechanism.
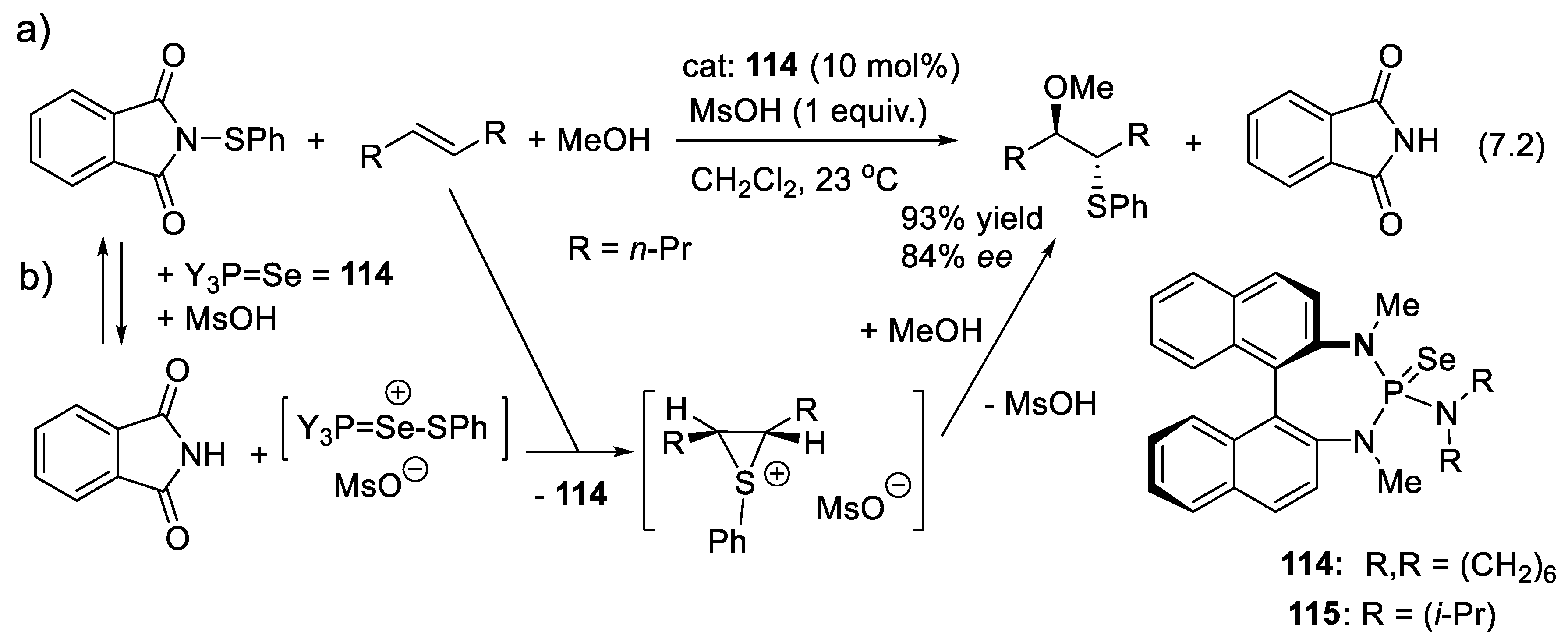
Figure 8.1.
Typical chiral precursors of NHC catalysts. For many of them both enantiomeric forms are available. a) The NHC generated by pre-catalysts 121, 123-133 induce enantioselectivity because of their shape (steric effects); b) The NHC issued from pre-catalysts such as 134-137 are polyfunctional and offer more than steric effects to the reactants (dipole/dipole interactions, H-bond formation, etc.) to realize enantioselective reactions.
Figure 8.1.
Typical chiral precursors of NHC catalysts. For many of them both enantiomeric forms are available. a) The NHC generated by pre-catalysts 121, 123-133 induce enantioselectivity because of their shape (steric effects); b) The NHC issued from pre-catalysts such as 134-137 are polyfunctional and offer more than steric effects to the reactants (dipole/dipole interactions, H-bond formation, etc.) to realize enantioselective reactions.
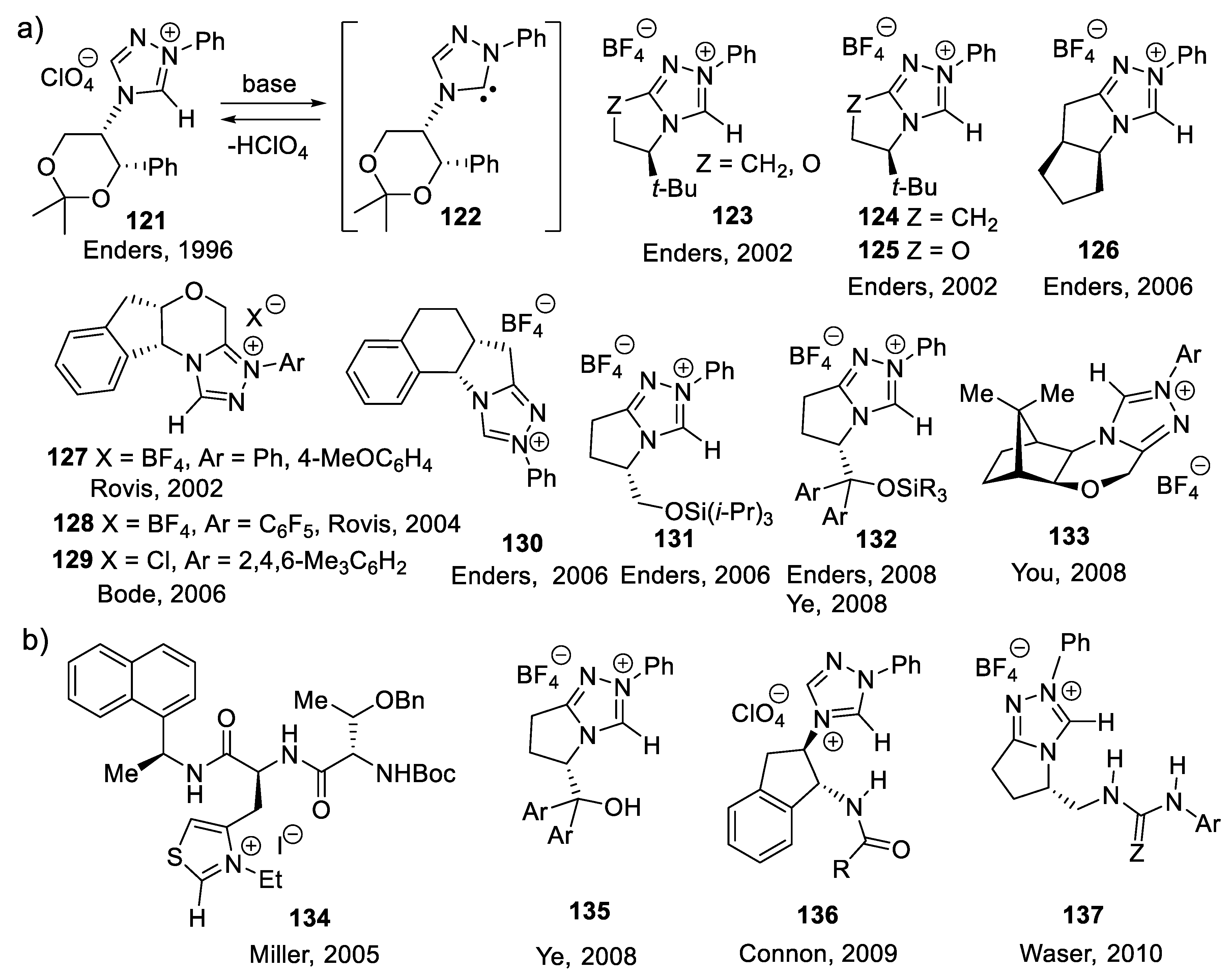
Figure 8.2.
Typical intermediates formed by reaction of NHCs with carbonyl functions.
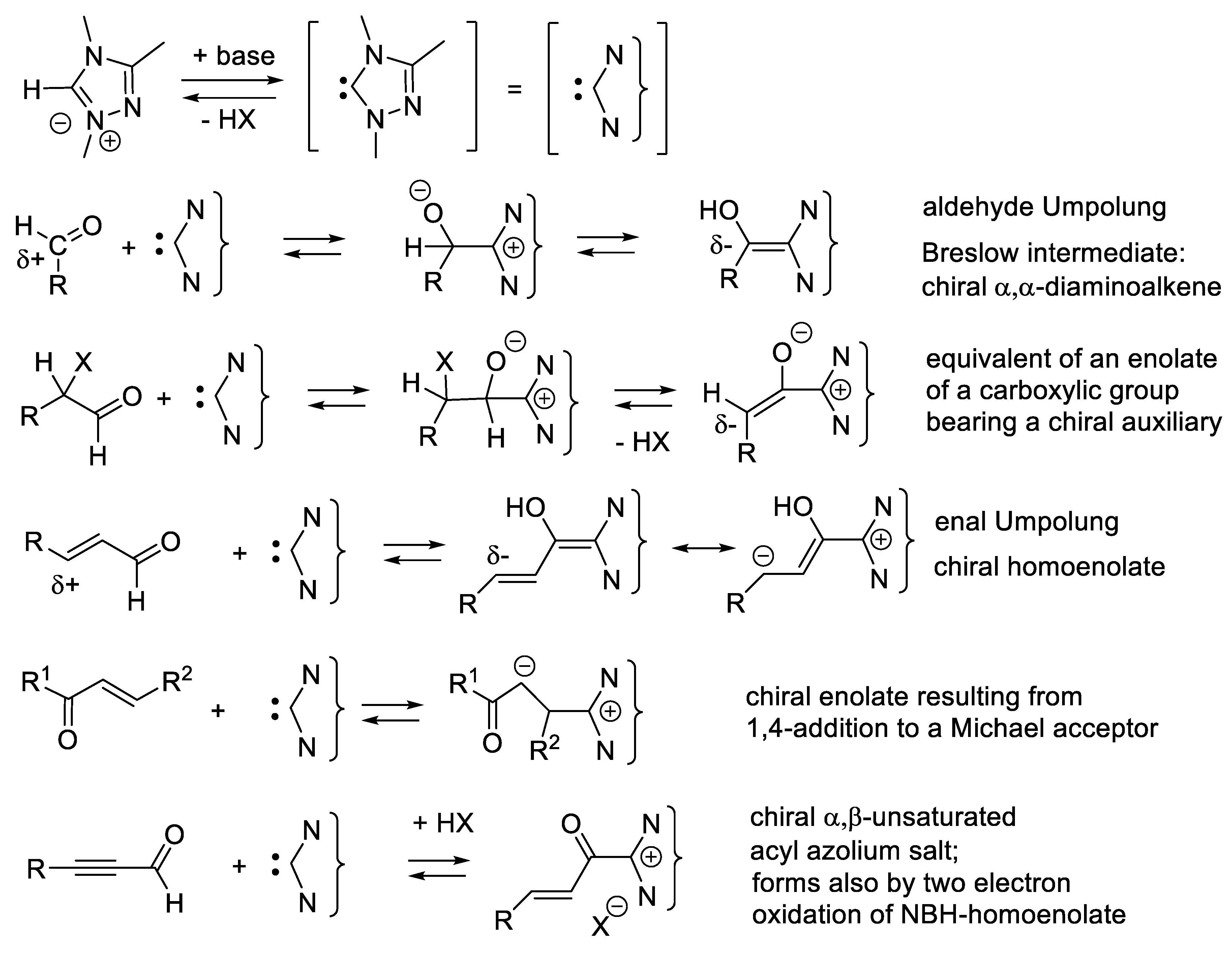
Figure 8.3.
a) Sheelan’s (1966) asymmetric benzoin condensation. b) Breslow’s (1958) mechanism: nucleophilic carbene intermediate converts the electrophilic carbon center of an aldehyde into an acyl anion equivalent; the electrophilic carbon center of the carbonyl group becomes a nucleophilic carbon center (Umpolung, polarity inversion).
Figure 8.3.
a) Sheelan’s (1966) asymmetric benzoin condensation. b) Breslow’s (1958) mechanism: nucleophilic carbene intermediate converts the electrophilic carbon center of an aldehyde into an acyl anion equivalent; the electrophilic carbon center of the carbonyl group becomes a nucleophilic carbon center (Umpolung, polarity inversion).
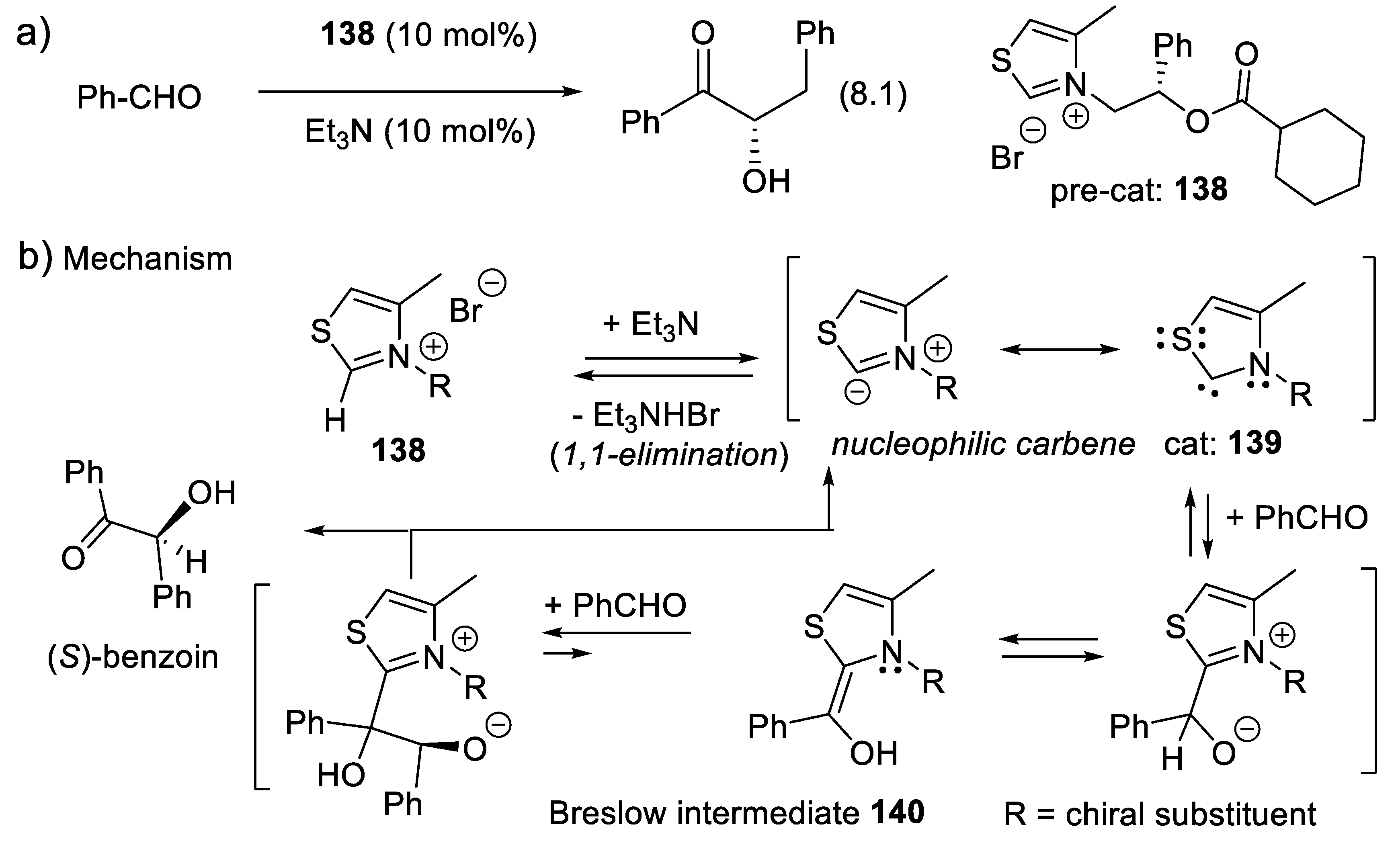
Figure 8.4.
a) Mechanism of the Stetter reaction; b,c) Examples of intramolecular asymmetric Stetter reactions by Rovis (2002; earlier examples by Enders, 1996).
Figure 8.4.
a) Mechanism of the Stetter reaction; b,c) Examples of intramolecular asymmetric Stetter reactions by Rovis (2002; earlier examples by Enders, 1996).
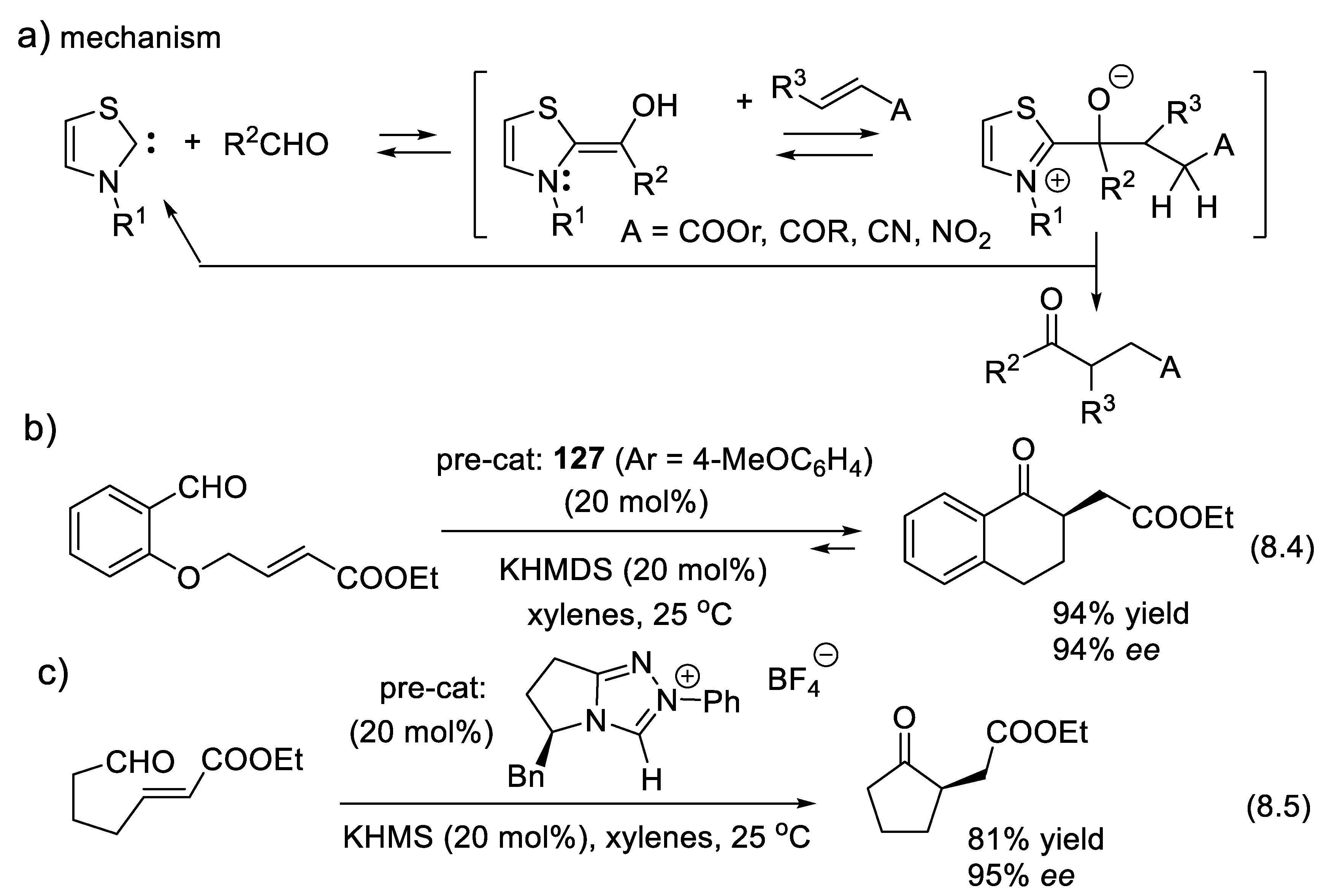
Figure 8.5.
a) Asymmetric synthesis of γ-iminolactones through NHC-catalyzed imino-Stetter reaction (Lupton, 2019); b) Possible mechanism.
Figure 8.5.
a) Asymmetric synthesis of γ-iminolactones through NHC-catalyzed imino-Stetter reaction (Lupton, 2019); b) Possible mechanism.
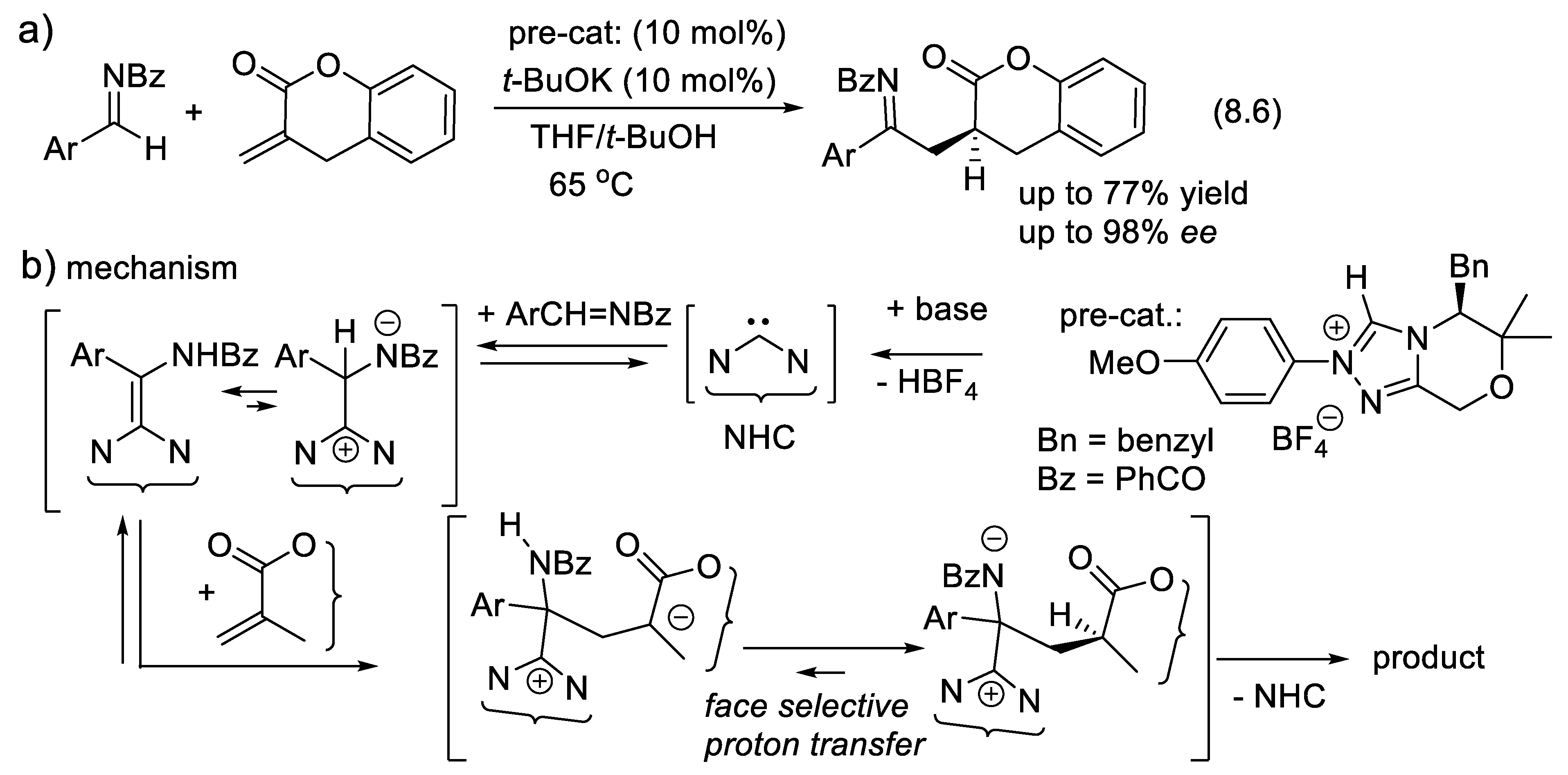
Figure 8.6.
a) Asymmetric aza-Diels Alder reaction catalyzed by NHC (Bode, 2006); b) Possible mechanism.
Figure 8.6.
a) Asymmetric aza-Diels Alder reaction catalyzed by NHC (Bode, 2006); b) Possible mechanism.
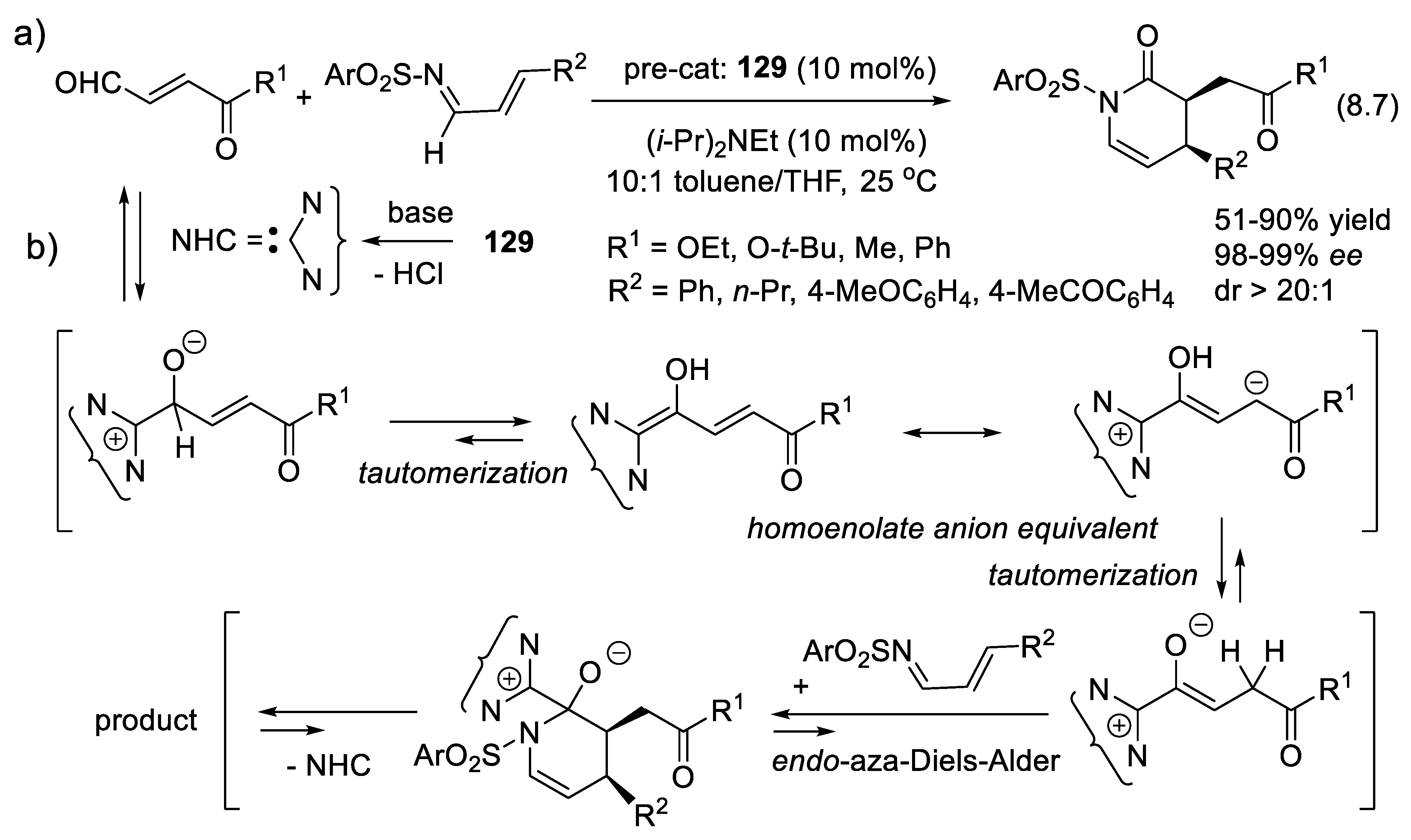
Figure 8.7.
a) Asymmetric NHC-catalyzed Staudinger cycloaddition (Ye, 2008); b) Possible mechanism.
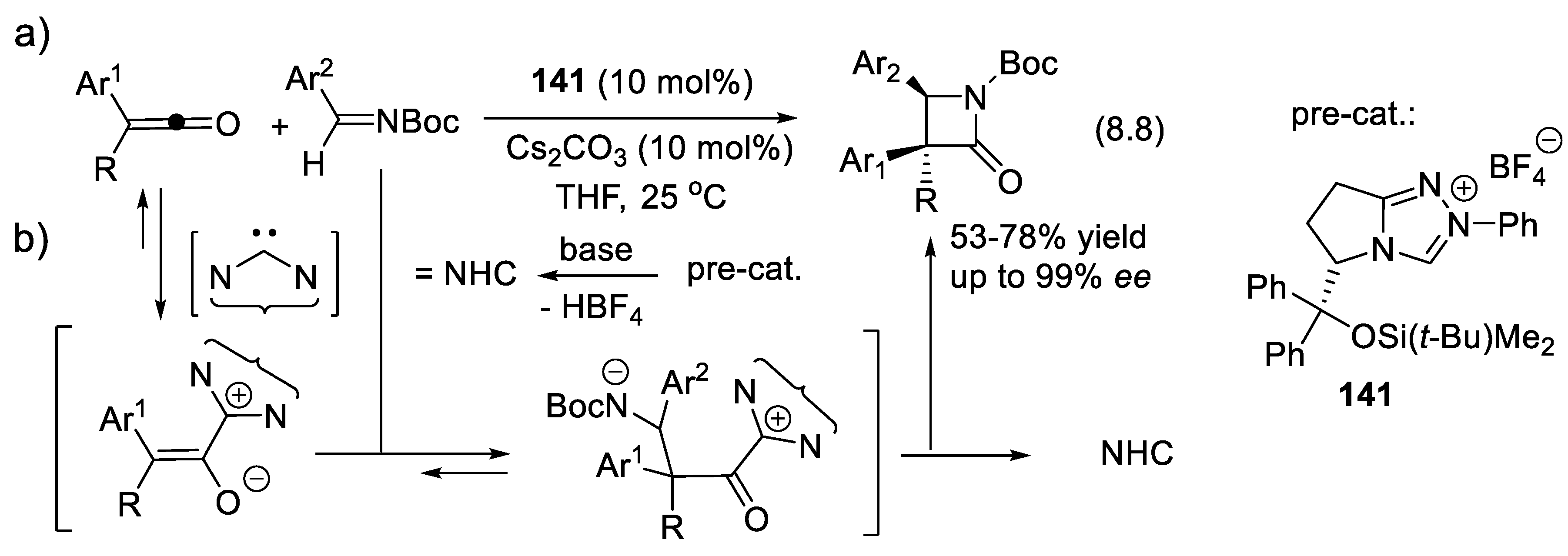
Figure 8.8.
a) Example of enantioselective oxidative annulation of conjugated enals and a sulfonylimine (Bode, 2012); b) Possible reaction mechanism.
Figure 8.8.
a) Example of enantioselective oxidative annulation of conjugated enals and a sulfonylimine (Bode, 2012); b) Possible reaction mechanism.
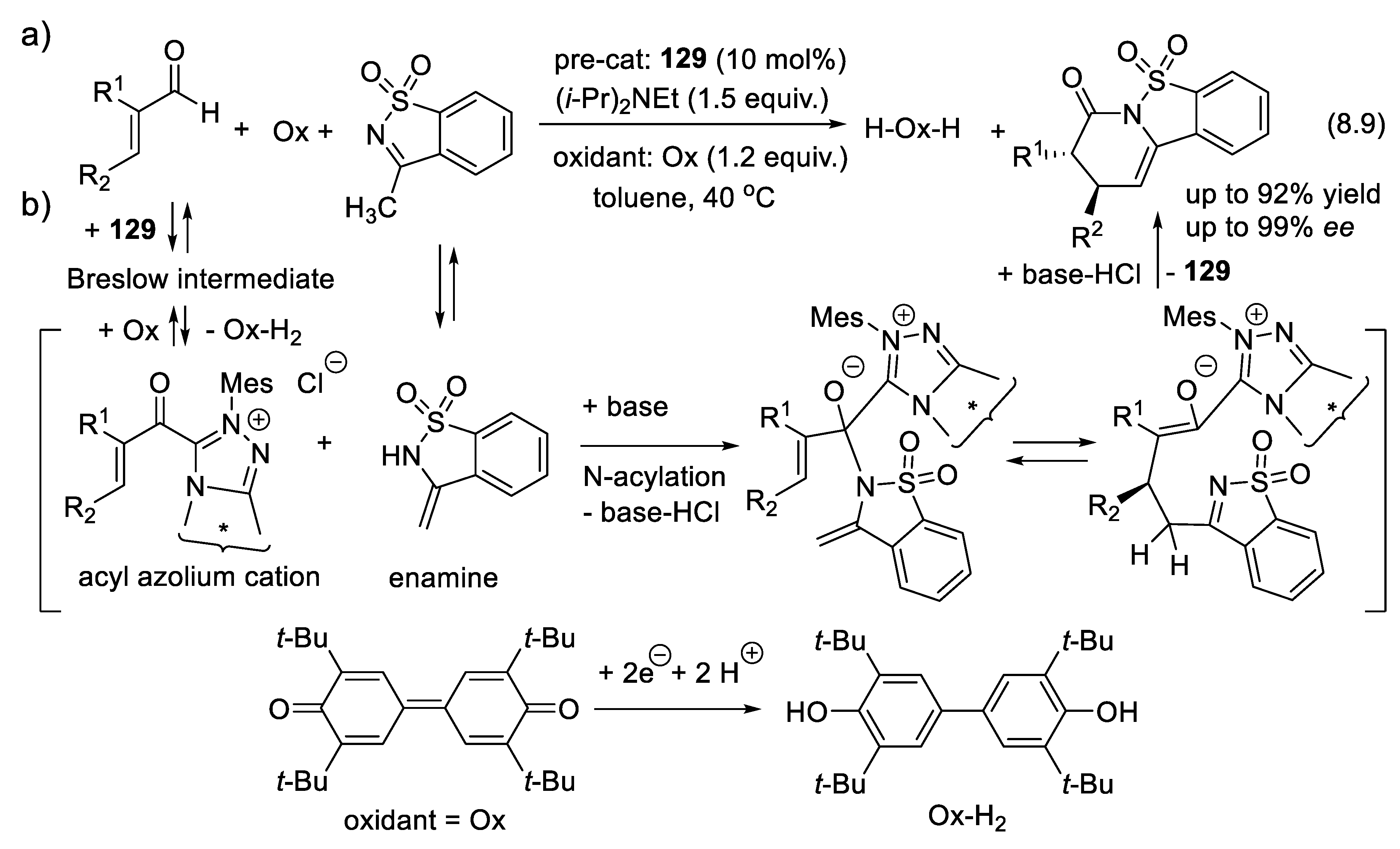
Figure 8.9.
a) NHC-catalyzed oxy-Cope rearrangement, asymmetric synthesis of 1,3,4-trisubstituted cyclopentenes (Bode, 2007); b) Possible mechanism.
Figure 8.9.
a) NHC-catalyzed oxy-Cope rearrangement, asymmetric synthesis of 1,3,4-trisubstituted cyclopentenes (Bode, 2007); b) Possible mechanism.
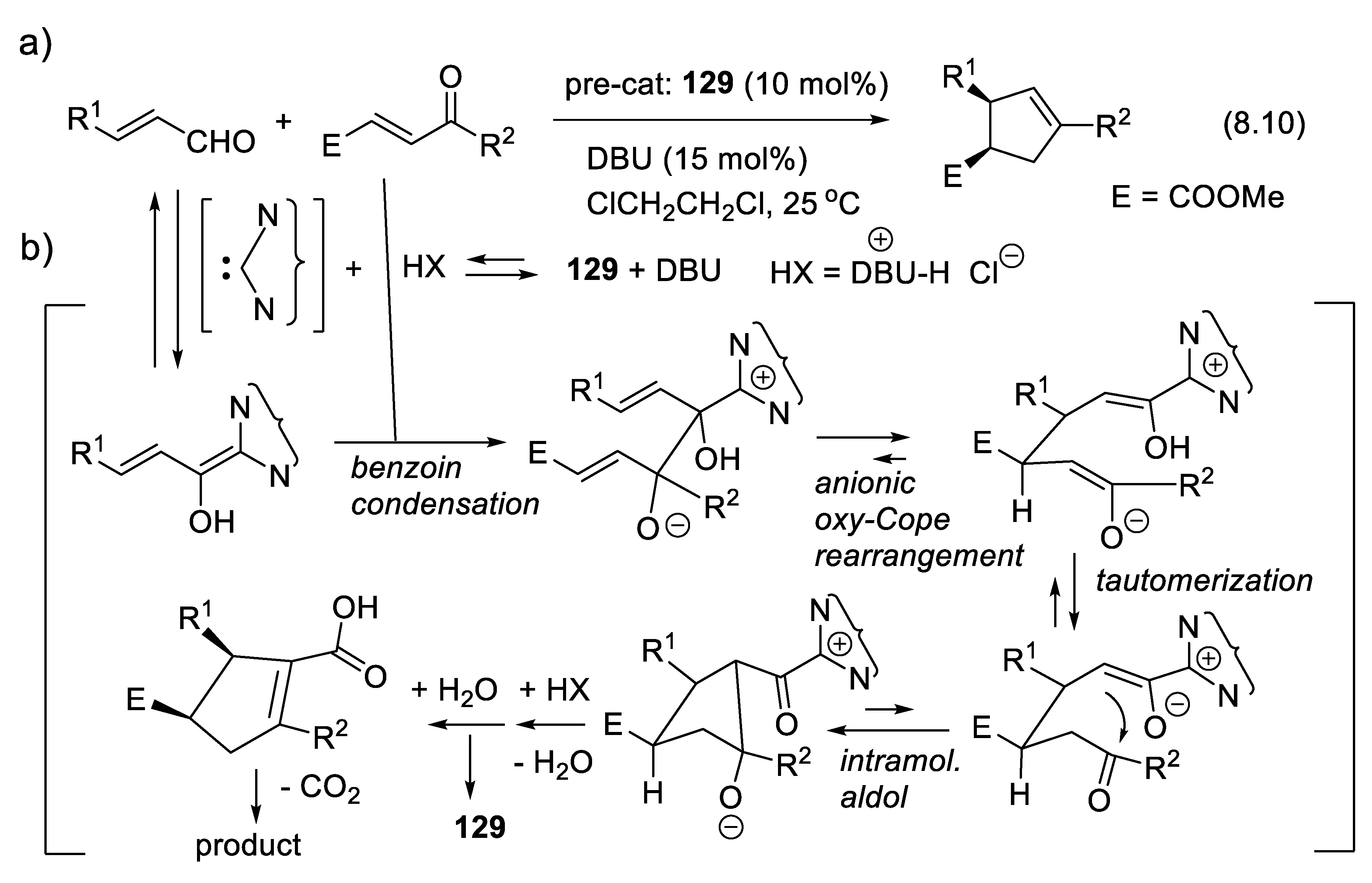
Figure 8.10.
a) Enantioselective synthesis of dihydropyranones (Bode, 2010); b) Possible mechanism.
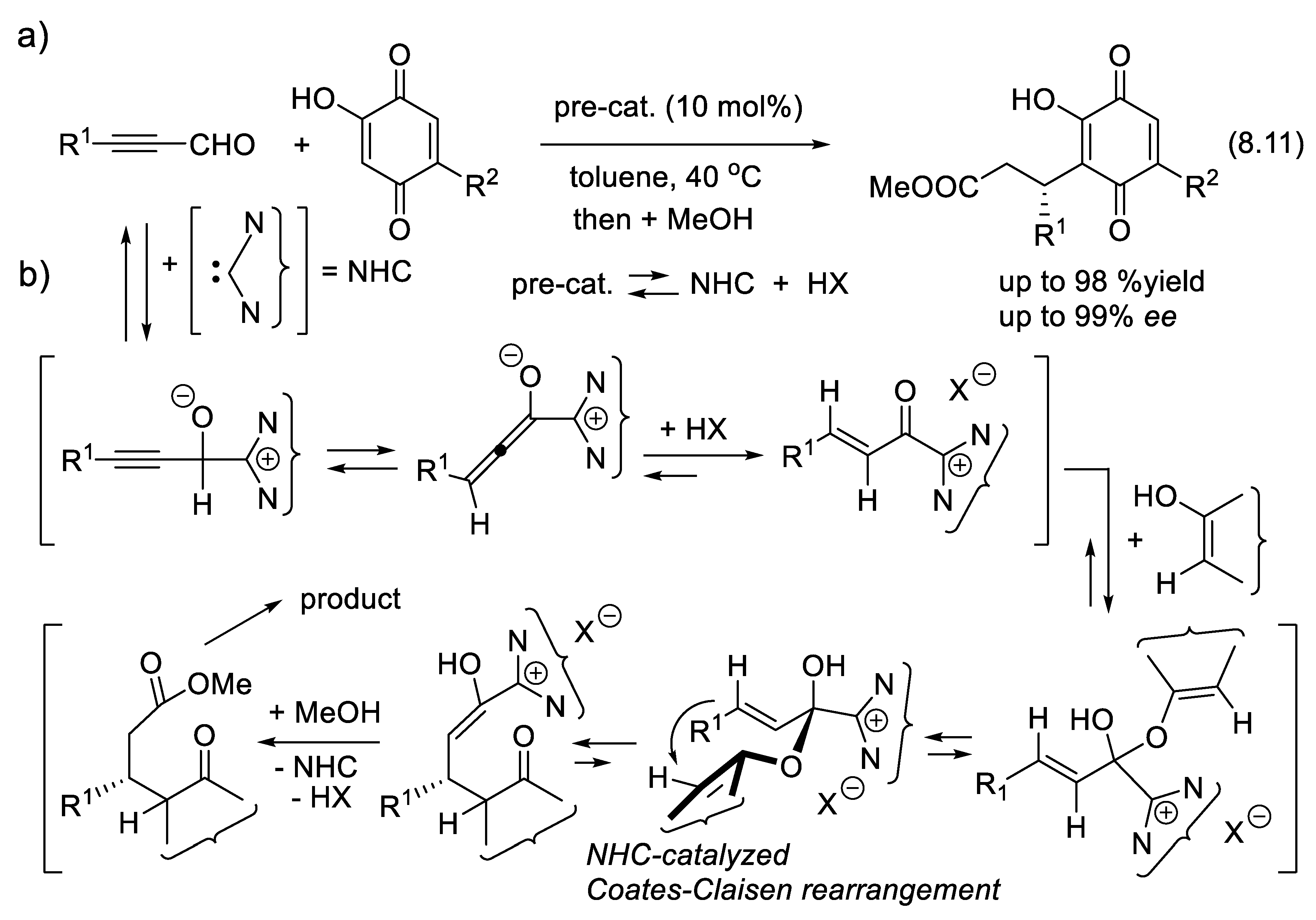
Figure 8.11.
a) Michael Umpolung forms homoenolates that add intramolecularly to a second Michael acceptor (Lupton, 2016). Asymmetric protonation of the resulting enolate follows. b) Possible mechanism.
Figure 8.11.
a) Michael Umpolung forms homoenolates that add intramolecularly to a second Michael acceptor (Lupton, 2016). Asymmetric protonation of the resulting enolate follows. b) Possible mechanism.
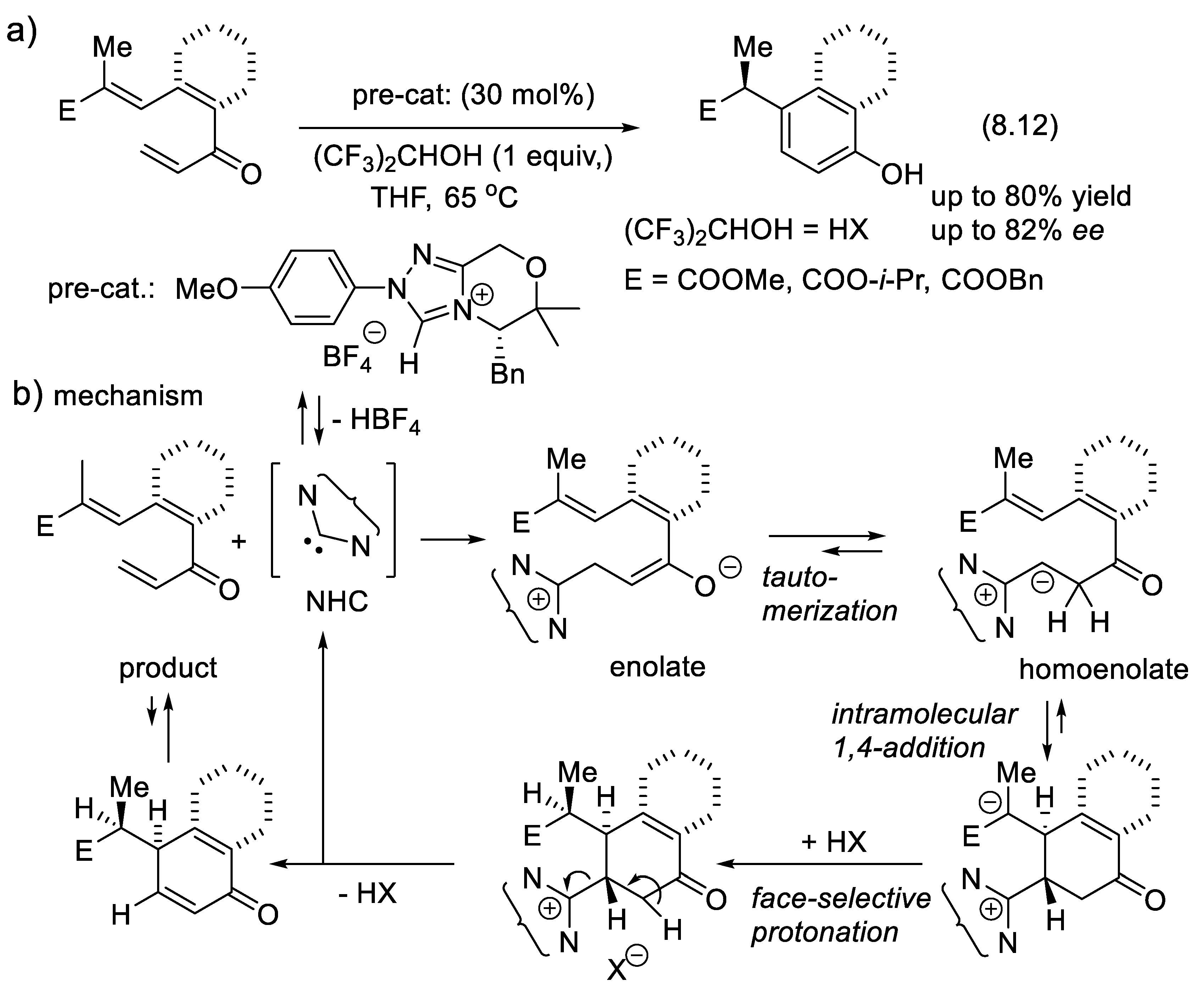
Figure 8.12.
a) Asymmetric oxidation of enals into β-hydroxyesters (Rovis, 2014); b) Possible mechanism.
Figure 8.12.
a) Asymmetric oxidation of enals into β-hydroxyesters (Rovis, 2014); b) Possible mechanism.
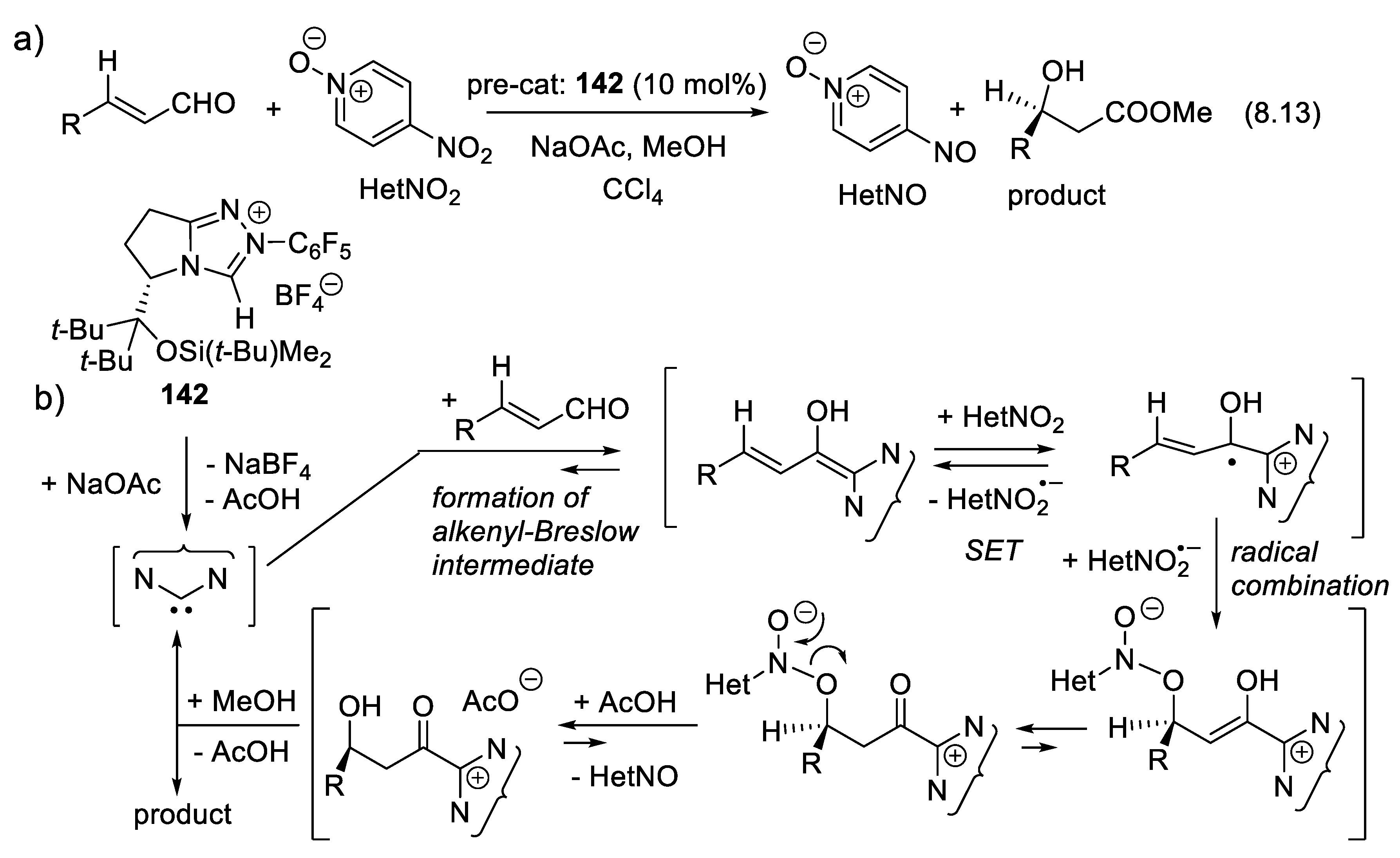
Figure 8.13.
a) Photocatalyzed enantioselective functionalization at the C(4) position of pyridinium salts through NHC catalysis (Hong, 2022); b) Possible reaction mechanism.
Figure 8.13.
a) Photocatalyzed enantioselective functionalization at the C(4) position of pyridinium salts through NHC catalysis (Hong, 2022); b) Possible reaction mechanism.
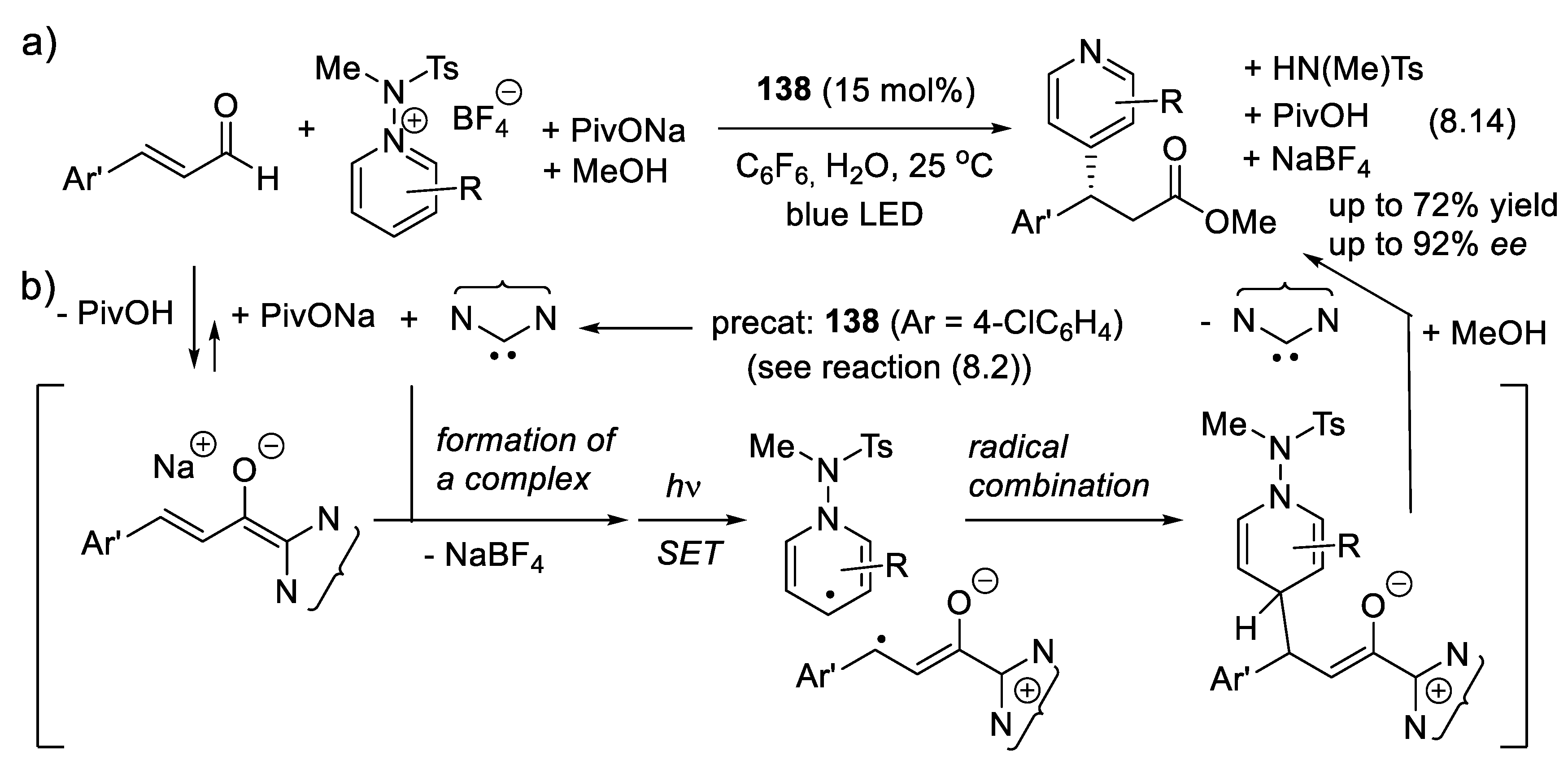
Figure 8.14.
Cooperative photo-redox and N-heterocyclic carbene catalysis. Enantioselective direct oxidative acylation of benzylic C-H’s (Ye, 2025).
Figure 8.14.
Cooperative photo-redox and N-heterocyclic carbene catalysis. Enantioselective direct oxidative acylation of benzylic C-H’s (Ye, 2025).
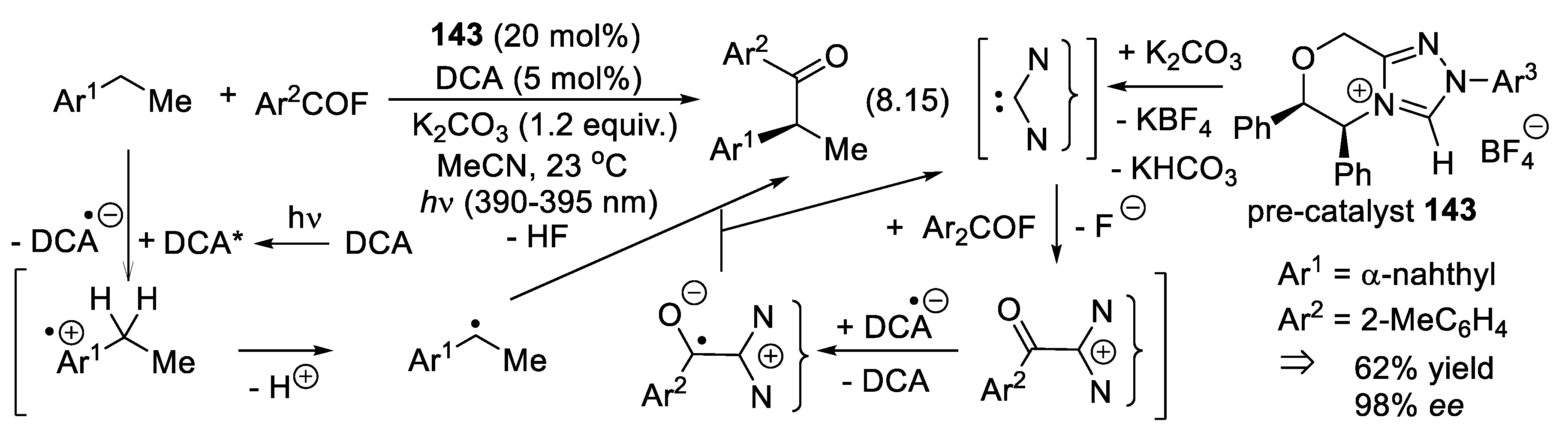
Figure 8.15.
Enantioselective (3+3)-annulation (Wang, 2918); b) Possible mechanism.
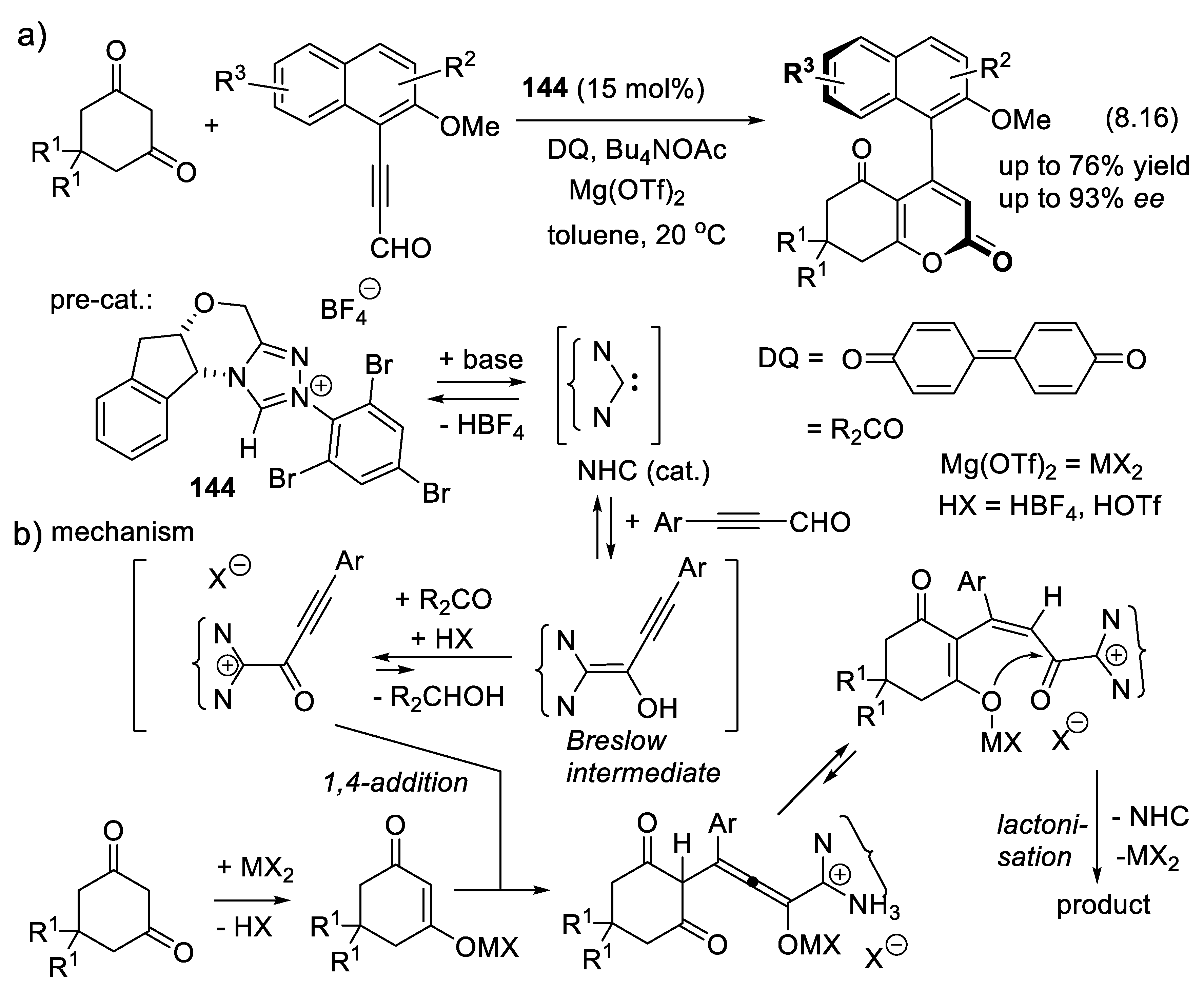
Figure 9.1.
Typical C2-symmetrical chiral diols. Both their enantiomeric forms are available.
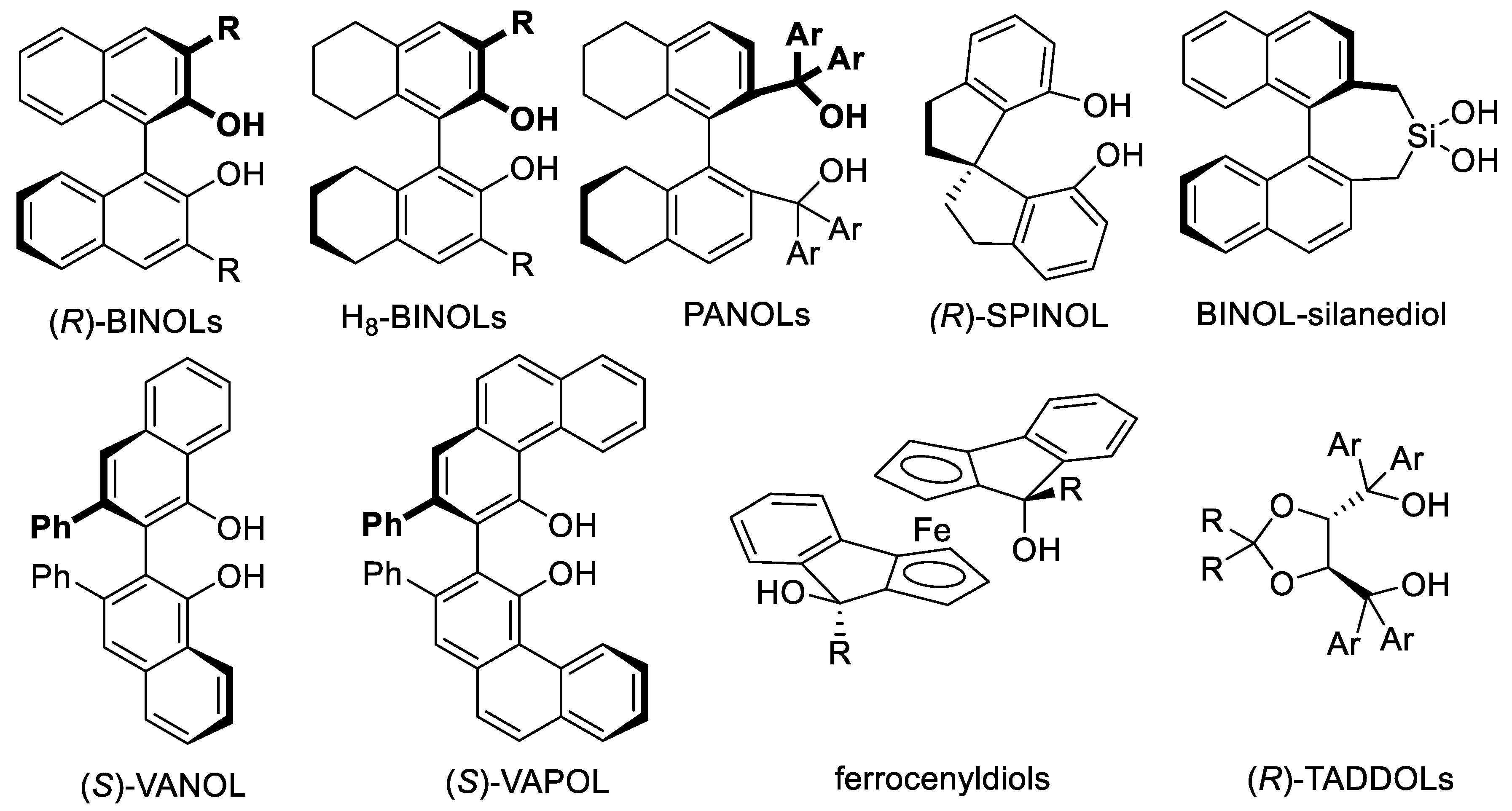
Figure 9.2.
a) Common chiral donors of two N-H bonds; b) Double activation of nucleophilic additions to carbonyl compounds or imines. The zwitterionic intermediate shown resembles the transition structures of the addition (Bell-Evans-Polanyi principle); c) formation of diastereoisomeric H-bonded complexes with anionic nucleophiles. If MX is a salt insoluble in the organic solvent, the role of the catalyst is to extract the anion X(-) from the salt by H-binding, generating a much larger ion-pair that is soluble in the organic solvent. One speaks of H-binding phase transfer catalysis or HB-PTC.
Figure 9.2.
a) Common chiral donors of two N-H bonds; b) Double activation of nucleophilic additions to carbonyl compounds or imines. The zwitterionic intermediate shown resembles the transition structures of the addition (Bell-Evans-Polanyi principle); c) formation of diastereoisomeric H-bonded complexes with anionic nucleophiles. If MX is a salt insoluble in the organic solvent, the role of the catalyst is to extract the anion X(-) from the salt by H-binding, generating a much larger ion-pair that is soluble in the organic solvent. One speaks of H-binding phase transfer catalysis or HB-PTC.
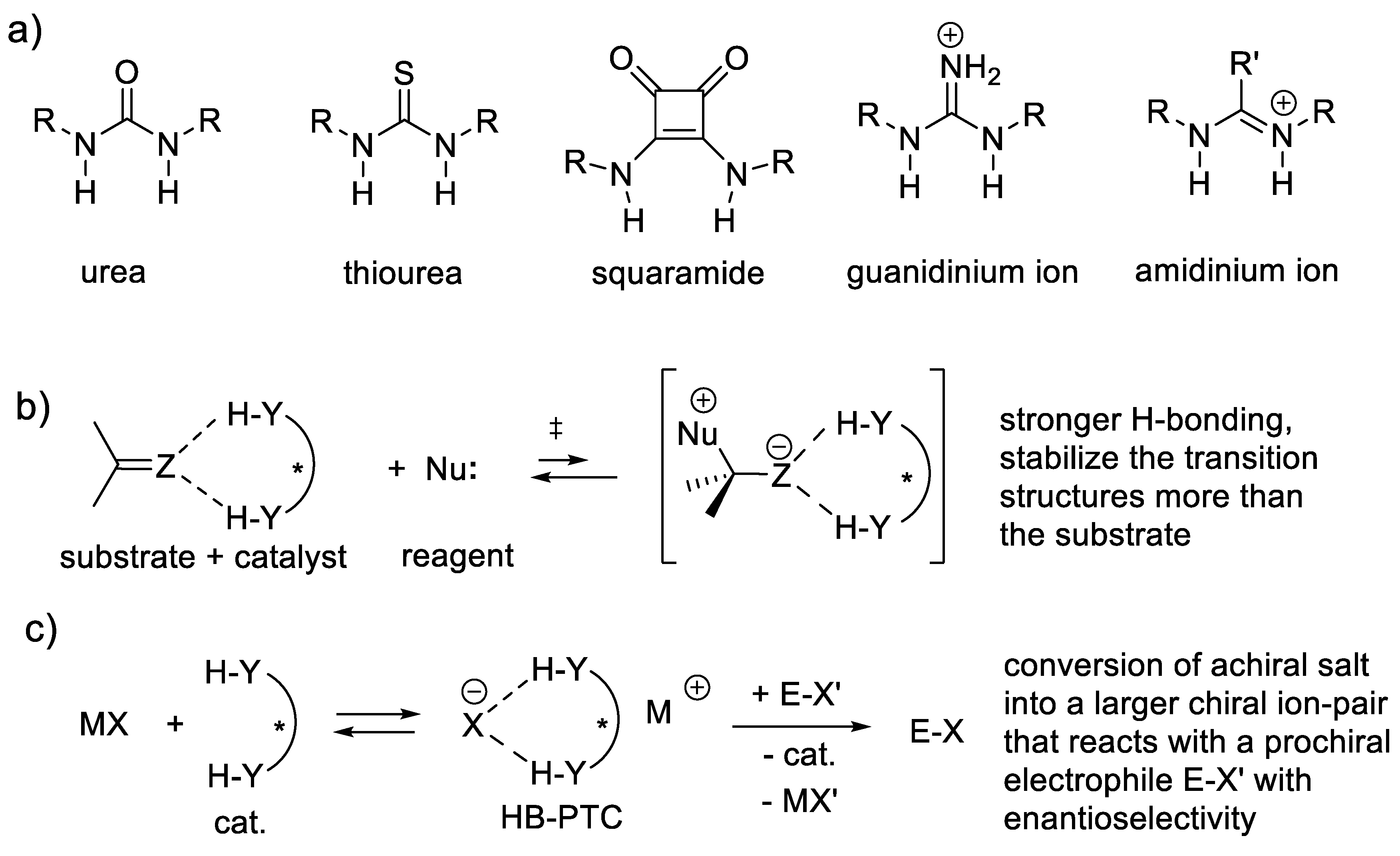
Figure 9.3.
a) Enantioselective anion-binding catalysis (Mattson, 2013); b) Possible mechanism.
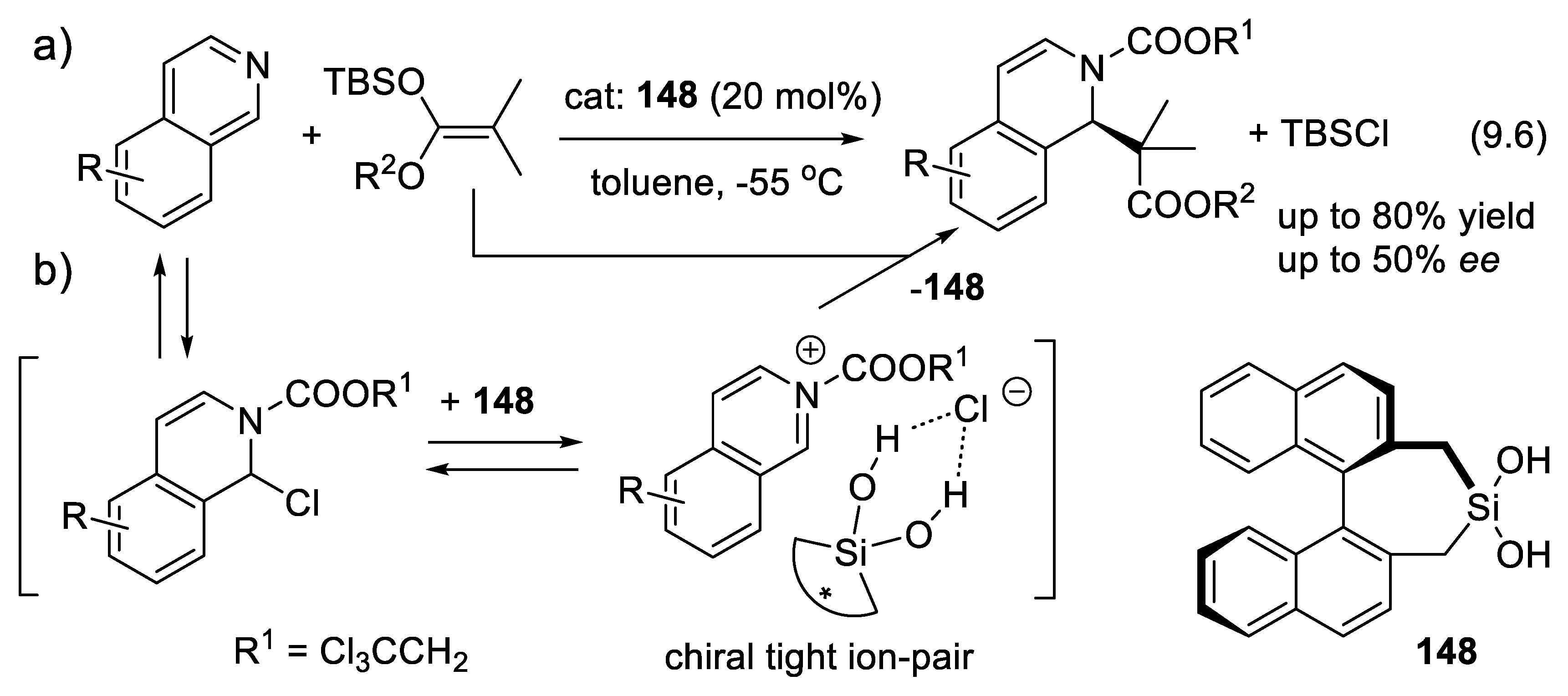
Figure 9.4.
Early examples of hydrogen bonding phase transfer catalysts (HB PTCs).
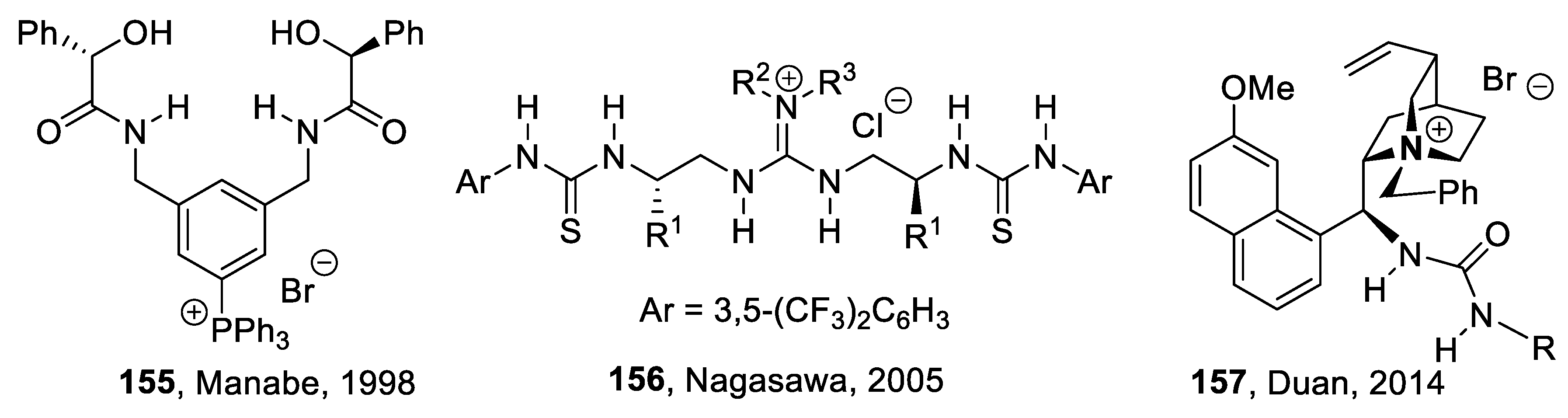
Figure 9.5.
An example of asymmetric hydrogen bonding phase transfer catalysis that does not contain an onium salt. a) Enantioselective nucleophilic fluorination (Gouverneur, 2018); b) Possible mechanism of the reaction that implies the formation of ion-pairs capable to exchange the bromide (in solution) with the fluoride anion of the solid salt (CsF).
Figure 9.5.
An example of asymmetric hydrogen bonding phase transfer catalysis that does not contain an onium salt. a) Enantioselective nucleophilic fluorination (Gouverneur, 2018); b) Possible mechanism of the reaction that implies the formation of ion-pairs capable to exchange the bromide (in solution) with the fluoride anion of the solid salt (CsF).
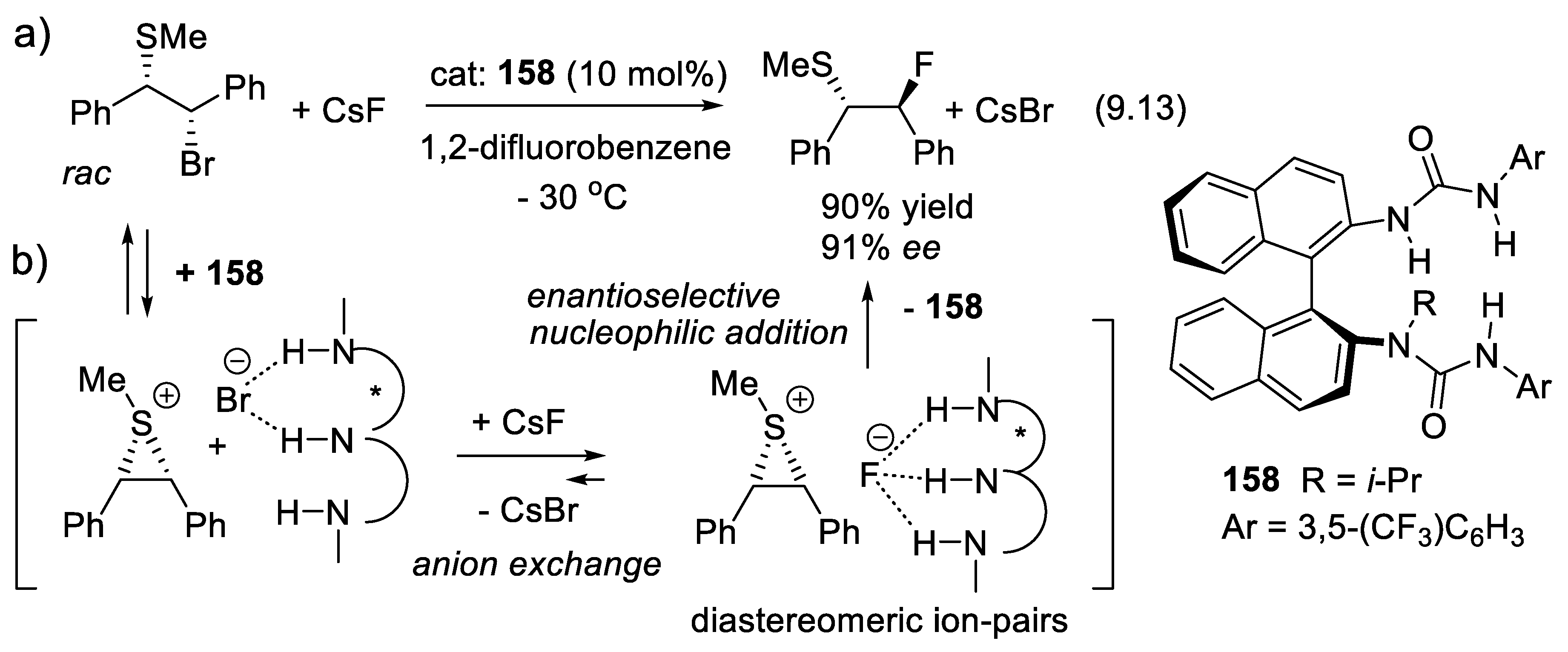
Figure 10.1.
Typical chiral diarylphosphates (CPAs).
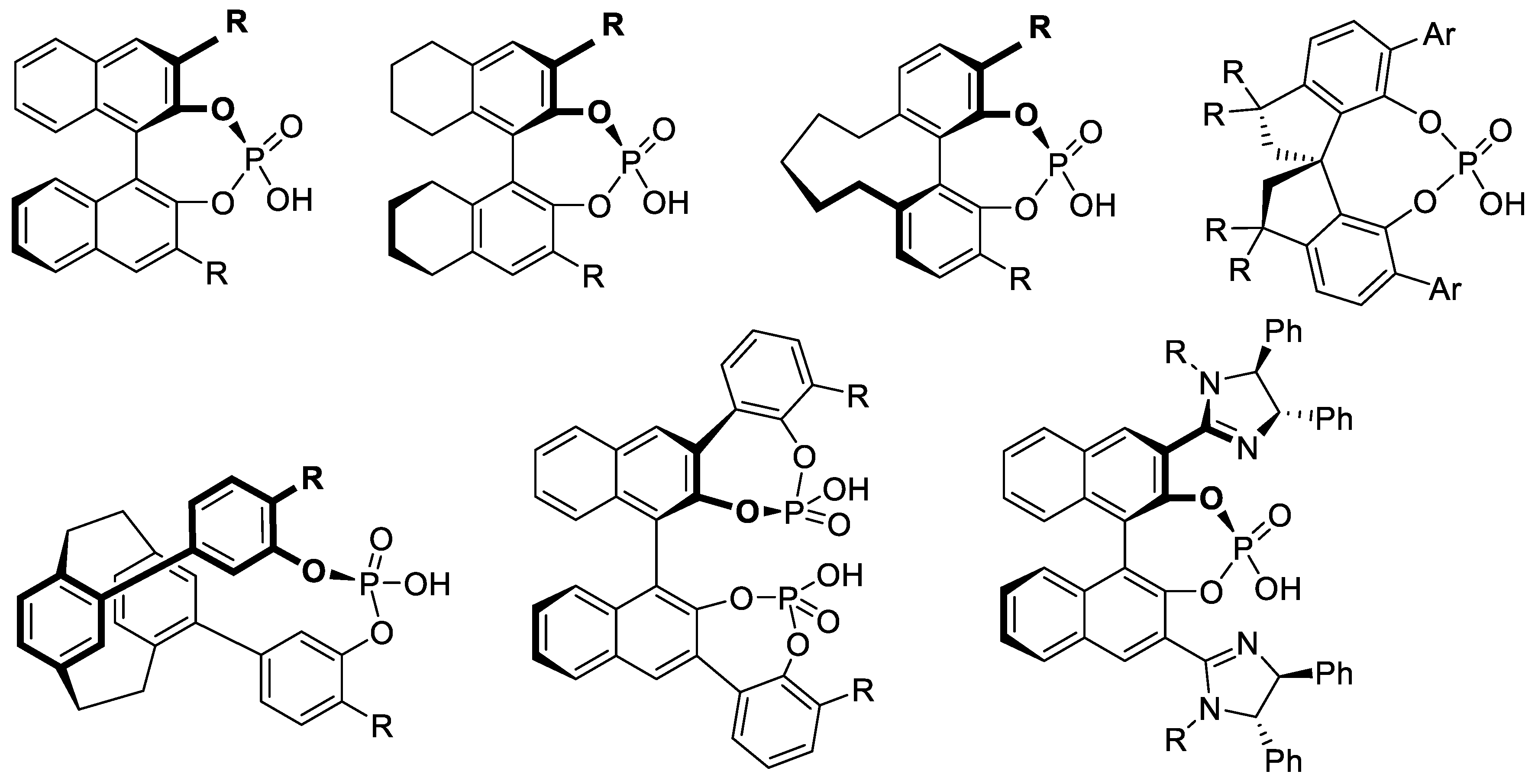
Figure 10.2.
Possible modes of activation by chiral diaryl phosphates.
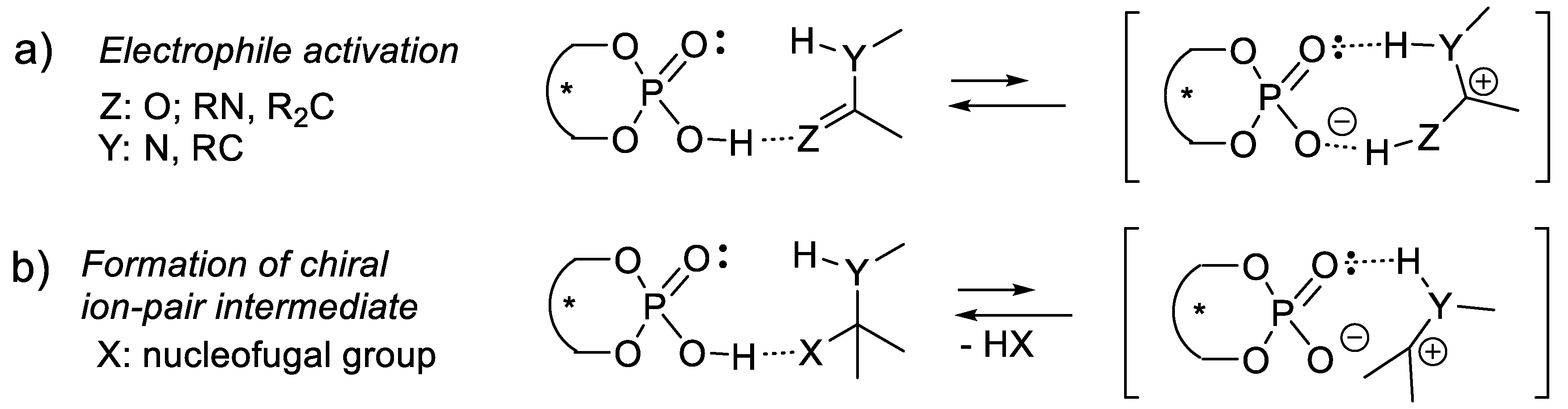
Figure 10.3.
Examples of asymmetric reactions catalyzed by BINOL-derived CPAs (see below for applications of catalysts 175-177).
Figure 10.3.
Examples of asymmetric reactions catalyzed by BINOL-derived CPAs (see below for applications of catalysts 175-177).
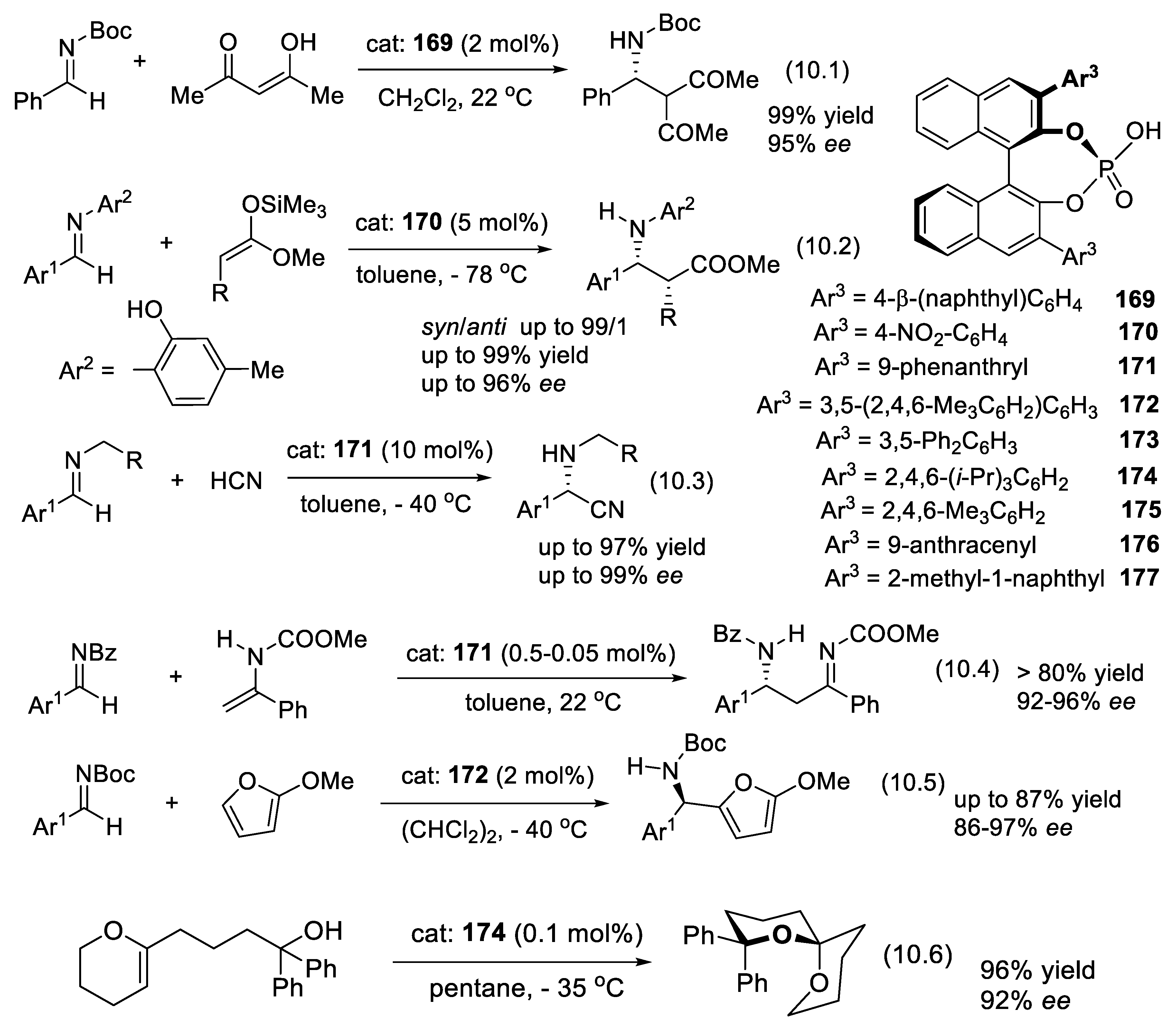
Figure 10.4.
a) Example of enantioselective bromocycloetherification by Lewis base/chiral Brønsted acid cooperative catalysis (Shi, Denmark, 2011); b) Proposed mechanism.
Figure 10.4.
a) Example of enantioselective bromocycloetherification by Lewis base/chiral Brønsted acid cooperative catalysis (Shi, Denmark, 2011); b) Proposed mechanism.
Figure 10.5.
a) CPA-catalyzed asymmetric addition of isocyanides to aldimines (Wang, Zhu, 2009); b) Proposed mechanism.
Figure 10.5.
a) CPA-catalyzed asymmetric addition of isocyanides to aldimines (Wang, Zhu, 2009); b) Proposed mechanism.
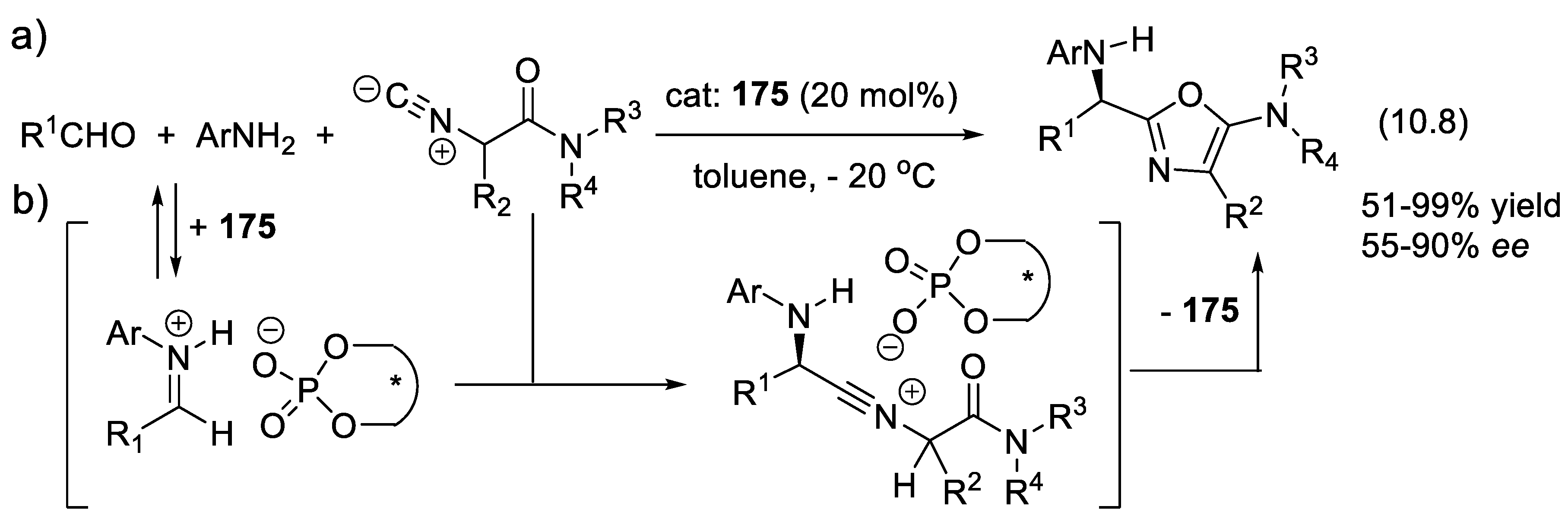
Figure 10.6.
CPA-catalyzed asymmetric Passerini three-component reaction (Tan, 2015).
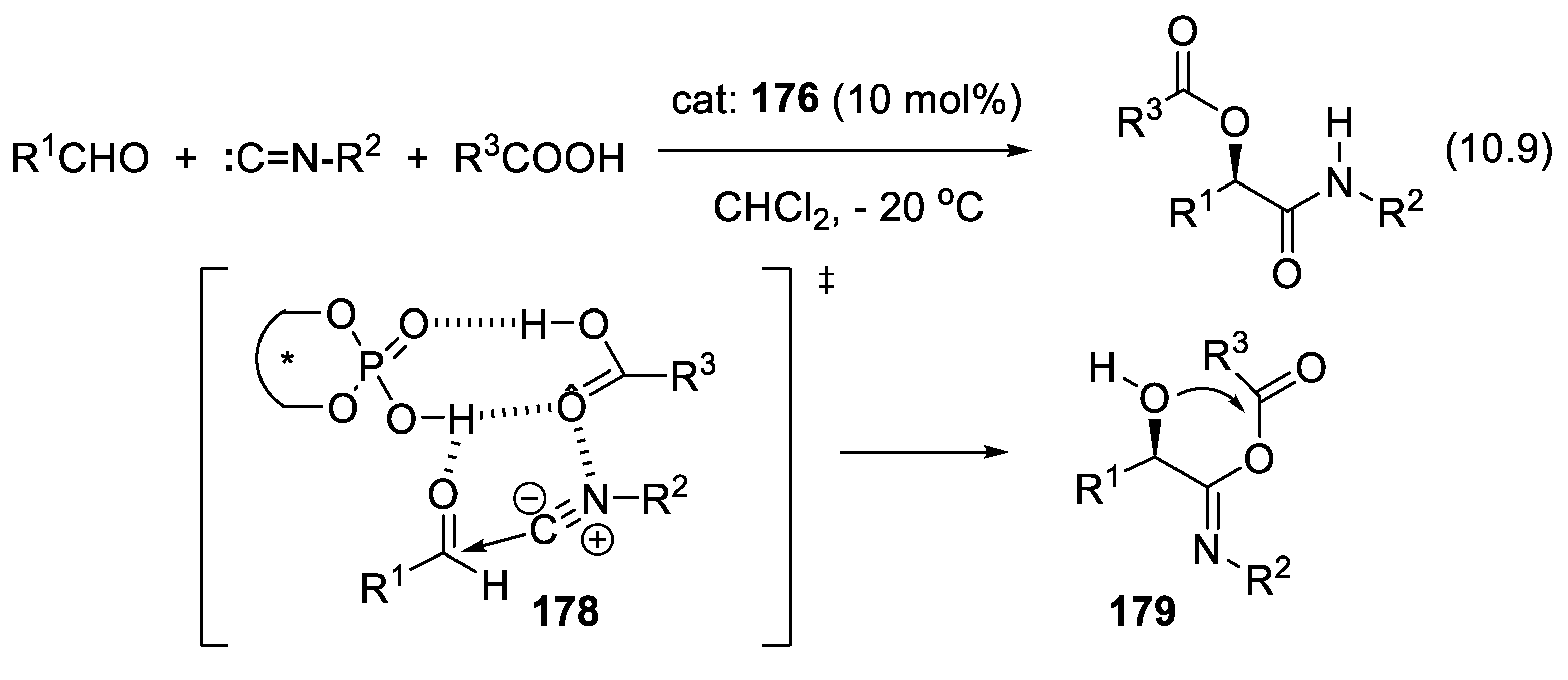
Figure 10.7.
CPA-catalyzed asymmetric Biginelli three-component reaction (Neto, 2018).
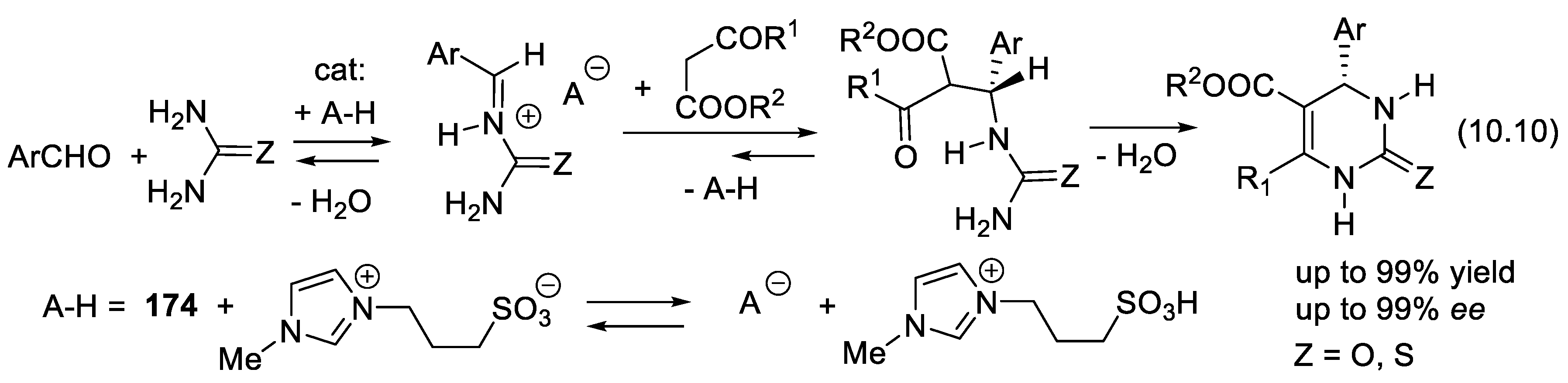
Figure 10.8.
a) CPA-catalyzed asymmetric 1,6-addition of α-arylketones to benzoquinone (List, 2018); b) Proposed mechanism involving proton-coupled electron transfer (PCET) between the enol of the ketone and the Michael acceptor.
Figure 10.8.
a) CPA-catalyzed asymmetric 1,6-addition of α-arylketones to benzoquinone (List, 2018); b) Proposed mechanism involving proton-coupled electron transfer (PCET) between the enol of the ketone and the Michael acceptor.
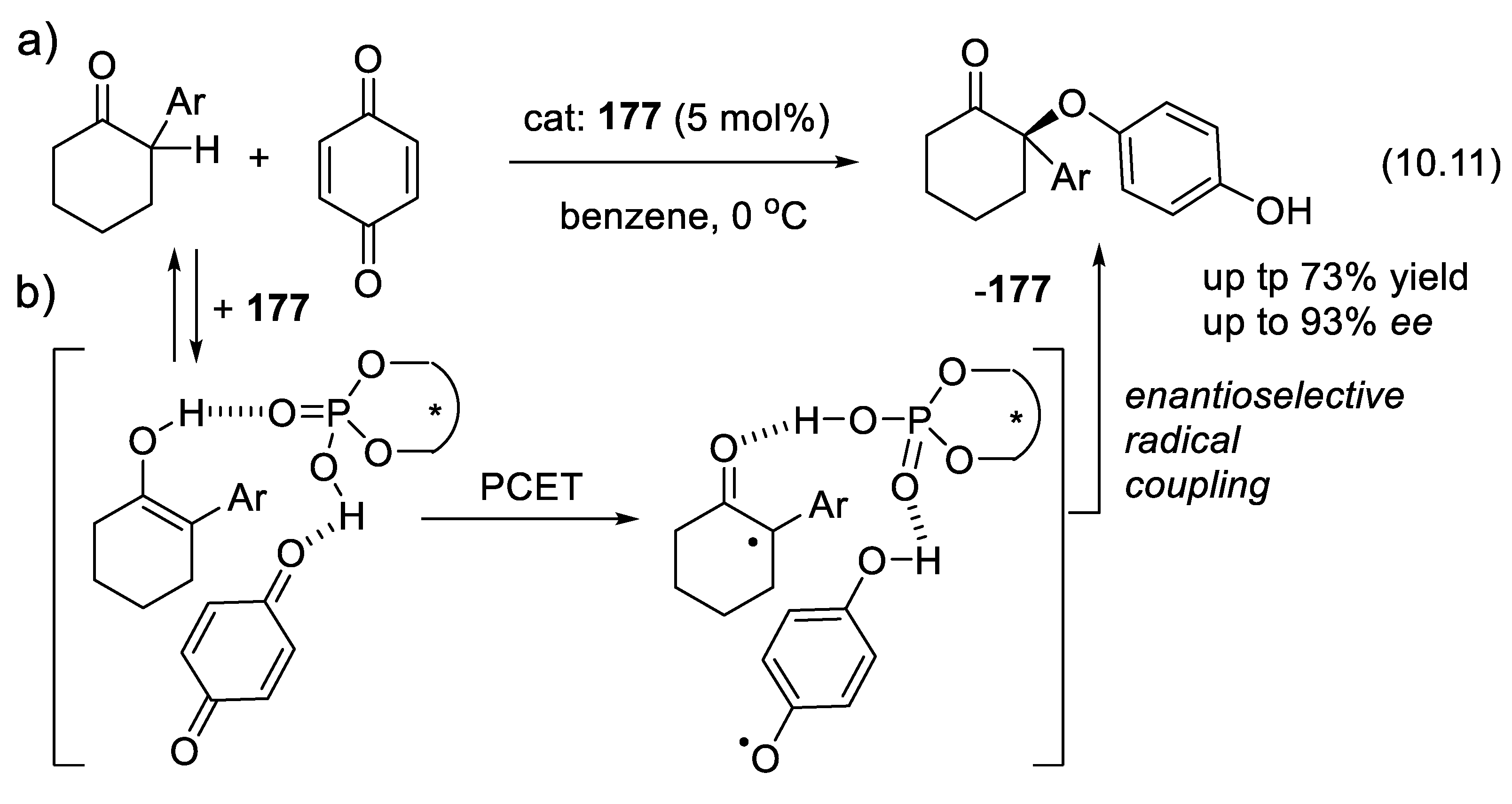
Figure 10.9.
a) Enantioselective (3+3)-annulation (Shi, 2024); b) Possible mechanism.
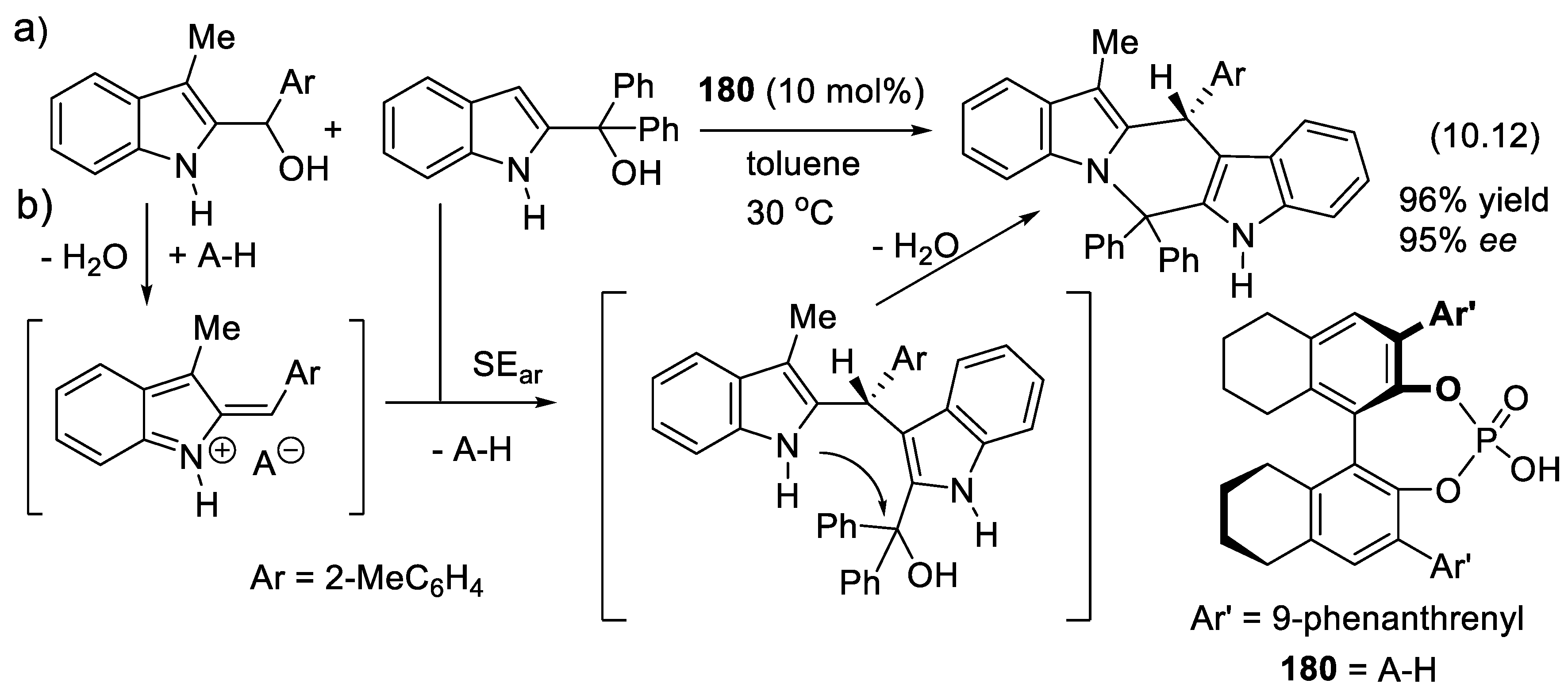
Figure 10.10.
a) Enantioselective intramolecular hydroalkoxylation (List, 2018); b) Proposed mechanism.
Figure 10.10.
a) Enantioselective intramolecular hydroalkoxylation (List, 2018); b) Proposed mechanism.
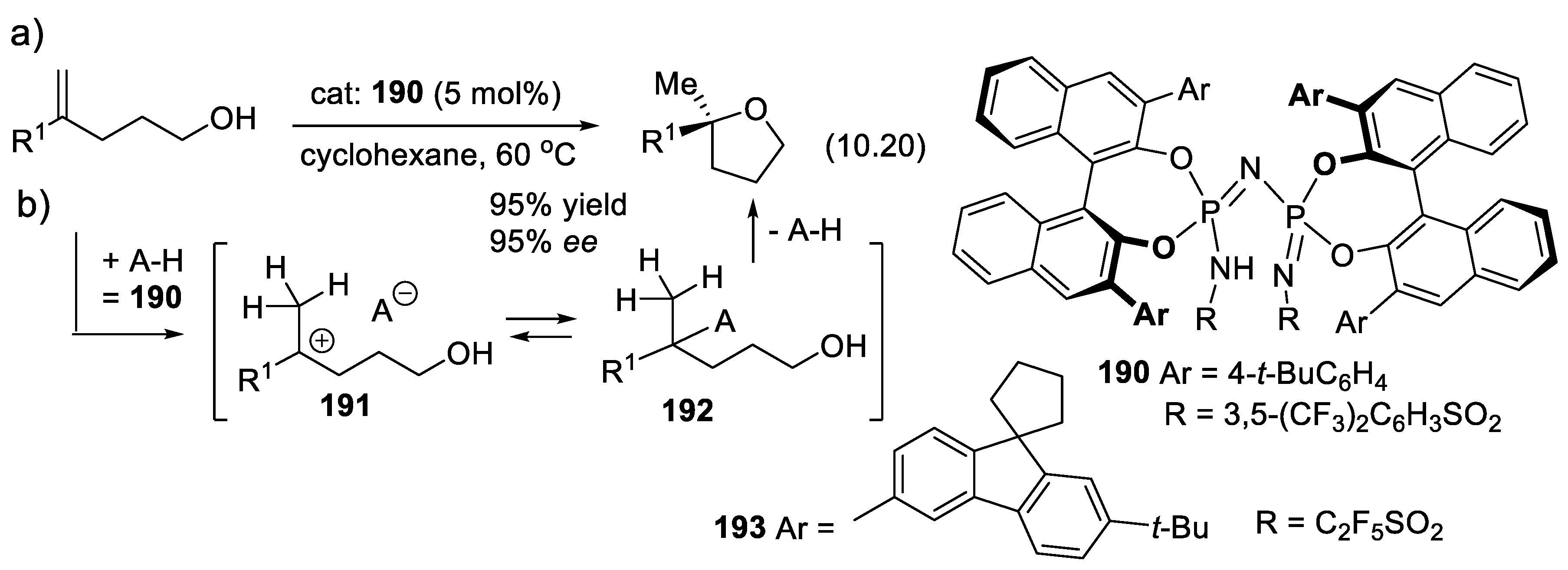
Figure 10.11.
a) Example of a photo-organo-catalyzed cyclopropanation reaction (List, 2025); b) Proposed mechanism.
Figure 10.11.
a) Example of a photo-organo-catalyzed cyclopropanation reaction (List, 2025); b) Proposed mechanism.
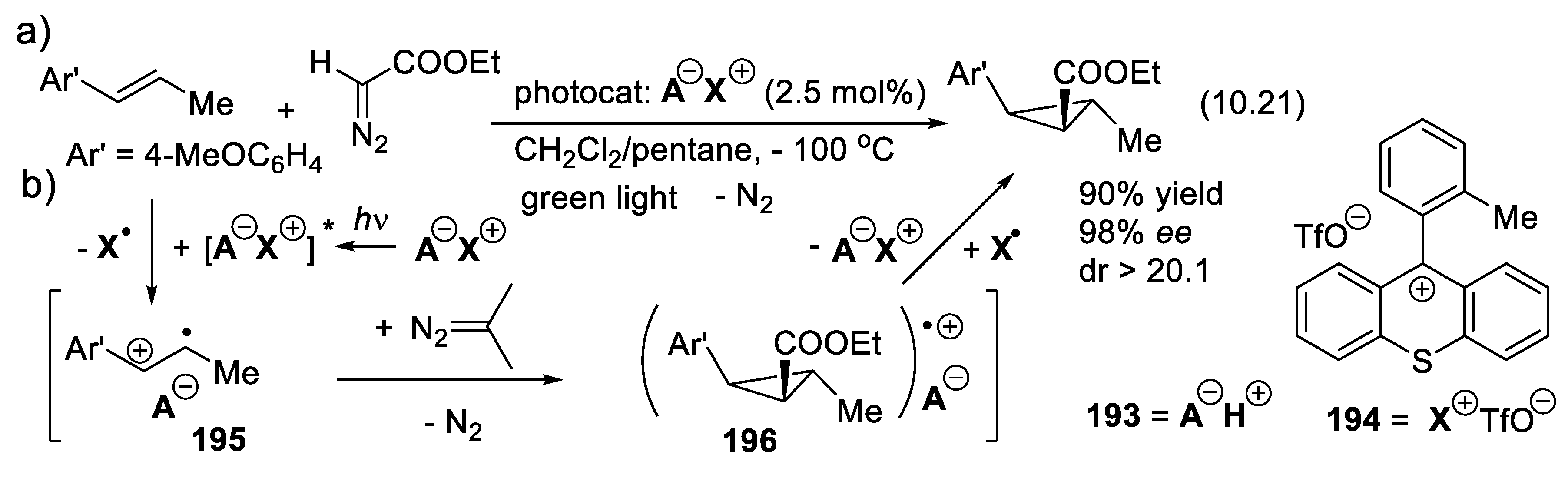
Figure 10.12.
a) Enantioselective three component dipolar cycloadditions (Gong, 2008); b) Possible mechanism.
Figure 10.12.
a) Enantioselective three component dipolar cycloadditions (Gong, 2008); b) Possible mechanism.
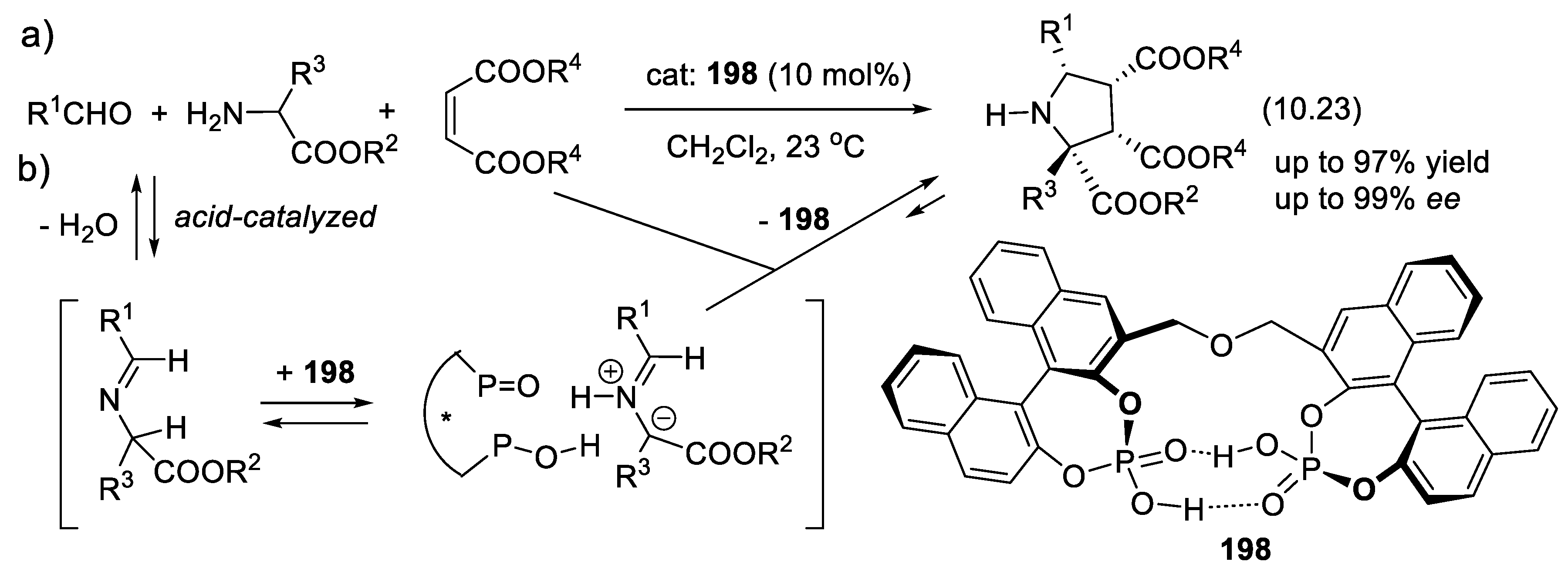
Figure 10.13.
Asymmetric electrophilic fluorination of alkenes (Toste, 2011).
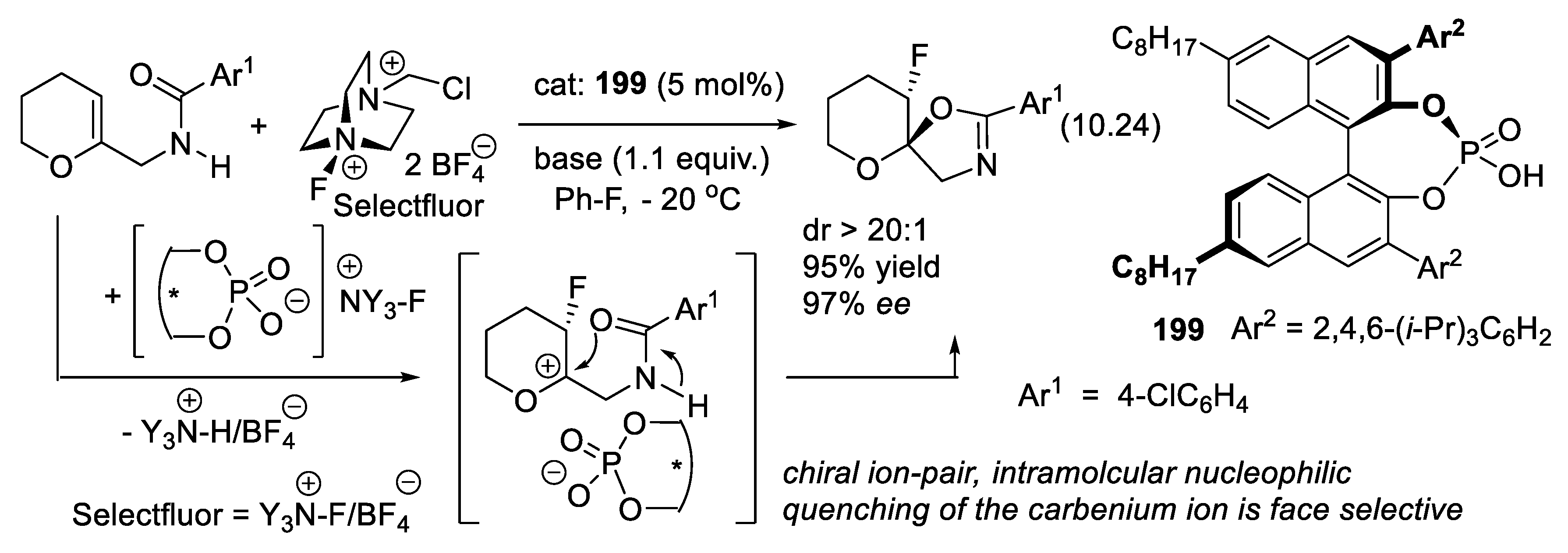
Figure 10.14.
a,c) Chiral disulfonimides are pre-catalysts of Mukaiyama aldol reactions and related reactions (List, 2009, 2013); c) The actual catalysts are N-silyl disulfonimides resulting from the reaction of the catalysts with the silyl enolates.
Figure 10.14.
a,c) Chiral disulfonimides are pre-catalysts of Mukaiyama aldol reactions and related reactions (List, 2009, 2013); c) The actual catalysts are N-silyl disulfonimides resulting from the reaction of the catalysts with the silyl enolates.
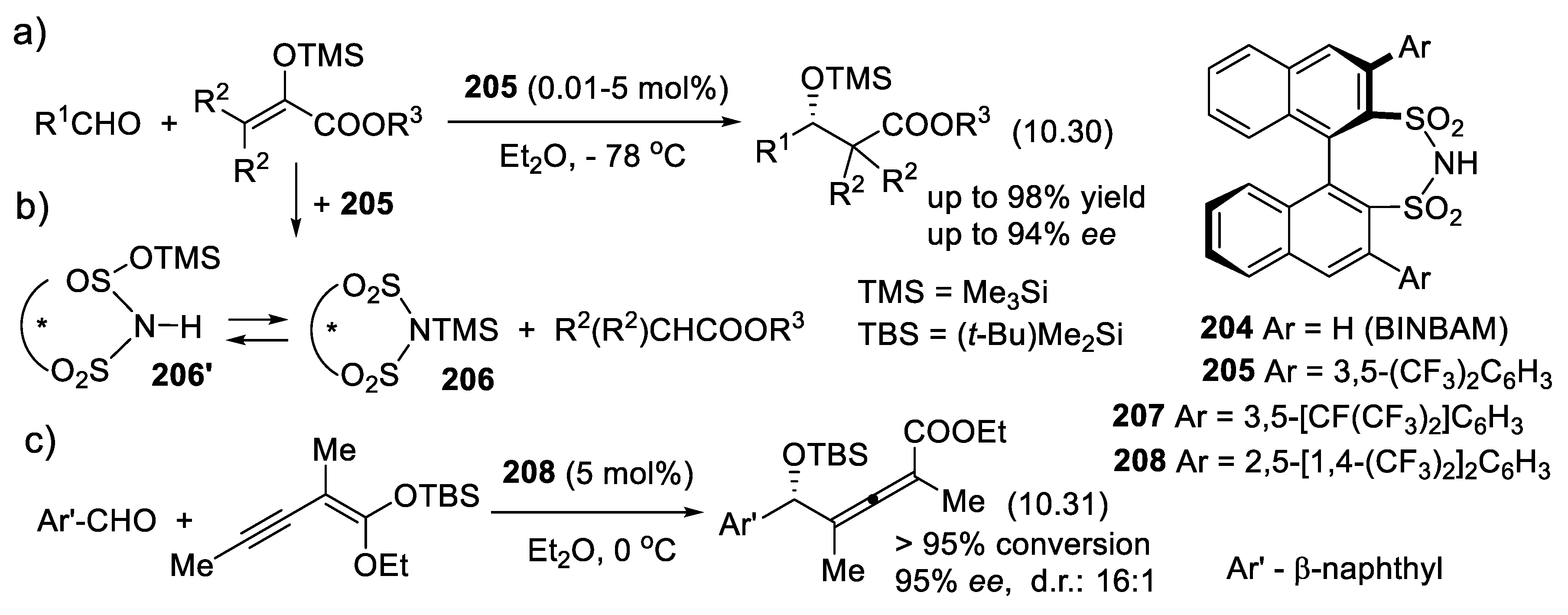
Figure 11.1.
a) Asymmetric epoxidation of alkenes (Shi, 1996). b) Mechanism proposed: formation of dioxirane intermediate.
Figure 11.1.
a) Asymmetric epoxidation of alkenes (Shi, 1996). b) Mechanism proposed: formation of dioxirane intermediate.
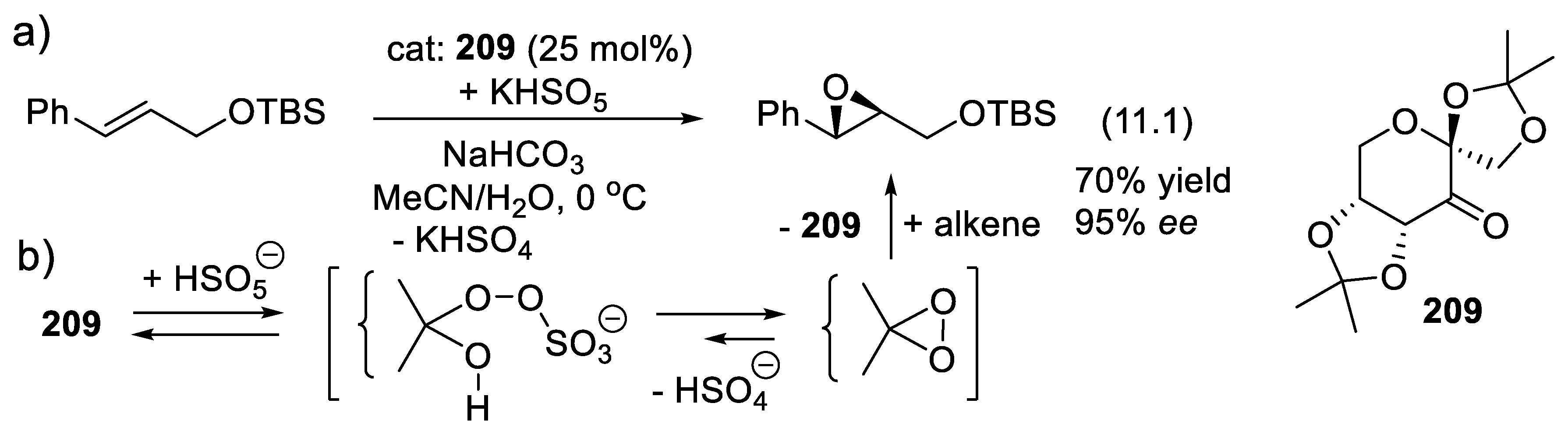
Figure 11.2.
Examples of chiral N-amine oxide catalysts applied to asymmetric allylation of aldehydes (Nakajima, 1998; Hayashi, 2002).
Figure 11.2.
Examples of chiral N-amine oxide catalysts applied to asymmetric allylation of aldehydes (Nakajima, 1998; Hayashi, 2002).
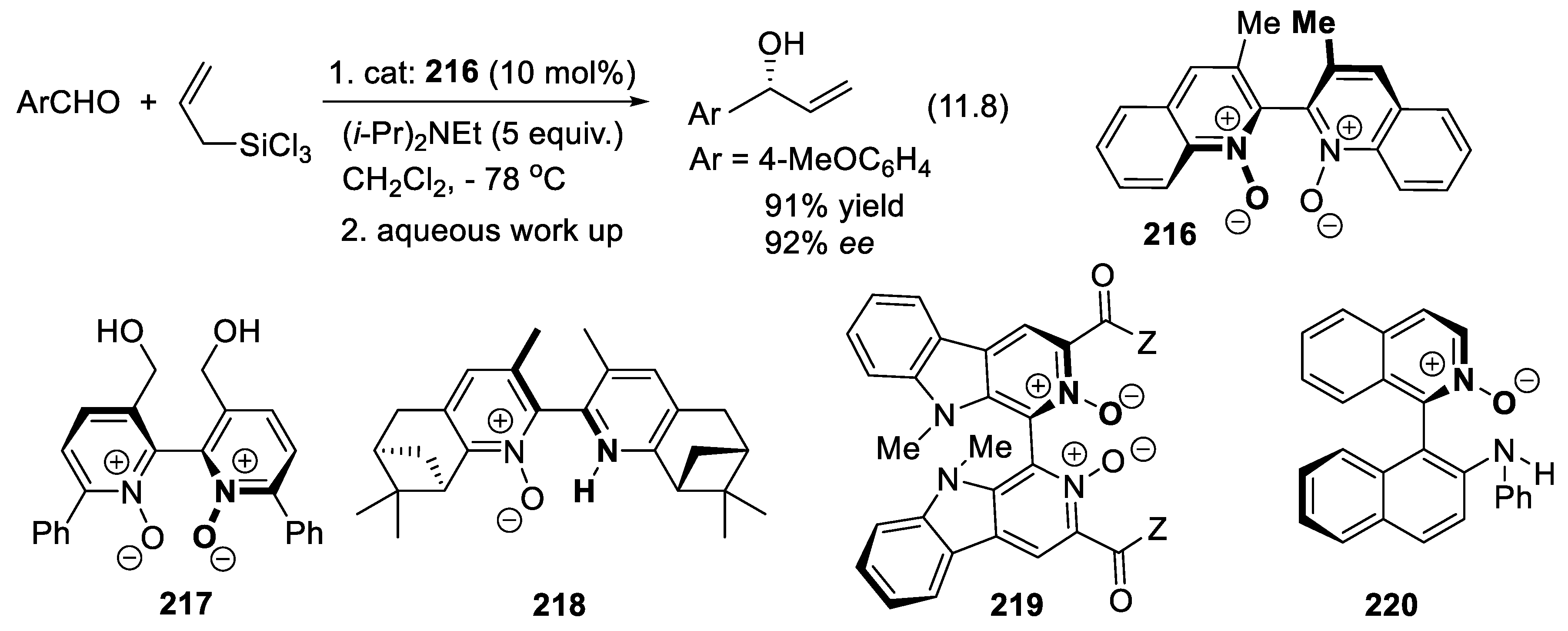
Figure 11.3.
Example of merging organo-catalysis and photocatalysis. Asymmetric Hoffmann-Löffler-Freytag-type of reaction (Lu, 2025).
Figure 11.3.
Example of merging organo-catalysis and photocatalysis. Asymmetric Hoffmann-Löffler-Freytag-type of reaction (Lu, 2025).
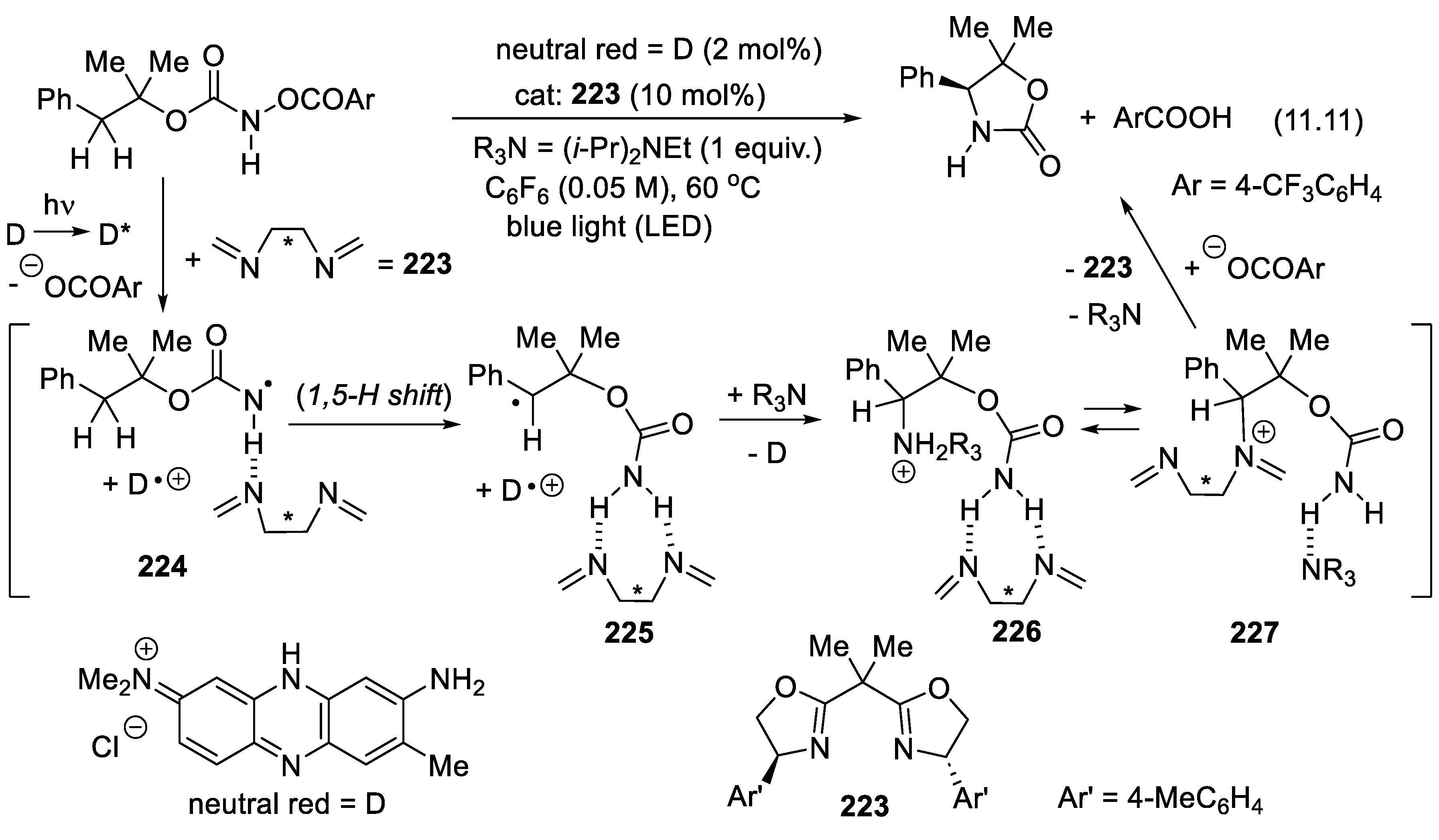
Figure 11.4.
Example of halogen-bonding enantioselective catalysis applying a neutral tetrakis iodotriazole as catalyst (García-Mancheño, 2023).
Figure 11.4.
Example of halogen-bonding enantioselective catalysis applying a neutral tetrakis iodotriazole as catalyst (García-Mancheño, 2023).
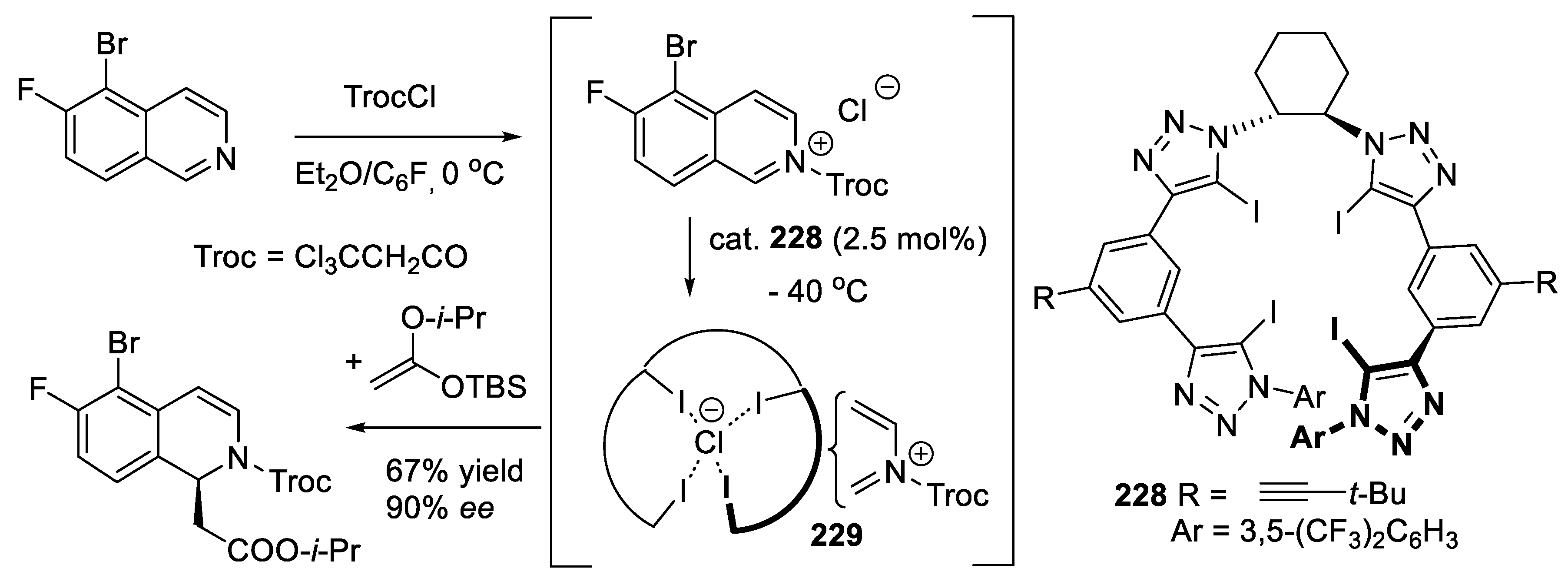
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



