Submitted:
10 October 2025
Posted:
15 October 2025
You are already at the latest version
Abstract
This review synthesizes the current evidence on glycolipid dysregulation in dementia, with a focus on the data generated by mass spectrometry (MS) and systems biology approaches. Glycolipids, key regulators of neuronal function and integrity, have emerged as important players in neurodegenerative processes. In this context, we ex-amine here their altered expression reported in major forms of dementia, including Alzheimer’s disease, frontotemporal dementia, dementia with Lewy body, Parkinson's disease dementia, Huntington’s disease, and mixed dementia. By comparing these conditions, we identified molecular signatures that appear broadly conserved, suggesting common pathways of neuronal vulnerability, while also outlining disease-specific changes that may cause the observed clinical diversity. A special consideration is given to methodological advances, particularly the development and application of high performance MS in glyolipidomics and integrative analytical workflows, which have significantly expanded our understanding of lipid-mediated mechanisms in the diseased brain. Situating these findings within the context of dementia research, we further discuss here how glycolipid biology intersects with protein aggregation, neuroinflammation, and synaptic dysfunction. The results assessed here highlight the major role of MS-based platforms in discovery of glycolipids associated to dementia pathogenesis and at the same time of species with potential biomarker role and/or as novel targets for therapeutic intervention. This review, thus, provides an integrative perspective on glycolipid research by modern MS and allied techniques, emphasizing its potential to influence future strategies for diagnosis and treatment of dementia.
Keywords:
glycosphingolipids
; dementia
; mass spectrometry
; systems biology approaches
; biomarkers
1. Introduction
Dementia encompasses a heterogeneous group of progressive neurodegenerative disorders that together represent one of the most critical global health challenges of the twenty-first century. Defined by cognitive decline severe enough to interfere with independent living, dementia is not a single disease but rather a syndrome with multiple etiologies and pathological substrates [1].
Dementia prevalence is rising inexorably. According to the most recent epidemiological estimates by World Health Organization (WHO), more than 55 million people live at present with dementia worldwide, and projections suggest this figure will exceed 135 million by 2050. This demographic trajectory reflects the combined effects of increased longevity, population aging, and the absence of treatments. The consequences extend beyond patients to caregivers, healthcare systems, and societies at large, with deep medical, social, and economic implications.
Despite the intense research efforts over the past decades, the development of effective therapies has been hampered by the remarkable clinical and molecular heterogeneity of dementia syndromes [2,3] such as Alzheimer’s disease (AD), dementia with Lewy body (DLB), frontotemporal dementia (FTD), Parkinson's disease dementia (PDD), Huntington’s disease (HD), and mixed dementia. Each of these disorders is characterized by a distinct constellation of clinical symptoms, neuropathological features and regional patterns of neurodegeneration (Table 1) [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. This diversity is a consequence of a set of convergent processes such as synaptic dysfunction, protein aggregation, mitochondrial impairment, oxidative stress and neuroinflammation. All these aspects suggest a shared molecular basis. The molecular basis of the major dementia types—highlighting both shared pathways and disease-specific features—are illustrated in Figure 1. Progressively, the dysregulation of lipid metabolism has emerged as a critical contributor to these processes [26], pointing toward a dimension of dementia biology that has been neglected to some extent.
Human brain is a particularly lipid-rich organ; brain lipids encompass nearly half of its dry weight and form the structural backbone of neuronal and glial membranes. Poorly explored and long regarded primarily as passive building blocks, lipids are nowadays recognized as dynamic regulators of brain physiology and pathology, linked with crucial structural and functional brain features [27]. They shape the membrane architecture, influence curvature and fluidity, organize signaling microdomains, and participate directly in intracellular communication, vesicle trafficking, and synaptic plasticity [26].
Among the diverse lipid classes, glycolipids represent one of the most functionally prominent groups in the nervous system [28]. These amphipathic molecules, defined by the covalent linkage of carbohydrate residues to ceramide (Cer) or glycerol lipid backbones, are highly enriched in neuronal membranes and localized in lipid rafts, where they coordinate receptor clustering, neurotransmission, and cell-cell recognition [29]. Glycolipids such as gangliosides (GGs) and sulfatides are integral to brain development, axonal guidance, and myelin stability [30,31]. Their importance is illustrated by lysosomal storage disorders such as Tay-Sachs disease and Gaucher disease in which inherited defects in glycolipid catabolism lead to catastrophic neurodevelopmental and neurodegenerative consequences [32,33]. These conditions provide compelling evidence that even subtle perturbations in glycolipid homeostasis can generate intense effects on neuronal survival and function.
Over the last two decades, the research has demonstrated that glycolipid dysregulation is not confined to rare genetic disorders, but also plays a role in common neurodegenerative diseases [32]. Alterations in glycolipid composition influence membrane biophysics and the organization of lipid rafts [29], thereby modulating the aggregation and toxicity of pathogenic proteins, including amyloid-β (Aβ), tau, α-synuclein, and huntingtin (HTT).
Specific GGs have been shown to seed Aβ fibrillogenesis in AD [34] while disruptions in sphingolipid-glycolipid balance contribute to α-synuclein misfolding in DLB [35]. In HD, impaired GG biosynthesis exacerbates excitotoxicity and mitochondrial dysfunction, accelerating neuronal loss [36,37].
Glycolipid metabolism is also tightly connected with neuroinflammatory processes; GGs and sulfatides modulate microglial activation, complement signaling, and peripheral immune responses, all of which are increasingly recognized as central to the pathophysiology of dementia [26.] These diverse lines of evidence highlight glycolipids not merely as passive bystanders of neurodegeneration but as active participants and amplifiers of disease progression.
The systematic study of glycolipids in neurodegeneration has long been hindered by technical challenges. Glycolipids are chemically diverse, encompassing a vast repertoire of species that differ in carbohydrate composition, Cer chain length, and degree of saturation. They are present in relatively low abundance compared to other lipids, and many exist as isobaric or closely related molecular species, making them difficult to resolve with conventional biochemical approaches [38]. Early studies relied on thin-layer chromatography (TLC) [39] and immunodetection [40] methods, which lacked the sensitivity and specificity to provide a comprehensive representation of glycolipid expression and metabolism.
In this context, the development of modern mass spectrometry (MS) has revolutionized the field of lipidomics. MS has become the most efficient method in contemporary glycolipid research, enabling the detection, de novo structural characterization, and quantification of hundreds of lipid species with unprecedented reliability. Advances in ionization methods, including electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI), have dramatically improved the sensitivity for glycolipid analysis. High-resolution platforms such as Fourier-transform ion cyclotron resonance and Orbitrap mass analyzers provide the mass accuracy required to distinguish isobaric species, while tandem mass spectrometry (MS/MS) allows for detailed structural elucidation of glycan moieties and lipid backbones [41]. Liquid chromatography coupled with MS (LC-MS) has further enhanced separation and quantitation, reducing ion suppression and enabling the analysis of complex biological mixtures such as cerebrospinal fluid and brain extracts [42,43,44]. On the other hand, MS imaging (MSI) techniques such as MALDI-MSI [44], desorption electrospray ionization (DESI) [45] and secondary ion MS (SIMS) [46] imaging allow spatially resolved detection and mapping of glycolipids directly in biological tissues. More recently, ion mobility spectrometry (IMS-MS) [47,48,49] and differential IMS (DMS) [50] have been integrated into MS workflows, offering an orthogonal dimension of separation based on molecular shape and size, thereby improving dramatically the detection and identification of single glycolipid species in highly complex mixtures, yielding at the same time valuable data on isomeric structures.
Table 2 represents a practical mapping of how different MS platforms and workflows compare for the discovery and validation of glycolipids species.
These technological advances have transformed our understanding of glycolipid biology in dementia. MS-based studies have revealed distinct alterations in glycolipid profiles across different dementia syndromes, some of which appear early in disease and may serve as biomarkers for diagnosis or disease progression. In AD, MS analyses of postmortem brain tissue have consistently reported reductions in sulfatides and alterations in GG composition, changes that correlate with amyloid pathology and cognitive decline [51]. In FTD, lipidomic profiling by MS has uncovered disruptions in glycosphingolipid (GSL) metabolism linked to progranulin deficiency and lysosomal dysfunction [52]. In DLB, MS has identified shifts in GSL species associated with α-synuclein aggregation [53], while in HD, altered GG metabolism detected by MS correlates with disease severity and neuronal vulnerability [54]. Importantly, these findings are not limited to postmortem studies. MS-based lipidomic profiling of CSF and plasma is able to reveal glycolipid alterations that mirror brain pathology, offering a potential window into disease processes during life and raising the possibility of minimally invasive biomarker assays.
Apart from classifying the changes in glycolipid expression and abundance, MS has enabled a deeper interrogation of lipid-protein and lipid-lipid structural and functional interactions. Techniques such as MSI allow for the spatial mapping of glycolipids within brain tissue, revealing region-specific alterations that correspond to neuropathological lesions [55]. Stable isotope labeling and flux analysis, when combined with MS provide insights into the dynamics of glycolipid metabolism, uncovering alterations in biosynthetic and catabolic pathways that might not be evident from static concentration measurements [56]. The integration of these high performance analytical methods with computational and systems biology approaches has opened new ways for understanding how glycolipid dysregulation fits into the broader network of dementia pathology. By combining lipidomic data with genomics, transcriptomics, proteomics, and metabolomics, researchers can construct multidimensional models that capture the interaction between glycolipid pathways, protein aggregation, mitochondrial function and immune activation. Network-based analyses have already begun to identify central nodes in glycolipid metabolism that exert disproportionate influence on disease trajectories, highlighting them as potential therapeutic targets.
The implications of this sustained research are highly important since, at present, GLSs are increasingly viewed as both mechanistic drivers of neurodegeneration and promising biomarkers for clinical application. The detection of disease-specific GSL signatures in accessible biofluids such as CSF and plasma is of high importance for improving early diagnosis, distinguishing between dementia subtypes, and monitoring the therapeutic response. At the same time, therapeutic strategies aimed at modulating GSL metabolism through enzyme inhibitors, substrate reduction therapies, or agents designed to normalize lipid-protein interactions are under investigation [57,58,59]. Although not yet fully developed these approaches illustrate the potential of GSL research for advancing translational applications.
In this review, we aim to provide a comprehensive synthesis of current knowledge on GSL alterations in major forms of dementia, with a particular emphasis on findings derived from MS and systems biology approaches. By examining the data across FTD, DLB, AD, PDD, HD, and the mixed pathology, we seek to emphasize both the shared molecular perturbations that may reflect convergent mechanisms of neurodegeneration and the disease-specific alterations that contribute to clinical heterogeneity. By placing glycolipid research within the broader landscape of dementia biology and by emphasizing the methodological advancements that have enabled its recent progress, this review highlights the major importance of GSLs in the pathogenesis of neurodegenerative disease and their potential as targets for biomarker discovery and therapeutic innovation.
2. Alzheimer's Disease (AD)
Every 20 years the number of people suffering of dementia worldwide doubles [34,60,61,62,63,64], with the most prevalent form being AD. This type of dementia is encountered in 60-70% of cases [41,57,63,64,65,66,67,68,69]. First described in 1906, this neurodegenerative condition which is most common in women [41,70], develops progressively starting around age 65 [41,65], the number of cases increasing from 4.078 million in 1992, to 9.837 million in 2021 [71]. Mainly characterized by impaired cognitive functions, AD impact the individual’s life from inability to carry out daily activities, to complete loss of independence, through symptoms like memory decline, impaired thinking and change in behavior [63,67,68,70,72,73].
AD prevalence increases dramatically depending on age, family history, inheritance of genetic mutations, environmental, metabolic, energetic and vascular factors [41,65,68,74]. It can be classified as familial/early onset and sporadic/late onset AD [34]. Although, a conclusive diagnosis of AD requires postmortem histopathology, current clinical diagnosis relies mainly on neuropsychological testing and neuroimaging. Macroscopically the neuronal and the synaptic loss in AD are marked by cerebral atrophy, most pronounced in the medial temporal lobe and hippocampus, with a posterior-anterior gradient. Structural imaging, computer tomography (CT) and magnetic resonance imaging (MRI) reveals temporal and parietal atrophy, while functional neuroimaging, single photon emission computer tomography (SPECT), demonstrates bilateral temporo-parietal hypoperfusion [41,75,76]. Microscopically, the hallmarks of AD are the abnormal protein aggregation within the brain, such as Aβ-peptide forming senile plaques (SPs), and phosphorylated tau (p-tau) protein which appears as neurofibrillary tangles (NFTs) and neuropil threads [57,67,68,69,72,73,75,77,78,79,80].
The aggregation of normally soluble, non-toxic Aβ peptide into large extracellular insoluble toxic fibrillar deposits (rich in β-sheet structures) within the brain, ultimately forms SP [34,57], through amyloid precursor protein (APP) sequential cleavage by β- and γ-secretases, being degraded by a wide range of proteases [57,72,75,79]. The development of SPs is also associated with astrogliosis, neuroinflammation and oxidative stress [69]. The mechanisms of Aβ are the following: (i) accumulation of SP, mainly composed of fibrillar Aβ in the cerebral cortex; (ii) early onset AD mutations within Aβ sequence or near single-membrane APP cleavage sites; and (iii) affection of neuronal cells by oligomers and fibrils [34]. In addition to its toxicity, the Aβ aggregated and deposited structures can also act as a potential storage site and/or producer of Aβ oligomers, which are the main synaptotoxic agents which lead to neuronal dysfunction and finally to brain tissue injury and death, resulting the AD pathology and progression [57,81,82].
NFTs are mainly composed of paired helical filament bundles with a small fraction of single straight filaments and appear in the cytoplasm of perinuclear cells [83,84]. The importance of NFT density is given by its correlation with the severity of diseases [84].
Based on these findings, up to date, advanced methods such as fluoro-deoxyglucose positron emission tomography (FDG-PET) and PET with amyloid- or tau-binding ligands are promising diagnostic methods, but inefficient and not widely used [41,75,76]. So far, US Food and Drug Administration (FDA) approved florbetapir, florbetaben and flutemetamol as amyloid PET ligands and fortaucipir as tau PET ligand. Moreover, FDA and the European Medicines Agency approved decreased Aβ42 and increased p-tau as CSF biomarkers. However, their use is invasive and expensive; therefore the discovery of blood biomarkers appears as a more promising diagnostic method. In this context, for brain amyloidosis detection, the ratio of Aβ42/Aβ40 with apolipoprotein E genotype from plasma, was validated. Also, C2N Diagnostic’s PrecivityADTM blood test and Roche’s Elecsys CSF phosphor-τ 181/Aβ42 assay received FDA approval for clinical use in AD diagnostic [64].
Studies suggest that clinical symptoms of AD appear long after neurodegeneration, which involve many molecular changes, which can be biochemically identified [75]. For example, AD neurodegenation implies cell membranes breakdown [75], therefore is characterized by dysregulated metabolism of lipids, affecting lipid rafts structure and function [29,67,69]. Many researchers studied the involvement of various lipids in AD development and progression, revealing different alterations in lipid classes base on disease phases [69,75]. Among the encounter classes which undergo functional and morphological alterations are glycerolipids, glycerophospholipids, sphingolipids, cholesterol [69,75].
For example, levels of glycerolipids such as monoacyl-, diacyl- and triacylglycerols were found elevated in AD brains. Contradictory, the levels of glycerophospholipids were reduced. Normally, this lipid class is critical for cell membranes, and since neuronal loss occurs in AD, the demand for these lipids decreases. In early stage AD, their levels remain relatively stable, but as the disease progresses, the concentration of glycerophospholipids such as phosphatidylinositol, phosphatidylethanolamine (PE), and phosphatidylcholine (PC) decreases. As for cholesterol, it contributes to the formation and regulation of fibrillation, transport, and elimination of Aβ plaques. Therefore, its excessive accumulation in the brain is also correlated with AD. With regard to sphingolipids, the results vary. For example, Cer levels are high in the early stages, contributing to neuroinflammation and cell death, and as the disease progresses, their levels decrease. Similarly, sulfatides levels decrease as the disease develops. On the other hand, high levels of sphingomyelin (SM) contribute to the formation of Aβ plaques, neuroinflammation, and cognitive impairment. GGs have also proven to be valuable, a distinct alteration in their composition, characterized by reduced levels of GM1, GD1a, GD1b and GT1b GGs, alongside increases in simpler species such as GM2, GM3 and GD3 and cholinergic neuron associated markers, like Chol-1α (GQ1bα) and GT1aα [29,69,76,79,85,86,87,88].
Technologies based on MS have become a popular approach for lipid analysis because of their high detection sensitivity and structural analysis capability. In a MALDI-MSI study were revealed significant reductions in sulfatide (SHexCer) and glycerophosphoinositol (GroPIn) species in specific brain regions of APP/PS1 transgenic mice, a model of AD, compared to age-matched wild-type (WT) controls. The authors identified significant reductions in eight SHexCer species (SHexCer 36:1; O2, SHexCer 38:1; O3, SHexCer 40:1; O2, SHexCer 40:1; O3, SHexCer 42:2; O2, SHexCer 42:1; O2, SHexCer 42:2; O3 and SHexCer 42:1; O3) and two GroPIn lipids (PI 36:4 and PI 38:4), particularly in brain regions involved in learning and memory such as cerebral cortex, hippocampus and cerebellum of APP/PS1 mice. Some lipid alterations were also observed in aged WT mice, suggesting that the decline in SHexCers and GroPIns reflects both aging and AD-related neurodegeneration [87]. Another study where glycolipids were analyzed, using a multimodal TOF-SIMS and MALDI-MSI approach in a transgenic mouse model (tgAPPArcSwe) was conducted by Michno et al. [79]. This work demonstrated a global depletion of cortical sulfatides and revealed plaque-specific lipid and isoforms of Aβ peptide. Cers, including [Cer d18:1/18:0-H]⁻, were found to localize within Aβ plaque-like structures, sulfatides exhibited a general depletion at plaques, most notably in long-chain species such as [ST d18:1/24:0-H]⁻ and [ST d18:1/22:0-H]⁻, with a lesser effect observed for [ST d18:1/20:0-H]⁻. MALDI-IMS further highlighted chemical heterogeneity among individual Aβ deposits, including altered GM1 to GM2/GM3 GG metabolism in the plaque periphery and enrichment of Aβ1-40 at the plaque core. Monosialogangliosides (GMs) containing C18:0 fatty acid moieties, including GM3 and GM2, also displayed plaque-associated localization, whereas GM1 was most prominently enriched at the central core of Aβ plaque-like structures. Moreover, arachidonic acid conjugated PI together with their degradation products, lysophosphatidylinositols, were localized to the periphery of plaques [79].
In order to identify lipid biomarkers associated with early onset AD, Xiao et al. [69]quantitatively analyze lipid changes in the hippocampus of APPswe/PS1dE9 (APP/PS1) transgenic mice in comparison to WT control, via ultra-high performance LC (UHPLC) coupled to MS/MS. Quantitative profiling revealed 43 lipid metabolites with significant alterations in the hippocampus of 7.5-month-old APP/PS1 transgenic mice compared to control. The major lipid classes affected were glycerolipids (36.36%), glycerophospholipids (29.55%), and sphingolipids (27.27%). Notably, in AD brain were found elevated levels of multiple triacylglycerol (TG) species including TG (56:4)_FA(20:4), TG (56:7)_FA(22:6), TG (54:4)_FA(20:4), TG (54:6)_FA(22:6) and TG (44:0)_FA(18:0), indicating altered energy storage or membrane dynamics. A novel finding in this study is the significant elevation of cholesteryl esters (CEs) CE (22:6) and CE (22:4), compounds linked to atherosclerosis. Their accumulation could promote Aβ1-42 aggregation, impair its vascular clearance, and disrupt cerebral blood flow, potentially exacerbating AD pathology. Elevated phospholipids, such as PE (O-16:0_22:4), PE (P-16:0_20:4), PC (18:1_16:1+AcO) and PC (16:0_20:3+AcO), were also observed, indicating ongoing membrane remodeling processes in the hippocampus of APP/PS1 mice. Also, elevated PCs, may serve as lipid biomarkers linked to Aβ accumulation and membrane remodeling. Conversely, significant decreases were found in monogalactosyldiacylglycerol (MGDG) species including MGDG (16:1_18:0), MGDG (16:0_16:1) and MGDG (14:0_16:0), which may compromise hippocampal antioxidant and anti-inflammatory functions in AD mice, suggesting a role for MGDG in neuroprotection. The majority of hexosylceramides (HexCers) and Cer, including HexCer (d18:0/20:0), HexCer (d18:1/16:0), Cer (d18:1/18:1), Cer (d18:1/16:1), Cer (d18:1/24:1) were significantly downregulated. The observed decline in HexCer, Cer and SM species supports a broader impairment of sphingolipid metabolism. These molecules are key components of myelin sheaths and neuronal membranes. Their loss may compromise membrane integrity, microglial phagocytic function, and overall neuronal viability, potentially contributing to cognitive impairment in AD models [69].
Using LC-MS, lipid profiles from 107 patient’s sera, including 39 AD patients, were observed significant differences in lipid metabolites among groups. Overall, the study identifies CE (16:3) and GM3 (d18:1/9Z-18:1) GG as promising serum biomarkers for the early clinical prediction of AD. Notably, levels of both metabolites showed a positive correlation with dementia severity, suggesting their potential utility in both early diagnosis and disease monitoring [89].
Among the multitude of studies conducted to date, GGs have been shown to be some of the most valuable lipids expressed in the AD affected brain, with their expression varying base on the affected area. In early onset AD, marked reductions in GGs occur in gray and frontal white matter, whereas in sporadic AD, declines are most pronounced in the temporal cortex, hippocampus, and frontal white matter [87].
Studies on GGs have shown that the monoclonal antibody A2B5 selectively stain neurons undergoing neurofibrillary degeneration and neuritic processes within SP in AD, recognizing c-series GG such as GQ1c, GT3 and O-Ac-GT3, which are normally abundant in embryonic brain development. Altered GG distribution, including the accumulation of c-series GGs (GQ1c and GP1c) in NFT, has been observed in specific AD brain regions [90].
MALDI-MSI has been applied to examine age-dependent alterations in a-series GG in the APP/PS1 transgenic mouse model of AD. The study revealed age-related modifications in GM1 and GD1a GGs, in both WT and transgenic mice, in white and grey matter, alongside significant increases in GM2 and GM3 in the cortex and dentate gyrus of APP/PS1 mice at 12 and 18 months. Notably, GM3 accumulation colocalized with Aβ plaques and correlated with HEXA gene expression, implicating GG degradation as a mechanism underlying its buildup [67].
A Q Exactive MS coupled with UltiMate 3000 LC system, was applied to characterize the GG species composition in the hippocampus of APPswe/PS1dE9 transgenic mice. 48 distinct GGs were associated with AD, including species acetylated and N-acetylgalactosaminylated. Also, di-O-Ac-GT1a (d36:1), O-Ac-GD1b (d36:1) and O-Ac-GD1b (d36:0) were proposed as biomarkers of AD progress, while O-Ac-GT1a (d36:2) as a non-progressive biomarker [91].
In another study, ESI-MS and MALDI-MSI were integrated to investigate GG composition and their alterations in the mice brain tissues. The results revealed striking differences in GG distribution between AD and control group. In particular, nearly 20 GG species were absent in the cerebellum of AD mice, while additional reductions were observed in the right cerebral hemisphere, validated histological by Aβ protein accumulation in the right cerebral hemisphere and a significant loss of neuronal cells in the cerebellum [92].
MALDI-MSI was also employed to examine the in situ distribution of a-series GGs with distinct sphingosine chain lengths (d18:1 vs. d20:1) in post-mortem human AD brain. This analysis revealed a marked reduction in the GM1 d20:1 to GM1 d18:1 ratio within the molecular leyer, dentate gyrus and entorhinal cortex of AD. Furthermore, GM3 and GM1 was found to colocalize with histologically confirmed Aβ plaques, with a possible change that causes metabolism to specifically target GM3 d20:1 species in these regions [86].
A TLC Blot/MALDI-TOF-MS platform was applied to human brain tissue of AD, Parkinson’s disease (PD) and control patients. AD brain exhibited significant reductions in GD1b and GT1b levels compared to PD and control cases. Molecular profiling further revealed higher levels of GG containing d18:1 ceramide in AD, compared to PD and control which expressed higher levels of d20:1 [93].
Caughlin et al. [94] used MALDI-MSI to characterize age and region specific GG alterations in WT and APP21 transgenic Fischer rats at birth, 3, 12 and 20 months. Compared to WT rats, APP21 animals showed a pathological shift toward elevated simple GGs and reduced complex GGs. This imbalance was particularly evident in white matter, which displayed a marked age-dependent decline of GD1 (d18:1) species, while d20:1 species were maintained or increased, resulting in the elevated d20:1/d18:1 ratio observed in aging and AD. Importantly, this ratio was clarified to be driven mainly by a loss of d18:1 rather than expansion of d20:1 form. GM1 levels increased with age in both genotypes, reflecting its role in neuroprotection, although APP21 rats exhibited a more pronounced depletion at 12 months. GM2 levels were modestly but consistently higher in APP21 rats, especially for d20:1 species, while GM3 was significantly elevated across multiple brain regions and from early life onward. GM3 d18:1 was enriched in medial and periventricular structures, and both d18:1 and d20:1 species showed stronger age- and region-dependent increases in APP21 rats, aligning with its known capacity to seed Aβ fibrils and promote neurodegeneration [94].
High spatial-resolution MALDI-MSI (10 μm/pixel) has been employed to investigate GG distributions within Aβ plaques in hippocampal and adjacent cortical regions of 12-month-old 5xFAD mouse brains. The analysis revealed brain region and long chain base (LCB) specific accumulations of GMs in hippocampal and cortical plaques. Distinct distributions of C18:1- and C20:1-LCB containing GMs were discoverd, with C18:1 species enriched in the subgranular zone of the dentate gyrus and C20:1 species localized to the molecular layer along the entorhinal hippocampal pathway. Also, the analysis demonstrated colocalized deposition of GM3 (d18:1/18:0), GM2 (d18:1/18:0), and GM1 (d18:1/18:0), with Aβ peptides (Aβ1-42 and Aβ1-40) in the subiculum, a region characterized by abundant Aβ [95].
Recent advances have combined small molecule MSI with MALDI-IHC to map GGs, Aβ peptides, and microglia in Aβ plaques of APP/PS1 mice at 5 μm spatial resolution. As revealed, GM1, GD1, and GT1 were observed as the most abundant GGs. GM3 (36:1);O2 and GT1 (36:1);O2 species were the most and least Aβ plaque discriminative species, GM3 (36:1);O2 having a distinct location in Aβ plaques, while GT1 (36:1);O2 being ubiquitous through the brain. Also, GM3 (36:1);O2 and GM2 (36:1);O2 were postulated as plaque-defining GGs. Notably, GG distributions varied by plaque location, with GD3 and GD2 enriched in hippocampal plaques and alternative-chain GGs such as GM3 (38:1);O2 more prominent in cortical plaques. Furthermore, GM3 (36:1);O2 and GM2 (36:1);O2 showed nearly identical distributions within plaques, but different relative abundances [96].
In a combined rat model of Aβ toxicity and ischemic stroke, Caughlin et al. [97] used MALDI-MSI to investigate how brain a-series GGs respond to dual injury. Rats were assigned to stroke alone, Aβ alone and combined stroke plus Aβ groups, and GG profiles were tracked over time. GM3 expression increases were restricted to the combined Aβ/stroke group at 3 days, while by day 21 elevated GM3 was evident in both stroke alone and combined group. Histological markers of neurodegeneration corresponded with regions of GM3 accumulation, confirming its association with degenerating cells. Both GM2 d18:1 and d20:1 species were elevated in the combined group at 3 days, while only the d20:1 species rose significantly in stroke alone. By 21 days, GM2 levels returned to baseline in stroke alone but remained elevated in the combined group, particularly for d20:1. GM1 d18:1 expression showed increases, only in the combined group, at 3 days, while GM1 d20:1 showed increases, also only in the combined group, but both at 3 and 21 days. GD1a responses were more complex, showing sodium adduct forms increased in the combined group at 3 days, which decreased to normal level at 21 days, being no differences between the groups. Whereas, potassium adduct forms decreased in stroke alone group, at 3 days, but normalized over time [97].
Using high-resolution MALDI-MSI, a group of researchers mapped the spatial distribution of GM1 molecular species in the hippocampus and identified region-specific alterations in AD. Notably, the normal localization of GM1 (d20:1/C18:0) at the edge of the dentate gyrus was lost in AD, indicating a selective vulnerability of this compartment. Also, regarding the ratio of GM1 (d20:1/C18:0) to GM1 (d18:1/C18:0) was significantly reduced in the outer molecular layer of the dentate gyrus in AD brains, while no differences were detected in other hippocampal subregions or in total hippocampal lipid content [73].
Neuronal degeneration in AD has also been linked to the presence of anti-GG antibodies in patient sera. In this context, using enzyme-linked immunosorbent assay and high performance TLC immunostaining, sera from individuals with AD, vascular dementia (VD) and age-matched controls were analyzed for anti-GSL antibodies. Patients with AD and VD showed significantly higher titers of anti-GSL antibodies, particularly of the IgM type, including antibodies against GM1, GD1b, GT1b, GQ1b, and notably GQ1bα (Chol-1α, a cholinergic-specific GG expressed in the cortex and hippocampus). While natural autoantibodies to brain GGs were present in all groups, demented patients showed substantially higher reactivity, especially to GM1 and GQ1bα. These two antibodies may contribute to neurodegeneration by disrupting cholinergic synaptic function, particularly in regions vulnerable to AD. Interestingly, IgG type antibodies, GT1b, GQ1b and GQ1bα were elevated in AD patients, especially anti-GT1b. Overall, the findings suggest that elevated anti-GSL antibody titers, especially against cholinergic-specific GGs, may serve as early immunological markers of neurodegeneration and aid in the differential diagnosis of dementia [70].
Several studies analyzed the role of GM1 in AD. Researchers demonstrate that higher levels of GM1 are associated to AD since this GG specie is capable of inducing conformational alterations in γ-secretase, enhancing Aβ production and SP accumulation, leading to aggravation of cognitive impairment [72]. Aβ can bound to GM1 GG, resulting GAβ, which leads to Aβ aggregation, GM1 enhancing the Aβ incorporation into the lipid membrane [81]. Studies have shown that cholesterol facilitates this interaction. In order to elucidate this mechanism, Fantini et al. [98] employed a combination of physicochemical analyses and molecular modeling. To isolate the specific influence of cholesterol on GM1, they used a minimal Aβ5-16 peptide, which retains the GM1-binding domain but lacks residues involved in cholesterol recognition. Thus, they demonstrated that cholesterol significantly accelerates Aβ5-16 binding to GM1. Molecular dynamics simulations revealed that this effect is mediated by a cholesterol-induced hydrogen bond between its hydroxyl group and the glycosidic linkage of GM1. These findings provide an insight into how cholesterol-rich lipid rafts promote Aβ-membrane interactions in AD pathology [98].
Also, Yahi and Fantini [99] presented how two major amyloid proteins, Aβ (in AD) and α-synuclein (in PD), recognize specific GG species by means of a common structural motif. They showed that although Aβ and α-synuclein differ in sequence, both use short loop-shaped 12-residue segments (positions 5-16 in Aβ and 34-45 in α-synuclein) to interact with cell-surface GGs, Aβ binding preferentially to GM1 and α-synuclein to GM3. Introducing two histidine residues (His-13 and His-14) from Aβ into the α-synuclein, the authors engineered a chimeric α-synucleyn/Aβ peptide that retains GM3 binding while gaining GM1 affinity, effectively combining the binding profiles of both parent proteins. Molecular dynamics modeling revealed selective recognition of GM1-cholesterol complexes typical of lipid rafts. The peptide also bound multiple brain GGs, but not neutral glycolipids, inhibited Aβ1–42 binding to neural cells, and showed no cytotoxicity. These findings led to the design of a minimal, non-toxic, GG-binding peptide with potential therapeutic relevance in neurodegenerative disease [99].
Consequently, alterations in lipid metabolism are increasingly recognized as key contributors to AD pathogenesis. Emerging evidence across multiple studies highlights that specific lipid changes, detected in tissue, blood, plasma, serum and CSF may serve as promising clinical biomarkers for the early detection and monitoring of AD. These findings underscore the potential of lipidomic profiling as a non-invasive approach to support timely diagnosis and therapeutic intervention.
3. Lewy Body Dementia (LBD)
LBD represents an umbrella term that includes both dementia with Lewy bodies (DLB), and Parkinson’s disease dementia (PDD). LBD is recognized as the second most widespread form of degenerative dementia in older people after AD [100].
3.1. Dementia with Lewy Bodies (DLB)
DLB is a progressive neurodegenerative disorder characterized by a combination of cognitive decline, parkinsonism, fluctuations in attention, visual hallucinations, rapid eye movement sleep behavior disorder, and other neuropsychiatric symptoms. Among all dementia cases, DLB account for about 20% of all cases [101].
Neuropathologically, DLB is characteried by abnormal accumulation of misfolded α-synuclein in neuronal inclusions termed Lewy bodies and Lewy neuritis [102]. This aggregation disrupts synaptic and neuronal function, impairs cholinergic and dopaminergic neurotransmission, and leads to widespread neurodegeneration involving cortical, limbic, basal ganglia, brainstem, olfactory, and autonomic pathways, contributing to clinical heterogeneity.
Although α-synuclein is widely abundant in the healthy brain—particularly at presynaptic terminals—its physiological role remains incompletely understood. In DLB, this abnormal aggregation disrupts synaptic and neuronal function, leading to widespread network dysfunction, including problems with thinking, movement, behavior, mood, and other body functions.
Currently, DLB diagnosis is primarily clinical due to the absence of a single accurate biomarker for DLB during life. Given the similarity of the symptoms within DLB, AD and PDD neuropsychological examinations, along with various blood tests are used to rely differential diagnosis. Supporting investigations include neuropsychological assessment and neuroimaging [20,103]. While CT and MRI may show relative preservation of medial temporal structures [104], FDG-PET can detect reduced occipital activity and/or the cingulate island sign, and dopaminergic imaging (DAT-SPECT/PET) typically demonstrates reduced striatal uptake. Amyloid and tau PET scans frequently reveal coexisting AD pathology, which is frequent in DLB, complicating therefore biomarker interpretation [105,106]. Despite these advances, definitive diagnosis remains possible only postmortem, underscoring the need for safe, non-invasive biomarkers for early and accurate differentiation of DLB from AD, which is essential for targeted management.
DLB remains underdiagnosed, with more than half of cases missed [107]. Disease progression typically spans five to seven years after diagnosis, although survival ranges widely, from two to 20 years, depending on age, comorbidities, and the rate of symptom progression [20,104]. Currently, there is no cure, but ongoing research continues to improve understanding, aiming to enable better/earlier diagnosis, enhanced care, and the development of new therapies.
Current biomarker research in DLB has focused on α-synuclein, the pathological characteristic of the disease, complementing AD-specific markers such as Aβ and tau. Earlier studies on serum α-synuclein showed potential for distinguishing DLB from AD and controls, but results seemed to be inconsistent, especially in PD cohorts. More recent analyses reported lower plasma α-synuclein levels in DLB and PDD compared with AD and controls; however, modest diagnostic performance (sensitivity ~58%, specificity ~85%) indicates that blood-based assays remain exploratory [108]. Similarly, conventional CSF α-synuclein quantification yielded inconsistent results (increased, decreased or unchanged in DLB) [109], likely due to contamination, methodological differences in CSF collection, processing and analysis, and frequent AD co-pathology [110]. A major advancement has been the development of real-time quaking-induced conversion assays, which amplify misfolded α-synuclein aggregates in CSF and have demonstrated high diagnostic accuracy, including effective discrimination of DLB from AD in both neuropathologically confirmed and clinical cohorts. Other investigational approaches include peripheral tissue assays (e.g., skin, gut biopsies) [111,112] and advanced imaging approaches [20,113].
Beyond α-synuclein, several CSF or plasma biomarkers have been evaluated for their potential to differentiate DLB from AD or PDD, including tau and p-tau [114,115], Aβ (Aβ42, Aβ42/40 ratio) [114,116], andneurofilament light chain(NfL) [117]. Additional candidates such as amino acids, neuropeptides [109], neurotransmitter metabolites [118], and even altered calcium and magnesium concentrations, have also been proposed as supportive markers. However, none has yet achieved sufficient specificity or robustness for routine clinical use.
An emerging and increasingly promising research direction involves MS characterization of lipid and glycolipid alterations in neurodegeneration. In AD, machine-learning approaches have already been applied to CSF [119] and plasma analytes [120], demonstrating utility in diagnostic prediction. In contrast, the application of machine learning (ML) to DLB has only recently begun to be explored.
Shen et al. [53] conducted the first comprehensive plasma lipidomic analysis of DLB, aiming to identify lipid signatures capable of discriminating DLB from healthy controlsand AD. Using UPLC-MS combined with machine-learning algorithms, the authors reported distinct alterations in lipid metabolism in DLB. Most notably, sphingolipid classes—including sphingoid bases, ceramides, and monohexosylceramides (Hex1Cers)—were differentially expressed in DLB compared to both healthy controls and AD. Several Hex1Cer species (e.g., Hex1Cer(d18:1_24:0), Hex1Cer(d18:1_23:0)) contributed to a 13-molecule classification panel that effectively distinguished DLB from AD, with models demonstrating high predictive accuracy. Across analyses, ceramides, sphingosines, PE and PC emerged as the most consistently dysregulated lipid classes in DLB. These findings demonstrate that plasma lipidomic profiling, combined with machine learning has strong potential as a non-invasive diagnostic tool able to effectively discriminate DLB from AD and healthy controls. Moreover, the results suggest that disruptions in sphingolipid signaling and membrane phospholipid remodeling may contribute to the pathophysiology of DLB [53].
Earlier investigations, such as Savica et al. [121], have assessed plasma sphingolipids in individual with autopsy-confirmed DLB and AD. Using ESI-MS, the authors quantified various sphingolipid species, including ceramides, sphinganine, sphingosine, sphingosine-1-phosphate, Hex1Cers, and free fatty acids in plasma collected approximately two years prior to death. They observed that plasma ceramides (C16:0, C18:1, C20:0, C24:1) and monohexosylceramides (C18:1, C24:1) were elevated in both AD and LB pathology groups compared with controls, but no considerable differences were detected between the AD and LB cohorts. Such findings suggest that sphingolipid changes are not sufficiently disease-specific to serve as plasma biomarkers for differentiating DLB from AD, although their alterations are indeed a feature of neurodegenerative dementias [121].
While plasma lipidomics captures broader DLB-associated dysregulation, supporting its potential as a source of clinically useful biomarkers for DLB, CSF instead reveal more subtle, disease-specific changes. Such an observation it belong to Lerche et al. [122], who investigated five key CSF d18:1 sphingolipid species—Cer(d18:1/18:0), GlcCer(d18:1/18:0), SM(d18:1/18:0), GlcSph(d18:1), and GalSph(d18:1)—in PD and DLB participants, with and without heterozygous GBA1 variants [122]. Using LC-MS/MS in positive multiple reaction monitoring mode, they observed lowergalactosylsphingosine (GalSph) and Cer levels in DLB compared with PD or controls. However, no significant differences in glucosylceramides (GlcCer) or GlcSph (glucosylsphingosine) were identified in heterozygous GBA1 carriers and wild-type participants, which indicates that heterozygous GBA1 variants do not substantially alter CSF d18:1 sphingolipid profiles.
Although the full potential of mass spectrometry-based GSL profiling in DLB remains underexplored, existing published studies illustrate its value in uncovering subtle disease-specific lipid alterations, and its promise as a non-invasive approach for differential diagnosis and pathophysiological understanding.
2.2. Parkinson’s Disease Dementia (PDD)
PD and HD are distinct conditions with different causes, pathologies, and clinical features. Identifying molecular biomarkers is essential for early diagnosis, disease monitoring, and treatment evaluation. MS, based approaches in proteomics, metabolomics, and lipidomics provide sensitive and specific tools for biomarker discovery and validation for PD.
PD is the second most common neurodegenerative disorder after AD. It is characterized clinically by motor symptoms like as bradykinesia, resting tremor, rigidity, postural instability as well as non-motor features e.g. autonomic dysfunction, sleep disorders, neuropsychiatric symptoms. Pathologically, the features include loss of dopaminergic neurons in the substantia nigra pars compacta and intracellular aggregation of α-synuclein, Lewy bodies. PD belongs to the family of α-synucleinopathies [123,124,125].
Because clinical diagnosis often occurs after significant neuronal loss, there is a pressing need for sensitive biomarkers that can detect PD at earlier preclinical or prodromal stages, stratify subtypes, monitor progression, or assess pharmacodynamic effects of therapies [126].
PDD is one of the most disabling non-motor complications of PD, affecting about 70% of patients in advanced stages. It is characterized by progressive decline across multiple cognitive domains: attention, executive function, visuospatial abilities, and later memory, which significantly reduces autonomy and quality of life [127,128].
Clinically, PDD is characterized by deficits in executive function, attention, visuospatial abilities, and later memory, as formalized in consensus diagnostic criteria [129].
Since the 1990s, Lewy bodies, the pathological hallmark of PD, have also served as the defining neuropathological substrate for a spectrum of related disorders, encompassing typical PD without dementia, PDD, and DLB. Consensus criteria established an important distinction between PDD and DLB based on the temporal sequence of symptom onset: DLB is diagnosed when dementia precedes or occurs concurrently when dementia is present prior to or alongside the motor symptoms of PD, in contrast to PDD, where dementia is diagnosed only if it appears more than one year later [130].
Dementia must be distinguished from DLB, although both share overlapping α-synuclein pathology and cortical involvement. Importantly, the development of dementia in PD is associated with faster progression, greater caregiver burden, and increased mortality [131].
High-throughput proteomics has already demonstrated that post-translational modifications (PTMs) of key neurodegenerative proteins such as tau, α-synuclein, Aβ, and TDP-43 can serve as disease-associated molecular signatures detectable by MS in both brain tissue and biofluids. Several phosphorylated, acetylated ubiquitinated, methylated, or truncated tau peptides have been reported in AD and corticobasal degeneration, and phosphorylated or C-terminal truncated tau species are also detectable in CSF and serum [132,133]. For α-synuclein, phosphorylated, ubiquitinated, and truncated proteoforms have been identified in brain tissue and biofluids of patients with DLB or PD [134]. In contrast, truncated forms of TDP-43 have been mainly studied in amyotrophic lateral sclerosis brain tissue [135], while Aβ PTMs have been primarily investigated in AD CSF [136]. These findings highlight the capacity of MS not only to capture global proteomic changes but also to resolve subtle proteoform diversity linked to disease mechanisms. However, despite these advances, PTM-based biomarker research has concentrated mainly on AD and DLB, with relatively limited exploration in PDD and Huntington’s disease dementia (HDD). This gap underscores the importance of extending MS-based biomarker discovery beyond classical protein targets toward lipidomic and glycolipidomic approaches, which may reveal complementary molecular mechanisms underlying cognitive decline.
In recent years, GSL metabolism has emerged as a promising area of biomarker research for PDD. Using targeted LC-MS/MS, found that plasma Cer and GlcCer levels were elevated in sporadic PD and that higher concentrations correlated with poorer cognitive performance [137,138]. Xing et al. [138]investigated the relationship between plasma ceramides and cognitive as well as neuropsychiatric manifestations in PDD using a validated HPLC-MS/MS platform. Using this rigorous analytical framework, the authors showed that although overall cognitive performance did not associate with Cer levels, specific glycoforms displayed strong negative correlations with verbal memory, in particular, C24:1 with immediate and delayed recall, and C14:0 with delayed recall and recognition.
In addition, certain ceramides correlated positively with neuropsychiatric manifestations: C22:0 with hallucinations, C20:0 with anxiety, and C18:0 with sleep behavior disturbances. These associations remained significant after controlling for relevant confounders, reinforcing the potential role of plasma Cers as biomarkers for domain-specific cognitive decline and neuropsychiatric symptoms in PDD.
High-resolution lipidomics has emerged as a powerful tool for the discovery of new insights into the transition from PD to dementia. Using serum samples analyzed by LC- QTOF-MS with both positive and negative ESI modes, Zardini Buzatto et al. [131] applied a high-sensitivity lipidomics workflow to assess whether baseline lipid profiles could predict cognitive outcomes over a 3-year follow-up. The authors reported that while thousands of lipid features were detected and classified into 36 subclasses, a focused set of lipid alterations distinguished PD patients, who remained cognitively stable, PD with no diagnosis of dementia, from those who later developed dementia, PD with incipient dementia. In particular, 24 ceramides, 24 diacylglycerols, and 17 triacylglycerols were increased in PDID, whereas 16 PCs, 14 bis(monoacyl)glycerophosphates, and 14 phosphatidylserines (PSs) were decreased. Multivariate models (PLS-DA, OPLS-DA, Random Forest) achieved excellent discrimination, and a five-lipid biomarker panel yielded an area under the curve (AUC) of 0.993 with 95% sensitivity and 100% specificity in training, and >95% accuracy in validation. These results indicate that serum lipidomic signatures measured years before diagnosis can robustly predict incipient dementia in PD, highlighting the promise MS based lipid panels for early risk stratification [131].
An important observation is that different Cer species appear to correlate with distinct cognitive and neuropsychiatric domains in PDD. For example, C24:1 and C14:0 are mainly associated with deficits in verbal memory, whereas C22:0 and C20:0 have been linked with hallucinations and anxiety [138]. This domain-specific relationship suggests that plasma Cers may not only serve as global markers of neurodegeneration but also as indicators of selective circuit dysfunction, which could inform precision approaches to diagnosis and treatment.
These molecular insights can be further expanded through artificial intelligence (AI) and ML, which are increasingly applied to PD to enhance diagnostic precision and predict cognitive outcomes. When combined with MS, based lipidomics, these approaches have shown particular promise for biomarker discovery and clinical prediction. Evidence from LC-MS serum lipidomics integrated with ML demonstrated that complex lipid panels can anticipate both motor and non-motor trajectories up to two years in advance, highlighting the prognostic potential of lipidomic signatures in PD [139]. In a complementary analysis, untargeted LC-MS lipidomics coupled with ML algorithms identified lipid signatures associated with clinical severity, further supporting the value of computational methods in biomarker research [140]. Collectively, these studies emphasize the potential of integrating MS-based lipidomics with AI-driven analytics to capture complex biomarker patterns linked to cognitive decline and dementia in PD, complementing proteomic and clinical approaches.
In PDD, MS-based sphingolipidomics has provided compelling evidence that specific Cer species and broader lipidomic signatures are closely associated with cognitive domains and neuropsychiatric symptoms [131,138]. Studies using LC-MS/MS and LC-QTOF-MS have shown that elevated plasma Cer forms such as C24:1 and C14:0 correlate with verbal memory impairment, while C22:0 and C20:0 are linked with hallucinations and anxiety [138]. Furthermore, longitudinal lipidomic analyses demonstrated that baseline lipid profiles can predict the transition to dementia, offering >95% accuracy in distinguishing patients at risk of cognitive decline [131]. These findings highlight GSL metabolism as a promising biomarker domain, complementing protein-based markers such as α-synuclein and NfL, and emphasize the translational potential of MS lipidomics for early stratification and therapeutic monitoring in PDD. More recently, the integration of lipidomics with artificial intelligence and machine learning has shown additional prognostic value, with machine-learning models applied to LC-MS data predicting motor and non-motor trajectories up to two years ahead and identifying lipid signatures correlated with disease severity [139,140].
4. Frontotemporal Dementia (FTD)
FTD, historically referred to as Pick’s disease, comprises a heterogeneous group of non-Alzheimer’s neurodegenerative dementias. Clinically, FTD comprises three major syndromes: behavioural variant FTD (bvFTD)—the most frequent type— progressive non-fluent aphasia, and semantic dementia. FTD is characterized by progressive deficits in behavior, language, and cognition, typically associated with atrophy of the frontal and temporal lobes [141,142]. Epidemiologically, FTD represents the third most common neurodegenerative dementia after AD and DLB [143], and is the second leading cause of dementia in individuals younger than 65 years of age [143].
A recent systematic review and meta-analysis reported a pooled incidence of 2.28 per 100,000 person-years and a prevalence of 9.17 per 100,000 [144]. However, these estimates are likely underestimated due to underdiagnosis and frequent misclassification—about 70% of patients are initially misdiagnosed [145]. While most FTD cases are sporadic, genetics contributions are significant: up to 30-40% of patients have a strong familial history [146,147] and about 15% exhibit an autosomal dominant inheritance pattern [147]. Recent advances in genetics have identified mutations in the C9orf72 [148,149], progranulin (GRN) [146], and tau [150] genes as contributors to DLB, with GRN mutations being the most frequently detected among them [13,147].
The clinical management of FTD focuses on two main objectives: (i) the development of new therapeutic strategies to prevent and/or reduce frontotemporal atrophy, and (ii) the identification of reliable fluid biomarkers to support diagnosis during lifetime and track progression. Definitive diagnosis remains challenging, as no single test exists. Therefore, clinical assessment typically involves a combination of neuropsychiatric evaluation to observe signs of slowly progressive dementia, blood or CSF laboratory testing [151,152] and neuroimaging techniques, such as MRI, CT, or PET, which revealed numerous biomarkers and helped clinicians in improving diagnostic accuracy. However, because brain atrophy in the early stages of the disease lacks in characteristic features, an average delay of about three years from symptom onset and the initial evaluation and diagnosis, longer than in other dementias, is reported [153,154]. Consequently, the current research direction, according to the growing literature in recent years, is the identification of fluid biomarkers capable of (i) differentiating FTD from other primary psychiatric or neurodegenerative disorders; (ii) monitoring disease progression and astrogliosis in FTD; (iii) distinguishing FTD subtypes and (iv) guiding therapeutic decisions.
Both blood and CSF represent a rich source of potential biomarkers, and researchers have employed a wide range of modern techniques to investigate changes of blood-based protein biomarker levels for FTD diagnosis, some of them more promising than others [155,156,157,158]. For instance, reduced progranulin (PGRN) levels are particularly relevant in FTD cases associated with GRN gene mutations [159], while elevated NfL levels can be correlated with disease severity, cognitive decline, brain atrophy, and help to differentiate FTD from AD [159,160]. In contrast, plasma phosphorylated tau (p-tau), particularly p-tau181 [157], and glial fibrillary acidic protein, indicative of astrocytosis, tend to be higher in AD compared to FTD [157], making them less useful as FTD-specific markers.
Beyond proteins, growing attention has turned to lipids—particularly GSLs-as promising biomarkers and mechanistic drivers of disease. GSLs such as GGs and HexCer (e.g. GlcCer and galactosylceramides, GalCer), play fundamental roles in synaptic stability, axonal function, and the maintenance of myelin integrity. Therefore, disruption of their metabolism has been consistently implicated in FTD pathogenesis [32,161,162], reflecting key disease mechanisms including lysosomal dysfunction, myelin breakdown, and impaired neuronal and glial lipid homeostasis.
Highly expressed in CNS, GGs are involved not just in physiological functions of the brain, playing essential roles in neuronal function, synaptic transmission, and cell signaling [163,164,165,166,167,168], but their dysregulation has been implicated in various diseases, such as including cancer [169,170,171,172,173,174,175], AD [34,91,176,177,178], and various lysosomal storage disorders [179,180]. Given the structural complexity of GGs and disease-specific alterations, detailed compositional and structural elucidation constitutes a fundamental requirement to understand their role in disease and to identify specific molecular species that may serve as biomarkers.
In this context, MS has become the method of choice for GSL analysis, due to its sensitivity, specificity and ability to provide structural and functional information. MS enables the detection and detailed characterization of individual GG species in complex biological mixtures, including those undetectable so far by any other method. More importantly, MS-based lipidomics has facilitated the exploration of region-specific expression in the brain and has extended detection to peripheral fluids such as CSF and plasma, thus directly supporting their application as minimally invasive biomarkers in FTD and related neurodegenerative disorders.
Recent lipidomics studies highlight diverse but converging mechanisms of lipid dysregulation in FTD, spanning both genetic and sporadic forms. In 2022, Boland et al. [181] demonstrated that GRN haploinsufficiency disrupt lysosomal homeostasis, leading reduced bis(monoacylglycero)phosphate (BMP) levels, and subsequent GG accumulation in human cells, murine brains, and in human frontal and occipital lobes [181]. Using MS-based lipidomics, Boland et al.[181] demonstrated that gangliosides such as GM1, GD3, and GD1 accumulate in GRN-associated FTD-TDP. Importantly, this was not due to impaired lysosomal enzyme activity, but to reduced BMP levels resulting from progranulin deficiency. While certain species were also elevated in sporadic FTD-TDP, GT1 was selectively reduced in GRN-FTD, highlighting both shared and mutation-specific lipid alterations. More importantly, functional experiments showed that exogenous BMP supplementation in PGRN-knockout cells normalized GM2 levels to those in control cells, suggesting therefore that PGRN in lysosomes helps maintain the BMP levels needed to prevent GGs from accumulating in brain cells — buildup that may contribute to FTD [181].
Complementary studies on patients have reported increased concentrations of specific GGs, such as GT1a and/or GD2, in Pick’s disease brains, indicating impaired degradation and clearance of GSLs [182]. Together, these findings highlight the central role of GG metabolism in FTD pathogenesis.
Expanding this theme, Kim et al. [183] used untargeted plasma lipidomics (LC-MS/MS) to investigate bvFTD, the most common clinical phenotype. They reported a widespread alteration in circulating lipid species, including increased TG and decreased PS and phosphatidylglycerol, consistent with hypertriglyceridemia and hypoalphalipoproteinemia in bvFTD. In contrast, monoglycerides, major sphingolipid subclasses, and sterols showed no significant changes. Noticeably, plant-derived lipids, such as monogalactosyldiacylglycerol and sitosteryl ester, were decresed in bvFTD, potentially reflecting altered dietary intake or altered absorption/metabolism related to eating behavior in bvFTD. Furthermore, specific individual lipid species —TG (16:0/16:0/16:0), diglyceride DG (18:1/22:0), PC (32:0), PS (41:5), and SM (36:4) — were identified and proposed by the authors as potential peripheral biomarkers, able to discriminated bvFTD from AD and healthy controls [183].
Given earlier evidence that GRN mutations interrupt lysosomal lipid catabolism, Marian et al. [184] further investigated lipid metabolism in FTD-GRN and FTD-C9orf72 subtypes, the most common genetic causes of FTD with TDP-43 pathology, using postmortem frontal (heavily-affected) and parietal (less-affected) brain regions. Their comprehensive lipidomics analyses by LC-MS/MS, enzyme activity assays (e.g. galactocerebrosidase), and Western blotting of myelin proteins complex assay revealed that both subtypes present abnormalities in myelin lipid metabolism. However, GRN carriers show more severe pathology, characterized by pronounced sphingolipid and myelin protein loss, along with a large accumulation of cholesterol esters in white matter and elevated acylcarnitines in frontal grey matter. These lipid disturbances observed in FTD-GRN are consistent with both myelin breakdown and altered fatty acid metabolism. Interestingly, both subtypes had increased markers of lysosomal and phagocytic activity, implicating microglial involvement in lipid/myelin clearance, while galactocerebrosidase activity – the enzyme responsible for GalCer and sulfatide catabolism – was selectively increased in FTD-GRN but not in C9orf72 cases [184]. Overall, their findings indicate a more severe disruption of myelin lipid homeostasis in FTD-GRN carriers compared with FTD-C9orf72, consistent with MRI evidence of pronounced white matter changes in FTD-GRN.
Extending these tissue-based findings to a peripheral biomarker context, Marian et al. [162] further demonstrated that plasma myelin-enriched glycolipids, particularly especially HexCer, are significantly reduced in familial bvFTD. By combining MRI-derived fiber tract density and cross-section with LC-MS lipidomics, the authors observed that very long-chain HexCer (C20–C24), especially C22:0 GlcCer and GalCer, are reduced in bvFTD and inversely correlated with disease duration. Notably, C22:0 GlcCer showed a stronger correlation with MRI-derived measures of white matter damage than C22:0 GalCer, suggesting that the reduced GSL levels in bvFTD reflect changes to peripheral lipid metabolism rather than direct demyelination, despite GalCer abundance in myelin. Importantly, plasma HexCer reductions appear specific to FTD, since no changes were observed in AD or multiple sclerosis. Overall, lower HexCer levels were correlated with frontal and temporal white matter integrity loss, longer disease duration, and cognitive decline, supporting therefore their potential as accessible blood biomarkers of neurodegeneration and disease severity in bvFTD.
At the molecular mechanistic level, Arrant et al. [185] investigated the effects of PGRN deficiency on β-glucocerebrosidase (GCase) activity and maturation in FTD-GRN brains. Their comprehensive assay, combining fluorogenic enzyme activity assays, Western blotting, IHC and HPLC-MS on inferior frontal gyrus tissue from postmortem FTD-GRN brain revealed reduced levels of the mature GCase protein, accumulation of improperly glycosylated and insoluble forms, and significantly diminished GCase activity in FTD-GRN brains. However, no evident accumulation of GCase substrates (GlcCer and GlcSph) was observed in the investigated brain regions. These enzymatic deficits, reversed by PGRN restoration in mouse models, emphasize a direct enzymatic deficit linking GRN mutations to impaired lysosomal sphingolipid catabolism. In the broader context, alongside the reported GG accumulation [181], myelin lipid loss in brain tissue [184], and plasma GSL reductions correlated with white matter damage [162], these findings [185] not only complement lipidomic studies by revealing the enzyme-level defects that drive the lipid composition alterations, but more importantly, establish a coherent picture in which GRN mutations disrupt lysosomal function, thereby driving both central lipid pathology and peripheral biomarker changes. Altogether, these insights explain why lipid pathology differs across brain and plasma and why is more severe in FTD-GRN than in FTD-C9orf72 and underscore lysosomal enzymes as both mechanistic drivers and potential therapeutic targets.
Adding further complexity, He et al. [186] extended lipidomic profiling to very long-chain fatty acid (VLCFA)–containing lipids, which, though relatively rare, have essential biological functions and whose accumulation is cytotoxic. Using discovery LC/MS in post-mortem frontal cortex tissue, He et al. [186] demonstrated that various phospholipid VLCFA species—particularly PC (30:5/18:1), PE (33:4/20:4) and PE (33:4/22:6)—are considerably elevated in FTD brains compared to controls, showing strong correlations with expression of the VLCFA-synthesizing enzyme ELOVL4. However, GlcCer were not significantly altered, suggesting that VLCFA- phospholipids, rather than containing VLCFA- glycolipids, are more directly implicated in the lipidomic signature associated with FTD [186].
Taken together, these convergent findings delineate that lipid dysregulation in FTD is multifaceted, spanning GG accumulation, sphingolipid and myelin lipid loss, lysosomal enzyme dysfunction, and VLCFA-containing phospholipid elevations. Importantly, both central (brain tissue) and peripheral (plasma) lipid changes show potential as diagnostic and prognostic biomarkers, while offering mechanistic insights into disease pathogenesis and therapeutic targets.
5. Huntington's Diseases (HD)
HD is a monogenic, autosomal dominant neurodegenerative disorder caused by expansion of a CAG (Cytosine–Adenine–Guanine) trinucleotide repeat in the HTT gene on chromosome 4, leading to an expanded polyglutamine tract in the HTT protein. Clinically, HD presents with a combination of movement disorders (chorea, dystonia, motor impairment), cognitive decline, and psychiatric/behavioral symptoms. Onset typically occurs in mid-adult life, though there is a prodromal phase [187].
Because the genetic mutation is known, HD offers a unique opportunity to study the early period before symptom onset. Biomarkers that reflect early molecular changes, disease burden, or response to therapies are actively sought [188].
Dementia is a defining outcome of HD, often developing years after subtle executive and memory deficits appear [189]; HD patients typically show a subcortical cognitive profile, characterized by attention deficits, cognitive slowing, impaired planning and problem solving, as well as visuoperceptual and constructional difficulties [190]. Deficits in psychomotor speed, attention, executive functions, and visuospatial abilities are consistently reported, and these impairments correlate with both functional decline and eventual dementia [189,191].
Untargeted UHPLC-MS/MS to plasma and CSF has identified broad lipidomic changes in HD biofluids [192,193], detecting alterations Cers, HexCers, SMs, diacylgly-cerols, and PCs. Importantly, these lipid classes correlated with cognitive scores from the Stroop and Verbal Fluency tests, linking systemic lipid changes to dementia trajectories [192,193].
In human brain tissue, MALDI-MSI allows mapping of lipid species across regions. Hunter et al. [194] demonstrated widespread sphingolipid dysregulation in HD caudate and cortex, areas essential for executive and memory functions. MALDI-MSI highlighted regional deficits in SMs and phospholipids consistent with disrupted neuronal membrane integrity and cognitive network failure [194]. Complementing this, Phillips et al. [195] showed chain-length remodeling of Cer and SMs in the caudate, with a shift from very-long-chain to long-chain species, providing a structural basis for dementia-related dysfunction [195,196].
GSLs, particularly GM1 ganglioside, have been studied with targeted LC-MS/MS. Maglione et al. [197] reported reduced GM1 in HD patient-derived cells, implicating impaired glycolipid metabolism in synaptic vulnerability [197]. Di Pardo et al. [36] demonstrated that GM1 deficiency contributes to synaptic dysfunction, while supplementation with exogenous GM1 improved motor and cognitive outcomes in HD mouse models. Alpaugh et al. [198] confirmed disease-modifying effects of GM1 across behavioral and cognitive domains, suggesting that GM1 deficiency is directly linked to dementia mechanisms in HD.
Beyond the established lipidomic alterations described in HDD, recent studies emphasize the need for a multi-omics framework. Protein biomarkers such as NfL and mutant huntingtin (mHTT) are robust measures of axonal damage and genetic burden, but they do not reflect the lysosomal and membrane lipid dysregulation uncovered by MS [194,199]. Integrating proteomic and lipidomic markers may therefore improve diagnostic accuracy and prognostic power for cognitive decline.
Mechanistically, lipids such as Cer and GSLs function not only as membrane constituents but also as bioactive signaling molecules that regulate apoptosis, synaptic activity, and neuroinflammation. Their dysregulation in HD links systemic metabolic stress to synaptic and cognitive dysfunction. MALDI-IMS and LC-MS studies of post-mortem brain tissue demonstrated that alterations in sphingolipid chain length and regional phospholipid depletion impair neuronal connectivity in circuits underlying executive and memory functions [195].
One major limitation is that most studies remain cross-sectional, which precludes establishing whether lipid profiles can predict the conversion from prodromal to demented stages. In contrast, longitudinal lipidomics in PD has successfully identified predictive biomarker panels for incipient dementia [131]. Applying similar approaches in HD would enable early stratification of gene-positive individuals and guide preventive interventions.
Although protein biomarkers such as NfL and mHTT provide robust measures of neuronal injury and disease burden [200,201,202], they primarily reflect global neurodegeneration rather than synaptic or metabolic dysfunction. By contrast, lipidomic and glycolipidomic studies have begun to uncover mechanisms of membrane instability and synaptic vulnerability in HDD [192,193,194]. Although most studies to date have been cross-sectional, they have already provided important insights into lipid dysregulation in HD and its link to cognitive decline. These findings establish a valuable foundation for future longitudinal MS-based research, which will be crucial to determine whether baseline lipidomic profiles can serve as reliable early predictors of dementia in gene-positive individuals.
AI and ML approaches are increasingly applied to HD research, offering new opportunities to complement MS-based biomarker discovery. Recent work has demonstrated that AI-driven pipelines integrating multimodal baseline biomarkers can improve prognostic enrichment in early-stage HD, supporting patient stratification for clinical trials and disease monitoring [203]. Other studies have applied AI and deep learning to neuroimaging and clinical datasets, showing potential for the early detection of cognitive decline and aiding in the development of predictive models for dementia in HD [204]. These approaches highlight the promise of combining multi-omics MS-based data with advanced computational methods to accelerate the translation of lipidomic and glycolipid biomarkers into clinically actionable tools.
In HDD, MS-based approaches, including LC-MS/MS, UHPLC-MS, and MALDI-IMS, have consistently revealed alterations in Cer, SMs, and GSLs, linking disrupted membrane lipid metabolism to cognitive and functional decline [194,195,197]. In particular, reductions in GM1 ganglioside have emerged as both mechanistic biomarkers of synaptic vulnerability and potential therapeutic targets, as exogenous GM1 supplementation has shown neuroprotective and disease-modifying effects in preclinical models [36,197,198]. Current lipidomic findings in HD highlight consistent alterations in sphingolipid and glycolipid pathways, establishing a strong basis for longitudinal studies that will determine their value as predictive biomarkers of cognitive decline. [192,193,194]. Future research should therefore focus on integrating MS-based multi-omicsi.e. lipidomic and proteomic data with advanced computational methods, including artificial intelligence and machine learning, to establish GSLs as specific and clinically actionable biomarkers of dementia in HD.
6. Mixed Dementia
Mixed dementia is increasingly recognized as a frequent, complex, clinically important and biologically heterogeneous form of cognitive impairment in older adults in which two or more neuropathological processes coexist often with additive or synergistic effects on cognitive decline; most typically these are AD proteinopathies i.e. Aβ accumulation and tau NFT, together with cerebrovascular lesions such as small-vessel disease, microinfarcts, white matter injury, and cerebral amyloid angiopathy [7]. Additional co-pathologies such as α-synuclein (Lewy body) or TDP-43 inclusions are frequent contributors that increase the clinical complexity and accelerate disease progression [205].
Autopsy series and population studies indicate that mixed pathologies become prevalent with advancing age and that coexisting vascular and neurodegenerative lesions account for a substantial fraction of dementia cases in older adults. Consequently, a significant proportion of clinically diagnosed AD or VD actually represent mixed forms when examined neuropathologically. For instance, in a population-based autopsy study, 12% of dementia cases were diagnosed as combined AD and VD, highlighting the overlap between these conditions [206]. Additionally, a review of the evidence reported that mixed vascular and Alzheimer’s disease is a very common pathological finding in the elderly, with a prevalence of about 22% [7].
Clinically, mixed dementia often presents features of both component disorders: the amnestic syndrome and episodic memory impairment characteristic of AD are frequently present; however, executive dysfunction, slowed processing speed, attention deficits, gait disturbance, focal neurological signs and stepwise or more fluctuating decline associated with vascular contributions are also common. When Lewy body or TDP-43 pathology is also present, supplementary signs such as visual hallucinations, parkinsonism or disproportionate temporal-hippocampal neuronal loss with hippocampal sclerosis may be seen, producing a phenotype that can shift over time as the different pathologies evolve [207].
Epidemiologically, mixed dementia incidence and prevalence rise steeply with age and are strongly associated with vascular risk factors [208] such as hypertension, diabetes, dyslipidaemia, smoking, obesity and physical inactivity, which not only promote cerebrovascular lesions but, also, interact with amyloid and tau pathways to amplify neurodegeneration. As a consequence, strategies that target vascular risk factors at the population level are a major public health method to reducing the overall burden of mixed pathology [209].
Modern diagnostic approaches attempt to deconvolute the contributions of degenerative and vascular processes in vivo by combining clinical assessment with (i) structural MRI in order to visualize infarcts, lacunes, white matter hyperintensities, microbleeds and atrophy [210]; (ii) PET imaging for amyloid and tau [211]; (iii) CSF assays [119] of Aβ42 or Aβ42/40, total and phosphorylated tau; and (iv) increasingly sensitive plasma biomarkers among which p-tau217, p-tau181, Aβ ratios, NfL that provide scalable screening options [212].
These molecular tools greatly increase specificity for Alzheimer pathology and, in conjunction with MRI, are able to identify cases where vascular lesions and AD biomarkers coexist, although important limitations still remain. For instance, MRI underestimates minor cortical microinfarcts and diffuse small-vessel damage, PET is costly and not universally available, CSF sampling is invasive and not always acceptable, plasma assays are influenced by peripheral factors and assay platform variability, and biomarker thresholds derived from pure disease cohorts may perform differently in mixed populations, thus interpretation requires careful integration of imaging, fluid, clinical and vascular data.
From a therapeutic perspective, treatment of mixed dementia must be multipronged. Aggressive management of vascular risk factors such as tight yet individualized blood-pressure control, glycemic management, lipid control, smoking cessation, antiplatelet therapy when indicated, and promotion of exercise and healthy diet has the clearest population-level evidence to limit further vascular injury and possibly to slow cognitive decline [213,214]. On the other hand, symptomatic pharmacotherapies used in AD such as cholinesterase inhibitors and memantine may provide some cognitive or functional benefit depending on phenotype [215]. The advent of disease-modifying anti-amyloid therapies has opened new possibilities for biomarker-confirmed AD, however, the efficacy and safety of these agents in patients with substantial vascular pathology or in truly mixed cohorts is not yet well established and necessitates focused trials because vascular lesions can both blunt clinical benefit and influence adverse-event profiles [216].
The current active research into tau-targeting agents, anti-inflammatory or immune-modulating strategies and therapies aims at preserving mitochondrial function and blood–brain barrier (BBB) integrity; overall, the consensus is that combinatorial and individualized treatment strategies addressing both neurodegenerative and vascular drivers are most plausible for mixed dementia [217].
At the molecular level, mixed dementia emerges from intersecting and mutually reinforcing pathogenic cascades. Canonical AD mechanisms include aberrant processing of the APP by β- and γ-secretases to produce Aβ peptides, which form oligomers and extracellular plaques that are synaptotoxic, provoke oxidative stress, activate glia and trigger neuroinflammation; tau protein becomes hyperphosphorylated, via kinases such as GSK-3β and CDK5, detaches from microtubules and aggregates into paired helical filaments and neurofibrillary tangles, disrupting axonal transport and neuronal integrity [218]. Vascular mechanisms, among which small-vessel arteriolosclerosis, lipohyalinosis, microinfarcts, chronic hypoperfusion, and cerebral amyloid angiopathy, produce ischemic injury, myelin loss, gliosis and energy deficits and compromise the BBB, leading to leakage of plasma proteins and immune cell infiltration that amplify neuroinflammation and oxidative damage.
Importantly, all these pathways interact: (i) vascular injury impairs perivascular and endothelial clearance of Aβ, promoting its accumulation; (ii) Aβ deposition in vessels directly injures endothelial cells and pericytes; and (iii) apolipoprotein E ε4, a major genetic risk factor, modulates both amyloid accumulation and vascular integrity since ε4 carriers exhibit earlier BBB breakdown and pericyte dysfunction that can precede overt amyloid or tau pathology, thereby creating an environment conducive to mixed pathology.
Downstream shared mechanisms across proteinopathies and ischemia include mitochondrial dysfunction, calcium dysregulation, chronic microglial and astrocytic activation, failure of proteostatic systems (ubiquitin-proteasome and autophagy-lysosome pathways), and metabolic shifts away from efficient oxidative phosphorylation toward impaired glycolysis, all of which converge to produce synaptic failure and neuronal death [219].
These molecular insights motivated an expanded biomarker agenda beyond the classical protein markers, to include measures of vascular injury such as CSF/plasma albumin ratio, pericyte or endothelial markers, neuroaxonal injury (NfL), inflammation indicated by the cytokine panels, metabolic and mitochondrial markers, and, importantly, lipidomic and glycolipidomic signatures that reflect membrane, synaptic and myelin integrity. Because glycolipids i.e. GSLs and GGs are abundant constituents of neuronal membranes and myelin, they participate in synapse formation and signal transduction, and modulate neuroinflammation and cell–cell interactions; their dysregulation plausibly reflects both degenerative and vascular/myelin damage and therefore holds promise as a biomarker set for mixed dementia [220].
In the last years, MS has been central to modern biomarker discovery and validation in neurodegenerative diseases because it enables sensitive, high-resolution, and relatively unbiased profiling of proteins, lipids, glycolipids and metabolites in brain tissue, CSF and plasma without relying solely on antibody reagents [221,222,223,224]. MS-based proteomics, including data-independent acquisition workflows, has expanded the CSF proteome and identified panels of proteins related to synaptic function, glycolysis, inflammation and BBB dysfunction that complement classical amyloid/tau/neurodegeneration (AT(N)) markers and improve diagnostic discrimination between AD, non-AD dementias and controls. Concurrently, advances in lipidomics including targeted LC-MS/MS, shotgun lipidomics and imaging mass spectrometry MALDI-MSI, DESI, SIMS coupled with high-resolving MS analyzers and ion mobility separation permit isomer-level separation, spatial mapping and quantitation of complex glycolipid species such as GGs, cerebrosides, sulfatides and Cers, revealing regionally specific lipid perturbations that associate with plaques, gliosis and white matter lesions in mixed and pure disease states.
Specific lipidomic studies of mixed dementia and related disorders illustrate these points. Comparative LC-MS lipidomic analysis of white and gray matter from temporal cortex of subcortical ischemic vascular dementia (SIVD), mixed dementia and controls found pronounced alterations in sphingolipid classes, Cers, SMs, GlcCers and GalCers, as well as increases in some GG species i.e. GM3 and markers of membrane breakdown in mixed dementia white matter compared with controls. These features are consistent with the combined effects of neuronal degeneration and myelin/axonal injury in mixed pathology [225]. Such tissue-level findings provide mechanistic plausibility for glycolipid markers in biofluids and suggest candidate species for further study.
Additional MS-based investigations mapped GG distributions to Aβ plaques and periplaque regions and demonstrated age- and disease-related dysregulation of ganglioside degradation pathways with increases in GM2/GM3 in plaque-rich areas [67]. The results linked lipidomic profiles to imaging indices of vascular injury and white matter lesion burden, evidencing that lipid changes reflect the spatial interplay of degenerative and vascular lesions.
Recent advances in MALDI-MSI and the related workflows, for example quantitative MSI combined with on-tissue extraction and LC-MS/MS, enable presently a near-cellular mapping of lipids and proteins, permitting the direct visualization of how glycolipid perturbations colocalize with plaques, microinfarcts or gliotic regions in human brain sections and model systems [226].
The importance of glycolipids as biomarkers derives from both biology and analytic feasibility: (i) GGs and GSLs directly report on membrane/synaptic composition and myelin health; (ii) are sensitive to enzymatic shifts in lipid metabolism that occur with aging, hypoxia and inflammation; (iii) some species can be measured in CSF and, with advanced techniques, even in plasma. In AD specifically, Noel et al. [227] discovered altered GG patterns, including changes in GM1, GM2, GM3, GD1a/b, GT1b and shifts in sulfatides and Cers that correlate with amyloid and tau pathology or cognitive decline. In vascular and mixed dementias, white matter lipid degradation products and altered sphingolipid ratios reflect ischemic/myelin injury and overlap with degenerative signatures in mixed cases, suggesting that panels of glycolipids together with protein biomarkers could enhance sensitivity and specificity for mixed pathology [228].
In the past years the modern and high performance MS technologies have made several concrete achievements in glycolipid biomarker discovery for mixed dementia: (i) methodological refinements in extraction, chromatographic separation i.e. reverse-phase, hydrophilic interaction chromatography, porous graphitized carbon for glycans, derivatization and ionization now permit reliable detection and partial isomer resolution of GG species differing by sialylation state, Cer backbone length and unsaturation; (ii) high-resolution TOF instruments combined with MS/MS fragmentation and IMS allow structural assignment of glycan headgroups and lipid moieties at previously unattainable sensitivity, reproducibility and wealth of compositional and structural data; (iii) MALDI-MSI and DESI imaging approaches enable spatially resolved lipidomics, linking molecular changes to histopathology: plaques, microinfarcts, white matter lesions in the same tissue section; and (iv) targeted LC-MS/MS assays developed for CSF and plasma enable quantitative panels suitable for larger cohort studies and longitudinal sampling, altogether moving glycolipid candidates from tissue discovery toward biofluid validation. Nevertheless, translation faces challenges such as the inter-laboratory standardization, the low abundance and peripheral dilution of brain-derived lipids in plasma, confounding by diet/peripheral metabolism and renal function, and the need for large, well-phenotyped mixed dementia cohorts with paired imaging and neuropathology for validation [26].
Below, recent glycolipid biomarker candidates compiled in the concise Table 3 from MS-based tissue and biofluid investigation of species that have been implicated across studies in AD, VD and mixed dementia are presented. Table 3 lists the representative glycolipid classes and specific molecular species that have appeared repeatedly as altered in disease cohorts; the table does not represent an exhaustive inventory of every molecular species reported, but rather a prioritized set based on reproducible reports and biological plausibility in mixed pathology.
In glycolipidomics of mixed dementia, the emerging guidance from methodological investigations and discovery studies supports the following practical strategies: (i) the use of untargeted high resolution IMS, LC-MS or MSI with IMS for initial discovery in well-phenotyped brain tissue, paired with histology and proteomics to connect lipid changes to plaques, gliosis and microinfarcts; (ii) prioritization of candidate species that are abundant enough and biochemically plausible for measurement in CSF, and then the development of targeted LC-MS/MS SRM assays with isotopically labelled internal standards for validation in CSF/plasma cohorts; (iii) performing, whenever possible, matched imaging by MRI and fluid biomarker inventory, i.e. Aβ/tau/NfL plus glycolipid panel, in order to determine whether glycolipid signals add diagnostic or prognostic value in mixed dementia beyond classical markers; and (iv) standardization of sample collection, extraction and instrument methods across centers to enable multi-site validation and regulatory qualification.
In conclusion, glycolipids i.e. GGs, sulfatides, glycosylceramides and related sphingolipids have strong mechanistic rationale as biomarkers of mixed dementia since they report on membrane/synaptic integrity, myelin health and lysosomal/autophagic flux. The advancements of mass spectrometry and allied systems biology techniques such as high-resolution LC-MS/MS, IMS-MS, MALDI-MSI and hybrid quantitative imaging workflows allow the reliable discovery of glycolipid biomarkers and targeted measurement.
The pathway forward requires large longitudinal cohorts with detailed clinical, imaging and neuropathological characterization, standardized MS workflows, multi-modal MS which pairs MSI and high resolution IMS or LC-MS [223] and multi-omics integration: proteomics, lipidomics, glycomics and genomics to validate glycolipid panels able to improve the diagnosis, prognostic and monitoring of mixed dementia and enable trials of combinatorial therapies that address both neurodegenerative and vascular mechanisms.
6. Conclusions and Perspectives
Research into the role of GSL in dementia is beginning to redesign our perspectives on the molecular bases of neurodegenerative diseases. Data collected from multiple studies now clearly indicate that GLSs are not just inactive compositional building blocks of neuronal membranes, but dynamic regulators of brain processes in health and disease. Their dysregulation was reported across many forms of dementia, a feature that points to both common threads and disease-specific differences. The recurring patterns indicate core weaknesses in neuronal GSL regulation, whereas the distinct changes cause the diverse symptoms and progression seen across different dementias. In this context, Table 4 presents a synthetic view on the altered GSL expression specific to each dementia type discussed.
Technological refinements in MS have been crucial in enabling the recent discoveries in glycolipid research. These progresses has not only provided the critical basis for discovering novel lipid signatures in dementia, but has also reshaped our broader understanding of glycolipid biology in the affected brain. By offering unprecedented sensitivity, resolving power, and detailed structural information, modern MS has created opportunities to map complex lipid alterations with high accuracy and depth. At the same time, cutting-edge developments in this field have served as a solid basis for integrating glycolipidomics into the wider context of dementia research, linking molecular findings to protein aggregation, inflammation, and synaptic dysfunction.
The up-to-date MS-based analytical platforms have the capability to detect and characterize a wide range of glycolipids with far greater precision than before, allowing to build detailed maps of lipid changes in brains affected by dementia. When these lipidomic datasets are connected with systems biology approaches, they can be placed into larger networks of the processes involving other biomolecules. This kind of integrative view is crucial since dementia is not driven by a single factor but emerges from the intersection of many disrupted pathways.
Several promising ways emerge when considering future directions. Hence, one key priority is to establish whether glycolipid changes occur early enough to serve as warning signals before clinical symptoms appear. This would provide the opportunity to use these glycoconjugates as biomarkers for risk assessment and early diagnosis. Equally critical is the alignment of methodologies and protocols across the laboratories and research groups involved in the study of dementia-associated glycolipids, in order to generate results, which are reliably compared, reproducible and validated in larger patient cohorts.
Beyond description, there is also a pressing need to move toward mechanistic studies that clarify how specific glycolipid changes influence neuronal survival, immune responses, and protein aggregation.
Equally noteworthy are the potential therapeutic implications and prospects, which warrant a particular attention. Hence, if certain glycolipids prove to be drivers rather than bystanders in dementia, they could become targets for interventions aimed at stabilizing neuronal membranes, modulating inflammation, or preventing toxic protein buildup. Even if they are not direct drivers, their measurable changes could complement other diagnostic tools, contributing to more precise personalized treatment strategies.
Moreover, the accelerating progress of AI presents unprecedented opportunities for glycolipid research in dementia, facilitating the integration and analysis of increasingly complex datasets. ML algorithms can uncover subtle patterns in high-dimensional lipidomics data that might elude conventional analysis, linking glycolipid changes to specific disease stages or phenotypes. AI-driven models can integrate lipidomic profiles with other omics layers, such as genomics, proteomics, and transcriptomics, to construct comprehensive systems-level maps of dementia pathophysiology.
When combined with MS, the potential of AI expands even further. MS produces vast, highly detailed datasets that capture the complexity and diversity of glycolipids in brain tissue and biofluids. AI can process these datasets at scale, revealing patterns and correlations that remain hidden to the traditional approaches. This synergy enables more precise mapping of glycolipid alterations across dementia types, enhancing biomarker discovery and deepening mechanistic insight. AI is also able to greatly improve MS workflows by automating data annotation, interpretation of spectra, and structural identification, while predicting the functional consequences of the detected changes in the glycolipid expression pattern.
In conclusion, MS and systems biology developed to the point where detailed molecular insights can be translated into practical clinical advances. By the ongoing research which aims to connect novel findings at the molecular level with the clinical outcomes, this research directions has the potential to significantly improve the methods of early dementia detection and its treatment.
Author Contributions
Conceptualization, A.D.Z., M.S. and L.D.; resources, M.S., R.I., M.R.B., L.D.; data curation, M.S., R.I., M.R.B., L.D.; writing—original draft preparation, M.S., R.I., M.R.B.; writing—review and editing, A.D.Z. and L.D.; supervision, A.D.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Romanian National Authority for Scientific Research, UEFISCDI, project PN-IV-P2-2.1-TE-2023-0175 and by Aurel Vlaicu University of Arad, Romania, grant number UAV-IRG-1-2025-8.
Institutional Review Board Statement
Manuscript approved for submission
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| AI | artificial intelligence |
| APP | amyloid precursor protein |
| AT(N) | amyloid/tau/neurodegeneration |
| AUC | area under the curve |
| Aβ | amyloid-β peptide |
| BBB | blood–brain barrier |
| BMP | bis(monoacylglycero)phosphate |
| bvFTD | behavioural variant FTD |
| CAG | Cytosine–Adenine–Guanine |
| CE | cholesteryl ester |
| Cer | ceramide |
| CSF | cerebrospinal fluid |
| CT | computer tomography |
| DESI | desorption electrospray ionization |
| DLB | dementia with Lewy body |
| DMS | differential ion mobility spectrometry |
| ESI | electrospray ionization |
| FDA | Food and Drug Administration |
| FDG-PET | fluoro-deoxyglucose positron emission tomography |
| FTD | frontotemporal dementia |
| GalCer | galactosylceramide |
| GalSph | galactosylsphingosine |
| GCase | β-glucocerebrosidase |
| GG | ganglioside |
| GlcCer | glucosylceramide |
| GlcSph | glucosylsphingosine |
| GM | monosialoganglioside |
| GRN | progranulin gene |
| GroPIn | glycerophosphoinositol |
| GSL | glycosphingolipid |
| HD | Huntington’s disease |
| HDD | Huntington’s disease dementia |
| Hex1Cers | monohexosylceramides |
| HEXA | Hexosaminidase Subunit Alpha gene |
| HexCers | hexosylceramides |
| HTT | huntingtin gene |
| IHC | immunohistochemistry |
| IMS-MS | ion mobility spectrometry |
| LacCer | lactosylceramide |
| LBD | Lewy body Dementia |
| LC-MS | liquid chromatography coupled with mass spectrometry |
| MALDI | matrix-assisted laser desorption/ionization |
| MGDG | monogalactosyldiacylglycerol |
| mHTT | mutant huntingtin |
| ML | machine learning |
| MRI | magnetic resonance imaging |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| MSI | mass spectrometry imaging |
| NfL | neurofilament light chain |
| NFT | neurofibrillary tangles |
| PC | phosphatidylcholine |
| PD | Parkinson's disease |
| PDD | Parkinson's disease dementia |
| PE | phosphatidylethanolamine |
| PGRN | progranulin |
| p-tau | phosphorylated tau |
| QTOF | quadrupol time of flight |
| SHexCer | sulfatide |
| SIMS | secondary ion mass spectrometry |
| SIVD | subcortical ischemic vascular dementia |
| SM | sphingomyelin |
| SP | senile plaques |
| SPECT | single photon emission computer tomography |
| SRM | selected reaction monitoring |
| TG | triglyceride |
| TLC | thin-layer chromatography |
| UHPLC MS/MS | ultra-high performance liquid chromatography coupled to tandem MS |
| VD | vascular dementia |
| VLCFA | very long-chain fatty acid |
| WHO | World Health Organization |
| WT | wild-type |
References
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med.2018, 131, 1161–1169. [CrossRef]
- Duara, R.; Barker, W. Heterogeneity in Alzheimer’s Disease Diagnosis and Progression Rates: Implications for Therapeutic Trials. Neurotherapeutics2022, 19, 8–25. [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent Advances in Alzheimer’s Disease: Mechanisms, Clinical Trials and New Drug Development Strategies. Signal Transduct. Target Ther.2024, 9, 211. [CrossRef]
- Gallagher, J.; Gochanour, C.; Caspell-Garcia, C.; Dobkin, R.D.; Aarsland, D.; Alcalay, R.N.; Barrett, M.J.; Chahine, L.; Chen-Plotkin, A.S.; Coffey, C.S.; et al. Long-Term Dementia Risk in Parkinson Disease. Neurology2024, 103, e209699.
- Hobbs, N.Z.; Barnes, J.; Frost, C.; Henley, S.M.; Wild, E.J.; Macdonald, K.; Barker, R.A.; Scahill, R.I.; Fox, N.C.; Tabrizi, S.J. Onset and Progression of Pathologic Atrophy in Huntington Disease: A Longitudinal MR Imaging Study. AJNR Am. J. Neuroradiol.2010, 31, 1036–1041. [CrossRef]
- Aarsland, D.; Kurz, M.W. The Epidemiology of Dementia Associated with Parkinson’s Disease. Brain Pathol.2010, 20, 633–639. [CrossRef]
- Custodio, N.; Montesinos, R.; Lira, D.; Herrera-Pérez, E.; Bardales, Y.; Valeriano-Lorenzo, L. Mixed Dementia: A Review of the Evidence. Dement. Neuropsychol.2017, 11, 364–370. [CrossRef]
- Attems, J.; Jellinger, K.A. The Overlap between Vascular Disease and Alzheimer’s Disease—Lessons from Pathology. BMC Med.2014, 12, 206. [CrossRef]
- Vollhardt, A.; Frölich, L.; Stockbauer, A.C.; Danek, A.; Schmitz, C.; Wahl, A.-S. Towards a Better Diagnosis and Treatment of Dementia: Identifying Common and Distinct Neuropathological Mechanisms in Alzheimer’s and Vascular Dementia. Neurobiol. Dis.2025, 208, 106638. [CrossRef]
- Mohandas, E.; Rajmohan, V. Frontotemporal Dementia: An Updated Overview. Indian J. Psychiatry2009, 51 (Suppl.1), S65–S69.
- Rabinovici, G.D.; Miller, B.L. Frontotemporal Lobar Degeneration: Epidemiology, Pathophysiology, Diagnosis and Management. CNS Drugs2010, 24, 375–398.
- Mollah, S.A.; Nayak, A.; Barhai, S.; Maity, U. A Comprehensive Review on Frontotemporal Dementia: Its Impact on Language, Speech and Behavior. Dement. Neuropsychol.2024, 18, e20230072. [CrossRef]
- Deleon, J.; Miller, B.L. Frontotemporal Dementia. In Handbook of Clinical Neurology; Geschwind, D.H., Paulson, H.L., Klein, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 409–430.
- Roberson, E.D.; Hesse, J.H.; Rose, K.D.; Slama, H.; Johnson, J.K.; Yaffe, K.; Forman, M.S.; Miller, C.A.; Trojanowski, J.Q.; Kramer, J.H.; Miller, B.L. Frontotemporal Dementia Progresses to Death Faster than Alzheimer Disease. Neurology2005, 65, 719–725. [CrossRef]
- Foxe, D.; Muggleton, J.; Cheung, S.C.; Mueller, N.; Ahmed, R.M.; Narasimhan, M.; Burrell, J.R.; Hwang, Y.T.; Cordato, N.J.; Piguet, O. Survival Rates in Frontotemporal Dementia and Alzheimer’s Disease. Neurodegener. Dis. Manag.2025, 15, 191–197. [CrossRef]
- Antonioni, A.; Raho, E.M.; Lopriore, P.; Pace, A.P.; Latino, R.R.; Assogna, M.; Mancuso, M.; Gragnaniello, D.; Granieri, E.; Pugliatti, M.; et al. Frontotemporal Dementia, Where Do We Stand? A Narrative Review. Int. J. Mol. Sci.2023, 24, 11732. [CrossRef]
- Manabe, T.; Fujikura, Y.; Mizukami, K.; Akatsu, H.; Kudo, K. Pneumonia-Associated Death in Patients with Dementia: A Systematic Review and Meta-Analysis. PLoS ONE2019, 14, e0213825. [CrossRef]
- Amoatika, D.A.; Absher, J.R.; Khan, M.T.F.; Miller, M.C. Dementia Deaths Most Commonly Result from Heart and Lung Disease: Evidence from the South Carolina Alzheimer’s Disease Registry. Biomedicines2025, 13, 1321. [CrossRef]
- Yao, J.; Liu, S.; Chen, Q. Mortality Rate of Pulmonary Infection in Senile Dementia Patients: A Systematic Review and Meta-Analysis. Medicine (Baltimore)2024, 103, e39816. [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and Management of Dementia with Lewy Bodies: Fourth Consensus Report of the DLB Consortium. Neurology2017, 89, 88–100. [CrossRef]
- Walker, Z.; Possin, K.L.; Boeve, B.F.; Aarsland, D. Lewy Body Dementias. Lancet2015, 386, 1683–1697. [CrossRef]
- Cycyk, L.M.; Wright, H.H. Frontotemporal Dementia: Its Definition, Differential Diagnosis, and Management. Aphasiology2008, 22, 422–444. [CrossRef]
- Tampi, R.R.; Tampi, D.J.; Parish, M. Easy to Miss, Hard to Treat: Notes on Frontotemporal Dementia. Psychiatric Times2020, 37, 37(10).
- Muangpaisan, W. Clinical Differences among Four Common Dementia Syndromes. Geriatrics Aging2007, 10, 425–429.
- Harciarek, M.; Jodzio, K. Neuropsychological Differences between Frontotemporal Dementia and Alzheimer’s Disease: A Review. Neuropsychol. Rev.2005, 15, 131–145. [CrossRef]
- He, S.; Xu, Z.; Han, X. Lipidome Disruption in Alzheimer’s Disease Brain: Detection, Pathological Mechanisms, and Therapeutic Implications. Mol. Neurodegener.2025, 20, 11. [CrossRef]
- Osetrova, M.; Tkachev, A.; Mair, W.; Guijarro Larraz, P.; Efimova, O.; Kurochkin, I.; Stekolshchikova, E.; Anikanov, N.; Foo, J.C.; Cazenave-Gassiot, A.; et al. Lipidome Atlas of the Adult Human Brain. Nat. Commun.2024, 15, 4455. [CrossRef]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The Role of Glycosphingolipid Metabolism in the Developing Brain. J. Lipid Res.2009, 50 (Suppl.), S440–S445. [CrossRef]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid Rafts and Neurodegeneration: Structural and Functional Roles in Physiologic Aging and Neurodegenerative Diseases. J. Lipid Res.2020, 61, 636–654. [CrossRef]
- Ledeen, R.; Wu, G. Gangliosides of the Nervous System. Methods Mol. Biol.2018, 1804, 19–55. [CrossRef]
- Blomqvist, M.; Zetterberg, H.; Blennow, K.; Månsson, J.E. Sulfatide in Health and Disease: The Evaluation of Sulfatide in Cerebrospinal Fluid as a Possible Biomarker for Neurodegeneration. Mol. Cell. Neurosci.2021, 116, 103670. [CrossRef]
- Wei, J.; Wong, L.C.; Boland, S. Lipids as Emerging Biomarkers in Neurodegenerative Diseases. Int. J. Mol. Sci.2023, 25, 131. [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl. Sci. Rare Dis.2017, 2, 1–71. [CrossRef]
- Matsuzaki, K. Aβ-Ganglioside Interactions in the Pathogenesis of Alzheimer's Disease. Biochim. Biophys. Acta Biomembr.2020, 1862, 183233. [CrossRef]
- Liang, J.; Li, R.; Wong, G.; Huang, X. Lewy Body Dementia: Exploring Biomarkers and Pathogenic Interactions of Amyloid β, Tau, and α-Synuclein. Mol. Neurodegener.2025, 20, 90. [CrossRef]
- Di Pardo, A.; Maglione, V.; Alpaugh, M.; Horkey, M.; Atwal, R.S.; Sassone, J.; Ciammola, A.; Steffan, J.S.; Fouad, K.; Truant, R.; Sipione, S. Ganglioside GM1 Induces Phosphorylation of Mutant Huntingtin and Restores Normal Motor Behavior in Huntington Disease Mice. Proc. Natl. Acad. Sci. U.S.A.2012, 109, 3528–3533. [CrossRef]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment Avenues in Neurodegenerative Disease. Front. Neurol.2019, 10, 859. [CrossRef]
- Zamfir, A.D. Neurological Analyses: Focus on Gangliosides and Mass Spectrometry. Adv. Exp. Med. Biol.2014, 806, 153–204. [CrossRef]
- Müthing, J. High-Resolution Thin-Layer Chromatography of Gangliosides Methods. J. Chromatogr. A. 1996, 720, 3–25. [CrossRef]
- Nishina, K.A.; Supattapone, S. Immunodetection of Glycophosphatidylinositol-Anchored Proteins Following Treatment with Phospholipase C. Anal. Biochem.2007, 363, 318–320. [CrossRef]
- Dehelean, L.; Sarbu, M.; Petrut, A.; Zamfir, A.D. Trends in Glycolipid Biomarker Discovery in Neurodegenerative Disorders by Mass Spectrometry. Adv. Exp. Med. Biol.2019, 1140, 703–729. [CrossRef]
- Touboul, D.; Gaudin, M. Lipidomics of Alzheimer's Disease. Bioanalysis2014, 6, 541–561. [CrossRef]
- Suzuki, A.; Suzuki, M.; Ito, E.; Nitta, T.; Inokuchi, J.I. Mass Spectrometry of Gangliosides. Methods Mol. Biol.2018, 1804, 207–221. [CrossRef]
- Jones, E.E.; Zhang, W.; Zhao, X.; Quiason, C.; Dale, S.; Shahidi-Latham, S.; Grabowski, G.A.; Setchell, K.D.R.; Drake, R.R.; Sun, Y. Tissue Localization of Glycosphingolipid Accumulation in a Gaucher Disease Mouse Brain by LC-ESI-MS/MS and High-Resolution MALDI Imaging Mass Spectrometry. SLAS Discov.2017, 22, 1218–1228. [CrossRef]
- Lanekoff, I.; Geydebrekht, O.; Pinchuk, G.E.; Konopka, A.E.; Laskin, J. Spatially Resolved Analysis of Glycolipids and Metabolites in Living Synechococcus sp. PCC 7002 Using Nanospray Desorption Electrospray Ionization. Analyst2013, 138, 1971–1978. [CrossRef]
- Shon, H.K.; Son, J.G.; Lee, S.Y.; Moon, J.H.; Lee, G.S.; Kim, K.S.; Lee, T.G. Comparison Study of Mouse Brain Tissue by Using ToF-SIMS within Static Limits and Hybrid SIMS Beyond Static Limits (Dynamic Mode). Biointerphases2023, 18, 031005. [CrossRef]
- Sarbu, M.; Fabris, D.; Vukelić, Ž.; Clemmer, D.E.; Zamfir, A.D. Ion Mobility Mass Spectrometry Reveals Rare Sialylated Glycosphingolipid Structures in Human Cerebrospinal Fluid. Molecules2022, 27, 743. [CrossRef]
- Biricioiu, M.R.; Sarbu, M.; Ica, R.; Vukelić, Ž.; Clemmer, D.E.; Zamfir, A.D. Human Cerebellum Gangliosides: A Comprehensive Analysis by Ion Mobility Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom.2024, 35, 683–695. [CrossRef]
- Ica, R.; Sarbu, M.; Biricioiu, R.; Fabris, D.; Vukelić, Ž.; Zamfir, A.D. Novel Application of Ion Mobility Mass Spectrometry Reveals Complex Ganglioside Landscape in Diffuse Astrocytoma Peritumoral Regions. Int. J. Mol. Sci.2025, 26, 8433. [CrossRef]
- Xu, H.; Boucher, F.R.; Nguyen, T.T.; Taylor, G.P.; Tomlinson, J.J.; Ortega, R.A.; Simons, B.; Schlossmacher, M.G.; Saunders-Pullman, R.; Shaw, W.; Bennett, S.A.L. DMS as an Orthogonal Separation to LC/ESI/MS/MS for Quantifying Isomeric Cerebrosides in Plasma and Cerebrospinal Fluid. J. Lipid Res.2019, 60, 200–211. [CrossRef]
- Michno, W.; Bowman, A.; Jha, D.; Minta, K.; Ge, J.; Koutarapu, S.; Zetterberg, H.; Blennow, K.; Lashley, T.; Heeren, R.M.A.; Hanrieder, J. Spatial Neurolipidomics at the Single Amyloid-β Plaque Level in Postmortem Human Alzheimer's Disease Brain. ACS Chem. Neurosci.2024, 15, 877–888. [CrossRef]
- Evers, B.M.; Rodriguez-Navas, C.; Tesla, R.J.; Prange-Kiel, J.; Wasser, C.R.; Yoo, K.S.; McDonald, J.; Cenik, B.; Ravenscroft, T.A.; Plattner, F.; et al. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep.2017, 20, 2565–2574. [CrossRef]
- Shen, H.; Yu, Y.; Wang, J.; Nie, Y.; Tang, Y.; Qu, M. Plasma Lipidomic Signatures of Dementia with Lewy Bodies Revealed by Machine Learning, and Compared to Alzheimer's Disease. Alzheimers Res. Ther.2024, 16, 226. [CrossRef]
- Galleguillos, D.; Zhao, Y.; Pan, B.; Vandermeer, B.; Zaidi, A.; Al Hamarneh, Y.N.; Sarna, J.; Suchowersky, O.; Curtis, J.; Sipione, S. Plasma Gangliosides Correlate with Disease Stages and Symptom Severity in Huntington’s Disease Carriers. bioRxiv2025. [CrossRef]
- Strnad, Š.; PraŽienková, V.; Holubová, M.; Sýkora, D.; Cvačka, J.; Maletínská, L.; Železná, B.; Kuneš, J.; Vrkoslav, V. Mass Spectrometry Imaging of Free-Floating Brain Sections Detects Pathological Lipid Distribution in a Mouse Model of Alzheimer's-like Pathology. Analyst2020, 145, 4595–4605. [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell2018, 173, 822–837. [CrossRef]
- Toprakcioglu, Z.; Jayaram, A.K.; Knowles, T.P.J. Ganglioside Lipids Inhibit the Aggregation of the Alzheimer's Amyloid-β Peptide. RSC Chem. Biol.2025, 6, 809–822. [CrossRef]
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid Pathway Dysfunction Is Prevalent in Patients with Parkinson's Disease. Brain2022, 145, 3472–3487. [CrossRef]
- Li, Q.; Jia, C.; Wu, H.; Liao, Y.; Yang, K.; Li, S.; Zhang, J.; Wang, J.; Li, G.; Guan, F.; et al. Nao Tan Qing Ameliorates Alzheimer's Disease-like Pathology by Regulating Glycolipid Metabolism and Neuroinflammation: A Network Pharmacology Analysis and Biological Validation. Pharmacol. Res.2022, 185, 106489. [CrossRef]
- Alzheimer’s Disease International. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 30.09.2025).
- Zhang, X.; Lu, H.; Xiong, J. Incidence trends and age-period-cohort analysis of Alzheimer's disease and other dementias in the world and China from 1990 to 2021: analyses based on the Global Burden of Disease Study 2021. Front. Neurol.2025, 16, 1628577. [CrossRef]
- Korczyn, A.D.; Grinberg, L.T. Is Alzheimer disease a disease? Nat. Rev. Neurol.2024, 20(4), 245–251. [CrossRef]
- Kamatham, P.T.; Shukla, R.; Khatri, D.K.; Vora, L.K. Pathogenesis, diagnostics, and therapeutics for Alzheimer's disease: Breaking the memory barrier. Ageing Res. Rev.2024, 101, 102481. [CrossRef]
- Oka, T.; Matsuzawa, Y.; Tsuneyoshi, M.; et al. Multiomics analysis to explore blood metabolite biomarkers in an Alzheimer's Disease Neuroimaging Initiative cohort. Sci. Rep.2024, 14(1), 6797. [CrossRef]
- Alzheimer’sAssociation. 2018 Alzheimer's disease facts and figures Alzheimer's & Dementia2018, 14, 367-429. [CrossRef]
- Zhang, H.; Tahami Monfared, A.A.; Zhang, Q.; Honig, L.S. Incidence and prevalence of Alzheimer's disease in Medicare beneficiaries. Neurol. Ther.2025, 14(1), 319–333. [CrossRef]
- Wang, W.; Myers, S.J.; Ollen-Bittle, N.; Whitehead, S.N. Elevation of ganglioside degradation pathway drives GM2 and GM3 within amyloid plaques in a transgenic mouse model of Alzheimer's disease. Neurobiol. Dis.2025, 205, 106798. [CrossRef]
- Cerasuolo, M.; Di Meo, I.; Auriemma, M.C.; Paolisso, G.; Papa, M.; Rizzo, M.R. Exploring the dynamic changes of brain lipids, lipid rafts, and lipid droplets in aging and Alzheimer's disease. Biomolecules2024, 14(11), 1362. [CrossRef]
- Xiao, S.; Wei, X.; Han, B.; et al. Quantitative analysis of targeted lipidomics in the hippocampus of APP/PS1 mice employing the UHPLC-MS/MS method. Front. Aging Neurosci.2025, 17, 1561831. [CrossRef]
- Ariga, T.; Kubota, M.; Nakane, M.; Oguro, K.; Yu, R.K.; Ando, S. Anti-Chol-1 antigen, GQ1bα, antibodies are associated with Alzheimer's disease. PLoS One2013, 8(5), e63326. [CrossRef]
- Xu, L.; Wang, Z.; Li, M.; Li, Q. Global incidence trends and projections of Alzheimer disease and other dementias: an age-period-cohort analysis 2021. J. Glob. Health2025, 15, 04156. [CrossRef]
- Wang, X.; Zhou, R.; Sun, X.; et al. Preferential regulation of γ-secretase-mediated cleavage of APP by ganglioside GM1 reveals a potential therapeutic target for Alzheimer's disease. Adv. Sci. (Weinh.)2023, 10(32), e2303411. [CrossRef]
- Hirano-Sakamaki, W.; Sugiyama, E.; Hayasaka, T.; Ravid, R.; Setou, M.; Taki, T. Alzheimer's disease is associated with disordered localization of ganglioside GM1 molecular species in the human dentate gyrus. FEBS Lett.2015, 589(23), 3611–3616. [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; et al. Alzheimer's disease. Lancet2021, 397(10284), 1577–1590. [CrossRef]
- Anand, S.; Barnes, J.M.; Young, S.A.; et al. Discovery and confirmation of diagnostic serum lipid biomarkers for Alzheimer's disease using direct infusion mass spectrometry. J. Alzheimers Dis.2017, 59(1), 277–290. [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J. Neurochem.2002, 82(4), 809–818. [CrossRef]
- Yuyama, K.; Sun, H.; Sakai, S.; et al. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem.2014, 289(35), 24488–24498. [CrossRef]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Metabolomic study of lipids in serum for biomarker discovery in Alzheimer's disease using direct infusion mass spectrometry. J. Pharm. Biomed. Anal.2014, 98, 321–326. [CrossRef]
- Michno, W.; Wehrli, P.M.; Zetterberg, H.; Blennow, K.; Hanrieder, J. GM1 locates to mature amyloid structures implicating a prominent role for glycolipid-protein interactions in Alzheimer pathology. Biochim. Biophys. Acta Proteins Proteom.2019, 1867(5), 458–467. [CrossRef]
- Sarbu, M.; Ica, R.; Zamfir, A.D. Gangliosides as biomarkers of human brain diseases: Trends in discovery and characterization by high-performance mass spectrometry. Int. J. Mol. Sci.2022, 23(2), 693. [CrossRef]
- Chi, E.Y.; Frey, S.L.; Lee, K.Y. Ganglioside GM1-mediated amyloid-beta fibrillogenesis and membrane disruption. Biochemistry2007, 46(7), 1913–1924. [CrossRef]
- Oikawa, N.; Matsubara, T.; Fukuda, R.; et al. Imbalance in fatty-acid-chain length of gangliosides triggers Alzheimer amyloid deposition in the precuneus. PLoS One2015, 10(3), e0121356. [CrossRef]
- Goux, W.J.; Rodriguez, S.; Sparkman, D.R. Analysis of the core components of Alzheimer paired helical filaments: a gas chromatography/mass spectrometry characterization of fatty acids, carbohydrates and long-chain bases. FEBS Lett.1995, 366(1), 81–85. [CrossRef]
- Goux, W.J.; Liu, B.; Shumburo, A.M.; Parikh, S.; Sparkman, D.R. A quantitative assessment of glycolipid and protein associated with paired helical filament preparations from Alzheimer's diseased brain. J. Alzheimers Dis.2001, 3(5), 455–466.
- Chakraborty, A.; Praharaj, S.K.; Prabhu, R.K.; Prabhu, M.M. Lipidomics and cognitive dysfunction–a narrative review. Turk. J. Biochem.2020, 45(2), 109–119. [CrossRef]
- Ollen-Bittle, N.; Pejhan, S.; Pasternak, S.H.; Keene, C.D.; Zhang, Q.; Whitehead, S.N. Co-registration of MALDI-MSI and histology demonstrates gangliosides co-localize with amyloid beta plaques in Alzheimer’s disease. Acta Neuropathol.2024, 147(1), 105. [CrossRef]
- Zhang, Q.; Li, Y.; Sui, P.; Sun, X.H.; Gao, Y.; Wang, C.Y. MALDI mass spectrometry imaging discloses the decline of sulfoglycosphingolipid and glycerophosphoinositol species in the brain regions related to cognition in a mouse model of Alzheimer's disease. Talanta2024, 266(2), 125022. [CrossRef]
- Blank, M.; Hopf, C. Spatially resolved mass spectrometry analysis of amyloid plaque-associated lipids. J. Neurochem.2021, 159(2), 330–342. [CrossRef]
- Zhang, L.; Li, L.; Meng, F.; et al. Serum metabolites differentiate amnestic mild cognitive impairment from healthy controls and predict early Alzheimer's disease via untargeted lipidomics analysis. Front. Neurol.2021, 12, 704582. [CrossRef]
- Ariga, T.; Jarvis, W.D.; Yu, R.K. Role of sphingolipid-mediated cell death in neurodegenerative diseases. J. Lipid Res.1998, 39(1), 1–16.
- Li, H.; Liu, Y.; Wang, Z.; et al. Mass spectrometry-based ganglioside profiling provides potential insights into Alzheimer's disease development. J. Chromatogr. A. 2022, 1676, 463196. [CrossRef]
- Zhang, Y.; Wang, J.; Liu, J.; et al. Combination of ESI and MALDI mass spectrometry for qualitative, semi-quantitative and in situ analysis of gangliosides in brain. Sci. Rep.2016, 6, 25289. [CrossRef]
- Taki, T. An approach to glycobiology from glycolipidomics: ganglioside molecular scanning in the brains of patients with Alzheimer’s disease by TLC-blot/matrix assisted laser desorption/ionization-time of flight MS. Biol. Pharm. Bull.2012, 35(10), 1642–1647.
- Caughlin, S.; Maheshwari, S.; Agca, Y.; et al. Membrane-lipid homeostasis in a prodromal rat model of Alzheimer's disease: characteristic profiles in ganglioside distributions during aging detected using MALDI imaging mass spectrometry. Biochim. Biophys. Acta Gen. Subj.2018, 1862(6), 1327–1338. [CrossRef]
- Kaya, I.; Jennische, E.; Dunevall, J.; et al. Spatial lipidomics reveals region and long chain base specific accumulations of monosialogangliosides in amyloid plaques in familial Alzheimer's disease mice (5xFAD) brain. ACS Chem. Neurosci.2020, 11(1), 14–24. [CrossRef]
- Good, C.J.; Bowman, A.P.; Klein, C.; et al. Spatial mapping of gangliosides and proteins in amyloid beta plaques at cellular resolution using mass spectrometry imaging and MALDI-IHC. J. Mass Spectrom.2025, 60(9), e5161. [CrossRef]
- Caughlin, S.; Hepburn, J.D.; Park, D.H.; et al. Increased expression of simple ganglioside species GM2 and GM3 detected by MALDI imaging mass spectrometry in a combined rat model of Aβ toxicity and stroke. PLoS One2015, 10(6), e0130364. [CrossRef]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer's β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol.2013, 4, 120. [CrossRef]
- Yahi, N.; Fantini, J. Deciphering the glycolipid code of Alzheimer's and Parkinson's amyloid proteins allowed the creation of a universal ganglioside-binding peptide. PLoS One2014, 9(8), e104751. [CrossRef]
- Prasad, S.; Katta, M.R.; Abhishek, S.; et al. Recent advances in Lewy body dementia: A comprehensive review. Dis. Mon.2023, 69(5), 101441. [CrossRef]
- McKeith, I.; Mintzer, J.; Aarsland, D.; et al. Dementia with Lewy bodies. Lancet Neurol.2004, 3(1), 19–28. [CrossRef]
- Kasuga, K.; Nishizawa, M.; Ikeuchi, T. α-Synuclein as CSF and blood biomarker of dementia with Lewy bodies. Int. J. Alzheimers Dis.2012, 2012, 437025. [CrossRef]
- Ferman, T.J.; Aoki, N.; Crook, J.E.; et al. The limbic and neocortical contribution of α-synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimers Dement.2018, 14(3), 330–339. [CrossRef]
- Armstrong, M.J. Advances in dementia with Lewy bodies. Ther. Adv. Neurol. Disord.2021, 14, 17562864211057666. [CrossRef]
- Scamarcia, P.G.; Agosta, F.; Caso, F.; Filippi, M. Update on neuroimaging in non-Alzheimer's disease dementia: a focus on the Lewy body disease spectrum. Curr. Opin. Neurol.2021, 34(4), 532–538. [CrossRef]
- Kantarci, K.; Lowe, V.J.; Chen, Q.; et al. β-Amyloid PET and neuropathology in dementia with Lewy bodies. Neurology2020, 94(3), e282–e291. [CrossRef]
- Palmqvist, S.; Hansson, O.; Minthon, L.; Londos, E. Practical suggestions on how to differentiate dementia with Lewy bodies from Alzheimer's disease with common cognitive tests. Int. J. Geriatr. Psychiatry2009, 24(12), 1405–1412. [CrossRef]
- Senanarong, V.; Wachirutmangur, L.; Rattanabunnakit, C.; Srivanitchapoom, P.; Udomphanthurak, S. Plasma alpha synuclein (α-syn) as a potential biomarker of diseases with synucleinopathy. Alzheimers Dement.2020, 16(S9), e044409.
- Mukaetova-Ladinska, E.B.; Monteith, R.; Perry, E.K. Cerebrospinal fluid biomarkers for dementia with Lewy bodies. Int. J. Alzheimers Dis.2010, 2010, 536538.
- Mollenhauer, B.; Schlossmacher, M.G. CSF synuclein: adding to the biomarker footprint of dementia with Lewy bodies. J. Neurol. Neurosurg. Psychiatry2010, 81(6), 590–591. [CrossRef]
- Donadio, V.; Incensi, A.; Rizzo, G.; et al. A new potential biomarker for dementia with Lewy bodies: Skin nerve α-synuclein deposits. Neurology2017, 89(4), 318–326. [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol.2016, 79(6), 940–949. [CrossRef]
- Kotzbauer, P.T.; Tu, Z.; Mach, R.H. Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clin. Transl. Imaging2017, 5(1), 3–14.
- Mavroudis, I.; Petridis, F.; Kazis, D. Cerebrospinal fluid, imaging, and physiological biomarkers in dementia with Lewy bodies. Am. J. Alzheimers Dis. Other Demen.2019, 34(7–8), 421–432. [CrossRef]
- Vrillon, A.; Bousiges, O.; Götze, K.; et al. Plasma biomarkers of amyloid, tau, axonal, and neuroinflammation pathologies in dementia with Lewy bodies. Alzheimers Res. Ther.2024, 16(1), 146. [CrossRef]
- Peña-Bautista, C.; Bolsewig, K.; Gonzalez, M.C.; et al. The association between plasma and MRI biomarkers in dementia with Lewy bodies. Alzheimers Res. Ther.2025, 17(1), 197. [CrossRef]
- Wang, S.Y.; Chen, W.; Xu, W.; et al. Neurofilament light chain in cerebrospinal fluid and blood as a biomarker for neurodegenerative diseases: A systematic review and meta-analysis. J. Alzheimers Dis.2019, 72(4), 1353–1361. [CrossRef]
- Lourenco, M.V.; Ribeiro, F.C.; Santos, L.E.; et al. Cerebrospinal fluid neurotransmitters, cytokines, and chemokines in Alzheimer's and Lewy body diseases. J. Alzheimers Dis.2021, 82(3), 1067–1074. [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: A systematic review and meta-analysis. Lancet Neurol.2016, 15(7), 673–684. [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; et al. Plasma P-tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat. Med.2020, 26(3), 379–386. [CrossRef]
- Savica, R.; Murray, M.E.; Persson, X.M.; et al. Plasma sphingolipid changes with autopsy-confirmed Lewy body or Alzheimer's pathology. Alzheimers Dement. (Amst.)2016, 3, 43–50. [CrossRef]
- Lerche, S.; Wurster, I.; Valente, E.M.; et al. CSF d18:1 sphingolipid species in Parkinson disease and dementia with Lewy bodies with and without GBA1 variants. NPJ Parkinsons Dis.2024, 10(1), 198. [CrossRef]
- Miglis, M.G.; Adler, C.H.; Antelmi, E.; et al. Biomarkers of conversion to α-synucleinopathy in isolated rapid-eye-movement sleep behaviour disorder. Lancet Neurol.2021, 20(8), 671–684. [CrossRef]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: teammates in neurodegeneration? Mol. Neurodegener.2014, 9, 43. [CrossRef]
- Gomperts, S.N. Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Continuum (Minneap Minn)2016, 22(2), 435–463. [CrossRef]
- Yamashita, K.Y.; Bhoopatiraju, S.; Silverglate, B.D.; Grossberg, G.T. Biomarkers in Parkinson’s disease: A state of the art review. Biomark. Neuropsychiatry2023, 9, 100074. [CrossRef]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov. Disord.2008, 23(6), 837–844. [CrossRef]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Kragh-Sørensen, P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch. Neurol.2003, 60(3), 387–392. [CrossRef]
- Emre, M.; Aarsland, D.; Brown, R.; et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov. Disord.2007, 22(12), 1689–1707. [CrossRef]
- Savica, R.; Knopman, D.S. Dementia with Lewy bodies. In: Schapira, A.; Wszolek, Z.; Dawson, T.M.; Wood, N., Eds. Neurodegeneration; Wiley-Blackwell: Hoboken, NJ, USA, 2017; pp. 83–92. [CrossRef]
- Zardini Buzatto, A.; Tatlay, J.; Bajwa, B.; et al. Comprehensive serum lipidomics for detecting incipient dementia in Parkinson's disease. J. Proteome Res.2021, 20(8), 4053–4067. [CrossRef]
- Azevedo, R.; Jacquemin, C.; Villain, N.; Fenaille, F.; Lamari, F.; Becher, F. Mass spectrometry for neurobiomarker discovery: the relevance of post-translational modifications. Cells2022, 11(8), 1279. [CrossRef]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell. Proteomics2013, 12(12), 3543–3558. [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem.2006, 281(40), 29739–29752. [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science2006, 314(5796), 130–133. [CrossRef]
- Wesseling, H.; Mair, W.; Kumar, M.; et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell2020, 183(6), 1699–1713.e13. [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One2013, 8(9), e73094. [CrossRef]
- Xing, Y.; Tang, Y.; Zhao, L.; et al. Associations between plasma ceramides and cognitive and neuropsychiatric manifestations in Parkinson's disease dementia. J. Neurol. Sci.2016, 370, 82–87. [CrossRef]
- Galper, J.; Mori, G.; McDonald, G.; et al. Prediction of motor and non-motor Parkinson's disease symptoms using serum lipidomics and machine learning: a 2-year study. NPJ Parkinsons Dis.2024, 10(1), 123. [CrossRef]
- Avisar, H.; Guardia-Laguarta, C.; Area-Gomez, E.; et al. Lipidomics prediction of Parkinson’s disease severity: a machine-learning analysis. J. Parkinsons Dis.2021, 11(3), 1141–1155. [CrossRef]
- Raz, L.; Knoefel, J.; Bhaskar, K. The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab.2016, 36(1), 172–186. [CrossRef]
- Peet, B.T.; Spina, S.; Mundada, N.; La Joie, R. Neuroimaging in frontotemporal dementia: heterogeneity and relationships with underlying neuropathology. Neurotherapeutics2021, 18(2), 728–752. [CrossRef]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol.2018, 10(4), a033118. [CrossRef]
- Urso, D.; Giannoni-Luza, S.; Brayne, C.; et al. Incidence and prevalence of frontotemporal dementia: a systematic review and meta-analysis. JAMA Neurol.2025, e253307. [CrossRef]
- Flavell, J.; Ahern, E.G.M.; Logan, B.; et al. Factors associated with true-positive and false-positive diagnoses of behavioural variant frontotemporal dementia in 100 consecutive referrals from specialist physicians. Eur. J. Neurol.2025, 32(1), e70036. [CrossRef]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology2009, 73(18), 1451–1456. [CrossRef]
- Wood, E.M.; Falcone, D.; Suh, E.; et al. Development and validation of pedigree classification criteria for frontotemporal lobar degeneration. JAMA Neurol.2013, 70(11), 1411–1417. [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron2011, 72(2), 245–256. [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron2011, 72(2), 257–268. [CrossRef]
- Lashley, T.; Rohrer, J.D.; Mead, S.; Revesz, T. An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol.2015, 41(7), 858–881. [CrossRef]
- Ahmed, R.M.; Hodges, J.R.; Piguet, O. Behavioural variant frontotemporal dementia: recent advances in the diagnosis and understanding of the disorder. Adv. Exp. Med. Biol.2021, 1281, 1–15. [CrossRef]
- Bott, N.T.; Radke, A.; Stephens, M.L.; Kramer, J.H. Frontotemporal dementia: diagnosis, deficits and management. Neurodegener. Dis. Manag.2014, 4(6), 439–454. [CrossRef]
- van Vliet, D.; de Vugt, M.E.; Bakker, C.; et al. Time to diagnosis in young-onset dementia as compared with late-onset dementia. Psychol. Med.2013, 43(2), 423–432. [CrossRef]
- Zhao, M.; Lv, X.; Tuerxun, M.; et al. Delayed help seeking behavior in dementia care: preliminary findings from the Clinical Pathway for Alzheimer's Disease in China (CPAD) study. Int. Psychogeriatr.2016, 28(2), 211–219. [CrossRef]
- Gendron, T.F.; Heckman, M.G.; White, L.J.; et al. Comprehensive cross-sectional and longitudinal analyses of plasma neurofilament light across FTD spectrum disorders. Cell Rep. Med.2022, 3(4), 100607. [CrossRef]
- Katisko, K.; Cajanus, A.; Huber, N.; et al. GFAP as a biomarker in frontotemporal dementia and primary psychiatric disorders: diagnostic and prognostic performance. J. Neurol. Neurosurg. Psychiatry2021, 92(12), 1305–1312. [CrossRef]
- Chouliaras, L.; Thomas, A.; Malpetti, M.; et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer's disease, frontotemporal dementia and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry2022, 93(6), 651–658. [CrossRef]
- Ntymenou, S.; Tsantzali, I.; Kalamatianos, T.; et al. Blood biomarkers in frontotemporal dementia: review and meta-analysis. Brain Sci.2021, 11(2), 244. [CrossRef]
- Liampas, I.; Kyriakoulopoulou, P.; Karakoida, V.; et al. Blood-based biomarkers in frontotemporal dementia: a narrative review. Int. J. Mol. Sci.2024, 25(21), 11838. [CrossRef]
- Gómez-Tortosa, E.; Agüero-Rabes, P.; Ruiz-González, A.; et al. Plasma biomarkers in the distinction of Alzheimer’s disease and frontotemporal dementia. Int. J. Mol. Sci.2025, 26(3), 1231. [CrossRef]
- Ambaw, Y.A.; Ljubenkov, P.A.; Singh, S.; et al. Plasma lipidome dysregulation in frontotemporal dementia reveals shared, genotype-specific, and severity-linked alterations. Alzheimers Dement.2025, 21(9), e70631. [CrossRef]
- Marian, O.C.; Matis, S.; Dobson-Stone, C.; et al. Reduced plasma hexosylceramides in frontotemporal dementia are a biomarker of white matter integrity. Alzheimers Dement. (Amst.)2025, 17(2), e70131. [CrossRef]
- Puljko, B.; Stojanović, M.; Ilic, K.; Kalanj-Bognar, S.; Mlinac-Jerkovic, K. Start me up: how can surrounding gangliosides affect sodium-potassium ATPase activity and steer towards pathological ion imbalance in neurons? Biomedicines2022, 10(7), 1518. [CrossRef]
- Vasques, J.F.; de Jesus Gonçalves, R.G.; da Silva-Junior, A.J.; et al. Gangliosides in nervous system development, regeneration, and pathologies. Neural Regen. Res.2023, 18(1), 81–86. [CrossRef]
- Schengrund, C.L. Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem. Sci.2015, 40(7), 397–406. [CrossRef]
- Sarbu, M.; Dehelean, L.; Munteanu, C.V.A.; Vukelić, Ž.; Zamfir, A.D. Assessment of ganglioside age-related and topographic specificity in human brain by Orbitrap mass spectrometry. Anal. Biochem.2017, 521, 40–54. [CrossRef]
- Mlinac, K.; Bognar, S.K. Role of gangliosides in brain aging and neurodegeneration. Transl. Neurosci.2010, 1(4), 300–307. [CrossRef]
- Ica, R.; Petrut, A.; Munteanu, C.V.A.; et al. Orbitrap mass spectrometry for monitoring the ganglioside pattern in human cerebellum development and aging. J. Mass Spectrom.2020, 55(5), e4502. [CrossRef]
- Sarbu, M.; Petrica, L.; Clemmer, D.E.; Vukelić, Ž.; Zamfir, A.D. Gangliosides of human glioblastoma multiforme: a comprehensive mapping and structural analysis by ion mobility tandem mass spectrometry. J. Am. Soc. Mass Spectrom.2021, 32(5), 1249–1257. [CrossRef]
- Ica, R.; Simulescu, A.; Sarbu, M.; Munteanu, C.V.A.; Vukelić, Ž.; Zamfir, A.D. High resolution mass spectrometry provides novel insights into the ganglioside pattern of brain cavernous hemangioma. Anal. Biochem.2020, 609, 113976. [CrossRef]
- Sarbu, M.; Clemmer, D.E.; Zamfir, A.D. Ion mobility mass spectrometry of human melanoma gangliosides. Biochimie2020, 177, 226–237. [CrossRef]
- Zamfir, A.D.; Fabris, D.; Capitan, F.; Munteanu, C.; Vukelić, Z.; Flangea, C. Profiling and sequence analysis of gangliosides in human astrocytoma by high-resolution mass spectrometry. Anal. Bioanal. Chem.2013, 405(23), 7321–7335. [CrossRef]
- Sasaki, N.; Toyoda, M.; Ishiwata, T. Gangliosides as signaling regulators in cancer. Int. J. Mol. Sci.2021, 22(10), 5076. [CrossRef]
- Groux-Degroote, S.; Delannoy, P. Cancer-associated glycosphingolipids as tumor markers and targets for cancer immunotherapy. Int. J. Mol. Sci.2021, 22(11), 6145. [CrossRef]
- Cavdarli, S.; Delannoy, P.; Groux-Degroote, S. O-acetylated gangliosides as targets for cancer immunotherapy. Cells2020, 9(3), 741. [CrossRef]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Ganglioside-mediated assembly of amyloid β-protein: roles in Alzheimer's disease. Prog. Mol. Biol. Transl. Sci.2018, 156, 413–434. [CrossRef]
- Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J.2015, 32(3–4), 87–91. [CrossRef]
- Fukami, Y.; Ariga, T.; Yamada, M.; Yuki, N. Brain gangliosides in Alzheimer’s disease: increased expression of cholinergic neuron-specific gangliosides. Curr. Alzheimer Res.2017, 14(6), 586–591. [CrossRef]
- Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of glycosphingolipids and their role in the pathophysiology of lysosomal storage disorders. Int. J. Mol. Sci.2020, 21(18), 6881. [CrossRef]
- Kim, J.; Byeon, S.K.; Oglesbee, D.; Schultz, M.J.; Matern, D.; Pandey, A. A multiplexed targeted method for profiling of serum gangliosides and glycosphingolipids: application to GM2-gangliosidosis. Anal. Bioanal. Chem.2024, 416(26), 5689–5699. [CrossRef]
- Boland, S.; Swarup, S.; Ambaw, Y.A.; et al. Deficiency of the frontotemporal dementia gene GRN results in gangliosidosis. Nat. Commun.2022, 13, 5924. [CrossRef]
- Kamp, P.E.; den Hartog Jager, W.A.; Maathuis, J.; de Groot, P.A.; de Jong, J.M.; Bolhuis, P.A. Brain gangliosides in the presenile dementia of Pick. J. Neurol. Neurosurg. Psychiatry1986, 49(8), 881–885. [CrossRef]
- Kim, W.S.; Jary, E.; Pickford, R.; et al. Lipidomics analysis of behavioral variant frontotemporal dementia: a scope for biomarker development. Front. Neurol.2018, 9, 104. [CrossRef]
- Marian, O.C.; Teo, J.D.; Lee, J.Y.; et al. Disrupted myelin lipid metabolism differentiates frontotemporal dementia caused by GRN and C9orf72 gene mutations. Acta Neuropathol. Commun.2023, 11(1), 52. [CrossRef]
- Arrant, A.E.; Roth, J.R.; Boyle, N.R.; Kashyap, S.N.; Hoffmann, M.Q.; Murchison, C.F.; Ramos, E.M.; Nana, A.L.; Spina, S.; Grinberg, L.T.; Miller, B.L.; Seeley, W.W.; Roberson, E.D. Impaired β-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol. Commun.2019, 7(1), 218. [CrossRef]
- He, Y.; Phan, K.; Bhatia, S.; Pickford, R.; Fu, Y.; Yang, Y.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Increased VLCFA-lipids and ELOVL4 underlie neurodegeneration in frontotemporal dementia. Sci. Rep.2021, 11(1), 21348. [CrossRef]
- Aqel, S.; Ahmad, J.; Saleh, I.; Fathima, A.; Al Thani, A.A.; Mohamed, W.M.Y.; Shaito, A.A. Advances in Huntington’s disease biomarkers: a 10-year bibliometric analysis and a comprehensive review. Biology2025, 14(2), 129. [CrossRef]
- Seeley, C.; Kegel-Gleason, K.B. Taming the Huntington's disease proteome: what have we learned? J. Huntingtons Dis.2021, 10(2), 239–257. [CrossRef]
- Peavy, G.M.; Jacobson, M.W.; Goldstein, J.L.; Hamilton, J.M.; Kane, A.; Gamst, A.C.; Lessig, S.L.; Lee, J.C.; Corey-Bloom, J. Cognitive and functional decline in Huntington's disease: dementia criteria revisited. Mov. Disord.2010, 25(9), 1163–1169. [CrossRef]
- Snowden, J.S.; Craufurd, D.; Thompson, J.; Neary, D. Psychomotor, executive, and memory function in preclinical Huntington's disease. J. Clin. Exp. Neuropsychol.2002, 24(2), 133–145. [CrossRef]
- Hamilton, J.M.; Salmon, D.P.; Corey-Bloom, J.; et al. Behavioural abnormalities contribute to functional decline in Huntington's disease. J. Neurol. Neurosurg. Psychiatry2003, 74(1), 120–122. [CrossRef]
- McGarry, A.; Gaughan, J.; Hackmyer, C.; Lovett, J.; Khadeer, M.; Shaikh, H.; Pradhan, B.; Ferraro, T.N.; Wainer, I.W.; Moaddel, R. Cross-sectional analysis of plasma and CSF metabolomic markers in Huntington's disease for participants of varying functional disability: a pilot study. Sci. Rep.2020, 10(1), 20490. [CrossRef]
- McGarry, A.; Gaughan, J.; Hackmyer, C.; et al. Author correction: Cross-sectional analysis of plasma and CSF metabolomic markers in Huntington's disease for participants of varying functional disability: a pilot study. Sci. Rep.2021, 11(1), 9947. [CrossRef]
- Hunter, M.; Demarais, N.J.; Faull, R.L.M.; Grey, A.C.; Curtis, M.A. An imaging mass spectrometry atlas of lipids in the human neurologically normal and Huntington's disease caudate nucleus. J. Neurochem.2021, 157(6), 2158–2172. [CrossRef]
- Phillips, G.R.; Saville, J.T.; Hancock, S.E.; Brown, S.H.J.; Jenner, A.M.; McLean, C.; Fuller, M.; Newell, K.A.; Mitchell, T.W. The long and the short of Huntington's disease: how the sphingolipid profile is shifted in the caudate of advanced clinical cases. Brain Commun.2021, 4(1), fcab303. [CrossRef]
- Phillips, G.R.; Hancock, S.E.; Brown, S.H.J.; Jenner, A.M.; Kreilaus, F.; Newell, K.A.; Mitchell, T.W. Cholesteryl ester levels are elevated in the caudate and putamen of Huntington's disease patients. Sci. Rep.2020, 10(1), 20314. [CrossRef]
- Maglione, V.; Marchi, P.; Di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired ganglioside metabolism in Huntington's disease and neuroprotective role of GM1. J. Neurosci.2010, 30(11), 4072–4080. [CrossRef]
- Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; Di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; Baker, G.B.; Fouad, K.; Sipione, S. Disease-modifying effects of ganglioside GM1 in Huntington's disease models. EMBO Mol. Med.2017, 9(11), 1537–1557. [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Johnson, E.B.; Wijeratne, P.A.; De Vita, E.; Alexander, D.C.; Palermo, G.; Czech, C.; Schobel, S.; Scahill, R.I.; Heslegrave, A.; Zetterberg, H.; Wild, E.J. Evaluation of mutant huntingtin and neurofilament proteins as potential markers in Huntington's disease. Sci. Transl. Med.2018, 10(458), eaat7108. [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; Wild, E.J. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol.2017, 16(8), 601–609. [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; et al. Corrections. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol.2017, 16(9), 683. [CrossRef]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; Mansbach, A.; Borowsky, B.; Konstantinova, P.; Mestre, T.A.; Panagoulias, J.; Ross, C.A.; Zauderer, M.; Mullin, A.P.; Romero, K.; Sivakumaran, S.; Turner, E.C.; Long, J.D.; Sampaio, C.; Huntington's Disease Regulatory Science Consortium (HD-RSC). A biological classification of Huntington's disease: the Integrated Staging System. Lancet Neurol.2022, 21(7), 632–644. [CrossRef]
- Ghofrani-Jahromi, M.; Poudel, G.R.; Razi, A.; Abeyasinghe, P.M.; Paulsen, J.S.; Tabrizi, S.J.; Saha, S.; Georgiou-Karistianis, N. Prognostic enrichment for early-stage Huntington's disease: an explainable machine learning approach for clinical trial. Neuroimage Clin.20242, 43, 103650. [CrossRef]
- Ganesh, S.; Chithambaram, T.; Krishnan, N.R.; Vincent, D.R.; Kaliappan, J.; Srinivasan, K. Exploring Huntington's disease diagnosis via artificial intelligence models: a comprehensive review. Diagnostics (Basel)2023, 13(23), 3592. [CrossRef]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 pathology in Alzheimer’s disease. Mol. Neurodegener.2021, 16, 84. [CrossRef]
- Knopman, D.S.; Parisi, J.E.; Boeve, B.F.; Cha, R.H.; Apaydin, H.; Salviati, A.; Edland, S.D.; Rocca, W.A. Vascular dementia in a population-based autopsy study. Arch. Neurol.2003, 60(4), 569–575. [CrossRef]
- Buciuc, M.; Whitwell, J.L.; Boeve, B.F.; Ferman, T.J.; Graff-Radford, J.; Savica, R.; Kantarci, K.; Fields, J.A.; Knopman, D.S.; Petersen, R.C.; Parisi, J.E.; Murray, M.E.; Dickson, D.W.; Josephs, K.A. TDP-43 is associated with a reduced likelihood of rendering a clinical diagnosis of dementia with Lewy bodies in autopsy-confirmed cases of transitional/diffuse Lewy body disease. J. Neurol.2020, 267(5), 1444–1453. [CrossRef]
- Song, R.; Pan, K.Y.; Xu, H.; Qi, X.; Buchman, A.S.; Bennett, D.A.; Xu, W. Association of cardiovascular risk burden with risk of dementia and brain pathologies: a population-based cohort study. Alzheimers Dement.20210, 17(12), 1914–1922. [CrossRef]
- Livingston, G.; Huntley, J.; Liu, K.Y.; Costafreda, S.G.; Selbæk, G.; Alladi, S.; Ames, D.; Banerjee, S.; Burns, A.; Brayne, C.; Fox, N.C.; Ferri, C.P.; Gitlin, L.N.; Howard, R.; Kales, H.C.; Kivimäki, M.; Larson, E.B.; Nakasujja, N.; Rockwood, K.; Samus, Q.; Mukadam, N. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet2024, 404(10452), 572–628. [CrossRef]
- Leocadi, M.; Canu, E.; Paldino, A.; Agosta, F.; Filippi, M. Awareness impairment in Alzheimer's disease and frontotemporal dementia: a systematic MRI review. J. Neurol.2023, 270(4), 1880–1907. [CrossRef]
- Rowley, P.A.; Samsonov, A.A.; Betthauser, T.J.; Pirasteh, A.; Johnson, S.C.; Eisenmenger, L.B. Amyloid and Tau PET imaging of Alzheimer disease and other neurodegenerative conditions. Semin. Ultrasound CT MR2020, 41(6), 572–583. [CrossRef]
- Manjavong, M.; Kang, J.M.; Diaz, A.; Ashford, M.T.; Eichenbaum, J.; Aaronson, A.; Miller, M.J.; Mackin, S.; Tank, R.; Weiner, M.; Nosheny, R. Performance of plasma biomarkers combined with structural MRI to identify candidate participants for Alzheimer's disease-modifying therapy. J. Prev. Alzheimers Dis.2024, 11(5), 1198–1205. [CrossRef]
- Mok, V.C.T.; Cai, Y.; Markus, H.S. Vascular cognitive impairment and dementia: mechanisms, treatment, and future directions. Int. J. Stroke2024, 19(8), 838–856. [CrossRef]
- Chang Wong, E.; Chang Chui, H. Vascular cognitive impairment and dementia. Continuum (Minneap Minn)2022, 28(3), 750–780. [CrossRef]
- Tisher, A.; Salardini, A. A comprehensive update on treatment of dementia. Semin. Neurol.2019, 39(2), 167–178. [CrossRef]
- Kapasi, A.; James, B.D.; Yu, L.; Sood, A.; Arvanitakis, Z.; Bennett, D.A.; Boyle, P.; Schneider, J.A. Mixed pathologies and cognitive outcomes in persons considered for anti-amyloid treatment eligibility assessment: a community-based study. Neurology2025, 105(5), e214004. [CrossRef]
- Yang, H.-M. Vascular dementia: from pathophysiology to therapeutic frontiers. J. Clin. Med.2025, 14, 6611. [CrossRef]
- Alrouji, M.; Alshammari, M.S.; Tasqeeruddin, S.; Shamsi, A. Interplay between aging and tau pathology in Alzheimer's disease: mechanisms and translational perspectives. Antioxidants (Basel)2025, 14(7), 774. [CrossRef]
- Mani, S.; Wasnik, S.; Shandilya, C.; Srivastava, V.; Khan, S.; Singh, K.K. Pathogenic synergy: dysfunctional mitochondria and neuroinflammation in neurodegenerative diseases associated with aging. Front. Aging2025, 6, 1615764. [CrossRef]
- Yu, W.; Ying, J.; Wang, X.; Liu, X.; Zhao, T.; Yoon, S.; Zheng, Q.; Fang, Y.; Yang, D.; Hua, F. The involvement of lactosylceramide in central nervous system inflammation related to neurodegenerative disease. Front. Aging Neurosci.2021, 13, 691230. [CrossRef]
- Teunissen, C.E.; Kimble, L.; Bayoumy, S.; Bolsewig, K.; Burtscher, F.; Coppens, S.; Das, S.; Gogishvili, D.; Fernandes Gomes, B.; Gómez de San José, N.; Mavrina, E.; Meda, F.J.; Mohaupt, P.; Mravinacová, S.; Waury, K.; Wojdała, A.L.; Abeln, S.; Chiasserini, D.; Hirtz, C.; Gaetani, L.; Vermunt, L.; Bellomo, G.; Halbgebauer, S.; Lehmann, S.; Månberg, A.; Nilsson, P.; Otto, M.; Vanmechelen, E.; Verberk, I.M.W.; Willemse, E.; Zetterberg, H.; MIRIADE consortium. Methods to discover and validate biofluid-based biomarkers in neurodegenerative dementias. Mol. Cell Proteomics2023, 22(10), 100629. [CrossRef]
- Yilmaz, A.; Ashrafi, N.; Ashrafi, R.; Akyol, S.; Saiyed, N.; Kerševičiūtė, I.; Gabrielaite, M.; Gordevicius, J.; Graham, S.F. Lipid profiling of Parkinson’s disease brain highlights disruption in lysophosphatidylcholines, and triacylglycerol metabolism. npj Parkinsons Dis.2025, 11, 159. [CrossRef]
- Kelley, A.R. Mass spectrometry-based analysis of lipid involvement in Alzheimer’s disease pathology—a review. Metabolites2022, 12, 510. [CrossRef]
- Cilento, E.M.; Jin, L.; Stewart, T.; Shi, M.; Sheng, L.; Zhang, J. Mass spectrometry: a platform for biomarker discovery and validation for Alzheimer's and Parkinson's diseases. J. Neurochem.2019, 151(4), 397–416. [CrossRef]
- Lam, S.M.; Wang, Y.; Duan, X.; Wenk, M.R.; Kalaria, R.N.; Chen, C.P.; Lai, M.K.; Shui, G. Brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol. Aging2014, 35(10), 2369–2381. [CrossRef]
- Hendriks, T.F.E.; Krestensen, K.K.; Mohren, R.; Vandenbosch, M.; De Vleeschouwer, S.; Heeren, R.M.A.; Cuypers, E. MALDI-MSI-LC-MS/MS workflow for single-section single step combined proteomics and quantitative lipidomics. Anal. Chem.2024, 96(10), 4266–4274. [CrossRef]
- Noel, A.; Ingrand, S.; Barrier, L. Ganglioside and related-sphingolipid profiles are altered in a cellular model of Alzheimer's disease. Biochimie2017, 137, 158–164. [CrossRef]
- Chua, X.Y.; Torta, F.; Chong, J.R.; Venketasubramanian, N.; Hilal, S.; Wenk, M.R.; Chen, C.P.; Arumugam, T.V.; Herr, D.R.; Lai, M.K.P. Lipidomics profiling reveals distinct patterns of plasma sphingolipid alterations in Alzheimer's disease and vascular dementia. Alzheimers Res. Ther.2023, 15(1), 214. [CrossRef]
- Zimmer, V.C.; Lauer, A.A.; Haupenthal, V.; Stahlmann, C.P.; Mett, J.; Grösgen, S.; Hundsdörfer, B.; Rothhaar, T.; Endres, K.; Eckhardt, M.; Hartmann, T.; Grimm, H.S.; Grimm, M.O.W. A bidirectional link between sulfatide and Alzheimer’s disease. Cell Chem. Biol.2024, 31(2), 265–283.e7. [CrossRef]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology2021, 31(11), 1416–1434. [CrossRef]
- Pujol-Lereis, L.M. Alteration of sphingolipids in biofluids: implications for neurodegenerative diseases. Int. J. Mol. Sci.2019, 20(14), 3564. [CrossRef]
- Koal, T.; Klavins, K.; Seppi, D.; Kemmler, G.; Humpel, C. Sphingomyelin SM(d18:1/18:0) is significantly enhanced in cerebrospinal fluid samples dichotomized by pathological amyloid-β42, tau, and phospho-tau-181 levels. J. Alzheimers Dis.2015, 44(4), 1193–1201. [CrossRef]
- Montine, T.J.; Morrow, J.D. Fatty acid oxidation in the pathogenesis of Alzheimer's disease. Am. J. Pathol.2005, 166(5), 1283–1289. [CrossRef]
Figure 1.
Molecular basis and halmarks of major dementia types (Schematic of neuron created in https://BioRender.com).
Figure 1.
Molecular basis and halmarks of major dementia types (Schematic of neuron created in https://BioRender.com).
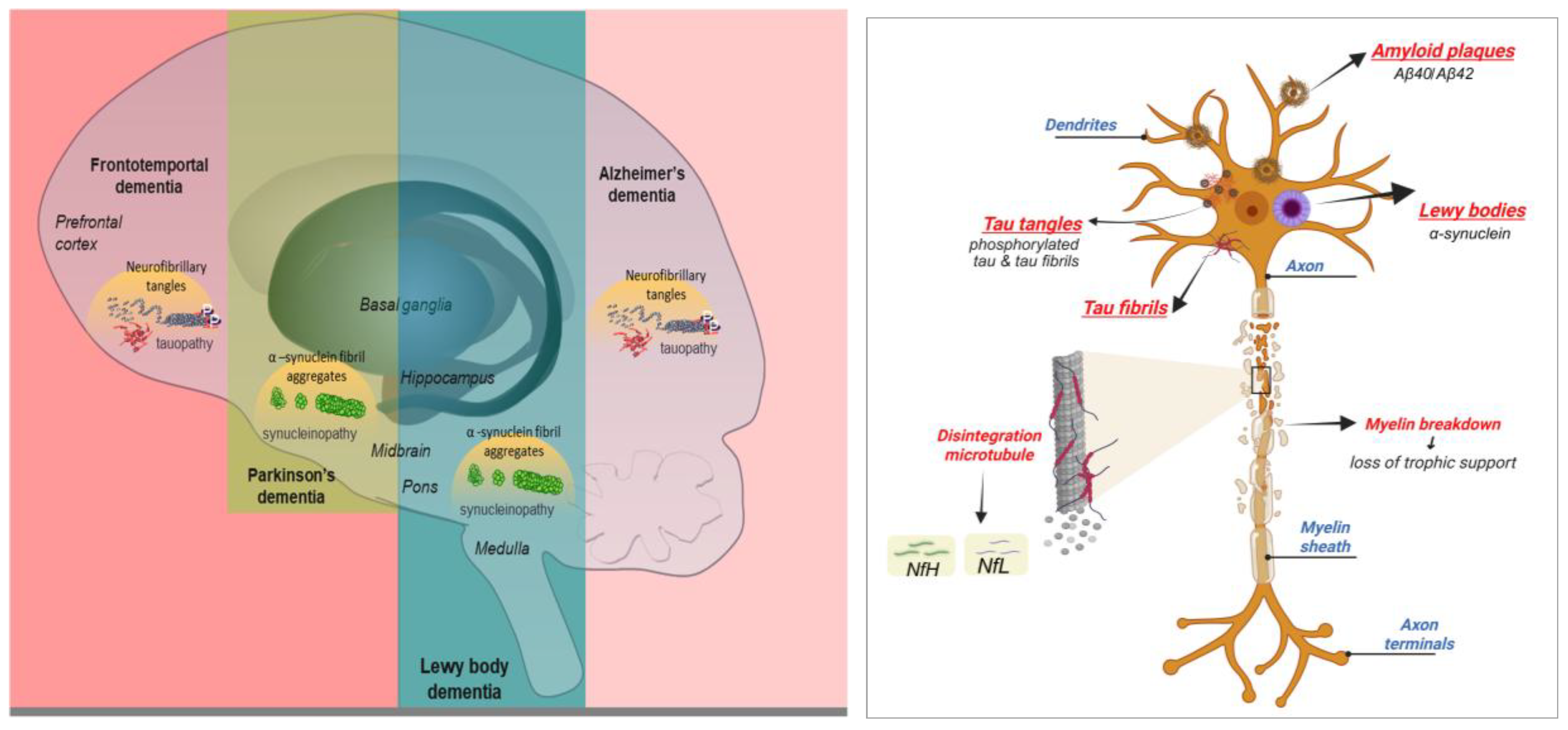

Table 1.
Key features of different dementia types [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25].
| Disorder Features |
Dementia of Alzheimer’s Type (AD) | Dementia with Lewy body (DLB) | Frontotemporal dementia (FTD) | Parkinson's disease dementia (PDD) | Huntington's disease (HD) | Mixed dementia |
|---|---|---|---|---|---|---|
| Onset | Presenile or senile | Senile | Presenile | Late onset, usually after Parkinson's diagnosis | Presenile | Senile onset |
| Age at diagnosis | < 65s or > 65s | 70s, but range 50s–80s | 40s and early 60s | >70s | 30s or 40s | >65 |
| Patient profile | Predominantly female | Slight male predominance | Predominantly male | Predominantly male, in early onset cases | Equal in males and females (autosomal dominant inheritance) | No gender preference |
| Brain Abnormalities | Accumulation of amyloid plaques and tau tangles throughout the brain, granulovacuolar degeneration in hippocampus | α-synuclein aggregation in cortical and subcortical Lewy bodies; often coexists with Alzheimer’s pathology | Buildup of abnormal tau and TDP-43 proteins in the frontal and temporal lobes | Accumulation of alpha-synuclein protein in Lewy bodies | Caused by a specific inherited gene mutation leading to neuron degeneration | Accumulation of tau and amyloid plaques, blood vessel damage and reduced blood flow |
| Cerebral damage | Diffuse cerebral atrophy, particularly in the posterior temporal hippocampus and parietal areas | Widespread Lewy body pathology affecting cortex, limbic regions, and brainstem; variable cortical atrophy | Severe atrophy of the frontal and anterior temporal lobes | Atrophy in subcortical regions and cortical Lewy body pathology | Neuronal loss in caudate nucleus and putamen | Combination of Alzheimer’s pathology and vascular lesions |
| Prominent symptom | Memory dysfunction | Fluctuating cognition, visual hallucinations, and parkinsonism (core clinical triad) | Personality and language disturbances | Impaired attention, executive dysfunction, memory issues | Cognitive decline with behavioral disturbances | Memory loss, cognitive decline, executive dysfunction |
| Motor signs | Less common | Parkinsonian motor features (rigidity, bradykinesia, tremor), usually milder than idiopathic Parkinson’s at onset | More common (in FTD with motor neuron disease). May include tremors, muscle stiffness, muscle spasms, poor coordination, swallowing difficulties, muscle weakness | Frequent; rigidity, bradykinesia, tremors | Chorea, dystonia, impaired voluntary movements | May include vascular-related motor symptoms (gait disturbance, weakness) |
| Visuospatial abilities | Severely impaired | Markedly impaired | Preserved | Moderately impaired | Impaired | Often impaired |
| Language problems | In late stages individuals lose the ability to understand or formulate words in a spoken sentence, or speaking is very hesitant, labored or ungrammatical | Mild word-finding difficulty possible, but language relatively preserved compared to Alzheimer’s and FTD | Trouble thinking of the right word or remembering names; Less difficulty making sense when they speak, understanding the speech of others, or reading | Word-finding difficulties, reduced fluency | Speech becomes slurred, eventual mutism | Variable; may mirror Alzheimer’s type difficulties |
| Mood | Depression, anxiety, suspiciousness | depression, anxiety, apathy as in Alzheimer's, plus anxiety secondary to confusional states | Marked irritability, lack of guilt, alexithymia (difficulties in understanding, processing, or describing emotions), euphoria, apathy | Depression, anxiety, apathy | Depression, irritability, aggression, apathy | Depression, anxiety, apathy |
| Intellectual deficit | Yes | Yes | No | Yes | Yes | Yes |
| Psychotic features | Usually have delusion of misidentification or prejudice secondary to memory impairment type and usually occur in the middle or late stage | Prominent visual hallucinations, delusions | Rare persecutory delusions, usually jealous, somatic, religious and bizarre behaviours | Visual hallucinations, paranoid delusions | Psychosis may occur in later stages | Possible delusions and hallucinations |
| Appetite, dietary change | Less common: anorexia and weight loss | Weight loss may occur | Increased appetite, carbohydrate craving 80%, weight gain | Weight loss more common | Weight loss despite high caloric intake | Variable; can include weight loss or gain |
| Prognosis | Progresses to death in 11.8 ± 0.6 years | Average survival 5–8 years after diagnosis | Progresses to death in 8.7 ± 1.2 years | Average survival 5–10 years after dementia onset | Survival 15–20 years after onset | Variable; progression faster than single dementia types |
| Cause of death | Aspiration pneumonia secondary to swallowing disorders | Aspiration pneumonia, complications of immobility, and infections | Physical changes that can cause skin, urinary tract and/or lung infections | Complications from immobility, aspiration pneumonia | Complications from immobility, infections, aspiration pneumonia | Cardiovascular disease, pneumonia, infections |
Table 2.
Mass spectrometry platforms for glycolipid biomarkers.
| Platform / Workflow | Key Features | Advantages | Limitations | Typical Use Cases |
|---|---|---|---|---|
| LC MS/MS with Orbitrap/quadrupol time of flight (QTOF) | Chromatographic separation of glycolipids; high-resolution MS/MS for structural information | Quantitative, robust, reduces isobaric interference; structural info on fatty acyl chains; sensitive for cerebrospinal fluid(CSF)/plasma; suitable for both discovery (untargeted) and validation (targeted selected reaction monitoring, SRM) | Requires optimized chromatography for polar glycans; some isomers need derivatization or specialized columns | Discovery (by untargeted high resolution LC MS) and validation (by targeted SRM) of candidate glycolipids |
| Shotgun Lipidomics (Direct infusion, Orbitrap/triple-TOF) | Rapid, high-throughput profiling without LC | Broad coverage; quick surveys; minimal preparation steps | Ion suppression; poor isomer/isobar separation; less quantitative | Initial screening before LC-MS validation |
| MALDI MSI | Spatial mapping of glycolipids on tissue; moderate to high resolution | Links molecular and anatomical information; enables regional distribution analysis; improved sensitivity with on-tissue derivatization and high resolution analyzers | Lower absolute quantitation than LC MS; matrix/analyte suppression | Mapping glycolipids in brain tissue; correlating with plaques, vessels, microinfarcts |
| DESI MSI and SIMS Imaging | Ambient ionization (DESI); ultra-high resolution imaging (SIMS) | Minimal sample preparation (DESI); sub-micron resolution (SIMS); powerful when combined with immunohistochemistry (IHC)/ fluorescence |
SIMS historically limited mass range and fragmentation; complex data analysis | Subcellular mapping of glycolipids; complementary spatial lipidomics |
| IMS MS | Separates isomeric/isobaric species by shape/size | Resolves gangliosides and their isomers in native complex and mixtures; improves confidence in structural identification; boosts discovery | Requires specialized instrumentation | Discovery of novel glycolipid biomarkers; detailed structural assignment of glycoforms with or without various modifications |
| Targeted Derivatization and Glycan-Specific Workflows | Chemical modifications (permethylation, sialic acid methylation); specialized columns | Improves chromatographic behavior and MS sensitivity for glycans; resolves isomers | Extra sample preparation; workflow complexity | Quantifying disease-relevant GG isomers |
| Quantitative MSI and LC MS Hybrid | Combines on-tissue MSI with microextraction and LC-MS/MS | Provides spatial localization and validated concentration data; emerging gold standard | Complex, multi-step workflow | Linking tissue glycolipid changes to histopathology; anatomical and quantitative mapping |
Table 3.
Glycolipid classes relevant to mixed dementia.
| Glycolipid Class | Representative Species | Why Relevant to Mixed Dementia | Representative Citation |
|---|---|---|---|
| Gangliosides (mono-/di-/tri-sialo) | GM1 (d18:1/18:0); GM2; GM3 (d18:1/16:0; d18:1/18:0); GD1a; GD1b; GT1b | Abundant in neuronal membranes and synapses; altered sialylation and acyl chain composition reflect membrane degradation, plaque association, and local inflammatory/ degenerative processes; GM3 and GM2 increased in near plaques and in white matter in several MS studies | Wang et al. [67] |
| Sulfatides (sulfated galactocerebrosides) | ST(d18:1/24:0) | Enriched in myelin; early sulfatide loss associated with AD and with white matter/myelin injury in vascular disease; sensitive to myelin degradation in mixed pathology | Zimmer et al. [229] |
| Galactosylceramides / Glucosylceramides (GalCer, GlcCer) | GalCer(d18:1/24:0); GlcCer species | Core myelin glycosphingolipids; shifts indicate demyelination and altered glycosphingolipid metabolism in ischemic white matter and mixed dementia | Reza et al. [230] |
| Ceramides (bioactive sphingolipids) | Cer(d18:1/16:0); Cer(d18:1/24:1) | Products of sphingomyelin breakdown; elevated ceramides associate with neurodegeneration, inflammation and vascular risk; link cell stress to apoptosis and promote Aβ production | Pujol-Lereis et al. [231] |
| Sphingomyelins (SM) | SM(d18:1/18:0); SM(36:1) | Structural membrane lipids; sphingomyelin/ceramide ratio changes indicate membrane changes and myelin injury; altered in mixed dementia tissue studies | Koal et al. [232] |
| Ganglioside Degradation Intermediates | GM2; lactosylceramides (LacCer) | Reflect increased glycosidase activity and disrupted catabolism around plaques and ischemic zones; accumulation indicates lysosomal/ autophagy perturbation common to mixed dementia |
Wang et al. [67] |
| Glycolipid Oxidation & Truncated Forms | Oxidized ceramides; truncated ganglioside species | Markers of oxidative stress and lipid peroxidation from ischemia/inflammation; likely elevated in tissue adjacent to microinfarcts and plaques | Montine et al. [233] |
Table 4.
Overview on GSL expression in various types of dementia.
| Type of dementia | Type | Sample | MS Platform | Glycolipid Findings | Study / Year – |
| AD | APP/PS1 transgenic mice | Brain tissue | MALDI MSI | ↓ShexCers and CroPIn in cerebral cortex, hippocampus and cerebellum | Zhang et al. 2024 [87] |
| APP/PS1 transgenic mice | Brain tissue | MALDI MSI | GM1 and GD1a modificationin white and grey matter ↑GM2 and GM3 in cortex and dentate gyrus, GM3 in Aβ plaques |
Wang et al. 2025 [67] | |
| APP/PS1 transgenic mice | Brain tissue | MALDI-IHC MSI | ↑GD3 and GD2 in hippocampal plaques, GM3 (38:1);O2 in cortical plaques GM3 (36:1);O2 and GM2 (36:1);O2 plaque-defining GGs |
Good et al. 2025 [96] | |
| APP21 transgenic Fischer rats | Brain tissue | MALDI MSI | ↑GM1, GM2, GM3, especially GM3 d18:1, d20:1/d18:1 ratio ↓complex GGs |
Caughlin et al. 2018 [94] | |
| TgAPPArcSwetransgenic mice | Brain tissue | TOF SIMS + MALDI MSI | ↑GM3 (C18:0) and GM2 (C18:0) inAβ plaque-like structures ↓[ST d18:1/24:0-H]⁻ and [ST d18:1/22:0-H]⁻ |
Michno et al. 2019 [79] | |
| APPswe/PS1dE9 (APP/PS1) | Hippocampal tissue | UHPLC-MS/MS | ↑TGs, CEs,PEs, PCs ↓MGDGs, HexCers |
Xiao et al. 2025 [69] | |
| APPswe/PS1dE9 | Hippocampal tissue | LC | di-O-Ac-GT1a (d36:1), O-Ac-GD1b (d36:1) and O-Ac-GD1b (d36:0) – biomarkers O-Ac-GT1a (d36:2) non-progressive biomarker |
Li et al. 2022 [91] | |
| 5xFAD mouse | Brain tissue | MALDI IMS | ↑GMs C18:1 in the subgranular zone of the dentate gyrus, GMs C20:1 in the molecular layer along the entorhinal hippocampal pathway Co-localized GM3 (d18:1/18:0), GM2 (d18:1/18:0), GM1 (d18:1/18:0), with Aβ peptides in the subiculum |
Kaya et al. 2020 [95] | |
| Dual injured mice (Aβ+stroke), stroke alone and Aβ alone | Brain tissue | MALDI IMS |
|
Caughlin et al. 2015 [97] | |
| AD patients | Hippocampal tissue | MALDI IMS | Loss of GM1 (d20:1/C18:0) at the edge of the dentate gyrus ↓GM1 (d20:1/C18:0) to GM1 (d18:1/C18:0) ratio in the outer molecular layer of the dentate gyrus No differences in other hippocampal subregions or in total hippocampal lipid content |
Hirano-Sakamaki et al. 2015 [73] | |
| AD patients | Serum | LC-MS | ↑CE (16:3) and GM3 (d18:1/9Z-18:1) – early clinical prediction and severity correlation | Zhang et al. 2021[89] | |
| AD patients | Brain tissue | MALDI MSI | GM3 and GM1 co-localizes with Aβ plaques ↓GM1 d20:1 to GM1 d18:1 in the molecular leyer, dentate gyrus and entorhinal cortex |
Ollen-Bittle et al. 2024 [86] | |
| AD patients | Brain tissue | TLC Blot/MALDI TOF MS | ↑GGs containing d18:1 compared to PD and control ↓GD1b and GT1b compared to PD and control |
Taki et al. 2012 [93] | |
| AD, VD and control patients | Serum | HPTLC + ELISA | ↑-GM1, -GD1b, -GT1b, -GQ1b, and anti-GQ1bα IgM type antibodies in AD and VD ↑ -GQ1b, -GQ1bα and anti-GT1b IgGtypeantibodies in AD |
Ariga et al. 2013 [70] | |
| DBL | Clinically diagnosed DLB | Plasma | untargeted UPLC–MS lipidomics + ML feature selection | sphingoid bases, ceramides, Hex1Cer differentially expressed in DLB vs controls and vs AD | Shen et al.2024 [53] |
| Autopsy-confirmed LB pathology | Plasma collected ~2 years before death | targeted LC-MS/MS | ↑HexCer/ceramide in both LB and AD groups; no significant difference between DLB and AD | Savica et al. 2016 [121] | |
| GBA1 variant and wild-type cohorts | CSF | targeted LC-MS/MS | no clear increase of Cer (d18:1/18:0), GlcCer (d18:1/18:0), SphM (d18:1/18:0), GlcSph (d18:1) and GalSph (d18:1); ↓GalSph and Cer vs controls/PD; no clear difference between GBA1 carriers and wild-type |
Lerche et al. 2024 [122] | |
| PDD | Clinically diagnosed Parkinson’s disease with dementia | Plasma | Targeted LC–MS/MS | ↑ Plasma ceramides&glucosylceramides; higher levels associated with cognitive impairment | Mielke et al. 2013 [137] |
| Clinically diagnosed Parkinson’s disease with dementia | Plasma | Targeted HPLC–MS/MS | C24:1 negatively correlated with immediate/delayed recall; C14:0 with delayed recall/recognition; C22:0 with hallucinations, C20:0 with anxiety, C18:0 with sleep disturbances | Xing et al. 2016 [138] | |
| Longitudinally followed PD patients without baseline dementia | Serum | Untargeted LC–QTOF–MS lipidomics with multivariate models | ↑ 24 ceramides, 24 diacylglycerols, 17 triacylglycerols; ↓ phosphatidylcholines, bis(monoacyl)glycerophosphates, phosphatidylserines; 5-lipid panel predicted dementia with >95% accuracy | ZardiniBuzatto et al. 2021 [131] | |
| FTD | GRN-mutation | Cells & human brain | MS–based lipidomics | ↑GM1, GD3, GD1 ↓ BMP levels (progranulin deficiency → BMP loss → GG accumulation) |
Boland et al. 2022 [181] |
| Pick presenile dementia | Brain tissue | TLC | ↑GT1a and/or GD2 ↓GalNAc-GDla |
Kamp et al. 1986 [182] | |
| bvFTD |
Plasma | untargeted LC-MS | largely unchangedsphingolipid profile | Kim et al. 2018 [183] | |
| GRN/C9orf72 FTD Subtypes | Brain tissue | LC-MS lipidomics+ enzymatic assays | ↓myelin sphingolipids; FTD-GRN shows more severe loss; consistent with MRI | Marian et al. 2023 [184] | |
| Familial bvFTD | Plasma | LC-MS lipidomics | ↓HexCers, especially C22:0 GlcCer and GalCer; reductions correlate with MRI measures of white matter damage and disease duration | Marian et al. 2025 [162] | |
| GRN-FTD Subtype | Frontal gyrus tissue | targeted HPLC-MS and biochemical assays | ↓mature GCase protein; accumulation of improperly processed forms; reduced GCase activity. No overt accumulation of GCase substrates (GlcCer, GlcSph) in examined regions. |
Arrant et al. 2019 [185] | |
| Not specified | Superior frontal cortex | quantitative discovery LC-MS lipidomics | ↑ VLCFA-lipid species ↑ ELOVL4 enzyme |
He et al. 2021 [186] | |
| HD | Clinically diagnosed Huntington’s disease | Plasma & CSF | Untargeted UHPLC-MS metabolomics | Altered ceramides, hexosylceramides, sphingomyelins, phosphatidylcholines; correlated with Stroop and Verbal Fluency | McGarry et al. 2020 [192] |
| Clinically diagnosed Huntington’s disease | Post-mortem brain (caudate, cortex) | MALDI-IMS | Regional sphingolipid and phospholipid dysregulation; linked to executive and memory circuits | Hunter et al. 2021 [194] | |
| Advanced clinical cases of Huntington’s disease | Post-mortem caudate | LC-MS | Ceramide/sphingomyelin chain-length shift: loss of very-long-chain, enrichment of long-chain species | Phillips et al. 2021 [195] | |
| Huntington’s disease patient-derived cells and animal models | Cultured cells and mouse brain tissue | Targeted LC-MS/MS gangliosides | ↓ GM1 ganglioside; supplementation with exogenous GM1 improved motor and cognitive phenotypes | Maglione et al. 2010 [197]; Di Pardo et al. 2012 [36]; Alpaugh et al. 2017 [198] | |
| Mixed dementia | AD + vascular pathology | Brain tissue | lipid pathway / histochemical analyses (study did not report MS platform in file) | ↑GM2 and GM3 within and around amyloid plaques; increased GG degradation pathway activity in plaque regions | Wang et al. 2025 [67] |
| SIVD, mixed dementia | White and gray matter from temporal cortex | comparative LC-MS lipidomic | ↑GM3 and markers of membrane breakdown in mixed dementia white matter; pronounced alterations in sphingolipid, ceramides, SM, GlcCer and GalCer classes | Lam et al. 2014 [225] | |
| VD | Plasma | LC-MS/MS | ↓sphingolipid d16:1 in VaD; ↑sphingolipid d18:1 in AD; Cer d16:1/24:0, Cer d18:1/16:0, Hex2Cer d16:1/16:0, HexCer d18:1/18:0, SM d16:1/16:0, SM d16:1/20:0, SM d18:2/22:0 - higher sensitivity and specificity for classifying VaD. |
Chua et al. 2023 [228] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



