Submitted:
11 October 2025
Posted:
13 October 2025
You are already at the latest version
Abstract
Neurodevelopmental disorders (NDDs), including autism spectrum disorder, intellectual disability, and attention deficit hyperactivity disorder, are increasingly recognized as disorders of early brain construction arising from defects in neural stem and progenitor cell (NSPC) proliferation. NSPCs are responsible for generating the diverse neuronal and glial lineages that establish cortical architecture and neural circuitry; thus, their expansion must be tightly coordinated by intrinsic cell cycle regulators and extrinsic niche-derived cues. Disruption of these mechanisms—through genetic mutations, epigenetic dysregulation, or environmental insults—can perturb the balance between NSPC self-renewal and differentiation, resulting in aberrant brain size and connectivity. Recent advances using animal models and human pluripotent stem cell–derived brain organoids have identified key signaling pathways, including Notch, Wnt, SHH, and PI3K–mTOR, as central hubs integrating proliferative cues, while transcriptional and chromatin regulators such as PAX6, CHD8, SETD5, and ANKRD11 govern gene expression essential for proper NSPC cycling. Furthermore, prenatal exposure to teratogens such as Zika virus infection, valproic acid, or metabolic stress in phenylketonuria can recapitulate proliferation defects and microcephaly, underscoring the vulnerability of NSPCs to environmental perturbation. This review summarizes emerging insights into the molecular and cellular mechanisms by which defective NSPC proliferation contributes to NDD pathogenesis, highlighting convergence among genetic and environmental factors on cell cycle control. A deeper understanding of these pathways may uncover shared therapeutic targets to restore neurodevelopmental trajectories and mitigate disease burden.
Keywords:
Neural stem and progenitor cells
; Neurodevelopmental disorders
; Cell cycle regulation
; Signal transduction
; Chromatin remodeling
; Microcephaly and macrocephaly
; Environmental teratogens
1. Introduction
Neurodevelopmental disorders (NDDs) encompass a broad spectrum of conditions including autism spectrum disorder (ASD), intellectual disability (ID), and attention deficit hyperactivity disorder (ADHD) [1]. These disorders affect approximately 1 in 20 children globally [2] and pose significant lifelong challenges for affected individuals and their families [3,4]. Despite their clinical heterogeneity, these disorders are believed to share a common underlying feature: the disruption of early neurodevelopmental processes critical for establishing proper brain architecture and function. [5,6].
Among the earliest processes in brain formation is the proliferation of embryonic cortical neural stem and progenitor cells (NSPCs), which give rise to the neurons and glial cells that populate the central nervous system [5,7,8]. In principle, neural stem cells generate neural progenitors that predominantly give rise to neurons, whereas glial progenitors are thought to exclusively produce astrocytes and oligodendrocytes (Figure 1). However, it has revealed that neural progenitor cells exhibit glial characteristics and are capable of producing not only neurons but also astrocytes and oligodendrocytes [9], thereby blurring the distinction between dedicated glial progenitors and multipotent neural progenitors. Notably, neurogenesis precedes gliogenesis, commencing at approximately embryonic day 10 in mice (gestational week 10 in humans) and shifting toward gliogenesis around embryonic day 16 in mice (gestational week 16 in humans) [10,11,12]. This temporal progression indicates a dynamic change in the developmental potential of neural progenitor cells. Although mounting evidence implicates glial abnormalities in NDDs [13,14], the specific contribution of aberrant gliogenesis remains less well defined. Accordingly, in this review, we highlight defective NSPC proliferation and its relevance to the pathogenesis of NDDs.
NSPCs are highly sensitive to both intrinsic and extrinsic cues that govern their proliferation, survival, and differentiation [15,16,17,18]. Tight regulation of NSPC expansion and lineage specification is indispensable for the proper establishment of brain size, cortical lamination, and neural circuitry [19,20,21,22]. Disruptions in these processes can yield profound neuroanatomical and functional abnormalities, exemplified by microcephaly and macrocephaly [23,24,25], as well as aberrant synaptic organization frequently observed in neurodevelopmental disorders [26] (Figure 2).
Leveraging animal models and pluripotent stem cell–derived NSPCs, recent studies have implicated a diverse spectrum of genetic factors—many of which are mutated in patients with NDDs—in the regulation of NSPC proliferation [27,28]. In parallel, NDD-associated environmental insults during gestation have also been shown to perturb NSPC proliferation [29,30,31,32]. Collectively, these findings underscore defective NSPC proliferation as a critical early pathogenic event in NDDs, emphasizing the necessity of elucidating this process to inform the development of targeted therapeutic strategies. In this review, we summarize current knowledge of the molecular mechanisms underlying aberrant NSPC proliferation, with particular focus on the regulation of the cell division cycle, a fundamental determinant of NSPC expansion.
2. Neural Stem and Progenitor Cells in Normal Brain Development
The earliest NSPCs in the developing brain are neuroepithelial cells, a population of neural stem cells that initially undergo symmetric divisions to expand the progenitor pool during early neurogenesis [7]. As the neural tube matures, these neuroepithelial cells progressively transform into radial glial cells—highly polarized progenitors that extend across the entire thickness of the embryonic cortex. Radial glia generate both neurons and intermediate progenitors through symmetric and asymmetric divisions [33]. Intermediate progenitors (IPs), residing predominantly within the subventricular zone, possess more restricted developmental potential and primarily give rise to neurons via neurogenic divisions [34,35] (Figure 3).
NSPC proliferation is orchestrated by a complex interplay of intrinsic and extrinsic mechanisms. Intrinsic regulation encompasses mitotic spindle orientation, chromatin remodeling, and the expression of lineage-determining transcription factors [36,37]. For instance, the balance between symmetric and asymmetric divisions, essential for self-renewal and differentiation, respectively, is critically shaped by the orientation of the mitotic spindle and the polarized distribution of cell fate determinants [20,38]. Extrinsic signals from the neural stem cell niche—including Notch, Wnt, Sonic Hedgehog (SHH), fibroblast growth factor (FGF), and insulin-like growth factor-I (IGF-I)—further modulate NSPC behavior [39,40,41,42,43,44]. In addition, interactions with the extracellular matrix and local metabolic states play pivotal roles in regulating proliferative capacity and lineage specification [45,46].
3. Cell Cycle Regulation in NSPC Proliferation
Because terminally differentiated cells have lost their proliferative capacity, disruptions in NSPC proliferation can arise through two principal mechanisms: direct perturbation of cell division or indirect perturbation mediated by aberrant differentiation timing (Figure 4). Consequently, abnormal NSPC proliferation often stems from mutations in genes that compromise the regulation of either cell cycle progression or differentiation.
Proper and timely cell division is tightly regulated through coordinated control of cell cycle progression. During the G1 phase, diploid cells prepare for DNA synthesis, which occurs in the S phase. The resulting tetraploid cells then enter the G2 phase, where they prepare for mitosis, ultimately producing two daughter cells in the M phase [37,47,48] (Figure 5). In response to growth factors described in Section 2, signaling cascades such as the MAPK and PI3K/mTOR pathways are activated, leading to the induction of Cyclin D. Cyclin D subsequently activates CDK4/6, which is required for entry into S phase and DNA replication. Cyclin D–CDK4/6 activity further induces Cyclin E, which activates CDK2 to drive G1 phase progression. Thereafter, the Cyclin A–CDK2 complex plays a pivotal role in initiating DNA replication by activating the replication machinery, while also coordinating the timely activation of the Cyclin B–CDK1 complex, which governs progression into mitosis.
The duration of the cell cycle is a critical determinant of NSPC proliferation and differentiation. As NSPCs progress toward neuronal differentiation, the overall cell cycle lengthens, predominantly due to an extended G1 phase. This prolonged G1 phase is thought to be essential, as it provides sufficient time for the accumulation and activity of fate-determining factors that promote differentiation [37,49]. Consistently, experimental elongation of the G1 phase via CDK inhibitors drives NSPC differentiation [50]. Conversely, artificial shortening of the G1 phase through overexpression of Cyclin D or Cyclin E delays differentiation and maintains NSPCs in a proliferative, undifferentiated state [51,52]. These findings indicate that inappropriate inhibition of cell cycle progression can prematurely trigger differentiation and deplete the NSPC pool, thereby contributing to the pathogenesis of NDDs.
4. Impairment of NSPC Proliferation in Neurodevelopmental Disorders
Abnormal cell cycle progression is largely driven by dysregulated expression of key cell cycle regulators, often as a consequence of disrupted signaling pathways [53,54], aberrant activity of transcription factors, and altered chromatin remodeling [55,56,57]. These perturbations collectively impair NSPC proliferation and compromise normal brain development.
4.1. Genetic Factors
Notch signaling is a master regulator of NSPC proliferation. Upon ligand activation (Jagged, Delta), Notch receptors undergo proteolytic cleavage to release the Notch intracellular domain (NICD), which associates with RBPJ and MAML to drive transcription of target genes, notably the Hes and Hey families [58] (Figure 6a). Hes and Hey proteins repress proneural genes, thereby maintaining NSPCs in an undifferentiated state and preventing premature neuronal differentiation [59]. Mutations in NOTCH2NL (encoding a Notch receptor) and DLL1 (encoding a Delta ligand) have been identified in NDD patients [60,61]. Importantly, NOTCH2NL deletions are associated with microcephaly, whereas duplications result in macrocephaly [60]. Similarly, DLL1 mutations have been linked to microcephaly [61], underscoring the essential role of Notch signaling in NSPC expansion.
The Wnt pathway is also pivotal in regulating NSPC proliferation. In the absence of Wnt ligands, β-catenin is continuously targeted for degradation by the destruction complex [62]. Wnt binding to Frizzled and LRP5/6 recruits Dishevelled (Dvl), sequestering the destruction complex at the membrane and preventing β-catenin degradation. Stabilized β-catenin then accumulates, translocates to the nucleus, and partners with TCF/LEF transcription factors to activate target genes such as Cyclin D, which promote NSPC proliferation [63,64] (Figure 6b). Pathogenic missense variants in ZNRF3, a negative regulator of Wnt signaling that ubiquitinates and inactivates Frizzled, have been identified in NDD patients [65]. Variants clustered in the RING domain impair Frizzled ubiquitination, leading to excessive Wnt activity, aberrant NSPC proliferation, and macrocephaly. Conversely, microcephaly-associated ZNRF3 variants localize to the RSPO-binding domain, conferring resistance to RSPO inhibition and resulting in hyperactive Frizzled degradation, diminished Wnt signaling, reduced NSPC proliferation, and microcephaly [65,66].
The SHH pathway likewise regulates NSPC proliferation. SHH binding to the PTCH1 receptor relieves its inhibition of Smoothened (SMO), thereby enabling SMO activation [67]. Activated SMO promotes nuclear translocation of GLI transcription factors, which induce expression of proliferation-associated genes, including Cyclin D and Cyclin E [67] (Figure 6c). Consistently, mutations in SHH or PTCH1 have been identified in patients with microcephalic NDDs [68,69].
FGF and IGF-I also critically regulate NSPC proliferation. These ligands bind to their receptors (FGFR and IGF-IR, respectively), activating shared downstream pathways—RAS-MAPK (RAS → RAF → MEK → MAPK) and PI3K-mTOR (PI3K → AKT → mTOR)—that are most extensively characterized [70,71,72]. The MAPK cascade phosphorylates transcription factors that upregulate cell cycle drivers such as Cyclin D [73,74], while mTOR promotes progression by degrading ARF [75], and repressing CDK inhibitors p21 and p27 [76] (Figure 6d). PI3K-mediated phosphorylation of PIP2 to PIP3 activates AKT, whereas PTEN antagonizes this pathway by dephosphorylating PIP3 back to PIP2, thereby acting as a brake on NSPC proliferation [77]. The pathological relevance of these signaling cascades to NDDs is underscored by numerous genetic findings: loss-of-function mutations in FGFR1 [78], FGFR3 [79], FGF8 [80,81], IGF-1 [82,83,84], IGF-IR [85,86], and AKT3 [87] have been linked to microcephalic NDDs. Conversely, loss-of-function mutations in PTEN [88,89,90,91], stabilizing mutations in Cyclin D2 [92], and activating mutations in PI3K [91,93,94], AKT3 [87,93,94], and mTOR [91,93,95] are consistently associated with macrocephalic NDDs.
In addition to the signaling pathways described above, several transcriptional regulators critically modulate NSPC proliferation. Loss of function of the transcription factor ARX has been associated with NDDs accompanied by microcephaly [96]. ARX deficiency leads to upregulation of p57, a Cip/Kip family CDK inhibitor (Figure 6e), which is normally repressed by ARX. This results in premature cell cycle exit and depletion of progenitor pools [97]. Moreover, ARX loss elevates expression of the transcription factor OLIG2 [98], which in turn reduces neurogenesis and contributes to overall brain size reduction [99]. Mechanistically, OLIG2 overexpression downregulates PAX6, a key transcription factor governing both proliferation and differentiation of cortical progenitors [98,99].
PAX6 positively regulates several cell cycle–promoting genes, including CDK2, CDK4, and Cyclin E, thereby enhancing NSPC proliferation during embryonic brain development [100] (Figure 6e). Consistently, compound heterozygous PAX6 mutations are associated with microcephaly in human patients [101,102]. Interestingly, PAX6 also functions as a repressor of NSPC cell cycle progression, as evidenced by studies in mice carrying Pax6 mutations [103] or conditional Pax6 deletions in developing neurons [104]. This inhibitory effect is likely mediated by suppression of CDK6 expression. Supporting evidence includes PAX6 binding to regulatory regions surrounding the Cdk6 locus [104], rapid upregulation of Cdk6 following acute Pax6 loss [105], and reduced NSPC proliferation upon CDK6 inhibition or gene knockout [104,106]. Consistently, CDK6 mutations have been linked to microcephaly [107].
Notably, PAX6 simultaneously promotes neuronal differentiation by activating neurogenic gene expression, suggesting a dual role in balancing NSPC proliferation and neurogenesis [100]. Indeed, experimental evidence from Pax6-mutant mice demonstrates that Pax6 loss reduces NSPC proliferation, whereas Pax6 overexpression accelerates neuronal differentiation at the expense of progenitor expansion [100]. Thus, both upregulation and downregulation of Pax6 ultimately converge on a common outcome—microcephaly. This sensitivity is partly managed by its antagonistic interactions within a network of neurogenic transcription factors such as Neurog2, Ascl1, Hes1 [100]. These findings suggest that the seemingly paradoxical roles of PAX6—in both promoting and restraining NSPC proliferation—are governed by context-dependent mechanisms, including gene dosage effects and interactions with other transcriptional regulators.
Mutations in CHD8, a gene encoding a chromatin remodeler, have been strongly associated with macrocephaly, suggesting that CHD8 acts as a transcriptional brake on NSPC proliferation. Consistently, haploinsufficiency of CHD8—commonly observed in NDD patients—shortens the cell cycle and enhances NSPC proliferation [108]. This phenotype has been attributed to the upregulation of Cyclin E, a key driver of S-phase entry [108] (Figure 6e). Similarly, germline Chd8 haploinsufficiency in mice accelerates NSPC proliferation, accompanied by dysregulated expression of cell cycle–associated genes [109]. Although these findings support a role for CHD8 as a negative regulator of cell proliferation, other studies have reported that CHD8 promotes NSPC proliferation at both cellular and organismal levels [110,111]. These discrepancies may reflect methodological differences, as studies reporting pro-proliferative roles primarily employed mRNA knockdown rather than genetic knockout models, suggesting that the degree of CHD8 reduction critically influences NSPC outcomes.
Gene dosage effects on chromatin remodeling also extend to components of the BAF (SWI/SNF) complex. Duplication of ARID1A (also known as BAF250A), a core subunit of the BAF complex, has been associated with microcephaly [112]. Conversely, haploinsufficiency of ARID1B (also known as BAF250B), a mutually exclusive paralog of ARID1A, similarly results in microcephaly [113,114,115], implying antagonistic roles in NSPC proliferation—ARID1A-BAF functioning as a repressor and ARID1B-BAF as an activator. Supporting this notion, conditional deletion of Arid1a in murine NSPCs enhances proliferation and upregulates cell cycle–related genes [116] (Figure 6e). By contrast, the role of ARID1B in NSPC proliferation remains largely unexplored. Nonetheless, Arid1b-deficient murine embryonic stem cells, as well as patient-derived fibroblasts with ARID1B haploinsufficiency, exhibit reduced proliferation accompanied by downregulation of CDC20, a key positive regulator of cell cycle progression [117,118]. Whether NSPC proliferation is similarly attenuated by ARID1B reduction remains an open question warranting further investigation.
Another gene whose mutation disrupts NSPC proliferation through transcriptional dysregulation is SETD5, an epigenetic regulator that modulates rDNA transcription and translation of key cell cycle proteins such as Cyclin D [119] (Figure 6e). Haploinsufficiency of SETD5 leads to reduced rRNA synthesis and impaired NSPC proliferation [119]. In humans, heterozygous loss-of-function mutations in SETD5 cause a neurodevelopmental disorder termed IDD23 (also known as MRD23) [120]. Intriguingly, pathogenic SETD5 variants have also been identified in patients presenting with KBG-like phenotypes—syndromes that phenotypically overlap with KBG syndrome [121,122].
Haploinsufficiency of another epigenetic regulator, ANKRD11, is recognized as the primary genetic cause of KBG syndrome, suggesting a functional interplay between ANKRD11 and SETD5 [120]. Supporting this notion, recent studies have shown that ANKRD11 positively regulates SETD5 expression, thereby sustaining translational capacity and neuronal proliferation—processes essential for mitigating NDD pathogenesis [123] (Figure 6e). Notably, both IDD23 and KBG syndrome patients frequently exhibit microcephaly, underscoring the crucial role of SETD5- and ANKRD11-mediated control of NSPC proliferation in maintaining proper brain development [124,125,126].
Key regulators that directly control NSPC proliferation also include centrosomal and spindle apparatus proteins such as ASPM, CDK5RAP2, WDR62, and MCPH1, mutations in which are causatively linked to microcephaly in humans [127,128]. Defects in these genes disrupt mitotic spindle orientation and impair symmetric cell divisions, resulting in premature depletion of the progenitor pool and consequent reduction in cortical size [127,128] (Figure 6f).
4.2. Environmental Factors
Environmental factors can also profoundly impact NSPC proliferation. Maternal infection with the Zika virus has been firmly established as a cause of congenital microcephaly and NDDs in offspring [129,130,131,132]. Consistent with these observations, Zika virus infection disrupts NSPC proliferation in developing mouse brains [133,134]], as well as in human three-dimensional brain organoids [135,136]. Although numerous cell cycle–related genes are downregulated [137,138], accumulating evidence indicates that premature activation of CDK1 and unscheduled mitotic entry, together with centrosomal amplification, lead to aberrant chromosome segregation, resulting in NSPC proliferation defects and cell death [137,138,139,140] (Figure 7a). This cascade is considered a primary mechanism underlying the proliferation defects observed in Zika virus–infected NSPCs.
In utero drug exposure also contributes to NDDs associated with microcephaly. Valproic acid (VPA), a widely used antiepileptic and mood stabilizer, has been shown to increase the risk of NDDs with microcephaly [141,142]. Experimental studies in mice have demonstrated that embryonic exposure to VPA induces microcephaly and behavioral phenotypes resembling NDDs [143,144,145], ruling out the possibility that maternal psychiatric conditions alone account for the observed abnormalities. Mechanistically, VPA exposure impairs NSPC proliferation in mice through upregulation of several cell cycle inhibitors [145] (Figure 7b). In human brain organoids, ARF—but not other CDK inhibitors—was significantly induced following VPA treatment, implicating ARF as a key mediator of NSPC proliferation defects [145] (Figure 7b).
Another environmental and metabolic risk factor for microcephalic NDDs is phenylketonuria (PKU), arising from either maternal or inborn deficiency [146,147]. Most PKU cases result from pathogenic loss-of-function variants in PAH, encoding phenylalanine hydroxylase, the enzyme responsible for converting phenylalanine to tyrosine. This defect leads to elevated phenylalanine levels in both blood and brain [146,147]. Studies using human iPSC-derived brain organoids have demonstrated that high phenylalanine concentrations selectively impair NSPC populations, while mature neurons and glia remain largely unaffected, highlighting the heightened vulnerability of NSPCs to metabolic stress [148]. Transcriptomic profiling further demonstrated a broad downregulation of genes essential for cell cycle progression, including CDT1 and MCMs involved in the S phase, as well as ASPM and CDK5RAP2 associated with the M phase [148] (Figure 7c). These findings suggest that impaired NSPC proliferation, driven by defective cell cycle regulation, directly contributes to microcephalic NDD in PKU.
5. Future Directions and Therapeutic Perspectives
The growing body of evidence implicating defective NSPC proliferation in the etiology of NDDs highlights the urgent need to translate mechanistic insights into therapeutic strategies. Future research should aim to delineate how distinct genetic mutations, epigenetic dysregulation, and environmental exposures converge on shared cell cycle and signaling pathways in NSPCs. Integration of single-cell transcriptomics, proteomics, and epigenomics with brain organoid models and in vivo systems will enable the identification of temporal and lineage-specific vulnerabilities that underlie aberrant proliferation. Such high-resolution approaches may reveal common “molecular bottlenecks” in NSPC regulation, offering tractable targets for intervention.
From a therapeutic standpoint, several avenues hold promise. Pharmacological modulation of core signaling cascades—such as Notch, Wnt, SHH, and PI3K–mTOR—could restore balanced NSPC proliferation, provided that interventions are carefully titrated to avoid exacerbating either hypo- or hyperplastic states. Small-molecule inhibitors of CDKs or pathway-specific agonists/antagonists represent potential candidates, some of which are already under clinical investigation for oncology and may be repurposed for NDDs. Epigenetic regulators such as CHD8, SETD5, and ANKRD11, whose dysfunction directly impairs NSPC proliferation, are also attractive therapeutic nodes, particularly as pharmacological modulators of chromatin structure and transcriptional machinery continue to expand.
Ultimately, future efforts will benefit from interdisciplinary approaches that link developmental neurobiology, stem cell biology, and translational neuroscience. By bridging the gap between mechanistic understanding and therapeutic application, these studies have the potential not only to uncover predictive biomarkers of disease severity but also to lay the groundwork for preventive and restorative interventions in NDDs. The challenge ahead is to transform knowledge of NSPC biology into tangible therapies that can improve neurodevelopmental trajectories and quality of life for affected individuals.
Author Contributions
Conceptualization, T.N. and M.N.; investigation, A.S.; E.N.; T.N. and M.N.; writing—original draft preparation, A.S.; E.N.; and T.N.; writing—review and editing, T.N.; M.N.; and T.H.; supervision, T.N.; project administration, T.N.; funding acquisition, T.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI (grant number: 23K06367).
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We would like to also thank lab members for the productive discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Morris-Rosendahl, D.J. and M.A. Crocq, Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci 2020, 22, 65–72. [Google Scholar] [CrossRef]
- Francés, L.; et al. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: a systematic review in accordance with the PRISMA criteria, in Child Adolesc Psychiatry Ment Health. 2022, © 2022. The Author(s). England. p. 27.
- Picardi, A.; et al. Parental Burden and its Correlates in Families of Children with Autism Spectrum Disorder: A Multicentre Study with Two Comparison Groups. Clin Pract Epidemiol Ment Health 2018, 14, 143–176. [Google Scholar] [CrossRef]
- Materula, D.; et al. Needs of children with neurodevelopmental disorders and medical complexity: Caregiver perspectives. Res Dev Disabil 2024, 153, 104815. [Google Scholar] [CrossRef]
- Zhou, Y. H. Song, and G.L. Ming, Genetics of human brain development. Nat Rev Genet 2024, 25, 26–45. [Google Scholar] [CrossRef]
- Dionne, O. S. Sabatié, and B. Laurent, Deciphering the physiopathology of neurodevelopmental disorders using brain organoids. Brain 2025, 148, 12–26. [Google Scholar] [CrossRef]
- Stiles, J. and T.L. Jernigan, The basics of brain development. Neuropsychol Rev 2010, 20, 327–48. [Google Scholar] [CrossRef]
- Ladran, I.; et al. Neural stem and progenitor cells in health and disease. Wiley Interdiscip Rev Syst Biol Med 2013, 5, 701–15. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A. and A. Alvarez-Buylla, The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 2009, 32, 149–84. [Google Scholar] [CrossRef]
- Sojka, C. and S.A. Sloan, Gliomas: a reflection of temporal gliogenic principles. Commun Biol 2024, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Vivi, E. and B. Di Benedetto, Brain stars take the lead during critical periods of early postnatal brain development: relevance of astrocytes in health and mental disorders. Mol Psychiatry 2024, 29, 2821–2833. [Google Scholar] [CrossRef]
- Blackshaw, S. and M. Cayouette, Timing neural development and regeneration. Curr Opin Neurobiol 2025, 91, 102976. [Google Scholar] [CrossRef]
- Molofsky, A.V.; et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 2012, 26, 891–907. [Google Scholar] [CrossRef]
- Séjourné, G. and C. Eroglu, Astrocyte-neuron crosstalk in neurodevelopmental disorders. Curr Opin Neurobiol 2024, 89, 102925. [Google Scholar] [CrossRef]
- Temple, S. ; The development of neural stem cells. Nature 2001, 414, 112–7. [Google Scholar] [CrossRef]
- Götz, M. and W.B. Huttner, The cell biology of neurogenesis. Nat Rev Mol Cell Biol 2005, 6, 777–88. [Google Scholar] [CrossRef]
- Urbán, N. and F. Guillemot, Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci 2014, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, H.; et al. Evaluation of the susceptibility of neurons and neural stem/progenitor cells derived from human induced pluripotent stem cells to anticancer drugs. J Pharmacol Sci 2019, 140, 331–336. [Google Scholar] [CrossRef]
- Farkas, L.M. and W.B. Huttner, The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Curr Opin Cell Biol 2008, 20, 707–15. [Google Scholar] [CrossRef] [PubMed]
- Homem, C.C. M. Repic, and J.A. Knoblich, Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 2015, 16, 647–59. [Google Scholar] [CrossRef] [PubMed]
- Packer, A. ; Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev 2016, 64, 185–95. [Google Scholar] [CrossRef]
- Ernst, C. ; Proliferation and Differentiation Deficits are a Major Convergence Point for Neurodevelopmental Disorders. Trends Neurosci 2016, 39, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012, 151, 1097–112. [Google Scholar] [CrossRef] [PubMed]
- Sun, T. and R.F. Hevner, Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci 2014, 15, 217–32. [Google Scholar] [CrossRef]
- Fei, J.F. C. Haffner, and W.B. Huttner, 3' UTR-dependent, miR-92-mediated restriction of Tis21 expression maintains asymmetric neural stem cell division to ensure proper neocortex size. Cell Rep 2014, 7, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H. D.V. Hansen, and A.R. Kriegstein, Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef]
- Li, M.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362(6420).
- Courchesne, E. V.H. Gazestani, and N.E. Lewis, Prenatal Origins of ASD: The When, What, and How of ASD Development. Trends Neurosci 2020, 43, 326–342. [Google Scholar] [CrossRef]
- Fujiki, R.; et al. A proapoptotic effect of valproic acid on progenitors of embryonic stem cell-derived glutamatergic neurons. Cell Death Dis 2013, 4, e677. [Google Scholar] [CrossRef]
- Fujimura, K.; et al. In Utero Exposure to Valproic Acid Induces Neocortical Dysgenesis via Dysregulation of Neural Progenitor Cell Proliferation/Differentiation. J Neurosci 2016, 36, 10908–10919. [Google Scholar] [CrossRef]
- Tsukada, T.; et al. Mid-pregnancy maternal immune activation increases Pax6-positive and Tbr2-positive neural progenitor cells and causes integrated stress response in the fetal brain in a mouse model of maternal viral infection. IBRO Neurosci Rep 2021, 11, 73–80. [Google Scholar] [CrossRef]
- McEwan, F. J.D. Glazier, and R. Hager, The impact of maternal immune activation on embryonic brain development. Front Neurosci 2023, 17, 1146710. [Google Scholar] [CrossRef]
- Casas Gimeno, G. and J. Paridaen, The Symmetry of Neural Stem Cell and Progenitor Divisions in the Vertebrate Brain. Front Cell Dev Biol 2022, 10, 885269. [Google Scholar] [CrossRef]
- Pontious, A.; et al. Role of intermediate progenitor cells in cerebral cortex development. Dev Neurosci 2008, 30(1-3), 24-32.
- Ashitomi, H.; et al. Cullin-RING Ubiquitin Ligases in Neurodevelopment and Neurodevelopmental Disorders. Biomedicines 2025, 13(4).
- Ahmed, S.; et al. Transcription factors and neural stem cell self-renewal, growth and differentiation. Cell Adh Migr 2009, 3, 412–24. [Google Scholar] [CrossRef]
- Hardwick, L.J.; et al. Cell cycle regulation of proliferation versus differentiation in the central nervous system. Cell Tissue Res 2015, 359, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Taverna, E. M. Götz, and W.B. Huttner, The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 2014, 30, 465–502. [Google Scholar] [CrossRef]
- Tropepe, V.; et al. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 1999, 208, 166–88. [Google Scholar] [CrossRef]
- Arsenijevic, Y.; et al. Insulin-like growth factor-I is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci 2001, 21, 7194–202. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A. and C.A. Walsh, Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–9. [Google Scholar] [CrossRef] [PubMed]
- Wen, S. H. Li, and J. Liu, Dynamic signaling for neural stem cell fate determination. Cell Adh Migr 2009, 3, 107–17. [Google Scholar] [CrossRef]
- Imayoshi, I.; et al. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci 2010, 30, 3489–98. [Google Scholar] [CrossRef]
- Yabut, O.R.; et al. The Neocortical Progenitor Specification Program Is Established through Combined Modulation of SHH and FGF Signaling. J Neurosci 2020, 40, 6872–6887. [Google Scholar] [CrossRef]
- Long, K.R.; et al. Extracellular Matrix Components HAPLN1, Lumican, and Collagen I Cause Hyaluronic Acid-Dependent Folding of the Developing Human Neocortex. Neuron 2018, 99, 702–719.e6. [Google Scholar] [CrossRef] [PubMed]
- Scandella, V.; et al. Neural stem cell metabolism revisited: a critical role for mitochondria. Trends Endocrinol Metab 2023, 34, 446–461. [Google Scholar] [CrossRef]
- Ohnuma, S. and W.A. Harris, Neurogenesis and the cell cycle. Neuron 2003, 40, 199–208. [Google Scholar] [CrossRef]
- Pellarin, I.; et al. Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct Target Ther 2025, 10, 11. [Google Scholar] [CrossRef]
- Calegari, F.; et al. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci 2005, 25, 6533–8. [Google Scholar] [CrossRef]
- Calegari, F. nd W.B. Huttner, An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 2003, 116(Pt 24), 4947–55. [Google Scholar] [CrossRef]
- Lange, C. W.B. Huttner, and F. Calegari, Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009, 5, 320–31. [Google Scholar] [CrossRef]
- Pilaz, L.J.; et al. Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proc Natl Acad Sci U S A 2009, 106, 21924–9. [Google Scholar] [CrossRef]
- Johansson, P.A. S. Cappello, and M. Götz, Stem cells niches during development--lessons from the cerebral cortex. Curr Opin Neurobiol 2010, 20, 400–7. [Google Scholar] [CrossRef]
- Solozobova, V. N. Wyvekens, and J. Pruszak, Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev Rep 2012, 8, 813–29. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; et al. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 2016, 17, 537–49. [Google Scholar] [CrossRef]
- Hota, S.K. and B.G. Bruneau, ATP-dependent chromatin remodeling during mammalian development. Development 2016, 143, 2882–97. [Google Scholar] [CrossRef]
- Lim, Y. ; Transcription factors in microcephaly. Front Neurosci 2023, 17, 1302033. [Google Scholar] [CrossRef] [PubMed]
- Sueda, R. and R. Kageyama, Regulation of active and quiescent somatic stem cells by Notch signaling. Dev Growth Differ 2020, 62, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N. D.S. Castro, and F. Guillemot, Proneural genes and the specification of neural cell types. Nat Rev Neurosci 2002, 3, 517–30. [Google Scholar] [CrossRef] [PubMed]
- Fiddes, I.T.; et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [CrossRef]
- Fischer-Zirnsak, B.; et al. Haploinsufficiency of the Notch Ligand DLL1 Causes Variable Neurodevelopmental Disorders. Am J Hum Genet 2019, 105, 631–639. [Google Scholar] [CrossRef]
- Mehta, S. S. Hingole, and V. Chaudhary, The Emerging Mechanisms of Wnt Secretion and Signaling in Development. Front Cell Dev Biol 2021, 9, 714746. [Google Scholar] [CrossRef]
- Shtutman, M.; et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 1999, 96, 5522–7. [Google Scholar] [CrossRef]
- Liu, J.; et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther 2022, 7, 3. [Google Scholar] [CrossRef]
- Boonsawat, P.; et al. Deleterious ZNRF3 germline variants cause neurodevelopmental disorders with mirror brain phenotypes via domain-specific effects on Wnt/β-catenin signaling. Am J Hum Genet 2024, 111, 1994–2011. [Google Scholar] [CrossRef]
- Hao, H.X.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Jing, J.; et al. Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies. Signal Transduct Target Ther 2023, 8, 315. [Google Scholar] [CrossRef]
- Heussler, H.S.; et al. Extreme variability of expression of a Sonic Hedgehog mutation: attention difficulties and holoprosencephaly. Arch Dis Child 2002, 86, 293–6. [Google Scholar] [CrossRef]
- Derwińska, K.; et al. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur J Hum Genet 2009, 17, 267–71. [Google Scholar] [CrossRef]
- Xie, Y.; et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther 2020, 5, 181. [Google Scholar] [CrossRef]
- Pandey, P.; et al. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed Pharmacother 2023, 161, 114491. [Google Scholar] [CrossRef]
- Werner, H. ; The IGF1 Signaling Pathway: From Basic Concepts to Therapeutic Opportunities. Int J Mol Sci 2023, 24(19).
- Zhang, W. and H.T. Liu, MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.E. H.J. Kim, and D.R. Kim, Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; et al. S6 Kinase- and β-TrCP2-Dependent Degradation of p19Arf Is Required for Cell Proliferation. Mol Cell Biol 2015, 35, 3517–27. [Google Scholar] [CrossRef]
- Mori, S.; et al. The mTOR pathway controls cell proliferation by regulating the FoxO3a transcription factor via SGK1 kinase. PLoS One 2014, 9, e88891. [Google Scholar] [CrossRef] [PubMed]
- Skelton, P.D. ; R.V. Stan, and B.W. Luikart, The Role of PTEN in Neurodevelopment. Mol Neuropsychiatry 2020, 5(Suppl 1), 60-71.
- Hong, S.; et al. Dominant-negative kinase domain mutations in FGFR1 can explain the clinical severity of Hartsfield syndrome. Hum Mol Genet 2016, 25, 1912–1922. [Google Scholar] [CrossRef]
- Toydemir, R.M.; et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet 2006, 79, 935–41. [Google Scholar] [PubMed]
- Arauz, R.F.; et al. A Hypomorphic Allele in the FGF8 Gene Contributes to Holoprosencephaly and Is Allelic to Gonadotropin-Releasing Hormone Deficiency in Humans. Mol Syndromol 2010, 1, 59–66. [Google Scholar] [CrossRef]
- McCabe, M.J.; et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab 2011, 96, E1709–E1718. [Google Scholar] [CrossRef]
- Woods, K.A.; et al. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 1996, 335, 1363–7. [Google Scholar] [CrossRef]
- Walenkamp, M.J.; et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab 2005, 90, 2855–64. [Google Scholar] [CrossRef]
- Netchine, I.; et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab 2009, 94, 3913–21. [Google Scholar]
- Abuzzahab, M.J.; et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med 2003, 349, 2211–22. [Google Scholar] [CrossRef]
- Yang, L.; et al. IGF1R Variants in Patients With Growth Impairment: Four Novel Variants and Genotype-Phenotype Correlations. J Clin Endocrinol Metab 2018, 103, 3939–3944. [Google Scholar] [CrossRef]
- Wang, X.; et al. The phenotypic and genetic spectrum of AKT3-related neurodevelopmental condition. Sci Rep 2025, 15 7484.
- Butler, M.G.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005, 42, 318–21. [Google Scholar] [PubMed]
- Varga, E.A.; et al. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 2009, 11, 111–7. [Google Scholar] [PubMed]
- Frazier, T.W.; et al. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry 2015, 20, 1132–8. [Google Scholar]
- Yeung, K.S.; et al. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol Autism 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Mirzaa, G.; et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet 2014, 46, 510–515. [Google Scholar]
- Lee, J.H.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012, 44, 941–5. [Google Scholar]
- Rivière, J.B.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012, 44, 934–40. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; et al. Association of MTOR Mutations With Developmental Brain Disorders, Including Megalencephaly, Focal Cortical Dysplasia, and Pigmentary Mosaicism. JAMA Neurol 2016, 73, 836–845. [Google Scholar] [CrossRef]
- Kato, M.; et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat 2004, 23, 147–159. [Google Scholar]
- Colasante, G.; et al. ARX regulates cortical intermediate progenitor cell expansion and upper layer neuron formation through repression of Cdkn1c. Cereb Cortex 2015, 25, 322–35. [Google Scholar] [CrossRef]
- Lim, Y.; et al. Arx Expression Suppresses Ventralization of the Developing Dorsal Forebrain. Sci Rep 2019, 9, 226. [Google Scholar] [CrossRef]
- Liu, W.; et al. Disruption of neurogenesis and cortical development in transgenic mice misexpressing Olig2, a gene in the Down syndrome critical region. Neurobiol Dis 2015, 77, 106–16. [Google Scholar] [CrossRef]
- Sansom, S.N.; et al. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Glaser, T.; et al. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 1994, 7, 463–71. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am J Med Genet A 2009, 149a, 2543–6. [Google Scholar] [CrossRef]
- Warren, N.; et al. The transcription factor, Pax6, is required for cell proliferation and differentiation in the developing cerebral cortex. Cereb Cortex 1999, 9, 627–35. [Google Scholar] [CrossRef] [PubMed]
- Mi, D.; et al. Pax6 exerts regional control of cortical progenitor proliferation via direct repression of Cdk6 and hypophosphorylation of pRb. Neuron 2013, 78, 269–84. [Google Scholar] [CrossRef]
- Mi, D.; et al. Pax6 Lengthens G1 Phase and Decreases Oscillating Cdk6 Levels in Murine Embryonic Cortical Progenitors. Front Cell Neurosci 2018, 12, 419. [Google Scholar] [CrossRef]
- Ferguson, K.L.; et al. The Rb-CDK4/6 signaling pathway is critical in neural precursor cell cycle regulation. J Biol Chem 2000, 275, 33593–600. [Google Scholar] [CrossRef]
- Hussain, M.S.; et al. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 2013, 22, 5199–214. [Google Scholar] [CrossRef]
- Coakley-Youngs, E.; et al. Autism-associated CHD8 keeps proliferation of human neural progenitors in check by lengthening the G1 phase of the cell cycle. Biol Open 2022, 11(9).
- Gompers, A.L.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci 2017, 20, 1062–1073. [Google Scholar] [CrossRef]
- Durak, O.; et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 2016, 19, 1477–1488. [Google Scholar] [CrossRef]
- Meert, L.; et al. A CHD8-TRRAP axis facilitates MYC and E2F target gene regulation in human neural stem cells. iScience 2025, 28, 111978. [Google Scholar] [CrossRef]
- Bidart, M.; et al. Microduplication of the ARID1A gene causes intellectual disability with recognizable syndromic features. Genet Med 2017, 19, 701–710. [Google Scholar] [CrossRef]
- Nagamani, S.C.; et al. Interstitial deletion of 6q25.2-q25.3: a novel microdeletion syndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur J Hum Genet 2009, 17, 573–81. [Google Scholar] [CrossRef] [PubMed]
- Santen, G.W.; et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 2012, 44, 379–80. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 2012, 44, 376–8. [Google Scholar] [CrossRef]
- Liu, X.; et al. Arid1a regulates neural stem/progenitor cell proliferation and differentiation during cortical development. Cell Prolif 2021, 54, e13124. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; et al. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 2008, 26, 1155–65. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.C.; et al. Expanding the phenotypic spectrum of ARID1B-mediated disorders and identification of altered cell-cycle dynamics due to ARID1B haploinsufficiency. Orphanet J Rare Dis 2014, 9, 43. [Google Scholar] [CrossRef]
- Nakagawa, T.; et al. The Autism-Related Protein SETD5 Controls Neural Cell Proliferation through Epigenetic Regulation of rDNA Expression. iScience 2020, 23, 101030. [Google Scholar] [CrossRef]
- Nakagawa, T.; et al. Neurobehavioral characteristics of mice with SETD5 mutations as models of IDD23 and KBG syndromes. Front Genet 2022, 13, 1022339. [Google Scholar] [CrossRef]
- Crippa, M.; et al. SETD5 Gene Haploinsufficiency in Three Patients With Suspected KBG Syndrome. Front Neurol 2020, 11, 631. [Google Scholar] [CrossRef]
- Pascolini, G.; et al. Clinical refinement of the SETD5-associated phenotype in a child displaying novel features and KBG syndrome-like appearance. Am J Med Genet A 2022, 188, 1623–1625. [Google Scholar] [CrossRef]
- Sashiyama, S.; et al. KBG syndrome-associated protein ANKRD11 regulates SETD5 expression to modulate rRNA levels and translation. iScience 2025, 28, 112699. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.; et al. The LOVD3 platform: efficient genome-wide sharing of genetic variants. Eur J Hum Genet 2021, 29, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Loberti, L.; et al. Natural history of KBG syndrome in a large European cohort. Hum Mol Genet 2022, 31, 4131–4142. [Google Scholar] [CrossRef]
- Ahsan, N.; et al. Expanding the Genotype and Phenotype of SETD5-Related Neurodevelopmental Syndrome. Pediatr Neurol 2023, 138, 25–26. [Google Scholar] [CrossRef]
- Jayaraman, D. B.I. Bae, and C.A. Walsh, The Genetics of Primary Microcephaly. Annu Rev Genomics Hum Genet 2018, 19, 177–200. [Google Scholar] [CrossRef]
- Saade, M.; et al. A centrosomal view of CNS growth. Development 2018, 145(21).
- Rasmussen, S.A.; et al. Zika Virus and Birth Defects--Reviewing the Evidence for Causality. N Engl J Med 2016, 374, 1981–7. [Google Scholar] [CrossRef]
- Wen, Z. H. Song, and G.L. Ming, How does Zika virus cause microcephaly? Genes Dev 2017, 31, 849–861. [Google Scholar] [CrossRef]
- Nielsen-Saines, K.; et al. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med 2019, 25, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; et al. Zika Virus Infection Associated with Autism Spectrum Disorder: A Case Report. Neuroimmunomodulation 2021, 28, 229–232. [Google Scholar] [CrossRef]
- Wu, K.Y.; et al. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 2016, 26, 645–54. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; et al. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–6. [Google Scholar] [CrossRef]
- Qian, X.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Cugola, F.R.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–71. [Google Scholar] [CrossRef]
- Onorati, M.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Souza, B.S.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 2016, 6, 39775. [Google Scholar] [CrossRef]
- Wolf, B.; et al. Zika virus causes supernumerary foci with centriolar proteins and impaired spindle positioning. Open Biol 2017, 7(1).
- Rychlowska, M.; et al. Zika Virus Induces Mitotic Catastrophe in Human Neural Progenitors by Triggering Unscheduled Mitotic Entry in the Presence of DNA Damage While Functionally Depleting Nuclear PNKP. J Virol 2022, 96, e0033322. [Google Scholar] [CrossRef] [PubMed]
- Ardinger, H.H.; et al. Verification of the fetal valproate syndrome phenotype. Am J Med Genet 1988, 29, 171–85. [Google Scholar] [CrossRef]
- Almgren, M. B. Källén, and C. Lavebratt, Population-based study of antiepileptic drug exposure in utero--influence on head circumference in newborns. Seizure 2009, 18, 672–5. [Google Scholar] [CrossRef]
- Kataoka, S.; et al. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int J Neuropsychopharmacol 2013, 16, 91–103. [Google Scholar] [CrossRef]
- Nicolini, C. and M. Exp Neurol 2018, 299(Pt A), 217–227. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; et al. Aberrant induction of p19Arf-mediated cellular senescence contributes to neurodevelopmental defects. PLoS Biol 2022, 20, e3001664. [Google Scholar] [CrossRef] [PubMed]
- Elhawary, N.A.; et al. Genetic etiology and clinical challenges of phenylketonuria. Hum Genomics 2022, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Rovelli, V. and N. Longo, Phenylketonuria and the brain. Mol Genet Metab 2023, 139, 107583. [Google Scholar] [CrossRef]
- Kim, J.; et al. Neurotoxicity of phenylalanine on human iPSC-derived cerebral organoids. Mol Genet Metab 2022, 136, 132–144. [Google Scholar] [CrossRef]
Figure 1.
Differentiation trajectory of neuron, astrocyte and oligodendrocyte. In principle, glial cells arise from glial progenitors; however, studies on neural stem and progenitor cells have demonstrated that neural progenitors themselves exhibit glial-like properties and can also function as glial progenitors. Three-quarter circular arrows denote self-renewal. Blue arrows indicate neurogenesis, purple arrows represent astrogenesis, and green arrows denote oligodendrogenesis.
Figure 1.
Differentiation trajectory of neuron, astrocyte and oligodendrocyte. In principle, glial cells arise from glial progenitors; however, studies on neural stem and progenitor cells have demonstrated that neural progenitors themselves exhibit glial-like properties and can also function as glial progenitors. Three-quarter circular arrows denote self-renewal. Blue arrows indicate neurogenesis, purple arrows represent astrogenesis, and green arrows denote oligodendrogenesis.
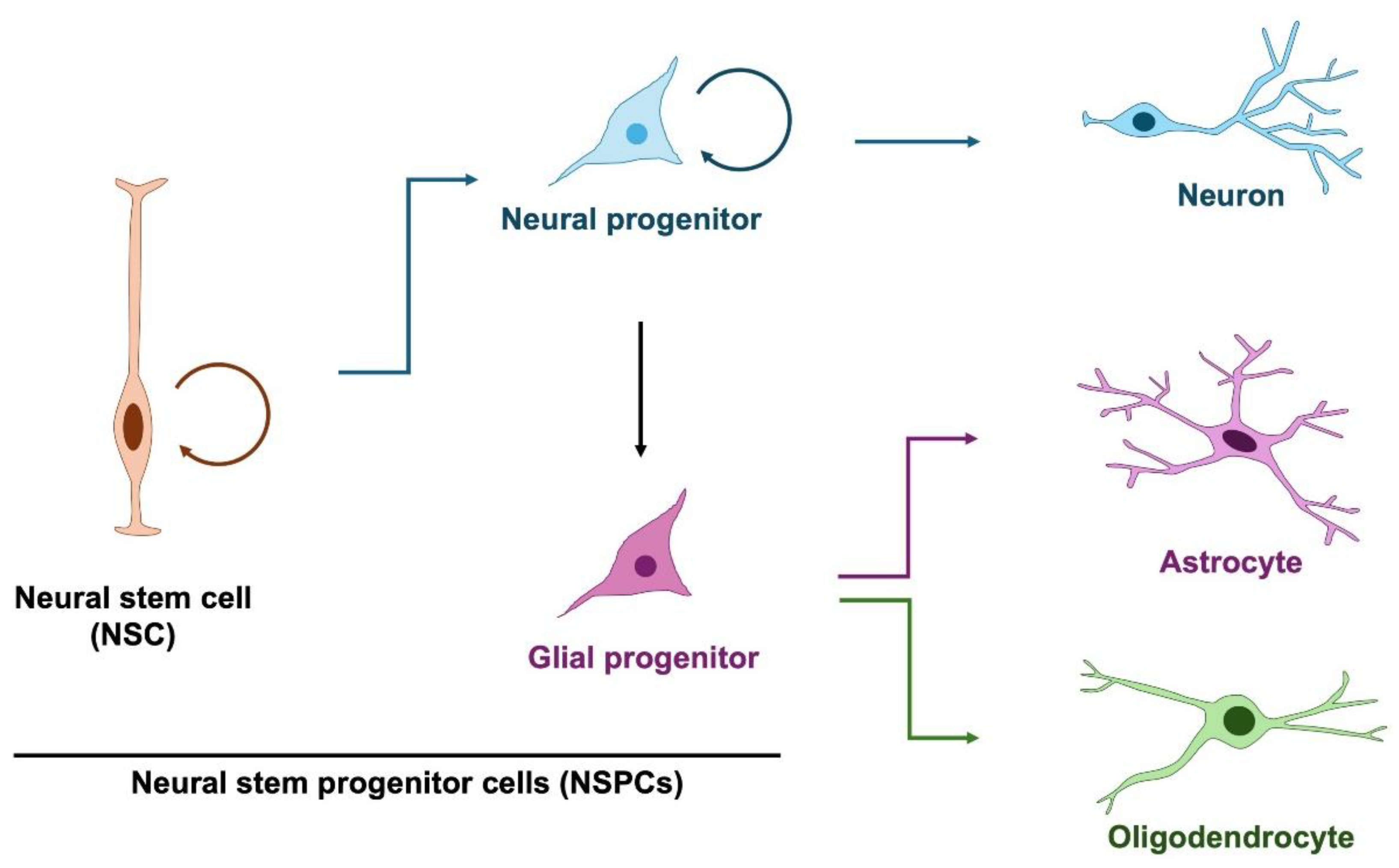
Figure 2.
Impact of NSPC proliferation on brain development. The extent of NSPC expansion through proliferative activity critically influences overall brain size. Macrocephaly and microcephaly are clinically defined as head circumferences exceeding or falling below two standard deviations from the population mean, respectively. Excessive proliferation of neural progenitors gives rise to macrocephaly, whereas insufficient proliferation results in microcephaly.
Figure 2.
Impact of NSPC proliferation on brain development. The extent of NSPC expansion through proliferative activity critically influences overall brain size. Macrocephaly and microcephaly are clinically defined as head circumferences exceeding or falling below two standard deviations from the population mean, respectively. Excessive proliferation of neural progenitors gives rise to macrocephaly, whereas insufficient proliferation results in microcephaly.
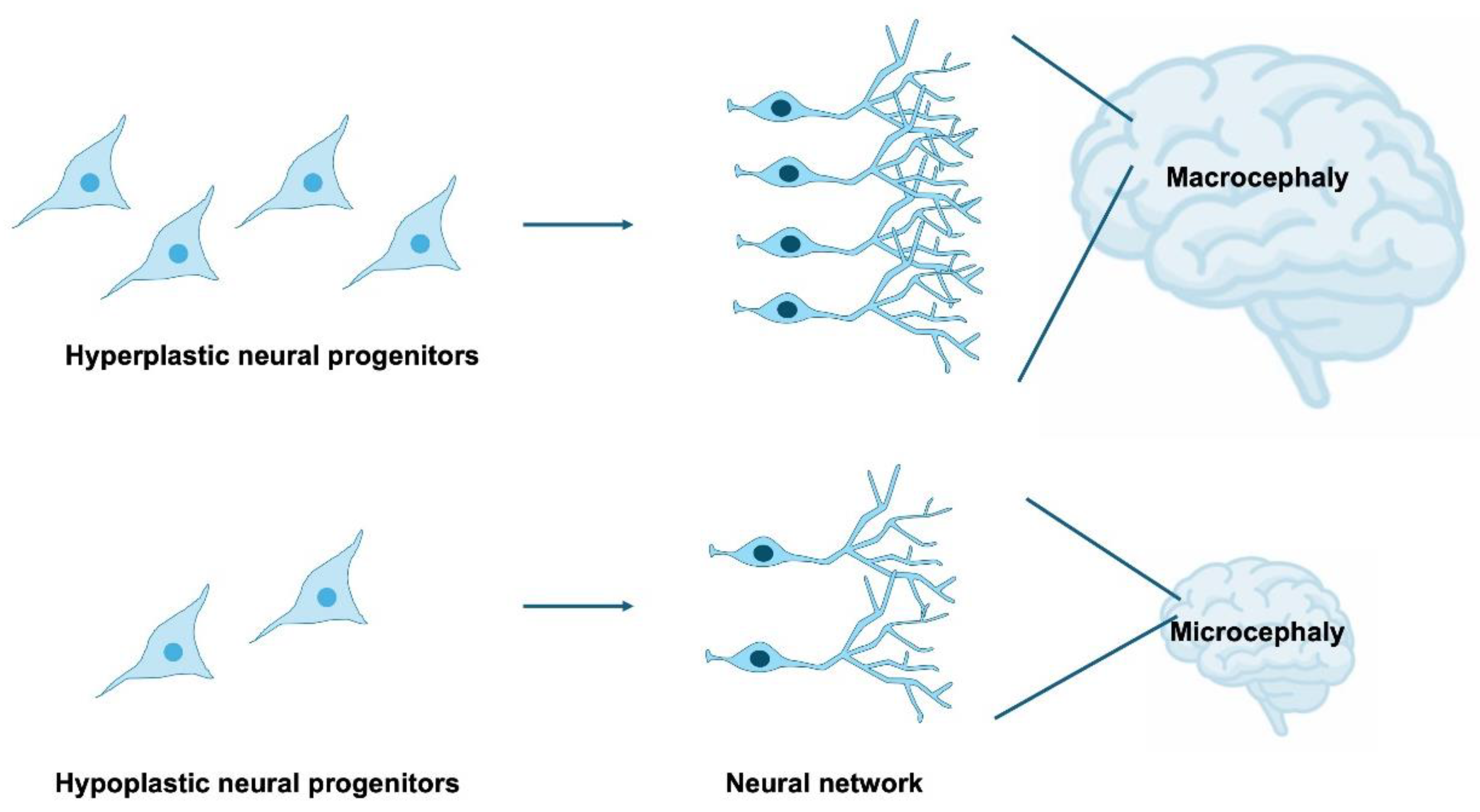
Figure 3.
Symmetric and asymmetric division of NSPCs. Symmetric division is essential for maintaining the NSPC pool through self-renewal, whereas asymmetric division drives the generation of differentiated neuronal lineages. IP, intermediate progenitor; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate.
Figure 3.
Symmetric and asymmetric division of NSPCs. Symmetric division is essential for maintaining the NSPC pool through self-renewal, whereas asymmetric division drives the generation of differentiated neuronal lineages. IP, intermediate progenitor; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate.
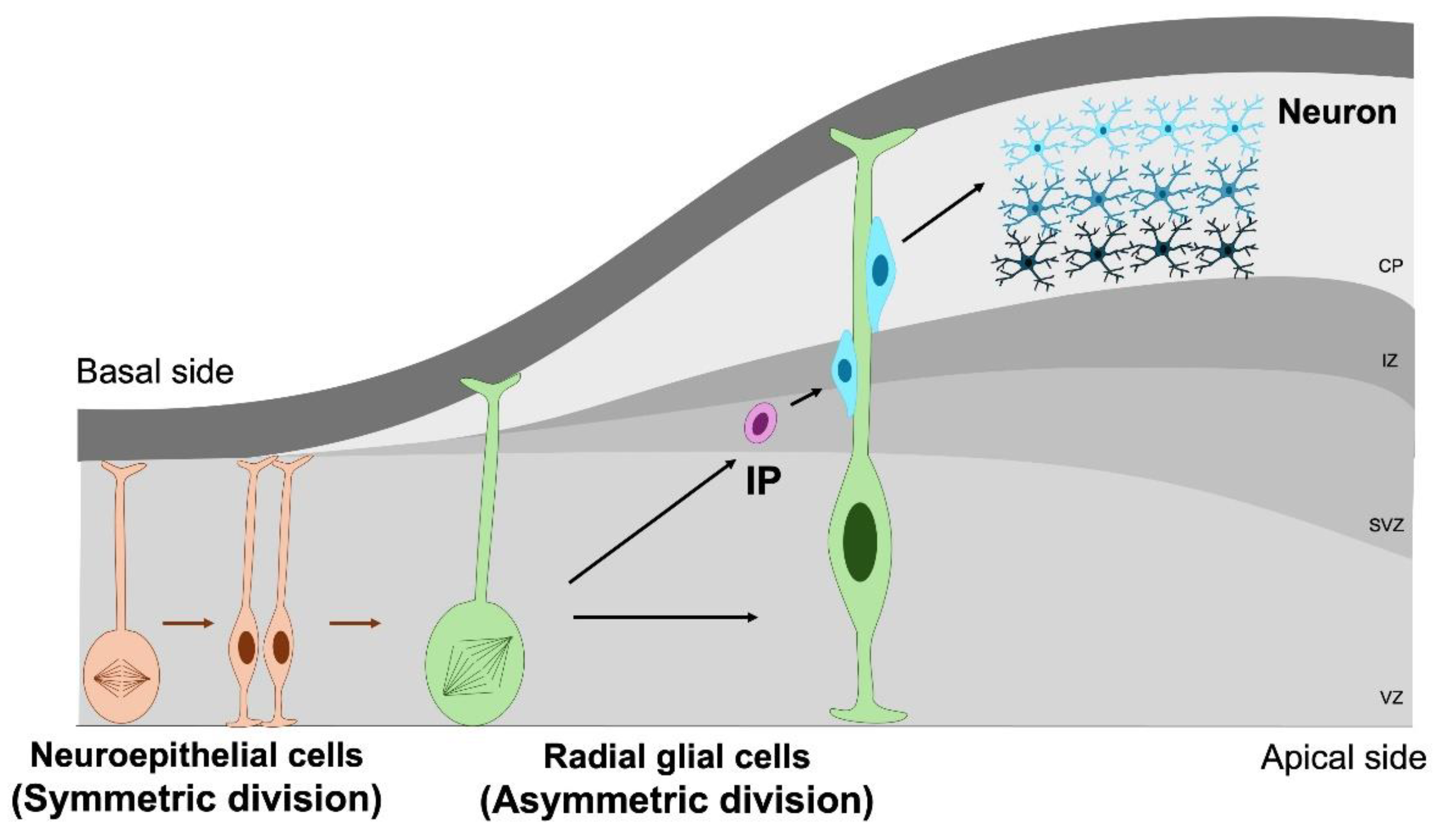
Figure 4.
Effects of NSPC proliferation and differentiation on neuronal number. Normal expansion of NSPCs ensures the proper production of mature neurons. In contrast, impaired proliferation or premature differentiation of NSPCs results in a marked reduction in neuronal numbers. NSPCs, neural stem and progenitor cells.
Figure 4.
Effects of NSPC proliferation and differentiation on neuronal number. Normal expansion of NSPCs ensures the proper production of mature neurons. In contrast, impaired proliferation or premature differentiation of NSPCs results in a marked reduction in neuronal numbers. NSPCs, neural stem and progenitor cells.
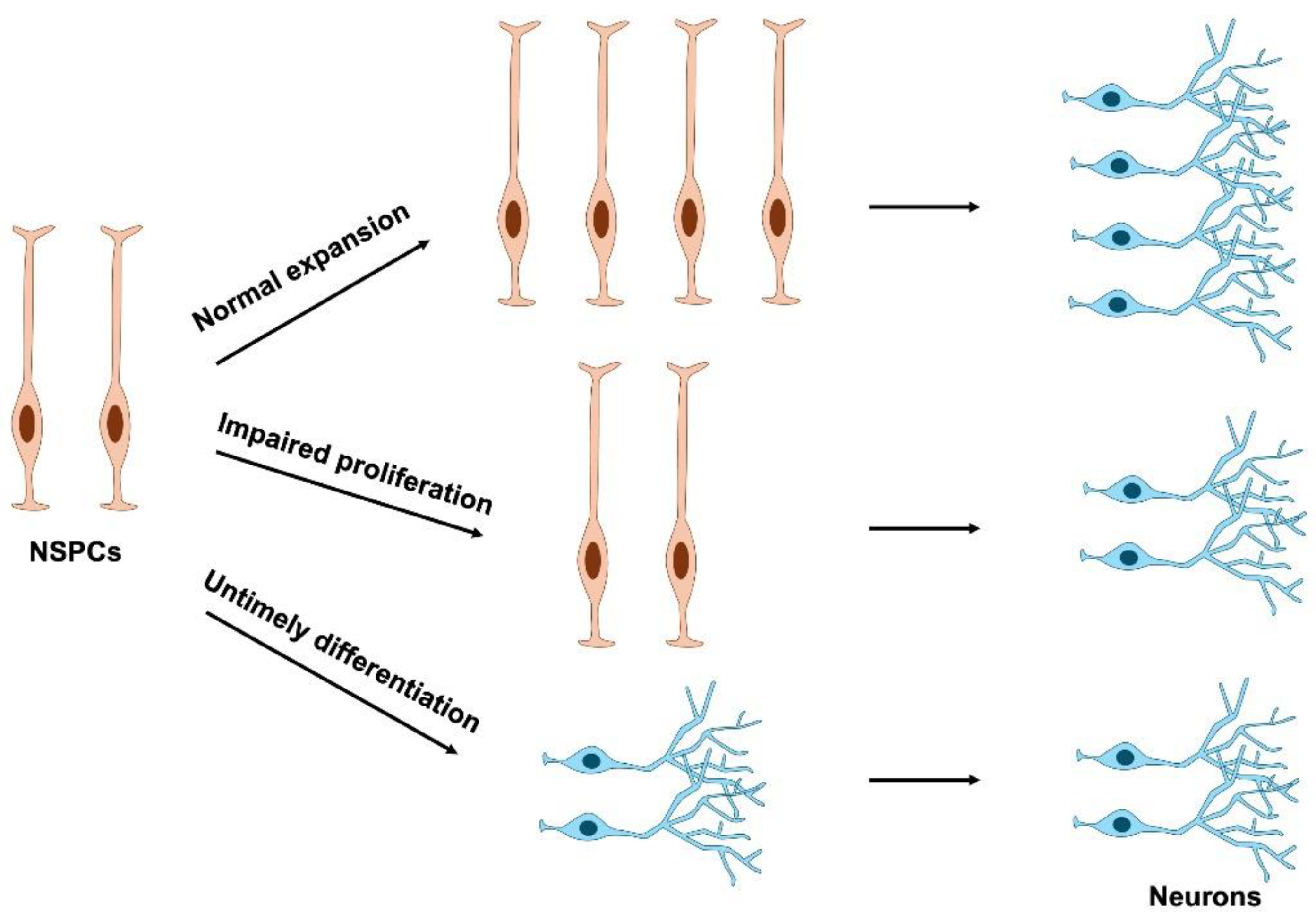
Figure 5.
Cell cycle regulation essential for NSPC expansion. During the G1 phase, Cyclin D and Cyclin E are sequentially expressed, resulting in the activation of CDK4/6 and CDK2. Growth factors promote G1 phase progression by inducing Cyclin D expression and suppressing Cip/Kip family CDK inhibitors (p21, p27, and p57) through the Ras-MAPK and PI3K-mTOR signaling pathways, respectively. ARF also acts as a negative regulator of G1 progression. In the S phase, Cyclin A expression activates CDK2, thereby initiating DNA replication. During the G2 phase, Cyclin B accumulation leads to CDK1 activation, which governs entry into and progression through the M phase, where cell division occurs. Black arrows denote activation, whereas blue T-shaped arrows represent inhibition.
Figure 5.
Cell cycle regulation essential for NSPC expansion. During the G1 phase, Cyclin D and Cyclin E are sequentially expressed, resulting in the activation of CDK4/6 and CDK2. Growth factors promote G1 phase progression by inducing Cyclin D expression and suppressing Cip/Kip family CDK inhibitors (p21, p27, and p57) through the Ras-MAPK and PI3K-mTOR signaling pathways, respectively. ARF also acts as a negative regulator of G1 progression. In the S phase, Cyclin A expression activates CDK2, thereby initiating DNA replication. During the G2 phase, Cyclin B accumulation leads to CDK1 activation, which governs entry into and progression through the M phase, where cell division occurs. Black arrows denote activation, whereas blue T-shaped arrows represent inhibition.
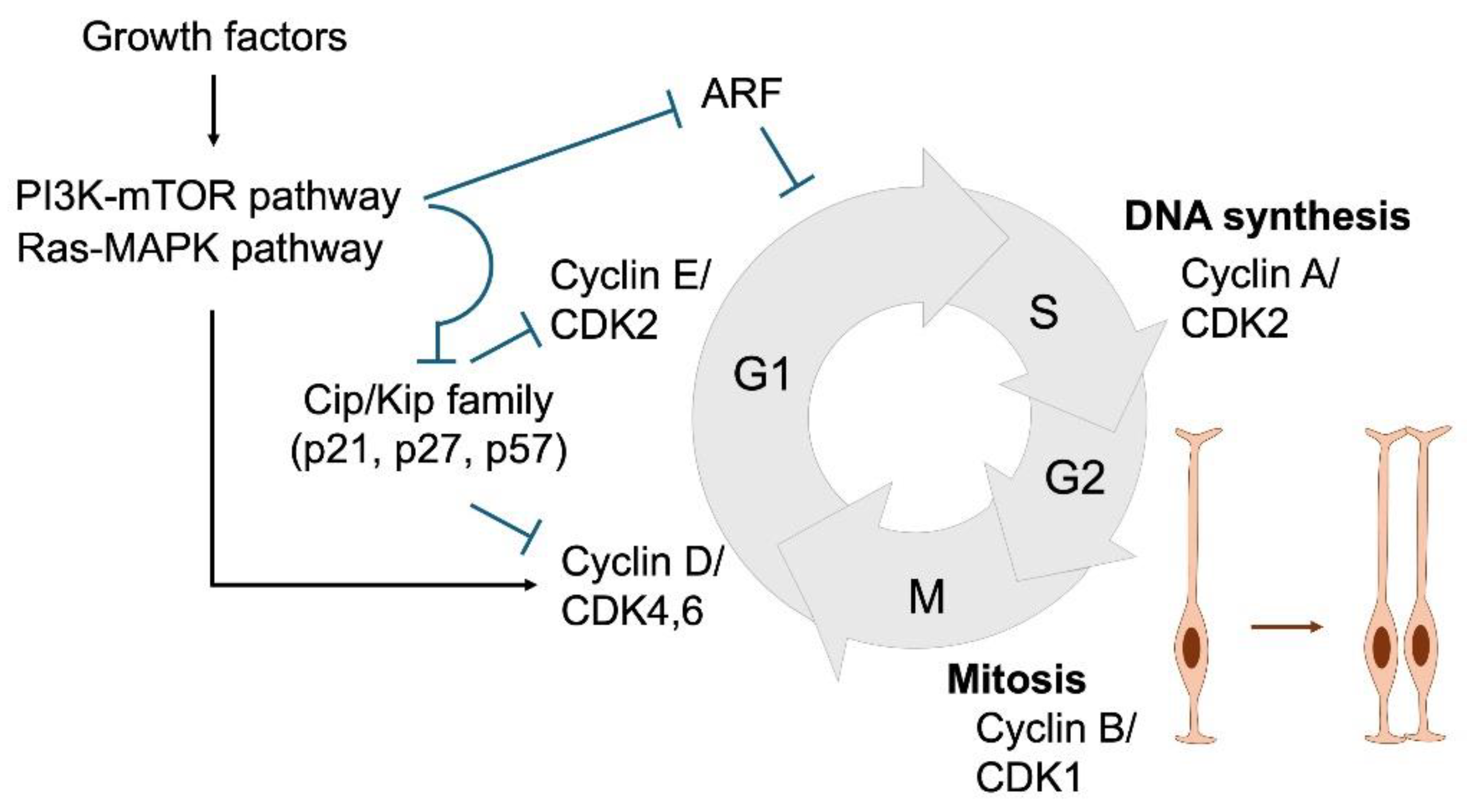
Figure 6.
Molecular regulators whose mutations contribute to impaired NSPC proliferation and NDDs. (a) Notch pathway. Ligands expressed on adjacent cells (Delta, Jagged) activate the Notch receptor, generating the Notch intracellular domain (NICD), which associates with RBPJ (not shown) and MAML to induce the transcription of Hes and Hey family genes. (b) Wnt pathway. Wnt ligands bind to the LRP5/6–Frizzled receptor complex, recruiting Dvl and stabilizing β-catenin, which translocates into the nucleus and binds to LEF1/TCF1 transcription factors to promote target gene expression, including Cyclin D. ZNRF3 acts as a negative regulator by ubiquitinating and degrading Frizzled receptors. (c) SHH pathway. Binding of SHH to PTCH1 relieves its inhibition of SMO, enabling GLI transcription factors to enter the nucleus and activate downstream genes such as Cyclin D and Cyclin E. (d) Growth factor pathway. Representative ligands such as FGF8 and IGF-I activate their respective receptors (FGFR and IGF-IR), initiating downstream cascades. The RAS–MAPK pathway (RAS → RAF → MEK → MAPK) promotes transcription of Cyclin D, whereas the PI3K–mTOR pathway (PI3K → AKT → mTOR) suppresses cell cycle inhibitors including ARF, p21, and p27. (e) Transcriptional factors beyond canonical signaling pathways. The CHD8 and ARID1A-containing BAF complex represses, whereas PAX6 enhances, the transcription of cell cycle activators such as CDK2, CDK4, and Cyclin E. ARX indirectly upregulates PAX6 expression through OLIG2 repression and concurrently inhibits the expression of the cell cycle inhibitor p57. Furthermore, ANKRD11 enhances rDNA transcription and subsequently induces Cyclin D expression via SETD5 upregulation. (f) Mitotic regulators. ASPM, CDK5RAP2, WDR62, and MCPH1 are essential for proper mitotic spindle assembly, thereby ensuring accurate neuroepithelial cell division.
Figure 6.
Molecular regulators whose mutations contribute to impaired NSPC proliferation and NDDs. (a) Notch pathway. Ligands expressed on adjacent cells (Delta, Jagged) activate the Notch receptor, generating the Notch intracellular domain (NICD), which associates with RBPJ (not shown) and MAML to induce the transcription of Hes and Hey family genes. (b) Wnt pathway. Wnt ligands bind to the LRP5/6–Frizzled receptor complex, recruiting Dvl and stabilizing β-catenin, which translocates into the nucleus and binds to LEF1/TCF1 transcription factors to promote target gene expression, including Cyclin D. ZNRF3 acts as a negative regulator by ubiquitinating and degrading Frizzled receptors. (c) SHH pathway. Binding of SHH to PTCH1 relieves its inhibition of SMO, enabling GLI transcription factors to enter the nucleus and activate downstream genes such as Cyclin D and Cyclin E. (d) Growth factor pathway. Representative ligands such as FGF8 and IGF-I activate their respective receptors (FGFR and IGF-IR), initiating downstream cascades. The RAS–MAPK pathway (RAS → RAF → MEK → MAPK) promotes transcription of Cyclin D, whereas the PI3K–mTOR pathway (PI3K → AKT → mTOR) suppresses cell cycle inhibitors including ARF, p21, and p27. (e) Transcriptional factors beyond canonical signaling pathways. The CHD8 and ARID1A-containing BAF complex represses, whereas PAX6 enhances, the transcription of cell cycle activators such as CDK2, CDK4, and Cyclin E. ARX indirectly upregulates PAX6 expression through OLIG2 repression and concurrently inhibits the expression of the cell cycle inhibitor p57. Furthermore, ANKRD11 enhances rDNA transcription and subsequently induces Cyclin D expression via SETD5 upregulation. (f) Mitotic regulators. ASPM, CDK5RAP2, WDR62, and MCPH1 are essential for proper mitotic spindle assembly, thereby ensuring accurate neuroepithelial cell division.
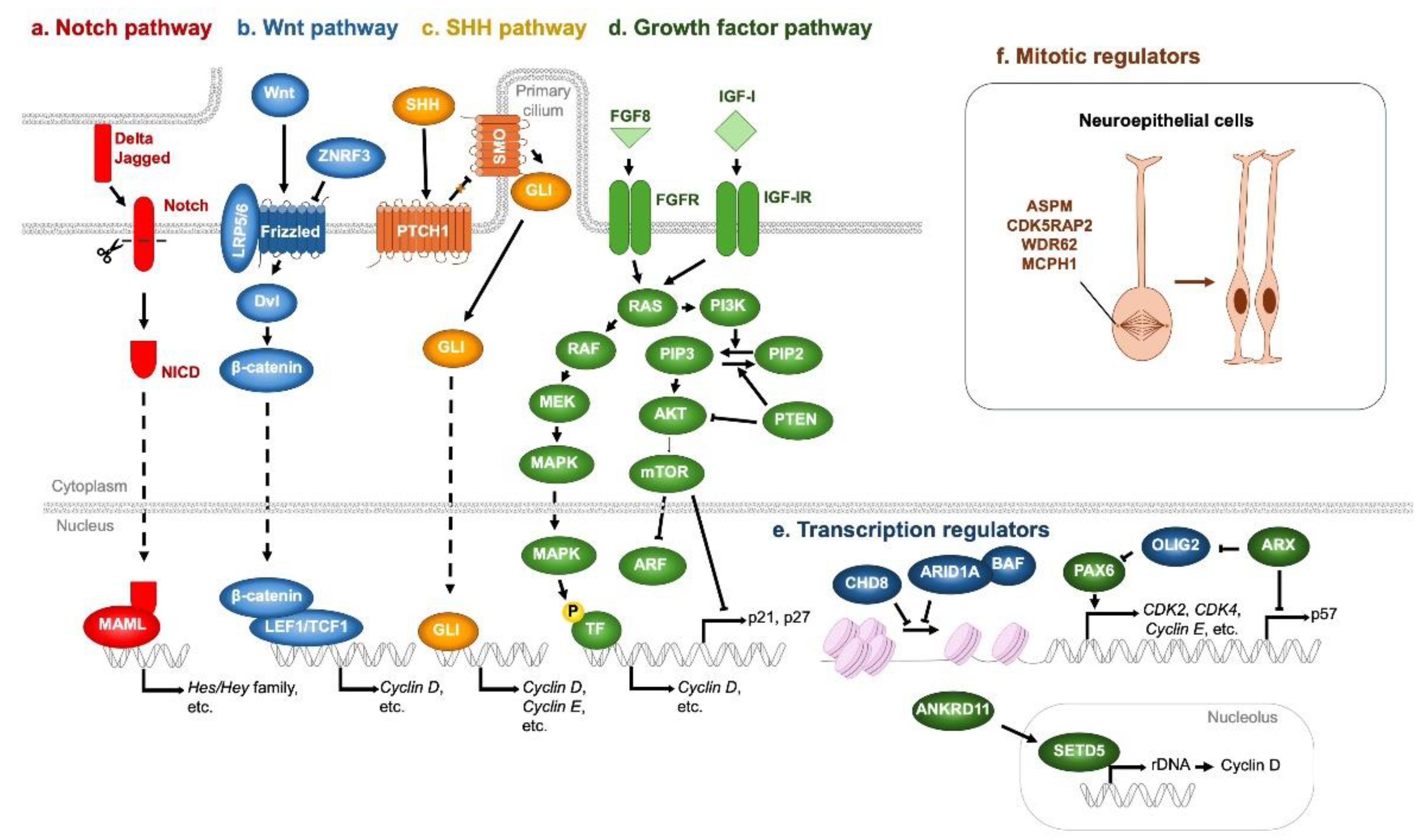
Figure 7.
Environmental factors leading to impaired NSPC proliferation and NDDs. (a) In both mouse embryonic brains and human cerebral organoids, Zika virus infection disrupts the expression of key cell cycle regulators. It also triggers premature CDK1 activation, unscheduled mitotic entry, and centrosomal amplification, resulting in aberrant chromosome segregation, and subsequent impairment of NSPC proliferation. (b) Valproic acid (VPA) exposure. In both mouse models and human brain organoids, exposure to VPA upregulates cell cycle inhibitors, thereby suppressing NSPC proliferation. (c) Phenylketonuria. In human brain organoids, elevated phenylalanine concentrations downregulate genes essential for cell cycle progression—such as CDT1 and MCM family members involved in the S phase, and ASPM and CDK5RAP2 associated with the M phase—ultimately leading to defective NSPC proliferation.
Figure 7.
Environmental factors leading to impaired NSPC proliferation and NDDs. (a) In both mouse embryonic brains and human cerebral organoids, Zika virus infection disrupts the expression of key cell cycle regulators. It also triggers premature CDK1 activation, unscheduled mitotic entry, and centrosomal amplification, resulting in aberrant chromosome segregation, and subsequent impairment of NSPC proliferation. (b) Valproic acid (VPA) exposure. In both mouse models and human brain organoids, exposure to VPA upregulates cell cycle inhibitors, thereby suppressing NSPC proliferation. (c) Phenylketonuria. In human brain organoids, elevated phenylalanine concentrations downregulate genes essential for cell cycle progression—such as CDT1 and MCM family members involved in the S phase, and ASPM and CDK5RAP2 associated with the M phase—ultimately leading to defective NSPC proliferation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



