
Submitted:
11 October 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
The only cytogenetic alteration defining a subtype of a myelodysplastic syndrome is represented by the deletion of the long arm of chromosome 5 (del(5q)), now classified as MDS with isolated del(5q). This subtype is associated with a peculiar phenotype mainly dependent by the haploinsufficiency of several genes located on the deleted arm of chromosome 5. These patients show a good prognosis and respond to treatment with lenalidomide, but some cases progress to acute myeloid leukemia. Molecular studies have in part elucidated th heterogeneity of MDS with isolated del(5q) mainly related to the association with different co-mutations that may affect leukemic transformation and survival.
Keywords:
leukemia
; myelodysplasia
; chromosome abnormalities
; karyotype
; mutational profile
; genome analysis
; molecular classification
1. Introduction
The discovery of 5q deletion associated with mDS was related to the characterization of three patients displaying a similar chromosomal abnormality in bone marrow and a similar hematological phenotype: the chromosomal abnormality consisted in a deletion at the level of the long arm of one chromosome 5; the hematological phenotype consisted in a macrocytic anemia refractory to conventional treatments, associated with normal or increased platelet counts and normal or decreased white blood cell count [1].
After this initial report, this hematological disorder associated with 5q chromosomal abnormality was observed in other patients and was defined as the 5q- syndrome. In the 2001 World Health Organization (WHO) classification of myeloid neoplasms this disorder was classified as a subtype of mDS and was termed MDS with isolated deletion 5q [2].
The diagnostic criteria for MDS associates with 5q deletion have been subsequently up dated in the more recent classifications of myeloid neoplasms [3,4]. 5q deletion is observed in MDS, not only as an isolated abnormality, but also in the context of other chromosomal abnormalities (complex karyotype, CK) and frequently in association with TP53 mutations. This review analyzes the recent developments in the understanding the pathogenesis and the prognostic impact of the different MDS subtypes bearing del(5q).
2. 5q Deletion in MDS
5q deletion represents one of the most frequent chromosomal abnormalities observed in MDS. MDS with 5q deletion can be subdivided into two groups: a group in which 5q deletion is either the sole cytogenetic abnormality or with one additional cytogenetic abnormality excluding -7/del(7q); a group in which del(5q) is associated with other cytogenetic abnormalities in the context of a complex karyotype (CK). The distinction of these two groups of MDS with del(5q) is fundamental both at biological and clinical level. These two groups of MDS correspond to 2 of the 16 molecular subgroups identified for their unique molecular profiles, clinical phenotypes and disease courses [5].
The del(5q) group of MDS with isolated 5q abnormality is usually characterized at the level of hematological phenotype by low bone marrow blast cell counts and lowered Hb levels (lower than in the rest of MDSs) and with increased or normal platelet levels (higher than in the rest of MDS); these MDSs are predominantly observed in females (75% of cases) and are associated with a favorable OS compared to the rest of MDS [5]. 84% of these patients had at least 1 gene mutation, the most frequent being SF3B1 (22%), DNMT3A (21%), TET2 (17%), ASXL1 (14%) and monoallelic TP53 mutations (13%) [5]. Interestingly, the mutations of three genes CSNK1A1 (10%), IRF1 (5.4%), RAD50 (4.1%) and NFE2 (3.6%), located on 5q, are enriched in del(5q) MDS [5]. Importantly, the study of variant allele frequency (VAF) of the additional mutations SF3B1, DNMT3A, TET2 and ASXl1 suggest that these are events secondary to del(5q) [5].
Meggendorfer et al explored a group of 123 MDS patients with isolated del(5q) for the presence of mutations using a panel of 27 genes frequently mutated in MDS and showed that about 40% of patients had no mutations and about 50% one mutation [6]. The genes most frequently mutated were SF3B1 (19%), DNMT3A (18%), TP53 (18%), TET2 (12%), CSNK1A1 (12%), ASXL1 (6%) and JAK2 (6%) [6]. The comparison of the mutation profile of various MDS subtypes showed that the mutation profile of MDS-del(5q) and MDS non-del(5q), TP53 mutations were more frequent in MDS with isolated del(5q) compared to the rest of MDS (18% vs 6%, respectively) [6]. In these patients, the presence of SF3B1 mutations correlated with the presence of ring sideroblasts, while JAK2 mutations correlated with elevated platelet counts [6]. At the level of the effect of mutations on prognosis, the presence of SF3B1 mutations was associated with shorter survival compared to patients without SF3B1; however, the survival of SF3B1-mutant MDS with isolated del(5q) was similar to that observed in SF3B1-mutant MDS without del(5q) [6].
A part of MDS patients with del(5q) display TP53 mutations. Montoro et al. analyzed a group of 628 patients with isolated del(5q); 18.9% of these patients displayed TP53 mutations: 24% of these patients with multi-hit TP53 alterations and 76% with mono-hit alterations [7]. Although the mOS was similar in TP53-WT and TP53-mut 5q-deleted MDS, TP53 multi-hit alterations and TP53 mono-hit alterations with VAF>20% were predictive of an increased risk of leukemia transformation [7]. Thus, the impact of VSF on monoallelic TP53 alterations was evident: VAF <20% displayed a behavior similar to TP53-WT and those with VAF >20% showed a behavior similar to multi-hit TP53 alterations [6]. Finally, MDS del(5q) with monoallelic TP53 alterations with VAF >20% displayed a significantly reduced mOS compared to MDS del(5q) TP53-WT [7]. The co-mutation profile of MDS del(5q) TP53-WT and TP53-mutated was similar [7].
Xie et al. evaluated the clinical correlation and the prognostic impact of cytogenetic clone size in MDs patients; their study included a cohort of 1001 MDS patients, comprising also 144 patients with isolated del(5q) [8]. In these patients, clone size exhibited a significant negative correlation with hemoglobin levels and a positive correlation between clone size and platelet levels [8]. Among the various MDS cytogenetic groups, those with isolated MDS del(5q) had the longest mOS (65.9 months) and mLFS (58.4 months) [8]. The observation that clonal sizes are associated with increased anemia and thrombocytosis support the view that clonal expansion contributes to the development of these disease manifestations [8].
The other group of del(5q) MDS is associated with TP53-complex MDS and CK MDS; the TP53-complex and CK MDS in large part overlap [9]. The TP53-complex MDS group comprises about 10% of total MDS; in this group there are 74% multi-hit TP53 and 26% mono-hit TP53; 91% of multi-hit TP53 MDS had CK [9]. CK group can be subdivided into two subgroups: one with TP53 mutations and with del(5q) in 87% of cases and the other without TP53 mutations and with del(5q) in 32% of cases [9]. The presence or not of TP53 mutations subdivides MDS with CK into two prognostic subgroups [9].
Figure 1.
Cytogenetic and mutational profile of MDS-del(5q). Top, left panel: Frequency of MDS-del(5q) as isolated del(5q) abnormality or in the context of a complex karyotype. Top, right panel: frequency of TP53 mutations in MDS-del(5q), analyzed as total, with isolated del(5q) or del(5q) in the context of CK. Middle panel: frequency of mono-hit and multi-hit TP53 alterations among MDS with isolated del(5q) or in the context of CK. Bottom panel: most recurrent mutations in MDS with isolated del(5q) or in the context of CK.
Figure 1.
Cytogenetic and mutational profile of MDS-del(5q). Top, left panel: Frequency of MDS-del(5q) as isolated del(5q) abnormality or in the context of a complex karyotype. Top, right panel: frequency of TP53 mutations in MDS-del(5q), analyzed as total, with isolated del(5q) or del(5q) in the context of CK. Middle panel: frequency of mono-hit and multi-hit TP53 alterations among MDS with isolated del(5q) or in the context of CK. Bottom panel: most recurrent mutations in MDS with isolated del(5q) or in the context of CK.
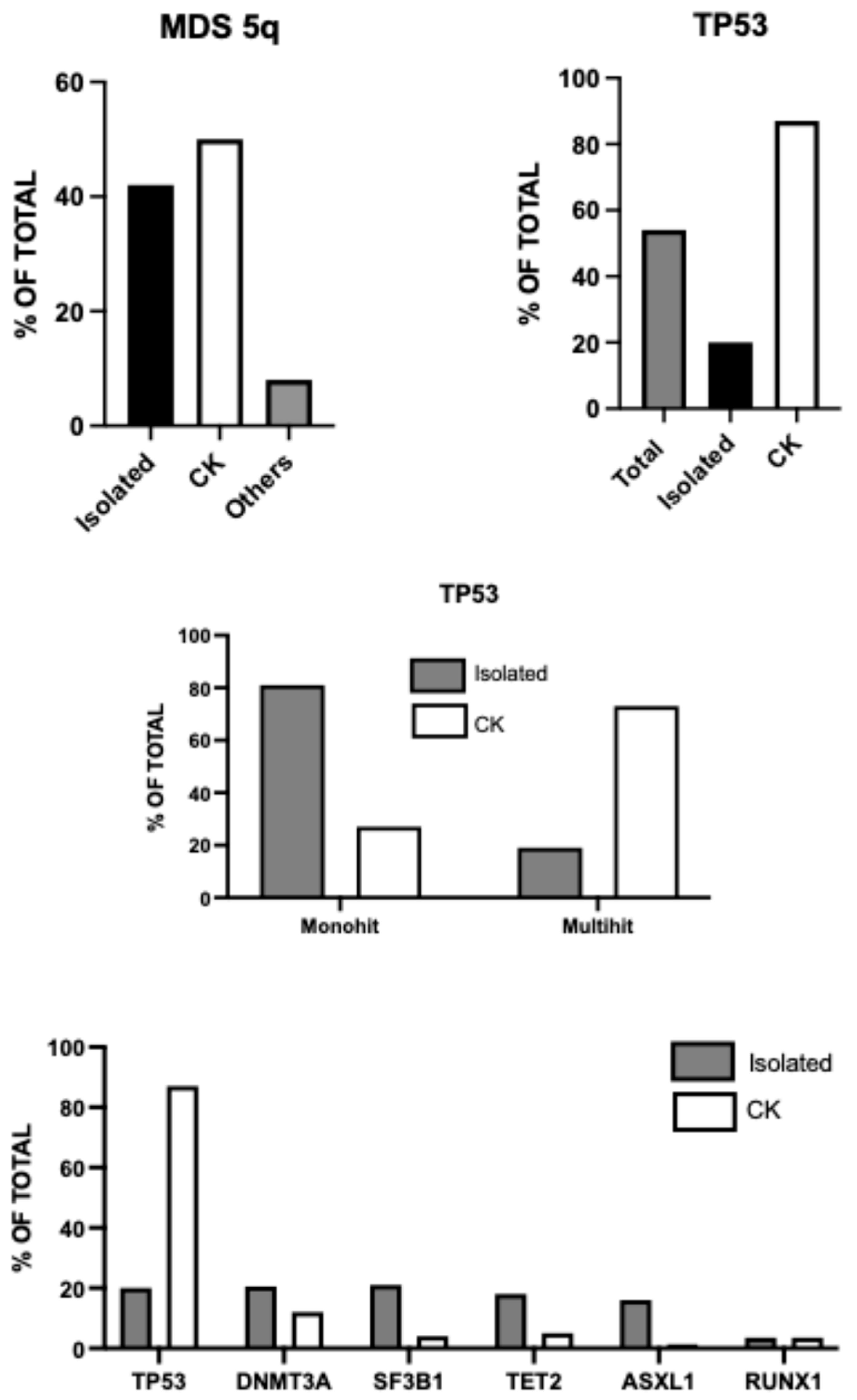
Huber and coworkers have analyzed a group of 789 MDS harboring del(5q); these MDS patients were extensively characterized at molecular level and 42% had isolated del(5q), 50% had CK and 8% was not classified as CK or MDS del(5q) [10]. TP53 mutations were detected in 54% of cases, with a higher frequency among patients with CK compared to those without CK (87% vs 20%, respectively) and were also more commonly multi-hit among MDS-CK compared to MDS del(5q) (81% vs 27%, respectively) [10]. Interestingly, the analysis of clonal hierarchy of del(5q) and TP53 mutations showed that the VAF of del(5q) was similar in both MDS del(5q) and MDS-CK, while the VAF of TP53 mutations was lower in MDS del(5q) than in MDS-CK [10]. These observations support a different hierarchical clonal origin of the two groups of MDS harboring del(5q): in MDS del(5q) the ancestral event is predominantly del (5q), while in MDS-CK is prevailingly a TP53 mutation [10]. However, in a consistent number of cases it is impossible to determine a hierarchy due to the similarity of VAF of del(5q) and TP53 mutations. In this study, a subset of 84 patients with MDS-del(5q) was analyzed in their evolution in the time with a median follow-up of 2.8 months. 4% of patients displayed a normal karyotype following transplantation or lenalidomide therapy; 4% of patients displayed clonal evolution with acquisition of one addition chromosomal abnormality; 24% developed CK (with a median time to progression of 2.8 years for patients receiving lenalidomide treatment and 1.1 years without this treatment) and 69% showed a stable karyotype [10]. The acquisition of TP53 mutations was more frequent among patients who developed CK compared with those who retained a stable karyotype (65% vs 38%, respectively) [10].
3. Classification of MDS Associated with del(5q)
As above discussed, MSA associated with 5q deletion must be subdivided into two different groups: one in which del(5q) ia an isolated chromosomal abnormality and a second in which del(5q) is associated with other chromosomal abnormalities in the context of complex karyotype.
According to the current ICC and WHO classifications of MDS with isolated del(5q), the MDSs are defined not only according to the presence of an isolated del(5q) chromosomal abnormality and the absence of chromosome 7 abnormalities or CK, but also by a number of blasts <5% [3,4].
Figure 2.
Main diagnostic criteria for MDS with isolated del(5q).
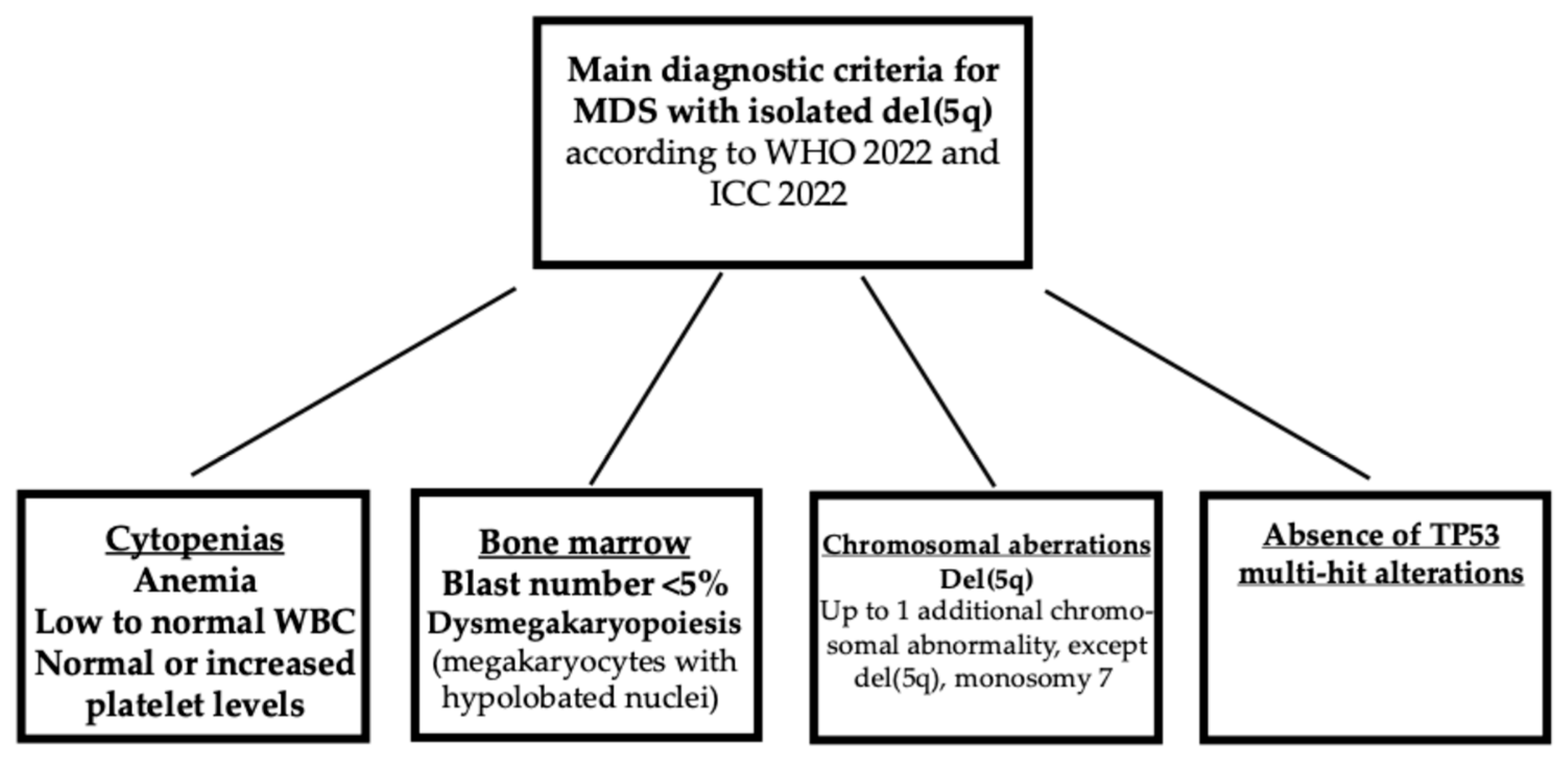
The taxonomy study showed that 22% of patients classified as del(5q) displayed a number of bone marrow blasts of >5% and should be excluded from this group according to ICC and WHO classifications [3,4]. However, the molecular taxonomy study proposes new criteria, where the driving and characterizing genetic alterations characterize a MDS group and the blast frequency represents a measure to evaluate the extent of disease stages between different molecular groups and within the same molecular group [5]. Thus, according to this view, MDS-del(5q) with blasts <5% represents an earlier stage and MDS-del(5q) with blasts between 5 and 19% a more advanced stage of disease evolution [5]. In this group of patients, mOS was inversely correlated with blasts cell counts in that MDS patients with del(5q) with 5-10% or >10% bone marrow blasts have a significantly shorter mOSA compared to those with <5% of blasts [5]. These observations are in line with a study by Kewan et al. proposing a classification of MDS based on molecular patterns identified using a machine learning approach; this approach identified 14 molecular distinct clusters and was not dependent on BM blast cell counts [11]. The observations made in many of some of these clusters showed a consistent intra-cluster heterogeneity of BM blast cell numbers, seemingly reflecting the stage of the disease rather than the molecular architecture [11].
Approximately 20% of MDS-del(5q) eventually progress to AML. Since the criteria of MDS-del(5q) of WHO 2022 and ICC2022 exclude the presence of >5% of BM blasts and of adverse cytogenetics, current prognostic scoring systems such the Revised International Prognostic System (IPSS-R) and its molecular counterpart (IPSS-M), fail to efficiently stratify the risk of MDS with isolated del(5q) [12,13]. The new score system for MDS-del(5q) was built as a weighed sum of six prognostic variables, including hemoglobin, sex, number of mutations, SF3B1 mutations and TP53 multi-hit/TP53 mono-hit with VAF >20% [14]. Using IPSS-del(5q) score, a group of 682 MDS-del(5q) patients was stratified as low/very-low risk (51.6%, with an EFS of 78.2 months), intermediate (31.3% with an EFS of 45.1 months) and high-risk (17.1%, with an EFS of 28.2 months) [14].
The International Consortium for Myelodysplastic Syndromes proposed a harmonized classification system for MDS, representing an evolution of WHO 2022 and ICC 2022 [14]. The new classification identified nine clusters with different genomic features. One of these clusters was represented by MDS with isolated del(5q). The criteria for appurtenance to this cluster were represented by the presence of del(5q), absence of -7/del(7q) or CK, absence of biallelic TP53 mutation, bone marrow blasts <5% [15]. It is important to note that 88% of MDS patients clusterized according to the above-mentioned criteria, excluding blast cell number, have bone marrow blasts <5%; the remaining 12% have blood number >5%, but were shifted in other clusters, such as in the cluster characterized by TP53 mutations [15].
The identification, definition and prognostic stratification of the other group opf MDS bearing del(5q) is largely related to the association with CK and TP53 mutations. A fundamental study by Bernard et al. reported the molecular analysis of 3234 MDS patients, including 3787 TP53 mutated patients: 72.5% with single mutation, 26.5% with two mutations and 1% with three mutations; allelic imbalance (due to focal deletions or regions of cnLOH) were observed in 178 patients [16]. According to the combination of mutations and allelic imbalances four groups of patients were observed: Monoallelic mutation (33% of total TP53-mutated), multiple mutations without deletion of cnLOH affecting the TP53 locus (24%); mutations and concomitant deletions (22%); mutations and concomitant cnLOH (21%) [16]. In subgroups 2 to 4 of patients there was multi-hit TP53 state (67%), while mono-hit TP53 state was observed in group 1 (33%); mono-hit cases were enriched in subclonal distribution (mVAF 13%), while multi-hit was predominantly clonal (mVAF 33%) [16]. Associations with complex karyotype, few co-occurring mutations, high-risk presentation and poor outcomes were specific to multi-hit patients; multi-hit status predicted a high risk of leukemic transformation [16]. Monoallelic TP53 MDS patients did not differ significantly from TP53-WT patients [16].
Stengel et al. have analyzed the interplay of TP53 allelic state, blast count, and complex karyotype on outcomes of MDS and AML patients [17[. This analysis showed that TP53-mutant MDS patients can be subdivided into three groups according to blast cell counts: <5% (24% with multi-hit TP53 alterations), 5-10% (67% multi-hit TP53 alterations) and 5-19% (91% multi-hit TP53 alterations, with a mOS of 17, 10 and 8 months, respectively [17]. In patients with single-hit TP53 alterations, the presence of CK considerably worsened mOS (46 months without CK vs 14 months with CK) [17]. A more recent analysis showed that karyotypic clonal fraction (evaluated as low ≤50% clonal cells and high with ≥50% clonal cells) and the presence of CK are determinant factors for predicting adverse outcome of TP53-mutant MDS [18].
Shah et al. reported the analysis of 580 myeloid neoplasia patients (mostly MDS) and explored the impact of VAF percentage, TP53 hit status, blast percentage and cytogenetic features on outcome [19]. Hierarchical analysis identified 4 risk groups with different survival: MDS-LB (low blast); MDS-EB1-EB2/AML VAF <10%; MDS-EB1-EB2 >10%; AML VAF >10% [19]. The significance of biallelic TP53 status was limited to MDS <5% of blasts and not extended to those with higher blast cell percentages; MDS-EB1 and -EB2 with VAF >10% had comparable survival; MDS EB-1 and EB-2 with VAF<10% and CK had a poor survival compared to those without CK and comparable to that observed for MDS EB-1 or EB-2 with VAF >10% [19]. The frequency of MDS-LB and monoallelic TP53 alterations with CK is markedly higher in cases with VAF >10% compared to those with VAF <10% [19].
4. Molecular Pathogenesis of MDS-del(5q)
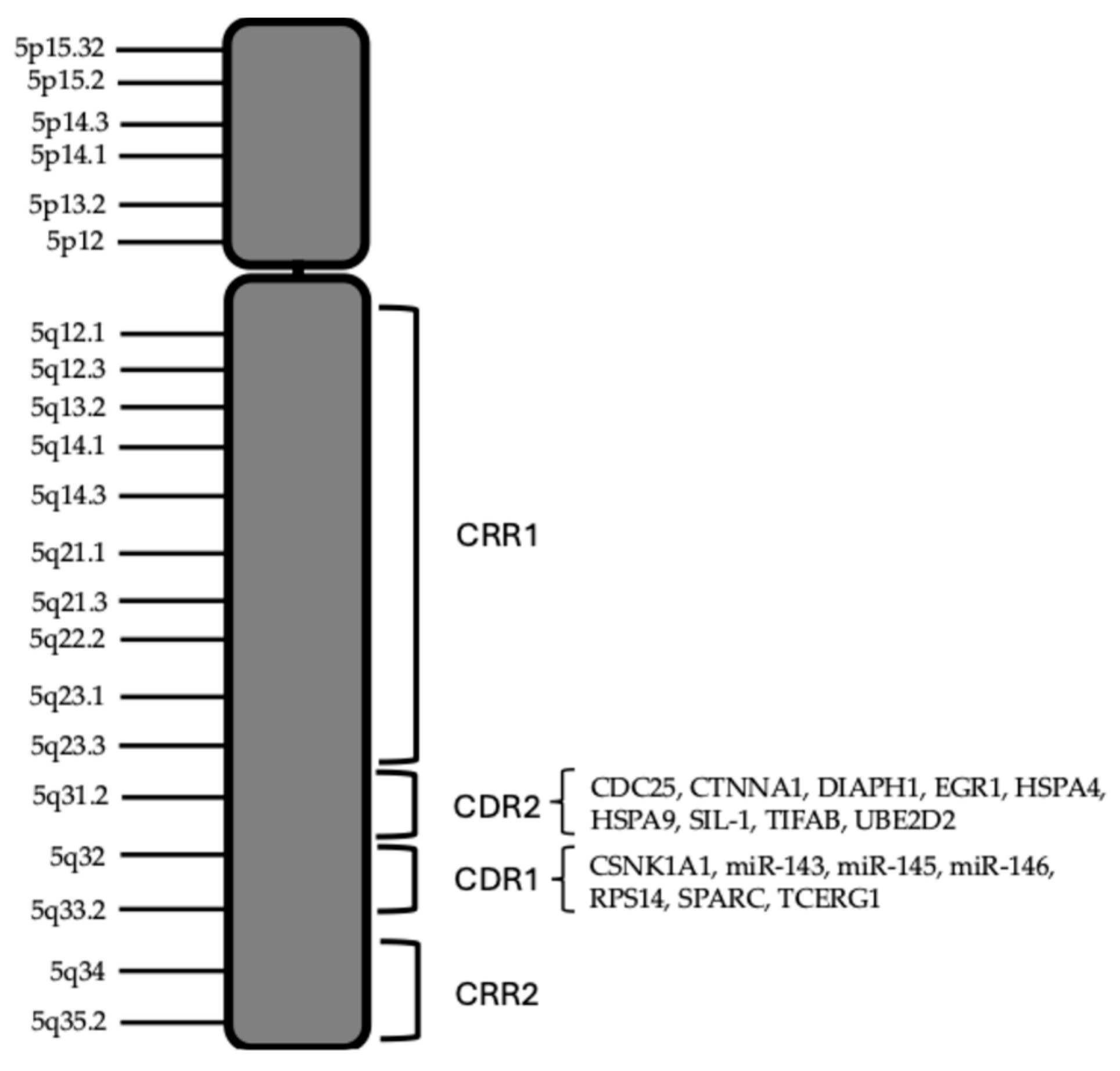
The molecular pathogenesis of MDS-del(5q) is mainly related to the regions of the long arm of chromosomes 5 deleted. Boultwood and coworkers identified two commonly deleted regions (CDR): a distal 1.5 Mb deletion encompassing 5q32-5q33 (CDR1), associated with MDS-del(5q) and a better prognosis; a proximal CDR at 5q31 (CDR2), associated with other types of MDS, AML, with CK and with worse prognosis [20,21,22]. It was estimated that the proximal CDR contains about 30 genes, while distal CDR contains about 41 genes: CNV studies indicate 405 genes in all 5q region, 41 in proximal CDR and 55 distal CDR. G-banding analyses, combined with FISH studies, showed that most of the deletions were large, extending from 5q13 to 5q33; the detailed characterization of 16 MDS-del(5q) patients showed a deletion of both CDRs in 15 of the 16 cases reported [21]. The study carried out by Jerez et al. showed a relevant role in MDS-del(5q) of some regions of 5q that are retained in the deletion process, called commonly retained regions (CRR): CRR1 for the proximal region, encompassing 81.7 Mb and ending at band 5q 14.2; CRR2 for the distal region (5q34) [23]. CRRs are not observed in other MDS subtypes or AML with chromosome 5 deletions. Non-isolated del(5q) MDS showed NPM1/5q 35.1 monoallelic loss in 42.5% of cases versus 2.3% in isolated MDS-del(5q); gross chromosome abnormalities and monosomies, as observed in high-risk MDS, were significantly related to NPM1 haploinsufficiency [24]. In non-isolated del(5q) MDS, centromeric breakpoints were significantly more frequently proximal to 5q14 than in cases of isolated del(5q) [25].
Figure 3.
Schematic representation of human chromosome 5. The CRRs and CDRs present in the 5q arm are outlined. The main functionally relevant genes located at the level of proximal and distal, CDRs are listed.
Figure 3.
Schematic representation of human chromosome 5. The CRRs and CDRs present in the 5q arm are outlined. The main functionally relevant genes located at the level of proximal and distal, CDRs are listed.
Chromosome analysis of the AML cases with isolated del(5q) showed a heterogeneous distribution of breakpoints involving del(5q); all AML cases with isolated del(5q) had deletions involving both CDRs at 5q31 and 5q33, 7/12 cases of AML with isolated del(5q) had deletions extending into the distal CRR beyond 5q33 [24]. The most common breakpoint in AML with del(5q) is del(5)(q22q35) [26]. Interestingly, in AML also, there are two groups of AMLs with del(5q) with distinct biologic features: one group with isolated del(5q), associated with frequent IDH1 or IDH2 mutations and with less frequent TP53 mutations; a second group with del(5q) observed in the context of CK and characterized by frequent TP53 mutations [26].
The type of structural abnormalities of del(5q) is different in MDS with isolated del(5q) and in MDS with del(5q) associated with CK. These differences are related to the size of 5q deleted regions and to the eventual translocation of the deleted fragment of chromosome 5. Thus, in MDs patients the size of the 5q deleted region was associated with the presence of TP53 mutations and of additional chromosomal alterations [27].
The analysis of structural abnormalities of 5q chromosome in MDS and AML patients showed two types of abnormalities: interstitial deletions and 5q loss due to unbalanced rearrangements. In unbalanced rearrangements partes of deleted chromosome 5q were fragmented and inserted elsewhere in the genome, the most recurrent partners being chromosomes 17, 3, 7 and 18 [28]. The unbalanced translocation der(5,17) involving chromosome 5q and 17 is a recurrent aberration in MDS and AML, resulting in TP53 loss; no fusion genes resulted from the unbalanced translocation [29]. This translocation is frequently observed in MDS-CK and AML-CK [29].
The analysis of a large cohort of MDS and AML patients with 5q deletions allowed to evaluate the frequency of the loss of the long arm of chromosome 5 due to unbalanced rearrangements [30]. Unbalanced rearrangements occurred more frequently in AML than in MDS (45.6% vs 32.8%, respectively) and were more frequent in MDS-CK than in MDS with isolated del(5q) (95.2% vs 32.2%, respectively) [31]. Furthermore, chromosome 5 unbalanced rearrangements were associated with TP53 mutations and shorter OS [30].
The pathogenesis of MDS-del(5q) seems to be related to a condition of haploinsufficiency involving the genes located in the chromosome 5q deleted regions. In line with this hypothesis, gene expression analysis of CD34+ cells derived from MDS-del(5q) patients showed that most of the genes located at 5q32-q33 (CDR1) displayed reduced expression levels [31].
Haploinsufficiency (HI) resulting from deletion of regions of the long arm of chromosome 5 and the accompanied loss of heterozygosity (LOH) are key pathogenic events in MDS-del(5q). Adema et al. analyzed genomic profiles at the level of gene alterations and gene expression and clinical phenotypes of 388 myeloid neoplasms (mostly MDS) with del(5q) [32]. The analysis of clonal architectures of MDS with deletions of 5q showed that not in all cases del(5q) is a primary hit; in fact, in some MDS, del(5q) is preceded by other mutations, such as TP53 mutations; in other MDS, del(5q) is codominant with TP53 mutations [32]. When del(5q) is dominant, in isolated MDS-del(5q), mutations in CSNK1A1 are the most common secondary hit, while in CK-MDS with del(5q) TP53 alterations are the primary hit [32]. The analysis of the genes involved in the deletion events at the level of 5q allowed to differentiate these genes at functional level into HI-driver genes which provide the support to promote clonal growth and HI-anti-driver genes promoting the phenotypic dysplasia and apoptosis of del(5q) [32]. HI-driver genes are CSNK1A1, CTNNA1 and TCERG1. HI-anti-driver genes include RPS14, HSPA4, SIL-1, UBE2D2, all promoting increased apoptosis of MDS-del(5q) cells. The balance between the effects of HI-driver and HI-anti-driver genes determines the growth features of MDS-del(5q). The growth of MDS-del(5q) is dictated by the capacity of some HI-drivers which provide the triggering for MDS clonal expansion, to enable a selective process to induce mechanisms suitable to overcome the pro-apoptotic effect of HI-anti-driver genes. This selective pressure triggers the acquisition of accelerator events, such as CSNK1A1 mutations, TP53 alterations and/or monosomy 7 [32].
The CDR is rich in genes functionally relevant, homing 40 genes, 33 of which were expressed in HSC/HPC CD34+ cells [33].
The haploinsufficiency of the various genes located in the 5q deleted regions is responsible for the phenotypic features of MDS-del (5q).

Table 1.
Some functionally relevant genes located in the distal and proximal CDRs deleted in MDS-del(5q), involved in the pathogenesis of this myelodysplasia.
Table 1.
Some functionally relevant genes located in the distal and proximal CDRs deleted in MDS-del(5q), involved in the pathogenesis of this myelodysplasia.
| Gene | Location | Biological Activity |
Gene Knockout Hematologic Phenotype |
| CDC25C | Proximal CDR (5q31.2) | It regulates the transition from G2 to the M phase of the cell cycle | CDC25 knockout mice are viable and display co alterations of cell cycle. CDC25 haploinsufficiency confers sensitivity to lenalidomide |
| CTNNA1 | Proximal CDR (5q31.2) | Catenin 1 alpha mediates the anchorage of actin filaments, signal transduction | Growth advantage to HSCs |
| DIAPH1 | Proximal CDR (5q31.3) | Cytoskeleton formation Tumor suppressor |
Development of age-dependent myelo- Proliferation or MDS. |
| EGR1 | Proximal CDR (5q31.2) | Transcription factor | Fitness advantage to HSCs |
| HSPA9 | Proximal CDR (5q31.2) | Control of cell proliferation and response to stress, inhibition of apoptosis | Apoptosis of hematopoietic progenitors. Block of erythroid maturation |
| TIFAB | Proximal CDR (5q31.1) | Inhibition of NF-kB signaling | Deregulation of TRAF6, NF-kB activation in HSCs, induction of ineffective hematopoiesis |
| CSNK1A1 | Distal CDR (5q32) | Serine/threonine kinase involved in multiple cellular processes and pathways | CSNK1A1 haploinsufficiency confers growth advantage to HSCs/HPCs |
| miR-145 | Distal CDR (5q33.1) | It targets various tumor-specific genes | miR-145 and miR-146a loss induces dysmegakaryopoiesis, thrombocytosis and innate immune signaling |
| miR-146a | Distal CDR (5q33.3) | It targets genes involved in the regulation of inflammation and innate immune system | miR-145 and miR-146a loss induces dysmegakaryopoiesis, thrombocytosis and innate immune signaling |
| RPS14 | Distal CDR (5q33) | 40S ribosomal protein | Macrocytic anemia |
| SPARC (Osteonectin) |
Distal CDR (5q32) | Glycoprotein that binds calcium | Thrombocytopenia Anemia (reduced erythroid progenitors) |
4.1. RPS14
Anemia refractory to standard treatments is one of the typical features of MDS-del (5q). The development of anemia in these patients seems to be related to haploinsufficiency and consequent reduced expression of RPS14. The RPS14 gene maps to the chromosome 5q33.1 and encodes a ribosomal protein that is a component of the 40S ribosome subunit. The expression of RPS14 was significantly reduced in CD34+ cells of MDS-del (5q) patients [31]. Ebert and coworkers showed through RNA interference experiments that deficient expression of RPS14 in MDS-del(5q) was responsible for the development of anemia; enforced expression of RPS14 rescues the disease phenotype in patient-derived bone marrow cells [34]. In bone marrow cells of MDS-del(5q) patients and in animal models, the impaired formation of the 40S ribosomal subunit induces an upregulation of the p53 pathway in erythroid cells [35-37].
Haploinsufficiency of RPS14 in patients with MDS-del(5q) syndrome, is associated with deregulated expression of ribosomal and translational-related genes, suggesting that 5q- syndrome represents a disorder of aberrant ribosome biogenesis [38].
Pellagatti et al. showed that haploinsufficiency of RPS14 and deregulation of ribosomal- and translational-related genes were equally observed in MDS with del(5q) associated with CK [39].
RPS14 deficiency in MDS is not limited only to MDS-del(5q) but is observed also in some MDS not associated with del(5q). In fact, a subset of low-risk MDs patients, without 5q deletion and RPS14 gene mutations, display a significantly low expression of RPS14 [39]. In these patients, low RPS14 expression was associated with a better prognosis compared to low-risk MDS patients with normal RPS14 expression [40]. The survival of non-5q MDS patients with low RPS14 expression seems to be improved by immune modulating drugs, thus suggesting that these MDSs may benefit as MDS-del(5q) of lenalidomide administration [41,42]. Adema et al. reported the genomic and expression profile of 995 MDS patients, 170 with MDS-del(5q) and 825 diploids for 5q; as it is expected, MDS-del(5q) patients displayed haploinsufficiency, associated with reduced expression of RPS14 [43]. The non-del(5q) NDS were grouped in 6 different clusters; cluster 2 displayed a normal karyotype, frequent ASXL1 and TET2 mutations, and marked downregulation of RPS14 expression in all the patients included in this cluster [43]. However, an important difference between these two groups of MDS with low RPS14 expression is that only MDS-del(5q) display frequent TP53 alterations and CSNK1A1 mutations [43].
Schneider et al. generated mice with conditional inactivation of RPS14 and showed in these mice a defect in erythroid differentiation dependent upon p53 activation and consisting in apoptosis occurring at transition from polychromatic to orthochromatic erythroblasts [44]. This defect was responsible for the development of a progressive anemic condition, megakaryocyte dysplasia and low of hematopoietic stem cell quiescence [44].
4.2. miR-145 and miR-146a
A typical feature of MDS-del(5q) is represented by normal or increased platelet levels, megakaryocyte hyperplasia associated with small, hypolobated and dysmorphic bone marrow megakaryocytes (many having a plasmocytoid appearance). These features seem to be related to the deletion of two miRNAs, miR-145 and miR-146a located in the 5q region deleted in 5q- syndrome [45]. The study of animal models showed that the loss of miR-145 and miR-146a induces dysmegakaryopoiesis, thrombocytosis and innate immune signaling [45]. Other studies showed that in mouse models the combined loss of miR-146 and RPS14 induces the generation of abnormalities of megakaryocytic differentiation similar to those observed in MDS-del(5q) patients [46].
4.3. CSNK1A1
MDSs are originated from the initial growth and clonal expansion of a hematopoietic stem cell (HSC) which acquired a somatic gene alteration conferring a selective growth advantage over normal HSCs. The study of some genes involved in del(5q) suggests that their haploinsufficiency could contribute to the proliferation and expansion of del(5q)-mutant hematopoietic cells. The reduced expression of the tumor suppressor gene Casein Kinase 1A1 (CSNK1A1) seems to play a relevant role in conferring a growth advantage to del(5q) cells, promoting their clonal expansion. The study of a murine model with conditional inactivation of CSNK1A1 showed that haploinsufficiency induced HSC expansion and a competitive repopulation advantage, whereas homozygous deletion induced HSC failure [47]. CSNK1A1 mutations occurring in the non-deleted allele occur frequently in MDS-del(5q) patients. Smith and coworkers retrospectively analyzed 250 MDS-del(5q) patients and observed that 16% of these patients had CSNK1A1 mutations, all missense mutations and occurring in a region of this protein highly conserved and involved in ATP catalysis; the presence of CSNK1A1 mutations was associated with reduced response to lenalidomide [48]. Heuser et al. reported CSNK1A1 mutations in 7.2% of MDS-del(5q) patients, all missense mutations occurring either at the level of glutamic acid E98 or at the level of aspartic acid D140; the presence of CSNK1A1 mutations was associated with significantly reduced OS compared to MDS-del(5q), CSNK1A1-WT patients [49]. Stolman and coworkers used a genetic barcoding strategy to compare genes implicated in pathogenesis of MDS-del(5q) in direct competition with each other and with WT cells and showed that CSNK1A1 haploinsufficient HSCs expand clonally and competed all other tested genes and combinations [50]. In mouse models, it was observed a cooperation between CSNK1A1 haploinsufficiency and TP53 mutations in promoting clonal advantage and leukemic transformation though MAPK and MYC pathways activation [51].
CSNK1A1 haploinsufficiency in MDS-del(5q) leads to increased platelet counts, while recurrent somatic mutations of CSNK1A1 within the del(5q) CDR in MDS determine a homozygous CSNK1A1 defect with concurrent thrombocytopenia in the affected patients [47]. In line with these observations, a recent study showed that CRISP3/Cas9-mediated genetic ablation of CSNK1A1 in human megakaryocytes resulted in a substantial defect in megakaryocyte maturation and platelet production [52]. These observations support an important biologic role of CSNK1A1 in megakaryocyte differentiation and maturation being required for cytoskeletal dynamics and polarization, proplatelet formation and polyploidization [52].
Mutations in CSNK1A1 have been observed at the level of E98 (E98K) and D140 (D140A) mutants are the two most frequently observed in MDS-del(5q). E98K and D140A mutants have a reduced capacity to promote phosphorylation of beta-catenin, thus inducing enhanced Wnt signaling; furthermore, E98K and D140A mutant displayed enhanced binding to the p53 inhibitor MDMX, increased MDMX-p53 binding and increased suppression of p21 expression [53]. These functional changes induced by CSNK1A1 mutants promote expansion of abnormal myeloid progenitors in MDS-del(5q).
4.4. HSPAP9 and SPARC
The haploinsufficiency of heat shock protein A9 (HSPA9) seems also to contribute to the erythroid maturation defect observed in MDS-del(5q). The HSPA9 gene encodes a protein called mortalin and is located to the 5q31.2 region (proximal CDR); this protein belongs to the HSP70 family and plays a role in various biologic processes, including control of cell proliferation, response to cell stress, inhibition of apoptosis [54]. Knockdown of HSPA9 in human hematopoietic cells significantly delayed the maturation of erythroid precursors, but not of myeloid or megakaryocytic precursors [55]. Knockdown of HSPA9 in a murine bone marrow transplantation model resulted in a decrease in hematopoietic progenitors, including a decrease of erythroid precursors [55]. Other studies have shown that knockdown of HSPA9 in human CD34+ cells induces apoptosis of HPCs via TP53 activation [56]. More recently, it was provided evidence that inhibitors of HPSA9 expression in human CD34+ cells resulted in an increased expression of TP53 in these cells and in a block of erythroid maturation; this block in erythroid maturation was in part inhibited by knockdown of TP53 [56]. These observations suggest that the reduced levels of HSPA9 expression may contribute to the anemia commonly observed in MDS-del(5q) patients [57].
The gene encoding secreted protein, acidic and rich in cysteine (SPARC) is located on human chromosome 5 at the level of 5q32; due to haploinsufficiency, its level of expression is significantly decreased in CD34+ cells of MDS-del(5q) patients [58]. The study of SPARC-null mice showed a hematologic phenotype, characterized by thrombocytopenia and reduced numbers of early erythroid progenitors (BFU-E) [58]. Another study confirmed that SPARC expression is required for the development of erythroid progenitors, but not for erythroid maturation [59].
4.5. CTNNA1
Liu et al have explored the expression in MDS-del(5q) of 12 genes present at the level of CDRs and normally expressed in HSCs; the analysis of the expression of these genes in leukemia-initiating stem cells of MDS-del(5q) patients [60]. Among these genes, the gene encoding α-catenin (CTNNA1), located at 5q32.1, is expressed at markedly lower levels in leukemia-initiating cells from MDS or AML patients with del(5q) than in AML or MDS patients without del(5q) or in normal HSCs [60]. Analysis of the gene promoter of the CTNN1A normal allele in del(5q) leukemic cells showed gene expression inhibition by methylation and histone deacetylation [60]. The loss of expression of α-catenin provides a growth advantage to AML or MDS cells with del(5q) [60].
4.6. EGR1
EGR1 gene encodes the transcription factor EGR1 located at the level of 5q31, a region frequently deleted in MDS and AML with del(5q). Studies of murine models with heterozygous or homozygous deletions of EGR1 gene support an important role of this gene in the control of hematopoiesis and as a tumor suppressor gene. EGR1-/- mice showed elevated white blood cell counts, elevated lymphocytes, and decreased neutrophil counts and incapacity to maintain normal RBC counts [61]. EGR1-/- or EGR1-/+ mice treated with phenylhydrazine develop anemia and are unable to rescue to their anemic condition [61-62]. EGR1-/- mice treated with the DNA alkylating agent, N-ethyl-nitrosourea, develop immature T-cell lymphomas or myeloproliferative disorders, characterized by elevated WBC, anemia with ineffective erythropoiesis and thrombocytopenia [62].
EGR1 binds genes critical for stem cell differentiation, inflammatory signaling, and the DNA damage response [63]. Haploinsufficiency of EGR1 biases HSCs/HPCs toward a self-renewal transcriptional signature, characterized by upregulation of MYC-driven proliferative signals, downregulation of p21, disrupted DNA damage response, and downregulated inflammation [63].
4.7. CDC25 and PP2A
Dual-specificity phosphatases cell division cycle 25 (CDC25) and protein phosphatase-2 (PP2) are encoded by genes located at 5q 31.2, a CDR in MDS-del(5q). Both these phosphatases are regulators of the cell cycle G2-M transition. Gene expression studies showed that CDC25 and PP2 expression is significantly reduced in MDS-del(5q), compared to MDS-5q-WT and normal controls [64]. Haploinsufficiency for CDC25 and PP2A genes does not seem to be involved in the generation of the peculiar hematologic phenotype of MDS-del(5q), but is essential for promoting selective sensitivity of MDS-del(5q) to lenalidomide-induced apoptosis [64]. Treatment of del(5q) leukemic cells with lenalidomide induces G2 arrest and apoptosis, whereas there was no effect in non-del(5q) leukemic cells [63]. Small interfering RNA suppression of CDC25 and PP2A gene expression recapitulates del(5q) susceptibility of MDS-del(5q) cells to lenalidomide with G2 arrest and induction of apoptosis [65].
4.8. DELE1
Death Ligand Signal Enhancer (DELE1) gene is located in 5q31.3 region and encodes for a protein associated with the inner mitochondrial membrane and is involved in death receptor-mediated apoptosis. DELE1 is one of these whose expression is most under-expressed in MDS-del(5q) [66]. Recent studies have shown that DELE1 protein is involved in the relay of mitochondrial stress to cytosol through the OMA1-DELE1-HRI pathway which leads to the activation of ATF4, the master transcription factor of the integrated stress response [64]. Partial loss of DELE1, as observed in MDS-del(5q) patients was sufficient to reduce the sensitivity to mitochondrial stress in leukemic cells [66].
4.9. DIAPH1
Formins are highly conserved proteins involved in the assembly of actini microfilament and microtubule cytoskeletons into cell architectures able to support cell adhesion and migration. The mammalian diaphanous-related formins are encoded by DIAPH genes; the three DIAPH gene isoforms are encoded by the DIAPH1 gene, located on chromosome 5q31.3. DIAPH1-deficient mice (in mice defined as mDia1) develop age-dependent myeloproliferative or myelodysplastic phenotypes, suggesting that DIAPH1 may act as a tumor suppressor [67]. The study of mice mDia1 heterozygous or homozygous showed that mDia1 deficiency led to a cell-autonomous overexpression of the membrane antigen CD14 and a hypersensitive innate immune response mediated by CD14/TLR4-like signaling; these mice develop age-dependent MDS that is accelerated by chronic stimulation of innate immunity [68].
An important role of of DIAPH1 in megakaryocyte proplatelet formation through accumulation of the actin and microtubule cytoskeletons was shown in another study [69].
A recent study reported the frequent occurrence of DIAPH1 mutations in these patients was correlated with lower megakaryocyte dysplasia in low-risk patients and higher megakaryocytes counts pre-transplant [68]. GP1BA and SETB1 mutations were positively and negatively associated with DIAPH1 mutations, respectively [70]. DIAPH1-mutated patients showed a favorable outcome [70].
4.10. TIFAB
TRAF-interacting protein with forkhead-associated domain B (TIFAB) is a TIFA family homolog lacking a phosphorylation site and a TRAF6 motif acting as a negative regulator of TIFA-TRAF6 signaling. Given these biochemical effects, TIFAB acts as an inhibitor of NF-kB signaling. TIFAB gene is located within the proximal CDR on band 5q31.1; consistent with haploinsufficiency, expression of TIFAB is decreased of about 50%$ in MDS-del(5q) compared to MDS without del(5q) or normal bone marrow [71]. Gene knockout studies provided evidence that TIFAB displays tumor suppressor-like functions, and its deletion induces an MDS-like phenotype in mice by modifying the dynamic range of the immune pathway reactivity in HSCs [72]. Verney et al. explored the mechanisms thorugh which loss of TIFAB affect hematopoiesis: TIFAB loss increases TRAF6 protein levels and the dynamic range of TLR4 (Toll-like receptor 4) signaling, contributing to ineffective hematopoiesis [73]. This effect of TIFAB is potentiated by the concomitant loss of miR-146a [73].
Another study showed that TIFAB regulates ubiquitin-specific peptidaes 15 (USP15) and consequently the USP15-mediated p53 signaling [74]. Deregulation of the TIFAB-USP15 complex, as observed in MDS-del(5q) modulates p53 activity and has critical functional consequences for HSCs under stress conditions [74].
4.11. NPM1
Nucleophosmin 1 is the most frequently mutated gene in AML; this gene is located on chromosome 5q35 and is lost in about 10% of MDS arising from large 5q deletions, mainly occurring in MDS with CK and in t-MDS [23,24]. NPM1+/- mice display an increased susceptibility to leukemia development and have been shown to generate hematologic syndromes with features similar to human MDS [75]. A mouse model of NPM1 knockout in HSCs showed premature ageing of HSCs and increased inflammatory response which favor the development of an MDS-like condition [76]. TP53 loss exacerbates the leukemic transition of NPM1-KO HSCs [76].
5. MDS-del(5q) as a Contiguous Gene Syndrome
The observations above reported suggest that MDS-del(5q) is a contiguous gene syndrome in which haploinsufficiency for several genes contributes to the hematologic phenotype observed in these patients. Some studies have explored whether the combined haploinsufficiency of several genes located on 5q and deleted in MDS-del(5q) induces a hematologic phenotype comparable to that observed in these patients. Thus, Ribezzo et al. have explored the combinatorial effect of haploinsufficiency for RPS14, SSNK1A1 and miR-145 using mice with genetically engineered, conditional heterozygous inactivation of RPS14 and CSNK1A1 and stable knockdown of miR145/mir146a [77]. These mice recapitulated all the main phenotypic features of MDS-del(5q) patients including severe anemia, dysmegakaryopoiesis with typical morphological abnormalities and increase of innate immune functions in macrophages (reflected by decreased phagocytic function and increased expression of S100A8 [77].
Another experimental model of MDS-del(5q) was based on a mouse model in which TIFAB and miR-146a were simultaneously deleted [78]. This model recapitulates several aspects of MDS-del(5q), such as defects of HSCs/HPCs mainly affecting the myeloid lineage, progressive peripheral blood cytopenias, myeloid dysplasia and altered cytokine production in the bone marrow microenvironment [78]. Deletion of TIFAB and miR-146a, as observed also in MDS-del(5q), induces the activation of IRAK2 and TRAF6, which determines an aberrant function of HSCs/HPCs. The study of TIFAB-/mir-146a- mice showed an impaired capacity to respond to an inflammatory condition, with reduced number of HSCs/HPCs and increased p53 expression [79]. This reduced proliferative capacity of HSCs/HPCs was restored by TP53 inactivation, thus indicating that inflammation confers a competitive advantage to functionally defective del(5q) cells upon loss of TP53 [79]. Thus, increased p53 activation in MDS-del(5q) HSC/HPCs due to inflammation triggers a selective pressure for genetic inactivation of p53 or expansion of pre-existing TP53-muant clone [79].
6. Therapy-Related MDS-del(5q)
Therapy-related MDS (t-MDS) are defined as MDS occurring after exposure to chemotherapy and/or radiation therapy and corresponds to about 15-20% of all MDS. T-MDS, compared to primary MDS (p-MDS) is characterized by a higher-risk clinical features, including more cytogenetic aberrations, higher frequency of high-risk mutations and a shorter overall survival. A nation-wide study confirmed that t-MDS display a significantly shorter mOS compared to p-MDS (15.8 months vs 31.1 months, respectively) [80].
Studies on large cohorts of t-MDS patients compared to p-MDS provided evidence about significant differences in their mutational profiles in that: TP53 and PPM1D mutations were clearly more frequent in t-MDS than in p-MDS; ASXL1, TET2, SRSF2 and SF3B1 mutations were less frequent in t-MDS than in p-MDS [12, 81-82]. T-MDS had shorte mOS than p-MDS even in patients who received allo-HSCT [82].
Chromosome 5 abnormalities are frequent in t-MDS, being observed in about 40% of patients [83]. However, isolated del(5q) was observed at a similar frequency in t-MDS and p-MDS [80].
The frequency of del(5q)/monosomy 5 abnormalities and of CK was significantly higher in t-MDS compared to p-MDS (for del(5q)/-5, 30% vs 14%; for CK, 28% vs 11%, respectively) [84]. Median survival for t-MDS patients was significantly shorter than for p-MDS within all risk group categories [84]. Furthermore, patients with t-MDS had a significantly higher hazard of death relative to p-MDS [84].
Hiwase et al. explored a group of 377 patients with therapy-related myeloid neoplasms (65% of t-MDS and 35% of t-AML) and observed that 34% of these tumors harbor TP53 mutations [85]. The frequency of chromosome 5 abnormalities (del(5q)/monosomy 5) was markedly higher in TP53-mutant MDS (81.4 in multi-hit and 66.7% in mono-hit) than in TP53-WT tumors (4.3% in TP53-WT); the same applied to CK (88.6% vs 11.5%) [85]. The analysis of the mOS in t-MDS patients showed that: TP53-mutant patients had a significantly shorter survival than TP53-WT patients; among TP53-mutant MDS, neither the allelic status nor the bone marrow percentage provides a significant prognostic information and 10% TP53 VAF is a clinically useful threshold to identify patients with poor survival [85].
In another study, Shah et al. have reported the analysis of a group of 488 t-MN patients (65% t-MDS and 35% t-AML), showing that 37% of these patients display TP53 alterations (multi-hit in 88% of cases) [86]. In tehse patients, del(5q) was observed in most cases in association with TP53 alterations [86]. The proportion of TP53 mutations increases from 4.5% in patients with normal karyotype to 17.3% in cases with two chromosomal abnormalities and 76.8% in cases with CK [86]. Importantly, the enrichment of TP53-mutant was observed in cases with del(5q) without CK; such enrichment of TP53 Mutations was not observed in cases with del(7q) without CK [86}. These findings support the view that TP53 mutation burden increases not only with the number but the type of chromosomal abnormalities [86]. TP53-mutant VAF ≥10% MDSs are associated with distinct presentation, profile of genomic instability and outcomes [86].
Buo explored a group of 138 t-MDS. 33% of these patients displayed TP53 abnormalities (73% multi-hit and 27% mono-hit) [87]. Del(5q) was observed in 30% of t-MDS compared to 10.9% in a group of p-MDS: CK in 39.2% of t-MDS and 15.7% of p-MDS [87}. In the group of t-MDS, del(5q)/-5 was observed in 57.5% of TP53-mutant and 17.8% of TP53-WT, while CK in 85% of TP53-mutant and 18.9% of TP53-WT [87]. In t-MDS, del(5q)/-5 was similarly associated with multi-hit or single-hit TP53 alterations (60% vs 50%, respectively) [87]. In t-MDS TP53-mutated, 100% of MDS with del(5q)/-5 are associated with CK, while in TP53-WT 72% of MDS with del(5q)/-5 are associated with CK [87]. In t-MDS, TP53-mutant MDS all none of the MDS with del(5q) display an isolated del(5q), while 28% of TP53-WT MDS display isolated del(5q) [87]. It is of interest to note that t-MDS exhibit a markedly higher frequency of TP53 and PPM1D mutations and a markedly lower frequency of ASXL1, U2AF1 and SRFSF2 mutations compared to p-MDS [87]. In t-MDS patients was significantly poorer for TP53-mutant compared to TP53-WT patients; among t-MDS, TP53-mutant patients, mOS does not seem to be affected by TP53 VAF or TP53 multi-hit or mono-hit status [87]. TP53 mutations in t-MDS are strongly associated with genomic instability; in fact, in TP53-mutant MDS, the frequency of patients with >3 cytogenetic abnormalities is very high, while in TP53-WT patients is low [87].
Lossard and coworkers explored the profile of chromosome abnormalities in 110 MDS-del(5q), 82 p-MDS and 28 t-MDS [88]. The breakpoints for 5q varied considerably in that the deletion size may be small (mainly 5q31), intermediate (with a size equivalent to half of the 5q arm) or large (corresponding to almost all the 5q arm) [88]. Among t-MDS patients, the frequency of small (21%), intermediate (18%) and large deleted fragments (61%) was similar to that observed for p-MDS patients [88].
As above reported, a minority of t-MDS displays a condition of isolated del(5q). Patients with isolated MDS-del(5q) show outcomes similar to those observed for p-MDS patients with isolated del(5q) for that concerns the response to lenalidomide treatment, mOS and the rate of leukemic transformation [89].
As above discussed, t-MDS can be subdivided into two groups different for mutational profile and prognosis: one group characterized by consistent genomic instability with frequent TP53 mutations and numerous cytogenetic abnormalities and associated with a poor prognosis; a second group with lower genomic instability with absent TP53 mutations, lower number of cytogenetic abnormalities and associated with a prognosis comparable to that of p-MDS. The longitudinal study of patients who developed t-MDS and t-AML suggests a pathogenetic mechanism based on the presence in these individuals of very minoritarian TP53 mutant clones of HSCs/HPCs that preferentially expanded after exposure to chemotherapy [81]. This interpretation is supported by the study of murine bone marrow chimeras containing both WT and TP53+/- HSCs/HPCs, the TP53+/- HSCs/HPCs preferentially expanded after exposure to chemotherapy [81]. These observations suggest that cytotoxic therapy does not directly induces TP53 alterations and that HSCs/HPCs bearing TP53 mutations are resistant to chemotherapy and expanded preferentially after treatment [81]. The early acquisition of TP53 mutations in the ancestral HSC/HPC clones could contribute to the accumulation of the frequent cytogenetic abnormalities.
7. Progression and Disease Evolution in MDSA-del(5q)
A high proportion of patients with isolated MDS-del(5q) respond to treatment with lenalidomide; however, 40% of these patients progress to AML by 5 years after starting treatment.
Several studies have documented treatment-emergent TP53 mutations in patients with MDS-(del5q) receiving lenalidomide therapy. In a first case-report study, Jadersten et al. reported the case of a patient with MDS-del(5q) without TP53 mutations at diagnosis, with complete erythroid and partial cytogenetic response to lenalidomide, who evolved to high-risk MDS with complex karyotype, associated with TP53 mutations [90].
Scharenberg and coworkers have reported the analysis of progression of 35 MDS patients with isolated del(5q) treated with lenalidomide (22 patients) or not (13 patients) and analyzed in the time for various clinical and laboratory parameters, including targeting sequencing for most relevant MDS-related mutations [91]. Progression was observed in 13 patients (4 to high-risk MDS and 9 patients to AML) and was associated with the detection of some new recurrent mutations, either occurring alone or in combination: TP53 in 9 cases, TET2 in 6 cases, RUNX1 in 3 cases, PTPN11 and SF3B1 in one case [91]. TP53 mutations were observed in 7 out 9 patients progressing to AML [91].
Mossner et al. have explored the adaptation and evolution of mutational hierarchies of MDS patients undergoing treatment. In their study they included 28 MDS MDS patients with del(5q), of which 21 were defined as isolated MDS-del(5q) according to WHO criteria [92]. Molecular analysis showed del(5q) was acquired as a secondary lesion or constituted a minor independent clone in 54% of all del(5q) MDS and in 62% of those with isolated del(5q); del(5q) was a founder event alone in 21.4% of the cases and with other founder lesions in 28.5% of cases [92]. Thus, del(5q() appeared to be the founder lesion in some patients and the secondary hit in other MDS patients. The longitudinal follow-up of these patients during their treatment with lenalidomide showed different patterns of molecular evolution. Some patients displayed a dynamic evolution of branching, with disappearance upon lenalidomide treatment of initially dominant subclones carrying del(5q); despite a significant improvement of hematological parameters, these patients displayed rapid emergence of previously undetectable branching subclones with new aberrations subsequent to lenalidomide administration [92]. Other patients showed rapid oligoclonal turnover following lenalidomide treatment, with cytogenetic remission of their del(5q) bearing subclones, early founding clones expanded in bone marrow [92]. The same authors reported the frequent detection of TP53 mutations in MDS-del(5q) with isolated 5q abnormality, negative for TP53 mutations at diagnosis and treated with lenalidomide (6 out of 15 patients) [93]. The study of 8 patients with TP53 mutations before the start of treatment with lenalidomide showed the negative impact of TP53 mutations on survival and lower sensitivity of TP53-mutant clones to lenalidomide treatment [93].
Lode and coworkers have reported the study of 24 MDS-del(5q) patients undergoing treatment, with 75% of patients reporting transfusion independence and 21% of complete cytogenetic responses [94]. 25% of these patients displayed TP53 mutations at diagnosis and 38% achieved TP53 mutations during follow-up [94]. A correlation was observed between acquisition of TP53 mutations and disease progression [94].
Sperling et al. reported the study of 416 patients who have developed therapy-related neoplasms (t-MN) and who have a detailed prior exposure history [95]. In these patients, TP53 mutations were significantly associated with thalidomide analogs, particularly with lenalidomide [95]. IN vitro and in vivo experimental studies supported that the effect of lenalidomide was specific to HSC/HPC with TP53 mutations [95]. This selective advantage of TP53-mutant HSCs/HPCs was conferred by lenalidomide and not by other thalidomide analogs, such as pomalidomide, this difference being related to the capacity of lenalidomide but not of pomalidomide to induce the degradation of CSNK1A1 [95]. These observations have suggested that lenalidomide induces the expansion of pre-existing TP53-mutant clones that are less sensitive to the suppressive effects exerted by this drug.
Abdallah and coworkers reported the analysis of 10 Mayo Clinic patients with MDS-del(5q), analyzed by NGS before and after treatment with lenalidomide [96]. Two of these patients had TP53 mutations at diagnosis and 3 acquired TP53 mutations (two of these patients displayed also SF3B1 mutations at diagnosis) [96]. In the tree patients acquiring TP53 mutations after therapy, TP53 mutations were monoallelic in one patient and biallelic in the other two patients [96]. The patients developing TP53 mutations after therapy apparently did not display and peculiar characteristics compared to the rest of patients [96].
Feurstein et al. have explored the routes of clonal evolution into CK in 1684 MDS patients with del(5q); 161 of these patients showed additional cytogenetic abnormalities that developed over time: 134 patients (8%) developed cytogenetic aberrations within the del(5q) clone and were defines as clonal evolution, while 27 patients developed independent clones not present within the del(5q) clone [97]. Two main pathways of cytogenetic clonal evolution have been identified: a more frequent (61% of cases) stepwise accumulation of cytogenetic events over time; a less frequent (39% of cases) catastrophic event, characterized by the occurrence of two or more aberrations occurring at the same time, determining the sudden development of clones bearing a CK [97]. The most frequent aberrations in the group with stepwise accumulation were trisomy 8 and trisomy 21; in the group with catastrophic events, del(7q)/-7 and del(17p)/-17 were the most recurrent chromosomal abnormalities [97]. Loss of 17p or monosomy 17 determine the loss of TP53 which could represent the driving force in MDS patients with del(5q) who undergo a sudden catastrophic event [97].
8. Treatment of MDS-del(5q)
The treatment of MDS-del(5q) is diversified according to the type of MDS associated with del(5q), being different for MDS with isolated del(5q) and for MDS with del(5q) associated with CK.
Table 2.
Comutations affecting outcomes of MDS-del(5q).
| Comutation |
Frequency in MDS-del(5q) |
Biologic and Clinical Implications |
| SF3B1 | 15-20% | Concomitant SF3B1 mutations are associated with lower response rate to lenalidomide, lower OS and increased arte of leukemic transformation. MDS-del(5q)/SF3B1-mutant are frequently associated with TP53 and RUNX1 mutations and display phenotypic properties of both SF3B1-mutant and MDS-del(5q) |
|
TP53 (monoallelic mutation) |
15-20% | Clinical impact of concomitant TP53 mutations depending on VAF frequency of mutant allele: <20% no effect on AML transformation rate and OS; >20% increased AML transformation rate and shorter OS. MDS-del(5q) with concomitant TP53 mutations have a trend to a reduced rate of response to lenalidomide. |
| RUNX1 | 1-3% | RUNX1 mutations are associated with reduced response to lenalidomide, reduced overall survival and a high risk of AML progression and the generation of t-MNs. |
| CSNK1A1 | 8-10% | CSNK1A1 mutation occurring at the level of the non-deleted CSNK1A1 allele is associated with reduced response to lenalidomide and increased risk of progression to a t-MN. |
Several recent studies have reviewer the treatment of low-risk MDS and, particularly, of MDS with isolated del(5q) [98,99,100]. Treatment goals for these patients include transfusion independence, increase in hemoglobin level, improvement of survival and maintenance or improvement of quality of life. Anemia observed in MDS-del(5q) was shown to be highly responsive to treatment with lenalidomide. According to the results observed in several studies, including randomized clinical trials, the FDA and EMA approved lenalidomide for the treatment of low- or intermediate-1 risk MDS with del(5q) and up to one additional cytogenetic abnormality, excluding chromosome 7 abnormalities. Usually, MDS patients with isolated del(5q) have a good prognosis. However, the presence of co-occurring SF3B1 or TP53 mutations may worsen this prognosis. In fact, a study of Meggendorfer et al. showed that MDS patients with isolated del(5q) and co-occurring SF3B1 mutations displayed a shorter OS compared to those without SF3B1 mutations (50 months vs not reached) [6]. On the other hand, Huber et al characterized a group of 231 MDS patients with SF3B1 mutations and observed that del(5q) and RUNX1 mutations were independent prognostic factors for overall survival [101]. Furthermore, among SF3B1-mutant MDS, those associated with del(5q) exhibited a higher frequency of TP53 and RUNX1 co-mutations compared to the SF3B1-mutant MDS without del(5q) [101].
Other studies have confirmed that SF3B1 co-mutations confer poor outcomes to MDS-del (5q). Thus, Chan et al reported that MDS-del(5q) patients had shorter mOS compared to WT (23.9 months vs 83.5 months) [102].
Other studies have provided evidence that MDS with concurrent del(5q) and SF3B1 mutations exhibit morphological, immunophenotypic and clinical properties mixed between those typically observed in MDS with isolated del(5q) and MDS with SF3B1 mutations, such as ringed sideroblasts and thrombocytosis [103-104].
A recent study reported a retrospective analysis on 77 MDS-del(5q) patients with SF3B1 mutations (SF3B1del5q); these patients received first-line treatment with ESAs (mostly with lenalidomide) [105]. The mOS of these patients was 66 months: 109 months for SF3B1del5q /TP53-WT and 64 months for SF3B1del5q/ TP53-mutant [105]. mOS was compared with a group of MDS-SF3B1 and MDS-del(5q) patients; mOS was 66,82 and 103 months, respectively for SF3B1del5q, MDS-del(5q) and MDS-SF3B1, with a rate of AML transformation in these three groups of 20%, 12% and 5%, respectively [105]. These observations support the conclusion that the mOS observed for SF3B1del5q patients was inferior to either isolated del(5q) patients or MDS-SF3B1 patients and may be driven by higher rate of concurrent TP53 and RUNX1 mutations [105].
Although these was some controversy about the prognostic impact of TP53 mutations in MDS with isolated del(5q), a large retrospective analysis carried out in 682 patients with isolated del(5q) ,defined according to Bernard et al [6], by Montoro et al showed that 18.9% of patients displayed TP53 mutations: the majority of these patients were monoallelic for TP53 alterations and only 4.5% with multi-hit TP53 alterations [106]. Patients with the TP53 monoallelic mutation with VAF >20% had a 32.2% risk of AML evolution, comparable to 40.4% risk observed for TP53 multi-hit patients and a shorter mOS [7]. In addition to these TP53-muant patients, also patients with SF3B1 and RUNX1 mutations displayed an increased risk of AML evolution [7].
RUNX1 is mutated in 1-3% of MDS with isolated del(5q) and its presence is associated with a reduced response to lenalidomide, shorter OS and increased tendency to AML progression [91,94,96]. Patinets who became resistant to lenalidomide harbor recurrent TP53 (53%) or RUNX1 (13%) mutations [106]. Experimental studies showed that lenalidomide upregulates RUNX1 protein and function in a CRBN- and TP53-dependent manner in del(5q) cells and mutation or downregulation of RUNX1 rendered cells resistant to lenalidomide [106]. It was shown that cell intrinsic innate immune signaling driven by miR-146a deletion, an event commonly occurring in MDS-del(5q), cooperates with mutant RUNX1 to initially induce marrow failure and MDS-like condition, progressing with the time to AML [107].
About 25-30% of MDS-del(5q) patients are refractory/resistant/ineligible to lenalidomide and will remain dependent on RBC transfusions; for these patients alternative treatment options are required.
One possible treatment could involve Luspatercept, a recombinant fusion protein that binds endogenous TGFβ superfamily ligands and promotes both early- and late-stage erythroid maturation. The COMMANDS phase II trial showed that Luspatercept achieved significantly greater rates of of RBC transfusion independence versus Epoietin alpha and these effects in responding patients were maintained at long-term [108,109,110]. However, this trial, as well as trial based on Luspatercept administration enrolled low-risk MDS patients, excluding MDS-del(5q) patients [111]. A single-arm, multicenter study is evaluating the efficacy of Luspatercept in reducing RBC transfusion-dependency in MDS patients with del(5q) refractory/resistant/intolerant to prior treatments [112]. Preliminary results on this trial showed a positive signal of efficacy and safety [112]. Patsialos et al. in a case report study showed the remarkable response to the treatment with Luspatercept in a MDS-del(5q) patient who was refractory to treatment with ESAs and Lenalidomide [113]. The patient achieved a remarkable erythroid response to Luspatercept after only five cycles of treatment [113]. Remarkable, the patients showed a trilineage response with normal hemoglobin levels and increased platelet and neutrophil counts, with no signs in the bone marrow of dyserythropoiesis, normally maturing megakaryocytes and granulocytes and <1% blasts, with a normal karyotype [113]. This response was maintained even fourteen months after Luspatercept discontinuation [113].
The treatment of MDS patients with del(5q) associated with CK alone or in association with TP53 alterations is highly challenging. The standard of care for these patients, as well for other high-risk MDS patients, is based on monotherapy is based on monotherapy with a hypomethylating agent (either azacitidine or decitabine). The AZA-001 phase III study showed that azacitidine prolonged survival of high-risk MDS patients ineligible for HSCT to 24.5 months compared with 15 months observed for patients treated with low-intensity conventional chemotherapy and supportive care regimens [114]. However, other studies have shown lower overall survival levels compared to those observed in the AZA-001 study [115,116].
A more recent study explored the safety and the efficacy of adding the BCL2 inhibitor Venetoclax to the hypomethylating agent Azacitidine. A single-arm phase Ib study showed in 107 high-risk MDS patients treated with Venetoclax+Azacitidine a CR rate of 29.9% and CR with incomplete count recovery (CRi) of 48.6%; the mOS at 26 months was good and 39% of patients went to receive HSCT [117]. In the phase III VERONA trial, the patients with high-risk MDS were randomized to Azacitidine and Venetoclax or Azacitidine alone; however, the results of this study recently presented at the Society of Hematologic Oncology (SOHO) Annual Meeting (Abstract MDS-1497) showed no benefit with addition of Venetoclax to Azacitidine compared to Azacitidine alone in high-risk MDS. The CRR was superior with Azacitidine plus Venetoclax.
The resistance to Venetoclax of MDS patients with biallelic TP53 alterations is in part related to the high expression of BCL-XL, an anti-apoptotic protein [118]. BCL-XL hyperexpression in these MDS is associated with an erythroid phenotype [118]. This observation suggests the potential use of BCL-XL inhibitors in the treatment of these patients.
As above discussed, the group of MDS associated with CK represents a heterogeneous entity subdivided according to the presence of TP53 mutations: the group CK-TP53-mutant had more frequent del(5q) and a significantly shorter mOS than the group CK-TP53-WT [9,10]. Considering these differences, Huber et al. in their MDS classification have proposed two distinct groups, one bearing TP53 biallelic alteration and the other bearing complex karyotype without TP53 alterations [119]. Both these MDS groups have a poor prognosis but mOS of biallelic TP53 MDS is shorter than that of MDS with CK [119]. As above reported, the molecular taxonomic study hierarchically classified MDS into molecular taxonomic groups according to mutational clusters; one of these clusters is multi-hit TP53 or CK [5]. Molina and coworkers evaluated the response to HMAs of a group 268 patients classified according to the molecular taxonomy; the TP53-multi-hit or CK and EZH2-ASXL1 groups exhibited the poorest OS (1.26 and 0.84 years compared to 4.74 years of the whole MDS cohort) and the highest risk of AML transformation [120]. A send study evaluated the prognostic impact of the molecular taxonomy classification in a group of 484 patients and observed that the TP53-multi-hit or CK group is heterogeneous in that TP53-mutated patients exhibited a significantly shorter OS than those with CK without TP53 mutations [121]. Importantly, in the TP53-multi-hit or CK group the survival probability of these patients was not affected by blast number either <5% or >5% [121]. Huber and coworkers reported blast cell numbers for various molecular subgroups of MDS patients and observed that blast cell number is highly variable in both the CK and biallelic TP53 groups, with a mean blast number higher for biallelic TP53 than for CK MDS [120]. The blast cell number could help to identify the stage of disease within these two MDS groups [122].
The relevance of blasts quantification in the classification of MDS isd questionable. Thus, while it is evident the relevance of blast cell quantification as a prognostic factor in the context of different MDS cases, the significance of blast counts is carriable for different MDS genetic groups [123].
The group of MDS with CK is heterogeneous and a recent study reported the identification of a subset of these MDSs, characterized by absent TP53 mutations or TP53 deletions due to chromosome 17 loss but exhibiting a dysfunction of p53 protein [124]. Thus, in a recent study Zampini et al. reported te molecular characterization of 6204 MDS patients with particular emphasis to the characterization of MDS exhibiting dysfunction of p53 related to genetic alterations or to altered expression/function [124]. In this study, the exploration of 42 MDS patients with TP53 mutations and displaying AML evolution showed that: 23 patients exhibiting monoallelic TP53 alterations at diagnosis, 18 progressed to a biallelic status at AML evolution; 19 patients displaying AML progression showed an increased TP53 mutation VAF and acquisition of additional chromosomal abnormalities [124]. These observations strongly support the view that mono-allelic and bi-allelic TP53 alterations represent different stages occurring through a process involving multiple hits during the natural evolution of MDS. These authors identified a subgroup of MDS characterized by TP53-WT and hyperexpression of p53 protein in bone marrow, constantly associated with CK; chromosome 5 abnormalities were observed in about half of these patients; TET2 (11%), ASXL1 (11.1%) and RUNX1 (12.2%) were the genes most frequently mutated [124]. Patients with TP53-WT and p53 protein hyperexpression displayed a dismal outcome, comparable to that observed in patients with biallelic TP53 alterations [124]. The increased p53 protein expression observed in these MDS was associated with absent activation of p53 targets, thus suggesting that p53 protein, although non-mutated and hyperrepressed, is abnormal and not functional [124]. These patients displayed several p53 upstream alterations at the level of PI3K cascade, RAS, WNT and NF-kB pathways and MDM2 gene amplification [124]. Furthermore, MDS with p53 hyperexpression and TP53-WT exhibited a peculiar immune dysregulation involving myeloid-derived inflammation and impaired antigen presentation [124].
Few studies have specifically explored the outcome of MDS with CK. Rasmussen et al. have reported the results of a randomized phase II study involving treatment with Azacitidine alone or Azacitidine+Lenalidomide, involving the enrollment of 72 MDS or AM (75% MDS) patients with a karyotype including del(5q) (83% with CK, 76% with TP53 mutations in 96% of cases multi-hit) [125]. The ORR in the treated cohorts was 39% for AZA and 44% for AZA+LEN, with a CRR of 17% and 28%, respectively [125]. The mOS was 115 months for the whole population and 13.6 months in the AZA arm and 10.8 months in the AZA+LEN arm [125]. In a subsequent study, the same authors have explored the response of these patients according to their karyotype defined by standard karyotyping analysis and FISH: the ORR did not differ between patients with <3 aberrations and patients with >3 aberrations; patients with >3 aberrations displayed shorter overall survival (9.9 months) compared to those with <3 aberrations (25.2 months) [126]. Patients with unbalanced translocations of 5q have significantly shorter OS than those with del(5q) (8.4 months vs 21.1 months, respectively) [126]. Unbalanced 5q translocations were more frequently associated with CK and multi-hit TP53 than del(5q) (for CK, 98% vs 67%; for multi-hit TP53, 88% vs 47%) [126]. Cytogenetic progression occurred at a similar frequency in patients treated with AZA or AZA+LEN [126]. Thus, according to these observations and other data present in the literature, the CK-MDS can be subdivided into four subgroups: a subgroup with biallelic TP53 alterations and frequent del(5q), a subgroup with monoallelic TP53 alterations and frequent del(5q); a subgroup with p53 hyperexpression without TP53 mutations or chromosome 17 aberrations and with less frequent del(5q); a subgroup without TP53 mutations but with frequent chromosome 17 deletions and less frequent del(5q).
9. Conclusions
The studies carried out in the last three decades have dramatically contributed to define the molecular spectrum of genetic abnormalities observed in MDSs associated with the presence of del(5q), with the identification of a MDS subtype associated with isolated del(5q) and of a heterogeneous group of MDS in which del(5q) is associated with TP53 alterations and/or with CK. It is fundamental to distinguish these two types of MDS with del(5q) for their different prognosis and treatments.
The molecular studies performed in patients with isolated del(5q) have contributed to define the functional role of the various genes present in the deleted regions of 5q, whose loss contributes to a pathogenetic mechanism of contiguous gene effect in which the final hematologic phenotype is dependent upon the collective effect of different gene deletions. However, although del(5q) is a key pathogenic event in 5q- syndrome, some co-occurring somatic mutations, including TP53, SF3B1, CSNK1A1 and RUNX1 mutations affect the outcomes of these patients reducing the response to treatment and/or increasing the risk of AML development. Given this heterogeneity of MDS with isolated del/5q), an integration of of morphologic, clinical, cytogenetic and genomic data for each patient is required to identify different clinical entities of patients and to monitor their response to treatment.
The other group of MDS with del(5q) associated with CK and TP53 alterations can be subdivided into four different subgroups: a subgroup with biallelic TP53 alterations and frequent del(5q), a subgroup with monoallelic TP53 alterations and frequent del(5q); a subgroup with p53 hyperexpression without TP53 mutations or chromosome 17 aberrations and with less frequent del(5q); a subgroup without TP53 mutations but with frequent chromosome 17 deletions and less frequent del(5q). The identification of these patients requires an accurate cytogenetic and molecular analysis. The prognosis of these patients is poor, particularly of those with biallelic TP53 alterations or with p53 hyperexpression. The treatment of these patients is challenging and is mainly based on hypomethylating agents, followed when possible by allogeneic SCT.
Author Contributions
G.C. and E.P.P. were involved in researching, writing, and editing the manuscript. U.T. was involved in conceptualization, organization, research, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Van den Berghe, H.; Cassiman, J.J.; Frins, J.P.; Michaux, J.L.; Sokal, G. Distinct hematological disorder with deletion of long arm of no. 5 chromosome. Nature 1974, 251, 437-438. [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J. World Health Organization classification of tumors. Pathology and genetics of tumors of hematopoietic and lymphoid tissues (ed 3rd) 2001 Lyon; IARC.
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvanicka, H.M.; Bagg, W.A.; Barbui, T.; Branford, S.; Beso-Ramos, C.E.; et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical and genomic data. Blood 2022, 140, 1200-1228. [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O; Akkori, Y.; Aleggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th edition of the world Health Organization Classification of hematolymphoid tumors: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022, 36, 1703-1719.
- Bernard, E.; Hasserjian, R.; Greenberg, P.L.; Ossa, J.E.; Creignou, M.; Tuechler, H.; Gutierrez-Abril, J.; Domenico, D.; Medina-Martinez, J.S.; Farmoud, N.; et al. Molecular taxonomy of myelodysplastic syndromes and its clinical implications. Blood 2024, 144, 1617-1632. [CrossRef]
- Meggendorfer, M.; Haferlach, C.; Kern, W.; Haferlach, T. Molecular analysis of myelodysplastic syndrome with isolated deletion of the long arm of chromosome 5 reveals a specific spectrum of molecular mutations with prognostic impact: a study on 123 patients and 27 genes. Haematologica 2017, 102, 1502-1510. [CrossRef]
- Montoro, M.J.; Palomo, L.; Haferlach, C.; Acha, P.; Chan, O.; Navarro, V.; Kubota, Y.; Schultz, F.I.; Meggendorfer, M.; Briski, R.; et al. Influence of TP53 gene mutations and their alleliuc status in myelodysplastic syndromes with isolated 5q deletion. Blood 2024, 144, 1722-1732. [CrossRef]
- Xie, Z.; Al Ali, N.; Zhang, L.; Papenhausen, P.; Volpe, V.O.; Chan, O.; Kuykendall, A.; Yun, S.; Walker, A.; Sweet, K.; et al. Clinical correlation and prognostic impact of cytogenetic clone size for myelodysplastic syndromes/neoplasm. Blood Neoplasia 2025, 2, 100062. [CrossRef] [PubMed]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Macjeweski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747-1758. [CrossRef] [PubMed]
- Huber, S.; Hutter, S.; Baer, C.; Meggendorfer, M.; Hoermann, G.; Kern, W.; Haferlach, T.; Haferlach, C. Two ways to complex karyotype in MDS-the role of del(5q) and TP53. Blood Cancer J 2025, 15, 96. [CrossRef] [PubMed]
- Kewan, T.; Durmaz, A.; Bahaj, W.; Gurnari, C.; Terkawi, L.; Awada, H.; Ogbue, O.; Ahmed, R.; Pagliuca, S.; Awada, H.; et al. Molecular patterns identify distinct subclasses of myeloid neoplasia. Nat Commun 2023, 14, 3136. [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 24534-2465. [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arongo Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid 2022, 1, EVIDoa22000008.
- Montero, M.J.; Palomo, L.; Haferlach, C.; Acha, P.; Chan, O.; Navarro, V.; Kubota, Y.; Schulz, F.; Briski, R.; Al Ali, N.; et al. Newly developed prognostic score for myelodysplastic syndrome (MDS) with isolated 5q deletion (IPSS-del(5q)). Blood 2024, 144 (suppl.1), 666-668. [CrossRef]
- Komrokji, R.S.; Lanino, L.; Ball, S.; Bewersdorf, J.P.; Marchetti, M.; Maggioni, G.; Travaglino, E.; Al Ali, N.; Fenaux, P.; Platzbecker, U.; et al. Data-driven, harmonized classification system for myelodysplastic syndromes: a consensus paper from the International Consortium for Myelodysplastic Syndromes. Lancer Hematol 2024, 11, e862-e872. [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Suki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med 2020, 26, 1549-1556. [CrossRef]
- Stengel, A.; Meggendorfer, M.; Walter, V.; Baer, C.; Nadarajah, N.; Hutetr, S.; Kern, W.; Haferlach, T.; Haferlach, C. Interplay of TP53 allelic state, blast count, and complex karyotype on survival of patients with AML and MDS. Blood Adv 2023, 7, 5540-5548. [CrossRef]
- Pandiri, M.; Stengel, A.; Zhang, J.; Wang, P.; Shao, H.; Velmurugan, S.; Jacob, A.; Symes, E.; Kaur, A.; Rojek, A.; et al. Karyotypic clonal fraction predicts adverse outcome in TP53-mutated myeloid neoplasms: an international TP53 investigators network (iTiN) study. J Clin Pathol 2025, 78, 629-635. [CrossRef] [PubMed]
- Shah, M.V.; Hung, K.; Baranwal, A.; Kutyna, M.; Al-Kali, A.; Toop, C.; Greipp, P.; Brown, A.; Shah, S.; Khanna, S.; et al. Evidence-based risk stratification of myeloid neoplasms harboring TP53 mutations. Blood Adv 2025, 9, 3370-3380. [PubMed]
- Boultwood J., Fidler C., Lewis S., Kelly S., Sheridan H., Littlewood T., Buckle V., Wainscoat J. Molecular Mapping of Uncharacteristically Small 5q Deletions in Two Patients with the 5q- Syndrome: Delineation of the Critical Region on 5q and Identification of a 5q- Breakpoint. Genomics. 1994; 19:425–432.
- Boultwood J., Fidler C., Strickson A.J., Watkins F., Gama S., Kearney L., Tosi S., Kasprzyk A., Cheng J.-F., Jaju R.J., et al. Narrowing and Genomic Annotation of the Commonly Deleted Region of the 5q- Syndrome. Blood. 2002; 99:4638–4641.
- Jaju R., Boultwood J., Oliver F., Kostrzewa M., Fidler C., Parker N., McPherson J., Morris S., Müller U., Wainscoat J., et al. Molecular Cytogenetic Delineation of the Critical Deleted Region in the 5q- Syndrome. Genes Chromosom. Cancer. 1998; 22:251–256.
- Jerez A., Gondek L.P., Jankowska A.M., Makishima H., Przychodzen B., Tiu R.V., O’Keefe C.L., Mohamedali A.M., Batista D., Sekeres M.A., et al. Topography, Clinical, and Genomic Correlates of 5q Myeloid Malignancies Revisited. JCO. 2012; 30:1343–1349.
- La Starza, R.; Matteucci, C.; Gorello, P.; Brandimarte, L.; Pierini, V.; Crescenzi, B.; Nofrini, V.; Rosati, R.; Gottardi, E.; Saglio, G.; et al. NPM1 deletion is associated with groos chromosomal rearrangements in leukemia. PLos One 2010, 5, e12855. [CrossRef]
- Nofrini, V.; La Starza, R.; Crescenzi, B.; Pierini, V.; Barba, G.; Mecucci, C. Different boundaries characterize isolated and non-isolated 5q deletions in myelodysplastic syndromes and acute myeloid leukemias. Haematologica 2012, 97, 792-794. [CrossRef]
- Rea, B.; Aggarwal, N.; Yetsenko, S.A.; Bailey, N.; Liu, Y.C. Acute myeloid leukemia with isolated del(5q) is associated with IDH1/IDH2 mutations and better prognosis when compared to acute myeloid leukemia with complex karyotype including del(5q). Modern Patology 2020, 33, 566-575.
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Haferlach, C. The 5q deletion size in myeloid malignancies is correlated with additional chromosomal aberrations and to TP53 mutations. Genes Chrom Cancer 2016, 55, 777-785. [CrossRef]
- Zemanova, Z.; Michalova, K.; Buryova, H.; Brezinova, J.; Lizcova, L.; Kostykova, K.; Sarova, I.; Izakova, S.; Rnasdorfova, S.; Krejcik, Z.; et al.
- Warnstorf, D.; Bawadi, R.; Schienke, A.; Starsser, R.; Schmidt, G.; Illig, T.; Tauscher, M.; Thol, F.; Heuser, M.; Steinemann, D.; et al. Unbalanced translocation del(5;17) resulting in TP53 loss as recurrent aberration in myelodysplastic syndrome and acute myeloid leukemia with complex karyotype. Genes Chrom Cancer 2021, 60, 452-457. [CrossRef] [PubMed]
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leiukemia and myelodysplastic syndrome. Genes Chrom Cancer 2014, 53, 402-410. [CrossRef] [PubMed]
- Boultwood,J.; Pellagatti, A.; Cattan, H. Gene expression profiling of CD34+ cells in patients with 5q- syndrome. Br J Haematol 2007, 139, 578-589.
- Adema, V.; Paloma, L.; Walter, W.; Mallo, M, Hutter, S.; La Franboise, T.; Arenillas, L.; Meggendorfer, M.; Radivoyevitch, T.; Xicoy, B.; et al. Pathophysiologic and clinical implications of molecular profiles resultant from deletion 5q. eBiomedicine 2022, 80, 104059. [CrossRef] [PubMed]
- Jaju, R.J.; Boutlwood, J.; Oliver, F.J.; Kostrezva, M.; Fidler, C.; Parker, N.; McPherson, J.D.; Morris, S.W.; Muller, U.; Wainscoat, J.S.; et al. Molecular cytogenetic delineation of the critical deleted region in the 5q- syndrome. Genes Chrom Cancer 1998, 22, 251-256. [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Dellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335-339. [CrossRef]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in huma erythroid progenitor cells. Blood 2011, 117, 2567-2576. [CrossRef]
- Pellagatti, A.; Marafioti, T. Paterson, J.C.;; Barlow, J.L.; Drynan, L.F.; Giagounidis, A.; Pileri, S.A.; Cazzola, M.; McKenzie, A.; Wainscoat, J.S.; et al. Induction of p53 and up-regulation of the p53 pathway in the human 5q- syndrome. Blood 2010, 115, 2721-2723.
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalkde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med 2010, 16, 59-66. [CrossRef]
- Pellagatti, A.; Hellstrom-Lindberg, E.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.; Jadersten, M.; Killick, S.; Fidler, C.; Cazzola, M.; et al. Haloinsufficiency of RPS14 in 5q- syndrome is associated with deregulation of ribosomal- and translation-related genes. Brit J Haematol 2008, 142, 57-64. [CrossRef]
- Pellagatti, A.; Hellstrom-Lindberg, E.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.; Jadersten, M.; Killick, S.; Sohal, D.; Verma, A.; et al. Haploinsufficiency of RPS14 and deregulation of ribosomal- and translation-related genes in MDS patients with del(5q). Blood 2008, 112 (suppl.1), 3641. [CrossRef]
- Cziberre, A.; Bruns, I.; Junge, B.; Kobbe, G.; Haas, R.; Germing, U. Low RPS14 expression is common in myelodysplastic syndromes without 5q- aberration and defines a subgroup of patients with prolonged survival. Haematologica 2009, 94, 1453-1455. [CrossRef]
- Wu, L.; Xu, F.; Zhang, Z.; Chang, C. Low Rps14 expression in MDS without 5q- aberration confers higher apoptosis rate of nucleated erythrocytes and predict prolonged survival and possible response to lenalidomide in lower risk non-5q- patients. Eur J Haematol 2013, 90, 486-493. [CrossRef]
- Linares, M.; Rapado, I.; Ruiz-Heredia, Y.; Cedena, M.T.; Quiroz, K.; Barrio, S.; Ayala, R.; Martinez, J. 5q+ MDS patients with low RPS14 expression are candidates to immune-modulating drugs. Blood 2017, 130, 5303.
- Adema, V.; Kongkiatkamon, S.; Palomo, L.; Walter, W.; Hutter, S.; LaFrambiose, T.; Diez-Campelo, M.; Mallo, M.; Xicoy, B.; Meggendorfer, M.; et al. Deficiency of RPs14 beyond the haploinsufficient loss in del(5q). Blood 2021, 138 Suppl.1), 2591. [CrossRef]
- Schneider, R.K.; Schenone, M.; Ventura Ferreira, M.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brummendorf, T.H.; Germin, L.; Pletzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med 2016, 22, 288-297. [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of MiR-145 and MiR-146a as Mediators of the 5q– Syndrome Phenotype. Nat. Med. 2010, 16, 49–58. [CrossRef]
- Kumar, M.S.; Narla, A.; Nonami, A.; Mullally, A.; Dimitrova, N.; Ball, B.; McAuley, J.R.; Poveromo, L.; Kutok, J.L.; Galili, N.; et al. Coordinate Loss of a MicroRNA and Protein-Coding Gene Cooperate in the Pathogenesis of 5q- Syndrome. Blood 2011, 118, 8. [CrossRef]
- Schneider R.K.; Adema, V.; Heckl, D.; Jaras, M.; Mallo, M.; Lord, A.; Chu, L.; McConkey, M.; Kramann, R.; Mullally, A.; et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 2014, 26, 509-520. [CrossRef]
- Smith, A.E.; Kulakaseraj, A.; Jiang, J.; Mian, S.; Mohamedali, A.; Gaken, J.; Ireland, R.; Czepulkowski, B.; Best, S.; Mufti, G.J.; et al. CSNK1A1 mutations and isolated del(5q) abnormality in myelodysplastic syndrome: a retrospective mutational analysis. Lancet Hematol 2015, 2, e212-e221. [CrossRef] [PubMed]
- Heuser, M.; Meggendorfer, M.; Cruz, M.M.; Fabisch, J.; Klesse, S.; Kohler, L.; Gohring, G.; Ganster, C.; Shirneshan, K.; Gutermuth, A.; et al. Frequency and prognostic impact of casein kinase 1A1 mutations in MDS patients with deletion of chromosome 5q. Leukemia 2015, 29, 1942-1945. [CrossRef] [PubMed]
- Stalmann, U.; Ticconi, F.; Snoeren, I.; Li, R.; Gleitz, H.; Coeley, G.; McConkey, M.; Wong, A.; Smitz, S.; Fuchs, S.; et al. Genetic barcoding systematically compares genes in del(5q) MDS and reveals a central role for CSNK1A1 in clonal expansion. Blood Adv 2022, 6, 1780-1786. [CrossRef]
- Fuchs, S.; Stalmann, U.; Snoeren, I.; Bindels, E.; Schmitz, S.; Banjamin, B.; Hoogenboezem, R.; van Herk, S.; Saad, M.; Walter, W.; et al. Collaborative effect of CSNK1A1 haploinsufficiency and mutant p53 in Myc induction can promote leukemic transformation. Blood Adv 2024, 8, 766-774. [CrossRef] [PubMed]
- Kollotzek, F.; Mott, K.; Fischer, M.; Findik, B.; Gob, V.; Manke, M.C.; Borst, C.E.; Polzin, A.; Burkhalter, M.D.; Eckly, A.E.; et al. Casein kinase 1α essentially regulates thrombopoiesis by driving megakaryocyte maturation and cytoskeleton organization. Blood 2025, in press. [CrossRef] [PubMed]
- Liu, X.; Huang, Q.; Chen, L.; Zhang, H.; Schobrunn, E.; Chen, J. Tumor-derived CK1α mutations enhance MDMX inhibition of p53. Oncogene 2020, 39, 176-186. [CrossRef] [PubMed]
- Esfahanian, N.; Knoblsch, C.D.; Bowman, C.A.; Rezvani, K. Mortalin: protein partners, biological impacts, pathological role, and therapeutic opportunities. Front Cell Dev Biol 2023, 11, 10228519. [CrossRef]
- Chen, T.H.; Kambai, A.; Krysiak, K.; Walhauser, M.A.; Raju, G.; Tibbitts, J.F.; Walter, M.J. Kockdown of Hspa9, a del(5q31.2), results in a decrease in hematopoietic progenitors in mice. Blood 2010, 117, 1530-1539.
- Liu, T.; Krysiak, K.; Shirai, C.L.; Kim, S.; Shao, J.; Ndonwi, M.; Walter, M.J. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PlOS ONE 2017, 12, e0170470. [CrossRef]
- Butler, C.; Dunmire, M.; Choi, J.; Szalai, G.; Johnson, A.; Lei, W.; Chen, X.; Liu, L.; Li, W.; Walter, M.J.; Liu, T. SSPA9/mortalin inhibition disrupts erythroid maturation through a TP53-dependent mechanism in human CD34+ hematopoietic progenitor cells. Cell Stress Chaperones 2024, 29, 300-311. [CrossRef]
- Lehmann, S.; O’Kelly, J.; Raynaud, S.; Funk, S.E.; Sage, E.H.; Koeffler, H.P. Common deleted genes in the 5q- syndrome: thrombocytopenia and reduced erythroid colony formation in SPARC null mice. Leukemia 2007, 21, 1931-1936. [CrossRef]
- Luo, Z.; Luo, P.; Yu, Y.; Zhao, Q.; Cheng, L. SPARC promotes the development of erythroid progenitors. Exp Hematol 2012, 40, 828-836. [CrossRef]
- Liu, T.X.; Becker, M.W.; Jelinck, J.; Wu, W.S.; Deng, M.; Mikhalkevich, N.; Hsu, K.; Blomfield, C.; Stone, R.M.; DeAngelo, D.J.; et al. Chromosome 5 deletion and epigenetic suppression of the gene encoding α-catenin (CTNNA1) in myeloid cell transformation. Nat Med 2007, 13, 78-83. [CrossRef]
- Joslin, J.M.; Fernald, A.A.; Qian, Z.; Crispino, J.D.; LeBeau, M. Egr1, a candidate gene within the commonly deleted segment of chromosome 5, plays a role in murine erythropoiesis and leukemogenesis. Blood 2005, 106 (suppl.1), 663. [CrossRef]
- Joslin, J.M.; Fernald, A.A.; Tennant, T.R.; Davis, E.M.; Kogan, S.C.; Anastasi, J.; Crispino, J.D.; LeBeau, M. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood 2007, 110, 719-726. [CrossRef]
- Stoddart, A.; Fernald, A.A.; Davis, E.M.; McNerney, M.E.; LeBeau, M. EGR1 haploinsufficiency confers a fitness advantage to hematopoietic stem cells following chemotherapy. Exp Hematol 2022, 115, 54-67. [CrossRef]
- Wei, S.; Rocha, K.; Williams, A.; Chen, X.; Burnette, P.K.; Djeu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; et al. Gene dosage of the cell cycle regulatory phosphatases Cdsc25C and PP2A determines sensitivity to lenalidomide in del(5q) MDS. Blood 2007, 110 (suppl.1), 118. [CrossRef]
- Wei, S.; Chen, X.; Rocha, K.; Epling-Burnette, P.K.; Dieu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; Pireddu, R.; et al. A critical role for phosphatase haploinsufficiency in the selective suppression of deletion 5q MDS by leniladomide. Proc Natl Acad Sci USA 2009, 106, 12974-12979. [CrossRef] [PubMed]
- Spinell, J.F.; Chanbgroul, J.; Moison, C.; Lavallée, V.P.; Bolin, I.; Gracias, D.; Lavallée, S.; Richard Carpentier, G.; Beliveau, F.; Hébert, J.; et al. DELE1 haploinsufficiency causes resistance to mitochondrial stress-induced apoptosis in monosomy 5/del(5q) AML. Leukemia 2024, 38, 530-537. [CrossRef] [PubMed]
- Peng, J.; Kitchen, S.M.; West, R.A.; Sigler, R.; Eisenmann, K.M.; Alberts, A.S. Myeloproliferative defects following targeting of the Drfg1 gene encoding the mammalian diaphanous-related formin mDia1. Cancer Res 2007, 67, 7675-7671. [CrossRef]
- Keerthivasan, G.; Mei, Y.; Zhao, B.; Zhang, L.; Harris, C.E.; Gao, J.; Basiorka, A.A.; Schipma, M.J.; McElherme, J.; Verma, A.K.; et al. Aberrant overexpression of CD14 on granulocytes sensitizes the innate immune response in mDia1 heterozygous del(5q) MDS. Blood 2014, 124, 780-790. [CrossRef]
- Pan, J.; Lodier, L.; Meyran, D.; Rameau, P.; Lecluse, Y.; Kitchen-Goosen, S.; Badirou, I.; Mokrani, H.; Narumya, S.; Alberts, A.S.; Vainchenker, W.; et al. The forming DIAPH1 (mDia1) regulates megakaryocyte proplatelet formation by remodeling the actin and microtubule cytoskeletons. Blood 2014, 124, 3967-3977. [CrossRef] [PubMed]
- Tang, Y.; Wang, H.; Zhang, Z.; Yao, Y.; Han, Y.; Wu, D. DIAPH1 mutations predict favorable outcome for de novo MDS. Cancer Letters 2024, 598, 217125. [CrossRef]
- Nakamura, T.; Ohyama, C.; Sakamoto, M.; Toma, T.; Tateishi, H.; Matsuo, M.; Chirifu, M.; Ikemizu, S.; Morioka, H.; Fujita, M.; et al. TIFAB regulates the TIFA-TRAF6 signaling pathway involved in innate immunity by forming a heterodimer complex with TIFA. Proc Natl Acad Sci USA 2024, 121, e2318794121. [PubMed]
- Varney, M.; Christie, S.; Niederkom, M.; Fang, J.; Jerez, A.; Maciejewski, D.T.; Inoue, J.; Starczynowski, D.T. Deletion of TIFAB, a novel candidate gene on chromosome 5q, results in hematopoietic defects by changing the dynamic range of innate immune pathway activation. Blood 2013, 122 (suppl.1), 102. [CrossRef]
- Varney, M.E.; Niederkon, M.; Konno, H.; Matsumara, T.; Gohdfa, J.; Yoshida, N.; Akiyama, T.; Christie, S.; Fang, J.; Miller, D.; et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J Exp Med 2015, 212, 1967-1985. [CrossRef]
- Niederkon, M.; Hueneman, K.; Choi, K.; Varney, M.E.; Romano, L.; Pujato, M.A.; Greis, K.D.; Inoue, J.; Meetei, R.; Starczynowski, D.T. TIFAB regulates USP15-mediated p53 signaling during stressed and malignant hematopoiesis. Cell Rep 2020, 30, 2776-2790. [CrossRef]
- Raval, A., Kusler, B.; Weissman, I.L.; Mitchell, B.S.; Park, C.Y. Effect of nucleophosmin 1 haploinsufficiency on hematopoietic stem cells. Leukemia 2012, 26, 853-855. [PubMed]
- Morganti, C.; Ito, K.; Yanase, C.; Verma, A.; Teruya-Feldstein, J.; Ito, K. NPM1 ablation induces HSC aging and inflammation to develop myelodysplastic syndrome exacerbated by p53 loss. EMBO Rep 2022, 23, e54262.
- Ribezzo, F.; Snoeren, I.; Ziegler, S.; Stoelben, J.; Olofsen, P.A.; Henic, A.; Ventura Ferreira, M.; Chen, S.; Stalmann, U,; et al. Rps14, Csnk1A1 and miR145/miR146a deficiency cooperate in the clinical phenotype and activation of the innate immune system in the 5q- syndrome. Leukemia 2019, 33, 1759-1772.
- Varney, M.E.; Choi, K.; Bolanos, L.; Epistasis between TIFAB and miRT-146a: neighboring genes in del(5q) myelodysplastic syndrome. Leukemia 2017, 31, 491-495.
- Muto, T.; Walker, C.S.; Agarwal, P.; Vick, E.; Sampson, A.; Choi, K.; Niederkon, M.; Ishikawa, C.; Hueneman, K.; Varney, M.; et al. Inactivation of p53 provides a competitive advantage to del(5q) myelodysplastic syndrome hematopoietic stem cells. Haematologica 2023, 108, 2715-2729. [CrossRef]
- Berggren, D.M.; Garelius, H.; Hjielm, P.W.; Nilsson, L.; Ramussen, B.; Elbult, C.E.; Lambe, M.; Lehmann, S.; Heelstrom-Lindberg, E.; Jadersten, M.; et al. Therapy-related MDAS dissected based in primary disease and treatment-a nation-wide perspective. Leukemia 2023, 37, 1103-1112.
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukemia. Nature 2015, 518, 552-555.
- Lindsley, R.C.; Saber, W.; Nar, B.G.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.H.; Spellman, R.R.; Lee, S.J.; Verneris, M.R.; et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med 2017, 376, 536-547. [CrossRef]
- Smith, S.M.; Le Beau, M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Varrdiman, J.W.; Rowley, J.D.; Lrason, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 2003, 102, 43-52. [CrossRef] [PubMed]
- Zeidan, A.M.; Al Ali, N.; Barnard, J.; Padron, E.; Lancet, J.E.; Sekeres, M.A.; Steensma, D.P.; DeZern, A.; Roboz, G.; Jabbour, E.; et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS clinical research consortium. Leukemia, 31, 1391-1397.
- Hiwase, D.; Hahn, C.; Tran, E.N.H.; Chhetri, R.; Baronwal, A.; Al-kali, A.; Sharplin, K.; Hillins, R.; Greipp, P.; et al. TP53 mutation in therapy-related myeloid neoplasms defines a distinct molecular subtype. Blood 2023, 141, 1087-1091. [CrossRef] [PubMed]
- Shah, M.V.; Tran, E.H.N.; Shah, S.; Chhetri, R.; Baranawl, A.; Ladon, D.; Shultz, C.; Al-kali, A.; Brown, A.L.; Chen, D.; et al. TP 53 mutation variant allele frequency of ≥10% is associated with poor prognosis in therapy-related myeloid neoplasms. Blood Cancer J 2023, 13, 51. [CrossRef] [PubMed]
- Bao, Z.; Li, B.; Qin, T.; Xu, Z.; Qu, S.; Jia, Y.; Li, C.; Pan, L.; Gao, Q.; Jiao, M.; et al. Molecular characteristics and clinical implications of TP53 mutations in therapy-related myelodysplastic syndromes. Blood Cancer J 2025, 15, 58. [CrossRef]
- Lessard, M.; Hélias, C.; Struski, S.; Perrusson, N.; Uetwiller, F.; Mozziconacci, M.J.; Lafage-Pochitaloff, M.; Dastugue, N.; Terré, C.; Brizard, F.; et al. Groupe francophone de cytogénétique hématologique. Fluorescence in situ hybridization analysis of 110 hematopoietic disorders with chromosome 5 abnormalities: do de novo and therapy-related myelodysplastic syndrome-acute myeloid leukemia actually differ? Cancer Genet Cytogenet 2007, 176, 1-21. [CrossRef]
- Fleti, F.; Singh, A.; Al-Kali, A.; Foran, J.M.; Elliott, M.A.; Begna, K.; Badar, T.; Khera, N.; Shah, M.V.; Alkhateeb, H.B.; et al. Therapy-related myelodysplastic syndromes with isolated del(5q): a comparative analysis of phenotype and long-term survival. Blood 2022, 140, 6940-6941. [CrossRef]
- Jadersten, M.; Saft, L.; Pellegatti, A.; Gohring, G.; Wainscopat, J.S.; Boutwood, J.; Porwit, A.; Schagelberger, B.; Hellstrom-Lindberg, E. Clonal heterogeneity in the 5q- syndrome: p53 expressing progenitors prevail during lenalidomide treatment and expand at disease progression. Haematologica 2009, 94, 1762-1766. [CrossRef]
- Schagelberger, B.; Giai, V.; Pellagatti, A.; Saft, L.; Dimitriou, M.; Jansson, M.; Jadersten, M.; Grandien, A.; Davagi, I.; Neuberg, D.S.; et al. Progression in patients with low- and intermediate-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 2017, 102, 498-508.
- Mossner, M.; Jann, J.C.; Witting, J.; Nolte, F.; Fey, S.; Nowak, V.; Oblander, J.; Pressler, J.; Palme, I.; Xanthopoulos, C.; et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood 2016, 128, 1246-1259. [CrossRef]
- Mossner, M.; Jann, J.C.; Nowak, D.; Platzbecker, U.; Giagounidis, A.; Gotze, K.; Prevalence, clonal dynamics and clinical impact of TP53 mutations in patients with myelodysplastic syndrome with isolated deletion (5q) treated with lenalidomide: results from a prospective multicenter study of the German MDS study group (GMDS). Leukemia 2016, 30, 1956-1959. [CrossRef] [PubMed]
- Lode, L.; Menard, A.; Flet, L.; Richebourg, S.; Loirat, M.; Eveillard, M.; et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 2018, 103, e143-e146. [CrossRef] [PubMed]
- Sperling, A.S.; Guerra, V.; Kennedy, J.A.; Yan, Y.; Hsu, J, Wang, F.; Nguyen, A.T.; Miller, P.G.; McConkey, M.; Quevedo Barrios, V.; et al. Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood 2022, 140, 1753-1763.
- Abdallah, M.; Reichard, K.; Gnagat, N.; Tefferi, A. Treatment-emergent mutations in myelodysplastic syndrome with del (5q)-lenalidomide related or disease-intrinsic clonal evolution. Blood Cancer J 2024, 14, 49. [CrossRef]
- Feurstein, S.; Thomay, K.; Hofmann, W.; Buesche, G.; Kreipe, H.; Thol, F.; Heuser, M.; Ganser, A.; Schlegelberger, B.; Gihring, G. Routes of clonal evolution into complex karyotypes in myelodysplastic syndrome patients with 5q deletion. Int Mol Sci 2018, 19, 3269. [CrossRef]
- Merz, A.M.A.; Platzbecker, U. Treatment of lower-risk myelodysplastic syndromes. Hamatologica 2025, 110, 330-338. [CrossRef]
- McMahon, C.; Raddi, M.G.; Mohan, S.; Santini, V. New approvals in low- and intermediate-risk myelodysplastic syndromes. Am Soc Clin Oncol Educ Book 2025, 45, 1-10. [CrossRef]
- Roncador, M.; Bernard, E.; Hasserjian, R.; Boulwood, J.; Elena, C.; Galli, A.; Gurnari, C.; Mecacci, C.; Michaux, L.; Mittelman, M.; et al. A precision medicine approach to the myelodysplastic syndrome with isolated deletion 5q, fifty years after its discovery. Blood 2025, in press.
- Huber, S.; Haferlach, T.; Meggendorfer, M.; Hutter, S.; Hoermann, G.; Baer, C.; Kern, W.; Haferlach, C. SF3B1 mutated MDS: blast count, genetic co-abnormalities and their impact on classification and prognosis. Leukemia 2022, 36, 2894-2902. [CrossRef]
- Chan, O.; Al Ali, N.; Sallman, D.A.; Padron, E.; Lancet, J.E.; Komrokji, R. SF3B1 mutations and not TP53 are associated with outcomes in patients with del(5q) myelodysplastic syndromes (MDS). Blood 2020, 136 (suppl.1), 25-26. [CrossRef]
- Duetz, C.; Westers, T.; Hout, F.; Cremers, E.; Alhan, C.; Venniker-Punt, B.; Visser-Wisselaar, B.; Chitu, D.; de Graaf, A.; Smit, L.; et al. DFistinct bone marrow immunophenotypic features define the splicing factor 3B subunit 1 (SF3B1)-mutant myelodysplastic syndromes subtype. Brit J Haematol 2021, 193, 798-803. [CrossRef] [PubMed]
- Sun, X.; Gao, Q.; Arcila, M.; Roshal, M.; Zhang, Y.; Xiao, W.; Chan, A. Diagnostic challenges and proposed classification of myeloid neoplasms with overlapping features of thrombocytosis, ring sideroblasts and concurrent del(5q) and SF3B1 mutations. Haematologica 2024, 109, 2676-2681.
- Komrokji, R.S.; Schwabkey, Z.I.; Al Ali, N.K.; Aguirre, L.E.; Stahl, M.; Ball, S.; Mason, E.F.; Savona, M.R.; Santini, V.; Consagra, A.; et al. Myelodysplastic syndromes with concomitant SF3B1 mutation and deletion of the long arm of chromosome 5 (SF3B1del5q): outcomes and response to treatment. Blood 2024, 144 (suppl.1), 1845-1847. [CrossRef]
- Marinez-Heter, S.; Deng, Y.; Parker, J.; Jiang, J.; Mo, A.; Decking, T.R.; Gharaee, N.; Li, J.; Umlandt, P.; Fuller, M.; et al. Loss of lenalidomide-induced megakaryocytic differentiation leads to therapy resistance in del(5q) myelodysplastic syndrome. Nat Cell Biol 2020, 22, 526-533.
- Barreyro, L.; Sampson, A.M.; Hueneman, K.; Choi, K.; Christie, S.; Ramesh, V.; Wyder, M.; Wang, D.; Pujato, M., Greis, K.D.; et al. Dysregulated innate immune signaling cooperates with RUNX1 mutations to transform an MDS-like disease to AML. I Science 2024, 27, 109809.
- Garcia-Manero, G.; Santini, V.; Zeidan, A.M.; Komrokoji, R.S.; Pozharskaya, V.; Rose, S.; Keeperman, K.; Lai, Y.; Karsekar, S.; Aggarwal, B.; et al. Long-term transfusion independence with Luspatercept versus epoietin alfa in erythropoiesis-stimulating agent-naïve, lower-risk myelodysplastic syndromes in the COMMANDS trial. Adv Ther 2025, 42, 3676-3689. [CrossRef] [PubMed]
- Della Porta, M.G.; Garcia-Manero, G.; Santini, V.; Zeidan, A.M.; Komrokji, R.S.; Shortt, J.; Valcarcel, D.; Jonasova, A.; Dimicoli-Salazar, S.; Tiong, I.S.; et al. Luspatercept versus epoietin alfa in erythropoiesis-stimulating agent-naïve, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): primary analysis of a phase 3, open-label, randomized, controlled trial. Lancet Hematol 2024, 11, e646-e659. [CrossRef] [PubMed]
- Platzbacker, U.; Della Porta, M.G.; Santini, V.; Zeidan, A.M.; Komrokji, R.S.; Shott, J.; Valcarcel, D.; Jonasova, A.; Dimicoli-Salazar, S.; Tiong, I.S.; et al. Efficacy and safety of luspatercept versus epoietin alfa in erythropoiesis-stimulating agent-naïve, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a phase 3, open-label, randomized controlled trial. Lancet 2023, 402, 373-385. [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckskin, R.; Santini, V.; Dier-Compelo, M.; Finelli, C.; Cazzola, M.; Ilhan, D.; et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med 2020, 382, 140-151. [CrossRef]
- Oliva, E.N.; Poloni, A.; Frairia, C.; Riva, M.; Capodanno, I.; Delfino, I.M.; D’Errigo, M.G.; Mammì, C.; Ianni, G.; Zini, G.; et al. Luspatercept for the treatment of transfusion-dependent anemia in patients with myelodysplastic neoplasms with del5q, refractory/resistant/intolerant to prior treatments (QOL_ONE Phoenix). Blood 2024, 144 (suppl.1), 6741-6742. [CrossRef]
- Patsialos, I.; Kontandreopoulou, C.N.; Vlachopoulou, D.; Safylidis, C.; Syriopoulou, S.; Kalala, F.; Anastasopoulou, A.; Mantzrourani, M; et al. A myelodysplastic neoplasm with del(5q) treated with luspatercept uncovers unexplored mechanisms of action for the drug. Brit J Haematol 2024, 205, 1641-1644.
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatments of myelodysplastic syndromes: a randomized, open-label, phase III study. Lancet Oncol 2009, 10: 223-232.
- Sekeres, M.A.; Othus, M.; List, A.F.; et al. Randomized phase II study of azacitidine alone or in combination with lenalidomide or verinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study sWOG S1117. J Cin Oncol 2017, 35, 2745-2753. [CrossRef]
- Bernal, T.; Martinez-Camblor, P.; Sanchez-Garcia, J.; et al. Effectiveness of azacitidine in unselected high-risk myelodysplastic syndromes: results from the Spanish registry. Leukemia 2015, 29, 1875-1881. [CrossRef]
- Garcia, J.S.; Platbecker, U.; Odenike, O.; Fleming, S.; Fong, C.Y.; Borate, U.; Jacoby, M.A.; Nowak, D.; Baer, M.R.; Petrlin, P.; et al. Efficacy and safety of venetoclax plus azacitidine for patients with treatment-naïve high-risk myelodysplastic syndromes. Blood 2025, 145, 1126-1135. [CrossRef]
- Bazinet, A.; Loghavi, S.; Wei, Y.; Bataller, A.; Sasaki, K.; Arani, N.; Darbaniyan, F.; Chien, K.; Hammond, D.; Bouligny, I.; et al. Leukemia 2025, 39, 2256-2265.
- Huber, S.; Haferlach, T.; Muller, H.; Meggendorfer, M.; Hutter, S.; Hoerman, G.; Haferlach, C. MDS subclassification-do we still have to count blasts? Leukemia 2023, 37, 942-945. [CrossRef]
- Molina, A.; Khanna, V.; Jensen, A.; Stehr, H.; Tan, B.; Yatsenko, S.; Greenberg, P.L. Molecular taxonomy of MDS/CMML patients influences responses to hypomethylating agents and clinical outcomes. Leuk Res 2025, 156, 107736. [PubMed]
- Patwardhan, P.P.; Al Amri, R.; Baloda, V.; Aarabi, M.; Aggarwal, N.; Djokic, M.; Monaghan, S.A.; Moore, E.M.; Rea, B.; Bailey, N.G. Validation of clinicopathologic features of a genetic myelodysplastic syndrome classification in an independent cohort. J Hematopathol 2025, 18, 42. [CrossRef]
- Huber, S.; Haferlach, T.; Hutter, S.; Hoermann, G.; Kern, W.; Haferlach, C. Relevance of blast counts for genetic subclassification in MDS. Leukemia 2025, 39, 271-273. [CrossRef]
- Al Amri, R.; Baloda, V.; Monaghan, S.A.; Rosado, F.G.; Moore, E.M.; Rea, B.; Diokic, M.; Aggarwal, N.; Yatsenko, S.A.; Bailey, N.G. Validation of independent prognostic significance of blast count in a large cohort of MDS patients. Leukemia 2024, 38, 2064-2067. [CrossRef]
- Zampini, M.; Riva, E.; Lanino, L.; Sauta, E.; Dos Reis, R.A.; Ejarque, R.M.A.; Maggioni, G.; Termanini, A.; Merlotti, A.; Campagna, A.; et al. Characterization and clinical implications of p53 dysfunction in patients with myelodysplastic syndromes. J Clin Oncol 2025, 43, 2069-2083.
- Rasmussen, B.; Gohring, G.; Bernard, E.; Nilsson, L.; Tobiasson, M.; Jadersten, M.; Garelius, H.; Dybedal, I.; Gronbaeck, K.; Ejerblad, E.; et al. Randomized phase II study of azacitidine ± lenalidomide in higher-risk myelodysplastic syndromes and acute myeloid leukemia with a karyotype including Del(5q). Leukemia 2022, 36, 1436-1439. [CrossRef]
- Rasmussen, B.; Nilsson, L.; Tobiasson, M.; Jadersten, M.; Garelius, H.; Dybedal, I.; Gronbaek, K.; Ejerblad, E.; Lorenz, F.; Flogegard, M.; et al. Influence of cytogenetics on the outcome of patients with high-risk myelodysplastic syndrome including deletion 5q treated with azacitidine with or without lenalidomide. Genes Chrom Cancer 2025, 64, e70029. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



